




Special delivery - extracellular vesicles released by commensal gut bacteria deliver bioactive protein to distal organs
 0
0  , , ...
, , ... Abstract
Aim: This study aims to investigate how the gut microbiota communicates with the host via bacterial extracellular vesicles (BEVs), given that direct contact between microbes and the healthy intestinal epithelium is prevented by a sterile mucin gel layer. Understanding these indirect interactions is critical because the specific pathways and mediators of microbiota-host interactions are incompletely understood. Tracking BEVs in vivo however is particularly challenging due to their nanoscale size and complex molecular composition.
Methods: To address these challenges, we developed a highly sensitive Nanoluciferase (NanoLuc) system for luminescence-based detection of BEVs produced by the model human commensal bacterium Bacteroides thetaiotaomicron. This approach was evaluated in germ-free and specific-pathogen-free mice, with comparisons between administration routes demonstrating the advantages of this system for in vivo BEV labelling over conventional lipophilic dyes.
Results: We report, for the first time, that BEVs endogenously produced in the gastrointestinal tract (GIT) of mice can deliver bioactive NanoLuc protein to multiple organ tissues, including the central nervous system. Our findings establish that naturally occurring BEVs in the GIT are capable of traversing multiple host barriers, including the intestinal epithelium, vascular endothelium, and the blood-brain-barrier, to access tissues such as the brain and eyes.
Conclusion: These findings advance our understanding of BEV-mediated microbe-host interactions and demonstrate the potential of BEVs as vehicles for long-distance delivery of bioactive therapeutics.
Keywords
INTRODUCTION
The intestinal microbiota is essential for maintaining human health, contributing significantly to digestion, nutrition, and immunity[1]. Increasing evidence shows that its influence extends beyond the gastrointestinal tract (GIT), affecting extra-intestinal organ systems, including the central nervous system (CNS) through bi-directional communication along the gut-brain axis[2]. However, the mechanisms by which specific microbiota constituents directly interact with host tissues and cells remain unclear, particularly in the lower GIT, where intestinal epithelial cells are protected by a sterile mucin gel layer that forms an impenetrable barrier to microbial contact[3]. Instead, interactions are more likely to occur via indirect mechanisms, including the release of secreted microbial products such as metabolites and bacterial extracellular vesicles (BEVs). These products can cross intestinal barriers, accessing underlying cells and the circulatory system[4]. BEVs, comprising DNA, RNA, and diverse bioactive compounds[5], represent a particularly promising mode of microbe-host communication. This mechanism is supported by animal studies demonstrating BEV biodistribution[6,7], and by the detection of GIT microbiota-derived BEVs in the bloodstream of both healthy individuals and patients with disease[8].
A single microbial species can generate multiple BEV subtypes that vary in both size (40-400 nm) and protein cargo (proteome)[5]. Bacteroides thetaiotaomicron (B. thetaiotaomicron), a well-characterised human GIT commensal, releases BEVs with distinct proteomes, capable of modulating epithelial, immune, and neuronal cell function[9,10]. However, progress in this field is constrained by the technical challenge of visualising and tracking BEVs due to their nanoscale size. In vivo studies are further complicated by the physical similarities of BEVs to eukaryotic vesicles (such as exosomes) and the lack of specific bacterial reporters. Conventional approaches for BEV labelling typically involve non-covalent attachment of lipophilic fluorescent dyes. However, these dyes present several limitations. These include an extended half-life, alteration of particle size and membrane structure, promotion of vesicle aggregation, and formation of free-dye micelles similar in size to BEVs, all of which can obscure accurate biodistribution analyses[11-14]. Furthermore, such studies often involve parenteral or oral delivery of purified, pre-labelled BEVs, which does not reflect the dynamics of BEVs produced naturally within the GIT. It also remains unclear whether fluorescent dyes remain associated with BEVs during lysosomal degradation, potentially resulting in non-specific fluorescence signals in tissue imaging.
To address these challenges, the present study uses bioengineering to generate Nanoluciferase (NanoLuc)-labelled BEVs (NanoLuc-BEVs), enabling sensitive luminescence-based detection of vesicles generated by
METHODS
Bacterial strains and culture conditions
B. thetaiotaomicron was cultured in Brain Heart Infusion medium (BO1230B; Oxoid, UK) supplemented with 4 µM hemin (CAS16009-13-5; Merck, UK) or in Bacteroides Defined Medium (BDM4[15]) at 37 °C in an anaerobic cabinet. Escherichia coli (E. coli) DH10β was cultured in Luria-Bertani (LB; Oxoid) medium at 37 °C with shaking. Antibiotics (Merck, UK) were used at the following concentrations: erythromycin
Molecular biology and cloning
A flexible plasmid vector system was developed for use in B. thetaiotaomicron (pBATH). In brief, the pGH044 shuttle plasmid was digested with restriction endonucleases Eco53kI/SfoI and re-ligated to excise a 331 bp fragment containing an NdeI restriction enzyme site. A Synthetic DNA fragment containing 5’ to 3’: BamHI restriction enzyme site; terminator BBa_B1001; BT_1830 promoter and ribosome binding site 7 (RBS7)[16]; protein coding sequence BT_3742 signal peptide (amino acids 1-32), tobacco etch virus (TEV) protease, mirRFP670nano near-infrared fluorescent protein, and two Strep-Tag II sequences; terminator BBa_B1007 and PciI restriction enzyme site (Supplementary Materials; Integrated DNA Technologies). This was inserted into the BamHI/PciI restriction enzyme site of the modified pGH044 vector, generating the pBATH.03 plasmid. For NanoLuc labelling, a synthetic fragment containing the coding sequence for NanoLuc replaced mirRFP670nano and the two Strep-Tag II sequences via insertion into the XhoI/SpeI restriction enzyme site of pBATH.03, generating the pBATH.51 plasmid (Supplementary Materials; Integrated DNA Technologies). A further pBATH.56 plasmid was generated by replacing the full coding sequence in pBATH.03 using the NdeI/SpeI restriction enzyme site. All cloning steps were performed in chemically competent E. coli DH10β cells.
Expression of NanoLuc was confirmed using the Nano-Glo Luciferase Assay System (N1110; Promega, UK) following the manufacturer’s guidelines, with 50 µL of sample and 50 µL of reagent combined. Luminescence was measured on a CLARIOstar Plate Reader (BMG Labtech, UK) using black, untreated polystyrene 96-well plates (Thermo Fisher Scientific, UK).
Isolation and characterisation of BEVs
B. thetaiotaomicron cultures (500 mL) were centrifuged at 7,000 × g for 45 min at 4 °C to pellet bacterial cells, and supernatants were filtered through 0.22 µm pore-size polyethersulfone (PES) membrane filters (Sartorius, UK) to remove residual debris. The supernatants were concentrated by crossflow ultrafiltration (100 kDa molecular weight cut-off (MWCO), Vivaflow 50R, Sartorius), and the retentate was rinsed once with 500 mL of phosphate-buffered saline (PBS, pH 7.4). BEV suspensions (0.5 mL) were loaded onto a qEV/35 nm size-exclusion chromatography (SEC) column (Izon Science Ltd., France), and fractions were collected according to the manufacturer’s instructions. Fractions 1-4, containing isolated BEVs, were pooled and filtered through 0.22 µm pore-size PES membrane filters (Sartorius).
BEV hydrodynamic diameters and concentrations were determined by nanoparticle tracking analysis (NTA) using a ZetaView TWIN instrument (Particle Metrix, Germany) according to the manufacturer’s instructions. Analyses were performed with ZetaView software (version 8.05.12 SP1) using a two-cycle, 11-position, high frame rate protocol at 25 °C. Samples were diluted in ultrapure water to fall within the optimal detection range. Camera control settings: sensitivity 80; frame rate 30; shutter 100. Post-acquisition parameters: min brightness 20; max area 2,000; min area 5; trace length 30; 5 nm/class; 64 classes/decade.
NanoLuc-BEV protease treatment
For TEV protease treatment, 90 µL of purified NanoLuc-BEVs (1012 particles/mL) were incubated with TEV protease (P8112; New England Biolabs, UK) under the manufacturer’s recommended conditions for 1 h at 30 °C. Fractions (0.5 mL) of treated and untreated NanoLuc-BEVs were collected using a qEVsingle 35 nm column (Izon Science Ltd.). For proteinase K treatment, 100 µL of purified NanoLuc-BEVs
In vitro assessment of NanoLuc-BEV cytotoxicity
The human colonic epithelial cell line Caco-2 (86010202; European Collection of Authenticated Cell Cultures, UK) was cultured at 37 °C in 5% CO2 in Eagle’s Minimal Essential Medium (EMEM) supplemented with 1% non-essential amino acids (M5650; Merck), 2 mM L-glutamine (G7513; Merck), 10% foetal bovine serum (F9665; Merck), and 1% penicillin-streptomycin (P4333; Merck).
