

Carbon-based materials for electrocatalytic energy conversion: from understanding to designing
 0
0 
Abstract
Renewable energy technologies are crucial for alleviating the energy crisis and pollution; electrocatalytic reactions such as oxygen reduction, hydrogen evolution, and oxygen evolution reactions are prospective energy conversion pathways. Although metal-based electrocatalysts are currently employed in electrochemical reactions, they encounter a series of issues with supply and price. Therefore, the development of new environmentally friendly, efficient, and low-cost electrochemical catalysts is imminent. Carbon-based materials such as amorphous carbons and nanostructured carbons have drawn extensive attention in electrocatalysis research due to their cost-effectiveness, environmental friendliness, and stability in acid and alkali media. In the initial stage, the heteroatoms embedded in the carbon skeleton (such as N, P, S, and B) were identified as active sites of carbon-based electrocatalysts. Subsequently, further investigations revealed that structural defects in carbon rings can disrupt the electronic conjugation system, which in turn affects the charge distribution and thereby enhances catalytic activity. Recently, our group has proposed a novel mechanism of defective carbon-based materials for electrochemical reactions, suggesting that the introduction of topological defects can boost electrocatalytic activity. Subsequently, extensive research has been carried out with direct evidence to prove different defects as active sites. Herein, we will emphasize the advancement of carbon-based electrocatalysts by a comprehensive understanding of catalyzing mechanisms. Then, the methodologies for controllably synthesizing doped carbons and carbons with defective structures will be summarized. Ultimately, we will outline the key challenges in designing intricate carbon active sites, particularly defect structures, provide insights into characterization techniques for investigating mechanisms, and importantly, look forward to future developments and opportunities.
Keywords
INTRODUCTION
The escalating global energy demand leads to extensive consumption of fossil fuels, heightening concerns regarding the finite nature of these resources and associated environmental challenges[1,2]. Modern energy transitions have progressively positioned clean energy technologies - particularly solar arrays, wind farms, and hydropower plants - as environmentally sound replacements for coal, oil, and natural gas systems[3-5]. Nevertheless, the inconsistent spatial and temporal availability of these renewable sources constrains their full utilization, driving an urgent need to engineer next-generation technologies for energy storage and conversion[6]. Hydrogen, with its superior mass energy density and environmental cleanliness, is a promising energy carrier and has been widely recognized as a leading candidate to substitute exhausting fossil fuels[7,8]. Concurrently, processes such as carbon capture and carbon dioxide reduction hold potential for mitigating the intensifying greenhouse effect and reducing energy consumption and carbon emissions, exemplified by the electrocatalytic production of ammonia[9]. In recent years, electrocatalytic processes, including oxygen reduction reaction (ORR)[10], hydrogen evolution reaction (HER)[11] and oxygen evolution reaction (OER)[12], have emerged as effective approaches to address both energy challenges and environmental concerns. The efficacy of these electrochemical processes is largely contingent upon electrode materials that exhibit high catalytic activity while being economically viable. Substantial advancements have been made in the development of highly active noble metal-based electrocatalysts for a variety of electrochemical reactions. Nevertheless, the scarcity and elevated expense of these materials impede their widespread industrial application[13,14]. To mitigate the cost of catalysts, researchers have innovated high-performance transition metal catalysts[15,16]. Nonetheless, the catalytic sites within these transition metal catalysts are often prone to degradation, which not only results in a marked decline in performance but may also lead to contamination of the reaction system[17]. Consequently, there is a pressing demand to create next-generation electrocatalysts that are not only highly efficient and affordable but also capable of maintaining stable and exceptional catalytic performance.
Carbon-based materials, encompassing amorphous carbon and nanostructured carbon, have garnered significant attention in the realm of electrocatalyst development owing to their environmental compatibility, economic efficiency, and robust stability in both acidic and alkaline environments[18-20]. Specifically, low-dimensional and multidimensional carbon nanostructures, including carbon nanotubes (CNTs), nanowires, and graphene, have emerged as highly promising candidates owing to their well-ordered graphitic architectures, extensive surface area, superior conductivity, and atomic-level structural tunability[21-23]. Initially, carbon materials primarily served as support matrices. Over time, electrocatalysts derived from both noble and non-noble metals have been developed, albeit with challenges related to diminished performance due to cost considerations and catalyst toxicity. The metal-nitrogen-carbon (M-N-C) framework, characterized by its relative affordability and excellent catalytic efficiency, has emerged as a leading contender to replace the currently prevalent noble metal-based catalysts[24,25]. As carbon-based materials continue to evolve, the concept of metal-free carbon materials, including nitrogen-doped CNTs with notable ORR activity, has been introduced. Initially, heteroatom dopants such as nitrogen, phosphorus, sulfur, and boron within the carbon matrix were identified as potential active sites for carbon-based electrocatalysts[26,27]. Further research revealed that heteroatom doping induces charge redistribution due to electronegativity mismatches and positional variations, thereby enhancing catalytic activity. However, distinguishing the contributions of heteroatoms, vacancies, or Stone-Wales defects remains challenging due to the defects and/or vacancies introduced by heteroatom doping[28,29].
Additionally, structural imperfections within the carbon ring can disrupt the conjugated electron framework, influencing charge distribution. Our research group, along with others, has proposed a new mechanism for defect carbon-based materials (DCMs), suggesting that the introduction of various topological defects can enhance electrocatalytic activity[30-32]. Comprehensive investigations have provided direct evidence of the impact of active sites formed by different defect types. Despite ongoing advancements in carbon-based material research, the catalytic mechanisms underlying the active centers of high-performance carbon-based catalysts require further elucidation. In diverse electrochemical reactions, the catalytic efficacy of M-N-C, heteroatom-doped, and DCMs can be explained by distinct mechanisms. Although the integration of density functional theory (DFT) calculations and experimental studies offers foundational insights into these mechanisms, a substantial gap persists between theoretical models and empirical data under practical conditions. Consequently, there remains a significant journey from predicting catalyst active site structures for various electrochemical reactions to the development and fabrication of materials.
This review summarizes the development history of carbon-based materials in the field of electrocatalytic ORR and the latest progress of other electrocatalytic reactions. It mainly discusses the development of carbon-based electrocatalysts and a deep understanding of the catalytic mechanisms and relevant theories. The details of the strategies and approaches to regulating the synthesis of M-N-C, doped carbon, and carbon with defect structures will be summarized, and the source of catalyst activity will be elaborated. In addition, we also introduce the utilization of carbon materials in HER and OER reactions. Finally, we will outline the primary challenges associated with designing intricate carbon active sites, particularly defect structures, providing guidance on characterization techniques for investigating mechanisms, and importantly, anticipate future development and opportunities.
ELECTROCATALYTIC REACTIONS OF CARBON MATERIALS AND BASIC PRINCIPLES
ORR: reaction pathway and mechanisms
Increased CO2 emissions and excessive consumption of fossil fuels ultimately lead to global warming. Therefore, scientists hope to address these issues by offering eco-friendly solutions for energy production. To achieve this goal, fuel cells, metal-air batteries constitute promising alternatives for energy production/storage. Electrocatalytic ORR has been considered as a basic reaction for sustainable fuel cells and metal-air batteries. ORR occurs in aqueous solution through two pathways: the direct 4e- reduction from O2 to H2O or the 2e- reduction pathway from O2 to H2O2[33]. The 2e- and 4e- reaction mechanisms are primarily controlled by reaction thermodynamics, involving the adsorption energies of intermediates and the energy barriers at the interface between the electrode, catalyst, and electrolyte[10]. The 4e- reaction mechanism is crucial for achieving high current efficiency in fuel cell technology, while the industrial synthesis of H2O2 depends on the 2e- pathway.
ORR process has different 4e- and 2e- transfer pathways in acidic and alkaline solutions. It is generally believed that in acidic solutions, ORR occurs as follows:
4e- pathway: O2 + 4H+ + 4e- → 2H2O
2e- pathway: O2* + 2H+ + 2e- → 2HOOH*
HOOH* + 2H+ + 2e- → 2H2O*
In alkaline solutions, the following reactions occur:
4e- pathway: O2* + 2H2O* + 4e- → 4OH*
2e- pathway: O2* + 2H2O* + 2e- → HO2* + OH*
HO2* + H2O* + 2e- → 3OH*
OER: reaction pathway and mechanisms
The OER is the reverse reaction of the ORR, involving electron loss to form O2, and is essential for energy conversion processes. The sluggish kinetics of the water oxidation reaction, entailing the transfer of four electrons for the generation of O2, necessitates a higher overpotential to achieve high current densities. The operating mechanism of OER is contingent upon the pH level of the electrolyte solution. The pathways and fundamental steps involved in OER can vary significantly between alkaline (high pH) and acidic (low pH) environments. Two predominant mechanisms have been proposed for OER: the adsorbate evolution mechanism (AEM) [Figure 1] and the lattice oxygen mechanism (LOM). In the AEM, reaction intermediates attach to the catalyst surface, where they undergo oxidation and subsequently release oxygen, and this mechanism is not influenced by pH. Conversely, the LOM involves the direct participation of lattice oxygen atoms from the catalyst material in the formation and desorption of oxygen[34]. In this mechanism, the lattice oxygens are replenished either from the electrolyte or through internal migration within the catalyst bulk. Carbon materials are less involved in the LOM during the OER.
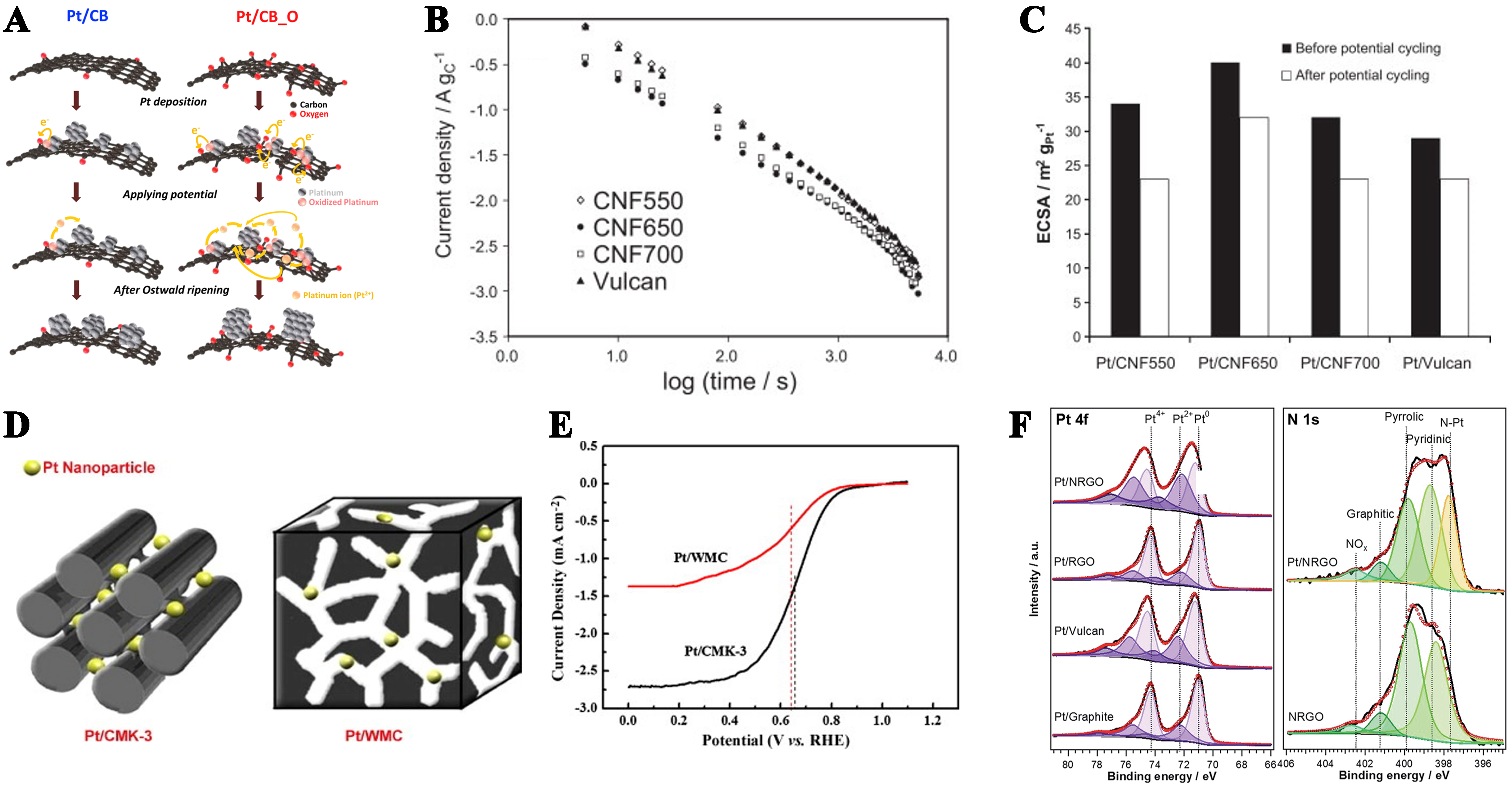
Figure 1. (A) Schematic diagram of the variations in Pt particle size distribution in Pt/CB and Pt/CB_O catalysts before and after accelerated durability tests. This figure is quoted with permission from Kim et al.[44]; (B) Current density-time curves illustrating the corrosion of carbon samples on a double logarithmic scale, conducted in a half-cell at 1.2 V vs. RHE; (C) Comparison of ECSA for the electrodes before and after Pt degradation tests. This figure is quoted with permission from Sebastián et al.[52]; (D) Schematic of carbon support pore morphology effect on Pt NP catalytic activity; (E) Polarization curves of Pt/WMC and Pt/CMK-3 catalysts. This figure is quoted with permission from Song et al.[55]; (F) Pt 4f and N 1s spectra deconvoluted into components for carbon supports and Pt/C catalysts. This figure is quoted with permission from Ma et al.[63]. RHE: Reversible hydrogen electrode; ECSA: electrochemical active surface area; NP: nanoparticle.
AEM pathways for the acidic electrolyte:
AEM pathways for the alkaline electrolyte:
HER: reaction pathway and mechanisms
Green hydrogen (H2), which can be generated through the electrolysis of water using renewable energy sources, represents a potential substitute for fossil fuels. The electrochemical HER is a process that enables the production of hydrogen via water cleavage within either an acidic or alkaline environment. This process encompasses several sequential steps, including adsorption, reduction, and desorption. Under acidic and alkaline conditions, the HER advances through two disparate pathways, specifically the Volmer-Heyrovsky mechanism and the Volmer-Tafel mechanism. Both of these pathways commence from the Volmer step, during which the electron transfer originating from the electrode is conjugated with the proton adsorption onto the catalyst, consequently leading to the formation of adsorbed hydrogen atoms:
In acidic and alkaline electrolytes, the hydronium ion (H3O+) and the water molecule serve as the respective sources of protons. Subsequently, within the framework of the Volmer-Tafel mechanism, the Tafel step facilitates the combination of two H* on adjacent sites, thereby leading to the formation of H2.
In the context of the Heyrovsky step of the Volmer-Heyrovsky mechanism, the formation of H2 occurs through the direct interaction of the H* atom with the proton in an acidic milieu and with the water molecule in an alkaline environment.
UNDERSTANDING OF CARBON-BASED ORR ELECTROCATALYSTS
It is widely acknowledged that supported metal catalysts demonstrate enhanced stability and increased activity compared to unsupported bulk metal catalysts. Carbon blacks (CBs), typically synthesized through the pyrolysis of hydrocarbons, are extensively employed as supports for platinum (Pt) and Pt-alloy catalysts in fuel cells[35,36]. Prior to their utilization as catalyst supports, CBs are often activated to improve metal dispersion and catalytic activity, primarily through physical and chemical activation techniques[37]. Common physical activation methods include thermal treatment in inert gas or air. Studies have shown that Pt/C catalysts prepared on Vulcan carbon substrates, which have been treated to modify or remove reactive species on the surface in an inert atmosphere, outperform untreated samples in ORR[38].
Our research group has demonstrated that catalysts synthesized from defective activated carbon, through nitrogen addition and removal, exhibit superior activity and stability[39,40]. Concurrently, chemical activation methods, such as the use of KOH[41], perfluoro sulfonic acid (Nafion)[42], and NH4F[43], can alter the surface characteristics and aggregation degree of CB, influencing the size and loading efficiency of metal nanoclusters. Despite the numerous advantages of CBs, it is crucial to consider the balance between catalytic activity and stability, even for Pt catalysts supported on modified CBs. Kim et al. highlighted that Pt catalysts supported on mildly oxygen-functionalized CB (Pt/CB_O) exhibit higher electrochemically active surface areas and ORR activity compared to Pt/CB catalysts. However, oxygen functionalization in Pt/CB_O catalysts partially oxidizes Pt nanoparticles (NPs), leading to rapid dissolution and Ostwald ripening, thereby accelerating the decline in ORR activity [Figure 1A][44]. The balance between catalytic activity and stability necessitates carbon substrates with larger surface areas, superior mechanical properties, and enhanced electron transfer capabilities. In response, various nanostructured carbon materials have been developed, including carbon nanofibers (CNFs), CNTs, ordered mesoporous carbon (OMC), and graphene. These materials differ from CB in terms of porosity and micromorphology. CNTs, formed by rolling single sheets of hexagonally arranged carbon atoms into cylindrical structures, are available as single-walled (SWCNTs) or multi-walled CNTs (MWCNTs). Their tubular structure, high surface area, superior electronic conductivity, and exceptional chemical stability make CNTs a promising alternative for catalyst support in fuel cells[45-48]. Wang et al. demonstrated that MWCNTs exhibit greater electrochemical stability compared to Vulcan XC-72[49].
In 2004, Novoselov et al. introduced graphene, a novel carbon material comprising an atomically thin sheet of hexagonally arranged carbon atoms, which can be considered as unrolled SWCNTs[50]. Graphene’s exceptional theoretical specific surface area, outstanding electrical conductivity, mechanical strength, and unique physical properties contribute to its enhanced catalytic activity in ORR compared to Pt/C[51]. However, carbon-based catalysts face critical challenges arising from carbon corrosion. This phenomenon not only causes the loss of active sites in ORR materials and undermines structural stability but also exacerbates detrimental effects, such as metal catalyst agglomeration/detachment, reduced electrical conductivity, hindered mass transport of reactants/products, and catalyst poisoning by corrosion-derived byproducts (e.g., CO). These interconnected issues collectively deteriorate ORR kinetics, accelerate performance degradation in electrochemical devices (e.g., decreased voltage output, power density, and service life), and pose safety risks (e.g., structural deformation and short-circuiting) due to compromised mechanical stability. As a result, they significantly impede the commercialization of energy systems such as proton exchange membrane (PEM) fuel cells. Therefore, Sebastián et al. prepared highly crystalline CNFs as electrocatalyst supports, which displayed better durability compared to CB-based catalysts due to their higher graphitic character and suitable Brunauer–Emmett–Teller (BET) surface area [Figure 1B and C][52].
The presence of micropores in CB is not conducive to its use as a catalyst support, whereas mesoporous carbons (MCs) offer higher specific surface areas and a reduced presence or absence of micropores[53,54]. The mesoporous structure facilitates efficient mass transport, leading to high limiting currents. The pore morphology of MC also impacts electrocatalytic activity, with a higher order of mesoporosity being more favorable for material transport. Song et al. investigated the effects of Pt NPs on ordered/disordered MC [Figure 1D], and showed that CMK-3-supported Pt has a higher density of electrochemically active sites and larger electrochemical surface area, which leads to improved catalytic performance [Figure 1E][55].
In alignment with previous modifications of CB, researchers have adapted these carbon-based materials to minimize the size of Pt particles, enhancing their dispersion and increasing the number of catalytic sites available. Orfanidi et al. chemically modified the sidewalls of MWCNTs by covalently attaching pyridine-based groups, thereby reducing the size of the Pt particles[56]. Kou et al. designed highly conductive functionalized graphene sheets (FGS) by high temperature thermal expansion method. After loading Pt, the catalytic performance was enhanced due to the size reduction and dispersion of Pt particles[57]. Beyond chemical surface functionalization of CNTs, graphene, and other carbon-based materials, the introduction of heteroatoms, particularly N elements, into the carbon structure has been explored. Nitrogen atoms contribute additional electrons, and doping with high nitrogen concentrations elevates conductivity by increasing the Fermi level guide band[58]. The incorporation of N atoms into the carbon structure enhances surface characteristics, including increased polarity, improved basicity, and the creation of diverse hydrophilic regions[59]. Thus, N-doped carbon materials serve as excellent catalyst substrates[60-62]. Ma et al. showed that N-doped reduced graphene oxide (NrGO) materials can establish Pt-N covalent linkages within Pt NPs, which enable efficient charge transfer across the catalyst-support interface [Figure 1F][63]. DFT calculations indicated that while nitrogen species in the support result in weaker O2 adsorption, they induce elongated O-O distances, indicating enhanced dissociation[64].
