


JOSD2 promotes cardiomyocyte apoptosis and exacerbates cardiac injury after myocardial infarction via LKB1-AMPK suppression
 0
0 Abstract
Aim: This study aimed to identify key ubiquitin-regulating molecules involved in cardiomyocyte survival under ischemic injury and to investigate the role of Josephin domain-containing protein 2 (JOSD2) in ischemic stress-induced cardiomyocyte death after myocardial infarction (MI).
Methods: A ubiquitin-related single-guide RNA (sgRNA) library targeting 903 genes was screened in AC16 cells under oxygen-glucose deprivation (OGD), followed by sgRNA sequencing. Integrated analysis of multiple transcriptomic datasets was performed to identify JOSD2 as a candidate. Stable JOSD2 knockdown and overexpression cell models were established. Reverse transcription quantitative polymerase chain reaction (RT-qPCR), Western blot, flow cytometry, terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining, and Cell Counting Kit-8 (CCK-8) assays were performed to evaluate OGD-induced cardiomyocyte apoptosis and survival. Transcriptomic analysis, co-immunoprecipitation (co-IP), and deubiquitination assays were performed to explore the underlying mechanism. Adeno-associated virus 9 (AAV9)-mediated cardiomyocyte-specific JOSD2-overexpressing mice were used to evaluate cardiac remodeling and function after MI.
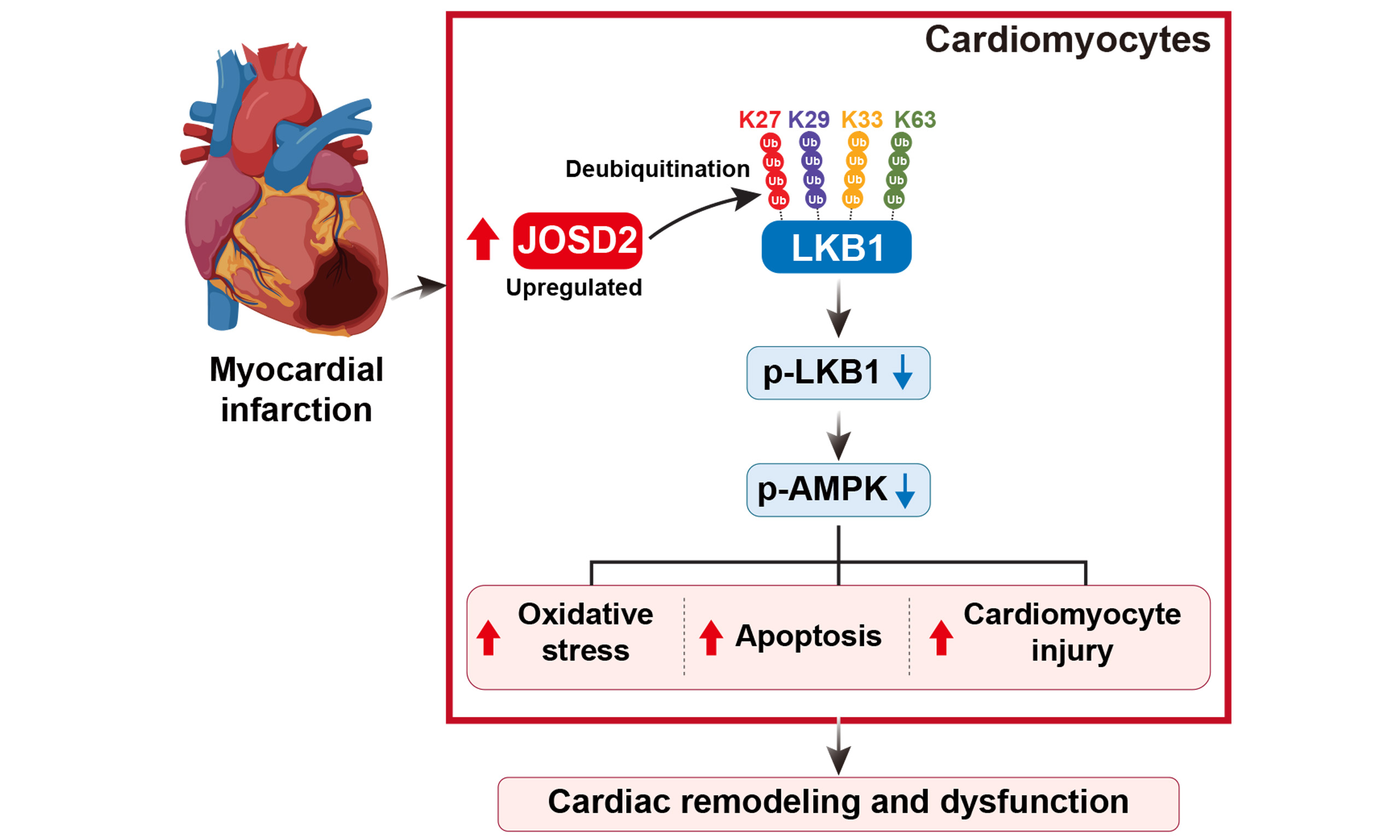
Results: The CRISPR-Cas9 screen identified a series of ubiquitin-regulating candidate genes associated with cardiomyocyte survival under ischemic stress. Combined transcriptomic and experimental analyses showed that JOSD2 was significantly upregulated in cardiomyocytes and mouse hearts after ischemic injury. JOSD2 knockdown attenuated OGD-induced cardiomyocyte apoptosis, whereas JOSD2 overexpression increased apoptosis. Mechanistically, JOSD2 reduced K27-, K29-, K33-, and K63-linked polyubiquitin chains on liver kinase B1 (LKB1), suppressed its activity, and inhibited downstream AMPK (AMP-activated protein kinase) phosphorylation. In vivo, cardiomyocyte-specific JOSD2 overexpression promoted cardiomyocyte death and aggravated cardiac remodeling and dysfunction after MI.
Conclusion: JOSD2 is upregulated in cardiomyocytes after MI and may promote post-infarction cell death and cardiac remodeling by suppressing the LKB1-AMPK pathway. Targeting JOSD2 may represent a potential therapeutic strategy for MI.
Keywords
INTRODUCTION
Myocardial infarction (MI) remains a major cause of disease burden worldwide, contributing substantially to morbidity and mortality. Despite the adoption of modern reperfusion strategies and guideline-directed medical therapy, the irreversible loss of cardiomyocytes and subsequent adverse cardiac remodeling continue to drive the progression to heart failure, imposing a long-term clinical burden on patients[1,2]. The sudden cessation of coronary artery perfusion during MI triggers a series of complex pathological processes, including oxidative stress, inflammation, disruption of cellular energy metabolism, mitochondrial dysfunction, and activation of multiple regulated cell death pathways in cardiomyocytes[3,4]. At the molecular level, ischemic stress activates a series of pro-apoptotic signaling events, including alterations in the BCL-2-associated X protein (BAX)/B-cell lymphoma 2 (BCL-2) balance and activation of the caspase-3 executioner caspase cascade, which contribute to programmed cardiomyocyte apoptosis and loss of contractile cells[5,6].
The pathological process of MI involves multiple signaling pathways, including phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt), AMP-activated protein kinase (AMPK), Janus kinase/signal transducers and activators of transcription (JAK/STAT), and nuclear factor kappa B (NF-κB)[7-9], each of which plays a key role in MI pathogenesis. However, direct intervention in these core pathways faces significant challenges in clinical translation, primarily due to the wide expression of regulatory molecules in these pathways and their involvement in maintaining various aspects of physiological homeostasis. Systemic modulation of these pathways often leads to unpredictable side effects, such as the potential weakening of host defense by widespread inhibition of NF-κB[10]. More importantly, in the dynamic repair process after MI, the same pathway may exert distinct or even opposing roles across different phases, as illustrated by the divergent roles of the inflammatory response in the acute and repair phases[11]. Therefore, identifying specific and precise regulatory targets under pathological conditions is essential for advancing post-MI therapeutic strategies.
Ubiquitination is a highly conserved and versatile post-translational modification that precisely regulates protein stability, localization, and function through spatiotemporally controlled substrate recognition and modification. E3 ubiquitin ligases and deubiquitinases selectively recognize and bind to specific target proteins, thereby regulating their fate and activity. A large body of evidence indicates that dysregulation of this system is closely associated with various cardiac pathological processes, including cardiac hypertrophy, heart failure, and ischemic myocardial injury[12-15]. In recent years, CRISPR-Cas9-based genetic screening has become a powerful tool for unbiased and systematic identification of key regulatory factors in complex biological processes. It has been widely applied in studies of stress responses, cell fate determination, and disease pathways, among others[16-21]. In the complex pathological environment of MI, numerous members of the ubiquitin system form intricate regulatory networks through synergistic or antagonistic actions[22]. To date, systematic analyses of key ubiquitination regulators that determine cardiomyocyte survival in the context of MI remain limited.
In this study, we employed a CRISPR-Cas9 loss-of-function screening strategy targeting ubiquitin-related genes to systematically screen cardiomyocytes using an in vitro ischemia model induced by oxygen-glucose deprivation (OGD). The screen identified the deubiquitinase Josephin domain-containing protein 2 (JOSD2) as a key candidate. JOSD2 expression is induced by ischemic stress both in vitro and in murine hearts post-MI. Functional studies showed that JOSD2 knockdown mitigates cardiomyocyte apoptosis and injury upon OGD, whereas its cardiomyocyte-specific overexpression worsens infarct size, impairs cardiac function, and aggravates post-MI remodeling. Mechanistically, we found that JOSD2 interacts with and deubiquitinates liver kinase B1 (LKB1), thereby attenuating downstream AMPK phosphorylation and activity. These findings may have translational relevance because direct targeting of core kinases such as AMPK is often constrained by their broad expression and central roles in cellular energy homeostasis, raising concerns regarding specificity and systemic effects. In contrast, JOSD2 may represent a more selective regulatory node, offering the possibility of modulating stress-responsive signaling with greater context specificity. Taken together, our results establish JOSD2 as a novel regulator of cardiomyocyte survival during ischemic stress and identify it as a promising upstream therapeutic target for MI.
MATERIALS AND METHODS
Cell culture
AC16 cells were maintained in a humidified incubator at 37 °C with 5% CO2. The cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Sigma, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Biosharp, China). The culture medium was replaced every 1-2 days.
OGD
To mimic ischemic stress, cells were rinsed with Phosphate Buffered Saline (PBS) and incubated in glucose-free DMEM (Procell, Cat. No. PM150270) in a tri-gas incubator maintained at 37 °C with 1% O2, 5% CO2, and 94% N2. For AC16 cells, OGD was performed for 6 or 9 h to induce stress responses or apoptosis/end-point assays, as indicated. For primary neonatal rat ventricular myocytes (NRVMs), OGD was performed under the same conditions for
Isolation and culture of NRVMs
NRVMs were isolated from 1-2-day-old Sprague-Dawley rats using a protocol adapted from previous studies. Briefly, ventricular tissues were minced and digested using a serial trypsinization procedure. First, 40 mL of 0.125% trypsin solution was prepared by mixing 15 mL of 0.25% trypsin (Gibco, USA) with 25 mL of 0.05% trypsin (Gibco, USA), the tissues were incubated at 4 °C in a solution containing 10 mL of 0.125% trypsin overnight, followed by repeated digestion at 37 °C for 7 min per cycle until complete tissue dissociation was achieved. The supernatants were collected and centrifuged at 1,500 rpm for 10 min. Red blood cells were removed using red blood cell lysis buffer (Biosharp, China), and the remaining cell pellet was resuspended in DMEM supplemented with 10% fetal bovine serum. To reduce fibroblast contamination, the cells were pre-plated for 90-120 min to allow preferential fibroblast attachment. Cardiomyocytes remaining in suspension were then collected, resuspended in F12 medium (Gibco, USA) containing 10% FBS, and seeded for subsequent experiments.
