


Advanced glycation endproduct-RAGE signaling fosters diabetic cardiomyopathy by enhancing cuproptosis and mitochondrial injury
 0
0 Abstract
Aim: Diabetic cardiomyopathy is characterized by altered myocardial structure and function, although the underlying mechanism remains unclear. This work evaluated the potential roles of advanced glycation end products (AGEs) and the AGE receptor (RAGE) in diabetes-evoked cardiac defects and the mechanisms involved, with a focus on cuproptosis.
Methods: C57BL/6 mice were induced with diabetes using a high-fat diet and streptozotocin. Diabetic and nondiabetic mice were treated with the RAGE blocker FPS-ZM1, followed by assessment of cardiac geometry, contractile performance, oxidative stress, apoptosis, and cuproptosis.
Results: RNA-seq analysis exhibited differentially expressed cuproptosis-related genes in diabetic hearts, implicating a cuproptosis signature. Levels of AGE and RAGE mRNA were elevated in diabetic hearts. Diabetes triggered cardiac hypertrophy, myocardial tissue fibrosis, impaired fractional shortening, ejection fraction, and cell shortening, alongside mitochondrial ultrastructural and functional defects. Diabetes promoted apoptosis (increased Bax and decreased Bcl2) and cuproptosis [copper transporter 1 (CTR1), ferredoxin 1 (FDX1), dihydrolipoamide S-acetyltransferase (DLAT), and S100 calcium-binding protein A13 (S100A13)], along with dampened Fe-S cluster proteins Aconitase 2 (ACO2) and NADH: ubiquinone oxidoreductase core subunit S8 (NDUFS8). These pathological changes were alleviated by FPS-ZM1, except for CTR1, plasma glucose, and lipid levels. Co-immunoprecipitation and domain mapping favored an interplay between RAGE and the Cu2+-binding protein S100A13. In vitro, methylglyoxal-derived AGE (MG-AGE) and high glucose levels prompted Cu2+ accumulation and/or compromised cardiomyocyte contractile function, reminiscent of in vivo diabetic cardiomyopathy. Inhibition of RAGE, cuproptosis, and S100A13 using FPS-ZM1, tetrathiomolybdate, and amlexanox, respectively, alleviated MG-AGE or high glucose-evoked Cu2+ and cardiomyocyte defects.
Conclusion: These results reveal a role for the RAGE-S100A13-cuproptosis signaling axis in AGE-driven myocardial anomalies in diabetes.
Keywords
INTRODUCTION
The economic burden of diabetes mellitus continues to escalate, driven by an aging global population, sedentary lifestyles, and the obesity epidemic. Recent estimates indicate that diabetes affects ~10.5% of adults aged 20 to 79 years globally (≈ 536.6 million people), with a projected total of 783 million by 2045[1,2]. Cardiovascular dysfunction remains the leading cause of morbidity and mortality in the diabetic population. Among these, heart failure represents a major, often underappreciated, clinical endpoint for both type 1 and type 2 diabetes[3-5]. Specifically, diabetic cardiomyopathy is increasingly recognized as a distinct clinical entity, characterized by structural remodeling and myocardial dysfunction occurring independently of hypertension, coronary artery disease, or primary valvular disease[3,6-9]. While population-based studies suggest that diabetic cardiomyopathy is commonly associated with significant adverse outcomes, the specific impact of these outcomes varies based on clinical definitions and screening strategies[10,11]. Mechanistically, diabetic cardiomyopathy reflects a complex interplay of metabolic derangements (gluco-lipotoxicity), oxidative stress, inflammation, Ca2+-handling abnormalities, mitochondrial dysfunction, and maladaptive remodeling[3,6], yet the proximal triggers that couple hyperglycemia to mitochondrial injury and cardiomyocyte death remain incompletely defined.
A major pathway linking hyperglycemia to myocardial damage is the accumulation of advanced glycation end products (AGEs). This process is triggered by elevated glucose and oxidative stress and is largely mediated by reactive dicarbonyl intermediates, specifically methylglyoxal (MGO). MGO levels rise sharply when glycolytic flux is high, and detoxification capacity is overwhelmed[12-14]. AGEs contribute to cardiac pathology through the modification and crosslinking of intracellular and extracellular proteins, as well as through receptor-mediated signaling - predominantly via the receptor for AGEs (RAGE)[15,16]. Engagement of the AGE-RAGE axis amplifies reactive oxygen species (ROS) production and inflammation, thereby promoting mitochondrial stress and tissue remodeling[15-17]. In cardiomyocytes, exposure to MGO-derived AGEs compromises contractile performance and intracellular Ca2+ handling, underscoring the cytotoxic role of dicarbonyl/AGE stress in diabetic myocardial dysfunction[18].
Regulated cell death programs are increasingly recognized as central drivers of myocardial loss and contractile decline in diabetes[3,6]. While apoptosis and other established death modalities occur in diabetic cardiomyopathy, emerging evidence suggests that metabolic-mitochondrial stress may also predispose cardiomyocytes to newly defined forms of cell death linked to organelle proteostasis[3,6]. Cuproptosis has recently emerged as a novel form of copper (Cu2+)-dependent programmed cell death. This pathway is characterized by the direct binding of Cu2+ to lipoylated components of the tricarboxylic acid (TCA) cycle, resulting in the proteotoxic aggregation of lipoylated enzymes and the subsequent depletion of iron-sulfur (Fe-S) cluster proteins[19-21]. As a result, electron transport chain complexes exhibit a significant reduction in redox properties, leading to the failure of cellular respiration. Furthermore, dysregulated mitochondria display elevated iron levels and ROS, exacerbating oxidative stress and functional impairment[21-24]. Given that diabetes is characterized by heightened oxidative stress, altered TCA cycle metabolism, and disturbed metal homeostasis[6,25], the present study aimed to examine the role of cuproptosis in diabetic myopathic changes, with a specific emphasis on the underlying signaling mechanisms mediated by the AGE-RAGE axis.
METHODS
Experimental diabetes mellitus and drug treatment
All animal studies performed in this work were conducted in accordance with the guidelines and approval of the Animal Care and Use Committees at Jinan University (No. 20241015-0025, Guangzhou, China) and Zhongshan Hospital, Fudan University (No. 20210220, Shanghai, China). Adult male C57BL/6J mice (8 weeks old, 22-24 g) were housed in a pathogen-free environment. The animals were kept on a 12-h light/dark cycle and provided with ad libitum access to water and food. To establish type 2 diabetes mellitus, mice were fed a 60% high-fat diet (percent fat calorie) for 28 days, followed by 5 consecutive days of intraperitoneal streptozotocin (STZ, 40 mg/kg/day, in citrate buffer, 0.1 M, pH 4.5) injections. Mice were maintained on a high-fat diet throughout STZ administration and for the remainder of the study. The following 4 animal groups were designed: (1) Control: standard chow diet and equivalent volume of citrate buffer for injection; (2) Diabetic: high-fat diet for 28 days, then STZ injection for 5 days and a continued high-fat diet for an additional 6 weeks; (3) Control-FPS-ZM1: standard chow diet plus the RAGE inhibitor FPS-ZM1 (10 mg/kg i.p., twice per week)[26] starting on day 33; and (4) Diabetic-FPS-ZM1: high-fat diet-STZ regimen as described, with concurrent FPS-ZM1 treatment (10 mg/kg i.p., twice per week) beginning on day 33 (end of STZ injection). Mice were weight-matched and randomized into 4 experimental groups (10 mice per group). Mice with overnight fasting blood glucose levels greater than 11.1 mM on two readings were considered diabetic[27]. Circulating levels of fasting glucose, triglycerides, cholesterol, and AGEs were measured at the completion of the entire high-fat diet intake regimen using commercial colorimetric assay kits (MyBioSource, San Diego, CA, USA). FPS-ZM1 is chosen primarily for its unparalleled specificity for the RAGE receptor compared to broader first-generation inhibitors. By targeting the V-domain of RAGE (Kd ≈ 25 nM), FPS-ZM1 effectively decouples the pathological signaling initiated by AGEs. Furthermore, its superior pharmacokinetic profile ensures adequate tissue distribution in vivo. In addition, intraperitoneal injections are the standard route for ensuring consistent systemic bioavailability[28,29].
Gene expression profiling (GEP) data processing and differential analysis
The GSE123975 dataset was retrieved from the Gene expression omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) using STZ-induced diabetic mice. Data were generated using the Agilent-028005 SurePrint G3 Mouse GE 8x60K Microarray platform on left ventricular tissues from 6 hyperglycemic mice and 6 normoglycemic mice. Raw data were processed in R using the affy package. Background correction and normalization were performed using the Oligo and robust multi-array average (RMA) algorithms, while missing values were imputed using the k-Nearest Neighbor method[30]. For genes represented by multiple probes, expression levels were collapsed to the median value. Differentially expressed genes (DEGs) were detected by the “limma” algorithm. Statistical significance was determined based on the following predefined criteria: |log2 fold change| greater than 0.7 (1.6-fold difference) and an adjusted P-value < 0.05[7].
Biological function enrichment
The clusterProfiler software was utilized to perform Gene Ontology (GO) functional annotation on the identified DEGs. Genes were categorized into three principal functional categories: Molecular Functions (MF), Cellular Components (CC), and Biological Processes (BP) for enrichment analysis. This hierarchical classification enabled the identification of significantly enriched functional clusters, providing a comprehensive overview of biological landscapes altered in experimental models[31]. Simultaneously, Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was performed using clusterProfiler to map DEGs to biochemical and signaling networks. This analysis was instrumental in elucidating the complex interactions of DEGs within established biological pathways and their coordinated roles in driving pathological phenotypes[32]. To complement these threshold-based methods, the fgsea package was used to perform Gene Set Enrichment Analysis (GSEA). Unlike traditional enrichment, GSEA assesses entire gene sets across an ordered list of all detected genes, allowing for detection of subtle yet biologically significant coordinated shifts. Enrichment scores were calculated to reveal functional pathways that were significantly correlated with the observed gene expression patterns between groups[33]. GO and KEGG enrichment analyses were performed using over-representation analysis based on Fisher’s exact test. To control the false discovery rate, P-values were adjusted using the Benjamini-Hochberg method, with a significance threshold set at adjusted P < 0.05. GSEA was conducted with 1,000 phenotype permutations, and significance was determined by normalized enrichment score (NES) with an false discovery rate (FDR) q value < 0.25. For differential expression analysis, between-group comparisons were evaluated using a two-sided Student’s t-test with Benjamini-Hochberg correction across all genes; an adjusted P < 0.05 was considered significant.
Echocardiographic evaluation
Cardiac structure and contractile function were evaluated in sedated mice (ketamine: 80 mg/kg; xylazine:
Histological evaluation
Following the induction of deep sedation, hearts were rapidly removed and fixed in 10% neutral-buffered formalin for 24 h. Subsequently, tissues underwent standard processing and were embedded in paraffin wax. Serial tissue sections (5 μm) were stained with hematoxylin and eosin (H&E) to evaluate cardiomyocyte morphology. Cross-sectional areas were quantified using ImageJ software (version 1.34S). In parallel, Masson’s trichrome staining was applied to evaluate the degree of myocardial interstitial fibrosis, expressed as the portion of the collagen-positive (blue colored) section within the entire microscopic field[7,35].