Although nitrogen doping of carbon supports can improve initial mass activities due to enhanced platinum dispersion, it does not necessarily enhance catalyst stability. Given the scarcity of precious metal Pt, identifying abundant substitutes is a critical task. Transition metals serve as alternatives to conventional catalysts, leading to the development of numerous carbon-based materials supported by transition metal compound catalysts[65,66]. Carbon-supported transition metals/nitrogen (M-Nx/C) are gaining attention owing to their favorable catalytic activity and the availability of low-cost precursor materials. As early as 1964, cobalt phthalocyanine (CoPc) was first demonstrated as an ORR electrocatalyst under alkaline conditions[67]. The ORR activity of various transition metal ion centers and macrocyclic molecular systems has been investigated[68-71], with carbon-based materials enhancing the electrical conductivity of the catalytic system[72-74]. Although macrocyclic molecules are unstable in acidic environments, pyrolysis can enhance their stability, albeit with potential alterations to the original catalyst configuration. Consequently, studies have proposed examining catalytic configuration changes through pyrolysis, suggesting that the M-N-C structure constitutes the catalyst’s active site[75-77]. The synthesis and ORR activity of M-N-C structures are further examined in Section “M-Nx/C catalysts”.
Since Gong et al. demonstrated that N-doped CNT arrays exhibit exceptional electrocatalytic performance for the ORR in alkaline environments, thereby outperforming Pt[78], the development of carbon-based metal-free catalysts (CMFCs) as substitutes for precious metal electrocatalysts has emerged as a promising research field. This has led to the creation of various N-doped carbon materials, including stacked N-doped CNT cups[79], CNTs[80], carbon nanocages[81], microporous carbon materials[82], and nanoporous carbon nanosheets[83]. Introducing N atoms into carbon materials avoids lattice mismatch due to the similar atomic radii of N and C, and the additional electron of N promotes electron-requiring reactions such as ORR[84]. Heteroatom doping in carbon materials, including species and placement, crucially modulates electrocatalytic activity, as discussed in Section “Heteroatom-doped carbon catalysts”.
In the synthesis of N-doped carbon materials, defective structures are often inadvertently introduced, and these defects can be difficult to control. When nitrogen atoms coexist with defective carbon, they complicate the exploration of catalytic mechanisms. The electronegativity of nitrogen affects the electronic structure of adjacent carbon atoms in doped carbon ORR. Similarly, vacancy defects introduce unsaturated carbon atoms with dangling bonds, potentially impacting the surrounding electronic structure and producing ORR activity akin to that of N-doped carbon. The synthesis methods for topological defects in carbon, along with defect identification and activity exploration, are discussed in Section “Defective carbon catalysts”.
DESIGN OF CARBON-BASED ORR ELECTROCATALYSTS
M-Nx/C catalysts
Synthesis
Pyrolysis strategy has always been the main preparation strategy of M-N-C catalyst. Early on, inexpensive precursors such as polyacrylonitrile were mixed with Co (II) or Fe (II) salts and Vulcan XC-72 CBs, then subsequently subjected to heat treatment, yielding catalysts with activity comparable to transition metal macrocycles and polypyrrole black-based catalysts[85]. However, the direct synthesis of M-Nx/C catalysts using CBs as precursor often lacks precise control of morphology and structure. To regulate the morphology and structure of the catalyst, the precursors of the carbon and nitrogen sources were changed. Co particles were synthesized and coated with graphite shells by pyrolysis of sucrose and urea[86]; alternatively, using 2D porous carbon nanosheets (PCNs) mixed with nitrogen sources, the metal ions were reduced to metal particles during the thermal treatment process, while facilitating the escape of nitrogen-containing gases released by cyanamide decomposition, thus achieving nitrogen doping in N-doped PCNs (NPCNs)[87]; The metal particles can also be encapsulated by synthetic CNTs and then N-doped in an ammonia atmosphere[25]. Although the carbon material encapsulation metal can control the material morphology and improve the catalytic activity, it is also difficult to conduct detailed analysis of its active sites because of the limited exposed active sites. At present, metal-organic framework (MOF) materials are the most widely used as precursors of oxygen reduction catalysts. Pyrolysis of MOF precursors can increase the specific surface area and pore distribution of materials, which is conducive to the exposure of catalytic active sites and enhances the electrocatalytic performance. In addition, due to the clear location and distribution of metal sites in MOF materials, good dispersion of metal sites is still maintained after pyrolysis[88]. In the carbonization process, MOF pyrolysis converts organic ligands to porous graphitic carbon matrices and metal nodes to oxide NPs, metallic NPs, or single-atom dopants. Pyrolysis conditions (atmosphere/temperature/precursor) significantly affect catalyst structure and properties[89,90]. The precursors of which are crucial. Recently, Huang et al. reported comprehensive chemical and morphological evolution and transformation of two commercial MOFs, ZIF-8 and ZIF-67, which share the same sodalite topology, using a combination of in situ and ex-situ techniques[91]. The reason why ZIF-67 loses the favorable porous structure after pyrolysis is carbon graphitization, which is catalyzed by Co clusters formed during pyrolysis; most of the metal in ZIF-8 evaporates initially and does not aggregate to form metal particles. In addition to MOF precursors, metal sources are also crucial in the pyrolysis process. Gao et al. reported that the Fe(acac)3 precursor results in exclusive formation of atomically dispersed Fe-Nx coordination, while the FeCl3 and Fe(NO3)3 lead to the formation of more inactive Fe3C NPs[92]. MOFs with precise porous structures and adjustable compositions are also considered ideal platforms for stabilizing SACs, enabling detailed structure-activity relationship studies to guide the design of high-performance catalysts. Wang et al. obtained a CoNx coordination catalyst with excellent electrochemical properties by pyrolysis co-doping MOF[93]. Notably, single-atom catalysts easily aggregate into clusters and particles, the formation of particles. In order to avoid such a situation, particles can be added to the MOF to support its structure and prevent its shrinkage during pyrolysis. Wu et al. successfully synthesized a Co monatomic catalyst using KCl particles as the supporting template[94].
Active sites and regions
Engineering active sites at the atomic/cluster scales
The central atom of the M-N-C catalyst is often considered to be the active center and directly affects the electron cloud arrangement. Choi et al. investigated the effect of different metal types on catalytic activity[95]. Pyrolysis of dicyandiamide with metal chlorides at 900 °C, using dicyandiamide as both the carbon and nitrogen sources, revealed that the type of metal seed can change the electrochemical and physical characteristics of the resulting N-doped carbon. N-doped carbon derived from Co demonstrated the highest ORR activity in 1M HClO4, exhibiting the most extensive sp2-carbon network compared to other metal types. However, due to the residual metal particles in the catalyst, it is not conducive to exploring the structure of the active site. Zitolo et al. synthesized Fe-N-C materials nearly devoid of crystallographic iron structures following pyrolysis in argon or ammonia atmospheres. Although Fe-N-C catalysts pyrolyzed in argon and ammonia have almost identical FeN4C12 structural units, their ORR activity is significantly different[96]. This difference is due to the highly basic N-groups that formed during the pyrolysis of NH3, which significantly enhanced the ORR activity of FeN4C12. According to the present studies, the order of ORR activities of transition metal-based M-N-C is (Fe > Co > C ≥ Mn > Ni) -N-C under alkaline conditions [Figure 2A]. Peng reported that the active N content, residual metal content, surface area and pore structure of M-N-C catalysts were influenced by the choice of central metals, and the improvement of ORR performance resulting from combined effect of these factors rather than any single factor [Figure 2B-D][97].
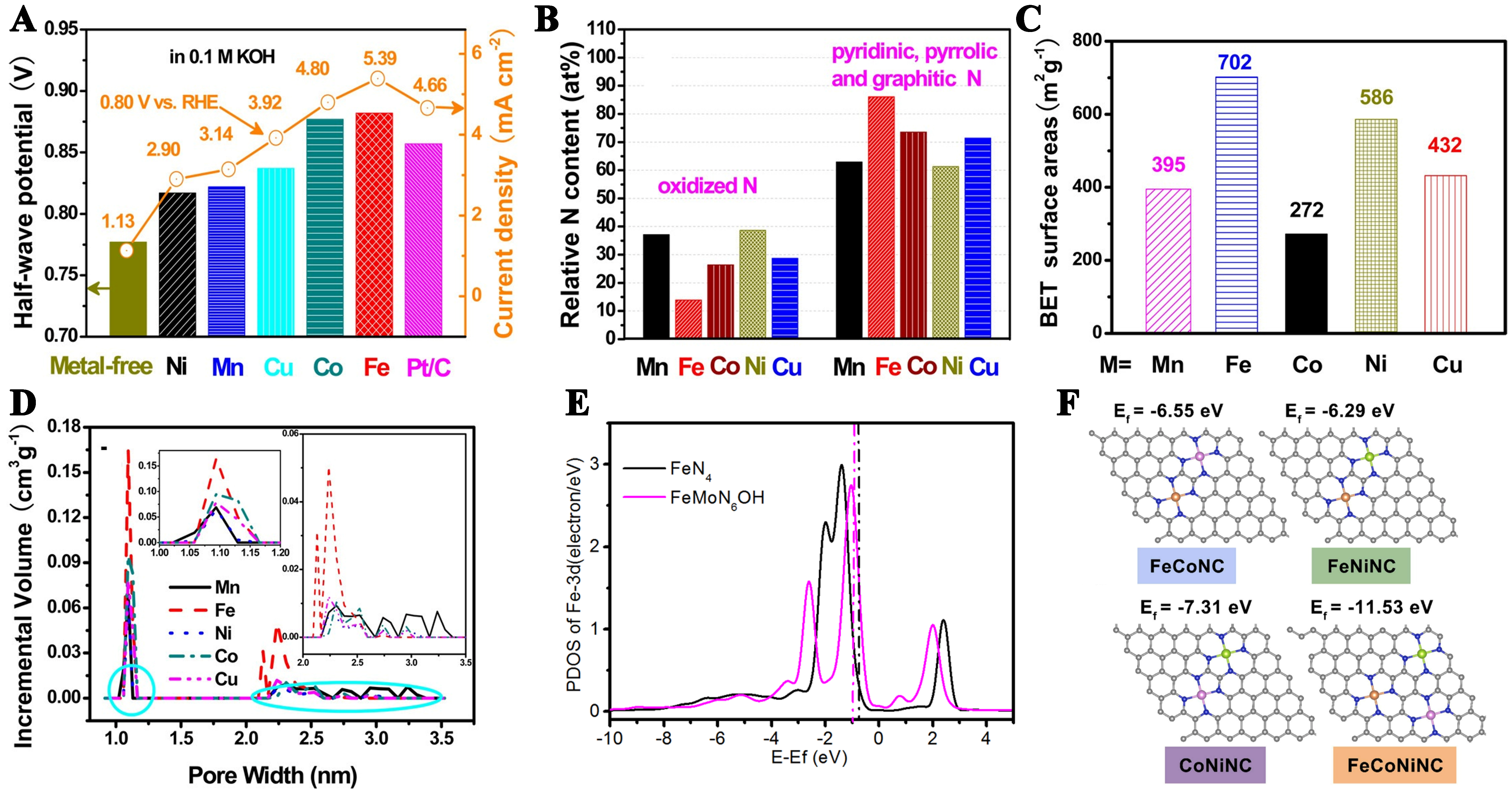
Figure 2. (A) Current density and half-wave potential of M-Pani /C-Mela (M = Co, Fe, Mn, Cu and Ni) and metal-free catalysts in 0.1M KOH; (B) N content (at%) of oxidized, Py-, Pyr-, and Gr-N; (C) BET surface areas of M-Pani /C-Mela; (D) Pore-size distribution of transition metal-codoped carbon catalysts. This figure is quoted with permission from Peng et al.[97]; (E) Projected density of states of Fe-3d orbitals of FeN4 and FeMoN6OH. This figure is quoted with permission from Zhu et al.[99]; (F) Atomic structures of electrocatalysts (blue: N, orange: Fe, purple: Co, and green: Ni). This figure is quoted with permission from Zhai et al.[101]. BET: Brunauer–Emmett–Teller.
The ORR performance of Fe-N-C catalyst is on par with that of the precious metal Pt, but the change of catalytic intensity of Fe-N-C catalyst intermediate makes it easy to produce Fenton effect and affect the stability of Fe-N-C catalyst. To mitigate this, introducing a second metal can alter the band center of Fe to optimize intermediate adsorption energy[98]. The Fe-Mo diatomic center within the N-doped carbon substrate can effectively downshift the d-band center of Fe by adjusting the electron configuration, thus reducing the adsorption of ORR intermediates and significantly improving the ORR performance of the Fe-Mo active site [Figure 2E][99]. Unlike the site where the diatomic center is combined, the introduction of Ni atoms near the FeN4 site can regulate the electronic structure and adsorption strength of ORR intermediates[100]. Zhai et al. reported the synthesis of a trimetallic monatomic catalyst (Fe3%Co3%Ni9%-NC) with excellent catalytic activity and stability[101]. Compared with monometallic and bimetallic single-atom structures, the d-band center of the FeCoNi-NC shifts toward the Fermi level [Figure 2F]. The upshift of d-band center can reduce the electron filling in the antibonding orbital, which is beneficial to the absorption of the reaction intermediates on the FeCoNi-NC surface. In addition to introducing other metal atoms into the structure, the introduction of metal clusters into the structure also generates synergistic effects[33]. Wan et al. reported an Fe-N-C catalyst featuring nitrogen-coordinated iron clusters and closely surrounding Fe-N4 active sites[102]. Due to unblocked electron transfer pathways and extremely short interaction distances, strong electronic interactions arise between the iron clusters and adjacent Fe-N4 sites. The iron clusters optimize the adsorption strength of oxygen reduction intermediates on Fe-N4 sites and shorten the bond amplitude of Fe-N4 with incoherent vibrations.
Coordination structure
The bond between the coordination atom and the metal atom produces the coordination bond. The bond length and the bond energy greatly affect the stability of the coordination bond. This alters the reactant’s adsorption behavior on the metal atom, ultimately influencing the catalyst’s performance. Additionally, the number of coordinating nitrogen atoms plays a critical role in modifying the charge distribution at metal sites, thereby adjusting the adsorption strength of oxygen-containing species at the active sites. Taking Fe-N-C catalyst as an example, its active site is N-coordinated iron atom FeNx (x = 1-5). FeN4 synthesized by high temperature pyrolysis is the ideal active site. DFT calculations show that the ORR activity and formation energies of FeNx species show an “inverted volcano” and “volcano” relationship with the Fe-N coordination number x, respectively[103]. The ORR activity order is calculated to be FeN4 > FeN3 > FeN2 > FeN1 > FeN5 [Figure 3A]. Among a series of FeNx (x = 1-5) and the configuration of FeN4 is the most stable. Xue et al. explained the high activity of FeN4 through the electron spin states[104]. The Fe active center of FeN5 (Fe-N5-LS) is in the low spin state (LS), and FeN4 (Fe-N4-HS) and FeN3 (Fe-N3-HS) are in the high spin state (HS) [Figure 3B-D]. The high spin Fe active center enables it to penetrate the antibonding π-orbital of oxygen more easily. The 3d-electronic structure of Fe-N4-HS is t2g3eg2, and this structure endows the catalyst with more suitable O2 adsorption capacity.
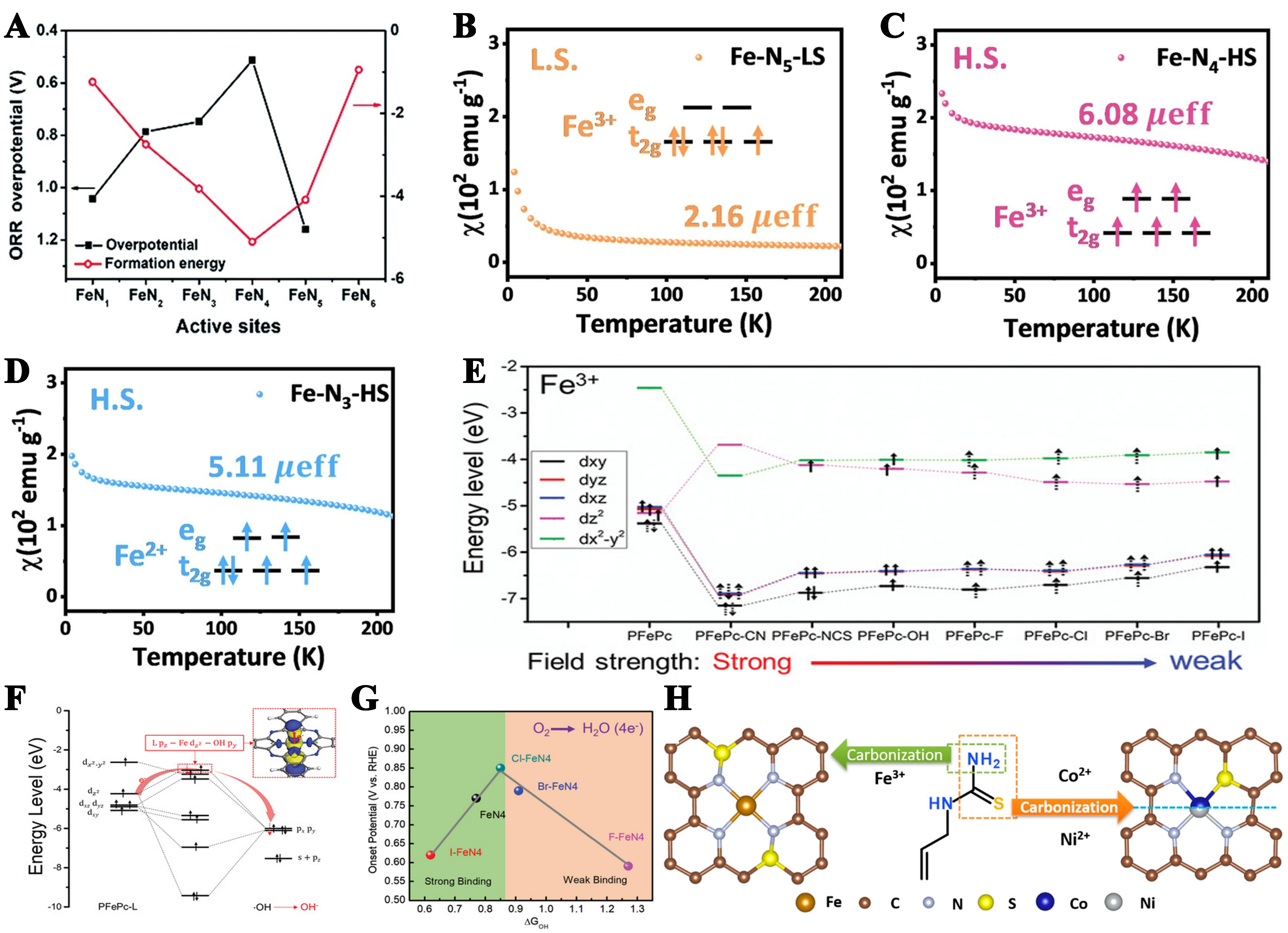
Figure 3. (A) DFT-calculated FeNx (x = 1-6) formation energies and FeNx (x = 1-5) ORR overpotentials. This figure is quoted with permission from Li et al.[103]; Temperature-dependent inverse susceptibility and electron configurations of (B) Fe-N5-LS, (C) Fe-N4-HS and (D) Fe-N3-HS. This figure is quoted with permission from Xue et al.[104]; (E) The Fe 3d orbital energy levels and the electron configurations of PFePc and PFePc-L were calculated; (F) Molecular orbitals of HO-PFePc-L. This figure is quoted with permission from Zhao et al.[109]; (G) ORR active volcano curve through the 4e- path (onset potential plotted as a function of ΔGOH). This figure is quoted with permission from Sabhapathy et al.[110]. (H) Schematic representation of the formation of Fe/Co/Ni-SAs/NS in different coordination environments. This figure is quoted with permission from Zhang et al.[111]. DFT: Density functional theory; ORR: oxygen reduction reaction; LS: low spin state; HS: high spin state.
In addition, some special coordination structures have been proposed, which have excellent catalytic activity and stability due to their unique catalytic mechanism. Different from the planar FeN4 structure, the planar Fe2N6 structure can greatly improve the catalytic activity and excellent stability. With optimized oxygen intermediate adsorption and synergistic effects from multiple active sites, the structure exhibits unique ORR redox behavior and strong O–O bond cleavage driving force, accelerating ORR kinetics and inhibiting side reactions[105]. Inspired by heme enzymes, the central iron ion in porphyrin rings coordinates with four nitrogen atoms and an axial ligand. In the FeN5 coordination structure, the axially oriented fifth nitrogen ligand is capable of exerting an electronic push-pull effect and/or a steric effect on the monoatomic site, thereby regulating the interaction energy between the active site and reaction intermediates[106,107]. Gong
Carrier effect
In addition to the above exploration of M-N-C coordination as the active center, it is also considered that the C structure around the N structure often affects the charge distribution of the active center, which indirectly affects the catalytic activity. The proposed configurations are FeN4-C8 with pyridine-N, FeN4-C10 with pyridine-N, pyrrolic and FeN4-C12 with pyrrolic[96,112]. Theoretical calculation proves that FeN4-C8 and FeN4-C12 exhibit better catalytic activity than FeN4-C10 in acidic media [Figure 4A]. In-situ Mössbauer spectroscopy results were conducted on electrodes subjected to 150 h of aging at varying potentials. The D1 signal is predominantly associated with either low-spin FeN4C8 or high-spin FeN4C12 configurations. Among these, FeN4C12 demonstrates superior activity and stability, while FeN4-C8’s instability is due to its experiencing more intense demetallation [Figure 4B][113]. However, other studies have suggested that the formation of FeN4-C10 sites from an intact graphene lattice is energetically favorable. In contrast, both FeN4-C12 and FeN4-C8 active sites require pre-existing suitable pore structures within the graphene layer to maintain their stability[114].
