


Reversing an obesogenic diet to control diet partially rescues pro-inflammatory lipid-immune memory in splenocardiac aging
 0
0 
Abstract
Introduction: Chronic exposure to an obesogenic diet (OBD) induces systemic inflammation and disrupts
Aim: This study examined whether switching from an OBD to a control diet can restore lipid-immune homeostasis during aging and evaluated associated changes in myocardial infarction (MI) response, macrophage activation, and immune cell heterogeneity.
Methods and Results: Male C57BL/6J mice were fed either a control or OBD diet for six months, followed by continued OBD or dietary reversal (OBD-R) for an additional four months. Cardiac function, splenic and cardiac lipid mediators, leukocyte profiles, and gene expression were assessed at baseline and post-MI. Lipopolysaccharide (LPS)-induced macrophage activation assays and single-cell RNA sequencing (scRNA-seq) of CD45+ cells were performed to characterize immune memory. Chronic OBD induced cardiac strain dysfunction, expansion of CCR2+ macrophages, elevated 12-HETE levels, and depletion of specialized pro-resolving mediators (SPMs). Dietary reversal restored SPMs, normalized certain immune parameters, and improved post-MI responses, but persistent activation of the 12-HETE-driven CCL2-CCR2/ALOX5-ALOX5AP axis maintained proinflammatory macrophage memory. scRNA-seq revealed diet-dependent transcriptional remodeling of immune populations, and LPS assays confirmed heightened inflammatory memory in macrophages after chronic OBD exposure.
Conclusions: Dietary intervention can potentially resolve OBD-induced inflammation and mitigate related cardiovascular consequences during aging, with the 12-HETE-induced CCL2-CCR2/ALOX5-ALOX5AP axis playing a critical role in shaping macrophage phenotype and sustaining inflamed immune memory.
Keywords
INTRODUCTION
Cardiovascular diseases (CVDs) remain a major global health concern, accounting for 19.05 million deaths annually and representing the leading cause of both mortality and morbidity[1]. Recent data suggest a 9.8% decline in CVD-related deaths during 2010-2019, followed by a 4.1% increase in 2020 across various demographic groups-spanning age, sex, race, and ethnicity[2]. Primordial lifestyle risk factors, such as consumption of processed foods, poor dietary quality, sleep deprivation, physical inactivity, and chronic stress, contribute to systemic inflammation, which clinically manifests as hypertension, diabetes, hyperlipidemia, and obesity- all key precursors to CVD[1,3].
Emerging evidence linking obesity, type 2 diabetes, and hypertension underscores systemic inflammation as a common predictor of future cardiovascular events, including heart failure (HF) with preserved or reduced ejection fraction, myocardial infarction (MI), and stroke[4]. This persistent, suboptimal inflammation is often termed “residual inflammatory risk” (RIR)[5,6]. While acute inflammation is essential for host defense, unresolved inflammation promotes sustained leukocyte trafficking at injury sites, resulting in HF[7,8]. In individuals with cardiometabolic conditions, aging is associated with a phenomenon termed inflammaging, marked by low-grade chronic systemic inflammation influenced by genetics and lifestyle-related dietary patterns. Impaired immune responses affect leukocyte plasticity, a characteristic of inflammaging that reduces the functional and polarization capacity of neutrophils and macrophages, depending on their surrounding microenvironment[9,10].
Dietary patterns play a critical role in modulating inflammation. Consumption of processed, high-fat
While the immune-metabolic effects of OBD have been extensively characterized in 2-6-month exposure models, the long-term (≥ 10 months) impact of OBD on immuno-lipidome memory in the context of aging and HF remains poorly understood.
Therefore, this study proposes a three-dimensional approach to investigate: (1) the long-term effects of OBD on the splenocardiac immune-lipid axis; (2) the reversibility of diet-induced immune reprogramming in key organs; and (3) whether switching from OBD to a control diet can restore the lipidome-immune microenvironment required for host defense and the resolution of post-MI inflammation.
MATERIALS AND METHODS
Animal care and compliance
Animal use and monitoring were conducted according to the “Guide for the Care and Use of Laboratory Animals” (8th Edition, 2011) and the AVMA Guidelines for the Euthanasia of Animals (2020 Edition). All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of South Florida, Tampa, USA, under protocol number 7371R. The study followed the ARRIVE 2.0 guidelines for experimental procedures[17].
Study design and diet intervention
A detailed description of the study design is provided in the Supplementary Materials.
Coronary artery ligation microsurgery
Mice were subjected to left anterior descending coronary ligation (MI surgery). Anesthesia was induced with 3%-4% isoflurane (ISOFLURANE, USP; Rx) and maintained at 1.5%-2% during the non-reperfused surgical occlusion[16].
Necropsy in control (no-MI) and post-MI mice
Detailed necropsy procedures are provided in the Supplementary Materials. Briefly, mice were initially anesthetized using 5% isoflurane and maintained at 3% isoflurane in 100% oxygen during sacrifice. Exsanguination was performed to ensure compliance with IACUC and ARRIVE 2.0 guidelines[16].
Statistical analysis
Data are expressed as mean ± Standard Error of the Mean (± SEM), and bar graphs include individual values. A minimum of 4-5
RESULTS
Transition from OBD to control diet regulates adipose size and partially restores cardiac strain
