


An activated CD8+ T cell-based signature stratifies prognosis and suggests differential sensitivity to immune checkpoint inhibitors in hepatocellular carcinoma
 0
0 Abstract
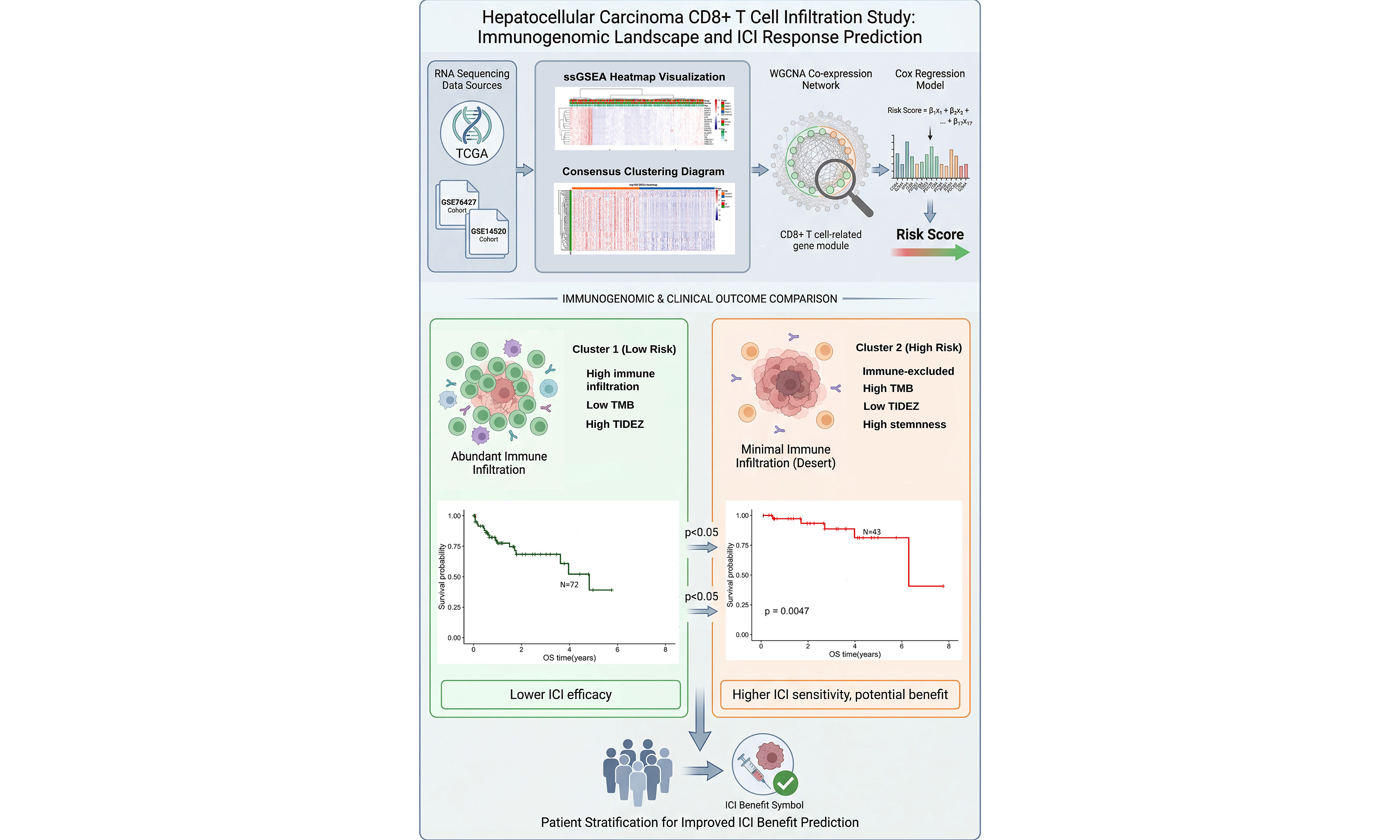
Aim: Hepatocellular carcinoma (HCC) remains a leading cause of cancer-related mortality, with heterogeneous responses to immune checkpoint inhibitors (ICIs). Activated CD8+ T cell plays pivotal role in antitumor immunity, yet their systematic integration into a prognostic and predictive molecular framework is lacking.
Methods: RNA-seq data from 363 HCC patients in The Cancer Genome Atlas (TCGA) and validation cohorts (GSE76427, GSE14520) were analyzed by single-sample gene set enrichment analysis (ssGSEA). We quantified 28 tumor-infiltrating immune cell subsets and performed consensus clustering based on activated CD8+ T cell infiltration. Weighted gene co-expression network analysis (WGCNA) identified a CD8+ T cell associated gene module. Cox regression analysis selected 17 hub genes, from which an immune infiltration-based prognostic model was constructed and externally validated. The two risk groups were compared for prognosis, molecular features, and immunotherapy sensitivity using immune, stromal, stemness, and tumor immune dysfunction and exclusion (TIDE) scores.
Results: We determined the proportion of activated CD8+ T lymphocytes associated with the coexpression network. Cluster 1 and low-risk groups exhibited higher immune infiltration and better prognosis (P < 0.05) than Cluster 2 and high-risk groups. Indicators of immunotherapy response - including lower mutation frequency, copy-number variations (CNVs), and stemness score, along with higher immune, stromal, and TIDE scores - suggested reduced ICI therapy efficacy in Cluster 1/low-risk groups, whereas the opposite pattern was observed in Cluster 2/high-risk groups.
Conclusion: We established a robust prognostic classifier based on activated CD8+ T cell-related genes. This model may aid in identifying HCC patients with poor prognosis who are likely to benefit from immune checkpoint inhibitor therapy.
Keywords
INTRODUCTION
Liver cancer ranks as the third leading cause of cancer-related mortality worldwide[1]. Hepatocellular carcinoma (HCC) accounts for approximately 90% of primary liver cancers. While surgery remains the primary curative option, many patients are diagnosed at advanced stages due to the insidious onset of HCC, thereby losing eligibility for resection[2].
Recent progress in immune checkpoint inhibition has opened new therapeutic possibilities for advanced HCC[3]. Activation of the PD-1 pathway suppresses T-cell activity and facilitates tumor immune escape[4]. Notably, activated CD8+ T cells - key effectors of antitumor immunity - are strongly associated with prognosis and response to immunotherapy[5]. These cells prominently express programmed cell death protein 1 (PD-1), and ICIs work by blocking PD-1/ligand interactions, thereby restoring CD8+ T cell-mediated tumor killing[6,7].
Despite these successes, many patients exhibit primary or acquired resistance to PD-1 blockade[8]. Mechanisms include Wingless/Integrated Beta-catenin (WNT/β-catenin) pathway activation leading to CD8+ T cell exclusion[9], altered antigen presentation, tumor mutational landscape, and dynamic shifts in the immune microenvironment[10]. To optimize ICI therapy, recent studies have explored immune biomarkers in HCC. For instance, a four-gene inflammatory signature (CD274, CD8A, LAG3, STAT1) was associated with improved response and overall survival in nivolumab-treated patients[11]. Further biomarker research may clarify key drivers of antitumor immunity and help stratify HCC patients for immunotherapy.
We hypothesize that molecular subtyping based on activated CD8+ T cell-related genes holds significant value for prognostic prediction and understanding heterogeneous ICI responses. Weighted gene co-expression network analysis (WGCNA) provides a powerful methodology for detecting co-expressed gene modules and hub genes associated with cancer phenotypes[12-14]. In this study, we applied WGCNA combined with unsupervised clustering and risk modeling to identify modules and hub genes associated with activated CD8+ T cell infiltration in HCC. We further validated their prognostic and immune relevance, aiming to establish a biomarker framework for guiding immunotherapy in HCC. Despite continuous advances in systemic therapy, early diagnosis remains key to improving the prognosis of HCC. The delays in follow-up and reductions in treatment caused by the COVID-19 pandemic have further highlighted the importance of maintaining standardized monitoring.
