


Immunometabolic drivers of renal fibrosis and potential therapeutic targets in diabetic kidney disease
 0
0 Abstract
Aim: Diabetic kidney disease (DKD) is a serious complication of diabetes, whose precise pathogenesis remains incompletely understood. Identifying therapeutic targets of DKD remains of great importance.
Methods: Four independent DKD microarray datasets were analyzed to identify differentially expressed genes. The expression quantitative trait locus (eQTL) data and DKD data from Genome-Wide Association Studies (GWAS) were utilized for Mendelian randomization (MR) analysis to pinpoint genes associated with DKD. The intersection of genes derived from two approaches was identified as key genes for DKD. Key genes were then subjected to enrichment analyses, immune infiltration assessment. Colocalization analysis, and quantitative polymerase chain reaction (qPCR) validation were used to identify core genes. A human DKD single-cell RNA sequencing dataset was analyzed to validate the cell-type-specific expression patterns of the core genes.
Results: We identified 275 up- and 184 downregulated genes. Combined with MR, seven key genes were determined. They were involved in lipid metabolism, protein secretion, signal transduction, immune response, and fibrosis. Cell-type identification by estimating relative subsets of RNA transcripts (CIBERSORT) analysis revealed the unique distribution of immune cells in DKD and the regulation of immune cells by key genes. Colocalization analysis indicated a strong association between cystatin A (CSTA), lipoprotein lipase (LPL), lysozyme (LYZ), transforming growth factor beta induced (TGFBI), interferon induced protein with tetratricopeptide repeats 1 (IFIT1), and DKD. The qPCR confirmed that CSTA, LYZ, and TGFBI were differentially expressed, serving as core genes. LYZ was the most crucial. Single-cell analysis further unmasked the specific upregulation of CSTA, LYZ, and TGFBI in macrophages, where simulated knockout suggested a regulatory role in restraining antigen presentation.
Conclusion: This study demonstrated the potential and underlying mechanisms of gene-targeted therapy for DKD, providing a foundation for future investigations.
Keywords
INTRODUCTION
Diabetic kidney disease (DKD) is a common and serious complication of diabetes that significantly increases mortality in diabetic patients[1]. As the incidence of diabetes increases, so does the prevalence of DKD, posing a heavy burden on public health and medical systems[2]. Although various treatments exist for DKD, the underlying mechanisms, especially at the genetic and molecular levels, remain incompletely understood[3]. This knowledge gap underscores the necessity of exploring potential pathogenic mechanisms and identifying key genes linked to DKD.
Related studies have shown that abnormal gene expression plays a significant role in the pathogenesis of DKD[3,4]. Abnormal gene expression in DKD often affects processes such as lipid metabolism, inflammatory responses, immune infiltration, and extracellular matrix remodeling, interacting synergistically to drive renal fibrosis[5,6]. Metabolic disorders are key drivers in initiating and maintaining the pathological process. Disruptions in glucose and lipid metabolism not only damage renal tubular epithelial cells (RTECs) but also exacerbate oxidative stress by generating advanced glycation end products (AGEs) and reactive oxygen species (ROS)[7]. The binding of AGEs to their receptors can trigger chronic inflammatory responses and upregulate transforming growth factor beta (TGF-β) to promote fibrosis[8]. Inflammatory responses and immune cell infiltration are significant forces driving pathological progression. Damaged RTECs release damage-associated molecular patterns, which attract immune cells to the site of injury and secrete pro-inflammatory cytokines, perpetuating a chronic inflammatory state. Immune cells contribute to the formation of a microenvironment favorable to fibrosis, known as the fibrotic niche[9]. In this environment, fibroblasts are activated and transformed into myofibroblasts, which produce large amounts of extracellular matrix proteins. The abnormal accumulation of extracellular matrix components is a hallmark of fibrosis, impairing blood perfusion and exacerbating hypoxia, oxidative stress, and inflammatory responses. Studies have found that extracellular matrix remodeling is also regulated by specific enzymes[10]. These interconnected processes form a complex pathological network in DKD through synergistic interactions and feedback loops, necessitating comprehensive interventions to disrupt this vicious cycle in clinical practice.
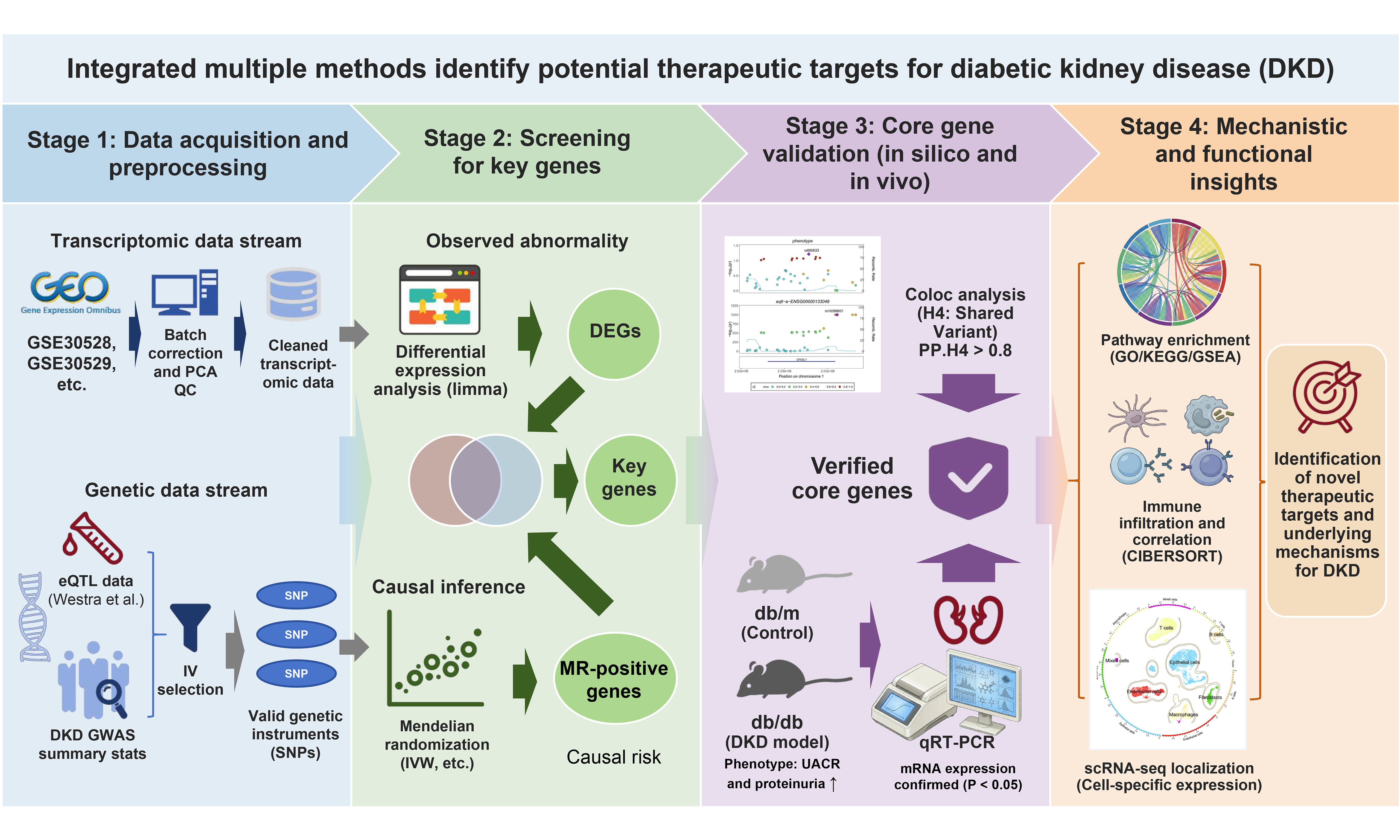
This study utilized a comprehensive range of methodologies, including microarray data analysis, Mendelian randomization (MR) analysis, multiple enrichment analyses, immune infiltration assessment, colocalization analysis, quantitative polymerase chain reaction (qPCR), and single-cell RNA sequencing (scRNA-seq) analysis, to comprehensively explore treatment targets associated with DKD. The strength of this integrative strategy lies in its capacity not only to identify genes significantly associated with DKD from extensive genomic datasets and evaluate causal relationships but also to validate their cell-type-specific expression patterns at high resolution. By elucidating how these genes modulate the renal fibrosis process in DKD via lipid metabolism and immunity, this research advances our understanding of the intrinsic mechanisms of DKD and offers valuable insights for the development of future therapeutic strategies.
