

Rational design of single-atom catalysts for efficient hydrogenation of nitro compounds
 0
0 Abstract
The catalytic reduction of nitro compounds to amines is crucial in the fine and bulk chemical industries. Single-atom catalysts (SACs), featuring high atomic utilization and unique unsaturated coordination structures, hold significant promise in this field and have garnered considerable attention in recent years. Despite notable advancements, their performance remains insufficient for industrial applications. Therefore, it is imperative to develop strategies to enhance their catalytic performance, particularly in terms of activity. This review comprehensively summarizes recent progress in the application of SACs for the hydrogenation of nitro compounds. Firstly, the synthesis and characterization of SACs are briefly discussed. Secondly, strategies to enhance the catalytic activity of SACs are highlighted, including the design of single-atom sites. This involves optimizing the metal center and its microenvironment to improve intrinsic activity, as well as increasing the loading and utilization efficiency of single-atom sites to enhance apparent activity. Key insights from the reviewed works are summarized. Third, the hydrogenation mechanism on some SACs is briefly discussed. Finally, the challenges and prospects of SAC applications in the hydrogenation of nitro compounds are discussed.
Keywords
INTRODUCTION
Aromatic amines and their derivatives are important chemical intermediates widely explored in synthesizing pesticides, pharmaceuticals, dyes, and pigments, thus holding great significance in the organic chemical industry. Reducing nitro compounds is a crucial method for synthesizing amines and their derivatives[1-4]. Traditionally, such a reduction process mainly relied on stoichiometric reducing agents, such as iron powder/HCl and sodium sulfide[5]. However, these traditional processes impose considerable environmental burdens and generate undesirable byproducts, which conflict with the principles of sustainable development.
Catalytic hydrogenation with molecular hydrogen is the principal method for reducing nitro compounds to aromatic amines and their derivatives. Catalysts are essential in this transformation because they provide active sites that optimize the reaction pathways, thereby reducing the activation energy required for the reduction of the nitro group. This enables the reaction to proceed under milder conditions. The conversion of nitro (-NO2) to amine (-NH2) groups proceeds via multiple intermediates, such as nitroso (-NO) and hydroxylamine (-NHOH) species. Additionally, when nitro compounds contain other functional groups on the aromatic ring (e.g., halogens, C=C, or C=O bonds), these may undergo competitive hydrogenation, leading to undesired saturated products. As such, catalysts enhance selectivity by preferentially facilitating the reduction of nitro groups to amino groups[6]. Consequently, developing high-performance catalysts that minimize energy consumption while maintaining selectivity and efficiency remains a central objective in both academic research and industrial practice.
Heterogeneous catalysts are the predominant catalytic systems for industrial hydrogenation of nitro compounds. These catalysts are classified as noble metal or non-noble metal-based, depending on the metal center. Noble metal catalysts, such as Pt/C and Pd/C, exhibit high catalytic activity and are widely utilized for the hydrogenation of nitro compounds; however, their high cost and limited sustainability remain significant drawbacks. Moreover, their selectivity is often inadequate for substrates containing halogens (-F, -Cl, -Br, -I) or unsaturated groups due to competing side reactions such as hydro dehalogenation and over-hydrogenation. In contrast, the supported non-noble metal catalysts generally possess low cost and high selectivity but limited activity. Additionally, traditional non-noble metal catalysts typically exist as nanoparticles (NPs), resulting in the inefficient utilization of metal atoms. To address the challenges faced by catalysts in practical applications, strategies that enhance the catalytic performance of supported heterogeneous catalysts are highly sought after[7-10].
Since Qiao et al. proposed the concept of single-atom catalysis, single-atom catalysts (SACs) have garnered significant attention across various fields[11]. When the size of the metal center is reduced to a single atom, the active metal is endowed with a unique geometry and electronic structure, affording the catalysts with behavior that markedly differs from that of the nanosized metal center[12]. SACs offer a promising avenue for improving the catalytic performance of supported catalysts in the catalytic hydrogenation of nitro compounds. On single-atom sites, hydrogen dissociates via a heterolytic manner, producing polar active hydrogen pairs (Hδ+ and Hδ-). These hydrogen pairs favor the preferential reduction of the polar group (e.g., -NO2), leading to enhanced catalytic selectivity[13]. The well-defined structure of SACs facilitates the investigation of reaction processes and intrinsic mechanisms in the hydrogenation of nitro compounds, providing insights for designing high-performance hydrogenation catalysts[14]. The major discoveries in the application of SACs for the hydrogenation of nitro compounds are illustrated in Figure 1.
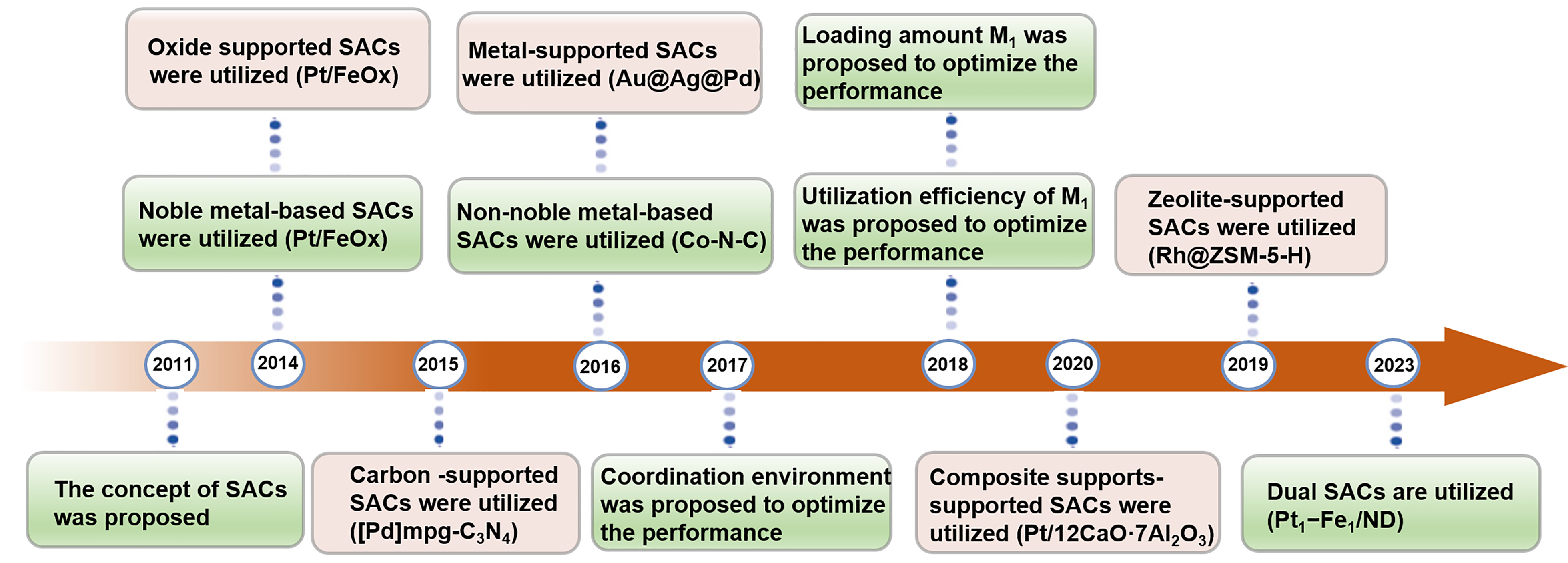
Figure 1. Timeline of major discoveries in SACs applications for nitro compound hydrogenation, 2011-2025; M1 represents the metal single atom. SAC: Single-atom catalyst.
Despite significant advancements in exploring catalysts (SACs) for the catalytic hydrogenation reduction of nitro compounds, their development is still in its infancy, and their catalytic performance is far from satisfactory[15-17]. Furthermore, the catalytic reduction mechanism of SACs remains unclear. Bearing the above-mentioned in mind, a timely review that summarizes the research progress of developing SACs for catalytic hydrogenation of nitro compounds is urgently required in terms of elucidating the hydrogenation reaction mechanisms on SACs and, more importantly, guiding the design of high-performance SACs for nitro compounds hydrogenation reduction, thereby together promoting their industrial applications[18]. While numerous reviews have been conducted on the synthesis, characterization, and application of SACs, there is a notable scarcity of reviews specifically focusing on their use in the catalytic hydrogenation reduction of nitro compounds. Specifically, previous work highlighted the potential of SACs in diverse fields, including photocatalysis, electrocatalysis, and organic transformations. As for their applications in organic transformations, the hydrogenation of nitro compounds was only taken as an example, and only a few examples were discussed. Moreover, the strategies for designing high-performance SACs for nitro compound hydrogenation reactions and the principles underlying their performance improvement have rarely been summarized[19-21].
Herein, we provide a comprehensive review of the recent development of SACs for the catalytic hydrogenation of nitro compounds. We start the review by briefly discussing the synthesis and characterization of SACs. Then, we systematically discussed the strategies proposed for constructing high-performance SACs for the catalytic reduction of nitro compounds, as well as the principles underlying these strategies. Generally, these strategies can be categorized into constructing high-performance single-atom sites and improving the loading amount and utilization efficiency of single-atom sites. In addition, we discussed some specific examples of reaction mechanisms on SACs. Finally, we presented a summary and outlook on the application of SACs in the catalytic hydrogenation of nitro compounds.
SYNTHESIS AND CHARACTERIZATION OF SACS
Synthesis of SACs
The synthesis of supported SACs is essential for investigating their structural, physicochemical, and functional properties. Due to their high surface free energy, single-atom sites are thermodynamically prone to aggregate into NPs with lower surface energy[12]. Consequently, SACs are typically prepared at low metal loadings to minimize the aggregation of metal sites, which limits their apparent catalytic activity. Therefore, the development of synthetic methods capable of producing SACs with well-defined structures and high metal loadings is critical[22,23]. Many synthesis strategies for SACs have been reported, and comprehensive reviews are available on this topic[24-33]. Accordingly, this section will only provide a brief overview of established synthesis methods. Generally, synthesis strategies for SACs are typically classified as bottom-up and top-down based on the metal site precursor. In bottom-up approaches, metal ions serve as precursors, whereas top-down strategies utilize bulk metals or their compounds as the source of metal centers[22]. The bottom-up strategy mainly includes wet chemical methods (e.g., impregnation, chemical deposition methods, such as electrochemical deposition and photochemical deposition), atomic layer deposition (ALD), in situ pyrolysis, and co-precipitation.
Wet chemical methods remain the most widely investigated strategies for loading precursors onto the as-prepared supports. Among them, impregnation is commonly used due to its simplicity and efficiency[34,35]. In this method, the metal precursor is mainly adsorbed onto the pre-treated supports via physical adsorption or weak chemical adsorption. This approach demonstrates excellent compatibility with a range of substrates, especially those with polar surfaces (e.g., metal oxides or heteroatom-doped carbon materials), whose functional groups can effectively anchor metal ions. For example, a Pt SAC supported on hierarchically nitrogen-doped carbon nanocages (Pt1/hNCNC) can be synthesized via an impregnation-adsorption method. Specifically, hNCNC is prepared by pyrolyzing an organic precursor containing pyridine and benzene using MgO as a template[36]. Then, H2PtCl6·6H2O was introduced into the hNCNC suspension (water as solvent) under stirring, finally affording single Pt atoms (Pt1)/hNCNC through the synergistic effect of anion solution adsorption, micropore trapping, and nitrogen-doped carbon anchoring. Notably, no thermal treatment was exerted during the formation of Pt1/hNCNC. Although impregnation applies to various metal centers and supports, it relies on weak interfacial interactions, which may lead to the aggregation of metal atoms into NPs. Furthermore, high-temperature annealing is typically required to eliminate ligands and enhance the interactions between the metal and the support. Nonetheless, elevated temperatures can cause metal migration and aggregation, thereby reducing the number of single-atom sites[37,38]. Therefore, developing suitable support structures and anchoring strategies that enable atomic dispersion of metal centers under mild conditions has become a key research focus in constructing stable SACs via the impregnation method.
As an alternative to impregnation, chemical deposition methods, such as electrochemical deposition and photochemical deposition, are extensively studied for loading metal precursors onto supports to fabricate SACs. In electrochemical deposition, metal ions are reduced to individual atoms at the defect sites of the support (such as carbon vacancies or oxide grain boundaries) under a controlled cathodic potential[39]. For example, using electrochemical deposition with various metal salts as precursors, more than 30 different SACs have been successfully obtained from both cathodic and anodic depositions. Specifically, all 4d and 5d metal species can be deposited and atomically dispersed on Co(OH)2 nanosheets. Interestingly, single atoms of the same metal exhibit distinct electronic states depending on whether they are deposited at the cathode or the anode [Figure 2A][39].
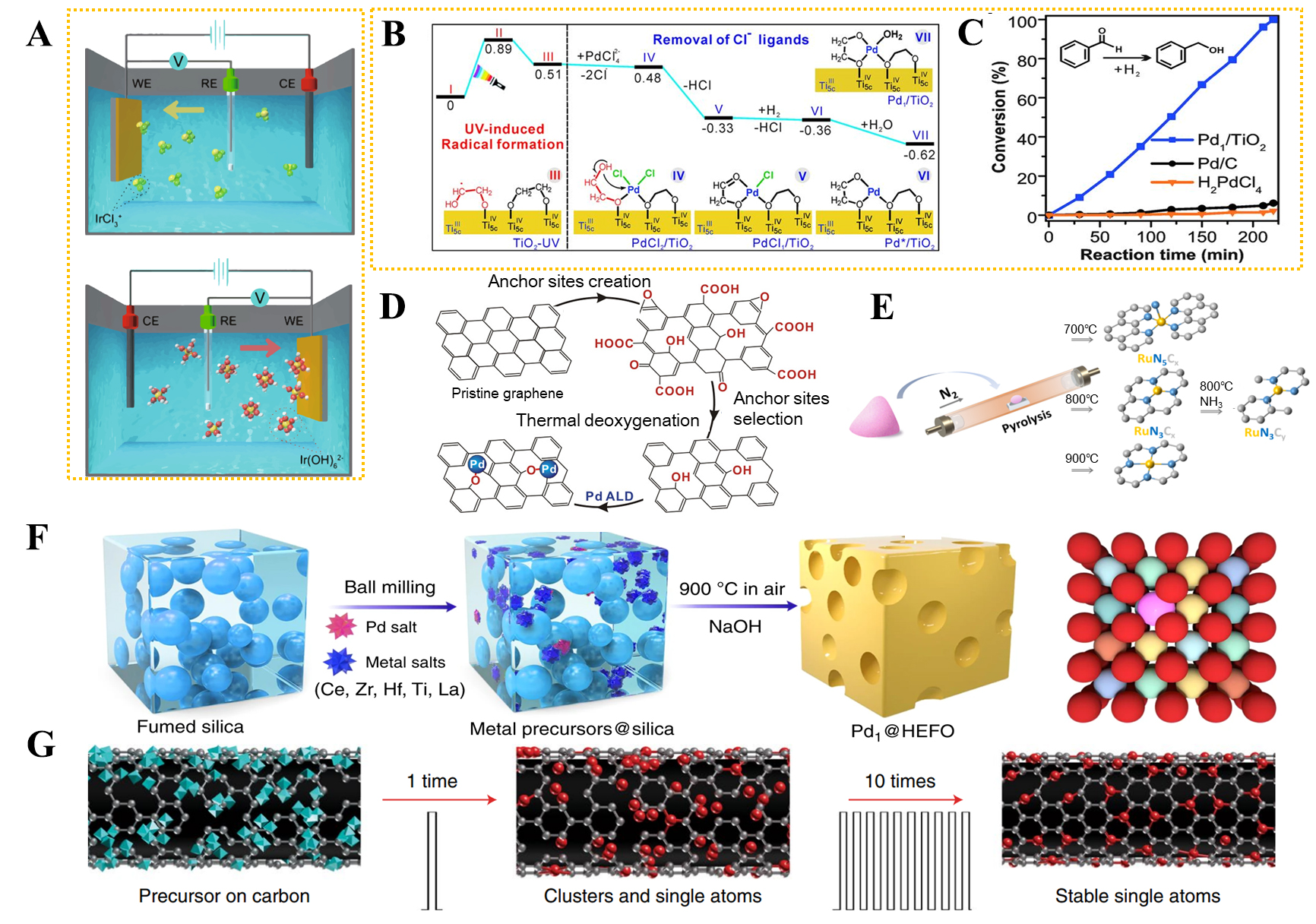
Figure 2. (A) Schematic illustration of cathodic and anodic deposition of Ir species[39]; (B) Energies and models of intermediates and transition states in the stepwise preparation mechanism of Pd1/TiO2[42]; (C) First-run reaction performances for Pd1/TiO2, Pd/C, and
Photodeposition involves irradiating semiconductor particles, typically utilized as supports, suspended in a metal salt solution. The photoexcited electrons from these semiconductor particles can reduce metal ions, promoting their deposition as single atoms on the semiconductor surface[40,41]. Atomically Pd1/TiO2 catalysts can be synthesized via photodeposition on ethylene glycol (EG)-stabilized ultrathin TiO2 nanosheets[42]. Upon ultraviolet irradiation on the TiO2 nanosheets in the presence of EG, EG radicals are generated through hydrogen transfer. These radicals reduce PdCl42- adsorbed on the TiO2 surface, releasing Cl- and forming Pd-O bonds, which stabilize the isolated Pd atoms and suppress NP aggregation [Figure 2B]. At room temperature, Pd1/TiO2 exhibits a turnover frequency (TOF) over 55 times higher than commercial Pd catalysts [Figure 2C].
Besides wet chemical methods, ALD is also widely utilized to introduce precursors onto the as-prepared supports. ALD is a vapor-phase technique based on sequential, self-limiting surface reactions, which enables the precise anchoring of metal species at the atomic scale, making it particularly suitable for synthesizing SACs[43]. Recently, ALD has demonstrated a promising strategy for fabricating SACs[44-46]. For instance, single-atom Pd/graphene catalysts can be prepared via ALD[47]. The process begins with the oxidation of pristine graphene nanosheets, followed by thermal deoxygenation to tailor the surface oxygen-containing functional groups, resulting in phenolic groups as the primary sites. Subsequently, alternating exposures to Pd(hfac)2 and formalin were carried out on the reduced graphene support at 150 °C. After one deposition cycle, the Pd loading amount reached 0.25 wt.%. Pd atoms could be uniformly dispersed on the graphene surface in the form of single metal atoms [Figure 2D].
In the bottom-up strategies for synthesizing SACs, the post-loading method, in situ synthesis has also been extensively studied. Co-precipitation and in situ pyrolysis are the most commonly used synthesis approaches. The co-precipitation strategy controls the interaction between metal ions and the support material by adjusting the pH of the solution, thereby ensuring a uniform distribution of metal ions. In such catalysts, metal ions can be stably embedded into the lattice of the support, forming a strong metal-oxygen framework that provides the necessary active sites for catalytic reactions[48,49]. For example, a Ru1/FeOx SAC can be synthesized using a co-precipitation strategy. This method enables the high dispersion of Ru atoms on the FeOx support, with Ru existing as isolated single atoms[50]. However, co-precipitation may embed metal atoms within the support aggregates or microcrystals, limiting their accessibility to reactants and thus compromising the catalytic performance of the resulting SACs[29].
The in situ pyrolysis method, a typical bottom-up approach, is primarily used for synthesizing carbon-supported SACs. During the synthesis process, precursors will first be prepared by mixing metal salts and organic ligands. After that, pyrolysis will be exerted, producing a carbon framework and atomically dispersed metal sites[51]. For example, a ruthenium (Ru) SAC was synthesized via an in situ pyrolysis strategy. Specifically, precursors composed of a ruthenium acetylacetonate complex and a support precursor were prepared, followed by thermal decomposition under an ammonia atmosphere. Although this method is straightforward, it suffers from drawbacks such as metal aggregation, limited control over the coordination environment of active sites, and low metal utilization efficiency [Figure 2E][52]. Overall, bottom-up synthesis strategies are widely used for the preparation of SACs; however, they are constrained by issues such as low metal loading and tedious post-treatment. Alternatively, top-down approaches could afford enhanced metal loading and dispersion by precisely atomizing metal particles and are gaining significant attention in the fabrication of SACs.
The top-down synthesis strategy mainly relies on external forces (heat, electricity, and light) to convert metal particles on the support into single atoms. These methods can be classified into thermochemical methods, electrochemical methods, and other approaches[22]. The thermal method primarily uses temperature fields to promote the breaking of metal-metal (or metal-nonmetal) bonds in metal particles or compounds, forming new bonds between metal atoms and the support. In thermochemical strategies, confined pyrolysis is widely investigated to achieve the atomic dispersion of metal centers, which involves assembling precursors and pyrolyzing them[53]. Typically, Ni NPs on defect-rich nitrogen-doped carbon could be transformed into isolated Ni atoms via surface thermodiffusion[54]. Under high-temperature conditions, Ni NPs on the carbon surface can help cleave surface C-C bonds, diffuse into the nitrogen-doped matrix, and ultimately anchor at nitrogen-rich defect sites. As a result, the NPs are gradually etched and ultimately converted into atomically dispersed species. As a unique thermochemical method, Joule heating has recently received particular attention in synthesizing SACs. Joule heating can generate temperatures exceeding 2,000 °C within seconds and is traditionally applied in ceramic sintering. A controllable high-temperature shockwave strategy has been developed, in which periodic on-off Joule heating is employed to synthesize and stabilize single atoms at temperatures ranging from 1,500 to 2,000 K. The transient thermal pulses promote metal atom dispersion via the formation of stable metal-defect bonds while preserving substrate integrity. This approach yields thermally stable single atoms with robust catalytic performance, offering a general, rapid, and scalable method applicable to various metals (e.g., Pt, Ru, Co) and supports (e.g., carbon, C3N4, TiO2), thereby addressing key challenges in SAC synthesis [Figure 2F][55].
