


Oral microbiota for diagnosis of etomidate abuse via machine learning
 0
0 Abstract
Aim: Illicit etomidate (ET) use in e-cigarettes has increased recently, but its effects on the oral microbiome remain unknown. This study investigates oral microbiota alterations in chronic ET users.
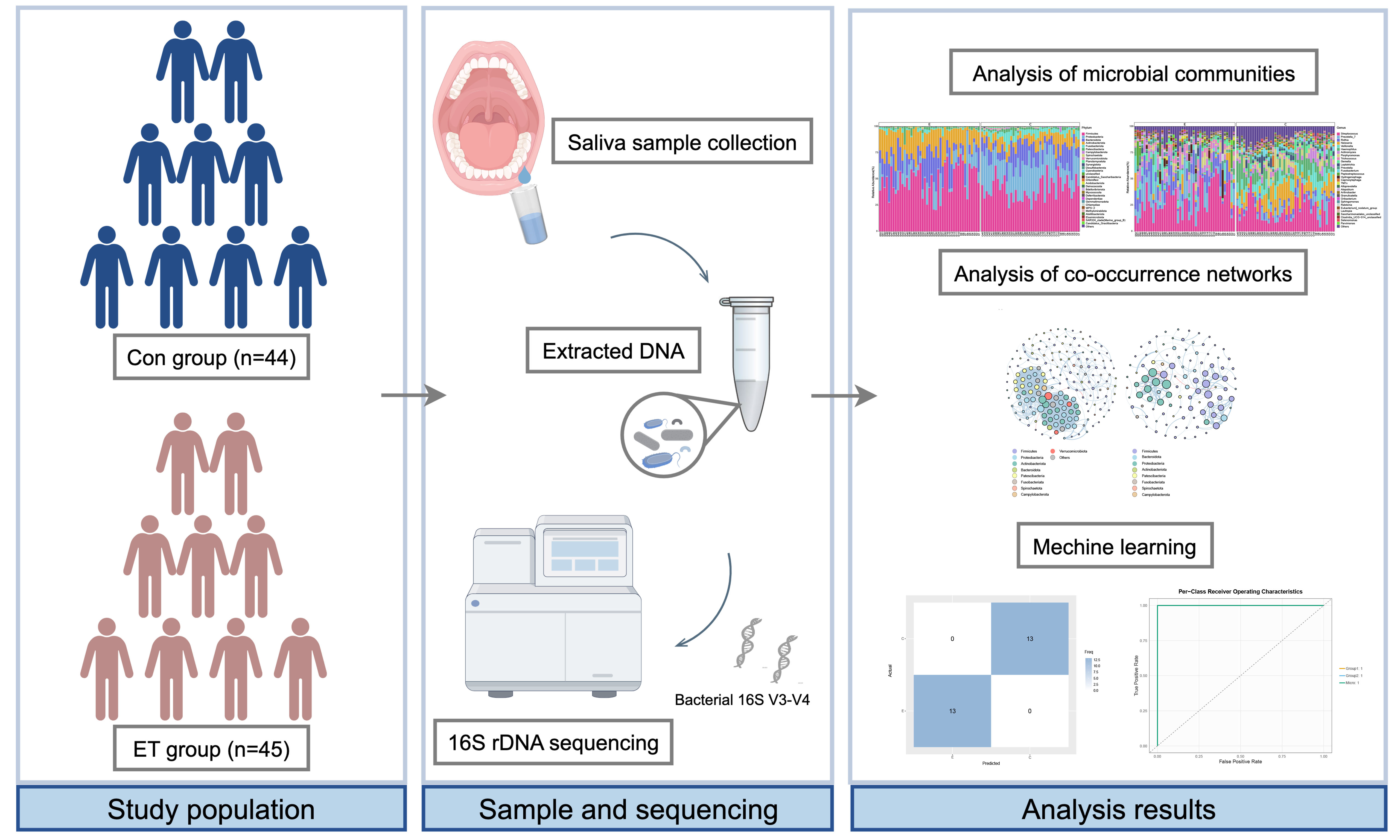
Methods: Saliva from 45 ET users and 44 controls underwent 16S ribosomal RNA (rRNA) sequencing. We compared microbial diversity, composition, predicted functions, and co-occurrence networks between groups, and developed 12 machine learning models to classify ET users based on microbial features.
Results: Chronic ET-containing e-cigarette use was strongly associated with significant oral microbial dysbiosis. Species richness and diversity were significantly lower in the ET group. ET users exhibited proliferation of taxa associated with oral diseases, including Actinomyces, Rothia, and Atopobium. PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2) predicted enhanced carbohydrate and amino acid metabolism in the ET group, while controls showed greater abundance of biotin and fatty acid metabolism pathways. Network analysis revealed reduced complexity and stability in the ET group. The ensemble model GLMBoost (generalized linear model boosting) + Random Forest achieved an area under the curve (AUC) of 1.00 and 100% accuracy on the test set. Seven key genera were identified as discriminative biomarkers: Prevotella_7, Rothia, Neisseria, Veillonella, Haemophilus, Actinomyces, and Fusobacterium.
Conclusion: Chronic use of ET-containing e-cigarettes is linked to altered homeostasis of the oral microbiota, providing a deeper understanding of how substance addiction impacts oral microbial ecology and highlights the potential of machine learning-derived microbial signatures as non-invasive tools for accurately distinguishing ET-containing e-cigarette users from non-users.
Keywords
INTRODUCTION
Etomidate (ET) belongs to the class of non-barbiturate sedative-hypnotic drugs and is primarily administered for the induction of anesthesia[1]. ET exerts anesthetic, sedative, and amnestic actions primarily through potentiation of γ-aminobutyric acid type A (GABAA) receptors[2]. Owing to its favorable hemodynamic stability profile, this intravenous anesthetic agent is widely employed for both general anesthesia induction and procedural sedation during painless endoscopic examinations[1]. Recent studies indicate a rising trend in the illicit use and trafficking of ET as an emerging substitute for controlled substances such as propofol[3,4]. Criminals took advantage of the narcotic effect of ET and began adding it to “e-cigarette oil” listed for sale on the Internet[5]. The proliferation of online commerce has facilitated more frequent and concealed abuse of ET by illicit users. Chronic or excessive administration can lead to serious health consequences, including adrenocortical suppression, myoclonus, and fatalities[6-8], thereby posing a substantial threat to public health, safety, and societal stability[6-8].
Approximately 700 bacterial species have been identified as residents of the human oral cavity, collectively essential for preserving oral health[9]. The oral cavity is a key site for environmental interactions with the human body[10]. It is well known that specific bacterial taxa that colonize the oral cavity are associated with environmental factors such as diet[3], substance use[11], smoking[12], oral health[13] and disease[9]. Drugs (e.g., methamphetamine[14], opioid[15], etc.) have been shown to affect the composition of the human oral microflora, which in turn affects oral health. Methamphetamine abusers often present with a spectrum of oral health issues, including xerostomia, dental caries, poor hygiene, excessive tooth wear, and a diminished quality of life related to oral health[14]. Smoking depletes beneficial oral bacteria, creating an ecological niche that facilitates the colonization of pathogens and increases disease susceptibility[12]. Oral dysbiosis, characterized by microbial community imbalance in the oral cavity, serves as a primary etiological factor for major dental pathologies[16]. However, the association between ET use and the oral microbiota has not been investigated to date.
This study was conducted to investigate differences in oral microbiota between ET users and non-users, with the aim of understanding how exposure to ET may adversely affect human health through oral microbial dysbiosis. To characterize the alterations in the oral microbiome associated with ET use, we employed 16S ribosomal RNA (rRNA) gene sequencing combined with bioinformatic analysis. Furthermore, we applied multiple machine learning algorithms to explore the potential of oral microbial taxa as non-invasive biomarkers for distinguishing ET users from healthy individuals.