METHODS
Data acquisition
Gene expression profiles and corresponding clinical data for HCC patients were obtained from The Cancer Genome Atlas (TCGA)-LIHC cohort (363 samples), which included 363 tumor samples, accessed through the UCSC Xena platform. Two additional microarray datasets (GSE76427 and GSE14520, comprising 115 and 220 HCC samples, respectively) were downloaded from the Gene Expression Omnibus (GEO) database. Comprehensive etiology data (e.g., viral status, metabolic disease) were not obtained in all cohorts and thus were not included as a primary stratifying variable in this analysis. A set of 3,187 immune-related genes (IRGs) was compiled from the InnateDB and ImmPort public repositories for subsequent analysis.
Estimation of tumor-infiltrating immune cells
The relative enrichment levels of 28 tumor-infiltrating immune cell (TIIC) subtypes were quantified via single-sample gene set enrichment analysis (ssGSEA) using the “GSVA” R package. This method generates a sample-wise enrichment score for each immune cell type based on the expression of a predefined gene signature, reflecting the relative abundance and activity of that cell population within the tumor microenvironment, rather than absolute cell counts. The gene signatures for the 28 immune cell types, including activated CD8⁺ T cells, were obtained from the comprehensive immune cell signature collection published by Charoentong et al.[15]. Specifically, the signature for “activated CD8⁺ T cells” was defined by genes associated with CD8⁺ T cell effector function and activation status, such as those defining effector and cytotoxic functions (e.g., GZMA, GZMB, PRF1, IFNG, CD8A, CD8B) as described in Charoentong et al.[15].
Co-expression network construction and hub module identification
A weighted gene co-expression network was built from the expression profiles of 3,187 IRGs using the WGCNA framework[12]. The module most strongly correlated with activated CD8+ T cell infiltration (|correlation| > 0.8, module significance > 0.6) was designated the hub module. Genes within this module were subjected to Cox regression analysis; those with P < 0.05 were defined as hub genes. Functional enrichment of hub genes was conducted through Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses utilizing the “clusterProfiler” and “gene set enrichment analysis (GSEA)” R packages.
Unsupervised clustering
HCC samples were clustered based on hub gene expression using the “ConsensusClusterPlus” R package. Clustering was performed with the partitioning around medoids (PAM) algorithm and Canberra distance, and stability was assessed over 100 iterations.
Risk score model construction
Principal component analysis (PCA) was used to analyze the hub genes. The first principal component was extracted as a PCA-based risk score for each patient, calculated as: Risk score = Σ (Expression of gene i × Coefficient i), where the coefficient represents the contribution of each hub gene to activated CD8+ T cell infiltration.
Copy number variation analysis
Recurrent copy number alterations were identified from SNP6 array data using the GISTIC 2.0 algorithm on the GenePattern platform (GenePattern module: GISTIC_2). Parameters were set as follows: significance threshold q ≤ 0.05, confidence level 0.90. Genomic segments with q < 0.05 were considered significantly recurrent.
Somatic mutation analysis
To identify genes with a significant burden of somatic mutations, we used the MutSigCV algorithm (version 2.0) as implemented on the GenePattern platform. MutSigCV compares the observed mutation rate of each gene to an expected background model, accounting for gene-specific mutational covariates such as expression and replication timing. Genes with a false discovery rate (FDR) q-value < 0.05 were considered significantly mutated.
Immune, stromal, and stemness scoring
Immune and stromal scores were calculated with the “ESTIMATE” R package to respectively quantify the relative presence of immune and stromal elements within the tumor microenvironment. A transcriptome-based stemness index, generated through a one-class logistic regression (OCLR) machine learning model, was used to evaluate tumor stemness.
Tumor immune dysfunction and exclusion (TIDE) prediction
The TIDE computational framework was applied to infer mechanisms of immune evasion and indicates potential responsiveness to immunotherapy. Elevated TIDE scores correlate with a higher probability of resistance to immune checkpoint inhibitors (ICIs).
Statistical analysis
Intergroup differences were compared using the Wilcoxon test. Kaplan-Meier survival analysis with log-rank testing was employed to assess prognostic disparities. The “pROC” package was utilized to plot receiver operating characteristic (ROC) curves and measure predictive accuracy. Somatic mutation profiles were visualized with the “maftools” package. All statistical computations were performed in R version 4.1.0, with a two-sided P-value < 0.05 considered statistically significant. For comparisons involving multiple groups, including the 28 TIIC subsets, P-values reported are nominal. Readers are advised to interpret findings with caution, noting that results with P < 0.01 represent more stringent, and thus more robust, differences.
RESULTS
Expression data of HCC
RNA expression data and clinical data of 363 HCC patients were obtained from TCGA-HCC cohort, via the UCSC Xena platform (https://xenabrowser.net/datapages/). GSE76427 containing 115 HCC samples, and GSE14520 containing 220 HCC samples were obtained from GEO database. Clinical information for each dataset is summarized in Supplementary Table 1. Expression profiles of 3,187 IRGs were obtained from InnateDB database and ImmPort database.
Evaluation of immune cell network and prognosis based on activated CD8+ T cell infiltration
Given the central role of activated CD8+ T cell in antitumor immunity, we first evaluated their infiltration level across HCC sample. Based on the estimated proportions of 28 TIIC subtypes in the TCGA cohort, we performed consensus clustering and univariate Cox proportional hazards analysis. An immune cell regulatory network illustrated the interactions among these cells and their correlation with clinical prognosis, revealing significant Spearman correlations [Figure 1A]. This network highlighted activated CD8+ T cells as a hub population with strong prognostic relevance.
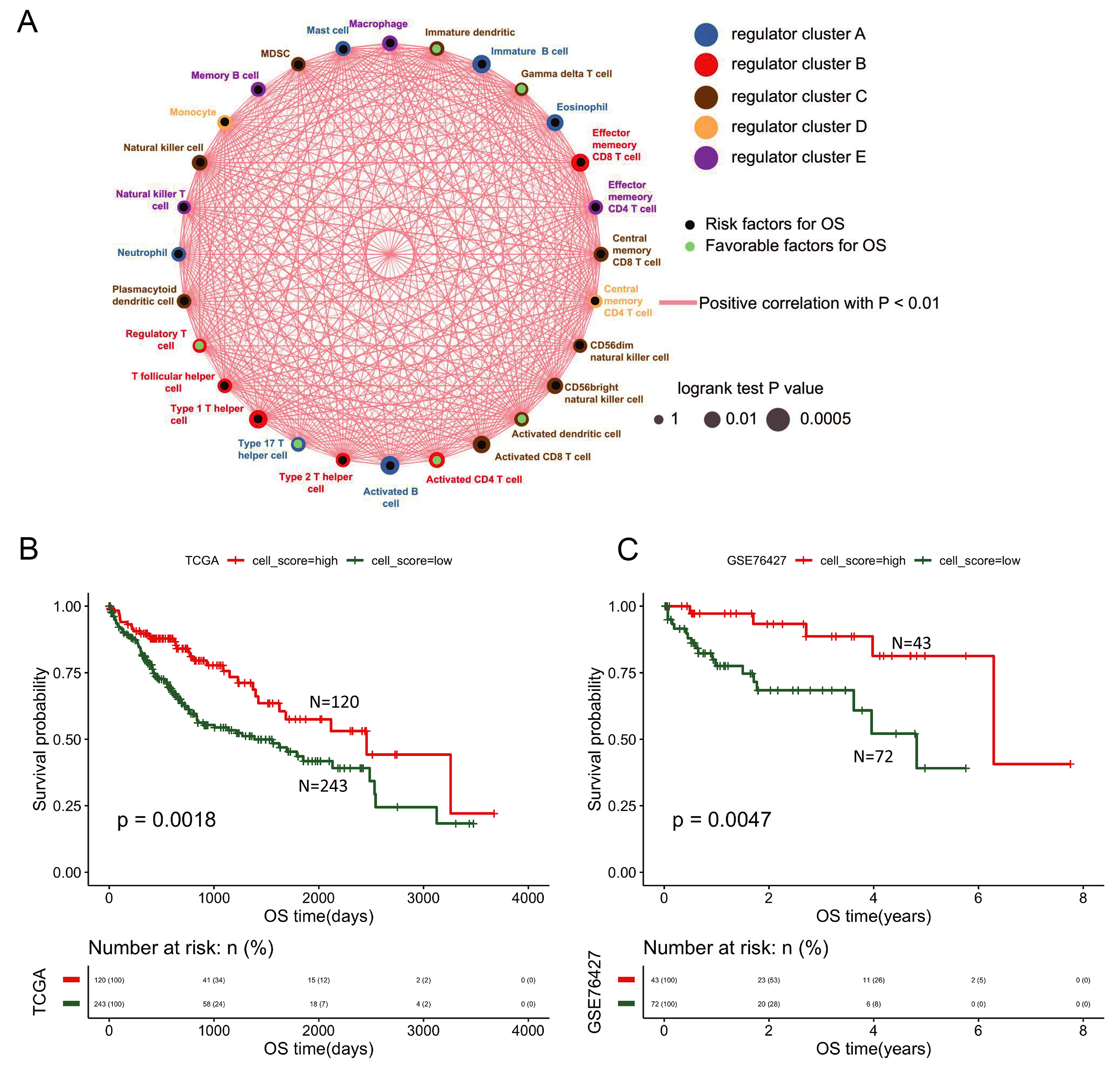
Figure 1. Immune cell network and activated CD8+ T cell infiltration group prognosis. (A) Network depicting interactions among 28 tumor-infiltrating immune cell types. Circle size represents the magnitude of association with OS; green and black nodes indicate favorable and unfavorable prognostic factors, respectively. Connecting lines denote interactions, with red lines representing positive correlations; (B) Prognostic survival curve of activated CD8+ T cell infiltration groups in the TCGA database; (C) Prognostic survival curve of activated CD8+ T cell infiltration groups in the GSE76427 database. OS: Overall survival; TCGA: the Cancer Genome Atlas.
