


Variation spectrum and reevaluation of selected VUS of ATP7B in a Chinese Wilson disease cohort
 0
0 
Abstract
Aim: The clinical diagnosis of Wilson disease (WD) remains challenging due to ethnic and phenotypic heterogeneity, as well as the high prevalence of variants of unknown significance (VUS). This study aimed to update the genetic variation spectrum in the Chinese population and reevaluate a subset of VUS to improve the accuracy of genetic diagnosis.
Methods: Patients suspected of having WD and known non-WD controls were selected and underwent ATP7B-targeted sequencing. The variant spectrum was analyzed. Recurrent VUS, particularly those associated with very low ceruloplasmin levels, were further evaluated based on mutational characteristics, including pathogenic variant linkage disequilibrium and zygosity.
Results: We analyzed a total of 679 WD suspects and 251 controls. Of these, 292 patients (282 cases) carried disease-causing variants or VUS. Demographic characteristics were comparable between groups. The most recurrent pathogenic variants - p.Arg778Leu, p.Pro992Leu, and p.Ala874Val - differed significantly from those observed in the Caucasian population. Among the 79 detected VUS, 26 were novel. The most frequent VUS included p.Leu770=, p.Val1297Ile, p.Val1106Ile, p.Ile929Val, p.Ala1003=, p.Ala476Thr, and p.Asp196Glu. Following reevaluation of 9 VUS, we classified p.Val1106Ile, the splice-site variant c.1543+40G>A, p.Ala1018Val, p.Leu1088Ser, p.Gly1335Arg, and the synonymous variants p.Ile1338= and p.Ala1003= as pathogenic candidates. Notably, p.Val1106Ile frequently co-occurred with pathogenic mutations, while p.Leu770= was reclassified as a tag-only variant.
Conclusion: Our study reveals a distinct mutation spectrum in the Chinese population, providing evidence supporting the classification of VUS. The unique mutational patterns observed at specific loci suggest their potential structural significance within the ATP7B protein.
Keywords
INTRODUCTION
Wilson's disease (WD) is a rare[1] autosomal recessive disorder of copper metabolism caused by mutations in the ATP7B gene, which is essential for biliary copper excretion. Early diagnosis is crucial for effective management. Despite being a relatively well-recognized rare condition, WD continues to pose significant diagnostic challenges[2], both clinically and genetically. The understanding of WD is evolving, with emerging features such as ceruloplasmin-normal WD and steatosis increasingly being recognized[3,4]. However, these newer clinical and biochemical manifestations have not yet been incorporated into the decades-old Leipzig[5] diagnostic scoring system, potentially leading to delayed or missed diagnoses.
Genetic testing is typically pursued only after clinical suspicion arises. Although it has become a cornerstone for diagnosing WD, particularly in patients with ambiguous biochemical profiles or atypical presentations, it has several limitations. These challenges stem partly from the lack of early clinical suspicion and the extensive allelic heterogeneity of the ATP7B gene. The Human Gene Mutation Database (HGMD) has documented numerous mutations, many of which are rare and population-specific, further complicating variant interpretation[6]. This genetic complexity, coupled with the high prevalence of variants of unknown significance (VUS), often results in diagnostic uncertainty and treatment delays. Consequently, while clinical and biochemical assessments remain essential, the accurate interpretation of genetic findings increasingly depends on robust variant classification. The ClinGen Expert Panels, a leading initiative in clinical genomics[7], have established Variant Curation Expert Panels (VCEPs) for numerous genes associated with monogenic disorders to ensure consistent and evidence-based classification of variants. Surprisingly, there is no dedicated VCEP for the ATP7B gene. This lack of centralized, up-to-date curation resources has hindered progress; for instance, the University of Alberta’s ATP7B database (www.medicalgenetics.med.ualberta.ca/wilson/index.php), once a valuable tool, was discontinued over a decade ago. This absence reflects a broader underrepresentation of rare disease genes in formal curation frameworks, even for disorders with relatively high clinical awareness and available treatment options.
Among the 3,469 variants submitted to ClinVar (as of October 27, 2025), 594 are confirmed pathogenic variants, and 92 are benign, while the clinical significance of the remaining variants, which constitute the majority, remains undetermined. Furthermore, the majority of confirmed pathogenic variants are not specific to Asian populations. In China[8], where the genetic landscape of WD may differ substantially from that of Western populations, this lack of population-specific data impedes
Recent efforts by Chinese researchers have provided both in silico and experimental evidence[10]. However, reevaluating VUS requires molecular case-control studies. Unfortunately, such studies are rare in the context of rare diseases. Therefore, analyses often rely on real-world data, which typically require extensive data cleaning and curation, a step that is frequently neglected. Moreover, even when controls are used, they are poorly defined or overlooked due to diagnostic challenges. The absence of well-matched controls can perpetuate a vicious cycle of uncertainty. Additionally, the current diagnostic system is not up-to-date[3,4,11]. Restricting analyses to only diagnosed cases risks false-negative results in identifying pathogenic variants.
To address these critical gaps, we conducted a molecular case-control study in a large Chinese cohort. By integrating population-specific data, biochemical profiles, and mutational characteristics, we aimed to reevaluate commonly observed and selected VUS, identify mutation-specific patterns, and ultimately enhance the clinical utility of genetic testing for WD.
METHODS
Study population
Cases were defined as individuals suspected of having subclinical WD without other definitive differential diagnoses. These individuals were expected to be enriched for pathogenic VUS, as they represent potential WD cases with incomplete phenotypic expression. Suspicions were primarily based on clinical manifestations (ocular, hepatic, or neurological), biochemical abnormalities (low serum ceruloplasmin, reduced copper levels, or elevated urinary copper excretion), a positive hepatocyte copper stain, or adherence to the Leipzig criteria and international guidelines for WD[12-16]. Additionally, unexplained or early-onset steatosis was considered a qualifying feature[4]. Briefly, patients without an alternative diagnosis who met at least one of the Leipzig criteria or exhibited unexplained steatosis were classified as cases. ATP7B genotype information was not available before genetic testing. The differential diagnosis included common viral hepatitis, hepatocellular carcinoma, alcohol-related liver disease, autoimmune disorders, and other neurological conditions.
Controls comprised randomly selected patients with diagnoses other than WD, including viral hepatitis, alcohol-related liver disease, autoimmune diseases, drug-induced liver injury, solid tumors, hematologic malignancies, Gilbert syndrome, hemochromatosis, Coombs-positive anemia, thalassemia, and other non-WD conditions. Expanding the case cohort further reduces the likelihood of undiagnosed cases in the control group. Randomization was performed using the RANDBETWEEN() function in Excel, with each control patient assigned a blinded identification number.
