




Adipose-derived stem cell extracellular vesicles attenuate liver fibrosis via restoration of gut barrier function and modulation of gut microbiota
 0
0 Abstract
Aim: Liver fibrosis (LF) is a major pathological stage that may progress to end-stage chronic liver injury but currently lacks effective treatment strategies. Previous studies have shown that adipose-derived stem cell extracellular vesicles (ADSC-EVs) play crucial roles in tissue repair, immune regulation, and anti-inflammatory effects. This study aims to elucidate the therapeutic effect of ADSC-EVs in LF and reveal their regulation mechanisms in gut-liver axis dysregulation.
Methods: The LF mouse model was established by intraperitoneal injection of diethylnitrosamine/CCl4. LF mice for ADSC-EV treatment received ADSC-EVs (200 μg per mouse) twice a week for three weeks. Then, hepatic function tests, liver and gut histopathology, and gut microbiota analyses were performed.
Results: ADSC-EVs effectively improved hepatic function, reduced collagen deposition and suppressed hepatic stellate cell activation, exhibiting potent anti-fibrotic potential in LF mice. Additionally, they significantly restored intestinal barrier integrity by reducing intestinal permeability and reinforcing the mucus barrier. Furthermore, ADSC-EV treatment regulated gut microbiota dysbiosis, increased the abundance of beneficial intestinal bacteria such as Akkermansia muciniphila. ADSC-EV intervention also elevated the level of butyric acid in cecal contents and significantly reduced systemic inflammation.
Conclusion: Our findings suggest that ADSC-EVs represent a promising novel therapeutic strategy for LF, promoting liver tissue repair, enhancing intestinal barrier function, and maintaining gut homeostasis to establish a virtuous circle within the liver-gut axis.

Keywords
INTRODUCTION
Liver fibrosis (LF) is an aberrant pathological wound-healing response to chronic liver injury, characterized by excessive deposition of extracellular matrix (ECM), which may subsequently develop into cirrhosis and hepatocellular carcinoma[1,2]. It is a major pathology status of chronic hepatitis, alcohol-related liver disease, and non-alcoholic fatty liver disease (NAFLD), with an increasing prevalence worldwide[3]. Its pathogenesis mainly involves the activation of hepatic stellate cells (HSCs), and the proliferation of ECM-producing myofibroblasts, driven by persistent inflammation and cytokines[4]. Currently, LF drugs primarily aim to eliminate pathogenic factors such as viruses, ethanol, and metabolic dysfunction, and restore tissue lesions[5,6]. Due to the complex etiology of LF, currently approved clinical drugs remain limited to resmetirom for NAFLD. Clinical candidate drugs such as irbesartan and cenicriviroc have reported failure in clinical trials[7]. Thus, effective therapeutic strategies for LF remain a critical unmet need.
Numerous studies indicated that the LF process generally had varying degrees of intestinal barrier dysfunction, such as abnormal tight junction protein expression, leading to altered intestinal permeability; and reduced goblet cell number or function, resulting in a thinning of the intestinal mucus layer, finally with a weakening of the physical barrier function. Alterations of the intestinal barrier might contribute to exacerbated immune responses in liver diseases[8]. Increased intestinal permeability permits metabolites or pathogen-associated molecular patterns (PAMPs) to translocate to the liver via the portal circulation, where interaction with pattern recognition receptors (PRRs) on hepatic-resident cells, potentially inducing or exacerbating detrimental immune responses[9]. Additionally, the progression of chronic liver disease is associated with gut microbiota imbalance. It has been reported that the gut microbiome in metabolic dysfunction-associated steatotic liver disease (MASLD) was featured with an enrichment of Proteobacteria, Escherichia, Clostridium and Streptococcus, along with a reduced abundance of beneficial commensals such as Firmicutes, Eubacterium and Faecalibacterium, implying a reduced capability of intestinal protection and anti-inflammatory function[10]. There was also gut microbial dysbiosis in LF, including dysregulated composition and function of the gut microbiome. Consequently, integrating intestinal modulatory strategies into anti-fibrotic protocols was critical for achieving maximal treatment benefits.
Extracellular vesicles (EVs), as nanoscale extracellular lipid bilayer vesicles, contain diverse bioactive molecules, such as proteins, nucleic acids, lipid secreted complexes, etc., and they play a crucial role in intercellular communication[11,12]. Several studies showed that mesenchymal stem cell-derived EVs (MSC-EVs) exhibited therapeutic potential in repairing tissue injuries, including renal failure[13], acute tubular injury[14,15] and skin damage[16]. Adipose-derived stem cells (ADSCs) offered the advantages of abundant sources, easy accessibility and higher proliferation in vitro, making them an ideal seed cell in regenerative medicine. Studies showed that MSC-EV therapy could reduce the hepatic fibrotic encapsulation, attenuate hepatic inflammation, and decrease collagen deposition in CCl4-induced fibrosis[17]. Our previous studies also demonstrated the anti-inflammatory, antioxidant, and tissue-repair functions of ADSC-EVs in an acute liver failure model[18].
In this research, we aimed to elucidate the therapeutic effect of ADSC-EVs in LF and revealed their regulation mechanisms in gut-liver axis dysregulation. Diethylnitrosamine (DEN)/CCl4 was used to establish the LF mouse model. Hepatic function, liver and gut histopathology, and gut microbiota of mice in each group were evaluated to assess the effects of ADSC-EVs. Our findings demonstrated that ADSC-EVs not only exhibited potent anti-fibrotic potential but also restored intestinal barrier integrity in LF mice. Additionally, ADSC-EV treatment improved gut microbiota dysbiosis by increasing beneficial bacteria such as Akkermansia muciniphila (A. muciniphila) and elevating butyric acid levels, significantly reducing systemic inflammation, making it a promising novel therapeutic strategy for LF.
METHODS
Reagents
CCl4 was purchased from MedChemExpress (MCE) Chemical Reagent (HY-Y0298, USA). N-Nitrosodiethylamine (DEN) was purchased from Meilunbio® Co., Ltd. (MB4816-1, Dalian, China).
Preparation of ADSCs and ADSC-EVs
Isolation and culture of primary ADSCs and isolation of ADSC-EVs were performed according to the established method in our previous study[19]. ADSCs were cultured in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM-F12, Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA), 10 ng/mL basic fibroblast growth factor (bFGF, Novoprotein, China), and 1% penicillin-streptomycin solution (10,000 U/mL, Gibco, USA) at 37 °C, 5% CO2. The basic culture medium was replaced with EV-free FBS complete medium 24 h before supernatant collection. The supernatant was then collected, and ADSC-EVs were isolated by differential centrifugation according to our established protocol.
Characteristic of isolated ADSC-EVs
EV concentration, particle size and distribution were quantified using nanoparticle tracking analysis (NTA, NanoSight300, Malvern Instruments Ltd, UK). Briefly, samples were diluted with 1 mL phosphate-buffered saline (PBS) to an optimal concentration, and then loaded into the cubicle. Particles were then tracked and illuminated by laser light under Brownian motion. Samples were measured with particle concentration, size distribution, scatter intensity and calculated based on the Stokes-Einstein equation.
The ultrastructure of EVs was characterized by transmission electron microscopy (TEM) using a Hitachi HT7800 electron microscope (Hitachi, Japan).
Fluorescent-labeled antibodies (FITC, PC5) targeting EVssomal surface markers CD63 (NHA063-A647, Fuliu, China), CD81 (NHA081-FITC, Fuliu, China), and CD9 (NHA009-A488, Fuliu, China) were incubated with EVs at 4 °C for 60 min in the dark. Control groups were established to exclude non-specific antibodies and auto-fluorescence. Processed samples were subjected to instrument detection at a flow rate of 35 nL/min (NanoFCM SNA-D1, Fuliu, China).
Animals
Specific pathogen-free C57/BL6 mice (Male, 3 weeks of age) were purchased from Shanghai Slack Laboratory Animal Co., Ltd. (Shanghai, China). All animal experiments and research protocols were approved by the Ethical Committee of Laboratory Animals Research Center of Tongji University (Approval No.: TJAA07624403).
Experimental design
To investigate ADSC-EV therapeutic effect in the LF mouse model, mice were randomly divided into three groups (n = 8 mice per group), including the Sham group (healthy controls without LF induction), the LF group (LF) and the liver fibrosis + ADSC-EV group (LF + EV). For the LF model, 3-week-old male mice were injected intraperitoneally (i.p.) with DEN (20 mg/kg) once a week for two weeks, followed by intraperitoneal injections of CCl4 (2 mL/kg) three times per week during weeks 3-6 and twice per week during weeks 7-9. Mice in the Sham group received dimethyl sulfoxide (DMSO) or oil injections instead of DEN or CCl4. Mice in the LF + EV group received ADSC-EVs (200 μg per mouse, intravenously via tail vein) twice a week for three weeks from weeks 7 to 9.
