


Macrophage STING activation induces cardiomyocyte hypertrophy
 0
0 Abstract
Background: Heart failure is a clinical syndrome caused by underlying cardiac structural or functional impairment, leading to insufficient cardiac output and abnormally elevated intracardiac pressures in both resting and stress conditions. The stimulator of interferon genes (STING), a transmembrane protein in the endoplasmic reticulum, plays a pivotal role in initiating and sustaining chronic inflammatory responses.
Aim: We investigated whether macrophage-derived STING contributes to the pathogenesis of heart failure with preserved ejection fraction (HFpEF).
Methods and Results: Activated STING was detected in heart tissues of HFpEF mice. To study macrophage-cardiomyocyte interactions, we constructed a co-culture system of bone marrow-derived macrophages (BMDMs) and HL-1 cardiomyocytes. STING activation in BMDMs, either via a gain-of-function mutation or an agonist, promoted hypertrophy of co-cultured HL-1 cells. Mechanistically, macrophage STING activation increased Z-DNA binding protein 1 (ZBP1) expression in HL-1 cells and facilitated ZBP1-mediated inflammasome assembly.
Conclusion: These findings provide novel insight into HFpEF pathogenesis and suggest that macrophage STING may serve as a potential therapeutic target.
Keywords
INTRODUCTION
Cardiovascular diseases have emerged as the predominant cause of mortality among both urban and rural populations in China, representing 44.26% and 46.74% of deaths, respectively[1]. When the heart is subjected to prolonged pressure, it undergoes adaptive remodeling processes. These include hypertrophy of cardiomyocytes, increased stress on the ventricular walls, and microvascular angiogenesis, all aimed at meeting its physiological demands[2]. Heart failure with preserved ejection fraction (HFpEF) is a highly prevalent clinical syndrome associated with substantial morbidity and mortality, representing nearly 50% of all heart failure-related hospitalizations[3]. Among them, HFpEF constitutes half of the number of heart failure patients admitted to hospitals, with a five-year survival rate of less than 50%, presenting an increasingly significant global medical burden. In HFpEF, macrophage infiltration into the myocardium adversely impacts left ventricular diastolic function, leading to interstitial fibrosis of the heart. Infiltrated macrophages directly induce cardiomyocyte hypertrophy and endothelial dysfunction through the imbalance of M1/M2 phenotypes, alongside the continuous release of inflammatory factors. Also, macrophages secrete profibrotic mediators, which stimulate cardiac fibroblasts to differentiate into myofibroblasts. This pathological process results in hypertrophy and stiffness of the cardiomyocytes by altering paracrine signaling in cardiomyocytes[4]. Emerging evidence indicates that the accumulation of monocytes/macrophages in the heart and their associated inflammatory responses are pivotal in the pathogenesis of hypertension-induced cardiac remodeling. Macrophages that infiltrate the myocardium promote the production of inflammatory cytokines, thereby exacerbating hypertension-induced diastolic dysfunction[5]. Thus, exploring novel mechanisms and targets in macrophages could provide promising therapeutic strategies for HFpEF.
The stimulator of interferon genes (STING) is an endoplasmic reticulum-anchored transmembrane protein, exerting a significant transduction role in innate immunity[6]. STING can be activated upon binding to cyclic dinucleotides, such as cyclic diadenosine monophosphate, which are secreted by intracellular bacteria[7]. This activates TANK binding kinase 1 (TBK1) and the downstream transcription factors [interferon regulatory factor 3 (IRF3) and nuclear factor kappa-B (NF-κB)], subsequently leading to an acceleration in the expression of type I interferons and pro-inflammatory factors, thereby stimulating adaptive immunity[8-10]. Numerous studies recently have disclosed that the activation of STING in macrophages exerts detrimental effects in cardiovascular diseases, autoimmune disorders, and non-alcoholic liver diseases[11-14]. Among patients with rheumatoid arthritis, decreased expression of Pol β in macrophages results in abnormal DNA accumulation. Subsequently, this activates the cyclic GMP-AMP synthase (cGAS)-STING-NF-κB pathway, upregulates the expressions of NOD-like receptor thermal protein domain associated protein 3 (NLRP3), interleukin (IL)-1β, and IL-18, exacerbates macrophage pyroptosis, and facilitates the progression of rheumatoid arthritis[11]. In aortic dissection, the DNA from smooth muscle cells and mitochondria leaks into the cytosol to initiate the STING-TBK1-IRF3 signaling cascade in macrophages, resulting in diminished vascular fibroelasticity and promoting the progression towards aortic dissection rupture[12]. Additionally, Cyclic GMP-AMP (cGAMP) accumulation in atherosclerotic plaque can induce STING-mediated inflammation and contribute to the pathogenesis and progression of atherosclerosis[15]. These findings demonstrate that STING plays a detrimental role in enhancing pro-inflammatory activation of macrophages. However, whether macrophage-derived STING participates in HFpEF remains unexplored.
In this study, we combined metabolic stress (high-fat diet-induced obesity) and mechanical stress [inhibition of nitric oxide (NO) synthase using Nω-nitro-L-arginine methyl ester (L-NAME)] to establish an HFpEF mouse model. Our findings showed activated STING in the heart tissues of HFpEF mice. Further, to focus on the interaction between macrophage and cardiomyocyte, we construct the co-culture system of bone marrow-derived macrophages (BMDMs) and HL-1 cells. STING activation in BMDMs, either by a gain-of-function mutation or by an agonist, promotes hypertrophy of HL-1 cells in co-culture. Mechanically, macrophage STING activation induced an increase in Z-DNA binding protein 1 (ZBP1) expression of HL-1 cells, which is closely associated with inflammasome activation. These findings offer a novel perspective on the pathogenesis of HFpEF and indicate that macrophage STING may serve as a promising therapeutic target for HFpEF.
METHODS
Animal experiments
Wild-type C57BL/6J male mice were procured from GemPharmatech Co., Ltd. (Jiangsu, China). Sting-/- mice (Stock#025805) of the C57BL/6J strain were obtained from Jackson Laboratory (Bar Harbor, ME, USA). The animals were maintained at the Experimental Animal Center of China Pharmaceutical University under standardized housing conditions. We confirm that all methods in experimental animals were carried out in compliance with the regulations of the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. All experimental protocols were approved by the Ethics Association of China Pharmaceutical University (approval no. 2020-11-007). Twelve Sting-/- and wild-type C57BL/6J mice were used to isolate BMDMs. The establishment of HFpEF mice was based on a previous study[16]. Twelve 8-week-old wild-type male C57BL/6J mice were randomly assigned to two groups: high-fat diet + L-NAME group and control group, n = 6 in each group. L-NAME was dissolved in sterilized water (0.5 g/L), and the reagent was completely dissolved using an ultrasonic instrument, with multiple preparations in small quantities[16]. The control group received standard chow and tap water ad libitum, whereas the model group was fed a high-fat diet and provided with drinking water supplemented with L-NAME for 10 weeks. After 10 weeks, the cardiac function of the mice was detected. The mice were anesthetized and sacrificed, and the cardiac tissues were immediately frozen or embedded in a -80 °C refrigerator.
Generation of myeloid STING gain-of-function mutation mice
The strategy of generating conditional STING N153S knock-in mice has been described in our previous study[9]. Crossing homozygous STING N153S floxed mice and LysM-Cre mice (Strain NO. T003822, procured from GemPharmatech Co., Ltd.) led to myeloid STING N153S mice (heterozygous STING N153S floxed; LysM-Cre mice, StingLysM N153S) and heterozygous STING N153S floxed mice (StingN153S floxed).