NanoLuc-BEVs (1010/mL) were diluted in cell culture medium to final concentrations of 109/mL (1:10) or 108/mL (1:100) and applied to confluent Caco-2 monolayers cultured in collagen-coated (125-50; Merck) white, flat-bottom, 96-well plates (Nunclon delta-treated, Thermo Fisher Scientific) for 24 h. Cell viability was measured using the CellTiterGlo 2.0 reagent (G9242; Promega) according to the manufacturer’s instructions, with luminescence measured after 10 min on a CLARIOStar plate reader (BMG Labtech).
Fluorescent DiD-BEV labelling
For DiD labelling, BEVs (1011/mL) were incubated with 5% (v/v) DiD Vybrant cell-labelling solution (V22887; Invitrogen Molecular Probes, UK) at 37 °C for 30 min. Unbound dye was removed by three washes with 0.5 mL PBS using centrifugal filters (100 kDa MWCO, Sartorius). DiD-labelled BEVs were further purified by SEC, checked for sterility, and characterised by NTA as above.
Animal studies
Male and female specific-pathogen-free (SPF; C57BL/6) and germ-free (GF; C57BL/6) mice (8-12 weeks old) were housed in individually ventilated cages (SPF) or sterile isolators (GF) in adjacent rooms at the University of East Anglia Disease Modelling Unit. All mice were maintained under a 12 h light/dark cycle and received autoclaved water and either Rodent Maintenance 3 (RM3) diet (SPF) or RM3-Autoclavable diet (GF; Special Diets Services, UK). Drinking water was supplemented with vancomycin [50 mg/kg (1709007, Merck)], neomycin [100 mg/kg (N6386, Merck)], metronidazole [100 mg/kg (M1547, Merck)], amphotericin B [1mg/kg (PHR1662, Merck)], and ampicillin [1 mg/mL (A8351, Merck)] for three days, followed by a two-day washout. After monocolonisation, faecal pellets were collected at 3, 6, 14, and 21 days and snap frozen. All animal experiments complied fully with the Animal Scientific Procedures Act (1986), were approved by the UK Home Office, and received ethical clearance from the local Animal Welfare and Ethical Review Body.
In vivo biodistribution imaging
To compare BEV labelling methods, GF mice received intravenous (IV) injections of DiD-labelled (n = 2), NanoLuc-labelled (n = 3), or dual-labelled DiD+NanoLuc-BEVs (n = 5) at a dose of 4 × 1010 particles, or PBS (200 µL/mouse). Animals were excluded from the study if full volume tail vein delivery was unsuccessful. Following injection, mice were housed in their original cage groupings to avoid cross-contamination of labelled BEVs via coprophagy. At 1 h post-administration, organs including eyes, brain, heart, lungs, liver, kidneys, spleen, mesenteric lymph nodes (MLNs), and thymus were excised. For monocolonisation, GF or SPF mice were administered a single oral gavage of 100 µL (OD600 = 0.72) of the relevant
Ex vivo images of organs were performed using an In vivo Xtreme multi-modal optical and X-ray small-animal imaging system (Bruker, UK) equipped with a back-illuminated 4 MP Charge-Coupled Device (CCD) detector with luminescence capability. Far-red DiD fluorescence (photons/s/mm2) was recorded using the following settings: excitation 650 nm, emission 700 nm, 19 cm field of view, 10 s exposure time, F-stop 1.1, and focal plane 0. Luminescence (Radiance: p/sec/cm2/sr) was recorded with no filter, 19 cm field of view, 30 s exposure, F-stop 1.1, and focal plane 0. Background images were captured by reflectance with
Ex vivo fluorescence imaging
Frozen brain and liver tissues from SPF mice intravenously administered DiD-BEVs (4 × 1010/mouse, 3 h) were embedded in optimal cutting temperature (OCT) compound (Agar Scientific, UK). Cryosections (6-
Ex vivo tissue luminescence assay
Frozen organs, faecal pellets, and serum from GF mice monocolonised with B. thetaiotaomicron strains for 3, 6, 14, or 21 days were thawed and resuspended in PBS at the following concentrations: faecal pellets
Statistical analysis
Data are presented as individual values and mean ± standard deviation (SD) with sample sizes indicated. GraphPad Prism 5 (v5.04) was used for statistical analyses. Paired t-tests were applied for ZetaView analysis, and two-way analysis of variance (ANOVA) with Bonferroni post-tests used for dual-labelling and monocolonisation biodistribution studies. Differences between means were considered statistically significant at *P ≤ 0.05.
RESULTS
Design of NanoLuc-containing B. thetaiotaomicron BEVs
To enable highly sensitive BEV detection, we incorporated NanoLuc[17] into B. thetaiotaomicron BEVs. NanoLuc-labelled BEVs offer distinct advantages over previous methods for in vivo BEV detection[18,19]. NanoLuc is incorporated into the membrane of bioengineered BEVs that are naturally produced by the parent bacteria in the GIT, without requiring additional modification or labelling. The NanoLuc enzyme emits an extremely bright, stable signal, allowing for detection at much lower concentrations than fluorescent protein labels[20]. NanoLuc is also not affected by host tissue autofluorescence or photobleaching as it does not require excitation[21]. Unlike chemically derived fluorescent dyes, which can accumulate in host cells, NanoLuc is predicted to be degraded in host cell lysosomes together with BEVs[22].
We designed a flexible plasmid system for B. thetaiotaomicron (pBATH) using a modified pGH044 vector backbone. The original coding region was replaced with a new promoter, ribosome binding site, and coding sequence flanked by two bidirectional terminators. This enables versatile genetic modification, such as N- and C-terminal tagging of the coding sequence.