Histological analysis
Fresh mouse liver or intestinal tissues were fixed in 4% paraformaldehyde (≥ 24 h), dehydrated through an ethanol gradient, embedded in paraffin, and sectioned at a thickness of 4 μm. Consecutive sections were processed for hematoxylin and eosin (H&E) staining (C0105S, Beyotime, China), Sirius Red staining (60415ES50, Yeasen Biotechnology, China), and Periodic Acid-Schiff (PAS) staining (C0142S, Beyotime, China) following standard protocols. Immunohistochemistry (IHC) staining for alpha smooth muscle actin (α-SMA), zonula occludens-1 (ZO-1), and myeloperoxidase (MPO) was also performed according to standard protocols. Primary antibodies included: Anti-α-SMA (ab124964, Abcam, USA), Anti-ZO-1 tight junction protein (ab221546, Abcam, USA), Anti-MPO (AG2657, Beyotime, China). Secondary antibodies included horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG) (H + L) (A0208, Beyotime, China).
Quantitative real-time polymerase chain reaction assay
Total RNA was extracted from freshly dissected mouse intestinal tissues using TRIzol® Reagent (Thermo Fisher Scientific, USA) according to the manufacturer’s protocol. Complementary DNA (cDNA) was synthesized from 1 µg of total RNA using the 1st Strand cDNA Synthesis Kit (Vazyme, China). Quantitative real-time polymerase chain reaction (PCR) (qRT-PCR) was performed using ChamQ Universal SYBR® quantitative PCR (qPCR) Master Mix (Vazyme, China) on a Light Cycler96 real-time PCR system. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) messenger RNA (mRNA) was used as an internal control. The primer sequences used in this experiment were as follows: mouse ZO-1 Forward, 5′-GCCGCTAAGAGCACAGCAA-3′; mouse ZO-1 Reverse, 5′-TCCCCACTCTGAAAATGAGGA-3′; mouse Occludin Forward, 5′-TTGAAAGTCCACCTCCTTACAGA-3′; mouse Occludin Reverse, 5′-CCGGATAAAAAGAGTACGCTGG-3′; mouse Claudin-3 Forward, 5′-ACCAACTGCGTACAAGACGAG-3′; mouse Claudin-3 Reverse, 5′-CAGAGCCGCCAACAGGAAA-3′; mouse Mucin (Muc)-1 Forward, 5′-GGCATTCGGGCTCCTTTCTT-3′; mouse Muc-1 Reverse, 5′-TGGAGTGGTAGTCGATGCTAAG-3′; mouse Muc-2 Forward, 5′-AGGGCTCGGAACTCCAGAAA-3′, mouse Muc-2 Reverse, 5′-CCAGGGAATCGGTAGACATCG-3′; mouse GAPDH Forward, 5′-CGGAGTCAACGGATTTGGTCGTAT-3′; and mouse GAPDH Reverse, 5′-GCCTTCTCCATGGTGGTGAAGAC-3.
16S rRNA sequencing
Intestinal contents were collected from the Sham group, LF-1W group, LF-1W-EV group, the LF-3W group, and the LF-3W-EV group for DNA extraction. The V3-V4 variable region of the 16S ribosomal RNA (rRNA) gene was amplified by PCR using specific primers (341F/806R). Then, the amplified product was purified using magnetic beads, and a sequencing library was constructed using Illumina TruSeq Nano DNA LT Library Prep Kit for further sequencing (Illumina MiSeq platform). The result was analyzed at the Omicsmart platform (https://www.omicsmart.com).
Detection of butyric acid level in intestinal contents
The butyric acid level in cecal contents was measured using a commercial enzyme-linked immunosorbent assay (ELISA) kit (HB142-SH, Hengyuan, China) via a competition method following the manufacturer’s protocol.
Western blot analysis
Protein extraction from both cultured cells and liver tissue homogenates was performed using radio-immunoprecipitation assay (RIPA) lysis buffer supplemented with 1% protease inhibitor (Epizyme, China). Protein concentration was quantified using a bicinchoninic acid (BCA) assay (Epizyme, China). Protein samples mixed with loading buffer (Beyotime, China) were denatured by boiling, separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA) via Western blotting. After blocking non-specific binding with 5% skim milk (Solarbio, China), membranes were incubated overnight at 4 °C with primary antibodies. Following washing with Tris-Buffered Saline and Tween 20 (TBST, Solarbio, China), membranes were incubated with secondary antibody for 1 h at room temperature, washed again, and bands were detected using electrochemiluminescence (ECL) reagent (Abcam, USA) and imaged. Primary antibodies included Anti-CD9 (A19027, Abclonal, China), Anti-CD81 (56039, CST, USA), Anti-ALIX (92880S, CST, USA), Anti-tumor susceptibility gene 101 (TSG101) (72312, CST, USA), Anti-Calnexin (A4846, Abclonal, China), Anti-α-SMA (ab124964, Abcam, USA), and Anti-GAPDH (60004-1-Ig, Proteintech, China). Secondary antibodies included HRP-conjugated goat anti-mouse IgG (H + L) (A0216, Beyotime, China) and HRP-conjugated goat anti-rabbit IgG (H + L) (A0208, Beyotime, China).
Serum biochemical analysis
Before sacrificing the mice, blood was collected and centrifuged at 4,000 × g for 15 min at room temperature to obtain serum for subsequent biochemical analysis. Serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) for hepatic function were measured using an automatic biochemical detection instrument (Rayto, China). Serum levels of D-Lactate and diamine oxidase (DAO), markers of intestinal barrier function, were determined by ELISA according to the manufacturer’s instructions. Commercial reagent kits for mouse serum D-Lactate (SBJ-M0727, Senbeijia, China) and DAO (SBJ-M0211, Senbeijia, China) were used.
Hepatic monocyte-derived macrophage analysis by flow cytometry
The detailed procedure followed the comprehensive analysis of liver macrophage composition by flow cytometry in murine non-alcoholic steatohepatitis (NASH)[20]. Briefly, the mice were perfused with PBS through the portal vein, and the liver was then transferred into a digestion buffer [consisting of DMEM medium supplemented with 0.75 mg/mL Collagenase A and 50 μg/mL deoxyribonuclease I (DNase I), Sigma, USA]. The tissue was incubated at 37 °C for 30 min to allow enzymatic digestion. After digestion, enzyme-containing DMEM was removed by centrifugation at 50 × g for 1 min, followed by 900 rpm for
Statistical analysis
All data are shown as the mean ± standard deviation (SD). Comparisons between two groups were performed using the unpaired Student’s t-test. P-value < 0.05 was considered statistically significant. Analyses were performed using GraphPad Prism 8.3.0 software. Flow cytometry data were analyzed using FlowJo 10.8.1.