Detection of cardiac function
Noninvasive assessment of cardiac performance was conducted by transthoracic echocardiography (VisualSonics, Toronto, Canada). M-mode echocardiograms acquired from long-axis sections were used to calculate left ventricular ejection fraction (EF), fractional shortening (FS), along with other indices indicative of diastolic function. Apical four-chamber views were obtained for diastolic function measurements, and the maximum blood flow velocity E peak in the early diastole of the left ventricle and the maximum blood flow velocity A peak in the atrial systole of the mitral valve were measured and analyzed.
BMDM’s isolation and culture
BMDMs were isolated using a well-established protocol adapted from a previous study[17]. To generate bone marrow-derived macrophages, femurs and tibias were collected from mice and flushed to obtain bone marrow cells. After filtration and red blood cell lysis, cells were centrifuged and the supernatant was discarded. The remaining cells were resuspended and cultured in the presence of recombinant mouse macrophage colony-stimulating factor (M-CSF) (Gibco, 315-02) at a final concentration of 10 ng/mL, which was continuously supplied to drive macrophage differentiation. Differentiated macrophages were then harvested for subsequent analyses.
Establishment of a co-culture system
Bone marrow cells were isolated from wild-type, Sting-/-, StingN153S floxed or StingLysM N153S mice and differentiated into BMDMs over 6 days. For co-culture experiments, 2 × 105 BMDMs were plated in the upper compartment of transwell inserts. BMDMs from wild-type and Sting-/- mice were treated with 1 μg/mL cGAMP following permeabilization with digitonin. In contrast, BMDMs from StingN153S floxed and StingLysM N153S mice were not treated. Subsequently, 5 × 104 HL-1 cells were seeded in the lower chamber of the transwell plates.
Transcriptome
After 24 h of co-culture with BMDMs, HL-1 cells were collected for transcriptomic analysis. Total RNA from each group (n = 3 biological replicates) was isolated using TRIzol reagent (Invitrogen) and subjected to RNA sequencing on the NovaSeq X Plus platform by LC-Bio Technology (Shanghai, China). For downstream analysis, genes with an adjusted P value below 0.05 were considered significantly differentially expressed based on Gene Ontology annotations (http://geneontology.org/).
Immunofluorescence staining
Following stimulation, HL-1 cells were briefly fixed with 4% paraformaldehyde (PFA) and subsequently incubated with primary antibodies targeting α-actinin (A7811, Sigma) at a dilution of 1:200. After washing, the cells were treated with corresponding secondary antibodies at room temperature. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) for 5 min. Finally, cell imaging was performed using a fluorescence microscope (Leica, Germany), and quantitative analysis was carried out with ImageJ software.
Wheat germ agglutinin staining
Tissue sections underwent a quick dewaxing and hydration process, followed by antigen retrieval using
Western blot analysis
According to a previously validated procedure, Western blot analysis was performed[17]. In brief, total protein was extracted from heart tissues or cells, and concentrations were quantified. Extracted proteins were then separated by gel electrophoresis and transferred onto nitrocellulose membranes. Following blocking with 5% non-fat dry milk in Tris Buffered Saline with Tween-20 (TBST), membranes were incubated overnight at 4 °C with primary antibodies diluted. Primary antibodies for STING (CST#13647), phospho-STING (CST#72971), phospho-TBK1 (CST#5483), TBK1 (CST#3504), phospho-IRF3 (CST#4947), and IRF3 (CST#4302) were purchased from Cell Signaling Technology (CST), Danvers, MA, USA. Primary antibodies for B-type Natriuretic Peptide (BNP) (GB11667) were purchased from Servicebio and primary antibodies for Atrial Natriuretic Peptide (ANP) (27426-1-AP), Myosin Heavy Chain 7 (MYH7) (22280-1-AP) and α-Tubulin (11224-1-AP) were purchased from Proteintech Group. Following this, membranes were then incubated with Horseradish Peroxidase (HRP)-conjugated secondary antibodies for 60 min at room temperature, and immunoreactive bands were visualized using an enhanced chemiluminescence (ECL) detection system.
Quantitative reverse-transcription polymerase chain reaction
Quantitative reverse-transcription polymerase chain reaction (RT-qPCR) was carried out using methods similar to those previously reported[9]. For normalization of quantitative polymerase chain reaction (qPCR) results, β-actin was used as the internal reference gene. The primer sequences, synthesized by Sangon Biotech Co., Ltd. (Shanghai, China), were as follows:
• Anp: forward AACCTGCTAGACCACCTGGA, reverse GGCAGTGTAACTCTTCTGCAT;
• Bnp: forward GTCAGTCGTTTGGGCTGTAAC, reverse AGACCCAGGCAGAGTCAGAA;
• Myh7: forward ACTGTCAACACTAAGAGGGTCA, reverse TTGGATGATTTGATCTTCCAGGG;
• β-actin: forward CCGTGAAAAGATGACCCAGA, reverse TACGACCAGAGGCATACAG.
Enzyme-linked immunosorbent assay
First, microplate wells were coated with 100 μL of an anti-Interferon-β (IFN-β) capture antibody diluted in coating buffer and incubated overnight at 4 °C. On the following day, wells were rinsed three times with 250 μL of wash buffer each. Afterward, the wells were blocked with 100 μL per well of enzyme-linked immunosorbent assay (ELISA) diluent for 1 h. Subsequently, 100 μL of samples or standards were added to each well and incubated for 2 h. The contents were aspirated, and the wells were washed before adding 100 μL per well of detection antibody diluted in buffer, followed by incubation for 1 h. After aspiration and washing, 100 μL of diluted streptavidin-HRP was added to each well and incubated for 30 min. Following washing, 100 μL of 1× 3,3’,5,5’-Tetramethylbenzidine (TMB) substrate solution was added to each well and incubated for 15 minutes in the dark. The reaction was stopped by adding 100 μL of stop solution per well, and absorbance was measured at 450 nm using a microplate spectrophotometer.
Statistical analysis
All data are shown as mean values with standard error of the mean (SEM). Parametric statistical analyses were performed using GraphPad Prism 9.0 (GraphPad, San Diego, CA, USA). One-way analysis of variance (ANOVA) with Bonferroni correction was applied when more than two groups were compared, whereas Student’s t-test was used for pairwise comparisons. Statistical significance was determined at P < 0.05.
RESULTS
STING was activated in the heart tissues of HFpEF mice
Systemic inflammation and NO imbalance are of vital importance for the development of HFpEF. We adopted the combined effect of metabolic stress (obesity induced by a high-fat diet) and mechanical stress (NO synthase inhibitor, L-NAME) to create the HFpEF mouse model[16]. To generate the HFpEF mouse model, a high-fat diet and 0.5 g/L L-NAME drinking water were selected and maintained for 10 weeks. Echocardiography was employed to detect changes in cardiac functions [EF, FS, early to late diastolic flow velocity ratio (E/A)] and structural indicators [Ventricular Internal Dimension in Systole (IVSd), Left Ventricular Internal Dimension in Diastole (LVIDd)]. Compared with the control (Ctrl) group, cardiac diastolic function indicators E/A in the L-NAME+high-fat diet (HFD) group were significantly reduced [Figure 1A and B]; however, the cardiac systolic function indicators EF and FS showed no significant changes [Figure 1C-E], suggesting reduced left ventricular diastolic function. In addition, the cardiac structural indicator IVSd in the L-NAME+HFD group was significantly increased [Figure 1F]; however, LVIDd showed no significant changes [Figure 1G]. The pathological analysis of WGA staining and Sirius Red staining also showed the increased cardiomyocyte size and myocardial fibrosis in the L-NAME+HFD group [Figure 1H and I]. These indicated left ventricular wall hypertrophy and abnormal diastolic function; the HFpEF model was successfully established. To investigate the relationship between the STING signaling pathway and HFpEF, we detected the phosphorylation of STING and downstream IRF3. The results demonstrated that the phosphorylation of STING and IRF3 in the cardiac tissue of the L-NAME+HFD group was significantly elevated [Figure 1J and K]. We also observed that the expression of pro-inflammatory factors, including tumour necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IFN-β, was markedly increased in the L-NAME+HFD group [Figure 1L]. Collectively, these results proved that in HFpEF, STING was activated in heart tissues.