A calorie-dense OBD induces widespread inflammation and exacerbates LV dysfunction[18]. However, the effects of transitioning from an OBD to a standard diet on heart health, function, and related physiological parameters remain poorly understood. To investigate the impact of dietary reversal on cardiac function, male C57BL/6J mice were fed an omega-6-rich diet (OBD, 10% w/w) for 6 months, followed by a switch to a standard chow diet containing 4%w/w linoleic acid for 4 months (OBD-R). Control (CON) mice and mice continuously fed the omega-6 diet (OBD) were maintained on their respective diets for the full 10 months [Figure 1A]. No significant differences in overall body weight were observed among the three groups at 10 months [Figure 1B]. Tissue analysis revealed no differences between groups in LV/body weight (LV/BW), LV/Tibia, spleen/BW, or spleen/tibia ratios [Figure 1C and D, Supplementary Figure 1A and B, Table 1]. Histomorphometry showed a shift toward larger adipocytes (> 7,000 μm2) in OBD mice, with partial normalization of adipose size distribution in OBD-R mice (< 5,000 μm2; Supplementary Figure 1C). These results indicate that an omega-6-rich diet induces visceral adipocyte hypertrophy, which is partly reversed upon dietary normalization.
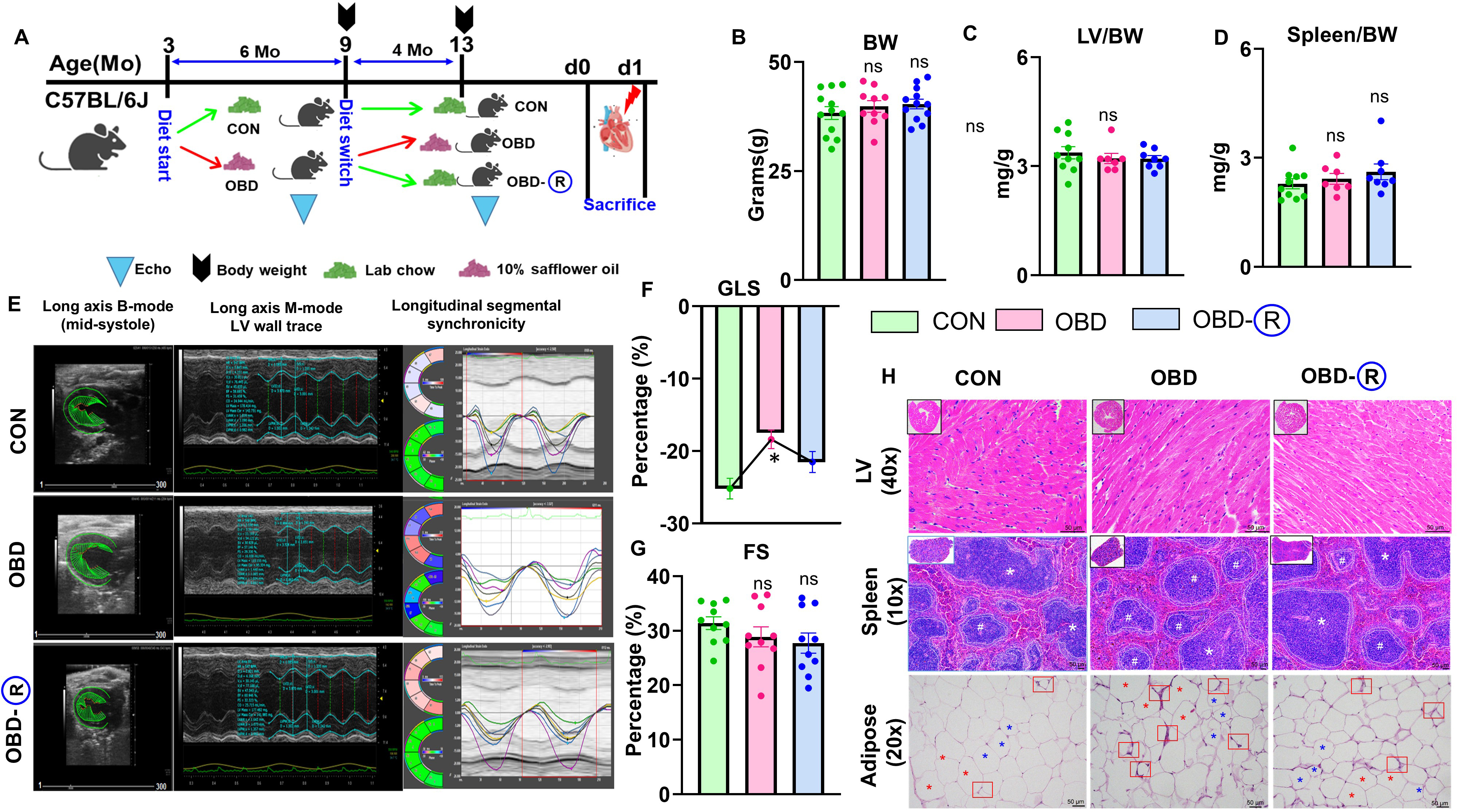
Figure 1. Chronic obesogenic diet impairs cardiac strain, which is partially reversed by a control diet. (A) Experimental schematic of the study design; (B) Body weight (BW) at study endpoint; (C) LV (Left Ventricle)/BW ratio; (D) Spleen/BW ratio; (E) Long-axis B-mode echocardiography showing LV wall trace and segmental strain changes in CON (10 months of lab chow), OBD (10 months of
Necropsy and echocardiography parameters pre- and post-MI following an obesogenic diet in mice
| Parameters | CON (No-MI) | OBD (No-MI) | OBD-CON (No-MI) | CON (MI-d1) | OBD (MI-d1) | OBD-CON (MI-d1) |
| n | 10 | 7 | 8 | 4 | 4 | 4 |
| Body weight (g) | 41 ± 2 | 40 ± 2 | 40 ± 1 | 37 ± 2 | 41 ± 1 | 42 ± 3 |
| LV (mg) | 135 ± 5 | 126 ± 4 | 127 ± 5 | 141 ± 9 | 132 ± 5 | 133 ± 12 |
| LV/BW (mg/g) | 3.4 ± 0.2 | 3.2 ± 0.1 | 3.2 ± 0.1 | 3.8 ± 0.4 | 3.3 ± 0.02 | 3.2 ± 0.3 |
| Right ventricle (mg) | 22 ± 2 | 20 ± 2 | 19 ± 1 | 21 ± 3 | 21 ± 3 | 21 ± 2 |
| RV/BW (mg/g) | 0.5 ± 0.05 | 0.5 ± 0.1 | 0.5 ± 0.02 | 0.6 ± 0.1 | 0.5 ± 0.1 | 0.5 ± 0.1 |
| Lung/BW(mg/g) | 6.4 ± 0.2 | 6.0 ± 0.5 | 7.0 ± 0.8 | 9.1 ± 2.4 | 8.7 ± 3 | 7.2 ± 1.1 |
| Tibia (mm) | 16 ± 0.4 | 16 ± 0.2 | 15 ± 0.3 | 17 ± 0.5 | 17 ± 07 | 16 ± 0.2 |
| Spleen (mg) | 93 ± 6 | 95 ± 5 | 103 ± 6 | 80 ± 6 | 81 ± 2 | 86 ± 5 |
| Spleen/BW (mg/g) | 2.3 ± 0.1 | 2.4 ± 0.1 | 2.6 ± 0.2 | 2.1 ± 0.1 | 2.0 ± 0.02 | 2.1 ± 0.3 |
| HR (bpm) | 451 ± 15 | 500 ± 50 | 473 ± 20 | 427 ± 36 | 499 ± 50 | 501 ± 24 |
| EDD (mm) | 4.1 ± 0.1 | 4.02 ± 0.1 | 4.24 ± 2.33 | 4.21 ± 0.3 | 4.2 ± 0.4 | 4.6 ± 0.2 |
| ESD (mm) | 2.8 ± 0.1 | 2.9 ± 0.1 | 3.1 ± 0.1 | 3.92 ± 0.3 | 3.91 ± 1$ | 4.32 ± 0.2$ |
| FS (%) | 31 ± 0.1 | 29 ± 2 | 28 ± 0.13 | 7.6 ± 2$ | 7.2 ± 1$ | 6.4 ± 1$ |
| LV PWTd (mm) | 0.88 ± 0.04 | 0.94 ± 0.04 | 0.9 ± 0.1 | 0.60 ± 0.13 | 0.73 ± 0.1 | 0.83 ± 0.1 |
| LV PWTs (mm) | 1.22 ± 0.04 | 1.18 ± 0.04 | 1.13 ± 0.02 | 0.7 ± 0.2 | 1 ± 0.1 | 0.8 ± 0.1$ |
| GLS (%) | -24 ± 2 | -17 ± 2* | -22 ± 2 | -7.7 ± 1$ | -6.3 ± 2$ | -7.7 ± 1$ |
Functional assessment using echocardiography showed no significant differences in LV wall thickness or speckle-tracking-derived segmental strain; however, global longitudinal strain (GLS)-a sensitive marker of systolic dysfunction-was significantly impaired in OBD mice and only partially restored in OBD-R mice, despite unchanged fractional shortening (FS) [Figure 1E-G]. Histological analysis confirmed that LV structure remained intact across all groups, while spleens from OBD mice exhibited a marked reduction in white pulp (WP) area, suggesting a disrupted immune landscape. The WP area was restored in OBD-R mice. In adipose tissue, crown-like structures (macrophages surrounding necrotic adipocytes) were prominent in OBD mice but reduced in OBD R mice [Figure 1H and Supplementary Figure 1D]. Collectively, these findings indicate that dietary reversal reduces adipocyte hypertrophy and partially improves cardiac strain and splenic immune architecture in male mice.
OBD-induced transient and reversible inflammation-resolution memory in the spleen
The spleen serves as a reservoir for immune cells and lipid mediators and may retain a transient
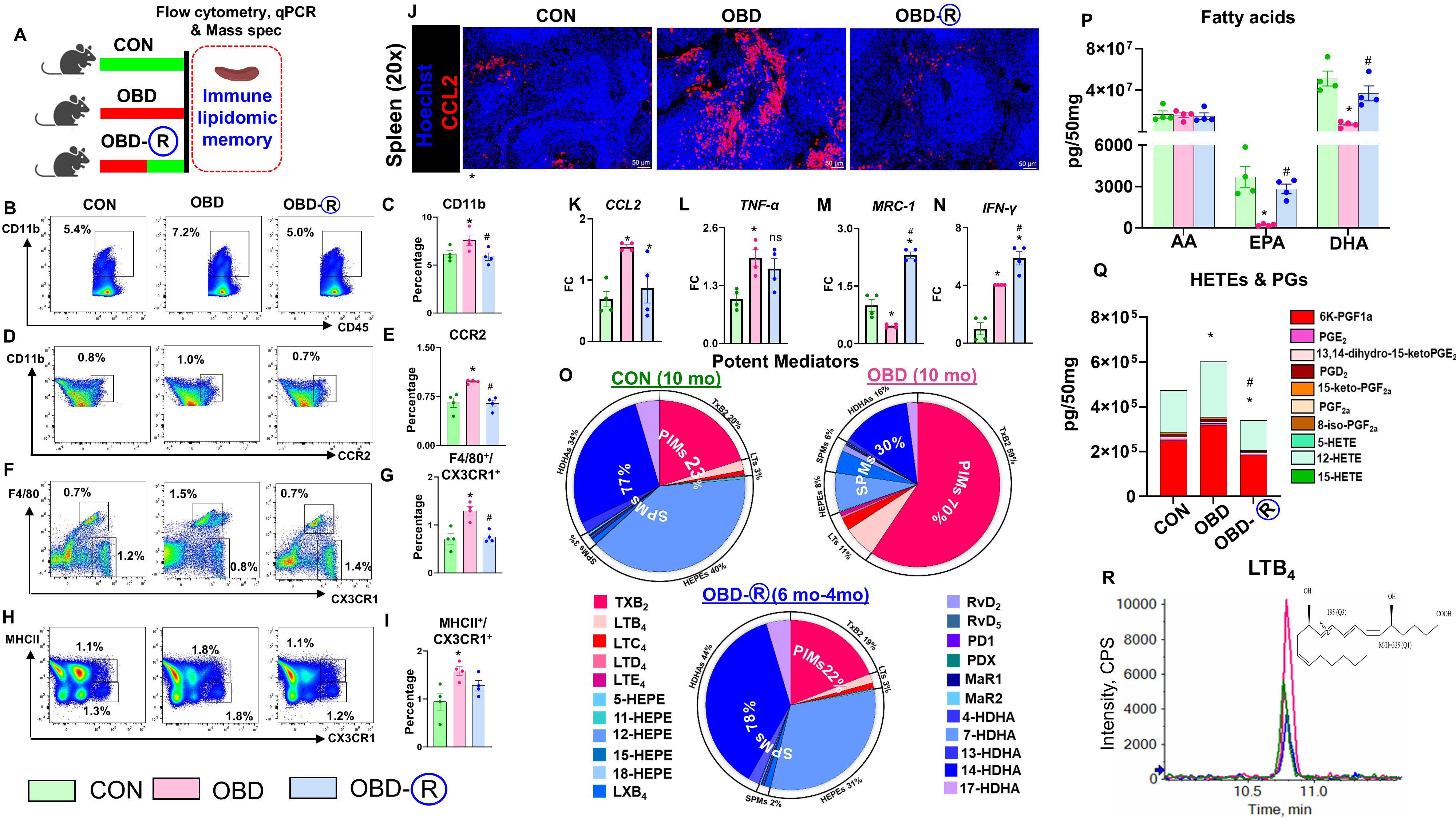
Figure 2. Prior chronic OBD supplementation retains a partial proinflammatory memory in the spleen. (A) Experimental schematic of the mouse study to assess immune-lipidome memory in the spleen; (B-I) Flow cytometry plots and quantification of CD11b+, CCR2+, F4/80+/CX3CR1+, and MHCII+/CX3CR1+ cells in CON (10 months of lab chow), OBD (10 months of omega-6-enriched diet), and
Pro-resolving markers (MRC-1, IFNγ, YM-1) increased in OBD-R spleens, while ARG-1 and IL-10 remained unchanged, indicating partial reprogramming toward a resolution phenotype [Figure 2M and N, Supplementary Figure 2D-F]. Alox12, a key lipoxygenase, was elevated in OBD mice but normalized after dietary reversal, whereas Alox15 and Alox5 were upregulated only in OBD-R mice compared with CON and OBD groups [Supplementary Figure 2G-I]. Both COX-1 and COX-2 were elevated in the spleens of OBD mice compared with controls; however, only COX-2 returned to control levels upon dietary reversal, while COX-1 remained elevated [Supplementary Figure 2J and K]. LC-MS/MS analysis showed a diet-dependent shift in splenic lipid mediator profiles: proinflammatory mediators (PIMs) increased from 23% in CON-fed mice to 70% in safflower oil-rich diet (OBD)-fed mice, while SPMs dropped from 77% to 30%. Reversal
These results highlight that dietary normalization reduces proinflammatory monocyte/macrophage populations and lipid mediators in the spleen, suggesting that immune-lipid memory is at least partially reversible in male mice.
