

Recent advances of single-atom catalysts in the selective catalytic reduction of NO by CO
 0
0 
Abstract
This review summarizes the advances and applications of single-atom catalysts (SACs) in the selective catalytic reduction of NO by CO (CO-SCR). Various types of SACs are discussed, including conventional SACs, negatively charged SACs, dual-atom catalysts, singly dispersed bimetallic sites catalysts, single-atom alloy catalysts, and single-atom-cluster/nanoparticle catalysts. The unique properties of each type of catalyst and their catalytic performance in CO-SCR are explored. SACs enhance the adsorption and activation of NO through synergistic interactions with the support, thereby improving catalytic activity and optimizing the reaction pathway. Furthermore, tuning atomic structure, coordination environment, and metal-support interactions can further enhance the catalytic performance. Despite showing excellent catalytic activity under laboratory conditions, SACs still face challenges in industrial applications, such as catalyst stability, scalability and resistance to poisoning. Future research will focus on improving SAC stability, increasing the density of active sites, and enhancing resistance to deactivation. Combining advanced synthesis methods, large-scale production techniques, and in-depth structural characterization will be crucial for the industrial application of SACs in environmental and energy-related fields.
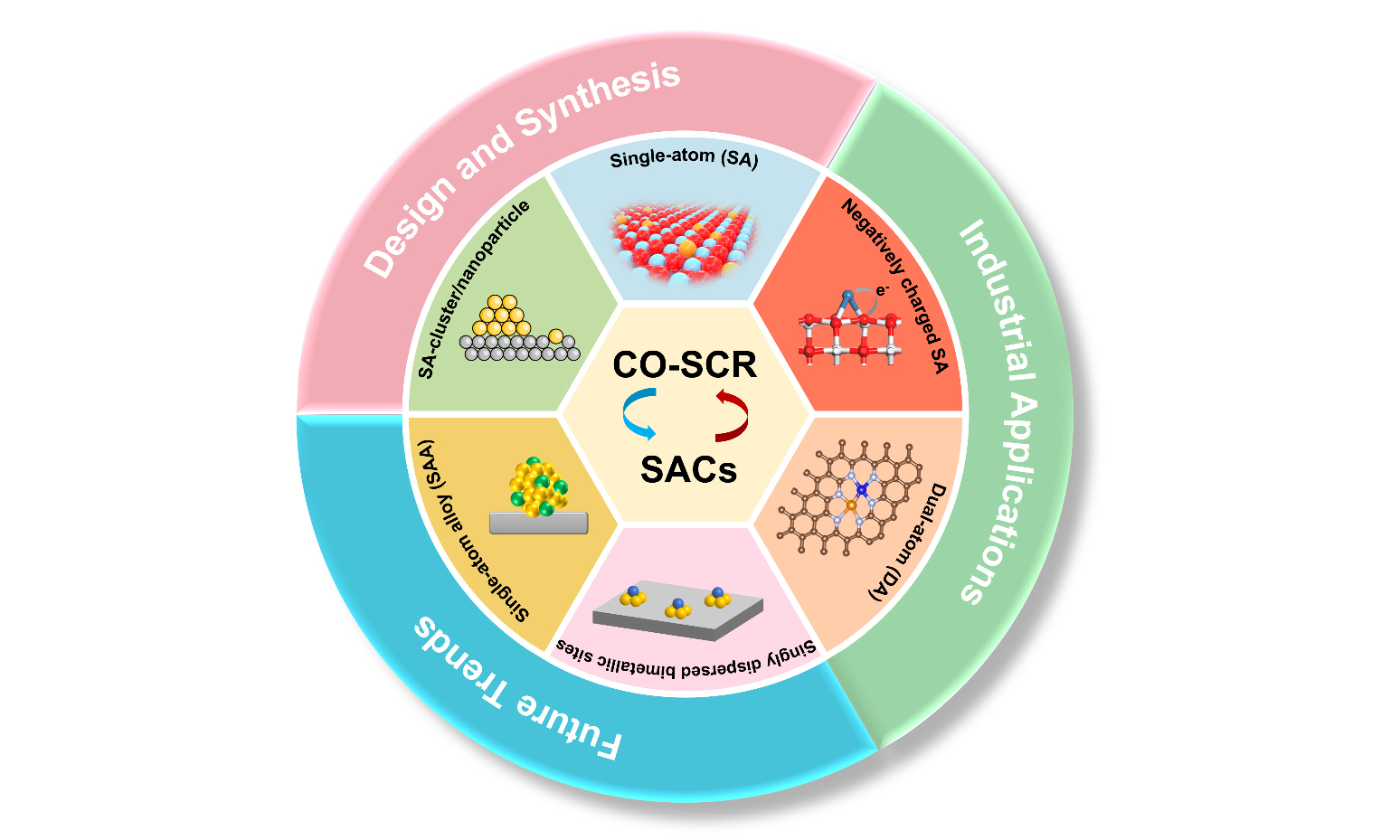
Keywords
INTRODUCTION
Nitrogen oxides (NOx) are major gaseous pollutants primarily generated from the combustion of fossil fuels, especially in industrial production and transportation. The primary components of NOx include nitric oxide (NO), which makes up over 95%, and nitrogen dioxide (NO2), along with other nitrogen oxides[1-3]. Depending on their sources, NOx emissions are generally categorized into stationary or mobile. Stationary sources mainly refer to power generation, chemical industries, and steel factories, while mobile sources primarily include vehicles, airplanes, and ships[4,5]. The harmful impact of NOx on the environment and human health is significant. As part of greenhouse gases, NOx contributes to global warming, acid rain, photochemical smog, and other environmental issues[1,4,6,7]. Moreover, NOx is highly toxic and can damage the respiratory and cardiovascular systems, and in severe cases, even cause death[8]. Therefore, controlling NOx emissions has become a pressing global issue, essential for improving air quality and protecting human health[9].
To address this challenge, various NOx removal technologies have been developed[4,10-12]. Among them, selective catalytic reduction (SCR) is the most mature and widely applied technology, particularly SCR of NOx with NH3 (NH3-SCR)[13-21]. However, ammonia reacts with SO2 in the flue gas to form ammonium sulfate, leading to catalyst deactivation[22-27]. In addition, the additional cost of ammonia and its potential leakage issues have not been fully addressed. In recent years, SCR of NOx by CO (CO-SCR) technology has gained increasing attention due to its economic advantages and higher reduction potential[28-36]. In CO-SCR, the toxic NO and CO in exhaust gases are simultaneously converted into non-toxic and non-polluting N2 and CO2, thus effectively removing pollutants.
Catalysts play a critical role in addressing environmental problems such as waste gas treatment and greenhouse gas reduction. Supported catalysts typically consist of active metal species dispersed on support materials, which help improve catalytic performance and durability[37-40]. However, they still face several challenges: (1) Limited active sites: Although increasing the dispersion of metal particles can increase the number of active sites, the utilization rate of active sites remains limited; (2) Poor selectivity: The reaction selectivity of supported catalysts may be poor in some reactions, especially in complex reaction systems where the active sites cannot precisely control the reaction pathways, leading to side reactions; (3) Limited long-term stability: Supported catalysts may suffer from deactivation, poisoning, and sintering over time[41].
In recent years, single-atom catalysts (SACs) have emerged as a promising new class of catalyst. Qiao et al.[42] first proposed the concept of “single-atom catalysis”, marking a breakthrough in understanding heterogeneous catalytic active sites [Figure 1A-C]. They reported that the Pt1/FeOx shows much higher activity in CO oxidation reaction compared to Au nanoparticle (NP) catalysts. This discovery revealed the unique catalytic properties of SACs and brought revolutionary progress to the field of catalysis. In SACs, metal atoms are individually anchored on the support surface, achieving nearly 100% atomic utilization. From traditional large metal NPs to small metal clusters and isolated single metal atoms, these structural changes dramatically alter the electronic properties of catalysts and greatly improve catalytic performance [Figure 1D and E][43,44]. This progress has also inspired the development of dual-atom catalysts (DACs), singly dispersed bimetallic sites catalysts, and single-atom alloy catalysts (SAACs)[45-47]. Currently, researchers are actively building a comprehensive “SACs toolbox”, encompassing SACs, dual- (multi-) atom catalysts, and single atom (SA)-cluster-NP catalysts, forming a multi-level catalytic system
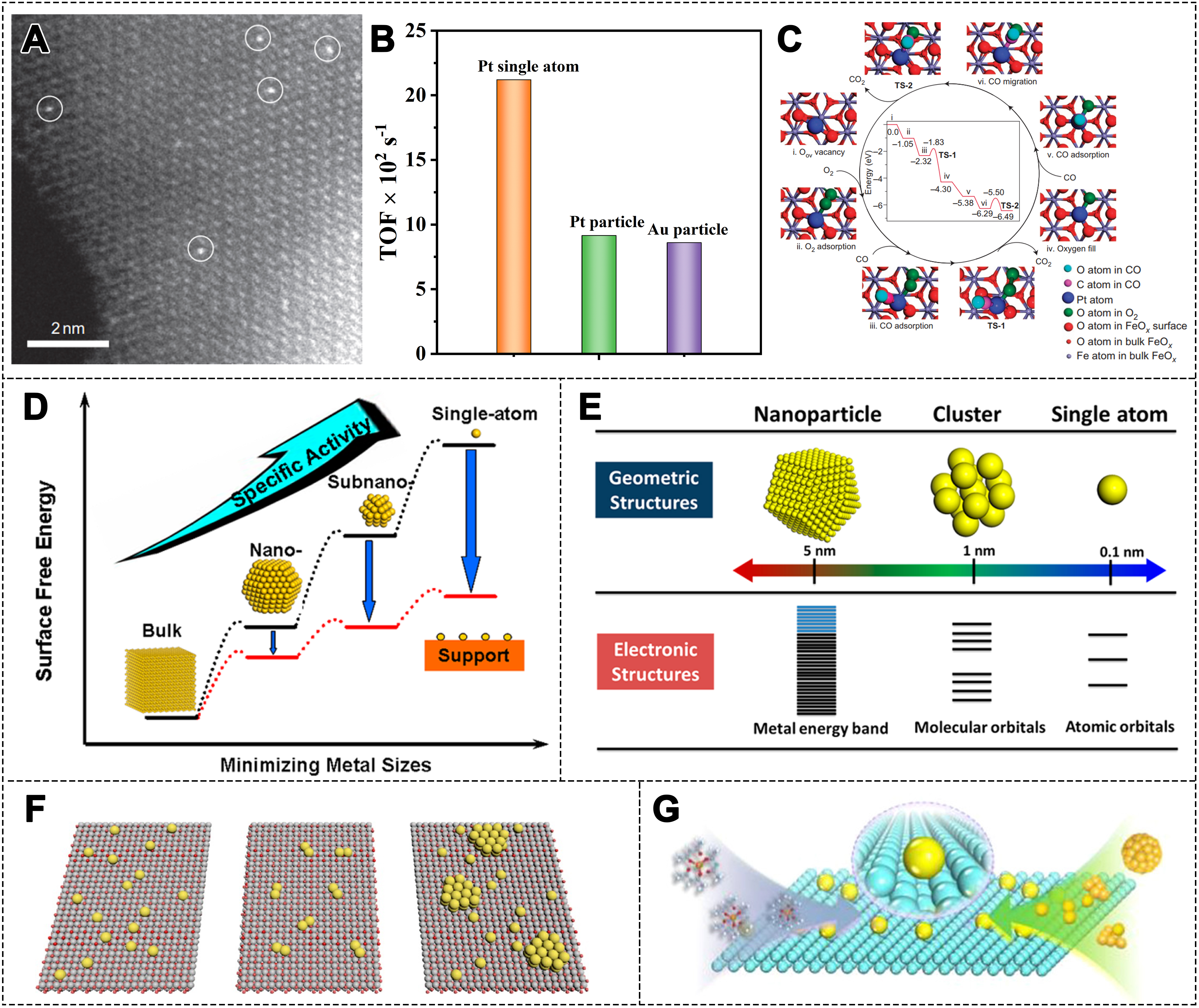
Figure 1. (A) HAADF-STEM image of the Pt1/FeOx catalyst where the Pt SAs occupy the positions of the Fe atoms; (B) Comparison of TOFs; (C) The proposed reaction pathways for CO oxidation on the Pt1/FeOx catalyst[42]. Copyright 2013, Springer Nature; (D) The changes of surface free energy and specific activity with metal particle size[43]. Copyright 2013, American Chemical Society; (E) The geometric and electronic structures of particles of several different sizes: SAs, clusters, and NPs[44]. Copyright 2018, American Chemical Society; (F) Schematic illustration of dispersion states of metal species on SACs, DACs and SA-cluster/NP catalysts[48]. Copyright 2022, American Chemical Society; (G) Schematic illustration of the controlled synthesis strategy for SACs[53]. Copyright 2020, American Chemical Society. HAADF-STEM: High angle annular dark field-scanning transmission electron microscopy; SA: single atom; TOF: turnover frequency; NP: nanoparticle; SAC: single-atom catalyst; DAC: dual-atom catalyst.
Research on SACs began in the early 21st century, but over the past decade, its application in various catalytic fields has attracted extensive global attention. Since the concept of single-atom catalysis was first proposed in 2011, the number of related research papers has surged dramatically, from approximately 100 papers per year to an exponential increase in recent years. According to Web of Science statistics, since 2011, the annual citation rate has steadily increased. By December 2024, over 8,000 related papers had been published [Figure 2][55,56]. In the multidisciplinary fields, the research on SACs has been widely reported, with the Chemistry field having the most related literature, followed by Physics, Materials Science, and Engineering. With ongoing research, SACs are gaining increasing application prospects in energy conversion, environmental protection, and chemical synthesis.