METHODS
Sample and individual information collection
The volunteers collected 2 mL unstimulated saliva in the morning without brushing their teeth. Sample collection was conducted between September 2023 and March 2024. For ET users, samples were collected within 24 h of their last reported ET use, representing a state of active use rather than withdrawal. These samples were immediately frozen at -80 °C for future microbiological assay. We recruited 45 individuals with ET abuse (range, 18-48 years) along with 44 sex-matched healthy control subjects who had never used ET or any other addictive drug (range, 20-45 years). ET was primarily abused via e-cigarettes, with subjects reportedly using it more than 5 times per month for a period of 6 to 12 months. Urine toxicology tests were positive for ET acid but negative for cocaine, amphetamines, barbiturates, opiates and benzodiazepines, etc. We classified participants into an ET user group and a healthy control group (Con). All subjects were of Han Chinese descent, resided in Guangdong Province, and followed comparable dietary regimens. The group of ET users is named the ET group, and the group of control subjects is named the Con group. None of the subjects had taken antibiotics, probiotics, or prebiotics in the 3 months prior to sample collection, and none were taking anti-inflammatory or antioxidant drugs. Individuals with a diagnosis of severe oral diseases (including severe periodontitis, oral cancer, or active oral candidiasis), or those who had undergone periodontal therapy within the last 6 months were also excluded from both the ET and Con groups. None of the subjects abused alcohol [Table 1]. All participants gave written informed consent. All procedures conformed to the ethical standards of the institutional and national responsible committee on human experimentation and to the Helsinki Declaration of 1975, as revised in 2008 (5). This study was approved by the Medical Ethics Committee of Southern Medical University, Guangzhou, China (No. 2023103).
The characteristics of the study population
| Demographic characteristics | Con (n = 44) | ET (n = 45) |
| Age (years) | 28 ± 6.24 | 26 ± 7.57 |
| Gender | ||
| Female Male | 10 (22.7%) 34 (77.3%) | 9 (20.0%) 36 (80.0%) |
| ET use frequency | - | > 10 times/month (18.4 ± 6.2 times/month) |
| Duration of ET use | - | Minimum 6 months of regular ET use (range: 6-12 months) |
| Smoking habit | ||
| With Without | 9 (20.5%) 35 (79.5%) | 14 (31.1%) 31 (68.9%) |
| Alcohol abuse | No | No |
| Basic disease | No | No |
DNA extraction and 16S rDNA amplicon sequencing
Saliva samples were processed for bacterial genomic DNA isolation using the E.Z.N.A. soil DNA Kit (Omega Bio-tek, USA) according to the manufacturer's protocol. The extracted DNA was quantified using a Qubit fluorometer (Invitrogen, USA) and subsequently amplified via polymerase chain reaction (PCR) with the universal bacterial primers 341F/805R. The resulting amplicons were purified with AMPure XT Beads (Beckman Coulter, USA), re-quantified, and quality-checked on an Agilent 2100 Bioanalyzer. Finally, libraries were constructed using Kapa Biosciences kits, pooled, and subjected to paired-end (PE250) sequencing on an Illumina NovaSeq 6000 platform at LC-Bio Technology Co., Ltd. (Hangzhou, China).
Data processing and 16S rDNA sequencing analysis
Sequencing primers were removed from de-multiplexed raw sequences using cutadapt (v1.9). Paired-end reads were then merged with FLASH (Fast Length Adjustment of SHort reads, v1.2.8). Quality control was performed using fqtrim (v0.94) with a sliding-window algorithm to discard reads with average quality scores below 20, length shorter than 100 bp, or containing over 5% ambiguous “N” bases. Chimeric sequences were filtered using Vsearch (v2.3.4). Denoising and inference of biological sequences into amplicon sequence variants (ASVs) were conducted with Divisive Amplicon Denoising Algorithm 2 (DADA2). Taxonomic annotation was achieved by aligning ASVs to the SILVA 16S rRNA database (v138) via the QIIME2 (Quantitative Insights Into Microbial Ecology version 2) feature-classifier plugin. Microbial diversity (alpha and beta) was assessed within QIIME2, while statistical analyses included Wilcoxon rank-sum tests (P < 0.05) for differential abundance and linear discriminant analysis effect size (LEfSe) (Linear Discriminant Analysis (LDA) ≥ 4.0, P < 0.05) for biomarker discovery. A Spearman correlation matrix was generated in R using the “hmisc”, “psych”, and “igraph” packages, with P-values adjusted by the Benjamini-Hochberg method. Following this adjustment, a co-occurrence network was generated from statistically significant pairwise correlations (adjusted P < 0.05, absolute Spearman correlation coefficient |r| > 0.6) and rendered using Gephi software (version 0.9.2).
Machine learning
A multi-step machine learning framework was employed to identify key microbial biomarkers. The dataset (n = 89) was randomly split into a training set (70%, n = 63) and a hold-out test set (30%, n = 26) using stratified random sampling. The stratification was performed based on the subject group (ET user vs. non-user) to ensure that the proportion of the two classes was preserved in both the training and test sets. Initially, three feature selection methods - Random Forest (RF), Stepglm, and glmBoost (Generalized Linear Model Boosting) - were applied to filter discriminative taxa. The selected features subsequently served as input for eight distinct classifiers (generalized linear model with elastic net regularization (glmnet), RF, support vector machine with radial basis function kernel (svmRadial), LDA, naive bayes (NB), partial least squares (PLS), redundancy analysis (RDA), TreeBag), which were systematically tuned under stratified 10-fold cross-validation. This process generated 24 unique model configurations. The final performance of each model configuration was assessed on the completely independent test set. Model performance was primarily assessed using the area under the curve (AUC), with the optimal model selected based on the highest mean AUC and accuracy on the test set. Feature importance was quantified via the Gini index and visualized using ggplot2 in R (v4.5.1), providing a measure of predictive contribution.
RESULTS
Sequencing data of microbial communities
We employed 16S rRNA gene amplicon sequencing to analyze the oral microbiota present in the saliva samples of participants (62,345 ± 8,921 sequences per sample). This analysis yielded a total of 5,381,585 high-quality sequences in total from 89 participants. When stratified by group, the Con group generated 2,812,456 sequences (63,919 ± 9,102 per sample), while the ET group generated 2,569,129 sequences (57,092 ± 8,743 per sample). All quality-filtered sequences were subsequently annotated, resulting in 7,090 Amplicon Sequence Variants (ASVs) that were taxonomically classified into 2 domains, 38 phyla, 100 classes, 226 orders, 382 families and 836 genera. The Con group harbored 5,197 ASVs, whereas the ET group harbored 3,614 ASVs, with 1,621 ASVs shared between groups.
Alpha diversity analysis in two groups
Microbial community richness and diversity in saliva samples were quantified using the Chao and Shannon indices. Both Chao and Shannon indices revealed significantly reduced species richness and community diversity in the ET group compared to controls [Figure 1A and B]. Additionally, the Shannon rarefaction curves for both groups reached a plateau with increasing sequencing depth [Figure 1C], indicating that the sequencing depth was sufficient to reflect the actual situation of oral microbes in the samples.