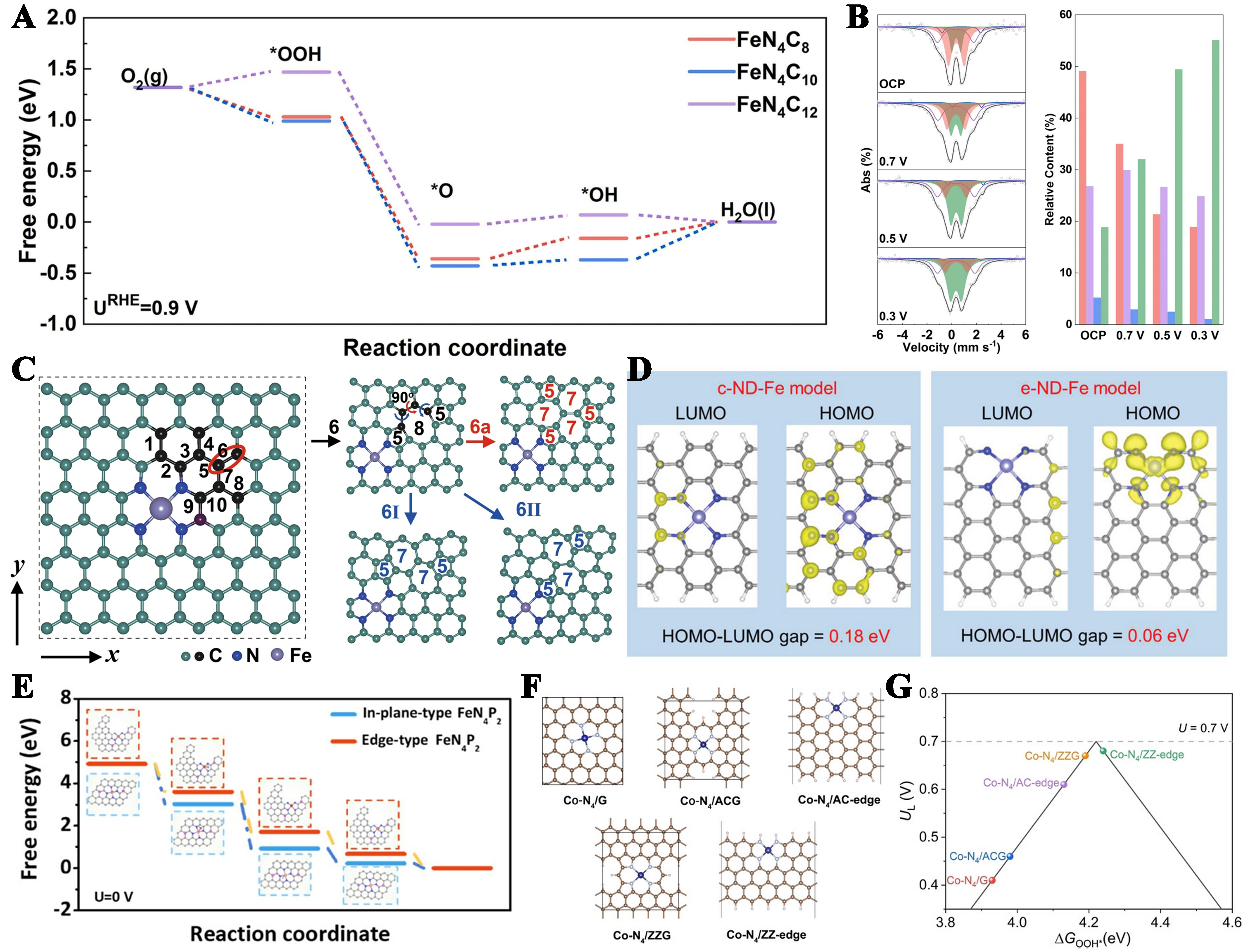
Figure 4. (A) The free energy diagram of typical ORR pathway of various FeN4 sites; (B) Fe-N-C in-situ Mössbauer spectra and the relative content of different types of FeN4. This figure is quoted with permission from Xu et al.[113]; (C) Optimized structure of a perfect FeN4 site and Divacancy defect models 6, 6a, 6I, and 6II. This figure is quoted with permission from Liu et al.[115]; (D) Molecular orbital distributions showing the HOMO and LUMO for c-ND-Fe and e-ND-Fe configurations. This figure is quoted with permission from Wang et al.[116]; (E) Calculated free energy diagrams for the ORR on in-plane-type FeN4P2 and edge-type FeN4P2 at zero electrode potential. This figure is quoted with permission from Yin et al.[117]; (F) The optimized structures of basal-plane Co-N4/G and edge-hosted Co-N4 configurations (brown: C; grey: N, blue: Co, and pink balls: H); (G) Computationally derived volcano plot for the 2e- ORR, illustrating the relationship between limiting potential (UL) and Gibbs free energy change for OOH* formation (ΔGOOH*). This figure is quoted with permission from Tian et al.[118]. ORR: Oxygen reduction reaction.
Because the ability of divacancy defects to modulate the electronic properties of carbon substrates and consequently alter the catalytic performance of Fe-N4 centers. Liu et al. engineered diverse divacancy configurations adjacent to the Fe-N4 site to investigate their influence on ORR activity. Various carbon defects are constructed near the FeN4 site, for example, 5-8-5 defects, 555-777 defects, 5-7-7-5 defects. DOS analysis showed divacancy defects (6, 6a, 6I) significantly altered Fe 3d orbital distribution, particularly 3dxz and 3dz2 states, compared to pristine FeN4 sites [Figure 4C][115]. The strong hybridization between the Fe 3dz2 and O 2pz orbitals results in substantial splitting of the O2 2pz orbital state, transferring electrons from the Fe site to *O2, reducing Δd and magnetic moments, and thus better activation of the O2 molecule. In our past studies, we believe that CoN4 and FeN4 have similar structures and good catalytic activity. When Co atoms are dispersed in N-doped carbon fibers (10Co-N@CNF) and defective carbon fibers (10Co-N@DCNF), the catalytic activity of 10Co-N@DCNF is close to that of Pt/C. Carbon deficiency is the root cause of enhanced ORR activity.
To further understand the role of nitrogen-modified divacancies (ND) configurations in M-N-C electrocatalysts, it is essential to create controllable ND trapped metal atoms and analyze how their location differences impact intrinsic activity. In DFT calculations, edge-trapped Fe sites (e-ND-Fe) and center-trapped Fe sites (c-ND-Fe) were modeled. The results reveal that e-ND-Fe shows greater electron redistribution than c-ND-Fe, leading to stronger electron density localization near the Fe-N4 active sites[116]. These results suggest that e-ND-Fe has a stronger electron-donating ability than c-ND-Fe, leading to improved catalytic reactivity. Additionally, the smaller highest occupied molecular orbital (HOMO)-lowest unoccupied molecular orbital (LUMO) gap in e-ND-Fe (0.06 eV) compared to c-ND-Fe (0.18 eV) facilitates more efficient electron transfer in catalytic reactions [Figure 4D]. We regulated the pore configuration by adjusting the amount of FeCl3 to regulate the exposure of e-ND-Fe species. Experimental results confirmed a strong link between ORR activity and the concentration of e-ND-Fe sites, aligning with theoretical predictions. Based on the edge effect, Yin et al. demonstrated that incorporation of long-range P at edge-type FeN4 can optimize the adsorption energy of the intermediate and reduce the energy barrier of the reaction [Figure 4E][117]. The introduction of P atoms into the carbon-base substrate can induce an interfacial charge density redistribution, which enhances the internal electron conduction. This optimization facilitates the adsorption of key intermediates on the charge-density-enriched surface Fe atoms, thus lowering the total energy barrier of the reaction. Highly efficient and selective ORR electrocatalysts are crucial for developing advanced energy storage systems that combine high performance, cost-effectiveness, and sustainable operation Tian et al. demonstrated different ORR pathways mediated by varying Co-N4 sites: edge-bearing sites (i.e., atomic Co-N4 partially located at the edge of the defects and in pockets) preferentially catalyze the 2e- ORR pathway, whereas the basal planar-bearing sites promote the 4e- ORR process by theoretical calculations [Figure 4F and G]. They synthesized a series of edge-to-bulk ratios carbon-supported Co-SACs catalysts and found that the exposed edges could be readily oxidized by oxygen functional groups during the ORR process, which enhances the kinetics of the 2e- ORR transfer pathway while consistently maintaining selectivity levels above 90%[118].
This extensive investigation into M-N-C catalysts underscores both the complexity and the promising potential of these materials for applications in the ORR. M-N-C catalysts present numerous advantages, making them viable alternatives to precious metal catalysts in ORR applications. These advantages include their cost-effectiveness, the ability to fine-tune their properties by modifying the central metal atom, coordination structure, and carrier effects, and their high catalytic activity, which is comparable to that of platinum-based catalysts, particularly in designs featuring iron (Fe) and cobalt (Co). Furthermore, M-N-C catalysts exhibit versatility, allowing for customization to suit specific applications, such as selective 2e- or 4e- ORR processes. These attributes position M-N-C catalysts as promising candidates for reducing costs and enhancing the performance of fuel cells and other electrochemical devices.
Metal-free carbon material
Carbon-based catalysts have been pivotal in ORR research. Early investigations predominantly focused on transition metal/oxide-doped carbon materials, in which metal-carbon active sites were demonstrated to enhance catalytic performance. However, the complexity associated with metal components and the inherent cost issues have significantly hindered their scalable application. In recent years, metal-free carbon-based catalysts have emerged as a transformative paradigm, offering distinct advantages such as abundant raw material sources, low cost, structural stability, and metal-free operational characteristics. Heteroatom doping strategies (e.g., N, S, P) have enabled precise modulation of the electronic structure and active site configuration of carbon materials, thereby achieving excellent ORR activity in both alkaline and acidic electrolyte environments. This transition from metal-dependent to carbon-intrinsic catalysis not only addresses long-standing challenges in the field but also paves the way for the development of sustainable and cost-effective ORR electrocatalysis systems.
Heteroatom-doped carbon catalysts
Synthesis
Pyrolysis: Thermal decomposition of organic compounds generally yields two primary products: volatile substances and carbon-enriched solid residues. Pyrolysis is a straightforward and commonly employed technique for producing diverse functional carbon materials. Heteroatom doping can be accomplished through thermal processing of combined carbon precursors and/or doping sources. The pyrolysis temperature, heating rate, and duration critically influence the composition, structure, and morphology of the resulting doped carbon materials. Carbon precursors are diverse, including chemical powders, polymers, covalent organic frameworks (COFs), covalent organic polymers (COPs), and MOFs. For example, furfuryl alcohol can be used to prepare N, O co-doped nanoporous carbon with a honeycomb structure, which has high uniformity and controllable pore size[119]; using self-assembled urea-formaldehyde (UF) resin as a single precursor can synthesize N-doped nanoporous carbon spheres composed of nanoplates[120]; The covalent organic framework (F-COF) can incorporate F and N into the precursor of COF in advance, and direct pyrolysis can prepare the dispersive F and N co-doped nanoporous carbon (F-NPC) catalyst[121]; Heteroatom-enriched covalent triazine polymers serve as effective self-doping precursors for the synthesis of nitrogen-phosphorus-fluorine tri-doped carbon nanospheres, simultaneously incorporating carbon, nitrogen, phosphorus, and fluorine elements[122]; direct carbonization of thin MIL-101-NH2 flakes and simple acid etching can construct an interesting porous carbon structure similar to brain platygyra coral, realizing the morphology control and uniform N atom doping of carbon nanostructures[123]. Currently, biomass, as an excellent substitute for fossil fuels, can be used to prepare carbon materials of different morphologies through pyrolysis, including porous carbon, layered porous carbon, carbon quantum dots, heteroatom-doped porous carbon, and carbon fibers. Using biomass as a precursor is also a method for synthesizing heteroatom-doped carbon materials that is environmentally sustainable and cost-effective. Garlic skin[124], tobacco[125], pine pollen[126] and other biomasses, as carbon and heteroatom precursors, make porous carbon and heteroatoms self-doped, without using any external heteroatom precursors, thus making the synthesis green and environmentally friendly. There is also the use of plant roots, stems, leaves, flowers, fruits mixed with urea as external N precursors to synthesize nitrogen/oxygen doped carbon materials with layered porosity[127].
Chemical vapor deposition (CVD): CVD differs from pyrolysis in that the process typically involves controlled exposure of the substrate to volatile precursor gases, followed by a reaction or decomposition process on the substrate surface. Kumar et al. annealed chitosan and graphene oxide simultaneously under the argon flow using CVD, they did not have any physical contact between them, chitosan pyrolysis generates volatile N-heterocycles that react with graphene oxide to synthesize NrGO[128].
Hydrothermal carbonization (HTC): HTC can convert organic compounds into carbon materials, commonly used for the production of nanostructured carbon. The HTC reaction occurs in a water medium, usually at temperatures above 100 °C and under high pressure. Song and co-worker reported using ultra-thin tellurium nanowires (Te NWs) as a template, nitrogen-carbon hydrate compounds as carbon sources, and a template-guided HTC process to synthesize nitrogen-doped carbonaceous nanofibers (N-CNFs) aerogels on a macro scale with diameters spanning from tens to hundreds of nanometers[129]. Biomass is also a promising precursor for the HTC method, such as lignin extracted from beech wood by alkaline hydrothermal treatment and subsequently functionalized by aromatic nitration, synthesizing microporous, mesoporous and macroporous N-doped carbon[130]; or using biomolecular guanine and various carbohydrate compounds (glucose, fructose, and cellulose) as carbon precursors to synthesize layered nitrogen-doped porous nano carbon[131].
Ball milling: Ball milling is a widely used large-scale mechanical technique for grinding and co-grinding, valued for its established methodology, solvent-free operation, and cost-effectiveness[132,133]. Recently, ball milling has also been researched and applied to the structural design of carbon materials, including intrinsic and extrinsic defects (such as heteroatoms, functional groups). The mechanical energy imparted by ball milling facilitates C=C bond cleavage and particle fragmentation, simultaneously generating abundant reactive sites including lattice defects, unsaturated bonds, and carbon-free radicals. Continuous mechanical energy input enables carbon atoms at these newly formed active sites to reorganize into stable graphite lattices through cold welding mediated by C–C and C–O–C bond formation, thereby facilitating defect healing and pore structure elimination[134]. In addition, some reaction sites will react with air (O2, CO2 or
Plasma treatment: Plasma-related technology offers an environmentally friendly and energy-efficient approach for both conventional etching procedures and advanced surface modification techniques in material engineering[137]. Plasma, consisting of ions, free radicals, electrons, and neutral atoms, can instantaneously generate free radicals, which are efficiently used for the synthesis of doped carbon materials. Based on four initial precursors (aniline, pyrrole, benzonitrile, and nitrobenzene), amino-N, pyrrolic-N, nitrile-N, and oxide-N can be successfully prepared through a room temperature plasma synthesis process[138]. Niu et al. synthesized nitrogen-doped graphene through solution plasma. Generated by discharge in the liquid, it has a single dielectric barrier that can reduce excessive current, thereby stabilizing the plasma and maintaining the overall temperature[139]. This promotes the preservation of nitrogen atoms during the synthesis process without evaporation, forming a graphite carbon skeleton. Additionally, precise control over nitrogen incorporation content in plasma-treated carbon materials can be synthesized by optimizing plasma exposure time and intensity, providing good flexibility for adjusting material properties[140].
Activity and selectivity
The arrangement of nitrogen doping is influenced by the chemical environment. N-doped graphitic structures exhibit multiple distinct configurations, primarily comprising pyridinic N, pyrrolic N, graphitic N, N oxides, and sp-hybridized N species [Figure 5A]. Different N configurations will affect the electronic properties of adjacent carbon atoms and regulate the adsorption energy on the surface of electrocatalysts, resulting in different catalytic performances. At present, research shows that the content of graphite nitrogen determines the limit current density, while the content of pyridine nitrogen increases the initial voltage of ORR[141], and the promotion of different types of N configurations for ORR reactions is inconsistent. Unfortunately, the synthesis of N-doped carbon catalysts results in heterogeneous mixtures of multiple N configurations, making the targeted fabrication of N-doped carbon materials with a single configuration a major scientific challenge. Therefore, how to evaluate the contribution of nitrogen configuration to ORR has certain research significance. N-doped highly oriented pyrolytic graphite (HOPG) was used to prepare specific types of N configurations. Guo et al. used this method to prepare pyridine-N-based HOPG (pyri-HOPG) and graphite-N-based HOPG (graph-HOPG)[142]. Under acidic conditions, thanks to the pairing of pyridine N with the lone pair of electrons on the carbon base plane, it can break the O–O bond, promoting ORR performance. Compared with graphite-N, the increase in pyridine N content can increase current density and initial potential [Figure 5B]. Interestingly, under alkaline conditions, graphite-N is considered to have a strong correlation with ORR performance. Wang et al. observed a strong correlation between the limiting current decay and the decrease in graphite N content, while identifying and quantitatively characterizing three different N-doping configurations. As the carbon atoms near the graphite nitrogen show smaller Gibbs free energy changes than those near the pyridine-N or pyrrole-N, graphite-N can be considered as a potential active site for the ORR reaction[143]. Furthermore, Zhao et al. reported and synthesized sp-N-doped graphene, and believed that the incorporated sp-N atoms possess significantly higher electron density compared to pyridinic and graphitic nitrogen species, which induces substantial positive charge accumulation on adjacent carbon atoms, creating a favorable chemical environment for O2 adsorption [Figure 5C][144]. Therefore, it has good application prospects in ORR.
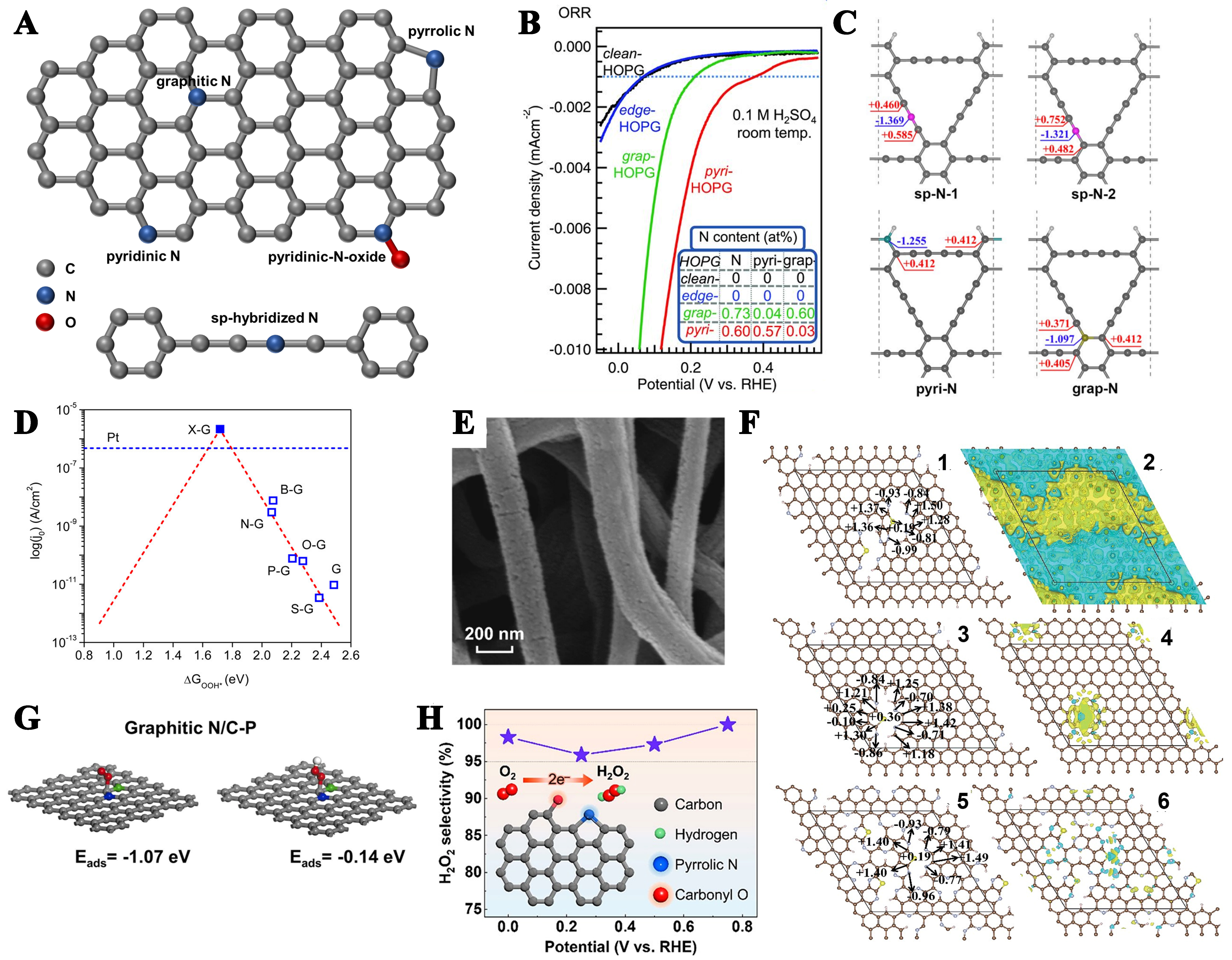
Figure 5. (A) Schematic diagram of different types of N; (B) The ORR performance of the model catalysts was evaluated, and the corresponding quantification of the nitrogen content is shown in the inset. This figure is quoted with permission from Guo et al.[142]; (C) Bader effective charges of N and adjacent C in N-doped graphdiyne (sp-N, pyri-N, grap-N). This figure is quoted with permission from Zhao et al.[144]; (D) Volcano plot from Tafel and DFT-calculated ΔGOOH* for doped graphene catalysts. This figure is quoted with permission from Jiao et al.[145]; (E) SEM images of N, F-MCFs. This figure is quoted with permission from Gong et al.[149]; (F) The impact of doping concentration on the electronic properties of a sulfur and pyridine nitrogen co-doped graphitic carbon system was studied for three compositions: (1,2) C:N:S = 112:8:2 (SN/C-900), (3,4) C:N:S = 120:4:1 (SN/C-1000), and (5,6) C:N:S = 91:23:3 (SN/C-400). The color coding represents the atomic species (dark brown: C, gray: N, yellow: S, pink: H). This figure is quoted with permission from Li et al.[151]; (G) Computationally optimized structural configurations and corresponding adsorption energies of O2 and *OOH intermediates on graphite N/C-P model systems. This figure is quoted with permission from Cheng et al.[156]; (H) Selectivity of the 2e- reaction of the coupling of pyrrolic N with carbonyl O. This figure is quoted with permission from Koh et al.[158]. ORR: Oxygen reduction reaction; DFT: density functional theory; SEM: scanning electron microscopy.