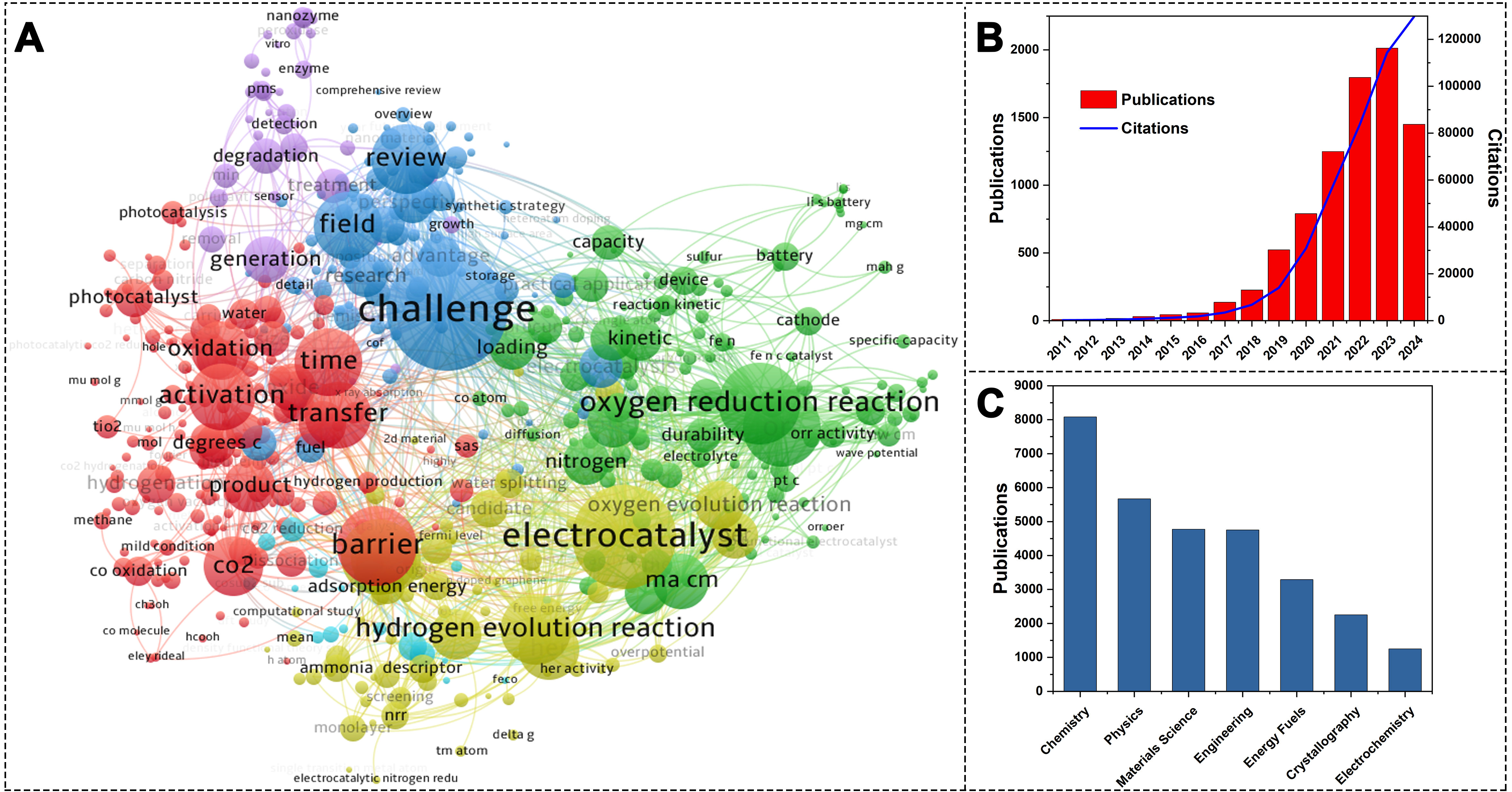
Figure 2. (A) Bibliometric map generated by VOSviewer based on the Web of Science All Databases[55,56]; (B) Publication and citation trends of single-atom catalysis-related articles over different years; (C) Publication distribution of single-atom catalysis-related articles across various academic disciplines. The keyword analyzed was “single-atom catalysis”. The size of each circle correlates with the occurrences in 8,444 Web of Science-indexed articles published from 1979 to 2025. The smallest circles represent 50 occurrences.
This review aims to systematically study the applications of SACs in the CO-SCR reaction and summarize the current research status and development trends in NOx removal. We classify CO-SCR catalysts into SACs, negatively charged SACs, DACs, singly dispersed bimetallic sites catalysts, SAACs, and single-atom-cluster/NP catalysts. We will explore the properties, catalytic mechanisms, and performance of these catalysts in the CO-SCR reaction [Table 1]. By introducing the advantages of different types of catalysts, we aim to provide theoretical and practical guidance for catalyst design and development. Finally, we will discuss future research directions and challenges, especially how to enhance catalyst stability and further promote the industrial application of SACs. Through this review, we hope to offer new insights for the development of SACs and the optimization of CO-SCR technology, contributing to the innovation and advancement of environmental pollution control technologies.
Comparison of catalytic performance of different single-atom catalysts
| Sample | Reaction conditions | T (°C) | Catalytic performance | Ref. | |||||
| NO (%) | CO (%) | O2 (%) | Others | GHSV or WHSV | NO (%) | N2 (%) | |||
| Single-atom catalysts | |||||||||
| Ir1/m-WO3 | 0.1 | 0.2 | 2 | / | 50,000 h-1 | 350 | 73 | 100 | [74] |
| 0.3Ag/m-WO3 | 0.1 | 0.4 | 1 | / | 50,000 h-1 | 250 | ~ 73 | 100 | [79] |
| 5Ag/m-WO3 | 0.1 | 0.4 | 1 | / | 50,000 h-1 | 250 | ~ 64 | 100 | [79] |
| Cr0.19Rh0.06CeOz | 0.5 | 1.0 | / | / | 6,500 h-1 | 120 | 100 | ~ 34 | [84] |
| Cr0.19Rh0.06CeOz | 0.5 | 1.0 | / | / | 6,500 h-1 | 200 | 100 | 100 | [84] |
| Fe1/CeO2-Al2O3 | 0.05 | 0.6 | / | / | 30,000 h-1 | 250 | 100 | 100 | [87] |
| Fe1/Al2O3 | 0.05 | 0.6 | / | / | 30,000 h-1 | 400 | 100 | ~ 100 | [87] |
| 0.2Rh/CeO2-Octahedron | 0.09 | 0.11 | / | 2.5% H2O | 225 L g-1 h-1 | 266 | 100 | / | [89] |
| 0.2Rh/CeO2-Rod | 0.09 | 0.11 | / | 2.5% H2O | 225 L g-1 h-1 | 350 | ~ 95 | / | [89] |
| Cu1- MgAl2O4 | 2.6 | 2.9 | / | / | 12 L g-1 h-1 | 300 | ~ 93 | ~ 92 | [90] |
| 0.05 wt.% Rh/γ- Al2O3 | 0.1 | 0.5 | / | / | - | 290 | 100 | / | [91] |
| 0.05 wt.% Rh/γ- Al2O3 | 0.1 | 0.5 | / | 2% H2O | - | 210 | 100 | / | [91] |
| 0.1wt.%Rh/CeO2 | 0.046 | 0.175 | / | / | 150 L g-1 h-1 | 100 | 100 | ~ 90 | [95] |
| 0.1wt.%Rh/CeO2 | 0.046 | 0.175 | / | 2.6% H2O | 150 L g-1 h-1 | 120 | 100 | ~30 | [95] |
| 0.5%Ru/CeO2 | 0.047 | 0.185 | / | ~ 3% H2O | 150 L g-1 h-1 | ~ 135 | ~ 95 | / | [96] |
| 0.05%Ru/CeO2 | 0.047 | 0.185 | / | ~ 3% H2O | 150 L g-1 h-1 | ~ 190 | ~ 95 | / | [96] |
| Rh1/CeO2 | 0.046 | 0.175 | / | 2.6% H2O | 150 L g-1 h-1 | 126 | 100 | ~ 40 | [97] |
| Ru1/CeO2 | 0.046 | 0.175 | / | 2.6% H2O | 150 L g-1 h-1 | 200 | 100 | ~ 18 | [97] |
| Pd1/CeO2 | 0.046 | 0.175 | / | 2.6% H2O | 150 L g-1 h-1 | 208 | 100 | ~ 67 | [97] |
| Pt1/CeO2 | 0.046 | 0.175 | / | 2.6% H2O | 150 L g-1 h-1 | 350 | 100 | ~ 70 | [97] |
| Ir1/CeO2 | 0.046 | 0.175 | / | 2.6% H2O | 150 L g-1 h-1 | ~ 270 | 50 | ~ 33 | [97] |
| 0.59% Rh/SiO2 | 1.5 | 4.5 | / | / | 540 L g-1 h-1 | 280 | 100 | / | [251] |
| 0.05% Rh/SiO2 | 1.5 | 4.5 | / | / | 540 L g-1 h-1 | 340 | 100 | / | [251] |
| 0.03% Rh/SiO2 | 1.5 | 4.5 | / | / | 540 L g-1 h-1 | 380 | 100 | ~ 50 | [251] |
| Negatively charged single-atom catalysts | |||||||||
| Pt-CuO/CoAlO | 0.1 | 0.2 | / | / | 10,000 h-1 | 200 | 100 | 100 | [105] |
| Pt-CuO/CoAlO | 0.1 | 0.2 | 3 | / | 10,000 h-1 | 200 | 91 | 80 | [105] |
| Pt-CuO/CoAlO | 0.1 | 0.2 | 1 | 200 ppm SO2 | 10,000 h-1 | 200 | 88.5 | 100 | [105] |
| IrW-WO3/KIT-6 | 0.1 | 0.4 | 1 | / | 50,000 h-1 | 250 | ~ 98 | 100 | [107] |
| IrW-WO3/KIT-6 | 0.1 | 0.4 | 3 | / | 50,000 h-1 | 250 | ~ 90 | 100 | [107] |
| IrW-WO3/KIT-6 | 0.1 | 0.4 | 1 | 100 ppm SO2 | 50,000 h-1 | 250 | ~ 83 | 100 | [107] |
| Ir1-IrW-WO3/KIT-6 | 0.1 | 0.4 | 1 | / | 50,000 h-1 | 250 | ~ 80 | 100 | [107] |
| Ir1-WO3/KIT-6 | 0.1 | 0.4 | 1 | / | 50,000 h-1 | 250 | ~ 40 | 100 | [107] |
| Pt(0.1)Co(1)/Al2O3 | 0.1 | 0.6 | 0.6 | 0.0555% C3H6 0.2% H2 13.9% CO2 | 60,000 h-1 | ~ 260 | ~ 65 | / | [252] |
| Dual-atom catalysts | |||||||||
| CuFe-N/C | 0.1 | 0.2 | / | / | 30,000 h-1 | 225 | 100 | 100 | [129] |
| Cu-N/C | 0.1 | 0.2 | / | / | 30,000 h-1 | 225 | ~ 92 | ~ 88 | [129] |
| Fe-N/C | 0.1 | 0.2 | / | / | 30,000 h-1 | 225 | ~ 75 | ~ 73 | [129] |
| Pt-Pd SACs-2 | 0.05 | 1 | 1.25 | 0.1% C3H6 | 120 L g-1 h-1 | 220 | 100 | / | [130] |
| Pd SAC & Pt SACs-2 | 0.05 | 1 | 1.25 | 0.1% C3H6 | 120 L g-1 h-1 | 290 | 100 | / | [130] |
| Pd SACs-2 | 0.05 | 1 | 1.25 | 0.1% C3H6 | 120 L g-1 h-1 | 270 | 100 | / | [130] |
| Pt SACs-2 | 0.05 | 1 | 1.25 | 0.1% C3H6 | 120 L g-1 h-1 | 270 | ~ 23 | / | [130] |
| Singly dispersed bimetallic sites catalysts | |||||||||
| Pd0.3Rh0.7 | 0.15 | 0.35 | 0.25 | 0.033% C3H6 10% H2O | - | 249 | 50 | / | [122] |
| Pd0.1Rh0.9 | 0.15 | 0.35 | 0.25 | 0.033% C3H6 10% H2O | - | 259 | 50 | / | [122] |
| Rh1Co3/CoO | 0.05 | 0.05 | / | / | 360 L g-1 h-1 | 100 | ~ 85 | ~ 100 | [135] |
| Rh1Co3/CoO | 0.05 | 0.05 | / | / | 360 L g-1 h-1 | 250 | 100 | 100 | [135] |
| Rh-Co alloy NPS/SiO2 | 0.05 | 0.05 | / | / | 360 L g-1 h-1 | 290 | 100 | ~ 94 | [135] |
| Single-atom alloy catalysts | |||||||||
| Cu5Pd/Al2O3 | 0.5 | 0.5 | / | / | 80,000 h-1 | 200 | 100 | 100 | [165] |
| Cu5Pd/Al2O3 | 0.5 | 0.5 | 0.5625 | / | 80,000 h-1 | 200 | ~ 95 | ~ 85 | [165] |
| Cu3Pd/Al2O3 | 0.5 | 0.5 | / | / | 80,000 h-1 | 250 | 100 | 100 | [165] |
| PdCu/Al2O3 | 0.5 | 0.5 | / | / | 240,000 h-1 | 225 | ~ 98 | ~ 36 | [167] |
| PdIn/Al2O3 | 0.5 | 0.5 | / | / | 240,000 h-1 | 225 | ~ 35 | ~ 100 | [167] |
| Pd(In0.33Cu0.67)/Al2O3 | 0.5 | 0.5 | / | / | 240,000 h-1 | 225 | ~ 78 | ~ 100 | [167] |
| PdIn/Al | 0.5 | 1.0 | 0.25 | / | 40,000 h-1 | 350 | 100 | 100 | [168] |
| Pd-Pt-In/Ce | 0.5 | 1.0 | 0.5 | / | 40,000 h-1 | 350 | 100 | 100 | [168] |
| Single-atom-cluster/NP catalysts | |||||||||
| CoSA + CoOxNC/CZO | 0.1 | 0.2 | 5 | / | 20,000 h-1 | 250 | 100 | 100 | [178] |
| CoSA + CoOxNC/CZO | 0.1 | 0.2 | 5 | 5% H2O | 20,000 h-1 | 300 | ~ 70 | 100 | [178] |
| CoSA + CoOxNC/CZO | 0.1 | 0.2 | 5 | 50 ppm SO2 | 20,000 h-1 | 300 | ~ 80 | 100 | [178] |
| CoSA/CZO | 0.1 | 0.2 | 5 | / | 20,000 h-1 | 250 | ~ 36 | ~ 100 | [178] |
| CoOxNC/CZO | 0.1 | 0.2 | 5 | / | 20,000 h-1 | 250 | ~ 56 | ~ 98 | [178] |
| 8Rh/TiO2/Ti | 0.06 | 0.3 | 0.15 | / | - | 210 | 100 | ~100 | [180] |
| 8Rh/TiO2/Ti | 0.06 | 0.3 | 0.3 | / | - | 175 | ~ 88 | / | [180] |
| D-04CuAl-HO | 2.5 | 5 | / | / | 12,000 h-1 | 375 | 100 | 100 | [183] |
| D-04CuAl-HW | 2.5 | 5 | / | / | 12,000 h-1 | 375 | ~ 55 | ~ 80 | [183] |
OVERVIEW OF THE CO-SCR REACTION
Mechanistic insights into the CO-SCR reaction
In the CO-SCR process, the reaction primarily involves three elementary steps: adsorption of the reactants, transformation of intermediate species, and desorption and diffusion of the products. During CO-SCR reaction, the main products are nitrogen (N2) and carbon dioxide (CO2). However, the formation of undesired by-products such as nitrous oxide (N2O) and NO2 may also occur, depending on factors including the catalyst composition, reaction temperature, and the concentrations of gases involved[57]. At relatively low temperatures, the reductive capacity of CO is weakened, which can lead to the formation of intermediate species such as N2O instead of the ideal N2 product. Enhancing the reducing environment can further promote the reduction of N2O to N2, thereby improving N2 selectivity[58,59]. Conversely, under conditions of high oxygen concentration and elevated temperature, NO may be oxidized to NO2, thereby negatively affecting the overall denitrification efficiency. The key transformation step centers on the cleavage of the N-O bond in NO and the subsequent recombination with CO to form CO2[3]. The overall CO-SCR reaction encompasses several fundamental pathways, as summarized below:
In the CO-SCR reaction, the catalytic mechanisms are generally categorized into three types: the Langmuir-Hinshelwood (L-H) mechanism, the Eley-Rideal (E-R) mechanism, and the Mars-van Krevelen (MvK) mechanism. In L-H mechanism, both NO and CO are first adsorbed onto the catalyst surface. NO interacts with the metal sites via its oxygen atom, leading to the elongation and cleavage of the N-O bond. The resulting nitrogen species may couple with another NO to form N2 or convert into N2O. Meanwhile, adsorbed CO reacts with surface oxygen species or oxygen derived from NO to generate CO2[60]. The E-R mechanism involves the reaction between a gas-phase molecule and a surface-adsorbed species. Typically, gaseous CO reacts with adsorbed NO. On catalysts such as H-CuCo-CeO2, NO undergoes N-O bond cleavage to produce lattice oxygen and nitrogen intermediates. The lattice oxygen is subsequently consumed by gaseous CO to form CO2 and oxygen vacancies (Ov), while the nitrogen species further react to produce N2[61]. The MvK mechanism involves CO reacting with lattice oxygen to form CO2, leaving behind Ov or oxygen-deficient sites. NO is then adsorbed at these vacancies, where its N-O bond is cleaved. The liberated oxygen restores the lattice structure, and the resulting nitrogen species couple to form N2[62].