Cardiac proinflammatory memory from chronic OBD persists despite dietary reversal
Obesity exacerbates heart failure by activating immune responses[19]. We investigated cardiac lipid-immune memory using gene expression analysis, LC-MS/MS, and flow cytometry [Figure 3A]. Analysis of fatty acids in mouse hearts revealed higher levels of arachidonic acid (AA) and lower levels of docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) in the OBD group [Figure 3B]. Hearts from OBD-R mice showed fatty acid levels similar to CON, suggesting that fatty acid alterations are partially reversible [Figure 3B]. Pathways for AA, DHA, and EPA are shown in Figure 3C. COX-mediated lipid mediators were elevated in OBD hearts but partially reversed in OBD-R [Figure 3D]. PIMs generated via 12/15LOX were increased in OBD but remained unchanged in OBD-R, with no change in Lipoxin B4 (LXB4) in all groups [Figure 3E]. No differences were observed in 5-LOX-generated PIMs [Figure 3F]. Hydroxy-DHA was decreased in OBD but remained high in OBD-R hearts [Figure 3G]. EPA-generated hydroxyicosapentaenoic acid (HEPEs) remained unchanged, except for 12-HEPE [Figure 3H]. Overall, PIM composition was increased in OBD hearts; in OBD-R hearts, PIM levels decreased but did not fully return to CON levels [Figure 3I, Supplementary Tables 2 and 3]. Pie charts of LMs indicated altered HETE levels in ODB, which decreased in the OBD-R group [Figure 3J]. Immunofluorescence data showed that CCL2 (red) expression was elevated in LV and adipose tissues of OBD mice, but reversed in OBD-R [Figure 3K]. Immune profiling revealed increased leukocyte populations in OBD hearts, which normalized to CON levels in OBD-R [Figure 3L and M]. Monocytes (CD11b+) were increased in OBD but decreased in OBD-R [Figure 3N and O], while proinflammatory monocytes
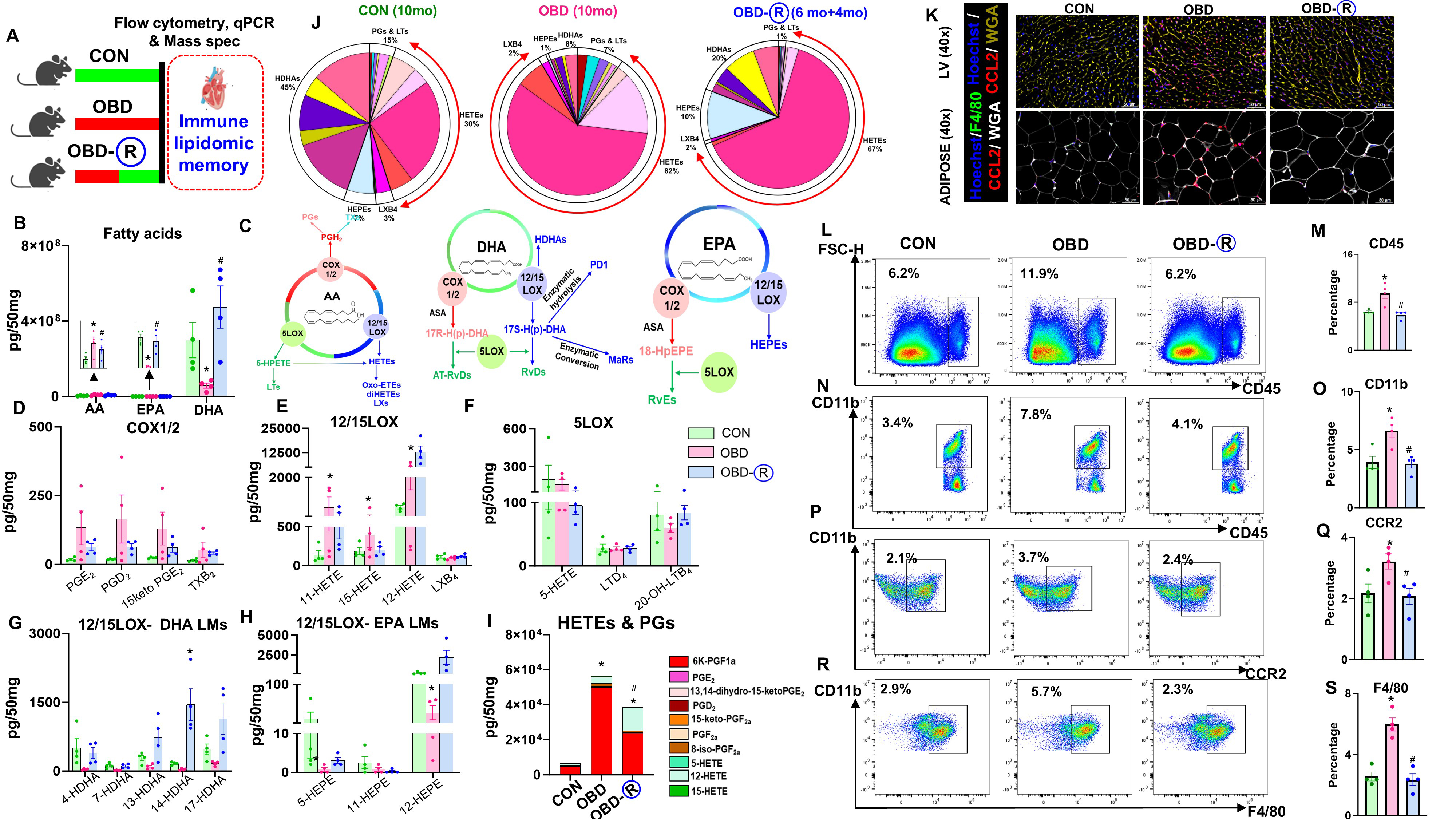
Figure 3. Residual inflammation is primed by prior obesity through partial maintenance of a proinflammatory profile in the heart. (A) Experimental schematic of the mouse study to assess immune-lipidome memory in naïve LV; (B) Fatty acids [arachidonic acid (AA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA)] in the LV of CON (10 months of lab chow), OBD (10 months of omega-6-enriched diet), and OBD-R (10 months of OBD diet + 4 months of CON diet) mice; (C) Schematic of AA, DHA, and EPA metabolic pathways; (D-F) Concentrations of (D) COX, (E) 12/15LOX, and (F) 5LOX products derived from AA in the LV; (G) Concentration of 12/15LOX products derived from DHA; (H) Concentration of 12/15LOX products derived from EPA. Quantification is reported as pg/50 mg LV tissue; detection limit ≈ 1 pg; (I) Stacked graphs showing average concentrations of prostaglandins (PGs) and hydroxyeicosatetraenoic acids (HETEs) in the LV; (J) Pie charts showing the distribution of AA, DHA, and EPA products. Percentages represent mean values for each identified lipid mediator; (K) Representative immunofluorescence images showing CCL2 (red) expression with cardiomyocyte area (WGA-yellow) in LV and CCL2 (red), F4/80 (green), and WGA (white) in adipocyte tissue. Nuclei are labeled with Hoechst (blue). Magnification, 40×. Scale bar, 50 μm; (L-S) Flow cytometry plots and quantification of CD45+, CD11b+, CCR2+, and F4/80+ populations in the LV. Comparisons were analyzed using one-way ANOVA with Tukey’s multiple comparisons test. *P < 0.05 vs. CON; #P < 0.05 vs. OBD. Data are presented as mean ± SEM; n = 4/group. LV: Left ventricle; CON: control diet; OBD: obesogenic diet; SEM: standard error of the mean; WGA: wheat germ agglutinin.