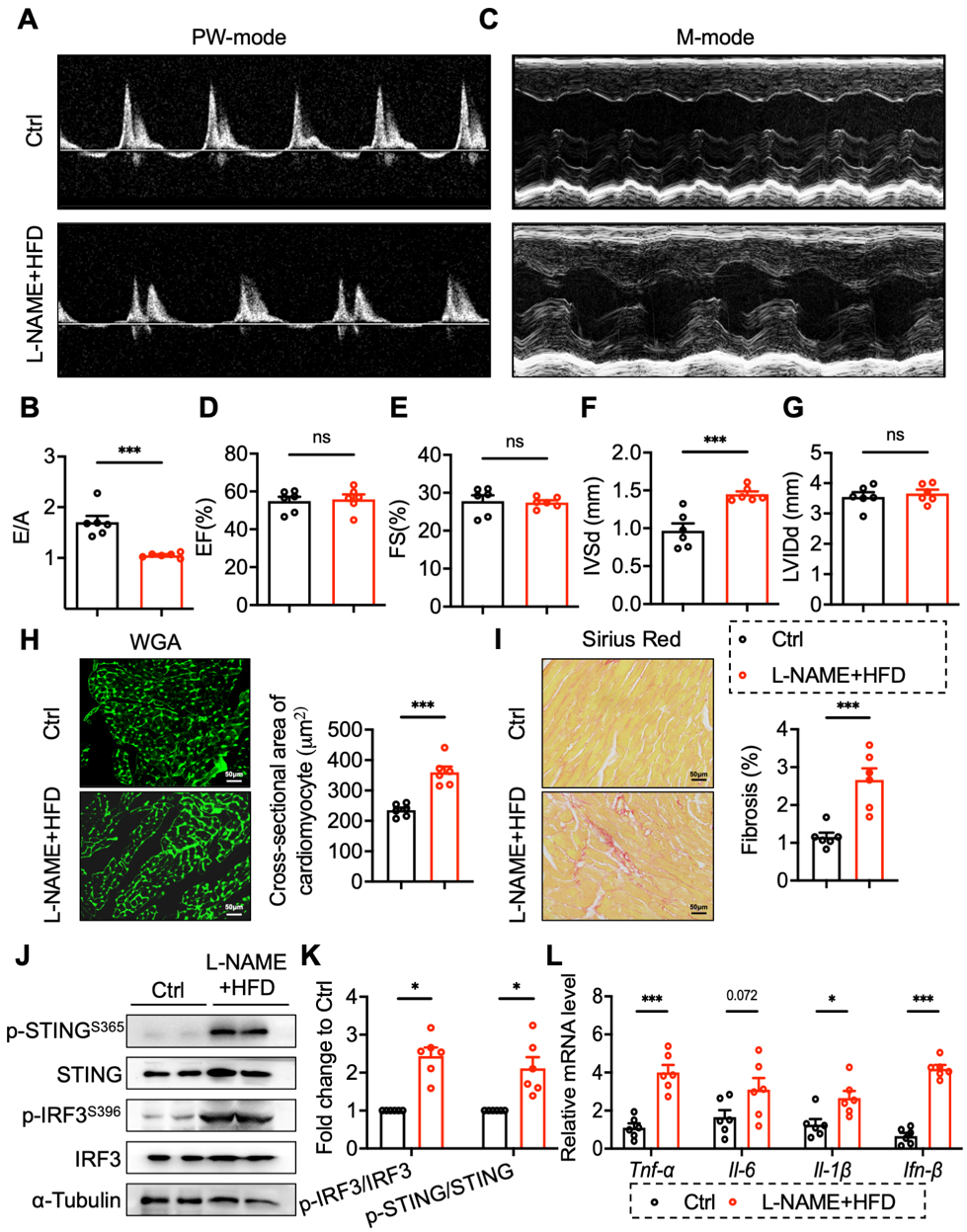
Figure 1. STING was activated in the heart tissues of HFpEF mice. (A and B) PW mode analysis of early to late diastolic flow velocity ratio (E/A); (C-G) M-mode analysis of left ventricular ejection fraction (EF) and fractional shortening (FS); (H) Representative image and analysis of WGA staining; (I) Representative image and analysis of Sirius Red staining; (J and K) Protein and phosphorylated levels of STING (Ser365) and IRF3 (Ser396) in cardiac tissues; (L) mRNA levels of TNF-α, IL-6, IL-1β and IFN-β in cardiac tissues. Mean ± SEM; n = 6 in each group, *P < 0.05, ***P < 0.001, ns, no significance. Student’s t-test for 1B-I, 1K-L. STING: Stimulator of interferon genes; HFpEF: heart failure with preserved ejection fraction; WGA: wheat germ agglutinin; mRNA: messenger RNA; SEM: standard error of the mean; TNF-α: tumour necrosis factor; IL-1β: interleukin-1β; IFN-β: interferon-β; IL-6: interleukin-6; PW: pulsed wave; IRF3: interferon regulatory factor 3; L-NAME: Nω-nitro-L-arginine methyl ester; HFD: high-fat diet.