Expression in B. thetaiotaomicron was driven by the BT_1830 promoter and RBS7, as defined by
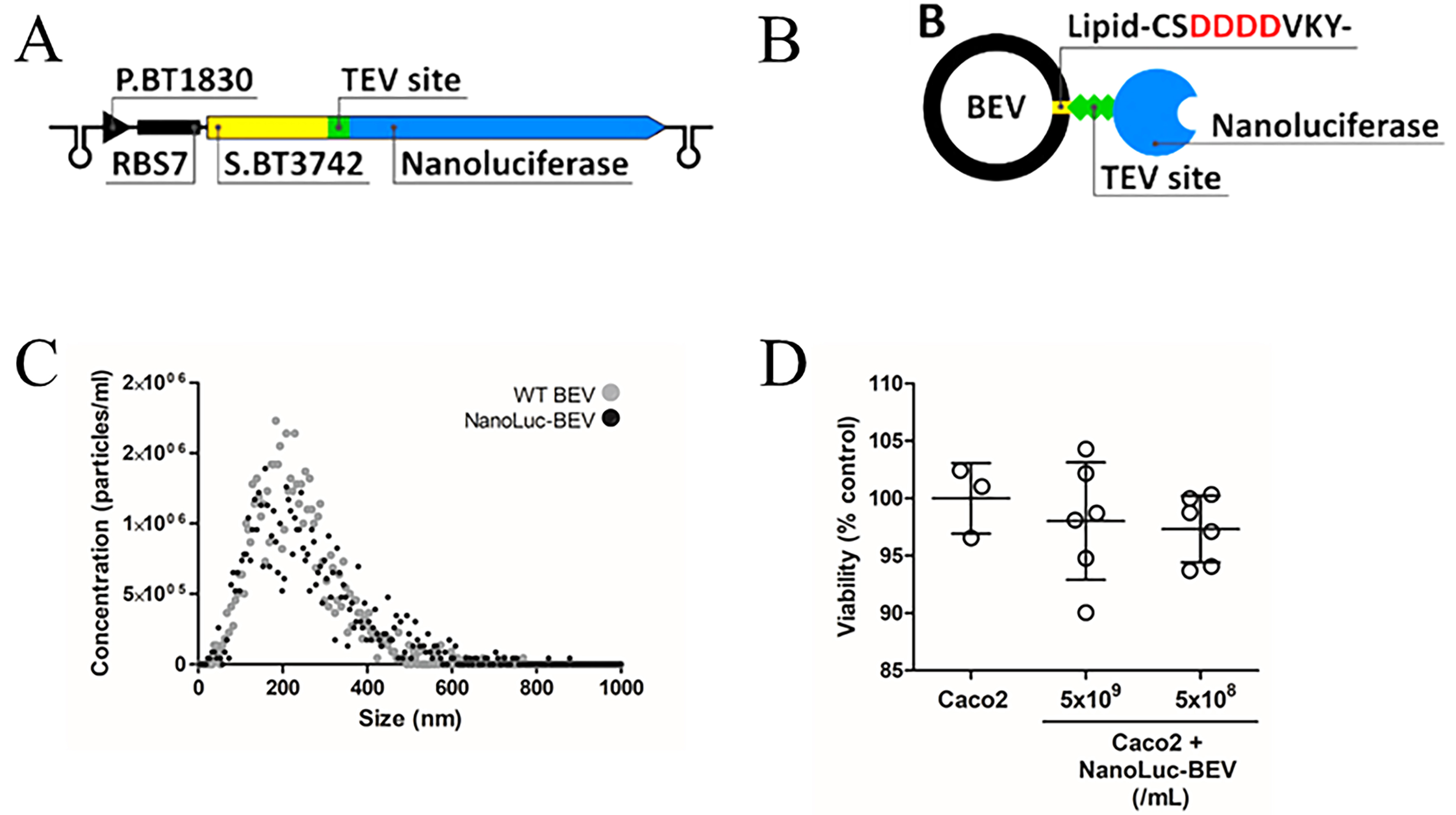
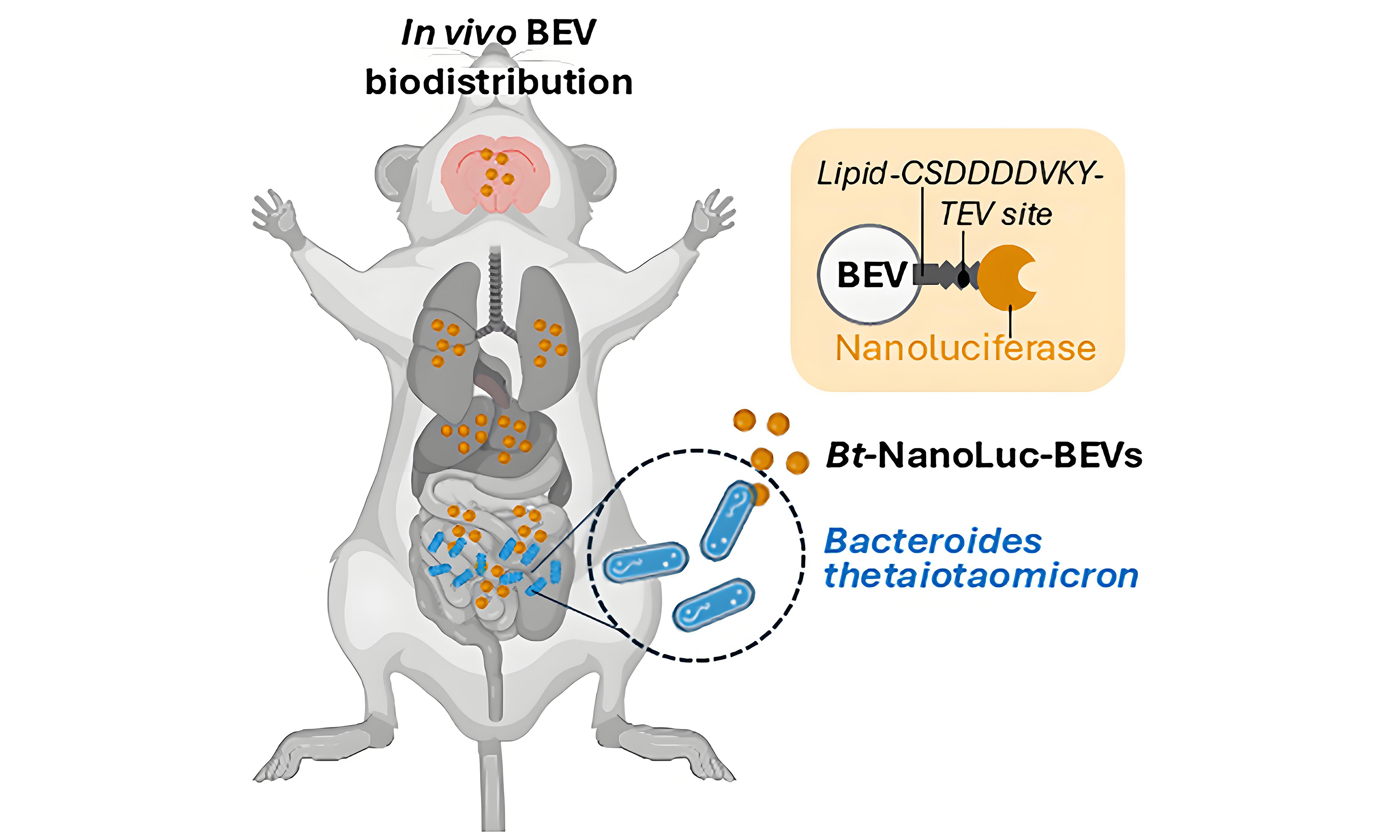
Figure 1. Design and characterisation of NanoLuc-BEVs. (A) BEV-targeted NanoLuc expression cassette cloned using the pBATH vector system. P.BT1830: promoter; RBS7: ribosome binding site; S.BT3742: signal peptide sequence; TEV site: TEV protease cleavage site. The cassette is flanked by bidirectional terminators; (B) Expected mature BEV-targeted NanoLuc structure. S.BT3742 directs the protein to the periplasm, where the signal peptide is cleaved and lipidated at Cys-24. The modified lipoprotein is inserted into the outer membrane, flipped to the outer leaflet, and incorporated into BEVs. Negatively charged aspartic acid residues upstream of the lipidation site facilitate this process (highlighted in the scheme); (C) Hydrodynamic diameter, distribution, and concentration of WT and NanoLuc-BEVs determined by nanoparticle tracking analysis; (D) Cytotoxicity of NanoLuc-BEVs to Caco-2 cells 24 h post-treatment, expressed as % viability relative to untreated control. Error bars represent mean ± SD. BEV: Bacterial extracellular vesicle; NanoLuc: Nanoluciferase; P.BT1830: promoter of BT1830; RBS7: ribosome binding site 7; S.BT3742: signal peptide of BT3742; TEV: tobacco etch virus; WT: wild type; SD: standard deviation.
Verification of NanoLuc incorporation into BEVs was performed by assaying the culture supernatants of pBATH.51-expressing B. thetaiotaomicron cells. Approximately 75% of the NanoLuc signal was localised to the supernatant fraction during late stationary phase (Total: 40,991,369 ± 286,496 Relative Light Units; RLU; supernatant: 30,938,113 ± 141,499 RLU). To confirm surface display, isolated NanoLuc-BEVs were subjected to TEV protease cleavage, which cleaves the engineered site between the lipidated signal peptide and NanoLuc. Following size-exclusion fractionation to separate BEVs from cleaved protein, 80% of the NanoLuc signal eluted in the protein fraction, while 20% remained associated with BEVs, with no further loss observed after additional TEV treatment [Supplementary Figure 1]. This result may reflect limited accessibility of the cleavage site or a fraction of NanoLuc anchored to the inner face of the BEV membrane. To investigate this, NanoLuc-BEVs were treated with non-specific proteinase K, which resulted in a > 98% reduction in NanoLuc signal (non-treated = 1,142,667 RLU; treated = 20,783 RLU), confirming that the majority of NanoLuc is displayed on the surface of bioengineered BEVs. ZetaView measurements indicated no significant difference in mean hydrodynamic diameter between wild-type (266.25 ± 35.5 nm) and NanoLuc-labelled BEVs (251.8 ± 1.1 nm) indicating the engineering did not alter vesicle biophysical characteristics [Figure 1C].
Impact of BEV labelling on in vivo biodistribution
Cytotoxicity assays in Caco-2 colonic epithelial cells demonstrated minimal impact on cell viability following treatment with NanoLuc-BEVs across all tested concentrations (5 × 109/mL = 98.02%; 5 × 108/mL = 97.33%; Figure 1D). These findings confirmed the suitability of NanoLuc-BEVs for biological administration.