The patient inclusion period spanned from January 2015 to October 2025. This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Shanghai Ruijin Hospital (approval number: 201617/201848).
Genetic analysis
Genomic DNA was extracted from peripheral whole blood samples. The entire coding and adjacent non-coding regions of the ATP7B gene were sequenced using either Sanger sequencing or next-generation exome sequencing. The choice was guided by cost-effectiveness, as detection rates were comparable between the two methods. In exome sequencing, the targeted regions achieved > 99.9% coverage, with a mean depth exceeding 200×. The percentage of the targeted area with a mean depth greater than 30× exceeded 99%. Key variants were validated by Sanger sequencing. Primer sequences are available in our previous publication[17].
Clinical and biochemical assessment
Ceruloplasmin levels, liver function tests, complete blood cell counts, and hemoglobin levels were assessed and included in the analysis due to their relative objectivity. This approach minimizes inter-operator variability compared to subjective clinical descriptions or examinations. Serum ceruloplasmin was measured using commercial reagents on an automatic biochemical analyzer (AU5800, Beckman Coulter, USA), with a lower limit of normal set at 200 mg/L.
Initial variant classification
Variants were initially classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines[18]. Automated variant classification was performed using a platform that integrates more than 20 databases to comprehensively annotate variants, including Human Phenotype Ontology (HPO)[19], ClinVar[20], OMIM[21,22], the 1000 Genomes Project[23], gnomAD (ExAC data assimilated)[24], and dbSNP[25]. These databases provide scoring modification functionality for 10 of the 28 manually scored evidence items in the ACMG guidelines. Based on the automated interpretation results, we conducted additional manual annotation.
Given the potential limitations of applying universal criteria to specific disorders[26], we adopted a more stringent classification approach: variants classified as unequivocally pathogenic by ACMG and as disease-causing mutations (DM) in the HGMD database were considered confirmed pathogenic variants. All other variants were classified as VUS. Variants not reported in public databases as of 2025 were deemed novel. In the absence of confirmed paternity and maternity, these novel variants were assigned the PM2 (Moderate) evidence criterion within the ACMG framework.
VUS selection and reevaluation
Variant-level selection was conducted in close alignment with ongoing developments in population databases, genomic curation, and bioinformatics, as well as our internal medical, allelic, and locus-specific knowledge. According to ACMG guidelines, variants with a significantly higher prevalence in affected individuals compared to controls provide strong evidence of pathogenicity (PS4 criterion). Similarly, the absence of a variant in controls (or, if recessive, at extremely low frequency) in major population datasets constitutes moderate evidence of pathogenicity. To enhance interpretive power, we incorporated in-house disease controls into our assessment.
To reevaluate WD-specific VUS, we established a threshold based on serum ceruloplasmin level. Among the various biochemical markers used in WD diagnosis, ceruloplasmin levels are relatively objective and well validated[12]. Given that levels below 100 mg/L are strongly indicative of WD[27], we included VUS associated with such low ceruloplasmin concentrations, as well as cases in which only VUS were identified (i.e., no pathogenic variants detected). Recurrent VUS were also considered. Each selected VUS was reviewed on a case-by-case basis. For in silico splicing prediction, SpliceAI (https://spliceailookup.broadinstitute.org/) was used.
Statistical analysis
Data analysis was performed using R version 4.5.2 (released October 2025) to assess associations and differences. All statistical tests were two-sided, and P-values < 0.05 were considered statistically significant. Normality of continuous variables was evaluated using the Shapiro-Wilk test, supplemented by visual inspection of histograms to assess distributional assumptions. Continuous variables were reported as mean ± standard deviation. Variables that deviated from normality were presented as medians (interquartile ranges, IQRs) and analyzed using non-parametric tests. Categorical variables were summarized as frequencies or percentages.
Differences in demographic and clinical characteristics between cases and controls, as well as within subgroups, were assessed using the Welch two-sample t-test or Mann-Whitney U test for continuous variables (age, protein levels, and red blood cell counts), as appropriate, and Pearson’s chi-square test with Yates’ continuity correction for categorical variables (sex). To compare the prevalence of molecular alterations between WD suspects and controls, logistic regression models were employed to assess the association between specific genetic variants and suspect status, adjusting for age and sex as confounders. Odds ratios (ORs) and corresponding 95% confidence intervals (CIs) were calculated to assess the strength of association between VUS in the case and control groups; results are presented in Supplementary Table 1. The detection of mutation only in cases and its absence in all controls, including both general and in-house study controls, was considered strong evidence of a potential disease association.
To confirm the linkage of tag variants, linkage disequilibrium (LD) between two loci was calculated using the coefficient of linkage disequilibrium (D), which quantifies the non-random association between alleles at two loci. D was calculated by comparing the observed frequency of haplotypes (allele combinations at the two loci) to the expected frequency under the assumption of random association. Normalized D' and r2 value were also computed, where D' adjusts for the maximum possible D given the allele frequencies, and r2 measures the correlation between alleles at the two loci, yielding a value between 0 and 1 that reflects the strength of LD. A positive D value and an r2 > than 0.8 indicate strong linkage disequilibrium.
RESULTS
Clinical characteristics of the cohort
A total of 930 patients were included in the study, comprising 679 suspected WD cases and 251 non-WD disease controls. Demographic and clinical characteristics, including age, sex, serum ceruloplasmin level, alanine aminotransferase (ALT), total bilirubin, red blood cell (RBC) count, and hemoglobin levels, were compared between suspected WD cases and non-WD controls, and further stratified by ATP7B genotype (wild-type vs. variant). Suspected WD cases and controls were comparable in terms of sex distribution and age, but ceruloplasmin levels were significantly lower in the suspected WD cohort. Among patients with suspected WD, those carrying ATP7B gene variants had significantly lower ceruloplasmin levels compared to those with the wild-type genotype. Significant differences were also observed in other biochemical and hematological markers between cases and controls, as well as between the ATP7B variant and wild-type groups. These findings highlight the distinctiveness of the suspected group from the controls and suggest that molecularly diagnosed cases differ from those with other underlying etiologies. Detailed demographic comparisons are presented in Table 1.