Methylglyoxal-derived advanced glycation end product (MG-AGE), cardiomyocyte isolation and drug treatment
MG-derived AGEs (MG-AGE) were generated by incubating 100 mM methylglyoxal with 7.2 mg/mL bovine serum albumin (BSA) in a 100 mM phosphate buffer (pH 7.4) at 37 °C for 48 h under sterile conditions. A control BSA solution was processed in parallel in the absence of methylglyoxal. Following incubation, residual unreacted carbonyl species were removed via extensive dialysis against ammonium bicarbonate buffer (30 mM, pH 7.9). Following preparation, MG-AGE and control BSA were sterilized using a 0.22 μm filter and stored at -20 °C until further use. Concentrations were determined based on the equivalent BSA levels supplied[18]. Adult murine cardiomyocytes were isolated via retrograde Langendorff perfusion, followed by enzymatic digestion using Liberase Blendzyme 4. Only cells exhibiting distinct striations and an absence of spontaneous twitches were selected for functional assessment. For assessment of RAGE, cuproptosis, and the S100 calcium-binding protein A13 (S100A13) Cu2+ binding protein in AGE- or diabetes-induced cardiomyopathy, adult C57BL/6 mouse cardiomyocytes were incubated with either MG-AGE (5 μM)[18] or high glucose (25.5 mM, to mimic hyperglycemia)[36] for 12 h with or without the RAGE blocker FPS-ZM1
Cell mechanics
Cardiomyocyte relengthening and shortening were examined using a SoftEdge MyoCam detection system (IonOptix, Milton, MA, USA) in a 1 mM CaCl2 environment. Indices evaluated encompassed maximal rate of cell relengthening (-dL/dt), shortening (+dL/dt), peak shortening (PS), time-to-90% relengthening (TR90), and time-to-peak shortening (TPS), in cardiomyocytes with 0.5 Hz field-stimulation[36,39].
Free radical generation
Isolated cardiomyocytes were incubated with intracellular fluorescence probe 5-(6)-chloromethyl-2’,7’-dichlorodihydrofluorescein diacetate (DCF; 5 μM; Molecular Probes, Eugene, OR, USA) at 37 °C for 30 min to detect intracellular ROS. After dye loading, fluorescence images were acquired using an Olympus fluorescence microscope under identical exposure settings for all groups[7,20].
Transmission electron microscopy (TEM)
For ultrastructural characterization, ventricular tissue was sectioned into small cubes (≤ 1 mm3) and fixed in 2.5% glutaraldehyde (0.1 M sodium phosphate buffer, pH 7.4) at 4 °C. After primary fixation, specimens were dehydrated through a graded series of ethanol and embedded in Epon-Araldite resin. Ultrathin sections (approximately 50 nm) were obtained using an Ultracut E ultramicrotome (Leica) and double-stained with lead citrate and uranyl acetate for contrast. Samples were visualized using a Hitachi H-7000 transmission electron microscope (Pleasanton, CA, USA) equipped with a high-resolution digital imaging system[20].
Mitochondrial membrane potential (MMP)
MMP was assessed using the ratiometric fluorescence dye JC-1 (5 μM). Isolated cells were exposed to JC-1 at 37 °C for 20 min in the dark. Following incubation, fluorescence intensities were quantified using a spectrofluorimeter (SpectraMax Gemini XS). The JC-1 monomer fluorescence (green, indicating mitochondrial depolarization) was recorded at an emission wavelength of 530 nm, while the JC-1 aggregate fluorescence (red, indicating polarized, healthy mitochondria) was recorded at 590 nm. The aggregate-to-monomer ratio was calculated and utilized as a sensitive index of MMP[20].
AGE detection
Myocardial AGE levels were evaluated via immunohistochemistry on 8 µm thick paraffin-embedded myocardial sections. Following deparaffinization and rehydration, sections underwent antigen retrieval with 0.05% proteinase K in phosphate-buffered saline (PBS) (pH 7.4) for 30 min. Background peroxidase activity was blocked with H2O2, followed by systematic PBS washes. To visualize AGE deposition, slides were incubated with the primary anti-AGE antibody 1H7G5 (1:500), followed by an anti-mouse fluorescein isothiocyanate (FITC)-conjugated immunoglobulin G (IgG) secondary antibody. Fluorescence signals (Excitation: 490 nm/Emission: 517 nm) were captured on an Olympus BX51 fluorescence microscope, coupled with a high-resolution cooled CCD camera. The intensity and distribution of FITC-labeled AGEs were quantified using Image-Pro Plus software[40].
RAGE reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from myocardial tissues utilizing TRIzol® reagent (Invitrogen, Carlsbad, CA, USA). cDNA was produced using the SuperScriptTM III system (Invitrogen) from 2 μg of RNA. Quantitative real-time PCR was performed using SYBR Green-based detection on an iCycler thermal cycler (Bio-Rad). Specific primers were used for RAGE (F: 5’-GGT CAT GCC GGA GTG TCT ATT-3’; R: 5’-GTG TAG AAC CCG TCG GTG AGG-3’) and the internal control glyceraldehyde-3-phosphate dehydrogenase (GAPDH; F: 5’-CTG GAA AGC TGT GGC GTG ATG-3’; R: 5’-GCC AGT GAG CTT CCC GTT CAG-3’). Relative mRNA levels were determined using the ΔCT protocol[39].
Immunoblotting analysis
Total protein was extracted from myocardial tissues, and concentrations were determined by a bicinchoninic acid (BCA) assay to ensure consistent loading. Protein extracts were resolved by sodium dodecyl sulfate (SDS) gel electrophoresis using the Mini-PROTEAN II system (Bio-Rad) and subsequently electrotransferred onto nitrocellulose membranes. After blocking, membranes were incubated with primary antibodies against uncoupling protein 2 (UCP2), peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), B-cell lymphoma 2 (Bcl2), Bcl-2-associated X protein (Bax), solute carrier family 31 member 1 (SLC31A1, CTR1), ATPase copper transporting beta (ATP7B), ferredoxin 1 (FDX1), dihydrolipoamide S-acetyltransferase (DLAT), aconitase 2 (ACO2), NADH dehydrogenase [ubiquinone] iron-sulfur protein 8 (NDUFS8), and S100A13, each at a dilution of 1:1,000. Housekeeping proteins, including GAPDH and α-tubulin (1:5,000), were used as internal loading controls to ensure accurate normalization and quantitative reliability. Immunoreactive bands were detected using enhanced chemiluminescence (ECL) and quantified via a Bio-Rad densitometry system. All experimental antibodies were sourced from Abcam or Cell Signaling Technology[41].
Intracellular Cu2+ measurement
Intracellular Cu2+ was quantified using a commercial detection kit (ab272528, Abcam, Boston, MA, USA). Briefly, cells were homogenized and diluted with ultrapure water to ensure consistent sample viscosity. A
Cell culture, transfection, and intracellular labile copper (Cu+) measurement
Mouse HL-1 atrial cardiomyocytes were cultured in medium supplemented with 1% penicillin-streptomycin and 10% fetal bovine serum in a humidified 5% CO2 atmosphere at 37 °C. For gene silencing, a specific small interfering RNA (siRNA) targeting murine S100A13 (sense: 5’-CGUGGGCUCUCUAGAUGAAtt-3’; antisense: 3’-UUCAUCUAGAGAGCCCACGtt-5’) was synthesized by Guangzhou IGE Biotechnology Co., Ltd. (Guangzhou, China). HL-1 cells were transfected with scrambled control or S100A13 siRNA using Lipofectamine 3000. Knockdown efficiency was rigorously validated by immunoblotting 48 h post-transfection. Following validation, HL-1 cells (scrambled control or S100A13-silenced) were incubated with 5 µM MG-AGE for 12 h with or without the S100A13 blocker AMX (20 μM) or FPS-ZM1 (25 μM) prior to assessment of intracellular Cu+ levels. HL-1 cells were incubated with the Cu+-selective fluorescent probe CS1 (5 μM, FocusBio, China) at 37 °C for 30 min. Fluorescence signals were captured using a laser scanning confocal microscope (Excitation: 543 nm; Emission: 550-650 nm). Mean fluorescence intensity, representing intracellular Cu+ level, was determined via ImageJ. All experimental groups were imaged under identical acquisition parameters to ensure the comparability of labile Cu+ levels[42].
RAGE-S100A13 interaction interface analysis
The putative complex structure between RAGE and S100A13 was predicted using AlphaFold3 (https://alphafoldserver.com/). Structure-based interaction interfaces were subsequently analyzed with PDBe-PISA (https://www.ebi.ac.uk/msd-srv/prot_int/pistart.html) and visualized using PyMOL (https://pymol.org/)[41].
Co-Immunoprecipitation (co-IP)
Physical protein-protein interactions were evaluated using the Thermo Fisher PierceTM Co-Immunoprecipitation Kit. Briefly, 50 μg of purified anti-RAGE, anti-S100A13, or anti-Flag antibodies were immobilized onto the provided amine-reactive resin. Myocardial protein lysates (1 mg total protein) were then incubated with the antibody-conjugated resin for 2 h to facilitate the formation of stable immune complexes. Following extensive washing, the captured protein complexes were eluted using an acidic elution buffer. The resulting eluates were neutralized and subjected to immunoblot analysis using complementary primary antibodies[39].
Data analysis
Results are expressed as the mean ± standard error of the mean (SEM). Statistical analyses were performed using GraphPad Prism software (GraphPad Software, San Diego, CA, USA). Differences between two groups were evaluated using a two-tailed Student’s t-test, while comparisons among multiple groups were conducted using one-way or two-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for multiple comparisons. Statistical significance was established at P < 0.05.
RESULTS
Cu2+-related DEGs and cuproptosis pathway enrichment in diabetic mouse hearts
GO analysis of the DEGs in diabetic hearts revealed a distinct functional landscape. MF analysis showed pronounced enrichment of metal ion transmembrane transporter activity, phospholipid binding, and GTPase regulator activity. Regarding CC, significant enrichment was observed in the monoatomic ion channel complex, mitochondrial protein-containing complex, and mitochondrial matrix. Furthermore, BP analysis identified key pathways related to the regulation of metal ion transport, response to metal ions, and response to oxidative stress [Figure 1A]. KEGG pathway analysis further revealed significant enrichment in metabolic and structural networks, including V-type ATPase, the citrate cycle (TCA/Krebs cycle), and inositol phosphate metabolism [Figure 1B]. To achieve a higher resolution of coordinated gene shifts, GSEA was performed, revealing a close correlation with the GOBP: cellular response to Cu2+ ion pathway in diabetic myocardial injury (enrichment Score = -0.677, NES = -1.833, P = 1.29e-03, adjusted P = 0.039) [Figure 1C]. To further delineate specific genes driving this Cu2+ dysregulation, we utilized a Venn diagram to identify overlapping DEGs and generated scatter plots to visualize the expression distribution of key regulators within the Cu2+ ion-binding pathway [Figure 1D and E].
