




Corosolic acid increases the therapeutic effect of cisplatin on gastric cancer by regulating Gpx4-dependent ferroptosis
 0
0 Abstract
Aim: Cisplatin serves as a primary chemotherapeutic agent in the treatment of gastric cancer (GC), but resistance to cisplatin-based chemotherapeutic regimens hampers its clinical application. Corosolic acid (CA), a natural triterpenoid, exhibits both anti-inflammatory and anti-cancer activities. However, the effect of CA on improving cisplatin resistance in GC remains unclear. The study primarily aimed to evaluate whether CA increases the therapeutic efficacy of cisplatin against GC and to reveal its underlying mechanism.
Methods: Cisplatin and CA were used to treat GC cells or cisplatin-resistant AGS cells (AGS-CR), and then cell viability, apoptosis, and growth were assessed using Cell Counting Kit-8, TdT-mediated dUTP nick end labeling, and clone formation assays, respectively. Glutathione peroxidase 4 (Gpx4) expression was measured through quantitative real-time PCR and western blotting assays.
Results: CA treatment induced a dose-dependent reduction in GC cell viability. The combination of cisplatin and CA resulted in enhanced cytotoxicity and pro-apoptotic effects compared to treatment with cisplatin alone. The effect of CA as a chemosensitizer in GC cells was damaged by a ferroptosis inhibitor, suggesting that CA decreased cisplatin chemoresistance by accelerating cancer cell ferroptosis. CA triggered cell ferroptosis by repressing Gpx4 expression in GC cells. Furthermore, elevated Gpx4 expression was significantly associated with poorer overall and disease-free survival.
Conclusion: CA has the potential to increase cisplatin chemosensitivity in GC, and Gpx4 may represent a promising therapeutic target for its treatment.
Keywords
INTRODUCTION
Gastric cancer (GC) ranks as the fifth most frequently diagnosed malignancy globally, with an estimated one million new cases in 2020. It is also the fourth leading cause of cancer-related mortality, with an estimated 770,000 deaths[1,2]. Owing to the deficiency of representative symptoms and effective markers, GC is typically diagnosed at an advanced stage, which results in a poor prognosis[3]. Over the past few decades, despite the great progress in molecular-targeted therapy, surgical resection and chemotherapy remain the mainstays of therapy for GC patients[4,5]. Cisplatin is an effective drug to treat GC, but resistance to cisplatin-based regimens hampers its clinical application. Numerous studies have investigated the molecular mechanisms underlying chemotherapy resistance by generating cisplatin-resistant GC cell lines, such as cisplatin-resistant AGS cells (AGS-CR). Studies on AGS-CR cells indicate a lower accumulation of cisplatin compared to sensitive cells, attributed to decreased drug uptake[6]. RNA-seq analysis of AGS-CR cells identified 189 differentially expressed genes (DEGs) primarily linked to cisplatin resistance-related molecular functions. DEG analysis further revealed enrichment across 23 metabolic pathways, with the inflammation-mediated chemokine and cytokine signaling pathway exhibiting the highest degree of enrichment[7].
Radix Actinidiae chinensis (RAC), a traditional Chinese medicine, possesses a significant anti-cancer property against different types of tumors, such as GC[8], colorectal cancer (CRC)[9], and lung cancer[10]. RAC treatment inhibits inflammatory response and cell proliferation in renal cell carcinoma (RCC) mice and accelerates cancer cell apoptosis[11]. Ethanol extract of RAC accelerates CRC cell apoptosis by repressing Notch signaling[12]. Importantly, several bioactive ingredients have been identified in RAC. Oleanolic acid (OA), a bioactive triterpenoid in RAC, exhibits effective anti-tumor activity in various types of tumors[13,14]. OA derivatives (CDDO, CDDO-me, K73-03, etc.) improve their anti-tumor activity through increasing water solubility[15-17]. Corosolic acid (CA), another bioactive triterpenoid in RAC, could effectively inhibit cell proliferation and tumor growth in many types of tumors, such as GC, prostate cancer (PC), and hepatocellular carcinoma (HCC)[18-20]. Furthermore, CA could increase the responsiveness of cancer cells to 5fluorouracil (5FU) treatment[21]. However, the potential of CA to increase the therapeutic effect of cisplatin on GC remains poorly characterized.
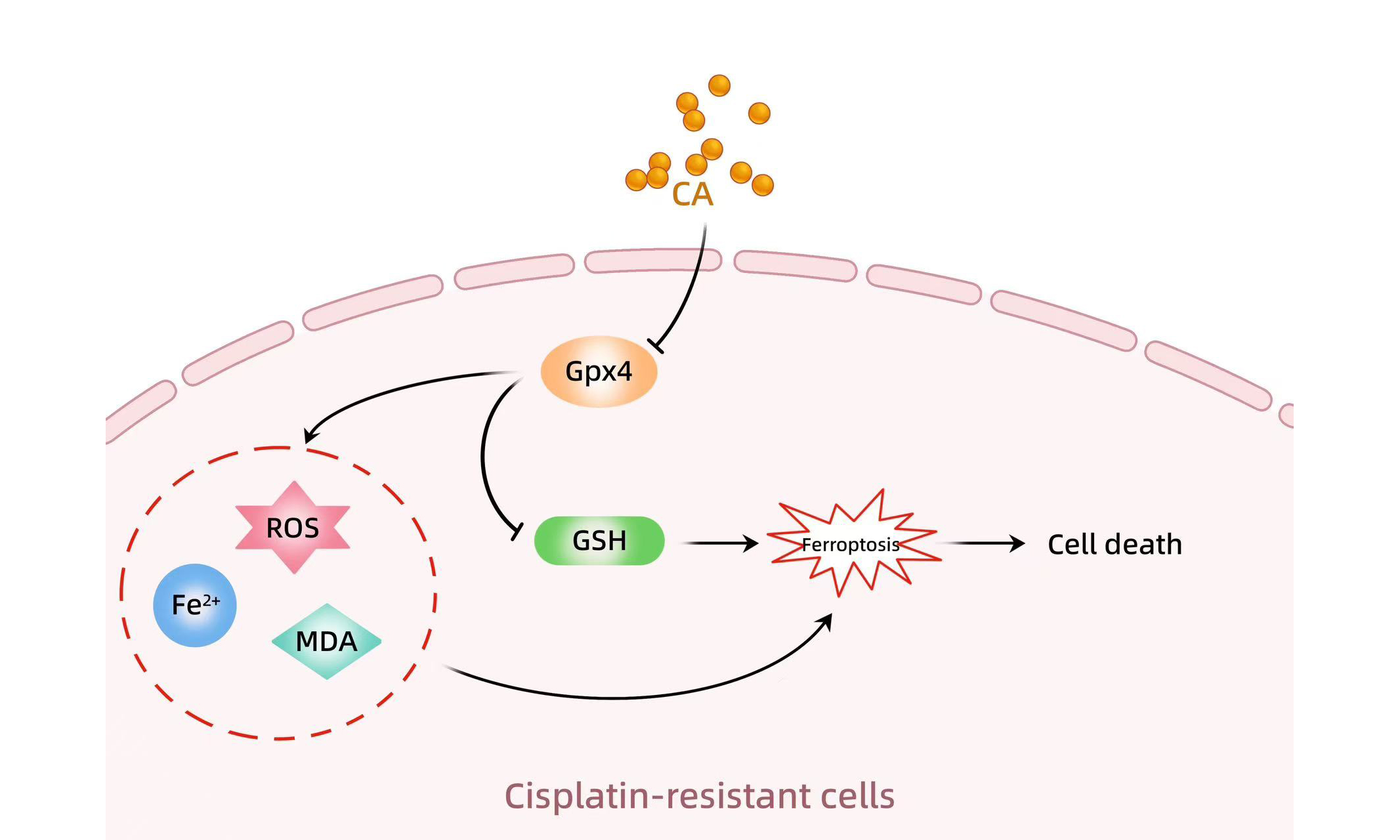
Ferroptosis is a kind of non-apoptotic cell death marked by iron-dependent lipid peroxidation[22]. Ferroptosis plays a vital role in regulating tumor progression and chemoresistance[23,24]. Wang et al. showed that overactivation of the Wnt/β-catenin pathway inhibits ferroptosis through increasing Gpx4 expression, resulting in GC cell chemoresistance[24]. A recent study has demonstrated that CA sensitizes HCC cells to ferroptosis through decreasing glutathione (GSH) synthesis[25]. At present, several critical genes involved in ferroptosis have been identified, such as prostaglandin-endoperoxide synthase 2 (Ptgs2), acyl-CoA synthetase, long-chain family member 4 (Acsl4), Gpx4, NADPH oxidase 1 (Nox1), Ferritin heavy chain 1 (Fth1), and solute carrier family 7 member 11 (Slc7a11)[26]. Increased expression of Acsl4[27], Ptgs2[28], and Nox1[29] contributes to inhibiting ferroptosis, while decreased expression of Gpx4[30], Slc7a11[31], and Fth1[32] promotes ferroptosis. The study aimed to assess the chemosensitizing potential of CA in GC cells and reveal its underlying mechanisms.