CRISPR-Cas9 ubiquitin gene knockout library screening
A focused CRISPR-Cas9 loss-of-function screen was performed to identify ubiquitin-related regulators of cardiomyocyte survival under OGD conditions. A pooled lentiviral human ubiquitin-related single guide RNA (sgRNA) library targeting 903 ubiquitin-related genes in the lentiCRISPR v2 backbone was used, with 10 sgRNAs per gene and 9,030 sgRNAs in total. AC16 cells (1.33 × 107) were transduced at a low multiplicity of infection (MOI < 0.3) to favor single-sgRNA integration in individual cells, achieving approximately 500-fold library coverage per sgRNA. 24 h after transduction, cells were selected with puromycin
Construction of stable JOSD2 knockdown and overexpression cell lines
For lentiviral shRNA-mediated knockdown, three independent shRNA constructs targeting human JOSD2 (sh-1, sh-2, and sh-3) and a scrambled shRNA control (sh-Scr) were cloned into lentiviral vectors. The shRNA target sequences were as follows: sh-1, 5’-CAGGTAGATGGCATCTACTAT-3’; sh-2, 5’-GCATCTACTATAATCTGGACT-3’; and sh-3, 5’-GCAACTATGATGTCAACGTGA-3’. HEK293T cells were transfected with the shRNA constructs together with the packaging plasmids psPAX2 and pMD2.G using polyethyleneimine (PEI; Yeasen, Cat. No. 40816ES03). Viral supernatants were collected at 48 and 72 h after transfection, filtered through a 0.45-µm filter, and used to infect AC16 cells in the presence of 5 µg/mL polybrene. Stable transductants were selected with puromycin, and knockdown efficiency was confirmed by reverse transcription quantitative polymerase chain reaction (RT-qPCR) and Western blotting before further experiments.
For lentiviral overexpression, full-length human JOSD2 cDNA (NM_001270639.2) was amplified by PCR and cloned into the pHAGE-3 × Flag overexpression vector. Virus production and transduction were performed as described above. Stably transduced cells expressing JOSD2 or the empty vector control were selected with puromycin, and overexpression was confirmed by RT-qPCR and Western blotting.
RNA extraction and RT-qPCR
Total RNA was extracted using TRIzol reagent (Vazyme, Cat. No. R411-01), and 1 µg of total RNA was reverse-transcribed into cDNA using the ReverTra Ace qPCR RT Kit (TOYOBO, Cat. No. FSQ-301) with random hexamers. Gene expression was measured by SYBR-based quantitative PCR and normalized to ACTB for human samples or Actb for mouse samples. Relative expression levels were calculated using the 2-ΔΔCt method. Primer sequences are listed in Supplementary Table 1.
Western blotting
Heart tissues or cultured cells were lysed in Radio Immunoprecipitation Assay (RIPA) buffer (Beyotime, China) supplemented with protease and phosphatase inhibitors. Protein concentrations were determined using a Bicinchoninic Acid (BCA) assay. Equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS PAGE) and transferred onto Polyvinylidene Fluoride (PVDF) membranes (Millipore, USA). After blocking with 5% skim milk, the membranes were incubated overnight at 4 °C with primary antibodies, followed by incubation with Horseradish peroxidase (HRP)-conjugated secondary antibodies. Antibody sources and dilutions are listed in Supplementary Table 2. Protein signals were detected using ECL reagent (Epizyme, China), and densitometric analysis was performed using ImageJ, with β-actin used as the loading control.
Annexin V-fluorescein isothiocyanate/propidium iodide flow cytometry
After the indicated treatments, cells were harvested, washed, and stained with Annexin Fluorescein Isothiocyanate (FITC) and propidium iodide (PI) according to the manufacturer’s instructions. Flow cytometric analysis was performed using a FACScan instrument, and the percentages of viable, early apoptotic, and late apoptotic/dead cells were quantified using FlowJo software.
TdT mediated dUTP nick end labeling staining
Cells grown on coverslips were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. Apoptotic cells were labeled using a Tetramethyl-Rhodamine-5-dUTP (TMR) Red TdT mediated dUTP Nick End Labeling (TUNEL) Detection Kit (Servicebio, Cat. No. MPC2503023) according to the manufacturer’s instructions. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Fluorescence images were acquired, and TUNEL-positive cells were quantified.
CCK-8 viability assay
Cells were seeded in 96-well plates at a density of 1 × 104 cells per well. After the indicated treatments, CCK-8 reagent (Dojindo) was added according to the manufacturer’s instructions. Following incubation at 37 °C for 4 h, absorbance at 450 nm was measured using a microplate reader.
RNA sequencing and bioinformatics
Total RNA was extracted, and poly(A)+ RNA was enriched for library preparation. Libraries were sequenced on a DNBSEQ (DNA Nanoball Sequencing) platform in 150-bp paired-end mode. Raw FASTQ reads were quality-trimmed using SOAPnuke and aligned to the human reference genome (GRCh38.p14) using HISAT2 v2.2.1. Gene-level read counts were obtained using RNA-Seq by Expectation Maximization (RSEM), and differential expression analysis was performed using DESeq2 v1.34.0. Differentially expressed genes were identified using thresholds of |log2 fold change| > 0.58 and an adjusted P value (Benjamini-Hochberg false discovery rate) < 0.05. All samples were prepared and sequenced in a single batch, and no obvious batch effects were observed. Principal component analysis (PCA), heatmaps, and volcano plots were generated from normalized counts. Gene Ontology (GO) enrichment analysis was performed to identify biological processes affected by JOSD2 silencing. The raw sequencing data have been deposited in the NCBI SRA under BioProject accession number PRJNA1422660.
Co-immunoprecipitation
Plasmids encoding Flag-tagged JOSD2, HA-tagged LKB1, or HA-tagged AMPK were transfected into HEK293T cells. At 24-48 h after transfection, cells were lysed in immunoprecipitation buffer supplemented with protease and phosphatase inhibitors, and the lysates were clarified by centrifugation. The clarified lysates were incubated with anti-Flag or anti-HA magnetic beads overnight at 4 °C. After washing, the bound proteins were eluted with SDS loading buffer and subjected to Western blot analysis. In addition, endogenous co-immunoprecipitation was performed in AC16 cells to confirm the interaction between endogenous JOSD2 and LKB1. Briefly, cell lysates were incubated with an anti-JOSD2 antibody, and the immune complexes were captured with Protein A/G beads, washed, and analyzed by Western blotting to detect co-precipitated LKB1. Input and IgG controls were included to verify specificity.
Ubiquitination assays
AC16 cells expressing Flag-tagged JOSD2, HA-tagged LKB1, and Myc-tagged ubiquitin or ubiquitin mutants were treated with 20 μM MG132 for 6 h. HA-LKB1 was immunoprecipitated using anti-HA magnetic beads, and ubiquitination was detected by Western blotting with an anti-Myc antibody. To determine the specific ubiquitin linkage types regulated by JOSD2, linkage-specific ubiquitination assays were performed using Myc-tagged wild-type ubiquitin and K-only mutants (K6O, K11O, K27O, K29O, K33O, K48O, and K63O), in which only the indicated lysine residue is retained and all other lysines are replaced with arginine. Linkage-specific ubiquitination of LKB1 was assessed by HA immunoprecipitation followed by anti-Myc immunoblotting.
siRNA-mediated LKB1 knockdown
To achieve transient LKB1 knockdown, AC16 cells were transfected with LKB1-specific or control siRNAs using Lipofectamine 2000 according to the manufacturer’s instructions. Three independent siRNAs targeting LKB1 were designed and tested. The siRNA target sequences were as follows: si-LKB1-1, 5’-GTGTAGAACAATCGTTTCTGTTG-3’; si-LKB1-2, 5’-AAGGGTTTTTCCCTTCCTTTTGG-3’; and si-LKB1-3, 5’-CCGTCAAGATCCTCAAGAAGAAG-3’. Knockdown efficiency was evaluated by RT-qPCR.
Seahorse mitochondrial respiration assay
Oxygen consumption rate (OCR) was measured using a Seahorse XFe24 Analyzer (Agilent Technologies) with the XF Cell Mito Stress Test Kit to assess mitochondrial respiration. Stable sh-Scr and sh-JOSD2 AC16 cells were seeded in XF24 plates at the optimal density, transfected with si-NC or si-LKB1, and subjected to OGD treatment. After OGD, the cells were incubated in Seahorse XF assay medium in a non-CO2 incubator before OCR measurement. Basal OCR was recorded first, followed by sequential injections of oligomycin
Mouse MI model
Male C57BL/6J mice aged 8-10 weeks were housed under specific pathogen-free (SPF) conditions with a 12 h light/dark cycle, controlled temperature (22 ± 2 °C), and humidity of 50%-60%, with free access to food and water. Mice were anesthetized with isoflurane and randomly assigned to the sham or MI groups. Unless otherwise indicated in the figure legends, six mice were included per group. MI was induced by permanent ligation of the left anterior descending (LAD) coronary artery with an 8-0 suture. Successful ligation was confirmed by immediate blanching of the anterior ventricular wall. Sham-operated mice underwent the same surgical procedure without coronary ligation. Postoperative analgesia was provided with buprenorphine (0.05 mg/kg, s.c.), and mice were closely monitored for 48 h.
At predetermined endpoints, mice were euthanized by intraperitoneal overdose of sodium pentobarbital
To minimize bias, investigators responsible for postoperative care, data acquisition, and data analysis were blinded to group allocation whenever feasible. All experimental procedures were approved by the Experimental Animal Welfare Ethics Committee of Zhongnan Hospital (Approval No. ZN2023239).
Adeno-associated virus 9-mediated cardiomyocyte-specific JOSD2 overexpression
Recombinant adeno-associated virus 9 (AAV9) vectors carrying JOSD2 or control ZsGreen under the control of the cardiac troponin T (cTnT) promoter were produced, purified, and titrated by qPCR. Mice were injected via the tail vein with 1 × 1011 viral genomes of AAV9-cTnT-JOSD2 or AAV9-cTnT-ZsGreen. Three weeks after injection, cardiac JOSD2 overexpression was confirmed by Western blotting.
Echocardiography
Transthoracic echocardiography was performed under light anesthesia with 2% isoflurane at the specified time points. Two-dimensional and M-mode images were acquired using a small animal ultrasound imaging system (Vevo 2100, VisualSonics, Toronto, Canada) equipped with a 30-MHz ultrasonic transducer. Left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were calculated from short-axis M-mode tracings and averaged over three consecutive cardiac cycles.
Triphenyltetrazolium chloride staining
Heart tissues were cut into approximately 2-mm-thick slices and incubated in 2% triphenyltetrazolium chloride (TTC) solution at 37 °C in the dark for 15 min. Viable myocardium was stained red, whereas infarcted areas remained white. The infarct size was quantified as the ratio of the infarct area to the total myocardial area.
Histology and immunofluorescence
Excised hearts were fixed, processed, and embedded for paraffin or frozen sectioning. For cardiomyocyte localization and protein expression analysis, sections were subjected to immunofluorescence staining with antibodies against JOSD2 and cTnT, followed by nuclear counterstaining with DAPI. Picrosirius Red (PSR) staining was performed to visualize and quantify interstitial fibrosis under polarized light, under which collagen fibers appear birefringent. For cardiomyocyte hypertrophy analysis, tissue sections were stained with wheat germ agglutinin (WGA) to visualize cell boundaries, and cardiomyocyte cross-sectional area was measured in defined regions.