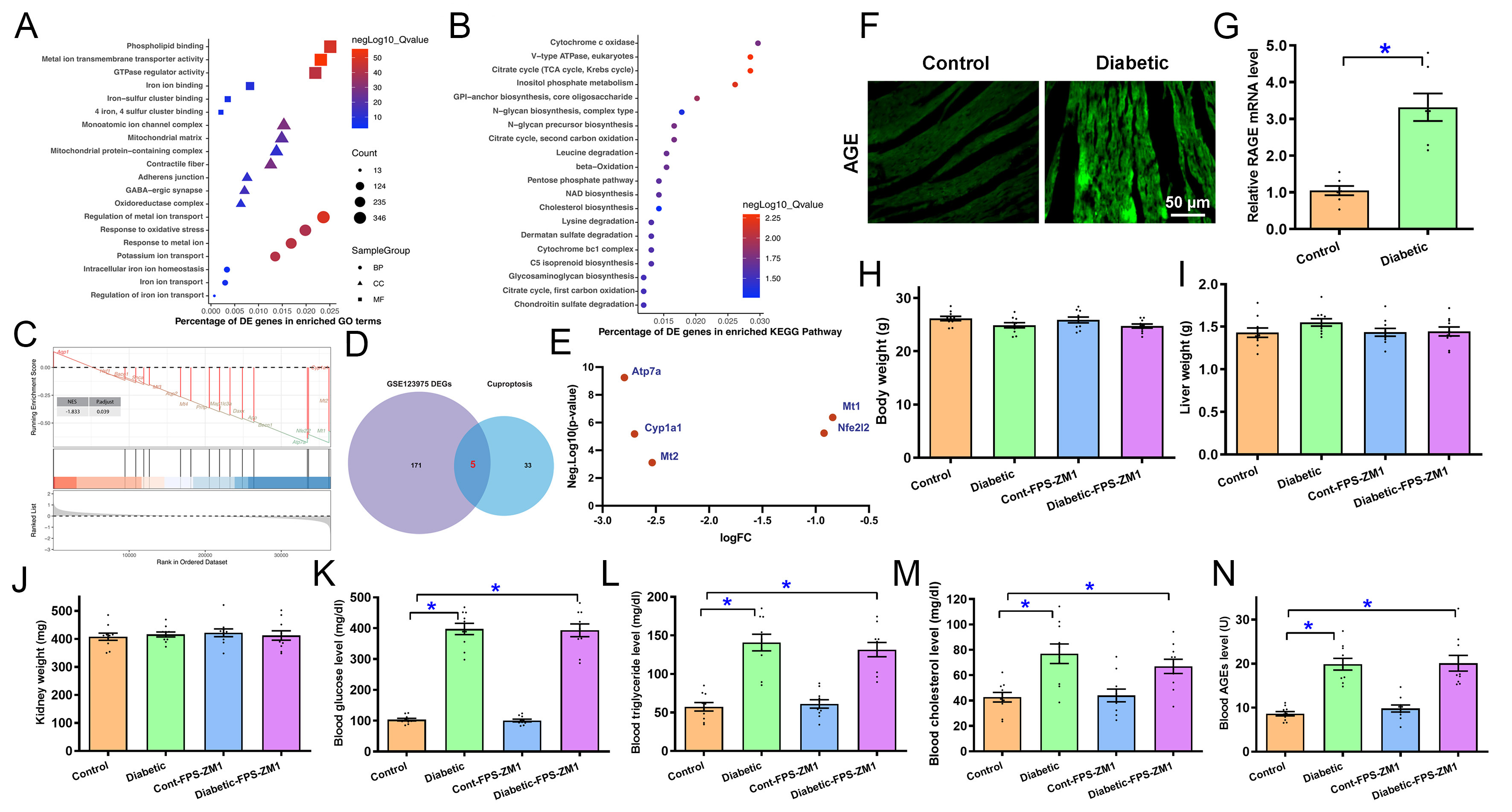
Figure 1. Biological enrichment and Cu2+ ion regulation-related pathway analysis, AGE/RAGE levels in diabetic hearts, and influence of RAGE inhibition on diabetes-instigated alterations in biometric and circulating profiling. (A) GO analysis of DEGs; (B) KEGG analysis of DEGs; (C) GSEA analysis quantifies GOBP: cellular response to Cu2+ ion pathway; (D) Crossover of cuproptosis-related genes with DEGs in GSE123975; (E) Levels of 5 cuproptosis DEGs from the intersection; (F) Representative immunofluorescent AGE staining in mouse hearts; (G) RAGE mRNA level in mouse hearts; (H-N) Effect of RAGE inhibition using FPS-ZM1 on diabetes-instigated alterations in biometric and circulating profiling; (H) Body weight; (I) Liver weight; (J) Kidney weight; (K) Blood glucose level; (L) Blood triglyceride; (M) Blood cholesterol; (N) Blood AGEs. Mean ± SEM, n = 6 (A-E), 7 (G) or 10 (H-N) mice per group. Bioinformatic analyses were described in detail in relevant method sections. Data in G were analyzed using two-sided Student’s t-test. Data shown in H-N were analyzed using two-way ANOVA followed by Tukey’s post hoc test, *P < 0.05 between labeled groups. AGE: Advanced glycation end products; RAGE: receptor for AGEs; GO: Gene Ontology; DEGs: differentially expressed genes; KEGG: Kyoto Encyclopedia of Genes and Genomes; GSEA: Gene Set Enrichment Analysis; ANOVA: analysis of variance; GOBP: gene ontology biological process; SEM: standard error of the mean.
Biometric and circulating profiles in experimental mice with or without RAGE inhibition
To determine if the AGE-RAGE axis, a primary driver of diabetic complications, acts as the upstream trigger for this observed cuproptosis signature in diabetic hearts, previously reported[43], we next evaluated the cardiac expression of AGE and RAGE in diabetic mouse hearts. Figure 1F and G revealed that diabetes increased AGE immunofluorescence by 1.86-fold and RAGE mRNA levels by 2.16-fold (P < 0.0001) compared with controls. In this context, we treated diabetic mice with the RAGE inhibitor FPS-ZM1
Echocardiographic features in experimental mice with or without RAGE inhibition
Echocardiographic evaluation revealed that diabetes enhanced septal thickness by 0.15-fold (P < 0.001), LVESD by 0.41-fold (P < 0.0001), LVEDD by 0.15-fold (P < 0.0001), absolute LV mass by 0.38-fold (P < 0.001), and normalized LV mass by 0.34-fold (P < 0.0001), and dampened ejection fraction by 0.12-fold (P < 0.0001) and fractional shortening by 0.23-fold (P < 0.0001) without affecting LV wall thickness or heart rate. Gross heart weight and size (normalized to body weight) were also elevated by 0.16-fold (P < 0.001) and 0.22-fold (P < 0.0001), respectively, in experimental diabetes. Although FPS-ZM1 alone failed to alter echocardiographic or heart parameters tested, it effectively mitigated diabetes-evoked changes in myocardial geometry and function [Figure 2A-L].
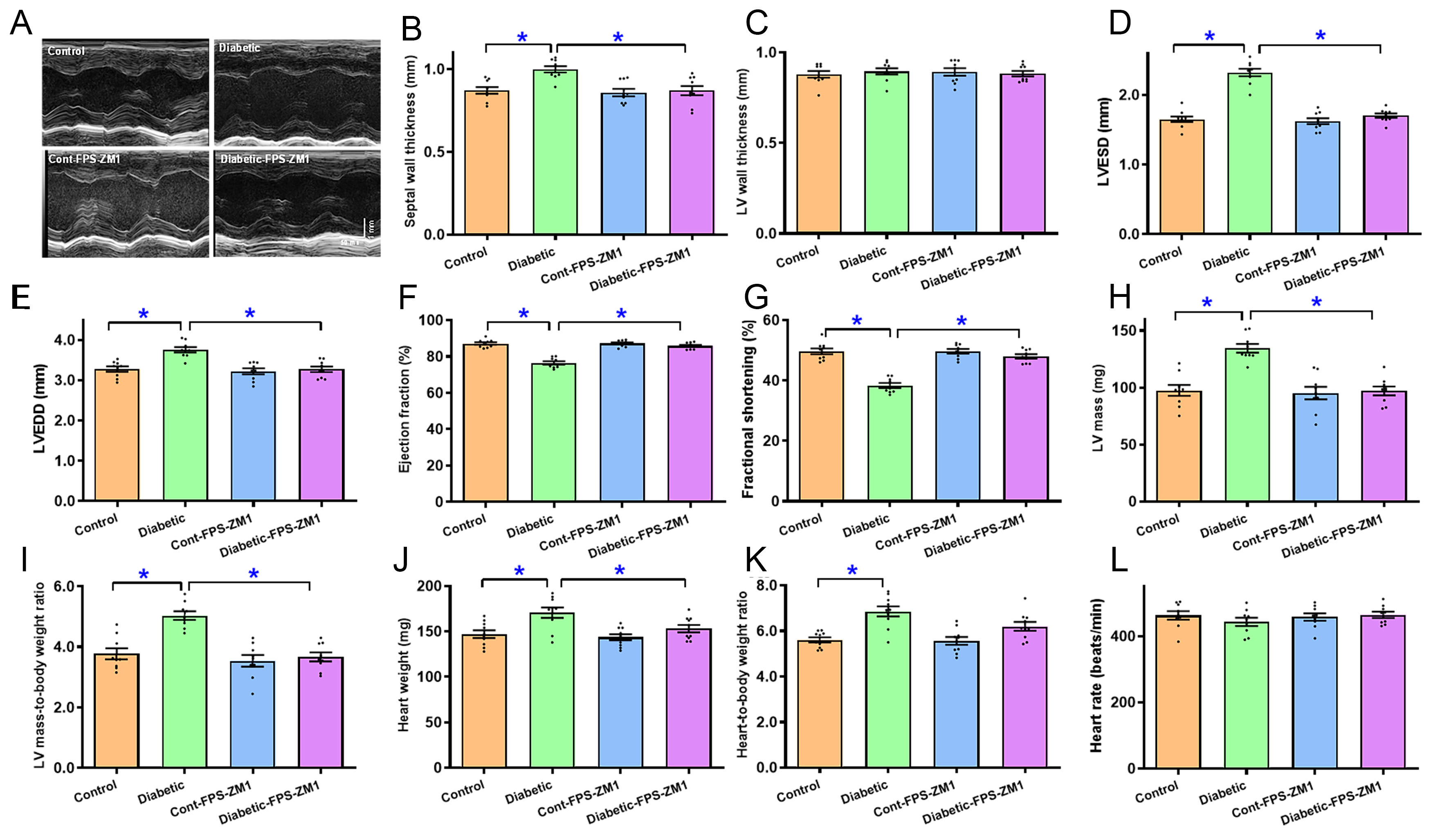
Figure 2. Echocardiographic properties of experimental mice treated with RAGE inhibitor FPS-ZM1 for 4 weeks. (A) M-mode echocardiographic traces from all groups; (B) septal thickness; (C) LV posterior wall thickness; (D) LVESD; (E) LVEDD; (F) Ejection fraction; (G) Fractional shortening; (H) LV mass; (I) Normalized LV mass (LV mass/body weight); (J) Heart weight; (K) Normalized heart weight (Heart weight/body weight); (L) Heart rate. Mean ± SEM, n = 9 mice per group, normal distribution was judged using the Shapiro-Wilk test, all experimental results were evaluated using one-way ANOVA and then Tukey’s post hoc test, *P < 0.05 between labeled groups. RAGE: Receptor for AGEs; LV: left ventricular; LVESD: LV end systolic diameter; LVEDD: LV end diastolic diameter; SEM: standard error of the mean; ANOVA: analysis of variance.
Cardiomyocyte shortening in experimental mice with or without RAGE inhibition
Sustained diabetes decreased cardiomyocyte shortening capacity, as manifested by a reduction in PS (0.52-fold, P < 0.001), +dL/dt (0.49-fold, P < 0.01), and -dL/dt (0.43-fold, P < 0.01), and prolongation of TR90 (0.41-fold, P < 0.0001) without altering TPS and resting cell length. FPS-ZM1 rescued diabetes-induced cardiomyocyte anomalies with little response itself [Figure 3A-F].