At present, single heteroatom doping cannot show good performance. Based on this, co-doping carbon materials with nitrogen and other heteroatoms have been widely explored to modify electron distribution and structural properties, thereby improving electrocatalytic performance. Jiao et al. confirmed through experiments and DFT calculations that non-metal heteroatoms such as B, O, S, P, etc., in graphene can obtain higher valence orbit energies of active atoms and cause stronger adsorption of *OOH and *OH intermediates, whose performance is comparable to that of Pt-based catalysts [Figure 5D][145]. The incorporation of electronegative heteroatoms, particularly N, into carbon matrices induces localized positive charge density on neighboring carbon atoms, which facilitates enhanced oxygen adsorption and improved charge transfer kinetics, ultimately boosting ORR activity. Moreover, the opposite strategy has been reported again, where the introduction of elements with lower electronegativity (e.g., boron or phosphorus) can also be effective in facilitating oxygen molecule adsorption and the subsequent reduction process. Choi et al. increased the ORR activity of N-doped carbon by enhancing the ratio of pyridine-N with additional B/P doping; P doping enhanced the charge delocalization of carbon atoms, and constructed a morphology with many splits and wrinkles at the open edge[146]. In addition, the S atom has a similar electronegativity to the C atom, and the individual S doping does not introduce additional unpaired electrons. However, the co-doped graphene model of S and graphite N has the highest carbon atom spin density. The *OOH intermediate is more inclined to adsorb carbon atoms with higher spin density in terms of energy[147].
The electronegativity of halogen elements is relatively high; carbon material samples doped with halogens (i.e., F, Cl, Br, and I) also showed improved electrocatalytic activity; for example, both Br-, and I-doped reduced graphene oxide (RGO) had more effective ORR than undoped RGO[148]. Notably, the high electronegativity of fluorine (χ = 4.0) makes it an efficient electron acceptor when forming ionic or semi-ionic bonds with carbon, which significantly enhances the charge transfer between the C–F bonds, thus dramatically increasing the electrical conductivity and altering the electronic properties of pristine carbon materials. The co-doping of nitrogen and fluorine atoms creates a synergistic effect that significantly boosts ORR catalytic performance. This enhancement stems from the combined influence of heteroatom doping and the distinctive nanofiber morphology featuring microporous walls, enabling N, F-MCFs to demonstrate both superior activity and remarkable stability in alkaline electrolytes [Figure 5E][149]. In addition, the dual doping of fluorine and sulfur on carbon fiber substrates produced synergistic effects via charge delocalization between molecules and spin state modulation, achieving catalytic performance rivaling conventional metal-based catalysts[150]. In our previous study, we developed a facile self-sponsored doping strategy to synthesize sulfur and nitrogen co-doped graphene, demonstrating that beyond the intrinsic effects of dopant elements, the catalytic performance is significantly influenced by the doping concentration[151]. At low doping concentrations, individual dopant sites exhibit pronounced electronic modifications, yet the overall catalytic impact remains limited due to insufficient active site density. Conversely, high doping levels increase the quantity of active centers but diminish the individual site effectiveness, ultimately compromising the overall catalytic performance [Figure 5F].
Although a large number of studies have demonstrated the reaction mechanism of N-doped carbon materials through experiments and theoretical tests, the selectivity of different configurations for 2e- reaction or 4e- reaction paths is still uncertain. Some studies have highlighted graphite N’s superiority over other N species in promoting the 2e- pathway, and some studies have emphasized the excellent selectivity of pyrrole N and pyridinic N for 2e- ORR, which may be due to the differences in the surrounding environment of the structure[152-154]. The addition of other heteroatoms based on nitrogen doping is very important to the catalytic mechanism. The carbon between pyridine-N and thiophene-S is considered to be the active site because it exhibits more positive charge distribution, and the larger the charge delocalization of this site, the stronger the absorption of O-intermediates, resulting in the O–O bond breaking in favor of 4e- ORR[155]. Cheng et al. synthesized N, P co-doped hollow MC spheres (N, P-HMCS) with rich graphitic N sites and rich electron domain C–P bonds as electron donors[156]. This configuration not only de-stabilizes the *OOH intermediate and desorbs H2O2 more easily [Figure 5G], but also induces significant distortions in the graphene sp2 lattice, leading to a synergistic catalysis of 2e- ORR. When reaction conditions are relatively mild, most defective carbon-based materials inevitably have surface oxygen groups that are one of the determinants of 2e- ORR in N-doped carbon catalysts. Chen et al. selectively targeted C=O, C-OH, and COOH functional groups by chemical titration to distinguish the catalytic contributions of different oxygenated species, and found that the carbonyl group (C=O) is the most active site in the two-electron ORR[157]. Moreover, it has been shown that the coupling effect of pyrrolic N and the O functional groups [including ether (O-C-O) and carbonyl (C=O)] tends to affect 2e- ORR, and the coupling of pyrrole N and carbonyl groups favors the 2e- ORR, with a H2O2 selectivity of 98.3% at 0.0 V (relative to reversible hydrogen electrode) in alkaline electrolytes [Figure 5H][158].
This section delves into the influence of nitrogen doping configurations and heteroatom co-doping on the activity and selectivity of carbon-based catalysts in the ORR. Various N configurations - such as pyridinic-N, pyrrolic-N, graphitic-N, oxide-N, and sp-hybridized N - affect catalyst performance in distinct manners. For example, graphitic-N is known to influence the limiting current density, whereas pyridinic-N contributes to an increased initial ORR voltage. Co-doping with additional heteroatoms, including boron (B), oxygen (O), sulfur (S), phosphorus (P), and halogens, can enhance electrocatalytic performance through synergistic interactions. The density of doping is a critical factor in determining catalyst activity and selectivity. Moreover, the selectivity for 2e- or 4e- ORR pathways is influenced by specific N configurations and their interactions with other functional groups.
However, during the evolution of N-doped carbon materials, researchers discovered that the observed enhancement in activity was not solely attributable to nitrogen doping. This realization has led to the exploration and development of carbon defect materials, which will be further discussed in the subsequent section.
Defective carbon catalysts
Synthesis and defect recognition
Essentially, carbon defects are a form of local disordered structure in the carbon lattice. The creation of carbon defects usually requires additional energy input to disrupt the long-range ordered carbon structure and undergo a structural reorganization process. In various carbon materials, there are many methods to create defects, such as heteroatom removal, thermal reduction of carbon, plasma etching, etc. Zhao et al. synthesized N-doped carbon and vacancy-deficient carbon by controlling different pyrolysis temperatures using N-enriched porous organic framework material (PAF-40)[159]. Thermal treatment induces carbonization of PAF-40, accompanied by partial nitrogen atom evolution, leading to the generation of monatomic vacancy defects in the interior. There is a monotonically decreasing trend of nitrogen content with increasing calcination temperature. Samples with lower nitrogen content (corresponding to higher defect densities) exhibit superior ORR performance. After the N atom was removed by heat treatment, the reconfiguration of carbon atoms caused by multiple monatomic vacancies forms various topological defects (pentagons, heptagons, and octagons), such as 585, 75585, and 5775. Raman analysis can clearly show the D band intensity of graphene is lower than the G band indicating that the carbon structure is highly regular and has relatively few defects [Figure 6A]. The removal of nitrogen-doped atoms leads to an increase in defects, and the increase in the ratio of ID/IG indicates a wider range of defect domains. And then they proposed that Zn-MOF was carbonized at 950 °C to synthesize a porous carbon material containing only carbon and oxygen, which showed excellent ORR activity despite the absence of any element doping, mainly due to the effect of defects arising from the removal of Zn atoms during the calcination process[160]. Tao et al. utilized the pristine surface of HOPG as the starting substrate and ideal model, successfully regulating defect levels via an efficient plasma etching strategy[161]. Through surface charge and potential analysis, it was found that the surface charge of HOPG increases with the increase of defects. Following Ar plasma etching, defect sites emerge on the HOPG surface. The partial removal of carbon atoms leads to an abundance of defects on the surface, which also becomes increasingly rough as the plasma irradiation time is extended. When the irradiation time exceeds 5 min, obvious nano-cones are produced on the HOPG surface. Post-etching, the I(D)/I(D′) remains approximately 2.4, suggesting that the defects produced by plasma irradiation are predominantly vacancy-type defects. Additionally, Kelvin probe force microscopy
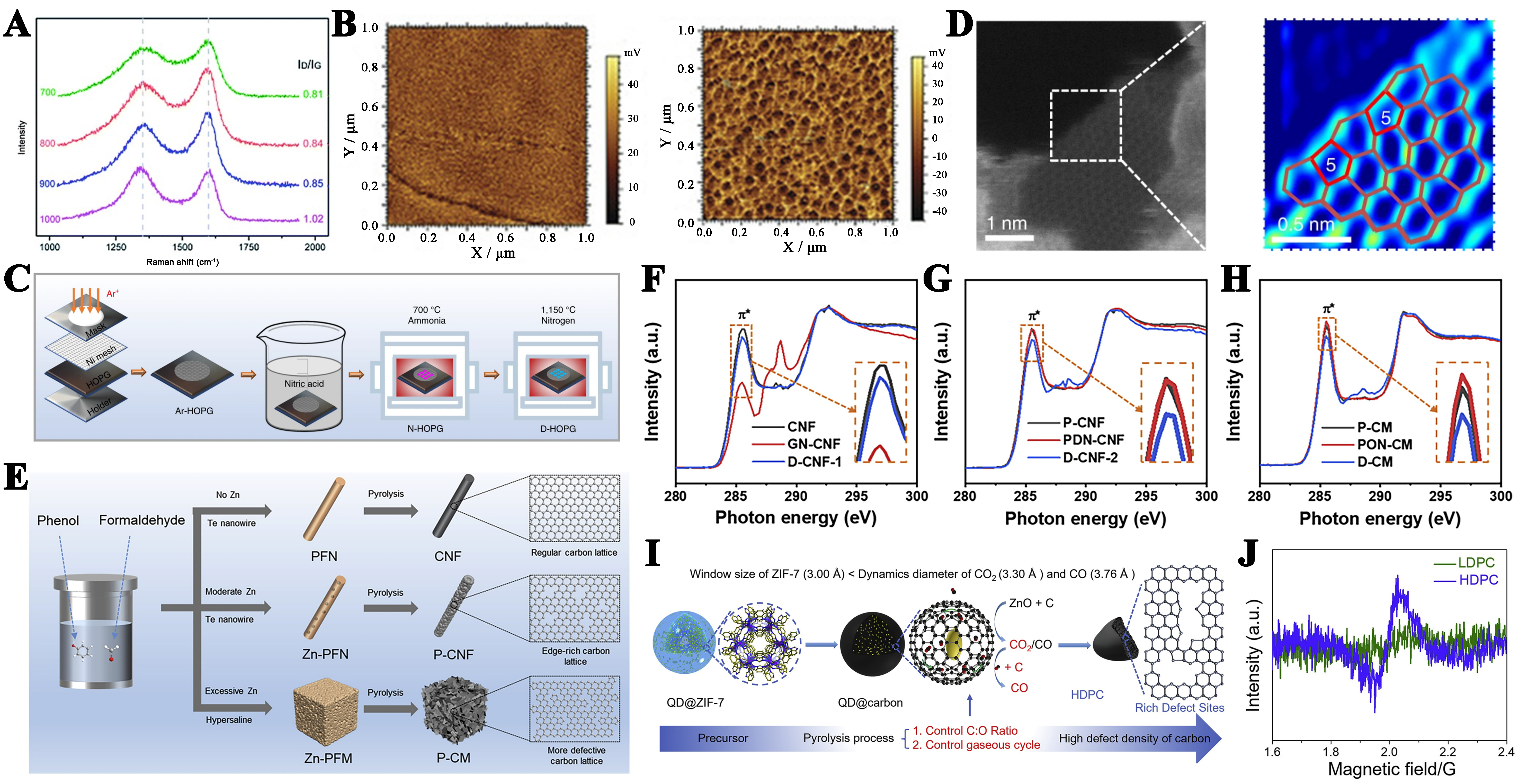
Figure 6. (A) Raman spectra of different types of catalysts. This figure is quoted with permission from Zhang et al.[159]; (B) KPFM test of HOPG before and after plasma etching. This figure is quoted with permission from Tao et al.[161]; (C) Synthetic scheme for the preparation of a D-HOPG sample; (D) The HAADF-STEM images derived from D-G [enlarged image of the dashed box in (D)]. This figure is quoted with permission from Jia et al.[162]; (E) Synthesis method of N-doped type carbon materials; (F-H) C K-edge XANES of different types CNF and CM. This figure is quoted with permission from Wang et al.[163]; (I) Schematic design and fabrication of HDPC; (J) EPR spectrum of LDPC and HDPC. This figure is quoted with permission from Wu et al.[165]. KPFM: Kelvin probe force microscopy; HOPG: highly oriented pyrolytic graphite; HAADF-STEM: high-angle annular dark field - scanning transmission electron microscopy; XANES: X-ray absorption near-edge structure; CNF: carbon nanofiber; CM: carbon monolith; EPR: electron paramagnetic resonance; LDPC: low density porous carbon; HDPC: high density porous carbon.
However, how to selectively synthesize a single type of carbon defect and evaluate its impact on ORR activity is a meaningful direction of research. Jia et al. first etched the original HOPG with a perfect graphite-carbon structure by argon plasma and formed a uniform concave through a nickel mesh with a square window [Figure 6C][162]. After removing the residual nickel from the surface, it is then annealed in an ammonia stream at 700 °C for 3 h. Thus, a specific N-doping (Pr-N) is formed at the edge of the N-HOPG groove. Finally, N-HOPG was annealed at 1,150 °C N2 for 2 h to remove the N atom and D-HOPG was obtained. Removing one Pr-N atom forms a pentagon on the edge of the graphene, and removing two adjacent Pr-N atoms confirms by calculation that the type of defect on the D-HOPG edge is most likely to be a reconstructed pentagon carbon defect. The defect areas of the materials can be visualized in practice through aberration-corrected high-resolution transmission electron microscopy (AC-HRTEM) [Figure 6D]. Structural defects primarily accumulate at pore boundaries, whereas the intrinsic hexagonal graphene configuration is predominantly preserved in the bulk regions of the material. To explore controlled synthesis of other configurations, edge engineering methods are available through one-to-one transformations of confirmative N configurations, such as graphite-N leads to C585, pyridine-N leads to S-C5, and pyrrole N leads to A-C5 [Figure 6E][163]. High-temperature pyrolysis converted zinc-free phenol-formaldehyde nanofiber (PFN) precursors into edge-deficient CNFs with desirable carbon network structures, while PFNS materials synthesized with a small amount of zinc ion doping yielded porous CNFs (P-CNFs) with abundant edge structures and hexagonal lattice configurations. CNF with graphite N (GN-CNF) and CNF with pyridine N (PDN-CNF) can be obtained by subsequent N doping process. The samples dominated by pyrrorole-N were prepared by the same N-doped phenol-formaldehyde monolith (PFM) containing a large number of Zn species prepared by the hypersaline polymerization method. For accurate detection of accurately detect the structural evolution of carbon materials, the characteristic peak at
Creating high-density defect sites in carbon materials is essential for further enhancing the activity of metal-free catalysts. A significant challenge remains in effectively introducing nitrogen heteroatoms to create numerous catalytically active pentagons following denitrification. It is therefore essential to investigate the key factors influencing the nitrogen doping process and to elucidate their correlation with the catalytic performance of carbon materials post-denitrification. Li et al. employed DFT calculations to reveal that native in-plane pentagonal defects in carbon materials alter the preferential nitrogen doping sites from edge hexagons to in-plane pentagons[164]. As the number of pentagons increases, the formation energy for nitrogen heteroatoms decreases progressively. Furthermore, post-denitrification, the nitrogen-containing in-plane pentagons transform into thermodynamically stable pentagon-heptagon-pentagon secondary defects, thereby increasing the density of pentagonal active sites. Inspired by these findings, they demonstrated that carbon materials derived from treated C60 exhibited excellent performance in alkaline oxygen reduction and zinc-air batteries, attributed to the abundance of secondary pentagonal defects, thus validating the theoretical predictions.
In contrast to traditional thermal decomposition and plasma etching methods, Wu et al. propose an interfacial self-etching method strategy by etching carbon bases with ZnO quantum dots [Figure 6I]. In this process, CO2 gas is generated inside the closed carbon cavity, which in turn for ultra-high density carbon defects[165]. The electron paramagnetic resonance (EPR) spectrum has confirmed the results of the Raman spectroscopy [Figure 6J]. The hyperdense defects in porous carbon (HDPC) formed through the interfacial self-corrosive strategy have stronger peak signals at the magnetic field value of 2.003G and higher ID/IG values, indicating that HDPC has a higher defect density in the bulk phase.
Despite the rapid development of defect characterization techniques, they are still insufficient in support of more fundamental and systematic research on the defects of electrocatalysts in the future. Therefore, it is necessary to develop more advanced characterization methods to study defects more accurately on time and spatial scales. Moreover, under harsh conditions (strong acid/base, high current density, long-term operation), the defect structure may be unstable and may undergo reconstruction or even oxidation. Therefore, it is necessary to develop in-situ characterization techniques for monitoring the dynamic evolution of defect structures during reaction processes and the actual interaction of reaction intermediates. For example, in-situ Raman spectroscopy technique can analyze intermediate products during the reaction process based on chemical structure, providing valuable information for exploring reaction mechanism. In-situ X-ray absorption spectroscopy (XAS) can reflect the chemical valence state and electronic structure of the measured elements during the reaction process well. By fitting the data, we can get important information such as the real spatial distribution of atoms, bonding conditions and coordination environment; In-situ XPS technology can analyze the valence changes and coordination environment of specific elements in real time accurately. However, the application of in-situ characterization techniques is often filled with challenges in future applications due to harsh operating environments or limited information obtained.
Cognition and design of active sites
Indeed, different types of carbon defects can effectively enhance the catalytic properties of materials. However, there is currently less research on the synthesis of single defects, so in order to deeply understand the role of single defects in ORR activity, it is particularly necessary to verify their importance with the help of theoretical calculations. Currently, exploring the impact of topological carbon defects, usually composed of one or more non-hexagonal rings, on catalytic activity through theoretical calculations [e.g., pentagon (C5), pentagon-octagon-pentagon (C5-8-5), pentagon-heptagon-heptagon-pentagon (C5-7-7-5)] can help develop catalysts with higher activity. Theoretical calculations show that the addition of nitrogen hinders the reduction of chemisorbed oxygen (O*), and that a large energy barrier of 1.03 eV is required for the rate-determining O* → OH* transition. The G585 defect facilitated the O2 → OOH* transition, reducing the Gibbs free energy change (ΔG) from 0.88 to 0.41 eV for pristine graphene, and all basic steps on G585 exhibited thermodynamic advantages approaching the performance of an ideal catalyst [Figure 7A][159]. It is found that different topological defect configurations show unique catalytic advantages in various electrochemical processes, e.g., the C5-1 defect shows excellent performance in ORR and OER reactions, while the C7-5-5-7 configuration performs better in HER [Figure 7B-E][166]. The HOMO and LUMO are mainly distributed on the edge atoms of graphene holes, and the introduction of C5-1, 585, and 7557 defects will produce HOMO/LUMO orbitals around the edge atoms of the defects, thus improving the catalytic activity.