Demographic profiles of WD suspects, controls, and subgroups
| Controls (n = 251) | WD suspects (n = 679) | P value | ||
| ATP7B Variations | ||||
| Positive ** (n = 282) | Negative (n = 397) | |||
| Age (year, median ± IQR*) | 38 ± 25 | 39 ± 26 | 0.62 | |
| 36 ± 23 | 43 ± 27 | 0.66 | ||
| Sex (Female/Male, Female%) | 100/151, 39.84% | 236/443, 34.57% | 0.1752 | |
| 108/174,38.30% | 128/269,32.24% | 0.1208 | ||
| Ceruloplasmin (mg/L, median ± IQR) | 232 ± 116 | 173 ± 90.3 | 2.1 × 10-10 | |
| 146 ± 110 | 189 ± 97.4 | 1.5 × 10-17 | ||
| ALT (IU/L, median ± IQR) | 26.5 ± 44.2 | 38 ± 64 | 0.00087 | |
| 35 ± 51.2 | 40 ± 73 | 0.08017 | ||
| Total Bilirubin (μmol/L, median ± IQR) | 20.5 ± 48.1 | 27.8 ± 59.6 | 0.0084 | |
| 21.4 ± 38 | 34.4 ± 66.5 | 0.00015 | ||
| RBC (×1012/L, median ± IQR) | 3.91 ± 1.44 | 4.11 ± 1.31 | 0.047 | |
| 4.08 ± 1.27 | 4.16 ± 1.36 | 0.8072 | ||
| Hemoglobin (g/L, median ± IQR) | 119 ± 46 | 128 ± 40 | 0.0048 | |
| 124 ± 38.5 | 129 ± 40 | 0.6636 | ||
Ceruloplasmin level between ATP7B wild-type and variants (pathogenic and VUS included)
Serum ceruloplasmin levels were compared across individuals with different ATP7B gene variants. Individuals with the wild-type ATP7B genotype exhibited significantly higher serum ceruloplasmin levels than those carrying any variant, whether pathogenic or a VUS. Variants were further categorized based on the co-occurrence of pathogenic variants, VUS, and the p.Leu770= variant (the most frequently observed VUS, discussed later in the mutational spectrum and VUS selection results). This classification yielded five subgroups: pathogenic-only, pathogenic with p.Leu770=, pathogenic with VUS, pathogenic with VUS and p.Leu770=, and VUS-only. All variant groups showed significantly lower median ceruloplasmin levels than the wild-type group. Notably, patients carrying VUS exhibited markedly reduced ceruloplasmin. Within the VUS subgroups, those with VUS in combination with known pathogenic mutations had lower median levels compared to those with VUS-only. This gradient of severity is consistent with the variable clinical impact of VUS - some likely pathogenic, others potentially benign or of intermediate effect. Figure 1 presents the detailed results. Significant differences in ceruloplasmin levels were observed in specific female subgroups, suggesting that the effect of ATP7B variants on ceruloplasmin may be modulated by sex.
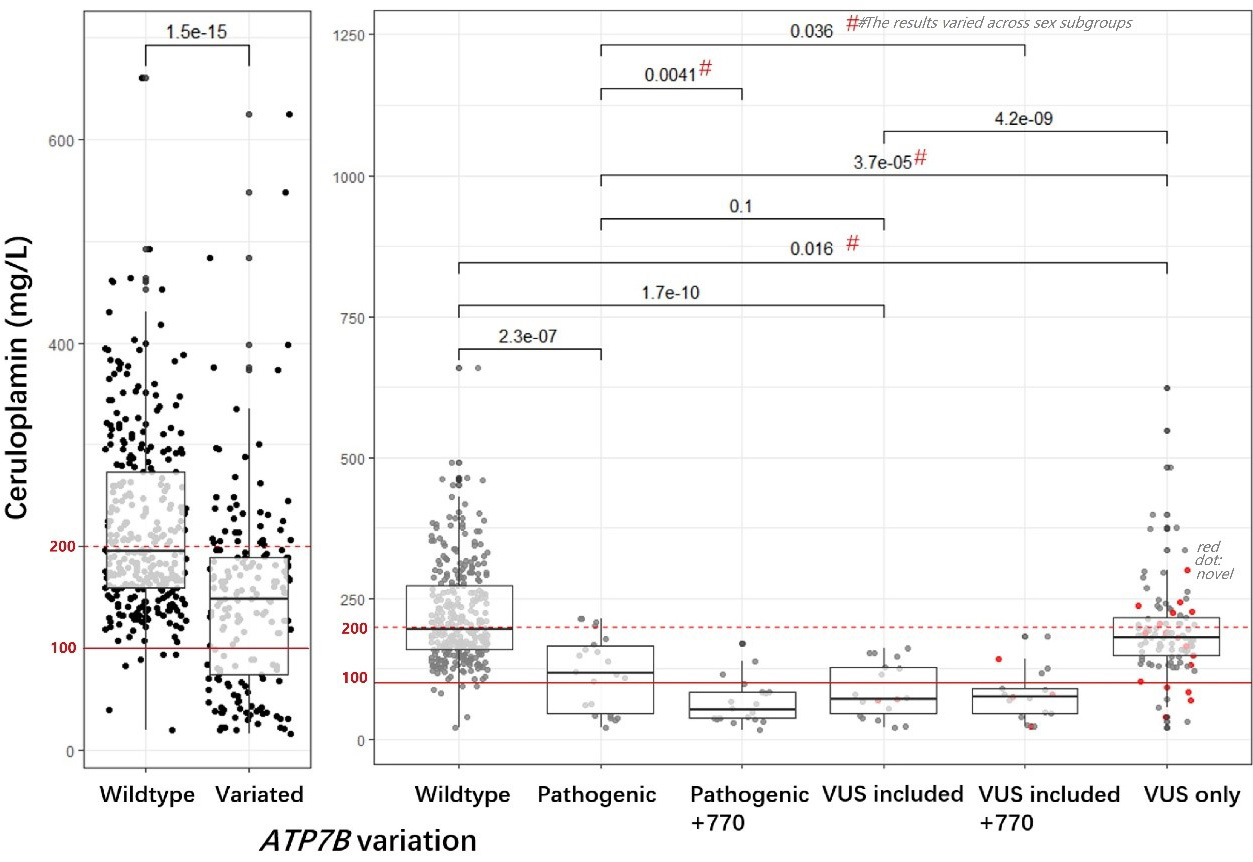
Figure 1. Comparisons of serum ceruloplasmin levels between ATP7B wild-type and variant carriers. The left panel compares ceruloplasmin levels between individuals with wild-type ATP7B and those with variants, showing significantly lower levels in the variant group. Further stratifying variants by pathogenicity and VUS status, the right panel compares individuals with wild-type alleles to those carrying various types of variants (from left to right: individuals with only known pathogenic variant, pathogenic plus p.Leu770=, pathogenic plus VUS, pathogenic plus both p.Leu770= and VUS, and VUS only). Red dots denote novel mutations identified in the cohort. All variant groups show significantly lower median ceruloplasmin levels than the wild-type group, as indicated by P-values (e.g., P = 0.016 for VUS-only vs. wild-type). The red dashed line indicates the normal reference range (200 mg/L), while the solid line marks 100 mg/L, below which ceruloplasmin levels are considered low and very low, respectively. The hashtag (#) signifies a statistically significant difference in ceruloplasmin levels between females and males within specific genotype groups. Notably, female VUS carriers had lower ceruloplasmin levels, whereas male VUS and wild-type carriers showed comparable levels (data not shown). A two-sample t-test was employed to assess the significance of mean differences. VUS: Variants of unknown significance.