Challenges and prospects of the CO-SCR reaction
However, CO-SCR still faces two major challenges in practical application: (1) Limited oxygen resistance: High concentrations of oxygen can oxidize the active sites, inhibit NO adsorption and dissociation, and thus reduce catalytic activity. Furthermore, oxygen may react with CO to form CO2, reducing the effectiveness of CO as a reducing agent, and further lowering the NO removal efficiency[63,64]; (2) Limited catalytic activity at low temperatures: At lower temperatures, particularly below 200 °C, the catalytic activity of CO-SCR is significantly reduced due to the weakened reductive capacity of CO. This impairs the activation and reduction of NO, promotes the formation of N2O, and ultimately leads to a decline in overall catalytic performance[63,65-67]. Therefore, developing catalysts with good low-temperature activity and oxygen tolerance is a key to advancing CO-SCR technology. In addition, industrial exhaust gases also contain SO2, H2O and other components, which can poison the catalyst and occupy active sites, thereby inhibiting the catalytic activity of the CO-SCR reaction and reducing catalytic efficiency[32,68-70].
SINGLE-ATOM CATALYTIC SYSTEM IN CO-SCR REACTION
Single-atom catalysts
In the CO-SCR reaction, noble metal catalysts exhibit high activity and selectivity[71,72]. Among them, noble metal SACs not only reduce costs but also further enhance catalytic performance. Among these metals, Iridium (Ir) typically demonstrates superior resistance to oxidation, which has been confirmed in CO-SCR reactions[69,73]. In this regard, the Ir1/m-WO3 catalyst was synthesized on mesoporous WO3 using a facile impregnation method[74]. In the presence of 2 vol.% O2, the Ir1/m-WO3 catalyst exhibited excellent catalytic performance, achieving a 73% NO conversion and 100% N2 selectivity at 350 °C [Figure 3A]. Notably, at 200 °C, the turnover frequency (TOF) value was 6 times higher than that of Ir NPs supported on mesoporous WO3. This superior performance can be attributed to the synergistic interaction between the isolated Ir atoms and the WO3, forming Ir-WO3 sites that effectively promote NO adsorption and activation[75-77].
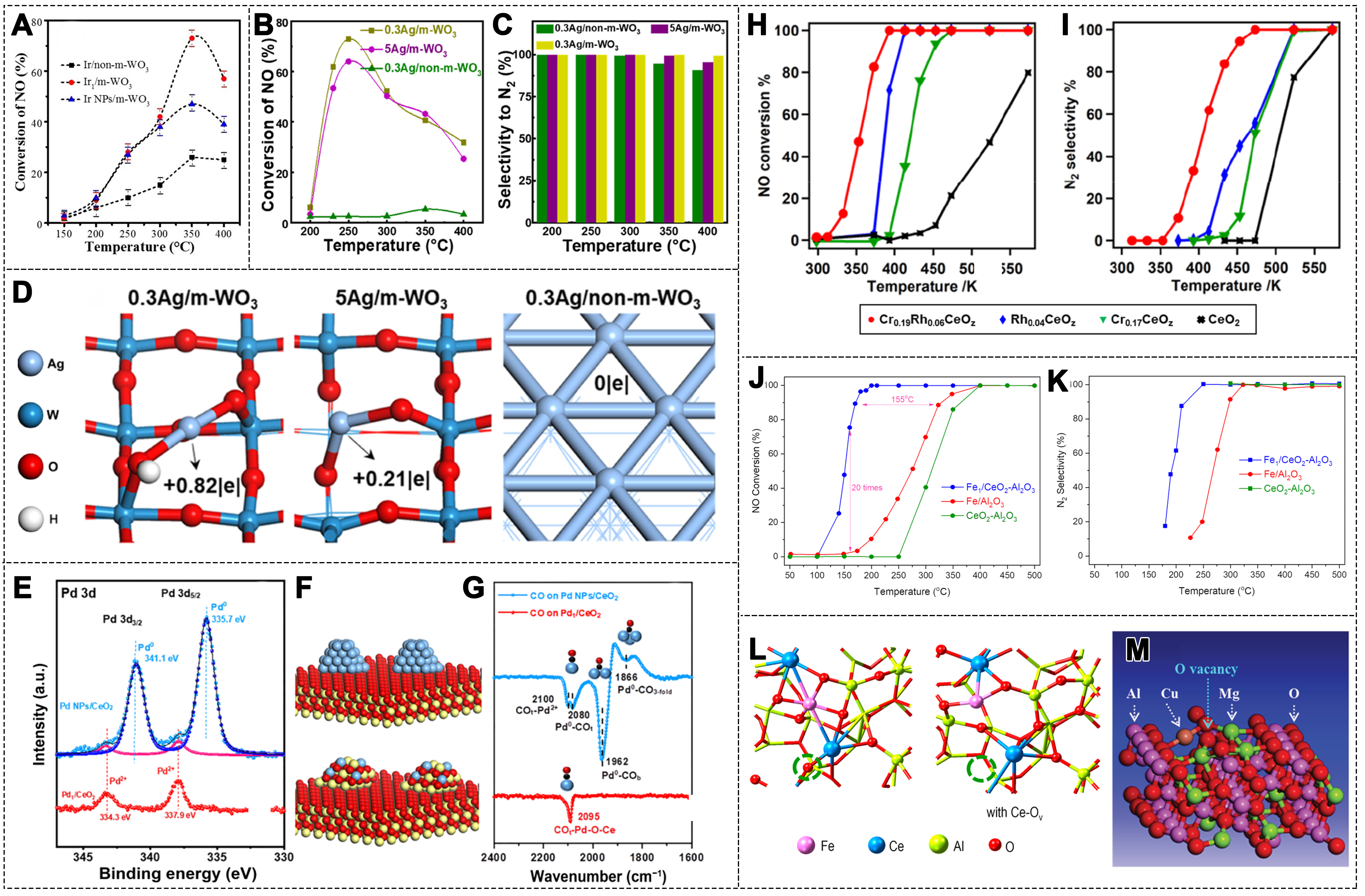
Figure 3. (A) NO conversion over different Ir catalysts (1000 ppm NO, 2,000 ppm CO, 2vol.% O2, N2 balance, GHSV = 50,000 h-1)[74]. Copyright 2021, Wiley-VCH; (B) NO conversion and (C) N2 selectivity over different Ag catalysts; (D) Bader charge values of Ag atoms calculated for different Ag catalysts[79]. Copyright 2023, American Chemical Society; (E) SRPES data for Pd1/CeO2 (red line) and Pd NPs/CeO2 (blue line) samples; (F) The proposed structural models of Pd NPs/CeO2 (top) and Pd1/CeO2 (bottom) catalysts; (G) CO IRAS spectra for Pd1/CeO2 (red line) and Pd NPs/CeO2 (blue line) samples. (color code: yellow, Ce; red, O; blue, Pd; and black, C)[83]. Copyright 2023, Springer; (H) NO conversion and (I) N2 selectivity over Cr0.19Rh0.06CeOz, Rh0.04CeOz, Cr0.17CeOz, and CeO2 (0.5% NO, 1.0% CO, 5% Ar, He balance, GHSV = 65,00 h-1)[84]. Copyright 2021, American Chemical Society; (J) NO conversion and (K) N2 selectivity over Fe1/CeO2-Al2O3, Fe1/Al2O3 and CeO2-Al2O3 (500 ppm NO, 0.6% CO, N2 balance, GHSV 30 000 h-1); (L) Model structure of the catalyst surface with or without Ov[87]. Copyright 2024, American Chemical Society; (M) Model structure of Mg11CuAl24O47 with Ov[90]. Copyright 2019, The Royal Society of Chemistry. Ir: Iridium; GHSV: gas hourly space velocity; SRPES: synchrotron radiation photoemission spectroscopy; Ov: oxygen vacancies.
SACs exhibit significant differences in catalytic activity due to variations in their local coordination environments and electronic structures[78]. Ji et al. successfully dispersed single Ag atoms on ordered mesoporous WO3 by varying the loading amount of the Ag precursor[79]. This resulted in Ag catalysts with different local structures (varying Ag-O coordination numbers), leading to significant differences in catalytic performance for CO-SCR. Notably, under conditions of 250 ℃ and 1% O2, the 5Ag/m-WO3 catalyst with a two-coordination structure exhibited 63% NO conversion and 100% N2 selectivity [Figure 3B and C]. Similarly, the 0.3Ag/m-WO3 catalyst with a six-coordination Ag-O structure achieved 73% NO conversion and 100% N2 selectivity at 250 °C. Density functional theory (DFT) results further indicated that the Bader charges of Ag atoms on 0.3Ag/m-WO3 and 5Ag/m-WO3 were +0.82|e| and +0.21|e|, respectively, suggesting that the Ag atoms in 0.3Ag/m-WO3 are in a more oxidized state with a higher d-band center [Figure 3D]. This promotes the adsorption of CO/NO and various intermediates, particularly the co-adsorption of key intermediates such as N2O* and CO*. By tuning the local structure of Ag SACs, CO can more effectively reduce the N2O* intermediate, thereby enhancing catalytic performance.
The small Pd NPs supported on CeO2 catalysts exhibit lower N2 selectivity at temperatures below 200 °C. In contrast, Pd atoms dispersed on Pd1/CeO2 catalysts achieve complete conversion of CO and NO below 200 °C, with 100% N2 selectivity[80-82].
To gain deeper insight into the reaction mechanisms of Pd NPs and Pd SAs in the CO-SCR reaction, dispersed Pd NPs and Pd SAs were prepared on CeO2 (111) thin films[83]. On the Pd1/CeO2 catalyst surface, only a peak at 337.9 eV was observed in synchrotron radiation photoemission spectroscopy (SRPES), which can be attributed to Pd2+ in Pd-O-Ce. In contrast, Pd NPs/CeO2 catalyst surfaces also showed the presence of Pd0, indicating that most of the deposited Pd remained in its metallic state [Figure 3E and F]. Additionally, the CO adsorption behavior on the two catalyst surfaces was clearly different [Figure 3G]. On the Pd NPs/CeO2 surface, N2 formation resulted from the binding of N atoms after the dissociation of NO, while N2O formation was likely due to the reaction between adsorbed NO and N atoms. The primary reaction products were CO2, N2, and N2O. On the Pd1/CeO2 catalyst surface, no desorption of N2O was detected, indicating nearly 100% N2 selectivity. This high selectivity can be attributed to the continuous N-O bond cleavage in the O-N-N-O intermediate formed on the Pd1/CeO2 catalyst surface.
To improve low-temperature activity and enhance N2 selectivity, recent studies have focused on tailoring the electronic structure of active sites and regulating metal–support interactions. The following examples demonstrate how these strategies boost CO-SCR performance under mild conditions. A ceria-based catalyst doped with Rh and Cr (Cr0.19Rh0.06CeOz) exhibits excellent redox activity at low temperatures[84]. This catalyst consists of dispersed Rh3 + δ and CeO2/CrO3-x, showing strong interactions between the components. The incorporation of Cr into Rh-containing ceria leads to the dispersion of positively charged Rh species. The Cr0.19Rh0.06CeOz catalyst displays significant NO reduction activity at 372 K, whereas the Rh0.04CeOz catalyst shows no activity at this temperature. The N2 selectivity of Cr0.19Rh0.06CeOz exceeds 99% at 473 K [Figure 3H and I]. Analysis indicates that the addition of Cr disperses the Rh species responsible for adsorbing and activating NO and CO, increasing the available adsorption sites for NO and CO. Chromium oxide species promote the dissociation of NO on Rh sites and facilitate the transfer of oxygen species from NO to CO, thereby lowering the activation energy for NO reduction.