OBD fails to activate physiological inflammation in the LV and spleen post-MI after reversal from OBD to CON diet
Although switching from an OBD to a CON diet partially reversed lipid and immune alterations, we further evaluated whether this reversal modulates the acute inflammatory response following cardiac injury. To this end, MI was induced by permanent coronary artery ligation in CON, OBD, and OBD-R mice. This design allowed us to assess early post-injury immune activation and explore the splenocardiac axis by profiling immune and lipid profiles in the heart under acute stress conditions. Following MI, the CON-MI group showed a reduction in FS (7.6 ± 2%) compared to CON-No-MI mice (31 ± 0.1%). No significant differences were observed among the CON, OBD, and OBD-R groups post-MI at day 1 [Figure 4A, Supplementary Figure 4A, and Supplementary Table 2]. Histological analysis of the LV confirmed comparable wall thinning and leukocyte infiltration in infarcted regions across all groups, indicating consistent coronary ligation
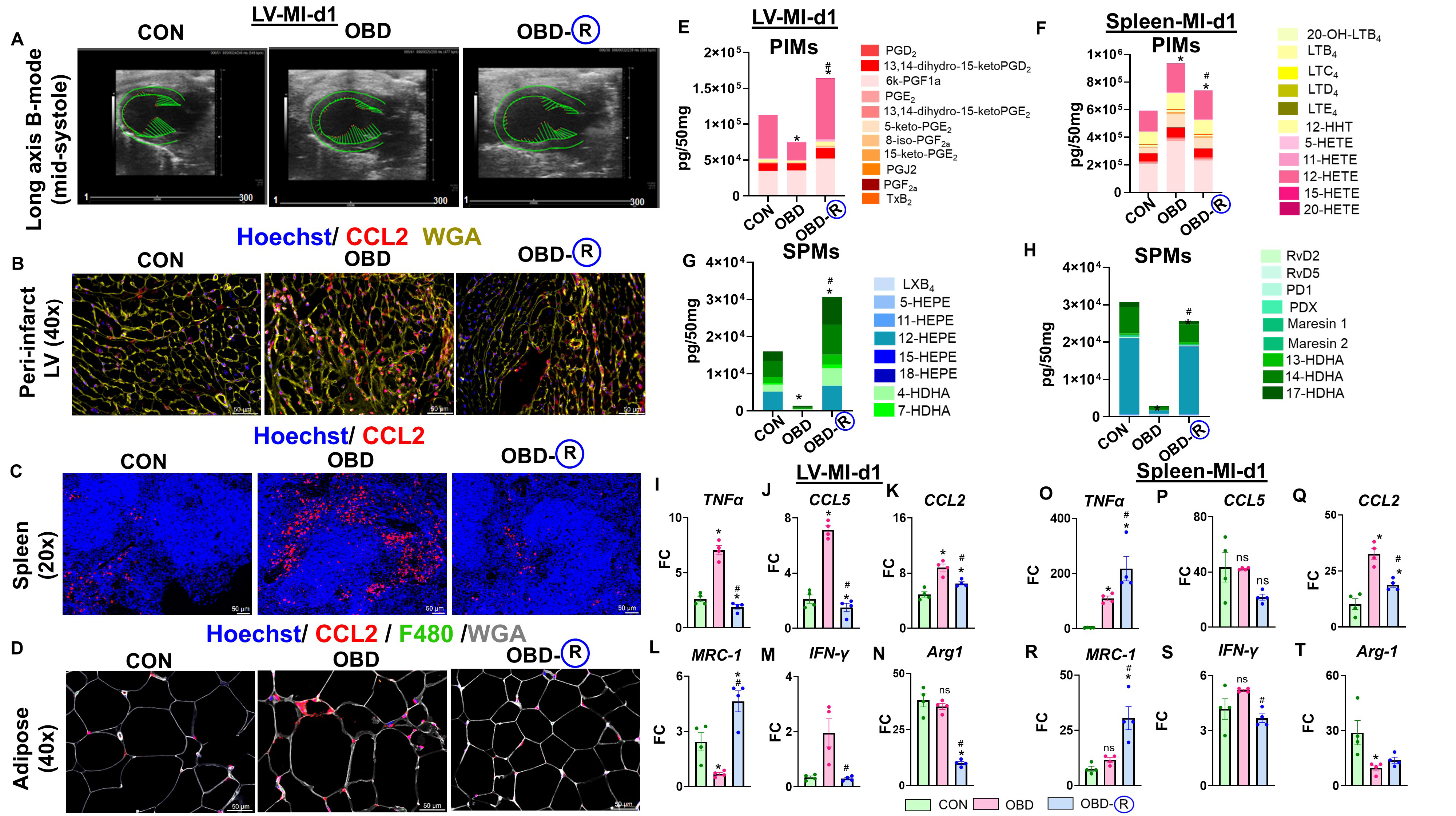
Figure 4. OBD intake fails to initiate physiological acute inflammation-resolution signaling in the spleen and infarcted LV post-MI. (A) Representative long-axis B-mode images showing longitudinal strain in the left ventricular (LV) of CON (10 months of lab chow), OBD (10 months of omega-6-enriched diet), and OBD-R (10 months of OBD diet + 4 months of CON diet) mice at day 1 post-MI. Representative immunofluorescence showing; (B) CCL2 expression (red) with cardiomyocyte area (WGA, yellow) in LV; (C) CCL2 (red) in spleen; (D) CCL2 (red), F4/80 (green), and WGA (white) in adipocytes of CON, OBD, and OBD-R mice. Nuclei were counterstained with Hoechst (blue); Magnification, 40× (LV, adipose) and 20× (spleen). Scale bar, 50 μm. Stacked graphs showing average (E) PIMs and (F) SPMs in LV at day 1 post-MI; Stacked graphs showing average (G) PIMs and (H) SPMs in the spleen at day 1 post-MI. Quantification values are expressed as pg/50 mg tissue (LV and spleen). Detection limit was ≈ 1 pg (mass spectrometry); mRNA expression of proinflammatory cytokines (I) TNF-α, (J) CCL5, (K) CCL2 in LV; pro-resolving cytokines (L) MRC-1; (M) IFN-γ, (N) Arg-1 in LV; proinflammatory cytokines (O) TNF-α, (P) CCL5, (Q) CCL2 in spleen; pro-resolving cytokines (R) MRC-1, (S) IFN-γ, (T) Arg-1 in LV at day 1 post-MI. Data are presented as fold change (FC) relative to CON-d0 (No-MI, FC = 1), with normalization to Hprt-1. Group comparisons were analyzed using one-way ANOVA with Tukey’s multiple comparisons test. *P < 0.05 vs. CON, #P < 0.05 vs. OBD. Data are presented as mean ± SEM; n = 4/group, ns: non-significant. MI: Myocardial infarction; SPMs: specialized pro-resolving mediators; PIMs: proinflammatory mediators; CON: control diet; OBD: obesogenic diet; SEM: standard error of the mean; WGA: wheat germ agglutinin.
Diet switch potentiates macrophage memory at local and hematopoietic levels
An omega-6-rich diet (OBD) enhances the proinflammatory response, particularly in macrophages. To determine whether this effect is driven by local tissue environments or hematopoietic programming, we isolated macrophages from bone marrow (BMDMs), spleen, and peritoneal cavities of CON, OBD, and OBD-R mice, differentiated them, and stimulated them with LPS for 4 h [Figure 5A]. At baseline, macrophages derived from OBD mice-regardless of origin-exhibited elevated expression of proinflammatory genes such as TNF-α and CCL2, along with increased expression of the pro-resolving marker Arg-1. In contrast, MRC-1 expression was reduced in OBD-derived macrophages. Notably, baseline TNF-α and CCL2 levels were lower in macrophages from the OBD-R group. Following LPS stimulation, macrophages (BMDMs, splenic, and peritoneal) from both CON and OBD groups showed increased expression of TNF-α, CCL2, MRC-1, and Arg-1. However, macrophages from the OBD-R group displayed attenuated proinflammatory cytokine responses, with minimal changes in MRC-1 and reduced Arg-1 expression [Figure 5B-M and Supplementary Figure 5A-F]. Immunofluorescence analysis further revealed heightened CCL2 expression in the OBD group and increased MRC-1 expression in the OBD-R group, both at baseline and post-LPS stimulation across all macrophage sources [Figure 5N-P]. To assess functional capacity, we performed a phagocytosis assay using pHrodo-labeled E.coli. Macrophages from OBD mice demonstrated reduced phagocytic activity, which was restored in the OBD-R group. Although LPS stimulation enhanced phagocytosis in the OBD group, activity remained lower compared with CON and OBD-R across all macrophage types [Figure 5Q-S].