Distinct mutation spectrum of ATP7B and VUS selection
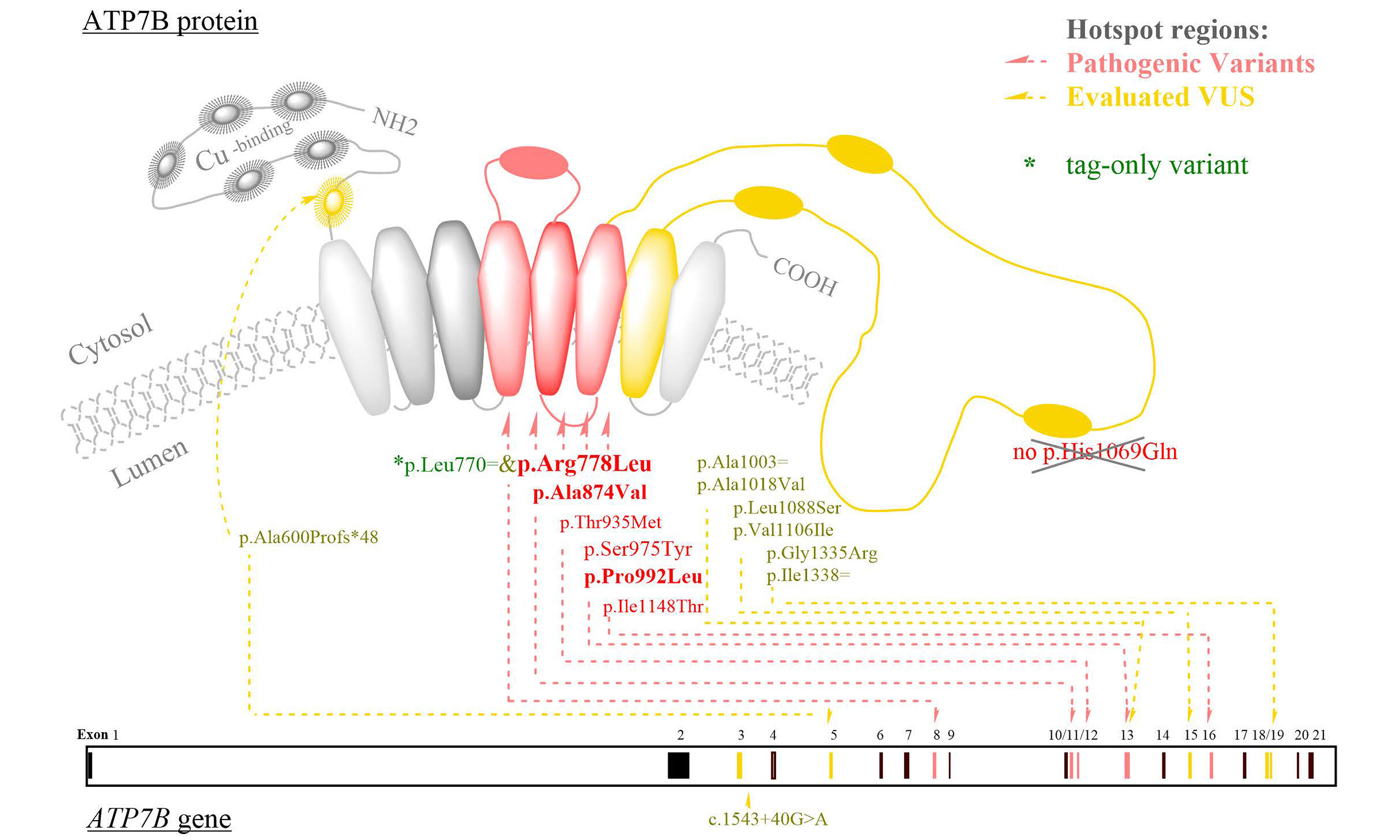
Among the 24 known pathogenic variants identified, the most frequently observed were p.Arg778Leu, p.Pro992Leu, and p.Ala874Val. The variant frequencies (in counts) were as follows: p.Arg778Leu (53), p.Pro992Leu (20), p.Ala874Val (15), p.Ser975Tyr (5), p.Ile1148Thr and p.Thr935Met (both 4), and p.Val1216Met (3). Variants occurred twice included p.Arg483fs, p.Arg919Gly, p.Glu332*, p.Gly869Arg, and p.Thr1178Ala. The remaining variants were detected only once: c.1543+1G>T, p.Asn1270Ser, p.Asn728Ser, p.Cys490*, p.Gly943Ser, p.Met769fs, p.Ser105*, p.Ser1365fs, p.Thr766Met, p.Thr888Pro, and p.Trp779*.
In addition, we identified 79 VUS, with the most common being p.Leu770=, p.Val1297Ile, p.Val1106Ile, p.Ile929Val, p.Ala1003=, p.Ala476Thr, and p.Asp196Glu. Furthermore, 26 novel variants were discovered. Detailed information on the detected variants – including chromosome coordinates, variant counts, HGVS[28] annotations, and functional categorizations - is provided in Figure 2.