METHODS
Research data sources
Microarray data (GSE30528, GSE30529, GSE1009, GSE46899) were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) [Table 1]. The expression quantitative trait locus (eQTL) data were derived from a comprehensive meta-analysis by Westra et al., which included eQTL data from peripheral blood samples of 5,311 individuals of European ancestry[11]. They were retrieved from the Integrative Epidemiology Unit (IEU) Genome-Wide Association Studies (GWAS) database (https://gwas.mrcieu.ac.uk/). R software (version 4.4.1) and the “TwoSampleMR” package were utilized to identify single nucleotide polymorphisms (SNPs). A genome-wide significance threshold of P < 5e-08 was applied. SNPs within a 10,000 kb window with an “r2 > 0.01” relative to the most significant SNP were removed to mitigate linkage disequilibrium. An F-statistic > 10 filter was applied to exclude weak instruments. Based on these criteria, 26,152 eQTL-associated SNPs were retained [Supplementary Table 1 (S1)]. DKD outcome data were also obtained from the IEU GWAS, with the ID ebi-a-GCST90018832, involving 1,032 disease cases and 451,248 controls of European descent, including 24,190,738 SNPs[12]. All data used were publicly available and ethically approved for use.
Information of four DKD GEO datasets
| GSE ID | Samples | Tissues | Platform | Experiment type | Last update date | PMID |
| GSE30528 | 9 cases and 13 controls | Renal glomerular tissue | GPL571 | Array | Dec 06, 2018 | 21752957 |
| GSE30529 | 10 cases and 12 controls | Renal tubular tissue | GPL571 | Array | Dec 06, 2018 | 21752957 |
| GSE1009 | 3 cases and 3 controls | Renal glomerular tissue | GPL8300 | Array | Dec 13, 2018 | 15042541 |
| GSE46899 | 4 cases and 2 controls | Peripheral blood | GPL6480 | Array | Jan 06, 2022 | 23835338 |
Identification of differentially expressed genes
The datasets from the GEO database were read and preprocessed using R (version 4.4.1) for dataset correction. Subsequently, these datasets were merged, and batch effect correction was performed. Principal component analysis (PCA) was performed using the “prcomp” function to eliminate batch effects and for visualization. The “limma” package was used to screen for differentially expressed genes (DEGs), with the significance threshold set at P < 0.05 and absolute value of log2 (Fold Change) (|logFC|) > 0.5. The “pheatmap” package was used to create volcano plots and heatmaps of DEGs.
MR analysis
Filtered eQTL data from the previous step were used for MR analysis with DKD. The instrumental variables (SNPs) used in this study were required to meet the following criteria: (1) strong association with the exposure factor; (2) influence on the outcome exclusively through the exposure; (3) no correlation with confounding factors[13]. This study adhered to the Strengthening the Reporting of Observational Studies in Epidemiology using MR (STROBE-MR) guidelines (https://www.strobe-mr.org/).
The inverse-variance weighted (IVW) method was primarily used to determine the causal relationship between genes and DKD, and preliminary screening was conducted for genes with P < 0.05 based on the results of this method. Four other methods, including MR Egger Regression (MR-Egger), simple mode, weighted median, and weighted mode, were used for additional validation. The false discovery rate (FDR) test was utilized to validate the IVW results.
These DKD-related genes were intersected with previously obtained DEGs to obtain key genes. Subsequently, two-sample MR analysis was performed again on the key genes and DKD to reconfirm their causal relationships. Heterogeneity testing, pleiotropy testing, and leave-one-out sensitivity analysis were included in this analysis to evaluate the robustness and reliability of the results. Scatter plots, forest plots, and funnel plots were drawn to visually present the results.
Colocalization analysis
Colocalization analysis was used to determine whether two phenotypes were driven by the same genetic variant in a specific region, thereby strengthening evidence for phenotype association. The “coloc” R package was used for this analysis to validate known associations between key genes and DKD. Bayesian analysis methods evaluated support for five hypotheses regarding SNPs within the locus. Results with posterior probability (PP) for H4 > 0.8 indicated evidence for colocalization[14]. The findings were visualized through Manhattan plots.
In vivo experimental validation
Experimental animals and feeding conditions
The C57BLKS/J background mice heterozygous for the spontaneous diabetes (db) mutation (hereafter db/m mice) and homozygous db/db littermates on the identical genetic background were purchased from Beijing Modelorg Technologies Company Limited (Beijing, China) and housed in the Animal Center of Changchun University of Chinese Medicine. The db/m mice are non-diabetic heterozygous mice and are often used as a control group for db/db mice. Male db/db and db/m mice at 8 weeks of age were included in the experiment and divided into two groups: the db/m group and the db/db group, with n = 5 in each group. All mice were housed under specific pathogen-free (SPF) conditions, with controlled environmental temperature (22 ±
The two groups of mice were raised until 20 weeks of age, and 24-hour urine samples were collected using metabolic cages for Urinary Albumin-to-Creatinine Ratio (UACR) and 24-hour urinary protein quantification. The results showed that the urinary protein excretion in db/db group mice was significantly higher than that in db/m group [Supplementary Table 1 (S2)]. Therefore, db/db mice can serve as a model for DKD. At the end of the experiment, all serum and tissue samples were stored at -80 °C for subsequent testing. All animal experiments were conducted in accordance with the regulations approved by the Animal Ethics Committee of Changchun University of Chinese Medicine (protocol number 2025363).
Quantitative real-time PCR
Total RNA was extracted from the kidney tissue using a total RNA extraction kit (Tiangen Biotechnology, China) following the manufacturer’s recommended protocol. The RNA concentration and purity (A260/A280 ratio > 1.8) were determined by NanoDrop 2000 spectrophotometry. The reverse transcription kit (Tiangen, Beijing) was used to prepare the reaction system (20 μL) for each group. Reverse transcription conditions: samples were incubated at 42 °C for 15 min and then inactivated at 95 °C for 3 min.
For real-time fluorescent qPCR, prepare the reaction system (20 μL) for each group according to the SYBR Green kit (Tiangen, Beijing). The Bio-Rad CFX96 real-time fluorescent qPCR instrument was used to detect messenger ribonucleic acid (mRNA), and set 40 cycles for amplification. Gene-specific primer pair designs are shown in Table 2. The primers exhibited amplification efficiency ≥ 90% and a calibration curve correlation coefficient (R2) ≥ 0.99, while melting-curve analysis showed a single specific peak, confirming primer specificity and amplicon homogeneity in line with the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines. Relative mRNA expression levels were quantified using the comparative 2-ΔΔCT method, with β-actin (Actb, Gene ID: 11461) serving as the endogenous normalization control. Statistical analyses were performed using unpaired Student’s t-tests in GraphPad Prism 10.5.0, with significance thresholds established at P < 0.05 vs. db/m control groups.
Primer sequences
| Gene | Forward primer (5′ → 3′) | Reverse primer (5′ → 3′) |
| CSTA | TGCTAACAAGGTCAGACCTCAG | CCATGGTTTTGTCAGTCTGGT |
| LYZ | GAGACCGAAGCACCGACTATG | CGGTTTTGACATTGTGTTCGC |
| TGFBI | AGCACGGCCCCAATGTAT | GGGACCTTTTCATATCCAGGACA |
| IFIT1 | CTGAGATGTCACTTCACATGGAA | GTGCATCCCCAATGGGTTCT |
| LPL | GGGAGTTTGGCTCCAGAGTTT | TGTGTCTTCAGGGGTCCTTAG |
| β-actin | CCAGCCTTCCTTCTTGGGTA | CAATGCCTGGGTACATGGTG |
GO, KEGG enrichment analysis
The Microbial Informatics Platform (https://www.bioinformatics.com.cn/) was used for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the key genes obtained above to understand potential functional pathways and intrinsic mechanisms of the impact on the development of DKD. Results meeting P < 0.05 were considered significant.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was used to determine whether functions or pathways associated with intersecting genes were enriched at the top or bottom of a ranked list, indicating up- or downregulation trends. This approach helps uncover biological significance from large gene expression datasets[15]. GSEA further explored potential mechanisms of genes with significant colocalization analysis results. In GSEA, P < 0.05 was considered statistically significant.