MATERIALS AND METHODS
Gene expression profiling interactive analysis
Gpx4 expression and its association with overall survival (OS) and disease-free survival (DFS) were analyzed using the gene expression profiling interactive analysis (GEPIA) platform based on data from the cancer genome atlas-stomach adenocarcinoma (TCGA-STAD), which comprises 408 tumor and 211 normal gastric tissue samples.
Cell culture
AGS and MKN-45 cells (The Shanghai Institute of Biochemistry and Cell Biology, Shanghai, China) were cultivated in DMEM/F12 (GIBCO, NY, USA) supplemented with 10% FBS (GIBCO) in a humidified 5% CO2 incubator. AGS-CR were established through persistent exposure of parental AGS cells to escalating concentrations of cisplatin for more than one year, as previously described[7]. AGS-CR cells were maintained in DMEM/F12 containing cisplatin to preserve their drug-resistant phenotype.
The pcDNA-Gpx4 recombinant plasmids, which encode full-length cDNA of Gpx4, were constructed to overexpress Gpx4 in GC cells. PolyFast Transfection Reagent (MedChem Express, NJ, USA) was applied to transfect the recombinant plasmids into cells when cells reached approximately 80% confluence.
To evaluate the cytotoxicity of cisplatin (MedChem Express) and CA (MedChem Express) to GC cells, AGS, MKN-45, or AGS-CR cells were treated with cisplatin (5, 15, 25, and 35 µm) and CA (0, 2, 5, 10, 20, 40, and 60 µm). Cell viability, clone formation ability, and cell apoptosis were evaluated through cell counting kit-8 (CCK-8), clone formation assay, TUNEL staining, and flow cytometry, respectively.
CCK-8
Cell viability was assessed by the CCK-8 assay. Cells were seeded in 96-well plates (5 × 103 cells per well) overnight, followed by treatment with cisplatin and CA for the specified duration. Subsequently, CCK-8 solution (10 μL, MedChem Express) was added to each well and incubated for 1 h, after which absorbance at 450 nm was measured using a VICTOR Nivo™ microplate reader (Univ-bio, Shanghai, China).
Clone formation assay
AGS or AGS-CR cells (400 cells per well) were treated with cisplatin (35 µm) and/or CA (5 µm) for 3 days. Then, cells were cultured in DMEM/F12 medium for two weeks, allowing colony formation to reach visible levels. After washing four times with phosphate buffer saline (PBS), cells were treated with 4% paraformaldehyde (PFA) for fixation, followed by staining with 0.1% crystal violet. Colonies were observed with an inverted microscope (VEDENG, Shanghai, China).
Apoptosis assay
Cell apoptosis was assessed using TUNEL and flow cytometry. In brief, AGS-CR cells were exposed to cisplatin (35 µm) and/or CA (5 µm) for 24 h, then fixed with 4% PFA and subsequently stained using TUNEL reagent (Solarbio, Beijing, China). TUNEL-positive cell quantification was performed using fluorescence microscopy (Leica). Apoptotic cell populations were additionally evaluated using the Annexin V-FITC/PI detection kit (MedChem Express) in conjunction with a flow cytometer (BD Biosciences Inc.)
Fluorescein diacetate staining
AGS-CR cells were exposed to cisplatin (35 µm) and/or CA (5 µm) or Ferrostatin-1 (Fer-1, a potent ferroptosis inhibitor, 1 μm) for 24 h, then fixed with 4% PFA and subsequently incubated with fluorescein diacetate (FDA) solution (Merck, MA, USA) for 20 min. FDA-positive cell quantification was performed using fluorescence microscopy
qRT-PCR
Total RNA was collected with Trizol (Solarbio), followed by first-strand cDNA synthesis with M-MLV and Oligo(dT) primers (TaKaRa, Tokyo, Japan). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was carried out with SYBR Green qPCR mix on the 7500 real-time qPCR system (Applied Biosystems). Relative mRNA level was quantified through the 2-ΔΔCt method[33]. β-actin was applied as a reference gene. All primers were listed in Supplementary Table 1.
Western blot
Total protein was collected from GC cells with RIPA buffer (Sigma-Aldrich, MO, USA) and quantitated with a BCA protein assay kit (Abcam, CA, USA). Approximately 20 µg of protein were separated on 12% SDS-PAGE at an unchanging voltage of 50 V (concentration gel) or 110 V (separation gel). Subsequently, protein was transferred to PVDF membranes (Merck), and the membranes were sealed with 5% skimmed milk solution, washed thrice with PBS, and incubated with primary antibodies for 1 h and HRP-conjugated secondary antibodies (1:5,000, ab6728 or ab6721, Abcam) for 1-h at room temperature. The primary antibodies used in the study were Gpx4 (1:4,000, ab125066, Abcam), Ptgs2 (1:2,000, #35-8200, Thermo Fisher Scientific, MA, USA), Acsl4 (1:10,000, #ab155282, Abcam), Fth1 (1:1,000, #ab75972, Abcam), Nox1 (1:2,000, #PA5-38031, Thermo Fisher Scientific), Slc7a11 (1:4,000, ab175186, Abcam), and β-actin (1:5,000, #ab8226, Abcam). The immunoblots were visualized with an ECL detection reagent (Solarbio) on the Tanon 5200 system (Tanon, Shanghai, China).
Reactive oxidative species assay
AGS and MKN-45 cells were treated with CA (5 µm). Twenty-four hours later, cells were incubated with C11-BODIPY (1 μm, Thermo Fisher Scientific) for 30 min at 37 °C. Lipid reactive oxidative species (ROS) level was tested with a flow cytometer (BD Biosciences, CA, USA).
Assessment of iron, malondialdehyde, and GSH levels
AGS and MKN-45 cells were exposed to CA (5 µm). Twenty-four hours later, ferrous iron concentration, malondialdehyde (MDA), and GSH content were evaluated using an iron assay kit (ab83366), a MDA assay kit (ab118970), and a GSH assay kit (ab65322), respectively.
Mouse xenograft tumor model
The work was approved by the Animal Ethics Committee of Shanghai Municipal Hospital of Traditional Chinese Medicine (No. 2023112) and conducted in compliance with the ARRIVE guidelines. BALB/c nude mice (Charles River, Shanghai, China) were maintained in pathogen-free rooms at 22-26 °C and 40%-60% humidity under a 12-h light-dark cycle. Mice had unrestricted access to standard chow and water. The GC xenograft model in mice was created by subcutaneously injecting AGS cells (5 × 106 cells in 100 μL PBS) into the right flank (n = 5 per group). Upon the tumors reaching roughly 100 mm3 in volume, the mice were administered according to the specified schedule and dosage: 1) intraperitoneal (i.p.) injection of vehicle, 2) i.p. injection of cisplatin once a day for 2 weeks (5 mg/kg), 3) i.p. injection of cisplatin once a day for 2 weeks (5 mg/kg) and CA once every 2 days (20 mg/kg). Tumor volume and body weight were surveyed at the specified intervals. Throughout the experiment, mice exhibiting severe illness or approaching death were humanely euthanized via cervical dislocation under deep anesthesia with pentobarbital sodium (100 mg/kg).
Statistics
Results from three biologically independent experiments were presented as mean ± standard deviation. Statistical comparisons were conducted through Student’s t test or one-way analysis of variance (ANOVA) tests via GraphPad Prism 7.0 (GraphPad Software, CA, USA), with P < 0.05 denoting significance.