Quantification and statistical analysis
Data are presented as the mean ± standard deviation (SD). Statistical analyses were performed using GraphPad Prism 10.0. For comparisons between two groups, statistical significance was assessed using an unpaired two-tailed Student’s t-test. For comparisons among multiple groups, one-way or two-way analysis of variance (ANOVA) was used, followed by Tukey’s multiple-comparisons test for post hoc analysis. A P value < 0.05 was considered statistically significant. Sample sizes for each experiment are provided in the figure legends. All experiments were independently repeated at least three times, and technical replicates, when performed, were not treated as independent biological replicates for statistical analysis.
RESULTS
CRISPR screen reveals ubiquitination gene candidates under OGD
To systematically identify ubiquitin-related regulators of cardiomyocyte survival under ischemic stress, we performed a pooled CRISPR-Cas9 loss-of-function screen in AC16 cells using a lentiviral human ubiquitin-related sgRNA library targeting 903 genes [Figure 1A]. Deep sequencing of two independent control libraries demonstrated near-complete sgRNA representation, with coverage rates of 99.89% and 99.93% [Figure 1B], and low Gini indices (0.0576 and 0.0595), consistent with uniform sgRNA abundance across the library [Figure 1C]. PCA of sgRNA abundance profiles showed clear separation between OGD-selected samples and controls, with tight clustering within each group, indicating a reproducible selection effect [Figure 1D]. Positive-selection analysis based on robust rank aggregation (RRA) prioritized candidate genes enriched in OGD-surviving cells relative to controls. The top-ranked candidates included ZBTB34, JOSD2, KLHL40, RNF185, UBE2L3, PSMB4, RAB40C, RING1, COMMD3, and KLHL26 [Figure 1E]; these candidates were subsequently subjected to integrated transcriptomic analysis and experimental validation.
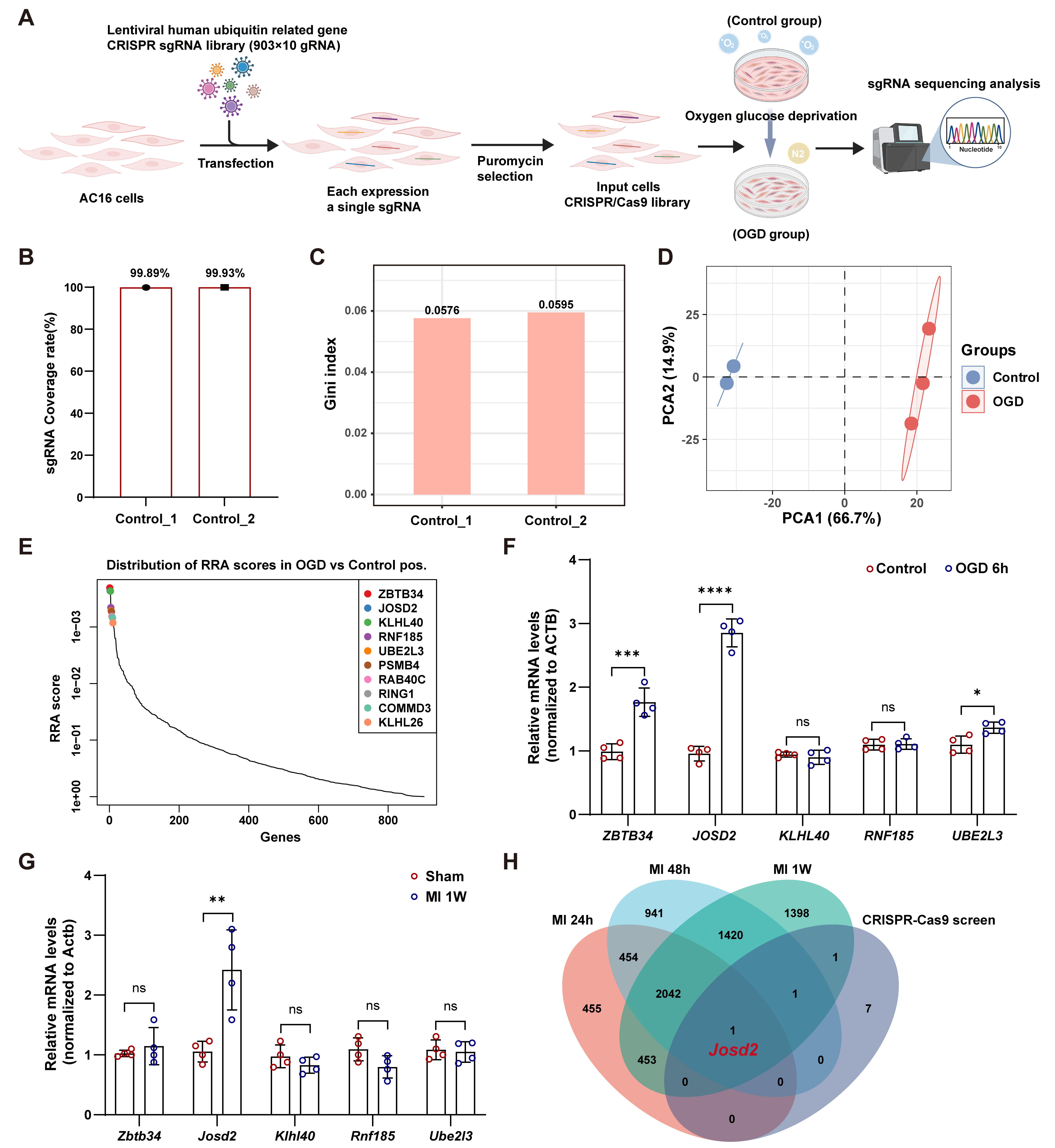
Figure 1. Ubiquitin-related pooled CRISPR-Cas9 screen under OGD. (A) Schematic of the pooled CRISPR-Cas9 screen in AC16 cells; (B) sgRNA coverage rate (%) in two independent control input libraries (Control_1 and Control_2); (C) Gini index of sgRNA read-count distribution in Control_1 and Control_2; (D) PCA of sgRNA abundance profiles in controls (n = 2) and OGD-selected samples (n = 3); (E) Rank-based distribution of RRA scores from the positive-selection CRISPR screen (OGD vs. control). Top-ranked candidate genes are highlighted; (F) RT-qPCR validation of the top five candidate genes in AC16 cells after 6 h of OGD vs control conditions; mRNA levels were normalized to ACTB; (G) RT-qPCR analysis of the corresponding genes in murine hearts 1 week after MI versus sham; mRNA levels were normalized to Actb; (H) Venn diagram showing overlap between genes from the CRISPR-Cas9 screen and genes identified from a published MI transcriptome time-course dataset at 24 h, 48 h, and 1-week post-MI (GSE775); Data in (F) and (G) are representative of four independent experiments and are presented as mean ± SD. Statistical significance was determined using an unpaired two-tailed Student’s t-test. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. OGD: Oxygen-glucose deprivation; SD: standard deviation; PCA: principal component analysis; sgRNA: single-guide RNA; RRA: robust rank aggregation; RT-qPCR: reverse transcription quantitative polymerase chain reaction; MI: myocardial infarction.
To determine whether top candidates were transcriptionally responsive to ischemic stress, we measured messenger RNA (mRNA) levels of the top five genes in AC16 cells after OGD. RT-qPCR analysis showed that ZBTB34, JOSD2, and UBE2L3 were significantly upregulated following OGD, with JOSD2 showing the most pronounced induction, whereas RNF185 and KLHL40 remained unchanged [Figure 1F]. To evaluate in vivo relevance, we next assessed these five genes in murine hearts at one week after MI. Among them, only Josd2 expression was significantly increased in MI hearts compared with sham controls [Figure 1G]. Consistently, integration of our CRISPR screen hits with a published MI transcriptome time-course dataset (24 h, 48 h, and 1-week post-MI; GSE775) identified Josd2 as the sole overlapping gene across datasets [Figure 1H], establishing JOSD2 as a convergent candidate for further mechanistic investigation.
JOSD2 is upregulated in response to cardiomyocyte ischemic stress in vitro and in vivo
To explore the potential involvement of JOSD2 in MI, we examined whether JOSD2 expression was altered in vitro under OGD conditions and in a mouse MI model. RT-qPCR and Western blot analyses revealed that both Josd2 mRNA and protein levels were significantly elevated with prolonged OGD exposure, in parallel with the upregulation of the cardiac stress marker natriuretic peptide A (Nppa) (encoding atrial natriuretic peptide, ANP) [Figure 2A-C]. In AC16 cells, OGD also markedly increased JOSD2 protein levels, accompanied by an increase in the apoptosis-associated protein BAX and a decrease in BCL-2 [Supplementary Figure 1A and B]. To assess the in vivo relevance of JOSD2 after MI, we evaluated regional expression in murine hearts at day 7 post-MI. Compared with sham controls, both the remote and border zones showed increased α-smooth muscle actin (α-SMA), a marker of myofibroblast activation and fibrotic remodeling, alongside elevated JOSD2, with the most prominent induction observed in the border zone [Figure 2D-F]. Moreover, in a post-MI time-course analysis, cardiac JOSD2 protein abundance increased significantly over time [Figure 2G and H]. Finally, immunofluorescence staining demonstrated substantial overlap between the JOSD2 signal and the cardiomyocyte marker cTnT, and revealed a marked increase in JOSD2 signal intensity in cardiomyocytes at one week post-MI compared with sham [Figure 2I].