Immune infiltration assessment
The CIBERSORT method and merged microarray data matrix obtained from previous studies were used to evaluate the infiltration levels of 22 immune cell phenotypes in DKD. Correlations between the identified key genes and various immune cell phenotypes were also examined to explore regulatory mechanisms.
scRNA-seq data analysis
The scRNA-seq dataset of DKD was retrieved from the GEO database (code: GSE209781), comprising single-cell profiles from 3 DKD patients and 3 healthy controls. Data processing was performed using the Seurat R package (version 5.0.3): Seurat objects were constructed from raw count matrices, followed by quality control filtering to retain cells with 300-7,000 detected genes (nFeature_RNA), mitochondrial gene percentage (percent.mt) < 10%, and red blood cell gene percentage (HB_percent) ≤ 3%.
Raw counts were normalized using the NormalizeData function. Highly variable genes (HVGs) were identified with the FindVariableFeatures function (variance-stabilizing transformation, vst method), selecting the top 2,000 HVGs for downstream analysis. Data scaling was performed using the ScaleData function with adjustment for percent.mt. Dimensionality reduction was conducted via PCA. The statistical significance of principal components (PCs) was evaluated using JackStraw and ScoreJackStraw tests, and the optimal number of PCs was determined by scree and elbow plots to retain relevant biological variance.
Batch effects were corrected using the Harmony method based on the first 20 PCs. Cell clustering was performed with FindClusters at a resolution of 0.8, and cell distribution was visualized via uniform manifold approximation and projection (UMAP). Cell types were annotated using canonical marker genes with reference to the CellMarker 2.0 database, enabling identification of distinct immune cell populations. Differential gene expression analysis within each cell type was conducted using the FindMarkers function, with thresholds set at |logFC| > 0.5 and adjusted P-value < 0.05.
In silico perturbation analysis
To interrogate the regulatory impact of core genes, we performed virtual knockout simulations using the scTenifoldKnk framework on the processed human renal scRNA-seq dataset. We constructed single-cell gene regulatory networks (scGRNs) from log-normalized expression matrices and compared the topological structures between the pseudo-knockout and wild-type manifolds. This analysis identified genes with significantly disrupted connectivity post-perturbation. Key parameters were set to ensure robustness: 500 HVGs, 10 subsampled networks (300 cells each), 3 PCs, and a minimum cluster size of 30 cells. Significant regulatory shifts were defined as an adjusted P < 0.05 and |log2FC| > 1.
Statistical analysis
Statistical analyses and data visualization were performed using R software (version 4.4.1) and GraphPad Prism 10.5.0. For in vivo experimental data, data are presented as mean ± standard deviation (SD). Comparisons between the two independent groups (db/m vs. db/db) were conducted using unpaired Student’s t-tests. For scRNA-seq data, differential expression markers were identified using the Wilcoxon rank-sum test, with P-values adjusted for multiple testing. Unless otherwise specified, a P-value < 0.05 was considered statistically significant.
RESULTS
Microarray datasets and differential gene analysis
The PCA method eliminated batch effects [Figure 1A and B]. After differential gene analysis on the merged microarray datasets, 275 upregulated genes and 184 downregulated genes were identified [Supplementary Table 1 (S3)]. Heatmaps and volcano plots were drawn to visualize the results [Figure 1C and D]. In the obtained results, the smaller the P-value, the higher the reliability of the differential gene expression.
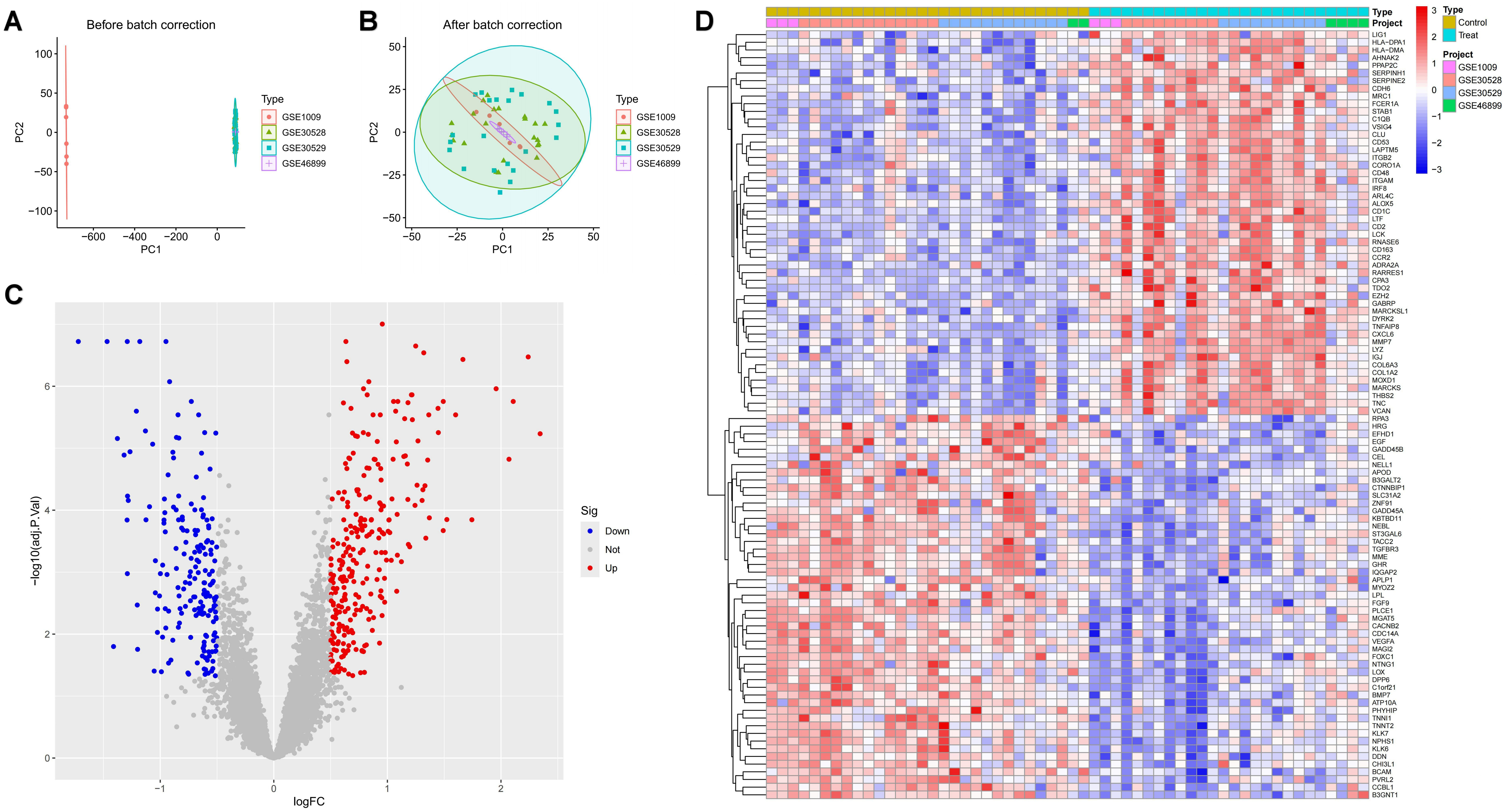
Figure 1. DKD microarray data and differential genes analysis. (A) Four GEO datasets before batch correction; (B) Four GEO datasets after batch correction; (C) Heatmap of top 50 upregulated and top 50 downregulated DEGs; (D) Volcano plot. DKD: Diabetic kidney disease; GEO: Gene Expression Omnibus; DEGs: differentially expressed genes.
MR analysis results
By using the two-sample MR method and the filtered eQTL data, 262 genes related to DKD were identified. These genes were used to intersect with DEGs, resulting in 7 key genes: chitinase 3-like 1 (CHI3L1), cystatin A (CSTA), interferon induced protein with tetratricopeptide repeats 1 (IFIT1), lipoprotein lipase (LPL), lysozyme (LYZ), testin LIM domain protein (TES), and transforming growth factor beta induced (TGFBI). The expression levels of CHI3L1, IFIT1, and LPL were downregulated, while those of CSTA, LYZ, TES, and TGFBI were upregulated.