RESULTS
Preparation and identification of ADSC-EVs
Human ADSCs were isolated and cultured as described in a previous study [21]. As shown in Figure 1A, ADSCs exhibited a similar typical morphology to mesenchymal stem cells (MSCs), characterized by adherent, fibroblast-like, and spindle-shaped appearance with elongated processes and a large central nucleus. Then, ADSC-EVs were isolated from conditioned media of Passage 3-Passage 5 ADSCs using differential ultracentrifugation, following established protocols with modifications [Figure 1B]. TEM analysis showed that ADSC-EVs were spherical with a lipid bilayer membrane and a typical cup-shaped or saucer-like concavity [Figure 1C]. The size distribution and concentration of EVs were quantified using NTA [Figure 1D]. The majority population of vesicles (93.4%) fell within the accepted size range
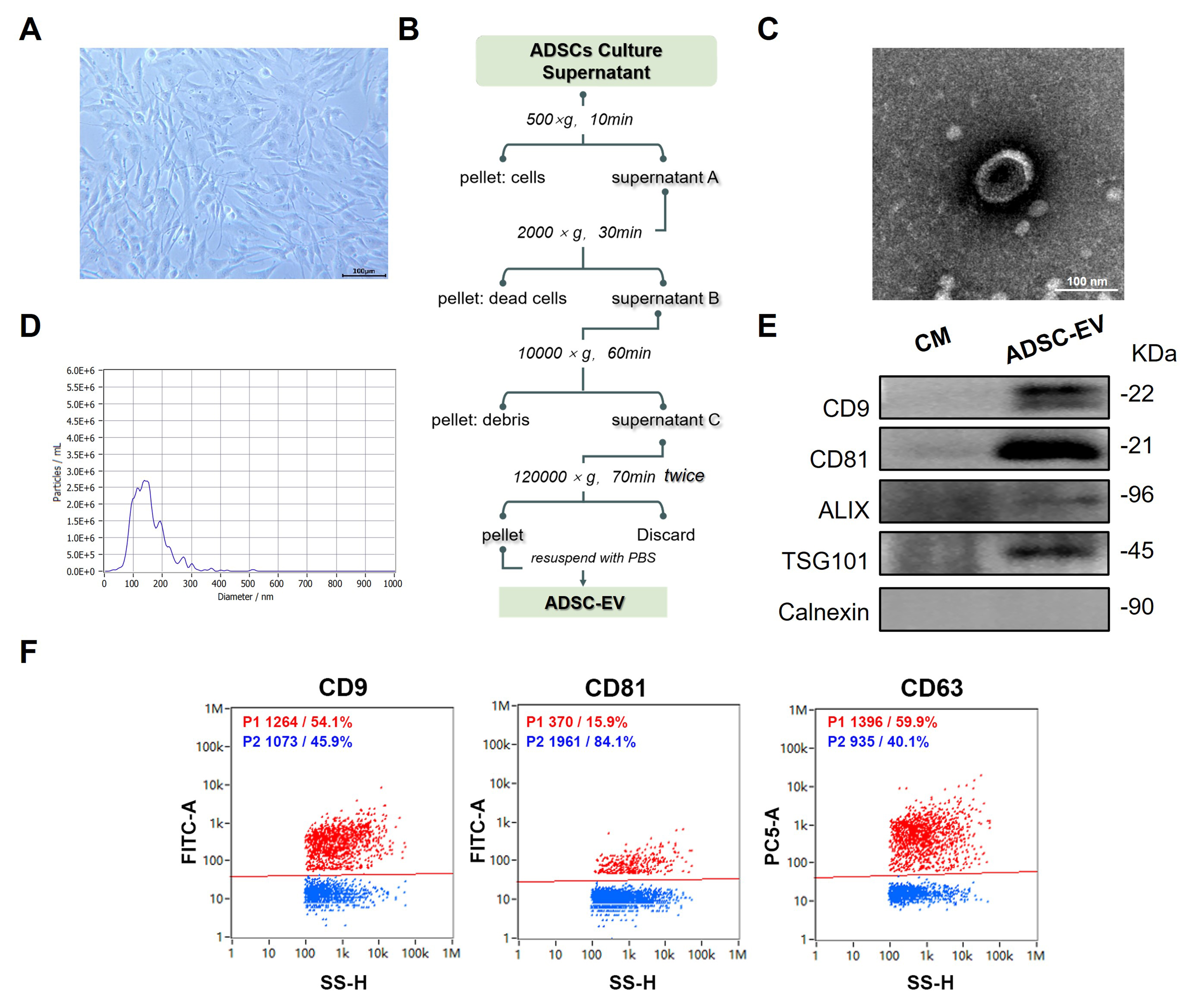
Figure 1. Preparation and characterization of ADSC-EV. (A) Morphology of primary ADSCs at passage 4 (P4) at 80% confluence (scale bar: 100 µm); (B) Schematic workflow for isolating ADSC-EV from the ADSC culture supernatant; (C) Representative TEM image of isolated ADSC-EV (scale bar: 100 nm); (D) NTA of ADSC-EV; (E) Immunoblots for typical EV markers (CD9, CD81, TSG101, Alix) and negative marker (Calnexin) in isolated ADSC-EV and conditional CM; (F) NanoFCM of isolated ADSC-EV for positive markers CD9, CD81, and CD63. ADSC-EV: Adipose-derived stem cell extracellular vesicle; TEM: transmission electron microscopy; NTA: nanoparticle tracking analysis; CM: culture medium; NanoFCM: nano-flow cytometry analysis.
ADSC-EVs alleviate DEN/CCl4-induced hepatic fibrosis in mice
To determine the therapeutic effect of ADSC-EVs in LF, we employed an LF model in which C57BL/6 mice received DEN (i.p.) once a week for two weeks, followed by CCl4 (2 mL/kg) three times a week for weeks 3-6 and 2 times a week for weeks 7-9, resulting in severe LF [Figure 2A]. Meanwhile, the LF + EV group was treated with ADSC-EVs (200 μg/mice) twice a week for three weeks. After the 3-week ADSC-EV treatment period, serum was collected from each group, and the mice were sacrificed to harvest liver tissue for further functional assessment.
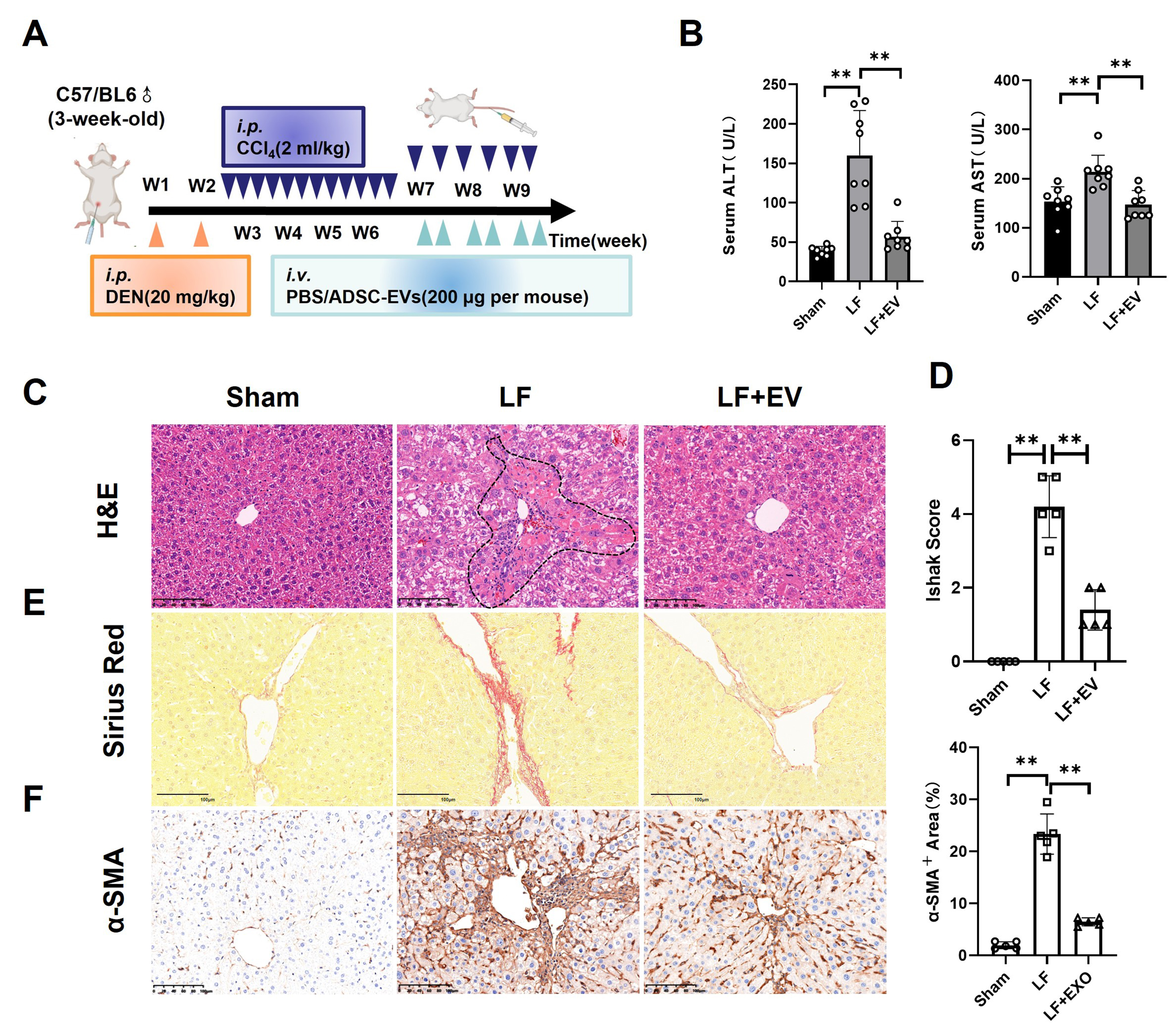
Figure 2. ADSC-EVs inhibit DEN/CCl4-induced hepatic fibrosis in mice. (A) Schematic diagram of DEN/CCl4-induced LF animal model establishment (i.p.) and ADSC-EV treatment strategy (i.v.); (B) The biochemical parameters measurement of ALT and AST in the mouse serum of each group (n = 8); (C) Representative histomorphology images of liver tissues by H&E staining (scale bar: 100 µm). The dotted line indicates the severely damaged area; (D) LF stage assessed using the Ishak score system (n = 5); (E) Representative images of liver tissues by Sirius Red staining (scale bar: 100 µm); (F) Expression of the activated HSCs marker α-SMA in liver tissues by IHC staining (scale bar: 100 µm). All data are shown as the mean ± SD. **P < 0.01. ADSC-EVs: Adipose-derived stem cell extracellular vesicles; DEN: diethylnitrosamine; LF: liver fibrosis; i.p.: intraperitoneal injection; i.v.: intravenous injection; ALT: alanine aminotransferase; AST: aspartate aminotransferase; H&E: hematoxylin and eosin; HSCs: hepatic stellate cells; α-SMA: alpha smooth muscle actin; IHC, immunohistochemistry; SD: standard deviation; PBS: phosphate-buffered saline; LF + EV: liver fibrosis + ADSC-EV group.
