


Advancing carbon dots research with machine learning: a comprehensive review
 0
0 
Abstract
Carbon-based nanomaterials, particularly carbon dots (CDs), have attracted growing attention due to their unique optical properties and cost-effective synthesis. Despite their promise, challenges remain in elucidating luminescence mechanisms and achieving controlled synthesis. Traditional trial-and-error approaches are inefficient, while machine learning (ML) offers powerful tools to accelerate materials discovery by capturing complex relationships. This review summarizes recent progress in applying ML to CDs, focusing on three key areas: enhancing the regulation of intrinsic properties, improving detection sensitivity and multicomponent recognition through the analysis of high-dimensional spectral data, and uncovering correlations between molecular features, experimental parameters, and CD performance with explainable ML. These advances enable more rational and efficient design of multifunctional CDs. Finally, we discuss future directions for CD informatics, including the development of structured data resources, the integration of large language models, interpretable ML techniques, and automated experimental platforms. These trends are expected to provide new insights and drive continued innovation in the multifunctional applications of CDs.
Keywords
INTRODUCTION
Carbon-based materials have attracted widespread interest due to their environmental friendliness and low cost. Traditional industrial carbon materials, such as activated carbon and carbon black, have long been focal points of materials science research[1,2]. However, bulk carbon materials generally lack a suitable bandgap in their electronic structure, hindering their ability to absorb or emit light at specific wavelengths efficiently. This limitation greatly restricts their effectiveness in optical and electronic applications. Consequently, the development of novel carbon-based materials with superior optical and electronic performance has become a prominent research focus, leading to the emergence of carbon nanomaterials.
Carbon dots (CDs), first identified in 2004 by Xu et al. during the arc-discharge synthesis of single-walled carbon nanotubes[3], are a new category of zero-dimensional carbon nanomaterials with diameters below
CDs can be synthesized by various methods, generally classified into two main approaches: “top-down” and “bottom-up”. The top-down method involves breaking down bulk carbon precursors (e.g., graphite, carbon nanotubes, or activated carbon) into nanoscale carbon structures using harsh conditions (e.g., strong oxidants, concentrated acids, or high temperatures) and physical processes such as laser ablation or arc discharge[10-14]. In contrast, the bottom-up approach is generally more flexible and straightforward, utilizing chemical or biological precursors that undergo dehydration/condensation, cross-linking, and carbonization reactions. Common synthesis methods include microwave-assisted synthesis, chemical oxidation, and hydrothermal methods[15-17].
Both strategies have distinct advantages and limitations. The bottom-up approach is economical and eco-friendly but often struggles to achieve precise control over the size of CDs[18]. Conversely, the top-down approach allows finer control of particle size but is more costly[19]. Regardless of the chosen route, the selection of precursors and precise control over synthesis parameters play pivotal roles in determining the structure and properties of CDs. The current consensus on the structure of CDs generally adopts a core-shell model, with the surface typically enriched with amino, epoxy, carbonyl, aldehyde, hydroxyl, and carboxyl groups. By tailoring these surface functionalities, properties such as solubility and surface charge of CDs can be modified[20]. Meanwhile, the carbon core comprises both sp2- and sp3-hybridized carbon atoms, and tuning the proportion of the conjugated (sp2) domains can enable tunable fluorescence behavior[21].
Despite the relative ease of obtaining starting materials and performing synthesis, the underlying mechanisms of the formation of CDs and methods for controllably producing CDs with application-specific properties remain under active investigation. Researchers face substantial challenges in understanding the inherent properties of CDs and enhancing their performance. On one hand, the diversity in carbon arrangements and the unique interfacial interactions between carbon cores and functional groups lead to the random distribution of morphologies and surface groups. On the other hand, quantum size effects, influenced by the structure and size of CDs, result in distinct optical characteristics[22]. These factors introduce significant unpredictability and complexity into the development and optimization of CDs.
Artificial intelligence (AI) is often defined as a system that simulates human behavior and cognitive processes by computational means. The 2024 Nobel Prizes in Chemistry and Physics jointly underscored AI’s interdisciplinary breakthroughs and profound influence on cutting-edge research. As a core methodology of AI, machine learning (ML) has become an indispensable tool in numerous fields. Its primary advantage lies in processing large datasets to build models that can uncover complex, latent relationships and optimize decision-making with minimal human intervention. With rapid advancements in data availability, computational power, and algorithmic innovation, ML is now extensively applied to speech recognition, machine translation, autonomous driving, and semantic segmentation, among other tasks[23-26].
In materials science, traditional research and development (R&D) often relies heavily on a “trial-and-error” approach, which is time-consuming and costly. This method falls short of meeting modern technology’s demands for rapid material iteration and optimization. By contrast, ML fosters what has been termed the “fourth paradigm” of materials research - a data-driven paradigm[27]. Through analyses of large volumes of experimental and simulation data, ML can guide material design and optimization [Figure 1]. Through the analyses of large volumes of experimental and simulation data, ML can guide material design and optimization. Applications of ML in materials science now range from materials discovery and structure-property prediction to performance optimization and the exploration of nanoparticle synthesis routes[28-30]. By leveraging ML, researchers can start from existing data to construct flexible, nonlinear models, thereby reducing reliance on purely physical experiments and complex mathematical fitting, ultimately accelerating the R&D process[31]. Because CDs exhibit diverse and intricate microstructures, conventional linear models often fail to capture their performance with sufficient accuracy. Introducing ML addresses this gap by enabling researchers to establish robust structure-property relationships, identify the key factors influencing performance, and optimize synthesis parameters. In doing so, ML not only enhances R&D efficiency but also improves the accuracy of performance predictions, effectively mitigating the unpredictability and complexity inherent in the development of CDs.
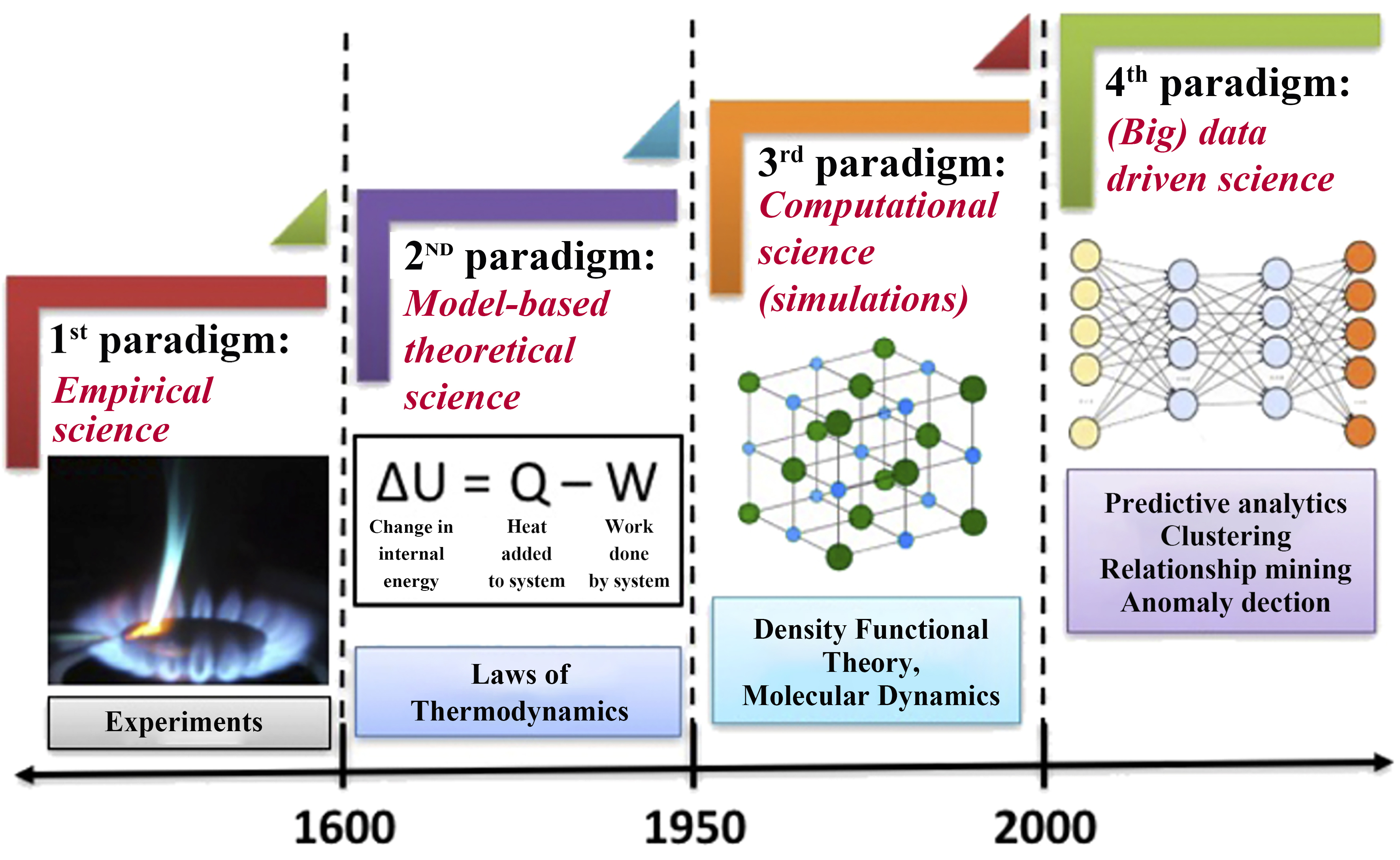
Figure 1. The four paradigms of science. This figure is quoted with permission from Agrawal et al.[27].
Despite ML’s growing role in materials science, there remains a relative scarcity of reviews focusing on how ML techniques can be applied to CDs. Existing publications generally adopt a simple classification from either an algorithmic or an application perspective, leaving a need for a more comprehensive overview of how ML can advance research on CDs[32-34]. In addition, current reviews offer limited discussion on addressing data scarcity. To fill this gap, this paper highlights two key directions for overcoming the challenges posed by small datasets in research on CDs: first, exploring data acquisition methods from multiple perspectives; and second, introducing algorithmic strategies such as imbalanced learning, transfer learning, and active learning to enhance model generalization under limited data conditions. To further support a clearer understanding of the practical application paths of ML in this field, existing studies are categorized into three areas: first, optimizing the intrinsic properties of CDs; second, improving data processing efficiency across diverse application domains; and third, enhancing the mechanistic interpretability of CDs. Finally, this paper discusses key challenges and emerging trends in this interdisciplinary area, aiming to offer valuable insights for researchers engaged in related studies.
STRATEGIES FOR DATA LIMITATIONS IN MATERIALS ML
In essence, ML employs algorithms and statistical methods to mimic the way humans draw inferences from experimental data. In the field of materials science, ML workflows typically encompass four key stages: data collection, data preprocessing, model training and optimization, and model evaluation and interpretation. However, the practical application of ML in research on CDs faces a persistent small-dataset dilemma, characterized primarily by limited data availability and imbalanced datasets distributions. These challenges not only impede accurate model training but also undermine the generalizability of models across different material systems. Although ML has been increasingly applied to the design and performance optimization of CDs, most existing studies have yet to explicitly address this critical issue. As research on CDs continues to advance, the small-dataset problem is expected to become even more pronounced, particularly in complex tasks such as precise structural regulation, elucidation of photoluminescence mechanisms, and performance prediction. Therefore, in this section, we summarize general strategies that have been successfully implemented in other domains of materials science, highlighting approaches that leverage diverse data collection and algorithmic optimization to alleviate the limitations imposed by small datasets. These strategies aim to provide a forward-looking methodological foundation and serve as a conceptual reference for future studies on CDs [Figure 2].
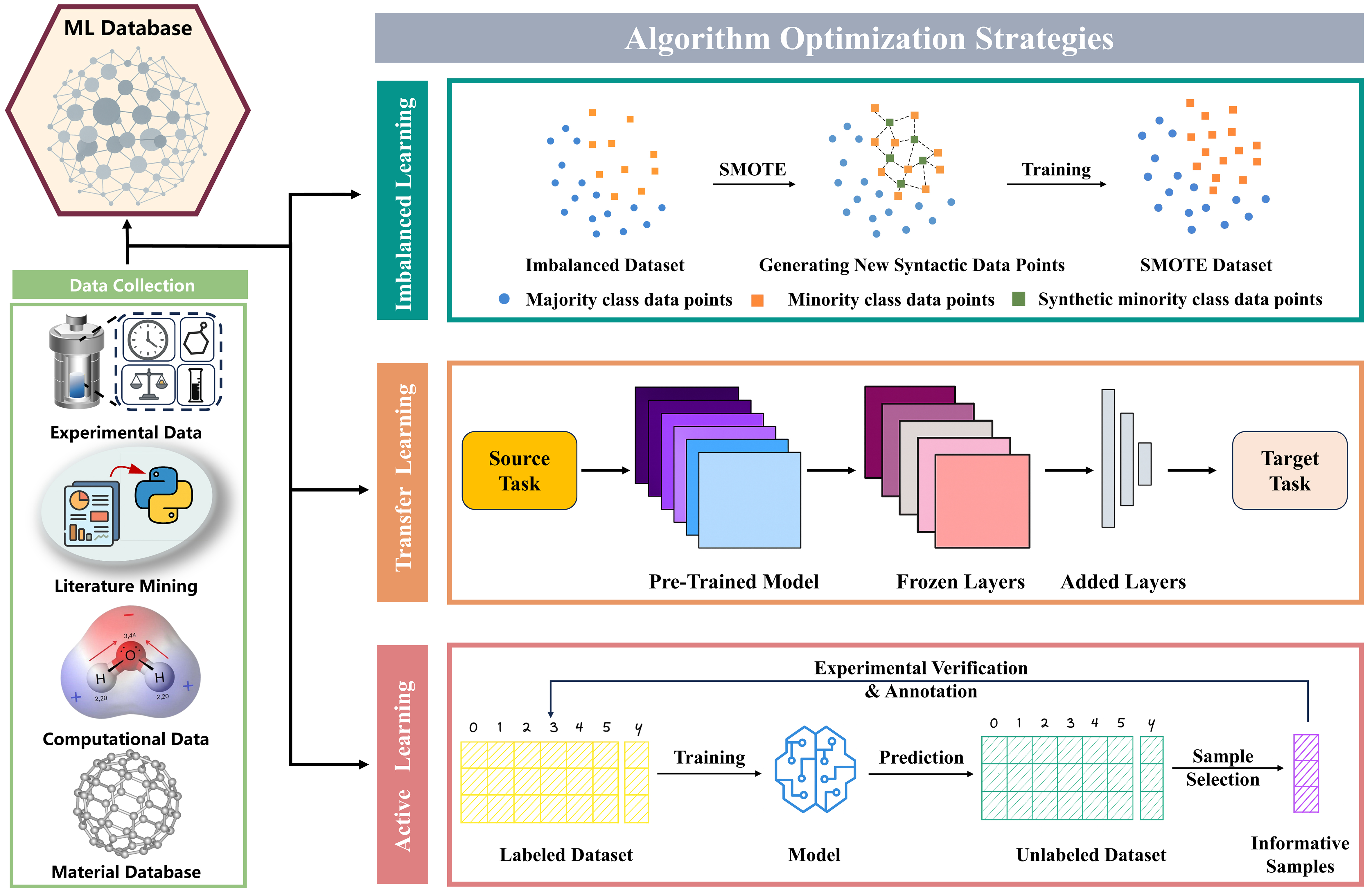
Figure 2. Strategies for data limitations in materials ML. ML: Machine learning; SMOTE: Synthetic Minority Over-sampling Technique.
Data collection
The application of ML in materials science is highly dependent on the quality and scale of the available data, and the same holds true for the design and optimization of novel functional materials such as CDs. The performance of CDs, including their fluorescence properties, quantum yield (QY), and application potential in fields such as energy, sensing, and bioimaging, requires the support of high-quality, large-scale datasets. In materials science, data can generally be categorized into product data and precursor data. Product data describe the target properties and experimental parameters of the final material, while precursor data provide physicochemical information about the starting materials, offering crucial inputs for enhancing model interpretability.
Product data
Product data primarily focus on the key properties of the target material and the corresponding experimental conditions. These data can be obtained through experimental characterization, literature mining, computational simulations, and materials databases.
Experimental characterization typically yields the most accurate and reliable data. However, the high cost and long-time requirements, particularly when dealing with noble metals or other expensive materials[35], significantly limit the size of the datasets, making it difficult to meet the large-scale data demands of ML models. The most direct and fundamental way to address this issue is to expand the dataset by collecting a larger amount of high-quality materials data. On this basis, this review explores a variety of data acquisition approaches to support the subsequent model development and optimization.
Literature data mining has recently gained widespread attention as a promising technique, primarily because the extracted data are closely aligned with the latest research progress. This approach has been widely used to guide the synthesis and application of CDs. However, mining data from the literature presents several challenges. It requires significant manual effort and time to search, extract, and organize relevant information. Moreover, even when similar synthesis and characterization methods are employed across studies, substantial discrepancies in the reported data may still occur, increasing uncertainty and the complexity of data preprocessing. To address these issues, advancements in text mining have provided new avenues by enabling the automatic extraction of useful information from unstructured scientific text[36]. This technique has been broadly applied in high-throughput experimental data analysis and the development of systematic materials databases[37,38]. Nevertheless, due to the inherent complexity of natural language, automated information extraction remains non-trivial. Consequently, text mining has evolved into an interdisciplinary field combining natural language processing (NLP) and ML, utilizing techniques such as named entity recognition, relation extraction, and topic modeling to identify and organize valuable information[39-41]. Since the discovery of CDs, a substantial number of experimental publications have accumulated. It is foreseeable that leveraging text mining techniques to systematically extract data related to the synthesis of CDs and performance characterization from this extensive body of literature will offer richer insights and stronger support for studying structure–property relationships and broadening the application scope of CDs.
First-principles calculations, such as density functional theory (DFT), represent another important source of data, offering notable advantages in the accuracy of simulations by enabling the description of the atomic-level electronic structure of materials[42,43]. By contrast, semiempirical methods provide higher computational efficiency[44], especially in the simulation of large systems or over extended time scales. These computational approaches have been increasingly applied to investigate the photoluminescence mechanisms, energy level structures, and surface states of CDs. However, the results of such simulations are often sensitive to model parameters and specific computational settings, necessitating careful evaluation of the associated errors[45]. Despite these limitations, computational data offer valuable alternatives in scenarios where experiments are costly or constrained, and they play a critical role in supporting the optimization of ML models by generating diverse and abundant datasets. Nevertheless, challenges such as model bias and strong dependence on computational conditions remain significant obstacles that need to be addressed.
Materials databases integrate large volumes of data generated through experiments or computational simulations, enabling researchers to access high-quality information in a short time. Several open-source databases have been established to date, such as the Harvard Clean Energy Project[46], the Open Quantum Materials Database (OQMD)[47], and the Materials Project[48]. These platforms help reduce the need for redundant experimental characterization or computational efforts. However, there are still limitations, including delays in data entry and review processes. Additionally, inconsistencies in data formats and reliability across different sources may arise. Therefore, researchers must perform data cleaning and validation before using these databases to ensure data quality and consistency.
Precursor data
Beyond material performance data, the physicochemical properties of precursors are equally crucial to constructing ML models. Precursor descriptors primarily characterize the physicochemical properties of precursor molecules, encompassing a wide range of features from basic elemental information to complex molecular structures and electronic properties. For CDs, precursor descriptors help ML models capture the intrinsic relationships between material performance and precursor characteristics.