RESULTS
CA sensitized GC cells to cisplatin treatment
To explore the effect of CA on increasing cisplatin sensitivity in GC cells, the cytotoxicity of CA to GC cells (AGS and MKN-45 cells) was first evaluated. Figure 1A reveals that AGS and MKN-45 cell viability were decreased by approximately 18% and 20%, respectively, at a concentration of 5 µm of CA and further decreased in a concentration-dependent manner. 5 µm of CA were not toxic to a normal gastric epithelial cell line [Supplementary Figure 1A] and thus were used in subsequent experiments. CA also decreased GC cell viability in a time-dependent manner in AGS and MKN-45 cells [Figure 1B]. Importantly, combination treatment with cisplatin and CA reduced AGS [Figure 1C] and MKN-45 [Supplementary Figure 1B] cell viability more effectively than cisplatin alone. Then the effect of CA on AGS-CR cell viability, proliferation, and apoptosis was assessed using CCK-8, clone formation, TUNEL, and flow cytometry assays, respectively. Combination treatment with cisplatin and CA decreased AGS-CR cell viability [Figure 1D] and clone formation capacity
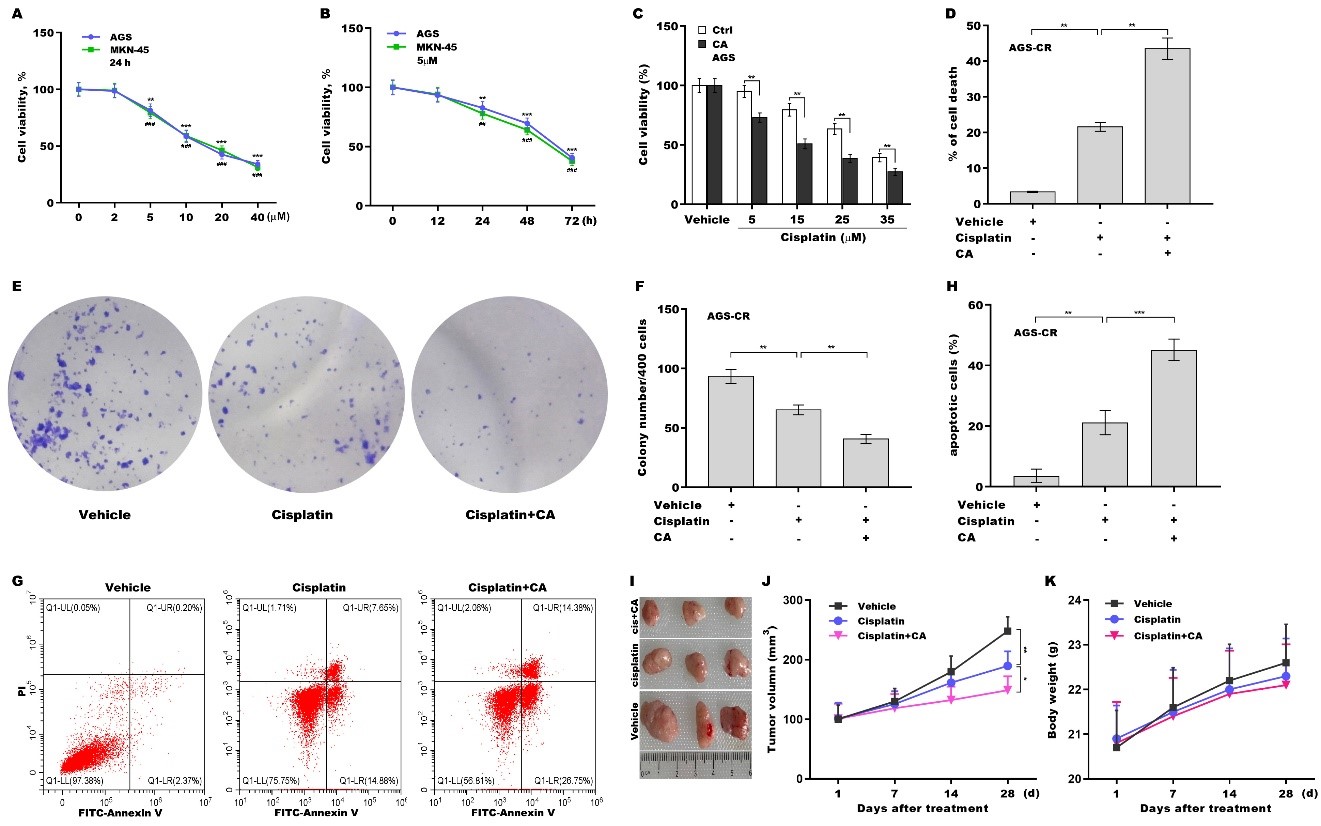
Figure 1. CA sensitized GC cells to cisplatin treatment. (A) AGS and MKN-45 cells were exposed to different concentrations of CA (0, 2, 5, 10, 20, and 40 μm) for 24 h, and cell viability was measured using CCK-8 (n = 3). Statistical significance was assessed using one-way ANOVA followed by Dunnett’s post-hoc test (AGS: **P < 0.01, ***P < 0.001. MKN-45: ##P < 0.01, ###P < 0.001); (B) AGS and MKN-45 cells were exposed to CA (5 µm) for different times (0, 12, 24, 48, and 72 h), and cell viability was assessed using CCK-8 (n = 3). Statistical significance was assessed using one-way ANOVA followed by Dunnett’s post-hoc test (AGS: **P < 0.01, ***P < 0.001. MKN-45: ##P < 0.01, ###P < 0.001); (C) AGS cells were exposed to CA (20 µm) and different concentrations of cisplatin (0, 5, 15, 25, and 35 µm) for 24 h, and cell viability was assessed using the CCK-8 assay (n = 3). Statistical significance was assessed using multiple t-tests; (D) AGS-CR cells were exposed to 5 µm of CA and 35 µm of cisplatin for 24 h, after which cell death was assessed using the CCK-8 assay (n = 3). Statistical significance was assessed using one-way ANOVA followed by Dunnett’s post-hoc test. AGS-CR cells were treated with 5 µm of CA and 35 µm of cisplatin for 3 days, and then clone formation assay (E) and quantitative analysis (F) were carried out (n = 3). Statistical significance was assessed using one-way ANOVA followed by Dunnett’s post-hoc test. AGS-CR cells were treated with 5 µm of CA and 35 µm of cisplatin for 24 h, after which flow cytometry (G) and quantitative analysis (H) were carried out (n = 3). Statistical significance was assessed using one-way ANOVA followed by Dunnett’s post-hoc test; (I-K) To assess the effect of CA on cisplatin resistance and its safety in vivo, a mouse xenograft model was developed by subcutaneously injecting AGS cells into nude mice. Once tumor volume approached 100 mm3, the mice with tumors were administered either vehicle, cisplatin, or combination treatment with cisplatin and CA, and tumor growth was monitored (I and J) and body weight was measured (K) (n = 3). Statistical significance was assessed using one-way ANOVA followed by Dunnett’s post-hoc test. *p < 0.05, **P < 0.01, ***P < 0.001. CA: Corosolic acid; GC: gastric cancer; CCK-8: cell counting kit-8; ANOVA: analysis of variance; CR: cisplatin-resistant.