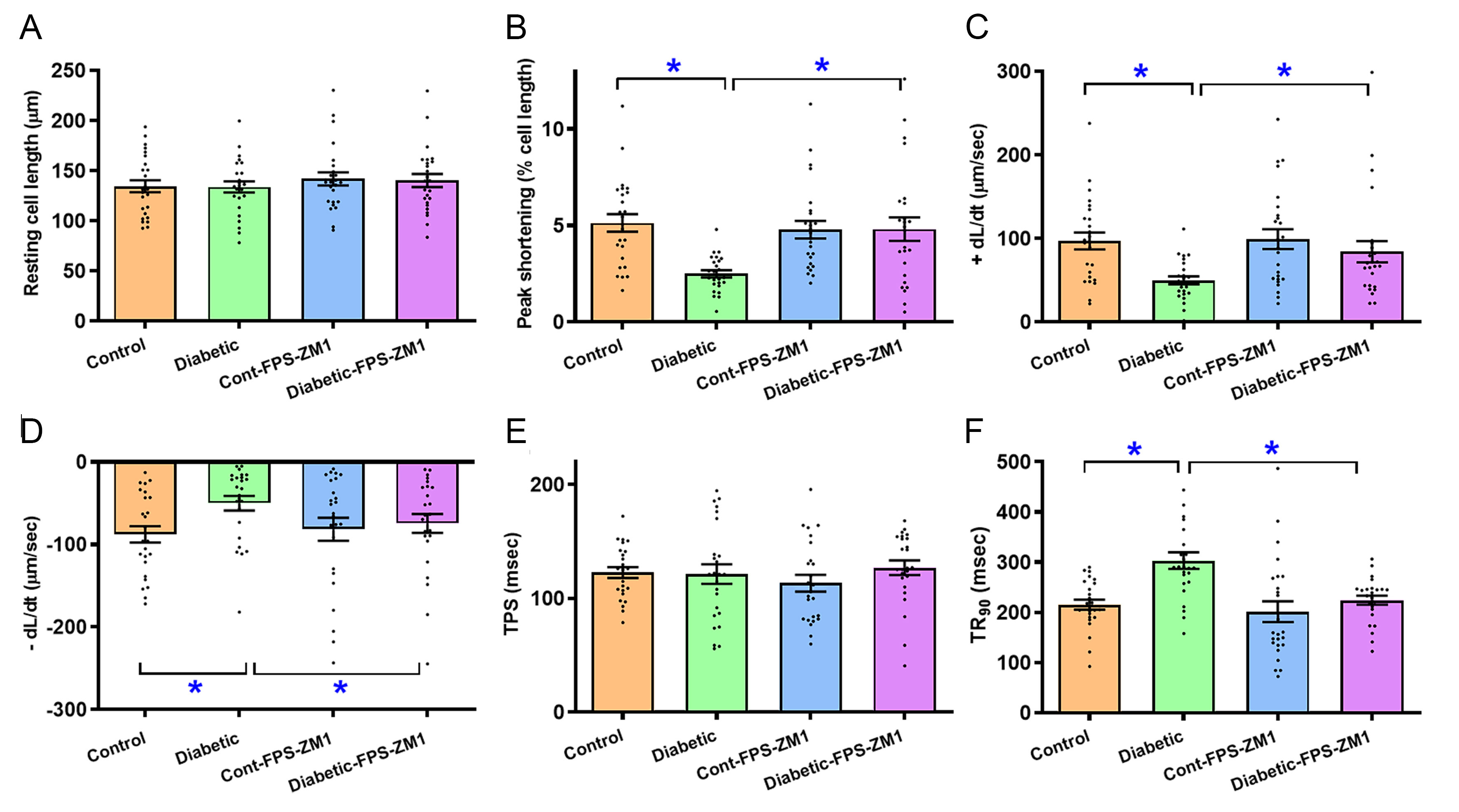
Figure 3. Cardiomyocyte contractile profiles in experimental mice treated with RAGE inhibitor FPS-ZM1 for 4 weeks. (A) Resting cell length; (B) PS; (C) Maximal velocity of shortening (+dL/dt); (D) Maximal velocity of relengthening (-dL/dt); (E) TPS; (F) TR90. Mean ± SEM, n = 25 cardiomyocytes (4 mice) per study group, normal distribution was evaluated using the Shapiro-Wilk test, data were assessed using one-way ANOVA and then Tukey’s post hoc test, *P < 0.05 between labeled groups. RAGE: Receptor for AGEs; PS: peak shortening; TPS: time-to-PS; TR90: time-to-90% relengthening; SEM: standard error of the mean.
Impact of RAGE inhibition on diabetes-elicited changes in cardiac structure and oxidative damage
H&E, Masson trichrome, and DCF staining were conducted to assess the possible effects of RAGE inhibition on diabetes-induced morphological pathology and oxidative damage. As shown in Figure 4A-F, diabetes provoked evident rises in cardiomyocyte cross-sectional area (by 0.19-fold, P < 0.0001), interstitial fibrosis (by 6.75-fold, P < 0.0001), and oxidative injury (by 2.53-fold, P < 0.0001, assessed using DCF staining), the effects of which were ablated by FPS-ZM1 with subtle effects itself.
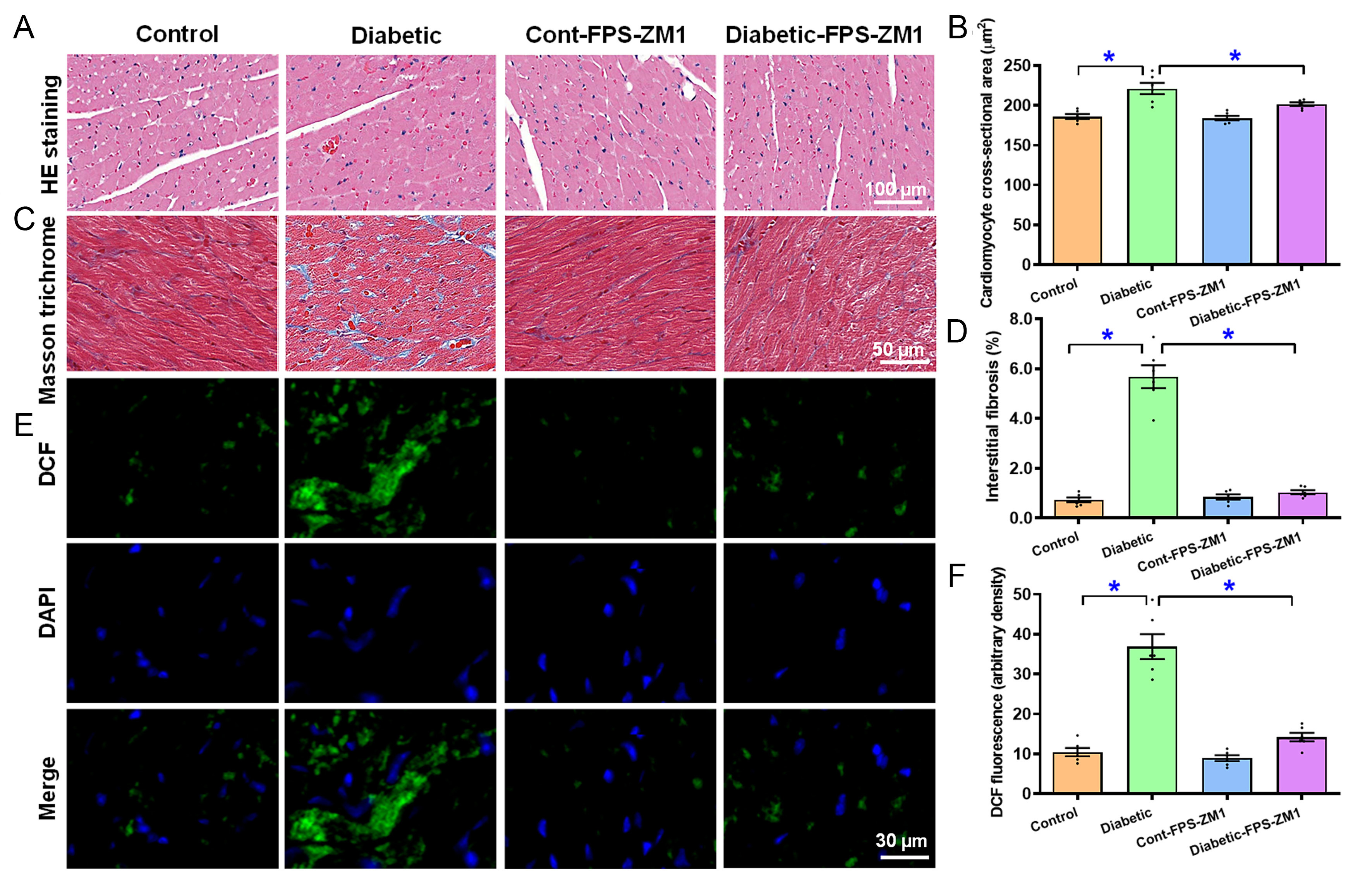
Figure 4. H&E, Masson trichrome, and DCF staining of the myocardium in experimental mice treated with RAGE inhibitor FPS-ZM1 for 4 weeks. (A) H&E images (representative); (B) Quantification of H&E area; (C) Masson trichrome image (representative); (D) Pooled data of Masson trichrome staining; (E) DCF staining images (first row: DCF staining; second row: DAPI staining; third row: Merged images); (F) Pooled DCF intensity. Mean ± SEM, n = 6 mice per group, normal distribution was evaluated using the Shapiro-Wilk test, experimental results were evaluated using one-way ANOVA and then Tukey’s post hoc test, *P < 0.05 between labeled groups. H&E: Hematoxylin and eosin; DCF: dichlorofluorofluorescein; DAPI: 4’,6-diamidino-2-phenylindole; SEM: standard error of the mean; ANOVA: analysis of variance.
Alteration in mitochondrial ultrastructure, function, and protein content in experimental mice with or without RAGE inhibition
TEM results showed that diabetes induced localized ultrastructural damage, characterized by increased mitochondrial area (0.19-fold, P < 0.0001) and circularity (0.23-fold, P < 0.0001), along with localized myofibrillar disarray. Consistent with myocardial functional changes, FPS-ZM1 canceled out diabetes-induced ultrastructural pathology in the absence of a notable effect itself [Figure 5A-C]. In addition, diabetes led to the collapse of mitochondrial membrane potential (MMP, 0.51-fold, P < 0.0001) and downregulation of mitochondrial proteins PGC1α (0.36-fold, P < 0.001) and UCP2 (0.32-fold, P < 0.05); the response was abolished by FPS-ZM1 without any effect of its own [Figure 5D-H].