Individually computing various descriptors for each precursor is not only labor-intensive but also complicated by the fact that structure-property relationships for certain materials remain insufficiently understood. This uncertainty makes it difficult to determine which precursor attributes are most critical to material performance, thereby increasing the complexity of descriptor selection. Consequently, obtaining a large number of precursor descriptors often relies on specialized cheminformatics tools, such as E-Dragon, PaDEL, and RDKit[49-51]. These tools efficiently generate a vast array of molecular descriptors, covering multidimensional features such as topological indices, shape factors, and electronic properties, thus providing a robust data foundation for ML models.
During model development, the granularity of descriptors can be dynamically adjusted based on research objectives and prediction requirements[52]. For exploring complex macroscopic phenomena, such as material stability or mechanical properties, relatively coarse descriptors (e.g., elemental composition and fundamental physical attributes) are often sufficient. By contrast, precise predictions of specific properties, such as fluorescence peak positions or QY values, require more detailed features at the molecular or atomic level. These refined descriptors facilitate capturing intricate structure-property relationships, thereby enhancing both the interpretability and predictive performance of ML in research on CDs.
Algorithm optimization strategies
Although data acquisition methods in materials science have become increasingly diverse in recent years, obtaining high-quality experimental data remains a significant challenge for material systems such as CDs, which exhibit complex structure–property relationships and high-dimensional characterization requirements. This difficulty is particularly pronounced in scenarios involving precise structural tuning and multi-parameter performance evaluation, where long experimental cycles and high costs hinder the effective expansion of dataset size.
Against this backdrop, improving the efficient use of limited samples has become a central challenge in materials-oriented ML research. As a result, algorithm-level optimization is regarded as a key pathway to enhancing model performance. When facing issues such as imbalanced data distribution and insufficient sample size, traditional algorithms often suffer from limited generalization ability, leading to a high risk of overfitting [Figure 3]. To address these challenges, this section focuses on three representative strategies - imbalanced learning, active learning, and transfer learning - to explore feasible approaches for improving model performance under small-sample conditions.
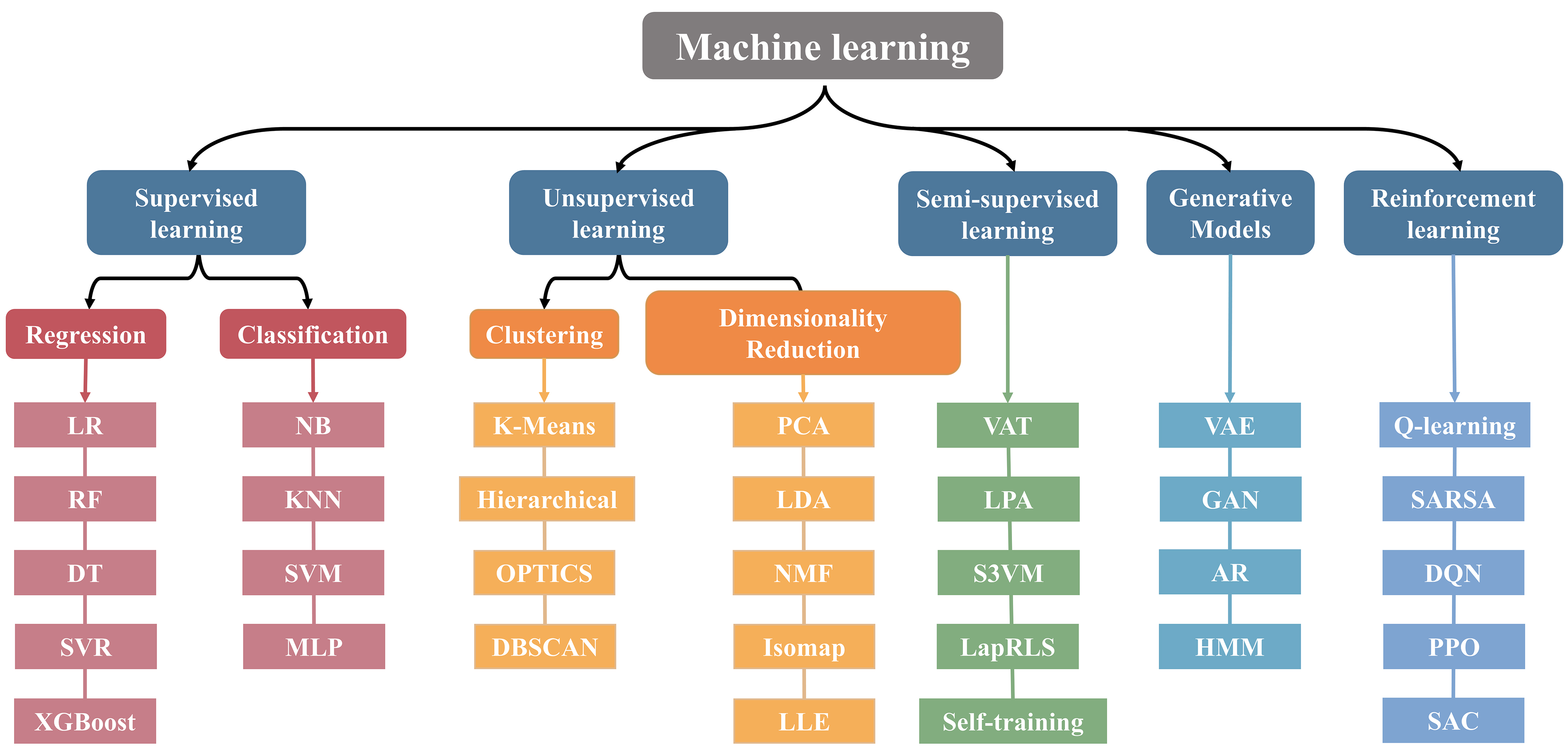
Figure 3. General classification of ML models. ML: Machine learning; LR: linear regression; RF: random forest; DT: decision tree; SVR: support vector regression; XGBoost: eXtreme gradient boosting; NB: naive Bayes; KNN: K-nearest neighbors; SVM: support vector machine; MLP: multilayer perceptron; OPTICS: ordering points to identify the clustering structure; DBSCAN: density-based spatial clustering of applications with noise; PCA: principal component analysis; LDA: linear discriminant analysis; NMF: non-negative matrix factorization; Isomap: isometric mapping; LLE: locally linear embedding; VAT: virtual adversarial training; LPA: label propagation algorithm; S3VM: semi-supervised SVM; LapRLS: laplacian regularized least squares; GAN: generative adversarial networks; VAE: variational autoencoders; AR: autoregressive models; HMM: hidden Markov model; SARSA: state-action-reward-state-action; DQN: deep Q-networks; PPO: proximal policy optimization; SAC: soft actor-critic.
Imbalanced learning
Imbalanced learning algorithms are primarily designed to address the problem of severe class distribution imbalance in classification tasks. Specifically, in many real-world datasets, certain classes are inherently underrepresented or exhibit a large disparity in sample size relative to majority classes, making it difficult for the model to effectively learn their features. In such cases, conventional ML methods tend to favor predicting majority classes, thereby overlooking minority classes that may be critical to the task. As a result, the trained classifier may appear to perform well in terms of overall accuracy, but often lacks sufficient discriminative power for minority classes, which significantly limits its practical utility in applications[53].
To address the aforementioned challenges, imbalanced learning methods are typically developed from both data-level and algorithm-level perspectives, aiming to improve the model’s ability to learn from underrepresented classes. The most fundamental data-level approach is sampling, which includes undersampling, oversampling, and hybrid sampling[54]. Undersampling reduces the dominance of majority class samples by randomly or systematically removing a portion of them, and is suitable for cases where the majority class has excessive redundancy[55]. Oversampling, on the other hand, expands the representation of minority classes by replicating existing samples or generating synthetic ones using techniques such as the Synthetic Minority Over-sampling Technique (SMOTE)[56]. Hybrid sampling combines the strengths of both approaches, offering a balance between class distribution and data diversity. This method has gained increasing popularity in recent years for materials classification tasks[57]. For example, Chen et al. proposed an ensemble learning classifier to screen efficient arsenene-based hydrogen evolution reaction (HER) catalysts with heteroatom doping[58]. In their study, the original dataset contained significantly fewer high-activity samples compared to low-activity ones, which caused conventional classifiers to favor the majority class and perform poorly in prediction. To mitigate this issue, the researchers employed SMOTE to generate synthetic samples for the minority class, thereby improving class balance. Comparative results showed that the model trained on the SMOTE-augmented dataset achieved substantially higher average accuracy than the model trained on the original imbalanced data, demonstrating the effectiveness of oversampling in handling imbalanced materials datasets[58]. This case highlights that data augmentation strategies can significantly enhance model performance and offer strong generalizability in addressing class imbalance problems. However, it should be noted that vanilla SMOTE assumes linear interpolation among nearest neighbors in a Euclidean feature space. For data subject to hard constraints - for example, spectral intensities that must satisfy non-negativity, band-limit and instrument-response consistency, and camera RGB (red, green, blue) values that are influenced by white balance, color-matrix transformations and gamma - this assumption may be violated, producing samples that do not conform to physical or measurement constraints; accordingly, direct use is not recommended. A more prudent approach is to employ modality-aware SMOTE variants, such as Borderline-SMOTE and SMOTE-ENN (ENN: edited nearest neighbors), and to pair them with group-aware resampling by source, device or batch to mitigate source leakage.
At the algorithmic level, various effective strategies have been proposed to enhance model performance under imbalanced data conditions. One such approach is clustering-assisted sampling, which incorporates spatial structural information by first clustering the data and then performing balanced sampling within each cluster. This method helps preserve the intrinsic distribution of the dataset[59]. Another widely adopted technique is cost-sensitive learning, which introduces class-specific weights into the loss function to increase the model’s sensitivity to minority class samples. This is particularly suitable for tasks where the cost of misclassification is highly asymmetric[60]. Additionally, the extreme learning machine (ELM) has emerged as an efficient tool for handling imbalanced classification problems in small-sample scenarios, owing to its fast-training speed and strong generalization capability. When used as a base classifier within ensemble frameworks, ELM has been shown to improve the recognition rate of minority classes[61].
Transfer learning
Transfer learning is a ML strategy that leverages knowledge acquired in a source domain and transfers it to a data-scarce target domain for model development [Figure 4A]. It is particularly suitable for applications where sample sizes are limited but task relevance between domains is high[63]. As a representative example of a small-data discipline, materials science often involves emerging material systems that are still in the early stages of exploration, where experimental data are extremely limited, making direct modeling challenging. Transfer learning offers a promising solution by utilizing the knowledge and data accumulated in well-studied material systems to improve modeling efficiency and predictive accuracy under data-scarce conditions.
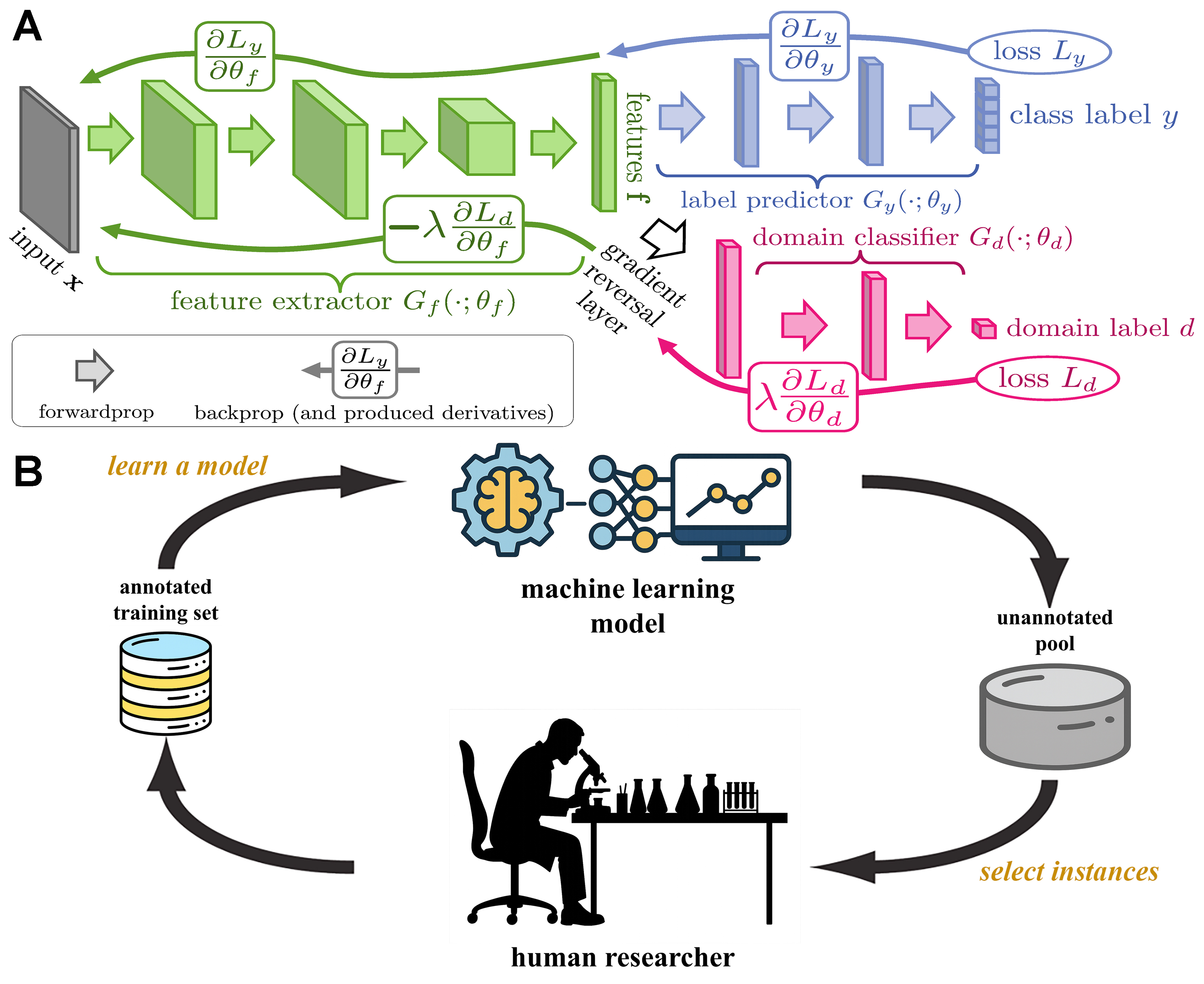
Figure 4. (A) Conceptual schematic of a deep-learning–based transfer-learning framework (illustrative only). This figure is quoted with permission from Yaroslav et al.[62], Copyright 2017, Springer International Publishing AG; (B) Schematic diagram of the active learning process.
As an effective strategy to address small-data challenges, model-based transfer learning has emerged as the most widely applied approach. In this method, parameters from a model pre-trained on a source domain are transferred to the target domain, and then adapted to the new task through fine-tuning or partial parameter freezing. This enables rapid model adaptation and significantly improves performance under limited data conditions. In contrast, relation-based transfer learning focuses on identifying structural relationships between source and target domain tasks, and is particularly suitable for applications such as reaction prediction, literature mining, and knowledge graph construction[64]. Instance-based transfer learning, on the other hand, mitigates distributional discrepancies between domains by reweighting samples from the source and target domains, making it effective when local similarities exist across different material systems[65]. While each of these strategies offers distinct advantages, model-based transfer learning has become the dominant approach in materials science due to its practicality and adaptability in small-sample modeling tasks.
As part of ongoing efforts to advance the field, researchers have begun exploring the integration of large language models (LLMs) with transfer learning to further enhance modeling performance. For example, Liu et al. proposed a method that combines LLMs with transfer learning to address the challenge of limited data in the design of circularly polarized phosphorescence (CPP) materials, which are characterized by complex molecular structures and scarce experimental data[66]. In their approach, LLMs were employed to screen candidate molecules from a vast body of literature, and a transfer learning model was then used to establish structure–property relationships based on existing data, enabling accurate CPP performance prediction and inverse design. Compared to models without transfer learning, this method significantly improved prediction accuracy, reducing the mean absolute error (MAE) from 0.24 to 0.14. This improvement demonstrates that latent material representations extracted through transfer learning can effectively capture the absorption properties of thin-film materials[66]. Despite limited experimental samples, the study successfully designed CPP materials with high luminescence dissymmetry factor (glum) values, narrow emission bandwidths, and customized optical properties, showcasing the strong potential and practical value of model-based transfer learning in small-sample materials modeling.
As an effective approach to addressing data scarcity in materials science, transfer learning has demonstrated strong adaptability and practical value across various subfields. Different types of transfer strategies - focusing respectively on model structures, task relationships, or sample distributions - enable efficient knowledge transfer and reuse tailored to specific problem scenarios. These methods offer robust technical support for tasks such as materials design and property prediction. When dealing with small or imbalanced datasets, it is crucial to evaluate model performance using multiple complementary metrics and to clearly define data-splitting strategies. This comprehensive evaluation approach helps avoid overly optimistic assessments caused by a single metric such as accuracy, leading to more reliable and scientifically rigorous conclusions.
Active learning
Active learning, as a strategy for improving sample efficiency, is increasingly being adopted in materials ML modeling[67]. It is particularly well-suited for early-stage research where data are limited and sample acquisition is challenging, making it one of the key techniques for addressing the small data problem [Figure 4B].
The core idea of active learning lies in selectively querying the most representative or informative samples from a large pool of unlabeled data, thereby enabling limited labeled data to effectively capture the overall structure and information distribution of the dataset[68]. In materials science, active learning is typically implemented as a cyclic, data-driven optimization process[69]. It begins with a small set of labeled samples used to train an initial ML model, which is then applied to assess the unlabeled data pool and identify candidates with the highest potential information gain or representativeness. These selected samples are subsequently labeled - through experiments, simulations, or other methods - and incorporated into the training set to update the model. By continuously iterating this cycle of sample selection, validation, and model retraining, active learning establishes a closed-loop framework between data generation and model refinement. This approach significantly enhances prediction accuracy and generalization, reduces data acquisition costs, and offers an efficient solution for new materials discovery under resource-limited conditions.
In the field of materials science, commonly used sampling strategies in active learning include uncertainty sampling, entropy-based ranking, and representativeness sampling[70-72]. Uncertainty sampling prioritizes labeling samples for which the model exhibits the least confidence, typically based on the predicted probability distribution. Entropy-based ranking selects samples with the highest information entropy in their predicted outcomes, targeting those that carry the most informative value. Representativeness sampling, on the other hand, considers the spatial distribution of samples in the feature space, aiming to select data points that best represent the overall dataset and prevent the model from converging prematurely to local optima. In practice, combinations of these strategies are often employed in materials research to strike a balance between exploration and efficiency.