Ferroptosis inhibition decreased the effect of CA as a chemosensitizer in GC cells
Given the vital role of ferroptosis in cisplatin resistance[24,34,35], the effect of ferroptosis on cisplatin- or CA-induced GC cell death was next assessed. For this purpose, a ferroptosis-specific inhibitor, Fer-1, was used to treat GC cells in the presence of CA, cisplatin, or both. Although Fer-1 alone did not affect cell viability, Fer-1 alleviated the cytotoxicity of cisplatin and CA to AGS cells [Figure 2A] and MKN-45 cels
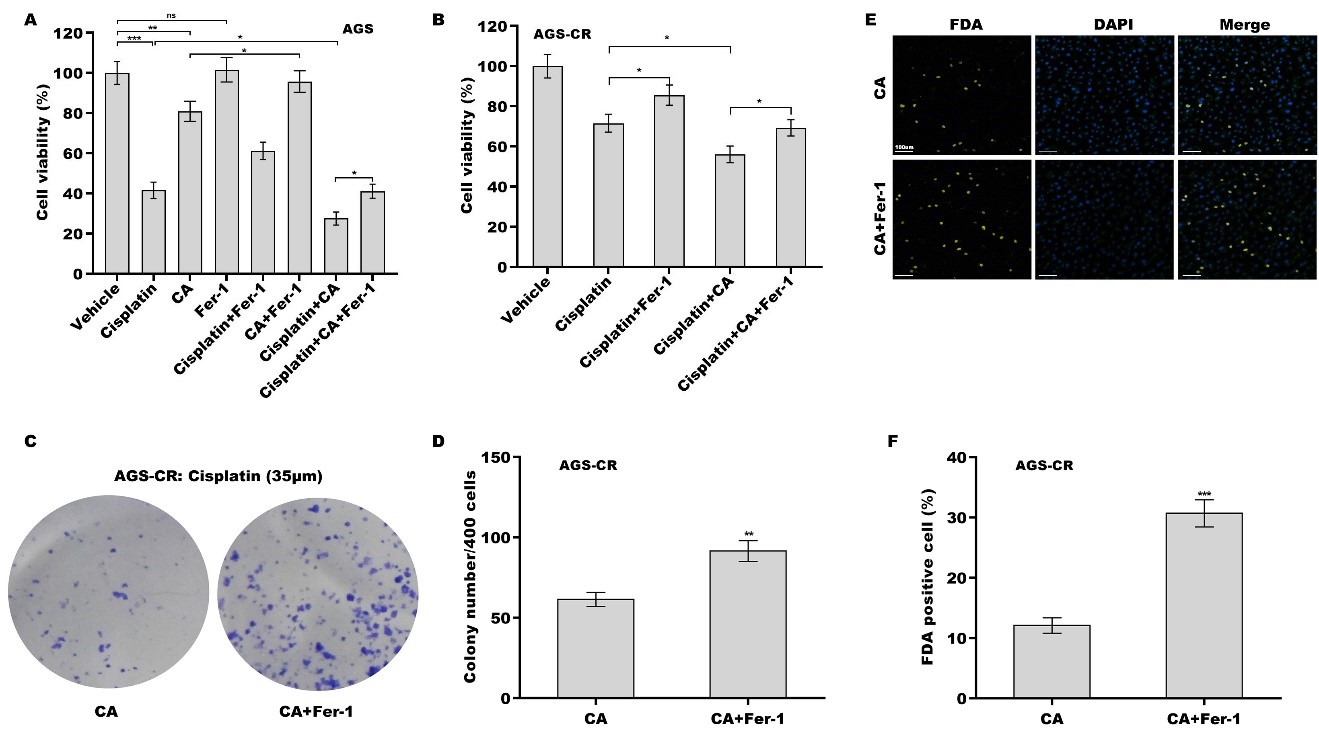
Figure 2. The effect of CA as a chemosensitizer in GC cells was damaged by Fer-1. AGS (A) and AGS-CR (B) cells were exposed to cisplatin (35 µm) and CA (5 µm) or Ferrostatin-1 (Fer-1, 1 μm) for 24 h, and then cell viability was measured using the CCK-8 assay (n = 3). Statistical significance was assessed using one-way ANOVA followed by Tukey’s post-hoc test. AGS-CR cells were exposed to cisplatin (35 µm) and CA (5 µm) in the presence or absence of Fer-1 (1 μm), and then clone formation assay (C) and quantitative analysis (D) were carried out (n = 3). Statistical significance was assessed using Student’s t test. AGS-CR cells were exposed to cisplatin (35 µm) and CA (5 µm) in the presence or absence of Fer-1 (1 μm), and then FDA staining (E) and quantitative analysis (F) were performed (n = 3). Statistical significance was assessed using Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001. CA: Corosolic acid; GC: gastric cancer; CR: cisplatin-resistant; CCK-8: cell counting kit-8; FDA: fluorescein diacetate.
CA triggered GC cell ferroptosis by inhibiting Gpx4 expression
To define the biological effect of CA on GC cell ferroptosis, CA was applied to treat GC cells, and then ferroptosis was evaluated by measuring iron, ROS, MDA, and GSH levels, as previously described[36]. Figure 3A-D reveals that CA treatment resulted in significantly increased levels of iron, ROS, and MDA and decreased levels of GSH in AGS and MKN-45 cells, indicating that CA promoted GC cell ferroptosis. To uncover the molecular mechanism underlying GC cell ferroptosis triggered by CA, the expression of key molecules involved in ferroptosis (Gpx4, Ptgs2, Acsl4, Fth1, Nox1, and Slc7a11)[26,27,30,37] was measured in GC cells following treatment with vehicle or CA. Compared to the vehicle control, CA treatment significantly downregulated Gpx4 mRNA expression while simultaneously upregulating Ptgs2 transcripts [Figure 3E]. At the protein level, CA markedly suppressed Gpx4 expression, while Ptgs2 levels remained unchanged [Figure 3F].
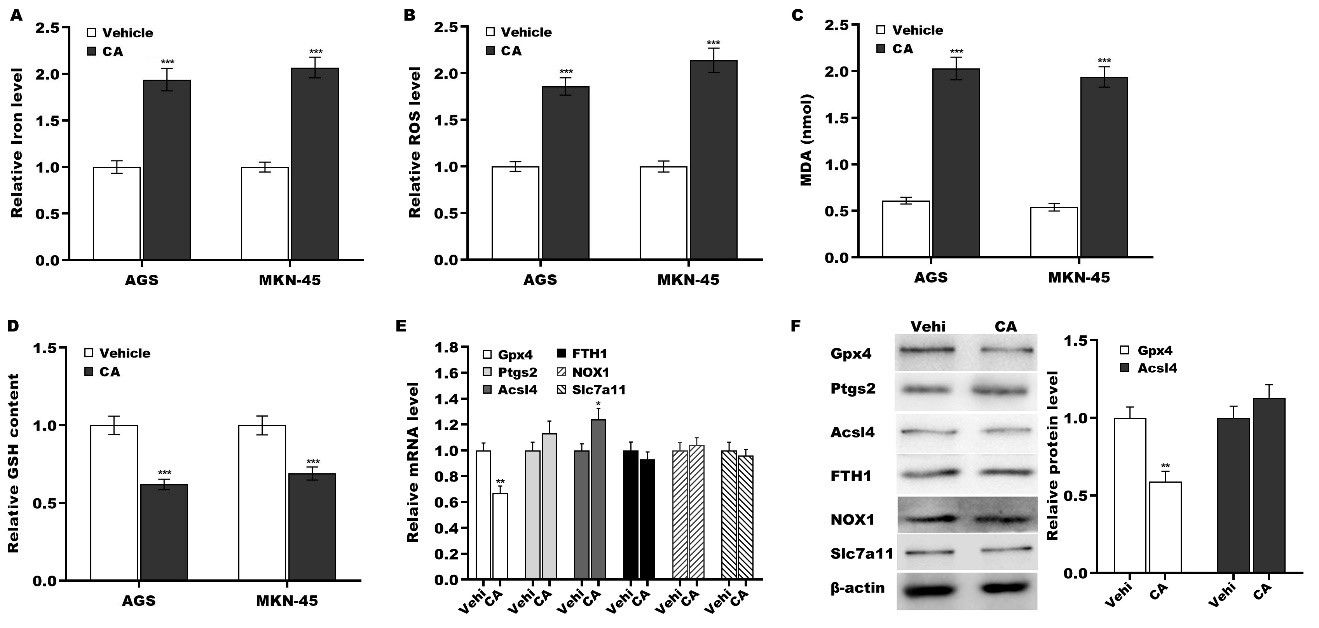
Figure 3. CA triggered GC cell ferroptosis by inhibiting Gpx4 expression. AGS and MKN-45 cells were exposed to 5 µm of CA for 24 h, and the levels of iron (A), ROS (B), MDA (C), and GSH (D) were assessed using a commercial assay kit. AGS cells were treated with 5 µm of CA for 24 h, and the mRNA (E) and protein (F) levels of Gpx4, Ptgs2, Acsl4, Fth1, Nox1, and Slc7a11 were assessed using qRT-PCR and western blot, respectively. All statistical analyses were assessed using Student’s t test (n = 3). **P < 0.01, ***P < 0.001. CA: Corosolic acid; GC: gastric cancer; Gpx4: glutathione peroxidase 4; Ptgs2: prostaglandin-endoperoxide synthase 2; Fth1: ferritin heavy chain 1; Nox1: NADPH oxidase 1; Slc7a11: solute carrier family 7 member 11; qRT-PCR: quantitative reverse transcription polymerase chain reaction.