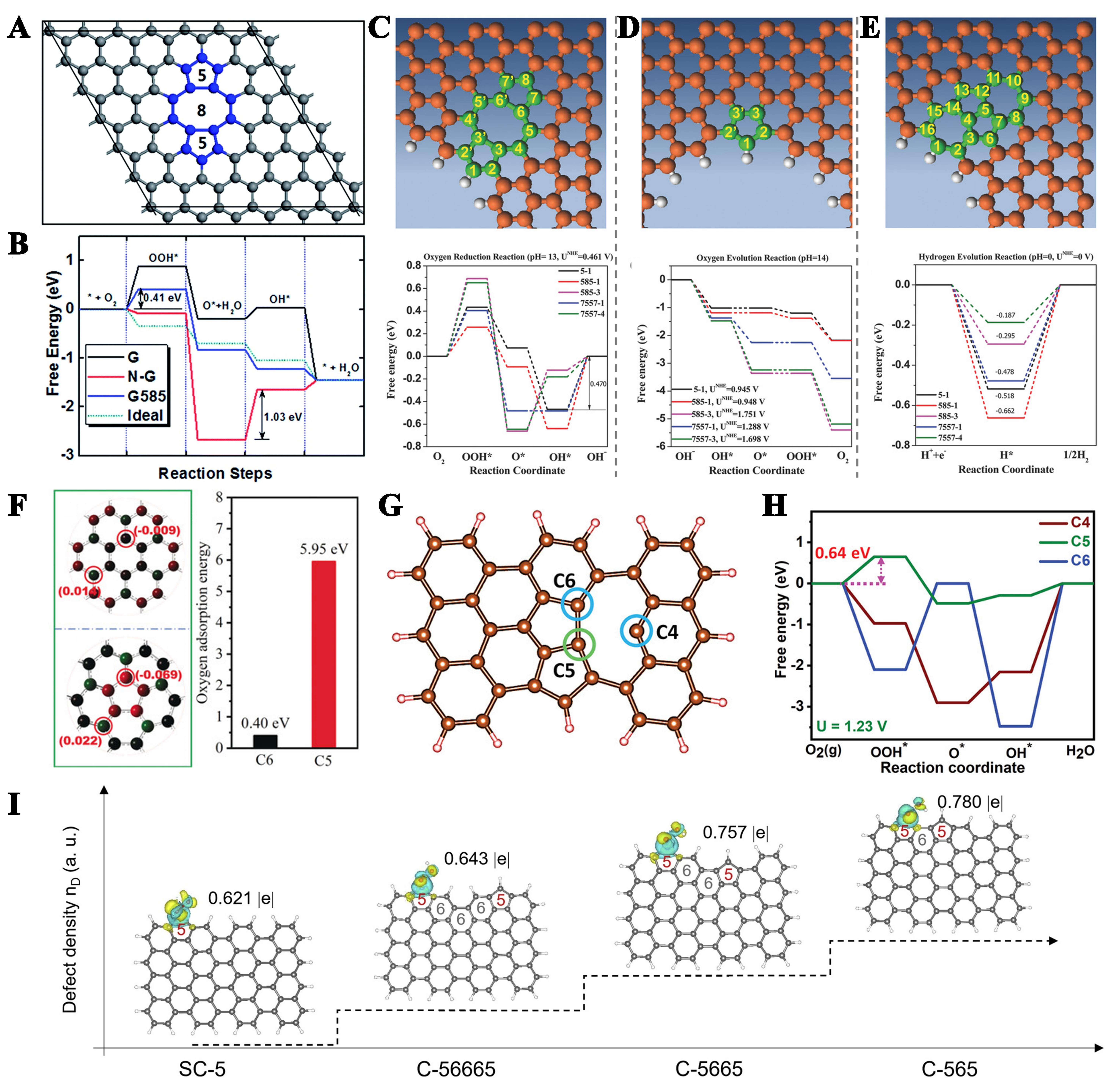
Figure 7. (A) Schematic diagram of G585 defects in graphene; (B) Computationally derived free energy profiles for ORR at equilibrium potential, comparing pristine monolayer graphene (G), NG, G585-defected graphene (G585), and an ideal catalyst (Ideal). This figure is quoted with permission from Zhao et al.[159]. Comparative energy maps of defect configurations of ORR, OER, and HER reaction pathways on DG under alkaline and acidic conditions: (C) edge pentagon, (D) 5-8-5 defect, (E) 7-55-7; This figure is quoted with permission from Jia et al.[166]; (F) Charge densities and oxygen adsorption energies of C6 and C5. This figure is quoted with permission from Zhu et al.[168]; (G) Theoretical models, and (H) the free energy diagram of typical ORR pathway of C4, C5 and C6. This figure is quoted with permission from Srinivas et al.[172]; (I) The electron density corresponding to adsorbed *OOH on different sites. This figure is quoted with permission from Wu et al.[165]. ORR: Oxygen reduction reaction; NC: nitrogen-doped graphene; OER: oxygen evolution reaction; HER: hydrogen evolution reaction; DG: defective graphene.
It is reported that carbon atoms with higher charge density are more likely to be active sites[167]. The HOMO of C5 has greater electron donor potential and better reactivity. The reduced HOMO-LUMO energy gap observed in C5 configurations, compared to C6 structures, facilitates more efficient electron transfer during electrochemical processes[168]. In addition, local electron redistribution results in a large difference in charge density between C6 and C5. When they are at the edge, the average charge density of C5 (-0.190) is still greater than that of C6 (-0.115) [Figure 7F]. Moreover, the adsorption energy of oxygen at the C6 and C5 sites shows that C6 has weaker adsorption capacity for oxygen molecules (0.40 eV), while C5 has much stronger adsorption capacity for oxygen (5.95 eV). Therefore, intrinsic pentagonal carbon has good chemical activity. When C5 and C7 defects are adjacently configured to form a curved C5+7 structure, the catalytic overpotential is significantly reduced. Oxygen species preferentially adsorb at the five-seven ring junction, where the inherent C5-C7 dipole effectively weakens the O–O bond in OOH intermediates through bond elongation, thereby facilitating oxygen reduction[169]. The generation and desorption of H2O2 in C57 are both thermodynamically uphill processes, with a total free energy change of 0.32 eV[170]. Conversely, the O–O cleavage pathway from *OOH to *O + OH- is exothermic, thus highly favorable. Similarly, the formation and desorption of H2O2 on the C7557 active site are also unfavorable. Therefore, at the pentagon/heptagon defect sites, the four-electron reduction to H2O is the preferred ORR pathway.
Besides pentagonal carbon defects, edge defects are also good ORR active sites. The different electronic configurations between the sawtooth-edge and armchair-edge structures induce different reaction patterns to the ORR intermediates, ultimately resulting in significantly different ORR performance characteristics. In sawtooth edge configurations, localized unpaired π electrons reside on individual edge carbon atoms, whereas armchair edge structures exhibit paired π electrons between adjacent carbon atoms, forming stable covalent bonding interactions. Jiang et al. found that the unpaired π electrons of the sawtooth edge are conducive to electron transfer to O2 and easy to form *OOH, with a free energy change of -0.222 eV[171]. In contrast, the armchair edge lacking unpaired π electrons must rearrange adjacent bonds to transfer an electron to O2 to form *OOH, thereby increasing the free energy by 1.089 eV. Comparative analysis of the vacancy configurations shows that the armchair pentagonal defect (C5-site) exhibits a pathway of reduced free energy required for the ORR process compared to the zigzag hexagonal (C4-site) and armchair hexagonal (C6-site) vacancy structures [Figure 7G][172]. From a thermodynamic perspective, in addition to zigzag edge carbon, the C5 site is also an actual active site for the ORR [Figure 7H]. Xia et al. proposed that the synergistic interaction between carbon vacancy defects and neighboring pentagonal structures enhances molecular oxygen adsorption and boosts catalytic performance[173]. Moreover, vacancies cause the redistribution of electrons in adjacent pentagons, thereby forming more conjugated π electrons. When conjugated π electrons are enriched near the Fermi surface, the *OH is easier to desorb due to the weakened binding strength of *OH. Wu et al. theoretically established single pentagon defects (SC5), two pentagon defects with three-hexagon (C-56665), two-hexagon (C-5665), and one-hexagon (C-565) intervals, representing reduced spatial distances (1.2-0.5 nm) between defects[165]. The results showed that the excellent electrocatalytic performance comes from the “proximity effect” generated by high-density carbon defects between carbon defect sites at different spatial distances. The smaller the spacing between pentagonal defects, the higher the number of transferred charges, reflecting the stronger interactions between defects and thus increasing ORR performance [Figure 7I]. In-plane defect models impose limitations on the target atomic structure. In contrast to planar configurations, local 3D curved structures induce more strained bonds due to their pronounced geometric curvature. These distinctive strain effects can alter the electronic structure of active sites and the adsorption-desorption dynamics of reaction intermediates, potentially enhancing catalytic efficiency. Wang et al. identified a correlation between the curvature of carbon defects and their activity in the ORR, highlighting the advantages of highly curved topological defects in carbon[174]. By augmenting the curvature of carbon materials, it is possible to modulate the electronic structure of edge pentagon defects, optimize the adsorption-desorption process of reaction intermediates, and lower the energy barrier for ORR.
Complex defects catalysts
A key advantage of carbon defect engineering is its extreme compatibility with a wide range of heteroatomic species, allowing the incorporation of non-metallic heteroatoms (N, P, S, and O) at the defect site. When the defect is combined with the dopant, the obtained catalyst usually exhibits the highest ORR activity, demonstrating a synergistic enhancement of electrocatalytic activity through combined defect-dopant interactions in nanocarbon materials. Zhang et al. proposed several possible combination models: pyridinic-N (PN6) + pentagon (EC5) carbon rings, PN6 + heptagon (EC7) carbon rings, pyrrolic-N (PN5) + EC5 and PN5 + EC7. The PN6 + EC5 composite exhibited the most favorable *OH adsorption characteristics, with a binding energy of 0.88 eV, suggesting significant potential for optimizing ORR performance [Figure 8A and B][175]. The improved catalytic performance in the synergistic region may stem from the high electronegativity of the PN6 dopant and induced redistribution of charge within the pentagonal carbon ring.
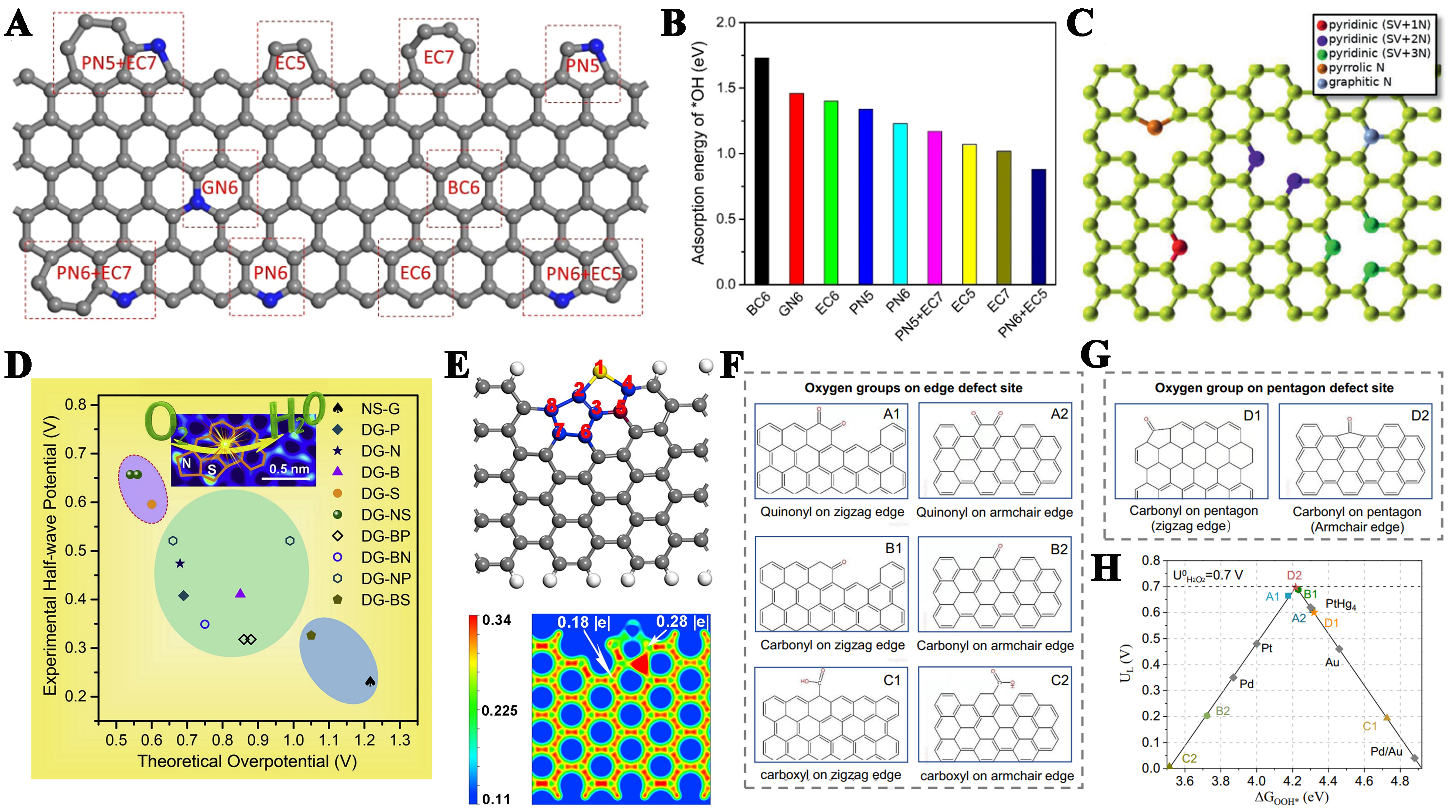
Figure 8. (A) Schematic representation of graphene at various potential active sites; (B) corresponding *OH adsorption energies for different reaction centers. This figure is quoted with permission from Zhang et al.[175]; (C) Schematic N-doped sites in graphene. This figure is quoted with permission from Fernandez-Escamilla et al.[176]; (D) Correlative analysis between experimentally determined half-wave potentials and computationally derived overpotentials for various samples under acidic conditions. This figure is quoted with permission from Yan et al.[177]; (E) The optimized structures of the N-S-D-G model; the electron density corresponding to N-S-D-G; the blue and red regions indicate electron-donating and electron-withdrawing areas, respectively. This figure is quoted with permission from Li et al.[178]; (F and G) The atomic structures of the examined O-groups on edge and pentagon defect sites; (H) The calculated activity volcano map This figure is quoted with permission from Wu et al.[179].
Charge redistribution resulting from defect-dopant interactions enhances binding affinity toward key reaction intermediates, consequently increasing adsorption energetics. This enhancement boosts the performance of ORR and alters the mechanism of ORR. The introduction of pyridine nitrogen into the basal plane of graphitic carbon substrates produces associated vacancy defects. Fernandez-Escamilla et al. employed a combined DFT and experimental approach to examine single vacancy (SV) configurations with various pyridinic nitrogen doping arrangements [Figure 8C][176]. The results showed that the ORR on graphite N and pyridine (SV + 3N) CNTs, which lack topological defects or dangling bonds, proceeds via a two-electron pathway through a physical adsorption mechanism. Yan et al. used computational simulations to explore the synergistic role of a variety of heteroatoms and carbon defects (C5) in ORR catalysis
The synergistic interaction between defects and dopants presents a promising avenue for further catalyst optimization [Table 1], potentially leading to materials that outperform traditional precious metal catalysts. When carbon defects are combined with heteroatom doping, two primary benefits arise. Firstly, carbon defects create additional active sites for the incorporation of heteroatoms, facilitating a more uniform distribution throughout the carbon matrix. Secondly, the presence of heteroatoms modulates the electronic structure and chemical properties of carbon defects, significantly enhancing their catalytic activity.
Comparison of activity and stability of catalysts with different defect types
| Defect type | Catalyst | Catalytic activity E1/2 | Stability | Ref. |
| C585 | DG | 0.76 V (0.1M KOH) | [167] | |
| C5 | PD-G | 0.78 V (0.1M KOH) | 96% 100,000 s | [169] |
| C5+7 | NGM | 0.77 V (0.1M KOH) | 98.5% 10 h | [170] |
| C7557 | HCNR-1000 | 0.72 V (0.1M KOH) | 95.5% 900 s | [171] |
| C955 | NR-900 | 0.837 V (0.1M KOH) | 92.72% 30,000 s | [173] |
| Vacancy-6556 | VP/CN | 1.08 V (0.1M KOH) | 96.8% 60 h | [174] |
| curvature of edge-C5 | HCHDC | 0.88 V (0.1M KOH) 0.74 V (0.5M H2SO4) | 99.2% 12 h | [175] |
| pyridinic-N + edge-C5 | ND-GLC | 0.875 V (0.1M KOH) | 84% 30,000 s | [176] |
| C5 + N + S | DG-NS | 0.68 V (0.1M HClO4) | [178] | |
| N-modified S-defect | NSCA-700-1000 | 0.76 V (0.5M H2SO4) | [179] | |
| Carbonyl on C5 | O-DG-30 | 0.75 V (0.1M KOH) | [180] |
ADVANCES IN ELECTROCATALYTIC CARBON-BASED MATERIALS
HER
HER based on hydrophobic CNTs was first reported in 2009. However, the original carbon material is electrochemically inert or has poor catalytic activity and is often used as a carrier for precious metals to enhance dispersibility and prevent aggregation during synthesis and reaction. Furthermore, the exceptional electrical conductivity of carbon-based materials facilitates efficient charge transfer between the support and catalytic surface. Their structural advantages, including high surface area and well-developed porosity, enhance active site accessibility and promote reactant/product mass transport. Moreover, carbon-based materials exhibit excellent electrochemical robustness within a wide potential window and can maintain structural stability during electrocatalytic reactions. The researchers have engineered diverse carbon-based materials with different morphologies as catalyst substrates, and according to size, the carbon carriers can be divided into 0D structures including carbon quantum dots, fullerenes, 1D materials such as CNTs, 2D materials such as graphene and 3D structures such as MC, graphite[180-182].
In addition to using carbon material as the catalytic substrate to support the NPs, the active single atom is anchored on the carbon-based material by using the coordination structure to reduce the utilization rate of precious metals. With tetra(4-tert-butyl-phenyl) porphyrinato platinum (PtTBPP) complexes as the precursor, the Pt-N4 coordination layer in PtTBPP molecules demonstrates remarkable thermal stability during high-temperature carbonization, enabling either complete or partial preservation of the molecular structure. It is beneficial to the formation of three-coordinated PtC2N1 stable structure and shows much better performance than HER reference electrocatalyst Pt/C (20 wt%) under alkaline environment due to the synergistic interaction between the close three-coordinated PtC2N1 groups[183]. In order to obtain a configuration favorable for HER, fixing Pt atoms at the defect sites of carbon can form a stable Pt-C coordination. Yang et al. synthesized N-doped graphene (NG) through pyrolysis of melamine-graphene composites at 700 °C for 2 h, followed by high-temperature annealing at 1,150 °C to generate DG via nitrogen removal [Figure 9A][184]. Then, Pt4+ cation is uniformly dispersed onto DG, and Pt@DG with a thickness of 0.3-0.5 nm is obtained by photoreduction. Defects in DG facilitate the formation of Pt-C3 configurations, where each platinum atom coordinates with approximately 2.9 carbon atoms. The stronger electron trapping ability of the 5d state of the Pt atom in the Pt-C3 configuration can effectively regulate the reduction of H+, the adsorption of H* intermediates, and ultimately accelerate the desorption of H2 under acidic and alkaline conditions [Figure 9B and C].
Figure 9. (A) Schematic representation of the Pt@DG synthesis process; Reaction potential energy of the HER process of different active sites under (B) acidic conditions and (C) alkaline conditions. This figure is quoted with permission from Yang et al.[184]; (D) EXAFS fitting analysis of Co-C3N4/rGO in R-space, with insets illustrating schematic models of Co-N1 and Co-N3 coordination environments. This figure is quoted with permission from Liu et al.[186]; (E) Schematic representation of the graphitic structure of A-CoPt-NC; pink: Co, white: Pt; (F and G) Side view and top view of the charge distribution of single-layered/double-layered model a(Co-Pt) @N8V4; (brown: C, green: N, white: Pt, blue: Co. the colors yellow and blue represent the increase and decrease of charge density, respectively). This figure is quoted with permission from Zhang et al.[187]; Universal formation of G5-7 topological rings as HER active sites. In situ Raman spectra of (H) N-GP, (I) P-GP, (J) S-GP, and (K) Se-GP during the HER process [The Inset in (H) shows the structure of H-G5-7; gray: C, blue: H]. This figure is quoted with permission from Liu et al.[189]. HER: Hydrogen evolution reaction; EXAFS: extended X-ray absorption fine structure.
So far, noble metal nanomaterials, especially Pt, remain the most effective electrocatalysts for HER, owing to their optimal hydrogen adsorption energetics and exceptional electrochemical stability. Regrettably, the limited natural abundance and prohibitive costs associated with precious metals have significantly hindered their large-scale implementation in commercial water electrolyzers. Consequently, the development of HER catalysts composed of inexpensive and abundant elements has emerged as a primary objective in recent years’ renewable energy research. Li et al. used a hard template-assisted and heat-treated synthesis strategy to anchor individual cobalt atoms to nitrogen-rich carbon nanocapsules (Co@CNB-N4)[185]. It has been shown that the incorporation of different transition metal elements into carbon materials leads to different N configurations (pyridine, pyrrole and graphite), which have different effects on the electrocatalytic properties. Cobalt incorporation in carbon matrices facilitates pyrrolic nitrogen (pyrr-N) formation, endowing Co@CNB-N4 with exceptional electrocatalytic activity for overall water splitting, and the material demonstrated a low HER overpotential of 45 mV, comparable to commercial Pt/C benchmarks. Studies have shown that the catalytic performance of the low-coordinated cobalt-nitrogen (Co-N) configuration was superior to that of the high-coordinated configuration, using graphite carbon nitride (g-C3N4) with rich lone pair electrons, providing several nitrogen coordinators for trapping single metals, synthesizing Co single atom and g-C3N4 coupling catalyst, containing 20% Co-N and 80% Co-3N [Figure 9D][186]. It is widely recognized that the coordination configuration of metal atoms is a key determinant of electronic properties and catalytic activity. However, recent studies have also emphasized the importance of interlayer synergistic effects in multilayer atomic metal catalysts. Zhang et al. successfully anchored Pt and Co atoms to multiple graphite layers, and DFT calculations showed that the interlayer Pt-Co interaction induces a significant charge polarization in the catalyst surface layer, exhibiting a crucial synergistic effect [Figure 9E][187]. The charge polarization surrounding Co atom modulates H* adsorption behavior, thereby reducing the Gibbs free energy barrier for intermediate formation [Figure 9F and G].