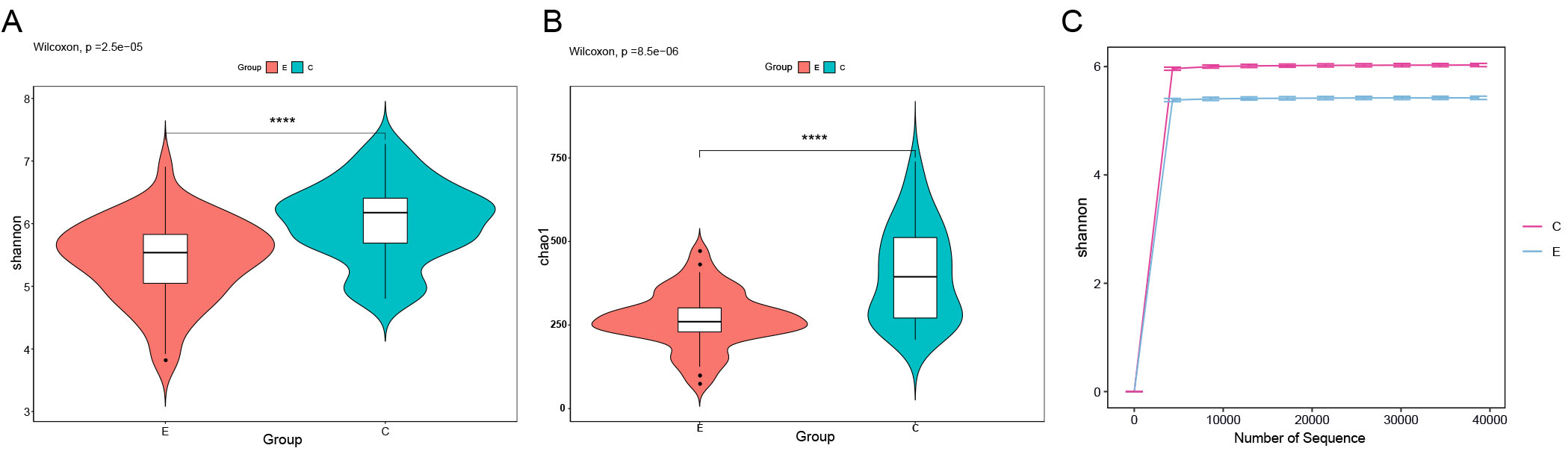
Figure 1. Alpha diversity analysis of oral microbial communities in Con group and ET group. (A) Shannon index of salivary microbiota in two groups. (B) Chao1 index of salivary microbiota in two groups. (C) Shannon rarefaction curve in two groups. Statistical significance between groups was determined using the Wilcoxon rank-sum test. Box plots display the median (center line), interquartile range (box bounds), and 1.5 × interquartile range (whiskers). Asterisks indicate significant differences between groups: ****P < 0.0001. Rarefaction curves reached a plateau, confirming sufficient sequencing depth.
Oral microbiota composition is altered in ET users
Beta diversity, assessed using Jaccard and Bray-Curtis distances, was used to analyze differences in the microbial community structure between groups. Both metrics revealed distinct clustering patterns that effectively separated the oral microbiota of the two groups [Figure 2A and B]. Furthermore, a Venn diagram showed how ASVs were distributed, identifying 1,621 common to both groups, with 3,576 and 1,993 ASVs unique to the Con and ET groups, respectively [Figure 2C].
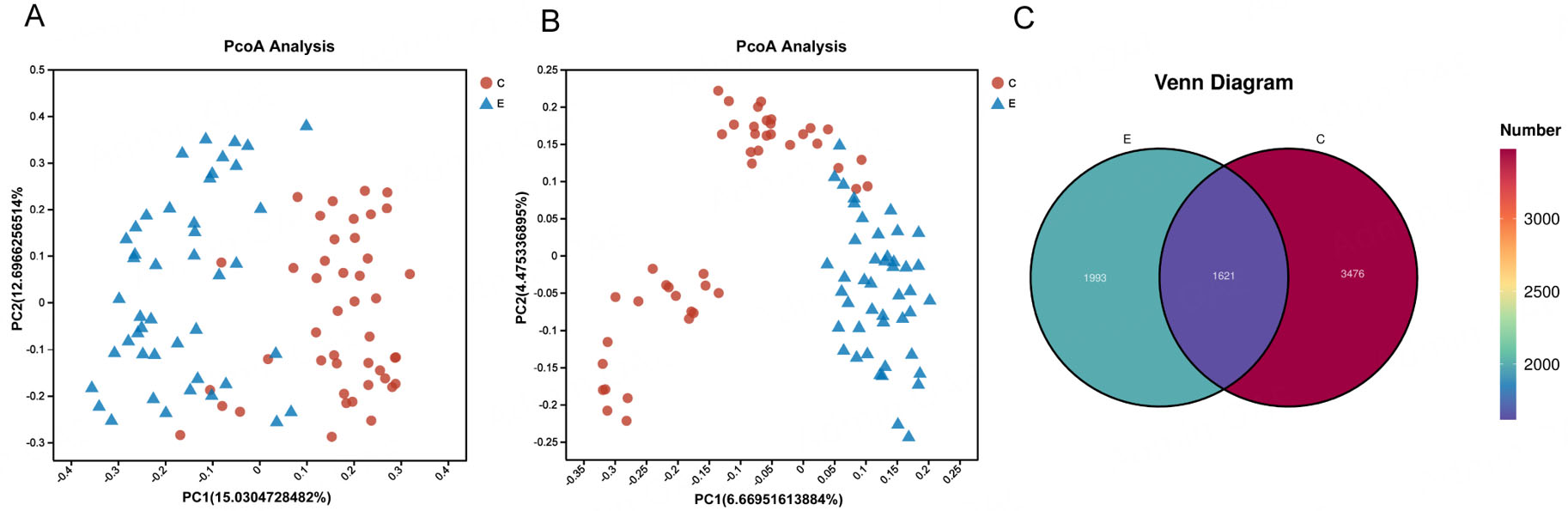
Figure 2. Oral microbiota composition is altered in group ET. (A) Principal coordinate analysis (PCoA) plot based on Bray-Curtis distance. (B) PCoA plot based on Jaccard distance. (C) Venn diagram of the observed bacterial ASVs in the two groups. Each point in (A) and (B) represents an individual sample. PERMANOVA (adonis) was used to test for significant differences in community structure between groups.
Different bacterial taxa were present in ET users and control subjects
Taxonomic profiling at the phylum and genus levels was performed to compare the oral microbiota between ET users and controls [Figure 3A and B], showing the microbial taxa with the average relative abundance > 1%. Relative abundance comparisons indicated distinct differences in the microbial community compositions between the Con and ET groups. At the phylum level, four dominant taxa (Firmicutes, Proteobacteria, Bacteroidota, and Actinobacteriota) accounted for an average of more than 80% of the sequences in both groups. At the genus level, the major taxa in both groups included Streptococcus, Prevotella_7, Rothia, Neisseria, Veillonella, Haemophilus, Actinomyces, Porphyromonas and Trichococcus. The relative abundances of Neisseria, Haemophilus, Leptotrichia, Fusobacterium, Capnocytophaga, Alloprevotella, Lautropia, Aggregatibacter, Campylobacter, Lachnoanaerobaculum genera were significantly reduced [Figure 3C], while the relative abundance of Rothia, Trichococcus, Atopobium, CHKCI001, Caulobacter, Leifsonia, Phenylobacterium, Turicibacter, Phreatobacter, Pannonibacter, Kocuria, Brevundimonas, Ruminococcus_torques_group, Hydrogenophaga, Comamonadaceae_unclassified, Cryptobacterium, Clostridium_sensu_stricto_1, Desulfovibrio, Actinomyces were significantly increased in the group of ET users compared to the Con group [Figure 3D].