The electrochemical method offers an environmentally friendly and energy-efficient alternative to the traditional thermal chemical approach. Using a simple electrode system, the electrochemical method enables precise control over the dispersion and stability of metal atoms under ambient conditions. By optimizing electrochemical parameters, such as current density, voltage, scan rate, and reaction time, one can regulate the formation of active sites and their electronic structure, thereby directly influencing catalytic performance. For example, a top-down approach has been proposed to convert metal NPs into uniformly dispersed single atoms via cathodic corrosion under mild electrochemical conditions[56]. In a three-electrode system, Metal NPs/N-doped carbon paper (NCP) functions as the working electrode, a graphite rod as the counter electrode, and an Hg/HgO electrode as the reference electrode. Cathodic corrosion was carried out in 10 M NaOH. Under a high negative potential, Pt NP precursors were converted into mobile Pt1, which were subsequently trapped and stabilized by nitrogen coordination sites on NCP. Such a strategy could be applied to other metals (e.g., Pd, Ir, Cu). However, a major challenge of this electrochemical strategy remains the precise control over the geometric configuration and electronic structure of the resulting SACs.
In addition to thermochemical and electrochemical methods, a series of novel synthesis strategies for SACs have been proposed in recent years[57-59]. Mechanochemical methods, for example, are defined as chemical reactions initiated by mechanical energy, typically without the use of solvents or with minimal solvent usage. Intense mechanical interactions between the grinding media and the precursor mixture (e.g., shear, friction, compression, and impact) promote uniform dispersion and induce significant changes in composition and structure[60-62]. For example, metal salt precursors (e.g., Ce, Zr, Hf, Ti, La, and Pd) were first mixed with fumed silica by ball milling. The resulting mixture was pyrolyzed in the air at 900 °C to obtain silica-templated metal oxide complexes. Finally, the M1(= Pd1)@high-entropy fluorite oxides (HEFO) catalyst was obtained after etching the silica with NaOH [Figure 2G][63].
Characterization of SACs
SACs have emerged as a prominent research focus in the field of catalysis. Understanding the structure and composition of SACs is essential for elucidating structure-property relationships, revealing reaction mechanisms, and guiding catalyst design and synthesis[64]. The characterization of SACs spans multiple scales, from atomic to macroscopic levels. At the atomic scale, insights into the electronic structure, geometric configuration, and coordination environment of the metal centers are crucial. At the nanoscale, it is equally important to assess the morphology, structure, and porosity of the support materials. Therefore, a comprehensive characterization of SACs requires integrating multiple analytical techniques, which are generally categorized into electron microscopy and spectroscopic methods. The former include transmission electron microscopy (TEM) and scanning tunneling microscopy (STM), while the latter include X-ray photoelectron spectroscopy (XPS), X-ray absorption spectroscopy (XAS), and Fourier transform infrared spectroscopy (FTIR), as well as other complementary techniques[65-67]. Additionally, theoretical calculations, such as density functional theory (DFT), have proven to be key methods for analyzing the structures of SACs. Notably, many in situ characterization techniques have recently been developed to reveal the structural evolution of SACs during reactions, providing a powerful strategy for unveiling active sites under working conditions. By integrating the advantages of these characterization techniques, a deeper understanding of the structure-activity relationships of SACs can be elucidated[67].
Electron microscopy techniques are essential tools for characterizing SACs, including TEM, STM, and aberration-corrected high-angle-annular-dark-field scanning transmission electron microscopy (AC-HAADF-STEM). TEM works by allowing an electron beam to traverse a sample, generating images from the scattering and absorption signals produced by the interaction between the electrons and the sample. In the study of SACs, TEM can not only help confirm the absence of NPs but also allow for the observation of the overall morphology of the catalyst[68]. For example, Guo et al. observed by TEM that no NPs were present in Fe@BC with a fibrous structure [Figure 3A][69]. High-resolution TEM (HR-TEM), with its ultra-high resolution, can achieve atomic-level clarity, providing strong support for understanding the material's microstructure[70].
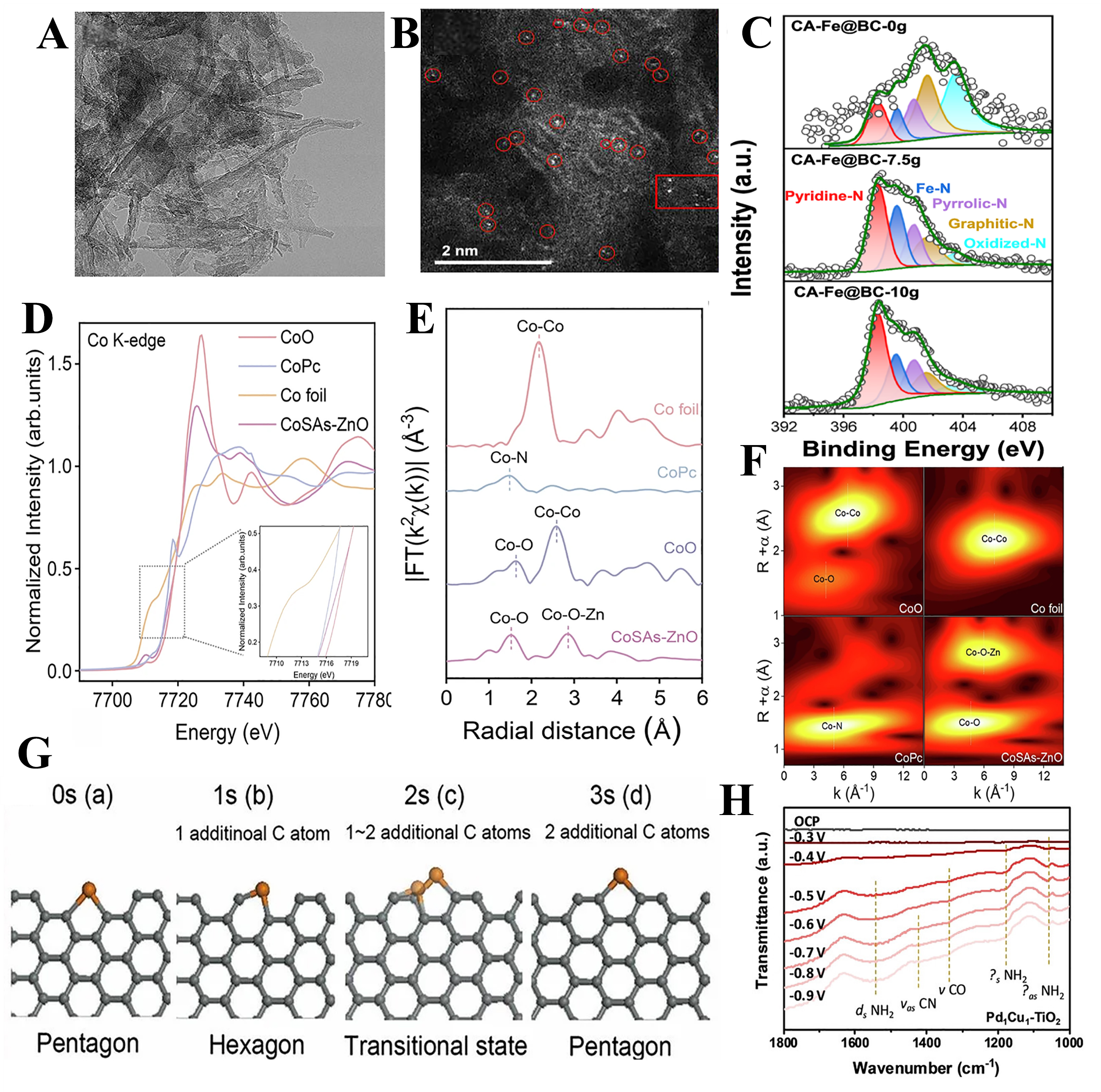
Figure 3. (A) TEM images of Fe@BC[69]; (B) AC-HAADF-STEM images of the Fe@BC catalyst (the red circles are atomic Fe species)[69]; (C) N1s XPS spectra of the CA-Fe@BC-ng catalyst[69]; (D) XANES spectra of CoSAs-ZnO at the Co K-edge (inset: magnification of local areas)[76]; (E) FT k3-weighted EXAFS spectra of CoSAs-ZnO and references[76]; (F) WT-EXAFS for K-edge for Co foil, CoO, CoPc, and CoSAs-ZnO[76]; (G) Schematic illustration for one cycle of catalytic growth of graphene edge[80]; (H) The in situ DRIFTS measurements during urea synthesis on Pd1Cu1-TiO2[82]. TEM: Transmission electron microscopy; AC-HAADF-STEM: aberration-corrected high-angle-annular-dark-field scanning transmission electron microscopy; XPS: X-ray photoelectron spectroscopy; XANES: X-ray absorption near-edge structure; FT: Fourier transform; WT-EXAFS: wavelet transform EXAFS; DRIFTS: diffuse reflectance infrared Fourier transform spectroscopy.
Additionally, STM is commonly employed in the analysis of SACs. STM offers atomic-level resolution, enabling the identification of single-atom sites and their binding sites on the surface of the support. STM allows in situ and dynamic observation of individual atoms under near-reaction conditions, capturing their migration, interactions, and structural changes. High-angle-annular-dark-field scanning transmission electron microscopy (HAADF-STEM) is an extension of STM technology, offering more comprehensive insights into the microscopic structure of materials. When electrons are accelerated at high voltage to interact with the catalyst surface, different interactions occur between the incident electrons and the atoms. Elastically scattered electrons are distributed over a wide scattering angle above the catalyst, as they cannot penetrate the material, whereas inelastically scattered electrons (transmitted electrons) are distributed at small angles. If only wide-angle electrons are detected, a dark-field image is obtained.
When metals are dispersed as individual atoms, traditional characterization techniques may not effectively identify them; this is where AC-HAADF-STEM becomes crucial. AC-HAADF-STEM uses aberration-correction technology to enhance the microscope’s resolution and image quality. Using this technique, researchers can observe individual metal atoms. When combined with energy-dispersive X-ray spectroscopy (EDS) or electron energy loss spectroscopy (EELS), the type of the metal center and its surrounding environment can be distinguished. Such a combination has become the standard configuration for analyzing SACs. For example, Guo et al. directly observed the presence of Fe atoms in the catalyst using AC HAADF-STEM, confirming the existence of isolated Fe atoms [Figure 3B][69]. Meanwhile, EDS elemental mapping revealed that Fe, S, N, and C atoms were uniformly distributed throughout the catalyst[71]. However, these techniques mainly focus on spatial structural information and are limited in revealing the oxidation state, coordination environment, and electronic interactions between metal atoms and the support. To better understand the single-atom sites, it is necessary to complement electron microscopy with spectroscopic techniques[72].
Spectroscopic tools primarily include XPS, XAS, and FTIR. X-ray diffraction (XRD) is a classic structural characterization tool commonly used to confirm the crystal structure of catalyst supports, rather than directly characterizing single-atom metal sites[73].
XPS is a powerful analytical technique for characterizing SACs, not only their support structures but also the chemical and electronic states of metal centers[74]. For example, the N1s spectra can be deconvoluted into pyridinic N (398.3 eV), Fe-N (399.3 eV), pyrrolic N (400.7 eV), graphitic N (401.5 eV), and oxidized N (403.5 eV), respectively. Further analysis reveals that CA-Fe@BC-10g exhibits a higher relative content of pyridinic N (43.03%) and Fe-N (19.61%) [Figure 3C][69].
XAS is a powerful technique for characterizing SACs, enabling precise identification of their electronic structure and local coordination environment. By integrating X-ray absorption near-edge structure (XANES), extended X-ray absorption fine structure (EXAFS), and advanced approaches such as Fourier-transform k2-weighted EXAFS (FT-EXAFS) or wavelet transform EXAFS (WT-EXAFS), a detailed analysis of single-atom centers is achieved. XAS also facilitates the qualitative assessment of the coordination environment of target atoms and, through fitting analysis, provides accurate predictions of bond lengths and coordination numbers (CNs)[75]. For example, to confirm the existence of the Co-O-Zn coordination structure in CoSAs-ZnO, detailed X-ray absorption fine structure (XAFS) analyses were conducted. The Co K-edge XANES spectrum showed that the absorption edge of Co atoms in CoSAs-ZnO closely resembled that of CoO, indicating that the Co atoms predominantly exist in the +2 oxidation state [Figure 3D]. Fourier transform EXAFS (FT-EXAFS) analysis revealed two prominent peaks at 1.5 Å and 2.8 Å, corresponding to Co-O and Co-O-Zn coordination, respectively [Figure 3E]. Notably, the absence of Co-Co peaks in the spectrum indicated the lack of Co clusters in the system, further confirming the atomic dispersion of Co species. Moreover, the WT-EXAFS results exhibited two distinct peaks at 4.7 Å and 5.9 Å, attributed to the Co-O and Co-O-Zn coordination shells, respectively [Figure 3F]. Again, no Co-Co signal was observed, reinforcing the atomically dispersed nature of the Co sites[76].
For certain iron-based SACs, Mössbauer Spectroscopy is a particularly distinctive characterization technique. This method can probe the hyperfine interactions of iron atoms, providing valuable information about the oxidation state, magnetic properties, and local environment of iron atoms[77]. For example, the Mössbauer effect can be employed to identify the types of Fe species. When γ-rays with specific energies irradiate the Fe-N-C catalyst sample, the iron nuclei in the sample can absorb the γ-ray energy and undergo energy-level transitions. Variations in the chemical environments of Fe species could alter the surrounding electron density and magnetic fields at the nuclei, resulting in distinct hyperfine parameters (such as isomer shift and quadrupole splitting) in the Mössbauer spectrum[78].
In addition, DFT is an important theoretical tool for analyzing SACs, providing strong support for catalyst design, performance optimization, and reaction mechanism analysis. It is widely used to study key information such as the electronic structure, surface properties, adsorption behavior, and reaction energy barriers of catalysts. DFT can predict the structural stability of catalysts, the electronic structure of active sites, energy changes during catalytic reactions, and reaction mechanisms[79].
While static characterization methods provide important information about the structure and composition of SACs, they fail to capture the dynamic evolution of catalytic sites during reactions. As such, an increasing number of in-situ characterization techniques were developed, encompassing electron microscopy methods [e.g., in situ TEM, STEM, and atomic force microscopy (AFM)] and spectroscopic techniques [e.g., in situ infrared (IR) spectroscopy, XRD, and XPS]. They can perform real-time observation of structural changes under working conditions, including metal atom migration, aggregation, and reconstruction, thereby advancing the understanding of catalytic mechanisms and supporting the optimization of catalysts. Aberration-corrected in situ TEM was used to directly capture the formation process of Fe atoms under electron irradiation[80]. The typical migration behavior of the single Fe atom along the graphene edge was demonstrated. Initially, the Fe atom was anchored on a pentagonal structure, then it adsorbed nearby carbon atoms and moved to the right. This movement of the single Fe atom formed a shadow line in the image and ultimately reformed a pentagonal structure [Figure 3G]. In single-atom catalytic reactions, in situ IR spectroscopy can monitor the adsorption and dissociation of reactants, the formation of surface intermediates, and the production of final products during the catalytic process. By dynamically tracking changes in IR peaks, the interaction mechanisms between the catalyst and reactants can be revealed[81]. In situ diffuse reflectance IR Fourier transform spectroscopy (DRIFTS) was employed to investigate the key intermediates involved in electrocatalytic urea synthesis[82]. Pd1Cu1-TiO2 were subjected to negative potential scanning from -0.3 V to -0.9 V (vs. RHE), and IR signals were collected in the range of 1,000 to 1,800 cm-1. The IR spectra confirmed that the peak at 1,545 cm-1 corresponds to the N-H bending vibration mode in NH3. In addition, the peaks at 1170 and 1,055 cm-1 were attributed to the N-H stretching and H-N-H stretching vibrations of the amidogen group in the product, confirming the formation of the *NH2CONH intermediate [Figure 3H].
DESIGN STRATEGIES OF SACs FOR EFFICIENT HYDROGENATION OF NITRO COMPOUNDS
Although SACs have demonstrated significant catalytic potential in the hydrogenation of nitro compounds, their catalytic performance, particularly in terms of catalytic activity, has yet to meet the requirements for industrial applications. In this section, we will systematically present the strategies that have been proposed for designing high-performance SACs in nitro compound hydrogenation reactions. These strategies mainly include designing single-atom sites (including metal centers and their microenvironment) to enhance their intrinsic activity and increasing the loading amount and utilization efficiency of single-atom sites to improve the apparent activity. Moreover, we will also explore the mechanisms behind these strategies, explaining how the structure and reaction environment of single-atom sites can be designed to promote hydrogenation reactions. Table 1 summarizes the latest representative research achievements of SACs in the field of nitroarene hydrogenation, along with their reaction systems and performance.