In recent years, with the rapid development of high-throughput experimental platforms and literature mining techniques, the application scope of active learning in materials science has continued to expand. This approach can be integrated with robotic experimental systems to form a closed-loop “experiment–learning” framework, and can also be combined with NLP techniques to automatically extract potentially unlabeled data from the literature, thereby establishing a low-cost, high-efficiency data acquisition mechanism. Active learning is particularly valuable in exploratory material systems, where it effectively guides experiments toward high-potential regions, significantly accelerating the materials discovery process. For instance, Noh et al. proposed a closed-loop optimization strategy that integrates high-throughput experimental platforms with active learning to improve the solubility of redox-active molecules in organic solvents[73]. Due to the scarcity of large-scale experimental data, the development of related electrolyte materials had long been constrained. In this study, by coupling a high-throughput robotic system with Bayesian optimization, the researchers successfully identified several high-performance solvent combinations while testing less than 10% of the candidate systems. The results demonstrate that active learning can optimize data acquisition pathways, improve sample efficiency, and, when combined with automated experimental systems, significantly enhance model predictive power and experimental guidance under small-data conditions, offering a powerful tool for efficient materials development[73].
In summary, this section highlights general strategies for addressing the small-dataset dilemma through diversified data collection and algorithmic optimization. While these methods are well established in other materials fields, their application to CDs remains limited. The next section turns to a systematic review of current studies that directly integrate ML with CDs, outlining recent progress in this rapidly developing area.
APPLICATIONS OF ML IN CDS
This section focuses on the multilevel applications and value of ML in research on CDs. Whether for parameter optimization of intrinsic properties, intelligent analysis of high-dimensional and nonlinear data, or further exploration of the underlying structure-property relationships, ML has demonstrated exceptional capabilities in data mining and interpretation. By integrating diverse datasets, uncovering hidden patterns, and enabling interpretable model analysis, researchers can efficiently explore and precisely design CDs with outstanding performance and specific functional objectives, laying a solid foundation for their applications across various fields. To enable a systematic, side-by-side comparison of the studies discussed in this section, the key parameters of each work are consolidated in Supplementary Table 1.
Optimizing CDs properties using ML
The intrinsic properties of CDs play a crucial role in bioimaging, sensing, energy, and environmental applications. However, these properties are often influenced by multiple factors, including precursor types, synthesis conditions, and post-treatment methods. In recent years, researchers have increasingly incorporated ML techniques to extract complex correlations between synthesis parameters and intrinsic properties from large-scale experimental datasets, enabling rapid prediction and efficient optimization[32]. This section summarizes recent advances in ML-driven intrinsic properties of CDs, covering optical property tuning, QY enhancement, multicolor emission, and biomedical applications, while discussing potential future developments in this field.
The optimization of optical properties is fundamental to the widespread application of CDs in bioimaging, fluorescent probes, and sensing. Enhancing photoluminescence performance and the QY significantly expands their applicability across various domains. Wang et al. employed a deep convolutional neural network (DCNN) to systematically predict and optimize the optical characteristics of CDs, including excitation-dependent and excitation-independent spectral features, as well as fluorescence color variations under different excitation wavelengths[74]. Using 170 published studies, the research extracted a comprehensive set of synthesis parameters (e.g., precursor types, reaction time, and reaction temperature) and, through DCNN modeling, uncovered the complex nonlinear relationships between these parameters and CDs’ optical properties. Experimental validation confirmed that the model accurately predicted and fine-tuned optical characteristics, with the optimized CDs exhibiting bright multicolor fluorescence in live HeLa cell imaging, demonstrating the strong potential of DCNN in guiding synthesis of CDs.
Building upon previous work, Hong et al. further applied an eXtreme gradient boosting (XGBoost) model to predict and optimize fluorescence intensity and emission center (λc) of CDs [Figure 5A][75]. Using 400 experimental datasets, principal component analysis (PCA) was incorporated to reduce dimensionality and refine feature selection. The model successfully guided the synthesis of CDs with high fluorescence intensity and tunable emission wavelengths (λem), achieving a maximum QY of 30.71%. The optimized CDs have been successfully employed in iron ion detection, controlled drug delivery, whole-cell imaging, and optical film fabrication.
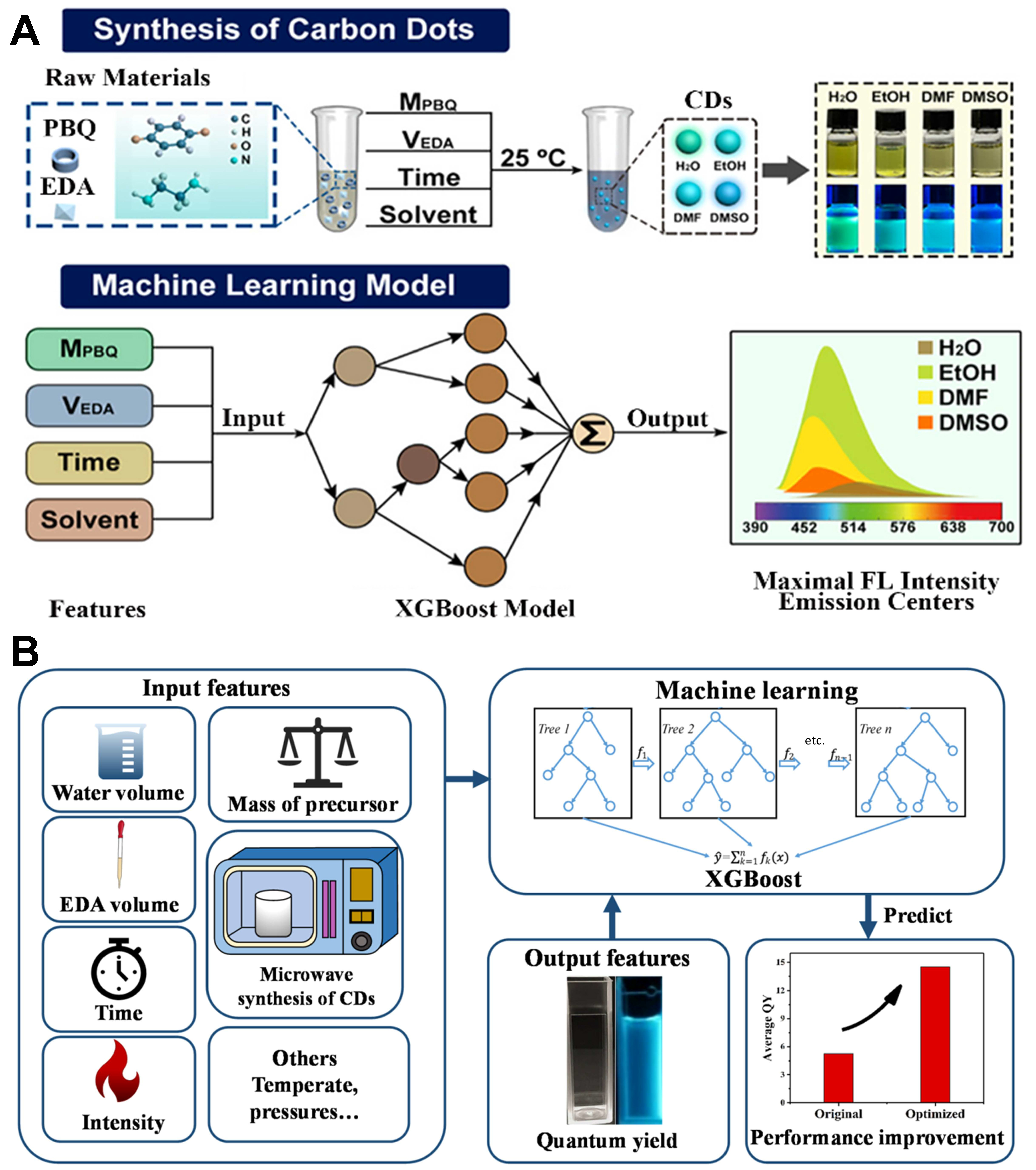
Figure 5. (A) Fluorescence intensity and the λc of CDs optimized using the XGBoost model. This figure is quoted with permission from Hong et al.[75], Copyright 2022, American Chemical Society; (B) The QY of CDs enhanced through XGBoost modeling. This figure is quoted with permission from Xu et al.[76], Copyright 2022, The Royal Society of Chemistry. CDs: Carbon dots; XGBoost: eXtreme gradient boosting; QY: quantum yield; PBQ: p-benzoquinone; EDA: ethylenediamine; DMF: N,N-dimethylformamide; DMSO: dimethyl sulfoxide.
Building on the advantages of XGBoost demonstrated in the study of Hong et al., subsequent research has further leveraged this model for QY optimization, achieving even greater performance improvements[75]. Xu
Similarly, in hydrothermal synthesis, Tang et al. experimentally validated that XGBoost modeling effectively uncovers the complex relationships between synthesis parameters and QY, identifying optimal synthesis conditions that yielded an impressive QY of 55.5%[77]. Han et al. conducted a systematic analysis of ethylenediamine (EDA) volume, precursor mass, and reaction temperature in hydrothermal synthesis, revealing that EDA volume is a key determinant in achieving high QY[78]. Under optimized conditions, the QY was increased to 39.3%, significantly surpassing that achieved by traditional trial-and-error methods. Furthermore, the optimized CDs demonstrated high sensitivity for Fe3+ detection, positioning them as effective fluorescent probes.
In addition to hydrothermal synthesis, Lan et al. extended ML-driven QY optimization to a low-temperature molten-salt route[79]. They first obtained kilogram-scale solid-state emissive CDs with a solid-state QY of ~90% and then, by training and selecting an XGBoost regressor to refine synthesis conditions, pushed the solid-state QY to 99.86%, enabling scalable production of high-efficiency solid-state fluorescent CDs.
While XGBoost excels in modeling nonlinear synthesis parameters, Polynomial regression and random forest (RF) have also shown noteworthy potential. Zhang et al. investigated nitrogen- and boron-co-doped CDs, utilizing polynomial regression to analyze the synergistic and antagonistic effects of synthesis temperature, H2O2 concentration, and reaction time on the 675/500 peak intensity ratio[80]. The study revealed that a higher H2O2 concentration combined with an optimal reaction time significantly improved optical performance, whereas excessively high synthesis temperatures led to fluorescence quenching, thereby suppressing optical properties. The optimized conditions resulted in an enhanced peak intensity ratio of 0.285, which was further validated by in vivo fluorescence imaging using animal models. Complementing these findings, Xing et al. constructed a large-scale dataset comprising 202 different precursor combinations and employed an RF model to predict and regulate fluorescence peak positions and the λem under 365 and 532 nm excitation[81]. The study uncovered complex nonlinear correlations between precursor composition and optical characteristics, further demonstrating the potential of RF in property optimization of CDs.
Expanding the predictive scope, Chen et al. also applied an RF model to establish a multidimensional prediction framework for the maximum λem, QY, and Stokes shift (Δλ) of CDs, integrating various synthesis conditions and solvent properties[82]. The model achieved an 80% prediction accuracy, providing a viable pathway for multicolor information encryption. Concurrently, Senanayake et al. combined an artificial neural network (ANN) with a classification-regression hybrid model, leveraging 407 literature datasets to significantly reduce prediction errors for the λem of CDs and color tuning (error reduced to 27 nm)[83]. Their findings further emphasized that multi-parameter synergistic effects have a far greater impact on emission performance than the traditionally considered reaction temperature and duration, thereby offering new avenues for precise optical tuning of CDs [Figure 6A].
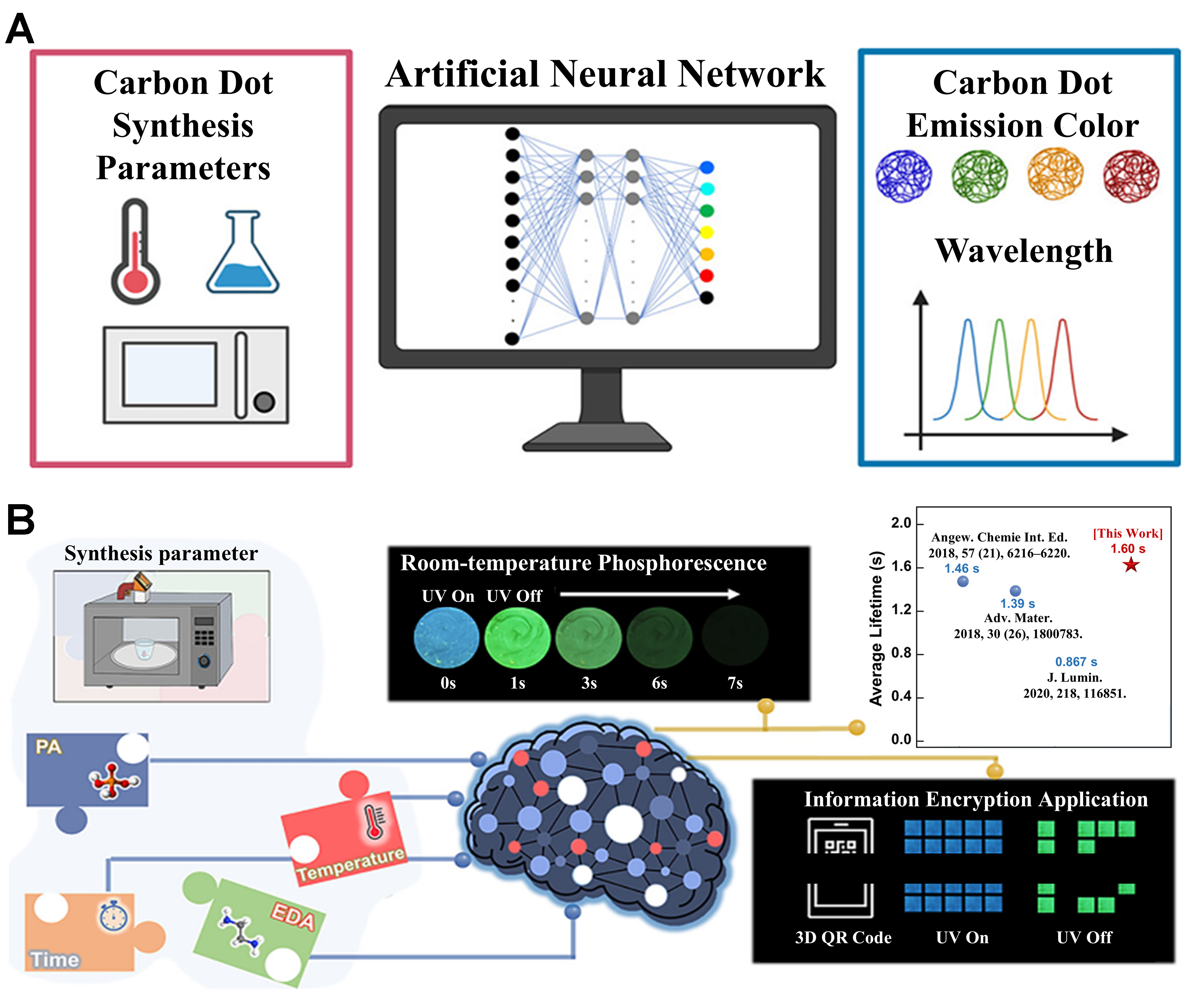
Figure 6. (A) Emission color and wavelength of CDs optimized via an ANN ensemble approach. This figure is quoted with permission from Senanayake et al.[83], Copyright 2022, American Chemical Society; (B) Phosphorescence lifetime of CDs improved using the XGBoost model. This figure is quoted with permission from Muyassiroh et al.[84], Copyright 2024, American Chemical Society. CDs: Carbon dots; ANN: artificial neural network; XGBoost: eXtreme gradient boosting; UV: ultraviolet light; PA: phosphoric acid; EDA: ethylenediamine; QR: quick response code.
The optimization of phosphorescence lifetime (τp) in CDs is crucial for advancing high-security applications, such as information encryption and anti-counterfeiting. Muyassiroh et al. investigated the relationship between EDA volume, phosphoric acid volume, reaction time, and final temperature with τp[84]. Using PCA for dimensionality reduction and selecting the XGBoost model for prediction and optimization [Figure 6B], they successfully identified key synthesis parameters. Experimental results demonstrated that under optimized conditions, the synthesized CDs achieved an average τp of 1.6 s, significantly outperforming traditional experimental methods. Furthermore, the study validated the effectiveness of this ML-driven strategy in identifying critical synthesis parameters and, for the first time, applied it to the optimized CDs in dynamic information encryption design, highlighting their potential value in multi-level anti-counterfeiting and data storage.
Building on this, Guo et al. constructed a dataset based on six synthesis parameters (precursor mass, NaOH mass, reaction temperature, heating rate, reaction time, and atmosphere) and trained an XGBoost classification model with the τp category (ns/μs/ms) as the prediction target[85]. Among the tested classifiers, XGBoost performed best on the held-out set, enabling efficient triage of a large combinatorial parameter space and guiding the synthesis of representative aqueous samples that matched the predicted τp categories.
Red-emissive CDs, owing to their long-wavelength absorption properties, exhibit optical advantages such as low photon scattering, high penetration depth, and minimal fluorescence background, providing crucial support for imaging and diagnostics in complex biological environments. Luo et al. employed various data processing techniques and classification models to systematically analyze the effects of key synthesis parameters - including temperature, reaction time, and precursor mass - on the optical properties of red-emissive CDs, ultimately identifying the optimal synthesis pathway [Figure 7][86]. By integrating 151 experimental datasets, the study combined the feature extraction capability of the XGBoost model with the classification power of PCA and logistic regression models, successfully predicting and verifying the synthesis conditions for CDs with red fluorescence characteristics. Experimental results demonstrated that the model-predicted synthesis conditions significantly improved the synthesis efficiency of red CDs, and real-world applications confirmed the optimized red CDs’ exceptional fluorescence labeling capability and stability in cell imaging, underscoring their practical potential in biomedical applications.