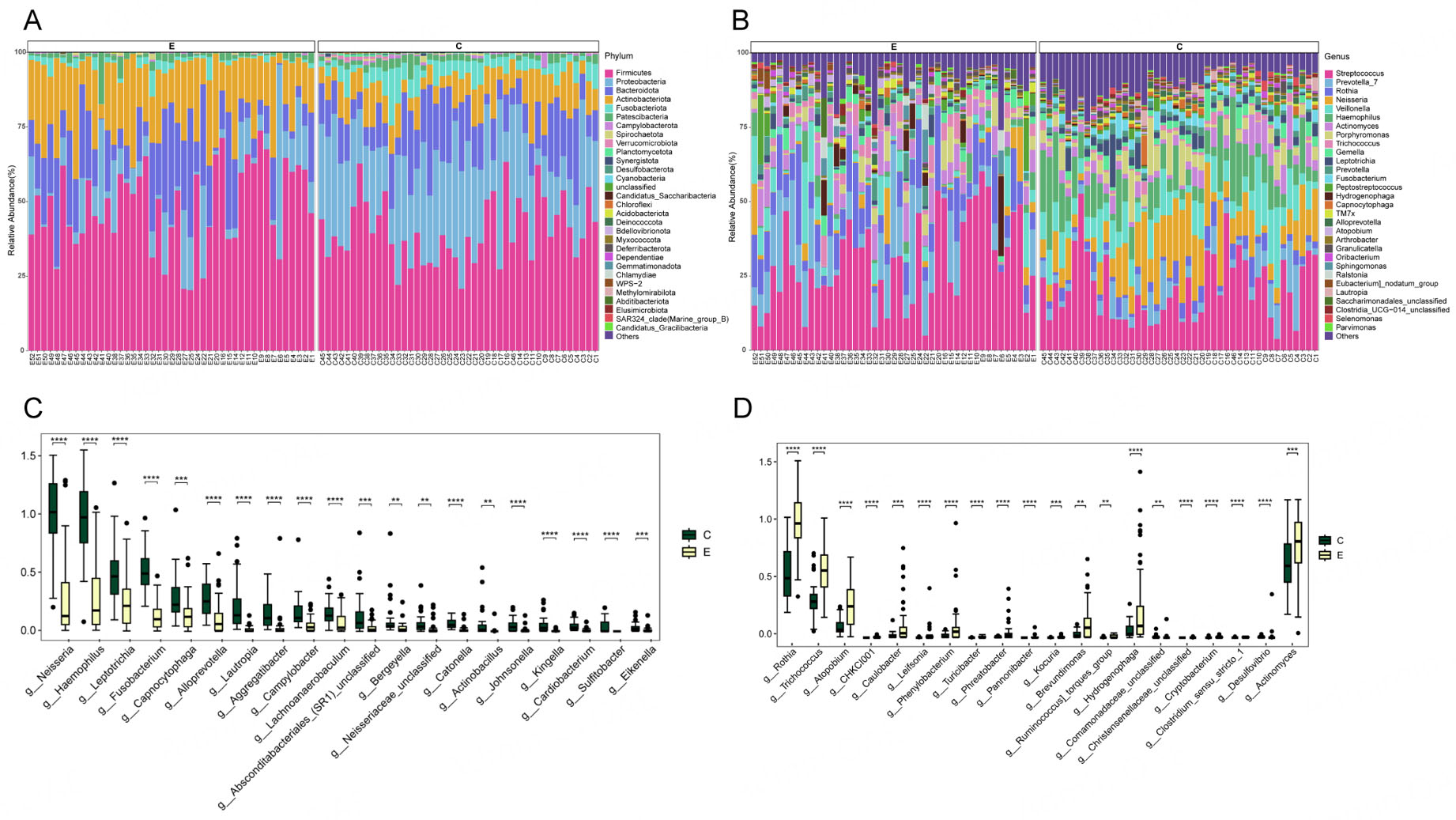
Figure 3. Compositional differences in the oral microbiota between Con and ET users. Microbial composition at the (A) phylum and (B) genus level (showing taxa with > 1% mean relative abundance). (C) Top 20 bacterial genera with significantly decreased relative abundance in the ET group. (D) Top 20 bacterial genera with significantly increased relative abundance in the ET group. Statistical analysis was performed using the Wilcoxon rank-sum test with Benjamini-Hochberg correction for multiple testing. Error bars represent the standard error of the mean (SEM). Asterisks indicate adjusted p-values: **P < 0.01, ***P < 0.001, ****P < 0.0001.
LEfSe analysis was conducted to identify differentially abundant taxa between the two groups. The LDA scores are shown in Figure 4A, and the taxonomic cladogram illustrating the community structure is presented in Figure 4B. In the ET group, taxa significantly enriched included Actinobacteriota, Actinobacteria, Bacilli, Rothia, Firmicutes, Lactobacillales, Streptococcaceae, Micrococcaceae, Streptococcus, Prevotella_7 and Actinomyces. In contrast, the Con group predominantly retained Gammaproteobacteria, Proteobacteria, Neisseriaceae, Neisseria, Enterobacterales, Pasteurellaceae, Haemophilus, Burkholderiales, Negativicutes, Veillonellaceae, Fusobacteriota. These markers have the potential to serve as distinct microbial signatures for differentiating between the ET users and healthy control subjects.
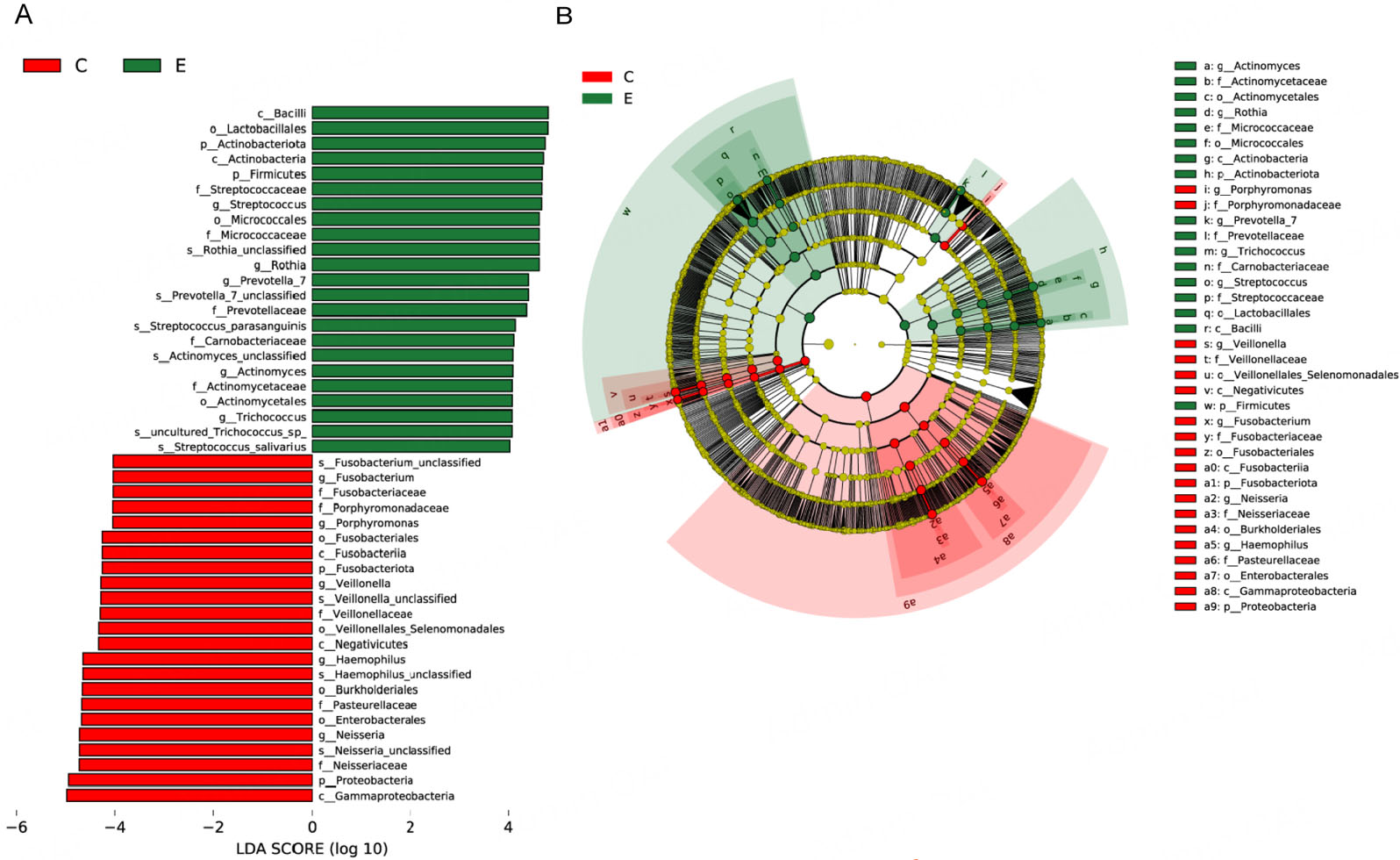
Figure 4. Taxa with significantly different abundance between the ET and Con groups. (A) LDA (LDA > 4.0, P < 0.05) for taxa distinguishing the groups. Positive and negative scores indicate enrichment in the ET and control groups, respectively. (B) Cladogram illustrating taxa with significant differences. Nodes in red and blue represent taxa enriched and depleted in the ET group, respectively. The concentric circles denote taxonomic levels from phylum to species, with node size corresponding to relative abundance. ET: Etomidate; LDA: linear discriminant analysis.