Consistent with the previous discussion, the incorporation of these heteroatoms with different electronegativities will change the spin density and charge distribution of the nearby carbon atoms, change the valence orbital energy levels, and thus activate the carbon atom to become an active site. In addition, the edge carbon atoms of the defects on graphene are originally in an unbalanced charge distribution state, so the defects are usually also active sites. Therefore, both doping and defect creation can improve the HER catalytic activity of graphene. Zhang et al. reported that due to the differences in atomic radius, the doped N, B, P, S heteroatoms and C atoms will have significantly different bond lengths when combined through chemical bonds[188]. The difference in bond length can change the lattice parameters of the catalyst, causing distortion of the carbon plane structure, destroying the integrity of the sp2 hybrid carbon atoms in the carbon plane, thereby adjusting its electronic structure, making these structural defects become centers of catalytic activity, to enhance its catalytic activity. In addition, the inherent defects of carbon materials (such as edges, vacancies, holes, topological defects, etc.) can also change the electronic structure and surface properties of the carbon base, thereby improving its HER catalytic ability. Furthermore, Lu et al. found that heteroatom-doped carbon materials undergo significant structural transformations during the HER process, ultimately forming defective structures[189]. The hydrogenation of N and C atoms in the HER process of N-doped graphite sheets (N-GP) and the subsequent reconstruction of the carbon skeleton significantly improved the activity of HER. Among them, the N-doping agent was gradually hydrogenated, causing the carbon skeleton to be reconstructed from a hexagonal ring to a 5,7-topological ring (G5-7) [Figure 9H]. Graphite doped with P, S, and Se also has the formation of G5-7 rings similar to the removal of doped impurity atoms [Figure 9I-K].
OER
OER serves as a crucial half-reaction in numerous electrochemical technologies, including water splitting systems and rechargeable metal-air battery applications[190]. The study of metal-free carbon-based materials as OER catalysts in alkaline media is increasingly extensive. In particular, nitrogen-doped nanostructured carbon catalysts (N/C) have been extensively studied. It is found that carbon cloth (CC) with high activity and excellent electrical conductivity has been widely reported as a catalyst carrier due to its cost-effectiveness, superior electrical conductivity, and remarkable mechanical flexibility. N-doped CC is highly realized by heat treatment of NH3 source, and its catalytic activity to OER is significantly improved due to the strong electron absorption capacity of pyridinic-N[191]. Based on N-doped C catalysts, a variety of heteroatomic-doped and even dopant-free carbons have been developed for OER. Due to the synergies between co-dopants leading to greater charge redistribution and more active sites, multi-doped/functionalized carbon NPs can exhibit higher catalytic activity. In 2015, Zhang et al. pioneered the development of dual-doped bifunctional catalysts, demonstrating that N and P co-doped MC NPs exhibit efficient ORR/OER electrocatalytic activity[192]. The most catalytically active structure has edge-localized N and P co-doped graphene sites, where the synergistic interaction between the N and P dopants significantly reduces the overpotential of the OER. Recent studies have doped carbon frameworks with N, O, S, P, and Se to create nonmetallic materials with tunable electronic structures. Lu et al. found that the impurity atoms in N-, P-, and Se-doped graphite flakes (GPs) were oxidized and dissolved in the OER process, but the catalyst maintained good electrochemical activity, so oxygen-rich residues rather than trace impurity atoms were shown to be responsible for the high OER activity [Figure 10A][193]. Different elemental dopings of GPs can generate unique ortho-quinone structures with distinct fused ring configurations. N-GP, P-DP, and Se-GP are converted to phenazone, benzoquinone, and naphthoquinone, respectively [Figure 10B-D]. DFT calculations show that the theoretical activity is in the order of phenanthraquinone > naphthoquinone > benzoquinone, not heteroatoms. However, the oxidation of heteroatoms in different coordination environments is uncertain, so it is very crucial to explore its impact on OER activity by directly introducing oxygen groups, such as β-CD. Based on this, Qiang et al. synthesized carboxyl-modified nitrogen-doped porous CNFs (NPCNFs-O) electrocatalyst by electrospinning[194]. The inclusion of carboxyl groups promotes the redistribution of local charges within the N-doped carbon framework, thus optimizing the intermediate adsorption energy [Figure 10E].
Figure 10. (A) Comparative analysis of catalytic activity in heteroatom-doped GPs pre- and post-activation; CV of (B) N-GP, (C) P-GP, and (D) Se-GP after the activation (Insets illustrate corresponding ortho-quinone structures). This figure is quoted with permission from Liu et al.[193]; (E) Gibbs free energy profiles for OER pathways on three distinct catalytic models at the thermodynamic equilibrium potential (U = 0.401 V). This figure is quoted with permission from Qiang et al.[194]; (F) Schematic illustration of CoCML and CoCMM nanocluster synthesis via thermal condensation at 800 °C using melamine and formamide as precursors. This figure is quoted with permission from Kumar et al.[196]; (G) Color-mapped contour plots illustrating the correlation between limiting potential and Gibbs free energy parameters (ΔG1 and ΔG4). This figure is quoted with permission from Lei et al.[197]; (H) Computational models of graphene incorporating various oxygen-containing functional groups; (I) Computationally derived overpotential volcano plots utilizing O* and OH* adsorption energies as catalytic descriptors. This figure is quoted with permission from Lu et al.[199]. GPs: Graphite flakes; CV: cyclic voltammetry; OER: oxygen evolution reaction.
Defect engineering and element doping in carbon materials not only provide additional freedom to explore synthesis strategies but also effectively regulate the electronic structure of the active site through strong metal-carrier interactions or interface effects, ultimately improving catalytic performance. Single-atom site catalysts, characterized by maximizing atom utilization and isolating active sites, have been a very prosperous branch of catalysis research in recent years. Zhang et al. fabricated highly stable atomically dispersed Ni catalysts on DG (A-Ni@DG) by initial wet impregnation and subsequent acid leaching, in which Ni atoms were captured by di-vacancy[195]. The interaction between Ni@Di-vacancy and the OER adsorption intermediate was optimal. In order to achieve higher catalytic activity, more active sites are often needed; that is, the development of high-density single-atom catalysts is required. The isolated atomic density of M-Nx-C SACs synthesized at high temperature is about 1%-1.5%, and the increase of metal content leads to agglomeration, which limits the availability of single atomic sites. Kumar et al. developed a synthetic strategy for fabricating high-density single-atom cobalt catalysts (10.6 wt%, 3.18 at%) with pyridinic coordination, embedded within nitrogen-enriched carbon matrices through condensation of CoPc tetramers and melem derivatives (CoMM) [Figure 10F][196]. Due to their unique electronic structure, uniform active centers, and nearly 100% atomic utilization rate, monatomic catalysts have shown excellent catalytic activity and great application prospects. In addition to increasing the density of monatomic, the use of multi-element active center monatomic can often provide more types of active centers, and the synergistic effect between adjacent atoms is also beneficial to improve its electrocatalytic activity. Lei et al. developed a new strategy to enhance the efficiency of OER by de-stabilizing high-entropic catalytic environments through π-networks for sustainable energy conversion [Figure 10G], successfully synthesizing a high entropy single atom (HESA) catalyst containing five transition metals (Fe, Mn, Co, Ni, and Cu) coordinated with dual N source[197]. The synergistic effect of entropy-stabilized architecture and robust interfacial coupling between monoatomic sites and the carbon matrix endows the HESA catalyst with exceptional stability and catalytic activity.
In acidic media, precious metals Ir, Ru, and carbon materials are often used as catalyst carriers. However, carbon-based support materials are easily oxidized under the harsh operating conditions of PEM electrolyzers. Graphite phase carbon nitride (g-C3N4) supported catalysts with high physicochemical stability under harsh acidic oxygen evolution conditions have attracted much attention. Incorporation of boron into CNx can improve thermal stability and oxidation resistance. Boron-containing carbon nitride (BCN) exhibits a lower charge transport impedance than g-C3N4, fixing Ru nanoclusters on highly stable BCN nanosheet carriers[198]. The higher electronegativity of boron-nitrogen (B–N) bonds compared to carbon-nitrogen (C–N) bonds enables boron-mediated activation of nitrogen sites, thereby strengthening Ru-N coordination interactions. This enhanced coordination contributes to improved durability of ruthenium-based catalysts under high-potential and corrosive operational environments. The oxidation of carbon materials in acidic electrolytes (0.5M H2SO4) is summarized as follows:
C + 2H2O → CO2 + 4H+ + 4e-, E0 = 0.207 V vs. RHE
C + H2O → CO2 + 2H+ + 2e-, E0 = 0.518 V vs. RHE
It can be seen that carbon materials are easily oxidized to CO2 in acidic OER at lower potential. The oxidation rate of carbon-based materials is related to their intrinsic properties, especially when surface oxidation produces oxygen-containing functional groups. Oxygen-containing functionalized carbon materials can greatly increase the theoretical oxidation potential of carbon. Lu et al. designed oxygen-containing functional groups on the surface of carbon materials by controlling the electrochemical oxidation process using concentrated nitric acid treatment and high-current-density electrochemical oxidation, where they precisely regulated the degree of oxidation and tailored the distribution of functional groups including carbonyl, hydroxyl, carboxyl, and epoxy groups [Figure 10H][199]. The oxygen-containing functional groups not only mitigate graphitic carbon oxidation but also enhance the kinetics of acidic OER, and it is confirmed theoretically and experimentally that the phenanthrenequinone group is the most active and stable group on graphite [Figure 10I].
This section underscores significant progress in engineering carbon-based electrocatalytic materials, with a particular emphasis on the HER and the OER. A key focus has been on reducing or eliminating the reliance on precious metals while preserving or enhancing catalytic activity, which is essential for improving the economic feasibility of these technologies. The investigation of single-atom catalysts, heteroatom doping, and defect engineering represents innovative strategies for enhancing catalytic performance. These advancements in carbon-based electrocatalysts hold significant promise for increasing the efficiency and cost-effectiveness of water splitting and other energy conversion technologies.
Future research in this area could prioritize the scaling up of these novel materials for practical applications and further optimization of their performance and stability in real-world conditions. This would involve addressing challenges related to the synthesis, integration, and long-term durability of these materials to ensure their viability for widespread industrial use.
CONCLUSION AND OUTLOOK
In summary, carbon-based electrocatalysts, due to their excellent activity and low cost, have great potential in replacing expensive rare precious metal catalysts. This article reviews the theoretical and experimental research progress of carbon electrocatalysts, including M-N-C, heteroatom-doped carbon, defective carbon materials, and synergistic heteroatom and defect on carbon materials. These electrocatalysts show good catalytic activity in different electrochemical reactions. M-N-C, heteroatom doping, topological carbon defects, and complex carbon defects significantly enhance electrocatalytic performance through electronic structure modulation, charge redistribution optimization, adsorption energy tuning and band structure, and conductivity improvement. Different synthesis strategies are used to control the production of electrocatalysts with specific active site types, such as pyrolysis, gas deposition, plasma irradiation, hydrothermal method, and ball milling. Although significant research progress has been made in electrocarbon-based catalysts for different reactions, several critical challenges remain to be addressed in future research endeavors.
1. Exploring new carbon materials and developing new electrocatalytic reactions: In addition to traditional materials such as graphene, CB, and CNTs, emerging carbon nanostructures such as graphene, fullerenes, and carbon dots can also be applied to electrocatalysis[200-202]. These new carbon materials have higher electronic conductivity and stability, positioning them as promising candidates for next generation of electrocatalytic materials[203]. Integrating machine learning with the design and preparation of carbon material catalysts presents a formidable approach to catalyst development. By employing model screening to identify optimal structures and preparation methods, this synergy can substantially inform and advance the design and development of catalysts. Machine learning algorithms can analyze vast datasets to discern patterns and predict the most effective configurations, thereby streamlining the research process and reducing the time and resources required for experimental trials. This integration not only enhances the precision of catalyst design but also accelerates the discovery of innovative materials with high catalytic performance. In addition to their applications in fuel cells and electrolysis of water, carbon materials can also be used in new electrocatalytic reactions, such as electrochemical reduction reactions in organic synthesis[204]. By studying the electrocatalytic properties of carbon materials, high-efficiency and low-energy organic synthesis can be realized.
2. Methodologies in precise synthesis and identification for carbon materials with a unique defect: A comprehensive understanding of the mechanisms underlying defect formation is essential, as is the exploration of novel synthesis pathways[31]. This necessitates an in-depth examination of the microstructure of carbon materials, including the bonding patterns between carbon atoms, lattice configurations, and electron density distributions. By combining theoretical calculations with experimental validation, it is possible to elucidate the thermodynamic and kinetic processes that govern defect formation. Such insights can inform and direct the development of innovative synthesis methods, ultimately enhancing the design and optimization of carbon-based materials for various applications. There is a need to develop dynamic observations of the electrocatalytic process, identify the actual active centers, and reveal the catalytic mechanism through operando characterization[205]. To comprehensively reveal the catalytic mechanism, dynamic observation of the electrocatalytic process is essential. This can be achieved through operando characterization techniques, which involve real-time monitoring of the catalyst’s structure and performance changes during the catalytic reaction. These techniques allow for direct visualization of active center evolution and the dynamic process of catalytic reactions, providing compelling evidence for elucidating the catalytic mechanism. In summary, the precise synthesis and identification of single-defect carbon materials is a complex, systematic process requiring the integration of various theoretical calculations and experimental characterization techniques. By deepening our understanding of defect formation mechanisms, exploring novel synthesis pathways, accurately identifying and locating individual defects, and conducting dynamic observations, we can establish a robust theoretical foundation and experimental support for developing high-performance electrocatalysts.
3. The relationship between the active sites of carbon materials and their electrocatalytic performance: The diverse types and densities of active sites exert varying influences on the electrocatalytic performance of carbon materials[13]. To bridge the gap between theoretical predictions and experimental observations, it is imperative to develop precise computational models. These models should accurately capture the atomic structure and electronic characteristics of active sites, as well as their interactions with the surrounding environment, including solvents and electrolytes[164]. By leveraging machine learning, an effective database can be established to evaluate the potential application value of different active sites and concentrations across various electrocatalytic reactions, such as oxygen reduction, hydrogen evolution, carbon dioxide reduction, and nitrogen reduction.
Furthermore, precise atomic-scale engineering of metal-coordination architectures, along with the synergistic effects of heteroatoms and topological defects in carbon-based materials, is crucial for the development of high-performance electrocatalysts. This approach will facilitate the design of catalysts with enhanced efficiency and stability, tailored to specific reaction conditions and applications.
4. Improvement of the durability in carbon materials: In practical applications, the stability and durability of carbon materials is an important issue[206]. In the electrocatalytic process, exploring how the active sites of carbon-based materials affect the reaction paths, intermediate products and the generation of final products will be the focus of research. Through in-depth study of these mechanistic issues, a theoretical basis can be provided for the design of more efficient and stable electrocatalysts made of carbon materials. Through in-depth study of the deactivation mechanism, structural changes and causes of performance decay of carbon materials during electrocatalytic processes, effective strategies can be provided to improve the durability of defective carbon materials[207,208].
5. Scale-up in preparation for practical application: To realize the practical application of carbon material catalysts in electrocatalysis applications, it is crucial to solve the problem of scale-up preparation[209]. This includes the development of synthesis methods suitable for large-scale production, ensuring the consistency and stability of products, and reducing production costs.
DECLARATIONS
Acknowledgments
This work is supported by the Ministry of Science and Technology (MOST) of China through the key project of research & development (2021YFF0500502) and Guangdong Province Introduced Innovative and Entrepreneurial Team Program (2023ZT10L061). Yao, X. thanks to Guangdong Basic Research Center of Ex-cellence for Functional Molecular Engineering. Fu, Y. thanks the support by Fundamental Research Funds for the Central Universities, Sun Yat-sen University (76280-31610003).
Authors’ contributions
Original draft writing: Mao, Z.; Fu, Y.
Manuscript modification: Long, X.; Li, C.; Li, M.; Zhu, F.; Ye, W.; Fan, Z.
Led the preparation of the manuscript: Fu, Y.; Yao, X.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the Ministry of Science and Technology (MOST) of China through the Key Project of Research & Development (2021YFF0500502); the Guangdong Province Introduced Innovative and Entrepreneurial Team Program (2023ZT10L061); and the Fundamental Research Funds for the Central Universities, Sun Yat-sen University (76280-31610003).
Conflicts of interest
Yao, X. is Executive Editors-in-Chief of the journal Chemical Synthesis. Yao, X. was not involved in any steps of editorial processing, notably including reviewers’ selection, manuscript handling and decision-making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Gasteiger, H. A.; Marković, N. M. Chemistry. Just a dream - or future reality? Science 2009, 324, 48-9.
3. Staszak-Jirkovský, J.; Malliakas, C. D.; Lopes, P. P.; et al. Design of active and stable Co-Mo-Sx chalcogels as pH-universal catalysts for the hydrogen evolution reaction. Nat. Mater. 2016, 15, 197-203.
5. Kittner, N.; Lill, F.; Kammen, D. M. Energy storage deployment and innovation for the clean energy transition. Nat. Energy. 2017, 2, 17125.
6. Xie, C.; Yan, D.; Chen, W.; et al. Insight into the design of defect electrocatalysts: from electronic structure to adsorption energy. Mater. Today. 2019, 31, 47-68.
7. Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294-303.
8. Jiao, Y.; Zheng, Y.; Jaroniec, M.; Qiao, S. Z. Design of electrocatalysts for oxygen- and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 2015, 44, 2060-86.
9. Han, N.; Wang, Y.; Ma, L.; et al. Supported cobalt polyphthalocyanine for high-performance electrocatalytic CO2 reduction. Chem 2017, 3, 652-64.
10. Wang, X.; Sun, H.; He, T.; et al. Precisely constructing pentagon trapped single-atomic iron sites in defective carbon for efficient oxygen electroreduction. Chem. Catal. 2024, 4, 101007.
11. Guo, F.; Macdonald, T. J.; Sobrido, A. J.; Liu, L.; Feng, J.; He, G. Recent advances in ultralow-Pt-loading electrocatalysts for the efficient hydrogen evolution. Adv. Sci. 2023, 10, e2301098.
12. Li, Y.; Chen, C.; Zhao, G.; et al. Unveiling the activity origin of electrochemical oxygen evolution on heteroatom-decorated carbon matrix. Angew. Chem. Int. Ed. Engl. 2024, 63, e202411218.
13. Wu, Q.; Yan, X.; Jia, Y.; Yao, X. Defective carbon-based materials: controllable synthesis and electrochemical applications. EnergyChem 2021, 3, 100059.
14. Wang, X.; Mao, Z.; Mao, X.; et al. Dual integrating oxygen and sulphur on surface of CoTe nanorods triggers enhanced oxygen evolution reaction. Adv. Sci. 2023, 10, e2206204.
15. Kawashima, K.; Márquez, R. A.; Smith, L. A.; et al. A review of transition metal boride, carbide, pnictide, and chalcogenide water oxidation electrocatalysts. Chem. Rev. 2023, 123, 12795-3208.
16. Jia, Y.; Zhang, L.; Gao, G.; et al. A heterostructure coupling of exfoliated Ni-Fe hydroxide nanosheet and defective graphene as a bifunctional electrocatalyst for overall water splitting. Adv. Mater. 2017, 29, 1700017.
17. Li, J.; Chen, M.; Cullen, D. A.; et al. Atomically dispersed manganese catalysts for oxygen reduction in proton-exchange membrane fuel cells. Nat. Catal. 2018, 1, 935-45.
18. Ding, S.; Yi, J.; Li, J.; et al. Nanostructure-based plasmon-enhanced Raman spectroscopy for surface analysis of materials. Nat. Rev. Mater. 2016, 1, 16021.
19. Yan, X.; Jia, Y.; Yao, X. Defects on carbons for electrocatalytic oxygen reduction. Chem. Soc. Rev. 2018, 47, 7628-58.
20. Ortiz-Medina, J.; Wang, Z.; Cruz-Silva, R.; et al. Defect engineering and surface functionalization of nanocarbons for metal-free catalysis. Adv. Mater. 2019, 31, e1805717.
21. Wang, J.; Kong, H.; Zhang, J.; Hao, Y.; Shao, Z.; Ciucci, F. Carbon-based electrocatalysts for sustainable energy applications. Prog. Mater. Sci. 2021, 116, 100717.
22. Fan, L.; Liu, P. F.; Yan, X.; et al. Atomically isolated nickel species anchored on graphitized carbon for efficient hydrogen evolution electrocatalysis. Nat. Commun. 2016, 7, 10667.
23. Jorge, A. B.; Jervis, R.; Periasamy, A. P.; et al. 3D carbon materials for efficient oxygen and hydrogen electrocatalysis. Adv. Energy. Mater. 2020, 10, 1902494.
24. Guo, J.; Huo, J.; Liu, Y.; et al. Nitrogen-doped porous carbon supported nonprecious metal single-atom electrocatalysts: from synthesis to application. Small. Methods. 2019, 3, 1900159.