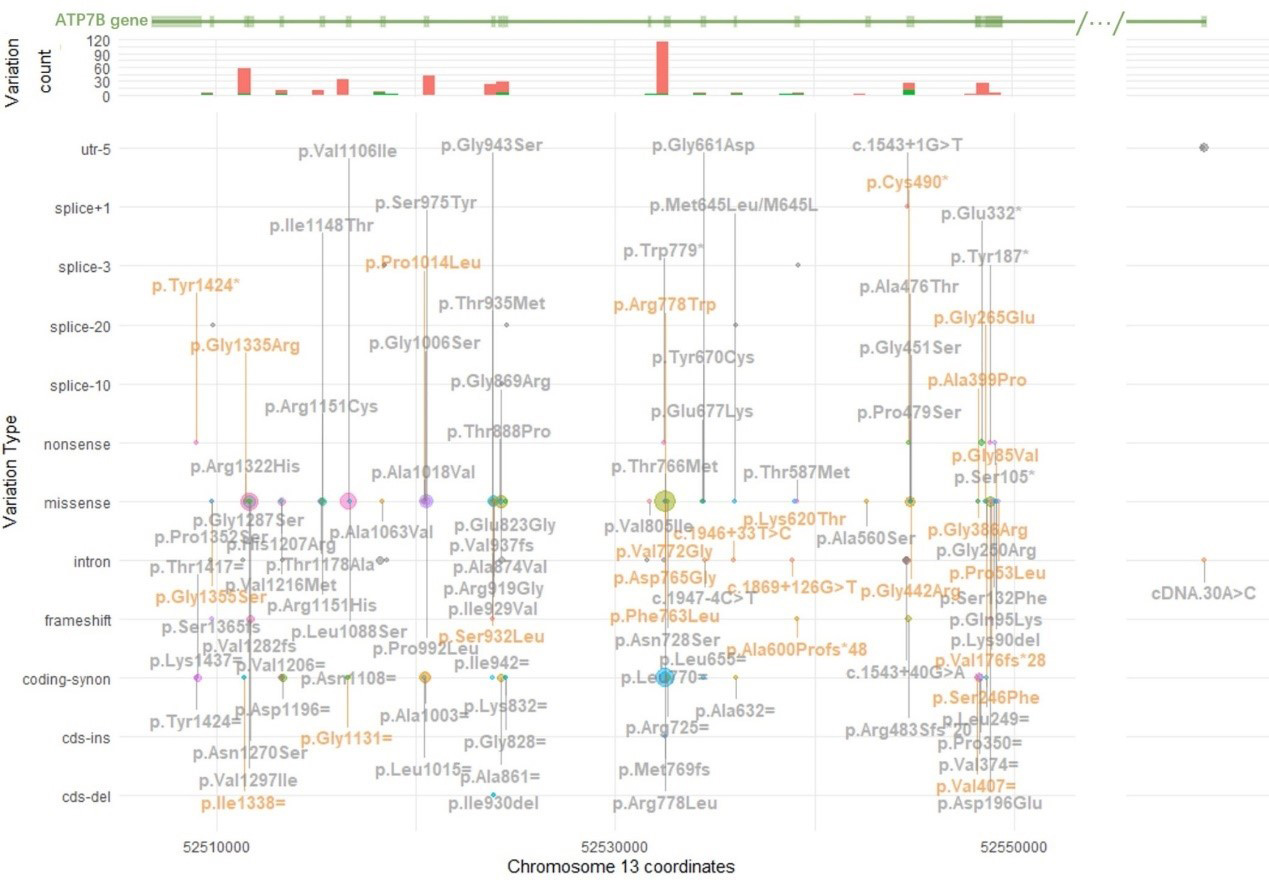
Figure 2. Variation spectrum, including counts and functional categorizations. Variation spectrum of ATP7B in this cohort. This figure illustrates the distribution and genomic locations of genetic variants in the ATP7B gene on chromosome 13. It consists of three panels. The x-axis represents chromosomal coordinates. The top panel, shown in green, depicts the structure of the ATP7B gene, with exons represented as boxes and introns, untranslated regions (UTRs), and non-coding regions as lines. Ellipses between slashes indicate omitted long intronic regions where no variants were detected. The second panel is a histogram, with the y-axis indicating the number of variants; red bars represent exonic variants, while green bars indicate intronic variants. The third and largest panel uses the y-axis to classify variation types, including 5’UTR (utr-5), splice sites (+1 to -20), nonsense, missense, intron, frameshift, synonymous coding (coding-synon), coding insertion (cds-ins), and coding deletion (cds-del). The number of each variation is represented by a circle, with size proportional to frequency - larger circles correspond to more frequent variants. HGVS annotations for each variant are shown in bold gray adjacent to the corresponding circle; when space is limited, vertical lines connect the labels to their respective circles. Novel variants are in orange. Due to space constraints, all 79 VUS, including allele counts, zygosity, and gnomAD East Asian frequencies, are listed in Supplementary Table 1. VUS: Variants of unknown significance.
Variants associated with very low ceruloplasmin levels were further evaluated for VUS reclassification. The distribution of these genetic variants was analyzed by functional categories (e.g., exonic, intronic, splice-site). Additionally, zygosity and novelty were assessed for each variant.
Reclassification of VUS based on integrated evidence
We selected nine VUS for detailed reevaluation. Among these, the splice site mutation c.1543+40G > A, along with the missense variants p.Val1106Ile, p.Ala1018Val, p.Leu1088Ser, p.Gly1335Arg, as well as the synonymous variant p.Ala1003=, and p.Ile1338=, were reclassified as likely pathogenic. The synonymous variant p.Leu770= was the most frequently observed VUS.
In the LD analysis between p.Leu770= and p.Arg778Leu, both variants were observed in 54 cases, including four homozygous individuals. All cases and controls were genotyped at both loci, and no individual carried p.Leu770= in the absence of p.Arg778Leu. Moreover, zygosity concordance was observed. The calculated LD parameters were as follows: D = 0.0291, D' = 1.0000, and r2 = 1.0000, indicating a strong linkage between the two loci. Consequently, the synonymous variant p.Leu770= - regardless of zygosity - should be regarded as a tag variant, and thus should not be designated as an independent pathogenic hit. Further studies in diverse populations are warranted before any recommendations can be made regarding modifications to diagnostic scoring systems, such as the Leipzig criterion.
Detailed descriptions and interpretations are provided in Table 2. Selected VUS, including the splice site variant c.1543+40G > A, are highlighted due to their novelty or presence in homozygosity in the VUS-only analysis. However, in-silico splicing prediction suggests that both c.1543+40G > A and the synonymous VUS are likely polymorphisms. A detailed visualization of these findings is presented in Figure 3.