Serum biochemical analysis revealed that exposure to DEN/CCl4 elevated ALT and AST levels in LF group mice, while ADSC-EV treatment significantly attenuated these elevations, indicating marked recovery of liver function [Figure 2B]. Then, the histopathological analysis of H&E staining demonstrated that, compared with the liver section in the LF group, LF + EV mice showed markedly reduced cellular edema, the areas of centrilobular necrosis, inflammatory cell infiltration, and ballooning degeneration [Figure 2C]. This improvement was a quantitative analysis using Ishak fibrosis scores, with significantly lower scores in ADSC-EV-treated mice [Figure 2D]. As is known, LF pathology was characterized by excessive deposition of fibrillar collagen and other ECM components, driven by activated HSCs that transformed into proliferative, contractile, fibrogenic myofibroblasts. Therefore, we evaluated the collagen deposition level by Sirius Red staining and HSC activation by IHC of α-SMA. Sirius Red staining showed a robust reduction in collagen deposition and bridging fibrosis, confirming the anti-fibrotic efficacy of ADSC-EVs [Figure 2E]. Critically, IHC staining revealed a significant decrease of α-SMA+ area portion compared to the LF control, indicating HSC activation [Figure 2F]. These findings suggested that ADSC-EVs could alleviate DEN/CCl4-induced hepatic fibrosis.
ADSC-EVs restore the intestinal barrier integrity through reduced permeability and reinforced mucus barrier in mice with LF
As shown in Figure 3A, PAS staining revealed that colon tissue in the Sham group was intact, with normal crypt architecture, preserved goblet cells, and only scattered inflammatory cells in the lamina propria. However, the colon tissue in the LF group exhibited significant damage to crypt structures, a reduction in the number of goblet cells, and a large number of inflammatory cells infiltrated between the mucosa and submucosa. By administering ADSC-EVs, the morphology of colon tissue was improved in terms of mucosal damage, crypt structure, the number of intestinal epithelium and goblet cells and inflammation. Then, we measured two critical biomarkers, D-lactate and DAO, to evaluate gut permeability. Generally, when the tight junctions of the intestinal epithelium are disrupted by factors such as portal hypertension or inflammation, D-lactate passively diffuses from the intestinal lumen into the bloodstream, resulting in increased serum levels[22]. DAO, mainly exists in the cytoplasm of mature intestinal epithelial cells, is responsible for catalyzing the oxidative deamination of biogenic amines such as histamine and putrescine[23]. When intestinal epithelial cells are in a state of necrosis or apoptosis, DAO is directly released into the blood. The serum biochemistry displayed that the abnormal increase of D-lactate and DAO in the LF group was suppressed by ADSC-EV treatment, indicating dual protection of both intestinal permeability and the integrity of the mucosal layer [Figure 3B].
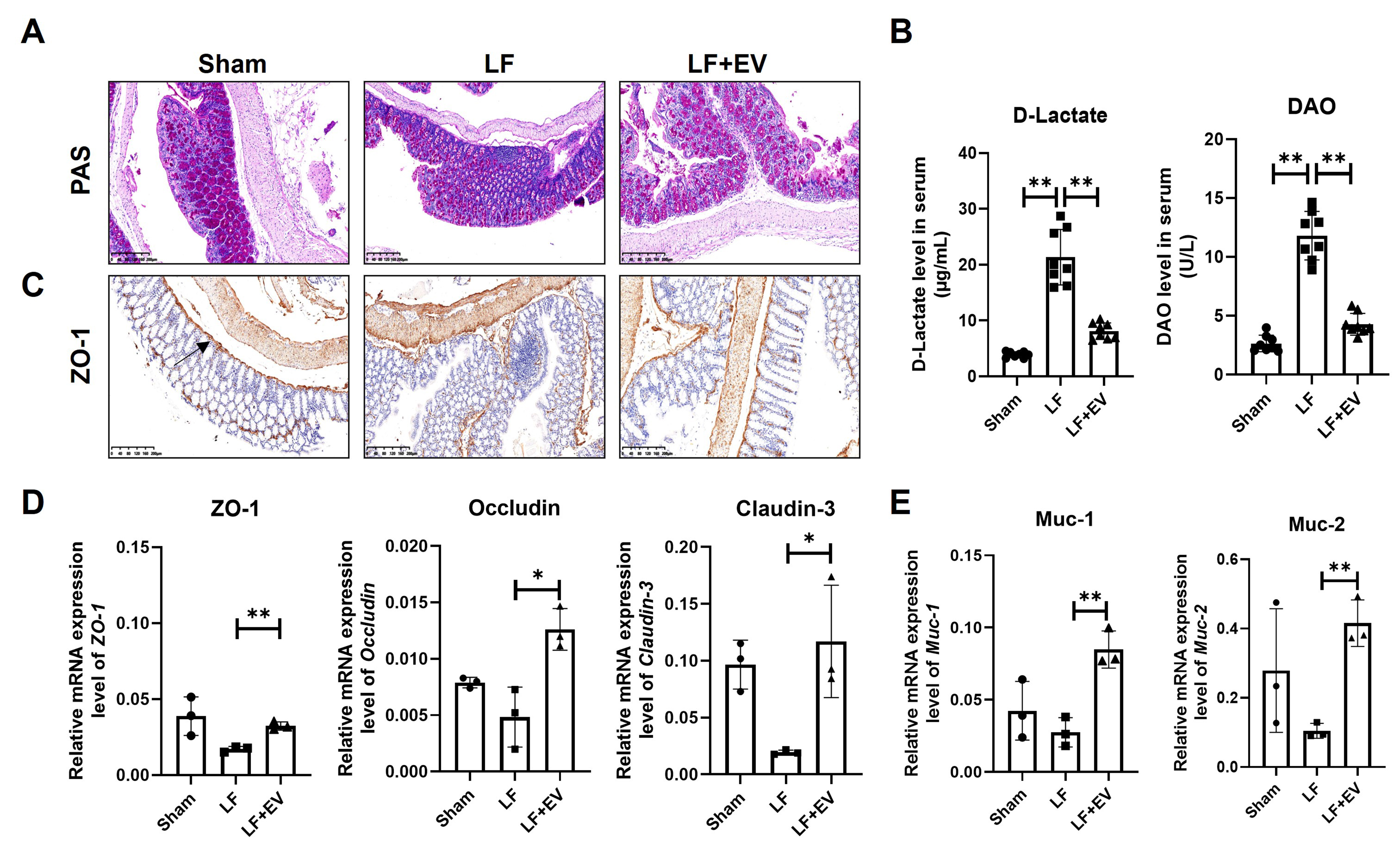
Figure 3. ADSC-EVs improve the intestinal barrier integrity by reducing intestinal permeability and reinforcing the mucus barrier in mice with LF. (A) PAS staining of colonic mucosa in Sham, LF and LF + EV group mice (scale bar: 200 µm); (B) The intestinal injury marker D-Lactate and DAO level in the serum of mice in each group (n = 8); (C) Representative images of ZO-1 expression in liver tissues by IHC staining (scale bar: 200 µm); (D) mRNA levels of tight junction associated genes ZO-1, Occludin, and Claudin-3 in colonic tissue were evaluated by q-PCR (n = 3); (E) The mRNA levels of mucosal barrier-associated genes Muc-1 and Muc-2 in colonic tissue were evaluated by q-PCR (n = 3). The expression was normalized to Gapdh. All data are shown as the mean ± SD. *P < 0.05; **P < 0.01. ADSC-EVs: Adipose-derived stem cell extracellular vesicles; LF: liver fibrosis; PAS: periodic acid Schiff; DAO: diamine oxidase; ZO-1: zonula occludens-1; IHC: immunohistochemistry; Occludin: tight junction protein Occludin; Claudin-3: tight junction protein 3; q-PCR: quantitative polymerase chain reaction; Muc-1: Mucin 1; Muc-2: Mucin 2; SD: standard deviation; LF + EV: liver fibrosis + ADSC-EV group.