Recent representative research achievements in the field of nitroarene hydrogenation catalyzed by SACs, along with their reaction systems and performance
| Number | Catalyst | Reaction conditions | Loading amount (w%) | Performance | Ref. | ||||||||
| Solvent | PH2 (MPa) | T (°C) | Time (h) | substrate | Dosagea | Dosageb | Con (%) | Sel (%) | |||||
| 1 | Pt/α-MoC | H2O | 2 | 35 | 2 | p-CNB | 1 mmol | 25 mg | 0.25 | 94 | 99.9 | [7] | |
| 2 | Pt/FeOx | Tol | 0.3 | 40 | 1 | 3-NS | 0.5 mmol | / | 0.08 | 88.8 | 91.9 | [88] | |
| 3 | Pt1/Fe2O3 | Tol | 1 | 40 | 0.1 | 3-NS | 0.5 mmol | 10 mg | 0.9 | 97.2 | 96.2 | [89] | |
| 4 | Pt1/Ni | MeOH | 0.3 | 40 | 1.6 | p-CNB | 0.5 mmol | 1 mg | 1 | 99.5 | 98.9 | [152] | |
| 5 | Pt1/h-NC | EtOH | 0.5 | 40 | 10 | 3-NS | 0.5 mmol | / | 1 | 97.1 | 77.8 | [127] | |
| 6 | Pt1/CeO2 | EtOH | 0.1 | 60 | 0.83 | 3-NS | 0.5 mmol | / | 1.1 | 100 | 100 | [181] | |
| 7 | Pt/C | EtOH | 0.1 | 40 | 60 | p-CNB | 0.5 mmol | 40 mg | 0.298 | 99 | 99 | [132] | |
| 8 | Pt/TiO2 | Tol | 0.3 | 40 | 45 | 3-NS | 0.5 mmol | / | 0.77 | 82.9 | 75.8 | [141] | |
| 9 | Pt/CeO2 | EtOH | 1 | 40 | 32 | p-CNB | 0.8 mmol | / | 0.6 | 100 | 100 | [140] | |
| 10 | Pt/C12A7 | MeOH | 0.5 | 60 | 1 | 4-CNB | 0.5 mmol | 5 mg | 0.1 | 100 | 100 | [142] | |
| 11 | Pt/g-C3N4 | CyH | 2 | 50 | 1 | NB | 10.8 mmol | / | 2.5 | 99 | 99 | [137] | |
| 12 | Pt1/N-MoO2 | H2O | 0.3 | 70 | 1.7 | NB | 0.2 mmol | 10 mg | 0.2 | 100 | 100 | [155] | |
| 13 | Pt-PMA/AC | EA | 1 | 50 | 1 | NB | 0.52 mmol | 5 mg | 0.91 | 99 | 99 | [169] | |
| 14 | Pd1/α-MoC | MeOH | 0.5 | 60 | 3 | p-CNB | 0.5 mmol | / | 0.5 | 100 | 100 | [98] | |
| 15 | Pd1/In2O3-x | H2O | 0.1 | 20 | 1 | NB | / | / | 0.34 | 99 | 99 | [143] | |
| 16 | RGO@AC/Pd | H2O | / | 25 | 0.03 | 4-NP | / | / | 0.3 | 99 | 99 | [135] | |
| 17 | CF@Pd1/NHG | / | / | / | 1 | 4-NP | 0.4 mmol | 650 mg | 0.13 | 99.5 | 96.8 | [182] | |
| 18 | Rh@S-1-H | MeOH/H2O | 25 | 1.5 | 11 | p-CNB | 0.1 mmol | / | 0.28 | 100 | 99 | [100] | |
| 19 | Rh@Al2O3@C | Tol | 0.2 | 40 | 2.5 | m-CNB | 0.5 mmol | 30 mg | 0.1 | 100 | 100 | [96] | |
| 20 | Ru-SAs/CeO2 | IPA | 3 | 100 | 8 | NB | 1.7 mmol | 40 mg | 1.6 | 100 | 100 | [97] | |
| 21 | Ag/Al2O3 | H2O | / | 25 | 0.25 | NB | 0.2 mmol | 20 mg | 0.2 | 95 | 100 | [101] | |
| 22 | Pt1-Fe1/ND | CyH | 1 | 80 | 0.33 | p-NB | 0.6 mmol | 5 mg | 1 | 100 | 100 | [16] | |
| 23 | Co-N-C | H2O | / | 145 | 24 | NB | 0.12 mmol | 50 mg | 1.12 | 99 | 99 | [20] | |
| 24 | Co-N-C | t-BuOH | 3 | 80 | 2.5 | NB | / | / | 3.6 | 97 | 99 | [103] | |
| 25 | Co@NX-C | EtOH | 1.5 | 120 | 1.5 | 4-CNB | 1 mmol | 40 mg | 1.1 | 100 | 100 | [109] | |
| 26 | Co1/G | THF/ H2O | / | 25 | 1 | p-NB | 0.1 mmol | / | 0.8 | 99 | 100 | [134] | |
| 27 | CoSAs/NCNS | THF | / | 100 | 2 | p-NB | 0.5 mmol | 5 mg | 4.57 | 99 | 99 | [130] | |
| 28 | CoSAs/NC | H2O | 0.5 | 120 | 2.5 | NB | 0.5 mmol | / | 1.06 | 99 | 99 | [183] | |
| 29 | Co1/NPC | EtOH/H2O | 3 | 110 | 3.5 | NB | 2 mmol | 5 mg | 0.45 | 99 | 99 | [160] | |
| 30 | Co1/NSC-AT | H2O | 0.5 | 85 | 3 | NB | 0.25 mmol | 20 mg | 0.85 | 100 | 100 | [162] | |
| 31 | Co-N-C | EtOH/H2O | 2 | 100 | 2 | p-NB | 0.5 mmol | 10 mg | 0.7 | 99 | 99 | [161] | |
| 32 | Co-N4P1 | EtOH/H2O | 0.5 | 80 | 2.5 | p-NB | 0.5 mmol | 10 mg | 8.93 | 99 | 99 | [165] | |
| 33 | Co-SACs | MeOH | 1 | 70 | 2 | p-CNB | 0.5 mmol | 80 mg | 1.8 | 93.8 | 99 | [166] | |
| 34 | Co@mesoNC | EtOH | 3 | 110 | 2 | NB | 1 mmol | / | 3.5 | 99 | 99 | [175] | |
| 35 | Co1/m-NC | EtOH | 2 | 120 | 0.4 | NB | 2.5 mmol | 120 mg | 1.2 | 99 | 99 | [180] | |
| 36 | Co SSCs@NG | IPA | / | 120 | 2 | NB | 0.5 mmol | 20 mg | 10.26 | 99 | 99 | [170] | |
| 37 | Co-NAC | MeOH/H2O | 2 | 80 | 4.5 | 3-NS | 0.1 mmol | 5 mg | 1.83 | 99 | 99 | [171] | |
| 38 | Co1/HOHC-M | IPA | / | 150 | 4 | NB | 0.25 mmol | / | 2.41 | 100 | 100 | [179] | |
| 39 | Co1/CSC | EtOH /H2O | / | 30 | 0.3 | NB | 0.1 mmol | 2 mg | 0.23 | 96 | 99 | [174] | |
| 40 | NiCo-MOF | MeOH | / | 25 | 0.17 | p-CNB | 2 mmol | 10 mg | Ni:1.7 Co:1 | 99.9 | 99.9 | [184] | |
| 41 | Ni-N-C | MeOH | 3 | 90 | 6 | NB | 0.5 mmol | 20 mg | 7.5 | 100 | 100 | [185] | |
| 42 | h-Ni1N4/NC | IPA | / | 90 | 3.5 | NB | 0.2 mmol | 10 mg | 3.3 | 93.8 | 99 | [111] | |
| 43 | Ni-N-C | MeOH | 3 | 80 | 36 | NS | 0.56 mmol | 20 mg | 2 | 81.3 | 99 | [186] | |
| 44 | Fe1/N-C | EtOH | / | 60 | 2 | 4-CNB | 0.2 mmol | / | 0.24 | 99 | 99 | [116] | |
| 45 | FeSA@NC | EtOH | / | / | 0.5 | NB | 3 mmol | 10 mg | 1 | 99 | 99 | [117] | |
| 46 | FeNC-2 | H2O | / | / | 0.05 | 4-NP | 0.2 mmol | 5 mg | 1.35 | 100 | 100 | [187] | |
| 47 | Fe-N/S-C | H2O | / | 25 | 1 | p-NP | 1 mmol | 2 mg | 0.96 | 100 | 100 | [163] | |
| 48 | Zn-N-C | EtOH | / | 50 | 2.7 | NB | 1 mmol | 20 mg | 6.8 | 100 | 100 | [118] | |
Designing single-atom sites
Design of the metal center
As stated in the Introduction Section, reducing the metal center to the scale of a single atom represents a new strategy for enhancing the hydrogenation efficiency of catalysts. Consequently, increasing attention has been directed toward this area[83]. Notably, single-atom sites typically consist of one type of single metal. Recent findings suggest that incorporating dual-metal centers into SACs can further enhance catalytic performance, underscoring their importance in developing high-performance catalysts for nitro compound reduction.
Monometallic SACs
Precious metal catalysts, such as platinum (Pt), gold (Au), palladium (Pd), and rhodium (Rh), exhibit high activity in the catalytic hydrogenation of nitro compounds; however, they face challenges including high cost, limited availability, and poor selectivity toward the -NO2 group. Reducing metal centers to the single-atom level is an effective strategy to overcome these limitations. SACs can achieve 100% metal-atom utilization, thereby maximizing efficiency while minimizing metal consumption[25]. Wei et al. conducted pioneering work in this area[84]. They synthesized a Pt SAC supported on FeOx (Pt/FeOx) via the co-precipitation method using chloroplatinic acid and ferric nitrate as precursors. The resulting precipitate was calcined to produce Pt/FeOx with an ultralow Pt loading of 0.08 wt.%. Compared to Pt NP catalysts, this SAC exhibited significantly higher activity and selectivity in the hydrogenation of nitro compounds, with a TOF that was up to 14.5 times higher than that of the NP catalyst. This enhancement is attributed to the positively charged Pt atoms and the absence of Pt-Pt metallic bonds, both of which promote preferential adsorption of the nitro group. Further studies revealed that modifying the Pt-O CN can optimize the hydrogenation activity of Pt centers. Specifically, reducing the Pt-O CN lowers the oxidation state of Pt, thereby significantly enhancing catalytic activity while maintaining high selectivity [Figure 4A and B][85].
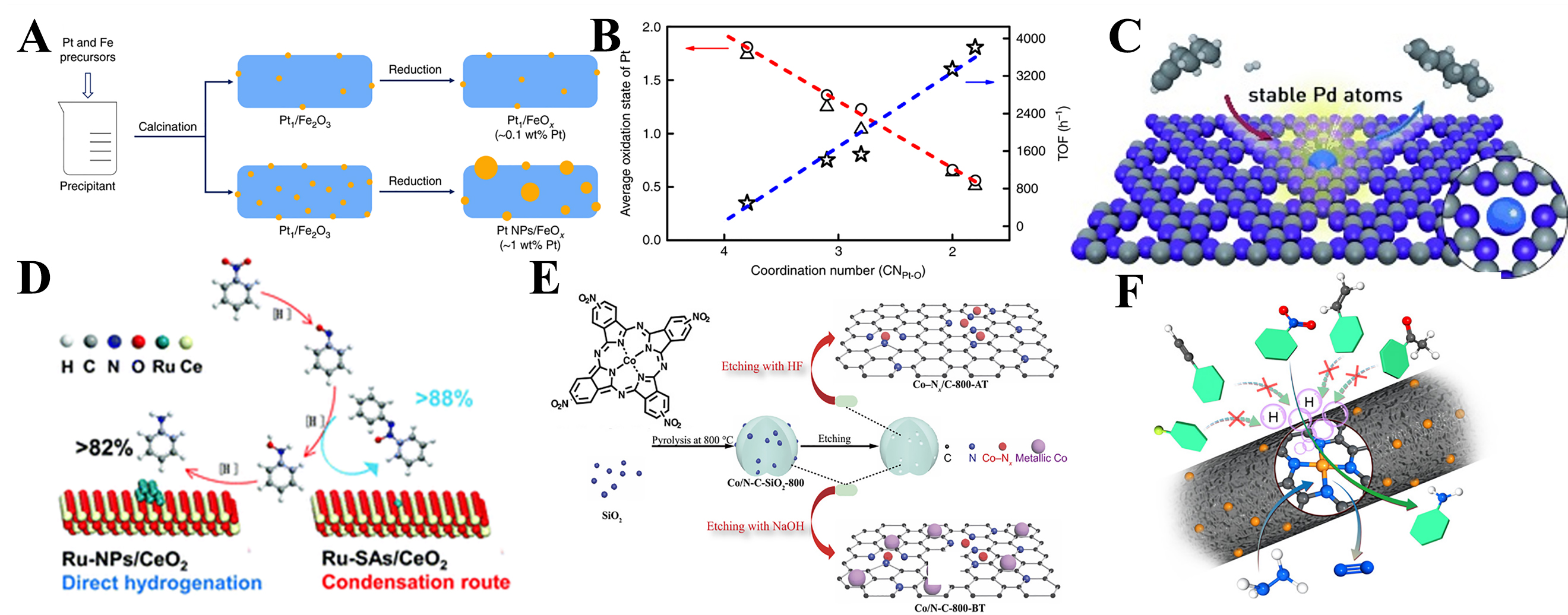
Figure 4. (A) Synthetic procedure of Pt1/FeOx[85]; (B) The correlation between coordination structure and catalytic performance. The star, triangle, and circle represent TOF, the average oxidation state of Pt determined from XPS and XANES, respectively. Linear correlations are obtained between the CN of Pt-O and the average oxidation state (red line), and between CN of Pt-O and the hydrogenation activity (blue line)[85]; (C) Schematic illustration for the Pd SACs[86]; (D) Structure of the Ru-SACs/CeO2[93]; (E) Schematic illustration for the preparation of the Co-Nx/C-800-AT catalyst[100]; (F) Schematic illustration for the Fe1/N-C[111]. Pt1: Single Pt atoms; TOF: turnover frequency; XPS: X-ray photoelectron spectroscopy; SAC: single-atom catalyst.
In addition to Pt, other noble metals such as Pd[86-89], Au[90], Rh[91,92], and Ru-based[93] SACs also demonstrated remarkable catalytic performance in the hydrogenation of nitro compounds. For instance, atomically dispersed Pd supported on mesoporous polymeric graphitic carbon nitride (mpg-C3N4) exhibits superior catalytic activity and selectivity in the hydrogenation of nitro compounds compared to conventional Pd NP-based catalysts. This performance enhancement is attributed to the unique electronic structure of single Pd atoms, which facilitates the adsorption of reactants and the desorption of products, thereby streamlining the reaction pathway and boosting overall efficiency [Figure 4C][86]. Similarly, in the hydrogenation of 4-nitrochlorobenzene (4-NCB) to 4-chloroaniline (4-CA), Pd1/α-MoC achieved nearly 100% selectivity for the target product, even after complete conversion or prolonged reaction times[94]. In contrast, supported Pd NPs tend to induce side reactions, leading to significantly lower selectivity for 4-CA (20%-56%).
Although Au is generally considered catalytically inert in bulk form[95], its catalytic potential increases dramatically when presented as single atoms. In the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP), Au SACs exhibit a TOF of as high as 22,075 h-1, five times higher than that of Au NPs under the same conditions. Further studies show that embedding Au single atoms into nitrogen-doped porous carbon derived from metal-organic frameworks (MOFs) not only stabilizes the isolated Au sites but also enhances their catalytic performance. Under identical conditions, this catalyst achieves 100% conversion of 4-NP within 30 min, whereas its NP counterpart, supported on the same substrate, yields only about 70% conversion[90].
Compared to other precious metals, Rh-based catalysts have been employed less frequently in the hydrogenation of nitro compounds due to their relatively high cost and limited intrinsic catalytic activity. Recent advances have demonstrated the potential of Rh SACs in this field. For example, single-atom Rh catalysts embedded within MFI-type zeolites have been synthesized, where isolated Rh atoms are stabilized by oxygen atoms of the zeolite[96]. These catalysts exhibited remarkable performance in shape-selective tandem hydrogenation of various nitroarenes, delivering amine products in yields exceeding 99%. However, it is worth noting that the reaction system relied on highly efficient hydrogen donors such as ammonia borane (AB). Following this work, subsequent research primarily focused on enhancing the intrinsic activity of the metal centers by tuning their electronic structure, thereby improving their ability to adsorb and activate reactant molecules[92].
Due to their cost-efficiency, Ru-based SACs are highly promising catalytic materials. For example, Ru single atoms anchored on the metal oxide support (e.g., CeO2) deliver a high performance in the hydrogenation of nitrobenzene[93]. In a continuous-flow reaction test at 120 °C, the Ru SACs catalyst maintained an azoxybenzene selectivity above 80% over 18 h without significant deactivation. This stability is primarily attributed to the strong interaction between Ru single atoms and the support [Figure 4D].
Besides the commonly studied precious metal SACs (e.g., Pt, Pd, Rh, and Ru), other elements such as iridium (Ir) and silver (Ag) have also shown promising perspectives in the hydrogenation of nitroarenes. For instance, the Ag/Al2O3 catalyst composed of electron-deficient silver single atoms supported on γ-Al2O3 exhibits a significantly enhanced reaction rate in the aqueous-phase hydrogenation of nitroaromatics compared to conventional silver NPs supported on the same supports[97].
Although precious metal-based SACs exhibit high specific activity and excellent catalytic selectivity, their practical application is limited by the large amount of precious metals required. As a result, non-precious metal catalysts have gained considerable attention due to their low cost and abundant availability. However, their hydrogenation performance, especially in terms of activity, still lags significantly behind that of precious metal catalysts. Junge et al. first demonstrated the potential of carbon-supported Fe-family metal (Fe, Co) as catalysts for the selective hydrogenation of nitro compounds into amino compounds. Specifically, nitro groups could be selectively reduced, even when the nitro compounds bear other substituted groups (e.g., -Cl, C=C)[98]. However, for carbon-supported Co-based catalysts, the hydrogenation reaction could only be completed under relatively harsh conditions (110 °C, 50 bar, 4-16 h). Recent advancements in SACs have significantly narrowed this gap. Liu et al. identified, for the first time, the structure of atomically dispersed Co-N-C catalyst as an excellent hydrogenation catalyst. The TOF of the Co-N-C catalyst in the chemoselective hydrogenation of nitroarenes to produce azo compounds was 35.9 h-1, only slightly lower than that of precious metals[99]. Further investigation shows that the single-atom sites are the primary contributors to catalytic activity compared to cobalt (Co) NPs[100,101]. Later, Zhou et al.[100] demonstrated that atomically dispersed Co-Nx are highly efficient active sites for nitroarene hydrogenation, achieving extremely high activity, chemical selectivity, and structural stability. Co-Nx catalyzed hydrogenation of nitro compounds could be performed under mild conditions (40 °C and 1 bar H2) [Figure 4E]. Since then, high-performance Co SACs have been continuously reported[102-105]. Overall, theoretical investigations reveal that single-atom sites possess a high capacity for H2 activation, which is recognized as the rate-determining step in the hydrogenation of nitro compounds. Moreover, Co SACs enable balanced adsorption and activation of substrates, thereby providing an optimized hydrogenation pathway with a relatively lower energy barrier. Compared to traditional Co NP catalysts, Co SACs exhibit superior resistance to acid/base corrosion and high-temperature deactivation and demonstrate reversible deactivation behavior.
In addition to Co, other non-noble metals such as iron (Fe)[106], nickel (Ni)[107], and copper (Cu)[108]-based SACs have attracted growing attention in the catalytic hydrogenation of nitro compounds. For instance, Raney nickel remains one of the most widely employed non-noble metal catalysts in industrial hydrogenation processes. However, despite its high activity, it suffers from limited selectivity and poor stability in the hydrogenation of nitro compounds. Recent studies have shown that nickel-based SACs deliver much better selectivity and stability while maintaining catalytic activity comparable to that of Raney nickel[109]. Notably, Ni-N-C SACs have demonstrated exceptional durability under harsh reaction conditions when compared to Ni NPs supported on activated carbon (Ni/AC)[110]. Mechanistic investigations indicate that Ni-N-C possesses a highly distorted octahedral coordination geometry. This, together with the fact that pyridinic nitrogen can function as a frustrated lewis pair, facilitates heterolytic H2 dissociation. Moreover, the strong interaction between Ni and N atoms contributes to the catalyst’s remarkable structural stability. Intriguingly, recent studies demonstrate that the single-atom Ni-N4 coordination structure outperforms Ni NP catalysts when isopropanol serves as both solvent and hydrogen donor[107].
Among non-noble metals, iron offers notable advantages due to its abundance and low cost, making it particularly suitable for large-scale catalytic applications. Fe-based SACs prepared via spatial confinement strategies have shown significantly enhanced activity in nitrophenol reduction reactions compared to their NP counterparts [Figure 4F][111]. Similarly, Fe SACs supported on carbon materials (Fe1/NC) have demonstrated outstanding catalytic performance in hydrogen transfer reactions. Fe1/NC possesses a TOF of 748 h-1, which exceeds that of Fe NPs (75 h-1) and many previously reported catalysts[112]. In addition, Wang, for the first time, applied a ZIF-derived Fe SAC to the hydrogenation of nitroarenes. By calcining aniline-modified ZIFs, they synthesized a gram-scale FeSA@NC-20A catalyst with a high loading amount of atomically dispersed Fe sites (2.4 wt.%)[113].
Cu has emerged as a highly promising metal center in SACs due to its economic viability and environmental friendliness. It, on the other hand, typically exhibits limited hydrogenation activity due to its poor H2 dissociation capability. However, the catalytic performance of Cu can be substantially improved through atomic dispersion. For example, anchoring Cu single atoms onto boron nitride supports can significantly enhance the adsorption of aromatic nitro compounds, leading to improved hydrogenation activity[108]. Nevertheless, both Fe and Cu SACs generally exhibit lower intrinsic hydrogenation activity compared to their Co-based counterparts. To achieve desirable performance, more reactive hydrogen donors, such as NaBH4 or N2H4·H2O are also required. Taken together, among 3d non-noble metal catalysts, Co SACs remain the most promising alternatives to noble metal catalysts, offering a superior combination of activity, selectivity, and stability in nitro compound hydrogenation reactions.
Beyond conventional non-precious metal SACs such as cobalt, iron, nickel, and copper, zinc has also exhibited certain activity in nitroarene hydrogenation[114]. For instance, a Zn-based SAC was synthesized via pyrolysis of a Zn-containing MOF (ZIF-8), yielding a Zn-N-C structure. The catalyst exhibited good performance, with a high TOF of 494 h-1, excellent selectivity for aniline, and good cycling stability, maintaining full activity even after seven consecutive runs[114].
Bimetallic SACs
Due to the isolated nature of their metal active centers, SACs typically require reaction intermediates to be adsorbed and transformed at the same site. This spatial constraint often results in elevated adsorption energy barriers, which suppress reaction kinetics and limit both the diversity of reaction pathways and the range of accessible products[115-117]. From a structural perspective, dual-atom catalysts can be broadly categorized into two types. The first type is dual-single-atom catalysts, in which two metal atoms are atomically dispersed on the catalyst surface without direct bonding between them. The second type is diatomic catalysts, where the metal centers are connected through either metal-metal bonds or heteroatom bridges. Despite their differences in spatial configuration, both types can exhibit strong synergistic effects through electronic modulation and spatial coupling. Specifically, the direct metal-metal bond or bridging configuration not only strengthens the interaction between metal atoms but also introduces the bridge itself as a potential active site, offering new dimensions to catalytic functionality[118]. Furthermore, when the distance between the two metal atoms is sufficiently reduced, adjacent active sites with cooperative characteristics can form, which facilitates the activation and cleavage of key chemical bonds. This can significantly lower the adsorption energy barriers, alter the reaction intermediate pathways, accelerate reaction kinetics, and ultimately broaden the scope of product regulation[119].
Recently, a Pt1-Fe1/nanodiamond (ND) dual SAC has been developed. In this system, Pt single atoms are anchored on the nanodiamond surface through Fe1-N4 sites, forming atomically dispersed bimetallic active centers [Figure 5A][16]. Subsequently, the hydrogen activation and surface hydrogenation of para-nitrobenzene (p-NB) were investigated. In Figure 5B, the hydrogen reaction orders over the Pt1-Fe1/ND and PtNP/ND catalysts were estimated to be 0.73 and 0.15, respectively. Compared to the nearly zero hydrogen reaction order (0.15) on the Pt NP surface, the higher reaction order (0.73) indicates that hydrogen activation is more difficult on the Pt1-Fe1 dual sites, which is also confirmed by the kinetic isotope effect (KIE) experiment [Figure 5C]. Upon replacing H2 with D2, the Pt1-Fe1/ND catalyst exhibited a much higher KIE value than PtNP/ND (5.2 vs. 2.3). Regarding the surface hydrogenation process, the p-NB substrate reaction orders for Pt1-Fe1/ND and PtNP/ND catalysts were 0.26 and 1.07, respectively, indicating at the Pt1-Fe1 dual sites more easily activate the nitro group [Figure 5D]. Subsequently, the apparent activation energy experiment was conducted [Figure 5E]. It demonstrated that the Pt1-Fe1/ND catalyst has a lower activation energy than PtNP/ND, which suggests that the Pt1-Fe1 dual sites significantly reduce the apparent activation energy for the hydrogenation of the nitro group.