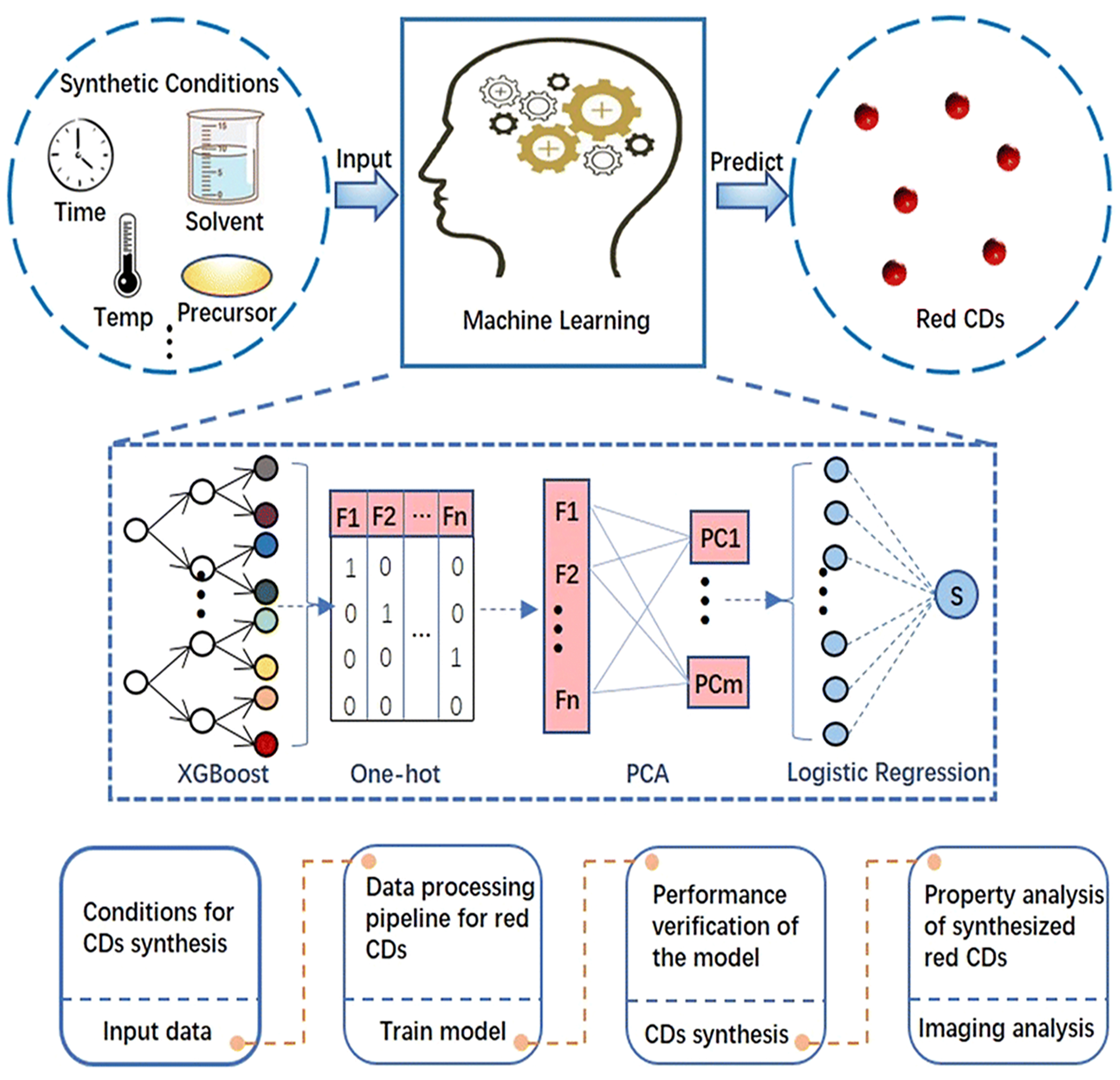
Figure 7. Synthesis efficiency of red CDs optimized using various data processing techniques and classification models. This figure is quoted with permission from Luo et al.[86], Copyright 2022, The Royal Society of Chemistry. CDs: Carbon dots; PCA: principal component analysis.
Building on this foundation, to explore the correlation between red and near-infrared (NIR) emissive CDs and synthesis parameters in a more quantitative manner, Tuchin et al. utilized multivariate linear regression and the K-nearest neighbors (KNN) model to systematically investigate the relationship between the optical properties of CDs in the red and NIR regions and their synthesis parameters[87]. They constructed a comprehensive database of 127 samples, providing in-depth insights into how different precursors and solvents influence the formation of the optical centers of CDs. Unlike previous ML-driven optimizations of red CDs, this study placed greater emphasis on precise prediction and quantitative evaluation of specific optical performance metrics, enabling fine-tuned control over the luminescent centers of CDs. Furthermore, the regression model demonstrated high accuracy and reproducibility across three independent laboratories, further validating the model’s robustness. These findings offer a new perspective for the precise modulation and optimization of red-emissive CDs across broader wavelength applications.
Green synthesis of CDs has emerged as a primary sustainable approach, as it effectively transforms low-value biowaste into high-value products. Chen et al. systematically investigated the key process parameters involved in the formation of CDs using biochar derived from ten types of agricultural waste, focusing on pyrolysis temperature, residence time, nitrogen content, and carbon-to-nitrogen ratio[88]. Their study successfully constrained the relative error of QY predictions within 4.6%, significantly enhancing the accuracy and reliability of performance control of CDs. This work provides a new perspective on high-value utilization of waste materials and the efficiency improvement of the synthesis of CDs.
To further explore the potential of biomass-derived CDs and develop efficient optimization strategies for large-scale production under the green chemistry framework, Pudza et al. combined response surface methodology (RSM) with an ANN-based approach to conduct a multidimensional parameter optimization of cassava-derived CDs[89]. This method enabled high-precision prediction of QY [coefficient of determination (R2) > 0.94] under nonlinear conditions, including temperature, reaction time, precursor dosage, and solvent ratio. The optimized CDs exhibited strong ultraviolet absorption, broad-band emission, and wavelength-dependent optical properties, demonstrating their potential for sustainable and scalable applications.
In the field of CDs, challenges such as high experimental costs, limited data availability, and the difficulty of comprehensively covering high-dimensional feature spaces have constrained research progress. Against this backdrop, active learning strategies are increasingly recognized as effective approaches for enhancing model generalization and accelerating the discovery of high-performance CDs. Yang et al. implemented an active learning framework based on the XGBoost model, combined with a Bayesian optimization algorithm, to efficiently screen materials with afterglow lifetimes reaching 3.43 s in just 103 iterations[90]. This approach enabled precise regulation of key performance metrics of CDs, even under data-limited experimental conditions, while significantly reducing experimental costs, thereby providing a novel strategy for exploring high-performance materials [Figure 8A].
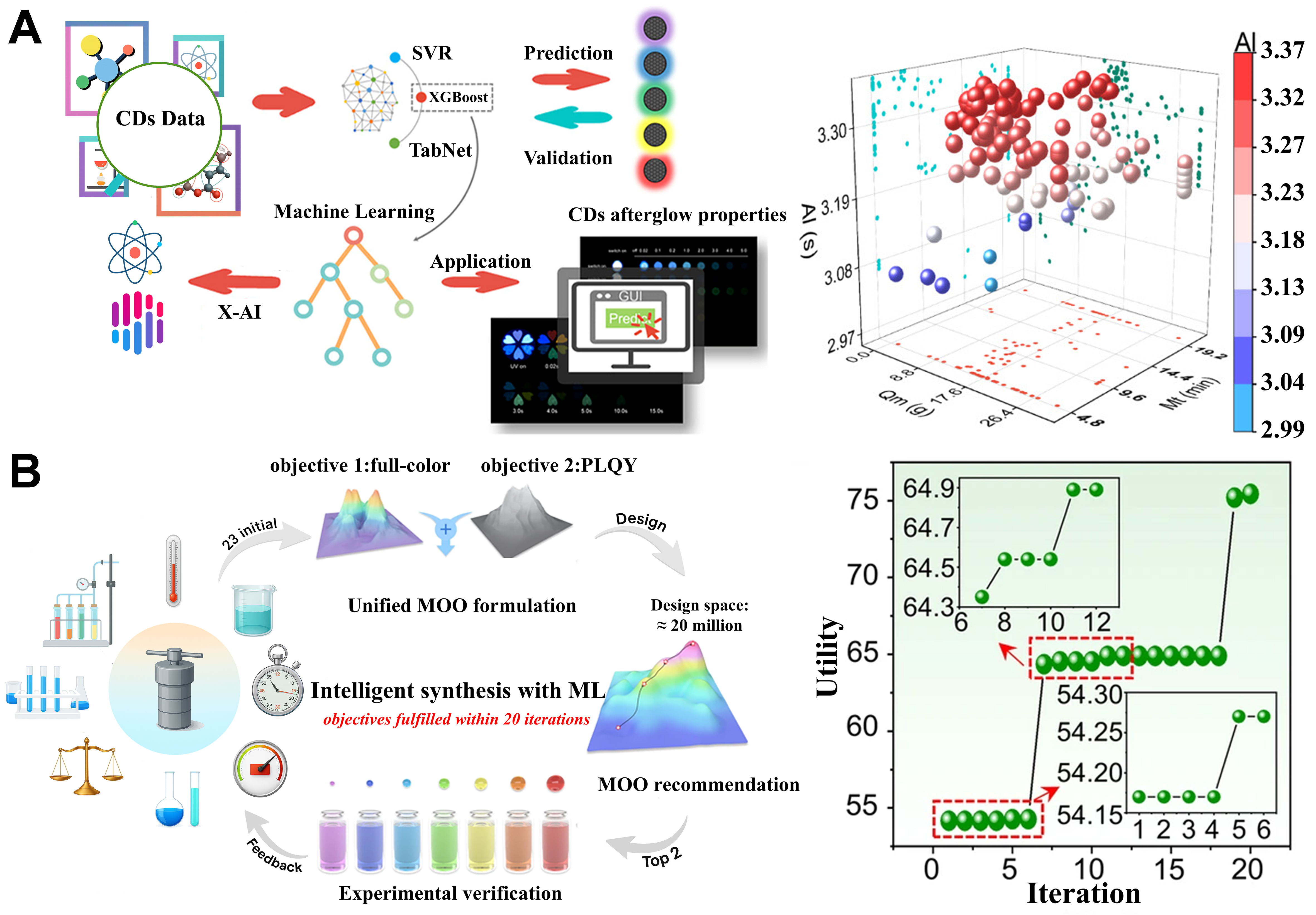
Figure 8. (A) After 103 iterations, the afterglow performance of CDs was improved using an active learning framework centered on the XGBoost model. The right panel shows the final search trajectory converging toward a high-lifetime region. These figures are quoted with permission from Yang et al.[90]; (B) Full-color CDs successfully synthesized with only 63 experimental datasets through a multi-objective active learning optimization strategy. The right panel presents the reported utility-vs-iteration curve, indicating progressive improvement and convergence within 20 iterations. These figures are quoted with permission from Guo et al.[91], Copyright 2024, American Chemical Society. CDs: Carbon dots; XGBoost: eXtreme gradient boosting; SVR: support vector regression; PLQY: photoluminescence quantum yield; MOO: multi-objective optimization; ML: machine learning.
To accommodate multi-objective requirements, Guo et al. proposed an innovative multi-objective active learning optimization strategy, which not only focused on the tuning of λem but also incorporated QY as a simultaneous optimization target[91]. By leveraging the XGBoost model, the study efficiently explored approximately 20 million synthesis conditions, yet required only 63 experimental datasets to successfully synthesize full-spectrum CDs with QY exceeding 60%, spanning the λem from violet to red [Figure 8B]. Compared with single-objective optimization strategies, multi-objective optimization not only substantially reduces experimental demand but also demonstrates greater adaptability and performance enhancement potential in complex material design. These findings underscore that, whether in deep exploration of single-performance characteristics or global searches for multi-objective optimization, active learning plays a crucial role in accelerating the discovery and optimization of new materials.
Despite the great potential of active learning methods in optimizing CDs, traditional approaches still face significant challenges, including imbalanced data distribution, unstable predictions, and inefficient use of experimental resources. To address these issues, Li et al. developed an efficient active learning optimization framework to enhance the nitric oxide (NO) release performance of iron-based CDs (Fe-Arg-CDs) [Figure 9][92]. This method first employs the sequential backward tree-classifier for Gaussian process regression (TCGPR) model, which uses a Gaussian radial basis function (RBF) kernel to partition the experimental data into multiple sub-datasets with enhanced internal consistency, thereby improving data consistency within each category. By combining the predictions of multiple submodels, the framework reduces the prediction uncertainty and enhances generalization. Furthermore, it explores 24,000 orthogonal synthesis routes through Monte Carlo sampling (1,000 simulations per candidate, totaling 24 million simulations) and applies a knowledge gradient (KG) strategy to efficiently identify the most promising experimental conditions. From this process, three top-ranked candidates were selected for experimental validation. Ultimately, the optimized framework successfully guided the synthesis of Fe-Arg-CDs, achieving a 20-fold increase in NO release capacity, which highlights the potential of ML-guided optimization for advancing nanozyme-based tumor therapies. Overall, this work demonstrates how ML-driven active learning can overcome discontinuities in complex, multi-distribution datasets, improve experimental design efficiency, and accelerate the discovery of high-performance CDs.
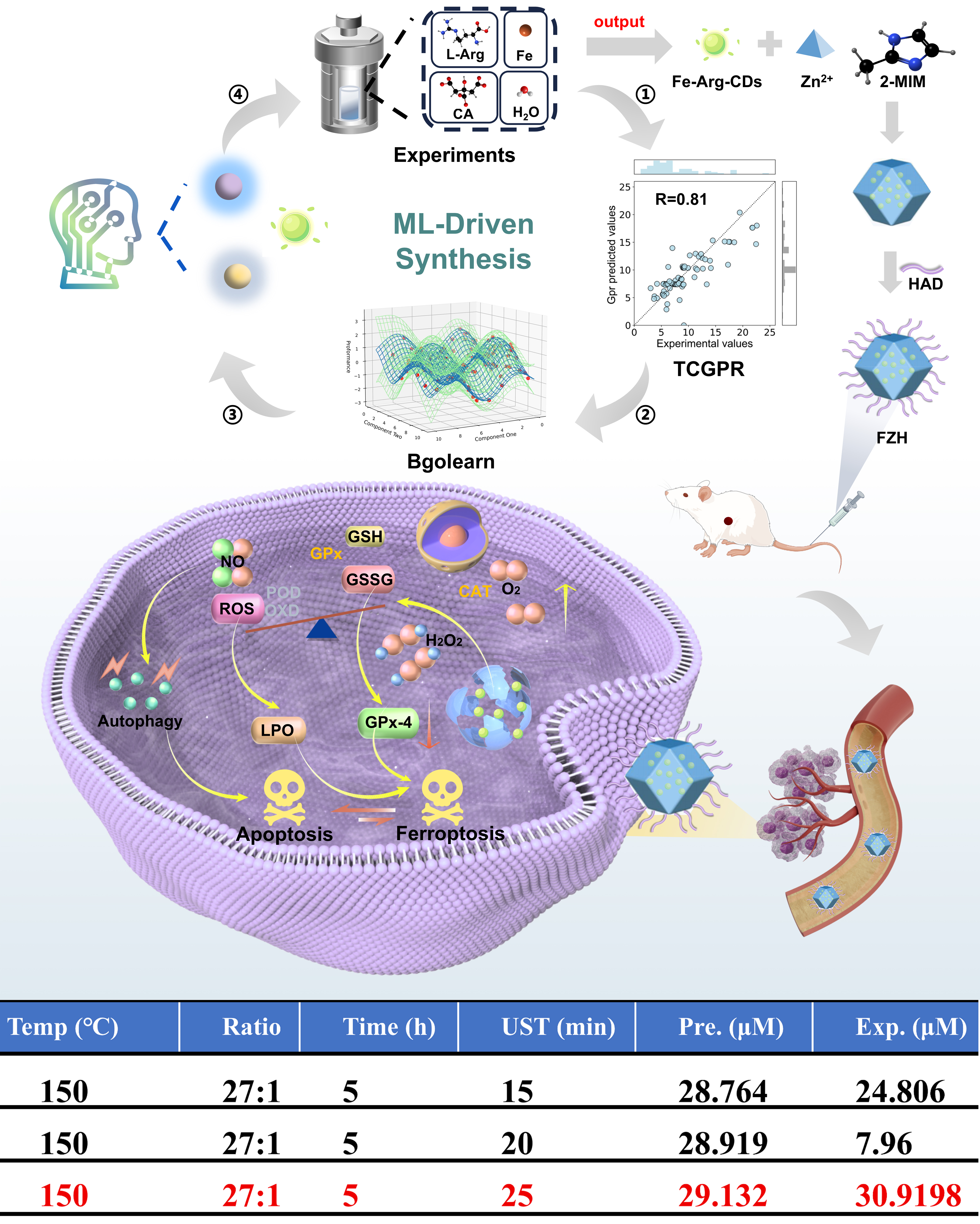
Figure 9. ML-engineered nanozyme system for synergistic anti-tumor ferroptosis/apoptosis therapy. The upper schematic illustrates the ML-driven synthesis and optimization cycle for Fe-Arg-CDs, showing the iterative interaction between experiments and model-guided predictions. The lower table compares predicted (Pre.) and experimental (Exp.) NO release values under different synthesis conditions, with the final optimal parameters highlighted in red. These figures are quoted with permission from Li et al.[92], Copyright 2024, Wiley-VCH GmbH. ML: Machine learning; CDs: carbon dots; NO: nitric oxide; TCGPR: sequential backward tree-classifier for Gaussian process regression; GSH: glutathione; GSSG: glutathione disulfide; CAT: catalase; ROS: reactive oxygen species; POD: peroxidase; OXD: oxidase; LPO: lipid peroxidation; UST: ultrasonic time.
Additionally, CDs are widely regarded as promising next-generation green corrosion inhibitors due to their exceptional anti-corrosion properties and environmental compatibility. He et al. systematically constructed and analyzed a dataset comprising 102 sets of synthesis parameters of CDs and their corresponding anti-corrosion performance data[93]. Using RF modeling, the study uncovered nonlinear relationships between hydrothermal synthesis parameters and anti-corrosion performance. Based on these findings, a genetic algorithm (GA) was employed to optimize the synthesis conditions. The optimized CDs exhibited an anti-corrosion efficiency of up to 92.3%, with an experimental error of less than 3% compared with the RF model predictions, validating the accuracy and reliability of the proposed approach[93].
Beyond the aforementioned categories, ML has also demonstrated significant potential in various functional applications, including optical devices and catalysis. Specifically, Wang et al. successfully achieved white light-emitting diodes (LEDs) with tunable correlated color temperatures by modulating the proportions of differently colored CDs, leveraging multiple ML models[94]. In their study, XGBoost and ANN models were used to process spectral data, enabling the optimization of the composition of CDs, which led to a significant enhancement in LED optical performance.
Building on this workflow, Wang et al. further employed ML techniques to systematically investigate the performance of CDs in cyclohexane catalytic oxidation reactions[95,96]. They developed an XGBoost-based analytical framework to explore the effects of various catalytic conditions, including the type of CDs, solvent selection, oxygen participation, and reaction duration, on catalytic efficiency. Using 275 experimental datasets, the model revealed complex nonlinear relationships between catalytic conditions, cyclohexane conversion rate, and adipic acid selectivity, enabling the optimization of catalytic conditions. The optimized results demonstrated that highly crystalline CDs, under the predicted optimal catalytic conditions (130 °C, 1.5 MPa, 10 h), achieved a cyclohexane conversion rate of 30.696% and an adipic acid selectivity of 92.52%[95]. These findings underscore the considerable potential of ML in catalysis and optical devices for precisely tuning the performance of CDs, providing novel strategies and methodologies for the efficient development and application of multifunctional materials.
Applying ML to tackle nonlinear and high-dimensional data in CDs analysis
In recent years, ML has been widely applied in fluorescence sensing data processing for systems of CDs, offering new opportunities to overcome the limitations of traditional methods in handling high-dimensional, nonlinear, and multi-interference conditions. By leveraging various ML approaches to process CDs-integrated data, researchers can not only achieve more precise quantitative detection, accurate classification, and pattern recognition in complex systems, but also expand the applicability of CDs for in situ analysis, rapid on-site detection, and high-throughput screening.
Compared with traditional methods that rely on single-parameter analysis or linear fitting, ML algorithms can extract more comprehensive features from complex spectral and multidimensional datasets, significantly enhancing model sensitivity, specificity, and robustness. To date, ML-driven strategies have demonstrated tremendous potential in image recognition, array sensing, spectral data processing, and portable applications, further broadening the scope of CDs-based fluorescence sensing technologies.
ML-driven multichannel array sensing
Array sensing methods employ multiple selective sensing elements with differential interactions, generating multidimensional “fingerprint” responses to precisely classify and differentiate structurally similar analytes. With the rapid advancement of ML techniques, integrating ML into multichannel data processing has become a key strategy for enhancing the sensing performance of CDs. Ostadhossein et al. utilized CDs prepared from five diaminopyridine (DAP) isomers and applied linear discriminant analysis (LDA) for dimensionality reduction and classification of oral microbial fluorescence data, achieving 100% classification accuracy in mixed bacterial samples [Figure 10A][97].