Subsequent in vivo experiments compared the biodistribution of NanoLuc-labelled BEVs and BEVs labelled with the lipophilic fluorescent dye DiD. Our previous studies showed systemic organ delivery following both oral and IV administration of DiD-labelled B. thetaiotaomicron BEVs, though IV delivery resulted in stronger organ signals[9,25]. Therefore, in the current study, IV administration of GF mice was used to deliver DiD-BEVs, NanoLuc-BEVs or dual-labelled BEVs to compare the specificity and sensitivity of labelling methods. After 1 h, both fluorescence and luminescence signals were quantified in excised tissues. The biodistribution pattern of DiD mirrored our previous findings, with the highest fluorescence signal observed in the liver, followed by the spleen, lungs, kidneys[9], and brain[25] [Figure 2A (left) and B (left axis)]. In contrast, NanoLuc-BEVs generated the highest luminescence in the spleen, followed by the kidneys, lungs, liver, heart, and brain [Figure 2A (right) and B (right axis)]. Notably, a detectable BEV signal was also observed in the eyes for both labelled BEVs [Figure 2A and B].
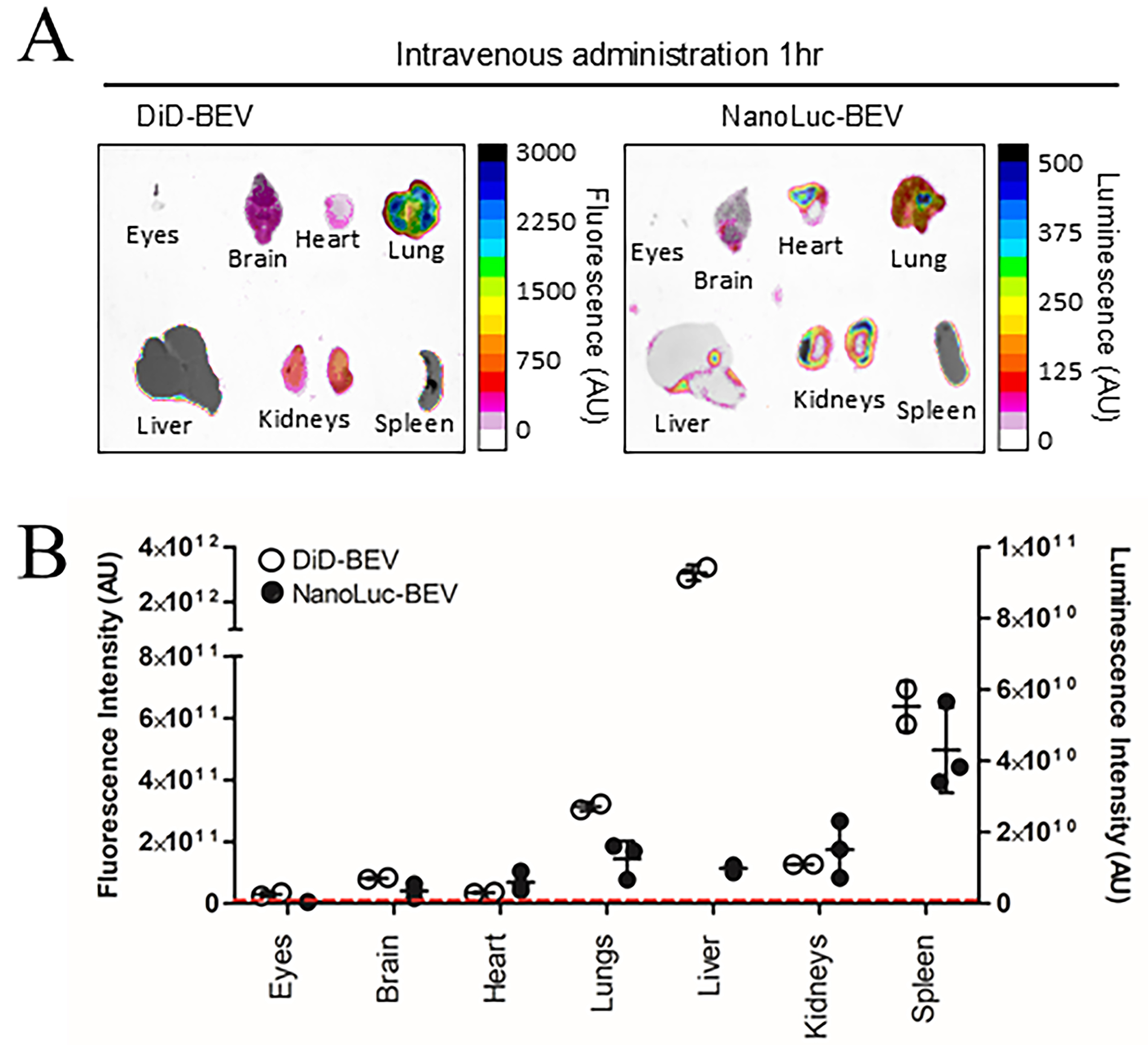
Figure 2. In vivo biodistribution of DiD- and NanoLuc-BEVs. (A) GF mice were intravenously administered DiD- or NanoLuc-BEVs (4 × 1010/mouse). Individual organs were excised 1 h post-administration and imaged using a Bruker In vivo Xtreme imaging system. The NanoGlo substrate was injected intraperitoneally 5 min prior to euthanasia and organ excision. Images represent n = 2 (DiD-BEV) or n = 3 (NanoLuc-BEV) mice per group; (B) Quantification of fluorescence (left axis) or luminescence (right axis) signals from each organ. Error bars represent mean ± SD. The red dotted line indicates the LOD. DiD: 1,1’-Dioctadecyl-3,3,3’,3’-tetramethylindodicarbocyanine; NanoLuc: Nanoluciferase; BEVs: bacterial extracellular vesicles; GF: germ-free; SD: standard deviation; LOD: limit of detection.
The single versus dual-labelled BEVs showed consistent DiD distributions, with no significant differences observed [Supplementary Figure 2A and B], while dual-labelling resulted in significantly increased NanoLuc
Building on this, further analysis revealed higher background fluorescence in tissues for DiD-labelled BEVs when compared to NanoLuc-BEVs [Supplementary Figure 2B and D], highlighting the increased sensitivity and reduced non-specific signal of NanoLuc-based tracing. Organ localisation differences between the two labelling methods were evident, with DiD signals favouring hepatic accumulation. To further visualise BEV localisation within excised organs, high-resolution confocal imaging of DiD-BEV tissues showed a diffuse liver fluorescence and a punctate peri-nuclear signal in the brain [Supplementary Figure 2E]. Due to reagent limitations, similar imaging of NanoLuc-BEVs could not be performed.
GIT bacteria-derived BEVs enter host circulation for delivery of bioactive proteins
Having established the capacity of IV administered BEVs to reach multiple organ tissues, we next evaluated whether BEVs naturally produced by colonising GIT bacteria could similarly enter the host circulation. GF mice were colonised with a B. thetaiotaomicron strain engineered to produce NanoLuc-labelled BEVs (pBATH.51). Organs including eyes, brain, heart, lungs, liver, kidney, and spleen were collected between 7 and 27 days post-colonisation and assessed for NanoLuc activity. Notably, luminescence was detected in all tissues, with a gradual decrease over time. The highest average signal occurred at 7 days post-inoculation and dropped below the LOD by day 27 [Figure 3A]. Luminescence in faecal pellet extracts also declined over 21 days [Figure 3B], which likely reflects the loss of the NanoLuc expression plasmid in the absence of antibiotic selection pressure and the metabolic burden of ongoing NanoLuc production. Minimal luminescence was observed in control animals receiving PBS or colonised with the cytoplasmic NanoLuc-expressing B. thetaiotaomicron strain (pBATH.56) [Supplementary Figure 3].
Having established the capacity of IV administered BEVs to reach multiple organ tissues, we next evaluated whether BEVs naturally produced by colonising GIT bacteria could similarly enter the host circulation. GF mice were colonised with a B. thetaiotaomicron strain engineered to produce NanoLuc-labelled BEVs (pBATH.51). Organs including eyes, brain, heart, lungs, liver, kidney, and spleen were collected between 7 and 27 days post-colonisation and assessed for NanoLuc activity. Notably, luminescence was detected in all tissues, with a gradual decrease over time. The highest average signal occurred at 7 days post-inoculation and dropped below the LOD by day 27 [Figure 3A]. Luminescence in faecal pellet extracts also declined over 21 days [Figure 3B], which likely reflects the loss of the NanoLuc expression plasmid in the absence of antibiotic selection pressure and the metabolic burden of ongoing NanoLuc production. Minimal luminescence was observed in control animals receiving PBS or colonised with the cytoplasmic NanoLuc-expressing B. thetaiotaomicron strain (pBATH.56) [Supplementary Figure 3].