In recent years, the synergistic effect of SAs and Ov has also attracted widespread attention[85,86]. Bai et al. prepared an Fe1/CeO2-Al2O3 catalyst using a co-precipitation method, where the Fe sites are atomically dispersed within the surface CeO2 lattice[87]. Compared to Fe1/Al2O3, the introduction of Ce atoms in Fe1/CeO2-Al2O3 causes charge transfer from Fe to Ce, resulting in the formation of electron-deficient Fe sites. Over a temperature range of 250-500 °C, Fe1/CeO2-Al2O3 achieves complete NO conversion and 100% N2 selectivity [Figure 3J and K]. In situ characterization and computational studies suggest that the high activity of Fe1/CeO2-Al2O3 can be attributed to the synergistic effect between Fe1 sites and Ce-Ov, where Ce-Ov enhances the adsorption of NO and N2O intermediates on Fe sites [Figure 3L]. The interaction and cooperation between the SAs and the local environment of the support promote the formation of Ov, thereby enhancing the catalytic activity[88,89]. Wu et al. confined Cu atoms within the MgAl2O4 spinel lattice to obtain Cu1-MgAl2O4, which was used in CO-SCR reactions [Figure 3M][90]. Its catalytic activity was found to be significantly higher than that of a 0.1 wt.% Pt/CeO2 noble metal catalyst. The high activity can be explained by a similar mechanism, where Cu atoms lower the activation energy for CO oxidation, while NO is adsorbed at the Ov, leading to the formation of N2O, which subsequently decomposes to N2.
To address the performance degradation caused by H2O, recent studies have focused on exploring SACs with superior H2O resistance. The following investigations present representative advances in understanding and improving the H2O tolerance of SACs, particularly Rh-based systems, in wet CO-SCR conditions. Asokan et al. synthesized Rh/γ-Al2O3 catalysts with Rh loadings ranging from 0.05 to 5 wt.%, which resulted in Rh species varying from SAs to clusters and NPs[91]. They characterized the Rh species using CO diffuse reflectance infrared Fourier transform spectroscopy (CO-DRIFTS)[92-94]. As the Rh loading increased, the proportion of atomically dispersed Rh species decreased [Figure 4A]. Under dry conditions, all catalysts started converting NO to N2 at approximately 210 °C. Among them, Rh clusters exhibited superior catalytic performance [Figure 4B]. The addition of H2O promoted the reactivity of low-Rh-loaded catalysts, while for catalysts with higher Rh content, NO conversion was inhibited [Figure 4C]. Notably, as the Rh content decreased, the concentration of NH3 increased. This suggests that as the proportion of atomically dispersed Rh species rose, the NH3 concentration also increased [Figure 4D and E]. This leads to the conclusion that the specific reaction sites in a wet environment are different: for Rh clusters, the reaction is 2NO + 2CO → N2 + 2CO2, while for Rh SAs: 2NO + 3H2O + 5CO → 2NH3 + 5CO2 [Figure 4F][91]. This indicates that atomically dispersed Rh species are active in NH3 formation at low temperatures, and Rh SAs also serve as active sites for the water-gas shift (WGS) reaction.
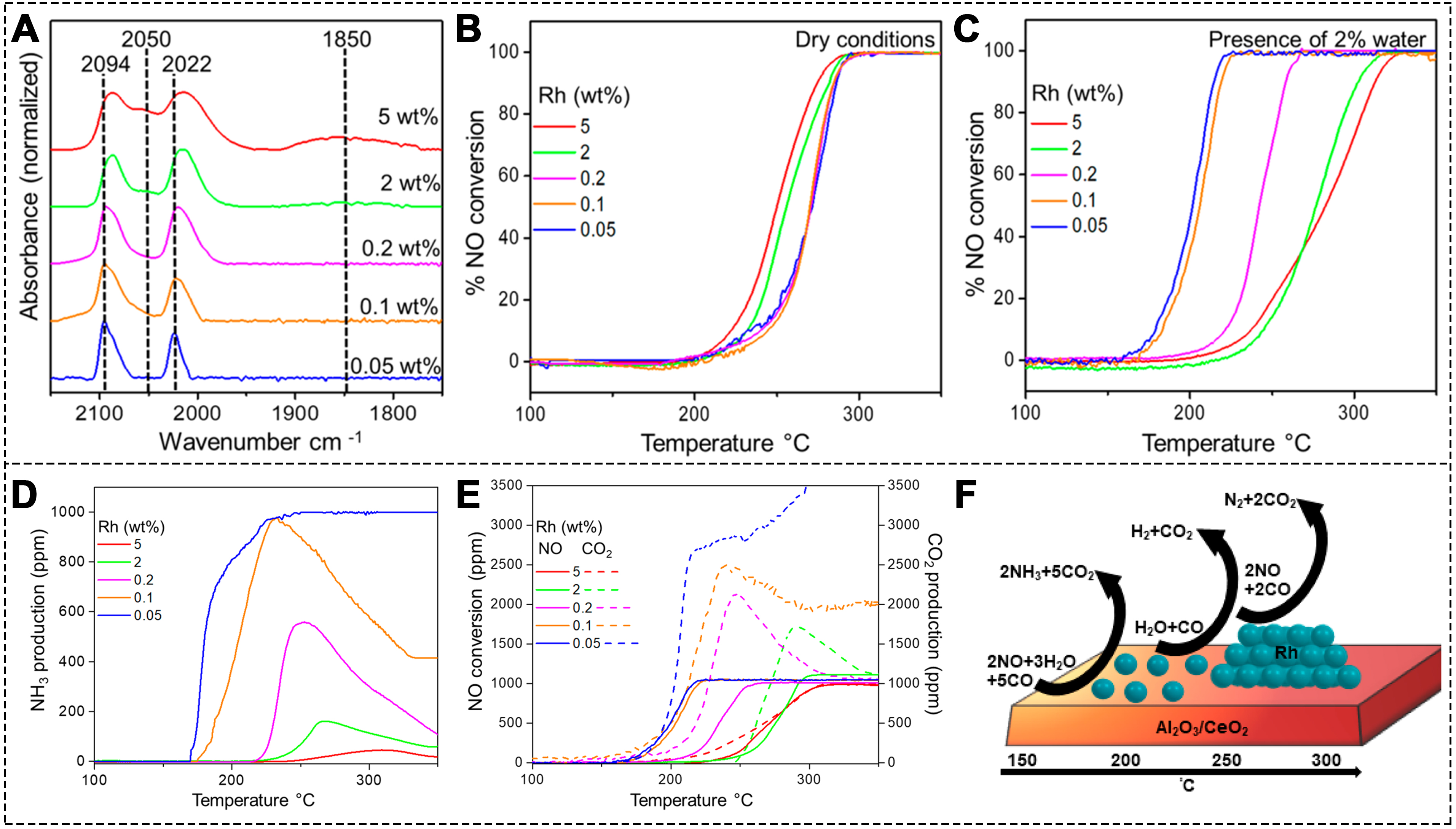
Figure 4. (A) CO-DRIFTS spectra for catalysts with different Rh wt.% contents; (B) NO conversion for different catalysts under dry and (C) wet conditions; (D) NH3 production; (E) NO conversion, and CO2 production for the series of catalysts (1000 ppm NO, 5,000 ppm CO, 2% H2O when used); (F) Schematic of product formation during NO reduction over catalysts with different Rh structures[91]. Copyright 2020, American Chemical Society. CO-DRIFTS: CO diffuse reflectance infrared Fourier transform spectroscopy.
Khivantsev et al. revealed that the dispersed Rh SAs in Rh1/CeO2 are highly active species for the CO + NO reaction[95]. Under dry conditions, the 0.1 wt.% Rh/CeO2 achieves complete NO conversion at 100 °C, with a small amount of N2O produced at 75 °C. Furthermore, under wet conditions, 0.1 wt.% Rh/CeO2 shows significant activity and selectivity, with complete NO removal occurring above 120 °C, and only a small amount of N2O produced above 125 °C [Figure 5A and B]. Notably, NH3 shows the highest selectivity (around 55%-60%) in the temperature range of 125-150 °C, indicating that both NH3 formation and the WGS reaction occur simultaneously at the same Rh atom. In this process, Rh-H complexes serve as intermediates in the hydrogenation of NO. The hydrogen stored on Rh atoms via the WGS pathway plays a crucial role in NH3 formation, with the isolated Rh atoms showing high WGS activity. The presence of Rh-H facilitates the hydrogenation of NO, leading to ammonia production. In their study, Ru1/CeO2 was also shown to have excellent reactivity at low temperatures. The 0.5 wt.% Ru/CeO2 achieved 100% NO conversion at approximately 150 °C under dry conditions[96]. Upon the addition of water to the gas flow, a significant amount of NH3 was generated, as Ru1/CeO2 is also an excellent WGS catalyst [Figure 5C and D].
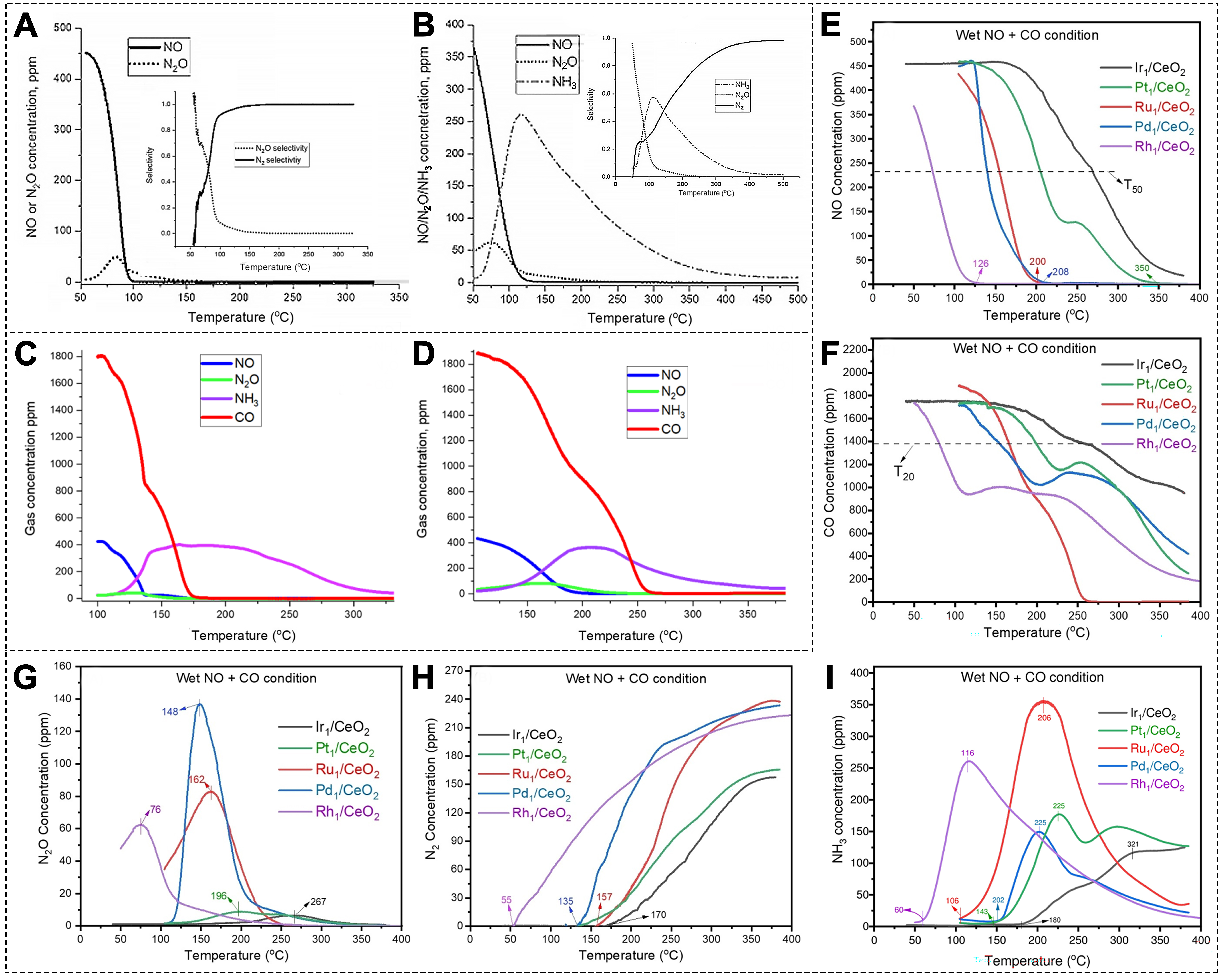
Figure 5. (A) Different gas concentrations and selectivity of reaction products under dry and (B) wet conditions (460 ppm NO, 1750 ppm CO, 2.6% H2O when used, N2 balance, 150 L g-1 h-1)[95]. Copyright 2021, Wiley-VCH; (C) Different gas concentrations during the reaction for 0.5 wt.% Ru/CeO2 and (D) 0.1 wt.% Ru/CeO2 (470 ppm NO, 1850 ppm CO, 3% H2O, N2 balance, 150 L g-1 h-1)[96]. Copyright 2023, American Chemical Society; (E-I) NO, CO, N2O, N2, and NH3 concentrations for wet reaction on M1/CeO2 (M = Ir, Pt, Ru, Pd, and Rh, 460 ppm NO, 1750 ppm CO, 2.6% H2O, N2 balance, 150 L g-1 h-1)[97]. Copyright 2021, Wiley-VCH.
Tian et al. synthesized different noble metal M1/CeO2 SACs (M = Rh, Ru, Pd, Pt, and Ir) and studied their catalytic activities under wet conditions[97]. All the catalysts were tested in the presence of 3% H2O, with 50% NO conversion as the activity indicator. The activity order of these catalysts was: Rh > Pd > Ru > Pt > Ir [Figure 5E]. The Rh1/CeO2 achieved complete NO conversion at 126 °C, whereas the Ir catalyst failed to achieve 100% NO conversion even near 400 °C. In addition, the presence of H2O promoted the conversion of both NO and CO compared to dry conditions [Figure 5F]. The maximum temperature for N2O production followed the order: Rh < Ru < Pd < Pt < Ir [Figure 5G]. For N2 selectivity, Pt and Ir exhibited poor performance [Figure 5H]. For the Rh1/CeO2 sample, NH3 started to form at 60 °C and reached a maximum (~ 260 ppm) at 116 °C, while the Ru1/CeO2 sample showed the highest NH3 production (~ 355 ppm). Other samples showed a maximum NH3 concentration around 150 ppm [Figure 5I]. This variation is due to the different catalytic activities of the SACs for the WGS reaction, which leads to different amounts of NH3 produced in the wet NO + CO reaction.
Negatively charged single-atom catalysts
The catalytic performance of metal catalysts fundamentally depends on the metal-reactant interaction (MRI) and the metal-support interaction (MSI). MSI not only stabilizes the catalyst but also influences interfacial processes, such as charge transfer and particle morphology [Figure 6A][98]. The charge transfer between the metal and support plays a critical role in determining catalytic performance. In 1978, Tauster et al. introduced the concept of strong metal-support interaction (SMSI), which has since been shown to significantly impact the activity, stability and selectivity[99,100]. In a detailed model, electrons are transferred from the oxide support to the metal atoms, leading to the formation of strong ionic bonds between negatively charged metal atoms and the surface cations[101].