To further evaluate the impact of ADSC-EVs on intestinal barrier integrity, we assessed the expression and localization of ZO-1, a critical tight junction protein, in intestinal tissues via IHC and q-PCR. IHC analysis revealed that intestinal tissues from the LF group exhibited disruption of the characteristic continuous linear ZO-1 expression along the apical membrane of intestinal epithelial cells. Instead, ZO-1 displayed a fragmented and discontinuous distribution, indicating the tight junction collapse and increased intestinal permeability. Administration of ADSC-EVs attenuated these pathological changes, effectively restoring the continuous linear expression of ZO-1 protein at the apical membrane of epithelial cells within both villi and crypts [Figure 3C]. Concomitantly, ADSC-EVs restored the mRNA expression of key tight junction-related genes, including ZO-1, Occludin, and Claudin-3 [Figure 3D]. Also, ADSC-EVs could upregulate Muc-1 and Muc-2 gene mRNA expression, which, both relative to goblet cells, constitute the mucus layer that physically segregates microbiota from the intestinal epithelium, forming a mucus barrier [Figure 3E]. Collectively, these EVs showed a positive effect in repairing intestinal barrier dysfunction by reinforcing tight junction protein expression and reconstructing the mucus barrier.
ADSC-EVs alter the gut microbiota disorder and increase the abundance of probiotic A. muciniphila in mice with LF
In addition to repairing the intestinal barrier, we speculated that ADSC-EV may also modulate the gut microbiota in LF mice. We therefore collected fecal samples from Sham, LF-Con, LF-1W, LF-1W-EV, LF-3W, and LF-3W-EV groups for 16s rRNA sequencing. The time points for sample collection in each group are illustrated in Figure 4A. Both the richness rarefaction curves (Sobs) and the Shannon rarefaction curves tend to flatten, indicating sufficient sequencing depth and sample capacity, which could cover most of the microbiota diversity [Figure 4B and C]. Then, we analyzed the sequencing data to evaluate microbiota composition. At the phylum level, the intestinal microbiota of mice in each group mainly include Bacteroidota, Firmicutes, Desulfobacterota, Patescibacteria, Verrucomicrobiota, etc. [Figure 4D], with Bacteroidetes and Firmicutes being the dominant bacterial phyla in the gut microbiota. We further characterized the effect of ADSC-EVs on the relative abundance of gut microbiota at the genus level. The general landscape of gut microbiota and differences in composition between ADSC-EV-treated and their control mice were shown at genus and species levels. At the genus level, the top five most abundant gut microbiota in mice included Akkermansia, Allobaculum, Lactobacillus, Lachnospiraceae_NK4A136_group and Alloprevotella [Figure 4E]. Compared with the Sham group, the relative abundance of Akkermansia was significantly decreased in the LF-Con, LF-1W and LF-3W groups; however, this reduction was reversed following one week and three weeks of ADSC-EV treatment. Also, ADSC-EV administration significantly increased the relative abundance of Lachnospiraceae-NK4A136-group in the gut microbiota compared with the LF-1W and LF-3W groups. By coincidence, both Lachnospiraceae NK4A136-group and Akkermansia are pivotal producers of short-chain fatty acid (SCFA), which maintain an acidic intestinal environment, inhibit the growth of pathogenic bacteria, and provide energy for intestinal epithelial cells[24]. To be specific, at the species level, the abundance of A. muciniphila was significantly decreased under LF stimulation, whereas it was significantly increased in ADSC-EV-treated mice [Figure 4F]. Some recent studies have shown that the decreased abundance of A. muciniphila is associated with the progression of LF and cirrhosis[25,26]. This bacterium exerts hepatoprotective effects by repairing the intestinal mucosal barrier, regulating immune inflammation, and improving metabolic homeostasis. Collectively, these results implied that ADSC-EVs restored the dysbiosis of the microbiota in LF mice by increasing the abundance of beneficial bacteria (e.g., Akkermansia, Lachnospiraceae NK4A136-group) and specifically elevating the levels of probiotic
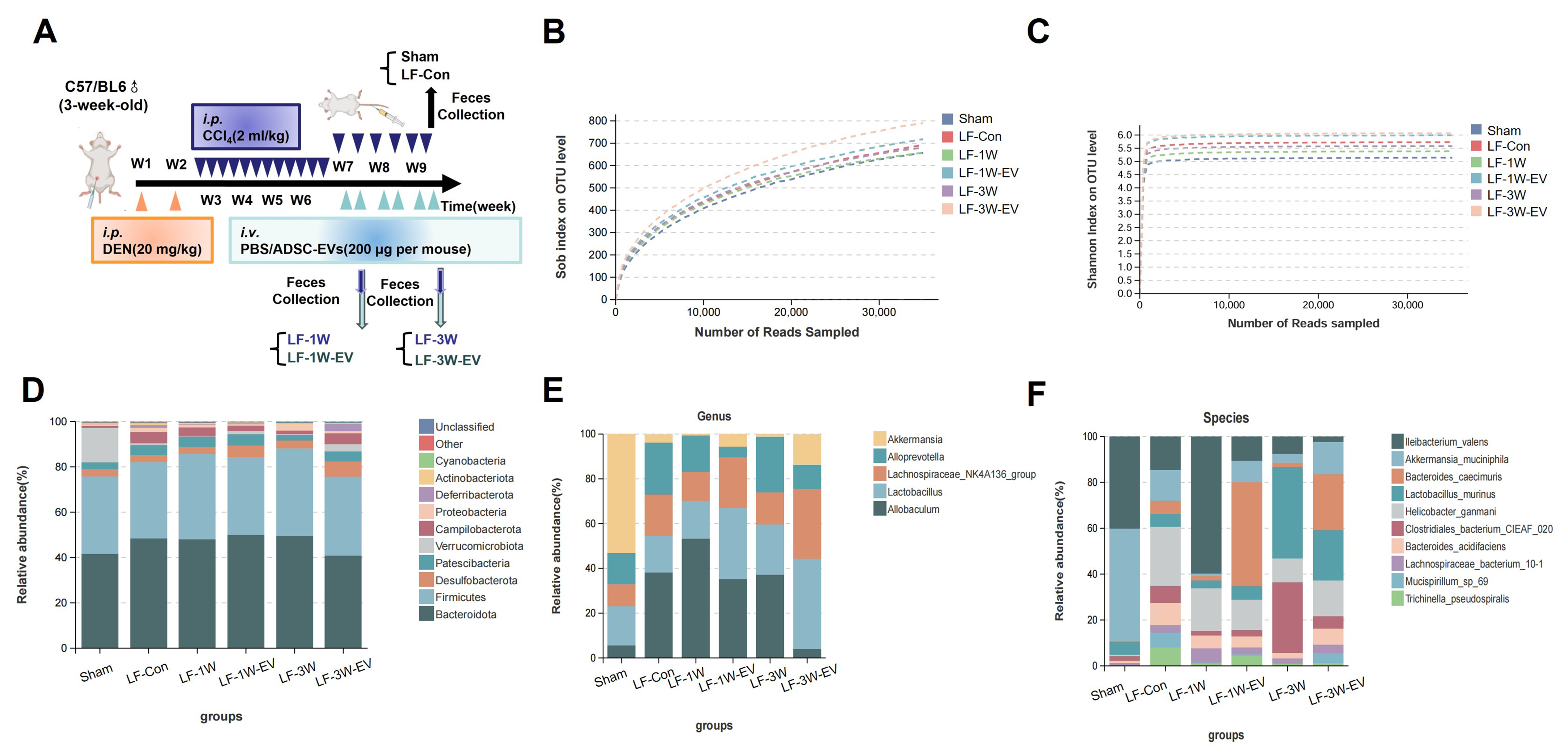
Figure 4. ADSC-EVs restore gut microbiota and increase the abundance of probiotic A. muciniphila in live fibrosis mice. (A) Schematic diagram of fecal samples collection for 16S rRNA sequencing, including the Sham group (n = 3), LF-Con (n = 5), LF-1W (n = 3), LF-1W-EV (n = 3), LF-3W (n = 3), LF-3W-EV (n = 3); Alpha diversity analysis based on (B) Sob index and (C) Shannon index, rarefaction curves indicate adequate sequencing depth; Relative abundance of gut microbiota at the (D) phylum, (E) genus and (F) species levels in each group of mice. All data are shown as the mean ± SD. *P < 0.05; **P < 0.01. ADSC-EVs: Adipose-derived stem cell extracellular vesicles; A. muciniphila: Akkermansia muciniphila; 16S rRNA: 16S ribosomal RNA; LF: liver fibrosis; SD: standard deviation; i.p.: intraperitoneal injection; DEN: diethylnitrosamine; i.v.: intravenous injection; PBS: phosphate-buffered saline; OTU: operational taxonomic units.