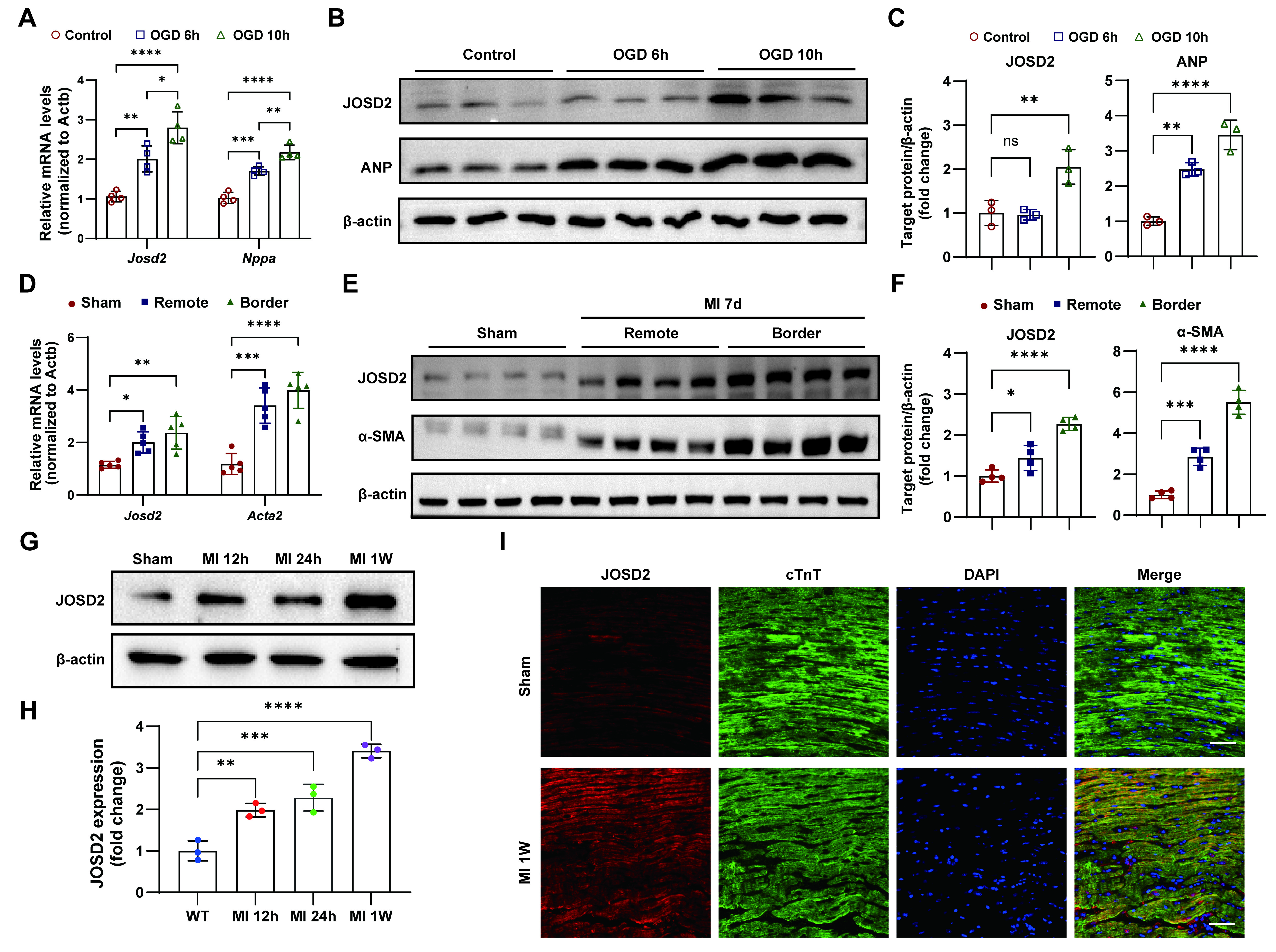
Figure 2. Ischemic injury-induced JOSD2 expression in cardiomyocytes and post-MI hearts. (A) RT-qPCR analysis of Nppa (encoding ANP) and Josd2 mRNA levels in NRVMs under control conditions and after OGD for 6 h or 10 h (n = 4); (B) Western blots showing ANP and JOSD2 protein levels in NRVMs after OGD for the indicated durations; (C) Densitometric quantification of ANP and JOSD2 immunoblots (n = 3); (D) RT-qPCR analysis of Acta2 (encoding α-SMA) and Josd2 expression in murine hearts at 7 days post-MI, comparing sham, remote zone, and border zone (n = 5); (E) Western blots showing α-SMA and JOSD2 protein abundance in sham hearts and in the remote zone and border zone of MI hearts at day 7; (F) Densitometric quantification of α-SMA and JOSD2 (n = 4 per group); (G) Western blots of JOSD2 in sham hearts and at 12 h, 24 h, and 1 week after MI (n = 3); (H) Densitometric quantification of JOSD2 (n = 3); (I) Representative immunofluorescence images of JOSD2, cTnT, and DAPI staining in sham and 1-week post-MI hearts. Scale bar, 50 μm. All results are representative of at least three independent experiments. Data are presented as mean ± SD, and statistical analysis in (A), (C), (D), (F), and (H) was performed using one-way ANOVA with Tukey’s post hoc test. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. JOSD2: Josephin domain-containing protein 2; RT-qPCR: reverse transcription quantitative polymerase chain reaction; MI: myocardial infarction; OGD: oxygen-glucose deprivation; ANP: atrial natriuretic peptide; NRVMs: neonatal rat ventricular myocytes; α-SMA: α-smooth muscle actin; ANOVA: analysis of variance; SD: standard deviation; cTnT: cardiac troponin T; WT: wild-type; DAPI: :4’,6-diamidino-2-phenylindole.
JOSD2 knockdown attenuates OGD-induced cardiomyocyte injury
To define the functional contribution of JOSD2 to ischemic stress-induced cardiomyocyte injury, we generated stable JOSD2-knockdown AC16 cells using lentiviral short hairpin RNAs (shRNAs). Among three shRNA constructs, sh-3 produced the strongest reduction in both JOSD2 mRNA [Figure 3A] and protein levels [Figure 3B and C] compared with sh-Scr, and was therefore selected for further experiments. We next assessed whether JOSD2 knockdown affects stress marker genes and apoptosis under basal conditions and after OGD. At baseline, JOSD2 knockdown did not alter the expression of cardiac stress markers NPPA and natriuretic peptide B (NPPB). Following OGD, however, JOSD2 knockdown significantly attenuated their induction compared to control [Figure 3D]. Similarly, although apoptosis-related proteins were unchanged at baseline, OGD triggered a clear pro-apoptotic shift in control cells that was reversed by JOSD2 knockdown, as shown by reduced BAX, cleaved caspase-3 (c-Cas-3), and cleaved caspase-9 (c-Cas-9), and increased BCL-2 [Figure 3E and F]. To directly quantify apoptosis, we performed flow cytometric analysis using sh-Scr and sh-JOSD2 AC16 cells under control conditions and after OGD for 6 h or 9 h. JOSD2 knockdown significantly decreased the apoptotic fraction at both OGD time points compared with sh-Scr cells [Figure 3G and H]. Consistently, TUNEL staining demonstrated that JOSD2 knockdown reduced the proportion of TUNEL+ cells under OGD [Figure 3I and J]. Moreover, Cell Counting Kit-8 (CCK-8) assays indicated improved cell viability in JOSD2-silenced cells under OGD stress [Supplementary Figure 2A]. To further characterize the transcriptional impact of JOSD2 silencing during ischemic stress, we performed RNA-seq. PCA showed clear separation between the two groups
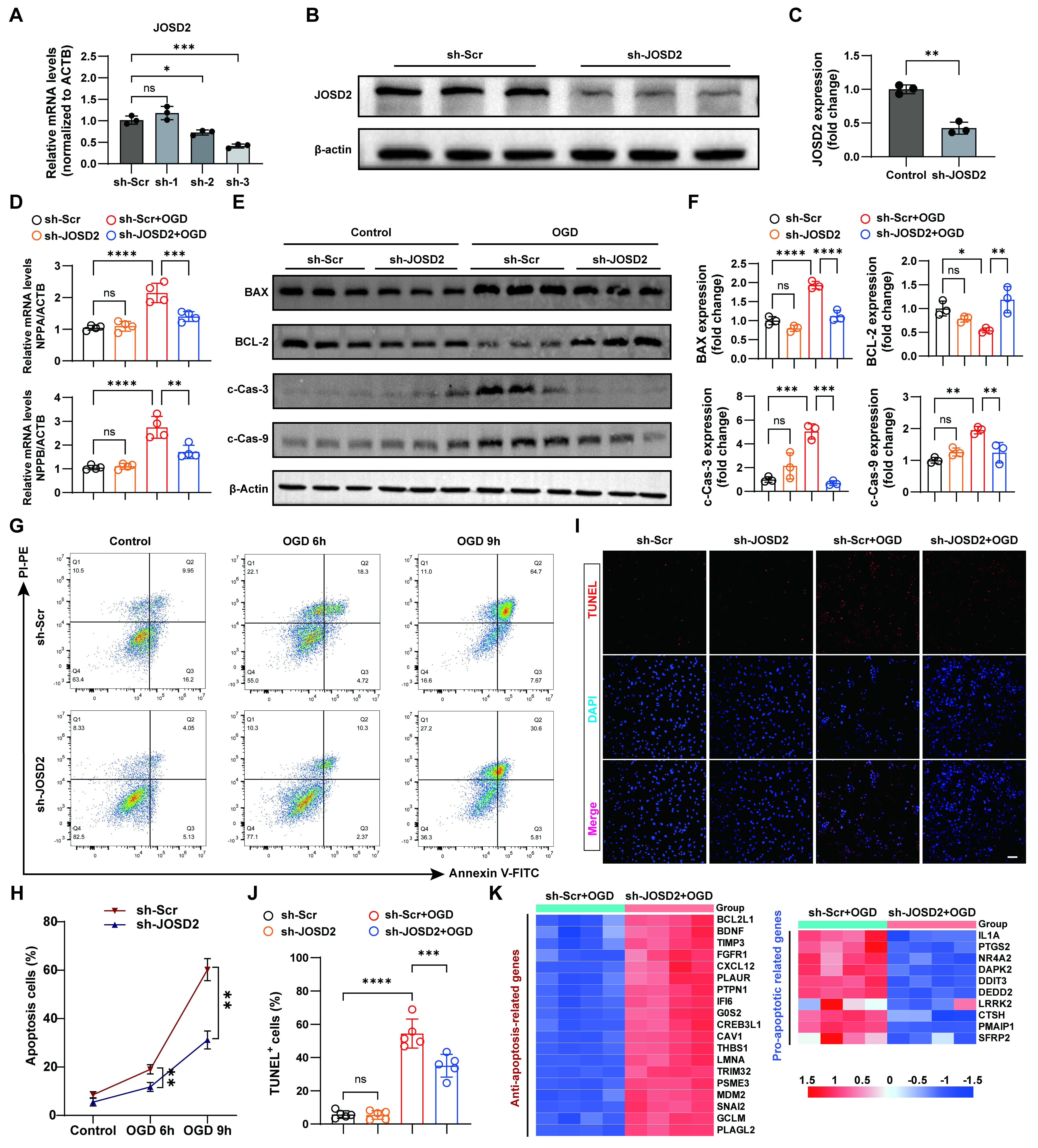
Figure 3. Protective effects of JOSD2 knockdown against OGD-induced apoptosis and injury-associated responses in AC16 cells. (A) RT-qPCR validation of JOSD2 mRNA levels in AC16 cells transduced with sh-Scr or three independent shRNA constructs targeting JOSD2 (n = 3 per group); (B) Western blots showing JOSD2 protein levels in sh-Scr and sh-JOSD2 (sh-3) stable AC16 cells; (C) Densitometric quantification of JOSD2 protein levels (n = 3); (D) RT-qPCR analysis of cardiac stress marker genes NPPA and NPPB in sh-Scr and sh-JOSD2 AC16 cells under basal conditions and after OGD (n = 4 per group); (E) Western blots of apoptosis-associated proteins (BAX, BCL-2, c-Cas-3, c-Cas-9) in sh-Scr and sh-JOSD2 AC16 cells under basal conditions and after OGD; (F) Densitometric quantification of apoptosis-associated proteins (n = 3); (G) Representative flow cytometry plots assessing apoptosis in sh-Scr and sh-JOSD2 AC16 cells under control conditions and after OGD for 6 h or 9 h; (H) Quantification of apoptotic cells from flow cytometry analyses (n = 3 independent biological replicate experiments); (I) Representative TUNEL staining images in sh-Scr and sh-JOSD2 AC16 cells under control conditions and after OGD. Scale bar, 50 μm; (J) Quantification of TUNEL+ cells (n = 5); (K) Heatmap showing the expression patterns of anti-apoptotic and pro-apoptotic genes derived from RNA-seq comparing sh-Scr+OGD versus sh-JOSD2+OGD. All results are representative of at least three independent experiments. Data are presented as mean ± SD. Statistical analysis was performed using one-way ANOVA with Tukey’s post hoc test for (A), an unpaired two-tailed Student’s t-test for (C), and two-way ANOVA with Tukey’s post hoc test for (D), (F), (H), and (J). ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. JOSD2: Josephin domain-containing protein 2; RT-qPCR: reverse transcription quantitative polymerase chain reaction; MI: myocardial infarction; OGD: oxygen-glucose deprivation; NPPB: natriuretic peptide B; TUNEL: transferase dUTP nick-end labelling; ANOVA: analysis of variance; ACTB: beta actin; NPPA: natriuretic peptide A; PI-PE: propidium iodide-P-phycoerythrin; Annexin V-FITC: Annexin V-fluorescein isothiocyanate.