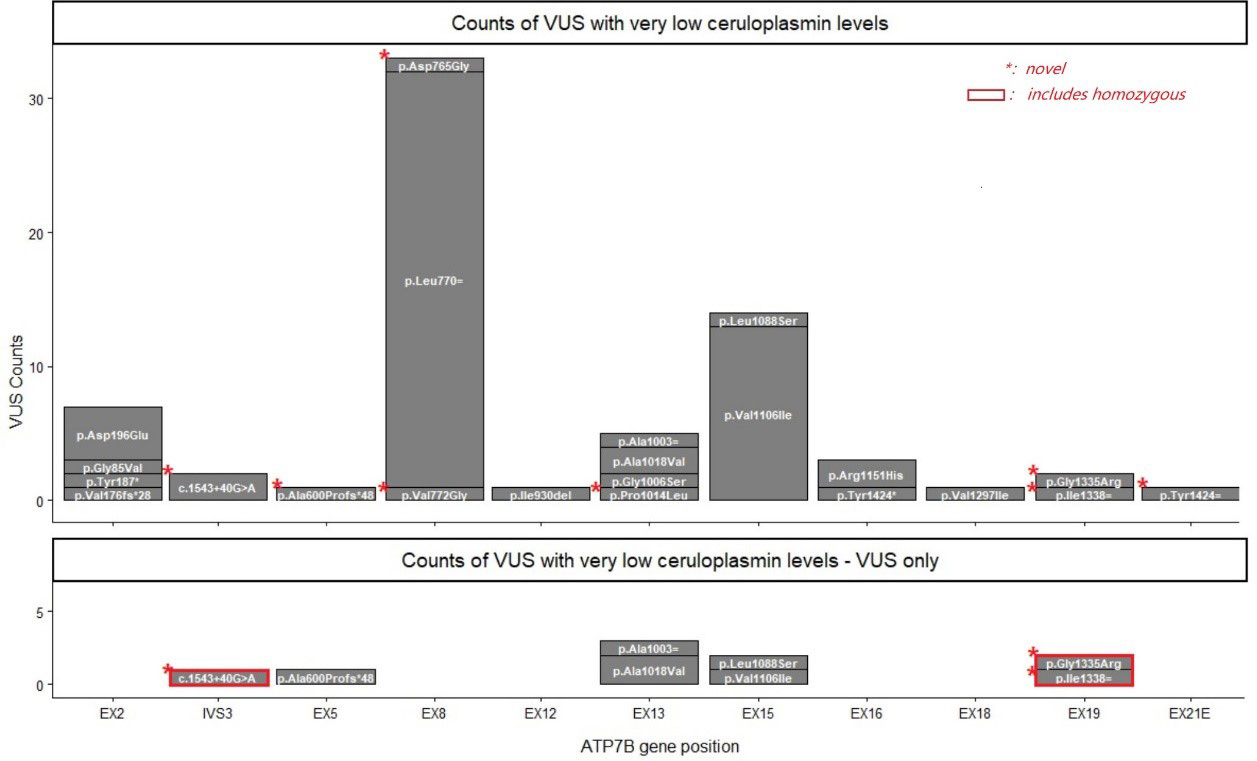
Figure 3. Occurrence of VUS associated with ceruloplasmin levels below 100 mg/L. Stacked bar plots comparing the distribution and frequency of selected ATP7B VUS. The x-axis represents gene positions, with labels such as EX (exon) and IVS (intron) indicating specific genomic regions. The y-axis shows the number of variants observed in each region in this study. Each bar consists of stacked segments, with the HGVS nomenclature for each variant labeled within its corresponding segment. Upper panel: all VUS; Lower panel: VUS occurring in isolation. Red rectangles represent variants in a homozygous state, while red asterisks in the upper left corner of a segment denote novel variants. VUS: Variants of unknown significance.
Reevaluation of selected VUSs
| Variant | Type | Case count | Control count | MAF GnomAD East Asian | Zygosity | ceruloplasmin range | Co-occurring pathogenic variants | ACMG criteria applied | final classification |
| p.Leu770= | synonymous | 53 | 0 | 0.001537502 | Het | 16-181 | p.Arg778Leu | PM2#/PS4 | Tag only (*r2 value = 1) |
| c.1543+40G>A | intronic | 3 | 0 | 0.004679144 | Het | 26 | p.Arg778Leu+p.Leu770= | PM2#/PS4 | Strong pathogenic candidate |
| Het | 206 | / | |||||||
| Hom | 40 | / | |||||||
| p.Ala1018Val | missense | 2 | 0 | 0 | Hom | 20-30 | / | PM2/PS4 | Strong pathogenic candidate |
| p.Val1106Ile | missense | 30 | 0 | 0.000891146 | Het | 22-297 | PM2/PS4 | Strong pathogenic candidate | |
| Ceruloplasmin level < 100 | |||||||||
| 13 patients | |||||||||
| 3 | Het | 46-68 | p.Arg778Leu+p.Leu770= | ||||||
| 1 | Het | 79 | p.Arg778Leu+p.Leu770= + p.Asp196Glu + p.Tyr1424= | ||||||
| 1 | Het | 89 | p.Arg778Leu+p.Leu770= + p.Asp196Glu | ||||||
| 1 | Het | 54 | p.Asp196Glu + p.Pro992Leu | ||||||
| 1 | Het | 22 | p.Asp196Glu | ||||||
| 1 | Het | 74 | p.Val176fs*28 | ||||||
| 1 | Het | 37 | p.Thr888Pro | ||||||
| 1 | Het | 66 | c.1543+1G>T | ||||||
| 1 | Het | 80 | p.Thr766Met | ||||||
| 1 | Het | 69 | p.Cys490* | ||||||
| 1 | Het | 70 | p.Ala600Profs*48 | ||||||
| Ceruloplasmin level ≥ 100 and had p.Val1106Ile only | |||||||||
| 8 patients | Het | 134-234, | / | ||||||
| (5 patient had low ceruloplasmin level) | |||||||||
| p.Ala600Profs*48 | frameshift | 1 | 0 | 0 | Het | 70 | with above p.Val1106Ile | PM2/PS4 | Strong pathogenic candidate |
| p.Ile1338= | synonymous | 2 | 0 | 0.000044565 | Het | / | PM2/PS4 | Strong pathogenic candidate | |
| p.Leu1088Ser | missense | 1 | 0 | 0 | Het | / | PM2/PS4 | Strong pathogenic candidate | |
| p.Gly1335Arg | missense | 1 | 0 | 0 | Het | / | PM2/PS4 | Strong pathogenic candidate | |
| p.Ala1003= | synonymous | 12 | 0 | 0.009550009 | Het | / | PM2#//PS4 | Strong pathogenic candidate | |
DISCUSSION
Using a large real-world dataset, we characterized the mutational spectrum specific to the Chinese population among WD suspects, extending beyond diagnosed cases. These individuals did not necessarily meet the Leipzig criteria of >4 and may have exhibited subclinical features[3,4,29,30]. This broadens the applicability of our findings to a more diverse and underrecognized segment of the WD patient population, enhancing our understanding of the functional consequences and penetrance of these mutations. Among our key findings, we observed that the mutational landscape and the most recurrent variants differed not only from those reported in Western populations[31,32] but also from those in geographically and ethnically proximate groups, including eastern Eurasians[33] and even Indian cohorts[34]. Notably, the p.His1069Gln variant, commonly reported in European and eastern Eurasian populations, was absent in all our cases. Similarly, the recurrent Eurasian variant p.Met769fs was observed in only one individual. These observations suggest an ethnic-level evolutionary drive.