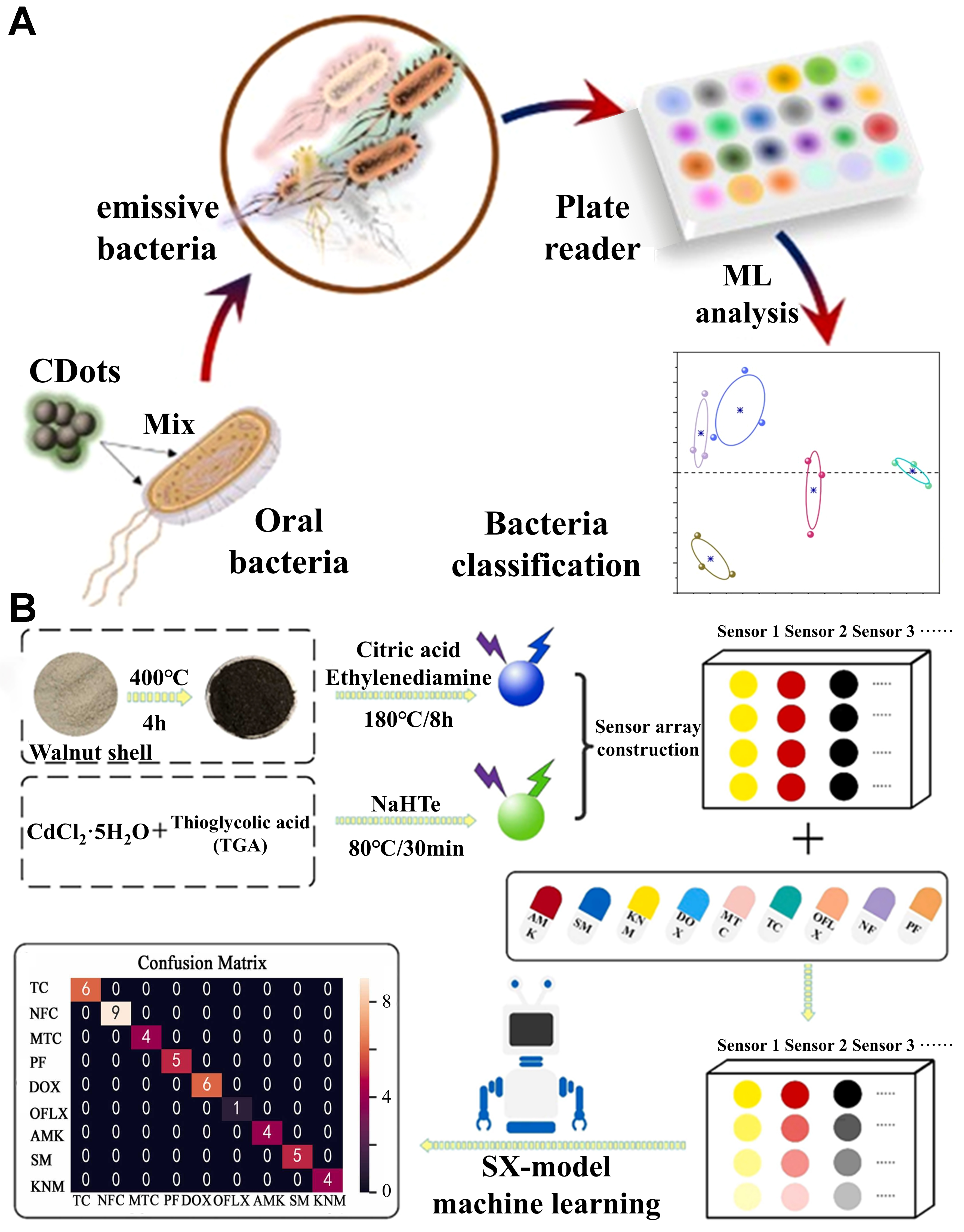
Figure 10. (A) Efficient classification of oral microbiota achieved using LDA. This figure is quoted with permission from Ostadhossein
However, relying solely on linear dimensionality reduction methods such as PCA or LDA may fail to capture nonlinear correlations among data points, limiting their effectiveness in certain complex classification tasks. To address this issue, Xu et al. developed a dual-channel fluorescence sensor array based on two types of CDs, integrating LDA and support vector machine (SVM) to analyze multidimensional response signals[98]. Their approach successfully differentiated four tetracycline antibiotics and binary mixtures, maintaining stable detection performance in real samples.
Building upon this foundation, Pandit et al. further employed gradient boosting trees, SVM, and other ML algorithms, improving protein detection classification accuracy from 83% (LDA) to 100%, while maintaining exceptional stability in high-noise environments[99]. Following a similar approach, Soares et al. developed a flexible sensor system based on curcumin CDs, incorporating capacitance responses at multiple frequencies[100]. By introducing a decision tree model to construct a multidimensional calibration space, their system achieved an average classification accuracy of 86.1% and a binary classification accuracy of 88.8% in the detection of Staphylococcus aureus in milk, significantly improving selectivity and sensitivity in complex sample analysis.
Shauloff et al. further leveraged the unique capacitance responses of CDs to different polarity gases and mixtures, combining the Rakel++ multi-label classification algorithm with an RF model to achieve precise identification of complex gas mixtures[101]. To advance the quantitative detection of target analytes, more systematic and efficient model optimization frameworks have garnered increasing attention. Xu et al. developed a dual-emission fluorescence sensor array based on high-quantum-yield CDs and CdTe quantum dots, significantly improving sensitivity and specificity in antibiotic detection by analyzing fluorescence intensity and the maximum λem as multidimensional response data[102]. This study first introduced a stepwise prediction strategy, integrating ML methods under the tree-based pipeline optimization technique (TPOT) framework, leading to the development of the eXperience (SX)-model for optimizing classification and concentration prediction performance [Figure 10B]. The SX-model first classifies the target analyte and then employs nine concentration models for quantitative prediction, enhancing its flexibility and generalization ability when analyzing unknown samples. Moreover, by converting fluorescence color into quantifiable RGB values, this study successfully achieved visual detection, offering valuable insights for on-site rapid screening and real-time monitoring using CD-based sensors.
Similarly, Liu et al. and Xu et al. expanded the SX-model’s applicability, employing fluorescence intensity ratios as input for stepwise prediction of heavy metal ion species and concentrations[103,104]. Under this strategy, the SX-model first identifies heavy metal ion types (e.g., Cr6+, Fe2+) using a classification model, followed by a regression model to estimate their concentrations, enabling high-precision analysis from single-metal to binary mixtures and complex environmental samples. The results demonstrate that the SX-model, combined with ML-driven optimization strategies, provides a feasible solution for transitioning from simple classification to high-dimensional multi-component prediction, laying a solid foundation for enhancing the versatility and sensitivity of CDs-based sensors in real-world complex systems. It should be noted that the two-stage cascade offers modularity and interpretability, but in practice it may introduce error propagation. Future studies may consider joint modeling (for example, sharing a feature extractor with separate classification and regression heads) and report side-by-side comparisons of joint vs. two-stage approaches under consistent data splits and evaluation metrics.
ML-driven strategies for high-dimensional and multimodal data fusion
In complex chemical sensing and biomedical detection, traditional data analysis methods typically rely on a limited number of spectral parameters for linear fitting or simple regression, which often fail to meet the demands for high sensitivity, broad applicability, and efficient processing of complex datasets. By leveraging ML, complete spectral and multidimensional data can be used for more precise feature extraction and classification, significantly enhancing detection sensitivity and selectivity while enabling self-calibration under multiple perturbation conditions.
Based on this, Zhang et al. developed CDs with concentration-dependent wavelength tunability and successfully classified Aspergillus flavus and Aspergillus fumigatus hyperspectral data with 99.6% sensitivity and specificity by integrating hyperspectral microscopy imaging with an SVM model [Figure 11A][105]. Furthermore, pseudo-color image analysis allowed for the quantitative assessment of fungal proportions in mixed samples, further validating the practicality of high-dimensional spectral information. Similarly, Cao
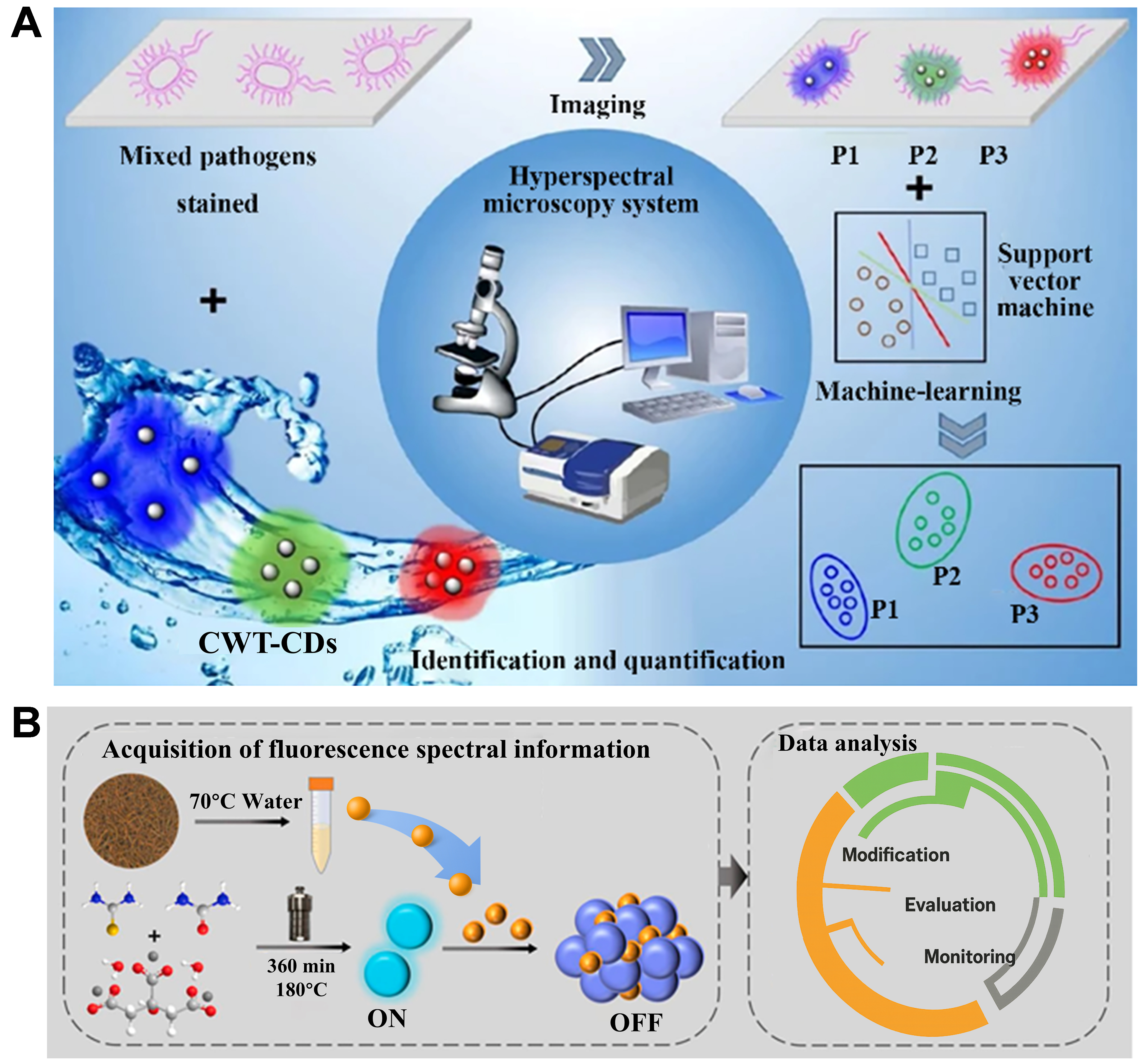
Figure 11. (A) Qualitative and quantitative analysis of two fungi performed via hyperspectral microscopic imaging combined with an SVM model. This figure is quoted with permission from Zhang et al.[105], Copyright 2023, SPIE; (B) Qualitative classification of black tea fermentation quality achieved using an LSSVM model. This figure is quoted with permission from Cao et al.[106], Copyright 2022, Elsevier B.V. SVM: Support vector machine; LSSVM: least squares support vector machines; CWT-CDS: concentration-dependent wavelength-tunable CDs.
Beyond single-spectrum deep analysis, multisource spectral data fusion enables a higher level of intelligent analysis. Döring et al. utilized the unique optical properties of CDs in ethanol concentration detection by integrating steady-state and time-resolved spectroscopy and applying a graph convolutional network (GCN) model to accurately predict ethanol concentration in ethanol-water mixtures and alcoholic beverages[107]. Compared to traditional linear regression, this approach achieved substantially lower MAE and improved robustness to complex backgrounds and noise, significantly reducing human intervention. Tuccitto et al. employed parallel factor analysis (PARAFAC) tensor decomposition to efficiently deconstruct multidimensional spectral data and incorporated an ANN-based classification model, achieving over 80% accuracy in mixed amino acid sample recognition, significantly improving data analysis convenience and accuracy[108]. Sarmanova et al. further applied multilayer perceptron (MLP) and nonlinear autoencoders for dimensionality reduction and modeling of multispectral fluorescence data, drastically reducing the mean squared error in the prediction of CDs and doxorubicin (Dox) concentrations[109]. Their superior performance far exceeded traditional PCA-based dimensionality reduction methods, laying a critical foundation for ML-based high-dimensional data processing in biomedical monitoring and diagnostics.
To address noise interference in complex matrix analysis, Liu et al. incorporated excitation wavelength as an additional dimension in a cross-reactive sensor array[110]. They used PCA and SVM for qualitative classification of single nitrophenol isomers, achieving up to 94.9% classification accuracy at ultra-low concentrations, and employed least-squares support vector regression and backpropagation artificial neural networks (BP-ANN) for quantitative prediction of concentrations and mixture proportions in binary and ternary samples, reporting the correlation coefficient (Rp) close to 1 with low root mean square error of the prediction set (RMSE). Similarly, Döring et al. prepared CDs from three o-phenylenediamine isomers and combined steady-state and time-resolved photoluminescence with a multiple linear regression model for temperature prediction[111]. Leveraging multi-feature inputs, the approach reduced the sensing error to
ML-based image data processing
In the field of image recognition, integrating computer vision with ML techniques for CDs-based detection can effectively overcome the limitations of human visual color discrimination, significantly enhancing detection accuracy and sensitivity across a broader range of applications. Zheng et al. developed a dual-emission fluorescence probe (CDs@Eu-MOF: CDs@europium metal-organic framework) and combined computer vision with a backpropagation neural network to comprehensively analyze fluorescence spectral data and RGB color values, establishing a highly linear correlation between Fe3+ concentration and fluorescence response [Figure 12A][112]. The detection model exhibited exceptional performance within a wide detection range (1-200 µM) and a low detection limit of 0.91 µM, with a R2 of 0.9964.
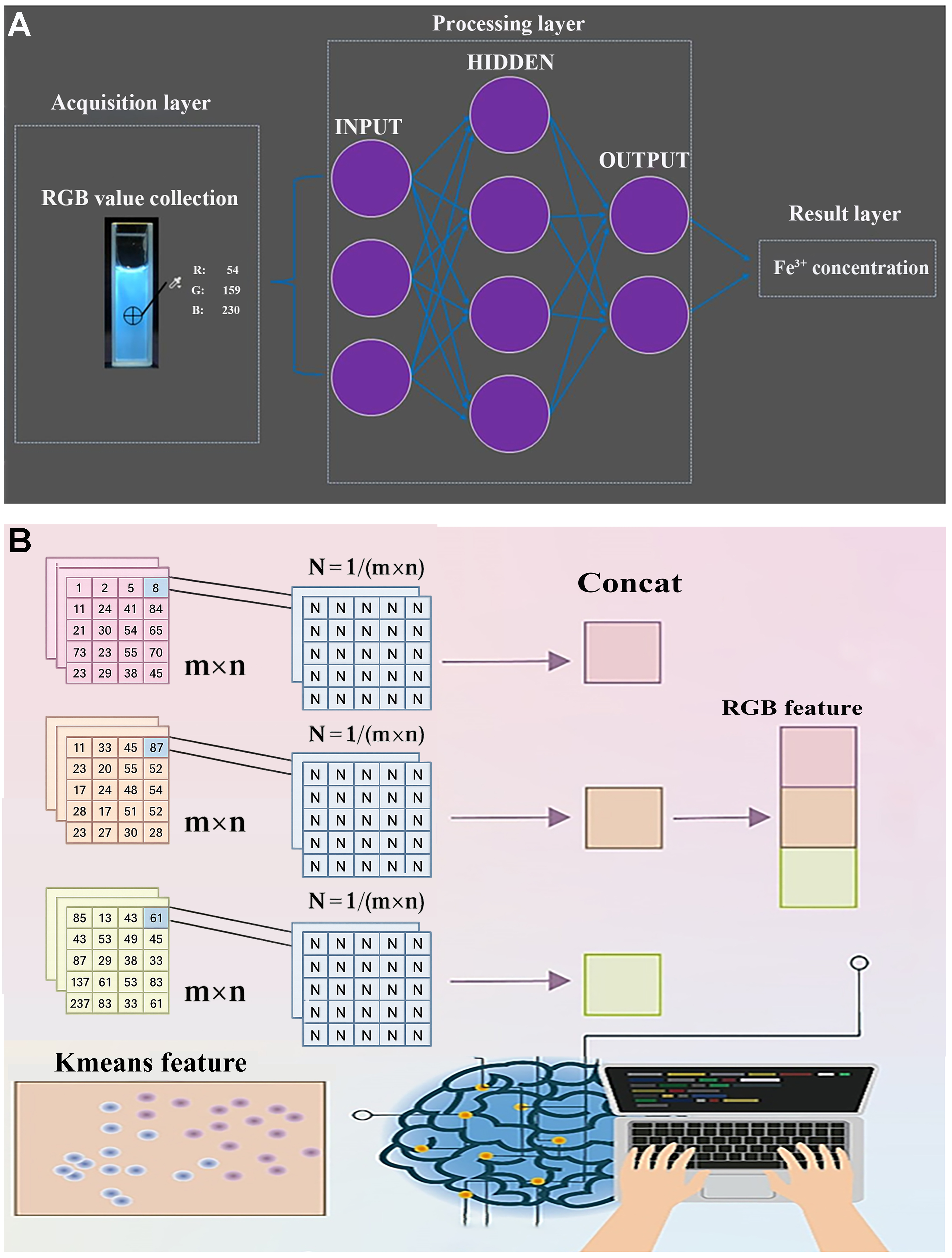
Figure 12. (A) Fe3+ concentration detected through integrated analysis of spectral data and RGB color values using computer vision. This figure is quoted with permission from Zheng et al.[112], Copyright 2021, Springer Nature; (B) Cr (VI) concentration detected via refined processing of RGB features with K-means clustering. This figure is quoted with permission from Zhang et al.[113], Copyright 2024, Elsevier Ltd. RGB: Red, green, blue.