Potential functional profiles of the oral microbiota
To elucidate the functional implications of the observed microbial differences, we performed a phylogenetic investigation of communities by reconstruction of unobserved states [PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2)] analysis based on the 16S rRNA gene data and referenced against the MetaCyc database. It is important to note that the following functional profiles are inferred from 16S rRNA gene data using PICRUSt2 and represent putative metabolic potentials, not directly measured microbial activities. Therefore, these predictions should be interpreted as hypothesis-generating and require validation through metagenomic or metatranscriptomic analyses in future studies. Differential enrichment analysis (Wilcoxon rank-sum test, P < 0.05) identified 277 MetaCyc pathways that were significantly altered between the two groups [Figure 5]. Specifically, the ET group exhibited enrichment in pathways primarily involved in carbohydrate metabolism (e.g., heterolactic fermentation, glycolysis III) and the biosynthesis of amino acids (e.g., L-lysine biosynthesis III) and nucleotides (e.g., 5-aminoimidazole ribonucleotide biosynthesis I/II). In contrast, the control group was characterized by higher abundances of pathways related to Guanosine 5'-diphosphate 3'-diphosphate biosynthesis, anhydromuropeptides recycling, biotin biosynthesis, L-lysine biosynthesis III, super pathway of glycol metabolism and degradation, super pathway of fatty acid biosynthesis initiation (Escherichia coli), 8-amino-7-oxononanoate biosynthesis.
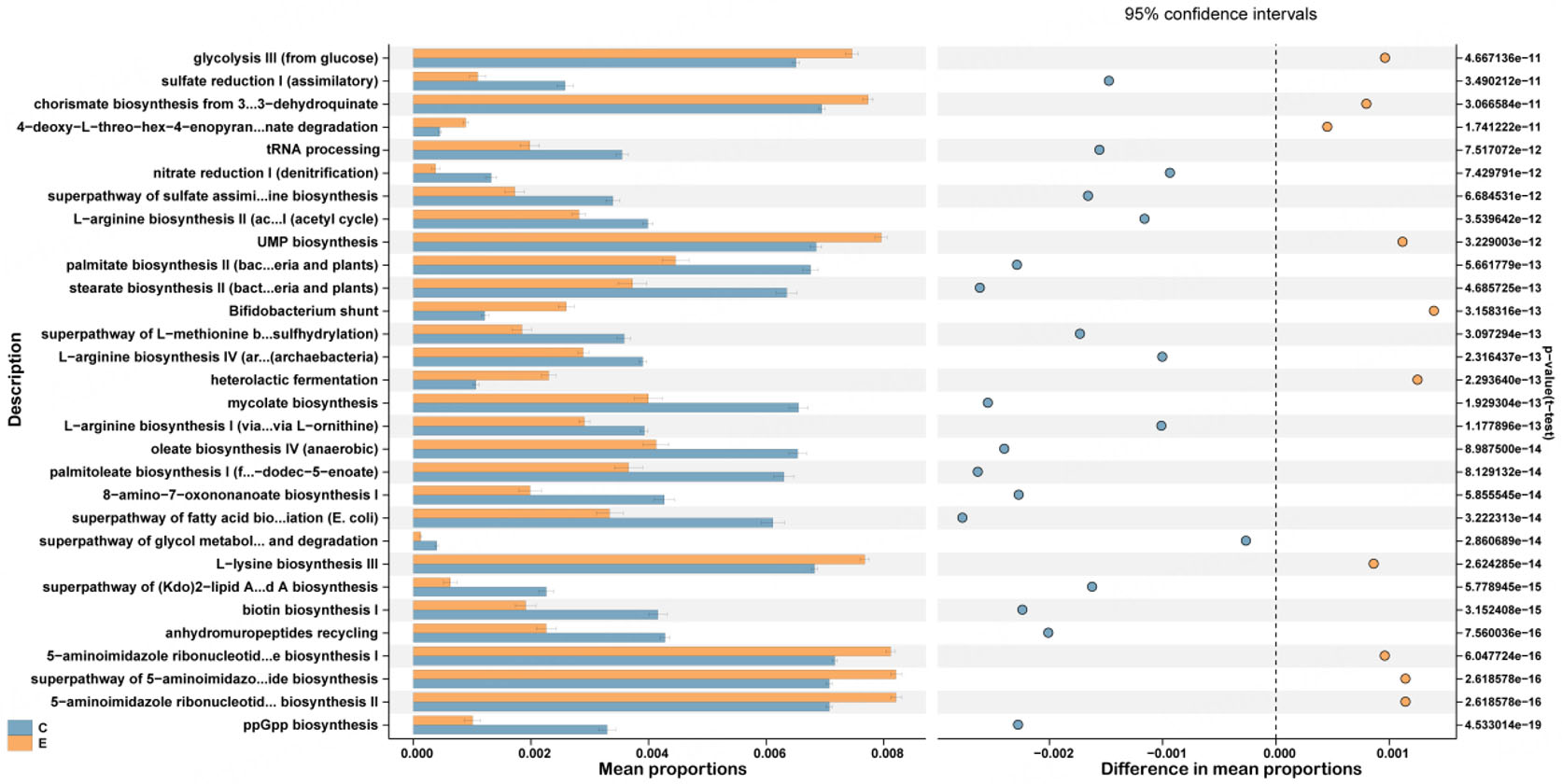
Figure 5. Functional prediction for the oral microbiome of the Con and ET groups. The top 30 pathways with significantly different abundances identified using PICRUSt2 are shown. ppGpp: Guanosine 5'-diphosphate 3'-diphosphate; E.coli: Escherichia coli; tRNA: transfer RNA; UMP: uridine monophosphate; PICRUSt2: Phylogenetic investigation of communities by reconstruction of unobserved states 2.
Cooccurrence networks of the oral microbial communities
To elucidate the interrelationships within the oral microbiota between the Con and ET groups, we constructed microbial co-occurrence networks based on ASV data. This approach enables inference of microbial co-abundance patterns, identification of potential symbiotic relationships, and characterization of taxonomic associations. The topological characteristics of the two groups of microbiota ecological networks are shown in Supplementary Table 1. Distinct topological differences were observed between the two networks. The Con network was more complex and connected, with 178 nodes and 1,025 edges, compared to the ET network, which contained 106 nodes and only 135 edges. The Con network also demonstrated higher network density (0.07 vs. 0.03) and a greater average degree (11.52 vs. 2.93), but lower modularity (0.53 vs. 0.78), consistent with a more cohesive and interdependent microbial architecture. Module analysis further underscored these structural disparities: 43 modules were identified in the Con network versus 53 in the ET group, indicating higher fragmentation and reduced functional integration in the latter [Figure 6]. Correspondingly, the ET network was characterized by reduced overall connectivity and a higher proportion of negative correlations, suggesting a transition towards a competitive and ecologically disrupted state.