Administration of ADSC-EVs elevates the butyric acid level of intestinal contents and decreases the systemic inflammation in mice with LF
We previously demonstrated a significant increase in the abundance of A. muciniphila in the gut contents of LF mice. Generally, A. muciniphila can degrade intestinal mucins by cleaving protective glycans, thereby releasing oligosaccharides that are further metabolized into acetic acid and propionic acid. These metabolites subsequently stimulate the proliferation of butyrate-producing bacteria[27]. Butyric acid, a key microbial metabolite, not only serves as the primary energy source for colonic epithelial cells but also reinforces intestinal barrier integrity by upregulating mucin synthesis and tight junction proteins. More importantly, butyrate also plays a positive role in inflammation suppression, which enables it to both maintain intestinal homeostasis and inhibit systemic inflammation[28]. Thus, we further measured the butyric acid level in intestinal content by the ELISA assay. We found that the butyric acid level in the LF group was significantly decreased compared with the Sham group while ADSC-EV treatment led to a higher production of butyric acid in LF mice [Figure 5A]. Given the change of butyrate levels, we hypothesized that concomitant changes in systemic inflammation may occur in LF mice. MPO staining of intestinal tissue revealed a marked increase in neutrophil accumulation in LF mice, with neutrophils predominantly clustered around crypt abscesses, indicating severe inflammatory infiltration of the intestinal mucosa. In contrast, ADSC-EV intervention significantly attenuated neutrophil infiltration, which might correlate with mucosal architecture and epithelial damage restoration [Figure 5B]. To clarify the characteristics of intrahepatic inflammation, we analyzed the subset distribution of infiltrated monocyte-derived macrophages (MoMFs) in liver tissue. Two functionally distinct macrophage subsets were focused on: CD45+-Ly6G--CD11c--CD3--CD11b+-
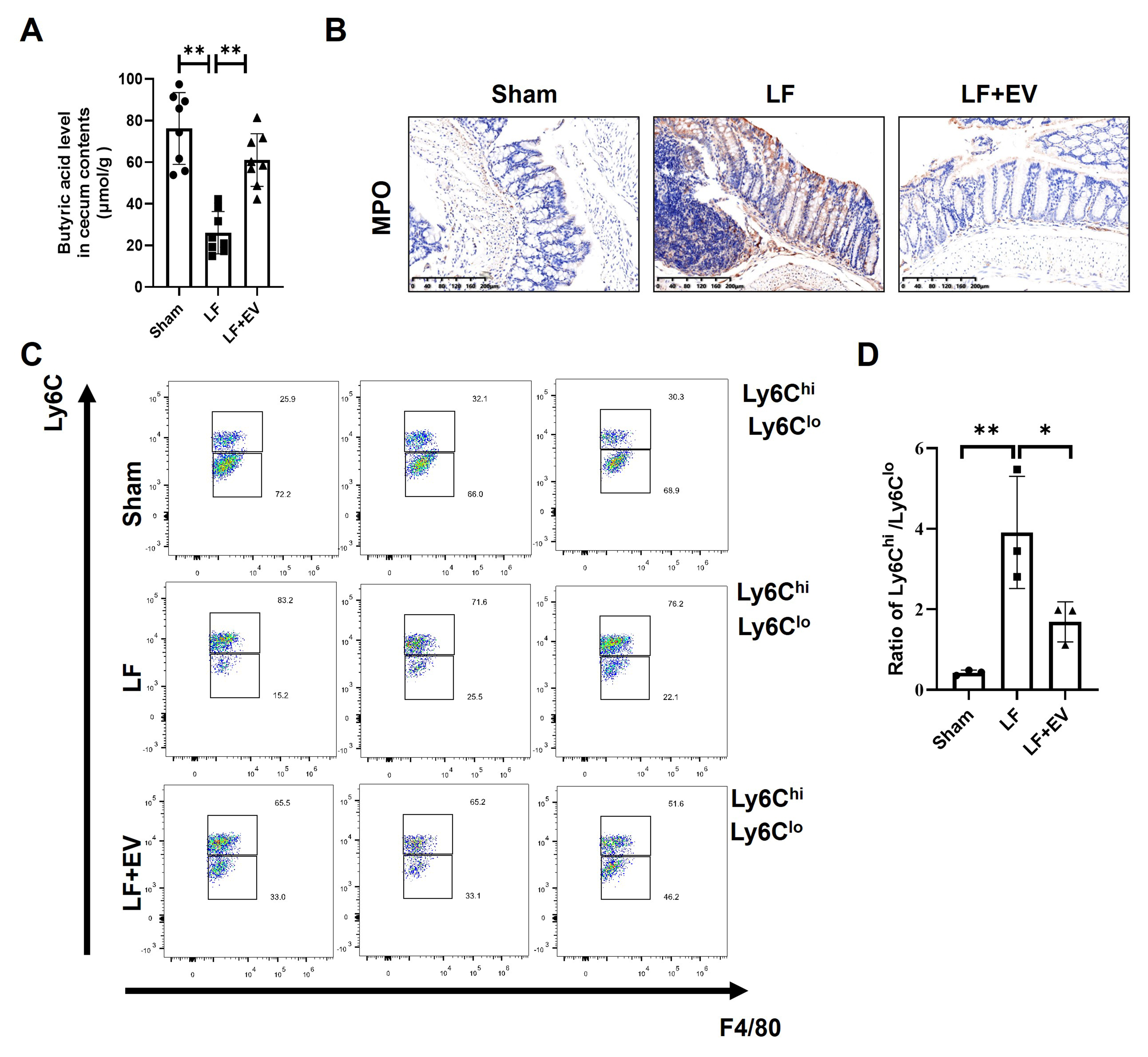
Figure 5. Administration of ADSC-EVs elevates the butyric acid level of intestinal contents and decreases the systemic inflammation in mice with LF. (A) Comparison of the butyric acid level in the cecal content from each group of mice (n = 8); (B) Representative images of MPO staining in the mouse colon tissues (scale bar: 200 µm); (C) The hepatic infiltrated MoMFs in mice with LF were analyzed by flow cytometry. The gating strategy for infiltrated inflammatory MoMFs was CD45+-Ly6G--CD11c--CD3--CD11b+-F4/80low-Ly6chigh and for infiltrated restorative MoMFs was CD45+-Ly6G--CD11c--CD3--CD11b+-F4/80low-Ly6clow. Also, (D) the result was quantified by the ratio of Ly6Chigh/Ly6Clow (n = 3). All data are shown as the mean ± SD. *P < 0.05; **P < 0.01. ADSC-EVs: Adipose-derived stem cell extracellular vesicles; LF: liver fibrosis; MPO: myeloperoxidase; MoMFs: monocyte-derived macrophages; SD: standard deviation; LF + EV: liver fibrosis + ADSC-EV group.
DISCUSSION
LF primarily arises from chronic hepatic injury triggered by persistent insults, including viral hepatitis [e.g., hepatitis B virus (HBV), hepatitis C virus (HCV)], excessive alcohol consumption, metabolic dysfunction-associated steatohepatitis (MASH), cholestatic diseases, and autoimmune disorders, which initiate a sustained wound-healing response[29]. These pathological characteristics of LF are the excessive accumulation of ECM, predominantly fibrillar collagens, driven by the activation and trans-differentiation of HSCs into myofibroblasts. This process leads to progressive architectural distortion, bridging fibrosis, and ultimately nodule formation in cirrhosis[30]. Globally, hundreds of millions of individuals are affected by LF to varying degrees. Significant fibrosis (≥ F2) is estimated to affect over 500 million people, while advanced fibrosis or cirrhosis affects more than 100 million[31]. The burden is still rising rapidly, due to the global epidemics of obesity, diabetes, and MASH.
At present, clinical management of LF remains reliant on etiology control (e.g., antiviral therapies, metabolic interventions). There is a lack of globally approved direct anti-fibrotic drugs. Pan-fibrotic agents such as pirfenidone are plagued by significant off-target toxicity and low liver-specific efficacy[32]. While the approval of resmetirom for MASH-associated fibrosis represents a pivotal advance, real-world data imply that liver biopsy is still required to screen eligible patients and assess its therapeutic potential and prognostic value[33,34]. Furthermore, glucagon-like peptide-1 (GLP-1)-based therapies (e.g., Semaglutide, Survodutide) are only beneficial for metabolism-associated fibrosis[35]. Therefore, effective and comprehensive anti-fibrotic strategies that address the heterogeneous etiologies are still lacking.
In chronic liver injury, intestinal barrier dysfunction is a common sequela. Disruption of tight junctions and destruction of mucus layer integrity are caused by factors such as bile acid (BA) deficiency, portal hypertension-induced edema, and systemic inflammation, increasing gut permeability, commonly known as intestinal leakage[36]. Increased intestinal permeability facilitates the translocation of PAMPs and damage-associated molecular patterns (DAMPs) into the portal circulation. These molecules can activate hepatic inflammatory pathways such as toll-like receptor 4 (TLR4)/nucleotide oligomerization domain-like receptor family, pyrin domain containing 3 (NLRP3), thereby inducing pro-inflammatory and pro-fibrotic cytokine release [e.g., tumor necrosis factor (TNF)-α, interleukin (IL)-1β, TGF-β] that facilitate HSC activation and ECM deposition, ultimately establishing a self-amplifying “gut-liver axis” of injury[37].