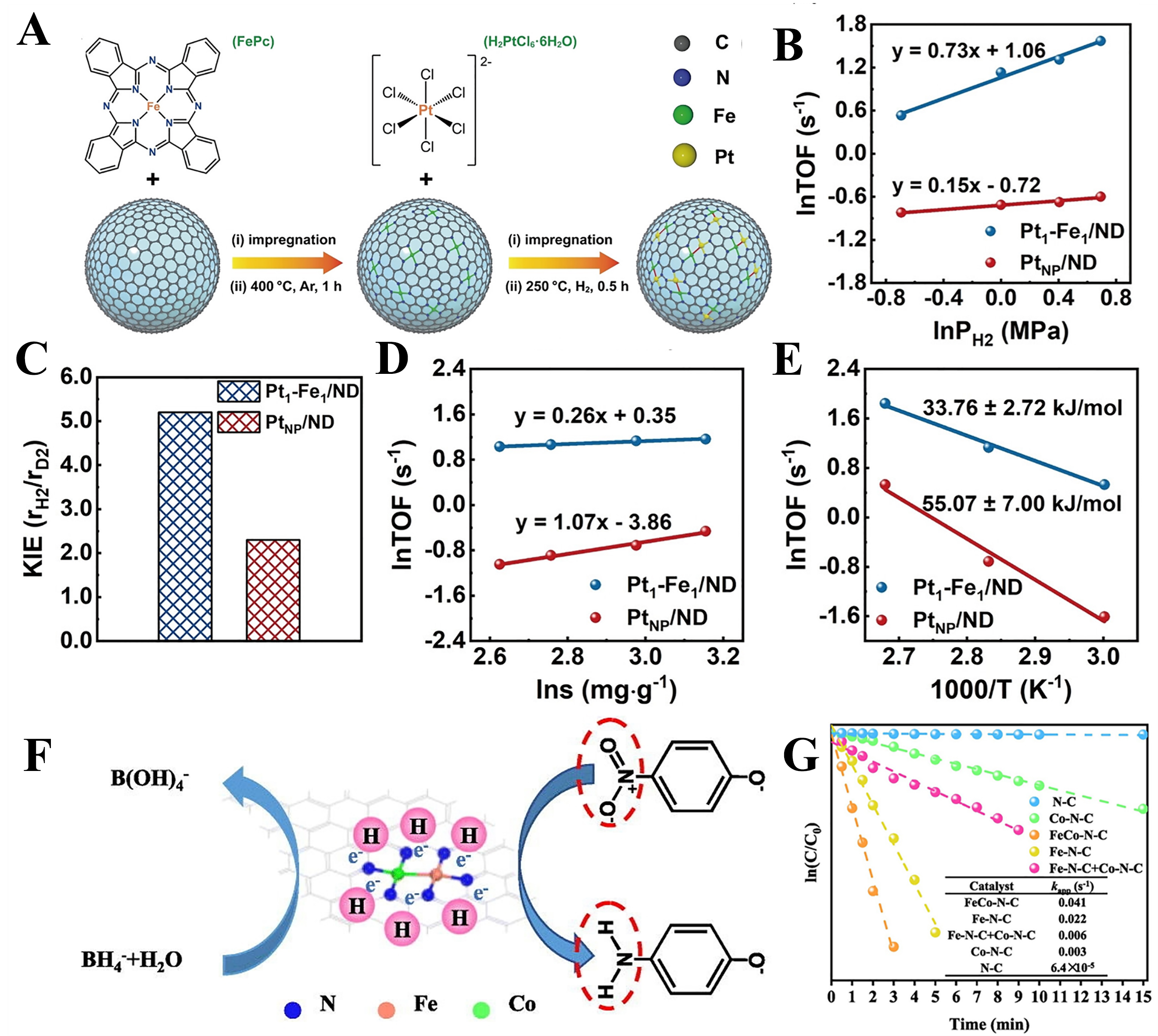
Figure 5. (A) The synthesis method for the Pt1-Fe1/ND catalyst[16]; Kinetics experiments of p-NB hydrogenation over Pt1-Fe1/ND and PtNP/ND catalysts: (B) H2 reaction order; (C) KIE value; (D) p-NB reaction order; and (E) apparent activation energy for p-NB hydrogenation[16]; (F) Structure of the FeCo-N-C[120]; (G) Degradation performance of FeCo-N-C and control samples N-C, Fe-N-C and Co-N-C in PNP reduction[120]. Pt1: Single Pt atoms; p-NB: p-nitrobenzene; NP: nanoparticle; ND: nanodiamond; KIE: kinetic isotope effect.
Similarly, a FeCo dual SAC was developed. The two metal centers show strong electronic interactions
Design of the metal center’s microenvironment
Design of support material
Support materials are crucial to the catalytic performance of SACs. The intrinsic properties of the support, such as electronic structure, electrical conductivity, and surface chemistry, can not only significantly influence the electron transfer between active sites and reactant molecules but also affect the adsorption behavior of reactants on the catalytic sites. These factors collectively determine the overall catalytic performance, including selectivity and activity. Through careful selection and optimization of the support material, it is possible to enhance the activity, stability, and selectivity of the catalyst, thereby providing valuable guidance for the rational design of advanced SACs[121,122]. Various supports, such as carbon materials, metal oxides, metal NPs, MOFs, and zeolites, have been developed to fabricate SACs used in the hydrogenation reduction of nitro compounds.
Carbon-based materials serve as excellent supports due to their unique structural and electronic properties, offering significant advantages in stabilizing single-atom metal centers. Porous carbon[123-125], carbon nanosheets[126], carbon nanotubes[127], graphite[128], and graphene[129-132] have been widely employed in the construction of high-performance SACs. For example, Pt SACs supported on nanocarbon were successfully synthesized simply via strong electrostatic adsorption[123]. The strong interaction between Pt atoms and edge/ or defect sites of nanocarbon could be utilized to regulate the electronic structure of the anchored Pt single atoms, thereby tuning the catalyst’s activity [Figure 6A]. However, when conventional carbon materials are used as supports, the dispersion and/or stability of metal atoms on the carbon support are often poor. Introducing heteroatoms into the carbon framework can alleviate such situations. On the one hand, it can create defects, benefiting metal site anchoring. Meanwhile, heteroatoms exhibit a strong interaction with metal sites through Lewis acid-base interactions, thereby enhancing the dispersion of the loaded metal. It was demonstrated that, in some cases, N-doped carbons are more effective in promoting the deposition and dispersion of Pt compared to N-free supports. As a result, under identical conditions, the N-doped materials exhibited a TOF of up to 41,193 h-1, whereas the catalyst supported on pure carbon was catalytically inert[133]. Interestingly, the interaction between nitrogen and Pt varies depending on the type of nitrogen species. Besides heteroatom doping, engineering carbon materials with specific structures can also enhance the performance of carbon-supported SACs in the catalytic hydrogenation of nitro compounds[134]. For example, a confined interfacial-oriented synthesis strategy was developed for preparing SACs, in which atomically dispersed Pd was formed within the confined interface between graphene oxide and activated carbon (RGO@AC/Pd SAC)[131]. Owing to the synergistic effect of well-dispersed atomic Pd and the presence of the RGO/AC interface, the catalyst exhibited exceptionally high activity and stability for the reduction of 4-NP. Notably, RGO@AC/Pd SAC has a TOF value more than 30 times higher than that of conventional Pd/carbon. Similarly, a graphite diacetylene/graphene (GDY/G) heterostructure was designed as a support for single-atom Pd (Pd1/GDY/G). Pd1/GDY/G exhibited high catalytic activity in the reduction of 4-NP[87]. Theoretical calculations revealed that the energy gap between the Fermi level of graphene and the minimum conduction band of GDY could facilitate electron transfer, thereby promoting the catalytic hydrogenation reaction [Figure 6B and C]. Carbon materials are generally inert and have weak interactions with single metal atoms, leading to metal atom aggregation. Although strategies such as heteroatoms doping and creating edges can help to stabilize the single atom, it is still challenging to precisely control the microenvironment of single-atom sites and improve their loading amount.
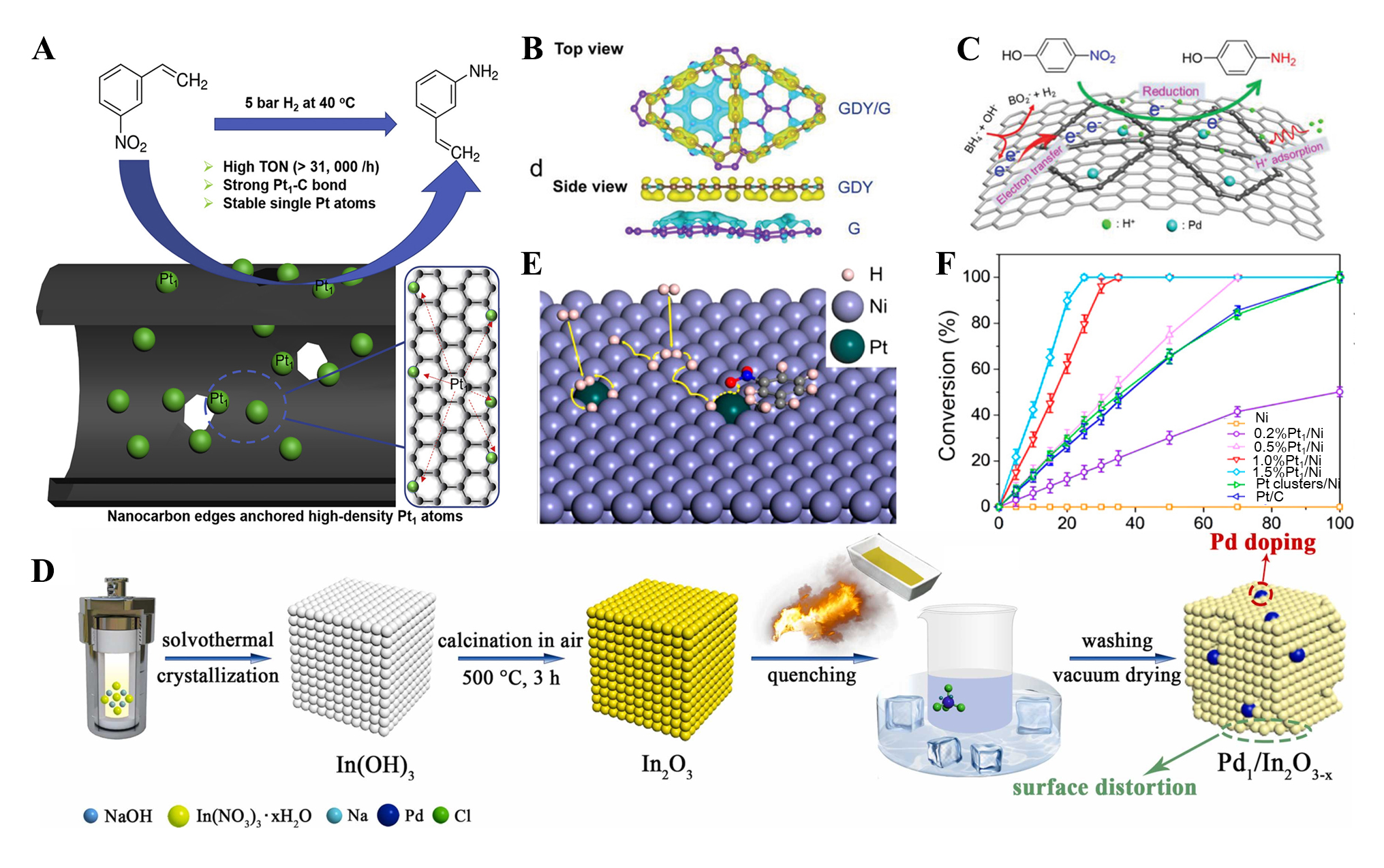
Figure 6. (A) A graphic illustration for designing nanocarbon edge anchored high-density Pt1 atoms for catalytic nitro compounds[123]; (B) Side views of real space electron redistribution between graphene and GDY[87]; (C) Schematic illustration of the proposed mechanism of 4-NP reduction catalyzed by Pd1/GDY/G[87]; (D) Schematic illustration for the construction of Pd1/In2O3-x[139]; (E) Schematic illustration of H2 dissociation and H diffusion on Pt1/Ni(111)[148]; (F) Time courses of the conversion for Ni nanocrystals, Pt1/Ni nanocrystals, Pt clusters/Ni nanocrystals, and Pt/C in selective hydrogenation of 3-nitrostyrene under 3 atm of H2 at 40 °C[148]. Pt1: Single Pt atoms; GDY/G: graphite diacetylene/graphene; NP: nanoparticle.
Metal oxides are widely employed as supports for SACs due to their unique polar surfaces and the abundance of coordinatively unsaturated sites, such as oxygen vacancies, metal vacancies, and lattice distortions[135-137]. These characteristics promote strong interactions with single-atom metals, enhancing dispersion and stability while suppressing the aggregation and migration of the metal species[138]. Metal oxide-supported SACs have garnered increasing attention in the hydrogenation of nitro compounds. For instance, a Pt-based single-atom and quasi-single-atom catalyst supported on FeOx demonstrated outstanding activity, chemoselectivity, and recyclability in the hydrogenation of substituted nitroarenes[84]. In the hydrogenation of 3-nitrostyrene, this catalyst exhibited a TOF value of approximately 1,500 h-1, which is 20 times higher than the best value previously reported, while achieving nearly 99% selectivity toward 3-aminostyrene. Similarly, Pd single atoms anchored on defect-rich indium oxide also demonstrated high activity and selectivity in the reduction of aromatic nitro compounds[139]. Theoretical calculations revealed that oxygen vacancies in the indium oxide support could optimize the electronic structure and oxidation state of Pd atoms, thereby enhancing their adsorption capacity for nitro compounds and optimizing the reaction pathway [Figure 6D].
Cerium dioxide (CeO2), due to its high density of oxygen vacancies, has been widely used in the field of catalysis[140]. These vacancies can selectively adsorb nitro groups. A representative example is the Ni/CeO2/SiO2 heterojunction catalyst. The electron transfers from Ni to CeO2, generating an internal electric field and thereby creating additional oxygen vacancies[141]. This built-in field promotes H2 dissociation, while the oxygen vacancies facilitate selective adsorption and activation of nitro groups, thereby suppressing undesired dehalogenation side reactions. The synergistic effects of the internal electric field and oxygen vacancies collectively enhance the catalytic performance.
More recently, composite support engineering has emerged as an effective strategy for tuning the catalytic behavior of SACs by harnessing the complementary properties of different support materials. For example, a highly efficient Rh SAC is supported on an alumina-carbon composite, in which atomically dispersed Rh sites are anchored on Al2O3 and further confined by an amorphous carbon layer[92]. This catalyst outperformed benchmark Rh/C and Rh/γ-Al2O3 catalysts in the selective hydrogenation of meta-chloronitrobenzene (m-CNB) to meta-chloroaniline (m-CAN). Specifically, it achieves an impressive TOF of 2,317 mol m-CNB·mol Rh-1·h-1 and maintains ~ 98% selectivity. Detailed characterization revealed that the improved catalytic activity and selectivity stemmed from the optimized electronic structure of Rh species and the enhanced exposure of acidic sites.
Additionally, metal[142], MOFs[143-145], zeolites[96], porous boron nitride[108], and organic polymers[146] supported SACs have also demonstrated excellent catalytic performance in the catalytic hydrogenation of nitro compounds. Among these, MOFs have attracted particular attention due to their tunable porosity, high surface area, and versatile coordination environments[147]. Additionally, zeolites offer rigid and thermally stable microporous environments that are equally favorable for stabilizing isolated metal atoms. The incorporation of metal species into zeolite matrices yields materials with well-defined active sites that facilitate selective hydrogenation. For example, single Rh atoms encapsulated within MFI zeolites achieved a high TOF of 699 molH2 (molRh)-1 min-1 at 298K for AB hydrolysis and demonstrated superior catalytic efficiency in shape-selective tandem hydrogenation of nitroarenes coupled with AB hydrolysis[96].
Non-metal-based supports, such as MOFs, zeolites, and carbon materials, generally promote hydrogen activation via a heterolytic cleavage mechanism, producing M-Hδ- and X-Hδ+ (where M represents metal atoms and X indicates non-metal atoms, such as N and O). While the energy barrier for X-Hδ+ bond cleavage often imposes thermodynamic limitations. As such, metal-supported SACs, particularly in the form of single-atom alloys (SAAs), provided a potential solution. By anchoring isolated metal atoms onto the surface of metallic substrates, such systems enable homolytic H2 dissociation, effectively bypassing the high-energy transition state associated with heterolytic cleavage. This modification of the activation pathway has led to significant enhancements in catalytic efficiency. For example, a SAC consisting of Pt single atoms supported on Ni nanocrystals exhibited the highest conversion in the hydrogenation of nitro compounds, significantly outperforming other catalysts [Figure 6E and F][148].
Design of the first coordination shell of metal centers
The first coordination shell of metal centers is a key factor influencing the catalytic performance of SACs. Rational modulation of the coordination environment can effectively regulate the electronic structure of active sites, thereby enhancing the adsorption and activation of reactants[149]. For example, it was found that reducing the Pt-N CN could enhance the electronic density of Pt single atoms and lower the hydrogen dissociation energy barrier, thereby markedly improving hydrogenation activity[150]. The TOF of Pt-N3 SACs was approximately six times that of Pt-N4 SACs and two orders of magnitude higher than that of Pt-N5 SACs, while selectivity remained nearly constant at 99%. At 35 °C, the TOF for Pt-N3, Pt-N4, and Pt-N5 was 1,926 h-1, 299.5 h-1, and 3.5 h-1, respectively, with the TOF of Pt-N3 SACs being about 550 times higher than that of Pt-N5 [Figure 7A and B].
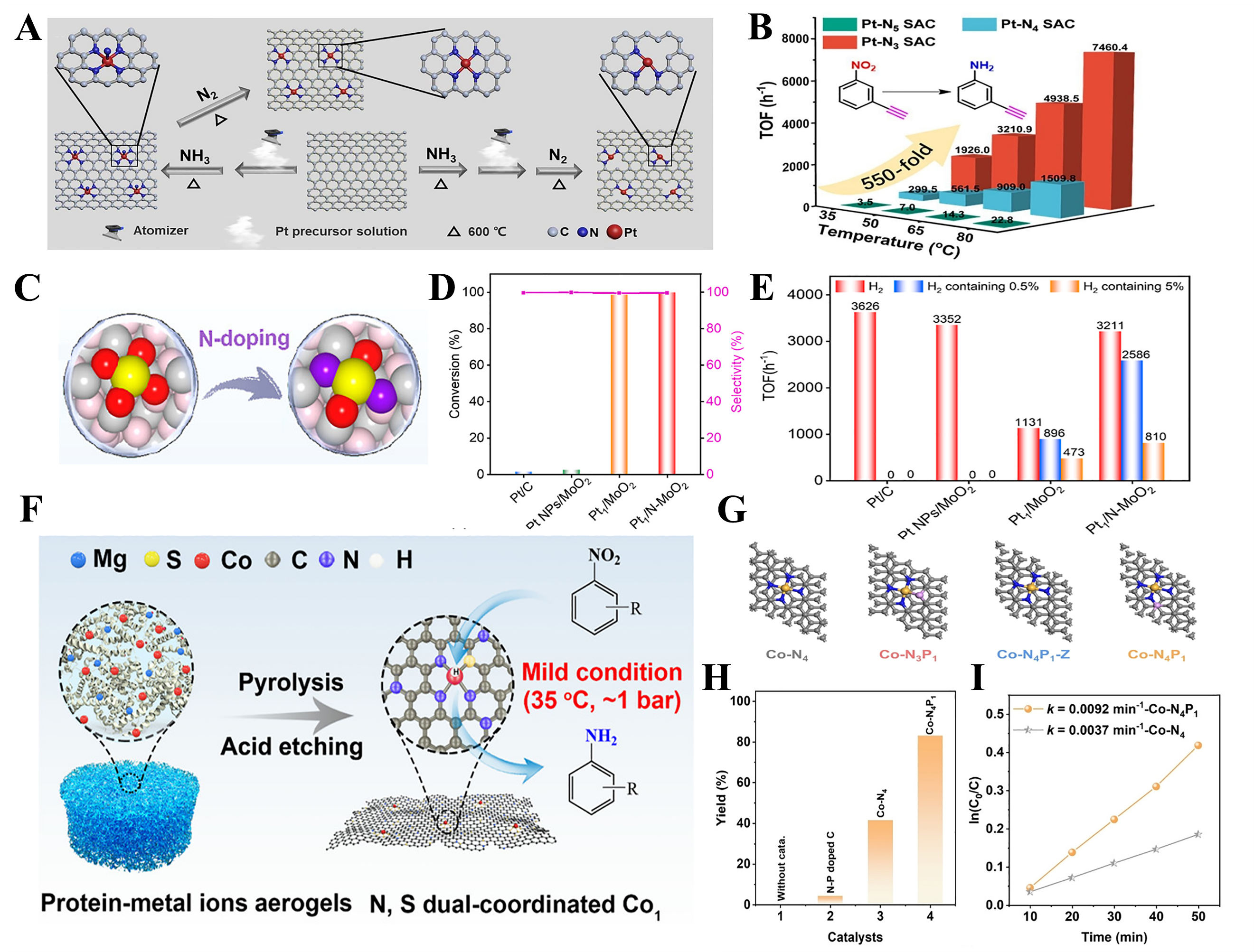
Figure 7. (A) Schematic diagram of the synthesis of Pt1/NC SACs[150]; (B) Catalytic activities of Pt1/NC SACs for hydrogenation of 3-nitrophenylacetylene at different temperatures[150]; (C) The structure diagram of Pt1/N-MoO2[151]; (D) Anti-CO poisoning ability over Pt/C, Pt NPs/MoO2, Pt1/MoO2, and Pt1/N-MoO2 under 20 bar H2 containing 0.5% CO[151]; (E) TOF value of per Pt sites for Pt/C, Pt NPs/MoO2, Pt1/MoO2, and Pt1/N-MoO2 H2, H2 containing 0.5% CO, and H2 containing 5% CO[151]; (F) Schematic diagram of the synthesis of Co1/NSC-AT[158]; (G) The structural models of Co-N4, Co-N3P1, Co-N4P1-Z and Co-N4P1[161]; (H) The catalytic performance of various catalysts for the p-NB selective hydrogenation[161]; (I) The reaction rates[161]. Pt1: Single Pt atoms; SAC: single-atom catalyst; NP: nanoparticle; TOF: turnover frequency; Co1/NSC-AT: Co single atoms on N,S-co-doped hierarchical porous carbon, after treatment.