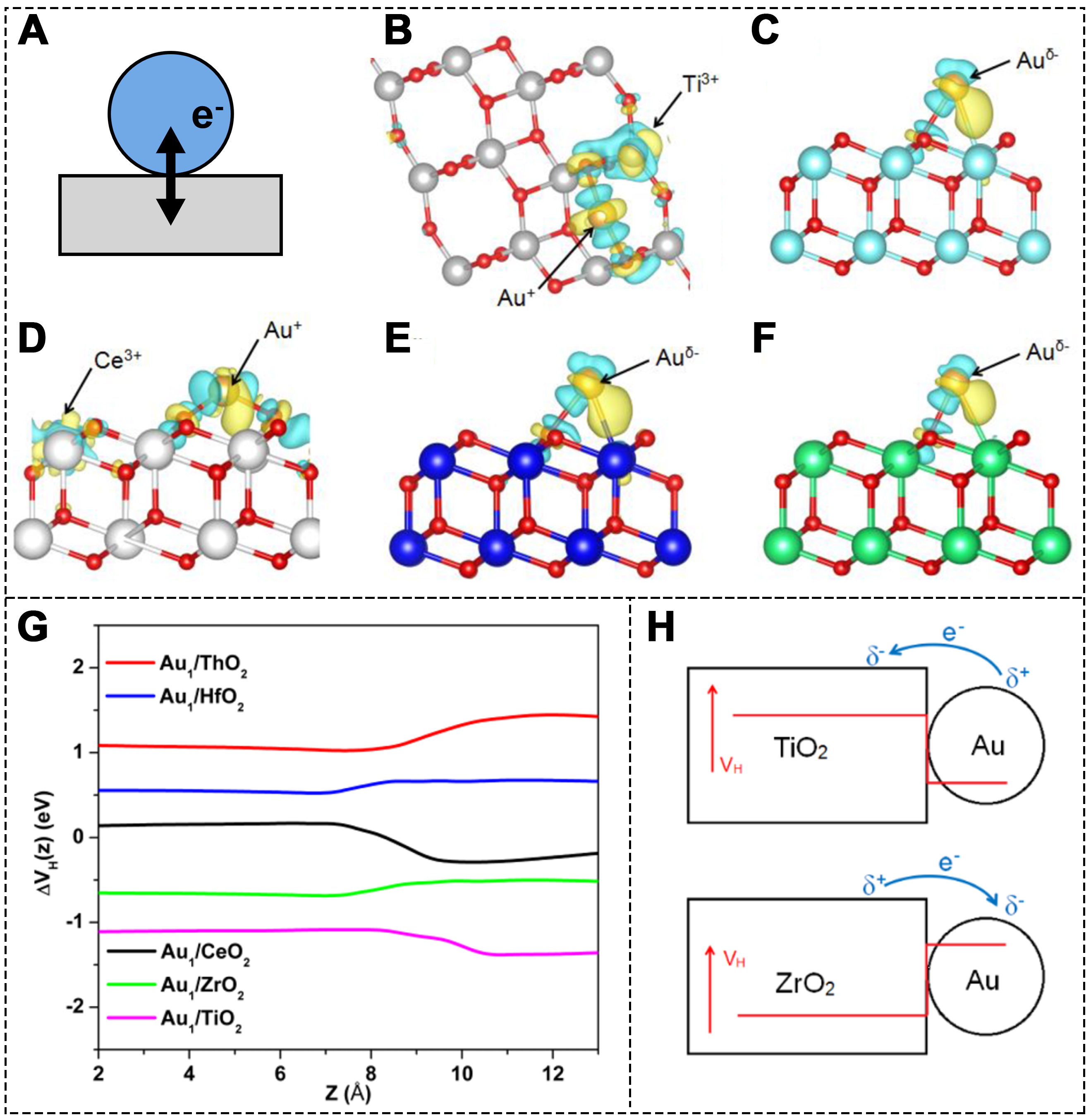
Figure 6. (A) Schematic of the electron transfer between metal and support; (B) Charge density differences of Au atoms on TiO2 (101); (C) c-ZrO2 (111); (D) CeO2 (111); (E) c-HfO2 (111); and (F) ThO2 (111) surfaces; (G) Charge transfer potentials of different SA model structures; (H) Schematic diagram of the relationship between Hartree potential (Vₕ) and charge transfer energy[102]. Copyright 2016, American Chemical Society.
Such negatively charged SAs, due to their unique electronic structure and charge distribution, can notably enhance the adsorption of reactants, thus improving the overall catalytic performance. The MSI effect is largely determined by the properties of the support material. Tang et al. used the DFT + U method to investigate the supporting role of different metal oxides (MO2, M = Ti, Zr, Ce, Hf, Th) in Au SACs[102]. They found that the oxidation state of Au atoms changes from Au(0) to Au(1) depending on the metal oxide support [Figure 6B-F]. Specifically, Au atoms on TiO2 and CeO2 are positively charged, while those on ZrO2, HfO2, and ThO2 are negatively charged [Figure 6G and H]. Adachi et al.[103] directly probed and understood the local environment and the charge state of the active sites on Au SACs, which also affects the adsorption of reactants[104]. On the rutile TiO2 surface, both positively and negatively charged Au SAs greatly promote CO adsorption, while neutral Au atoms show no noticeable adsorption. This demonstrates that negatively charged SACs, stabilized by strong MSIs, exhibit enhanced catalytic behavior, particularly in the adsorption of key reactants, which is critical in catalytic reactions.
High oxygen levels and SO2 inhibit CO-SCR by oxidizing active sites and competing for adsorption. To overcome these issues, negatively charged SACs enhance resistance via electron transfer regulation. Ji et al. prepared Pt-CuO/CoAlO catalysts and negatively charged Pt SAs were embedded into the CuO surface, which was supported on the CoAlO nanosheets[105]. The Pt 4f X-ray photoelectron spectroscopy (XPS) spectrum indicated that the Pt0 peak in the Pt-CuO/CoAlO shifted significantly towards the lower binding energy side, suggesting the formation of negatively charged Pt [Figure 7A]. X-ray absorption fine structure (XAFS) analysis confirmed the SA dispersion and negatively charged state of the Pt atoms
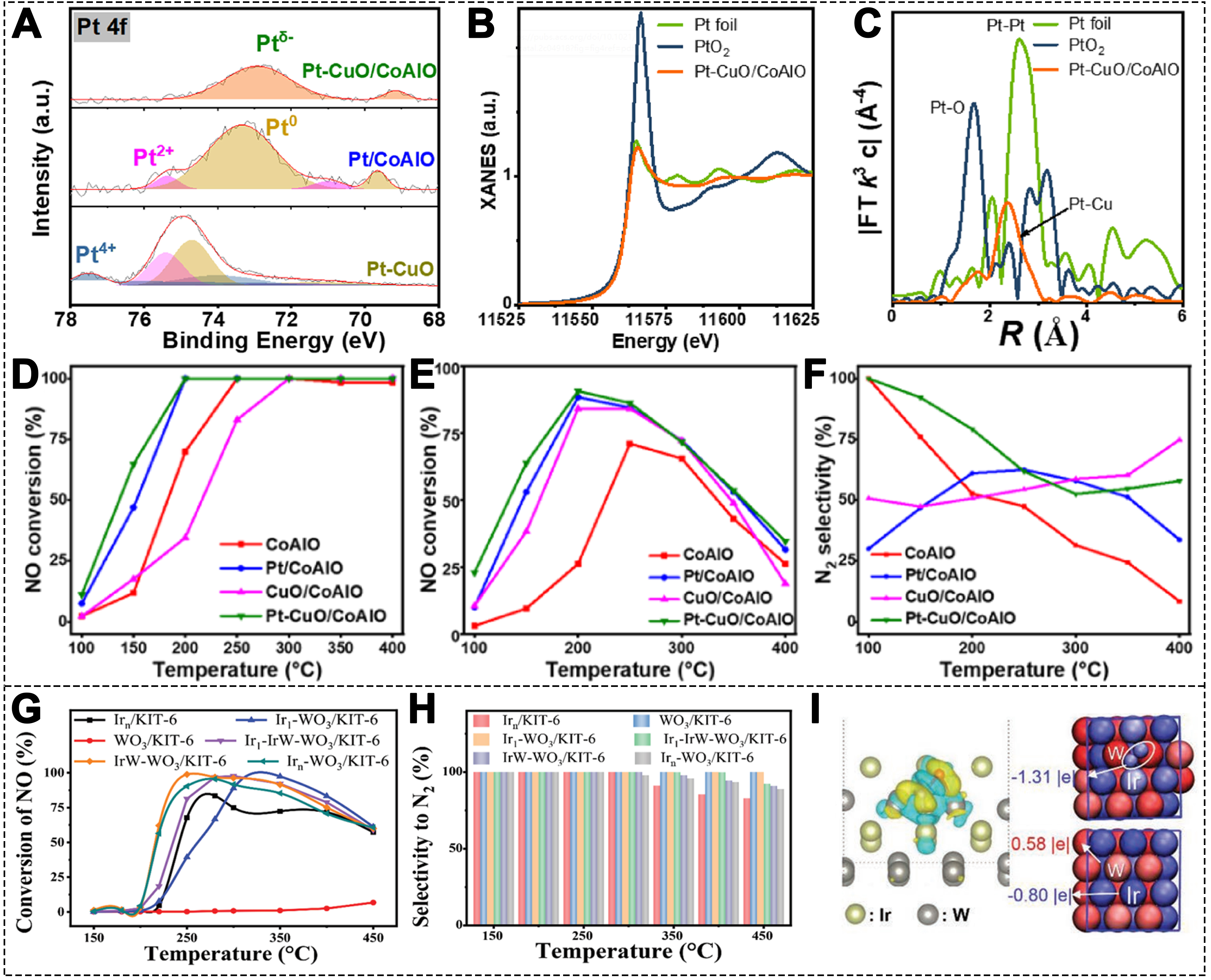
Figure 7. (A) Pt 4f XPS spectra of various catalysts; (B) Normalized Pt L3-edge XANES and (C) FT k3-weighted EXAFS spectra of Pt-CuO/CoAlO; (D) NO conversion in the absence of O2; (E) NO conversion and (F) N2 selectivity in the presence of 3% O2 for various catalysts (1000 ppm NO, 2000 ppm CO, 3% O2 when used, Ar balance, 10000 h-1)[105]. Copyright 2023, American Chemical Society; (G) NO conversion and (H) N2 selectivity of different catalysts (1000 ppm NO, 4000 ppm CO,1% O2, Ar balance, 50000 h-1); (I) charge density differences (blue and yellow represent electron accumulation and loss, respectively) and Bader charge map (red and blue represent positive and negative charges, respectively)[107]. Copyright 2021, Wiley-VCH. XPS: X-ray photoelectron spectroscopy; XANES: X-ray absorption near edge structure; FT: fourier transforms; EXAFS: extended X-ray absorption fine structure.
Dual-atom catalysts
SACs, while offering high atom utilization and well-defined active sites, often fall short in complex multi-molecule catalytic reactions due to their limited structural diversity and absence of cooperative interactions between active sites[66,108]. In contrast, DACs largely retain the advantages of SACs while introducing two adjacent or separated metal atoms as active sites, which can be either homometallic or heterometallic[109-114]. Even when not directly bonded, metal pairs can exhibit strong electronic interactions if positioned within appropriate interatomic distances[115-117]. Generally, DACs have a higher metal loading than SACs, as metal atoms are more likely to form atom pairs under higher loadings[118-121]. The second metal atom not only enhances the adsorption of reactant molecules but also tunes the local electronic structure of the dual-metal catalyst, thus altering the interface electron transfer[122-126]. DACs composed of different metal atoms exhibit higher structural complexity, playing a more significant role and holding greater potential in regulating reaction pathways and improving reaction selectivity[127,128].
Ding et al. developed a novel N-coordinated bimetallic catalyst (CuFe-N/C)[129]. The CuFe-N/C catalyst achieved nearly 100% NO conversion and excellent N2 selectivity within the temperature range of 225-400 °C [Figure 8A and B]. Two NO molecules adsorb at the Cu-Fe sites and couple to form the ONNO intermediate. CO then adsorbs and reacts with ONNO to form CO2 and N2O intermediates. N2O subsequently dissociates into N2 and O atoms, where the O atoms attached to Fe react with CO adsorbed on Cu to generate a second CO2 [Figure 8C]. The Cu-Fe coupling facilitates electron transfer from Fe to Cu, making the electrons around Cu more delocalized and electron-donor-like. The electrons are then transferred to the ONNO intermediate and activate it. The Fe → Cu → ONNO → Fe electron transfer forms a complete cycle, improving electron transfer efficiency during the reaction.