Regarding VUS, we observed a high prevalence in our cohort, highlighting the challenges in genetic diagnosis and the need for functional and population-specific reevaluation. As sequencing technologies expand, diagnostic yield increases, but so does the detection of VUS. Over time, as additional evidence on variant pathogenicity accumulates, many VUS are reclassified, necessitating routine reevaluation. ACMG has recommended that laboratories develop standardized protocols for variant reevaluation and maintain up-to-date internal databases of reported variants. This process involves integrating new evidence and reassessing the entire body of supporting data[35]. While the ACMG criteria for classifying pathogenic variants use the general population as a control (e.g., in the PS4 and PM2 criteria), our study took a more targeted approach by incorporating ethnically matched controls and patient-specific data, thereby strengthening the pathogenicity assessment. Nevertheless, it is important to note that while ethnically matched controls complement global population data for ACMG population-based criteria, they do not replace them.
In practice, both variant-level and case-level reanalysis are typically initiated and periodically repeated once the knowledge base has matured, underscoring the importance of accumulating clinical cases. Aligning with this, our case-by-case integrated approach - combining generic classification methods - led to the reclassification of several VUS:
The reassignment of c.2310C>G:p.Leu770= to tag-only variation prevents false-positive genetic diagnoses in this locus and avoids misclassifying carriers as patients. This variation should not be counted separately from c.2333G>T:p.Arg778Leu, as the two are strongly linked in the Chinese population to date.
The revelation of the pathogenic potential of some synonymous mutations is also intriguing. This finding corroborates a recent Chinese case report[36]. While their case involved compound heterozygosity, our study identifies some synonymous variants in the heterozygous state, suggesting a potentially more pronounced impact on protein function. These mutations may induce functional deficiency through mechanisms such as exon skipping[37,38] or other splicing alterations[10]. Our study uncovers and highlights concerns about these seemingly harmless variants, such as the c.1543+40 variant in the “dark matter” intronic region[39], again emphasizes the need for expanded, continuous screening and annotation. The observed haplotype associated with the p.Val1106Ile mutation suggests that this site may be structurally or functionally important. Collectively, these findings underscore the critical role of population-specific databases in refining regional genetic testing protocols.
Therefore, we recommend that in Chinese patients suspected of WD, when no known pathogenic variants are identified, the variants listed in Table 2 - particularly c.3316G>A: p.Val1106Ile and the synonymous c.3009G>A: p.Ala1003= - should be thoroughly screened. If the results remain negative but clinical suspicion persists, Supplementary Table 1 should also be examined. Additionally, note that c.2310C>G: p.Leu770= should not be counted if it co-occurs with c.2333G>T:p.Arg778Leu.
Limitations
The primary limitation of this study stems from the rarity of WD. As with most rare disease research, this field faces inherent challenges, including limited awareness, diagnostic delays, and insufficient funding. Furthermore, the study relies predominantly on ceruloplasmin levels as an objective biomarker for ATP7B dysfunction. While ceruloplasmin is among the first-line and most widely used clinical parameters, it does not directly reflect the functional status of the ATP7B protein. Emerging biomarkers - such as the measurement of ATP7B peptides[40] and serum non-ceruloplasmin-bound copper[41] - are currently under development and evaluation, and hold promise for more accurate assessment of ATP7B function in the future.
Another concern relates to the use of a very low ceruloplasmin cutoff for reclassifying VUS. This approach led to the exclusion of cases with ceruloplasmin levels above 100 mg/L and, critically, extremely rare WD cases with normal ceruloplasmin levels. As such, the reclassification strategy may be most applicable to variants associated with severe ceruloplasmin deficiency and may fail to capture milder or atypical disease presentations. Future case-control studies may help address this limitation - either by excluding ceruloplasmin from the analysis or by incorporating novel biomarkers that better reflect ATP7B function.
Additionally, the study lacks sufficient functional validation (e.g., in vitro copper transport assays) for the reclassified variants, limiting confidence in their pathogenicity assessment. The use of same-ethnic controls in applying ACMG population criteria may introduce potential bias. The possible misclassification within the “suspect” and “control” groups raises additional concerns.
Our study underscores the critical need to establish a dedicated ATP7B Gene Curation Expert Panel to ensure ongoing review and refinement of variant interpretations, encompassing both coding and non-coding regions. The identification of recurrent variants will further support the development of ethnically targeted screening strategies. Constant communication is of pivotal importance. Clinical laboratories should make concerted efforts to prioritize the reporting of any changes made that may affect clinical management. Particularly, the reclassification of any VUS should be prioritized and promptly communicated to clinicians, as it carries greater clinical impact than reclassifications such as from likely pathogenic to pathogenic.
DECLARATIONS
Acknowledgments
The graphical abstract was created with ChemDraw Professional (Version 25.5.0.5789) © 1998-2025 Revvity Signals Software, Inc.
Authors’ contributions
Made substantial contributions to conception and design of the study and performed data analysis and interpretation: Han Y; Provided administrative support: Zhang X
Performed data acquisition, as well as provided technical and material support: Gu L, Yao B, Xu Q, Han Y
Availability of data and materials
All data generated or analyzed during this study are available from the corresponding author upon reasonable request.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (No. 82002126).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Informed consent to participate in the study was obtained from participants. This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Shanghai Ruijin Hospital (approval number: 201617/201848).
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Roberts EA, Schilsky ML. Current and emerging issues in Wilson’s disease. N Engl J Med. 2023;389:922-38.
2. Calinas F, Cardoso H, Carvalhana S, et al. Practical and multidisciplinary review on Wilson disease: the Portuguese perspective. GE Port J Gastroenterol. 2024:1-17.