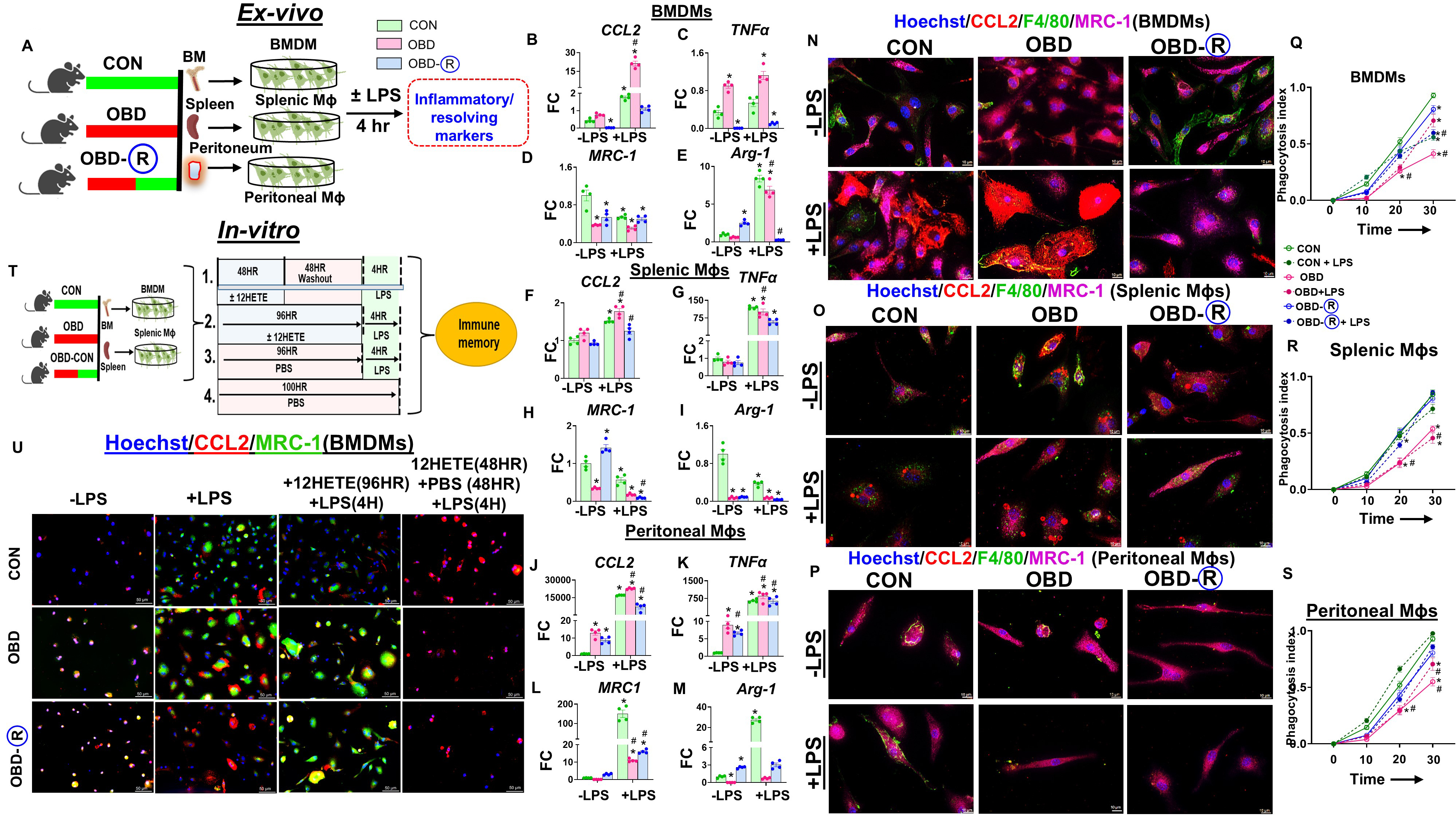
Figure 5. Diet switch to control diet activates macrophage memory and function at hematopoietic and local levels. (A) Experimental schematic of in vitro LPS-induced immune memory. BMDMs, splenic MΦ, and peritoneum MΦ isolated from CON (10 months of lab chow), OBD (10 months of omega-6-enriched diet), and OBD-R (10 months of OBD diet + 4 months of CON diet) mice, with or without LPS (100 ng/mL, 4 h), to examine MΦ memory; BMDM mRNA expression of (B) CCL2, (C) TNF-α, (D) MRC-1, and (E) ARG-1 in the presence or absence of LPS from CON, OBD, and OBD-R mice. Splenic MΦ mRNA expression of (F) CCL2, (G) TNF-α, (H) MRC-1, and (I) ARG-1; Peritoneum MΦ mRNA expression of (J) CCL2, (K) TNF-α, (L) MRC-1, and (M) ARG-1. Representative immunofluorescence images showing MRC-1 (purple), CCL2 (red), F4/80 (green), and nuclei (Hoechst, blue) in (N) BMDMs, (O) splenic MΦs, and (P) peritoneal MΦs with or without LPS from CON, OBD, and OBD-R mice. Magnification, 100×. Scale bar, 10 μm. Phagocytosis index quantified by the percentage of pHrodo-Red-positive macrophages at 0, 15, 30, and 60 min in (Q) BMDM, (R) splenic MΦs, (S) peritoneal MΦs; (T) Experimental schematic of the in vitro 12-HETE and LPS-induced immune memory model; (U) Representative immunofluorescence images showing expression of MRC-1 (green), CCL2 (red), and nuclei (Hoechst, blue) depicting 12-HETE-induced immune memory in BMDMs isolated from CON, OBD, and OBD-R mice. Magnification, 40×. Scale bar, 50 μm. Comparisons between groups were analyzed using two-way ANOVA with Sidak’s multiple comparisons test for -LPS and +LPS samples. *P < 0.05 vs. CON
Upon LPS stimulation, macrophages from different tissue sources exhibited distinct eicosanoid enzyme profiles. Splenic, peritoneal, and bone marrow macrophages from OBD mice consistently showed upregulated COX-1, COX-2, and Alox12. In contrast, Alox15 and Alox5 were markedly reduced in splenic and peritoneal macrophages, but not in BMDMs. Reversion to the control diet (OBD-R) partially normalized COX-2 and Alox12 expression in splenic macrophages, with minimal effects on other enzymes
OBD amplifies, and OBD-R retains, lipid milieu-dependent inflammatory immune memory after removal of the obesity stressor
To investigate immune profiles and gene expression patterns under control (CON), obesity-inducing diet (OBD), and diet-reversal (OBD-R) conditions, CD45+ immune cells were isolated from the hearts and spleens of CON, OBD, and OBD-R mice under naïve (non-injured) conditions using fluorescence-activated cell sorting. Single-cell transcriptional profiling was performed using the 10x Genomics Chromium platform, and only cells passing stringent quality control filters were included in downstream analysis. Using the 10x Genomics cloud platform and Loupe analysis tools, we found that chronic OBD exposure significantly altered the single-cell transcriptomic landscape.
At baseline (no-MI), LVs in CON mice exhibited four major immune cell clusters: macrophages (MΦs), B cells, T cells, and natural killer (NK) cells (very low). In contrast, OBD mice displayed five distinct clusters: monocytes (Mo), macrophages (MΦs), neutrophils (Neu), T cells, and B cells, with a notable absence of NK cells. Following dietary reversal (OBD-R), the immune landscape shifted toward a CON-like profile, showing four clusters consisting of monocytes, macrophages, neutrophils, and T cells [Figure 6A]. In the context of acute MI at day 1, CON-MI-d1 myocardium exhibited seven distinct clusters. By comparison, OBD and OBD-R groups showed reduced cluster diversity (4 and 6, respectively), although neutrophils persisted in both groups [Figure 6B]. Subsequent sub-clustering revealed heterogeneous subtypes within major lineages, distinguished by proinflammatory and pro-resolving markers. Marker expression profiles varied significantly by diet, indicating diet-induced changes in gene regulation and immune activation. Differentialexpression of markers such as CCR2, TNF-α, IL1β, CXCR2, Arg-1, MRC-1, C1qa, CCL5, Alox5, Alox5ap, Ptgs-1, Ptgs-2, and Lta4h highlighteddiet-dependent variations across immnue cell types
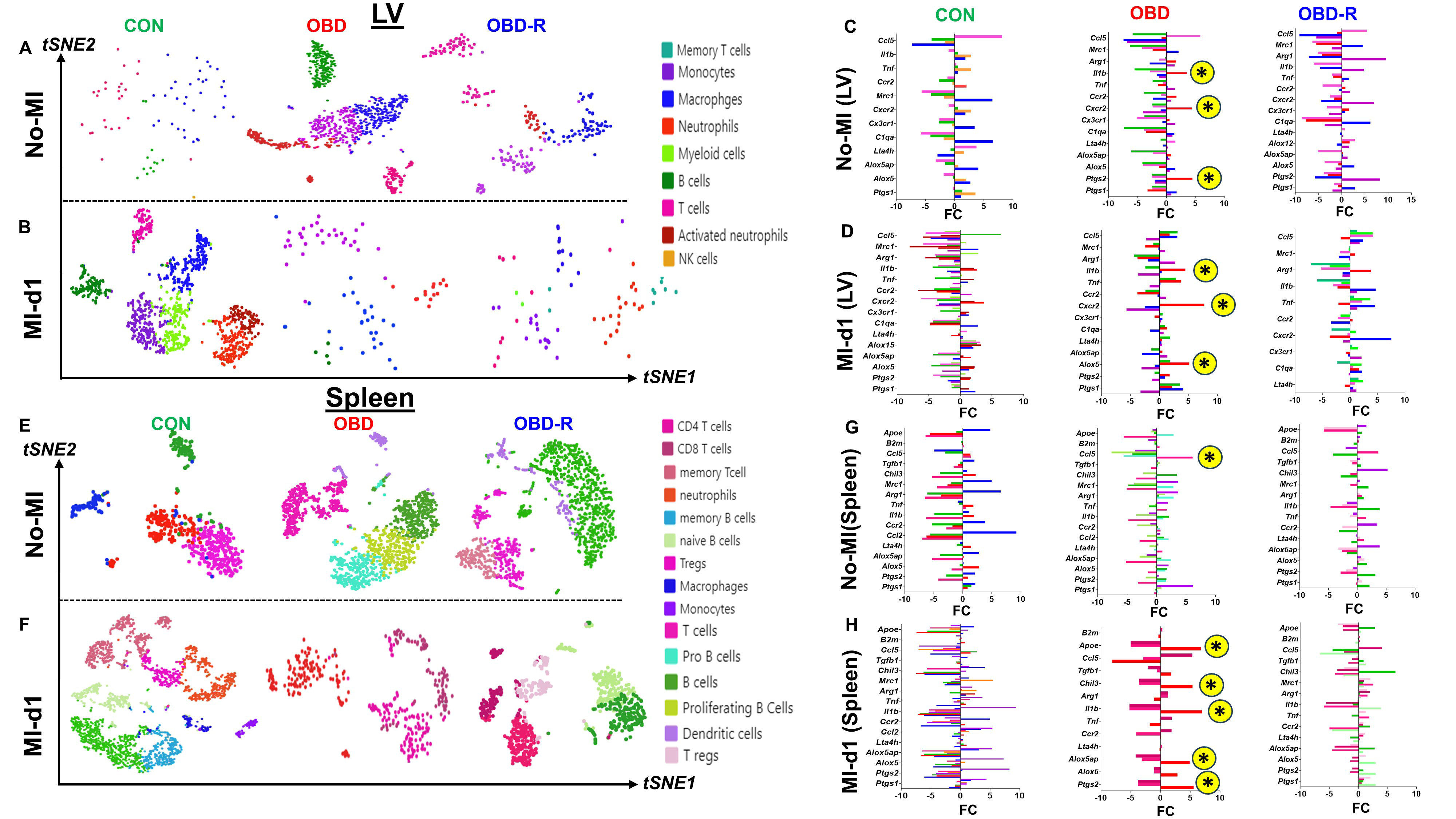
Figure 6. Impact of diet switch on CD45+ immune cells identified by scRNA-seq in the heart and spleen under homeostatic (no-MI) and infarcted (post-MI) conditions. (A) t-SNE plot of immune cells in LV of CON (10 months of lab chow), OBD (10 months of
Obesity-driven reprogramming of macrophages correlates with CCR2 and ALOX5AP activation
ALOX5AP (FLAP) is a key enzyme in the biosynthesis of proinflammatory lipid mediators such as leukotriene B4 (LTB4)[20]. To explore how chronic dietary exposure might influence immune gene expression, single-cell RNA sequencing was performed on FACS-sorted CD45+ cells from the heart and spleen of CON, OBD, and OBD-R mice under naïve conditions. This analysis revealed that monocytes and neutrophils from OBD mice displayed a transcriptional profile consistent with activation, including increased expression of CCR2 and ALOX5AP, even in the absence of injury or exogenous stimulation (e.g., 12-HETE). Although these observations suggest a link between chronic dietary intake and pre-activated immune states, they remain correlative in nature.