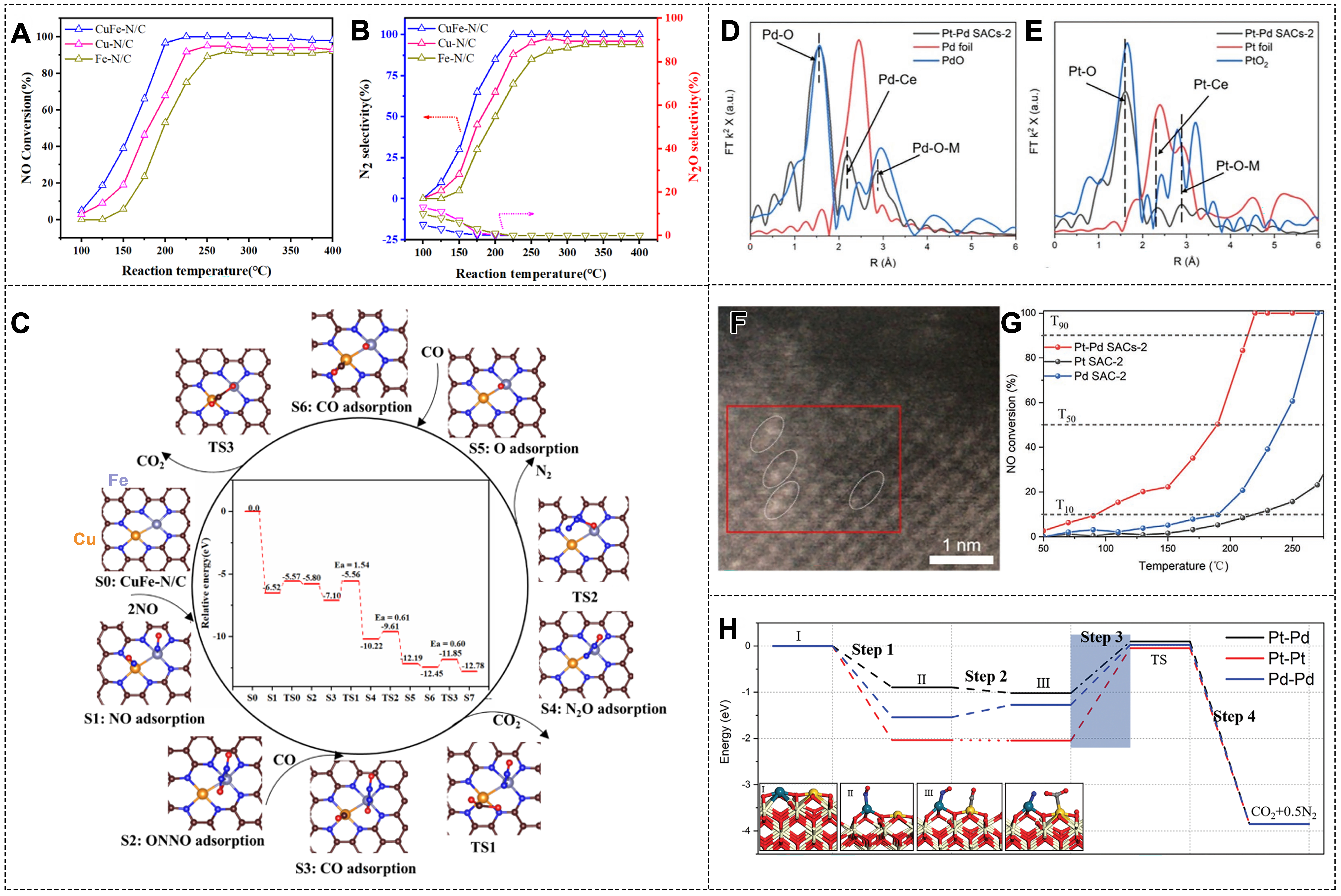
Figure 8. (A) NO conversion and (B) N2 and N2O selectivity of CuFe-N/C, Cu-N/C, and Fe-N/C (1,000 ppm NO, 2,000 ppm CO, Ar balance, 30,000 h-1). (C) Reaction pathway for NO reaction with CO on CuFe-N/C[129]. Copyright 2021, American Chemical Society. The Fourier transform of extended X-ray absorption fine structure (EXAFS) spectra in the R face obtained from (D) Pd K-edge and (E) Pt L-edge adsorption spectra of Pt and Pd species; (F) HAADF-STEM image of Pt-Pd SAC-2. (G) NO conversion of Pt-Pd SAC-2, Pt SAC-2 and Pd SAC-2 (500 ppm NO, 10,000 ppm CO, 1,000 ppm C3H6, 1.25% O2, He balance, 120 L g-1 h-1). (H) Free energy profiles for reactions in Pt-Pd, Pt-Pt, and Pd-Pd dual-site structures[130]. Copyright 2022, Wiley-VCH. EXAFS: Extended X-ray absorption fine structure; HAADF-STEM:
Researchers prepared a Pt-Pd DAC on vacancy-enriched CeO2 support [Figure 8D-F][130]. The Pt-Pd DAC exhibited excellent catalytic performance, with a T90 (the temperature at which 90% of NO is removed) as low as 210 °C, much lower than Pd SAC and Pt SAC physical mixtures (Pd SAC + Pt SAC) [Figure 8G]. DFT calculations indicated that adjacent Pt and Pd atoms serve as active sites for CO and NO adsorption, respectively [Figure 8H]. The energy barrier at Pt-Pd was 1.03 eV, lower than those for Pd-Pd and Pt-Pt, suggesting that the catalytic process is easier at the Pt-Pd site. The Pt-Pd DAC facilitates the migration of active intermediates, effectively lowering the energy barrier and accelerating the reaction.
Singly dispersed bimetallic sites catalysts
Catalytic sites typically consist of one or more atoms on the catalyst surface, arranged in a configuration that provides a specific electronic structure for the adsorption or dissociation of reactant molecules, thereby catalyzing the reaction by lowering the activation energy[131]. Compared to monometallic catalysts, bimetallic catalysts exhibit enhanced catalytic performance due to the inclusion of a second metal, which alters the electronic structure and modulates the catalytic properties of the monometallic catalyst[132,133]. Singly dispersed bimetallic sites refer to isolated catalytic sites composed of two different metal atoms distributed at the atomic scale, typically stabilized on a support. In a bimetallic site M1An, where M and A are the elements present, M refers to the active center for adsorption or bond activation, while A can modulate the local electronic environment, thereby enhancing catalytic performance[46,134]. In bimetallic NPs, the M1An site on the surface of the NPs exists in the metallic state, whereas on non-metallic supports, the M1An site is typically in a cationic form [Figure 9A].
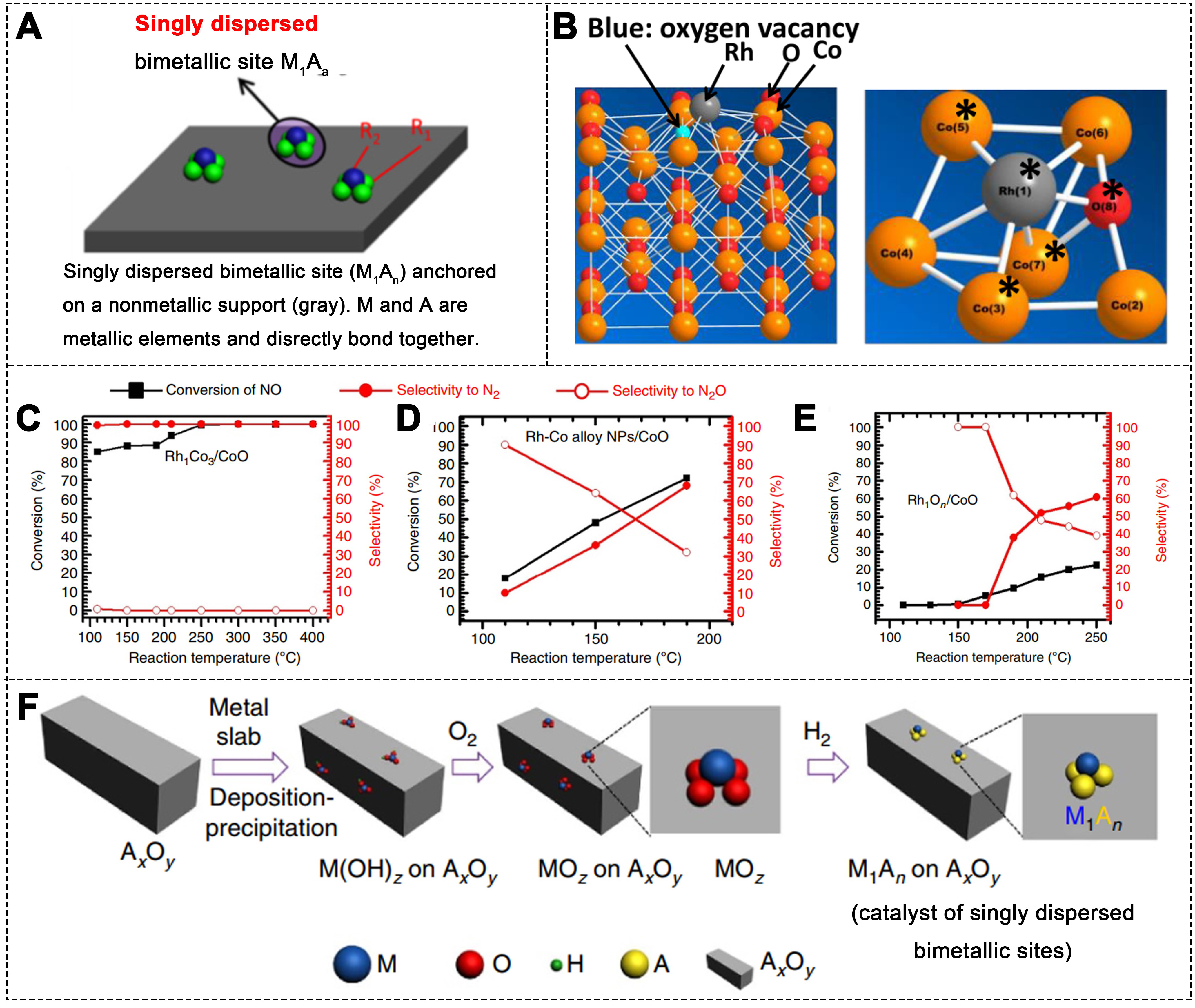
Figure 9. (A) Schematic of singly dispersed bimetallic sites[46]. Copyright 2023, Elsevier; (B) Optimized Rh1Co3 sites and Rh atoms on the CoO surface, and the bonding environment of Rh1Co3; (C) Catalytic activity and selectivity of Rh1Co3/CoO; (D) Rh-Co alloy NPs/CoO and (E) Rh1On/CoO (500 ppm NO, 500 ppm CO, Ar balance); (F) Schematic of the synthesis of singly dispersed bimetallic sites catalysts[135]. Copyright 2015, Springer Nature.
For example, Co3O4 was chosen as an oxide support (AxOy), with Rh as the metal M, resulting in the Rh1Co3/CoO catalyst, which was employed in CO-SCR reaction [Figure 9B][135]. At temperatures as low as 110 °C, the Rh1Co3/CoO catalyst exhibited 100% N2 selectivity. In contrast, the Rh-Co alloy NPs/CoO catalyst displayed much lower N2 production activity and selectivity, with values of 20% and 10%, respectively
The successful development of a catalyst with singly dispersed bimetallic sites, Rh1Co3, provides a new approach to achieve highly selective bimetallic catalysts. This concept can be applied to design catalysts where isolated bimetallic sites M1An are constructed by having metal atoms M occupy the O vacancies of metal oxides A-O[136]. A similar preparation method [Figure 9F] can also be used to synthesize catalysts composed of isolated bimetallic sites, such as Pt1Con or PdmCom’[137]. Additionally, many transition metal elements can serve as either M or A in the construction of isolated bimetallic sites M1An, enabling the development of a wide range of bimetallic catalysts with such sites[136,138]. Notable examples include previously reported Pt1Con/Co3O4[139], Ir1Znn/ZnO[140], Pd1Znn/ZnO[134] and Ir1Ti3/TiO2[141]. The extension of M1An catalysts to other catalytic reactions shows that the charge-buffering capability of metal M and the synergistic effect of metal A in catalytic reactions are key factors for achieving high catalytic efficiency[142-144].
Single-atom alloy catalysts
SAACs typically consist of two different metal atoms, one in a SA state and the other in an alloyed state[132,145-149]. SAACs offer several significant advantages. Firstly, the alloying effect enables these catalysts to retain the high catalytic activity of SACs while enhancing their structural and thermal stability[150,151]. Secondly, by precisely tuning the composition and spatial configuration of metal atoms, SAACs can achieve superior control over catalytic reactions, thereby improving both selectivity and efficiency[152,153]. Additionally, SAACs exhibit exceptional thermal stability and selectivity[154,155]. Moreover, co-adsorption at the active sites of SAACs can alter the energy distribution of the entire reaction, significantly boosting catalytic activity by several orders of magnitude[156].
The advantages of alloy-based catalysts in the CO-SCR reaction are increasingly evident. In the case of IrRu alloy catalysts, the incorporation of Ru can suppress the agglomeration of Ir particles and provide more active sites for NO adsorption[157,158]. Ru also facilitates the CO oxidation process at lower activation energy barriers while enhancing the oxidative resistance of Ir[157,159]. The synergistic effect between Ir and Ru is attributed to the inherent properties of the IrRu alloy phase, rather than to the individual activities of isolated Ir or Ru species[160]. In SAAs, small amounts of active metals are well distributed on the surface of another metal, enhancing catalytic activity via stronger metal-metal bonds or metal-NO interactions[161,162]. In studies of Ir-doped Ni (110), the doping of Ir significantly reduced the formation barrier of N2 and increased the barrier for N2O formation, thereby improving N2 selectivity[163].