Gain-of-function mutation of STING in macrophage promotes cardiomyocyte hypertrophy in co-culture
Because global expression of the STING N153S mutation resulted in embryonic lethality, Cre-inducible myeloid-specific STING N153S mutant mice (LysM;STING N153S, hereafter STINGLysM N153S) were generated using Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 (CRISPR/Cas9) technology [Figure 2A]. The activation of the STING pathway was verified [Figure 2B]. To explore the impact of macrophage-specific STING activation on cardiomyocytes, BMDMs and HL-1 cells were co-cultured in a transwell system [Figure 2C]. After 24 h of co-culture, the culture medium was collected and total RNA was extracted from HL-1 cells. The results showed that IFN-β secretion [Figure 2D] and the messenger RNA (mRNA) expression levels of ANP, BNP, and MYH7 [Figure 2E-G] were elevated
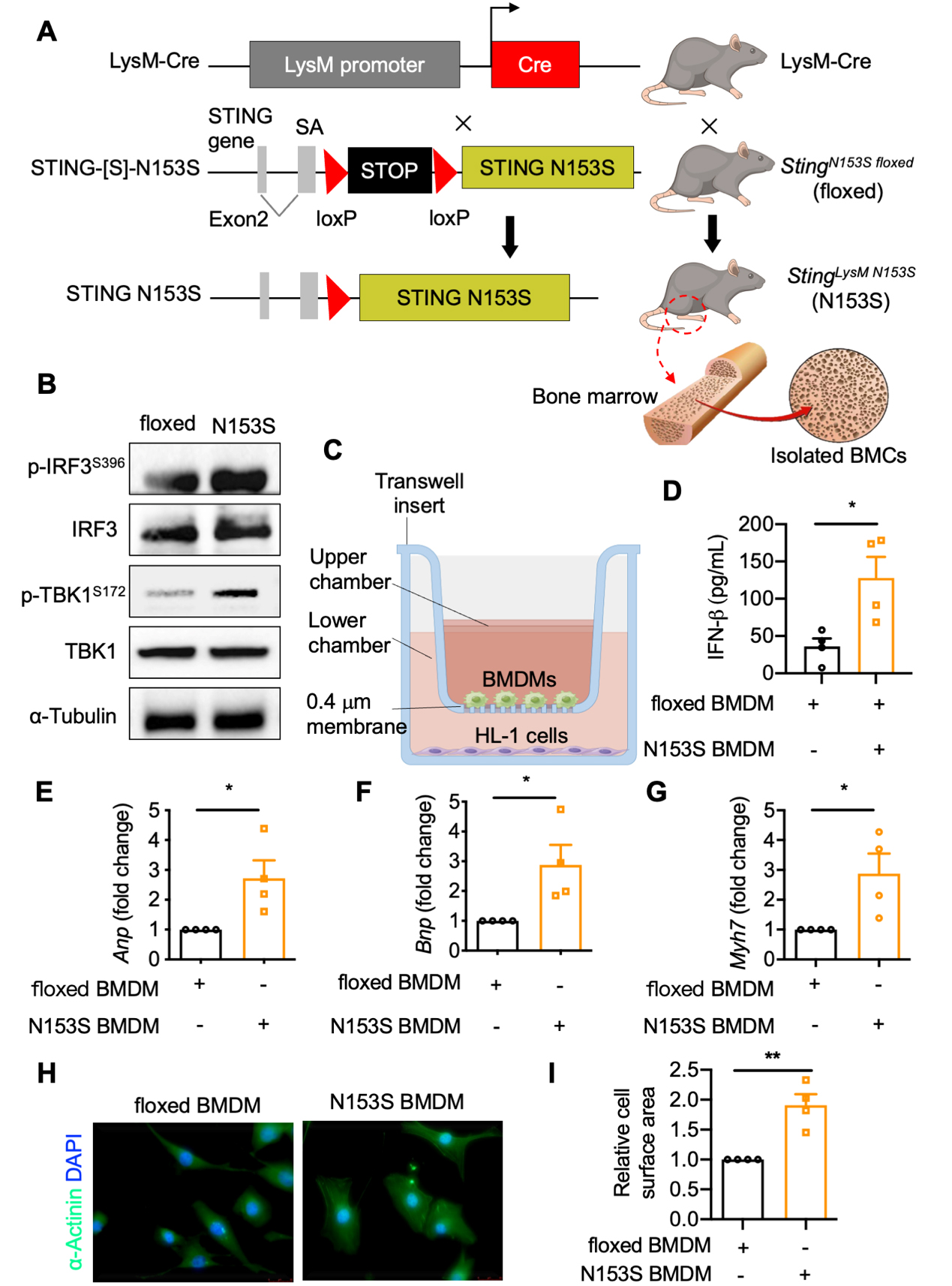
Figure 2. Gain-of-function mutation of STING in macrophage promotes cardiomyocyte hypertrophy in co-culture. (A) StingN153S floxed mice were crossed with LysM-Cre mice to render the myeloid STING N153S mice (StingLysM N153S). Then isolate the BMCs from bone marrow (generated by FigDraw, ID: UPTYU88837); (B) Protein and phosphorylated levels of IRF3 (Ser396) and TBK1 (Ser172) in BMDMs of StingN153S floxed and StingLysM N153S mice; (C) Schematic diagram of co-culture of HL-1 cells and StingN153S floxed and StingLysM N153S BMDMs (generated by FigDraw, ID: OITPS2a3a3); (D) Secreted IFN-β levels in medium of the above co-culture system (n = 4 in each group); (E-G) mRNA levels of ANP, BNP, and MYH7 in HL-1 cells co-cultured with StingN153S floxed and StingLysM N153S BMDMs (n = 4 in each group); (H and I) Cell surface area of HL-1 cells co-cultured with StingN153S floxed and StingLysM N153S BMDMs (n = 4 in each group). Mean ± SEM; *P < 0.05, **P < 0.01. Student’s t-test for 2D-G, 2I. STING: Stimulator of interferon genes; BMDMs: bone marrow-derived macrophages; mRNA: messenger RNA; SEM: standard error of the mean; BMCs: bone marrow cells; IRF3: interferon regulatory factor 3; TBK1: TANK binding kinase 1; IFN-β: interferon-β; ANP: atrial natriuretic peptide; BNP: B-type natriuretic peptide; MYH7: myosin heavy chain 7.
The activation of STING in macrophages induced by cGAMP promotes cardiomyocyte hypertrophy in co-culture
Based on the previous findings of increased activation of the STING pathway in the HFpEF mice, we hypothesized that macrophage STING activation and cardiac injury might have a certain relationship. We used cGAMP as a STING agonist to stimulate BMDM and observe the hypertrophy of HL-1 cells co-cultured with BMDMs. After co-culturing cGAMP-treated BMDMs with HL-1 cells for 24 h, the secretion of IFN-β [Figure 3A] in BMDMs and the mRNA expression of ANP and MYH7 in HL-1 cells [Figure 3B-C] were elevated in the cGAMP group. It was also observed that the protein expression of ANP and BNP was increased in the cGAMP group [Figure 3D]. Furthermore, the size of cardiomyocytes in the cGAMP group was significantly larger than that in the untreated group [Figure 3E and F]. Thus, it can be concluded that macrophage STING activation can promote the cardiomyocyte hypertrophy.
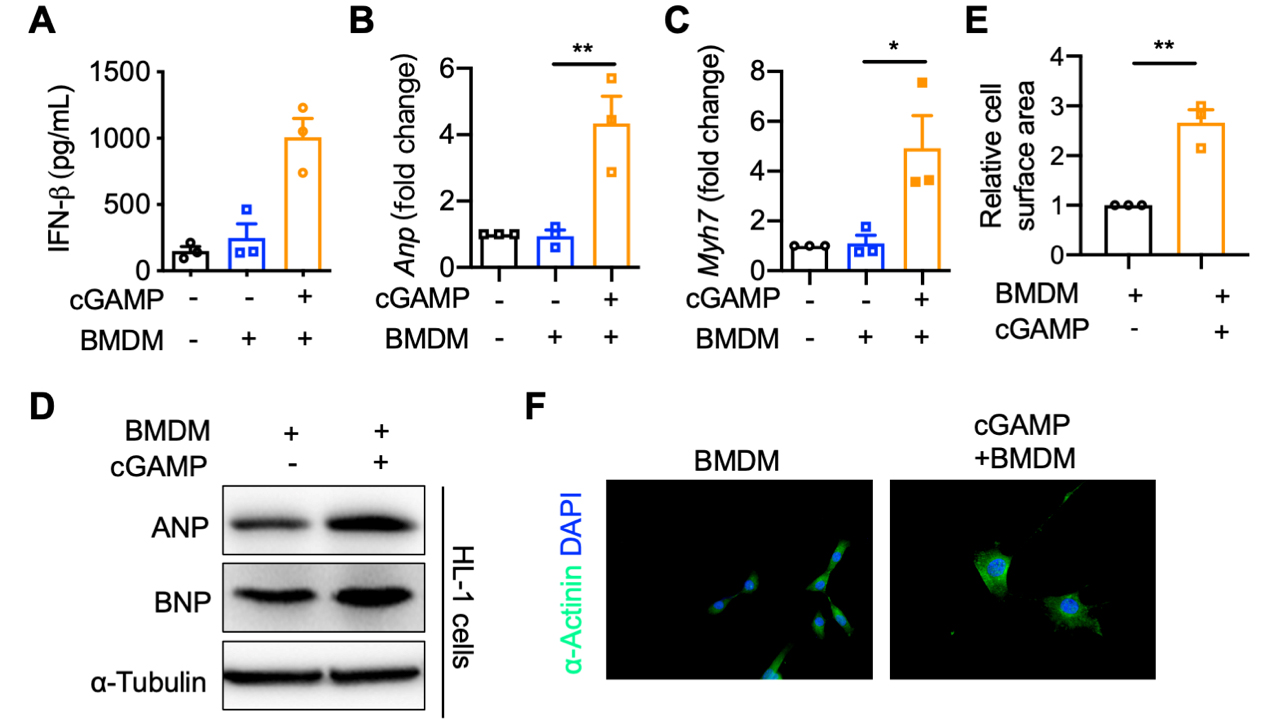
Figure 3. cGAMP-induced STING activation promotes cardiomyocyte hypertrophy in co-culture. BMDMs were pretreated with or without cGAMP (1 μg/mL) for 15 min of permeabilization. Then, HL-1 cells were co-cultured with BMDMs. (A) Secreted IFN-β levels in medium of co-culture system of HL-1 cells and BMDMs (n = 3 in each group); (B and C) mRNA levels of ANP and MYH7 in HL-1 cells co-cultured with BMDMs (n = 3 in each group); (D) Protein levels of ANP and BNP in HL-1 cells co-cultured with BMDMs; (E and F) Cell surface area of HL-1 cells co-cultured with BMDMs (n = 3 in each group). Mean ± SEM; *P < 0.05, **P < 0.01. Student’s t-test for 3A-C, 3E. STING: Stimulator of interferon genes; BMDMs: bone marrow-derived macrophages; mRNA: messenger RNA; SEM: standard error of the mean; cGAMP: cyclic GMP-AMP; IFN-β: interferon-β; ANP: atrial natriuretic peptide; MYH7: myosin heavy chain 7; BNP: B-type natriuretic peptide; DAPI: 4’,6-diamidino-2-phenylindole.