High Gpx4 expression predicted poor OS and DFS
To define the function of Gpx4 on GC progression, Gpx4 expression was analyzed in the TCGA-STAD database. Figure 4A shows that Gpx4 levels were prominently upregulated in GC tissues compared with normal tissues. Moreover, high Gpx4 expression predicted poor 10-year OS [Figure 4B] and DFS [Figure 4C]. Gpx4 expression was also assayed in 19 GC tissues and matched normal tissues. Figure 4D reveals that Gpx4 expression was also upregulated in most GC tissues compared with matched normal tissues.
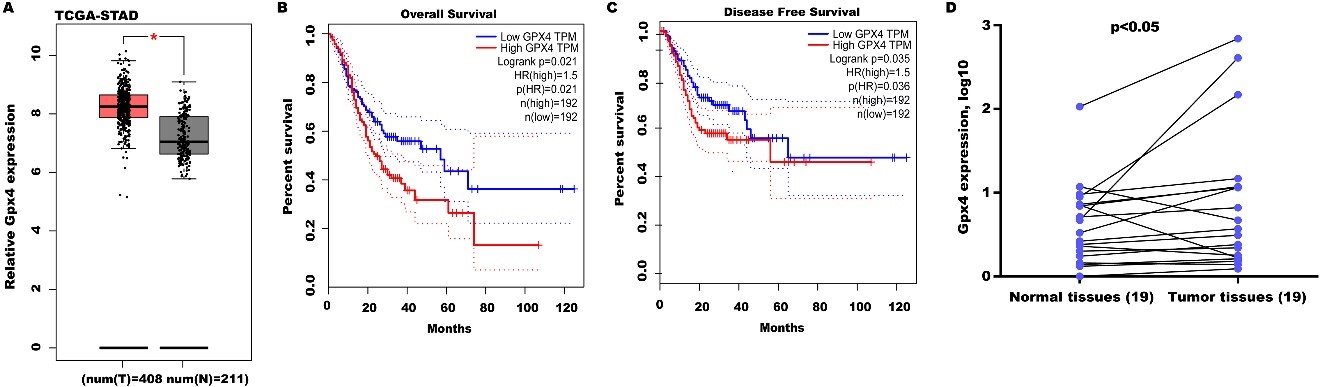
Figure 4. High Gpx4 expression predicted a poor OS and DFS. (A) Gpx4 expression was analyzed in the TCGA-STAD database through the GEPIA tool. STAD tissues (n = 408) were displayed as red and normal tissues (n = 211) were displayed as grey. The GEPIA tool was applied to analyze the correlation of Gpx4 expression with OS (B) and DFS (C) in the TCGA-STAD database; (D) qRT-PCR analysis of Gpx4 mRNA level in 19 pairs of GC tissues and normal tissues. Statistical significance was assessed using Student’s t test (n = 19). *P < 0.05, **P < 0.01. Gpx4: Glutathione peroxidase 4; OS: overall survival; DFS: disease-free survival; TCGA-STAD: the cancer genome atlas-stomach adenocarcinoma; GEPIA: gene expression profiling interactive analysis; qRT-PCR: quantitative reverse transcription polymerase chain reaction.
CA increased cisplatin sensitivity by regulating Gpx4
Finally, we investigated whether CA inhibited Gpx4 expression and thus increased cisplatin sensitivity in GC cells. Figure 5A-C shows that treatment with either cisplatin or CA alone obviously decreased Gpx4 expression at the mRNA and protein levels, and the inhibitory effect was reinforced through combination treatment with cisplatin and CA. Gpx4 expression level in AGS-CR cells was enhanced compared with AGS cells, whereas CA repressed Gpx4 expression in AGS-CR cells [Figure 5D-F], indicating that CA sensitized GC cells to cisplatin, possibly by inhibiting Gpx4 expression. To clarify Gpx4’s involvement in CA-mediated enhancement of cisplatin sensitivity, Gpx4 was overexpressed in AGS-CR cells, followed by evaluation of cell viability before and after its overexpression. As expected, Gpx4 overexpression in AGS-CR cells
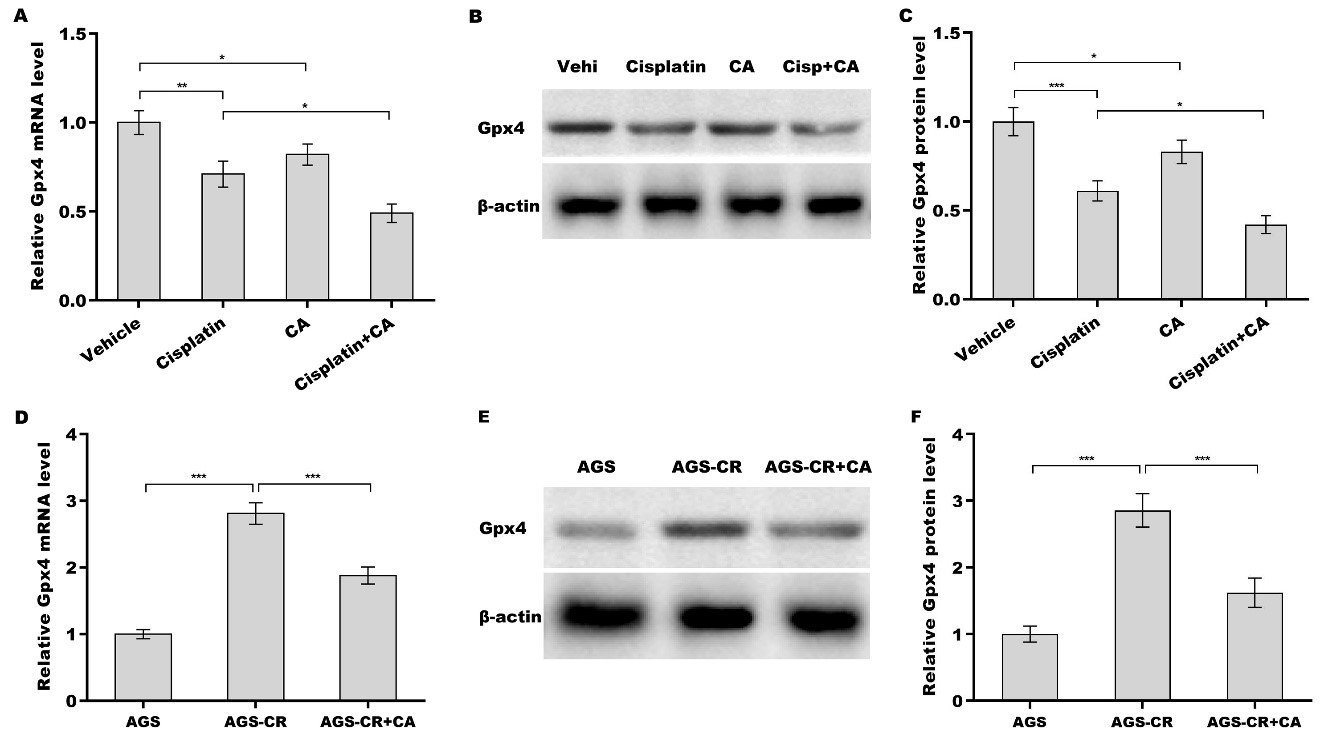
Figure 5. CA decreased Gpx4 expression. After treatment with cisplatin (35 µm) alone, CA (5 µm) alone, or combination treatment with cisplatin (35 µm) and CA (5 µm) in AGS cells, Gpx4 mRNA (A) and protein (B and C) levels were assessed using qRT-PCR and western blot, respectively. qRT-PCR (D) and western blot (E and F) analyses of Gpx4 expression in AGS, AGS-CR, and CA-treated AGS-CR cells. All statistical analyses were assessed using one-way ANOVA followed by Tukey’s post-hoc test (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001. CA: Corosolic acid; Gpx4: glutathione peroxidase 4; qRT-PCR: quantitative reverse transcription polymerase chain reaction; CR: cisplatin-resistant; ANOVA: analysis of variance.