Non-metal-based catalysts often suffer from CO poisoning. Metal oxide-supported SACs exhibit enhanced resistance to CO poisoning due to their relatively weak adsorption of CO molecules. However, these catalysts often face limitations in H2 activation, as hydrogen dissociation on such surfaces generally proceeds via a heterolytic pathway, generating M-Hδ+ and O-Hδ- species. The cleavage of the O-Hδ- bond is frequently the rate-determining step and typically requires a high activation energy, which restricts the intrinsic catalytic activity. Coordination environment engineering was demonstrated to be feasible to simultaneously enhance both CO tolerance and hydrogen activation of metal oxides supported SACs [Figure 7C]. Nitrogen-doped MoO2 supported Pt atoms (Pt1/N-MoO2) demonstrated not only superior CO resistance compared to Pt NPs supported on MoO2 or commercial Pt/C, but also significantly improved catalytic activity. Taking the hydrogenation of nitrobenzene to aniline as an example, the anti-CO poisoning ability of Pt1/N-MoO2 and the comparison samples was evaluated under hydrogen atmospheres with and without CO. Under a H2 atmosphere containing 0.5% CO, the catalytic activities of Pt/C and Pt NPs/MoO2 were almost completely suppressed [Figure 7D]. In contrast, Pt1/MoO2 and Pt1/N-MoO2 were scarcely affected and achieved 100% conversion of nitrobenzene to aniline. In addition, under the presence of 0.5% CO, Pt1/N-MoO2 also exhibited excellent performance in the hydrogenation of furfural (FF) and cinnamaldehyde (CALD), with conversion rates of FF to FFA and CALD to HCALA reaching as high as 94.1% and 97.8%, respectively [Figure 7E][151].
Coordination environment engineering could also be utilized to enhance the catalytic efficiency of non-precious metal-based SACs[152,153]. In particular, modulating the CN and heteroatom composition around the metal center can significantly alter the electronic structure and reaction parameters. For example, among different Zn-Nx species, Zn-N3 sites perform better in nitrobenzene hydrogenation, achieving complete conversion to aniline with a TOF of 494 h-1. This performance exceeds that of Zn-N4 sites (TOF of 439 h-1) and Zn-N2 sites (TOF of 57 h-1), highlighting the importance of the local coordination environment on catalytic behavior[114]. Besides the CN, coordination atom types can also influence the catalytic performance of SACs. Specifically, introducing electron-rich heteroatoms into N-coordinated frameworks has emerged as an effective approach to modulating the electron density at the metal center, thereby enhancing its catalytic activity[154,155]. For instance, Co single atoms possessing a Co1-N3P1 motif exhibit higher electron density than conventional Co1-N4 configurations. KIE measurements and DFT calculations indicate that increased electron density may enhance molecular hydrogen dissociation. Particularly, Co1-N3P1 possesses a TOF value in nitrobenzene hydrogenation that is 60 times higher than for Co1-N4[156]. Subsequent studies show that adjusting the P atom ratio can provide Co-N-P sites with an optimized electronic structure that promotes both aromatic substrate adsorption and H2 activation. As the optimum configuration motifs, CoN2P2 achieves a TOF of 241.5 h-1, 7.8 times higher than that of a conventional Co-N-C catalyst (30.8 h-1)[157]. Besides P, sulfur and oxygen have also been demonstrated to modulate the coordination environment of N-coordinated SACs[158]. Asymmetric configurations such as M1-N3S1 have proved a pronounced effect on H2 activation and the reduction of nitrogen-containing substrates[159,160]. Particularly, a hierarchically porous carbon-supported N, S dual-coordinated cobalt SAC (Co1/NSC-AT) was developed for the hydrogenation of nitro compounds. The unique coordination environment of the Co center, combined with the hierarchical porous structure of the carbon support, endows Co1/NSC-AT with excellent catalytic performance; the hydrogenation reaction can proceed under mild conditions (35 °C, 1 bar H2) with high conversion and selectivity [Figure 7F][158].
Design of other coordination shells of metal centers
Beyond the coordination environment (first coordination shell) of single-atom centers, the peripheral environment, often referred to as the second or more distant coordination sphere, can also influence the catalytic behavior of SACs. Modulating such a peripheral environment has emerged as an effective strategy to further optimize catalytic performance, particularly in the selective hydrogenation of nitro compounds.
One representative approach involves tuning the second coordination sphere through heteroatom doping. The incorporation of phosphorus atoms into the local environment of Co-based SACs, for instance, has been shown to break the symmetry of the Co–N coordination and increase electron localization around the metal center [Figure 7G]. This electronic restructuring could promote H2 activation and facilitate product desorption, thereby enhancing both activity and selectivity in the hydrogenation of halogenated nitroaromatic compounds[161]. The selective hydrogenation of p-NB to para-chloroaniline (p-CA) was employed as a model reaction to evaluate the catalytic performance. As shown in Figure 7H, under the conditions of 80 °C and 0.5 MPa H2 for 2 h, no reaction occurred in the absence of a catalyst. Notably, the Co-N4P1 catalyst exhibited an 83.2% conversion and > 99% selectivity under the same conditions, indicating that the phosphorus-induced asymmetric SAC structure plays a key role in promoting H2 activation and product desorption. In addition, no product was detected under an inert Ar atmosphere, highlighting the essential role of H2. Furthermore, as shown in Figure 7I, the Co-N4P1 catalyst achieved a reaction rate constant (k) of 0.0092 min-1 for the selective hydrogenation of p-NB, which is 2.5 times higher than that of the Co-N4 catalyst (0.0037 min-1). Similarly, introducing sulfur atoms into the second coordination shell of Co single atoms has been demonstrated to be effective in improving the catalytic performance of SACs[162].
In addition to non-metallic dopants, alkali metal cations have also been employed to modulate the local electronic structure of SACs. It has been reported that alkali metals such as Li+, Na+, and K+ can convert Pt/FeOx catalysts into highly selective single-atom systems[163]. Specifically, sodium cations were found to coordinate with Pt and Fe through bridging oxygen atoms, forming surface Pt-O-Na-O-Fe complexes. This interaction not only suppresses Pt NP aggregation but also stabilizes positively charged, low-coordination Pt species.
Design of the loading amount of single-atom sites
The catalytic performance of SACs is fundamentally governed by both the intrinsic activity of individual metal centers and the surface density of accessible active sites[164]. Although isolated metal atoms exhibit high specific activity, the low loading amount of conventional SACs limits the total number of active sites, thereby constraining their practical applicability. Achieving both high loading amounts and atomic dispersion remains challenging. In response, multiple strategies have been proposed to construct stable, high-loading SACs (typically in the range of 1-5 wt.%), and their influence on hydrogenation performance has been systematically explored in nitro compound reduction reactions. For instance, the modification of activated carbon supports with phosphomolybdic acid (PMA) enables the incorporation of 1 wt.% Pt atoms while maintaining atomic dispersion[165]. The Keggin-type structure of PMA provides a quasi-planar square coordination environment that could stabilize Pt atoms [Figure 8A]. The hydrogenation reaction of the
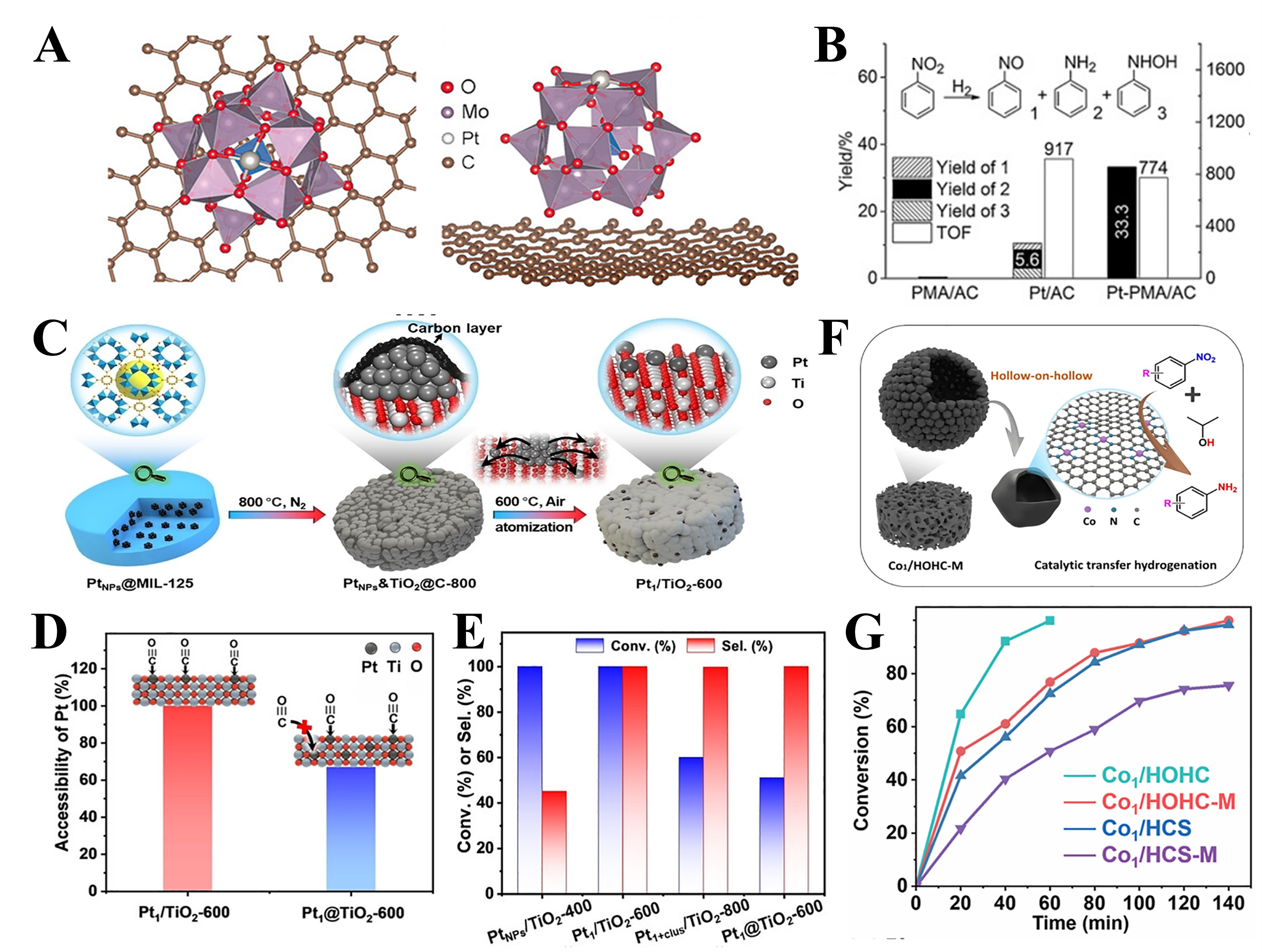
Figure 8. (A) Top view and side view of the most stable configuration of Pt1 on PMA/graphene, based on DFT calculations[165]; (B) Catalytic performance of PMA/AC, Pt/AC, and Pt-PMA/AC in hydrogenation reactions[165]; (C) Illustration for the construction of Pt1/TiO2-600[169]; (D) Accessibility of Pt1 sites determined by CO pulse chemisorption of Pt1/TiO2-600 and Pt1@TiO2-600[169]; (E) Catalytic performance of different catalysts in the hydrogenation of p-CNB at 80 °C under 15 bar H2 within 1.5 h[169]; (F) Structure of the Co1/HOHC-M[175]; (G) Time courses of NB conversion over Co1/HOHC, Co1/HOHC-M, Co-HCs, and Co-HCs-M[175]. Pt1: Single Pt atoms; PMA: phosphomolybdic acid; DFT: density functional theory; p-CNB: para-chloronitrobenzene; NB: nitrobenzene.
In non-noble metal systems, increasing metal loading is particularly attractive due to cost considerations. A spatial confinement strategy involving hard-template-assisted pyrolysis of g-C3N4 was successfully employed for the synthesis of SACs with a Co loading amount exceeding 10 wt.%[166]. In this system, a gradient anchoring strategy was proposed to prevent metal aggregation and ensure the isolated dispersion of Co atoms. The catalyst achieved 99% conversion and 96.3% selectivity toward aniline in the transfer hydrogenation of nitrobenzene using formic acid as the hydrogen source. The excellent catalytic activity was attributed to efficient HCOOH activation and strong Co-N coordination interactions, which also contributed to the outstanding cycling stability.
Further progress in improving metal loading amounts while maintaining atomic dispersion has also been achieved through the use of chelating precursors. Co SAC supported on nitrogen-assembled carbon (Co-NAC) was developed for the selective hydrogenation of nitroaromatics under mild reaction conditions (80 °C, 2 MPa H2). The catalysts deliver a high catalytic efficiency (TOF = 19.7 h-1) and > 99% selectivity toward 3-nitrostyrene hydrogenation[167]. The superior performance was attributed to the combination of a high density of atomically dispersed Co sites, a large surface area with an ordered mesoporous structure, and a high nitrogen content, all of which could be tuned by adjusting the precursor composition and coordination environment[168].
Besides Co, Ni SACs with high Ni loading amounts were also developed for the catalytic hydrogenation of nitro compounds. For example, by introducing a high proportion (72.2%) of pyridinic nitrogen into the support, a Ni-N-C SAC with a Ni loading of 7.5 wt.% was synthesized[110]. The high density of Ni single-atom sites enhanced the adsorption and polarization of nitroarenes, resulting in a reduction of the reaction energy barrier by approximately 0.3 eV compared to conventional Ni NPs.
Enhancing the utilization of single-atom sites
The catalytic performance of SACs largely depends on the spatial distribution of active sites. Unfortunately, single-atom sites are often hosted within a micropore structure or, in most cases, are buried within the carbon layer. Such situations pose a significant challenge for reactants, especially substrates with large molecular sizes and limited solubility. Consequently, SACs exhibit low efficiency in utilizing their active sites. Therefore, enhancing the accessibility of single-atom sites to reactants is crucial for optimizing SAC performance. This factor is particularly important for nitroarenes, as their large molecular size and limited solubility hinder effective interaction with single-atom sites.
On the one hand, the use of support materials with high specific surface areas, such as two-dimensional nanosheets, ultrathin layers, or hollow structures, can effectively enhance the exposure of single-atom sites on the outside surface of catalysts and reduce the likelihood of their being buried within the support. However, precisely controlling the spatial distribution of individual atoms remains a significant challenge, as some metal atoms may still become deeply embedded and thus inactive. To address this situation, a "sequential thermal transformation" strategy has been employed to preferentially anchor Pt single atoms on the outer surface of TiO2 [Figure 8C]. CO pulse chemisorption experiments show that the proportion of Pt sites capable of adsorbing CO in Pt1/TiO2-600 reaches nearly 100%, indicating the complete exposure of Pt sites on the TiO2 surface [Figure 8D]. In the hydrogenation of para-chloronitrobenzene, Pt1/TiO2-600 catalyst exhibited a high selectivity of 99% toward p-CA, significantly outperforming Pt NP-based catalysts (45%) [Figure 8E][169]. Mechanistic studies revealed that Pt1/TiO2-600 achieved full accessibility of Pt single atoms and preferentially adsorbed the nitro (-NO2) group, while showing weaker interaction with the chlorine (-Cl) groups in both the reactant and the product. Such selective adsorption behavior contributed synergistically to enhanced activity and product selectivity.
A pre-coordination strategy was developed to expose single-atom sites. By introducing specific functional groups onto a layered Mg(OH)2 template, metal precursors are pre-coordinated before pyrolysis. Because of the strong d-p orbital hybridization between metal and ligands, a highly dispersed metal center was obtained after pyrolysis, which was anchored within a two-dimensional honeycomb-like carbon nanomesh. This structure not only promotes active site exposure but also facilitates reactant enrichment and mass transport during the catalytic process. Despite an extremely low cobalt loading of only 0.12 wt.%, the resulting Co1/NC catalyst exhibited remarkable activity and stability in nitroarene reduction, with a TOF of 73,668 h-1, outperforming most previously reported metal-based catalysts[170].
On the other hand, introducing mesopores or micropores into the catalyst structure offers an alternative, effective route for enhancing single-atom site utilization, as such a strategy could improve the exposure degree of single-atom sites on the inside surface of catalysts. Compared to micropores, mesopores and macropores not only increase the accessibility of single-atom sites but also facilitate the transport of reactants and products, thereby accelerating the kinetics of hydrogenation. For example, atomically dispersed Co catalysts supported on mesoporous nitrogen-doped carbon matrices have been synthesized via high-temperature pyrolysis followed by leaching. In the hydrogenation of nitrobenzene to aniline, these catalysts demonstrated superior performance under mild conditions[171-173]. Compared with microporous structures, the mesoporous framework provided enhanced mass transport, improved accessibility of active sites, and reduced catalyst deactivation, collectively contributing to its outstanding hydrogenation performance.
Building on this, hierarchical porous structures could balance the loading amount of single-atom sites and their accessibility, synergistically enhancing the catalytic performance of SACs. In a typical case, atomically dispersed metals were precisely anchored onto carbon nanospheres through an in situ etching using NH3 and an anchoring strategy. The NH3 etching step significantly increased the specific surface areas and introduced hierarchical porosity, greatly enhancing the accessibility of active centers and strengthening the interactions between active sites and reactants. As a result, this catalyst achieved over 99% conversion of highly concentrated 4-NP within just 4 minutes, far surpassing catalysts with buried active sites[174]. Moreover, hollow-in-hollow structure motifs were also developed to improve the utilization of single-atom sites, thereby enhancing catalytic performance. This architecture featured a large internal cavity (~ 290 nm in diameter, generated by the decomposition of polystyrene nanospheres) and a thin shell with hollow spherical mesopores (~ 10 nm in diameter, formed in situ via ZnO NP volatilization during the pyrolysis of α-cellulose in the presence of CO2) [Figure 8F]. The catalytic performance of the transfer hydrogenation of nitrobenzene to aniline using isopropanol as the hydrogen donor was systematically investigated. A full conversion of nitrobenzene with a selectivity of 96% toward aniline was achieved over Co/HOHC-M within 140 min. In comparison, the conversion of nitrobenzene over Co/HCS-M (derived from pyrolysis of PS@ZnCo-ZIFs pellets) and HOHC-M (derived from pyrolysis of PS@Zn-ZIFs/a-cellulose) was only 76% and 37%, respectively, under the same reaction conditions and duration [Figure 8G][175]. Similar structural advantages have also been confirmed in other systems[176].
STUDY ON REACTION MECHANISM
The hydrogenation of nitroarenes to anilines has long attracted significant attention and constitutes one of the core topics in catalytic hydrogenation research. As early as 1898, Haber proposed the classical macroscopic mechanism for nitrobenzene hydrogenation, which suggests that the reaction typically proceeds via a stepwise hydrogenation pathway involving several key intermediates, including nitrosobenzene (PhNO*), phenylhydroxylamine (PhNHOH*), azobenzene (PhNNPh*), azoxybenzene [PhN(O)NPh*], and hydrazobenzene (PhNHNHPh*)[177]. In the classical Haber mechanism, the reaction can proceed through two main pathways. The first is the direct hydrogenation route, where nitrosobenzene (PhNO) and phenylhydroxylamine (PhNHOH) are formed sequentially, eventually producing aniline (PhNH2) via continuous hydrogenation. The second pathway, known as the indirect or condensation route, involves the condensation of nitrosobenzene with phenylhydroxylamine to generate azoxy intermediates, which are subsequently hydrogenated to form azo and hydrazo species before yielding aniline [Figure 9][178].