Macrophage STING knockout alleviates the hypertrophic effect of HL-1 cardiomyocytes co-cultured with cGAMP-treated macrophages
To further elucidate the role of macrophage STING in cardiomyocyte hypertrophy, BMDMs were isolated from Sting-/- mice and differentiated in vitro for 5 to 7 days. The experiment was conducted using four groups: WT, WT + cGAMP, Sting-/-, Sting-/-+ cGAMP. A co-culture system was employed, with BMDMs in the upper chamber and HL-1 cells in the lower chamber. The mRNA expression of ANP, BNP, and MYH7 in HL-1 cells was reduced by macrophage STING knockout [Figure 4A-C]. Similar results were also observed in the protein expression of MYH7, ANP, and BNP in HL-1 cells [Figure 4D]. Immunofluorescence staining demonstrated that cardiomyocyte surface area in the Sting-/-+ cGAMP group was significantly smaller than in the WT + cGAMP group, with no significant difference compared to the WT group [Figure 4E and F]. These findings suggest that macrophage STING knockout protects against cardiomyocyte hypertrophy.
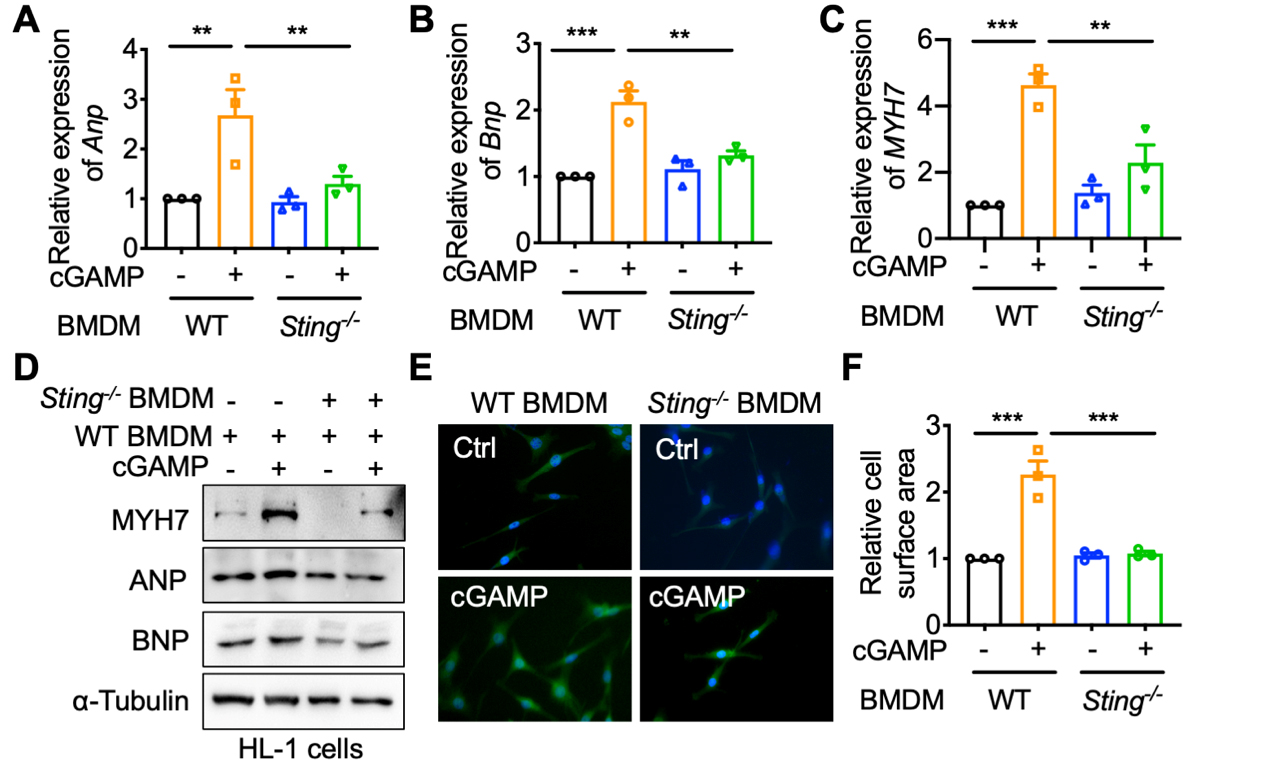
Figure 4. Macrophage STING knockout alleviates the hypertrophic effect of HL-1 cardiomyocytes co-cultured with cGAMP-treated macrophages. WT or Sting-/- BMDMs were pretreated with or without cGAMP (1 μg/mL) for 15 min permeabilization. Then, HL-1 cells were co-cultured with BMDMs. (A-C) mRNA levels of ANP, BNP, and MYH7 in HL-1 cells co-cultured with WT or Sting-/- BMDMs (n = 3 in each group); (D) Protein levels of ANP, BNP, and MYH7 in HL-1 cells co-cultured with WT or Sting-/- BMDMs; (E and F) Cell surface area of HL-1 cells co-cultured with WT or Sting-/- BMDMs (n = 3 in each group). Mean ± SEM; **P < 0.01, ***P < 0.001. One-way ANOVA with Bonferroni correction for 4A-C, 4F. STING: Stimulator of interferon genes; BMDMs: bone marrow-derived macrophages; mRNA: messenger RNA; SEM: standard error of the mean; cGAMP: cyclic GMP-AMP; WT: Wild-type; ANP: atrial natriuretic peptide; BNP: B-type natriuretic peptide; MYH7: myosin heavy chain 7; ANOVA: analysis of variance.