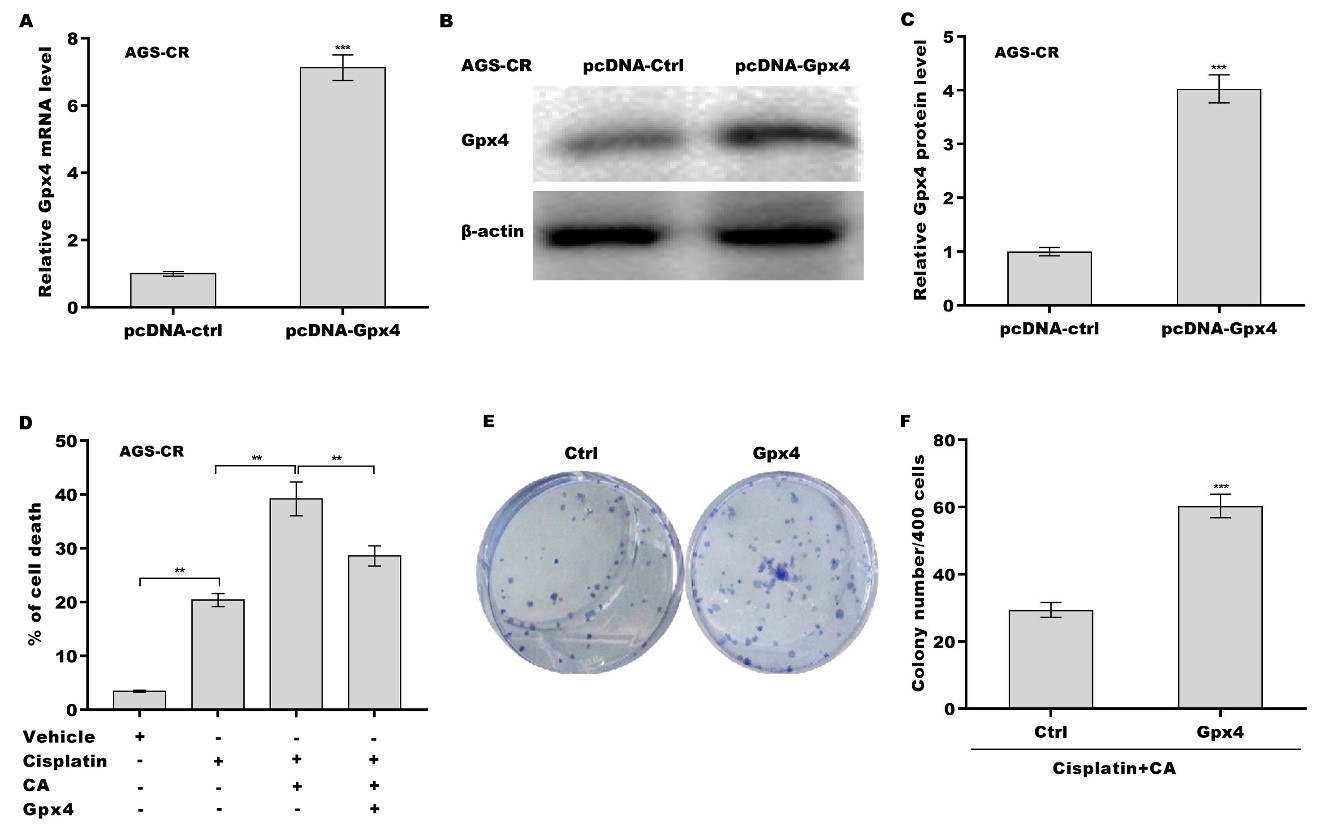
Figure 6. CA increased cisplatin sensitivity by regulating Gpx4. AGS-CR cells were transfected with recombinant plasmids of pcDNA-Gpx4 (1.5 µg) for 48 h, and the Gpx4 mRNA (A) and protein (B and C) levels were assessed using qRT-PCR and western blot, respectively; (D) AGS-CR cells were treated with cisplatin (35 µm), CA (5 µm), and pcDNA-Gpx4 and cell death was assessed using CCK-8; (E and F) AGS-CR cells were treated with cisplatin (35 µm) and CA (5 µm) in the presence or absence of Gpx4 overexpression and clone formation ability was assessed. All statistical analyses, except for panel D (analyzed using one-way ANOVA followed by Tukey’s post-hoc test), were performed using Student’s t-test (n = 3). **P < 0.01, ***P < 0.001. CA: Corosolic acid; Gpx4: glutathione peroxidase 4; qRT-PCR: quantitative reverse transcription polymerase chain reaction; CR: cisplatin-resistant; CCK-8: cell counting kit-8; ANOVA: analysis of variance.
DISCUSSION
GC is typically diagnosed at an advanced stage, which leads to a poor prognosis[3,38]. Cisplatin-based chemotherapy is one of the most broadly applied regimens to treat advanced patients with GC[39,40]. Unfortunately, almost all patients ultimately develop acquired resistance during the course of chemotherapy[41]. The mechanisms underlying cisplatin resistance are gradually being elucidated. For example, Cisplatin induces cytotoxicity by forming Platinum (Pt)-DNA adducts, which interfere with DNA replication and transcription. However, cancer cells may develop resistance by enhancing DNA repair mechanisms, especially nucleotide excision repair[42,43]. Cisplatin enters cells through copper transporters(CTR1), but resistant cells may downregulate CTR1, decreasing drug uptake[44]. An increasing number of studies highlight the potential of combination therapy in cancer treatment. In the study, we demonstrated that, (1) CA sensitized GC cells to cisplatin treatment; (2) Ferroptosis inhibition damaged the effect of CA as a chemosensitizer; (3) CA triggered GC cell ferroptosis by repressing Gpx4 expression; (4) High Gpx4 expression predicted a poor OS and DFS in patients with GC; and (5) CA increased cisplatin sensitivity by regulating Gpx4. These results reveal that CA alleviates cisplatin resistance in GC by regulating Gpx4-dependent ferroptosis.
Plants have been a credible source of medication for thousands of years because of their efficiency and low toxicity in the treatment of diseases[45,46]. Several bioactive monomers in medicinal plants have been identified as effective antineoplastic drugs, such as paclitaxel, vincristine, homoharringtonine, and cytarabine[47,48]. RAC is a traditional Chinese medicine, possessing a significant anti-tumor property. Ethanol extract of RAC inhibits cancer cell proliferation and invasion in many types of tumors[11,12]. CA is a bioactive triterpenoid in RAC and exhibits an effective anti-tumor property in GC, PC, and HCC[18-20]. Cheng et al. demonstrated that CA accelerates GC cell apoptosis by inhibiting nuclear factor kappa B (NF-κB) p65 expression and NF-κB signaling activation[18]. Moreover, CA could increase the chemosensitivity of GC cells to 5-FU by inhibiting the mammalian target of rapamycin (mTOR) and activating adenosine monophosphateactivated protein kinase (AMPK) signaling[21,49]. A recent study revealed that CA promotes HCC cell ferroptosis, a ROS-dependent cell death form, by inhibiting GSH synthesis[25]. CA also promotes oxidative stress in non-small cell lung cancer[50]. Nevertheless, most natural ingredients including CA[51-54] in medicinal plants possess antioxidative activity[55-57]. Perhaps the effects of natural ingredients on regulating oxidative stress and cell viability may vary depending on drug concentration or cell type. It is very meaningful to investigate the role of CA in regulating cisplatin sensitivity and oxidative stress in GC cells.
Ferroptosis is an iron-dependent cell death mechanism characterized by ROS overproduction[22]. Upregulated Gpx4 inhibits ferroptosis in cancer cells to acquire cisplatin resistance[58]. Inhibition of nuclear factor erythroid 2-related factor 2 (Nrf2) upregulates ROS levels, potentiating ferroptosis and enhancing cisplatin sensitivity[59]. Therefore, initiating ferroptosis is emerging as a potential strategy for cancer therapy[60,61]. An increasing number of ferroptosis inducers are being identified and developed. As classical ferroptosis inducers, erastin and RLS3, possess potent anti-cancer capacity in different types of tumors, such as HCC[62], melanoma[63], GC[64], and breast cancer[65]. Moreover, ferroptosis inducers can also overcome resistance to several first-line chemotherapy drugs. For example, RSL3 increases the anti-cancer effectiveness of cisplatin by regulating Gpx4-dependent ferroptosis[66]. Erastin reverses the resistance of head and neck cancer cells to cisplatin by inducing ROS overproduction[67]. Previous studies have demonstrated that CD44 inhibits ferroptosis by stabilizing Slc7a11, maintaining cystine uptake and GSH levels, and suppressing lipid ROS accumulation[68-70]. CD44-positive gastric cancer stem cells (CSCs) are less susceptible to ferroptosis, contributing to their survival and resistance to treatment[71]. Therefore, it is of interest to investigate whether CA regulates CD44 expression to inhibit ferroptosis resistance-mediated chemoresistance.