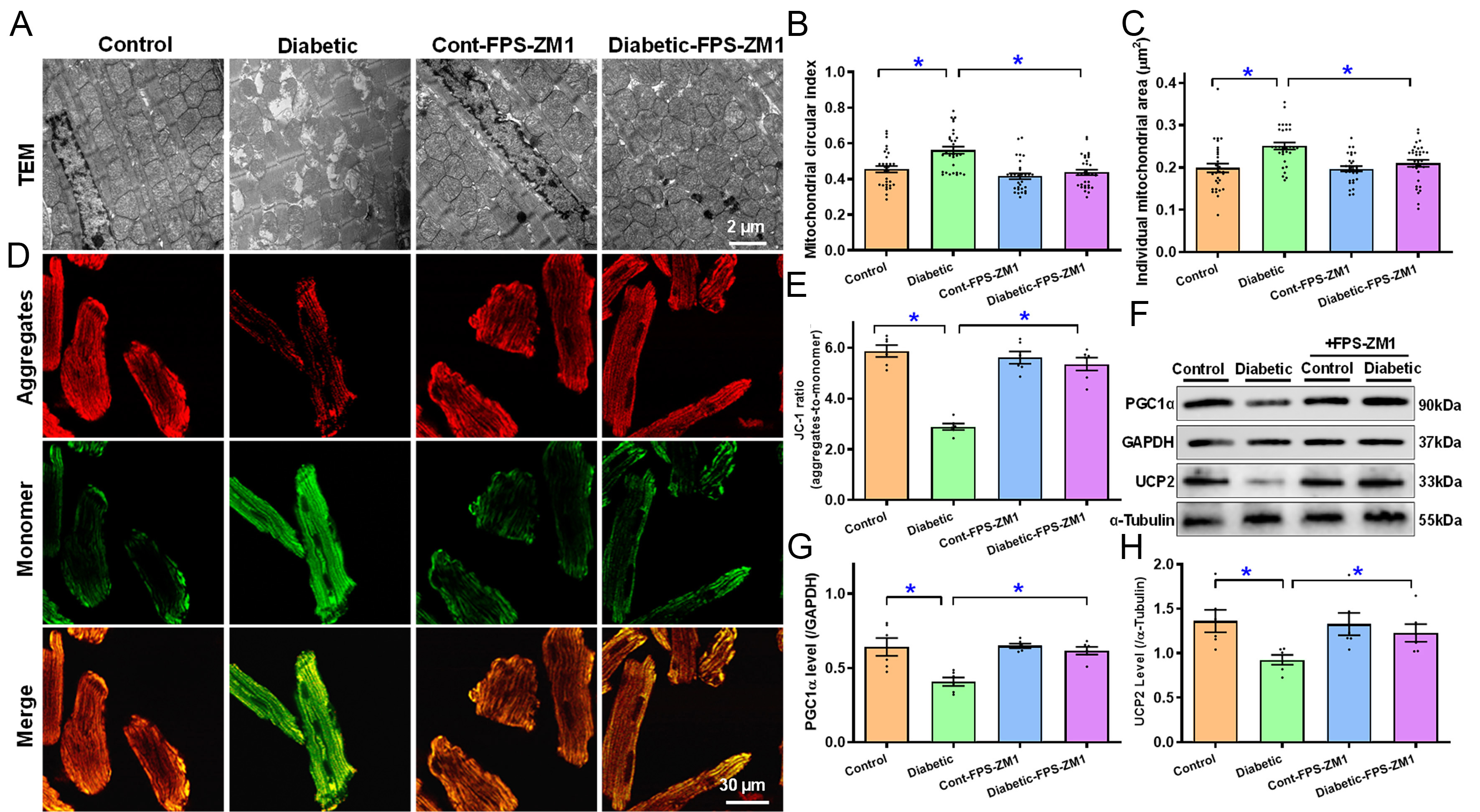
Figure 5. TEM ultrastructure and MMP of myocardial tissues or cardiomyocytes from experimental mice treated with RAGE inhibitor FPS-ZM1 for 4 weeks. (A) TEM ultrastructure micrographs (representative); (B) Short-to-long axis ratio for mitochondrial circularity; (C) Individual mitochondrial size; (D) JC-1 aggregates (red, top row), monomer (green, middle row) and merged (bottom row) fluorescence signals (representative); (E) Summarized data for JC-1 aggregate-to-monomer ratio; (F) Gel blotting depicting UCP2 and PGC1α levels (representative) using specific antibodies (α-tubulin or GAPDH as loading controls); (G) PGC1α; (H) UCP2. Mean ± SEM, n = 30-33 mitochondria [5 pictures/group, (B and C)] or 6 mice (E, G and H) per group, normal distribution was determined via the Shapiro-Wilk test, results were assessed via one-way ANOVA and then Tukey’s post hoc test, *P < 0.05 between labeled groups. MMP: Mitochondrial membrane potential; RAGE: receptor for AGEs; TEM: transmission electron microscope; UCP2: uncoupling protein 2; PGC1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; SEM: standard error of the mean; ANOVA: analysis of variance.
Change in apoptosis, cuproptosis and iron-sulfur cluster proteins in experimental mice with or without RAGE inhibition
Given the bioinformatics finding of prominent cell death features, including cuproptosis, in diabetic hearts, protein markers of apoptosis, cuproptosis and intercellular Cu2+ were evaluated. Our results revealed evident apoptosis [increased Bax (0.37-fold, P < 0.05) and decreased Bcl2 (0.54-fold, P < 0.01)], cuproptosis [increased Cu2+ importer SLC31A1 (CTR1, 0.55-fold, P < 0.0001), FDX1 (0.99-fold, P < 0.01) and DLAT (1.33-fold, P < 0.0001), with unchanged Cu2+ exporter ATP7B], Cu2+ binding protein S100A13, loss of Fe-S cluster proteins (NDUFS8 and ACO2) along heightened intracellular Cu2+ levels (by 0.65-fold, P < 0.0001) following diabetic insult. FPS-ZM1 reversed diabetes-induced alterations in Bax, Bcl2, FDX1, DLAT, ACO2, NDUFS8, S100A13, and intracellular Cu2+ levels, without affecting SLC31A1 (CTR1) and ATP7B. FPS-ZM1 did not affect any of the apoptotic, Cu2+-related proteins or intracellular Cu2+ levels [Figure 6A-K].
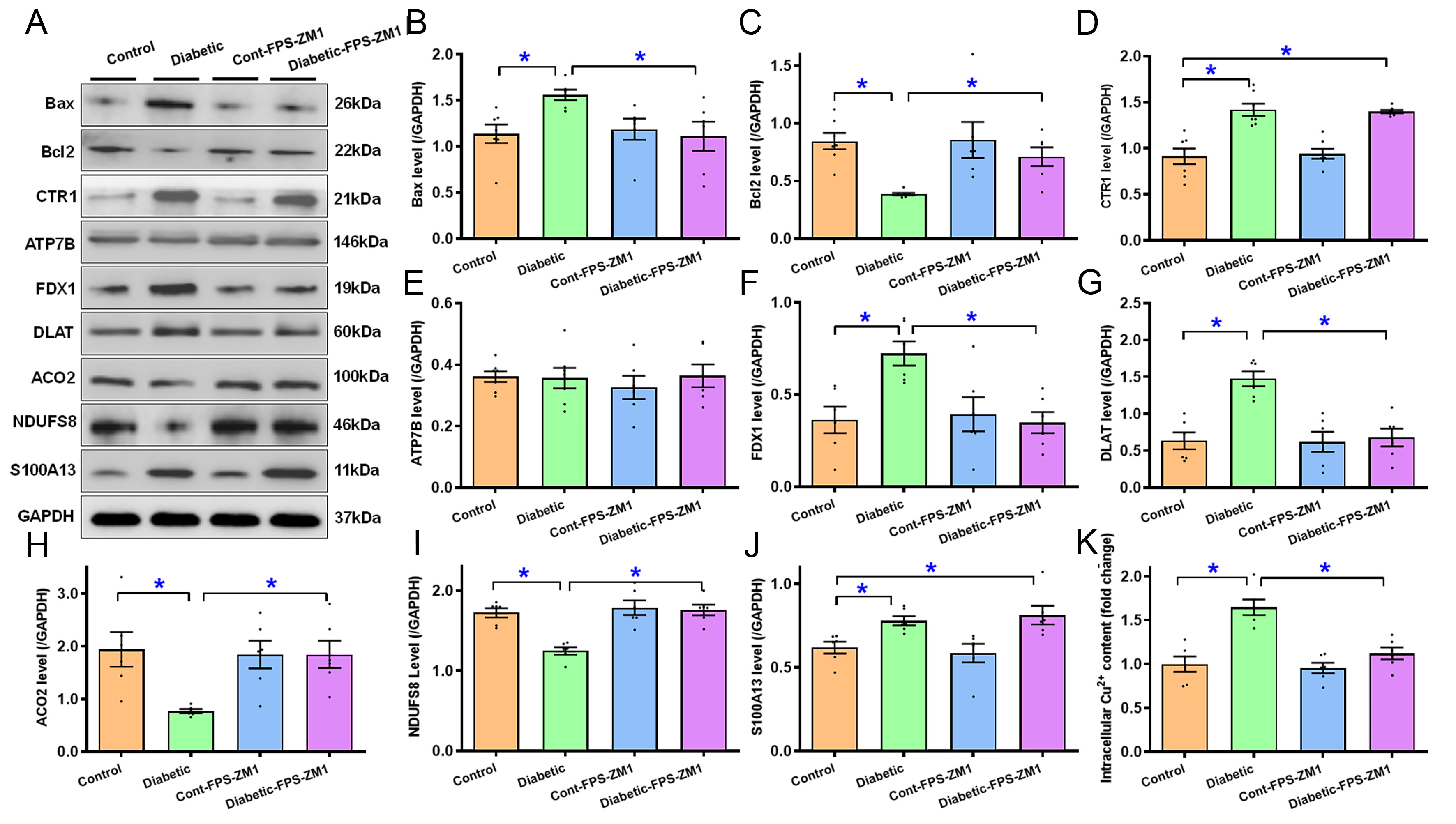
Figure 6. Apoptosis, cuproptosis and Fe-S protein markers, and intracellular Cu2+ of ventricular tissues from experimental mice treated with RAGE inhibitor FPS-ZM1 for 4 weeks. (A) Gel blots displaying Bax, Bcl2, CTR1 (SLC31A1), ATP7B, FDX1, DLAT, ACO2, NDUFS8, and S100A13 using specific antibodies (GAPDH for loading); (B) Bax; (C) Bcl2; (D) CTR1; (E) ATP7B; (F) FDX1; (G) DLAT; (H) ACO2; (I) NDUFS8; (J) S100A13; (K) Intracellular Cu2+. Mean ± SEM, n = 6-7 mice per group, normal distribution was determined via the Shapiro-Wilk test, results were assessed via one-way ANOVA and then Tukey’s post hoc test, *P < 0.05 between labeled groups. RAGE: Receptor for AGEs; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; Bcl2: B-cell lymphoma 2; Bax: Bcl-2-associated X protein; CTR1: copper transporter 1; ATP7B: ATPase copper transporting beta; FDX1: ferredoxin 1; ACO2: aconitase 2; NDUFS8: NADH: ubiquinone oxidoreductase core subunit S8; S100A13: S100 calcium-binding protein A13; SEM: standard error of the mean; ANOVA: analysis of variance.
Role of RAGE, cuproptosis and S100A13 in AGE precursor-induced cardiomyocyte functional aberrations
Diabetes results in elevated levels of methylglyoxal (MG-AGE), the precursor of AGE, prompting pathogenesis of diabetic complications[43]. To decipher the possible involvement of RAGE, cuproptosis and Cu2+ binding protein S100A13 in diabetic cardiomyopathy, C57BL/6 mouse cardiomyocytes were exposed to MG-AGE (5 μM) for 12 h in vitro with or without the RAGE blocker FPS-ZM1 (25 μM), the cuproptosis blocker TTM (20 μM)[43], and the S100A13 inhibitor AMX (20 μM)[37]. Our findings displayed in Figure 7A-F revealed that MG-AGE overtly compromised cardiomyocyte mechanical properties, manifested as decreased PS (by 0.38-fold, P < 0.05) and ±dL/dt (by 0.35- and 0.38-fold, respectively, P < 0.05 for both) along with prolonged TR90 (by 0.55-fold, P < 0.0001) without influencing resting cell length and TPS. Intriguingly, these MG-AGE-evoked cardiomyocyte mechanical anomalies were effectively mitigated by FPS-ZM1, TTM, and amlexanox, with no discernable response from the inhibitors themselves. These results suggest a role for RAGE, cuproptosis, and S100A13 in MG-AGE-elicited cardiomyocyte dysfunction.