In these SAACs, isolated noble metal atoms exhibit unique reactivity, while interactions with surrounding metal atoms contribute to enhanced catalytic performance in a variety of reactions[164]. Xing et al. prepared a series of CuPd alloy NPs supported on Al2O3 and evaluated their catalytic performance in the NO reduction reaction[165]. The Cu and Pd atoms were uniformly distributed, with Pd atoms isolated by Cu atoms
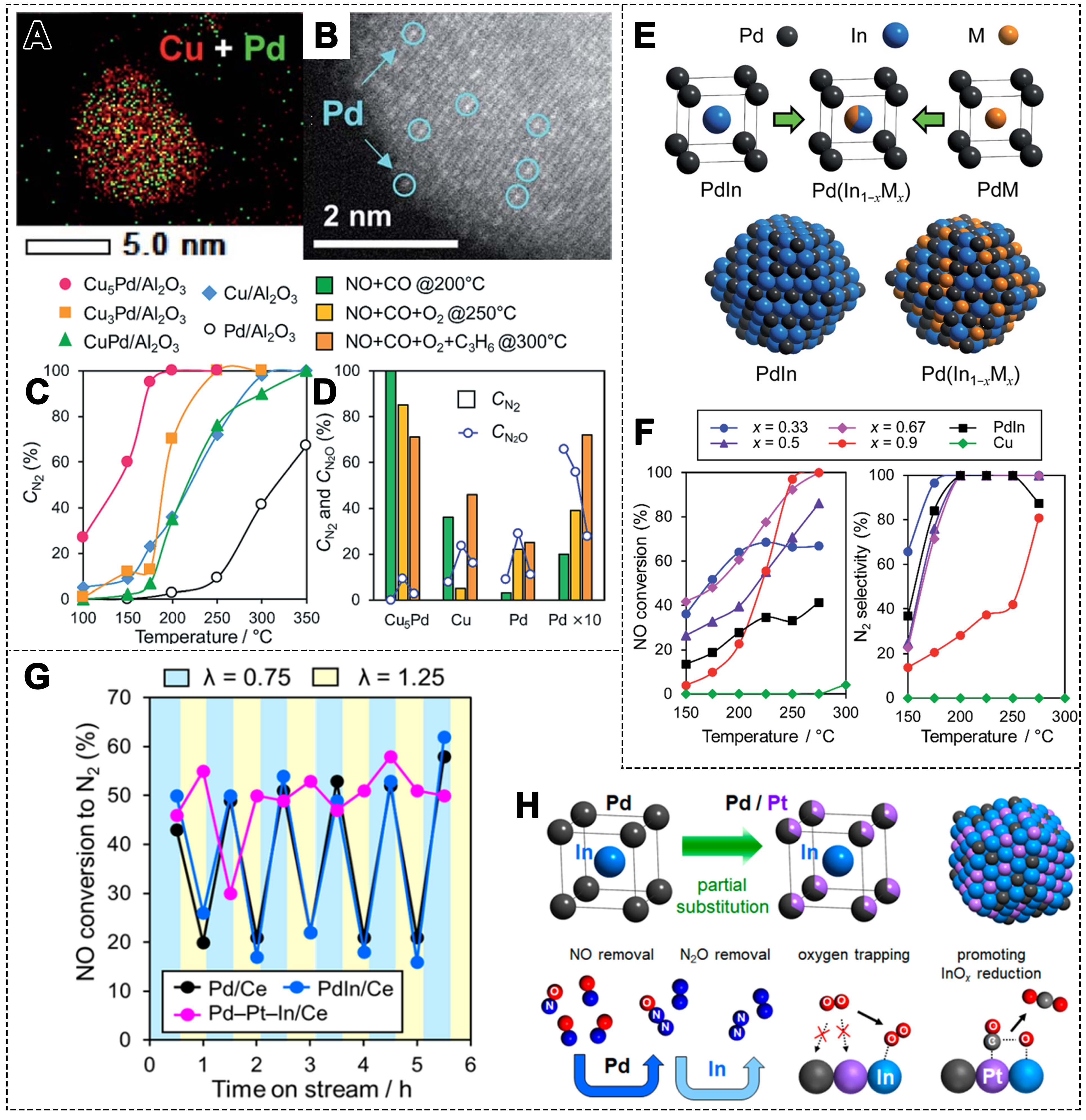
Figure 10. (A) Mapping of Pd and Cu elements in Cu5Pd/Al2O3 catalyst; and (B) high-resolution image of a single NP; (C) NO conversion to N2 over Pd-Cu catalysts and (D) comparison of NO reduction in the presence of O2 and C3H6 (5,000 ppm NO, 5,000 ppm CO, He balance, 80,000 h-1)[165]. Copyright 2019, The Royal Society of Chemistry; (E) Models of NP PdIn and Pd(In1-xMx); (F) NO conversion and N2 selectivity on different Pd-In1-xCux/Al2O3 catalysts (5000 ppm NO, 5,000 ppm CO, He balance, 240,000 h-1)[167]. Copyright 2019, The Royal Society of Chemistry; (G) NO-CO-O2 reaction on Pd-M/CeO2 catalysts after five rich-lean cycles at 300 °C (5,000 ppm NO, 10,000 ppm CO, 0-7,500 ppm O2, He balance, 40,000 h-1); (H) Model structure of (Pd1-xPtx)In and the role of each metal in the catalysis[168]. Copyright 2020, American Chemical Society. NP: Nanoparticle.
Alloying different metals can alter the electronic and coordination environment of active metals, thereby enhancing both catalytic activity and selectivity[166]. In Pd-based bimetallic catalysts (PdM/ Al2O3: M = Cu, In, Pb, Sn, and Zn), PdCu exhibited higher NO conversion, while PdIn showed superior N2 selectivity[167]. The PdIn/ Al2O3 catalyst demonstrated excellent N2 selectivity at 200 °C. By replacing a portion of In with Cu, a Pd(In1-xCux) pseudo-binary alloy structure was formed, further enhancing the catalytic performance [Figure 10E]. The optimized Pd(In0.33Cu0.67)/Al2O3 catalyst achieved complete NO conversion to N2 at 200 °C [Figure 10F]. Cu substitution improved the adsorption and dissociation of NO, enhancing high-temperature activity. In the presence of O2, CO reacted more readily, but residual NO reduced the NO conversion [Figure 10G]. By partially substituting Pd in PdIn with Pt, the metallic state of In could be retained [Figure 10H][168]. During the rich-lean cycle (air-fuel equivalence ratios λ = 0.75/1.25),
Single-atom-cluster/nanoparticle catalysts
In the case of complex reactions or systems, SACs may have limited capability to regulate catalytic activity and selectivity due to their structural simplicity and the absence of necessary synergistic active sites. Additionally, variations in the adsorption energies of different reactants may promote certain reaction steps, while subsequent step may require distinct active sites, thereby limiting the overall catalytic efficiency[169]. To overcome these limitations, integrating SAs, clusters, and NPs into a unified heterogeneous catalyst can enhance performance through synergistic effects and electronic structure tuning[170-172]. This approach can involve either the combination of SAs and NPs of the same element[169,173-175], or the combination of SAs and NPs from different elements [Figure 11A-C][176]. The NPs can be metal particles or metal oxides. When SAs, clusters, and NPs are combined, the active metal components distribution in terms of size, morphology, local coordination environment, and the surrounding electronic states of active sites can be adjusted through coupled synergistic interactions, thus influencing catalytic performance [Figure 11D][177]. The heterogenization of the surface structure offers distinct advantages in complex multiphase catalytic reactions.
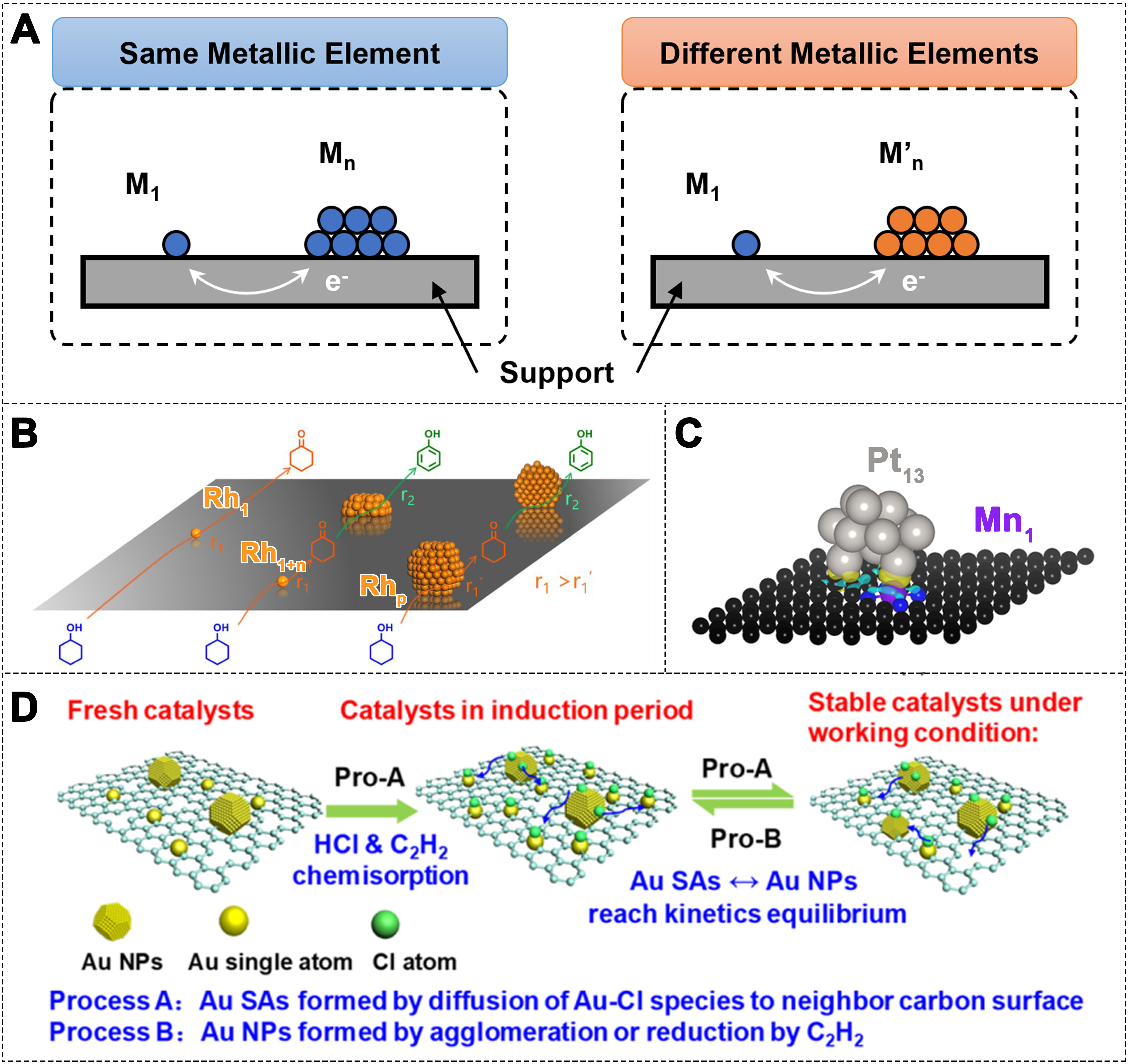
Figure 11. (A) Schematic model of single atom-cluster/NP structures; (B) Illustration of the hydrogen production behavior from cyclohexanol over Rh1/ND@G, Rh1+n/ND@G, and Rhp/ND@G catalysts[169]. Copyright 2022, American Chemical Society; (C) Optimized atomic structure of Pt13 clusters in graphene layers[176] Copyright 2023, American Chemical Society; (D) Schematic diagram of the self-dispersion mechanism of Au NPs on defective carbon[173]. Copyright 2020, American Chemical Society. NP: Nanoparticle.
Liu et al. developed a dual-active-center catalyst, which integrated Co SAs and CoOx nanoclusters (NCs) on Ce0.75Zr0.25O2 (CZO) support [Figure 12A and B][178]. The DFT calculations showed that Co SAs primarily act as adsorption sites for CO, while CoOx clusters preferentially adsorb and activate NO. This catalyst demonstrated over 99.7% NO conversion and 100% N2 selectivity at 250-400 °C under 5 vol.% O2 conditions [Figure 12C and D]. Moreover, the Co SAs and CoOx clusters exhibit stronger adsorption to NO than to H2O and SO2, providing excellent resistance to H2O and SO2 poisoning. The unique synergistic effect between Co SAs and CoOx clusters was identified as the key factor for the high CO-SCR performance of the CoSA + CoOxNC/CZO catalyst.
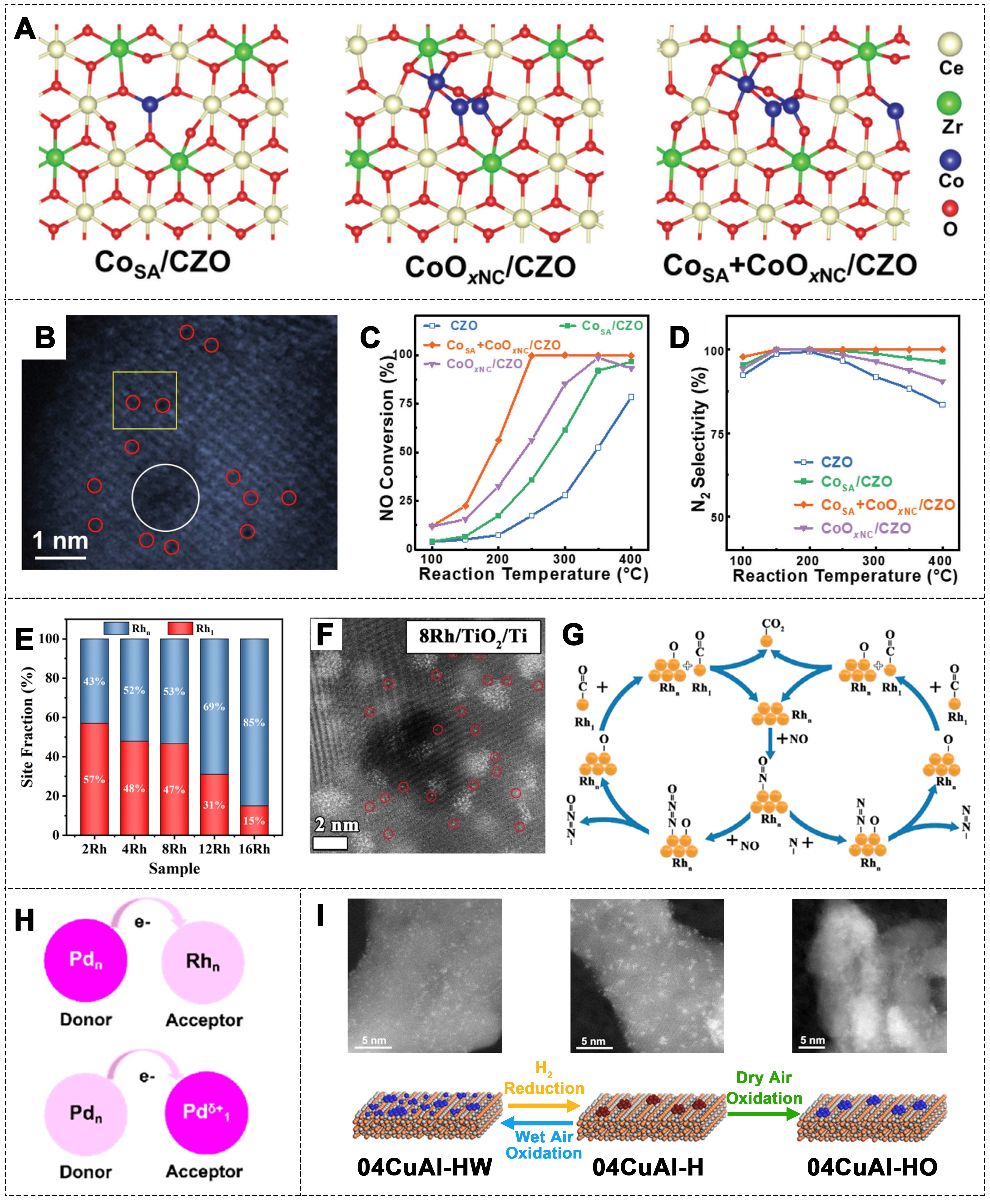
Figure 12. (A) Computational structural models of CoSA/CZO, CoOxNC/CZO, and CoSA CoOxNC/CZO; (B) AC HAADF-STEM image of CoSA+CoOxNC/CZO; (C) NO conversion and (D) N2 selectivity of different catalysts (1,000 ppm NO, 2,000 ppm CO, 5% O2, N2 balance, 20,000 h-1)[178]. Copyright 2023, Wiley-VCH; (E) Site fraction (%) of Rh1 and Rhn for all Rh/TiO2/Ti catalysts; (F) HAADF-STEM images of 8Rh/TiO2/Ti. (G) Schematic diagram of the possible CO-SCR reaction mechanism in Rh/TiO2/Ti catalyst[180]. Copyright 2023, Wiley-VCH; (H) Schematic diagram of electron acceptor and donor between PdNP/RhNP pairs and Pd1/PdNP pairs[48]. Copyright 2022, American Chemical Society; (I) HR-STEM images and structural transformation diagram of the 04CuAl sample after various treatments[183]. Copyright 2019, American Chemical Society. HAADF-STEM: CO-SCR: selective catalytic reduction of nitrogen oxides by CO.