Macrophage STING promotes ZBP1-mediated inflammasome activation in cardiomyocytes
STING activation by the agonist 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) causes the death of BMDMs in a dose- and time-dependent manner[18]. Further, to eliminate the potential influence of cytoplasmic DNA released as a result of cell death, we employed Deoxyribonuclease I (DNase I) to degrade DNA present in the culture supernatant. The results showed that in the co-culture system, STING activation in BMDMs - by gain-of-function mutation or cGAMP - was capable of inducing injury in HL-1 cells, irrespective of whether DNase I treatment was administered. [Figure 5A-C]. These findings indicated cardiomyocyte hypertrophy does not originate from the DNA released by cGAMP-induced macrophage death. Subsequently, HL-1 cells co-cultured with BMDM underwent transcriptome sequencing. The volcano plot results indicated that the transcript expression of 122 genes was significantly upregulated, while that of 22 genes was significantly downregulated [Figure 6A]. KEGG enrichment [Figure 6B] and heatmap analyses [Figure 6C] revealed that genes related to inflammatory signaling pathways, such as nucleotide-binding oligomerization domain (NOD)-like receptors, Tumour necrosis factor (TNF), Toll-like receptors, cytosolic DNA sensing, necroptosis, and Janus kinase-signal transducer and activator of transcription (JAK-STAT), were enriched. It was discovered that ZBP1, which is closely associated with NLRP3 inflammasome activation, exhibited a significant upregulation in cardiomyocytes [Figure 6C]. Subsequently, we verified the expression alterations of ZBP1 in cardiomyocytes in the co-culture of BMDMs and HL-1 cells through the utilization of gain-of-function mutation and cGAMP, respectively [Figure 6D and E]. Further, we employed Search Tool for the Retrieval of Interaction Gene/Proteins (STRING) to predict the interaction between ZBP1 and NOD-like receptors signaling pathway. The interaction network showed that ZBP1 might interact with NLRP3 or Melanoma-associated serum protein 2 (AIM2) [Figure 6F], thereby promoting inflammasome assembly. Thus, it is evident that ZBP1-mediated inflammasome activation is likely the key mechanism by which macrophage STING promotes cardiomyocyte hypertrophy.
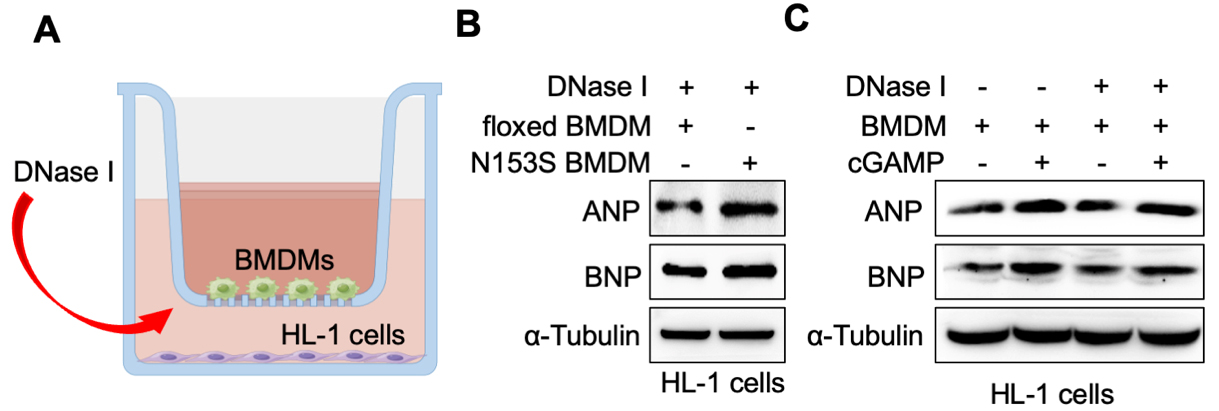
Figure 5. Cardiomyocyte hypertrophy does not originate from the DNA released by cGAMP-induced macrophage death. (A) Schematic diagram of DNase I treatment in a co-culture system (generated by Figdraw, ID: OITPS2a3a3); (B) Protein levels of ANP and BNP in HL-1 cells co-cultured with StingN153S floxed and StingLysM N153S BMDMs; (C) Protein levels of ANP and BNP in HL-1 cells co-cultured with cGAMP-stimulated BMDMs. BMDMs: Bone marrow-derived macrophages; ANP: atrial natriuretic peptide; BNP: B-type natriuretic peptide; cGAMP: cyclic GMP-AMP.
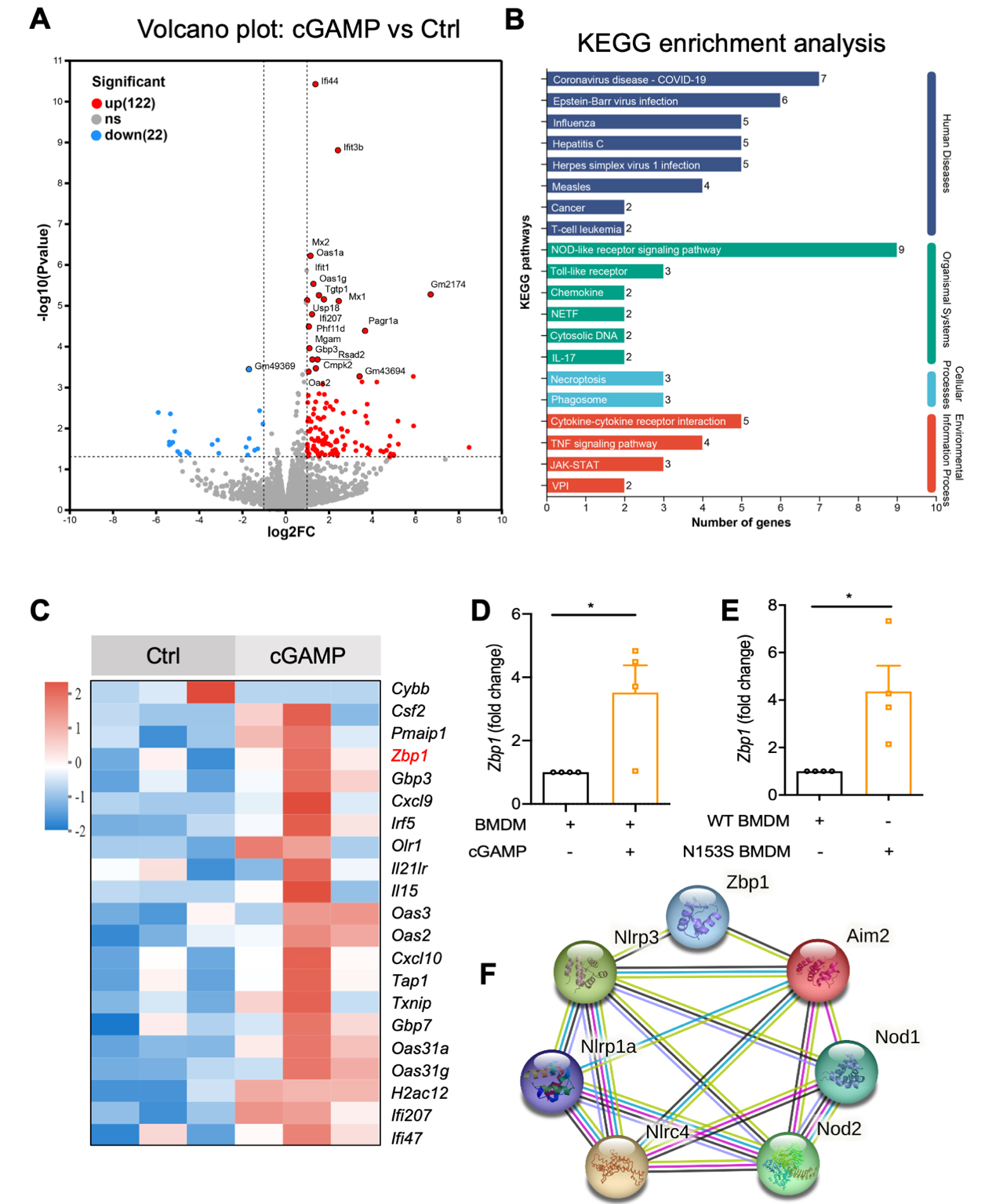
Figure 6. Macrophage STING promotes ZBP1-mediated NLRP3 inflammasome activation in cardiomyocytes. HL-1 was co-cultured with BMDMs for 24 h. HL-1 cells from each group (n = 3 biological replicates) were collected for RNA extraction using TRIzol reagent (Invitrogen), followed by RNA-seq. (A and B) Volcano plot, KEGG enrichment analysis from RNA-seq of HL-1 cells; (C) Heatmap analysis of inflammatory signaling pathway-related genes; (D and E) mRNA levels of ZBP1 in HL-1 cells co-cultured with BMDMs (n = 4 in each group); (F) Prediction of the interaction between ZBP1 and the NOD-like receptors signaling pathway from STRING. Mean ± SEM; *P < 0.05. STING: Stimulator of interferon genes; ZBP1: Z-DNA binding protein 1; BMDMs: bone marrow-derived macrophages; SEM: standard error of the mean; NLRP3: NOD-like receptor thermal protein domain associated protein 3; KEGG: Kyoto Encyclopedia of Genes and Genomes; NOD: nucleotide-binding oligomerization domain; STRING: Search Tool for the Retrieval of Interacting Genes/Proteins; TNF: tumor necrosis factor; NETF: neutrophil extracellular trap formation; JAK-STAT: Janus kinase-signal transducer and activator of transcription; VPI: viral protein interaction.