The study by Zhang et al. further highlights the potential of ML in RGB color feature extraction and optimization [Figure 12B][113]. They utilized an N-doped blue-emitting system of CDs and applied K-means clustering to refine RGB feature processing, thereby improving the signal-to-noise ratio and achieving a more ideal linear correlation between Cr (VI) concentration and fluorescence color variation. Compared to traditional RGB extraction techniques, this approach significantly improved the detection accuracy of linear models and achieved a lower detection limit in real water samples, strongly demonstrating the synergistic potential of ML and technologies based on CDs for complex environmental monitoring.
Beyond fluorescence color recognition and quantitative detection, ML techniques have also been successfully applied to expand the use of CDs in fingerprint recognition. Yadav et al. developed a novel fluorescent fingerprint powder based on N-S co-doped CDs, which, when combined with an ML algorithm, enabled high-precision identification of latent fingerprints[114]. Their study employed a three-stage digital image processing workflow (grayscale conversion, normalization, and binarization) to process latent fingerprint images, extracting multiple feature points and constructing a similarity matching model using Euclidean distance.
On-site analysis using smartphone platforms and deep learning
While ML-based image data processing has proven highly effective for RGB feature extraction and recognition, its typical use has been limited to offline analysis. Building on the same fundamental principles, integrating deep learning algorithms with portable smartphone platforms enables real-time, automated, and more accessible CD-based sensing. Huang et al. developed a multimodal deep learning model incorporating CNN and fully connected networks, utilizing multicolor fluorescent CDs for simultaneous high-precision processing of spectral and image data [Figure 13A][115]. This model achieved rapid on-site detection of illicit drugs while ensuring precise laboratory analysis in complex backgrounds. For spectral data tasks, the model achieved 99.9% (qualitative) and 99.6% (semi-quantitative, five-class) classification accuracies, whereas for image data it achieved 98.4% (qualitative) and 84.4% (semi-quantitative, five-class) classification accuracies. Even under strong interference conditions, such as artificial urine, the model maintained outstanding stability and robustness, validating deep learning’s potential in enhancing multimodal data processing accuracy for CD-based sensing.
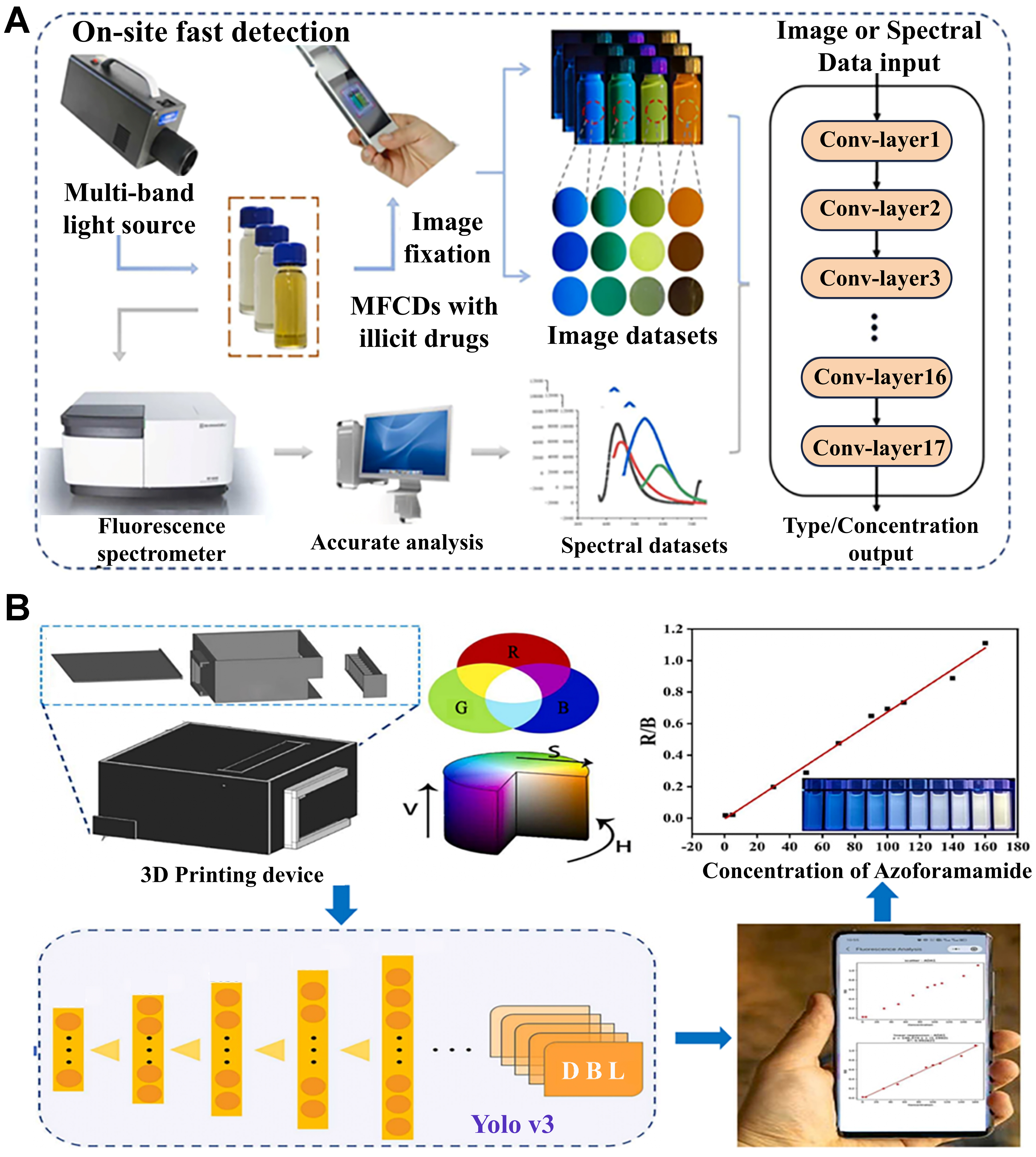
Figure 13. (A) Multimodal detection of illicit drugs realized by integrating CNN with a fully connected network. This figure is quoted with permission from Huang et al.[115], Copyright 2023, Elsevier B.V; (B) Real-time analysis of GSH and ADA achieved by integrating YOLO v3 with a smartphone. This figure is quoted with permission from Liu et al.[116], Copyright 2021, Elsevier B.V. CNN: Convolutional neural network; GSH: glutathione; ADA: azodicarbonamide; YOLO: You Only Look Once; MFCDs: multicolor fluorescent CDs.
The Rao research group has further validated the feasibility of combining deep learning with smartphone platforms for real-time and convenient detection in practical applications. For instance, Liu et al. integrated You Only Look Once (YOLO) v3 deep learning with a smartphone-based platform to enable high-sensitivity, high-precision, and real-time detection of glutathione (GSH) and azodicarbonamide (ADA) using CDs [Figure 13B][116]. The YOLO v3 model was used for target detection and segmentation in fluorescence images, accurately extracting RGB and hue, saturation, and value (HSV) from the test tube fluorescence region. Subsequently, linear fitting analysis was employed to establish the relationship between fluorescence signal ratios (e.g., R/G and R/B) and analyte concentrations, achieving high sensitivity and precision. Compared with traditional laboratory fluorescence analysis methods, this platform leverages the portability of smartphones and the powerful data processing capabilities of deep learning, significantly simplifying data acquisition and analysis while enhancing real-time detection accuracy[116].
Following a similar approach, Lu et al. combined YOLO v3 deep learning algorithms with a tricolor fluorescence sensing system based on CDs for high-precision detection of Cu2+ and thiabendazole[117]. In another study[118], a dual-color ratiometric fluorescence probe based on CDs and Fe/Zr-MOF was developed for real-time qualitative analysis of meat freshness. To further enhance detection accuracy and data processing flexibility, their subsequent studies introduced least squares regression and Lasso regression into the YOLO v3 target detection framework, improving the sensitivity and anti-interference capability for detecting tetracycline antibiotics and ions (e.g., Hg2+ and S2-)[119,120].
Similarly, Wang et al. applied LDA, decision trees, and Naïve Bayes algorithms to analyze a CDs-based paper sensor array functionalized with three antibiotic modifications, achieving 100% classification accuracy for bacterial concentrations ranging from 1.0 × 103 to 1.0 × 107 colony-forming units per milliliter (CFU/mL) [Figure 14A][121]. Thonghlueng et al. employed an RF model to analyze the RGB fluorescence response of nitrogen-doped CDs, enabling high-precision detection of cytosine and 5-methylcytosine with an average deviation of only 9.68%, providing a powerful tool for epigenetics research and early disease diagnosis[122]. Yen et al. designed a multifunctional sensing array combining CDs, gold nanoclusters, silver nanoclusters, and Marquis reagents, integrating a YOLO v4 deep learning platform for smartphone-based remote detection of five common illicit drugs, achieving 100% accuracy at low concentrations[123].
Figure 14. (A) Real-time bacterial analysis performed using smart recognition combined with multiple ML algorithms. This figure is quoted with permission from Wang et al.[121], Copyright 2023, Elsevier B.V; (B) High-precision real-time detection of food spoilage realized by embedding the RF model into a smartphone application. This figure is quoted with permission from Doğan et al.[124]. ML: Machine learning; RF: random forest; UV: ultraviolet light; FG: fish gelatin; ARCE: anthocyanins rich red cabbage extract; CD: carbon dot; TVB-N: total volatile basic nitrogen.
Additionally, to reduce operational costs and enable offline analysis, Doğan et al. further expanded the boundaries of ML applications on smartphones [Figure 14B][124]. By embedding an RF model into a smartphone application, they utilized anthocyanin-loaded CDs to enhance fish gelatin membranes, enabling real-time, high-precision detection of food spoilage. Without relying on cloud computing, this system completed colorimetric analysis within approximately 0.1 s, achieving 98.8% classification accuracy in general tests and 99.6% accuracy in real fish samples.
The preceding two sections indicate that integrating RGB and image features with ML holds substantial promise. To further ensure cross-device and cross-batch comparability and reproducibility, one can standardize the color baseline through color-chart calibration and color management via conversion from RGB to CIE XYZ and subsequently to CIE L*a*b* color space. In addition, reserving one device as an external-domain test set enables direct assessment of generalization to unseen hardware, thereby markedly improving the comparability of results across studies.
Decoding CDs mechanisms with ML
In the process of optimizing and expanding the performance of CDs, ML not only provides an efficient approach for parameter prediction and regulation, but also lays a solid foundation for elucidating the structure–performance relationship and unraveling the intrinsic mechanisms governing the properties of CDs. However, traditional data-driven strategies often emphasize prediction accuracy, making it challenging to comprehensively interpret the internal logic of models and their fundamental correlations.
At the same time, DFT calculations offer insights into the reaction pathways of CDs, active sites, and energy distribution at the electronic structure level, playing a crucial role in understanding the microscopic mechanisms underlying their luminescence and catalytic properties. For instance, a recent review summarized the applications of DFT in quantum size effects, heteroatom doping, molecular states, and environmental effects within the field of fluorescence of CDs, providing a more intuitive and controllable theoretical foundation for elucidating the fundamental mechanisms of CDs[125]. However, due to the high computational cost, system size limitations, and extended simulation times, DFT alone struggles to systematically explore a wide range of structures and complex environments, hindering a comprehensive understanding of CDs’ mechanisms.
In light of this, researchers have increasingly adopted explainable ML strategies to deepen insights into the causal relationships and intrinsic logic governing the performance regulation of CDs. By leveraging explainability methods to identify key features and influential parameters in the model training and prediction processes, researchers can not only understand how and why a specific prediction is generated but also extract universal mechanistic principles across large-scale and diverse systems of CDs. Compared to relying solely on DFT-based microscopic mechanism inference, explainable ML does not require extensive computational resources and remains highly adaptable to complex datasets and strongly nonlinear factors. By balancing prediction accuracy and interpretability, ML introduces a novel perspective for deciphering the structure–performance relationships of CDs, offering a more flexible and efficient research pathway for subsequent mechanistic studies and performance optimization.
In recent studies on explainability in research of CDs, extensive efforts have focused on using ML methods to identify and quantify how experimental conditions (such as precursor mass, heating intensity, pH, reaction temperature, and reaction time) influence the mechanisms underlying the properties of CDs. For instance, Xu et al. applied Pearson correlation coefficients and grey relational analysis to confirm the pivotal roles of precursor mass and microwave intensity in influencing the growth kinetics of CDs [Figure 15A][76]. Their findings suggest that these two factors regulate nucleation and crystal growth by controlling reactant concentration in solution and heating rate, respectively. Tang et al. employed an XGBoost model coupled with feature importance analysis to systematically characterize key factors that influence the QY of CDs[77]. Their results show that pH substantially affects QY by modulating the solubility of CDs, whereas reaction temperature exerts a decisive influence on the formation of CDs by controlling molecular kinetic energy and collision frequency. Meanwhile, reaction time influences quantum confinement effects by regulating particle size. Guo et al. conducted an interpretable analysis of the τp of CDs using an XGBoost model coupled with SHapley Additive exPlanations (SHAP)[85]. They found that precursor mass, reaction temperature, and NaOH amount contributed most to τp. Increasing precursor mass showed a negative contribution, likely attributable to aggregation-induced increases in nonradiative pathways, whereas raising reaction temperature, increasing NaOH amount, and moderately extending reaction time produced positive contributions that shifted τp from the ns to μs and ultimately to the ms regime, enabling the aqueous-phase synthesis of long-lived phosphorescent CDs. By quantitatively assessing feature importances in the model, these three studies not only deepened the mechanistic understanding of the optical properties of CDs but also provided robust data support and a theoretical basis for further optimization of their intrinsic properties.
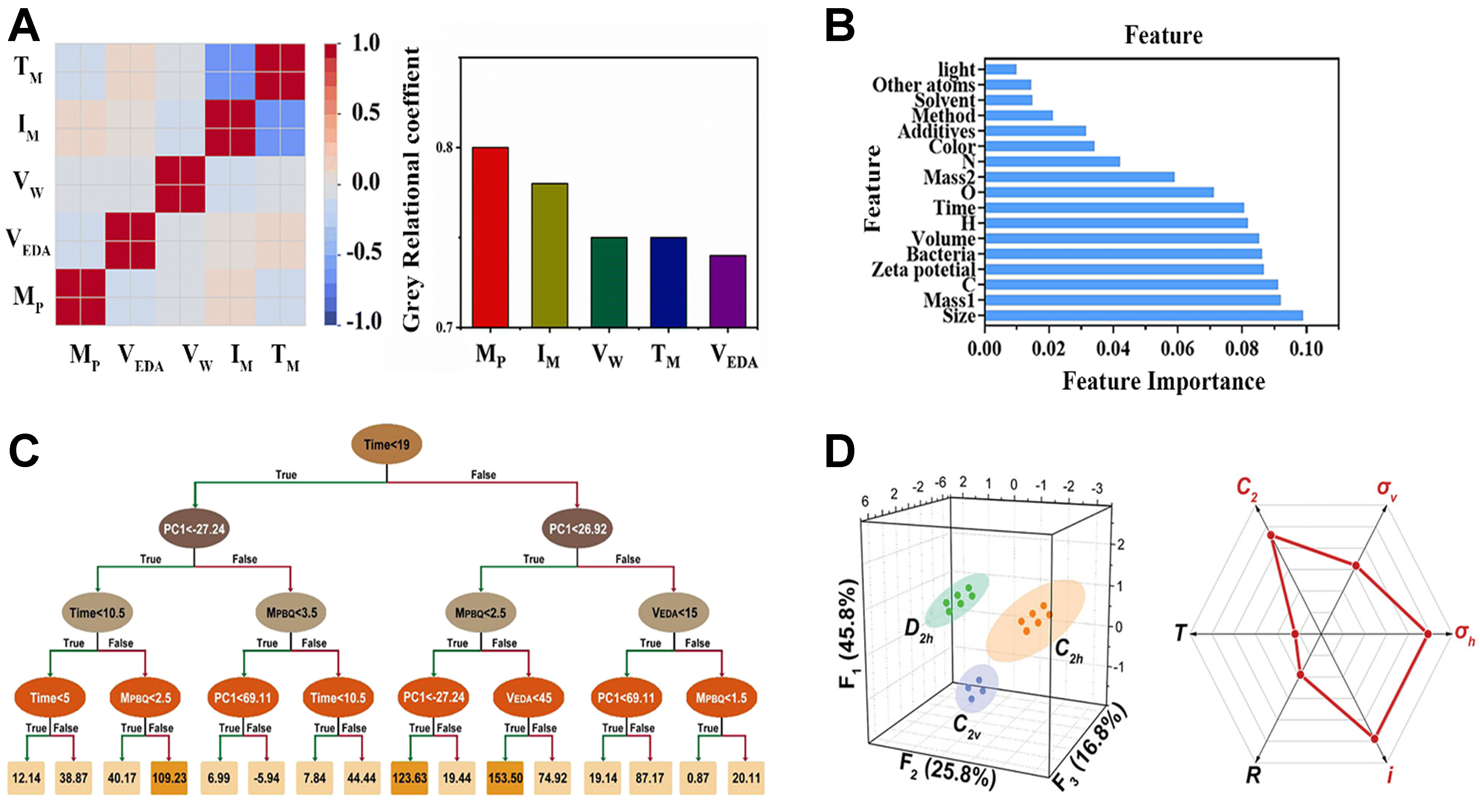
Figure 15. (A) Interpretability of CDs synthesis conditions enhanced using Pearson correlation coefficients and grey relational analysis. This figure is quoted with permission from Xu et al.[76], Copyright 2022, The Royal Society of Chemistry; (B) Contribution of CDs’ physicochemical properties to antibacterial performance quantified through Gini importance analysis. This figure is quoted with permission from Bian et al.[128]; (C) Dual role mechanisms of solvents systematically analyzed using tree models. This figure is quoted with permission from Hong et al.[75], Copyright 2022, American Chemical Society; (D) The QY mechanisms newly interpreted by integrating ML with molecular symmetry theory. This figure is quoted with permission from Chen et al.[130], Copyright 2024, Wiley-VCH GmbH. CDs: Carbon dots; QY: quantum yield; ML: machine learning.
Unlike explanations derived solely from experimental conditions, quantitative structure-property relationship (QSPR) approaches reveal deeper connections between the underlying physicochemical nature of a material and its target properties. By integrating ML with QSPR modeling, a direct correlation between macroscopic performance and microscopic physicochemical traits can be established. Salahinejad et al. screened ionic descriptors via a GA and an enhanced replacement method (ERM), then employed multiple linear regression (MLR) and SVM models to elucidate the key mechanisms underlying the fluorescence quenching of cysteine-based CDs by heavy metal ions[126]. To further investigate how intrinsic attributes of CDs influence this process, Roozbahani et al. applied MLR and SVM to systematically explore how the physicochemical characteristics of amino acids, used as precursors of CDs, affect Hg(II)-driven fluorescence quenching[127]. Their results reveal that the hydrophobic properties of amino acids dominate interactions between N-doped CDs (N-CDs) and Hg (II) ions, whereas hydrogen bond donor and acceptor characteristics play ancillary roles in photoluminescence regulation. This work provides a systematic account of the interplay between precursor molecular features and the fluorescence quenching mechanisms of N-CDs, offering data-backed, theoretical rationales for performance optimization and highlighting ML’s potential in elucidating and regulating the mechanisms of CDs.