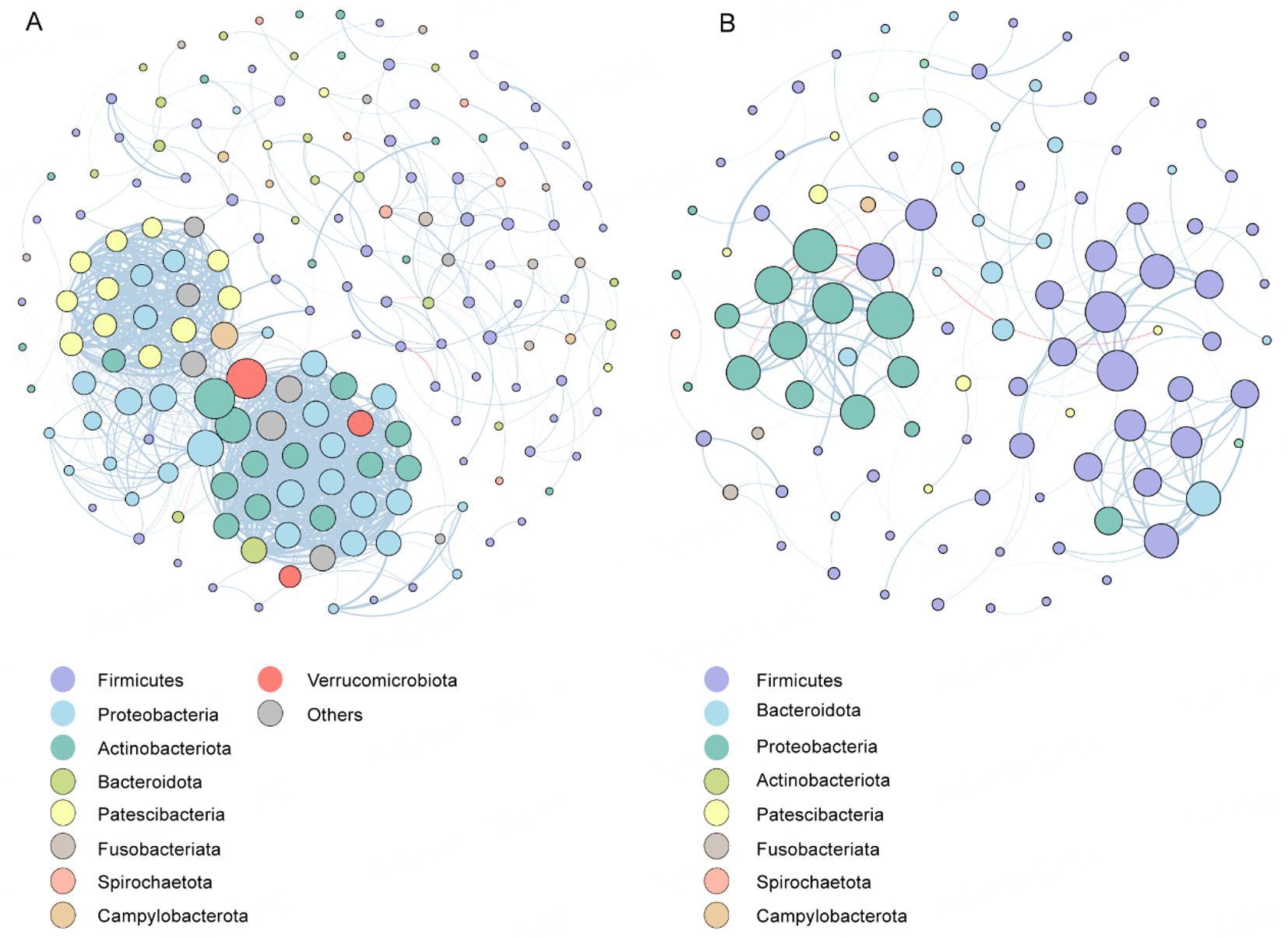
Figure 6. Co-occurrence networks in Con and ET groups. (A) Correlation-based network analysis in the Con group. (B) Correlation-based network analysis in the ET group. The node colors and sizes indicate degree. Edges represent positive (blue) or negative (red) co-occurrence relationships. Edge width is proportional to the absolute value of the correlation coefficient. All correlations shown are statistically significant (adjusted P < 0.05, |r| > 0.6).
Machine learning identification of key microbial biomarkers
LEfSe analysis (LDA > 4.0, P < 0.05) identified ten bacterial genera as potential biomarkers. To refine these into optimal biomarkers and construct a machine learning model for discriminating between ET abusers and non-users, feature selection was performed on the training set using three independent methods: RF, Stepglm, and glmBoost. These methods yielded three distinct feature sets: RF selected ten genera, Stepglm selected five genera, and glmBoost selected seven genera. Rather than taking a consensus approach, we evaluated all three feature sets by training them with eight different classifiers, generating 24 unique model configurations. All models were assessed using stratified 10-fold cross-validation on the training set and final performance was evaluated on the independent test set. The optimal model - a RF classifier trained on the glmBoost-selected feature set - was selected based on the highest cross-validation AUC and test set accuracy [Supplementary Table 2]. Consequently, the seven genera from the glmBoost feature set were designated as the final biomarker panel, which demonstrated highly accurate classification with a cross-validation AUC of 1 on the training set and achieved an AUC of 1.00 and 100% accuracy on the held-out test set within this study cohort [Figure 7A]. A Naive Bayes model built on RF-selected features also exhibited strong performance (AUC = 1.00, accuracy = 0.92) [Figure 7A]. Based on overall performance, the RF model with glmBoost feature selection was chosen as the final model. The seven selected biomarker genera included: Prevotella_7, Rothia, Neisseria, Veillonella, Haemophilus, Actinomyces, and Fusobacterium [Figure 7B]. The confusion matrix for the RF model on the test set [Figure 7C] demonstrated accurate classification, with 13 true positives and 13 true negatives.
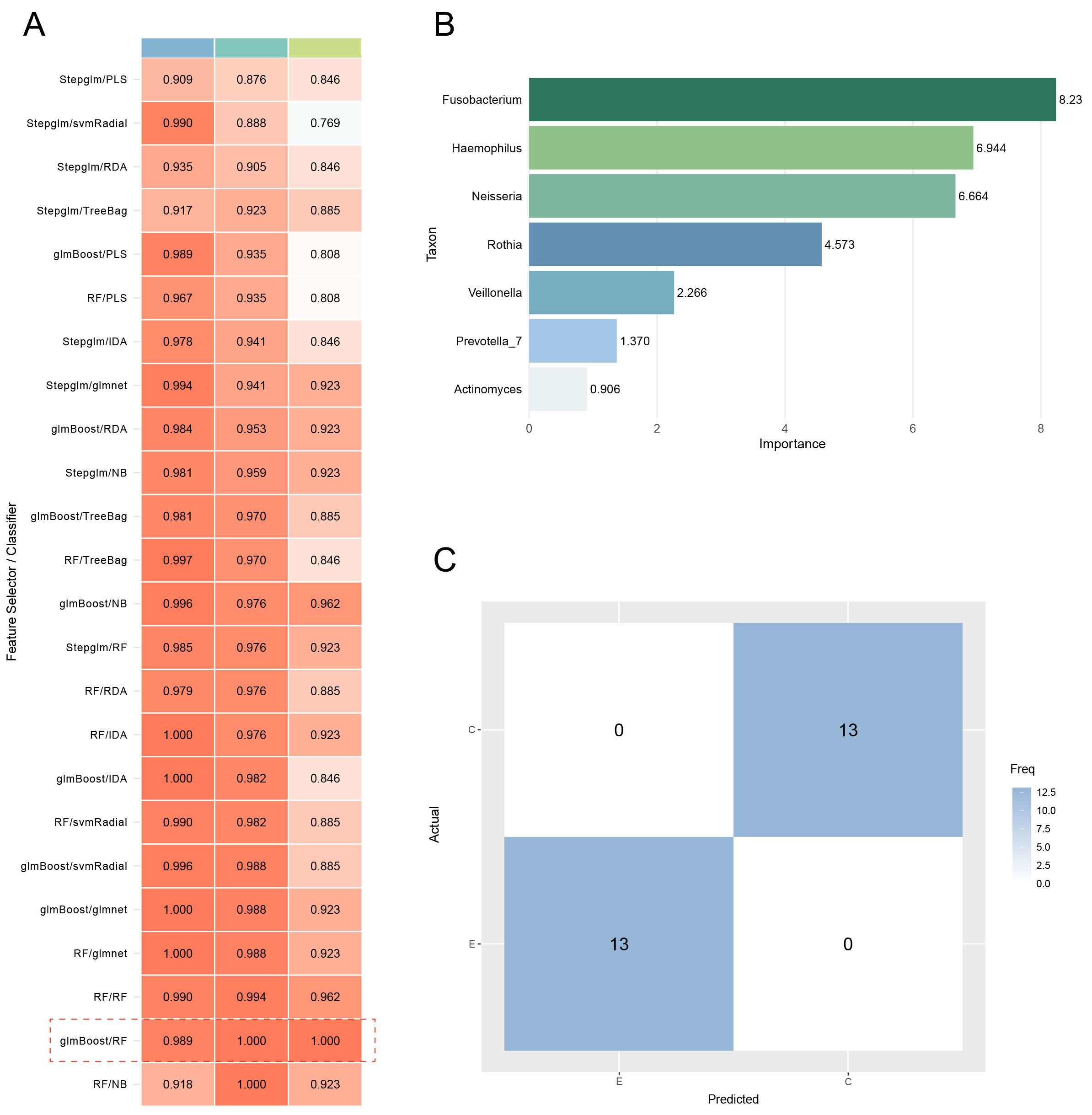
Figure 7. The evaluation of the predictive model. (A) The AUC values of multiple machine learning algorithms combined on two datasets. (B) Feature importance bar plot. (C) Confusion matrices for RF model. AUC: Area under the curve; RF: random forest; GLMBoost: generalized linear model boosting; LDA: linear discriminant analysis; NB: naive bayes; PLS: partial least squares; RDA: regularized discriminant analysis; CV: cross-validation; glmnet: generalized linear model with elastic net regularization; svmRadial: support vector machine with radial basis function kernel; NB: naive bayes; Stepglm: stepwise generalized linear model.