JOSD2 overexpression exacerbates OGD-induced cardiomyocyte injury
To further assess the functional consequences of JOSD2 during ischemic injury, we generated stable JOSD2-overexpressing AC16 cells using lentiviral transduction. Successful overexpression of JOSD2 was confirmed by both RT-qPCR and Western blot analysis [Figure 4A-C]. We then examined the impact of JOSD2 overexpression on cardiomyocyte stress markers NPPA and NPPB under basal conditions and after OGD. In the absence of OGD, JOSD2 overexpression did not significantly alter the expression of these markers. However, following OGD, JOSD2 overexpression significantly exacerbated the induction of NPPA and NPPB compared to control cells [Figure 4D]. Next, we assessed apoptosis-related proteins BAX, c-Cas-3, c-Cas-9, and the anti-apoptotic protein BCL-2 across the experimental groups. Western blotting revealed that JOSD2 overexpression significantly upregulated pro-apoptotic proteins and downregulated BCL-2 expression under OGD, suggesting a shift toward a pro-apoptotic phenotype [Figure 4E and F]. To quantify apoptosis, we performed flow cytometric analysis of these cells after OGD. JOSD2 overexpression significantly increased the apoptotic fraction compared to control [Figure 4G and H]. Consistent with these findings, TUNEL staining also revealed a higher proportion of TUNEL+ cells in the JOSD2 overexpression groups under OGD [Figure 4I and J]. Additionally, CCK-8 assays demonstrated that while JOSD2 overexpression did not significantly affect cell viability under control conditions, it significantly suppressed cell viability under OGD [Figure 4K]. Taken together, these results indicate that JOSD2 overexpression exacerbates cardiomyocyte injury and apoptosis, particularly under ischemic stress, underscoring its potential role in promoting ischemic cell death.
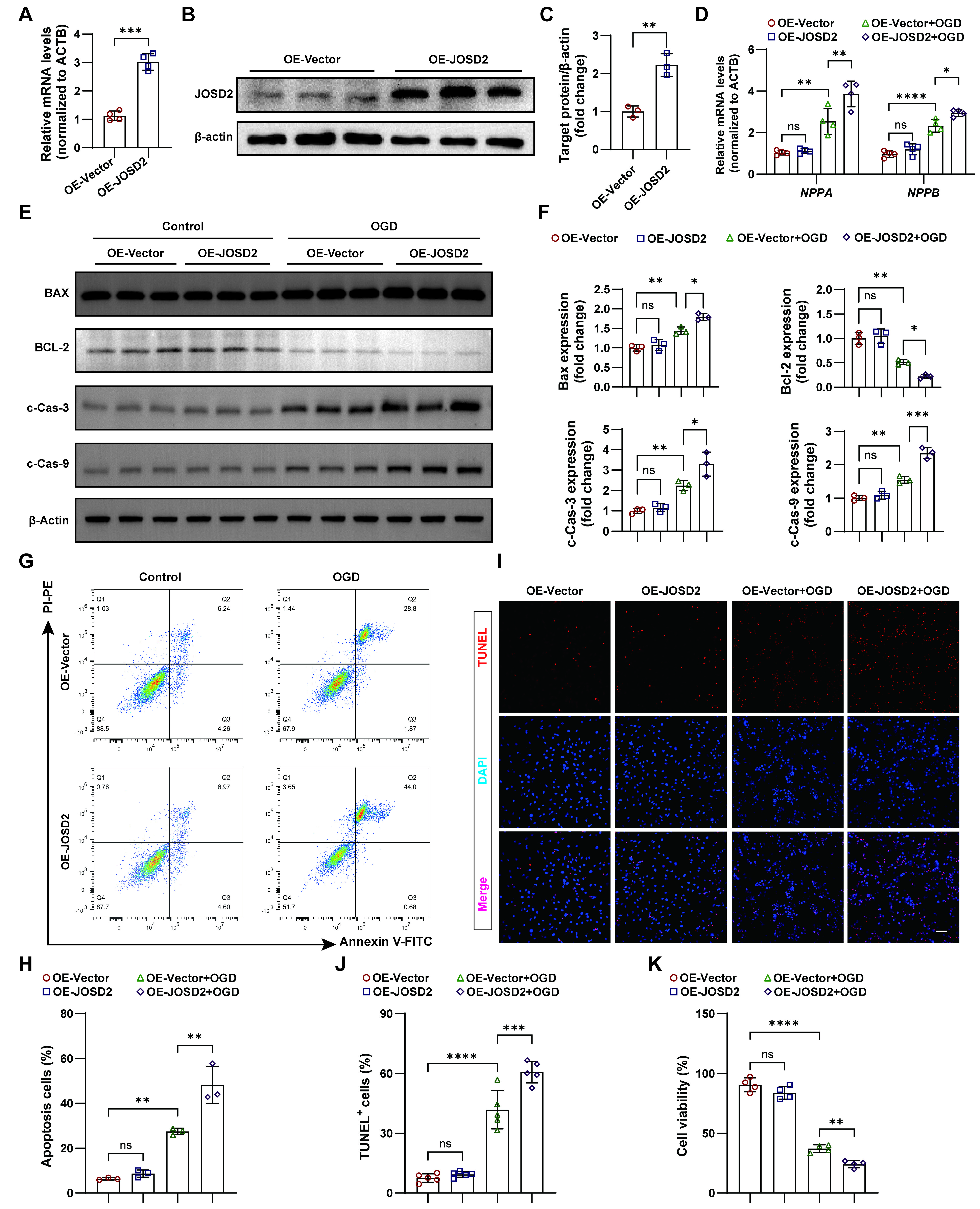
Figure 4. Exacerbation of OGD-induced cardiomyocyte injury and apoptosis by JOSD2 overexpression. (A) RT-qPCR validation of JOSD2 mRNA levels in AC16 cells transduced with JOSD2-overexpressing lentiviral vectors (OE-JOSD2) or control vector (OE-Vector) (n = 4 per group); (B) Western blots showing JOSD2 protein levels in OE-JOSD2 and OE-Vector stable AC16 cells; (C) Densitometric quantification of JOSD2 protein levels (n = 3); (D) RT-qPCR analysis of cardiac stress marker NPPA and NPPB in OE-Vector, OE-JOSD2, OE-Vector+OGD, and OE-JOSD2+OGD cells (n = 4 per group); (E) Western blots of apoptosis-associated proteins (BAX, BCL-2, c-Cas-3, c-Cas-9) in OE-Vector and OE-JOSD2 cells with and without OGD; (F) Densitometric quantification of apoptosis-associated proteins in (E) (n = 3); (G) Representative flow cytometry plots assessing apoptosis in OE-Vector and OE-JOSD2 cells under control conditions and after OGD; (H) Quantification of apoptotic cells from flow cytometry analyses (n = 3 independent biological replicates); (I) Representative TUNEL staining images in OE-Vector and OE-JOSD2 AC16 cells under control conditions and after OGD. Scale bar, 50 μm; (J) Quantification of TUNEL+ cells (n = 5); (K) CCK-8 assay assessing cell viability in OE-Vector and OE-JOSD2 cells under control conditions and after OGD (n = 4). All results are representative of at least three independent experiments. Data are presented as mean ± SD. Statistical analysis was performed using an unpaired two-tailed Student’s t-test for (A) and (C), and two-way ANOVA with Tukey’s post hoc test for (D), (F), (H), (J), and (K). ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. JOSD2: Josephin domain-containing protein 2; RT-qPCR: reverse transcription quantitative polymerase chain reaction; MI: myocardial infarction; OGD: oxygen-glucose deprivation; TUNEL: transferase dUTP nick-end labelling; SD: standard deviation; NPPB: natriuretic peptide B; ANOVA: analysis of variance; NPPA: natriuretic peptide A; PI-PE: propidium iodide-P-phycoerythrin; Annexin V-FITC: annexin V-fluorescein isothiocyanate; ACTB: beta actin.
JOSD2 deubiquitinates LKB1 to suppress AMPK activation
To explore the mechanism by which JOSD2 regulates ischemic cell death, we analyzed transcriptomic profiles of JOSD2-knockdown cells after OGD. Transcriptomic profiling revealed that JOSD2 expression was closely associated with alterations in genes involved in canonical AMPK-downstream pathways, including lipid and glucose metabolism, cell growth, and autophagy, suggesting that JOSD2 may function by modulating AMPK activity [Figure 5A]. In line with this inference, Western blot analysis demonstrated that JOSD2 knockdown markedly increased phosphorylation of AMPK (p-AMPK) and its key upstream regulator LKB1 (p-LKB1) compared with control cells [Figure 5B and C]. Further co-immunoprecipitation experiments confirmed that JOSD2 interacts with LKB1, whereas no interaction was detected between JOSD2 and AMPK under the same conditions [Figure 5D and E]. Furthermore, endogenous co-immunoprecipitation (co-IP) experiments in AC16 cells confirmed that endogenous JOSD2 interacts with endogenous LKB1 under basal conditions [Figure 5F].