MR analysis results showed that CSTA and IFIT1 were positively associated with the onset of DKD using the IVW method. CHI3L1, LPL, LYZ, TES, and TGFBI were all negatively associated with the onset of DKD [Figure 2A]. Four other methods consistently indicated that CSTA and IFIT1 increased the risk of DKD [odds ratio (OR) > 1], while CHI3L1, LPL, LYZ, TES, and TGFBI decreased the risk of DKD (OR < 1). The FDR test results for the IVW method all met P < 0.05, indicating high credibility of the findings
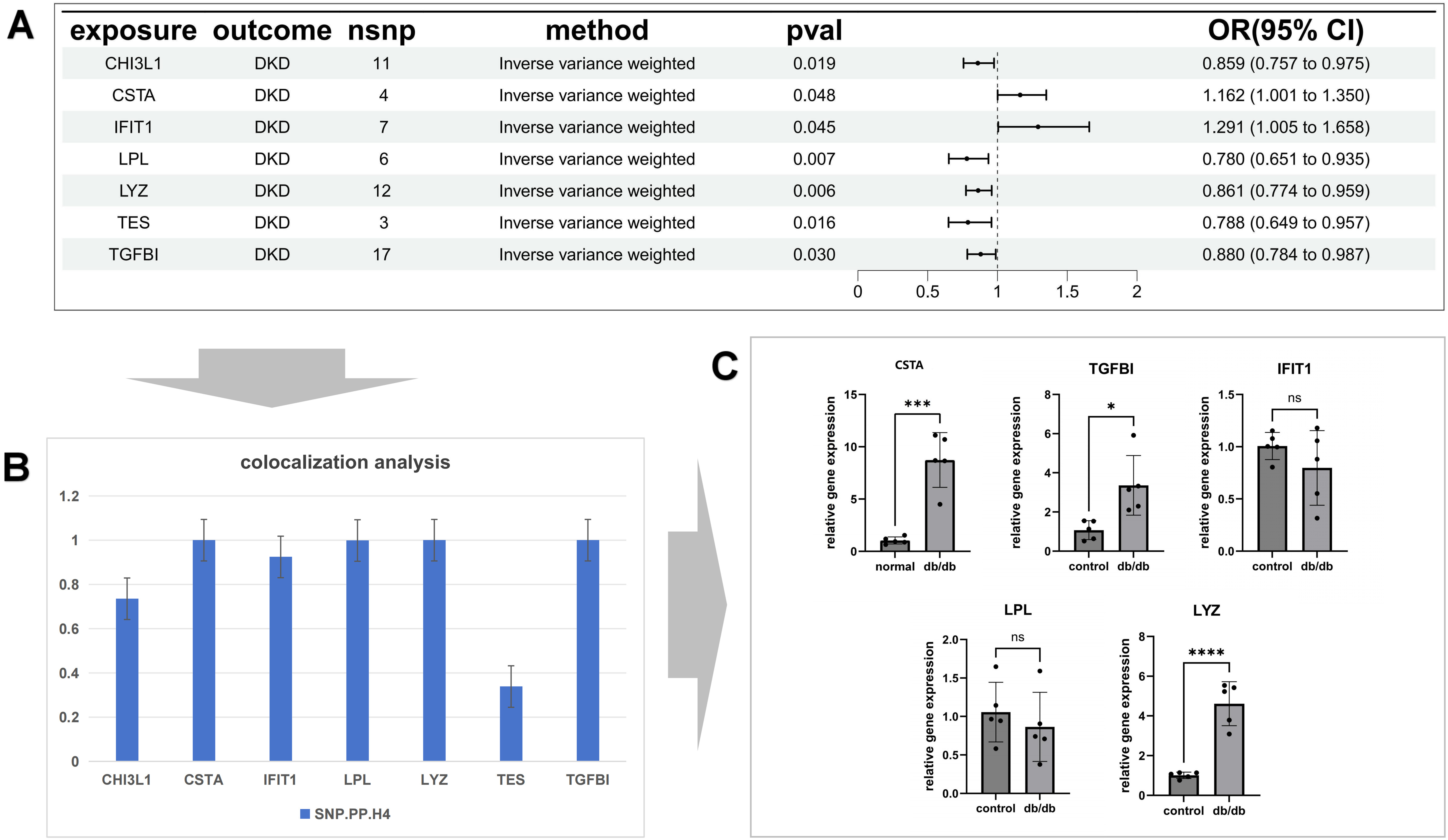
Figure 2. Identification of potential causal proteins for DKD via MR and experimental validation. (A) Forest plot of MR analysis showing the causal effects of seven plasma proteins on DKD risk using the IVW method. The OR and 95%CI are presented; (B) Colocalization analysis evaluating the probability of shared causal variants between protein expression and DKD. The y-axis represents the PP.H4; (C) Validation of relative gene expression levels for candidate targets (CSTA, TGFBI, IFIT1, LPL, and LYZ) in kidney tissues of db/db mice compared with controls (normal). Data are presented as mean ± SD. Statistical significance was determined using unpaired Student’s t-tests (*P < 0.05, ***P < 0.001, ****P < 0.0001; ns, not significant). DKD: Diabetic kidney disease; MR: Mendelian randomization; IVW: inverse-variance weighted; OR: odds ratio; CI: confidence interval; CSTA: cystatin A; TGFBI: transforming growth factor beta induced; IFIT1: interferon induced protein with tetratricopeptide repeats 1; LPL: lipoprotein lipase; LYZ: lysozyme; SD: standard deviation; CHI3L1: chitinase 3-like 1; TES: testin LIM domain protein; SNP: single nucleotide polymorphism.
By using the Cochran Q test based on the IVW method and MR-Egger intercept analysis, all results of tests for heterogeneity and horizontal pleiotropy showed P > 0.05, indicating no statistical significance and negligible heterogeneity and horizontal pleiotropy for the results [Supplementary Table 1 (S5) and (S6)]. The scatter plot results were used to show that the regression lines for the analysis of the impact of key genes on DKD by five methods remain consistent. The funnel plot results were used to demonstrate the basic symmetric distribution of causal effects, unaffected by potential biases. Leave-one-out sensitivity analysis was used to reveal that no single SNP had a significant impact on the overall results, thereby proving the robustness [Supplementary Figures 1-3].
Validation of the obtained key genes
Results of colocalization analysis
This study conducted colocalization analysis on seven key genes, further excluding the effects of potential linkage disequilibrium or other confounding factors. Results were detailed in Supplementary Figures 4 and 5 and Supplementary Table 2. The results indicated that, apart from CHI3L1 and TES, the remaining five genes met PP.H4 > 0.8, and thus may be potential therapeutic targets for DKD. Among these genes, CSTA, LPL, LYZ, and TGFBI were considered the strongest candidates for DKD treatment targets (PP.H4 > 0.99), with LYZ being particularly noteworthy (PP.H4 = 1). Additionally, IFIT1 was also considered to be closely related to the risk of DKD onset (PP.H4 = 0.92) [Figure 2B].
Results of qPCR validation
The qPCR analysis revealed that LYZ, CSTA, and TGFBI were significantly upregulated in DKD tissues compared to the db/m control group (LYZ: ****P < 0.0001; CSTA: ***P < 0.001; TGFBI: *P < 0.05) [Figure 2C]. The expression levels of IFIT1 and LPL showed a downward trend, but these changes did not reach statistical significance. The upregulation or downregulation trends of all genes were consistent with the previous differential gene expression analysis results. The qPCR results for LYZ (****P < 0.0001) and the colocalization analysis results (PP.H4 = 1) provided corroborative evidence.
Follow-up research results
GO, KEGG enrichment analyses
GO and KEGG enrichment analyses were performed to explore the potential roles of 7 key genes. The biological processes (BP) were enriched in very-low-density lipoprotein (VLDL) particle remodeling, regulation of helicase activity, and triglyceride-rich lipoprotein particle remodeling [Figure 3A]. The cellular components (CC) were enriched in specific granule lumen, specific granule, and secretory granule lumen [Figure 3B]. The molecular functions (MF) were enriched in extracellular matrix structural constituent, 1-acyl-2-lysophosphatidylserine acylhydrolase activity, and phosphatidylserine 1-acylhydrolase activity [Figure 3C]. KEGG enrichment analysis identified pathways including cholesterol metabolism, glycerolipid metabolism, and the peroxisome proliferator-activated receptor (PPAR) signaling pathway [Figure 3D and Supplementary Table 1 (S7)].