Based on this pattern, we hypothesized that the CCR2-ALOX5AP axis contributes to lipid-immune memory in the context of an omega-6-rich diet. We therefore profiled CCR2 and ALOX5AP expression in the LV and spleen. CCR2 mRNA levels were elevated in both OBD and OBD-R groups compared with CON at baseline, consistent with a chronic inflammatory state. Following MI, CCR2 expression increased across all groups [Figure 7A and B]. Immunofluorescence analysis further revealed that CCR2 expression in the OBD group extended to both cardiomyocytes and non-cardiomyocytes, whereas in the CON group it was restricted to non-cardiomyocytes. In OBD-R mice, CCR2 expression persisted at baseline, suggesting a residual inflammatory imprint. Post-MI, CCR2 expression increased further in all groups [Figure 7C]. In the spleen, OBD mice exhibited widespread CCR2 expression at baseline, which declined in OBD-R mice but remained elevated post-MI [Figure 7D].
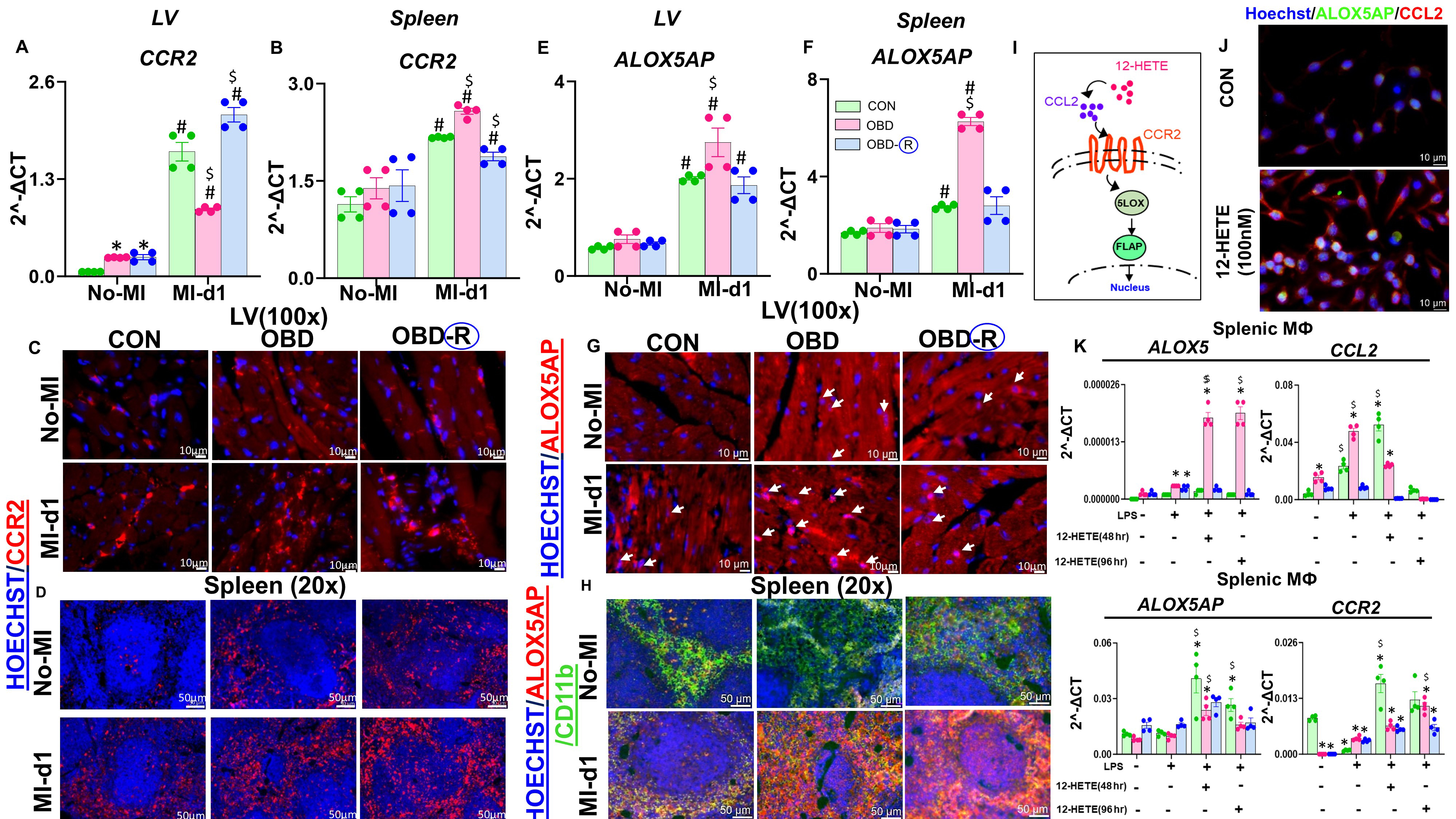
Figure 7. Obesogenic diet reprograms macrophages through CCR2 and ALOX5AP activation. (A and B) CCR2 mRNA expression in LV and spleen samples from CON (10 months of lab chow), OBD (10 months of omega-6-enriched diet), and OBD-R (10 months of OBD diet + 4 months of CON diet) groups under no-MI and post-MI-d1 conditions; (C) Representative immunofluorescence images showing CCR2 (red) expression in the LV of CON, OBD, and OBD-R mice under no-MI and post-MI-d1 conditions. Nuclei are counterstained with Hoechst (blue). Magnification, 100×. Scale bar, 10 μm; n = 3/group; (D) Representative immunofluorescence images showing CCR2 (red) expression in spleens of CON, OBD, and OBD-R mice under no-MI and post-MI-d1 conditions. Nuclei are counterstained with Hoechst (blue). Magnification, 20×. Scale bar, 50 μm; n = 3/group; (E and F) ALOX5AP mRNA expression in LV and spleen samples from CON, OBD, and OBD-R mice under no-MI and post-MI-d1 conditions; (G) Representative immunofluorescence images showing ALOX5AP (red) expression in the LV of CON, OBD, and OBD-R mice under no-MI and post-MI-d1 conditions. Nuclei are counterstained with Hoechst (blue). White arrows indicate nuclear localization (pink). Magnification, 100×. Scale bar, 10 μm; n = 3/group; (H) Representative immunofluorescence images showing ALOX5AP (red) and CD11b (green) expression in spleens of CON, OBD, and OBD-R mice under no-MI and post-MI-d1 conditions. Nuclei are counterstained with Hoechst (blue). Magnification, 20×. Scale bar, 50 μm; n = 3/group; (I) Graphical representation of the ALOX5AP pathway activated by 12-HETE; (J) Representative immunofluorescence images showing ALOX5AP (green) and CCL2 (red) expression in RAW 264.7 cells, untreated or treated with
ALOX5AP mRNA levels did not differ significantly between groups at baseline but were markedly increased in the LV and spleen of OBD mice post-MI compared to CON and OBD-R mice [Figure 7E and F]. Imaging showed nuclear translocation of ALOX5AP in the OBD group, an effect attenuated in the OBD-R group. Following MI, nuclear translocation of ALOX5AP was also observed in the CON group but occurred to a lesser extent than in OBD mice [Figure 7G]. In the spleen, co-localization of ALOX5AP with CD11b+ monocytes was observed across all no-MI control groups; after MI, this co-localization intensified in OBD mice and was moderately elevated in OBD-R mice [Figure 7H].
To further investigate potential upstream lipid mediators, RAW 264.7 macrophages were stimulated with 12-HETE for 4 hours. This treatment upregulated CCL2 and ALOX5AP and induced nuclear translocation of ALOX5AP [Figure 7I-J and Supplementary Figure 13]. To examine memory-like responses, macrophages were subjected to continuous or discontinuous 12-HETE exposure. Continuous 12-HETE treatment increased expression of ALOX5, CCL2, ALOX5AP, and CCR2 in OBD-derived cells, whereas discontinuous treatment selectively upregulated ALOX5AP and CCR2 across all groups [Figure 7K]. Taken together, these in vivo and in vitro findings suggest an association between elevated 12-HETE and enhanced CCR2 and ALOX5AP expression under OBD conditions. While these findings support a role for the CCL2-CCR2 and ALOX5-ALOX5AP axes in macrophage priming and lipid-immune reprogramming, they remain correlative, and functional studies are needed to establish causality.