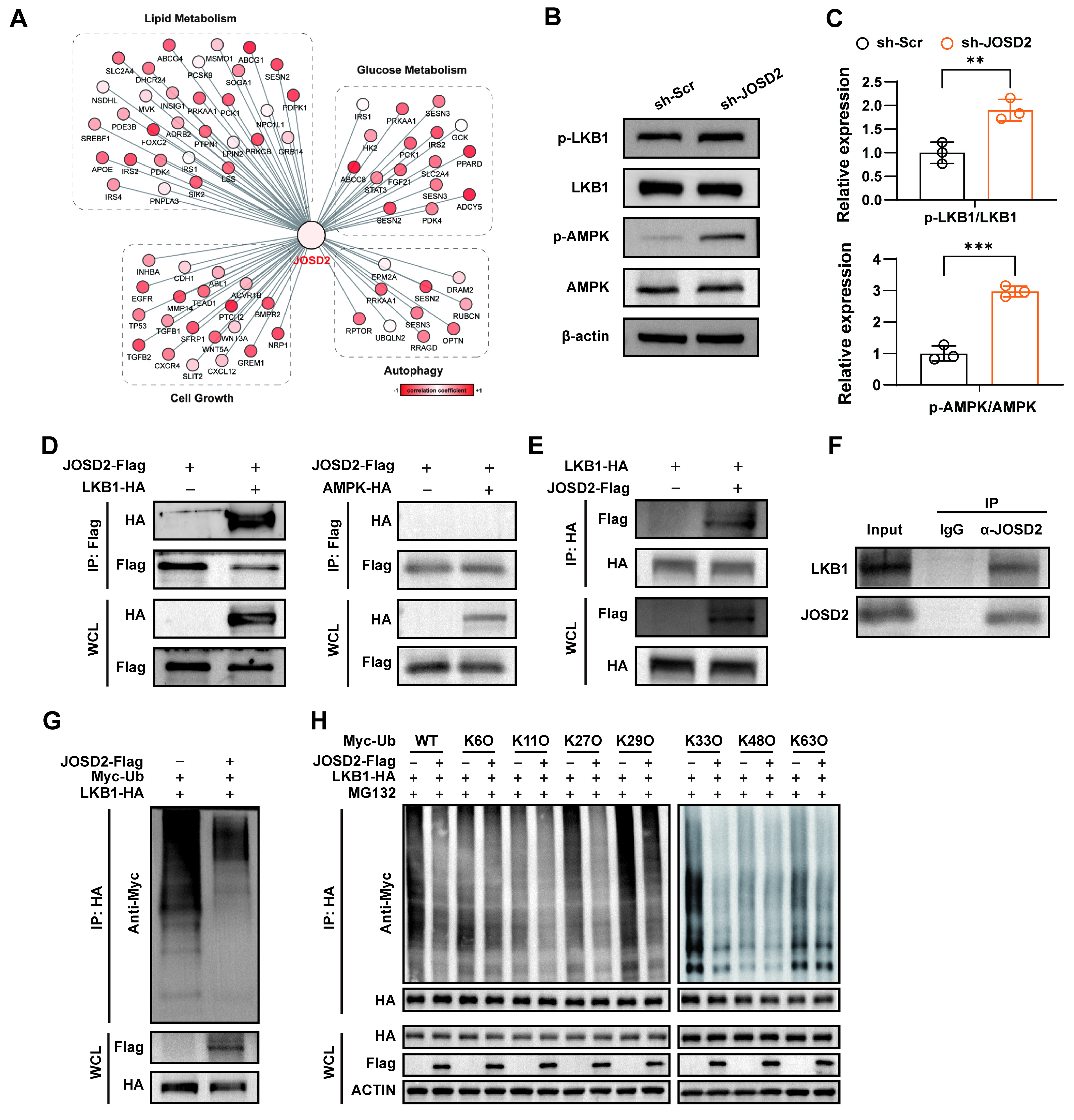
Figure 5. Interaction of JOSD2 with LKB1 and promotion of LKB1 deubiquitination. (A) Correlation of JOSD2 expression with the expression profiles of genes related to lipid metabolism, glucose metabolism, cell growth, and autophagy based on RNA-seq data; (B) Western blots showing levels of phosphorylated and total LKB1 and AMPK in sh-Scr and sh-JOSD2 AC16 cells; (C) Quantification of p-LKB1/LKB1 and p-AMPK/AMPK ratios (n = 3); (D) Co-immunoprecipitation of JOSD2-Flag with LKB1-HA or AMPK-HA in HEK293T cells; (E) Reciprocal co-IP of LKB1-HA with JOSD2-Flag in HEK293T cells; (F) Endogenous co-IP of JOSD2 and LKB1 in AC16 cells; (G) Ubiquitination assay of LKB1 in AC16 cells transfected with LKB1-HA and Myc-Ub, with or without JOSD2 overexpression; (H) Linkage-specific ubiquitination assay of LKB1 in AC16 cells. Cells were co-transfected with LKB1-HA and the indicated Myc-tagged ubiquitin mutants (WT, K6O, K11O, K27O, K29O, K33O, K48O, or K63O), with or without JOSD2-Flag, and treated with MG132. All results are representative of three independent experiments. Data are presented as mean ± SD. Statistical analysis was performed using an unpaired two-tailed Student’s t-test for (C). **P < 0.01, ***P < 0.001. JOSD2: Josephin domain-containing protein 2; LKB1: liver kinase B1; SD: standard deviation; AMPK: AMP-activated protein kinase; p-AMPK: phosphorylation of AMPK; p-LKB1: phosphorylation of LKB1; WT: wild type; HA: human influenza hemagglutinin; WCL: whole cell lysate.
To examine whether JOSD2 regulates LKB1 through deubiquitination, we performed ubiquitination assays. JOSD2 overexpression reduced the overall ubiquitination level of LKB1 [Figure 5G], indicating that JOSD2 post-translationally regulates LKB1. To identify the types of ubiquitin chains removed by JOSD2, we performed linkage-specific ubiquitination assays. The results showed that JOSD2 preferentially reduced K27-, K29-, K33-, and K63-linked polyubiquitination of LKB1, whereas no obvious effect was observed on K6-, K11-, and K48-linked ubiquitination under the conditions tested [Figure 5H]. Collectively, these data demonstrate that JOSD2 interacts with and deubiquitinates LKB1, primarily at K27-, K29-, K33-, and K63-linked chains, thereby suppressing downstream AMPK phosphorylation and activation.
JOSD2 knockdown-mediated protection against OGD-induced injury is dependent on LKB1
To further determine whether the protective effect of JOSD2 knockdown depends on LKB1, we designed three siRNAs targeting LKB1. RT-qPCR and Western blot analysis confirmed that siLKB1-1 achieved effective knockdown of LKB1 [Supplementary Figure 3A-C]. We then assessed apoptosis-related protein expression in sh-Scr and sh-JOSD2 AC16 cells with or without LKB1 silencing under OGD. Western blot analysis showed that JOSD2 knockdown reduced the expression of pro-apoptotic markers (BAX, cleaved caspase-3) and increased anti-apoptotic BCL-2, and these protective changes were largely reversed by LKB1 silencing [Figure 6A and B]. To evaluate the metabolic relevance of this LKB1-dependent effect, we performed Seahorse analysis to measure the OCR as an indicator of mitochondrial function. JOSD2 knockdown significantly increased OCR, basal respiration, maximal respiration, and ATP production after OGD exposure, indicating improved mitochondrial respiratory function, whereas this protective metabolic effect was largely abolished by LKB1 silencing [Figure 6C and D].
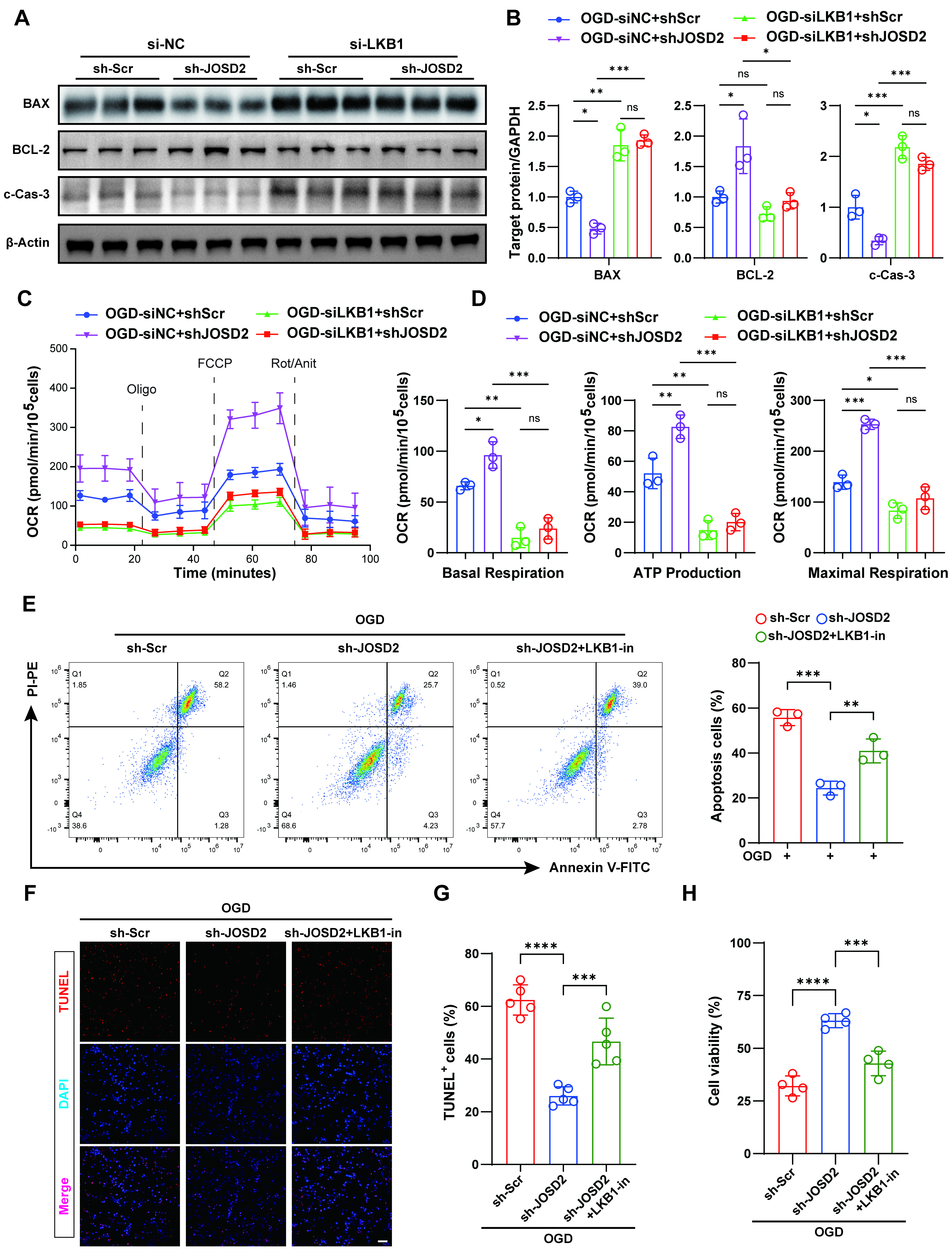
Figure 6. Requirement of LKB1 for the protective effect of JOSD2 knockdown under OGD. (A) Western blots of apoptosis-associated proteins (BAX, BCL-2, cleaved caspase-3) in siNC + sh-Scr, siNC + sh-JOSD2, siLKB1 + sh-Scr, and siLKB1 + sh-JOSD2 AC16 cells under OGD; (B) Densitometric quantification of apoptosis-associated proteins (n = 3); (C and D) OCR measurements and quantification by Seahorse analysis in siNC + sh-Scr, siNC + sh-JOSD2, siLKB1 + sh-Scr, and siLKB1 + sh-JOSD2 AC16 cells following OGD exposure; (E) Representative flow cytometry plots and quantification of apoptotic cells in sh-Scr, sh-JOSD2, and sh-JOSD2 + LKB1-in AC16 cells under OGD (n = 3); (F) Representative TUNEL staining images in sh-Scr, sh-JOSD2, and sh-JOSD2 + LKB1-in AC16 cells under OGD. Scale bar,
As a complementary pharmacological approach, we treated OGD-stressed cells with Pim1/AAK1-IN-1 (LKB1-in; also known as an LKB1/AAK1 dual inhibitor), which has reported inhibitory activity against LKB1[23]. In line with the genetic silencing results, LKB1-in treatment largely reversed the anti-apoptotic effects of JOSD2 knockdown, as evidenced by restored expression of pro-apoptotic markers and decreased anti-apoptotic BCL-2 levels [Supplementary Figure 3D and E]. Consistently, flow cytometry, TUNEL staining, and CCK-8 viability assays all showed that the reduction in apoptosis and improvement in cell viability conferred by JOSD2 knockdown under OGD were abolished by LKB1 inhibition with LKB1-in [Figure 6E-H]. Collectively, these genetic, metabolic, and pharmacological data demonstrate that LKB1 is functionally required for the cardioprotective effect of JOSD2 depletion under ischemic stress.