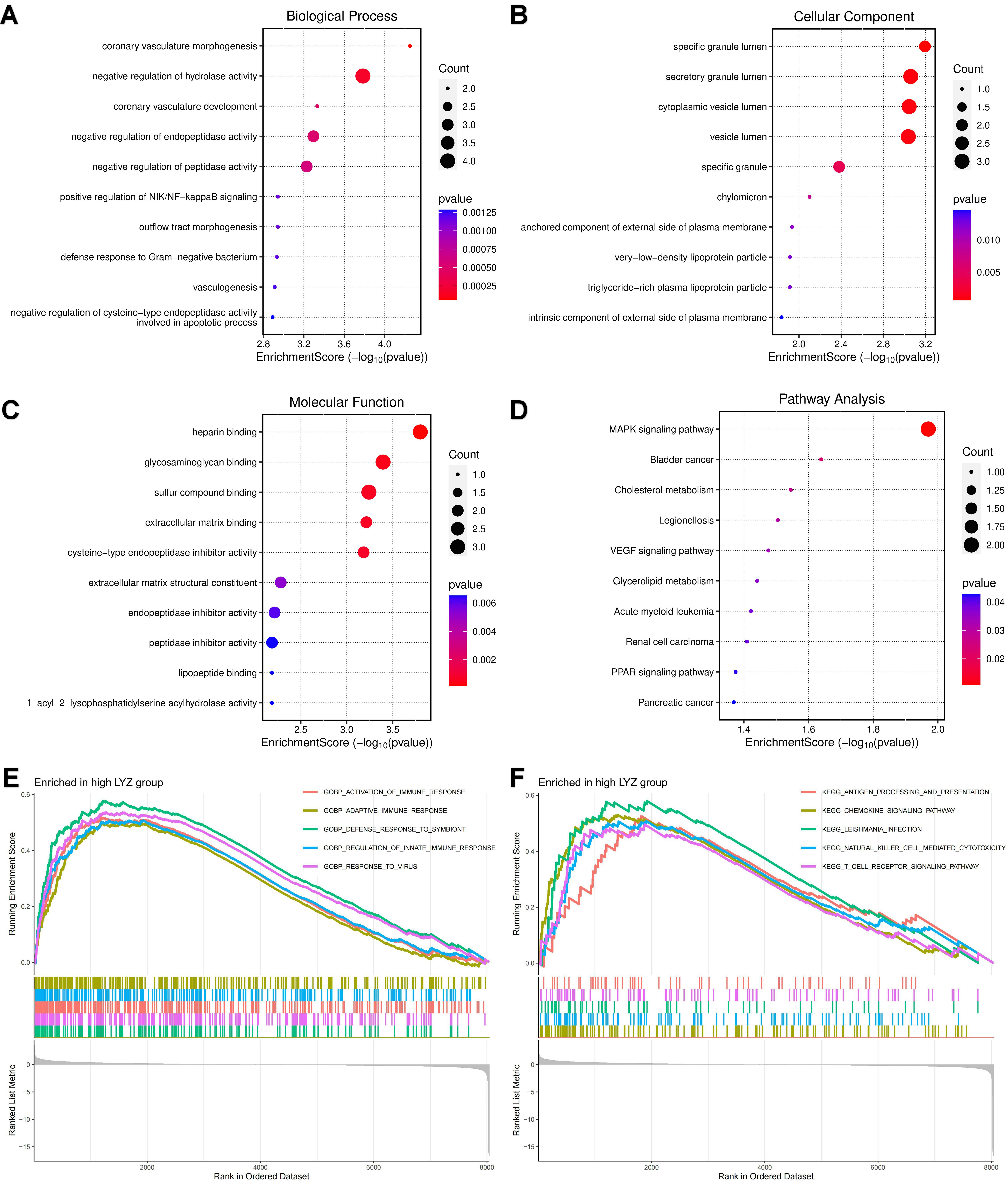
Figure 3. Enrichment analysis visualization. (A) Biological process enrichment results; (B) CC enrichment results; (C) MF enrichment results; (D) Pathway enrichment results; (E and F) LYZ’s different effects on the biological functions and pathways of DKD. CC: Cellular components; MF: molecular functions; LYZ: lysozyme; DKD: diabetic kidney disease; NIK: nuclear factor kappa-light-chain-enhancer of activated B cells-inducing kinase; NF-kappaB: nuclear factor kappa-light-chain-enhancer of activated B cells; VEGF: vascular endothelial growth factor; PPAR: peroxisome proliferator-activated receptor.
Results of GSEA on LYZ
GSEA was used to further explore the potential mechanisms of LYZ in DKD. The results showed that in DKD samples with high expression of LYZ, there were enrichments of pathways such as antigen processing and presentation, chemokine signaling pathway, Leishmania infection pathway, natural killer cell-mediated cytotoxicity pathway, and T cell receptor signaling pathway, and enrichments of BPs such as immune response activation, adaptive immune response, defense response to symbionts, regulation of innate immune response, and response to viruses [Figure 3E and F].
Assessment of immune cell infiltration in DKD
GO and KEGG enrichment analyses revealed associations between key genes and inflammatory/immune responses. The locations of key genes were marked on the chromosomes [Figure 4A]. The results of the CIBERSORT algorithm were used to analyze the phenotypes of 22 immune cells in DKD samples, as well as the associations between these 7 key genes and the immune cell phenotypes.
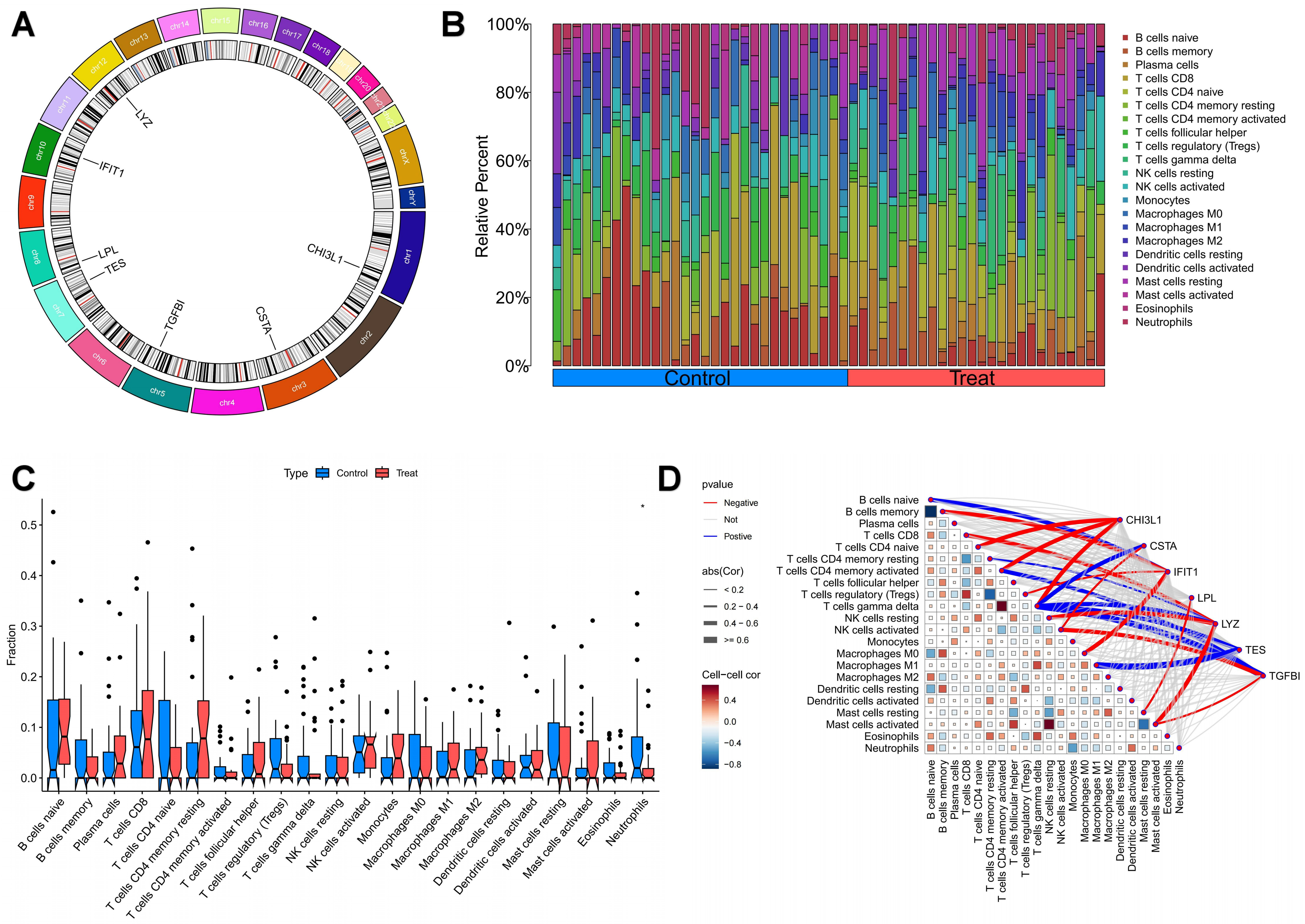
Figure 4. Visualization of immune infiltration assessment for key genes. (A) Circos plot of intersecting genes; (B) Stacked histogram of immune cell proportions between DKD groups and control groups; (C) Box plot comparing 22 immune cells between DKD groups and control groups; (D) Heatmap of correlations between 22 types of immune cells and key genes. Differences in immune cell infiltration between groups (C) were analyzed using the Wilcoxon rank-sum test. Correlations between key genes and immune cells in DKD samples (D) were assessed using Spearman’s rank correlation analysis. DKD: Diabetic kidney disease; LYZ: lysozyme; IFIT1: interferon induced protein with tetratricopeptide repeats 1; LPL: lipoprotein lipase; TES: testin LIM domain protein; TGFBI: transforming growth factor beta induced; CSTA: cystatin A; CHI3L1: chitinase 3-like 1; NK: natural killer.