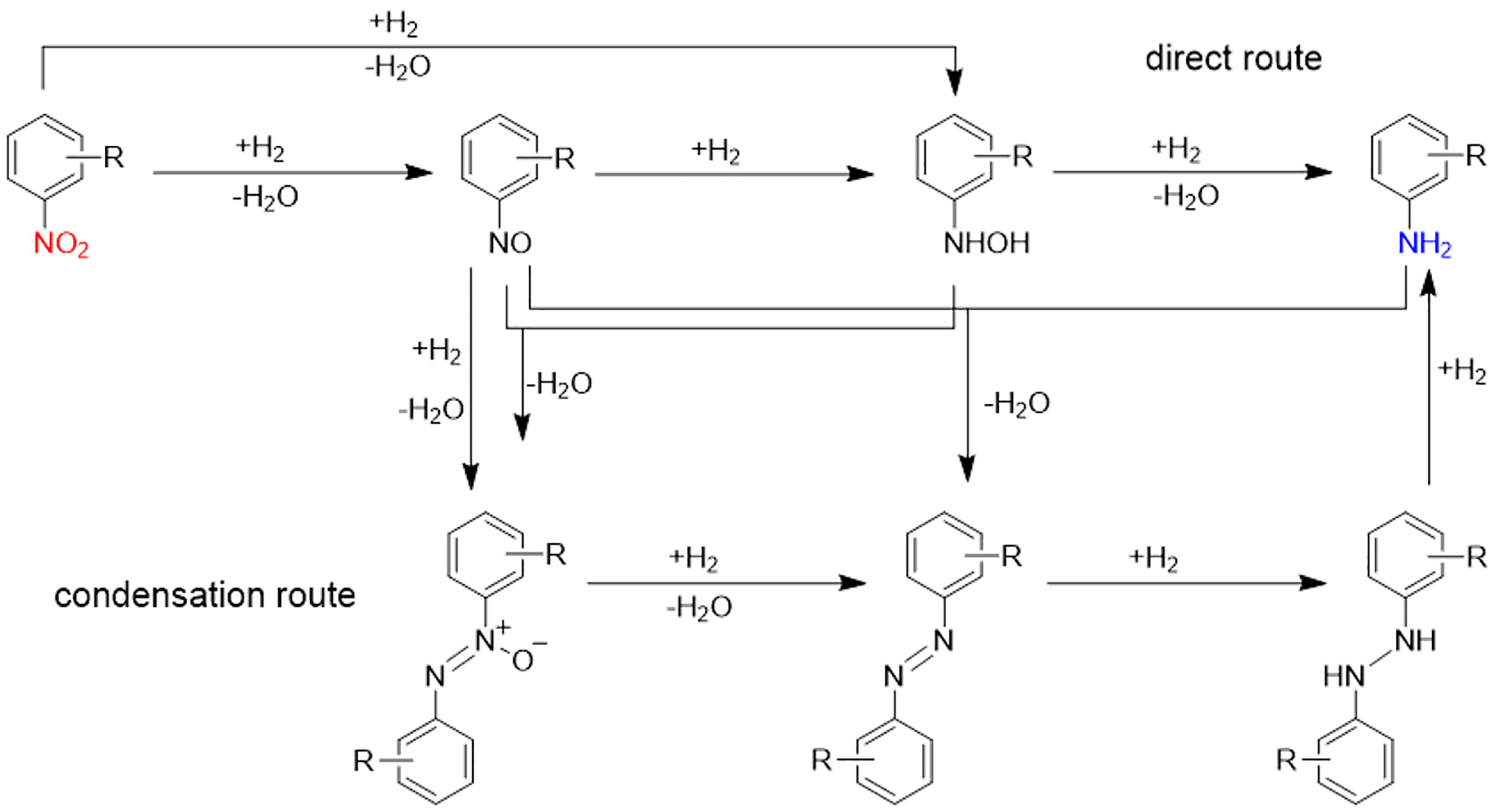
Although the above concept is widely accepted as a foundational framework for understanding the hydrogenation of nitroarenes, few experimental or theoretical studies have identified alternative pathways. For example, Jackson and colleagues proposed a mechanism bypassing the nitrosobenzene (PhNO*) intermediate, in which nitrobenzene (PhNO2*) is directly hydrogenated to form hydroxy-nitrosobenzene (PhNOH*), which then reacts with surface hydrogen to yield phenylhydroxylamine (PhNHOH*), eventually dehydrating and hydrogenating to form aniline[179]. However, when PhNO* is formed, the subsequent PhNHOH* intermediate tends to dehydrate, resulting in undesired azoxy side products, thereby lowering the selectivity toward aniline. Corma et al. experimentally validated that this direct hydrogenation pathway could also lead to high aniline selectivity on catalysts such as Au/TiO2[180]. It is worth noting that the reaction pathway is influenced by both catalyst properties and reaction conditions. For example, accumulation of the nitrosobenzene intermediate has been observed on Raney Ni surfaces under low hydrogen pressure[180].
The classical Haber pathway (Ph-NO2 → Ph-NO → Ph-NHOH → Ph-NH2) is followed by most SACs catalyzed nitro compounds hydrogeaniton. However, in contrast to traditional nanosized metal sites, single-atom sites possess limited spatial dimensions, preventing the simultaneous adsorption of H2 and substrates. Moreover, substrates tend to adopt different adsorption modes on single-atom sites relative to those observed on nanosized metal surfaces[42,181]. As a result, the overall hydrogenation reaction pathway, rate-determining step, and activation barrier may differ significantly from those associated with nanosized metal sites. For example, Du conducted an in-depth investigation into the catalytic performance of Pt single atoms supported on graphitic carbon nitride (Pt@g-C3N4) for the hydrogenation of nitrobenzene[182]. Adsorption configuration analysis demonstrated that nitrobenzene could adsorb onto Pt single atoms either through the benzene ring or the nitro group, with the former being more stable. This adsorption mode is accompanied by significant charge transfer, indicating a strong interaction between the substrate and the catalytic site, which facilitates subsequent hydrogenation steps. Further analysis shows that the reaction follows a typical “direct hydrogenation pathway,” with the most favourable route proceeding as: Ph-NO2 → Ph-NOOH → Ph-NO → Ph-NOH → Ph-N → Ph-NH → Ph-NH2. Among these steps, the cleavage of the second N-O of
Figure 10. (A) Energy profiles of the optimal reaction pathway for the hydrogenation of nitrobenzene into aniline on single atom Pt@g-C3N4 catalyst[182]; (B) Proposed possible reaction pathways for the reduction of nitrobenzene into aniline through direct routes (the reaction pathway in blue is the optimal one)[182]; (C) Top and side views of (A) parallel adsorption via nitro N, (B) vertical adsorption via nitro N, (C) parallel adsorption via nitro O, and (D) vertical adsorption via nitro O of nitrobenzene on the Co-N4/C surface[5]; (D) The adsorption and dissociation energies of H2 on different active sites on Ni1/NSPC, NiNPs/NSPC, and Ni1+NPs/NSPC[5]; (E) Energy barriers for H-transfer at different positions on Ni1+NPs/NSPC[5]; (F) Relationship between the Ea of the dominant H2 dissociation and the second N-O bond breaking step of PhNOH* + H* → PhN* + H2O in nitrobenzene hydrogenation[13]. NP: Nanoparticle.
The mechanism of the nitrobenzene hydrogenation reaction over the non-noble SACs was also investigated. Wang et al. found that the reaction preferentially proceeds via the direct pathway on Co single atoms supported on nitrogen-doped carbon (Co-N4/C)[5]. Specifically, nitrobenzene can adsorb on the Co-N4/C surface in various configurations, with vertical adsorption through the nitro oxygen atoms being dominant [Figure 10C]. Further analysis indicated that H2 dissociation is the rate-determining step on the Co-N4/C surface, with an energy barrier of 1.10 eV. In the subsequent hydrogenation steps, the N-O bond cleavage of nitrobenzene could proceed via two mechanisms: single-hydrogen or dual-hydrogen models. From an energy barrier perspective, the single-hydrogen pathway (PhNO2* → PhNOOH* → PhNO* → PhNOH* → PhN* → PhNH* → PhNH2*) was more favorable. Similarly, Liu systematically compared the hydrogenation process on two types of catalytic sites - single-atom Ni (Ni1/NSPC) and Ni NPs (NiNPs/NSPC) - in terms of their adsorption behavior toward H2 and p-CNB[181]. The results showed that H2 adsorption energy on NiNPs (-0.75 eV) was significantly lower than on Ni1 (-0.07 eV), indicating that H2 preferentially adsorbs and dissociates on NiNPs. In contrast, p-CNB exhibited a much stronger adsorption on Ni1 (-1.47 eV) compared to NiNPs (-0.04 eV), suggesting that the substrate molecule tends to bind more readily to the Ni single-atom sites. These findings reveal a spatial and functional separation between hydrogen activation and substrate adsorption, providing theoretical support for a bifunctional site synergistic catalytic mechanism
Similarly, detailed mechanistic investigations have been conducted on SACs supported on metal oxides. For example, in the case of a Ni1/CeO2-x(111) SAC, H2 exhibits distinct dissociation behaviors at different active sites[183]. At the B site (a Ni-Ce4-Ce5 hollow site) on the CeO2(111) surface, H2 readily undergoes heterolytic cleavage to produce H-Ni and H-O species, followed by a hydride-transfer step that induces the formation of oxygen vacancies. In contrast, at the Ni site on the Ni1/CeO2-x(111) surface, H2 preferentially dissociates homolytically to generate two H-O species. For the hydrogenation of nitrobenzene to aniline over this catalyst, the direct pathway is both thermodynamically and kinetically more favorable than the condensation pathway. Mechanistic calculations further reveal that, compared with single-hydrogen or double-hydrogen induced dissociation routes, the mixed single–double hydrogen-induced dissociation pathway (PhNO2 → PhNOOH* → PhNO* → PhNHO* → PhNHOH* → PhNH* → PhNH2*) is the most favorable. The rate-determining step of this route is the fourth hydrogen-transfer reaction (PhNHO* + H* → PhNHOH*), with an energy barrier of 1.24 eV. Moreover, the presence of water molecules also plays an important role in the reduction of nitrobenzene to aniline. In the presence of nitrobenzene, the Co-N4/C catalyst can efficiently activate hydrogen via the Co-N activation mode with an energy barrier of 1.10 eV[5]. For the single-H-induced N-O bond dissociation mechanism under water-participating conditions, the fourth hydrogen transfer step, PHNOH* + H2O + H* → PHN* + 2H2O, exhibits the highest energy barrier of 0.87 eV. In contrast, under water-free conditions, the fourth hydrogen transfer step, i.e., PHNOH* + H* → PHN* + H2O, has the highest barrier of 1.00 eV. Similarly, in the double-H-induced N–O bond dissociation mechanism, the fourth hydrogen transfer step under water-participating conditions (PhNOH* + H2O + H* → PhNHOH* + H2O) is the most difficult step, with an energy barrier of 0.93 eV, which is 0.34 eV lower than that under water-free conditions (PhNOH* + H* → PhNHOH*). This indicates that water molecules play an important role in the reduction of nitrobenzene to aniline. This mechanistic insight elucidates the preferential use of aqueous solvents in SAC-catalyzed hydrogenation of nitro compounds. For this reaction, the single-H-induced N-O bond dissociation mechanism (PhNO2* → PhNOOH* → PhNO* → PhNOH* → PhN* → PhNH* → PhNH2*) is more favorable than the double-H-induced mechanism (PhNO2* → PhNOOH* → PhN(OH)2* → PhNOH* → PhNHOH* → PhNH* → PhNH2*). The rate-determining step of the entire reaction is hydrogen activation, with an energy barrier of 1.10 eV.
Recently, Lei et al. systematically studied, via DFT calculations, the hydrogenation of nitrobenzene over twelve late transition metal SACs supported on nitrogen-doped carbon (M1-N4/C, M = Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, Au)[13]. The results revealed that H2 dissociation modes are significantly influenced by the nature of the catalytic sites - including the metal center (M1), M1-N coordination structures, and M1-N-C ternary configurations - thereby modulating the entire reaction pathway. Specifically, Fe, Ru, Os, Ir, and Pt favor homolytic dissociation at the M1; Co, Ni, Cu, Rh, Ag, and Au tend toward heterolytic dissociation at the M1-N site; while Pd prefers heterolytic cleavage at the M1-N-C site. Further studies revealed that co-adsorption of PhNO2 and H2 in some systems can significantly lower the energy barrier for H2 dissociation, demonstrating a pronounced synergistic promotional effect. Overall, the cleavage of the second N-O bond in the PhNOH* intermediate was identified as a key step across various catalysts. Upon comparing the energy barriers of H2 dissociation and this critical N-O bond cleavage among different catalyst systems, it was found that Au, Cu, Ni, Pd, Pt, and Co-based catalysts are primarily limited by H2 activation, while Ru, Ag, Fe, Ir, Rh, and Os-based catalysts are mainly controlled by N-O bond cleavage. Notably, Ru1-N4/C exhibited the lowest activation energies for both key steps (0.11 eV and 0.99 eV, respectively) [Figure 10F].
In summary, the vast majority of SACs in the hydrogenation of nitrobenzene still follow the classical Haber mechanism, with reaction pathways largely consistent with those of traditional nanosized metal catalysts. However, differences in size effects and electronic structures lead to variations in specific reaction pathways. For instance, due to spatial constraints at single-atom active sites, simultaneous efficient adsorption of H2 and substrate molecules, as observed on NP surfaces, is often not possible. This results in H2 activation and substrate adsorption occurring at different sites, forming a spatial division of labor or a bifunctional synergistic mechanism. Moreover, single-atom catalytic systems are highly sensitive to the reaction microenvironment, where factors such as water molecules have significant impacts on their catalytic performance, and also their regulation strategies differ from those of NP catalysts. For example, the presence of water molecules not only lowers the energy barrier for the critical N-O bond cleavage but also accelerates the reaction by stabilizing transition states or facilitating proton transfer. Meanwhile, defect structures on the support (such as oxygen vacancies in CeO2) dynamically form and heal during the reaction, modulating the mode of H2 activation (heterolytic or homolytic cleavage) and the stability of intermediates. For SACs, such microenvironmental regulation often directly influences the priority of reaction pathways.
CONCLUSIONS AND OUTLOOK
SACs, characterized by atomically dispersed metal centers and unsaturated coordination structure, possess high specific activity and hold great promise in the catalytic hydrogenation of nitroarenes. Still, they can activate hydrogen via heterolytic dissociation, generating polarized hydrogen species (Hδ+ and Hδ-), which facilitates the selective reduction of polar functional groups such as -NO2. In addition, the well-defined structure of SACs provides an ideal model for elucidating hydrogenation mechanisms and reaction pathways, thereby informing the rational design of efficient hydrogenation catalysts. Although notable progress has been made in recent years, the catalytic performance and mechanisms of SACs in nitro compound reduction remain inadequately understood, necessitating more systematic investigation.
This review comprehensively summarizes recent advances in SACs for the hydrogenation of nitro compounds. It begins by outlining the structural features and inherent advantages of SACs over NP catalysts in the catalytic hydrogenation of nitro compounds. Subsequently, the synthesis methods and characterization techniques are briefly discussed. The synthesis strategies are categorized into top-down and bottom-up approaches. Characterization techniques are divided into static and dynamic methods, primarily including electron microscopy, spectroscopic techniques, and other analytical tools. Thirdly, the main strategies for optimizing the catalytic performance of SACs are discussed, with a focus on the effects of the single-atom sites (e.g., metal species, local environment), metal atom loading, and utilization efficiency on the hydrogenation of nitroarenes. In detail, the electronic structure and geometrical configuration of the metal center could exert a significant influence on the activity and selectivity of the catalytic process. In nitro compound hydrogenation reactions, single-atom sites exhibit distinct advantages over their nanoscale counterparts across various metal species. Moreover, compared with conventional SACs, dual-atom catalysts not only retain the benefits of isolated metal sites but also introduce various synergistic interactions between different metal atoms, exhibiting a “1 + 1 > 2” effect. Besides, rational modulation of the microenvironment of metal sites - such as tuning the support material or coordination sphere - can enhance selectivity and effectively suppress atom migration and aggregation, thereby improving reaction kinetics and enhancing catalyst stability. In addition, moderate increases in metal loading can improve the catalytic activity per unit mass; however, excessive loading may lead to the agglomeration of metal atoms, compromising their catalytic performance. Therefore, achieving a synergistic balance between high metal loading and atomic dispersion remains a key challenge in designing high-performance SACs. Ultimately, optimizing both the support architecture and the interfacial architecture can enable strong anchoring and well-ordered dispersion of metal atoms on supports, thereby improving the loading amounts and utilization of single-atom sites. Finally, we discussed the mechanistic studies of nitro compound hydrogenation catalyzed by SACs. Typically, the reaction proceeds via direct hydrogenation; however, the specific pathway varies with the metal center and reaction environment. H2 undergoes heterolytic cleavage at single-atom sites with a higher energy barrier due to geometric constraints, unlike at nanosized metal sites. Nitro compounds generally adsorb at single-atom sites in a vertical or tilted configuration. Moreover, simultaneous adsorption of H2 and nitro compounds at single-atom sites is unlikely owing to steric constraints. In SAC systems, H2 dissociation is usually rate-determining, although hydrogenation of the first N-O bond can be rate-limiting in some instances, particularly for non‑noble metals. Despite significant progress in designing high-performance SACs for the transformation of nitro compounds, the development of practical catalysts with enhanced activity and cost-effectiveness remains a major challenge.
First, for those catalysts that can be applied to nitro compound hydrogenation, several key challenges in their synthesis should be taken into consideration. On the one hand, among non-noble SACs, carbon-supported iron group metals (Fe, Co, Ni), particularly Co, are the most promising alternatives to noble metal catalysts due to their catalytic efficiency and lower cost. However, most Co SACs are prepared by in situ pyrolysis, which affords low carbon yields and limited single-atom site loadings, thus increasing preparation costs. For example, imidazole-based precursors typically deliver ~ 10% carbon yield, indicating substantial precursor loss as gaseous products during pyrolysis and failure to form the carbon support.
Moreover, micropore-hosted single-atom sites are often encapsulated by carbon layers, which compromises their utilization efficiency. Still, these micropores will become blocked by nitro compounds during reactions, embedding the active sites and thereby reducing the recyclability of SACs. These factors increase the cost of using carbon-supported iron group metals (SACs) in nitro compound hydrogenation, in some cases even exceeding that of widely used Pt/C catalysts. This is particularly the case when considering the complete lifecycle of catalysts. For noble-metal-based catalysts, e.g., Pt/C, the primary expense is platinum metals, which can be recovered by burning off the carbon support. In contrast, for non-noble metal SACs synthesized via in situ pyrolysis, the primary cost arises from the carbon support, which is not readily recyclable. Considering the above, there is an urgent need for synthetic strategies that enable the low-cost preparation of carbon-supported non-noble SACs with high site utilization and loading capacity, thereby advancing practical applications. As such, post-loading methods offer promising solutions by dispersing single-atom sites on the surface of carbon supports, a strategy widely employed for noble-metal catalysts using inexpensive, pre-synthesized carbon materials. More significantly, this method facilitates large-scale catalyst production, a limitation of in situ pyrolysis approaches. However, applying post-loading methods to non-noble metals remains challenging, as metals such as Co are prone to oxidation and aggregation, which limits the formation of high-efficiency active sites. To address this issue, gas-assisted migration strategies can be employed - for example, by introducing ammonia (NH3) or ammonium salts (e.g., NH4+). This approach not only introduces nitrogen into the carbon support to create anchoring sites for the metal atoms, but also promotes the dispersion of active sites. Furthermore, it helps prevent the formation of carbon overcoats derived from the decomposition of traditional carbon-containing small molecules at high temperatures, thereby favoring the generation of more exposed active sites.
On the other hand, noble metal-based catalysts, especially Pt/C, remain the most widely used catalysts for the hydrogenation of nitro compounds in industry. However, the catalytic performance of Pt SACs is still unsatisfactory due to the relatively low loading amount, as there is a weak interaction between Pt and the carbon matrices. Although polar supports, such as metal oxides, can accommodate more Pt single atoms due to the stronger metal-support interactions, their catalytic performance in gas-liquid-solid hydrogenation reactions still falls short of the requirements for industrial applications. This is partly attributable to the fact that H2 undergoes heterolytic cleavage at single-atom sites, which requires a significantly higher energy barrier than the homolytic cleavage observed at nanoscale counterparts. Additionally, the recovery of metal oxide-supported SACs also poses challenges. Besides, the excellent catalytic performance in gas-liquid-solid systems arises from their small size, making it difficult to separate them from reactants using conventional methods such as filtration or just static settlement in industrial applications. To address these challenges, designing catalysts with reduced Pt content and a combination of single-atom and nanoscale metal sites offers a promising approach. The synergistic interaction between distinct active sites can enhance the catalytic performance of noble metal catalysts supported on metal oxides. Additionally, composite supports that integrate metal oxides and carbon matrices further improve catalyst properties by combining the advantages of both components. Finally, regarding the recycling experiment, enhancing the catalyst volume or density is a key direction for improving recyclability. Of course, developing advanced separation techniques is also highly alluring.
Secondly, non-protic solvents, such as toluene and dimethylbenzene, are commonly employed in the hydrogenation of nitro compounds and their derivatives to facilitate product separation and increase substrate solubility. However, hydrogenation catalyzed by non-noble metal SACs typically requires protic solvents, such as water, due to reduced activity in non-protic environments. This is because the hydrogenation reaction in gas-liquid-solid three-phase systems proceeds via a classical proton-coupled electron transfer mechanism. Protic solvents, such as water, facilitate such a process, thereby accelerating the hydrogenation reaction. Thus, enhancing the performance of carbon-supported non-noble metal SACs in non-polar solvents remains a significant research challenge. Introducing polar components or functional groups may offer a solution by promoting hydrogen spillover and increasing the coverage of active hydrogen on the surface of carbon-supported catalysts.
Thirdly, the majority of recently developed SACs are nanoscale materials, with their performance predominantly evaluated in batch reactor systems. While these small-sized SACs demonstrate exceptional catalytic activity in such environments due to their high surface area and rich, accessible active sites, their suitability for continuous-flow reactors - an essential aspect for industrial applications - mains ambiguous. In continuous-flow systems, such as fixed-bed reactors, SACs typically undergo a molding process to facilitate recyclability, a procedure that frequently compromises their catalytic performance. Consequently, advancing SACs for industrial applications requires greater emphasis on reaction engineering and catalyst shaping methodologies.