Figure 3. Time course of in vivo luminescence following colonisation with NanoLuc-BEV-producing B. thetaiotaomicron. GF mice were monocolonised at day 0. Samples were collected at 7, 10, 14, and 27 days post-colonisation. (A) Quantification of luminescence from organ tissues of animals administered PBS (n = 6) or the engineered B. thetaiotaomicron strain on day 7 (n = 6), 10 (n = 3), 14 (n = 2) and 27 (n = 4) post-colonisation. Each point represents the average luminescence for the eyes, brain, heart, lungs, liver, kidney, or spleen. Individual organs were excised for imaging using a Bruker In vivo Xtreme system. Substrate was injected intraperitoneally 5 min prior to organ excision; (B) Luminescence in the soluble fraction of faecal pellets from monocolonised GF mice. All samples were adjusted to 100 mg/mL prior to analysis (n = 12 for days 3, 6, and 14; n = 6 for day 21). Error bars represent mean ± SD. Red dotted line indicates LOD. NanoLuc-BEV: Nanoluciferase-bacterial extracellular vesicles; B. thetaiotaomicron: Bacteroides thetaiotaomicron; GF: germ-free; PBS: phosphate-buffered saline; SD: standard deviation; LOD: limit of detection.
Previous research suggests BEV biodistribution in GF mice can be influenced by increased barrier permeability in intestinal epithelial cells and the blood-brain-barrier (BBB) due to reduced tight junction protein expression[26,27]. To control for these variables, SPF mice were evaluated alongside GF mice 7 days after colonisation with NanoLuc-BEV-producing B. thetaiotaomicron. Both SPF and GF groups displayed organ-level NanoLuc signals with no overall statistical differences, however, the highest luminescence signals were observed in the eyes and brains of some GF animals [Figure 4]. Compared to IV inoculation, monocolonised mice showed a significantly higher luminescence signal in the eyes (*P ≤ 0.05; Figure 5).
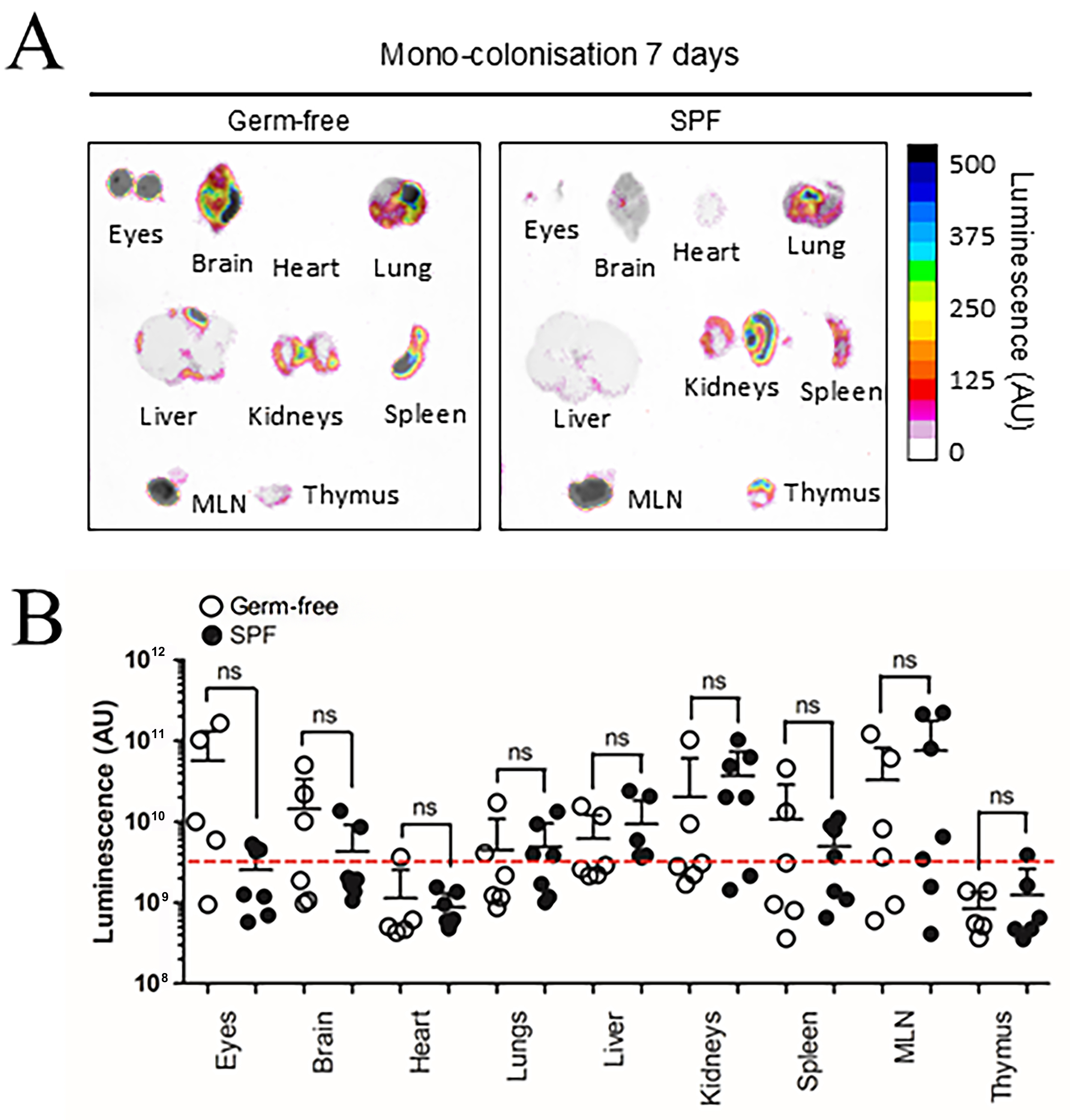
Figure 4. BEV distribution in vivo following colonisation with NanoLuc-BEV-producing B. thetaiotaomicron. (A) GF or SPF mice were monocolonised for 7 days, after which individual organs were excised and imaged using the Bruker In vivo Xtreme system. Substrate was injected intraperitoneally 5 min prior to euthanasia and organ excision. Representative images from GF (n = 6) or SPF (n = 7) mice are shown; (B) Quantification of luminescence from each organ. Error bars represent mean ± SD. Red dotted line indicates LOD. ns: Not significant; BEV: bacterially derived extracellular vesicle; NanoLuc: Nanoluciferase; B. thetaiotaomicron: Bacteroides thetaiotaomicron; GF: germ-free; SPF: specific pathogen-free; MLN: mesenteric lymph node; SD: standard deviation; ns: not significant; LOD: limit of detection.