CIBERSORT analysis revealed a distinct profile of immune activation in the DKD group compared to controls. Specifically, the proportions of M1 macrophages, activated CD4+ T cells, plasma cells, and activated dendritic cells were significantly elevated, while the abundance of naive and resting lymphocytes was correspondingly reduced [Figure 4B and C]. The proportion of resting CD4 memory T cells was positively correlated with the expression of LYZ. Activated CD4 memory T cells were positively correlated with the expression of TGFBI, and negatively correlated with the expression of CHI3L1. Gamma delta T cells were influenced by the largest number of key genes, activated by the expression of CSTA, LYZ, TES, and TGFBI, but inhibited by the expression of CHI3L1. Natural killer (NK) cells activated were negatively correlated with the expression of CSTA and TGFBI. Macrophages M1 were positively correlated with the expression of TES. Mast cells activated were negatively correlated with the expression of LYZ and TGFBI [Figure 4D and Supplementary Table 3].
Single-cell resolution unmasks cell-type-specific signatures of CSTA, LYZ, and TGFBI in DKD
After rigorous quality control, the GSE209781 dataset yielded 17,620 high-quality cells (8,830 Control; 8,790 DKD). Unsupervised clustering identified 22 distinct subclusters [Figure 5A]. UMAP visualization revealed density shifts between groups, indicating significant cellular remodeling in DKD [Figure 5B]. The subclusters were annotated into seven major lineages: macrophages, T cells, B cells, epithelial cells, endothelial cells, fibroblasts, and mixed cells [Figure 5C]. Quantitative analysis [Figure 5D] demonstrated an expansion of immune populations (macrophages, T cells, and B cells) in DKD, reflecting renal inflammation. Conversely, a reduction in endothelial cells was observed, consistent with DKD-associated microvascular injury.

Figure 5. Single-cell transcriptomic landscape and cell-type-specific expression validation of DKD-associated causal proteins. (A and B) UMAP plots of single-cell transcriptomes integrated from DKD and normal kidney samples, colored by unsupervised clusters (A) and sample group (B); (C) Circle packing visualization showing the annotation of major cell lineages, including T cells, B cells, epithelial cells, fibroblasts, macrophages, and mixed cells; (D) Stacked bar plot representing the relative proportion of each cell type in DKD vs. normal tissues; (E) Aggregate expression levels of candidate genes (CSTA, LYZ, and TGFBI) comparing DKD and normal groups across all cells; (F) Feature plots illustrating the spatial distribution and abundance of CSTA, LYZ, and TGFBI on the UMAP embedding, split by condition; (G-I) Cell-type-specific differential expression analysis of CSTA (G), LYZ (H), and TGFBI (I). Box plots display the normalized expression levels in DKD (blue) and normal (red) groups across identified cell types. Statistical significance was determined using the Wilcoxon rank-sum test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, not significant). DKD: Diabetic kidney disease; UMAP: uniform manifold approximation and projection; CSTA: cystatin A; LYZ: lysozyme; TGFBI: transforming growth factor beta induced.
We next investigated the expression heterogeneity of core genes (CSTA, LYZ, and TGFBI). While only CSTA exhibited significant differential expression at the bulk tissue level [Figure 5E], cell-specific analysis unmasked distinct regulatory patterns masked by global averaging [Figure 5F]. CSTA was significantly upregulated in DKD macrophages and epithelial cells [Figure 5G]. LYZ showed elevated expression in macrophages, epithelial, and mixed cells [Figure 5H], suggesting a pro-inflammatory macrophage phenotype. Global TGFBI expression showed no significant difference. However, it was specifically upregulated in endothelial cells, macrophages, fibroblasts, and epithelial cells [Figure 5I]. These findings implicate TGFBI as a broad driver of fibrosis and extracellular matrix (ECM) remodeling in the DKD microenvironment.
Results of simulated gene knockout
The study simulated the knockout of CSTA, LYZ, and TGFBI in DKD macrophages to investigate their regulatory roles [Supplementary Table 4]. Strikingly, silencing any of these genes consistently triggered a significant upregulation of major histocompatibility complex (MHC) class II molecules, such as human leukocyte antigen (HLA)-DQB1 and HLA-DPB1 [Figure 6]. This convergent pattern indicates that these genes naturally function to restrain antigen presentation. Consequently, their disruption in DKD may reprogram macrophages towards an active immunogenic phenotype by releasing these checkpoints.
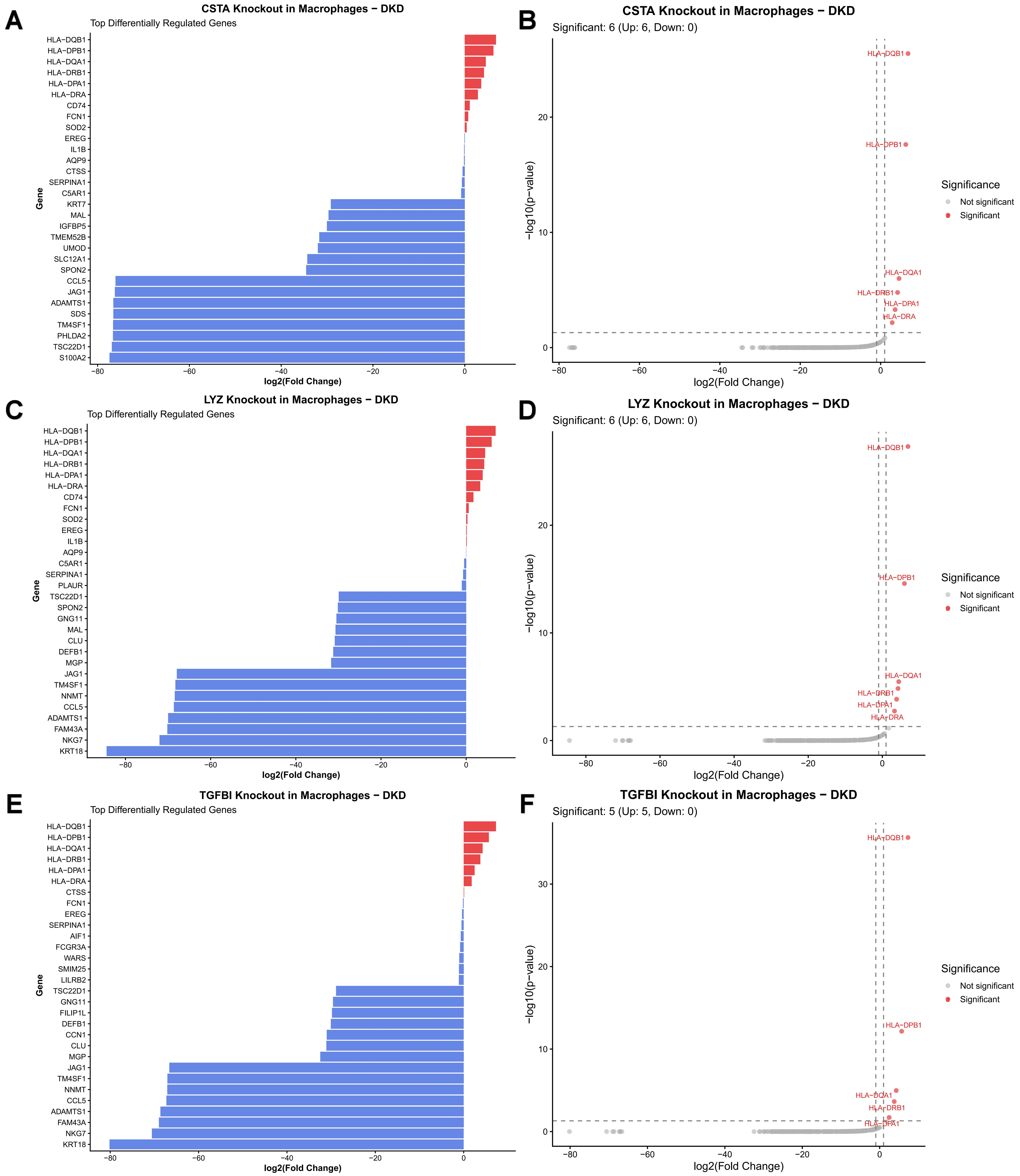
Figure 6. Transcriptional changes following simulated knockout of candidate genes in macrophages. (A and B) Simulated knockout of CSTA in macrophages of DKD patients, showing top DEGs ranked by fold change (A) and the corresponding volcano plot highlighting significant DEGs (B); (C and D) Simulated knockout of LYZ in macrophages of DKD patients, DEGs up- and down-regulation (C) and volcano plot (D); (E and F) Simulated knockout of TGFBI in macrophages of DKD patients, DEGs up- and down-regulation (E) and volcano plot (F). In the bar plots, red bars indicate upregulation and blue bars indicate downregulation. In the volcano plots, red dots represent statistically significant genes. Significance was determined using the scTenifoldKnk algorithm based on one-sided Chi-square tests (P < 0.05 and |log2FC| > 1). CSTA: Cystatin A; DKD: diabetic kidney disease; DEGs: differentially expressed genes; LYZ: lysozyme; TGFBI: transforming growth factor beta induced.