Samples were stratified into high- (n = 120) and low-infiltration (n = 243) groups based on activated CD8+ T cell proportion using the “surv_cutpoint” function (“survminer” package). Kaplan-Meier analysis with log-rank test showed that the high-infiltration group had significantly better prognosis in both TCGA and GSE76427 datasets (P < 0.05, Figure 1B and C), underscoring the prognostic value of CD8+ T cell infiltration in HCC.
WGCNA identifies modules associated with activated CD8+ T cell infiltration
To systematically identify gene modules co-expressed with activated CD8+ T cell infiltration, we performed WGCNA. Among 3,187 IRGs, 2308 were present in the TCGA dataset and used to construct a weighted gene co-expression network via the “WGCNA” package. After outlier removal (cutHeight = 65, minSize = 10; Supplementary Figure 1), 352 samples were retained. A soft-threshold power β = 3 was selected to achieve a scale-free topology (R2 > 0.85). Genes were clustered by average-linkage hierarchical clustering based on topological overlap, and modules containing fewer than 30 genes were merged, resulting in seven distinct modules (Figure 2A; gene lists in Supplementary Table 2).
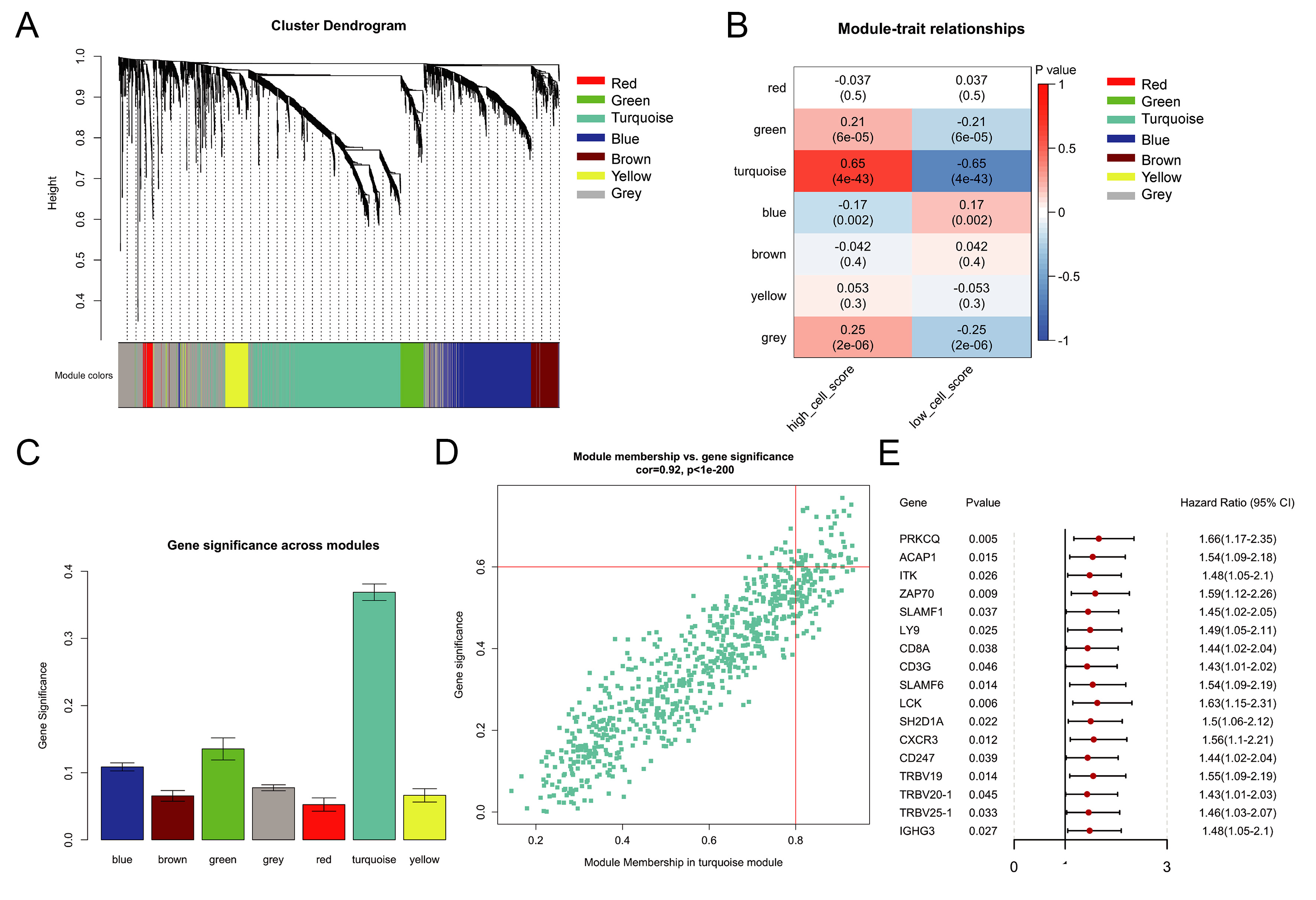
Figure 2. WGCNA co-expression module. (A) Gene dendrogram and module colors; (B) Heatmap of module-trait correlations. Rows represent MEs, columns represent clinical traits. Color intensity reflects Pearson correlation coefficients (red: positive; blue: negative); each cell shows the correlation coefficient and corresponding P-value; (C) GS value for every module; (D) MM value and GS value of turquoise module genes; (E) Forest plot displaying 17 hub genes significantly associated with prognosis (P < 0.05) identified by univariate Cox regression. MEs: Module eigengenes; GS: gene significance; MM: Module Membership.
We computed the Pearson correlation coefficients between each module eigengene (ME) and the phenotypic traits of the samples, with a higher coefficient indicating stronger module relevance. Each row corresponds to an ME, and each column represents a trait. Color intensity from red to blue reflects correlation strength (high to low), and each cell displays both the correlation coefficient and its associated P-value [Figure 2B]. We further determined the gene significance (GS) value for each module. A larger GS value signifies a closer association between the module and the activated CD8⁺ T cell infiltration group [Figure 2C]. According to the results of the relationship between the phenotype and module assessed by the two methods, the most relevant module for activated CD8+ T cell immune infiltration was the turquoise module. Within this module, 51 hub genes were selected using the following criteria: module membership > 0.8, gene significance > 0.6, and P < 0.05 [Figure 2D]. Subsequent univariate Cox regression further identified 17 of these genes as significantly associated with prognosis (P < 0.05; Figure 2E). These 17 hub genes formed the basis for subsequent clustering and risk modeling.