DISCUSSION
This study aimed to identify alterations in the oral microbiome associated with ET abuse by comparing affected individuals to healthy controls through 16S rRNA gene sequencing. Significant differences in microbial community structure were observed, characterized by significantly reduced species richness and diversity alongside a marked expansion of putative pathobionts in the ET group. These results suggest that exposure to ET is associated with disrupted oral homeostasis, thereby increasing the risk of oral diseases in abusers.
As a highly heterogeneous ecological niche, the oral cavity harbors diverse microbial communities whose composition is shaped by both host-related factors and external environmental influences[17]. Under pathological conditions or environmental stressors, metabolic byproducts and immune mediators modify the oral microenvironment, thereby contributing to oral dysbiosis[18]. The association between substance abuse and poor oral health is well-documented in terms of dental caries and periodontal diseases. Methamphetamine users have been shown to exhibit significantly higher rates of untreated caries, any periodontitis, and severe periodontitis compared to non-users[19]. Alterations in the oral microbiome of ET abuse could result from impairment of the different constituents involved in maintaining these communities in a commensal state. In our study, oral microbiota of ET users demonstrated pronounced proliferation of putative pathobionts and caries-associated taxa, including Actinomyces[20], Rothia[21], Atopobium[22], Cryptobacterium[23], and Streptococcus. Rothia plays a pathological role in the development of tooth decay and may provide essential substrates for pro-inflammatory oral bacteria[21]. Atopobium was more abundant in the supragingival microbiota of children with dental caries than in those without dental caries[22]. Our findings demonstrate that the altered bacterial taxa in ET users are frequently associated with periodontal disease and dental caries, suggesting that ET abuse may lead to oral diseases. This microbial profile resembles patterns observed in populations at higher risk for oral diseases, suggesting a potential vulnerability that warrants future clinical investigation with direct assessment of oral health outcomes.
The observed oral microbial shifts associated with chronic ET use may be mechanistically linked to the pharmacological action of ET on GABAA receptors. ET potentiates GABAA receptor function, leading to sedative and anesthetic effects[1]. Beyond its central nervous system activity, GABAergic signaling is also present in the peripheral nervous system, including parasympathetic pathways that regulate salivary gland function[24]. ET-induced hyposalivation would lead to increased oral pH (reduced buffering capacity), decreased clearance of dietary carbohydrates, and reduced delivery of antimicrobial factors - collectively creating a dysbiotic oral microenvironment that favors pathogenic overgrowth and disrupts the commensal microbial community, contributing to oral ecological imbalance[25,26]. Future mechanistic studies should directly measure salivary flow rate, pH, and inflammatory markers in ET users to test these hypotheses.
Studies have shown that tobacco and cannabis smoke exposure significantly reduced Neisseria, Leptotrichia, Capnocytophaga, Fusobacterium and Aggregatibacter within the oropharyngeal microbiota[27-29]. This pattern was echoed in our comparison between ET users and non-users. The taxonomic profile parallels observations in tobacco and opium users, where comparative analyses revealed decreased Proteobacteria and elevated Actinobacteria and Firmicutes abundance[11,30]. Methamphetamine use has been linked to increased abundances of caries-associated taxa such as Negativicutes, Veillonella[14], while opioid use has been associated with shifts in Prevotella and Veillonella[31]. These results suggest that long-term exposure to ET may also be associated with alterations in the oral flora, potentially through modification of the oral microenvironment, which in turn affects oral health. Based on its potential role in health and disease, the salivary microbiome harbors a great potential for being used as a health monitor or disease diagnostic tool.
In terms of the overall trend of functional prediction, ET users showed substantial changes in metabolic pathways and significant changes in the ability to metabolize and synthesize substances in the dental plaque microecology compared to healthy controls. The enrichment of pathways associated with carbohydrate fermentation, amino acid biosynthesis, and nucleotide metabolism in the ET group suggests a microbial community adapted to a more nutrient-competitive environment. Ecological network analysis was employed to investigate potential ecological interactions within the oral microbiota and to assess the impact of ET exposure. Ecological network analysis can be used to illustrate the effects of ET exposure on oral microbial communities and understand potential ecological associations between them. Compared to the ET group, the Con group was characterized by a greater complexity, mainly in terms of total nodes, total links, and average degree. These topological differences carry ecological significance. In microbial ecology, higher network complexity (more nodes and edges) and greater connectivity (higher average degree) are often associated with increased community stability and resilience to environmental perturbations, as they represent a greater number of metabolic interdependencies and cooperative interactions among taxa. Conversely, the higher modularity observed in the ET network indicates a more fragmented community structure, where subgroups of microbes (modules) operate more independently with fewer inter-module connections. This fragmentation is frequently interpreted in ecology as a sign of ecosystem stress or degradation, potentially reflecting a loss of key symbiotic interactions that maintain community stability in healthy states. These results suggest that ET exposure is associated with a destabilized microbial community, whereas the control group exhibited a more robust ecological network.
Machine learning, when integrated with microbiome analysis, holds significant promise for forensic applications[32]. Previous studies have suggested that individuals with substance use disorders have distinct patterns in their oral microbiome, which could be used to differentiate them from healthy subjects[33].
This study still has several limitations. Firstly, the relatively small sample size from a single geographic and ethnic cohort may restrict how widely the findings can be generalized. Secondly, and most critically, it is not possible to fully disentangle the specific effect of ET from that of its delivery vehicle. Although we statistically adjusted for conventional tobacco smoking, liquids in illegal e-cigarettes also contain substances such as propylene glycol, glycerol, and added nicotine, which may influence the oral microbiome independently or synergistically. Consequently, our findings should be interpreted as characterizing the oral microbiota associated with "ET-containing e-cigarette use" rather than ET abuse per se. Thirdly, the cross-sectional nature of our research precludes any causal inference or investigation into the temporal dynamics of dysbiosis. Our ability to explore dose-response relationships was limited as we relied on predefined inclusion criteria regarding frequency and duration of abuse rather than collecting precise, continuous exposure data from individuals. Subsequent research should incorporate detailed quantitative measures of use to elucidate these relationships. Finally, while the machine learning model demonstrated flawless discrimination in our cohort, this exceptional performance requires validation in larger, independent, multicenter populations to confirm its generalizability and practical utility. Future studies should prioritize longitudinal designs, incorporate well-matched control groups (including e-cigarette use without ET), correlate microbial shifts with quantitative measures of exposure, recruit from diverse populations to establish causality, isolate the specific effect of the drug from its vehicle, and validate the diagnostic model. In addition, future investigations should integrate multi-omics approaches (metabolomics, metagenomics) to validate the predicted functional alterations and elucidate the underlying mechanisms. Furthermore, future studies should incorporate body mass index, dietary habits and oral hygiene practices as a matching criterion to further strengthen the robustness of group comparisons.