In addition, the elucidation of hydrogenation mechanisms over SACs remains limited. The current understanding is largely extrapolated from Haber’s classical macroscopic framework, which is based on electrochemical measurements[177]. Whether these principles apply to SACs - particularly in thermally driven, solvent-mediated reactions - remains unverified due to the complexity and heterogeneity of isolated active sites. Resolving these uncertainties is crucial for the rational design of high-performance SACs and the development of advanced hydrogenation techniques. This requires not only the synthesis of SACs with well-defined active sites but also the advancement of characterization techniques capable of resolving the structures and dynamical changes of single-atom sites under working conditions. However, accurately identifying single-atom active sites at the atomic scale remains challenging. For example, while XAFS spectroscopy is widely used to probe the local coordination environment, its limited ability to differentiate elements with similar atomic numbers hinders the precise determination of coordination structures and elemental identity. Consequently, complementary characterization methods with higher spatial and elemental resolution are necessary.
Finally, the development of high-performance, low-cost catalysts remains a primary goal in catalysis research. Traditional experiment-driven approaches are often inefficient and resource-intensive. In recent years, advances in materials informatics, integrating DFT, machine learning, and experimental data, have enabled high-throughput screening and theoretical prediction in the synthesis and application of SACs. The future development of SACs can be advanced through several specific approaches. Leveraging artificial intelligence (AI) and machine learning techniques, models can be constructed based on large-scale DFT calculation data to accurately predict the electronic structure and geometric structure of single-atom sites. This, together with AI-driven molecular dynamics simulation, mass transfer process fitting, etc., can help to evaluate the overall catalytic performance of single-atom active sites, thereby efficiently screening catalyst candidates with superior activity and stability. By integrating automated high-throughput synthesis and characterization platforms, a closed loop between theoretical calculation and experimental validation can be established to accelerate the discovery and optimization of novel SACs. Meanwhile, the introduction of microchannel or fluidized-bed reactors significantly enhances mass transfer efficiency and catalytic activity. In addition, combining advanced online monitoring techniques (such as IR spectroscopy and electrochemical sensors) with intelligent control systems allows real-time monitoring and dynamic adjustment of the reaction process, ensuring sustained catalytic activity and selectivity.
DECLARATIONS
Authors’ contributions
Writing-original draft: Cui, X.
Validation, review and editing: Liu, K.; Han, Y.; Wang, L.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (Nos. 22478363, 22238013, 22008221, and 22404043), the Excellent Young Scientists Fund of Henan Province (Grant No. 252300421177), the Key Scientific Research Projects of Colleges and Universities in Henan Province (25A530001), the Postdoctoral Fellowship Program of CPSF (GZC20240417) and the Start-up Grant of Henan University of Technology (2023BS023).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Formenti, D.; Ferretti, F.; Scharnagl, F. K.; Beller, M. Reduction of nitro compounds using 3d-non-noble metal catalysts. Chem. Rev. 2019, 119, 2611-80.
3. Blaser, H.; Steiner, H.; Studer, M. Selective catalytic hydrogenation of functionalized nitroarenes: an update. ChemCatChem 2009, 1, 210-21.
4. Yu, M.; Ouyang, D.; Wang, L.; Liu, Y. N. Catalytic reduction of aromatic nitro compounds to phenylhydroxylamine and its derivatives. Molecules 2024, 29, 4353.
5. Wang, H.; Zhang, W.; Liu, Y.; Pu, M.; Lei, M. First-Principles study on the mechanism of nitrobenzene reduction to aniline catalyzed by a N-doped carbon-supported cobalt single-atom catalyst. J. Phys. Chem. C. 2021, 125, 19171-82.
6. Corma, A.; Serna, P. Chemoselective hydrogenation of nitro compounds with supported gold catalysts. Science 2006, 313, 332-4.
7. Lin, L.; Yao, S.; Gao, R.; et al. A highly CO-tolerant atomically dispersed Pt catalyst for chemoselective hydrogenation. Nat. Nanotechnol. 2019, 14, 354-61.
8. Gao, R.; Guo, H.; Wang, B.; Qiu, P.; Sun, M.; Chen, L. Co based N, S co-doped carbon hybrids for catalytic hydrogenation: role of cobalt salt and doped S. Appl. Catal. A-Gen. 2019, 579, 99-105.
9. Gao, R.; Xu, J.; Wang, J.; et al. Pd/Fe2O3 with electronic coupling single-site Pd-Fe pair sites for low-temperature semihydrogenation of alkynes. J. Am. Chem. Soc. 2022, 144, 573-81.
10. Liu, F.; Gao, R.; Shi, C.; et al. Avoiding Sabatier’s limitation on spatially correlated Pt-Mn atomic pair sites for oxygen electroreduction. J. Am. Chem. Soc. 2023, 145, 25252-63.
11. Qiao, B.; Wang, A.; Yang, X.; et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634-41.
12. Yang, X. F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740-8.
13. Wang, H.; Shi, F.; Pu, M.; Lei, M. Theoretical study on nitrobenzene hydrogenation by N-doped carbon-supported late transition metal single-atom catalysts. ACS. Catal. 2022, 12, 11518-29.
14. Zhang, L.; Zhou, M.; Wang, A.; Zhang, T. Selective hydrogenation over supported metal catalysts: from nanoparticles to single atoms. Chem. Rev. 2020, 120, 683-733.
15. Greeley, J.; Mavrikakis, M. Alloy catalysts designed from first principles. Nat. Mater. 2004, 3, 810-5.
16. Deng, P.; Duan, J.; Liu, F.; et al. Atomic insights into synergistic nitroarene hydrogenation over nanodiamond-supported Pt1-Fe1 dual-single-atom catalyst. Angew. Chem. Int. Ed. Engl. 2023, 62, e202307853.
17. Macino, M.; Barnes, A. J.; Althahban, S. M.; et al. Tuning of catalytic sites in Pt/TiO2 catalysts for the chemoselective hydrogenation of 3-nitrostyrene. Nat. Catal. 2019, 2, 873-81.
18. Li, X.; Yang, X.; Huang, Y.; Zhang, T.; Liu, B. Supported noble-metal single atoms for heterogeneous catalysis. Adv. Mater. 2019, 31, e1902031.
19. Deelen TW, Hernández Mejía C, de Jong KP. Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat. Catal. 2019, 2, 955-70.
20. Xu, D.; Liu, R.; Li, J.; Zhao, H.; Ma, J.; Dong, Z. Atomically dispersed Co-N4 sites anchored on N-doped carbon for aqueous phase transfer hydrogenation between nitroarenes and saturated N-heterocycles. Appl. Catal. B. Environ. 2021, 299, 120681.
22. Xing, L.; Jin, Y.; Weng, Y.; et al. Top-down synthetic strategies toward single atoms on the rise. Matter 2022, 5, 788-807.
23. Chen, R.; Chen, S.; Wang, L.; Wang, D. Nanoscale metal particle modified single-atom catalyst: synthesis, characterization, and application. Adv. Mater. 2024, 36, e2304713.
24. He, T.; Chen, S.; Ni, B.; et al. Zirconium-porphyrin-based metal-organic framework hollow nanotubes for immobilization of noble-metal single atoms. Angew. Chem. Int. Ed. Engl. 2018, 57, 3493-8.
25. Xi, J.; Jung, H. S.; Xu, Y.; Xiao, F.; Bae, J. W.; Wang, S. Single-atom catalysts: synthesis strategies, catalytic applications, and performance regulation of single-atom catalysts Adv Funct Mater 2021;31:2170081.
26. Huang, Y.; Xiong, J.; Zou, Z.; Chen, Z. Emerging strategies for the synthesis of correlated single atom catalysts. Adv. Mater. 2025, 37, e2312182.
27. Xue, K.; Mo, Y.; Long, B.; et al. Single-atom catalysts supported on ordered porous materials: Synthetic strategies and applications. InfoMat 2022, 4, e12296.
28. Ding, S.; Hülsey, M. J.; Pérez-ramírez, J.; Yan, N. Transforming energy with single-atom catalysts. Joule 2019, 3, 2897-929.
29. Cheng, N.; Zhang, L.; Doyle-davis, K.; Sun, X. Single-atom catalysts: from design to application. Electrochem. Energ. Rev. 2019, 2, 539-73.
30. Li, L.; Chang, X.; Lin, X.; Zhao, Z. J.; Gong, J. Theoretical insights into single-atom catalysts. Chem. Soc. Rev. 2020, 49, 8156-78.
31. Xue, Z.; Luan, D.; Zhang, H.; Lou, XW(D). Single-atom catalysts for photocatalytic energy conversion. Joule 2022, 6, 92-133.
32. Xiong, H.; Datye, A. K.; Wang, Y. Thermally stable single-atom heterogeneous catalysts. Adv. Mater. 2021, 33, e2004319.
33. Singh, B.; Gawande, M. B.; Kute, A. D.; et al. Single-atom (iron-based) catalysts: synthesis and applications. Chem. Rev. 2021, 121, 13620-97.
34. Ye, Y.; Xu, J.; Gao, L.; et al. CuO/CeO2 catalysts prepared by modified impregnation method for ethyl acetate oxidation. Chem. Eng. J. 2023, 471, 144667.
35. Li, Y.; Yang, L.; He, H.; et al. In situ photodeposition of platinum clusters on a covalent organic framework for photocatalytic hydrogen production. Nat. Commun. 2022, 13, 1355.
36. Zhang, Z.; Chen, Y.; Zhou, L.; et al. The simplest construction of single-site catalysts by the synergism of micropore trapping and nitrogen anchoring. Nat. Commun. 2019, 10, 1657.
37. Ji, D.; Fan, L.; Li, L.; et al. Atomically transition metals on self-supported porous carbon flake arrays as binder-free air cathode for wearable zinc-air batteries. Adv. Mater. 2019, 31, e1808267.
38. Sun, G.; Zhao, Z. J.; Mu, R.; et al. Breaking the scaling relationship via thermally stable Pt/Cu single atom alloys for catalytic dehydrogenation. Nat. Commun. 2018, 9, 4454.
39. Zhang, Z.; Feng, C.; Liu, C.; et al. Electrochemical deposition as a universal route for fabricating single-atom catalysts. Nat. Commun. 2020, 11, 1215.
40. Wenderich, K.; Mul, G. Methods, mechanism, and applications of photodeposition in photocatalysis: a review. Chem. Rev. 2016, 116, 14587-619.
41. Oros-ruiz, S.; Pedraza-avella, J. A.; Guzmán, C.; et al. Effect of gold particle size and deposition method on the photodegradation of 4-chlorophenol by Au/TiO2. Top. Catal. 2011, 54, 519-26.
42. Liu, P.; Zhao, Y.; Qin, R.; et al. Photochemical route for synthesizing atomically dispersed palladium catalysts. Science 2016, 352, 797-801.
44. Libera, J.; Elam, J.; Pellin, M. Conformal ZnO coatings on high surface area silica gel using atomic layer deposition. Thin. Solid. Films. 2008, 516, 6158-66.
45. Elam, J. W.; Routkevitch, D.; Mardilovich, P. P.; George, S. M. Conformal coating on ultrahigh-aspect-ratio nanopores of anodic alumina by atomic layer deposition. Chem. Mater. 2003, 15, 3507-17.
46. Lu, J.; Fu, B.; Kung, M. C.; et al. Coking- and sintering-resistant palladium catalysts achieved through atomic layer deposition. Science 2012, 335, 1205-8.
47. Yan, H.; Cheng, H.; Yi, H.; et al. Single-atom Pd1/graphene catalyst achieved by atomic layer deposition: remarkable performance in selective hydrogenation of 1,3-butadiene. J. Am. Chem. Soc. 2015, 137, 10484-7.
48. Zwiener, L.; Girgsdies, F.; Brennecke, D.; et al. Evolution of zincian malachite synthesis by low temperature co-precipitation and its catalytic impact on the methanol synthesis. Appl. Catal. B. Environ. 2019, 249, 218-26.
49. Behrens, M. Coprecipitation: an excellent tool for the synthesis of supported metal catalysts - from the understanding of the well known recipes to new materials. Catal. Today. 2015, 246, 46-54.
50. Sun, L.; Cao, L.; Su, Y.; Wang, C.; Lin, J.; Wang, X. Ru1/FeOx single-atom catalyst with dual active sites for water gas shift reaction without methanation. Appl. Catal. B. Environ. 2022, 318, 121841.
51. Peng, J.; Yang, W.; Jia, Z.; Jiao, L.; Jiang, H. Axial coordination regulation of MOF-based single-atom Ni catalysts by halogen atoms for enhanced CO2 electroreduction. Nano. Res. 2022, 15, 10063-9.
52. Qi, H.; Yang, J.; Liu, F.; et al. Highly selective and robust single-atom catalyst Ru1/NC for reductive amination of aldehydes/ketones. Nat. Commun. 2021, 12, 3295.
53. Deng, D.; Yu, L.; Chen, X.; et al. Iron encapsulated within pod-like carbon nanotubes for oxygen reduction reaction. Angew. Chem. 2013, 52, 371-5.
54. Yang, J.; Qiu, Z.; Zhao, C.; et al.
55. Yao, Y.; Huang, Z.; Xie, P.; et al. High temperature shockwave stabilized single atoms. Nat. Nanotechnol. 2019, 14, 851-7.
56. Li, R.; Xu, J.; Zhao, Q.; et al. Cathodic corrosion as a facile and universal method for the preparation of supported metal single atoms. Nano. Res. 2022, 15, 1838-44.
57. Deng, D.; Chen, X.; Yu, L.; et al. A single iron site confined in a graphene matrix for the catalytic oxidation of benzene at room temperature. Sci. Adv. 2015, 1, e1500462.
58. Cui, X.; Xiao, J.; Wu, Y.; et al. A graphene composite material with single cobalt active sites: a highly efficient counter electrode for dye-sensitized solar cells. Angew. Chem. Int. Ed. Engl. 2016, 55, 6708-12.
59. Khan, K.; Liu, T.; Arif, M.; et al. Laser-irradiated holey graphene-supported single-atom catalyst towards hydrogen evolution and oxygen reduction. Adv. Energy. Mater. 2021, 11, 2101619.
60. James, S. L.; Adams, C. J.; Bolm, C.; et al. Mechanochemistry: opportunities for new and cleaner synthesis. Chem. Soc. Rev. 2012, 41, 413-47.
61. Yang, J.; Li, W.; Wang, D.; Li, Y. Electronic Metal-support interaction of single-atom catalysts and applications in electrocatalysis. Adv. Mater. 2020, 32, e2003300.
62. Ouyang, R.; Liu, J. X.; Li, W. X. Atomistic theory of Ostwald ripening and disintegration of supported metal particles under reaction conditions. J. Am. Chem. Soc. 2013, 135, 1760-71.
63. Xu, H.; Zhang, Z.; Liu, J.; et al. Entropy-stabilized single-atom Pd catalysts via high-entropy fluorite oxide supports. Nat. Commun. 2020, 11, 3908.
64. Liu, J.; Bunes, B. R.; Zang, L.; Wang, C. Supported single-atom catalysts: synthesis, characterization, properties, and applications. Environ. Chem. Lett. 2018, 16, 477-505.
65. Wu, M.; Zhang, G.; Wang, W.; et al. Electronic metal-support interaction modulation of single-atom electrocatalysts for rechargeable zinc-air batteries. Small. Methods. 2022, 6, e2100947.
66. Qi, P.; Wang, J.; Djitcheu, X.; He, D.; Liu, H.; Zhang, Q. Techniques for the characterization of single atom catalysts. RSC. Adv. 2021, 12, 1216-27.
67. Li, X.; Yang, X.; Zhang, J.; Huang, Y.; Liu, B.
68. Jiang, J.; Tong, M.; Shen, D.; et al. Utilizing molybdenum to tailor the electronic structure of iron through electron complementary effect for promoting oxygen reduction activity. Adv. Funct. Mater. 2025, 35, 2500065.
69. Guo, W.; Pan, M.; Xie, Q.; et al. Achieving pH-universal oxygen electrolysis via synergistic density and coordination tuning over biomass-derived Fe single-atom catalyst. Nat. Commun. 2025, 16, 2920.
70. Zhang, L.; Zong, L.; Lu, F.; et al. Metal-saloph complexes pre-coordination for Fe single atom catalyst towards oxygen reduction reaction in rechargeable quasi-solid-state Zn-air battery. Appl. Catal. B. Environ. 2025, 370, 125189.
71. Zhang, S.; Sun, B.; Liao, K.; et al. Boosting oxygen reduction reaction performance of Fe single-atom catalysts via precise control of the coordination environment. Adv. Funct. Mater. 2025, 35, 2425640.
72. Li, S.; Kan, Z.; Wang, H.; et al. Single-atom photo-catalysts: synthesis, characterization, and applications. Nano. Mater. Sci. 2024, 6, 284-304.
73. Dai, H.; Hu, T.; Zhu, S.; Zhang, Y.; Zhou, W. Regulating the Fe-Nx coordination structure of Fe single-atom catalysts for efficient catalytic degradation of methylparaben. Chem. Eng. J. 2025, 507, 160462.
74. Lan, L.; Wu, Y.; Pei, Y.; et al. High-density accessible iron single-atom catalyst for durable and temperature-adaptive laminated zinc-air batteries. Adv. Mater. 2025, 37, e2417711.
75. Wang, C.; Chen, B.; Ren, H.; et al. Stabilizing Ni single-atom sites through introducing low-valence Ni Species for durably efficient electrochemical CO2 reduction. Appl. Catal. B. Environ. 2025, 368, 125151.
76. Zhang, Z. Q.; Duan, P. J.; Bai, C. W.; Chen, X. J.; Wang, J.; Chen, F. Surface-hydroxylated single-atom catalyst with an isolated Co-O-Zn configuration achieves high selectivity in regulating active species. Nat. Commun. 2025, 16, 2376.
77. Gallenkamp, C.; Kramm, U. I.; Proppe, J.; Krewald, V. Calibration of computational Mössbauer spectroscopy to unravel active sites in FeNC catalysts for the oxygen reduction reaction. Int. J. Quantum. Chem. 2021, 121, e26394.
78. Bates, J. S.; Martinez, J. J.; Hall, M. N.; et al. Chemical kinetic method for active-site quantification in Fe-N-C catalysts and correlation with molecular probe and spectroscopic site-counting methods. J. Am. Chem. Soc. 2023, 145, 26222-37.
79. Wu, B.; Yang, H.; Li, L.; et al. Integrating PtCo intermetallic with highly graphitized carbon toward durable oxygen electroreduction in proton exchange membrane fuel cells. Adv. Mater. 2025, 37, e2500096.
80. Zhao, J.; Deng, Q.; Avdoshenko, S. M.; Fu, L.; Eckert, J.; Rümmeli, M. H. Direct in situ observations of single Fe atom catalytic processes and anomalous diffusion at graphene edges. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 15641-6.
81. Guo, X.; Qi, W.; Liu, W.; et al. Oxidative dehydrogenation on nanocarbon: revealing the catalytic mechanism using model catalysts. ACS. Catal. 2017, 7, 1424-7.
82. Pan, L.; Wang, J.; Lu, F.; et al. Single-atom or dual-atom in TiO2 nanosheet: which is the better choice for electrocatalytic urea synthesis? Angew. Chem. Int. Ed. Engl. 2023, 62, e202216835.
83. Li, X.; Liu, L.; Ren, X.; Gao, J.; Huang, Y.; Liu, B. Microenvironment modulation of single-atom catalysts and their roles in electrochemical energy conversion. Sci. Adv. 2020, 6.
84. Wei, H.; Liu, X.; Wang, A.; et al. FeOx-supported platinum single-atom and pseudo-single-atom catalysts for chemoselective hydrogenation of functionalized nitroarenes. Nat. Commun. 2014, 5, 5634.
85. Ren, Y.; Tang, Y.; Zhang, L.; et al. Unraveling the coordination structure-performance relationship in Pt1/Fe2O3 single-atom catalyst. Nat. Commun. 2019, 10, 4500.
86. Vilé, G.; Albani, D.; Nachtegaal, M.; et al. A stable single-site palladium catalyst for hydrogenations. Angew. Chem. Int. Ed. Engl. 2015, 54, 11265-9.
87. Li, J.; Zhong, L.; Tong, L.; et al. Atomic Pd on graphdiyne/graphene heterostructure as efficient catalyst for aromatic nitroreduction. Adv. Funct. Mater. 2019, 29, 1905423.
88. Dasgupta, A.; He, H.; Gong, R.; et al. Atomic control of active-site ensembles in ordered alloys to enhance hydrogenation selectivity. Nat. Chem. 2022, 14, 523-9.
89. Yang, J.; Yang, L.; Zhang, L.; et al. Hydrogenation reactions with synergistic catalysis of Pd single atoms and nanoparticles under near-ambient conditions. Chemistry 2023, 29, e202203108.
90. Shen, X.; Liu, K. Discrete Au1(0) stabilized by 15-crown-5 for high-efficiency catalytic reduction of nitrophenol and nitroaniline. Catalysts 2023, 13, 776.
91. Fu, H.; Zhang, H.; Yang, G.; et al. Highly dispersed rhodium atoms supported on defect-rich Co(OH)2 for the chemoselective hydrogenation of nitroarenes. New. J. Chem. 2022, 46, 1158-67.
92. Wang, W.; Lin, L.; Qi, H.; et al. MIL-53 (Al) derived single-atom Rh catalyst for the selective hydrogenation of m-chloronitrobenzene into m-chloroaniline. Chinese. J. Catal. 2021, 42, 824-34.