Functional annotation of the 17-gene prognostic signature
GO and KEGG enrichment analyses performed with the “clusterProfiler” package demonstrated that the 17 hub genes were significantly enriched in immune-associated pathways. These included “T cell receptor signaling,” “programmed death-ligand 1 (PD-L1)/PD-1 checkpoint pathway in cancer,” and “Th17 cell differentiation” [Supplementary Figure 2], reinforcing their functional relevance in T-cell activation processes. This enrichment supports their biological relevance in T-cell activation and immune checkpoint regulation, aligning with our focus on immunotherapy response.
Unsupervised clustering based on hub genes
The heatmap was analyzed with Z‐Score using the log2 [fragments per kilobase of transcript per million mapped reads (FPKM)] expression of the 17 hub genes. The heat scale shows the Z-score-normalized hub gene expression [Figure 3A]. Unsupervised cluster analysis was scaled with the expression profiles of the 17 hub genes. A total of 363 patients in the TCGA dataset were divided into two different subtypes, which were named Cluster 1 and Cluster 2 [Figure 3B]. There was a significant difference in prognosis between those two clusters [Figure 3C]. The average silhouette width is a popular cluster validation index used to estimate the number of clusters[16]. The average silhouette width (0.36) confirmed optimal clustering [Figure 3D]. Differential expression analysis (“limma” package, |logFC| > 0.585, FDR < 0.01) identified 648 differentially expressed genes (DEGs) between clusters [top 100 shown in Figure 3E]. These subtypes served as a foundation for exploring molecular and immune differences relevant to therapy response.
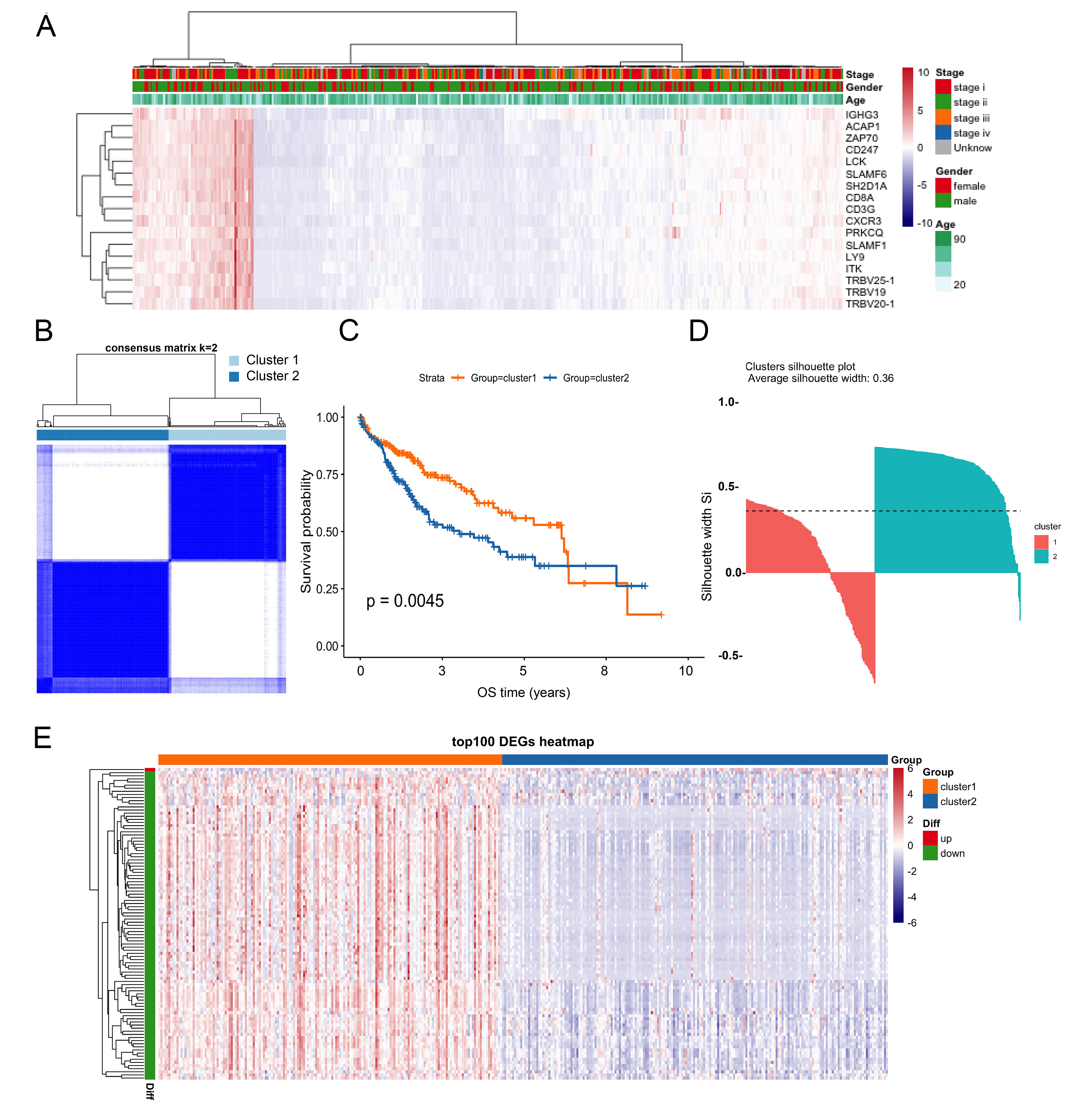
Figure 3. Classification and subtypes based on the hub genes. (A) The heat scale shows the Z-score-normalized hub gene expression; (B) Consensus clustering identified two robust subtypes: Cluster 1 and Cluster 2; (C) Kaplan-Meier survival curves comparing Clusters 1 and 2; (D) Assessment of clustering stability using average silhouette width; (E) The heat scale shows the Z-score-normalized to the top 100 DEGs. DEGs: Differentially expressed genes.
Functional enrichment of DEGs
The “ClusterProfiler” and “GSEA” R packages were used to perform functional enrichment analysis of these 648 DEGs. KEGG pathway analysis revealed significant enrichment in “chemokine signaling pathway” and “cytokine-cytokine receptor interaction” [Supplementary Figure 3A]. GO analysis identified key biological processes such as “leukocyte migration” and “immune response-activating cell surface receptor signaling pathway” [Supplementary Figure 3B-D]. GSEA further indicated that DEGs were involved in immune system regulation and T-cell differentiation [Supplementary Figure 3E and F].
Molecular differences between clusters
Using Wilcoxon test to compare the proportions of infiltrating immune cells between the two clusters. Results showed that there were significant differences in all 28 TIIC types [Figure 4A and B]. Somatic mutation analysis (“maftools” R package) showed higher mutation burden in Cluster 2 (P < 0.05, Figure 4C). Among the top 20 mutated genes, 16 overlapped between clusters; TTN and CTNNB1 exhibited notably higher mutation frequencies in Cluster 2 [Figure 4D and E], which have been linked to immune exclusion. Copy-number variation (CNV) landscapes also differed between clusters [Figure 4F and G]. TIDE webtool[17] was used to evaluate the clinical effect of ICI therapy on the two clusters, and the TIDE score of Cluster 1 was significantly higher than that of Cluster 2 (P < 0.05, Figure 4H), suggesting lower ICI response - a finding consistent with its lower mutation burden and immune-excluded phenotype.