DISCUSSION
DKD is a severe complication of diabetes and a major driver of morbidity and premature mortality in people with diabetes. Despite its clinical burden, the mechanisms that initiate and sustain DKD are not fully understood, and current therapeutic options remain limited. This underscores the need to identify new therapeutic targets and to clarify the pathways that link metabolic stress to renal inflammation and fibrosis. In this study, we integrated differential gene expression analysis, MR, colocalization, complementary bioinformatics analyses, and qPCR validation. We leveraged microarray datasets together with GWAS and eQTL summary statistics. We identified several key genes (CHI3L1, CSTA, IFIT1, LPL, LYZ, TES, and TGFBI). Among them, CSTA, LYZ, and TGFBI emerged as the most plausible therapeutic targets for DKD. Colocalization and experimental validation consistently highlighted LYZ as the most significant candidate. These genes likely function as a coordinated pathogenic network rather than as isolated effectors. This network appears to connect disordered lipid metabolism with immune cell reprogramming and ECM remodeling.
A notable observation concerns the context-dependent role of LYZ. MR suggested a protective causal effect on DKD risk, yet LYZ was markedly upregulated in clinical samples and in animal models. This apparent discrepancy may reflect a biphasic response. Early induction of LYZ could be compensatory and protective under diabetic stress, including heightened susceptibility to infection. LYZ is a core effector of innate host defense. It cleaves bacterial peptidoglycan and also exerts broad immunomodulatory effects[16]. In line with this biology, our enrichment results implicated LYZ in antigen processing and presentation, as well as in activation of CD8+ T cells and NK cells. In early DKD, LYZ induction may therefore support innate defense and stress tolerance in the kidney[17]. However, sustained overexpression may become maladaptive. Prior studies indicate that LYZ can activate the Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) axis, exacerbate tubular epithelial injury (including apoptosis- and senescence-related phenotypes), and promote renal fibrogenesis[18]. Persistent innate immune activation can also maintain a “sterile inflammatory” milieu, with ongoing cytokine signaling and immune cell recruitment. This process can drive progressive tubular damage and accelerate DKD progression[17,19]. Consistent with this model, urinary LYZ-related mRNA signatures have been associated with tubulointerstitial injury and fibrosis, and they can predict renal function decline in DKD[20]. Collectively, these findings support a stage- and context-dependent role for LYZ: protective when transiently induced, but pathogenic when chronically elevated during sustained metabolic stress.
Metabolic reprogramming and lipid dysmetabolism are central features of DKD onset and progression[21]. In our data, LPL was strongly downregulated in DKD tissues, consistent with its inferred causal association with disease progression. LPL is a key triglyceride hydrolase. Together with GPIHBP1, it hydrolyzes triglycerides in chylomicrons and VLDL, releasing free fatty acids[22]. Reduced LPL expression in DKD could therefore contribute to intrarenal lipid deposition and lipid droplet accumulation. Such lipid overload has been linked to lipotoxic injury in tubular and glomerular compartments and to downstream coupling between inflammation and fibrosis[21,23]. At the systemic level, impaired LPL activity is closely associated with insulin resistance and hypertriglyceridemia, which may further increase metabolic stress on renal resident cells[22]. Increasing evidence also indicates that tissue lipid overload can trigger innate immune activation and fibrogenic remodeling in chronic kidney disease, including DKD[17,21]. In addition, genetic variation in LPL has been associated with DKD susceptibility and faster kidney function decline in type 2 diabetes cohorts[24]. These observations support the hypothesis that restoring LPL expression or correcting LPL pathway function could reduce renal lipotoxicity and potentially blunt the upstream trigger for inflammatory amplification[21,22].
The transition from inflammation to fibrosis is driven by ECM remodeling, and our analyses implicated CSTA and TGFBI in this process[19,25]. CSTA is an endogenous type I cystatin that inhibits lysosomal cysteine cathepsins and helps maintain the protease–antiprotease balance[26]. Because cysteine cathepsins contribute to extracellular proteolysis and matrix turnover, excessive cathepsin inhibition may impair physiological ECM degradation and promote matrix accumulation and sclerosis[25,27]. Beyond matrix biology, cystatin–cathepsin systems within immune cells also influence antigen processing/presentation and inflammatory signaling. CSTA upregulation may therefore reprogram macrophage function in ways that extend beyond proteolysis[26]. Notably, our data suggest that CSTA, LYZ, and TGFBI act in concert to suppress macrophage MHC-II molecules (HLA-DQB1 and HLA-DPB1). This pattern is compatible with an immunoregulatory and/or pro-fibrotic macrophage state that prioritizes tissue remodeling over antigen presentation and immune surveillance[19]. However, the functional consequences of altered MHC-II signaling are likely cell-type dependent. For example, MHC-II expression by proximal tubules can promote CD4+ T-cell activation and renal fibrosis in experimental models[28]. These observations emphasize the need to interpret MHC-II changes in a cell-resolved manner.
TGFBI emerged as another key regulator of fibrotic remodeling in our integrative analyses[29,30]. TGFBI is a secreted ECM protein that binds ECM components and integrins, thereby influencing cell adhesion, matrix organization, and tissue remodeling[31]. Under chronic injury, TGFBI may amplify TGF-β-driven profibrotic programs, a central pathway governing myofibroblast activation and matrix deposition[25,31]. TGFBI has also been linked to fibroblast activation and matrix remodeling enzyme programs [including matrix metalloproteinase (MMP)-related pathways] in multiple disease contexts[31]. In DKD, TGFBI [often together with fibronectin 1 (FN1)] has been proposed as a macrophage-associated injury marker and correlates with worse renal function[30]. Co-expression of CHI3L1 with TGFBI may further reinforce a profibrotic niche. CHI3L1 regulates apoptosis-associated responses, inflammasome-related inflammation, macrophage polarization, and ECM remodeling[32]. In kidney injury models, breast regression protein 39 (BRP-39)/CHI3L1 has been shown to promote macrophage–myofibroblast crosstalk and profibrotic signaling during maladaptive repair[33].
The identification of IFIT1 as a risk gene adds complexity to DKD pathogenesis. IFIT1 is an interferon-stimulated gene best known for its role in antiviral defense[34]. In human macrophages, however, IFIT1 can exert context-dependent effects. It can restrain subsets of pro-inflammatory genes while supporting interferon-stimulated gene programs[34]. Thus, IFIT1-associated genetic risk in DKD may reflect dysregulated interferon–inflammatory crosstalk and an exaggerated “antiviral-like” response to endogenous danger signals during chronic metabolic injury[17,34].