25. Zhao, W.; Yuan, P.; She, X.; et al. Sustainable seaweed-based one-dimensional (1D) nanofibers as high-performance electrocatalysts for fuel cells. J. Mater. Chem. A. 2015, 3, 14188-94.
26. Hu, C.; Dai, L. Doping of carbon materials for metal-free electrocatalysis. Adv. Mater. 2019, 31, e1804672.
27. Singh, S. K.; Takeyasu, K.; Nakamura, J. Active sites and mechanism of oxygen reduction reaction electrocatalysis on nitrogen-doped carbon materials. Adv. Mater. 2019, 31, e1804297.
28. Hong, J.; Jin, C.; Yuan, J.; Zhang, Z. Atomic defects in two-dimensional materials: from single-atom spectroscopy to functionalities in opto-/electronics, nanomagnetism, and catalysis. Adv. Mater. 2017, 29, 1606434.
29. Hu, C.; Paul, R.; Dai, Q.; Dai, L. Carbon-based metal-free electrocatalysts: from oxygen reduction to multifunctional electrocatalysis. Chem. Soc. Rev. 2021, 50, 11785-843.
30. Zhu, J.; Mu, S. Defect engineering in carbon-based electrocatalysts: insight into intrinsic carbon defects. Adv. Funct. Mater. 2020, 30, 2001097.
31. Jia, Y.; Yao, X. Defects in carbon-based materials for electrocatalysis: synthesis, recognition, and advances. Acc. Chem. Res. 2023, 56, 948-58.
32. Guo, K.; Li, N.; Bao, L.; Zhang, P.; Lu, X. Intrinsic carbon structural imperfections for enhancing energy conversion electrocatalysts. Chem. Eng. J. 2023, 466, 143060.
33. Wang, X.; Huang, R.; Mao, X.; et al. Coupling Ni single atomic sites with metallic aggregates at adjacent geometry on carbon support for efficient hydrogen peroxide electrosynthesis. Adv. Sci. 2024, 11, e2402240.
34. Xu, X.; Pan, Y.; Zhong, Y.; et al. New undisputed evidence and strategy for enhanced lattice-oxygen participation of perovskite electrocatalyst through cation deficiency manipulation. Adv. Sci. 2022, 9, e2200530.
35. Mcbreen, J.; Olender, H.; Srinivasan, S.; Kordesch, K. V. Carbon supports for phosphoric acid fuel cell electrocatalysts: alternative materials and methods of evaluation. J. Appl. Electrochem. 1981, 11, 787-96.
36. Watanabe, M.; Sei, H.; Stonehart, P. The influence of platinum crystallite size on the electroreduction of oxygen. J. Electroanal. Chem. Interfacial. Electrochem. 1989, 261, 375-87.
37. Antolini, E. Carbon supports for low-temperature fuel cell catalysts. Appl. Catal. B. Environ. 2009, 88, 1-24.
38. Pinheiro, A. L. N.; Oliveira Neto, A.; Souza, E.; et al. Electrocatalysis on noble metal and noble metal alloys dispersed on high surface area carbon. J. New. Mater. Electrochem. Syst. 2003, 6, 1-8. http://hdl.handle.net/11449/224471. (accessed 2 Jul 2025).
39. Yan, X.; Jia, Y.; Zhang, L.; Yao, X. Platinum stabilized by defective activated carbon with excellent oxygen reduction performance in alkaline media. Chin. J. Catal. 2017, 38, 1011-20.
40. Yan, X.; Jia, Y.; Chen, J.; Zhu, Z.; Yao, X. Defective-activated-carbon-supported Mn-Co nanoparticles as a highly efficient electrocatalyst for oxygen reduction. Adv. Mater. 2016, 28, 8771-8.
41. Kim, S.; Park, S. Effect of acid/base treatment to carbon blacks on preparation of carbon-supported platinum nanoclusters. Electrochim. Acta. 2007, 52, 3013-21.
42. Luo, F.; Liao, S.; Chen, D. Platinum catalysts supported on Nafion functionalized carbon black for fuel cell application. J. Energy. Chem. 2013, 22, 87-92.
43. Sun, X.; Zhang, Y.; Song, P.; et al. Fluorine-doped carbon blacks: highly efficient metal-free electrocatalysts for oxygen reduction reaction. ACS. Catal. 2013, 3, 1726-9.
44. Kim, J. H.; Cheon, J. Y.; Shin, T. J.; Park, J. Y.; Joo, S. H. Effect of surface oxygen functionalization of carbon support on the activity and durability of Pt/C catalysts for the oxygen reduction reaction. Carbon 2016, 101, 449-57.
45. Che, G.; Lakshmi, B. B.; Fisher, E. R.; Martin, C. R. Carbon nanotubule membranes for electrochemical energy storage and production. Nature 1998, 393, 346-9.
46. Tang, H.; Chen, J.; Huang, Z.; et al. High dispersion and electrocatalytic properties of platinum on well-aligned carbon nanotube arrays. Carbon 2004, 42, 191-7.
47. Wen, Z.; Wang, Q.; Li, J. Template synthesis of aligned carbon nanotube arrays using glucose as a carbon source: Pt decoration of inner and outer nanotube surfaces for fuel-cell catalysts. Adv. Funct. Mater. 2008, 18, 959-64.
48. Liu, C.; Wang, C. C.; Kei, C. C.; Hsueh, Y. C.; Perng, T. P. Atomic layer deposition of platinum nanoparticles on carbon nanotubes for application in proton-exchange membrane fuel cells. Small 2009, 5, 1535-8.
49. Wang, X.; Li, W.; Chen, Z.; Waje, M.; Yan, Y. Durability investigation of carbon nanotube as catalyst support for proton exchange membrane fuel cell. J. Power. Sources. 2006, 158, 154-9.
50. Novoselov, K. S.; Geim, A. K.; Morozov, S. V.; et al. Two-dimensional gas of massless Dirac fermions in graphene. Nature 2005, 438, 197-200.
51. Yoo, E.; Okata, T.; Akita, T.; Kohyama, M.; Nakamura, J.; Honma, I. Enhanced electrocatalytic activity of Pt subnanoclusters on graphene nanosheet surface. Nano. Lett. 2009, 9, 2255-9.
52. Sebastián, D.; Ruíz, A. G.; Suelves, I.; et al. Enhanced oxygen reduction activity and durability of Pt catalysts supported on carbon nanofibers. Appl. Catal. B. Environ. 2012, 115-6, 269-75.
53. Joo, S. H.; Choi, S. J.; Oh, I.; et al. Ordered nanoporous arrays of carbon supporting high dispersions of platinum nanoparticles. Nature 2001, 412, 169-72.
54. Chen, X.; He, J.; Dang, W.; et al. Synthesis and electrocatalytic performance of ordered mesoporous carbons produced by a hard templating method using phenolic resol as carbon precursor. Carbon 2009, 47, 354.
55. Song, S.; Liang, Y.; Li, Z.; et al. Effect of pore morphology of mesoporous carbons on the electrocatalytic activity of Pt nanoparticles for fuel cell reactions. Appl. Catal. B. Environ. 2010, 98, 132-7.
56. Orfanidi, A.; Daletou, M.; Neophytides, S. Preparation and characterization of Pt on modified multi-wall carbon nanotubes to be used as electrocatalysts for high temperature fuel cell applications. Appl. Catal. B. Environ. 2011, 106, 379-89.
57. Kou, R.; Shao, Y.; Wang, D.; et al. Enhanced activity and stability of Pt catalysts on functionalized graphene sheets for electrocatalytic oxygen reduction. Electrochem. Commun. 2009, 11, 954-7.
58. Czerw, R.; Terrones, M.; Charlier, J.; et al. Identification of electron donor states in N-doped carbon nanotubes. Nano. Lett. 2001, 1, 457-60.
59. Maiyalagan, T.; Viswanathan, B. Template synthesis and characterization of well-aligned nitrogen containing carbon nanotubes. Mater. Chem. Phys. 2005, 93, 291-5.
60. Balgis, R.; Anilkumar, G. M.; Sago, S.; Ogi, T.; Okuyama, K. Ultrahigh oxygen reduction activity of Pt/nitrogen-doped porous carbon microspheres prepared via spray-drying. J. Power. Sources. 2013, 229, 58-64.
61. Zhang, S.; Chen, S. Enhanced-electrocatalytic activity of Pt nanoparticles supported on nitrogen-doped carbon for the oxygen reduction reaction. J. Power. Sources. 2013, 240, 60-5.
62. Melke, J.; Peter, B.; Habereder, A.; et al. Metal-support interactions of platinum nanoparticles decorated N-doped carbon nanofibers for the oxygen reduction reaction. ACS. Appl. Mater. Interfaces. 2016, 8, 82-90.
63. Ma, J.; Habrioux, A.; Luo, Y.; et al. Electronic interaction between platinum nanoparticles and nitrogen-doped reduced graphene oxide: effect on the oxygen reduction reaction. J. Mater. Chem. A. 2015, 3, 11891-904.
64. Galeano, C.; Meier, J. C.; Soorholtz, M.; et al. Nitrogen-doped hollow carbon spheres as a support for platinum-based electrocatalysts. ACS. Catal. 2014, 4, 3856-68.
65. Zhang, L.; Yang, X.; Cai, R.; et al. Air cathode of zinc-air batteries: a highly efficient and durable aerogel catalyst for oxygen reduction. Nanoscale 2019, 11, 826-32.
66. Odedairo, T.; Yan, X.; Ma, J.; et al. Nanosheets Co3O4 interleaved with graphene for highly efficient oxygen reduction. ACS. Appl. Mater. Interfaces. 2015, 7, 21373-80.
68. Gisselbrecht, J. P.; Gross, M.; Koecher, M.; Lausmann, M.; Vogel, E. Redox properties of porphycenes and metalloporphycenes as compared with porphyrins. J. Am. Chem. Soc. 1990, 112, 8618-20.
69. Li, W.; Yu, A.; Higgins, D. C.; Llanos, B. G.; Chen, Z. Biologically inspired highly durable iron phthalocyanine catalysts for oxygen reduction reaction in polymer electrolyte membrane fuel cells. J. Am. Chem. Soc. 2010, 132, 17056-8.
70. Baran, J. D.; Grönbeck, H.; Hellman, A. Analysis of porphyrines as catalysts for electrochemical reduction of O2 and oxidation of H2O. J. Am. Chem. Soc. 2014, 136, 1320-6.
71. Kadish, K. M.; Frémond, L.; Ou, Z.; et al. Cobalt(III) corroles as electrocatalysts for the reduction of dioxygen: reactivity of a monocorrole, biscorroles, and porphyrin-corrole dyads. J. Am. Chem. Soc. 2005, 127, 5625-31.
72. Nguyen-Thanh, D.; Frenkel, A. I.; Wang, J.; O’brien, S.; Akins, D. L. Cobalt–polypyrrole–carbon black (Co–PPY–CB) electrocatalysts for the oxygen reduction reaction (ORR) in fuel cells: composition and kinetic activity. Appl. Catal. B. Environ. 2011, 105, 50-60.
73. Morozan, A.; Campidelli, S.; Filoramo, A.; Jousselme, B.; Palacin, S. Catalytic activity of cobalt and iron phthalocyanines or porphyrins supported on different carbon nanotubes towards oxygen reduction reaction. Carbon 2011, 49, 4839-47.
74. Tang, H.; Yin, H.; Wang, J.; Yang, N.; Wang, D.; Tang, Z. Molecular architecture of cobalt porphyrin multilayers on reduced graphene oxide sheets for high-performance oxygen reduction reaction. Angew. Chem. Int. Ed. Engl. 2013, 52, 5585-9.
75. Lefèvre, M.; Dodelet, J. P.; Bertrand, P. Molecular oxygen reduction in PEM fuel cells: evidence for the simultaneous presence of two active sites in Fe-based catalysts. J. Phys. Chem. B. 2002, 106, 8705-13.
76. Lefèvre, M.; Dodelet, J. P.; Bertrand, P. Molecular oxygen reduction in PEM fuel cell conditions: ToF-SIMS analysis of co-based electrocatalysts. J. Phys. Chem. B. 2005, 109, 16718-24.
77. Faubert, G.; Lalande, G.; Côté, R.; et al. Heat-treated iron and cobalt tetraphenylporphyrins adsorbed on carbon black: physical characterization and catalytic properties of these materials for the reduction of oxygen in polymer electrolyte fuel cells. Electrochim. Acta. 1996, 41, 1689-701.
78. Gong, K.; Du, F.; Xia, Z.; Durstock, M.; Dai, L. Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction. Science 2009, 323, 760-4.
79. Tang, Y.; Allen, B. L.; Kauffman, D. R.; Star, A. Electrocatalytic activity of nitrogen-doped carbon nanotube cups. J. Am. Chem. Soc. 2009, 131, 13200-1.
80. Sharifi, T.; Hu, G.; Jia, X.; Wågberg, T. Formation of active sites for oxygen reduction reactions by transformation of nitrogen functionalities in nitrogen-doped carbon nanotubes. ACS. Nano. 2012, 6, 8904-12.
81. Chen, S.; Bi, J.; Zhao, Y.; et al. Nitrogen-doped carbon nanocages as efficient metal-free electrocatalysts for oxygen reduction reaction. Adv. Mater. 2012, 24, 5593-7.
82. Zhao, X.; Zhao, H.; Zhang, T.; et al. One-step synthesis of nitrogen-doped microporous carbon materials as metal-free electrocatalysts for oxygen reduction reaction. J. Mater. Chem. A. 2014, 2, 11666-71.
83. Chen, P.; Wang, L.; Wang, G.; et al. Nitrogen-doped nanoporous carbon nanosheets derived from plant biomass: an efficient catalyst for oxygen reduction reaction. Energy. Environ. Sci. 2014, 7, 4095-103.
84. Yang, L.; Shui, J.; Du, L.; et al. Carbon-based metal-free ORR electrocatalysts for fuel cells: past, present, and future. Adv. Mater. 2019, 31, e1804799.
85. Gupta, S.; Tryk, D.; Bae, I.; Aldred, W.; Yeager, E. Heat-treated polyacrylonitrile-based catalysts for oxygen electroreduction. J. Appl. Electrochem. 1989, 19, 19-27.
86. Zhang, G.; Lu, W.; Cao, F.; Xiao, Z.; Zheng, X. N-doped graphene coupled with Co nanoparticles as an efficient electrocatalyst for oxygen reduction in alkaline media. J. Power. Sources. 2016, 302, 114-25.
87. Hou, Y.; Cui, S.; Wen, Z.; Guo, X.; Feng, X.; Chen, J. Strongly coupled 3D hybrids of N-doped porous carbon nanosheet/CoNi alloy-encapsulated carbon nanotubes for enhanced electrocatalysis. Small 2015, 11, 5940-8.
88. Wang, H. F.; Chen, L.; Pang, H.; Kaskel, S.; Xu, Q. MOF-derived electrocatalysts for oxygen reduction, oxygen evolution and hydrogen evolution reactions. Chem. Soc. Rev. 2020, 49, 1414-48.
89. Meng, J.; Niu, C.; Xu, L.; et al. General oriented formation of carbon nanotubes from metal-organic frameworks. J. Am. Chem. Soc. 2017, 139, 8212-21.
90. Xie, X.; Peng, L.; Yang, H.; Waterhouse, G. I. N.; Shang, L.; Zhang, T. MIL-101-derived mesoporous carbon supporting highly exposed Fe single-atom sites as efficient oxygen reduction reaction catalysts. Adv. Mater. 2021, 33, e2101038.
91. Huang, Y.; Chen, Y.; Xu, M.; et al. Catalysts by pyrolysis: transforming metal-organic frameworks (MOFs) precursors into metal-nitrogen-carbon (M-N-C) materials. Mater. Today. 2023, 69, 66-78.
92. Gao, L.; Xiao, M.; Jin, Z.; et al. Correlating Fe source with Fe-N-C active site construction: guidance for rational design of high-performance ORR catalyst. J. Energy. Chem. 2018, 27, 1668-73.
93. Wang, X. X.; Cullen, D. A.; Pan, Y. T.; et al. Nitrogen-coordinated single cobalt atom catalysts for oxygen reduction in proton exchange membrane fuel cells. Adv. Mater. 2018, 30, 1706758.
94. Wu, J.; Zhou, H.; Li, Q.; et al. Densely populated isolated single Co-N site for efficient oxygen electrocatalysis. Adv. Energy. Mater. 2019, 9, 1900149.
95. Choi, C. H.; Park, S. H.; Woo, S. I. N-doped carbon prepared by pyrolysis of dicyandiamide with various MeCl2·xH2O (Me = Co, Fe, and Ni) composites: effect of type and amount of metal seed on oxygen reduction reactions. Appl. Catal. B. Environ. 2012, 119-20, 123-31.
96. Zitolo, A.; Goellner, V.; Armel, V.; et al. Identification of catalytic sites for oxygen reduction in iron- and nitrogen-doped graphene materials. Nat. Mater. 2015, 14, 937-42.
97. Peng, H.; Liu, F.; Liu, X.; et al. Effect of transition metals on the structure and performance of the doped carbon catalysts derived from polyaniline and melamine for ORR application. ACS. Catal. 2014, 4, 3797-805.
98. Ye, C.; Xu, L. Recent advances in the design of a high performance metal–nitrogen–carbon catalyst for the oxygen reduction reaction. J. Mater. Chem. A. 2021, 9, 22218-47.
99. Zhu, P.; Xiong, X.; Wang, X.; et al. Regulating the FeN4 moiety by constructing Fe-Mo dual-metal atom sites for efficient electrochemical oxygen reduction. Nano. Lett. 2022, 22, 9507-15.
100. Wang, B.; Tang, J.; Zhang, X.; et al. Nitrogen doped porous carbon polyhedral supported Fe and Ni dual-metal single-atomic catalysts: template-free and metal ligand-free sysnthesis with microwave-assistance and d-band center modulating for boosted ORR catalysis in zinc-air batteries. Chem. Eng. J. 2022, 437, 135295.
101. Zhai, W.; He, Y.; Duan, Y.; et al. Densely populated trimetallic single-atoms for durable low-temperature flexible zinc-air batteries. Appl. Catal. B. Environ. 2024, 342, 123438.
102. Wan, X.; Liu, Q.; Liu, J.; et al. Iron atom-cluster interactions increase activity and improve durability in Fe-N-C fuel cells. Nat. Commun. 2022, 13, 2963.
103. Li, Y.; Liu, X.; Zheng, L.; et al. Preparation of Fe–N–C catalysts with FeNx (x = 1, 3, 4) active sites and comparison of their activities for the oxygen reduction reaction and performances in proton exchange membrane fuel cells. J. Mater. Chem. A. 2019, 7, 26147-53.
104. Xue, D.; Yuan, P.; Jiang, S.; et al. Altering the spin state of Fe-N-C through ligand field modulation of single-atom sites boosts the oxygen reduction reaction. Nano. Energy. 2023, 105, 108020.
105. Zhang, N.; Zhou, T.; Ge, J.; et al. High-density planar-like Fe2N6 structure catalyzes efficient oxygen reduction. Matter 2020, 3, 509-21.
106. Zhang, L.; Jin, N.; Yang, Y.; et al. Advances on axial coordination design of single-atom catalysts for energy electrocatalysis: a review. Nanomicro. Lett. 2023, 15, 228.
108. Gong, L.; Zhang, H.; Wang, Y.; et al. Bridge bonded oxygen ligands between approximated FeN4 sites confer catalysts with high ORR performance. Angew. Chem. Int. Ed. Engl. 2020, 59, 13923-8.
109. Zhao, K.; Liu, S.; Li, Y.; et al. Insight into the mechanism of axial ligands regulating the catalytic activity of Fe–N4 sites for oxygen reduction reaction. Adv. Energy. Mater. 2022, 12, 2103588.
110. Sabhapathy, P.; Raghunath, P.; Sabbah, A.; et al. Axial chlorine induced electron delocalization in atomically dispersed FeN4 electrocatalyst for oxygen reduction reaction with improved hydrogen peroxide tolerance. Small 2023, 19, e2303598.
111. Zhang, J.; Zhao, Y.; Chen, C.; et al. Tuning the coordination environment in single-atom catalysts to achieve highly efficient oxygen reduction reactions. J. Am. Chem. Soc. 2019, 141, 20118-26.
112. Lefèvre, M.; Proietti, E.; Jaouen, F.; Dodelet, J. P. Iron-based catalysts with improved oxygen reduction activity in polymer electrolyte fuel cells. Science 2009, 324, 71-4.
113. Xu, X.; Zhang, X.; Kuang, Z.; et al. Investigation on the demetallation of Fe-N-C for oxygen reduction reaction: the influence of structure and structural evolution of active site. Appl. Catal. B. Environ. 2022, 309, 121290.
114. Liu, K.; Wu, G.; Wang, G. Role of local carbon structure surrounding FeN4 sites in boosting the catalytic activity for oxygen reduction. J. Phys. Chem. C. 2017, 121, 11319-24.
115. Liu, K.; Fu, J.; Lin, Y.; et al. Insights into the activity of single-atom Fe-N-C catalysts for oxygen reduction reaction. Nat. Commun. 2022, 13, 2075.
116. Wang, X.; Jia, Y.; Mao, X.; et al. Edge-rich Fe-N4 active sites in defective carbon for oxygen reduction catalysis. Adv. Mater. 2020, 32, e2000966.
117. Yin, H.; Yuan, P.; Lu, B.; et al. Phosphorus-driven electron delocalization on edge-type FeN4 active sites for oxygen reduction in acid medium. ACS. Catal. 2021, 11, 12754-62.