Beyond detection performance, some researchers have also employed QSPR methods to examine the correlations between the physicochemical properties of CDs and their biological applications[128]. Bian et al., using Gini importance analysis, quantified the relative contributions of different features to antibacterial performance, revealing that particle size and surface charge (zeta potential) are central parameters affecting the antibacterial efficacy of CDs [Figure 15B][128]. The study points out that smaller-sized CDs can penetrate bacterial cell membranes and interact with respiratory chains, thereby compromising bacterial metabolic functions. In addition, positively charged CDs significantly enhance binding efficiency through electrostatic interactions with negatively charged bacterial cell walls, further promoting reactive oxygen species (ROS) generation and boosting antibacterial activity.
Building on these findings, researchers have extended their scope to solvent environments during synthesis, aiming to bridge microscopic environments and macroscopic performance. Hong et al. were the first to systematically analyze the dual regulatory mechanisms of reaction solvents on the optical properties of CDs by integrating tree-based models with feature importance analysis[75]. On the one hand, solvent effects notably influence the position and intensity of fluorescence emission peaks by modulating interactions of CDs in the ground and excited states. On the other hand, the physicochemical properties of reaction solvents dictate carbonization levels and surface chemical states of the carbon core, thereby determining the photoluminescence behavior and spectral characteristics of CDs [Figure 15C]. To further investigate how different solvent parameters affect the optical properties of CDs, Chen et al. used feature importance analysis to demonstrate the critical reaction influence of solvent characteristics[82]. Specifically, reaction solvent boiling point affects the λem of CDs by regulating precursor carbonization and dehydration; heat capacity indirectly controls the reaction temperature, thus influencing QY; and dielectric constant modulates charge distribution and influences the magnitude of the Δλ.
The profound influence of the precursors’ intrinsic structural properties on the performance of CDs cannot be fully elucidated by studying only experimental conditions and solvent parameters. Li et al. addressed this issue using a graph neural network (GNN) approach to propose a quantitative measure of structural disorder in CDs, focusing on the physical link between precursor molecular symmetry and the phosphorescence lifetimes of CDs[129]. In that study, the authors extracted simplified graph structural features of CDs via GNN, establishing direct mappings from the structural features to descriptors, namely structural variability (Ω) and porosities (V), and subsequently defined a comprehensive structural disorder parameter (S) for CDs. Incorporating experimental data, they uncovered multiple layers of relationships: first, a qualitative correlation between precursor structural symmetry and S of CDs; next, a linear fit between the disorder of CDs and phosphorescence lifetime. Building on this work, Chen et al. combined ML and molecular symmetry theory to link molecular vibrational modes to nonradiative transitions in CDs, thereby offering novel insights into QY mechanisms [Figure 15D][130]. By using PCA and the vibrational descriptor χvib, they identified the key regulators of QY in CDs based on point-group symmetry and symmetry elements. The study specifically underscores the core roles of vertical mirror plane (σv) and horizontal mirror plane (σh) symmetry elements in enhancing QY, and, through incorporating external variables such as reaction temperature and precursor concentration ratios, further validates the central function of these symmetry elements in nonradiative transition mechanisms. This research marks the first successful use of ML to elucidate the physics underlying QY in CDs, addressing previous gaps in the physical interpretability of ML-driven studies. It not only provides theoretical guidance for data-driven optimization of the performance of CDs but also indicates a new pathway for fabricating high-performance CDs via precise modulation of precursor symmetry.
In addition to the above approaches, to overcome the limitations of traditional spectroscopic analysis that often fails to fully reveal intrinsic characteristics of the emission state, Dager et al. introduced ML-based methods to systematically parse the spectral data of CDs, thereby proposing a robust toolkit and methodology for studying emission states[131]. PCA was employed to quantify the variance contributions of different excitation wavelengths, facilitating dimensionality reduction and clustering of spectral data. This approach enabled the isolation of key excitation wavelength intervals, offering insights into wavelength-dependent emission behavior.
CHALLENGES AND FUTURE DIRECTIONS
Despite significant progress in ML-driven materials research, the application of ML to CDs remains at an early exploratory stage, presenting both substantial challenges and exciting opportunities. Among the most critical bottlenecks is the efficient integration of multisource heterogeneous data. Experimental data related to CDs are often highly multidimensional and heterogeneous in format, encompassing spectral, electrical, and microscopic structural information. Accordingly, the intelligent fusion of these multimodal datasets, together with the construction of more comprehensive and high-quality characterization databases, will be essential for accurately deciphering the properties of CDs. In this context, adaptive data fusion models can help researchers assign appropriate weights and integration strategies to different data modalities, thereby improving generalization across diverse data sources. Equally important, the establishment of standardized databases for CD synthesis, property characterization, and testing methodologies will be crucial for minimizing data bias and noise, enhancing the reliability of ML training, and facilitating automated experimental platforms for rapid data generation and processing.
Beyond data integration, the growing range of potential application scenarios further highlights the promise of ML-driven CDs research in areas such as biomedical imaging, sensing, energy storage, and optoelectronic devices. From deep learning-assisted optimization of spectral responses in biosensing to the prediction of conductivity and stability for flexible electronic applications, these emerging directions rely heavily on multimodal data interpretation and model explainability. At the same time, however, ML applications in CDs research are still constrained by limited data quantity, inconsistent data quality, and an incomplete mechanistic understanding of structure formation and performance evolution. To address these limitations while maintaining accurate and reliable predictions, future efforts should focus on three closely connected priorities: the construction of standardized databases, the development of automated experimental platforms, and the deeper integration of cheminformatics tools. Collectively, these advances will provide a more effective research framework for elucidating the synthesis mechanisms and structural regulation of CDs.
At the present stage, where data accumulation remains limited, the small-sample problem continues to be a major obstacle to both model accuracy and scalability. High-quality experimental data are not only time- and resource-intensive to obtain, but are also vulnerable to inconsistencies arising from manual annotation and measurement error. To mitigate these challenges, strategies such as transfer learning, data augmentation, semi-supervised learning, and active learning have been increasingly explored. In parallel, incorporating physically meaningful constraints derived from quantum chemistry calculations into ML models can partially compensate for data scarcity and improve predictive performance. Active learning is particularly attractive because it identifies the most informative experimental directions in each iteration, thereby maximizing information gain with limited data. This “machine-guided experimentation” paradigm can reduce redundant trials while substantially improving research efficiency. Furthermore, continued advances in text mining and automated experimental systems may accelerate data accumulation and enable more effective use of fragmented yet valuable information embedded in the literature, further alleviating the limitations imposed by small datasets.
As predictive performance improves, interpretability is becoming an equally important requirement. In practice, materials scientists need more than accurate black-box predictions; they also require mechanistic insight into the key factors governing the properties of CDs. For this reason, the integration of domain knowledge with explainable AI methods will be pivotal for unraveling the structure–performance relationships of CDs. Methods such as symbolic regression and SHAP-based analysis can quantify the contributions of different features to CD performance, thereby clarifying the intrinsic relationships among precursor selection, molecular structure, and optical or electrical behavior. More importantly, if molecular databases and cheminformatics tools can be leveraged to establish automated correlation frameworks linking molecular structures to the macroscopic properties of CDs, they may provide valuable molecular-level guidance for synthesis optimization and performance tuning. In this way, interpretability not only enhances the practical applicability of ML models in real experimental settings, but also deepens our fundamental understanding of the physicochemical nature of CDs.
Alongside these developments, LLMs are showing increasing potential in chemistry and materials science, opening new possibilities for CDs research. By extracting and organizing knowledge from vast scientific literature, these models can support a range of task-specific research activities. When coupled with automated synthesis platforms, LLMs may assist researchers during real-time experiments by offering immediate recommendations for refining synthesis parameters or characterization protocols, thereby accelerating the exploration of CD performance boundaries. As these models continue to evolve, domain-specific training and fine-tuning tailored to CDs research may further improve experimental efficiency and help accelerate innovative breakthroughs.
Looking further ahead, the convergence of these methodologies and tools is gradually bringing the concept of AI-driven autonomous laboratories closer to reality. Within this paradigm, ML and high-throughput automation can form a closed-loop system in which the synthesis, characterization, and optimization of CDs proceed with minimal human intervention. High-throughput screening can rapidly identify promising candidates across a vast parameter space, while ML continuously analyzes experimental outcomes, updates predictive models in real time, and autonomously proposes subsequent experimental steps for robotic execution. Such an iterative workflow could markedly accelerate the discovery of novel CDs while improving reproducibility and scalability, thereby laying a stronger foundation for their large-scale synthesis and broader practical deployment.
CONCLUSION
In conclusion, this review provides a systematic overview of the application of ML in CDs research, with particular emphasis on practical methodologies and recent advances across different stages of investigation. Rather than revisiting commonly discussed ML algorithms, it focuses on data collection and data processing within the broader ML workflow in materials science, thereby underscoring the central role of data in ML-enabled materials innovation. The review also highlights representative applications of ML in CDs research, especially in intrinsic property optimization, data analysis, and mechanistic interpretation, illustrating the substantial potential of ML to advance this field.
Taken together, the existing body of work suggests that ML is poised to become an increasingly important driver of progress in CDs research. At the same time, realizing this potential will require continued advances in several interconnected areas, including heterogeneous data integration, improved data availability and quality, stronger model interpretability, and the deeper incorporation of automation and intelligent tools. Progress on these fronts will be essential for building more robust and mechanistically grounded research frameworks, ultimately enabling the rational design, scalable synthesis, and broader application of CDs.
DECLARATIONS
Acknowledgments
We express our gratitude for the financial support.
Authors’ contributions
Writing - review and editing, writing - original draft, visualization, validation, methodology, investigation, formal analysis, data curation, conceptualization: Ren, Y.
Writing - original draft, visualization, data curation, formal analysis: Yan, X.
Writing - original draft, data curation: Fang, R.
Visualization, data curation: Deng, H.
Project administration, methodology, funding acquisition: Chen, Y.
Supervision, resources, project administration, conceptualization: Li, Z.
Writing - review and editing, supervision, resources, project administration, methodology, investigation, funding acquisition, formal analysis, data curation, conceptualization: Feng, L.; Qu, X.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the Program for Distinguished Professor of Shanghai Universities (Oriental Scholars), Tracking Plan (GZ2022009), the National Natural Science Foundation of China (22177067) and the Shanghai Rising-Star Program (20QA1403400).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Wong, S.; Ngadi, N.; Inuwa, I. M.; Hassan, O. Recent advances in applications of activated carbon from biowaste for wastewater treatment: a short review. J. Clean. Prod. 2018, 175, 361-75.
2. Fan, Y.; Fowler, G. D.; Zhao, M. The past, present and future of carbon black as a rubber reinforcing filler - a review. J. Clean. Prod. 2020, 247, 119115.
3. Xu, X.; Ray, R.; Gu, Y.; et al. Electrophoretic analysis and purification of fluorescent single-walled carbon nanotube fragments. J. Am. Chem. Soc. 2004, 126, 12736-7.
4. Hou, Y.; Lu, Q.; Deng, J.; Li, H.; Zhang, Y. One-pot electrochemical synthesis of functionalized fluorescent carbon dots and their selective sensing for mercury ion. Anal. Chim. Acta. 2015, 866, 69-74.
5. Das, A.; Arefina, I. A.; Danilov, D. V.; et al. Chiral carbon dots based on L/D-cysteine produced via room temperature surface modification and one-pot carbonization. Nanoscale 2021, 13, 8058-66.
6. Wang, L.; Li, B.; Xu, F.; et al. Visual in vivo degradation of injectable hydrogel by real-time and non-invasive tracking using carbon nanodots as fluorescent indicator. Biomaterials 2017, 145, 192-206.
7. Boakye-Yiadom, K. O.; Kesse, S.; Opoku-Damoah, Y.; et al. Carbon dots: applications in bioimaging and theranostics. Int. J. Pharm. 2019, 564, 308-17.
8. Tao, S.; Zhou, C.; Kang, C.; et al. Confined-domain crosslink-enhanced emission effect in carbonized polymer dots. Light. Sci. Appl. 2022, 11, 56.
9. Liu, J.; Li, R.; Yang, B. Carbon dots: a new type of carbon-based nanomaterial with wide applications. ACS. Cent. Sci. 2020, 6, 2179-95.
10. Tao, H.; Yang, K.; Ma, Z.; et al. In vivo NIR fluorescence imaging, biodistribution, and toxicology of photoluminescent carbon dots produced from carbon nanotubes and graphite. Small 2012, 8, 281-90.
11. Sun, Y. P.; Zhou, B.; Lin, Y.; et al. Quantum-sized carbon dots for bright and colorful photoluminescence. J. Am. Chem. Soc. 2006, 128, 7756-7.
12. Su, Y.; Xie, M.; Lu, X.; et al. Facile synthesis and photoelectric properties of carbon dots with upconversion fluorescence using arc-synthesized carbon by-products. RSC. Adv. 2014, 4, 4839-42.
13. Zhao, Q. L.; Zhang, Z. L.; Huang, B. H.; Peng, J.; Zhang, M.; Pang, D. W. Facile preparation of low cytotoxicity fluorescent carbon nanocrystals by electrooxidation of graphite. Chem. Commun. 2008, 5116-8.
14. Qiao, Z. A.; Wang, Y.; Gao, Y.; et al. Commercially activated carbon as the source for producing multicolor photoluminescent carbon dots by chemical oxidation. Chem. Commun. 2010, 46, 8812-4.
15. Wu, Z. L.; Gao, M. X.; Wang, T. T.; Wan, X. Y.; Zheng, L. L.; Huang, C. Z. A general quantitative pH sensor developed with dicyandiamide N-doped high quantum yield graphene quantum dots. Nanoscale 2014, 6, 3868-74.
16. Zhu, H.; Wang, X.; Li, Y.; Wang, Z.; Yang, F.; Yang, X. Microwave synthesis of fluorescent carbon nanoparticles with electrochemiluminescence properties. Chem. Commun. 2009, 5118-20.
17. Yan, X.; Cui, X.; Li, L. S. Synthesis of large, stable colloidal graphene quantum dots with tunable size. J. Am. Chem. Soc. 2010, 132, 5944-5.
18. Chae, A.; Choi, Y.; Jo, S.; et al. Microwave-assisted synthesis of fluorescent carbon quantum dots from an A2/B3 monomer set. RSC. Adv. 2017, 7, 12663-9.
19. Shi, W.; Han, Q.; Wu, J.; et al. Synthesis mechanisms, structural models, and photothermal therapy applications of top-down carbon dots from carbon powder, graphite, graphene, and carbon nanotubes. Int. J. Mol. Sci. 2022, 23, 1456.
20. Yan, F.; Jiang, Y.; Sun, X.; Bai, Z.; Zhang, Y.; Zhou, X. Surface modification and chemical functionalization of carbon dots: a review. Mikrochim. Acta. 2018, 185, 424.
21. Wang, B.; Yu, J.; Sui, L.; et al. Rational design of multi-color-emissive carbon dots in a single reaction system by hydrothermal. Adv. Sci. 2020, 8, 2001453.
22. Zhu, S.; Song, Y.; Wang, J.; et al. Photoluminescence mechanism in graphene quantum dots: quantum confinement effect and surface/edge state. Nano. Today. 2017, 13, 10-4.
23. Dhyani, M.; Kumar, R. An intelligent Chatbot using deep learning with Bidirectional RNN and attention model. Mater. Today. Proc. 2021, 34, 817-24.
24. Xu, X.; Deng, J.; Cummins, N.; Zhang, Z.; Zhao, L.; Schuller, B. W. Exploring zero-shot emotion recognition in speech using semantic-embedding prototypes. IEEE. Trans. Multimedia. 2021, 24, 2752-65.
25. Ouni, A.; Royer, E.; Chevaldonné, M.; Dhome, M. Leveraging semantic segmentation for hybrid image retrieval methods. Neural. Comput. Appl. 2022, 34, 21519-37.
26. Birchler, C.; Khatiri, S.; Bosshard, B.; Gambi, A.; Panichella, S. Machine learning-based test selection for simulation-based testing of self-driving cars software. Empirical. Software. Eng. 2023, 28, 71.
27. Agrawal, A.; Choudhary, A. Perspective: Materials informatics and big data: realization of the “fourth paradigm” of science in materials science. APL. Mater. 2016, 4, 053208.
28. Wahl, C. B.; Aykol, M.; Swisher, J. H.; Montoya, J. H.; Suram, S. K.; Mirkin, C. A. Machine learning-accelerated design and synthesis of polyelemental heterostructures. Sci. Adv. 2021, 7, eabj5505.
29. Dai, Y.; Zhang, Z.; Wang, D.; et al. Machine-learning-driven G-quartet-based circularly polarized luminescence materials. Adv. Mater. 2024, 36, e2310455.
30. Li, Y.; Zhu, R.; Wang, Y.; Feng, L.; Liu, Y. Center-environment deep transfer machine learning across crystal structures: from spinel oxides to perovskite oxides. npj. Comput. Mater. 2023, 9, 109.
31. Chen, Z.; Liu, Y.; Kang, Z. Diversity and tailorability of photoelectrochemical properties of carbon dots. Acc. Chem. Res. 2022, 55, 3110-24.
32. Kakhki, R. M.; Mohammadpoor, M. Machine learning-driven approaches for synthesizing carbon dots and their applications in photoelectrochemical sensors. Inorg. Chem. Commun. 2024, 159, 111859.
33. Duman, A. N.; Jalilov, A. S. Machine learning for carbon dot synthesis and applications. Mater. Adv. 2024, 5, 7097-112.
34. Tang, Y.; Xu, Q.; Zhu, P.; Zhu, R.; Wang, J. Utilizing machine learning to expedite the fabrication and biological application of carbon dots. Mater. Adv. 2023, 4, 5974-97.
35. Brust, M.; Kiely, C. J. Some recent advances in nanostructure preparation from gold and silver particles: a short topical review. Colloids. Surf. A. 2002, 202, 175-86.
36. Allahyari, M.; Pouriyeh, S.; Assefi, M.; et al. A brief survey of text mining: classification, clustering and extraction techniques. arXiv 2017, arXiv:1707.02919. Available online: https://doi.org/10.48550/arXiv.1707.02919. (accessed 27 Feb 2026).
37. Shahriari, B.; Swersky, K.; Wang, Z.; Adams, R. P.; De Freitas, N. Taking the human out of the loop: a review of Bayesian optimization. Proc. IEEE. 2016, 104, 148-75.
38. Zhang, R.; Zhang, J.; Chen, Q.; et al. A literature-mining method of integrating text and table extraction for materials science publications. Comput. Mater. Sci. 2023, 230, 112441.
39. Zhang, Y.; Xiao, G. Named entity recognition datasets: a classification framework. Int. J. Comput. Intell. Syst. 2024, 17, 71.
40. Veena, G.; Hemanth, R.; Hareesh, J. Relation extraction in clinical text using NLP based regular expressions. In 2019 2nd International Conference on Intelligent Computing, Instrumentation and Control Technologies (ICICICT), Kannur, India, July 05-06, 2019; IEEE; 2019. pp. 1278-82.
41. Abdelrazek, A.; Eid, Y.; Gawish, E.; Medhat, W.; Hassan, A. Topic modeling algorithms and applications: a survey. Inf. Syst. 2023, 112, 102131.
42. Segall, M. D.; Lindan, P. J. D.; Probert, M. J.; et al. First-principles simulation: ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter. 2002, 14, 2717-44.