DISCUSSION
Cardiac pressure overload-induced cardiomyocyte hypertrophy and reduced myocardial contractility are regarded as the primary triggers of heart failure. The pathological characteristics of HFpEF typically manifest as elevated expression of pro-inflammatory cytokines, augmented infiltration of neutrophils and macrophages, maintained normal EF, and impaired diastolic function[19]. Currently, the clinical treatment outcome is unsatisfactory, and the molecular mechanism of HFpEF remains ambiguous. There is an urgent demand to explore new targets and therapeutic approaches.
We established a mouse model of HFpEF by subjecting the mice to a 10-week high-fat diet in combination with the dual action of the Endothelial nitric oxide synthase (eNOS) inhibitor L-NAME. In vivo experiments demonstrated that STING activation was implicated in the progression of HFpEF. The above experiments validated our initial conjecture that STING signaling pathways are involved in the development of HFpEF. Cardiomyocytes are intrinsically capable of sensing mechanical forces and converting them into intracellular signals, a phenomenon referred to as mechanotransduction. Through this process, mechanical cues are translated into biochemical signaling cascades that drive changes in cardiac structure and function. Initially, mechanotransduction-mediated responses function as adaptive and compensatory mechanisms that enable the myocardium to accommodate increased mechanical demands. However, sustained exposure to pressure or volume overload leads to dysregulation of this process, shifting it from a protective to a maladaptive state, ultimately contributing to impaired cardiac function, pathological hypertrophy, and the progression to heart failure[20]. Hence, uncovering the mechanisms of cardiomyocyte hypertrophy can prevent and reverse the occurrence of HFpEF. Cardiomyocyte hypertrophy is regarded as an adaptive remodeling process through which the heart maintains normal function and efficiency. Accumulated studies have proven that Elevated levels of STING, Interferon-α (IFN-α), and IFN-β have been found in cardiac tissues of hypertrophic cardiomyopathy patients and mouse models of myocardial hypertrophy, indicating that STING was involved in cardiomyocyte hypertrophy[8,9]. Our previous study showed that STING N153S mutation promoted STING activation and cardiac hypertrophy[9]; on the contrary, cardiomyocyte-specific STING depletion protects against cardiac dysfunction upon pressure overload[8]. On the other hand, single-cell RNA sequencing revealed that cardiac-resident macrophage-derived STING was involved in myocardial infarction (MI)-induced cardiac remodeling[21]. However, the role of macrophage-derived STING in HFpEF remains uncertain. We extracted BMDMs and co-cultured them with HL-1 cells to investigate the effects of specific activation and knockout of STING in macrophages on cardiomyocyte hypertrophy. Treatment of wild-type mouse BMDMs with the STING agonist cGAMP resulted in significant hypertrophy of HL-1 cells. It has been reported that among 6 STING-associated vasculopathy with onset in infancy (SAVI) patients, 4 patients presented the STING N154S mutation (STING N153S in mice), and it was verified that this mutation resulted in the upregulation of type I interferon expression in 293T cells[22,23]. The STING N153S mutant mice constructed by CRISPR/Cas9 technology manifested pulmonary inflammation, polycythemia, immune cell infiltration, and skin ulcers when they grew to 4-6 weeks of age[23]. In this study, Cre-inducible myeloid-specific STING N153S mutant mice were generated, and co-culture experiments using BMDMs from these mice induced hypertrophy in HL-1 cells. In contrast, co-culture with Sting gene knockout BMDMs significantly attenuated cardiomyocyte hypertrophy compared to wild-type controls. These findings robustly demonstrate that macrophage STING activation promotes cardiomyocyte hypertrophy and contributes to myocardial injury.
Macrophages are a critical component of the mammalian heart and exhibit extensive infiltration in response to various internal or external stimuli. Following sustained pressure overload, the accumulation of cardiac macrophages via local proliferation and monocyte migration profoundly influences the progression of myocardial hypertrophy and remodeling[24]. Our study revealed that abnormally activated macrophages could affect the normal physiological function of cardiomyocytes. To elucidate the mechanisms by which macrophage STING activation promotes cardiomyocyte hypertrophy, we performed RNA sequencing and identified the enrichment of multiple inflammatory signaling pathways in cardiomyocytes. Notably, the expression of ZBP1, a key regulator of inflammatory cell death, was significantly upregulated. ZBP1 was initially identified as an IFN-induced Z-nucleic acid (Z-NA) binding protein, capable of recognizing Z-NAs produced during viral replication in the cytoplasm via its Zα domain[25,26]. Recent studies have revealed that during myocardial ischemic perfusion, ZBP1 can initiate PANoptosome assembly, leading to cardiomyocyte PANoptosis[27]. Moreover, ZBP1 stabilizes Z-DNA to promote cGAS activation and doxorubicin-induced cardiomyopathy[28]. Heat stress has also been shown to induce ZBP1-mediated PANoptosis, ultimately resulting in multiple organ failure[29]. We found that the NOD-like receptor signaling pathway was enriched, and ZBP1 may interact with NLRP3 or AIM2. Collectively, these findings suggest that ZBP1 likely plays a critical role in this process.
Limitations
Although our study provides new insights into the pathogenesis and therapeutic targets of HFpEF, several limitations remain that warrant further improvement. Primary ventricular myocytes maintain their natural phenotype, contractile function, and physiological responses to microenvironmental stimuli, closely resembling the pathophysiological conditions of HFpEF. The phenotypes and functions of HL-1 cells differ from those of primary cardiomyocytes in vivo, which may hinder their ability to accurately reflect the responses of cardiomyocytes under physiological conditions. On the other hand, cardiac macrophages are classified into tissue-resident macrophages (TRMs) and bone marrow-derived recruited macrophages (MoMs). The BMDMs used in this study are more representative of bone marrow-derived recruited macrophages and therefore do not reflect the effects of STING activation in resident macrophages. Macrophages interact with cardiomyocytes through physical contact, paracrine signaling, and molecular transport via extracellular vesicles[30]. We believe that macrophage STING activation promotes cardiomyocyte hypertrophy primarily via a paracrine mechanism (through the release of pro-inflammatory factors), but a limitation of the study is that the existing data remain insufficient. Moreover, although our study proposed a mechanism involving STING-ZBP1 axis-mediated inflammasome activation, direct verification is lacking. Our previous study demonstrated that the N153S mutation in cardiomyocytes can spontaneously induce myocardial hypertrophy and cardiac dysfunction[9], with mice showing a hypertrophic phenotype at 16 weeks of age. We are also interested in investigating whether the N153S mutation in macrophages can spontaneously lead to HFpEF. However, despite generating STING mutant and knockout mice, a main limitation is that HFpEF was not assessed following intervention in macrophage-derived STING via the N153S mutation or knockout.