It is worth noting that ROS plays important roles in cancer cell survival because ROS overproduction is a key factor responsible for apoptosis[72], ferroptosis[73], and pyroptosis[74]. Paradoxically, low ROS levels are essential to maintaining cell and tissue homeostasis and cancer stem cell survival[75,76], which is a major cause of tumor chemotherapy resistance. Therefore, CA increases the therapeutic effect of cisplatin on GC, at least in part, by decreasing Gpx4 expression and resulting in ROS overproduction and ferroptosis.
This study has three major limitations that can be addressed in future research: (1) Given the roles of ROS in multiple cell death modalities (apoptosis, ferroptosis, pyroptosis, autophagy, etc.), it is necessary to investigate whether CA regulates other cell death mechanisms besides ferroptosis; (2) Although current findings have demonstrated that CA treatment significantly downregulates Gpx4 expression at both the mRNA and protein levels, elucidating the molecular mechanism underlying CA-mediated suppression of Gpx4 expression remains of considerable importance; (3) In many experiments, cell viability and apoptosis were assessed 24 h after drug exposure. Expanding the analysis to additional time points (e.g., 48 or 72 h) could further enhance the reliability and persuasiveness of the findings.
Conclusion
CA increases cisplatin sensitivity in GC by regulating Gpx4-dependent ferroptosis, indicating its therapeutic potential in augmenting cisplatin efficacy and establishing Gpx4 as a promising molecular target for GC treatment.
DECLARATIONS
Authors’ contributions
Performed the research study and collected the samples and data: Lin L, Wang J, Sheng S, Shen Y, Liu X, Yong Li
Analyzed the data: Xu R
Designed the research study: Lin L, Xu R
Wrote the paper: Lin L, Wang J, Xu R, Li Y
Prepared all the tables: Lin L, Xu R
All authors contributed significantly to this work.
All authors have read and approved the final manuscript to be published.
Availability of data and materials
The GEPIA tool (http://gepia.cancer-pku.cn/) was applied to analyze the correlation of Gpx4 expression with OS and DFS in the TCGA-STAD database. The data supporting the results of this study are available from the corresponding author (Yong Li) upon reasonable request.
Financial support and sponsorship
This work was supported by a grant from Traditional Chinese Medicine Science and Technology Development Project of Shanghai Medical Innovation & Development Foundation (WL-HBQN-2021008K), Traditional Chinese Medicine Science and Technology Development Project of Shanghai Medical Innovation & Development Foundation (WL-HBQN-2021005K), Scientific Research Project of Traditional Chinese Medicine of Shanghai Municipal Health Commission (No.2022QN051), Scientific Research Project of Traditional Chinese Medicine of Shanghai Municipal Health Commission (No.2022QN083), and the National Natural Science Foundation of China (No. 82304932).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethics approval and consent to participate
The work was approved by the Animal Ethics Committee of Shanghai Municipal Hospital of Traditional Chinese Medicine (No. 2023112) and conducted in compliance with the ARRIVE guidelines.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
Supplementary Materials
REFERENCES
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209-49.
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424.
3. Jiang Y, Zhang Q, Hu Y, et al. ImmunoScore signature: a prognostic and predictive tool in gastric cancer. Ann Surg. 2018;267:504-13.
4. Pereira C, Park JH, Campelos S, et al. Comparison of east-Asia and west-Europe cohorts explains disparities in survival outcomes and highlights predictive biomarkers of early gastric cancer aggressiveness. Int J Cancer. 2022;150:868-80.
5. Jin X, Liu Z, Yang D, Yin K, Chang X. Recent progress and future perspectives of immunotherapy in advanced gastric cancer. Front Immunol. 2022;13:948647.
6. Basu A, Krishnamurthy S. Cellular responses to cisplatin-induced DNA damage. J Nucleic Acids. 2010;2010:201367.
7. Mora-Lagos B, Cartas-Espinel I, Riquelme I, et al. Functional and transcriptomic characterization of cisplatin-resistant AGS and MKN-28 gastric cancer cell lines. PLoS One. 2020;15:e0228331.
8. Huo J, Qin F, Cai X, et al. Chinese medicine formula “Weikang Keli” induces autophagic cell death on human gastric cancer cell line SGC-7901. Phytomedicine. 2013;20:159-65.
9. Huang J, Jiang T, Kang J, et al. Synergistic effect of Huangqin decoction combined treatment with radix Actinidiae chinensis on DSS and AOM-induced colorectal cancer. Front Pharmacol. 2022;13:933070.
10. Lv J, Wang L, Shen H, Wang X. Regulatory roles of OASL in lung cancer cell sensitivity to Actinidia chinensis Planch root extract (acRoots). Cell Biol Toxicol. 2018;34:207-18.
11. Liu B, Zhang L. Radix Actinidia chinensis suppresses renal cell carcinoma progression: network pharmacology prediction and in vivo experimental validation. Anal Cell Pathol (Amst). 2022;2022:3584445.
12. Hu W, Wu C, Yuan C, Chen M, Jin C, Zheng C. Ethanol extracted from radix of Actinidia chinensis inhibits human colon tumor through inhibiting notch-signaling pathway. J Cancer. 2021;12:622-9.
13. Xu QF, Peng HP, Lu XR, Hu Y, Xu ZH, Xu JK. Oleanolic acid regulates the Treg/Th17 imbalance in gastric cancer by targeting IL-6 with miR-98-5p. Cytokine. 2021;148:155656.
14. Lu X, Li Y, Yang W, et al. Inhibition of NF-κB is required for oleanolic acid to downregulate PD-L1 by promoting DNA demethylation in gastric cancer cells. J Biochem Mol Toxicol. 2021;35:e22621.
15. Akuetteh PDP, Huang H, Wu S, et al. Synthetic oleanane triterpenoid derivative CDDO-Me disrupts cellular bioenergetics to suppress pancreatic ductal adenocarcinoma via targeting SLC1A5. J Biochem Mol Toxicol. 2022;36:e23192.
16. Rogers LJ, John T, Park J, et al. Growth inhibition and apoptosis of human multiple myeloma cells induced by 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid derivatives. Anticancer Drugs. 2020;31:806-18.
17. Zhou Z, Dong Y, Li N, et al. An oleanolic acid derivative, K73-03, inhibits pancreatic cancer cells proliferation in vitro and in vivo via blocking EGFR/Akt pathway. Cell Biol Int. 2022;46:1801-13.
18. Cheng QL, Li HL, Li YC, Liu ZW, Guo XH, Cheng YJ. CRA (Crosolic Acid) isolated from Actinidia valvata Dunn. Radix induces apoptosis of human gastric cancer cell line BGC823 in vitro via down-regulation of the NF-κB pathway. Food Chem Toxicol. 2017;105:475-85.
19. Yang J, Wu R, Li W, et al. The triterpenoid corosolic acid blocks transformation and epigenetically reactivates Nrf2 in TRAMP-C1 prostate cells. Mol Carcinog. 2018;57:512-21.
20. Jia M, Xiong Y, Li M, Mao Q. Corosolic acid inhibits cancer progress through inactivating YAP in hepatocellular carcinoma. Oncol Res. 2020;28:371-83.
21. Park JB, Lee JS, Lee MS, Cha EY, Kim S, Sul JY. Corosolic acid reduces 5‑FU chemoresistance in human gastric cancer cells by activating AMPK. Mol Med Rep. 2018;18:2880-8.
22. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72.
23. Xiao J, Zheng L, Liu J. Comprehensive analysis of the aberrance and functional significance of ferroptosis in gastric cancer. Front Pharmacol. 2022;13:919490.
24. Wang Y, Zheng L, Shang W, et al. Wnt/beta-catenin signaling confers ferroptosis resistance by targeting Gpx4 in gastric cancer. Cell Death Differ. 2022;29:2190-202.
25. Peng Y, Li N, Tang F, et al. Corosolic acid sensitizes ferroptosis by upregulating HERPUD1 in liver cancer cells. Cell Death Discov. 2022;8:376.