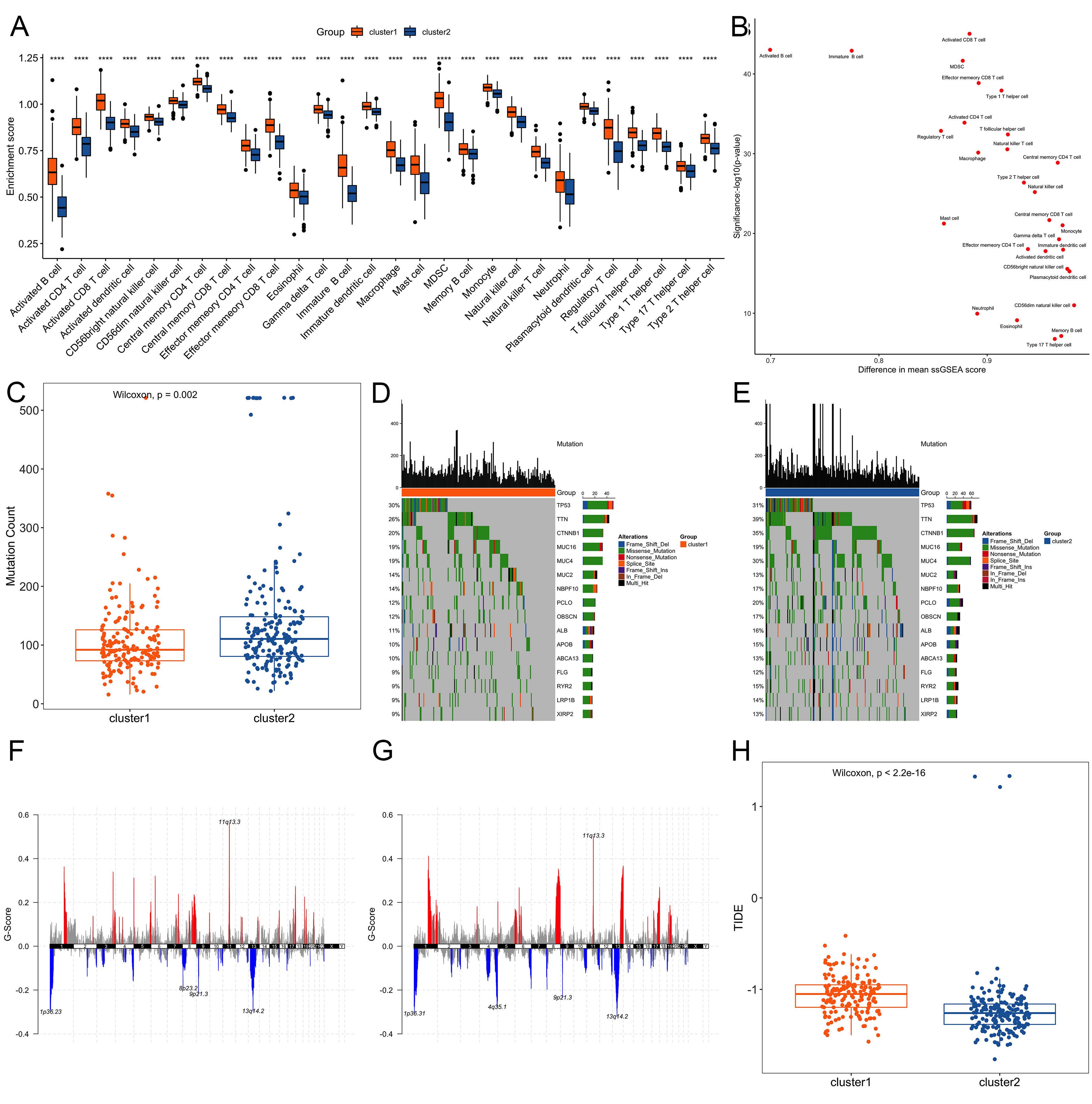
Figure 4. Comparative molecular profiling between Cluster 1 and Cluster 2. (A) Distribution difference of immune cell infiltration ratio between the two clusters. Significance markers are based on nominal P-values from Wilcoxon tests. ***P < 0.001, **P < 0.01, *P < 0.05; (B) Difference distribution scatter plot of the immune cell infiltration ratio in the two clusters; (C) Number of gene mutations between the two clusters; (D and E) Distribution of mutations of 16 intersecting genes in the two clusters; (F and G) Distribution of copy number variation regions in the two clusters; (H) TIDE score of Cluster 1 was significantly higher than that of Cluster 2. TIDE: Tumor immune dysfunction and exclusion.
Risk model construction and validation
We performed PCA of the 17 hub genes in the TCGA. The PCA-based risk score came from the first principal component of the 17 hub genes [Figure 5A]. The proportion of variance explained by each PCA was shown in the Supplementary Figure 4 and Supplementary Table 3. Using the “surv_cutpoint” method, patients were stratified into high- and low-risk groups, which showed significantly distinct prognostic outcomes (P < 0.05; Figure 5B). This model is primarily used for population-level risk stratification rather than precise individual prediction, and its area under the ROC curve (AUC, 0.55) indicates moderate predictive capability [Figure 5C]. A Sankey diagram illustrated relationships among risk groups, clusters, and clinical features [Figure 5D]. Importantly, the risk model also stratified patients by TIDE scores and mutation burden, suggesting utility in predicting immunotherapy response. External validation in GEO datasets (GSE76427, GSE14520) confirmed prognostic stratification [Figure 5E and F].
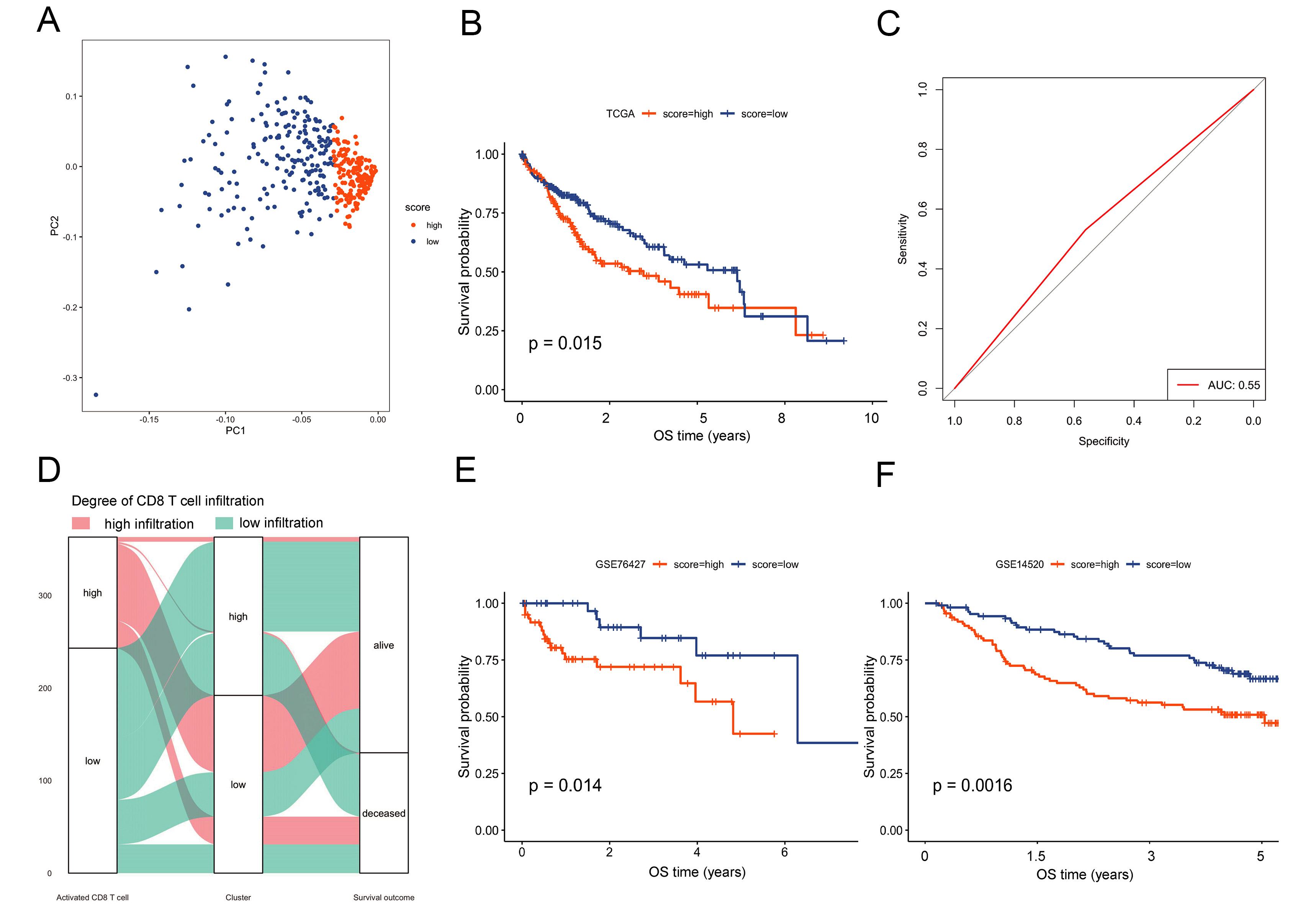
Figure 5. Construction and verification of the risk model. (A) PCA of the 17 hub genes; (B) Kaplan-Meier survival curves for high-risk vs. low-risk groups in the TCGA cohort; (C) ROC curve reached 0.55; (D) Sankey diagram illustrating relationships among molecular clusters, risk groups, and key clinical features; (E and F) External validation of the risk model in GEO datasets GSE76427 and GSE14520. PCA: Principal component analysis; TCGA: the Cancer Genome Atlas; ROC: receiver operating characteristic; GEO: the Gene Expression Omnibus; OS: overall survival; AUC: area under the ROC curve.