118. Tian, Y.; Li, M.; Wu, Z.; et al. Edge-hosted atomic Co-N4 sites on hierarchical porous carbon for highly selective two-electron oxygen reduction reaction. Angew. Chem. Int. Ed. Engl. 2022, 61, e202213296.
119. Liu, Y.; Zou, K.; Zhang, T.; et al. Novel honeycomb-like carbons with tunable nanopores as metal-free N, O-codoped catalysts for robust oxygen reduction. Chem. Eng. J. 2022, 433, 133560.
120. Ren, G.; Chen, S.; Zhang, J.; et al. N-doped porous carbon spheres as metal-free electrocatalyst for oxygen reduction reaction. J. Mater. Chem. A. 2021, 9, 5751-8.
121. Li, W.; Wang, J.; Jia, C.; Chen, J.; Wen, Z.; Huang, A. Covalent organic framework-derived fluorine, nitrogen dual-doped carbon as metal-free bifunctional oxygen electrocatalysts. J. Colloid. Interface. Sci. 2023, 650, 275-83.
122. Zheng, Y.; Song, H.; Chen, S.; et al. Metal-free multi-heteroatom-doped carbon bifunctional electrocatalysts derived from a covalent triazine polymer. Small 2020, 16, e2004342.
123. Li, J.; Shen, C.; Luo, J.; Pan, T.; Deng, J.; Cao, Z. Metal-organic framework-derived brain platygyra coral-like porous carbon architectures for real-time monitoring of hydrogen peroxide in biological matrices. Chem. Eng. J. 2023, 471, 144805.
124. Kanagavalli, P.; Pandey, G. R.; Bhat, V. S.; Veerapandian, M.; Hegde, G. Nitrogenated-carbon nanoelectrocatalyst advertently processed from bio-waste of Allium sativum for oxygen reduction reaction. J. Nanostruct. Chem. 2021, 11, 343-52.
125. Li, M.; Liu, Z.; Wang, F.; Xuan, J. The influence of the type of N dopping on the performance of bifunctional N-doped ordered mesoporous carbon electrocatalysts in oxygen reduction and evolution reaction. J. Energy. Chem. 2017, 26, 422-7.
126. Kim, J.; Park, J.; Lee, J.; Lim, W.; Jo, C.; Lee, J. Biomass-Derived P, N self-doped hard carbon as bifunctional oxygen electrocatalyst and anode material for seawater batteries. Adv. Funct. Mater. 2021, 31, 2010882.
127. Li, X.; Guan, B. Y.; Gao, S.; Lou, X. W. A general dual-templating approach to biomass-derived hierarchically porous heteroatom-doped carbon materials for enhanced electrocatalytic oxygen reduction. Energy. Environ. Sci. 2019, 12, 648-55.
128. Kumar, S.; Gonen, S.; Friedman, A.; Elbaz, L.; Nessim, G. D. Doping and reduction of graphene oxide using chitosan-derived volatile N-heterocyclic compounds for metal-free oxygen reduction reaction. Carbon 2017, 120, 419-26.
129. Song, L.; Wu, Z.; Liang, H.; et al. Macroscopic-scale synthesis of nitrogen-doped carbon nanofiber aerogels by template-directed hydrothermal carbonization of nitrogen-containing carbohydrates. Nano. Energy. 2016, 19, 117-27.
130. Graglia, M.; Pampel, J.; Hantke, T.; Fellinger, T. P.; Esposito, D. Nitro lignin-derived nitrogen-doped carbon as an efficient and sustainable electrocatalyst for oxygen reduction. ACS. Nano. 2016, 10, 4364-71.
131. Huang, B.; Liu, Y.; Xie, Z. Biomass derived 2D carbons via a hydrothermal carbonization method as efficient bifunctional ORR/HER electrocatalysts. J. Mater. Chem. A. 2017, 5, 23481-8.
132. Burmeister, C. F.; Kwade, A. Process engineering with planetary ball mills. Chem. Soc. Rev. 2013, 42, 7660-7.
133. Shen, X.; Zhou, Q.; Han, M.; et al. Rapid mechanochemical synthesis of polyanionic cathode with improved electrochemical performance for Na-ion batteries. Nat. Commun. 2021, 12, 2848.
134. Yuan, R.; Dong, Y.; Hou, R.; et al. Structural transformation of porous and disordered carbon during ball-milling. Chem. Eng. J. 2023, 454, 140418.
135. Morawa Eblagon, K.; Rey-Raap, N.; Figueiredo, J. L.; Pereira, M. F. R. Relationships between texture, surface chemistry and performance of N-doped carbon xerogels in the oxygen reduction reaction. Appl. Surf. Sci. 2021, 548, 149242.
136. Yang, N.; Zhu, X.; Wang, G.; et al. Pyrolysis-free mechanochemical conversion of small organic molecules into metal-free heteroatom-doped mesoporous carbons for efficient electrosynthesis of hydrogen peroxide. ACS. Mater. Lett. 2023, 5, 379-87.
137. Wang, Y.; Yu, F.; Zhu, M.; et al. N-Doping of plasma exfoliated graphene oxide via dielectric barrier discharge plasma treatment for the oxygen reduction reaction. J. Mater. Chem. A. 2018, 6, 2011-7.
138. Li, O. L.; Prabakar, K.; Kaneko, A.; Park, H.; Ishizaki, T. Exploration of Lewis basicity and oxygen reduction reaction activity in plasma-tailored nitrogen-doped carbon electrocatalysts. Catal. Today. 2019, 337, 102-9.
139. Niu, J.; Chokradjaroen, C.; Saito, N. Graphitic N-doped graphene via solution plasma with a single dielectric barrier. Carbon 2022, 199, 347-56.
140. Khan, A.; Goepel, M.; Colmenares, J. C.; Gläser, R. Chitosan-based N-doped carbon materials for electrocatalytic and photocatalytic applications. ACS. Sustain. Chem. Eng. 2020, 8, 4708-27.
141. Lai, L.; Potts, J. R.; Zhan, D.; et al. Exploration of the active center structure of nitrogen-doped graphene-based catalysts for oxygen reduction reaction. Energy. Environ. Sci. 2012, 5, 7936.
142. Guo, D.; Shibuya, R.; Akiba, C.; Saji, S.; Kondo, T.; Nakamura, J. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 2016, 351, 361-5.
143. Wang, N.; Lu, B.; Li, L.; et al. Graphitic nitrogen is responsible for oxygen electroreduction on nitrogen-doped carbons in alkaline electrolytes: insights from activity attenuation studies and theoretical calculations. ACS. Catal. 2018, 8, 6827-36.
144. Zhao, Y.; Wan, J.; Yao, H.; et al. Few-layer graphdiyne doped with sp-hybridized nitrogen atoms at acetylenic sites for oxygen reduction electrocatalysis. Nat. Chem. 2018, 10, 924-31.
145. Jiao, Y.; Zheng, Y.; Jaroniec, M.; Qiao, S. Z. Origin of the electrocatalytic oxygen reduction activity of graphene-based catalysts: a roadmap to achieve the best performance. J. Am. Chem. Soc. 2014, 136, 4394-403.
146. Choi, C. H.; Park, S. H.; Woo, S. I. Binary and ternary doping of nitrogen, boron, and phosphorus into carbon for enhancing electrochemical oxygen reduction activity. ACS. Nano. 2012, 6, 7084-91.
147. Chao, G.; Zhang, L.; Wang, D.; et al. Activation of graphitic nitrogen sites for boosting oxygen reduction. Carbon 2020, 159, 611-6.
148. Wu, K.; Wang, D.; Zeng, Q.; Li, Y.; Gentle, I. R. Solution phase synthesis of halogenated graphene and the electrocatalytic activity for oxygen reduction reaction. Chin. J. Catal. 2014, 35, 884-90.
149. Gong, T.; Qi, R.; Liu, X.; Li, H.; Zhang, Y. N, F-codoped microporous carbon nanofibers as efficient metal-free electrocatalysts for ORR. Nanomicro. Lett. 2019, 11, 9.
150. Xiang, F.; Zhao, X.; Yang, J.; et al. Enhanced selectivity in the electroproduction of H2O2 via F/S dual-doping in metal-free nanofibers. Adv. Mater. 2023, 35, e2208533.
151. Li, Y.; Zhang, H.; Wang, Y.; et al. A self-sponsored doping approach for controllable synthesis of S and N co-doped trimodal-porous structured graphitic carbon electrocatalysts. Energy. Environ. Sci. 2014, 7, 3720-6.
152. Liu, J.; Song, P.; Ruan, M.; Xu, W. Catalytic properties of graphitic and pyridinic nitrogen doped on carbon black for oxygen reduction reaction. Chin. J. Catal. 2016, 37, 1119-26.
153. Li, L.; Tang, C.; Zheng, Y.; et al. Tailoring selectivity of electrochemical hydrogen peroxide generation by tunable pyrrolic-nitrogen-carbon. Adv. Energy. Mater. 2020, 10, 2000789.
154. Iglesias, D.; Giuliani, A.; Melchionna, M.; et al. N-doped graphitized carbon nanohorns as a forefront electrocatalyst in highly selective O2 reduction to H2O2. Chem 2018, 4, 106-23.
155. Zhang, T.; Wang, H.; Zhang, J.; et al. Carbon charge population and oxygen molecular transport regulated by program-doping for highly efficient 4e-ORR. Chem. Eng. J. 2022, 444, 136560.
156. Cheng, J.; Lyu, C.; Li, H.; et al. Steering the oxygen reduction reaction pathways of N-carbon hollow spheres by heteroatom doping. Appl. Catal. B. Environ. 2023, 327, 122470.
157. Chen, S.; Luo, T.; Chen, K.; et al. Chemical identification of catalytically active sites on oxygen-doped carbon nanosheet to decipher the high activity for electro-synthesis hydrogen peroxide. Angew. Chem. Int. Ed. Engl. 2021, 60, 16607-14.
158. Koh, K. H.; Kim, Y. J.; Mostaghim, A. H. B.; Siahrostami, S.; Han, T. H.; Chen, Z. Elaborating nitrogen and oxygen dopants configurations within graphene electrocatalysts for two-electron oxygen reduction. ACS. Mater. Lett. 2022, 4, 320-8.
159. Zhao, H.; Sun, C.; Jin, Z.; et al. Carbon for the oxygen reduction reaction: a defect mechanism. J. Mater. Chem. A. 2015, 3, 11736-9.
160. Zhao, X.; Zou, X.; Yan, X.; et al. Defect-driven oxygen reduction reaction (ORR) of carbon without any element doping. Inorg. Chem. Front. 2016, 3, 417-21.
161. Tao, L.; Qiao, M.; Jin, R.; et al. Bridging the surface charge and catalytic activity of a defective carbon electrocatalyst. Angew. Chem. Int. Ed. Engl. 2019, 58, 1019-24.
162. Jia, Y.; Zhang, L.; Zhuang, L.; et al. Identification of active sites for acidic oxygen reduction on carbon catalysts with and without nitrogen doping. Nat. Catal. 2019, 2, 688-95.
163. Wang, X.; Jia, Y.; Mao, X.; et al. A directional synthesis for topological defect in carbon. Chem 2020, 6, 2009-23.
164. Li, N.; Li, M.; Guo, K.; et al. Deciphering the role of native defects in dopant-mediated defect engineering of carbon electrocatalysts. Adv. Energy. Mater. 2024, 14, 2401008.
165. Wu, Q.; Jia, Y.; Liu, Q.; et al. Ultra-dense carbon defects as highly active sites for oxygen reduction catalysis. Chem 2022, 8, 2715-33.
166. Jia, Y.; Zhang, L.; Du, A.; et al. Defect graphene as a trifunctional catalyst for electrochemical reactions. Adv. Mater. 2016, 28, 9532-8.
167. Zhu, J.; Li, W.; Li, S.; et al. Defective N/S-codoped 3D cheese-like porous carbon nanomaterial toward efficient oxygen reduction and Zn-Air batteries. Small 2018, 14, e1800563.
168. Zhu, J.; Huang, Y.; Mei, W.; et al. Effects of intrinsic pentagon defects on electrochemical reactivity of carbon nanomaterials. Angew. Chem. Int. Ed. Engl. 2019, 58, 3859-64.
169. Tang, C.; Wang, H. F.; Chen, X.; et al. Topological defects in metal-free nanocarbon for oxygen electrocatalysis. Adv. Mater. 2016, 28, 6845-51.
170. Shi, P.; Si, D.; Yao, M.; et al. Spiral effect of helical carbon nanorods boosting electrocatalysis of oxygen reduction reaction. Sci. China. Mater. 2022, 65, 1531-8.
171. Jiang, Y.; Yang, L.; Sun, T.; et al. Significant contribution of intrinsic carbon defects to oxygen reduction activity. ACS. Catal. 2015, 5, 6707-12.
172. Srinivas, K.; Liu, D.; Ma, F.; et al. Defect-engineered mesoporous undoped carbon nanoribbons for benchmark oxygen reduction reaction. Small 2023, 19, e2301589.
173. Xia, H.; Pang, R.; Dong, X.; et al. Boosting oxygen reduction reaction kinetics by designing rich vacancy coupling pentagons in the defective carbon. J. Am. Chem. Soc. 2023, 145, 25695-704.
174. Wang, X.; Han, C.; Han, Y.; et al. Highly curved defect sites: how curvature effect influences metal-free defective carbon electrocatalysts. Small 2024, 20, e2401447.
175. Zhang, J.; Sun, Y.; Zhu, J.; et al. Defect and pyridinic nitrogen engineering of carbon-based metal-free nanomaterial toward oxygen reduction. Nano. Energy. 2018, 52, 307-14.
176. Fernandez-Escamilla, H. N.; Guerrero-Sanchez, J.; Contreras, E.; et al. Understanding the selectivity of the oxygen reduction reaction at the atomistic level on nitrogen-doped graphitic carbon materials. Adv. Energy. Mater. 2021, 11, 2002459.
177. Yan, X.; Liu, H.; Jia, Y.; et al. Clarifying the origin of oxygen reduction activity in heteroatom-modified defective carbon. Cell. Rep. Phys. Sci. 2020, 1, 100083.
178. Li, D.; Jia, Y.; Chang, G.; et al. A defect-driven metal-free electrocatalyst for oxygen reduction in acidic electrolyte. Chem 2018, 4, 2345-56.
179. Wu, Q.; Zou, H.; Mao, X.; et al. Unveiling the dynamic active site of defective carbon-based electrocatalysts for hydrogen peroxide production. Nat. Commun. 2023, 14, 6275.
180. Song, H.; Wu, M.; Tang, Z.; Tse, J. S.; Yang, B.; Lu, S. Single atom ruthenium-doped CoP/CDs nanosheets via splicing of carbon-dots for robust hydrogen production. Angew. Chem. Int. Ed. Engl. 2021, 60, 7234-44.
181. Lv, Y.; Wu, X.; Lin, H.; et al. A novel carbon support: few-layered graphdiyne-decorated carbon nanotubes capture metal clusters as effective metal-supported catalysts. Small 2021, 17, e2006442.
182. Li, D.; Cheng, H.; Hao, X.; et al. Wood-derived freestanding carbon-based electrode with hierarchical structure for industrial-level hydrogen production. Adv. Mater. 2024, 36, e2304917.
183. Liu, W.; Ji, J.; Yan, X.; et al. A cascade surface immobilization strategy to access high-density and closely distanced atomic Pt sites for enhancing alkaline hydrogen evolution reaction. J. Mater. Chem. A. 2020, 8, 5255-62.
184. Yang, Q.; Liu, H.; Yuan, P.; et al. Single carbon vacancy traps atomic platinum for hydrogen evolution catalysis. J. Am. Chem. Soc. 2022, 144, 2171-8.
185. Li, T.; Ren, S.; Zhang, C.; et al. Cobalt single atom anchored on N-doped carbon nanoboxes as typical single-atom catalysts (SACs) for boosting the overall water splitting. Chem. Eng. J. 2023, 458, 141435.
186. Liu, X.; Deng, Y.; Zheng, L.; Kesama, M. R.; Tang, C.; Zhu, Y. Engineering low-coordination single-atom cobalt on graphitic carbon nitride catalyst for hydrogen evolution. ACS. Catal. 2022, 12, 5517-26.
187. Zhang, L.; Jia, Y.; Liu, H.; et al. Charge polarization from atomic metals on adjacent graphitic layers for enhancing the hydrogen evolution reaction. Angew. Chem. Int. Ed. Engl. 2019, 58, 9404-8.
188. Zhang, Y.; Yun, S.; Dang, J.; et al. Defect engineering via ternary nonmetal doping boosts the catalytic activity of ZIF-derived carbon-based metal-free catalysts for photovoltaics and water splitting. Mater. Today. Phys. 2022, 27, 100785.
189. Lu, S.; Cheng, C.; Shi, Y.; Wu, Y.; Zhang, Z.; Zhang, B. Unveiling the structural transformation and activity origin of heteroatom-doped carbons for hydrogen evolution. Proc. Natl. Acad. Sci. U. S. A. 2023, 120, e2300549120.
190. Zhang, K.; Zou, R. Advanced transition metal-based OER electrocatalysts: current status, opportunities, and challenges. Small 2021, 17, e2100129.
191. Fan, X.; Pang, Q.; Yi, S.; et al. Intrinsic-structural-modulated carbon cloth as efficient electrocatalyst for water oxidation. Appl. Catal. B. Environ. 2021, 292, 120152.
192. Zhang, J.; Zhao, Z.; Xia, Z.; Dai, L. A metal-free bifunctional electrocatalyst for oxygen reduction and oxygen evolution reactions. Nat. Nanotechnol. 2015, 10, 444-52.
193. Lu, S.; Shi, Y.; Zhou, W.; Zhang, Z.; Wu, F.; Zhang, B. Dissolution of the heteroatom dopants and formation of ortho-quinone moieties in the doped carbon materials during water electrooxidation. J. Am. Chem. Soc. 2022, 144, 3250-8.
194. Qiang, F.; Feng, J.; Wang, H.; et al. Oxygen engineering enables N-doped porous carbon nanofibers as oxygen reduction/evolution reaction electrocatalysts for flexible zinc–air batteries. ACS. Catal. 2022, 12, 4002-15.
195. Zhang, L.; Jia, Y.; Gao, G.; et al. Graphene defects trap atomic Ni species for hydrogen and oxygen evolution reactions. Chem 2018, 4, 285-97.
196. Kumar, P.; Kannimuthu, K.; Zeraati, A. S.; et al. High-density cobalt single-atom catalysts for enhanced oxygen evolution reaction. J. Am. Chem. Soc. 2023, 145, 8052-63.
197. Lei, X.; Tang, Q.; Zheng, Y.; et al. High-entropy single-atom activated carbon catalysts for sustainable oxygen electrocatalysis. Nat. Sustain. 2023, 6, 816-26.
198. Bai, X.; Zhang, X.; Sun, Y.; et al. Low ruthenium content confined on boron carbon nitride as an efficient and stable electrocatalyst for acidic oxygen evolution reaction. Angew. Chem. Int. Ed. Engl. 2023, 62, e202308704.
199. Lu, S.; Zhou, W.; Shi, Y.; Liu, C.; Yu, Y.; Zhang, B. Phenanthrenequinone-like moiety functionalized carbon for electrocatalytic acidic oxygen evolution. Chem 2022, 8, 1415-26.
200. Xie, L.; Liang, C.; Wu, Y.; et al. Isomerization engineering of oxygen-enriched carbon quantum dots for efficient electrochemical hydrogen peroxide production. Small 2024, 20, e2401253.
201. Zhang, C.; Shen, W.; Guo, K.; Xiong, M.; Zhang, J.; Lu, X. A pentagonal defect-rich metal-free carbon electrocatalyst for boosting acidic O2 reduction to H2O2 production. J. Am. Chem. Soc. 2023, 145, 11589-98.
202. Zhu, D.; Wu, H. W.; Fong, W. K.; Tabor, R. F.; Zhang, J. The ratio of sp2 and sp3 hybridized carbon determines the performance of carbon-based catalysts in H2O2 electrosynthesis from O2. Angew. Chem. Int. Ed. Engl. 2025, 64, e202500145.
203. Wang, H.; Chen, K.; Lu, Z.; et al. Nonmetallic high-entropy-engineered nanocarbons for advanced ORR electrocatalysis. Angew. Chem. Int. Ed. Engl. 2025, 64, e202501290.
204. Lu, X.; Yao, Z. C.; Ma, X.; et al. Multiple secondary bond-mediated C-N coupling over N-doped carbon electrocatalysts. J. Am. Chem. Soc. 2025, 147, 19342-52.
205. Luo, X.; Wei, W.; Xu, Y.; et al. Neighboring carbon defects enhanced molecular oxygen activation of cobalt single atom catalysts toward efficient aerobic alcohols oxidation. Angew. Chem. Int. Ed. Engl. 2025, 64, e202502430.
206. Zhang, Y.; Yu, C.; Song, X.; et al. Defect-enabled local high-temperature field within carbon to promote in-plane integration of an electrocatalyst for CO2-to-CO conversion. Energy. Environ. Sci. 2025, 18, 1331-42.
207. Huang, Z.; Yao, Y.; Pang, Z.; et al. Direct observation of the formation and stabilization of metallic nanoparticles on carbon supports. Nat. Commun. 2020, 11, 6373.
208. Ma, R.; Chen, Y.; Zhang, B.; et al. Defect-repair in carbon for fast and stable potassium and sodium storage. Chem. Eng. J. 2025, 512, 162509.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].