CONCLUSIONS
In conclusion, this study comprehensively delineated associations between ET use and oral microbiota differences, providing inaugural characterization of oral microbiota variations specific to ET users. Our findings reveal that the compositional structure of the oral bacterial community may have potential as a non-invasive biomarker for effectively distinguishing ET users from healthy individuals. This study highlights the promising potential forensic utility of the human microbiome in revealing individual characteristics.
DECLARATIONS
Acknowledgments
The graphical abstract was drawn by Figdraw (www.figdraw.com) and exported with the image ID: AASUI90b27. We are grateful to all colleagues and volunteers who contributed support for this study.
Authors' contributions
Conceptualization, Data curation, Methodology, Writing - original draft: He M
Data curation, software, visualization: Huang L
Investigation, methodology: Ye L
Investigation, conceptualization: Yang M
Software, visualization: Zhang X
Resources: Liu X, Yang M
Conceptualization, writing - review & editing: Liu C
Supervision, writing - review & editing, funding acquisition: Chen L
All the authors approved the submitted manuscript for publication and other aspects of the work.
Availability of data and materials
Not applicable.
AI and AI-assisted tools Statement
During the preparation of this manuscript, the AI tool ChatGPT was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This study was supported by the National Key Research and Development Program of China (2024YFC3306605), the National Natural Science Foundation of China (32301063), and the Natural Science Foundation of Guangdong Province (2023A1515010598). We thank all volunteers who contributed samples to this study.
Conflicts of interest
Chen L is a Junior Editorial Board member of the Journal of Translational Genetics and Genomics. Chen L was not involved in any steps of the editorial process, notably including reviewer selection, manuscript handling, or decision-making, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008 (5). Informed consent was obtained from all patients for being included in the study. Approval was granted by the Medical Ethics Committee of Southern Medical University (No. 2023103).
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Valk BI, Struys MMRF. Etomidate and its Analogs: a review of pharmacokinetics and pharmacodynamics. Clin Pharmacokinet. 2021;60:1253-69.
2. Desai R, Ruesch D, Forman SA. Gamma-amino butyric acid type A receptor mutations at beta2N265 alter etomidate efficacy while preserving basal and agonist-dependent activity. Anesthesiology. 2009;111:774-84.
3. Sun S, Zhang H, Ye L, et al. Combined analysis of the microbiome and metabolome to reveal the characteristics of saliva from different diets: a comparison among vegans, seafood-based omnivores, and red meat (beef and lamb) omnivores. Front Microbiol. 2024;15:1419686.
4. Uhm J, Hong S, Han E. The need to monitor emerging issues in etomidate usage: the misuse or abuse potential. Forensic Sci Med Pathol. 2024;20:249-60.
5. Xie W, Zhou L, Liu J, et al. How to trace etomidate in illegal E-cigarettes from authentic human hair: identification, quantification and multiple-factor analysis. Forensic Toxicol. 2025;43:74-85.
6. Kim MG, Park SW, Kim JH, et al. Etomidate versus propofol sedation for complex upper endoscopic procedures: a prospective double-blinded randomized controlled trial. Gastrointest Endosc. 2017;86:452-61.
7. Chen Y, Liu J, Song T, et al. A case of fatal poisoning caused by etomidate: evidence from pathological and toxicological analyses. Forensic Sci Med Pathol. 2024;20:1453-7.
8. Ding S, Li K, Han X, et al. Long-term use of etomidate disrupts the intestinal homeostasis and nervous system in mice. Toxicology. 2024;504:153802.
9. Krishnan K, Chen T, Paster BJ. A practical guide to the oral microbiome and its relation to health and disease. Oral Dis. 2017;23:276-86.
10. Peng X, Cheng L, You Y, et al. Oral microbiota in human systematic diseases. Int J Oral Sci. 2022;14:14.
11. Wu Z, Han Y, Caporaso JG, et al. Cigarette smoking and opium use in relation to the oral microbiota in Iran. Microbiol Spectr. 2021;9:e0013821.
12. Nociti FH Jr, Casati MZ, Duarte PM. Current perspective of the impact of smoking on the progression and treatment of periodontitis. Periodontol 2000. 2015;67:187-210.
13. Bostanghadiri N, Kouhzad M, Taki E, et al. Oral microbiota and metabolites: key players in oral health and disorder, and microbiota-based therapies. Front Microbiol. 2024;15:1431785.
14. Yang Y, Yu X, Yang X, et al. Oral microbiota profile of individuals who abuse methamphetamine. Front Cell Infect Microbiol. 2021;11:706961.
15. Jenkins WD, Beach LB, Rodriguez C, Choat L. How the evolving epidemics of opioid misuse and HIV infection may be changing the risk of oral sexually transmitted infection risk through microbiome modulation. Crit Rev Microbiol. 2020;46:49-60.
16. Rajasekaran JJ, Krishnamurthy HK, Bosco J, et al. Oral microbiome: a review of its impact on oral and systemic health. Microorganisms. 2024;12:1797.
17. Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005;43:5721-32.
18. Lamont RJ, Koo H, Hajishengallis G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol. 2018;16:745-59.
19. Hegazi F, Alhazmi H, Abdullah A, et al. Prevalence of oral conditions among methamphetamine users: NHANES 2009-2014. J Public Health Dent. 2021;81:21-8.
20. Zang T, Zhang Z, Liu W, et al. Structural and functional changes in the oral microbiome of patients with craniofacial microsomia. Sci Rep. 2025;15:5400.
21. Vieira AR, Hiller NL, Powell E, et al. Profiling microorganisms in whole saliva of children with and without dental caries. Clin Exp Dent Res. 2019;5:438-46.
22. Al-Hebshi NN, Baraniya D, Chen T, et al. Metagenome sequencing-based strain-level and functional characterization of supragingival microbiome associated with dental caries in children. J Oral Microbiol. 2019;11:1557986.
23. Jiang W, Ling Z, Lin X, et al. Pyrosequencing analysis of oral microbiota shifting in various caries states in childhood. Microb Ecol. 2014;67:962-9.
24. Okubo M, Kawaguchi M. Rat submandibular gland perfusion method for clarifying inhibitory regulation of GABAA receptor. J Pharmacol Sci. 2013;122:42-50.
25. Dawes C, Pedersen AM, Villa A, et al. The functions of human saliva: a review sponsored by the World Workshop on Oral Medicine VI. Arch Oral Biol. 2015;60:863-74.
26. Pei XM, Zhou LX, Tsang MW, Tai WC, Wong SC. The oral microbial ecosystem in age-related xerostomia: a critical review. Int J Mol Sci. 2024;25:12815.
27. Wang D, Feng Y, Yang M, et al. Variations in the oral microbiome and metabolome of methamphetamine users. mSystems. 2024;9:e0099123.
28. Luo Z, Fitting S, Robinson C, et al. Chronic cannabis smoking-enriched oral pathobiont drives behavioral changes, macrophage infiltration, and increases β-amyloid protein production in the brain. EBioMedicine. 2021;74:103701.
29. Charlson ES, Chen J, Custers-Allen R, et al. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS ONE. 2010;5:e15216.
30. Wu J, Peters BA, Dominianni C, et al. Cigarette smoking and the oral microbiome in a large study of American adults. ISME J. 2016;10:2435-46.
31. Barkus A, Baltrūnienė V, Barkienė L, et al. Exploring the gut and oral microbiomes in psychoactive substance use: a scoping review of clinical studies. J Neurochem. 2025;169:e70165.
32. Metcalf JL, Xu ZZ, Bouslimani A, Dorrestein P, Carter DO, Knight R. Microbiome tools for forensic science. Trends Biotechnol. 2017;35:814-23.
33. Kosciolek T, Victor TA, Kuplicki R, et al. Individuals with substance use disorders have a distinct oral microbiome pattern. Brain Behav Immun Health. 2021;15:100271.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].