Molecular differences between risk groups
To further explore the molecular differences between the two risk groups, we used the “maftools” R package to analyze the differences in somatic mutations between the two risk groups. There was a significant difference in the number of gene mutations between the two risk groups (P < 0.05, Figure 6A). In these two groups, of the genes with the top 20 highest mutation frequencies [Figure 6B and C]. The high-risk group showed distinct CNV profiles [Figure 6D and E]. We also compared the differences in the immune score, stromal score and stemness score between the two groups. Immune and stromal scores were lower, while stemness scores were higher in the high-risk group (P < 0.05, Figure 6F-H). TIDE was used to evaluate the clinical effect of ICI treatment in the two groups, and the TIDE score of the high-risk group was significantly lower than that of the low-risk group (P < 0.05, Figure 6I), indicating lower TIDE score points toward a potentially better response to ICIs - a finding aligned with their higher tumor mutational burden (TMB) and immune-activated gene signatures. Furthermore, infiltration levels of all 28 TIIC types showed significant differences between the risk groups (Wilcoxon test, Figure 6J and K).
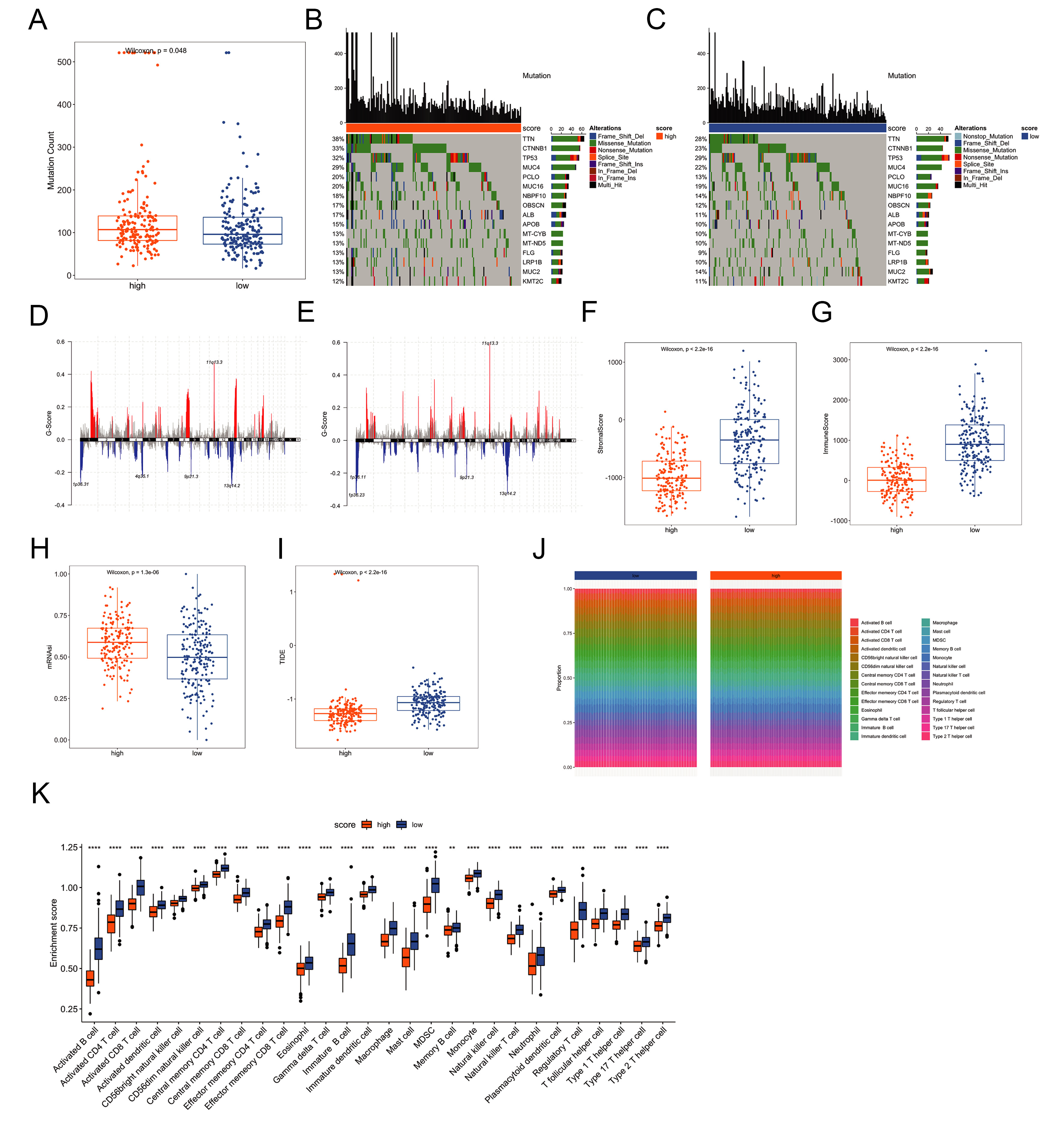
Figure 6. Molecular differences between risk groups. (A) Comparison of mutation counts between risk groups; (B and C) Mutation spectra of the 16 most frequently altered genes in each group; (D and E) CNV profiles across risk groups; (F and G) Immune and stromal scores estimated using the ESTIMATE algorithm; (H) Stemness scores derived from transcriptomic data; (I) TIDE scores predicting immune checkpoint inhibitor response; (J and K) Differential immune-cell infiltration patterns between risk groups. Significance markers are based on nominal P-values from Wilcoxon tests. ***P < 0.001, **P < 0.01, *P < 0.05. CNV: Copy-number variation; TIDE: tumor immune dysfunction and exclusion.
Stability of the risk model
To assess the robustness of the risk model, we performed subgroup analyses according to clinical characteristics in the TCGA cohort (n = 363). Within each clinical stratum, patients were classified into high- or low-risk categories using the previously defined cutoff from the “surv_cutpoint” function (“survminer” package). Kaplan-Meier survival curves were then generated to evaluate prognostic differences between risk groups. Results showed that significant survival distinctions persisted across multiple clinical subsets - including M0, N0, T1, T2-4, male, female, and stage I-II subgroups [Figure 7A-J]. These findings confirm that the predictive capability of the risk model remains stable across varied clinical contexts and support its utility in prognostic assessment for HCC patients.