43. Pribram-Jones, A.; Gross, D. A.; Burke, K. DFT: a theory full of holes? Annu. Rev. Phys. Chem. 2015, 66, 283-304.
44. Gale, J. D. Semi-empirical methods as a tool in solid-state chemistry. Faraday. Discuss. 1997, 106, 219-32.
45. Parker, R. M.; Guidetti, G.; Williams, C. A.; et al. The self-assembly of cellulose nanocrystals: hierarchical design of visual appearance. Adv. Mater. 2018, 30, e1704477.
46. Hachmann, J.; Olivares-Amaya, R.; Atahan-Evrenk, S.; et al. The Harvard Clean Energy Project: large-scale computational screening and design of organic photovoltaics on the world community grid. J. Phys. Chem. Lett. 2011, 2, 2241-51.
47. Kirklin, S.; Saal, J. E.; Meredig, B.; et al. The open quantum materials database (OQMD): assessing the accuracy of DFT formation energies. npj. Comput. Mater. 2015, 1, 15010.
48. Jain, A.; Ong, S. P.; Hautier, G.; et al. Commentary: The materials Project: a materials genome approach to accelerating materials innovation. APL. Mater. 2013, 1, 011002.
49. Zhang, K.; Zhang, H. Predicting solute descriptors for organic chemicals by a deep neural network (DNN) using basic chemical structures and a surrogate metric. Environ. Sci. Technol. 2022, 56, 2054-64.
50. Tetko, I. V.; Gasteiger, J.; Todeschini, R.; et al. Virtual computational chemistry laboratory - design and description. J. Comput. Aided. Mol. Des. 2005, 19, 453-63.
51. Beckner, W.; Mao, C. M.; Pfaendtner, J. Statistical models are able to predict ionic liquid viscosity across a wide range of chemical functionalities and experimental conditions. Mol. Syst. Des. Eng. 2018, 3, 253-63.
52. Ramprasad, R.; Batra, R.; Pilania, G.; Mannodi-Kanakkithodi, A.; Kim, C. Machine learning in materials informatics: recent applications and prospects. npj. Comput. Mater. 2017, 3, 54.
53. Sun, Y.; Wong, A. K. C.; Kamel, M. S. Classification of imbalanced data: a review. Int. J. Pattern. Recognit. Artif. Intell. 2009, 23, 687-719.
54. Wang, L.; Han, M.; Li, X.; Zhang, N.; Cheng, H. Review of classification methods on unbalanced data sets. IEEE. Access. 2021, 9, 64606-28.
55. Sun, Z.; Ying, W.; Zhang, W.; Gong, S. Undersampling method based on minority class density for imbalanced data. Expert. Syst. Appl. 2024, 249, 123328.
56. Chawla, N. V.; Bowyer, K. W.; Hall, L. O.; Kegelmeyer, W. P. SMOTE: synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321-57.
57. Li, D. C.; Liu, C. W.; Hu, S. C. A learning method for the class imbalance problem with medical data sets. Comput. Biol. Med. 2010, 40, 509-18.
58. Chen, A.; Cai, J.; Wang, Z.; Han, Y.; Ye, S.; Li, J. An ensemble learning classifier to discover arsenene catalysts with implanted heteroatoms for hydrogen evolution reaction. J. Energy. Chem. 2023, 78, 268-76.
59. Xu, P.; Ji, X.; Li, M.; Lu, W. Small data machine learning in materials science. npj. Comput. Mater. 2023, 9, 42.
60. Ghazikhani, A.; Monsefi, R.; Yazdi, H. S. Online cost-sensitive neural network classifiers for non-stationary and imbalanced data streams. Neural. Comput. Appl. 2013, 23, 1283-95.
61. Wang, J.; Lu, S.; Wang, S. H.; Zhang, Y. D. A review on extreme learning machine. Multimedia. Tools. Appl. 2022, 81, 41611-60.
62. Ganin, Y.; Ustinova, E.; Ajakan, H.; et al. Domain-adversarial training of neural networks. In Domain Adaptation in Computer Vision Applications, Springer, 2017; pp. 189-209.
63. Yu, F.; Xiu, X.; Li, Y. A survey on deep transfer learning and beyond. Mathematics 2022, 10, 3619.
64. Liu, Z.; Wu, C. T.; Koishi, M. Transfer learning of deep material network for seamless structure - property predictions. Comput. Mech. 2019, 64, 451-65.
65. Liu, X.; Liu, Z.; Wang, G.; Cai, Z.; Zhang, H. Ensemble transfer learning algorithm. IEEE. Access. 2018, 6, 2389-96.
66. Liu, X.; Zhang, Y.; Xie, Y.; et al. Design of circularly polarized phosphorescence materials guided by transfer learning. Nat. Commun. 2025, 16, 4970.
67. Settles, B. Active learning literature survey. University of Wisconsin-Madison Department of Computer Sciences, 2009. http://digital.library.wisc.edu/1793/60660. (accessed 2026-02-27).
68. Doolittle, P.; Wojdak, K.; Walters, A. Defining active learning: a restricted systemic review. Teach. Learn. Inq. 2023, 11.
69. Koizumi, A.; Deffrennes, G.; Terayama, K.; Tamura, R. Performance of uncertainty-based active learning for efficient approximation of black-box functions in materials science. Sci. Rep. 2024, 14, 27019.
70. Tian, Y.; Xue, D.; Yuan, R.; et al. Efficient estimation of material property curves and surfaces via active learning. Phys. Rev. Mater. 2021, 5, 013802.
71. Allotey, J.; Butler, K. T.; Thiyagalingam, J. Entropy-based active learning of graph neural network surrogate models for materials properties. J. Chem. Phys. 2021, 155, 174116.
72. He, T.; Zhang, S.; Xin, J.; et al. An active learning approach with uncertainty, representativeness, and diversity. ScientificWorldJournal 2014, 2014, 827586.
73. Noh, J.; Doan, H. A.; Job, H.; et al. An integrated high-throughput robotic platform and active learning approach for accelerated discovery of optimal electrolyte formulations. Nat. Commun. 2024, 15, 2757.
74. Wang, X. Y.; Chen, B. B.; Zhang, J.; et al. Exploiting deep learning for predictable carbon dot design. Chem. Commun. 2021, 57, 532-5.
75. Hong, Q.; Wang, X. Y.; Gao, Y. T.; et al. Customized carbon dots with predictable optical properties synthesized at room temperature guided by machine learning. Chem. Mater. 2022, 34, 998-1009.
76. Xu, Q.; Tang, Y.; Zhu, P.; et al. Machine learning guided microwave-assisted quantum dot synthesis and an indication of residual H2O2 in human teeth. Nanoscale 2022, 14, 13771-8.
77. Tang, B.; Lu, Y.; Zhou, J.; et al. Machine learning-guided synthesis of advanced inorganic materials. Mater. Today. 2020, 41, 72-80.
78. Han, Y.; Tang, B.; Wang, L.; et al. Machine-learning-driven synthesis of carbon dots with enhanced quantum yields. ACS. Nano. 2020, 14, 14761-8.
79. Lan, Y.; Zheng, G. S.; Song, R. W.; et al. Low-temperature molten-salt enabled synthesis of highly-efficient solid-state emitting carbon dots optimized using machine learning. Nat. Commun. 2025, 16, 8167.
80. Zhang, Q.; Tao, Y.; Tang, B.; et al. Graphene quantum dots with improved fluorescence activity via machine learning: implications for fluorescence monitoring. ACS. Appl. Nano. Mater. 2022, 5, 2728-37.
81. Xing, C.; Chen, G.; Zhu, X.; et al. Synthesis of carbon dots with predictable photoluminescence by the aid of machine learning. Nano. Res. 2024, 17, 1984-9.
82. Chen, J.; Luo, J. B.; Hu, M. Y.; Zhou, J.; Huang, C. Z.; Liu, H. Controlled synthesis of multicolor carbon dots assisted by machine learning. Adv. Funct. Mater. 2023, 33, 2210095.
83. Senanayake, R. D.; Yao, X.; Froehlich, C. E.; et al. Machine learning-assisted carbon dot synthesis: prediction of emission color and wavelength. J. Chem. Inf. Model. 2022, 62, 5918-28.
84. Muyassiroh, D. A. M.; Permatasari, F. A.; Hirano, T.; Ogi, T.; Iskandar, F. Machine learning-guided synthesis of room-temperature phosphorescent carbon dots for enhanced phosphorescence lifetime and information encryption. ACS. Appl. Nano. Mater. 2024, 7, 5465-75.
85. Guo, R.; Song, S. Y.; Cao, Q.; et al. Machine learning-driven achieving efficient phosphorescent carbon nanodots in aqueous solution by suppressing triplet electron leakage. Adv. Mater. 2025, 37, e2505925.
86. Luo, J. B.; Chen, J.; Liu, H.; Huang, C. Z.; Zhou, J. High-efficiency synthesis of red carbon dots using machine learning. Chem. Commun. 2022, 58, 9014-7.
87. Tuchin, V. S.; Stepanidenko, E. A.; Vedernikova, A. A.; et al. Optical properties prediction for red and near-infrared emitting carbon dots using machine learning. Small 2024, 20, e2310402.
88. Chen, J.; Zhang, M.; Xu, Z.; Ma, R.; Shi, Q. Machine-learning analysis to predict the fluorescence quantum yield of carbon quantum dots in biochar. Sci. Total. Environ. 2023, 896, 165136.
89. Pudza, M. Y.; Abidin, Z. Z.; Rashid, S. A.; Yasin, F. M.; Noor, A. S. M.; Issa, M. A. Sustainable synthesis processes for carbon dots through response surface methodology and artificial neural network. Processes 2019, 7, 704.
90. Yang, H.; Ran, Z.; Luo, Y.; et al. Exploration and design of carbon dot-based long afterglow materials using active machine learning and quantum chemical simulations. ACS. Nano. 2024, 18, 29203-13.
91. Guo, H.; Lu, Y.; Lei, Z.; et al. Machine learning-guided realization of full-color high-quantum-yield carbon quantum dots. Nat. Commun. 2024, 15, 4843.
92. Li, T.; Cao, B.; Su, T.; et al. Machine learning-engineered nanozyme system for synergistic anti-tumor ferroptosis/apoptosis therapy. Small 2025, 21, e2408750.
93. He, H.; Shuang, E.; Ai, L.; et al. Exploiting machine learning for controlled synthesis of carbon dots-based corrosion inhibitors. J. Clean. Prod. 2023, 419, 138210.
94. Wang, X.; Wang, B.; Wang, H.; et al. Carbon-dot-based white-light-emitting diodes with adjustable correlated color temperature guided by machine learning. Angew. Chem. Int. Ed. 2021, 60, 12585-90.
95. Wang, X.; Bian, W.; Zhang, T.; et al. Highly crystalline core dominated the catalytic performance of carbon dot for cyclohexane to adipic acid reaction. Nano. Res. 2022, 15, 7662-9.
96. Wang, X.; Chen, S.; Ma, Y.; et al. Continuous homogeneous catalytic oxidation of C-H bonds by metal-free carbon dots with a poly(ascorbic acid) structure. ACS. Appl. Mater. Interfaces. 2022, 14, 26682-9.
97. Ostadhossein, F.; Moitra, P.; Alafeef, M.; et al. Ensemble and single-particle level fluorescent fine-tuning of carbon dots via positional changes of amines toward “supervised” oral microbiome sensing. J. Biomed. Opt. 2023, 28, 082807.
98. Xu, Z.; Wang, Z.; Liu, M.; Yan, B.; Ren, X.; Gao, Z. Machine learning assisted dual-channel carbon quantum dots-based fluorescence sensor array for detection of tetracyclines. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2020, 232, 118147.
99. Pandit, S.; Banerjee, T.; Srivastava, I.; Nie, S.; Pan, D. Machine learning-assisted array-based biomolecular sensing using surface-functionalized carbon dots. ACS. Sens. 2019, 4, 2730-7.
100. Soares, A. C.; Soares, J. C.; Dos Santos, D. M.; et al. Nanoarchitectonic E-tongue of electrospun zein/curcumin carbon dots for detecting Staphylococcus aureusin milk. ACS. Omega. 2023, 8, 13721-32.
101. Shauloff, N.; Morag, A.; Yaniv, K.; et al. Sniffing bacteria with a carbon-dot artificial nose. Nanomicro. Lett. 2021, 13, 112.
102. Xu, Z.; Wang, K.; Zhang, M.; et al. Machine learning assisted dual-emission fluorescence/colorimetric sensor array detection of multiple antibiotics under stepwise prediction strategy. Sens. Actuators. B. 2022, 359, 131590.
103. Liu, Y.; Chen, J.; Xu, Z.; et al. Detection of multiple metal ions in water with a fluorescence sensor based on carbon quantum dots assisted by stepwise prediction and machine learning. Environ. Chem. Lett. 2022, 20, 3415-20.
104. Xu, Z.; Chen, J.; Liu, Y.; Wang, X.; Shi, Q. Multi-emission fluorescent sensor array based on carbon dots and lanthanide for detection of heavy metal ions under stepwise prediction strategy. Chem. Eng. J. 2022, 441, 135690.
105. Zhang, Y.; Liu, K.; Yu, J.; et al. Single stain hyperspectral imaging for accurate fungal pathogens identification and quantification. Nano. Res. 2022, 15, 6399-406.
106. Cao, S.; Dong, S.; Chen, Y.; et al. Rapid fluorescence detection of black tea fermentation degree based on cobalt ion mediated carbon quantum dots. Food. Control. 2024, 165, 110610.
107. Döring, A.; Rogach, A. L. Utilizing deep learning to enhance optical sensing of ethanol content based on luminescent carbon dots. ACS. Appl. Nano. Mater. 2022, 5, 11208-18.
108. Tuccitto, N.; Fichera, L.; Ruffino, R.; et al. Carbon quantum dots as fluorescence nanochemosensors for selective detection of amino acids. ACS. Appl. Nano. Mater. 2021, 4, 6250-6.
109. Sarmanova, O. E.; Kudryashov, A. D.; Laptinskiy, K. A.; et al. Applications of fluorescence spectroscopy and machine learning methods for monitoring of elimination of carbon nanoagents from the body. Opt. Mem. Neural. Netw. 2023, 32, 20-33.
110. Liu, S.; Zhang, J.; Liu, X.; et al. Excitation wavelength as additional dimension in cross-reactive sensor arrays. Sens. Actuators. B. 2021, 344, 130183.
111. Döring, A.; Qiu, Y.; Rogach, A. L. Improving the accuracy of carbon dot temperature sensing using multi-dimensional machine learning. ACS. Appl. Nano. Mater. 2024, 7, 2258-69.
112. Zheng, Y.; Wang, X.; Guan, Z.; et al. Application of CD and Eu3+ dual emission MOF colorimetric fluorescent probe based on neural network in Fe3+ detection. Part. Part. Syst. Charact. 2022, 39, 2200124.
113. Zhang, M.; He, H.; Huang, Y.; et al. Machine learning integrated high quantum yield blue light carbon dots for real-time and on-site detection of Cr(VI) in groundwater and drinking water. Sci. Total. Environ. 2023, 904, 166822.
114. Yadav, N.; Mudgal, D.; Mishra, A.; Shukla, S.; Malik, T.; Mishra, V. Harnessing fluorescent carbon quantum dots from natural resource for advancing sweat latent fingerprint recognition with machine learning algorithms for enhanced human identification. PLoS. One. 2024, 19, e0296270.
115. Huang, R.; Zhou, Y.; Hu, J.; Peng, A.; Hu, W. Deep learning-assisted multicolor fluorescent probes for image and spectral dual-modal identification of illicit drugs. Sens. Actuators. B. 2023, 394, 134348.
116. Liu, T.; Chen, S.; Ruan, K.; et al. A handheld multifunctional smartphone platform integrated with 3D printing portable device: on-site evaluation for glutathione and azodicarbonamide with machine learning. J. Hazard. Mater. 2022, 426, 128091.
117. Lu, Z.; Chen, M.; Li, M.; et al. Smartphone-integrated multi-color ratiometric fluorescence portable optical device based on deep learning for visual monitoring of Cu2+ and thiram. Chem. Eng. J. 2022, 439, 135686.
118. Lu, Z.; Li, M.; Chen, M.; et al. Deep learning-assisted smartphone-based portable and visual ratiometric fluorescence device integrated intelligent gel label for agro-food freshness detection. Food. Chem. 2023, 413, 135640.
119. Lu, Z.; Chen, S.; Chen, M.; et al. Trichromatic ratiometric fluorescent sensor based on machine learning and smartphone for visual and portable monitoring of tetracycline antibiotics. Chem. Eng. J. 2023, 454, 140492.
120. Lu, Z.; Chen, M.; Liu, T.; et al. Machine learning system to monitor Hg2+ and sulfide using a polychromatic fluorescence-colorimetric paper sensor. ACS. Appl. Mater. Interfaces. 2023, 15, 9800-12.
121. Wang, F.; Xiao, M.; Qi, J.; Zhu, L. Paper-based fluorescence sensor array with functionalized carbon quantum dots for bacterial discrimination using a machine learning algorithm. Anal. Bioanal. Chem. 2024, 416, 3139-48.
122. Thonghlueng, J.; Ngernpimai, S.; Chuaephon, A.; et al. Dual-responsive carbon quantum dots for the simultaneous detection of cytosine and 5-methylcytosine interpreted by a machine learning-assisted smartphone. ACS. Appl. Mater. Interfaces. 2023, 15, 40141-52.
123. Yen, Y. T.; Lin, Y. S.; Chang, Y. J.; Li, M. T.; Chyueh, S. C.; Chang, H. T. Nanomaterial-based sensor arrays with deep learning for screening of illicit drugs. Adv. Mater. Technol. 2022, 7, 2200243.
124. Doğan, V.; Evliya, M.; Nesrin Kahyaoglu, L.; Kılıç, V. On-site colorimetric food spoilage monitoring with smartphone embedded machine learning. Talanta 2024, 266, 125021.
125. Yu, J.; Yong, X.; Tang, Z.; Yang, B.; Lu, S. Theoretical understanding of structure-property relationships in luminescence of carbon dots. J. Phys. Chem. Lett. 2021, 12, 7671-87.
126. Salahinejad, M.; Sadjadi, S.; Abdouss, M. Investigating fluorescence quenching of cysteine-functionalized carbon quantum dots by heavy metal ions: experimental and QSPR studies. J. Mol. Liq. 2021, 334, 116067.
127. Roozbahani, A.; Salahinejad, M.; Gholipour, V. An exploratory in N-doped carbon dots as green fluorescence probes for Hg(II) ions detection. Environ. Technol. 2024, 45, 3612-20.
128. Bian, Z.; Bao, T.; Sun, X.; et al. Machine learning tools to assist the synthesis of antibacterial carbon dots. Int. J. Nanomedicine. 2024, 19, 5213-26.
129. Li, Y.; Chen, L.; Yang, S.; et al. Symmetry-triggered tunable phosphorescence lifetime of graphene quantum dots in a solid state. Adv. Mater. 2024, 36, e2313639.
130. Chen, L.; Yang, S.; Li, Y.; et al. Precursor symmetry triggered modulation of fluorescence quantum yield in graphene quantum dots. Adv. Funct. Mater. 2024, 34, 2401246.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].