Several limitations should be considered. First, our MR and eQTL analyses primarily used summary statistics from European cohorts. Genetic heterogeneity across populations may limit generalizability to non-European groups. In addition, although we validated key targets in a db/db mouse model, species differences in immune and metabolic regulation warrant confirmation in human-relevant systems and diverse clinical cohorts. Second, our genetic instruments were derived from blood eQTLs, which capture systemic regulatory effects but may miss kidney-specific cis-regulatory mechanisms shaped by the renal microenvironment. Third, immune cell composition was estimated using CIBERSORT. While informative, these in silico results require experimental validation (e.g., flow cytometry or multiplex immunohistochemistry). Fourth, our in vivo validation focused on transcriptional changes in a small cohort (n = 5 per group). We did not perform protein-level validation (e.g., Western blot) or detailed histological assessments, which limits pathological inference. Finally, both the public microarray datasets and our validation cohort had modest sample sizes, which may reduce power to detect subtle effects despite the integrative design.
In summary, we propose a model in which metabolic lipotoxicity driven by LPL downregulation initiates innate immune activation (LYZ and IFIT1). This response is subsequently shaped by protease inhibition (CSTA) to favor a fibrotic, immune-remodeling microenvironment (TGFBI and CHI3L1). This metabolic–immune–fibrotic axis highlights actionable nodes for precision therapeutic development. However, the molecular links between these modules remain incompletely defined. Future work should clarify how LPL-associated lipid accumulation and lipid droplet dysregulation engage innate immune signaling pathways that culminate in LYZ induction. It will also be essential to determine whether macrophage MHC-II downregulation primarily reflects impaired antigen presentation or instead marks polarization toward immunoregulatory and pro-fibrotic states, while accounting for cell-type-specific roles of MHC-II in renal fibrosis. Large, multicenter studies will be required to validate the robustness, clinical utility, and translational potential of these targets.
DECLARATIONS
Acknowledgments
We thank the eQTLGen consortium and other GWAS researchers and participants for providing publicly available data for this study.
Authors’ contributions
Conceptualized and designed the study: Hu D, Zhang L, Zhao L
Wrote the original draft: Hu D, Tang R
Experimentally validated the obtained results through qPCR assays: Wang Y
Participated in the image sorting: Hang X
Participated in the collection and organization of the literature: Tang R, Wei Y, Zhou L, Lin R, Wang R
Responsible for reviewing manuscripts and securing funding: Zhang L, Zhao L
All authors have read and approved the final manuscript.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This study was supported by the National Science and Technology Major Project (2023ZD0509300), the Natural Science Foundation of Beijing Municipality (7244497), the Clinical and Basic Research on Shen Zhuo Formula for the Treatment of DKD Based on the Theory of “State Target Differentiation and Treatment” (EF-YS-002), the Clinical Research Fund of the Central High-Level Hospital of Traditional Chinese Medicine (No. HLCMHPP2023084), the Young Elite Scientists Sponsorship Program by CACM (CACM-2023-QNRC2-A08), and the Escort Project of Guang’anmen Hospital, China Academy of Chinese Medical Sciences (Backbone Talent Training Project) (Grant No. GAMHH9324025).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
This experiment was approved by the Animal Ethics Committee of the Changchun University of Chinese Medicine (approval No. 2025363) and strictly followed animal ethics. There were no human subjects in this article, and informed consent was not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Gaddy A, Elrggal M, Madariaga H, Kelly A, Lerma E, Colbert GB. Diabetic kidney disease. Dis Mon. 2025;71:101848.
2. Hu H, Liang W, Ding G. Ion homeostasis in diabetic kidney disease. Trends Endocrinol Metab. 2024;35:142-50.
3. Jin H, Kim YA, Lee Y, et al. Identification of genetic variants associated with diabetic kidney disease in multiple Korean cohorts via a genome-wide association study mega-analysis. BMC Med. 2023;21:16.
4. Dwivedi OP, Barreiro K, Käräjämäki A, et al.; iBEAt. Genome-wide mRNA profiling in urinary extracellular vesicles reveals stress gene signature for diabetic kidney disease. iScience. 2023;26:106686.
5. Jiang WJ, Xu CT, Du CL, et al. Tubular epithelial cell-to-macrophage communication forms a negative feedback loop via extracellular vesicle transfer to promote renal inflammation and apoptosis in diabetic nephropathy. Theranostics. 2022;12:324-39.
6. Zhang JQ, Li YY, Zhang XY, et al. Cellular senescence of renal tubular epithelial cells in renal fibrosis. Front Endocrinol. 2023;14:1085605.
7. Guo M, He F, Zhang C. Molecular therapeutics for diabetic kidney disease: an update. Int J Mol Sci. 2024;25:10051.
8. Wang N, Zhang C. Oxidative stress: a culprit in the progression of diabetic kidney disease. Antioxidants. 2024;13:455.
9. Li L, Fu H, Liu Y. The fibrogenic niche in kidney fibrosis: components and mechanisms. Nat Rev Nephrol. 2022;18:545-57.
10. Chen Y, Zou H, Lu H, Xiang H, Chen S. Research progress of endothelial-mesenchymal transition in diabetic kidney disease. J Cell Mol Med. 2022;26:3313-22.
11. Westra HJ, Peters MJ, Esko T, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238-43.
12. Sakaue S, Kanai M, Tanigawa Y, et al.; FinnGen. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet. 2021;53:1415-24.
14. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383.
15. Wang H, Fang F, Zhang M, et al. Nuclear receptor 4A1 ameliorates renal fibrosis by inhibiting vascular endothelial growth factor A induced angiogenesis in UUO rats. Biochim Biophys Acta Mol Cell Res. 2024;1871:119813.
16. Ragland SA, Criss AK. From bacterial killing to immune modulation: recent insights into the functions of lysozyme. PLoS Pathog. 2017;13:e1006512.
18. Ren Y, Yu M, Zheng D, He W, Jin J. Lysozyme promotes renal fibrosis through the JAK/STAT3 signal pathway in diabetic nephropathy. Arch Med Sci. 2023;20:233-47.
19. Tang PM, Nikolic-Paterson DJ, Lan H. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol. 2019;15:144-58.
20. Lee YH, Seo J, Kim M, et al. Urinary mRNA signatures as predictors of renal function decline in patients with biopsy-proven diabetic kidney disease. Front Endocrinol. 2021;12:774436.
21. Mitrofanova A, Merscher S, Fornoni A. Kidney lipid dysmetabolism and lipid droplet accumulation in chronic kidney disease. Nat Rev Nephrol. 2023;19:629-45.
22. Kurooka N, Eguchi J, Wada J. Role of glycosylphosphatidylinositol‐anchored high‐density lipoprotein binding protein 1 in hypertriglyceridemia and diabetes. J Diabetes Investig. 2023;14:1148-56.
23. Folestad E, Mehlem A, Ning FC, et al. Vascular endothelial growth factor B-mediated fatty acid flux in the adipose-kidney axis contributes to lipotoxicity in diabetic kidney disease. Kidney Int. 2025;107:492-507.
24. Wu Y, Cheng S, Gu H, et al. Variants within the LPL gene confer susceptility to diabetic kidney disease and rapid decline in kidney function in Chinese patients with type 2 diabetes. Diabetes Obes Metab. 2023;25:3012-9.
25. Meng X, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325-38.
26. Kopitar-Jerala N. The role of cystatins in cells of the immune system. FEBS Lett. 2006;580:6295-301.
27. Wang H, Inoue A, Lei Y, Wu H, Hong L, Cheng XW. Cathepsins in the extracellular space: focusing on non-lysosomal proteolytic functions with clinical implications. Cell Signal. 2023;103:110531.
28. Zhou Y, Luo Z, Liao C, et al. MHC class II in renal tubules plays an essential role in renal fibrosis. Cell Mol Immunol. 2021;18:2530-40.
29. Zhang W, Ma L, Zhou Q, Gu T, Zhang X, Xing H. Therapeutic targets for diabetic kidney disease: proteome-wide mendelian randomization and colocalization analyses. Diabetes. 2024;73:618-27.
30. Dou F, Liu Q, Lv S, et al. FN1 and TGFBI are key biomarkers of macrophage immune injury in diabetic kidney disease. Medicine. 2023;102:e35794.
31. Huang H, Tang Q, Li S, Qin Y, Zhu G. TGFBI: a novel therapeutic target for cancer. Int Immunopharmacol. 2024;134:112180.
32. Zhao T, Su Z, Li Y, Zhang X, You Q. Chitinase-3 like-protein-1 function and its role in diseases. Sig Transduct Target Ther. 2020;5:201.
33. Montgomery TA, Xu L, Mason S, et al. Breast regression protein–39/chitinase 3–like 1 promotes renal fibrosis after kidney injury via activation of myofibroblasts. J Am Soc Nephrol. 2017;28:3218-26.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].