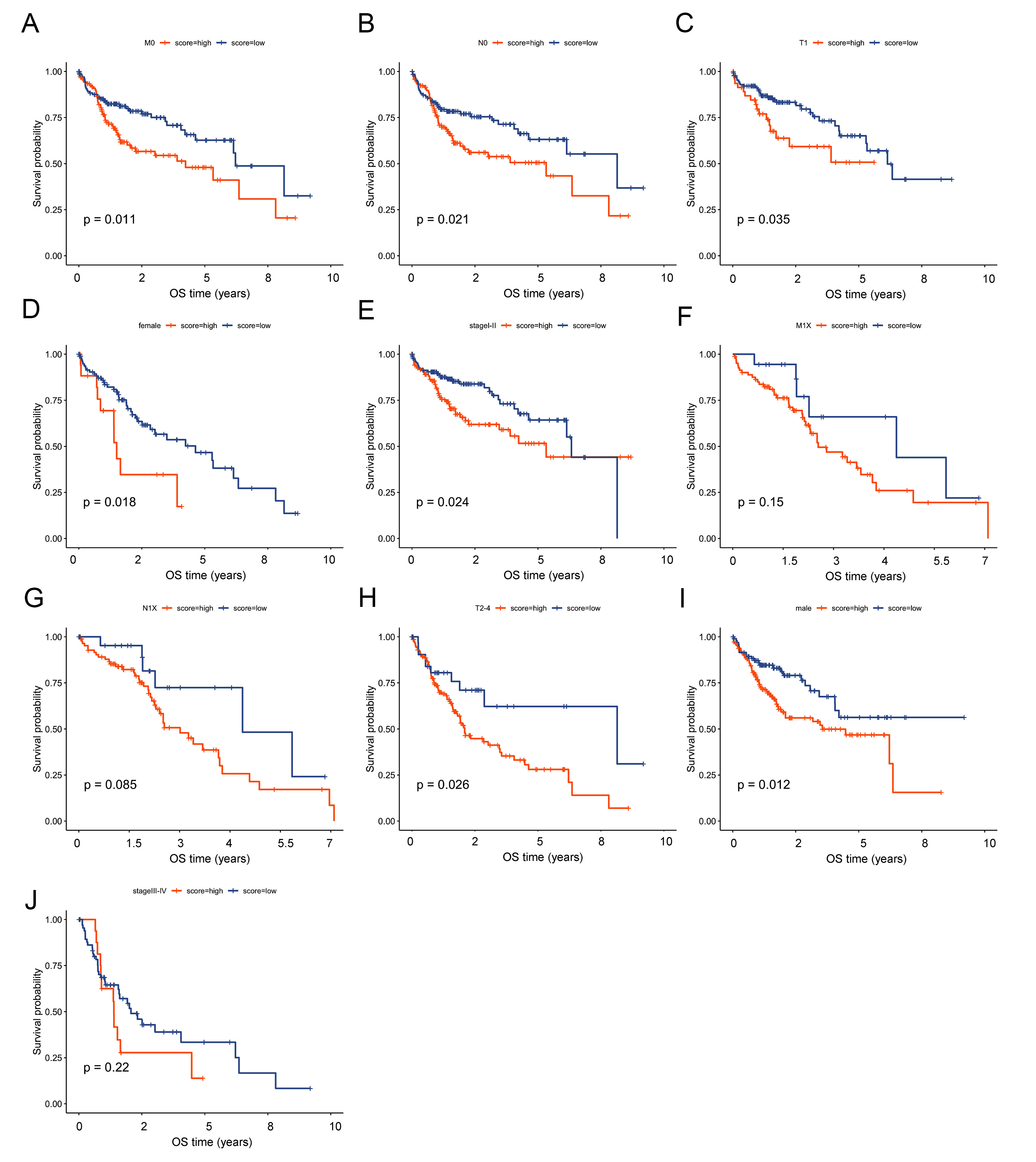
Figure 7. Subgroup analyses confirm stable predictive performance of the risk model across clinical strata. Kaplan-Meier survival analysis showed that the predictive ability of the risk model was stable under clinical characteristics such as M0, N0, T1, T2-4, male, female, and stage I-II. OS: Overall survival.
DISCUSSION
Immunotherapy based on ICIs has revolutionized oncology practice and shown encouraging efficacy in advanced HCC[18]. Clinical trials have further substantiated the therapeutic potential of PD-1 inhibitors, including pembrolizumab and camrelizumab[19,20]. However, objective response rates remain limited in most HCC patients [9], underscoring the need for predictive biomarkers to identify individuals more likely to benefit from ICI treatment. In this study, we established an immune-related prognostic classifier based on activated CD8+ T cell-associated genes. Our integrated analytical framework not only stratifies patient prognosis but also provides compelling evidence linking molecular subtypes to differential immunotherapy susceptibility, thereby offering a dual-utility tool for clinical decision-making in HCC.
Our findings reinforce the established prognostic significance of tumor-infiltrating lymphocytes in HCC. both unsupervised clustering and risk-model consistently indicated that enriched immune infiltration, notably of activated CD8+ T cells, CD4+ T cells, B cells, and NK cells, correlated with improved prognosis. These findings align with prior studies linking dense tumor-infiltrating lymphocytes to favorable outcomes across multiple malignancies[21-27]. Our study extends these observations by systematically linking CD8+ T cell-centric gene expression patterns not only to survival but also to genomic and microenvironment features that modulate immunotherapy response.
At the genomic level, Cluster 2 and high-risk groups exhibited higher TMB, consistent with reports that elevated TMB may predict enhanced response to immune checkpoint blockade[28]. In particular, we observed notable enrichment of mutations in TTN and CTNNB1 within these subgroups. CTNNB1 mutations are known to impair antitumor immunity and promote immune exclusion[29]. The co-occurrence of high TMB and CTNNB1 alterations in the same subgroup presents a nuanced genomic landscape that may partly explain the heterogeneous clinical outcomes and immunotherapy responses observed in HCC.
Furthermore, higher CNV burden and elevated stemness scores characterized the high-risk group. Both features are hallmarks of genomic instability and are associated with aggressive tumor phenotypes, therapeutic resistance, and poor prognosis[30-36]. Conversely, lower immune and stromal scores in this subgroup indicated a more immune-suppressed and stroma-poor microenvironment, which corresponded with inferior survival - a pattern consistent with prior studies linking low immune/stromal infiltration to poor prognosis[37-39].
A pivotal finding of our study is the inverse relationship between the risk score and TIDE score. Lower TIDE scores in the high-risk group suggest a reduced likelihood of immune evasion mechanisms and a potentially greater probability of responding. This prediction is mechanistically supported by same group’s higher TMB and enrichment of immune-active gene signatures. Importantly, our risk model demonstrated comparable predictive performance to TIDE in stratifying immunotherapy response, while also offering robust prognostic value over longer follow-up. The model maintained stable predictive power across diverse clinical subgroups stratified by tumor grade, stage, and sex, supporting its potential clinical applicability.
The functional relevance of our 17-gene signature is underscored by the known roles of its key constituents in antitumor immunity. For instance, CXCL10 encodes a chemokine crucial for recruiting effector T cells to the tumor site[40]. GZMB encodes granzyme B, a key cytotoxic molecule released by activated CD8⁺ T cells to induce tumor cell apoptosis. Furthermore, the inclusion of PDCD1 (encoding PD-1) directly links our signature to immune checkpoint biology, as PD-1 is the canonical target of ICI therapy[41]. In contrast, PDCD1 (encoding PD-1 protein) represents the direct target of ICIs; its elevated expression is frequently associated with T-cell exhaustion, whereas blockade of the PD-1/PD-L1 axis can reverse this suppression and restore T-cell antitumor activity[42]. The co-enrichment of these genes within our prognostic module provides a mechanistic basis for its association with both patient survival and predicted immunotherapy response.
Several previous studies have sought to identify immune-based signatures in HCC. For instance,
Several limitations of this study should be noted. First, our analysis was conducted using publicly available retrospective datasets. It is important to note that the etiology of underlying liver disease (e.g., viral vs. non-viral) is a major source of heterogeneity in HCC and can influence the tumor immune microenvironment. The cohorts used in this study lacked uniformly annotated etiology data, which precluded a stratified analysis by cause. Therefore, while our model identifies a prognostically relevant immune axis, its applicability across all HCC etiologies requires validation in future, well-annotated prospective cohorts where etiology is a predefined covariate. Second, while we incorporated genomic and transcriptomic features, proteomic and spatial multi-omics data could further refine the model’s biological interpretability. Third, although the risk model constructed in this study can significantly stratify prognosis, its discriminative ability is limited (AUC ≈ 0.55), making it more suitable as a supplementary tool rather than an independent diagnostic indicator. Future work may integrate parameters such as clinical stage and etiology to build a composite model. Fifth, the P-values for the comparison of the 28 tumor-infiltrating immune cell subsets were not adjusted for multiple comparisons. Although we employed a stringent nominal threshold (P < 0.01) to highlight the most robust differences and have transparently reported this methodological point, future studies should apply formal correction methods (e.g., Benjamini-Hochberg FDR control) to confirm these associations. Tools such as TIDE are merely hypothesis-generating computational predictors and require validation in prospective ICI-treated cohorts. Finally, although the model’s risk groups correlate with computational predictors of ICI response TIDE and genomic features associated with immunotherapy outcomes, these results remain hypothesis-generating. Direct validation in independent, prospectively collected cohorts of ICI-treated HCC patients is required to establish the model’s clinical utility in guiding immunotherapy decisions.