93. Wu, B.; Lin, T.; Yang, R.; et al. Ru single atoms for efficient chemoselective hydrogenation of nitrobenzene to azoxybenzene. Green. Chem. 2021, 23, 4753-61.
94. Ma, Y.; Ren, Y.; Zhou, Y.; et al. High-density and thermally stable palladium single-atom catalysts for chemoselective hydrogenations. Angew. Chem. Int. Ed. Engl. 2020, 59, 21613-9.
95. Campbell, C. T. Physics. The active site in nanoparticle gold catalysis. Science 2004, 306, 234-5.
96. Sun, Q.; Wang, N.; Zhang, T.; et al. Inside cover: zeolite-encaged single-atom rhodium catalysts: highlyefficient hydrogen generation and shape-selective tandem hydrogenation of nitroarenes. Angew. Chem. Int. Ed. 2019, 58, 18300.
97. Li, H.; Sun, Z.; Fan, Y.; et al. Enhancing hydride formation and transfer for catalytic hydrogenation via electron-deficient single-atom silver. J. Colloid. Interface. Sci. 2025, 682, 751-9.
98. Junge, K.; Wendt, B.; Shaikh, N.; Beller, M. Iron-catalyzed selective reduction of nitroarenes to anilines using organosilanes. Chem. Commun. (Camb). 2010, 46, 1769-71.
99. Liu, W.; Zhang, L.; Yan, W.; et al. Single-atom dispersed Co-N-C catalyst: structure identification and performance for hydrogenative coupling of nitroarenes. Chem. Sci. 2016, 7, 5758-64.
100. Zhou, P.; Jiang, L.; Wang, F.; Deng, K.; Lv, K.; Zhang, Z. High performance of a cobalt-nitrogen complex for the reduction and reductive coupling of nitro compounds into amines and their derivatives. Sci. Adv. 2017, 3, e1601945.
101. Cheng, T.; Yu, H.; Peng, F.; Wang, H.; Zhang, B.; Su, D. Identifying active sites of CoNC/CNT from pyrolysis of molecularly defined complexes for oxidative esterification and hydrogenation reactions. Catal. Sci. Technol. 2016, 6, 1007-15.
102. Wei, X.; Hu, Z.; Li, C.; et al. High-density atomically dispersed CoNx catalysts supported on nitrogen-doped mesoporous carbon materials for efficient hydrogenation of nitro compounds. Catal. Today. 2022, 405-406, 92-100.
103. Sheng, Y.; Wang, X.; Yue, S.; Cheng, G.; Zou, X.; Lu, X.
104. Li, M.; Chen, S.; Jiang, Q.; et al. Origin of the activity of Co-N-C catalysts for chemoselective hydrogenation of nitroarenes. ACS. Catal. 2021, 11, 3026-39.
105. Liu, X.; Wang, C.; Meng, J.; et al. Single-atom cobalt catalysts for chemoselective hydrogenation of nitroarenes to anilines. Chin. Chem. Lett. 2023, 34, 108745.
106. Li, A. Y.; Pedersen, A.; Feng, J.; et al. From haemoglobin to single-site hydrogenation catalyst. Green. Chem. 2022, 24, 7574-83.
107. Zhang, T.; Xie, Z.; Jiang, L.; et al. Selective transfer hydrogenation coupling of nitroaromatics to azoxy/azo compounds by electron-enriched single Ni-N4 sites on mesoporous N-doped carbon. Chem. Eng. J. 2022, 443, 136416.
108. Liang, J.; Song, Q.; Wu, J.; et al. Anchoring copper single atoms on porous boron nitride nanofiber to boost selective reduction of nitroaromatics. ACS. Nano. 2022, 16, 4152-61.
109. Yang, F.; Wang, M.; Liu, W.; et al. Atomically dispersed Ni as the active site towards selective hydrogenation of nitroarenes. Green. Chem. 2019, 21, 704-11.
110. Liu, W.; Chen, Y.; Qi, H.; et al. A durable nickel single-atom catalyst for hydrogenation reactions and cellulose valorization under harsh conditions. Angew. Chem. Int. Ed. Engl. 2018, 57, 7071-5.
111. Qin, L.; Chen, W.; Lai, C.; et al. Highly efficient reduction of nitrophenols by Fe-N-C single-atom catalyst: performance and mechanism insights. J. Environl. Chem. Eng. 2023, 11, 110278.
112. Cheong, W. C.; Yang, W.; Zhang, J.; et al. Isolated iron single-atomic site-catalyzed chemoselective transfer hydrogenation of nitroarenes to arylamines. ACS. Appl. Mater. Interfaces. 2019, 11, 33819-24.
113. Lu, G.; Sun, K.; Lin, Y.; et al. Single-atomic-site iron on N-doped carbon for chemoselective reduction of nitroarenes. Nano. Res. 2022, 15, 603-11.
114. Song, Y.; Guo, R.; Feng, B.; et al. Coordination number engineering of Zn single-atom sites for enhanced transfer hydrogenation performance. Chem. Eng. J. 2023, 465, 142920.
115. Yang, X.; Xu, L.; Li, Y. Do we achieve “1 + 1 > 2” in dual-atom or dual-single-atom catalysts? Coord. Chem. Rev. 2024, 516, 215961.
116. Zhou, D.; Zeng, K.; Wang, L.; Tang, F. The d-p electron coupling over the unsaturated oxygen coordinated CuCo alloy surface for enhanced N-heteroarenes hydrogenation under mild conditions. J. Energy. Chem. 2025, 106, 671-80.
117. Yang, Y.; Zhang, W.; Wu, G.; et al. Electronic structure tuning in Cu-Co dual single atom catalysts for enhanced COOH* spillover and electrocalytic CO2 reduction activity. Angew. Chem. Int. Ed. Engl. 2025, 64, e202504423.
118. Liu, H.; Zhu, P.; Yang, D.; et al. Pd-Mn/NC dual single-atomic sites with hollow mesopores for the highly efficient semihydrogenation of phenylacetylene. J. Am. Chem. Soc. 2024, 146, 2132-40.
119. Park, D.; Hong, S.; Han, J.; et al. Insights into the synergy effect in dual single-atom catalysts on defective CeO2 under CO2 hydrogenation. Appl. Catal. B. Environ. 2025, 365, 124987.
120. Gong, L.; Qiu, L.; Xing, X.; et al. Coupling Fe-Co atomic pair to promote the selective reduction of nitroaromatics under mild conditions. Sci. Total. Environ. 2024, 912, 169161.
121. Mu, Y.; Wang, T.; Zhang, J.; Meng, C.; Zhang, Y.; Kou, Z. Single-atom catalysts: advances and challenges in metal-support interactions for enhanced electrocatalysis. Electrochem. Energy. Rev. 2022, 5, 145-86.
122. Lin, L.; Chen, Z.; Chen, W. Single atom catalysts by atomic diffusion strategy. Nano. Res. 2021, 14, 4398-416.
123. Lou, Y.; Wu, H.; Liu, J. Nanocarbon-edge-anchored high-density Pt atoms for 3-nitrostyrene hydrogenation: strong metal-carbon interaction. iScience 2019, 13, 190-8.
124. Zhang, G.; Tang, F.; Wang, X.; An, P.; Wang, L.; Liu, Y. Co,N-codoped porous carbon-supported Coy ZnS with superior activity for nitroarene hydrogenation. ACS. Sustain. Chem. Eng. 2020, 8, 6118-26.
125. Wei, X.; Zhang, Z.; Zhou, M.; Zhang, A.; Wu, W. D.; Wu, Z. Solid-state nanocasting synthesis of ordered mesoporous CoNx-carbon catalysts for highly efficient hydrogenation of nitro compounds. Nanoscale 2018, 10, 16839-47.
126. Li, H.; Cao, C.; Liu, J.; et al. Cobalt single atoms anchored on N-doped ultrathin carbon nanosheets for selective transfer hydrogenation of nitroarenes. Sci. China. Mater. 2019, 62, 1306-14.
127. Li, J.; Chen, S.; Liu, M.; et al. Self-template construction of high-performance Co, N-decorated carbon nanotubes from a novel cobalt dicyandiamide molecule. ChemCatChem 2021, 13, 2609-17.
128. Yan, X.; Duan, P.; Zhang, F.; et al. Stable single-atom platinum catalyst trapped in carbon onion graphitic shells for improved chemoselective hydrogenation of nitroarenes. Carbon 2019, 143, 378-84.
129. Gu, Y.; Wu, A.; Wang, L.; et al. A “competitive occupancy” strategy toward Co-N4 single-atom catalysts embedded in 2D TiN/rGO sheets for highly efficient and stable aromatic nitroreduction. J. Mater. Chem. A. 2020, 8, 4807-15.
130. Yan, H.; Zhao, X.; Guo, N.; et al. Atomic engineering of high-density isolated Co atoms on graphene with proximal-atom controlled reaction selectivity. Nat. Commun. 2018, 9, 3197.
131. Xi, J.; Sun, H.; Wang, D.; et al. Confined-interface-directed synthesis of Palladium single-atom catalysts on graphene/amorphous carbon. Appl. Catal. B. Environ. 2018, 225, 291-7.
132. Ma, T.; Tan, X.; Zhao, Q.; et al. Template-oriented synthesis of Fe-N-codoped graphene nanoshells derived from petroleum pitch for efficient nitroaromatics reduction. Ind. Eng. Chem. Res. 2020, 59, 129-36.
133. Wang, C.; Ye, B.; Zhou, R.; Jiang, Y.; Hou, Z. Structure-activity relationship for the catalytic hydrogenation of nitrobenzene by single platinum atoms supported on nitrogen-doped carbon. ACS. Appl. Nano. Mater. 2022, 5, 13601-11.
134. Chen, J.; Yao, Y.; Zhao, J.; et al. A highly active non-precious metal catalyst based on Fe-N-C@CNTs for nitroarene reduction. RSC. Adv. 2016, 6, 96203-9.
135. Jones, J.; Xiong, H.; DeLaRiva, A. T.; et al. Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science 2016, 353, 150-4.
136. Wang, C.; Mao, S.; Wang, Z.; et al. Insight into single-atom-induced unconventional size dependence over CeO2-supported Pt catalysts. Chem 2020, 6, 752-65.
137. Han, B.; Guo, Y.; Huang, Y.; et al. Strong metal-support interactions between Pt single atoms and TiO2. Angew. Chem. Int. Ed. Engl. 2020, 59, 11824-9.
138. Ye, T. N.; Xiao, Z.; Li, J.; et al. Stable single platinum atoms trapped in sub-nanometer cavities in 12CaO·7Al2O3 for chemoselective hydrogenation of nitroarenes. Nat. Commun. 2020, 11, 1020.
139. Li, Z.; Zhang, M.; Dong, X.; et al. Strong electronic interaction of indium oxide with palladium single atoms induced by quenching toward enhanced hydrogenation of nitrobenzene. Appl. Catal. B. Environ. 2022, 313, 121462.
140. Shi, X.; Wang, X.; Shang, X.; Zou, X.; Ding, W.; Lu, X. High performance and active sites of a ceria-supported palladium catalyst for solvent-free chemoselective hydrogenation of nitroarenes. ChemCatChem 2017, 9, 3743-51.
141. Wang, J.; Zhang, Y.; Xu, X.; Bao, M. Oxygen vacancy-rich Ni-CeO2 heterojunction catalyst for hydrogenating halogenated nitroarenes with high activity and selectivity. ACS. Appl. Mater. Interfaces. 2023, 15, 8149-56.
142. Liu, R.; Zhang, L. Q.; Yu, C.; Sun, M. T.; Liu, J. F.; Jiang, G. B. Atomic-level-designed catalytically active palladium atoms on ultrathin gold nanowires. Adv. Mater. 2017, 29.
143. Ji, P.; Manna, K.; Lin, Z.; et al. Single-site cobalt catalysts at new Zr12(μ3-O)8(μ3)-OH)8(μ2-OH)6 metal-organic framework nodes for highly active hydrogenation of nitroarenes, nitriles, and isocyanides. J. Am. Chem. Soc. 2017, 139, 7004-11.
144. Antil, N.; Kumar, A.; Akhtar, N.; et al. Aluminum metal-organic framework-ligated single-site nickel(II)-hydride for heterogeneous chemoselective catalysis. ACS. Catal. 2021, 11, 3943-57.
145. Sun, X.; Olivos-suarez, A. I.; Oar-arteta, L.; et al. Metal-organic framework mediated cobalt/nitrogen-doped carbon hybrids as efficient and chemoselective catalysts for the hydrogenation of nitroarenes. ChemCatChem 2017, 9, 1854-62.
146. Zhang, M.; Liu, Y.; Zhao, H.; et al. Pd anchored on a phytic acid/thiourea polymer as a highly active and stable catalyst for the reduction of nitroarene. ACS. Appl. Mater. Interfaces. 2021, 13, 19904-14.
147. He, J.; Li, N.; Li, Z.; et al. Strategic defect engineering of metal-organic frameworks for optimizing the fabrication of single-atom catalysts. Adv. Funct. Mater. 2021, 31, 2103597.
148. Peng, Y.; Geng, Z.; Zhao, S.; et al. Pt single atoms embedded in the surface of Ni nanocrystals as highly active catalysts for selective hydrogenation of nitro compounds. Nano. Lett. 2018, 18, 3785-91.
149. Chugh, V.; Chatterjee, B.; Chang, W. C.; et al. An adaptive rhodium catalyst to control the hydrogenation network of nitroarenes. Angew. Chem. Int. Ed. Engl. 2022, 61, e202205515.
150. Zhang, H.; Zhang, X.; Sun, Q.; He, Q.; Ji, H.; He, X. Boosting hydrogenation properties of Pt single-atom catalysts via tailoring the electronic structures by coordination number regulation. Chem. Eng. J. 2023, 455, 140808.
151. Zhao, X.; Wang, L.; Zhang, G.; et al. Breaking the trade-off between CO tolerance and intrinsic activity in hydrogenation on metal oxide-supported noble metal single atoms through coordination environment engineering. ACS. Catal. 2024, 14, 15325-35.
152. Feng, B.; Guo, R.; Cai, Q.; et al. Construction of isolated Ni sites on nitrogen-doped hollow carbon spheres with Ni-N3 configuration for enhanced reduction of nitroarenes. Nano. Res. 2022, 15, 6001-9.
153. Wang, L.; Zhu, C.; Xu, M.; et al. Boosting activity and stability of metal single-atom catalysts via regulation of coordination number and local composition. J. Am. Chem. Soc. 2021, 143, 18854-8.
154. Tian, S.; Hu, M.; Xu, Q.; et al. Single-atom Fe with Fe1N3 structure showing superior performances for both hydrogenation and transfer hydrogenation of nitrobenzene. Sci. China. Mater. 2021, 64, 642-50.
155. Li, X.; She, W.; Wang, J.; Li, W.; Li, G. Highly efficient N-doped carbon supported FeSx-Fe2O3 catalyst for hydrogenation of nitroarenes via pyrolysis of sulfurized N,Fe-containing MOFs. Appl. Organomet. Chem. 2021, 35, e6294.
156. Jin, H.; Li, P.; Cui, P.; et al. Unprecedentedly high activity and selectivity for hydrogenation of nitroarenes with single atomic
157. Cao, F.; Ni, W.; Zhao, Q.; et al. Precisely manipulating the local coordination of cobalt single-atom catalyst boosts selective hydrogenation of nitroarenes. Appl. Catal. B. Environ. 2024, 346, 123762.
158. Zhang, G.; Tang, F.; Wang, X.; Wang, L.; Liu, Y. Atomically dispersed Co-S-N active sites anchored on hierarchically porous carbon for efficient catalytic hydrogenation of nitro compounds. ACS. Catal. 2022, 12, 5786-94.
159. Zhao, Q.; Xu, D.; Wang, L.; et al. A core-shell confinement strategy towards single-atom Fe-N/S-C bifunctional catalyst for selective nitroarene reduction and olefin epoxidation. J. Alloy. Compd. 2025, 1012, 178488.
160. Tang, F.; Zhang, G.; Wang, L.; Huang, J.; Liu, Y. Unsymmetrically N, S-coordinated single-atom cobalt with electron redistribution for catalytic hydrogenation of quinolines. J. Catal. 2022, 414, 101-8.
161. Chen, G.; Gu, J.; Gong, W.; et al. Precisely tailoring the second coordination sphere of a cobalt single-atom catalyst for selective hydrogenation of halogenated nitroarenes. Angew. Chem. Int. Ed. Engl. 2025, 64, e202421277.
162. Duan, Y.; Xia, Y.; Ling, Y.; et al. Regulating second-shell coordination in cobalt single-atom catalysts toward highly selective hydrogenation. ACS. Nano. 2024, 18, 21326-35.
163. Wei, H.; Ren, Y.; Wang, A.; et al. Remarkable effect of alkalis on the chemoselective hydrogenation of functionalized nitroarenes over high-loading Pt/FeOx catalysts. Chem. Sci. 2017, 8, 5126-31.
164. Iihama, S.; Furukawa, S.; Komatsu, T. Efficient catalytic system for chemoselective hydrogenation of halonitrobenzene to haloaniline using PtZn intermetallic compound. ACS. Catal. 2016, 6, 742-6.
165. Zhang, B.; Asakura, H.; Zhang, J.; Zhang, J.; De, S.; Yan, N. Stabilizing a platinum1 single-atom catalyst on supported phosphomolybdic acid without compromising hydrogenation activity. Angew. Chem. Int. Ed. Engl. 2016, 55, 8319-23.
166. Zhang, F.; Li, J.; Liu, P.; et al. Ultra-high loading single CoN3 sites in N-doped graphene-like carbon for efficient transfer hydrogenation of nitroaromatics. J. Catal. 2021, 400, 40-9.
167. Lin, Y.; Nie, R.; Li, Y.; et al. Highly efficient and anti-poisoning single-atom cobalt catalyst for selective hydrogenation of nitroarenes. Nano. Res. 2022, 15, 10006-13.
168. Wang, C.; Dang, J.; Han, Y.; Zeng, Q.; Wang, L. Carbon nanotube-supported Co-N-C with enriched mesopores for hydrogenation of nitro compounds. J. Catal. 2025, 447, 116152.
169. Guo, M.; Meng, Q.; Gao, M. L.; et al. Single-atom Pt loaded on MOF-derived porous TiO2 with maxim-ized Pt atom utilization for selective hydrogenation of halonitro-benzene. Angew. Chem. Int. Ed. Engl. 2025, 64, e202418964.
170. Cao, F.; Zhao, Q.; Tan, X.; et al. A pre-coordinated strategy precisely tailors the coordination structure of single-atom sites toward efficient catalysis. Adv. Funct. Mater. 2025, 35, 2423398.
171. Sun, X.; Olivos-suarez, A. I.; Osadchii, D.; Romero, M. J. V.; Kapteijn, F.; Gascon, J. Single cobalt sites in mesoporous N-doped carbon matrix for selective catalytic hydrogenation of nitroarenes. J. Catal. 2018, 357, 20-8.
172. Lian, L.; Zhang, G.; Zhao, X.; Huang, J.; Wang, L.; Liu, Y. Protein-metal ion networks coated carbon matrix as a precursor: to construct carbon-supported Mo-based catalysts with highly exposed active sites for hydrogenation of nitro compounds. New. J. Chem. 2024, 48, 8331-7.
173. Chen, R.; Wang, X.; Dang, J.; et al. Shedding light on the reversible deactivation of carbon-supported single-atom catalysts in hydrogenation reaction. Nano. Res. 2024, 17, 4807-14.
174. Huang, H.; Han, X.; Zhao, Q.; et al. An in-situ etching strategy toward fully exposed Fe-N5 active sites for boosted nitroaromatic reduction. Appl. Surf. Sci. 2025, 689, 162592.
175. Gong, X.; Li, D.; Zhang, Q.; et al. Cobalt single atoms supported on monolithic carbon with a hollow-on-hollow architecture for efficient transfer hydrogenations. Nano. Res. 2023, 16, 11358-65.
176. Huang, R.; Cao, C.; Liu, J.; et al. Integration of metal single atoms on hierarchical porous nitrogen-doped carbon for highly efficient hydrogenation of large-sized molecules in the pharmaceutical industry. ACS. Appl. Mater. Interfaces. 2020, 12, 17651-8.
177. Haber, F. Über stufenweise Reduktion des Nitrobenzols mit begrenztem Kathodenpotential. Zeitschrift. für. Elektrochemie. 1898, 4, 506-14. (in German).
178. Millán, R.; Liu, L.; Boronat, M.; Corma, A. A new molecular pathway allows the chemoselective reduction of nitroaromatics on non-noble metal catalysts. J. Catal. 2018, 364, 19-30.
179. Gelder, E. A.; Jackson, S. D.; Lok, C. M. The hydrogenation of nitrobenzene to aniline: a new mechanism. Chem. Commun. (Camb). 2005, , 522-4.
180. Corma, A.; Concepción, P.; Serna, P. A different reaction pathway for the reduction of aromatic nitro compounds on gold catalysts. Angew. Chem. Int. Ed. Engl. 2007, 46, 7266-9.
181. Yu, M.; Zhang, G.; Li, K.; Tang, F.; Wang, L.; Liu, Y. Ni nanoparticles decorated Ni-N-C catalyst: dual-site synergy for enhanced catalytic hydrogenation. AIChE. J. 2025, 71, e18830.
182. He, T.; Zhang, C.; Zhang, L.; Du, A. Single Pt atom decorated graphitic carbon nitride as an efficient photocatalyst for the hydrogenation of nitrobenzene into aniline. Nano. Res. 2019, 12, 1817-23.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].