Cardiomyocyte-specific JOSD2 overexpression worsens post-MI injury in vivo
To validate the pathological relevance of JOSD2 function in vivo, we established a cardiomyocyte-targeted overexpression model using tail-vein delivery of AAV9 under the control of the cTnT promoter (AAV-JOSD2). Cardiomyocyte-specific overexpression of JOSD2 was confirmed by immunoblotting three weeks after AAV9 injection [Supplementary Figure 4A-C]. We then subjected AAV-treated mice to MI or sham surgery following the protocol outlined in Figure 7A. At 14 days post-MI, AAV-JOSD2 mice exhibited a significantly greater reduction in LVEF and LVFS compared with AAV-ZsGreen controls, indicating worsened systolic dysfunction [Figure 7B and C]. Consistent with impaired early myocardial preservation, TTC staining at 1 day post-MI revealed that cardiomyocyte-specific JOSD2 overexpression significantly increased infarcted area after MI [Figure 7D and E]. Molecular assessment at 14 days post-MI further supported the presence of aggravated cardiac remodeling. RT-qPCR analysis showed that AAV-JOSD2 did not upregulate heart failure markers (Nppa and Nppb) in sham hearts, whereas JOSD2 overexpression markedly enhanced the induction of these markers after MI compared with AAV-ZsGreen controls [Figure 7F]. Histological analyses at 14 days post-MI further demonstrated more severe adverse remodeling in AAV-JOSD2 mice, characterized by increased infarct size, greater interstitial fibrosis, and enlarged cardiomyocytes [Figure 7G and H].
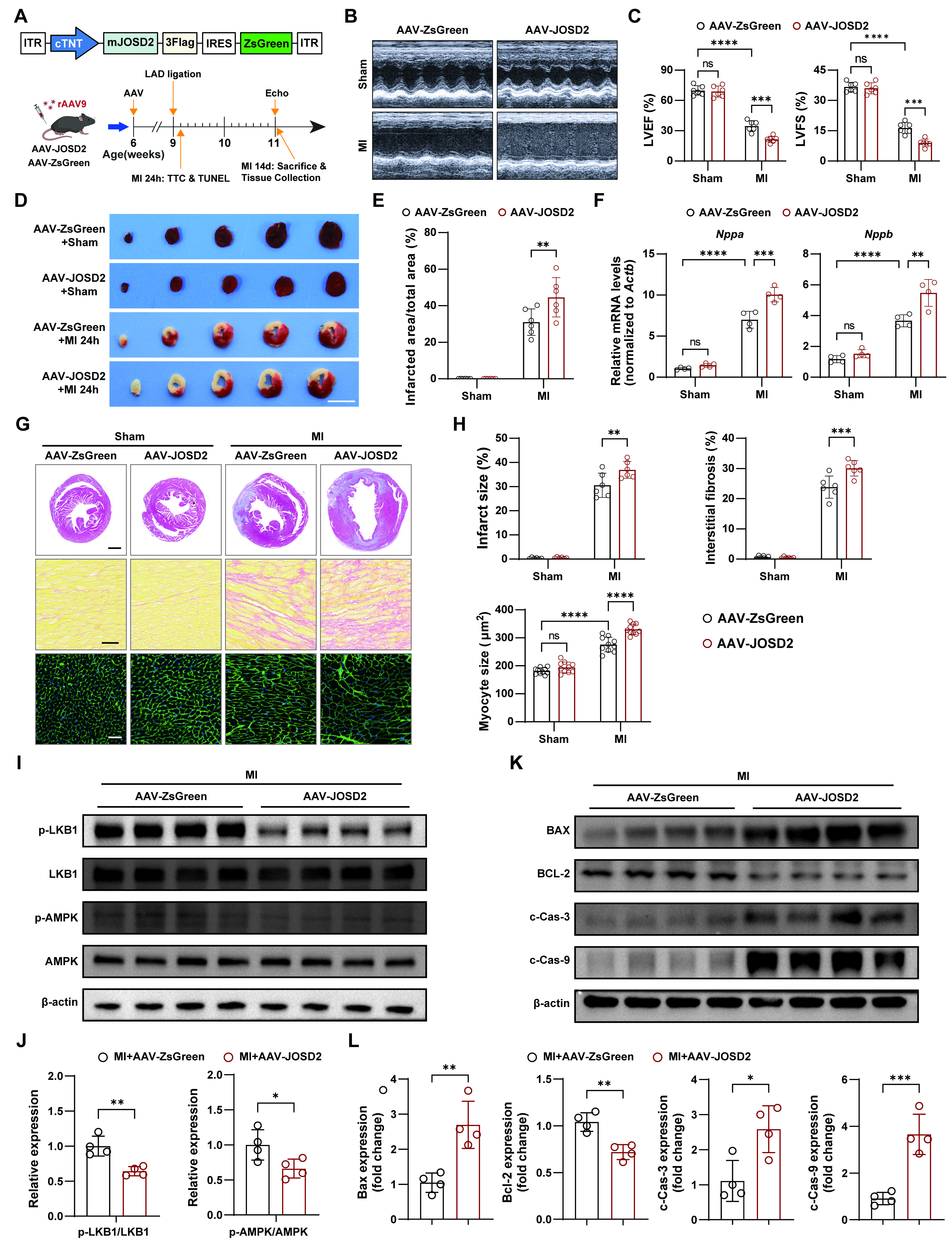
Figure 7. Aggravation of infarction and adverse remodeling after MI by cardiomyocyte-specific JOSD2 overexpression. (A) Schematic of AAV9-mediated cardiomyocyte-specific JOSD2 overexpression, MI/sham surgery, and tissue collection timelines; (B) Representative echocardiograms from C57BL/6J mice treated with AAV-ZsGreen or AAV-JOSD2 and subjected to MI/sham surgery; (C) Quantification of LVEF (%) and LVFS (%) at 14 days post-MI (n = 6 mice per group); (D) Representative TTC-stained heart sections at 1 day post-MI or sham. Scale bar, 5 mm; (E) Quantification of infarcted area/total area (%) from TTC staining (n = 6 mice per group); (F) RT-qPCR analysis of heart failure markers Nppa and Nppb in hearts at 14 days post-MI or sham (n = 4 mice per group); (G) Representative photomicrographs showing infarct size (scale bar, 1 mm), interstitial fibrosis (scale bar, 50 μm), and cardiomyocyte hypertrophy (scale bar, 50 μm) in AAV-ZsGreen- and AAV-JOSD2-treated mice subjected to sham or MI; (H) Quantitative evaluation of infarct size, interstitial fibrosis, and myocyte cross-sectional area (n = 6 mice per group); (I) Western blots of phosphorylated and total LKB1 and AMPK in hearts from the indicated groups; (J) Densitometric quantification of p-LKB1/LKB1 and p-AMPK/AMPK ratios (n = 4); (K) Western blots of apoptosis-associated proteins (BAX, BCL-2, c-Cas-3, and c-Cas-9); (L) Densitometric quantification of apoptosis-associated proteins in (K) (n = 4). Data are presented as mean ± SD. Data were analyzed by two-way ANOVA with Tukey’s post hoc test (C, E, F, and H) and unpaired two-tailed Student’s t-test (J and L). ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. JOSD2: Josephin domain-containing protein 2; RT-qPCR: reverse transcription quantitative polymerase chain reaction; MI: myocardial infarction; OGD: oxygen-glucose deprivation; TUNEL: transferase dUTP nick-end labelling; SD: standard deviation; LVEF: left ventricular ejection fraction; LVFS: left ventricular fractional shortening; TTC: triphenyltetrazolium chloride; AAV: adeno-associated virus; AMPK: AMP-activated protein kinase; LKB1: liver kinase B1; ANOVA: analysis of variance; ITR: inverted terminal repeat; p-AMPK: phosphorylation of AMPK; p-LKB1: phosphorylation of LKB1; BAX: BCL-2-associated X protein; BCL-2: B-cell lymphoma 2.
Mechanistically, consistent with our in vitro findings, JOSD2 overexpression in vivo suppressed activation of the LKB1-AMPK axis after MI, as evidenced by decreased p-LKB1/LKB1 and p-AMPK/AMPK ratios [Figure 7I and J]. In parallel, JOSD2 overexpression promoted a pro-apoptotic protein signature in post-MI hearts, increasing BAX, c-Cas-3, and c-Cas-9 while decreasing BCL-2 [Figure 7K and L]. Collectively, these data indicate that cardiomyocyte JOSD2 overexpression exacerbates infarction and adverse remodeling after MI, at least in part by suppressing LKB1-AMPK activation and enhancing ischemic cell death in vivo.
DISCUSSION
The irreversible loss of cardiomyocytes after MI is a central pathological event leading to the progression of heart failure. There is a major unmet need for therapies that protect the myocardium[24]. Protein ubiquitination, as a key post-translational regulatory mechanism, can precisely control a series of signaling pathways, including stress responses and cell death[25,26]. However, the specific components of the ubiquitin system that determine the fate of myocardial cells under ischemic conditions remain unclear[27].
To systematically address this gap, we applied a CRISPR-Cas9 screening strategy to identify ubiquitin-related genes involved in cardiomyocyte survival under OGD conditions. This unbiased screening strategy successfully identified JOSD2 as a key candidate regulator for the survival of myocardial cells under ischemic stress.
The core of the JOSD family consists of a catalytic Josephin domain, which can cleave K63 and K48-linked polyubiquitin chains and plays a crucial role in regulating substrate protein stability and function[28]. Notably, JOSD2 functions in various pathological conditions by acting on different substrates. For example, in models of heart failure caused by pressure overload, JOSD2 deubiquitinates and enhances the phosphorylation of calcium/calmodulin-dependent protein kinase IIδ (CaMKIIδ), thereby exacerbating pathological myocardial hypertrophy, apoptosis, and fibrosis[29]. These findings highlight the context-dependent nature and substrate specificity of JOSD2 function. However, the role and mechanism of JOSD2 in acute myocardial ischemic injury have not been explored.
Our validation data indicate that JOSD2 is specifically upregulated by ischemic stress in vitro and in vivo. Functionally, JOSD2 knockdown significantly reduces ischemia-induced myocardial cell apoptosis and damage, whereas JOSD2 overexpression exacerbates these adverse effects. In vivo experiments further confirm that cardiomyocyte-specific JOSD2 overexpression aggravates cardiac dysfunction, infarct area expansion, and adverse remodeling in mice after MI. Mechanistically, we demonstrated that JOSD2 interacts with LKB1 and, through its deubiquitinating enzyme activity, inhibits LKB1 activity, thereby attenuating the phosphorylation and activation of the downstream AMPK.
Consistent with our observations, recent studies in non-small-cell lung cancer have also shown that JOSD2 drives tumor progression by inhibiting the tumor suppressor LKB1[30]. We further demonstrated the importance of JOSD2 as a deubiquitinating enzyme in responding to MI by modulating LKB1 activity. These data suggest that JOSD2-mediated suppression of LKB1 contributes to ischemic cardiomyocyte injury and adverse post-MI remodeling.
AMPK plays a central role in maintaining myocardial cell energy balance and resisting ischemic injury, partly through regulating processes such as autophagy[31,32]. Activation of AMPK is a well-established cardioprotective strategy[33,34]. However, directly targeting AMPK or LKB1 faces significant challenges due to their broad roles in systemic metabolic processes[35,36]. Therefore, identifying a more pathophysiology-specific upstream regulator is important for achieving precise therapeutic intervention[37,38]. Recent advances in cardiovascular disease research have highlighted the importance of identifying novel therapeutic nodes beyond conventional strategies. For example, MrgD-directed intervention has been proposed as a receptor-targeting strategy for pulmonary hypertension[39], whereas bioengineered human pluripotent stem cell-derived cardiac tissue/microtissue approaches have shown promise for myocardial repair in preclinical MI models[40-42]. In this context, our findings identify JOSD2 as another potential mechanistic and therapeutic node to explore for MI intervention. Inhibiting JOSD2 may offer a “condition-dependent” therapeutic strategy that restores protective signaling under pathological conditions without causing sustained systemic activation of the pathway, potentially providing a more favorable therapeutic window.