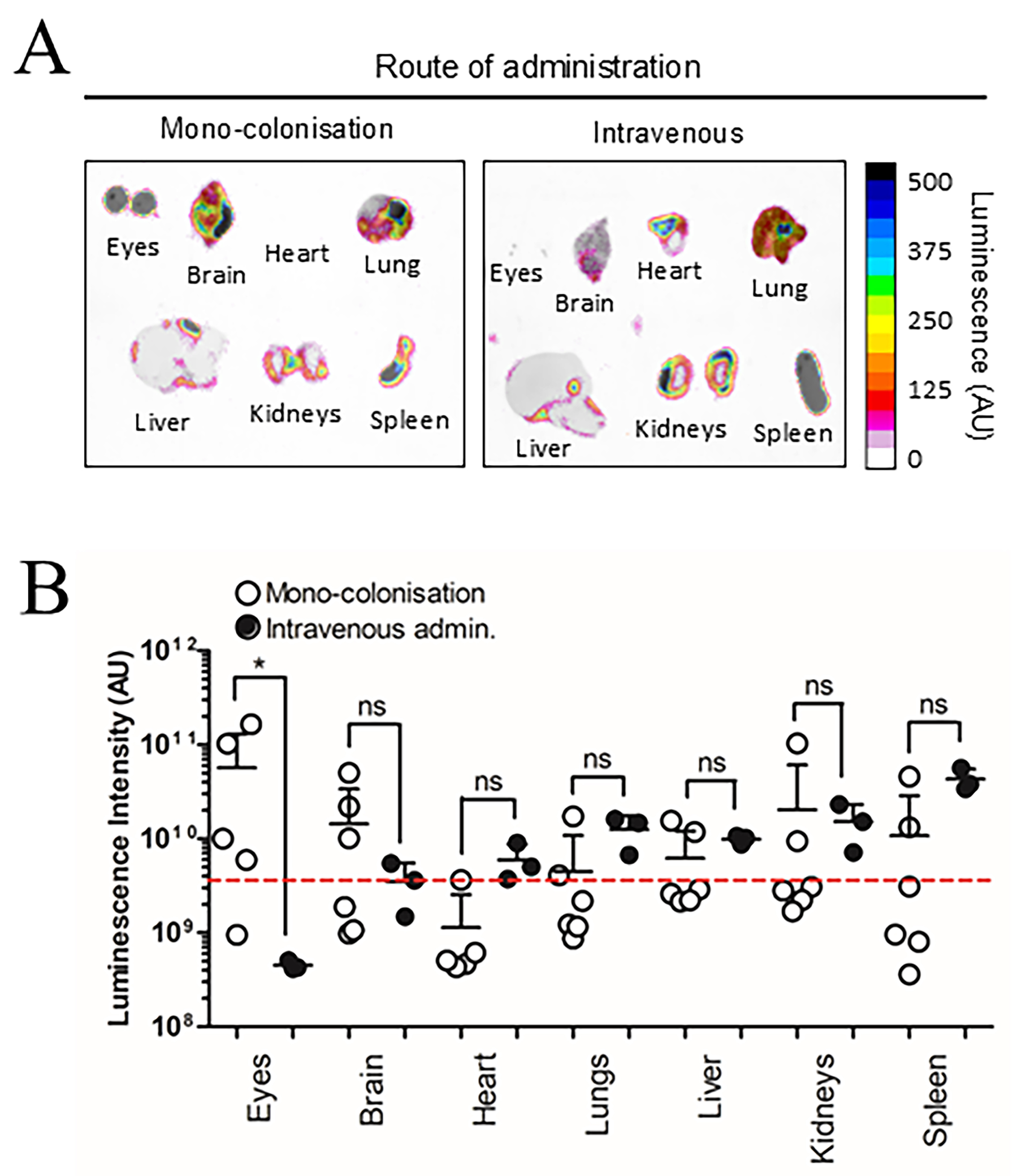
Figure 5. Route of administration alters NanoLuc-BEV biodistribution. GF mice were either monocolonised with NanoLuc-BEV-producing B. thetaiotaomicron or intravenously administered NanoLuc-BEVs (4 × 1010/mouse). (A) Organs were excised 7 days post-colonisation or 1 h post-IV administration and imaged using the Bruker In vivo Xtreme system. Substrate was injected intraperitoneally 5 min before euthanasia. Representative images from monocolonised (n = 6) or intravenously administered (n = 3) mice are shown; (B) Quantification of organ luminescence. Error bars represent mean ± SD. Red dotted line indicates LOD. *P ≤ 0.05. ns: Not significant; NanoLuc-BEV: Nanoluciferase-bacterial extracellular vesicle; GF: germ-free; B. thetaiotaomicron: Bacteroides thetaiotaomicron; IV: intravenous; SD: standard deviation; LOD: limit of detection.
To further confirm entry of NanoLuc-BEVs into the host circulation, soluble fractions from tissue homogenates and serum were assayed for luminescence. Following freeze-thawing, all tissue extracts displayed detectable NanoLuc activity, with GF mice showing the highest signals, in agreement with ex vivo whole-organ imaging results [Figure 6]. High variability was observed among samples, which we attribute to the freeze-thaw procedures required for sample preparation and transport prior to analysis.
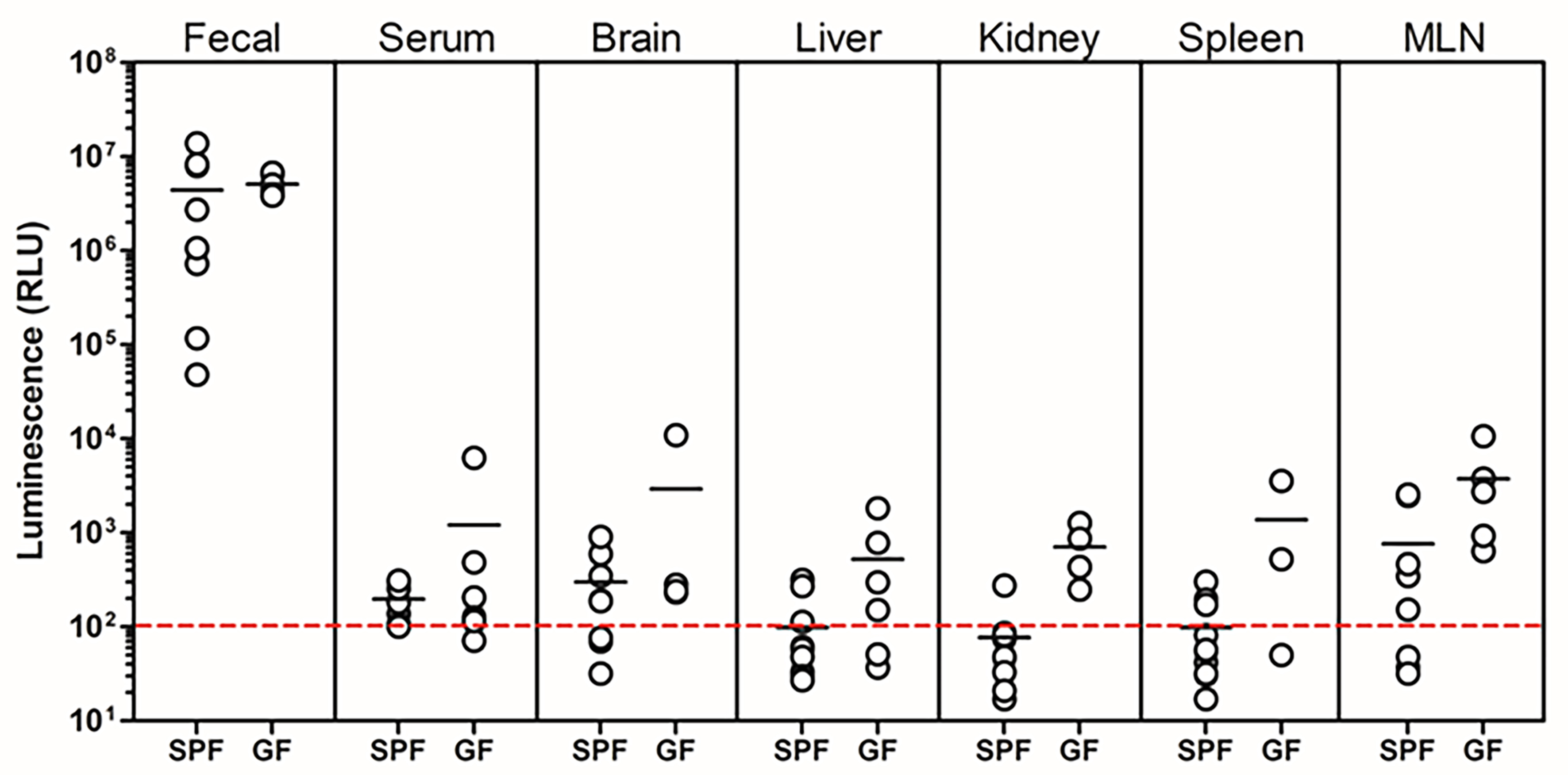
Figure 6. Organ distribution of NanoLuc activity following colonisation with NanoLuc-BEV-producing B. thetaiotaomicron. GF or SPF mice were monocolonised for 7 days. Individual organs were excised, snap-frozen, lysed, and luminescence quantified in vitro. Data represent 3-5 GF or 7-10 SPF mice per group. Error bars represent mean ± SD. NanoLuc: Nanoluciferase; BEV: bacterial extracellular vesicles;
DISCUSSION
This study demonstrates that bioengineered NanoLuc-BEVs produced by the human commensal bacterium B. thetaiotaomicron can enter the systemic, hepatic, and lymphatic circulatory systems in vivo. They are capable of traversing multiple physical host barriers, including the intestinal epithelium, vascular endothelium, and BBB, and localise to various organ tissues, including those of the CNS. Critically, the NanoLuc protein remained catalytically active within organ tissues, highlighting the potential of BEVs as bioactive, long-distance therapeutic delivery vehicles within the host[28]. Prior studies of BEV trafficking primarily relied on administering pre-labelled BEVs via oral or IV routes, approaches that bypass the physiological dissemination mechanisms of BEVs that are naturally produced by the GIT microbiota.