Conclusions
In conclusion, our study demonstrates that activation of the STING signaling pathway in macrophages significantly influences HFpEF. Further investigations show that macrophage STING promotes cardiomyocyte hypertrophy, an effect likely mediated by ZBP1 in cardiomyocytes. These findings provide novel insights into the pathogenesis of HFpEF and suggest that macrophage STING may represent a potential therapeutic target.
DECLARATIONS
Acknowledgments
We are grateful to Liuliu Feng from the Experimental Animal Center, China Pharmaceutical University, for her technical support.
Authors’ contributions
Methodology, writing - original draft, investigation: Du Y
Methodology, investigation: Han Y
Methodology: Shi Y, Xie H
Investigation, Funding Acquisition: Qiu X
Resources: Qi Y
Investigation: He L
Conceptualization, methodology, writing - review and editing, resources: Wang L
Writing - Review And Editing: Kang L, Xu B
Availability of data and materials
All data included within the article are available from the corresponding authors upon reasonable request.
Financial support and sponsorship
This study was supported by the Natural Science Foundation of Jiangsu Province (No. BK20240226 to Wang L), the Key Project of the Medical Science and Technology Development Foundation, Nanjing Department of Health (No. YKK24067 to Wang L), the Zhejiang Provincial Project of Medical and Health Technology (No. 2020KY457 to Qi Y), and the Wenzhou Basic Public Welfare Scientific Research Project (No. Y20220194 to Qiu X).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethics approval and consent to participate
All animal experiments were conducted in accordance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. Experimental protocols were approved by the Animal Ethics Committee of China Pharmaceutical University (Approval No. 2020-11-007).
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. National Center for Cardiovascular Disease TWCotRoCHaDiC. Report on cardiovascular health and diseases in China 2021: an updated summary. Chin Circ J. 2022. Available from https://www.chinacirculation.org/#/browse_details?year=2022&issue=6&issuecid=280067 [accessed 26 February 2026].
2. Camici PG, Tschöpe C, Di Carli MF, Rimoldi O, Van Linthout S. Coronary microvascular dysfunction in hypertrophy and heart failure. Cardiovasc Res. 2020;116:806-16.
3. Campbell P, Rutten FH, Lee MM, Hawkins NM, Petrie MC. Heart failure with preserved ejection fraction: everything the clinician needs to know. Lancet. 2024;403:1083-92.
4. Wang B, Jankauskas SS, Mone P, Varzideh F, Santulli G. Immunology of heart failure with preserved ejection fraction. Expert Rev Clin Immunol. 2025;21:1725-39.
5. Zhang N, Ma Q, You Y, et al. CXCR4-dependent macrophage-to-fibroblast signaling contributes to cardiac diastolic dysfunction in heart failure with preserved ejection fraction. Int J Biol Sci. 2022;18:1271-87.
6. Chai S, Xu H, Liu R, Cai C. STING-inflammasome axis in autoimmune diseases and inflammation-related disease. Autoimmun Rev. 2025;24:103898.
7. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol. 2020;21:501-21.
8. Zhang Y, Chen W, Wang Y. STING is an essential regulator of heart inflammation and fibrosis in mice with pathological cardiac hypertrophy via endoplasmic reticulum (ER) stress. Biomed Pharmacother. 2020;125:110022.
9. Wang L, Zhang S, Liu H, et al. STING activation in cardiomyocytes drives hypertrophy-associated heart failure via NF-κB-mediated inflammatory response. Biochim Biophys Acta Mol Basis Dis. 2024;1870:166997.
10. Fan X, Han J, Zhong L, et al. Macrophage-derived GSDMD plays an essential role in atherosclerosis and cross talk between macrophages via the mitochondria-STING-IRF3/NF-κB Axis. Arterioscler Thromb Vasc Biol. 2024;44:1365-78.
11. Gu L, Sun Y, Wu T, et al. A novel mechanism for macrophage pyroptosis in rheumatoid arthritis induced by Pol β deficiency. Cell Death Dis. 2022;13:583.
12. Luo W, Wang Y, Zhang L, et al. Critical role of cytosolic DNA and its sensing adaptor STING in aortic degeneration, dissection, and rupture. Circulation. 2020;141:42-66.
13. Oduro PK, Zheng X, Wei J, et al. The cGAS-STING signaling in cardiovascular and metabolic diseases: future novel target option for pharmacotherapy. Acta Pharm Sin B. 2022;12:50-75.
14. Han J, Dai S, Zhong L, et al. GSDMD (Gasdermin D) mediates pathological cardiac hypertrophy and generates a feed-forward amplification cascade via mitochondria-STING (stimulator of interferon genes) axis. Hypertension. 2022;79:2505-18.
15. Pham PT, Fukuda D, Nishimoto S, et al. STING, a cytosolic DNA sensor, plays a critical role in atherogenesis: a link between innate immunity and chronic inflammation caused by lifestyle-related diseases. Eur Heart J. 2021;42:4336-48.
16. Schiattarella GG, Altamirano F, Tong D, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019;568:351-6.
17. Wang L, Luo W, Zhang S, et al. Macrophage-derived FGFR1 drives atherosclerosis through PLCγ-mediated activation of NF-κB inflammatory signalling pathway. Cardiovasc Res. 2024;120:1385-99.
18. Wu J, Liu Q, Zhang X, Wu X, Zhao Y, Ren J. STING-dependent induction of lipid peroxidation mediates intestinal ischemia-reperfusion injury. Free Radic Biol Med. 2021;163:135-40.
19. Zhang L, Chen J, Yan L, He Q, Xie H, Chen M. Resveratrol ameliorates cardiac remodeling in a murine model of heart failure with preserved ejection fraction. Front Pharmacol. 2021;12:646240.
20. Bazgir F, Nau J, Nakhaei-Rad S, et al. The microenvironment of the pathogenesis of cardiac hypertrophy. Cells. 2023;12:1780.
21. King KR, Aguirre AD, Ye YX, et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. 2017;23:1481-7.
22. Bennion BG, Ingle H, Ai TL, et al. A human gain-of-function STING mutation causes immunodeficiency and gammaherpesvirus-induced pulmonary fibrosis in mice. J Virol. 2019;93:10.1128/jvi.01806-18.
23. Warner JD, Irizarry-Caro RA, Bennion BG, et al. STING-associated vasculopathy develops independently of IRF3 in mice. J Exp Med. 2017;214:3279-92.
24. Martini E, Kunderfranco P, Peano C, et al. Single-cell sequencing of mouse heart immune infiltrate in pressure overload-driven heart failure reveals extent of immune activation. Circulation. 2019;140:2089-107.
25. Karki R, Kanneganti TD. ADAR1 and ZBP1 in innate immunity, cell death, and disease. Trends Immunol. 2023;44:201-16.
26. Mishra S, Dey AA, Kesavardhana S. Z-nucleic acid sensing and activation of ZBP1 in cellular physiology and disease pathogenesis. Immunol Rev. 2025;329:e13437.
27. Zhang X, Song S, Huang Z, et al. Z-DNA-binding protein 1 exacerbates myocardial ischemia-reperfusion injury by inducing noncanonical cardiomyocyte PANoptosis. Signal Transduct Target Ther. 2025;10:333.
28. Lei Y, VanPortfliet JJ, Chen YF, et al. Cooperative sensing of mitochondrial DNA by ZBP1 and cGAS promotes cardiotoxicity. Cell. 2023;186:3013-3032.e22.
29. Yuan F, Cai J, Wu J, et al. Z-DNA binding protein 1 promotes heatstroke-induced cell death. Science. 2022;376:609-15.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].