Because the intestine and liver are closely connected through the “gut-liver axis”, the substances absorbed by the intestine (including the microbiota and its metabolites) are directly transported to the liver via the portal vein. Consequently, the imbalance of gut microbiota and the dysregulated changes in their metabolites have also emerged as core mechanisms driving the progression of LF. Firstly, during LF progression, the gut microbiota exhibits an overall decrease in species richness and evenness. Secondly, it undergoes compositional alterations. The microbial dysbiosis manifests as a depletion of beneficial bacteria, including Lactobacillus, Bifidobacterium, etc., while an increase in the abundance of conditionally pathogenic bacteria with pro-inflammatory potential, such as Enterobacteriaceae, Streptococcaceae, etc. Several studies have reported that the decrease of Firmicutes/Bacteroidetes (F/B) and Firmicutes/Proteobacteria (F/P) ratios is associated with poor prognosis in patients with LF and cirrhosis[38,39]. Thirdly, microbial metabolic function is disrupted, primarily due to a decline in SCFAs produced by microbial communities. The levels of crucial SCFAs such as acetate, propionate and butyrate with anti-inflammatory and barrier protective functions were decreased in the intestine. Conversely, excessive proliferation of conditional pathogens such as Enterobacteriaceae, Enterococcus, and Streptococcus may exacerbate liver inflammation by directly or indirectly increasing lipopolysaccharide (LPS), cytolysin and reactive oxygen species (ROS) levels. Therefore, effective therapeutic strategies for LF not only exert direct hepato-protective and anti-fibrotic effects but also restore intestinal barrier integrity and maintain microbial homeostasis to disrupt the pathological gut-liver axis cycle. There is an urgent need for a novel strategy to target this dual pathway.
EVs, as a promising cell-free therapy, critically mediate intercellular communication by transferring bioactive molecules such as miRNAs, proteins, and lipids. During liver injury, these molecules can modulate the crosstalk among hepatocytes, immune cells, HSCs and endothelial cells, thereby regulating key pathological processes including inflammation, apoptosis, oxidative stress, and fibrogenesis. In our previous study, we found that ADSCs-derived EVs significantly improved the survival rate from 25% to over 70% in the acute liver failure rat model by releasing the lncH19 and inhibiting hepatocytes apoptosis[18]. A recent study also demonstrated that ADSC-EVs with augmenter of liver regeneration (ALR) over-expressed could protect the injured liver from apoptosis, regeneration suppression, cellular mitochondrial structure and function dysregulation[40]. Another research reported that ADSC-EVs could reduce the pyroptosis in injured liver and promote liver regeneration-associated factors expression[41]. These studies supported that ADSC-EVs represent a promising cell-free therapeutic strategy for combating liver injury and its progression to fibrosis or cirrhosis.
In this study, we isolated and purified ADSC-EVs that exhibited classical morphology, size and EV markers. Subsequent experiments demonstrated that ADSC-EV treatment exerted significant therapeutic effects in the DEN/CCl4-induced LF mouse model, including improving liver function, reducing excessive ECM deposition and suppressing HSC activation. To further elucidate the protective potential of ADSC-EVs against gut-liver axis dysregulation, we then evaluated intestinal function and gut microbiota in LF mice. Our findings showed that ADSC-EV treatment significantly restored intestinal barrier integrity by reducing intestinal permeability and reinforcing the mucus barrier. More importantly, ADSC-EV treatment modulated the gut microbiota dysbiosis, characterized by an increase in beneficial genera, including Akkermansia and Lachnospiraceae NK4A136-group, with a particularly significant elevation in the abundance of A. muciniphila. We believed that the intestinal barrier repair might effectively limit bacterial translocation, thereby disrupting the pathogenic gut-liver crosstalk. Additionally, ADSC-EV-induced alterations in the microbiota may further contribute to a better intestinal microenvironment to support gut barrier health, forming a virtuous cycle. Based on this hypothesis, we detected increased butyric acid levels in cecal content and a significant reduction in systemic inflammation. Based on current data, we tended to believe that the improvement of microbial homeostasis was the sum of direct and indirect effects of ADSC-EVs. Improved liver function and an intact intestinal barrier could reduce bacterial translocation by reducing systemic inflammation, altering BA profiles, and creating a favorable intestinal microenvironment for beneficial microorganisms, then indirectly promoting the recovery of microbial homeostasis. A quantitative biodistribution and pharmacokinetics study of exosomes labeled with 89Zr radioisotope in mice and rats reported that 89Zr-labeled exosomes (89Zr-Exo) were rapidly internalized by cells and tissues within 15 min and mainly distributed in the liver and spleen. At approximately 6 h of administration, 89Zr-Exo appeared in the intestine and stomach with a peak intensity[42].
Recent studies suggested that A. muciniphila played a vital role in strengthening intestinal barrier integrity, mitigating inflammation through multiple potential mechanisms such as inhibiting the adenosine 5’-monophosphate (AMP)-activated protein kinase (AMPK) pathway, nucleotide-binding domain (NOD)-like receptor family and NLRP3 activation[25]. Emerging evidence has also demonstrated that a reduced abundance of A. muciniphila is significantly correlated with the progression of LF. A. muciniphila acted as a beneficial commensal bacterium in liver injury disease[43]. Crucially, several studies have shown that A. muciniphila can promote the growth of butyrate-producing bacteria by supplying essential metabolites that facilitate their growth and metabolic activity within the gut environment[44,45]. Furthermore, butyric acid serves as the primary energy source for colonic epithelial cells, directly enhancing tight junction assembly, stimulating mucus production by goblet cells, and exerting anti-inflammatory effects. Thus, the increased butyric acid levels synergize with the enriched A. muciniphila to facilitate gut barrier restoration.
Therefore, ADSC-EVs orchestrated a multi-faceted restoration of gut homeostasis, modulating the gut microbiota by enriching barrier-promoting commensal bacteria A. muciniphila, then increasing beneficial metabolites butyric acid, and consequently enhancing intestinal barrier integrity. This collective action disrupts the vicious cycle of the liver-gut axis, ultimately contributing significantly to the alleviation of LF. For other intestinal diseases such as inflammatory bowel disease (IBD), Crohn’s disease (CD), and ulcerative colitis (UC), natural and modified therapeutic EVs can also be taken up by intestinal cells and bacteria to relieve gastrointestinal injury through immunomodulation and restoration of intestinal homeostasis, which may provide a broad clinical application prospect[46]. Notably, this study has several limitations that should be acknowledged. Although we demonstrated the beneficial effects of ADSC-EVs on liver function, gut barrier integrity, and the microbial community, our findings are still limited to correlative analyses of the “ADSC-EVs–gut–liver” axis. In this research, our data suggested an upregulatory effect of ADSC-EVs on the probiotic A. muciniphila in gut microbiota; however, the direct influence of ADSC-EVs on A. muciniphila colonization and its metabolic mechanism remains to be fully elucidated by a germ-free mice model and metabolomics. In particular, in future research, we would use gas chromatography-mass spectrometry (GC-MS) or liquid chromatography-mass spectrometry (LC-MS) for absolute quantification of SCFAs (including butyric acid, acetic acid, propionic acid, etc.) in intestinal contents. This will support the precise study of causal relationships and molecular mechanisms. Moreover, the therapeutic ADSC-EVs administered in this study were native and non-targeted EVs, resulting in a liver-dominated biodistribution profile with limited direct delivery to intestinal tissue. Therefore, it is crucial to develop gut-specific, engineered EVs to maximize local effects and functional potency in future studies.
DECLARATIONS
Acknowledgments
The authors would like to acknowledge the following agencies for funding this research: the China Postdoctoral Science Foundation, the National Natural Science Foundation of China, and the Shanghai Municipal Education Commission. We also acknowledge the public technology platforms of the College of Life Science and Technology and the School of Medicine at Tongji University for instrument use. The authors declare that they have not used AI-generated work in this manuscript.
Authors’ contributions
Performed the major experiments and participated in data analysis: Wu B, Guo J
Wrote the manuscript, prepared the figures, and were responsible for data compilation and integration: Wu B, Guo J
Participated in the animal experiments and tissue collection: Wu B, Guo J, Wang J
Contributed to manuscript revision and additional experiments: Wu B, Guo J
Led the entire study as co-corresponding authors: Xu J, Feng J
All authors reviewed and approved the manuscript.
Availability of data and materials
Additional data related to this article may be obtained from the corresponding author upon reasonable request.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This research was funded by the National Key Research and Development Program of China (2024YFA1107305), the China Postdoctoral Science Foundation (GZC20231944), the National Natural Science Foundation of China (32170986), the Innovation Program of the Shanghai Municipal Education Commission (2023ZKZD31), and Xu Jun’s Expert Work Station of Yunnan (202005AF150050). We also acknowledge the public technology platforms of the College of Life Science and Technology and the School of Medicine at Tongji University for instrument use.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Animal studies were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health. All animal experiments were approved by the Ethical Committee of the Laboratory Animals Research Center, Tongji University, for the period from 28/11/2024 to 31/12/2026. The approval number was TJAA07624403 (dated 28/11/2024).