DISCUSSION
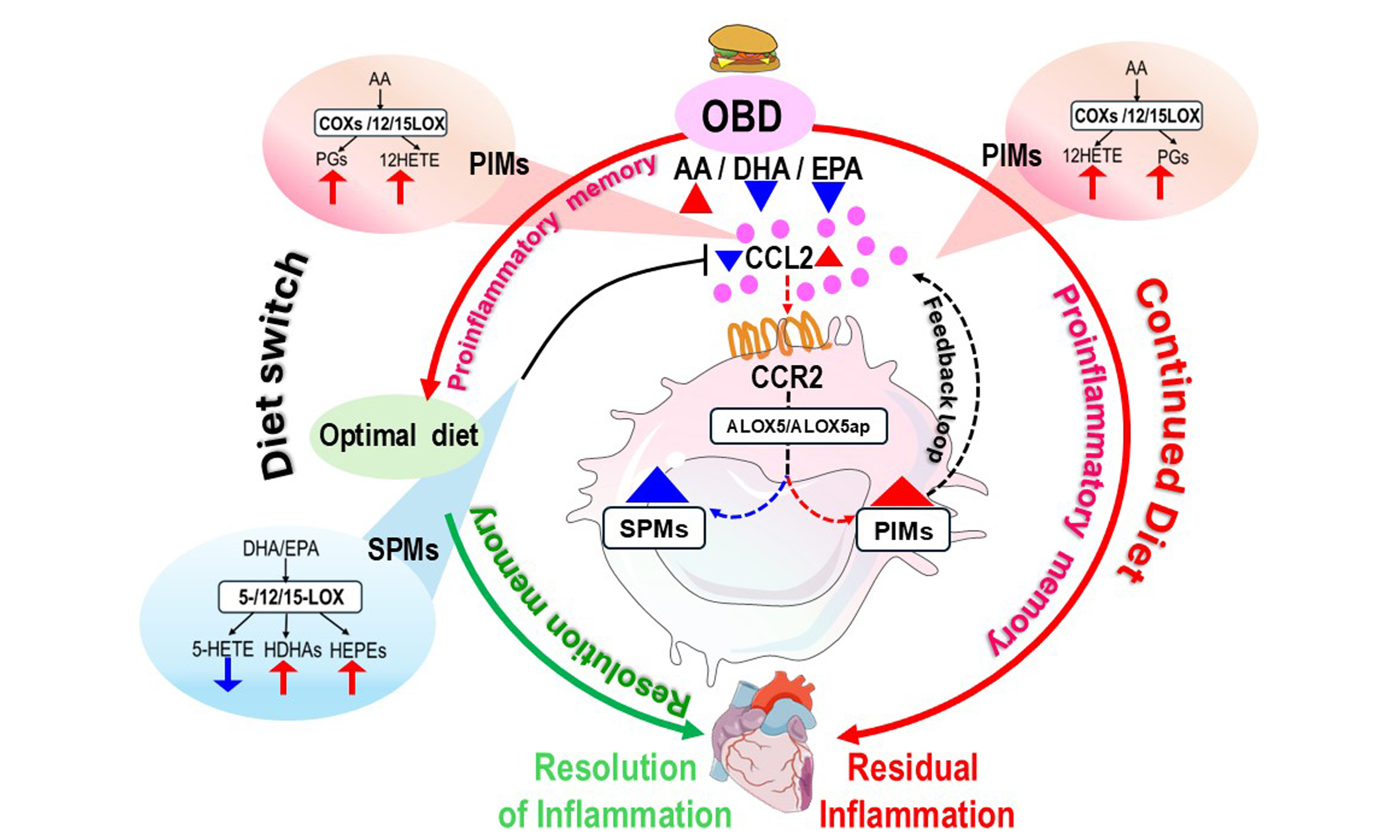
A diet enriched in saturated fats, refined carbohydrates, and processed foods promotes chronic inflammation and non-communicable diseases, affecting multiple immune cell types[21]. The health effects of fatty acids differ between youth and aging, influencing cardiovascular health and immune memory[13]. Overall, diet regulates lipid metabolism and transcriptomes related to immunity and inflammation, with implications for cardiac disease and cancer[22-24]. In this study, we tested whether prolonged supplementation with an omega-6 fatty acid-enriched diet induces lasting changes in innate immunity and enhances proinflammatory responses in the myocardium. Reversion to a regular diet partially normalized fatty acid profiles and immune signatures toward a pro-resolving phenotype, supporting the concept that diet modulates both inflammation and immune memory (as summarized in the graphical abstract). Our data indicate that: (1) a safflower oil-rich diet (OBD) drives a proinflammatory lipidome-immune memory without overt impairment of cardiac systolic function, though subtly affecting cardiac strain; (2) OBD reduces SPMs while expanding PIMs, particularly hydroxyeicosatetraenoic acids (HETEs), which activate the CCL2-CCR2 and ALOX5-ALOX5ap axes, particularly in macrophages; (3) switching from chronic OBD to a control diet restores fatty acid levels associated with lipid memory, but does not completely reverse the proinflammatory lipidome, leaving a residue “inflamed” immune memory.
Lipid mediators - both PIMs and SPMs - are essential for safe resolution of inflammation and regulation of the immune response[25,26]. A diet high in omega-6 fatty acids (OBD) increases AA levels while reducing omega-3 fatty acids, specifically DHA and EPA. This shift in the omega-6/omega-3 ratio promotes proinflammatory responses in the myocardium and spleen[13,27]. AA-derived molecules foster inflammation, vasoconstriction, and platelet aggregation[28]. Elevated proinflammatory HETEs (e.g., 12-HETE), together with cytokines such as IL-1β and TNF-α, compromise immune cell health[29]. Although OBD had only modest effects on heart function, it lowered cardiac strain, indicating a higher risk of future heart disease. Returning to a regular diet reduced AA levels, restored omega-3 fatty acids, and partially improved cardiac strain, underscoring the influence of fatty acid quality and quantity on cardiometabolic health.
Dietary reversal after OBD preserved elements of immune memory, reflected in sustained transcriptional immune activation despite normalization of immune cell numbers. OBD appears to reprogram myeloid cells both locally and systemically, influencing their phenotypic and functional characteristics[30]. Specifically, OBD triggered CCL2 expression in the heart, spleen, and adipose tissue, alongside CCR2 in CD45+ myeloid cells. Together, CCL2 and CCR2 drive inflammation and immune cell recruitment to injury sites. Switching to a control diet reduced CCL2 expression across the heart, spleen, and bone marrow, suggesting partial restoration of immune memory toward a homeostatic state resembling that of CON-fed mice. These results point to the involvement of innate immune memory, potentially maintained by stable epigenetic and metabolic reprogramming of monocytes and macrophages. Our findings are consistent with studies demonstrating that Western diets induce sterile inflammation and imprint long-lasting innate immune memory. In chronic OBD, this memory manifested as persistent expression of proinflammatory genes-including TNF-α, CCL5, CCL2, and IL-6-particularly in cardiac myeloid cells[31-33]. Cardiac inflammation remained more pronounced than splenic inflammation, where TNF-α and CCL2 also remained active in myeloid cells. Thus, dietary reversal retains aspects of proinflammatory lipid mediator “memory”, sustaining inflamed immune cell programming.
Excess circulating saturated fatty acids further exacerbate inflammation in obesity induced by a high-fat diet[34]. Excessive omega-6 fatty acid intake depletes pro-resolving mediators and their precursors[13]. Both heart and spleen exhibited significant reductions in SPMs before and after myocardial infarction, indicating disruption of the transition from inflammation to resolution[16]. Dietary reversal partially restored SPMs in the heart and fully in the spleen, suggesting that fatty acid availability can be recovered post-OBD, but the memory of bioactive lipid mediator production may not be fully reversible. Prior studies show that diet strongly influences macrophage activation, with elevated CCL2 expression across macrophage subsets[35,36]. Consistently, 12-HETE levels rose after MI, indicating ongoing cardiac injury. Clinical studies have similarly reported elevated 12-HETE in patients with cardiovascular disease, implicating it in pathogenesis. Moreover, 12-HETE can induce macrophage proinflammatory responses independent of LPS, supporting its role as an initiator of innate immune memory[37]. Single-cell RNA sequencing and in vitro memory activation assays confirmed that unsaturated fat-derived proinflammatory mediators directly affect macrophage phenotype and gene pathways. 12-HETE appears to indirectly activate ALOX5AP through the CCL2-CCR2 axis, suggesting involvement of a CCL2-CCR2-ALOX5-ALOX5AP signaling pathway in response to OBD, potentially via a feed-forward loop. Although ALOX5AP lacks intrinsic enzymatic activity, it facilitates ALOX5 function during leukotriene biosynthesis from AA, underscoring its role in promoting proinflammatory lipid mediator production. By contrast, DHA and EPA generate SPMs in response to ischemic injury. However, only limited studies have addressed the role of FLAP agonists in SPM formation, which may depend on the PUFA substrate[38]. This aligns with our findings that dietary reversal from OBD to a control diet with reduced omega-6 content shifted the ALOX5-ALOX5AP axis toward SPM biosynthesis, thereby promoting resolution-phase immune memory in macrophages. Dietary changes also altered transcriptional and functional diversity within innate immune cells of the heart and spleen. Single-cell RNA sequencing showed that OBD increased expression of specific genes in neutrophils, monocytes, and macrophages, some of which - such as ptgs-2, cxcr2, ccr2, and IL1β- remained irreversibly elevated despite dietary reversal. These results highlight the need for further investigation into the resolution of meta-inflammation, particularly in the context of reversing omega-6-rich diets. Obesity is a known contributor to several hallmarks of accelerated and potentially irreversible aging[39]. The persistent elevation of senescence-associated markers-Cdkn1a, JUNB, and GOX4-despite dietary intervention, supports the notion that OBD accelerates aging processes not fully reversible by nutritional correction alone.