Limitations
First, the main mechanistic validation of JOSD2-mediated regulation of the LKB1-AMPK signaling pathway in this study was conducted primarily in vitro using AC16 cells. Although AC16 cells can partially model cardiomyocyte injury and apoptosis under ischemic stress, they do not fully recapitulate the complex pathological and physiological environment of MI in the heart, including inflammatory infiltration, cell-cell interactions, microenvironmental changes, and metabolic remodeling. Therefore, more rigorous in vivo studies, particularly in animal models with cardiomyocyte-specific conditional gene modification, are needed to clarify the role and mechanism of JOSD2 in ischemic heart injury. Second, this study focused on the acute and relatively early stages of MI and did not include systematic evaluation of long-term repair and chronic remodeling processes beyond four weeks post-MI. Whether JOSD2 continues to participate in the progression of chronic heart failure and whether it affects long-term ventricular remodeling and cardiac function outcomes remain to be investigated. Such studies would help more comprehensively evaluate the therapeutic potential of targeting JOSD2 in chronic heart failure. Third, this study lacks clinical samples and clinical data. The expression pattern of JOSD2 in myocardial tissues or peripheral blood from patients with MI, its correlation with disease severity and prognosis, and its clinical translational relevance as a potential intervention target all need to be further evaluated in clinical samples. In summary, although this study provides functional and mechanistic evidence that JOSD2 is an important negative regulator of ischemic myocardial injury, its long-term pathological significance and clinical translational value require further investigation.
Conclusion
This study identifies the deubiquitinase JOSD2 as a previously unrecognized mediator of myocardial ischemic injury through CRISPR screening. JOSD2 exerts its detrimental effects by inhibiting the cardioprotective LKB1-AMPK pathway, highlighting JOSD2 as a promising upstream therapeutic target.
DECLARATIONS
Acknowledgments
We thank the staff at the Research Center for Medicine and Structural Biology of Wuhan University for their technical assistance. The graphical schematics shown in Figures 1A, 7A, and Supplementary Figure 4A were created using BioRender.com.
Authors’ contributions
Writing - original draft, writing - review & editing, conceptualization, data curation, formal analysis, investigation, methodology, project administration, software, supervision, validation, visualization: Huang YD
Data curation, investigation, methodology, software, writing - original draft, writing - review & editing: Nie MH
Methodology, software, writing -review & editing: Xia LH
Methodology, writing - original draft: Duan P
Writing - review & editing: Yang BS, Zhang SH, Qin PY
Methodology, writing -review & editing: Jiang J, Gu Y, Li G
Data curation, investigation, writing -original draft: Wang H
Data curation, writing - original draft, writing -review & editing: conceptualization, data curation, funding acquisition, project administration, resources, supervision, validation, visualization, writing - original draft, writing - review & editing: Gao R
Conceptualization, data curation, funding acquisition, project administration, resources, supervision, validation, visualization, writing - original draft, and writing - review and editing: Ji YX
All authors read and approved the final manuscript.
Availability of data and materials
The raw RNA-seq data from sh-Scr+OGD and sh-JOSD2+OGD AC16 cells have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject accession number PRJNA1422660, and the raw sgRNA sequencing data from the CRISPR-Cas9 loss-of-function screen have been deposited under BioProject accession number PRJNA1423684. All processed data are available from the corresponding author upon reasonable request.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool DeepSeek (version V3.2, released 2025-12-01) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (82370395 and 82070079); Chinese Academy of Medical Science Innovation Fund for Medical Sciences (2021-I2M-1-009), and the Joint Fund for Translational Medicine and Interdisciplinary Research of Zhongnan Hospital, Wuhan University (ZNJC202306).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
This animal study was approved by the Experimental Animal Welfare Ethics Committee, Zhongnan Hospital of Wuhan University (Approval No. ZN2023239). All experimental procedures were performed in accordance with the relevant guidelines and regulations of the committee.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Zhu M, Li Y, Xu Q, Wang W, Liu Y, Liu Y. Acute myocardial infarction: molecular pathogenesis, diagnosis, and clinical management. MedComm. 2025;6:e70418.
2. Zhang T, Deng W, Deng Y, et al. Mechanisms of ferroptosis regulating oxidative stress and energy metabolism in myocardial ischemia-reperfusion injury and a novel perspective of natural plant active ingredients for its treatment. Biomed Pharmacother. 2023;165:114706.
3. Tsurusaki S, Kizana E. Mechanisms and therapeutic potential of multiple forms of cell death in myocardial ischemia-reperfusion injury. Int J Mol Sci. 2024;25:13492.
4. Xiang Q, Yi X, Zhu X, Wei X, Jiang D. Regulated cell death in myocardial ischemia-reperfusion injury. Trends Endocrinol Metab. 2024;35:219-34.
5. Liu Y, Li L, Wang Z, Zhang J, Zhou Z. Myocardial ischemia-reperfusion injury; molecular mechanisms and prevention. Microvasc Res. 2023;149:104565.
6. Qin Z, Kong B, Zheng J, Wang X, Li L. Alprostadil injection attenuates coronary microembolization-induced myocardial injury through GSK-3β/Nrf2/HO-1 signaling-mediated apoptosis inhibition. Drug Des Devel Ther. 2020;14:4407-22.
7. Yao M, Wang Z, Jiang L, et al. Oxytocin ameliorates high glucose- and ischemia/reperfusion-induced myocardial injury by suppressing pyroptosis via AMPK signaling pathway. Biomed Pharmacother. 2022;153:113498.
8. Timmermans AD, Balteau M, Gélinas R, et al. A-769662 potentiates the effect of other AMP-activated protein kinase activators on cardiac glucose uptake. Am J Physiol Heart Circ Physiol. 2014;306:H1619-30.
9. Zhang Q, Wang L, Wang S, et al. Signaling pathways and targeted therapy for myocardial infarction. Sig Transduct Target Ther. 2022;7:78.
10. Silverman N, Maniatis T. NF-κB signaling pathways in mammalian and insect innate immunity. Genes Dev. 2001;15:2321-42.
11. Matter MA, Paneni F, Libby P, et al. Inflammation in acute myocardial infarction: the good, the bad and the ugly. Eur Heart J. 2024;45:89-103.
12. Han J, Lin L, Fang Z, et al. Cardiomyocyte-derived USP28 negatively regulates antioxidant response and promotes cardiac hypertrophy via deubiquitinating TRIM21. Theranostics. 2024;14:6236-48.
13. Ye B, Zhou H, Chen Y, et al. USP25 ameliorates pathological cardiac hypertrophy by stabilizing SERCA2a in cardiomyocytes. Circ Res. 2023;132:465-80.
14. Tong G, Chen Y, Chen X, et al. FGF18 alleviates hepatic ischemia-reperfusion injury via the USP16-mediated KEAP1/Nrf2 signaling pathway in male mice. Nat Commun. 2023;14:6107.
15. Zhong L, Dai S, Yu F, et al. Cardiomyocyte‐enriched USP20 ameliorates pathological cardiac hypertrophy by targeting STAT3 deubiquitination. Adv Sci. 2025;12:2416478.
16. Roberts MA, Deol KK, Mathiowetz AJ, et al. Parallel CRISPR-Cas9 screens identify mechanisms of PLIN2 and lipid droplet regulation. Dev Cell. 2023;58:1782-1800.e10.
17. Wang W, Zheng Y, Sun S, et al. A genome-wide CRISPR-based screen identifiesKAT7 as a driver of cellular senescence. Sci Transl Med. 2021;13:eabd2655.
18. Mircetic J, Camgöz A, Abohawya M, et al. CRISPR/Cas9 screen in gastric cancer patient‐derived organoids reveals KDM1A‐NDRG1 axis as a targetable vulnerability. Small Methods. 2023;7:2201605.
19. Tyagi A, Kaushal K, Chandrasekaran AP, et al. CRISPR/Cas9-based genome-wide screening for deubiquitinase subfamily identifies USP1 regulating MAST1-driven cisplatin-resistance in cancer cells. Theranostics. 2022;12:5949-70.
20. Ma X, Jia S, Wang G, et al. TRIM28 promotes the escape of gastric cancer cells from immune surveillance by increasing PD-L1 abundance. Sig Transduct Target Ther. 2023;8:246.
21. Arroyo JD, Jourdain AA, Calvo SE, et al. A genome-wide CRISPR death screen identifies genes essential for oxidative phosphorylation. Cell Metab. 2016;24:875-85.
22. Zhang Y, Qian H, Wu B, et al. E3 Ubiquitin ligase NEDD4 family-regulatory network in cardiovascular disease. Int. J. Biol. Sci. 2020;16:2727-40.
23. Bamborough P, Drewry D, Harper G, Smith GK, Schneider K. Assessment of chemical coverage of kinome space and its implications for kinase drug discovery. J Med Chem. 2008;51:7898-914.
25. Komander D, Clague MJ, Urbé S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550-63.
26. Wang ZV, Hill JA. Protein quality control and metabolism: bidirectional control in the heart. Cell Metab. 2015;21:215-26.
27. Willis MS, Patterson C. Proteotoxicity and cardiac dysfunction - alzheimer’s disease of the heart? N Engl J Med. 2013;368:455-64.
28. Seki T, Gong L, Williams AJ, Sakai N, Todi SV, Paulson HL. JosD1, a membrane-targeted deubiquitinating enzyme, is activated by ubiquitination and regulates membrane dynamics, cell motility, and endocytosis. J Biol Chem. 2013;288:17145-55.
29. Xu J, Liang S, Wang Q, et al. JOSD2 mediates isoprenaline-induced heart failure by deubiquitinating CaMKIIδ in cardiomyocytes. Cell Mol Life Sci. 2024;81:18.
30. Yuan T, Zeng C, Liu J, et al. Josephin domain containing 2 (JOSD2) promotes lung cancer by inhibiting LKB1 (Liver kinase B1) activity. Sig Transduct Target Ther. 2024;9:11.
31. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251-62.
32. Dyck JRB, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally? J Physiol. 2006;574:95-112.
33. Potenza MA, Sgarra L, Nacci C, Leo V, De Salvia MA, Montagnani M. Activation of AMPK/SIRT1 axis is required for adiponectin-mediated preconditioning on myocardial ischemia-reperfusion (I/R) injury in rats. PLoS ONE. 2019;14:e0210654.
34. Ikeda Y, Sato K, Pimentel DR, et al. Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J Biol Chem. 2009;284:35839-49.
35. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563-75.
36. Steinberg GR, Hardie DG. New insights into activation and function of the AMPK. Nat Rev Mol Cell Biol. 2022;24:255-72.
37. Rossi FA, Rossi M. Emerging role of ubiquitin-specific protease 19 in oncogenesis and cancer development. Front. Cell Dev Biol. 2022;10:889166.
38. Li J, Johnson JA, Su H. Ubiquitin and ubiquitin-like proteins in cardiac disease and protection. Curr Drug Targets. 2018;19:989-1002.
39. Zhong H, Yao L, An H, et al. MrgD as a novel modeling and treatment target for pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2025;45:e164-83.
40. Li J, Minami I, Shiozaki M, et al. Human Pluripotent Stem Cell-Derived Cardiac Tissue-like Constructs for Repairing the Infarcted Myocardium. Stem Cell Rep. 2017;9:1546-59.
41. Gao L, Gregorich ZR, Zhu W, et al. Large cardiac muscle patches engineered from human induced-pluripotent stem cell-derived cardiac cells improve recovery from myocardial infarction in swine. Circulation. 2018;137:1712-30.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].