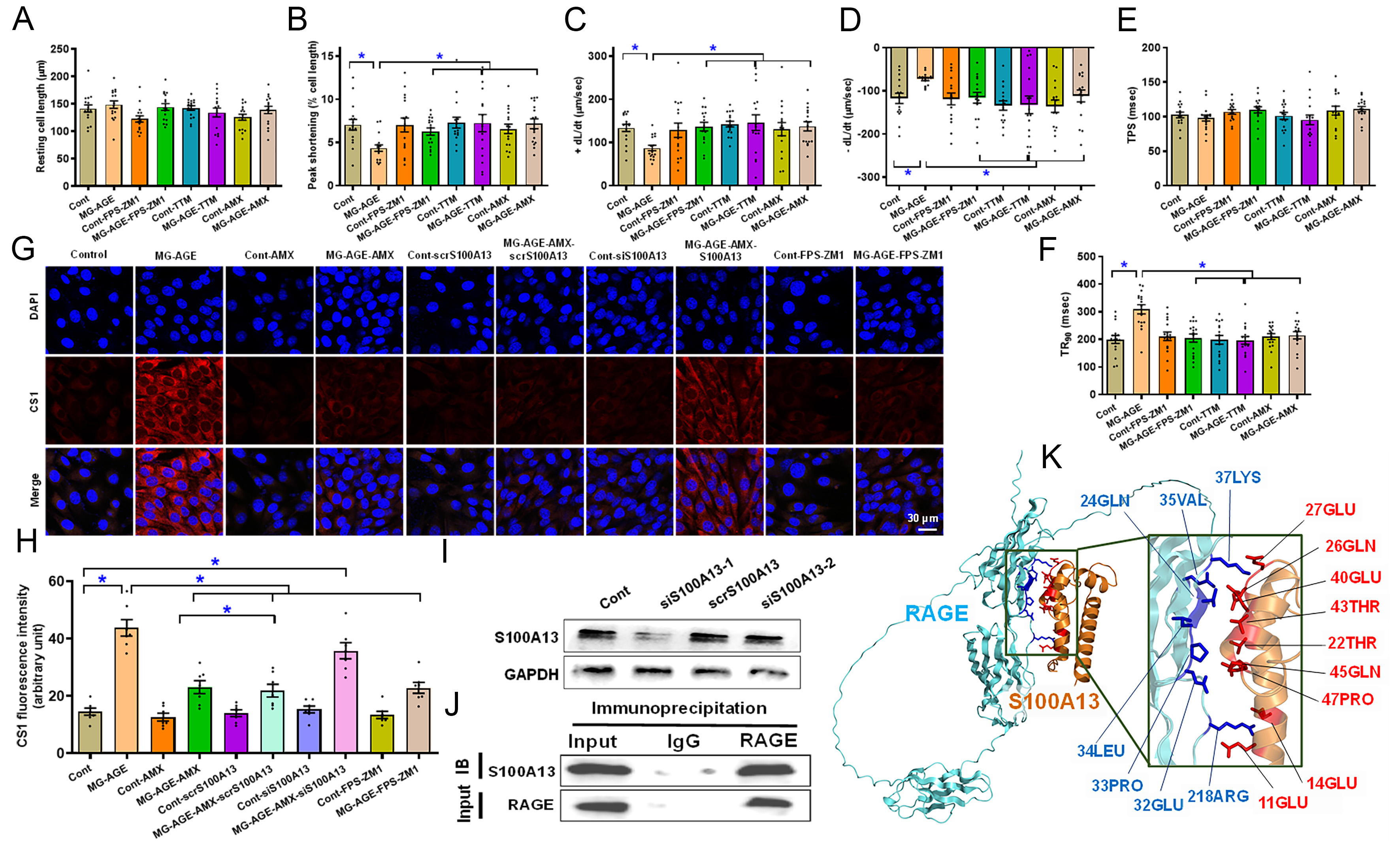
Figure 7. Impact of S100A13 inhibition/silencing on cardiomyocyte function and intracellular labile Cu+, co-IP, and the structure-based protein interaction interface between RAGE and S100A13. (A-F) Effect of RAGE inhibitor FPS-ZM1, cuproptosis inhibitor TTM and S100A13 inhibitor amlexanox (AMX) on MG-AGE-evoked cardiomyocyte defect in vitro. Freshly isolated C57BL/6 mouse cardiomyocytes were challenged with 5 µM MG-AGE for 12 h with or without 25 μM FPS-ZM1, 20 μM TTM, or 20 μM AMX prior to evaluation of cardiomyocyte mechanics. (A) Resting cell length; (B) Peak shortening normalized to cell length; (C) +dL/dt; (D) -dL/dt; (E) TPS; (F) TR90; (G and H) Representative images and pooled summary exhibiting effect of S100A13 silencing on AMX-induced response against MG-AGE-evoked rise in intracellular labile Cu+. HL-1 cells transfected with scramble or S100A13 siRNA were exposed to MG-AGE (5 µM) for 12 h in the absence or presence of AMX (20 μM) or FPS-ZM1 (25 μM) prior to assessment of labile Cu+ levels; (I) Validation of S100A13 silencing (using 2 batches of plasmids); (J) Co-IP analysis between RAGE and S100A13 in myocardial tissues from C57BL/6 mice; (K) Predicted interaction interface between RAGE and S100A13. Structural representation of the predicted RAGE-S100A13 complex. The RAGE and S100A13 subunits are depicted in cyan and orange, respectively. Interface residues involved in the interaction are highlighted in blue (RAGE) and red (S100A13) to delineate the binding pocket. Mean ± SEM, n = 16 cardiomyocytes from 3 mice (A-F) or 7 independent HL-1 cell cultures (H) per group, normal distribution was determined via the Shapiro-Wilk test, results were evaluated via two-way ANOVA and then Tukey’s post hoc test, *P < 0.05 between indicated groups. RAGE: Receptor for AGEs; Co-IP: co-immunoprecipitation; S100A13: S100 calcium-binding protein A13; TTM: tetrathiomolybdate; AMX: amlexanox; MG-AGE: methylglyoxal-derived advanced glycation end product; TPS: time-to-peak shortening; TR90: time-to-90% relengthening; SEM: standard error of the mean; ANOVA: analysis of variance; IgG: immunoglobin G; LEU: leucine; GLN: glutamine; LYS: lysine; ARG: arginine; PRO: proline; THR: threonine; GLU: glutamate/glutamic acid; VAL: valine.
Role of S100A13 in amlexanox-induced response against AGE precursor MG-AGE-evoked labile Cu+ accumulation
To determine whether S100A13 is the functional target of amlexanox, we conducted a gene-silencing study in HL-1 cells. Cells transfected with either scramble or S100A13 siRNA were incubated with 5 μM MG-AGE for 12 h, with or without amlexanox (20 μM) or the RAGE inhibitor FPS-ZM1 (25 μM). As shown in Figure 7G and H, MG-AGE challenge significantly increased intracellular labile Cu+ accumulation (1.93-fold, P < 0.0001), a response that was effectively mitigated by amlexanox. Notably, siRNA knockdown of S100A13, but not scramble RNA, essentially abrogated the protective effect of amlexanox against MG-AGE-induced labile Cu+ buildup. S100A13 silencing [efficiency validated in Figure 7I] failed to induce a discernible effect itself without MG-AGE exposure. Furthermore, pharmacological inhibition of RAGE with FPS-ZM1 obliterated the rise in intracellular labile Cu+, phenocopying the effect of amlexanox. These findings demonstrate that S100A13 is likely a requisite mediator for amlexanox-induced benefit against MG-AGE-driven Cu+ dysregulation.
Interaction between RAGE and Cu2+ binding protein S100A13
To understand the processes underlying AGE-RAGE-instigated governance of cuproptosis, interactions between RAGE and cuproptosis regulatory components were evaluated. Co-IP assay demonstrated pronounced physical interaction between RAGE and S100A13, a known Cu2+ binding protein [Figure 7J]. The interaction interface was modeled by AlphaFold3 software[44] and a detectable protein interaction interface between RAGE and S100A13 was identified in the model, as evaluated by PDBe-PISA software (interaction interface of 650.3 square angstrom, Figure 7K).
Inhibition of RAGE, cuproptosis or S100A13 on high glucose-provoked cardiomyocyte defects
To discern the possible roles of RAGE, cuproptosis and S100A13 in high glucose (mimicking hyperglycemia in diabetes)-induced myocardial mechanical anomalies, a high glucose-supplemented culture medium was employed as an in vitro model for diabetes[36]. Freshly isolated murine cardiomyocytes were exposed to normal glucose (NG, 5.5 mM) or high glucose (HG, 25.5 mM) for 12 h with or without the RAGE blocker FPS-ZM1 (25 μM), the cuproptosis blocker TTM (20 μM)[43], the S100A13 blocker amlexanox (20 μM)[37,38] or the Cu2+ ionophore elesclomol (40 nM)[43]. Our results showed that high glucose disrupted cardiomyocyte mechanical properties, shown as compromised PS (by 0.44-fold, P < 0.001) and ±dL/dt (by 0.44- and 0.51-fold, respectively, P < 0.01 for both) in conjunction with prolonged TR90 (by 0.46-fold, P < 0.0001) and unchanged TPS and resting cell length. Intriguingly, the high glucose-instigated cardiomyocyte anomalies were rescued by FPS-ZM1, TTM, and amlexanox. Moreover, Cu2+ ionophore elesclomol canceled out the FPS-ZM1-elicited beneficial effect against high glucose concentrations. None of these pharmacological agents had any notable effect on cardiomyocyte function under normal glucose culture conditions [Figure 8A-F]. These observations suggest a vital role for RAGE, cuproptosis and the Cu2+-binding protein S100A13 in high glucose culture-elicited myocardial pathologies.