26. Sun LL, Linghu DL, Hung MC. Ferroptosis: a promising target for cancer immunotherapy. Am J Cancer Res. 2021;11:5856-63.
27. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91-8.
28. Miyauchi A, Watanabe C, Yamada N, et al. Apomorphine is a potent inhibitor of ferroptosis independent of dopaminergic receptors. Sci Rep. 2024;14:4820.
29. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107-25.
30. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by Gpx4. Cell. 2014;156:317-31.
31. Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455-8.
32. Nishizawa H, Matsumoto M, Shindo T, et al. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH1. J Biol Chem. 2020;295:69-82.
33. Winer J, Jung CK, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem. 1999;270:41-9.
34. Zhang H, Deng T, Liu R, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer. 2020;19:43.
35. Fu D, Wang C, Yu L, Yu R. Induction of ferroptosis by ATF3 elevation alleviates cisplatin resistance in gastric cancer by restraining Nrf2/Keap1/xCT signaling. Cell Mol Biol Lett. 2021;26:26.
36. Nguyen KT, Mun SH, Yang J, et al. The MARCHF6 E3 ubiquitin ligase acts as an NADPH sensor for the regulation of ferroptosis. Nat Cell Biol. 2022;24:1239-51.
37. Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369-79.
38. Allemani C, Weir HK, Carreira H, et al; CONCORD Working Group. Global surveillance of cancer survival 1995-2009: analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet. 2015;385:977-1010.
39. Brown A, Kumar S, Tchounwou PB. Cisplatin-based chemotherapy of human cancers. J Cancer Sci Ther. 2019;11:97.
40. Kim HK, Choi IJ, Kim HS, et al. DNA microarray analysis of the correlation between gene expression patterns and acquired resistance to 5-FU/cisplatin in gastric cancer. Biochem Biophys Res Commun. 2004;316:781-9.
41. Liu S, Jiang Z, Xiao P, et al. Hsa_circ_0005576 promotes osimertinib resistance through the miR-512-5p/IGF1R axis in lung adenocarcinoma cells. Cancer Sci. 2022;113:79-90.
42. Fu R, Zhao B, Chen M, et al. Moving beyond cisplatin resistance: mechanisms, challenges, and prospects for overcoming recurrence in clinical cancer therapy. Med Oncol. 2023;41:9.
43. Li F, Sun H, Ren J, et al. A nuclease-mimetic platinum nanozyme induces concurrent DNA platination and oxidative cleavage to overcome cancer drug resistance. Nat Commun. 2022;13:7361.
44. Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869-83.
45. Taiwo BJ, Fatokun AA, Olubiyi OO, Bamigboye-Taiwo OT, van Heerden FR, Wright CW. Identification of compounds with cytotoxic activity from the leaf of the Nigerian medicinal plant, Anacardium occidentale L. (Anacardiaceae). Bioorg Med Chem. 2017;25:2327-35.
46. Li Y, Wang J, Li L, et al. Natural products of pentacyclic triterpenoids: from discovery to heterologous biosynthesis. Nat Prod Rep. 2023;40:1303-53.
47. Gu Z, Lin S, Yan W, et al. Enhanced water solubility and anti-tumor activity of oleanolic acid through chemical structure modification. Int J Mol Sci. 2022;23:13291.
48. Ye M, Xiu LJ, Ji QQ, et al. Research progress in targeted therapies for gastric cancer. Int J Clin Pharmacol Ther. 2022;60:509-14.
49. Lee HS, Park JB, Lee MS, Cha EY, Kim JY, Sul JY. Corosolic acid enhances 5-fluorouracil-induced apoptosis against SNU-620 human gastric carcinoma cells by inhibition of mammalian target of rapamycin. Mol Med Rep. 2015;12:4782-8.
50. Jin M, Wu Y, Lou Y, et al. Corosolic acid reduces A549 and PC9 cell proliferation, invasion, and chemoresistance in NSCLC via inducing mitochondrial and liposomal oxidative stress. Biomed Pharmacother. 2021;144:112313.
51. Zhang JX, Feng WJ, Liu GC, et al. Corosolic acid attenuates hepatic lipid accumulation and inflammatory response via AMPK/SREBPs and NF-κB/MAPK signaling pathways. Am J Chin Med. 2020;48:579-95.
52. Peng M, Qiang L, Xu Y, Li C, Li T, Wang J. Inhibition of JNK and activation of the AMPK-Nrf2 axis by corosolic acid suppress osteolysis and oxidative stress. Nitric Oxide. 2019;82:12-24.
53. Woo SM, Seo SU, Min KJ, et al. Corosolic acid induces non-apoptotic cell death through generation of lipid reactive oxygen species production in human renal carcinoma caki cells. Int J Mol Sci. 2018;19:1309.
54. Li Y, Zhou ZH, Chen MH, et al. Inhibition of mitochondrial fission and Nox2 expression prevent NLRP3 inflammasome activation in the endothelium: the role of corosolic acid action in the amelioration of endothelial dysfunction. Antioxid Redox Signal. 2016;24:893-908.
55. Bjørklund G, Shanaida M, Lysiuk R, et al. Natural compounds and products from an anti-aging perspective. Molecules. 2022;27:7084.
56. Carsono N, Tumilaar SG, Kurnia D, Latipudin D, Satari MH. A review of bioactive compounds and antioxidant activity properties of piper species. Molecules. 2022;27:6774.
57. Cheng G, Ma T, Deng Z, et al. Plant-derived melatonin from food: a gift of nature. Food Funct. 2021;12:2829-49.
58. Zhang X, Gu G, Li X, Zhang C. Lidocaine alleviates cisplatin resistance and inhibits migration of MGC-803/DDP cells through decreasing miR-10b. Cell Cycle. 2020;19:2530-7.
59. Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254-62.
60. Luo Y, Bai XY, Zhang L, et al. Ferroptosis in cancer therapy: mechanisms, small molecule inducers, and novel approaches. Drug Des Devel Ther. 2024;18:2485-529.
61. Ma W, Hu N, Xu W, Zhao L, Tian C, Kamei KI. Ferroptosis inducers: a new frontier in cancer therapy. Bioorg Chem. 2024;146:107331.
62. Nie J, Lin B, Zhou M, Wu L, Zheng T. Role of ferroptosis in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2018;144:2329-37.
63. Yang Y, Luo M, Zhang K, et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat Commun. 2020;11:433.
64. Wang J, Jia Q, Jiang S, Lu W, Ning H. POU6F1 promotes ferroptosis by increasing lncRNA-CASC2 transcription to regulate SOCS2/SLC7A11 signaling in gastric cancer. Cell Biol Toxicol. 2024;40:3.
65. Desterke C, Xiang Y, Elhage R, Duruel C, Chang Y, Hamaï A. Ferroptosis inducers upregulate PD-L1 in recurrent triple-negative breast cancer. Cancers (Basel). 2023;16:155.
66. Zhang X, Sui S, Wang L, et al. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J Cell Physiol. 2020;235:3425-37.
67. Roh JL, Kim EH, Jang HJ, Park JY, Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016;381:96-103.
68. Ando T, Yamasaki J, Saya H, Nagano O. CD44: a key regulator of iron metabolism, redox balance, and therapeutic resistance in cancer stem cells. Stem Cells. 2025:43.
69. Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19:387-400.
70. Jogo T, Oki E, Nakanishi R, et al. Expression of CD44 variant 9 induces chemoresistance of gastric cancer by controlling intracellular reactive oxygen spices accumulation. Gastric Cancer. 2021;24:1089-99.
71. Ni Z, Nie X, Zhang H, et al. Atranorin driven by nano materials SPION lead to ferroptosis of gastric cancer stem cells by weakening the mRNA 5-hydroxymethylcytidine modification of the Xc-/Gpx4 axis and its expression. Int J Med Sci. 2022;19:1680-94.
72. Zaidieh T, Smith JR, Ball KE, An Q. ROS as a novel indicator to predict anticancer drug efficacy. BMC Cancer. 2019;19:1224.
73. Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10:822.
74. Wu J, Wang L, Xu J. The role of pyroptosis in modulating the tumor immune microenvironment. Biomark Res. 2022;10:45.
75. Choi HJ, Jhe YL, Kim J, et al. FoxM1-dependent and fatty acid oxidation-mediated ROS modulation is a cell-intrinsic drug resistance mechanism in cancer stem-like cells. Redox Biol. 2020;36:101589.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].