In summary, we developed and validated an activated CD8+ T cell-related risk model that effectively stratifies HCC patients into distinct prognostic subgroups. The developed classifier combines features of immune infiltration, genomic instability, and tumor stemness. It demonstrates potential as a non-invasive biomarker for predicting both patient prognosis and probable response to immunotherapy. Further prospective studies are warranted to translate this model into clinical practice for personalized treatment planning in HCC. Moreover, immunophenotypic features not only help predict the efficacy of first-line ICIs but may also provide clues for selecting second-line treatment options after ICI failure. For example, the recent REFINE study demonstrated that regorafenib continues to exhibit favorable efficacy and safety in patients who have failed ICI therapy[43]. Therefore, future studies could explore the correlation between our model and second-line treatments such as tyrosine kinase inhibitors (TKIs), offering a basis for personalized sequential therapy.
DECLARATIONS
Acknowledgement
The Graphical Abstract was created using Adobe Illustrator and Photopea.
Authors’ contributions
Contributed to the conception of the study: Zhang Y, Wang L
Performed the experiment: Wu Z, Wang Y
Contributed significantly to analysis and manuscript preparation: Xu W, Pan Q
Performed the data analyses and wrote the manuscript: Zhang Y, Wang Y
Helped perform the analysis with constructive discussions: Xu W, Pan Q
Availability of data and materials
The datasets TCGA-HCC for this study can be found at http://cancergenome.nih.gov/. The datasets GSE76427 and GSE14520 in this study can be found at http://www.ncbi.nlm.nih.gov/geo/. These data can be obtained from the corresponding author upon reasonable request.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This study was supported by grants from the Clinical Research Plan of SHDC (SHDC2020CR4018), the National Natural Science Foundation of China (82503795), China Postdoctoral Science Foundation (BX20240082, 2024M750536), and the Shanghai Rising Star Program Sailing Project (24YF2705600, 24YF2738200).
Conflicts of Interest
Zhang Y is a Junior Editorial Board Member of Hepatoma Research. He was not involved in any steps of the editorial process for this manuscript, including reviewer selection, manuscript handling, or decision-making. The other authors declare no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229-63.
3. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275-87.
4. Queirolo P, Boutros A, Tanda E, Spagnolo F, Quaglino P. Immune-checkpoint inhibitors for the treatment of metastatic melanoma: a model of cancer immunotherapy. Semin Cancer Biol. 2019;59:290-7.
5. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1-10.
6. Seo JS, Kim A, Shin JY, Kim YT. Comprehensive analysis of the tumor immune micro-environment in non-small cell lung cancer for efficacy of checkpoint inhibitor. Sci Rep. 2018;8:14576.
7. Yau T, Kang YK, Kim TY, et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the CheckMate 040 randomized clinical trial. JAMA Oncol. 2020;6:e204564.
8. Macek Jilkova Z, Aspord C, Decaens T. Predictive factors for response to PD-1/PD-L1 checkpoint inhibition in the field of hepatocellular carcinoma: current status and challenges. Cancers. 2019;11:1554.
9. Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol. 2019;20:25-39.
10. Wang Z, Wu X. Study and analysis of antitumor resistance mechanism of PD1/PD‐L1 immune checkpoint blocker. Cancer Med. 2020;9:8086-121.
11. Sangro B, Melero I, Wadhawan S, et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J Hepatol. 2020;73:1460-9.
12. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
13. Niemira M, Collin F, Szalkowska A, et al. Molecular signature of subtypes of non-small-cell lung cancer by large-scale transcriptional profiling: identification of key modules and genes by weighted gene co-expression network analysis (WGCNA). Cancers. 2019;12:37.
14. Liu H, Sun Y, Tian H, et al. Characterization of long non-coding RNA and messenger RNA profiles in laryngeal cancer by weighted gene co-expression network analysis. Aging. 2019;11:10074-99.
15. Charoentong P, Finotello F, Angelova M, et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18:248-62.
16. Lengyel A, Botta‐Dukát Z. Silhouette width using generalized mean - a flexible method for assessing clustering efficiency. Ecol Evol. 2019;9:13231-43.
17. Jiang P, Gu S, Pan D, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24:1550-8.
18. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19:133-50.
19. Qin S, Ren Z, Meng Z, et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: a multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 2020;21:571-80.
20. Zhu AX, Finn RS, Edeline J, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19:940-52.
21. Goehrig D, Nigri J, Samain R, et al. Stromal protein βig-h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut. 2019;68:693-707.
22. Varn FS, Tafe LJ, Amos CI, Cheng C. Computational immune profiling in lung adenocarcinoma reveals reproducible prognostic associations with implications for immunotherapy. OncoImmunology. 2018;7:e1431084.
23. Isaeva OI, Sharonov GV, Serebrovskaya EO, et al. Intratumoral immunoglobulin isotypes predict survival in lung adenocarcinoma subtypes. J Immunother Cancer. 2019;7:279.
24. Stiff A, Trikha P, Mundy-Bosse B, et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing Fc receptor-mediated natural killer cell function. Clin Cancer Res. 2018;24:1891-904.
25. Kang JY, Kim KE. Prognostic value of interleukin-32 expression and its correlation with the infiltration of natural killer cells in cutaneous melanoma. J Clin Med. 2021;10:4691.
26. Chung W, Eum HH, Lee HO, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. 2017;8:15081.
27. Ganesan AP, Clarke J, Wood O, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol. 2017;18:940-50.
28. Chan TA, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30:44-56.
29. Wei L, Delin Z, Kefei Y, Hong W, Jiwei H, Yange Z. A classification based on tumor budding and immune score for patients with hepatocellular carcinoma. OncoImmunology. 2019;9:1672495.
31. Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell. 2019;24:25-40.
32. Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. 2017;16:41.
33. Liu S, Zhou Z, Jia Y, et al. Identification of portal vein tumor thrombus with an independent clonal origin in hepatocellular carcinoma via multi-omics data analysis. Cancer Biol Med. 2019;16:147-70.
34. Mamlouk S, Childs LH, Aust D, et al. DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat Commun. 2017;8:14093.
35. Zhang C, Chen T, Li Z, et al. Depiction of tumor stemlike features and underlying relationships with hazard immune infiltrations based on large prostate cancer cohorts. Brief Bioinform. 2021;22:bbaa211.
36. Miranda A, Hamilton PT, Zhang AW, et al. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc Natl Acad Sci U S A. 2019;116:9020-9.
37. Wang Z, Xu H, Zhu L, He T, Lv W, Wu Z. Establishment and evaluation of a 6‐gene survival risk assessment model related to lung adenocarcinoma microenvironment. Biomed Res Int. 2020;2020:6472153.
38. Song C, Guo Z, Yu D, et al. A prognostic nomogram combining immune-related gene signature and clinical factors predicts survival in patients with lung adenocarcinoma. Front Oncol. 2020;10:1300.
39. Zhang C, Cheng W, Ren X, et al. Tumor purity as an underlying key factor in glioma. Clin Cancer Res. 2017;23:6279-91.
40. Shigeta K, Matsui A, Kikuchi H, et al. Regorafenib combined with PD1 blockade increases CD8 T-cell infiltration by inducing CXCL10 expression in hepatocellular carcinoma. J Immunother Cancer. 2020;8:e001435.
41. Zöphel D, Angenendt A, Kaschek L, et al. Faster cytotoxicity with age: increased perforin and granzyme levels in cytotoxic CD8+ T cells boost cancer cell elimination. Aging Cell. 2022;21:e13668.
42. Cosgrove D, Tan R, Osterland AJ, et al. Atezolizumab plus bevacizumab in patients with unresectable hepatocellular carcinoma: real-world experience from a US community oncology network. J Hepatocell Carcinoma. 2025;12:791-804.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].