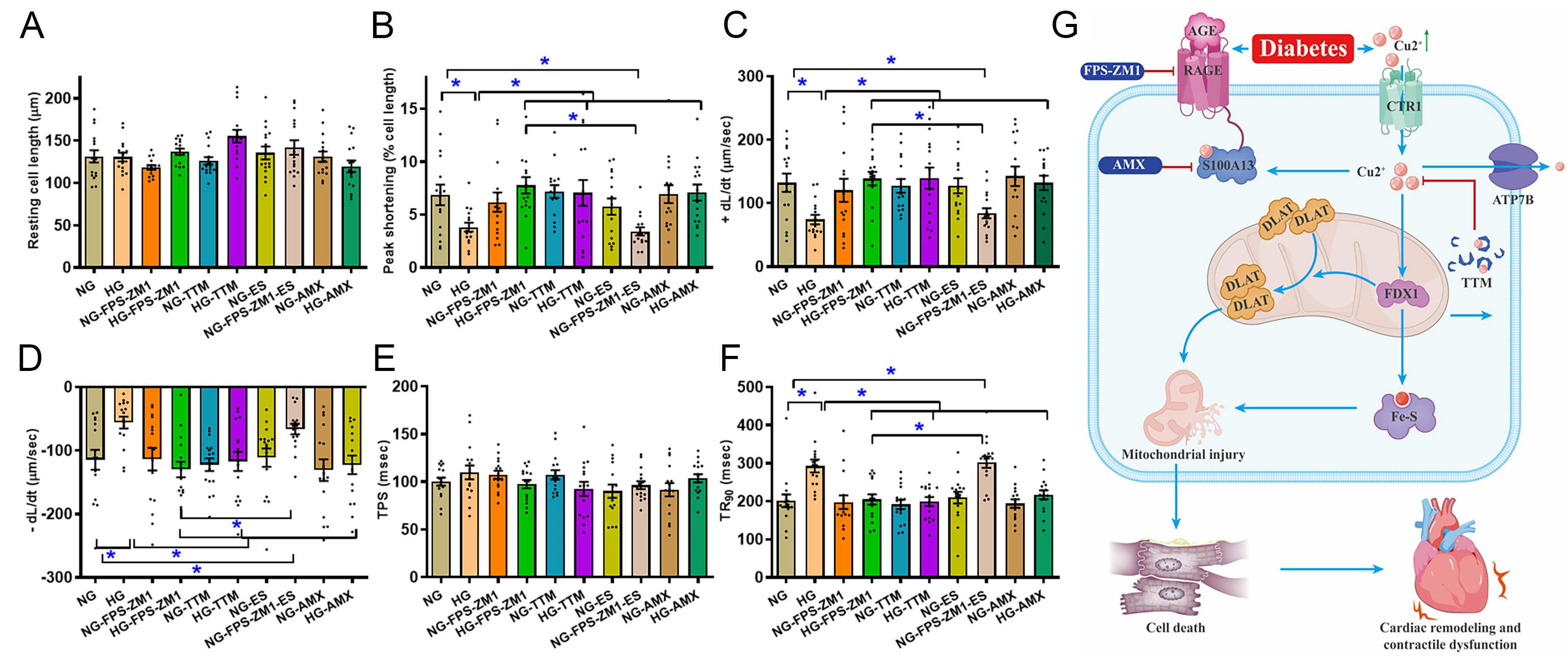
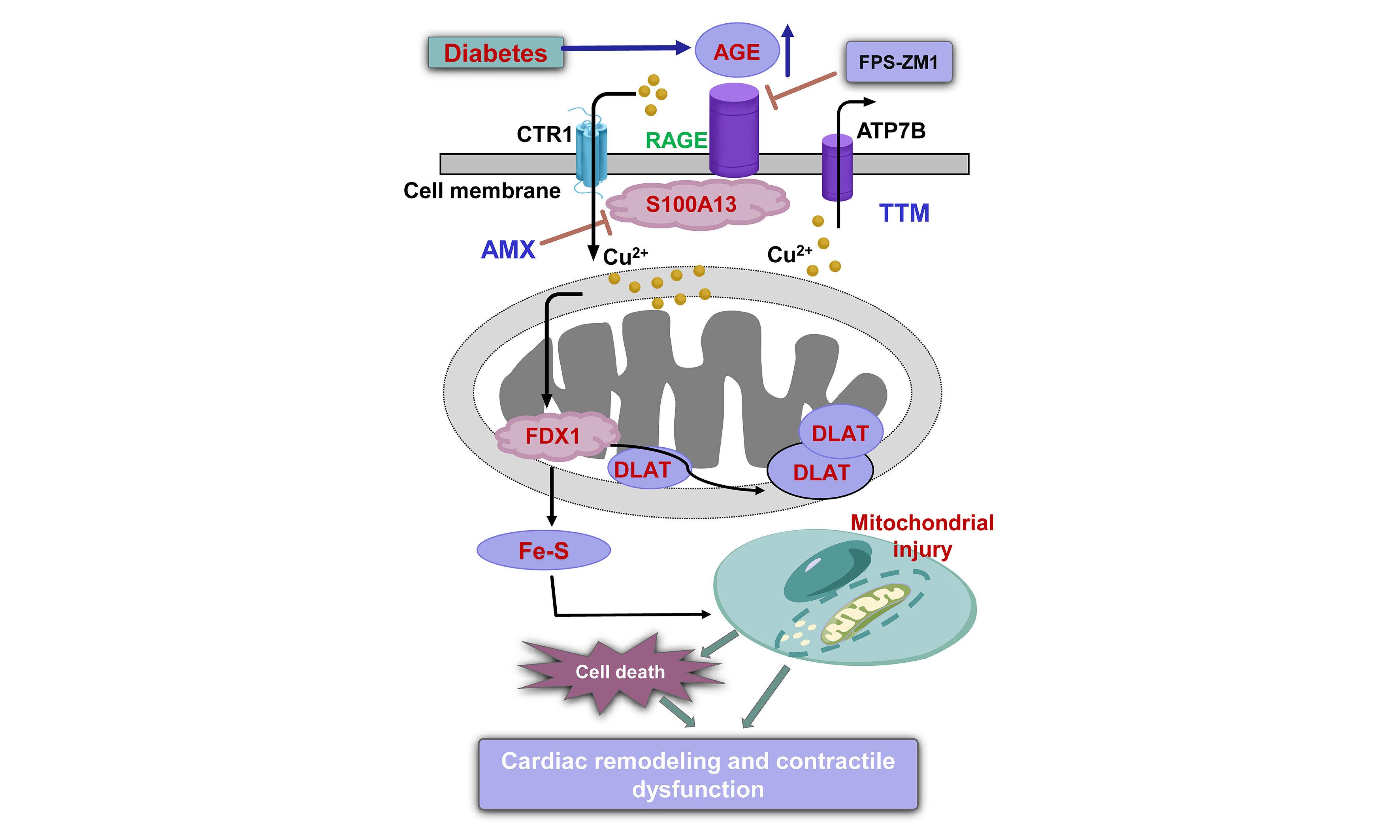
Figure 8. Impact of RAGE blocker FPS-ZM1, cuproptosis blocker TTM, Cu2+ ionophore ES and S100A13 inhibitor AMX on high glucose-evoked cardiomyocyte contractile anomalies in vitro. Adult C57BL/6 mouse cardiomyocytes were maintained in a DMEM medium supplemented with either HG (25.5 mM) or NG (5.5 mM) for 12 h with or without FPS-ZM1 (25 μM), TTM (20 μM), AMX (20 μM), or ES (40 nM) before determination of cardiomyocyte mechanics. (A) Resting cell length; (B) PS; (C) Maximal velocity of shortening (+dL/dt); (D) Maximal velocity of relengthening (-dL/dt); (E) TPS; (F) TR90; (G) Scheme depicting proposed mechanism behind RAGE inhibition-induced benefits against diabetes-induced mitochondrial damage, cuproptosis, and mechanical defect. Diabetes upregulates AGEs, prompting RAGE-dependent RAGE-S100A13 interaction, which facilitates Cu2+ binding, favoring cuproptosis and mitochondrial damage, ultimately cardiac remodeling and contractile anomalies. Mean ± SEM, n = 16 cells from 3 mice (A-F) per group, normal distribution was judged via the Shapiro-Wilk test, results were assessed using two-way ANOVA and then Tukey’s post hoc test, *P < 0.05 between indicated groups. RAGE: Receptor for AGEs; ES: elesclomol; HG: high glucose; NG: normal glucose; PS: peak shortening; TPS: time-to-peak shortening; TR90: time-to-90% relengthening; AGEs: advanced glycation end products; ANOVA: analysis of variance; TTM: tetrathiomolybdate; S100A13: S100 calcium-binding protein A13; AMX: amlexanox; DMEM: Dulbecco’s Modified Eagle Medium; SEM: standard error of the mean; FDX1: ferredoxin 1; DLAT: dihydrolipoamide S-acetyltransferase; CTR1: copper transporter 1; ATP7B: ATPase copper transporting beta.
DISCUSSION
In the present study, we identified AGE-RAGE activation as a key upstream driver of diabetic cardiomyopathy and provide evidence that RAGE inhibition with FPS-ZM1 robustly protects diabetic hearts from damage. Diabetes mellitus markedly increased myocardial AGE accumulation and RAGE expression, and RAGE inhibition using FPS-ZM1 reversed diabetes-induced myocardial dysfunction despite leaving systemic hyperglycemia/hyperlipidemia and circulating AGE levels unchanged, supporting a heart-intrinsic mechanism downstream of AGE-RAGE signaling. In parallel, our protein and functional profiling revealed a diabetes-associated “Cu2+/cuproptosis signature” (e.g., FDX1, DLAT, S100A13, loss of Fe-S cluster proteins), together with mitochondrial injury and apoptosis, all of which were mitigated by FPS-ZM1. These findings favor a cuproptosis-mediated mechanism in AGE-RAGE signaling-driven pathology in diabetic hearts [Figure 8G].
Functionally, experimental diabetes led to adverse cardiac remodeling and contractile impairment across tissue and cellular readouts, including increased LV dimensions/mass and reduced fractional shortening and ejection fraction, as well as compromised cardiomyocyte mechanics. Morphologically, diabetes increased cardiomyocyte size, ventricular tissue fibrosis, and oxidative injury, consistent with previous findings from our labs and others[6-8,25]. Importantly, FPS-ZM1 alleviated diabetes-evoked abnormalities, including cardiac remodeling, myocardial and cardiomyocyte mechanics, and oxidative stress. These convergent benefits suggest that RAGE inhibition interrupts diabetes-associated AGE-RAGE signaling anomaly to combat diabetes-associated structural remodeling and contractile failure. Beyond classical paradigms centered on oxidative stress and cardiac remodeling, our findings place mitochondrial injury as a central downstream consequence of AGE-RAGE signaling. In particular, diabetes elicited mitochondrial ultrastructural derangements (enlarged mitochondrial area/circularity and myofibrillar disarray) and impaired mitochondrial function (MMP collapse, loss of UCP2 and PGC1α), which were effectively rescued by FPS-ZM1. In parallel, diabetes increased pro-apoptotic signaling (elevated Bax, decreased Bcl2) and activated a Cu2+-linked, cuproptosis-related program characterized by increased CTR1, FDX1, DLAT, and Cu2+-binding molecule S100A13, unaltered Cu2+ exporter ATP7B, elevated intracellular Cu2+, and loss of Fe-S cluster proteins (ACO2, NDUFS8). Notably, while FPS-ZM1 rescued diabetic hearts from Cu2+-associated injury and intracellular Cu2+/Cu+ overload, it failed to normalize the elevated Cu2+ importer CTR1 alongside unaltered ATP7B. This discrepancy may indicate that RAGE activation primarily influences the labile Cu+ pool and its intracellular sequestration/redistribution rather than the initial uptake phase. By regulating the capacity of internal buffers, such as S100A13, metallothionein, or Cu2+-chaperone networks, RAGE inhibition using FPS-ZM1 may lessen intracellular Cu2+ binding (likely through S100A13) to neutralize imported Cu2+ and its accumulation within the mitochondrial matrix. Consequently, the protective effects of FPS-ZM1 likely stem from its ability to modulate intracellular Cu2+ partitioning and bioavailability, thereby decoupling high import pressure from mitochondrial proteotoxic stress. Collectively, these results support a mechanistic model in which AGE-RAGE signaling destabilizes mitochondrial proteostasis and bioenergetics, thereby lowering the threshold for Cu2+-driven proteotoxicity and cell death, which manifests as both contractile dysfunction and adverse remodeling.
At the cellular level, MG-AGE exposure recapitulated the diabetic cardiomyocyte phenotype (decreased
Several limitations warrant acknowledgment. First, while FPS-ZM1 effectively reduces mitochondrial injury, these effects may be partially indirect. Given that RAGE inhibition broadly mitigates oxidative stress and apoptosis[16-18], the observed reduction in cuproptosis might be a secondary consequence of improved global cellular proteostasis rather than a result of direct pathway blockade alone. Second, while our in vitro HL-1 cellular data established the functional relevance of S100A13, future investigations utilizing cardiomyocyte-specific S100A13 gain- or loss-of-function mouse models are essential to confirm whether S100A13 is an indispensable mediator of AGE-RAGE-dependent Cu2+ stress in vivo. Third, a significant technical hurdle remains the lack of direct evidence for protein lipoylation and the characteristic aggregation of specific mitochondrial proteins, largely due to technical challenges in preserving high-molecular-weight aggregates during protein extraction. Future studies employing non-reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and specialized solubility assays are required to definitively confirm the presence of these proteotoxic aggregates and establish their causal link to lipoylated mitochondrial proteins. Fourth, while our pharmacologic tools, including FPS-ZM1, TTM, amlexanox, and Cu2+ ionophores, provide robust evidence for pathway involvement, these agents may exert context-dependent off-target effects. For instance, TTM is a potent copper chelator whose benefits extend beyond the specific inhibition of cuproptosis to a generalized reduction in Cu2+ availability for diverse cellular processes. Moreover, RAGE inhibition corrects downstream Cu2+ mishandling (e.g., via S100A13) although its effect on the upstream Cu2+ import remains elusive. Also, levels of CTR1 were unaffected by FPS-ZM1, we cannot rule out possible post-translational modification of CTR1 at this time. In addition, complementary genetic approaches and precise Cu2+ flux measurements would significantly strengthen the mechanistic resolution of the RAGE-S100A13 axis. Additional work is required to define the exact temporal sequence of events, moving from AGE accumulation and RAGE activation to Cu2+ dysregulation and eventual mitochondrial injury. Crucially, determining the hierarchical order of these events, specifically, whether mitochondrial dysfunction precedes or follows the onset of cuproptosis activation, would be highly insightful. It remains to be determined whether the early deficits observed in Fe-S cluster proteins (such as ACO2 and NDUFS8) are a primary driver of disrupted membrane potential or a secondary consequence of the lipoylated protein aggregation characteristic of cuproptosis. Validating the translatability of these findings within clinically relevant settings and across diverse biological variables, including sex, age, and diabetes duration, is essential for assessing the therapeutic potential of targeting this pathway in humans. Finally, our cardiomyocyte mechanical analysis utilized a large cell-based sample size from a cohort of four mice per group. While capturing cellular heterogeneity, treating individual cells as independent entities can lead to pseudoreplication, potentially overestimating statistical power. We thus cross-referenced these results with tissue-level data to mitigate this concern. Nonetheless, future studies should employ nested or mixed-effect models to more rigorously account for animal-to-animal variability and the true biological unit of analysis.