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Karsdal MA, Detlefsen S, Daniels SJ, Nielsen MJ, Krag A, Schuppan D. Is the total amount as important as localization and type of collagen in liver fibrosis attributable to steatohepatitis? Hepatology. 2020;71:346-51.
2. Villesen IF, Daniels SJ, Leeming DJ, Karsdal MA, Nielsen MJ. Review article: the signalling and functional role of the extracellular matrix in the development of liver fibrosis. Aliment Pharmacol Ther. 2020;52:85-97.
3. Zamani M, Alizadeh-Tabari S, Ajmera V, Singh S, Murad MH, Loomba R. Global prevalence of advanced liver fibrosis and cirrhosis in the general population: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2025;23:1123-34.
4. Ferdek PE, Krzysztofik D, Stopa KB, et al. When healing turns into killing - the pathophysiology of pancreatic and hepatic fibrosis. J Physiol. 2022;600:2579-612.
5. Chen S, Zhou J, Wu X, et al. Comparison of fibrosis regression of entecavir alone or combined with pegylated interferon alpha2a in patients with chronic hepatitis B. Hepatol Int. 2021;15:611-20.
6. Cai X, Liu X, Xie W, et al. Hydronidone for the treatment of liver fibrosis related to chronic hepatitis B: a phase 2 randomized controlled trial. Clin Gastroenterol Hepatol. 2023;21:1893-901.e7.
7. Rotman Y, Sanyal AJ. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut. 2017;66:180-90.
8. Di Zeo-Sánchez DE, Díaz-Alberola I, Pinazo-Bandera JM, et al. Intestinal permeability and immune-inflammatory markers in patients with idiosyncratic drug-induced liver injury, drug-induced steatosis and metabolic dysfunction-associated steatotic liver disease (MASLD). Br J Pharmacol. 2025;182:5303-16.
9. Chopyk DM, Grakoui A. Contribution of the intestinal microbiome and gut barrier to hepatic disorders. Gastroenterology. 2020;159:849-63.
10. Lau HC, Zhang X, Yu J. Gut microbiome in metabolic dysfunction-associated steatotic liver disease and associated hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2025;22:619-38.
11. He C, Zheng S, Luo Y, Wang B. Exosome theranostics: biology and translational medicine. Theranostics. 2018;8:237-55.
12. Jeppesen DK, Fenix AM, Franklin JL, et al. Reassessment of exosome composition. Cell. 2019;177:428-45.e18.
13. Zhou Y, Xu H, Xu W, et al. Exosomes released by human umbilical cord mesenchymal stem cells protect against cisplatin-induced renal oxidative stress and apoptosis in vivo and in vitro. Stem Cell Res Ther. 2013;4:34.
14. Gatti S, Bruno S, Deregibus MC, et al. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol Dial Transplant. 2011;26:1474-83.
15. Bruno S, Grange C, Collino F, et al. Microvesicles derived from mesenchymal stem cells enhance survival in a lethal model of acute kidney injury. PLoS One. 2012;7:e33115.
16. Zhang B, Wang M, Gong A, et al. HucMSC-exosome mediated-Wnt4 signaling is required for cutaneous wound healing. Stem Cells. 2015;33:2158-68.
17. Li T, Yan Y, Wang B, et al. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cells Dev. 2013;22:845-54.
18. Jin Y, Wang J, Li H, et al. Extracellular vesicles secreted by human adipose-derived stem cells (hASCs) improve survival rate of rats with acute liver failure by releasing lncRNA H19. EBioMedicine. 2018;34:231-42.
19. Zhao S, Xiu G, Wang J, et al. Engineering exosomes derived from subcutaneous fat MSCs specially promote cartilage repair as miR-199a-3p delivery vehicles in Osteoarthritis. J Nanobiotechnology. 2023;21:341.
20. Daemen S, Chan MM, Schilling JD. Comprehensive analysis of liver macrophage composition by flow cytometry and immunofluorescence in murine NASH. STAR Protoc. 2021;2:100511.
21. Wu B, Feng J, Guo J, et al. ADSCs-derived exosomes ameliorate hepatic fibrosis by suppressing stellate cell activation and remodeling hepatocellular glutamine synthetase-mediated glutamine and ammonia homeostasis. Stem Cell Res Ther. 2022;13:494.
22. Li C, Deng L, Pu M, Ye X, Lu Q. Coptisine alleviates colitis through modulating gut microbiota and inhibiting TXNIP/NLRP3 inflammasome. J Ethnopharmacol. 2024;335:118680.
23. Liu Y, Luo R, Sun Z, et al. Synergistic toxicity of combined exposure to acrylamide and polystyrene nanoplastics on the gut-liver axis in mice. Biology. 2025;14:523.
24. Aimaier D, Bai W, Zhang Y, et al. Susceptibility factor TNF-α synergizes with Polygonum multiflorum to drive idiosyncratic liver injury in mice by disrupting gut microbiota composition and hepatic metabolite homeostasis. J Inflamm Res. 2025;18:9477-94.
25. Ahmadi Badi S, Tavakoli Aval H, Moradi HR, et al. Comparative study of liver injury protection by Akkermansia muciniphila and Faecalibacterium prausnitzii interventions in live and cell-free supernatant forms via targeting the hepcidin - ferroportin axis in mice with CCl4-induced liver fibrosis. Gut Pathog. 2025;17:54.
26. Oguri N, Miyoshi J, Nishinarita Y, et al.
27. Ding Y, Hou Y, Lao X. The role of Akkermansia muciniphila in disease regulation. Probiotics Antimicrob Proteins. 2025;17:2027-38.
28. Mandaliya DK, Patel S, Seshadri S. Postbiotic potential of SCFAs on metaflammation and gut microbiota alteration in diabetes. J Biosci. 2025;50:57.
32. Gan C, Wei W, Xue T, et al. Focal adhesion kinase inhibitors in fibrotic diseases therapy: development and therapeutic potential. Eur J Med Chem. 2025;296:117882.
33. Kaya E, Aksoy S, Oruc N, et al. Non-invasive tests for resmetirom treatment fail to accurately define the target population: evidence from a biopsy-proven MASLD cohort. Hepatol Forum. 2025;6:111-5.
34. Adali G, Akdogan RA. Non-invasive tests fail to ensure therapeutic precision for resmetirom in MASLD. Hepatol Forum. 2025;6:89-90.
35. Wang W, Gao Y, Chen Y, et al. TGF-β inhibitors: the future for prevention and treatment of liver fibrosis? Front Immunol. 2025;16:1583616.
36. Dumitru A, Tocia C, Bădescu AC, et al. Linking gut permeability to liver steatosis: noninvasive biomarker evaluation in MASLD patients - a prospective cross-sectional study. Medicine. 2025;104:e42476.
37. Cui C, Gao S, Shi J, Wang K. Gut-liver axis: the role of intestinal microbiota and their metabolites in the progression of metabolic dysfunction-associated steatotic liver disease. Gut Liver. 2025;19:479-507.
38. Jasirwan COM, Muradi A, Hasan I, Simadibrata M, Rinaldi I. Correlation of gut Firmicutes/Bacteroidetes ratio with fibrosis and steatosis stratified by body mass index in patients with non-alcoholic fatty liver disease. Biosci Microbiota Food Health. 2021;40:50-8.
39. Mantovani A, Longo L, Thoen RU, et al. Firmicutes/Bacteroidetes and Firmicutes/Proteobacteria ratios are associated with worse prognosis in a cohort of Latin American patients with cirrhosis. Clinics. 2024;79:100471.
40. Masihi KN, Brehmer W, Lange W, Werner H, Ribi E. Trehalose dimycolate from various mycobacterial species induces differing anti-infectious activities in combination with muramyl dipeptide. Infect Immun. 1985;50:938-40.
41. Piao C, Sang J, Kou Z, et al. Effects of exosomes derived from adipose-derived mesenchymal stem cells on pyroptosis and regeneration of injured liver. Int J Mol Sci. 2022;23:12065.
42. Choi H, Kim MY, Kim DH, et al. Quantitative biodistribution and pharmacokinetics study of GMP-grade exosomes labeled with 89Zr radioisotope in mice and rats. Pharmaceutics. 2022;14:1118.
43. Yu JX, Wu J, Chen X, et al. Gut microbiota in liver diseases: initiation, development and therapy. Front Med. 2025;12:1615839.
44. Zhang YL, Li ZJ, Gou HZ, Song XJ, Zhang L. The gut microbiota-bile acid axis: a potential therapeutic target for liver fibrosis. Front Cell Infect Microbiol. 2022;12:945368.
45. Pisarello MJL, Marquez A, Chaia AP, Babot JD. Targeting gut health: probiotics as promising therapeutics in alcohol-related liver disease management. AIMS Microbiol. 2025;11:410-35.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].