The use of NanoLuc as a sensitive detection system has been well described for tracing mammalian extracellular vesicles (EVs) in vivo[29]. Here we utilised the technology to enable tracking of BEVs in situ, allowing direct comparison with fluorescent dyes. Lipophilic dyes, such as DiD, have long half-lives, which can lead to dye accumulation, non-specific signals, and altered nanoparticle biodistribution[19,30]. The advantages of NanoLuc include higher specificity, minimal background signal, reduced autofluorescence, and negligible photobleaching. Our findings show that DiD-labelled BEVs disproportionately accumulate in the liver, consistent with prior studies[9,31]. This is likely due to uptake by highly pinocytic liver sinusoidal endothelial cells (LSECs), where slow blood flow facilitates uptake[32,33], or from trafficking of free DiD dye released after BEV degradation in host-cell lysosomes[34,35]. In contrast, NanoLuc labelling does not require membrane integration and is less likely to affect the natural tissue targeting of BEVs.
Our dual-labelling experiments revealed that BEV organ tropism is influenced by the choice of tracking method, highlighting that label selection can introduce experimental artefacts. While DiD-labelling casts light on common clearance pathways such as liver uptake, NanoLuc-labelling offers higher sensitivity and a greater signal-to-noise ratio. Nevertheless, NanoLuc-based tracking is not without limitations, including the requirement for luminescent FFz substrate diffusion in tissues, as well as rapid in vivo clearance, and increased NanoLuc signal in lungs and spleen[22,36]. Such challenges may reflect altered BEV surface protein composition due to bacterial bioengineering or degradation of NanoLuc during uptake and endo-lysosomal trafficking within host cells[22]. Overall, our findings emphasise that labelling strategies strongly influence
We observed that monocolonisation of mice with NanoLuc-BEV-producing B. thetaiotaomicron resulted in signals in the eyes, MLN, kidneys, brain, spleen, liver, lungs, heart, and thymus in both SPF and GF animals, the latter known to have more permeable intestinal and epithelial and blood-brain barriers[27]. To our knowledge, this is the first study to evaluate the biodistribution of endogenously produced, GIT microbiota-derived BEVs and to report their entry into the CNS in vivo.
Previous work, using pathogenic or pathobiont-derived labelled BEVs, has demonstrated that orally or intravenously administered vesicles access the brain via the vagus or trigeminal nerves[37,38], or via circulation and crossing of the BBB[39-42], potentially mediated by CNS monocytes/macrophages within the meninges[43]. Huang et al. also examined the biodistribution of bioengineered E. coli NanoLuc-BEVs, but these were delivered subcutaneously, and CNS trafficking was not evaluated[44]. Our study provides direct evidence that BEVs generated in vivo by the microbiota can also mediate CNS access. Supporting this, our brain imaging data and earlier in vitro gut-brain axis model findings show that B. thetaiotaomicron BEVs can traverse intestinal epithelial and BBB endothelial cell monolayers and be internalised by neuronal cells[25]. However, the precise pathways by which commensal BEVs transit from the gut to the brain in vivo remain to be clarified and will be vital for advancing their potential therapeutic application.
The biological consequences of commensal BEV uptake by neuronal cells require further investigation. Evidence from studies of pathogen-derived BEVs suggests that such interactions can contribute to cognitive impairment or behavioural changes in rodents[45], potentially via neuronal damage[39] or amyloid plaque accumulation and neurotoxicity[40,46]. By contrast, our in vitro gut-brain axis model indicates that commensal-derived B. thetaiotaomicron BEVs may instead drive protective anti-inflammatory and regulatory responses[25].
Similarly, our observation of NanoLuc-BEV trafficking to the MLN suggests an immunomodulatory role, in line with their established function in filtering microbial products from intestinal lymph[47], where transmigrating BEVs can interact with immune cells[48,49]. Indeed, 16S ribosomal RNA (rRNA)-based bacterial DNA signatures have been detected in MLNs of rats, likely in part derived from BEVs[50]. These findings provide further evidence that GIT microbiota-derived BEVs naturally disseminate through lymphatic pathways in hosts with intact intestinal barriers.
Although our study establishes proof-of-concept for NanoLuc as a reporter of BEV trafficking in vivo, several limitations should be acknowledged. Current in vivo imaging approaches do not resolve the cellular and subcellular distribution of BEVs in recipient tissues. Although confocal imaging confirmed punctate, perinuclear localisation consistent with previous in vitro models[9,25], and diffuse hepatic distribution, higher-resolution imaging modalities and cell-type markers will be required to visualise BEVs at the single-vesicle level and to assess their intracellular fate. For clinical translation, understanding BEV internalisation, trafficking, degradation, and potential escape from endo-lysosomal compartments will be crucial to evaluate their utility for therapeutic cargo delivery.
Furthermore, the small study sample size, while sufficient for feasibility and consistent with the 3Rs (Replacement, Reduction and Refinement) principles, limits statistical power so future studies with larger cohorts will be needed. Additionally, the work here focused exclusively on BEVs from a single commensal species; it remains to be defined how BEVs from diverse microbial communities may alter host targeting, cargo delivery and functional impact.
Despite these limitations, this proof-of-concept study establishes the suitability of NanoLuc as a sensitive and physiologically relevant tool for tracking microbiota-derived BEVs in vivo, offering significant advantages over dye-based methods including higher sensitivity, lower background, and minimal disruption of natural targeting. Moving forward, further development of non-disruptive, high-sensitivity labelling techniques, together with expanded animal studies and nanoscale imaging, will be essential for fully realising the potential of BEVs as precision delivery vehicles for host-targeted therapies. Given the likelihood that BEVs may exploit neuronal or lymphatic routes, focused studies of these pathways will be essential for understanding their therapeutic potential.
In conclusion, this work provides the first direct evidence that BEVs endogenously produced by the gut commensal B. thetaiotaomicron have the capacity to access multiple organ systems and the CNS, including the brain and eye, when generated in situ within the GIT. These findings illuminate new routes of microbe-host communication and underscore the potential of BEVs as platforms for next-generation bioactive therapeutic delivery[28].
DECLARATIONS
Acknowledgements
The authors thank the staff of the Disease Modelling Unit at the University of East Anglia for their support with animal husbandry and for ensuring animal welfare.
Authors’ contributions
Supervised the research: Blackshaw LA, Carding SR
Conducted the experimental work: Jones EJ, Parker A, Juodeikis R, Brion A
Performed image and statistical analyses: Jones EJ
All authors contributed to data interpretation.
All authors contributed to the conception and design of the experiments.
All authors revised, read, and approved the final manuscript.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the Biological Sciences Research Council (BBSRC) Institute Strategic Program Grant Gut Microbes and Health (BB/R012490/1) and its constituent projects (BBS/E/F/000PR10353, BBS/E/F/000PR10355, and BBS/E/F/000PR10356; Jones EJ, Parker A, Juodeikis R, Blackshaw LA and Carding SR), as well as by the BBSRC Core Capability Grant (BB/CCG1860/1; Blackshaw LA).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval
All animal experiments were conducted in strict accordance with the Animal Scientific Procedures Act (1986) at a licensed facility, under an active project licence, and with approval from the local Animal Welfare and Ethical Review Board (AWERB) at the University of East Anglia. All procedures were performed by experienced practitioners who hold a UK Home Office personal licence.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
Supplementary Materials
REFERENCES
2. Cryan JF, O’Riordan KJ, Cowan CSM, et al. The microbiota-gut-brain axis. Physiol Rev. 2019;99:1877-2013.
3. Song C, Chai Z, Chen S, Zhang H, Zhang X, Zhou Y. Intestinal mucus components and secretion mechanisms: what we do and do not know. Exp Mol Med. 2023;55:681-91.
4. Ghosh S, Whitley CS, Haribabu B, Jala VR. Regulation of intestinal barrier function by microbial metabolites. Cell Mol Gastroenterol Hepatol. 2021;11:1463-82.