3. Wang M, Wang F, Tao Z, Yang W. Analysis of factors affecting early hepatic steatosis in pediatric patients with Wilson’s disease in China: a retrospective study. Medicine (Baltimore). 2025;104:e45501.
4. Najimi Z, Elsaboni L, Mtegha M, et al. Analysis of tissue copper levels as a reliable diagnostic tool in paediatric liver disease. Eur J Pediatr. 2025;185:23.
5. Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease1. Liver Int. 2003;23:139-42.
6. Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207.
7. Rehm HL, Berg JS, Brooks LD, et al. ClinGen - the clinical genome resource. N Engl J Med. 2015;372:2235-42.
8. Zhang SJ, Yang WM, Yang YL, Wang JX, He W. Analysis of clinical phenotype and ATP7B variants characteristics of 337 patients with hepatolenticular degeneration in Anhui province. Zhonghua Yi Xue Za Zhi. 2025;105:1172-6.(in Chinese).
9. Mishra AK, Sen Sarma M, Moirangthem A, Sait H. Nuances in ATP7B Genetic Testing and Interpretation in India. J Clin Exp Hepatol. 2026;16:103205.
10. Xu W, Wang R, Dong Y, Wu Z. Pathogenicity of intronic and synonymous variants of ATP7B in Wilson Disease. J Mol Diagn. 2023;25:57-67.
11. Han M, Yang Z. A rare presentation of Wilson disease with normal levels of serum ceruloplasmin and copper and MODY: A case report. Medicine (Baltimore). 2025;104:e43080.
12. Socha P, Jańczyk W, Zanetto A, et al. EASL-ERN clinical practice guidelines on Wilson’s disease. J Hepatol. 2025;82:690-728.
13. Palumbo CS, Schilsky ML. Clinical practice guidelines in Wilson disease. Ann. Transl. Med. 2019;7 Suppl:S65.
14. Cao H, Chen Y, Fan J. Wilson’s disease: from clinical practice to guidelines. Zhonghua Gan Zang Bing Za Zhi. 2014;22:570-2.
15. Tang S, Hou W, Zheng SJ. Recommendations from the European Association for the study of the liver and the European reference network for rare liver diseases clinical practice guidelines for hepatolenticular degeneration. Zhonghua Gan Zang Bing Za Zhi. 2025;33:988-992. (in Chinese).
16. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol. 2012;56:671-85.
17. Li X, Lu Y, Ling Y, et al. Clinical and molecular characterization of Wilson’s disease in China: identification of 14 novel mutations. BMC Med Genet. 2011;12:6.
18. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24.
19. Gargano MA, Matentzoglu N, Coleman B, et al. The Human Phenotype Ontology in 2024: phenotypes around the world. Nucleic Acids Res. 2024;52:D1333-46.
20. Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062-7.
21. Francomano CA. Victor Almon McKusick: in the footsteps of Mendel and Osler. American J of Med Genetics Pt A. 2021;185:3193-201.
22. Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789-98.
23. Auton A, Abecasis GR, Altshuler DM, et al. ; Writing group. A global reference for human genetic variation. Nature. 2015;526:68-74.
24. Karczewski KJ, Francioli LC, Tiao G, et al. ; Genome Aggregation Database Consortium. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434-43.
25. Phan L, Zhang H, Wang Q, et al. The evolution of dbSNP: 25 years of impact in genomic research. Nucleic Acids Res. 2025;53:D925-31.
26. Niehaus A, Azzariti DR, Harrison SM, et al. A survey assessing adoption of the ACMG-AMP guidelines for interpreting sequence variants and identification of areas for continued improvement. Genet Med. 2019;21:1699-701.
27. Ryan A, Nevitt SJ, Tuohy O, Cook P. ; Cochrane Cystic Fibrosis and Genetic Disorders Group. Biomarkers for diagnosis of Wilson’s disease. Cochrane Database Syst Rev. 2019;2019.
28. Hart RK, Fokkema IFAC, Distefano M, et al. HGVS Nomenclature 2024: improvements to community engagement, usability, and computability. Genome Med. 2024;16:149.
29. Ghosh U, Sen Sarma M, Samanta A. Challenges and dilemmas in pediatric hepatic Wilson’s disease. World J Hepatol. 2023;15:1109-26.
30. Stättermayer AF, Entenmann A, Gschwantler M, Zoller H, Hofer H, Ferenci P. The dilemma to diagnose Wilson disease by genetic testing alone. Eur J Clin Investig. 2019;49:e13147.
31. Møller LB, Horn N, Jeppesen TD, et al. Clinical presentation and mutations in Danish patients with Wilson disease. Eur J Hum Genet. 2011;19:935-41.
32. Couchonnal E, Bouchard S, Sandahl TD, et al. ATP7B variant spectrum in a French pediatric Wilson disease cohort. Eur J Med Genet. 2021;64:104305.
33. Garbuz M, Ovchinnikova E, Ovchinnikova A, et al. Spectrum of pathogenic variants of the ATP7B gene and genotype-phenotype correlation in eastern Eurasian patient cohorts with Wilson’s disease. Biomedicines. 2024;12:2833.
34. Kumar M, Sharma S, Pandey S, et al. The genomic landscape of Wilson disease in a pan India disease cohort and population-scale data. Movement Disord Clin Pract. 2024;12:185-95.
35. Deignan JL, Chung WK, Kearney HM, Monaghan KG, Rehder CW, Chao EC. Points to consider in the reevaluation and reanalysis of genomic test results: a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2019;21:1267-70.
36. Zhang Q, Xie X, Li H, Li Y, Huang H. Silent but significant: Functional elucidation of a synonymous ATP7B mutation in Wilson’s disease pedigrees. Front. Genet. 2025;16:1604683.
37. Panzer M, Viveiros A, Schaefer B, et al. Synonymous mutation in adenosine triphosphatase copper‐transporting beta causes enhanced exon skipping in Wilson disease. Hepatol Commun. 2022;6:1611-9.
38. Zhou X, Zhou W, Wang C, et al. A comprehensive analysis and splicing characterization of naturally occurring synonymous variants in the ATP7B gene. Front. Genet. 2021;11:592611.
39. Girardini KN, Olthof AM, Kanadia RN. Introns: the “dark matter” of the eukaryotic genome. Front. Genet. 2023;14:1150212.
40. Collins CJ, Yi F, Dayuha R, et al. Direct measurement of ATP7B peptides is highly effective in the diagnosis of Wilson disease. Gastroenterology. 2021;160:2367-2382.e1.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].