In summary, long-term consumption of an omega-6-rich diet induces a persistent lipid-immune memory in mice. Although dietary reversal can partially normalize fatty acid composition, proinflammatory lipid memory within innate immune cells persists, driving sustained cardiac inflammation. These OBD-driven alterations reflect systemic immune reprogramming that links spleen and heart. This study underscores the importance of dietary composition in reshaping immune memory and cardiac health, reinforcing the central principle of lifestyle medicine: “Optimal food is prime for health”[40].
This study has limitations. The exclusive use of male mice may limit generalizability, given the
DECLARATIONS
Acknowledgments
The authors acknowledge the support from the University of South Florida cardiac physiology and lipidomics cores. For the summary sketch, the authors acknowledge the use of heart and spleen images from Servier Medical Art.
Author’s contributions
Conceived the study design: Halade GV
Developed the experimental strategy and implemented the experimental plans: Halade GV, Kain V
Performed specific experiments, as well as data processing and analysis: Halade GV, Ma Y, Yeatman TJ, Kain V, Upadhyay G, Marimuthu M
All authors contributed to discussions, participated in manuscript drafting, and approved the final version for submission.
Availability of data and materials
A detailed description of the materials and methods is provided in the Supplemental Material. Single-cell sequencing data are submitted to the NCBI BioProject database (BioProject ID: PRJNA1026923). Data supporting the findings of this study are available from the corresponding author upon reasonable request.
Financial support and sponsorship
Authors acknowledge support from University of South Florida and National Institutes of Health HL164446 to YM, and U01CA293474 to TJY and GVH.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Animal use and monitoring were conducted according to the “Guide for the Care and Use of Laboratory Animals” (8th Edition, 2011), and AVMA Guidelines for the Euthanasia of Animals (2020 Edition) were approved by the Institutional Animal Care and Use Committee at the University of South Florida, Tampa, USA (Protocol No. 7371R). The study followed the ARRIVE 2.0 guidelines for experimental procedures.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
Supplementary Materials
REFERENCES
1. Tsao CW, Aday AW, Almarzooq ZI, et al; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2023 update: a report from the American Heart Association. Circulation. 2023;147:e93-e621.
2. Woodruff RC, Tong X, Jackson SL, Loustalot FV, Vaughan AS. Abstract 9853: trends in national death rates from heart disease in the United States, 2010-2020. Circulation. 2022;146:A9853.
3. Babalola OO, Akinnusi E, Ottu PO, et al. The impact of ultra-processed foods on cardiovascular diseases and cancer: epidemiological and mechanistic insights. Asp Mol Med. 2025;5:100072.
4. Kittleson MM, Benjamin EJ, Blumer V, et al. 2025 ACC scientific statement on the management of obesity in adults with heart failure: a report of the American College of Cardiology. J Am Coll Cardiol. 2025.
5. Nishio R, Dohi T, Takeuchi M, et al. Combined impact of residual inflammatory risk and chronic kidney disease on long-term clinical outcomes in patients undergoing percutaneous coronary intervention. J Cardiol. 2022;79:509-14.
6. Yudkin JS, Stehouwer CD, Emeis JJ, Coppack SW. C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler Thromb Vasc Biol. 1999;19:972-8.
7. Halade GV, Norris PC, Kain V, Serhan CN, Ingle KA. Splenic leukocytes define the resolution of inflammation in heart failure. Sci Signal. 2018;11:eaao1818.
8. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11:255-65.
9. Aprahamian T, Takemura Y, Goukassian D, Walsh K. Ageing is associated with diminished apoptotic cell clearance in vivo. Clin Exp Immunol. 2008;152:448-55.
10. Gasparoto TH, Dalboni TM, Amôr NG, et al. Fcγ receptors on aging neutrophils. J Appl Oral Sci. 2021;29:e20200770.
11. Hata M, Andriessen EMMA, Hata M, et al. Past history of obesity triggers persistent epigenetic changes in innate immunity and exacerbates neuroinflammation. Science. 2023;379:45-62.
12. Mente A, Dehghan M, Rangarajan S, et al. Diet, cardiovascular disease, and mortality in 80 countries. Eur Heart J. 2023;44:2560-79.
13. Halade GV, Kain V, Black LM, Prabhu SD, Ingle KA. Aging dysregulates D- and E-series resolvins to modulate cardiosplenic and cardiorenal network following myocardial infarction. Aging. 2016;8:2611-34.
14. Lopez EF, Kabarowski JH, Ingle KA, et al. Obesity superimposed on aging magnifies inflammation and delays the resolving response after myocardial infarction. Am J Physiol Heart Circ Physiol. 2015;308:H269-80.
15. Kain V, Van Der Pol W, Mariappan N, et al. Obesogenic diet in aging mice disrupts gut microbe composition and alters neutrophil:lymphocyte ratio, leading to inflamed milieu in acute heart failure. FASEB J. 2019;33:6456-69.
16. Halade GV, Kain V, De La Rosa X, Lindsey ML. Metabolic transformation of fat in obesity determines the inflammation resolving capacity of splenocardiac and cardiorenal networks in heart failure. Am J Physiol Heart Circ Physiol. 2022;322:H953-70.
17. Percie du Sert N, Hurst V, Ahluwalia A, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. BMJ Open Sci. 2020;4:e100115.
18. Larsen TS, Jansen KM. Impact of obesity-related inflammation on cardiac metabolism and function. J Lipid Atheroscler. 2021;10:8-23.
19. Powell-Wiley TM, Poirier P, Burke LE, et al; American Heart Association Council on Lifestyle and Cardiometabolic Health; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Epidemiology and Prevention; and Stroke Council. Obesity and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2021;143:e984-e1010.
20. Byrum RS, Goulet JL, Griffiths RJ, Koller BH. Role of the 5-lipoxygenase-activating protein (FLAP) in murine acute inflammatory responses. J Exp Med. 1997;185:1065-76.
21. Giugliano D, Ceriello A, Esposito K. The effects of diet on inflammation: emphasis on the metabolic syndrome. J Am Coll Cardiol. 2006;48:677-85.
22. Fritsche LG, Igl W, Bailey JN, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016;48:134-43.
23. Olsen L, Thum E, Rohner N. Lipid metabolism in adaptation to extreme nutritional challenges. Dev Cell. 2021;56:1417-29.
24. Broadfield LA, Pane AA, Talebi A, Swinnen JV, Fendt SM. Lipid metabolism in cancer: new perspectives and emerging mechanisms. Dev Cell. 2021;56:1363-93.
25. Basil MC, Levy BD. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol. 2016;16:51-67.
26. Serhan CN, Chiang N, Dalli J, Levy BD. Lipid mediators in the resolution of inflammation. Cold Spring Harb Perspect Biol. 2014;7:a016311.
27. Tulk HMF, Robinson LE. Modifying the n-6/n-3 polyunsaturated fatty acid ratio of a high-saturated fat challenge does not acutely attenuate postprandial changes in inflammatory markers in men with metabolic syndrome. Metabolism. ;2009:58:1709-16.
28. Badimon L, Vilahur G, Rocca B, Patrono C. The key contribution of platelet and vascular arachidonic acid metabolism to the pathophysiology of atherothrombosis. Cardiovasc Res. 2021;117:2001-15.
29. Kayama Y, Minamino T, Toko H, et al. Cardiac 12/15 lipoxygenase-induced inflammation is involved in heart failure. J Exp Med. 2009;206:1565-74.
30. Braune J, Weyer U, Hobusch C, et al. IL-6 Regulates M2 polarization and local proliferation of adipose tissue macrophages in obesity. J Immunol. 2017;198:2927-34.
31. Christ A, Günther P, Lauterbach MAR, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. 2018;172:162-175.e14.
32. Netea MG, Domínguez-Andrés J, Barreiro LB, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. 2020;20:375-88.
33. Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab. 1998;83:847-50.
34. Frohnert BI, Jacobs Jr DR, Steinberger J, Moran A, Steffen LM, Sinaiko AR. Relation between serum free fatty acids and adiposity, insulin resistance, and cardiovascular risk factors from adolescence to adulthood. Diabetes. 2013;62:3163-9.
35. Dommel S, Blüher M. Does C-C motif chemokine ligand 2 (CCL2) link obesity to a pro-inflammatory state? Int J Mol Sci. 2021;22:1500.
36. Upadhyay G, Gowda SGB, Mishra SP, et al. Targeted and untargeted lipidomics with integration of liver dynamics and microbiome after dietary reversal of obesogenic diet targeting inflammation-resolution signaling in aging mice. Biochim Biophys Acta Mol Cell Biol Lipids. 2024;1869:159542.
37. Chen M, Yang ZD, Smith KM, Carter JD, Nadler JL. Activation of 12-lipoxygenase in proinflammatory cytokine-mediated beta cell toxicity. Diabetologia. 2005;48:486-95.
38. Dahlke P, Peltner LK, Jordan PM, Werz O. Differential impact of 5-lipoxygenase-activating protein antagonists on the biosynthesis of leukotrienes and of specialized pro-resolving mediators. Front Pharmacol. 2023;14:1219160.
39. Correa-Burrows P, Burrows R, Albala C, et al. Multiple events case-control study in a prospective cohort to identify systemic, cellular, and molecular biomarkers of obesity-induced accelerated aging in 30-years-olds: the ObAGE study protocol. BMC Geriatr. 2022;22:387.
40. Volpp KG, Berkowitz SA, Sharma SV, et al; American Heart Association. Food is medicine: a presidential advisory from the American Heart Association. Circulation. 2023;148:1417-39.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].