5. Toyofuku M, Schild S, Kaparakis-Liaskos M, Eberl L. Composition and functions of bacterial membrane vesicles. Nat Rev Microbiol. 2023;21:415-30.
6. Melo-Marques I, Cardoso SM, Empadinhas N. Bacterial extracellular vesicles at the interface of gut microbiota and immunity. Gut Microbes. 2024;16:2396494.
7. Zou C, Zhang Y, Liu H, Wu Y, Zhou X. Extracellular vesicles: recent insights into the interaction between host and pathogenic bacteria. Front Immunol. 2022;13:840550.
8. Schaack B, Hindré T, Quansah N, Hannani D, Mercier C, Laurin D. Microbiota-derived extracellular vesicles detected in human blood from healthy donors. Int J Mol Sci. 2022;23:13787.
9. Jones EJ, Booth C, Fonseca S, et al. The uptake, trafficking, and biodistribution of Bacteroides thetaiotaomicron generated outer membrane vesicles. Front Microbiol. 2020;11:57.
10. Carvalho AL, Fonseca S, Miquel-Clopés A, et al. Bioengineering commensal bacteria-derived outer membrane vesicles for delivery of biologics to the gastrointestinal and respiratory tract. J Extracell Vesicles. 2019;8:1632100.
11. Dehghani M, Gulvin SM, Flax J, Gaborski TR. Systematic Evaluation of PKH labelling on extracellular vesicle size by nanoparticle tracking analysis. Sci Rep. 2020;10:9533.
12. Yi YW, Lee JH, Kim SY, et al. Advances in analysis of biodistribution of exosomes by molecular imaging. Int J Mol Sci. 2020;21:665.
13. Takov K, Yellon DM, Davidson SM. Confounding factors in vesicle uptake studies using fluorescent lipophilic membrane dyes. J Extracell Vesicles. 2017;6:1388731.
14. Simonsen JB. Pitfalls associated with lipophilic fluorophore staining of extracellular vesicles for uptake studies. J Extracell Vesicles. 2019;8:1582237.
15. Fonseca S, Carvalho AL, Miquel-Clopés A, et al. Extracellular vesicles produced by the human gut commensal bacterium Bacteroides thetaiotaomicron elicit anti-inflammatory responses from innate immune cells. Front Microbiol. 2022;13:1050271.
16. Whitaker WR, Shepherd ES, Sonnenburg JL. Tunable expression tools enable single-cell strain distinction in the gut microbiome. Cell. 2017;169:538-46.e12.
17. Hall MP, Unch J, Binkowski BF, et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol. 2012;7:1848-57.
18. England CG, Ehlerding EB, Cai W. NanoLuc: a small luciferase is brightening up the field of bioluminescence. Bioconjug Chem. 2016;27:1175-87.
19. Gupta D, Liang X, Pavlova S, et al. Quantification of extracellular vesicles in vitro and in vivo using sensitive bioluminescence imaging. J Extracell Vesicles. 2020;9:1800222.
20. Troy T, Jekic-McMullen D, Sambucetti L, Rice B. Quantitative comparison of the sensitivity of detection of fluorescent and bioluminescent reporters in animal models. Mol Imaging. 2004;3:9-23.
21. Schaub FX, Reza MS, Flaveny CA, et al. Fluorophore-NanoLuc BRET reporters enable sensitive in vivo optical imaging and flow cytometry for monitoring tumorigenesis. Cancer Res. 2015;75:5023-33.
22. Lázaro-Ibáñez E, Faruqu FN, Saleh AF, et al. Selection of fluorescent, bioluminescent, and radioactive tracers to accurately reflect extracellular vesicle biodistribution in vivo. ACS Nano. 2021;15:3212-27.
23. Valguarnera E, Scott NE, Azimzadeh P, Feldman MF. Surface exposure and packing of lipoproteins into outer membrane vesicles are coupled processes in Bacteroides. mSphere. 2018;3:e00559-18.
24. Juodeikis R, Martins C, Saalbach G, et al. Differential temporal release and lipoprotein loading in B. thetaiotaomicron bacterial extracellular vesicles. J Extracell Vesicles. 2024;13:e12406.
25. Modasia AA, Jones EJ, Martel LM, et al. The use of a multicellular in vitro model to investigate uptake and migration of bacterial extracellular vesicles derived from the human gut commensal Bacteroides thetaiotaomicron. J Extracell Biol. 2023;2:e93.
26. González-González M, Díaz-Zepeda C, Eyzaguirre-Velásquez J, González-Arancibia C, Bravo JA, Julio-Pieper M. Investigating gut permeability in animal models of disease. Front Physiol. 2018;9:1962.
27. Braniste V, Al-Asmakh M, Kowal C, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med. 2014;6:263ra158.
28. Ji N, Wang F, Wang M, Zhang W, Liu H, Su J. Engineered bacterial extracellular vesicles for central nervous system diseases. J Control Release. 2023;364:46-60.
29. Shi Y, Wang X, Zhang S, et al. Research progress in in vivo tracing technology for extracellular vesicles. Extracell Vesicles Circ Nucl Acids. 2023;4:684-97.
30. Álamo P, Pallarès V, Céspedes MV, et al. Fluorescent dye labeling changes the biodistribution of tumor-targeted nanoparticles. Pharmaceutics. 2020;12:1004.
31. Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332-48.
32. Hosseini-Kharat M, Bremmell KE, Prestidge CA. Why do lipid nanoparticles target the liver? Understanding of biodistribution and liver-specific tropism. Mol Ther Methods Clin Dev. 2025;33:101436.
33. He Y, Wang Y, Wang L, Jiang W, Wilhelm S. Understanding nanoparticle-liver interactions in nanomedicine. Expert Opin Drug Deliv. 2024;21:829-43.
34. Parthasarathy G, Hirsova P, Kostallari E, Sidhu GS, Ibrahim SH, Malhi H. Extracellular vesicles in hepatobiliary health and disease. Compr Physiol. 2023;13:4631-58.
35. Gandek TB, van der Koog L, Nagelkerke A. A comparison of cellular uptake mechanisms, delivery efficacy, and intracellular fate between liposomes and extracellular vesicles. Adv Healthc Mater. 2023;12:e2300319.
36. Boudna M, Campos AD, Vychytilova-Faltejskova P, Machackova T, Slaby O, Souckova K. Strategies for labelling of exogenous and endogenous extracellular vesicles and their application for in vitro and in vivo functional studies. Cell Commun Signal. 2024;22:171.
37. Lee KE, Kim JK, Han SK, et al. The extracellular vesicle of gut microbial Paenalcaligenes hominis is a risk factor for vagus nerve-mediated cognitive impairment. Microbiome. 2020;8:107.
38. Ma X, Shin YJ, Yoo JW, Park HS, Kim DH. Extracellular vesicles derived from Porphyromonas gingivalis induce trigeminal nerve-mediated cognitive impairment. J Adv Res. 2023;54:293-303.
39. Palacios E, Lobos-González L, Guerrero S, et al.
40. Park AM, Tsunoda I.
41. Xie J, Cools L, Van Imschoot G, et al.
42. Bittel M, Reichert P, Sarfati I, et al. Visualizing transfer of microbial biomolecules by outer membrane vesicles in microbe-host-communication in vivo. J Extracell Vesicles. 2021;10:e12159.
43. Ha JY, Choi SY, Lee JH, Hong SH, Lee HJ. Delivery of periodontopathogenic extracellular vesicles to brain monocytes and microglial IL-6 promotion by RNA cargo. Front Mol Biosci. 2020;7:596366.
44. Huang Y, Beringhs AO, Chen Q, et al. Genetically engineered bacterial outer membrane vesicles with expressed nanoluciferase reporter for in vivo bioluminescence kinetic modeling through noninvasive imaging. ACS Appl Bio Mater. 2019;2:5608-15.
45. Wei S, Peng W, Mai Y, et al. Outer membrane vesicles enhance tau phosphorylation and contribute to cognitive impairment. J Cell Physiol. 2020;235:4843-55.
46. Meng D, Lai Y, Zhang L, et al. Helicobacter pylori outer membrane vesicles directly promote Aβ aggregation and enhance Aβ toxicity in APP/PS1 mice. Commun Biol. 2024;7:1474.
47. Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313-23.
48. Durant L, Stentz R, Noble A, et al.
49. Weinberger V, Darnhofer B, Thapa HB, et al. Proteomic and metabolomic profiling of extracellular vesicles produced by human gut archaea. Nat Commun. 2025;16:5094.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].