In summary, our findings support a model in which diabetes-associated AGE accumulation activates myocardial RAGE signaling, promotes mitochondrial injury, and creates a Cu2+-permissive environment that engages a cuproptosis-linked proteotoxic program, culminating in cardiomyocyte dysfunction and adverse myocardial remodeling in diabetes. Pharmacological RAGE inhibition using FPS-ZM1 confers robust cardioprotection, and mechanistic experiments implicate Cu2+ bioavailability and Cu2+-binding protein S100A13 as actionable nodes within this pathway [Figure 8G]. Moving forward, important priorities should include establishing causal roles for S100A13 and specific Cu2+ transport/handling steps in vivo and in vitro, mapping how AGE-RAGE signaling rewires mitochondrial proteostasis and TCA/Fe-S cluster protein biology to sensitize to cuproptosis, and finally evaluating whether combined targeting of AGE formation, RAGE signaling, and Cu2+ stress yields additive benefit beyond single-node intervention. Such an approach may provide complementary strategies to ameliorate diabetic heart disease beyond conventional glycemic management, a hypothesis ripe for preclinical study.
DECLARATIONS
Authors’ contributions
Performed the study: Li F, Zhan Y, Lan X, Huang W, Li B, Ma H, Liang X, Xu Y, Ren J, Zhou Y
Drafted the initial draft of the manuscript: Ren J, Zhang X, Liang X
Edited the manuscript: Reiter RJ, Haidara MA
Conceived the study and provided the manuscript: Ren J, Zhang X
Availability of data and materials
All original data will be made available from the corresponding authors upon reasonable request.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool Gemini (version 3.0, released 2025-11-18) was used solely for language editing. The tool did not influence study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was funded in part by the National Natural Science Foundation of China (82470273), Guangzhou Science and Technology Project (2025A04J3720), the Scientific Research project of Guangdong Provincial Bureau of Traditional Chinese Medicine (20251085) and Guangdong Provincial Medical Science Foundation (B2025669).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
All animal protocols used in our study were approved by the Animal Care and Use Committees at Jinan University (20241015-0025, Guangzhou, China) and Zhongshan Hospital Fudan University (20210220, Shanghai, China).
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Sun H, Saeedi P, Karuranga S, et al. IDF diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183:109119.
2. Li YY, Yu Y, Jing HB, et al. Global epidemiology of diabetes and prediabetes in lean or non-obese patients with NAFLD: a systematic review and meta-analysis. Ann Med. 2026;58:2602995.
3. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. 2018;122:624-38.
4. Gebretsadik GG, Yang B, Glenn AJ, et al. Dietary manganese, type 2 diabetes, and cardiovascular disease: a UK Biobank cohort study and meta-analysis of over 270,000 individuals. J Nutr Health Aging. 2026;30:100754.
5. Wang M, Zhang SY, Zhai XR, et al. Long-term clinical outcomes in elderly patients with chronic total occlusion and type 2 diabetes: the impact of coronary collateralization following successful recanalization. Cardiology Plus. 2024;9:80-90.
6. Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. 2020;17:585-607.
7. Zhuang Z, Zhu Y, Tao J, et al. UCF101 Rescues against Diabetes-Evoked Cardiac Remodeling and Contractile Anomalies through AMP-activated protein kinase-mediated induction of mitophagy. Pharmacology. 2025;110:127-40.
8. Zhang Y, Zou R, Abudureyimu M, et al. Mitochondrial aldehyde dehydrogenase rescues against diabetic cardiomyopathy through GSK3β-mediated preservation of mitochondrial integrity and Parkin-mediated mitophagy. J Mol Cell Biol. 2024;15:mjad056.
9. Jiang MY, Man WR, Zhang XB, et al. Adipsin inhibits Irak2 mitochondrial translocation and improves fatty acid β-oxidation to alleviate diabetic cardiomyopathy. Mil Med Res. 2023;10:63.
10. Dandamudi S, Slusser J, Mahoney DW, Redfield MM, Rodeheffer RJ, Chen HH. The prevalence of diabetic cardiomyopathy: a population-based study in Olmsted County, Minnesota. J Card Fail. 2014;20:304-9.
11. Segar MW, Khan MS, Patel KV, et al. Prevalence and prognostic implications of diabetes with cardiomyopathy in community-dwelling adults. J Am Coll Cardiol. 2021;78:1587-98.
12. Schalkwijk CG, Stehouwer CDA. Methylglyoxal, a highly reactive dicarbonyl compound, in diabetes, its vascular complications, and other age-related diseases. Physiol Rev. 2020;100:407-61.
13. Berdowska I, Matusiewicz M, Fecka I. Methylglyoxal in cardiometabolic disorders: routes leading to pathology counterbalanced by treatment strategies. Molecules. 2023;28:7742.
14. Lai SWT, Lopez Gonzalez EJ, Zoukari T, Ki P, Shuck SC. Methylglyoxal and its adducts: induction, repair, and association with disease. Chem Res Toxicol. 2022;35:1720-46.
15. Mota KO, de Vasconcelos CML, Kirshenbaum LA, Dhalla NS. The role of advanced glycation end-products in the pathophysiology and pharmacotherapy of cardiovascular disease. Int J Mol Sci. 2025;26:7311.
16. Vianello E, Beltrami AP, Aleksova A, et al. The advanced glycation end-products (AGE)-receptor for age system (RAGE): an inflammatory pathway linking obesity and cardiovascular diseases. Int J Mol Sci. 2025;26:3707.
17. Wang B, Jiang T, Qi Y, et al. AGE-RAGE axis and cardiovascular diseases: pathophysiologic mechanisms and prospects for clinical applications. Cardiovasc Drugs Ther. 2025;39:1489-506.
18. Zhang L, Huang D, Shen D, et al. Inhibition of protein kinase C βII isoform ameliorates methylglyoxal advanced glycation endproduct-induced cardiomyocyte contractile dysfunction. Life Sci. 2014;94:83-91.
19. Yang L, Yang P, Lip GYH, Ren J. Copper homeostasis and cuproptosis in cardiovascular disease therapeutics. Trends Pharmacol Sci. 2023;44:573-85.
20. Liu Y, Shen M, Zhu S, et al. Metallothionein rescues doxorubicin cardiomyopathy via mitigation of cuproptosis. Life Sci. 2025;363:123379.
21. Li P, Li Y, Meng Q, Wang J, Wang K, Yang S. Copper dyshomeostasis and cardiovascular disease: Molecular mechanisms and new strategies for targeted intervention with cuproptosis (Review). Int J Mol Med. 2026;57:19.
22. Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254-61.
23. Saha PP, Kumar SKP, Srivastava S, Sinha D, Pareek G, D’silva P. The presence of multiple cellular defects associated with a novel G50E iron-sulfur cluster scaffold protein (ISCU) mutation leads to development of mitochondrial myopathy. J Biol Chem. 2014;289:10359-77.
24. Wang Z, Wu C, Li R, et al. Cuproptosis and cardiovascular diseases: mechanisms, pathophysiology, and therapeutic strategies - a narrative review. Rev Cardiovasc Med. 2025;26:38833.
25. Wold LE, Ceylan-Isik AF, Ren J. Oxidative stress and stress signaling: menace of diabetic cardiomyopathy. Acta Pharmacol Sin. 2005;26:908-17.
26. Liu C, Zhang H, Guan R, et al. Inhibition of the HMGB1-RAGE axis attenuates microglial inflammation and ameliorates hypoxia-induced cognitive impairment. Int J Mol Sci. 2025;26:8782.
27. Chen L, Yin Z, Qin X, et al. CD74 ablation rescues type 2 diabetes mellitus-induced cardiac remodeling and contractile dysfunction through pyroptosis-evoked regulation of ferroptosis. Pharmacol Res. 2022;176:106086.
28. Shen C, Ma Y, Zeng Z, et al. RAGE-specific inhibitor FPS-ZM1 attenuates AGEs-induced neuroinflammation and oxidative stress in rat primary microglia. Neurochem Res. 2017;42:2902-11.
29. Shen L, Zhang T, Yang Y, Lu D, Xu A, Li K. FPS-ZM1 alleviates neuroinflammation in focal cerebral ischemia rats via blocking ligand/RAGE/DIAPH1 pathway. ACS Chem Neurosci. 2021;12:63-78.
30. Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy - analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307-15.
31. Ashburner M, Ball CA, Blake JA, et al. Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25-9.
32. Kanehisa M, Goto S, Hattori M, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354-7.
33. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545-50.
34. Peng H, Fu S, Wang S, et al. Ablation of FUNDC1-dependent mitophagy renders myocardium resistant to paraquat-induced ferroptosis and contractile dysfunction. Biochim Biophys Acta Mol Basis Dis. 2022;1868:166448.
35. Chen Y, Zhao J, Ye H, et al. Beneficial impact of cardiac heavy metal scavenger metallothionein in sepsis-provoked cardiac anomalies dependent upon regulation of endoplasmic reticulum stress and ferroptosis but not autophagy. Life Sci. 2024;336:122291.
36. Ren J, Dominguez LJ, Sowers JR, Davidoff AJ. Metformin but not glyburide prevents high glucose-induced abnormalities in relaxation and intracellular Ca2+ transients in adult rat ventricular myocytes. Diabetes. 1999;48:2059-65.
37. Quan MY, Song XJ, Liu HJ, et al. Amlexanox attenuates experimental autoimmune encephalomyelitis by inhibiting dendritic cell maturation and reprogramming effector and regulatory T cell responses. J Neuroinflammation. 2019;16:52.
38. Rani SG, Mohan SK, Yu C. Molecular level interactions of S100A13 with amlexanox: inhibitor for formation of the multiprotein complex in the nonclassical pathway of acidic fibroblast growth factor. Biochemistry. 2010;49:2585-92.
39. Ren J, Sun M, Zhou H, et al. FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci Adv. 2020;6:eabc8561.
40. Blackburn NJR, Vulesevic B, Mcneill B, et al. Methylglyoxal-derived advanced glycation end products contribute to negative cardiac remodeling and dysfunction post-myocardial infarction. Basic Res Cardiol. 2017;112:57.
41. Li F, Hu H, Wu L, et al. Ablation of mitophagy receptor FUNDC1 accentuates septic cardiomyopathy through ACSL4-dependent regulation of ferroptosis and mitochondrial integrity. Free Radic Biol Med. 2024;225:75-86.
42. Chen N, Guo L, Wang L, Dai S, Zhu X, Wang E. Sleep fragmentation exacerbates myocardial ischemia-reperfusion injury by promoting copper overload in cardiomyocytes. Nat Commun. 2024;15:3834.
43. Huo S, Wang Q, Shi W, et al. ATF3/SPI1/SLC31A1 signaling promotes cuproptosis induced by advanced glycosylation end products in diabetic myocardial injury. Int J Mol Sci. 2023;24:1667.
44. Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024;630:493-500.
45. Sivaraja V, Kumar TK, Rajalingam D, Graziani I, Prudovsky I, Yu C. Copper binding affinity of S100A13, a Key Component of the FGF-1 nonclassical copper-dependent release complex. Biophys J. 2006;91:1832-43.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].