In single-atom-cluster/NP catalysts, the electron-deficient SA sites and the electron-rich NP sites can selectively adsorb different reactants and cooperate catalytically in the reaction[48,179]. Yang et al.[180] successfully synthesized Rh SAs and clusters on Rh/TiO2/Ti catalysts [Figure 12E]. The 8Rh/TiO2/Ti catalyst achieved 100% NO conversion in the presence of O2 at 190 °C. Additionally, it maintained a certain catalytic activity under different O2 concentrations. The analysis revealed that the reactants NO and CO were selectively adsorbed on the Rhn clusters and Rh1 SAs, respectively, alleviating the competitive adsorption between NO and CO [Figure 12F and G]. The electronic interaction between SA and NP/cluster sites plays an important role in modulating the electronic structure[181,182].
In three-way catalyst (TWC), Rh acts as an electron donor, while Pd serves as an electron acceptor. This electron transfer between the two metals influences the catalytic performance[48]. In single-metal-based catalysts, systems with Pd SAs and Pd NPs can be used. The difference in electronegativity can drive electron transfer from Pdn to Pd1 sites, enabling the TWC reactions as Pd-Rh catalysts while reducing the use of valuable Rh [Figure 12H]. Moreover, the structure of single-atom-cluster/NP catalysts can transform under different reaction conditions, and such dynamic changes can significantly influence catalytic performance. Liu et al. systematically studied the dynamic structural changes of CuO species during redox processes[183]. The 04CuAl sample, after treatment with hydrogen, showed a large number of Cu NCs. After oxidation in dry air, the 04CuAl-HO sample retained a large number of Cu clusters. However, oxidation in wet air resulted in most of the Cu being present as atomically dispersed Cu2+ in the 04CuAl-HW sample, with a few CuO clusters remaining [Figure 12I].
Comparison of different single-atom catalyst systems
Different types of SAC systems exhibit distinct structural features, electronic properties, and catalytic behaviors in CO-SCR reactions. A clear understanding of their respective advantages and limitations is essential for rational catalyst design [Table 2].
Comparison of different types of SAC systems
| SAC type | Key features | Advantages | Disadvantages |
| Conventional SACs | Isolated metal atoms on supports | High atom utilization; well-defined sites; enables detailed mechanistic study | Lack of cooperative sites; limited performance in complex multi-step reactions |
| Negatively charged SACs | Charge transfer from support to metal | Enhanced NO adsorption; good O2/SO2 resistance; better low-temp activity | Difficult to control uniform charge distribution across catalyst surface |
| DACs | Homonuclear or heteronuclear metal pairs | Synergistic effects; improved multi-step reaction performance | Challenges in precise characterization and control of dual-atom active sites; complex synthesis; limited site stability under reaction conditions |
| Singly dispersed bimetallic sites | Two different atoms co-dispersed and isolated on support | Tunable electronic structure; enhanced NO activation and selectivity | Complex synthesis process; challenging atomic-scale characterization |
| SAACs | Single atoms embedded in alloy NPs | Combined activity and thermal stability; alloy effects optimize reactivity | Performance highly dependent on alloy composition and surface structure |
| Single-atom-cluster/NP catalysts | Atomically dispersed species combined with clusters or nanoparticles | High activity in complex systems; broadened adsorption and activation scope | Difficult to control interface structure; potential for sintering or performance inconsistency |
Conventional SACs provide high atomic utilization and uniform active sites, offering an ideal platform for mechanistic investigations. Their well-defined structures allow for precise control over reaction pathways. However, the isolated nature of SAs limits their ability to activate multiple reactants cooperatively, which may reduce performance under complex conditions. Negatively charged SACs, formed through electron transfer between the metal and support, enhance NO adsorption and promote low-temperature activity. These catalysts also demonstrate improved resistance to O2 and SO2 poisoning due to their modified electronic structure. Nevertheless, maintaining a uniform charge distribution across the surface remains challenging. DACs introduce synergistic effects between two neighboring atoms - either of the same or different elements - thereby facilitating multi-step reactions and improving metal loading. The main limitations lie in their complex synthesis and structural instability under harsh reaction conditions. Singly dispersed bimetallic site catalysts integrate two different metal atoms into isolated sites, tuning local electronic environments and adsorption energies. This leads to improved NO activation and higher selectivity. However, the synthesis process is typically more demanding, and thermal stability is a concern. SAACs embed SAs into alloy NPs, balancing high activity with enhanced stability. The alloying effect optimizes intermediate adsorption/desorption behavior, though the catalyst’s performance can be highly sensitive to the precise alloy composition and surface arrangement. Single-atom-cluster/NP catalysts combine atomically dispersed species with clusters or NPs to broaden the range of active sites and improve reaction selectivity in complex gas environments. The major drawback is the difficulty in controlling the interfacial interactions between SAs and larger metal domains, which may lead to performance inconsistency or partial sintering.
In summary, each SAC system has its own strengths tailored to specific operating conditions. The choice of system should be based on the targeted reaction environment, temperature, gas composition, and durability requirements. Continued exploration of synthetic strategies, stability control, and MSIs will be key to advancing the design of next-generation CO-SCR catalysts.
DISCUSSION AND OUTLOOK
To comprehensively promote the development and application of SACs, this section discusses their key progress and challenges from three perspectives: (1) the design and synthesis of SACs, with emphasis on atomic-level control and scalable production; (2) the industrial applications and limitations of SACs, particularly under complex operating environments; and (3) future research trends, including innovative catalyst structures, advanced characterization, and theoretical insights. These aspects collectively provide guidance for the practical and sustainable advancement of SAC technologies.
Design and synthesis of single-atom catalysts
SACs offer a platform for precise catalysis, enabling the design of catalysts at the atomic level. The main focus is to achieve atomic-scale control over active sites to optimize reaction activity and stability[184].
Design strategies for SACs
Effective SAC design hinges on the precise manipulation of active sites. This can be achieved through manipulating the spatial configuration of active atoms[185-187], fine-tuning the distances between active atoms[188], and modifying the coordination environment of these atoms [Figure 13A][189]. In recent years, interface adaptation strategies have emerged, such as designing dual-atom structures with specific metal distances to optimize catalytic performance[190,191]. The chemical environment of metal atoms in SACs, including coordination structures and interactions with support, significantly influences their electronic properties and catalytic activity[78,192-195]. Developing methods to precisely regulate these environments and achieve long-term stability of surface species and active sites is a key direction in catalyst design.
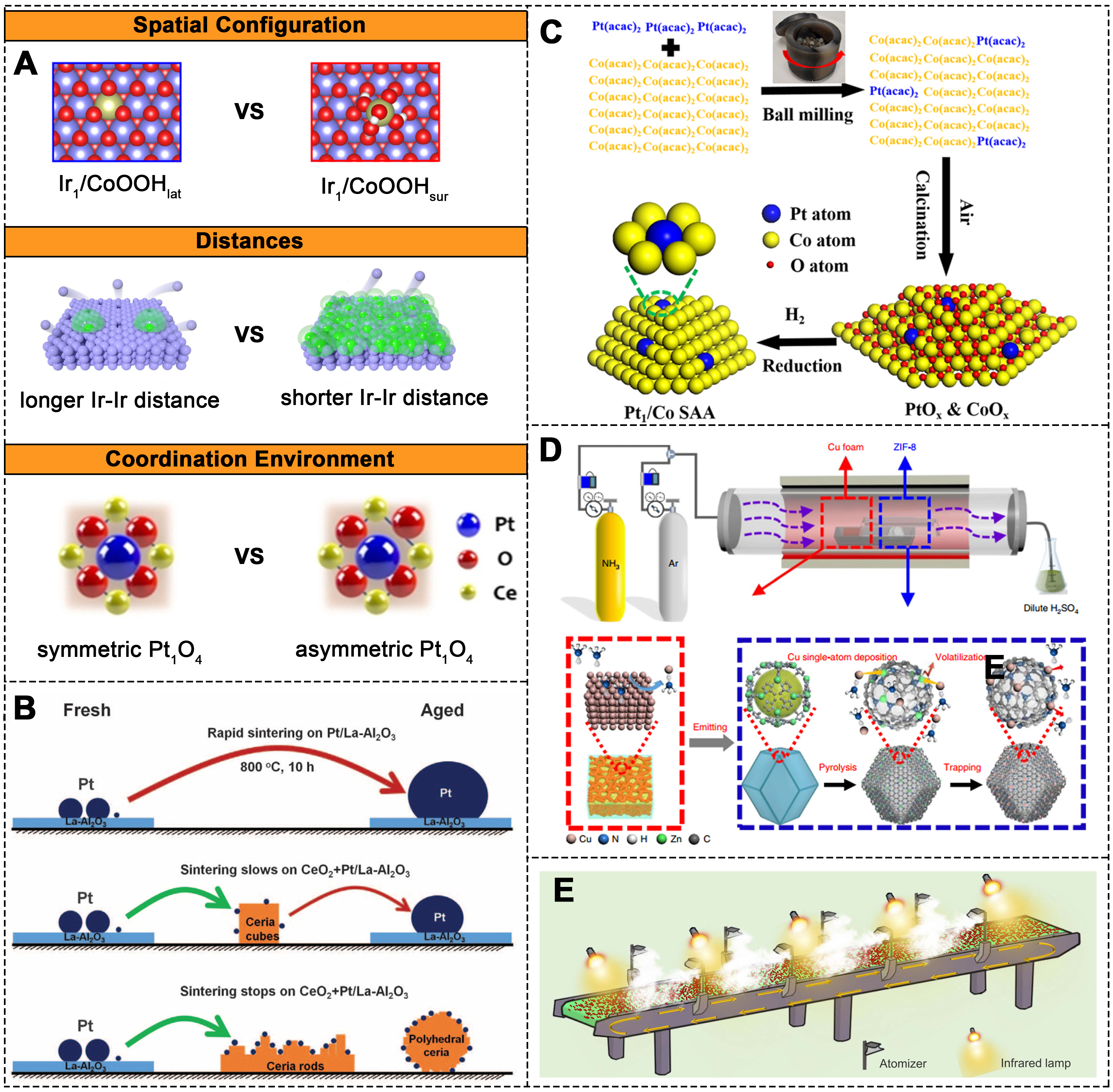
Figure 13. (A) SAC regulation strategies: spatial configuration, distances, and coordination environment of active atoms. Schematic structure models of Ir1/CoOOHlat and Ir1/CoOOHsur. The white, red, purple, and yellow spheres represent H, O, Co, and Ir atoms, respectively[189]. Copyright 2022, American Chemical Society. Illustration of the effect of Ir atomic distance on the stability of cobalt oxide catalysts. The blue and green spheres represent Co and Ir atoms[190]. Schematic representation of symmetric (near-perfect) and asymmetric (distorted square-planar) Pt1O4 coordination[78]. Copyright 2021, Wiley-VCH; (B) Schematic of the sintering process of Pt NPs after physical mixing of ceria with Pt/La-Al2O3[202]. Copyright 2016, AAAS; (C) Schematic of the preparation of Pt1/Co SAACs via ball milling[204]. Copyright 2020, American Chemical Society; (D) Schematic of the experimental device and preparation of Cu-SAs/N-C catalysts[208]. Copyright 2020, Springer Nature; (E) SAC: synthesis production line based on precursor aerosol strategy[214]. SAC: Single-atom catalyst.
Synthesis Methods for SACs
The synthesis of SACs typically uses techniques such as impregnation[196], ALD[51,197], dissolution-and-carbonization[198] and cation exchange[199]. In terms of catalyst stability, dispersion and scalability, each method presents distinct advantages and limitations. Bottom-up synthesis methods, such as chemical adsorption, co-deposition, and high-temperature treatments, establish strong MSIs that promote the dispersion and stabilization of SAs[200]. Top-down methods, on the other hand, convert bulk metals or NPs into stable SAs[201]. For example, mixing ceria powders of different forms with Pt/La-Al2O3 catalysts and treating them at high temperatures results in the formation of Pt SAs on the ceria support [Figure 13B][202]. Reported synthesis methods have achieved kilogram-scale production. A widely used method is mechanical chemical synthesis, which uses mechanical energy to facilitate the physicochemical transformation and chemical reactions between precursors[203]. This method is particularly attractive due to its scalability, high catalyst yield, and broad applicability. For example, Gan et al. synthesized kilogram-scale Pt1/Co SAACs using ball milling [Figure 13C][204]. By altering the metal precursors, they demonstrated the preparation of different co-loaded noble metal SAACs (M1/Co, M = Pd, Ru, Ir, Rh). Kilogram-scale SACs, such as
Increasing metal loading in SACs often causes atom aggregation and NP formation, which compromises atomic dispersion and catalytic efficiency. This challenge arises from limited anchoring sites and the inherent instability of isolated atoms during synthesis and operation. To address this, advanced strategies such as nitrogen-rich coordination environments[198,209,210] and negative-pressure annealing treatments[211,212] have been employed to stabilize high-density SAs. For instance, Xiong et al. synthesized SAS-Fe catalysts with a loading of up to 30 wt.%[213], and Chang et al. reported 17 SACs with loadings exceeding 20 wt.%, enabled by high levels of nitrogen doping and optimized local coordination environments[52]. However, high metal loading is not the ultimate objective - catalytic performance and long-term stability are of greater importance. Without metal-support interactions, high-loading SACs may suffer from serious aggregation and reduced durability under practical conditions.
Large-scale production for SACs
To enable large-scale, continuous production, extensive efforts have been made to establish scalable and reliable fabrication processes for SACs. One strategy involves atomizing precursor solutions, which are then sprayed onto supports and annealed to produce SACs [Figure 13E][214]. This method is highly versatile and has been successfully used to synthesize 19 types of SACs, including SAACs, DACs, and bimetallic SACs, all in a continuous production setting. Emerging synthesis methods, such as pyrolysis[213] and photochemical methods[215], are also under development. These techniques offer new possibilities for achieving efficient, cost-effective and environmentally friendly production of SACs.
Although current synthesis methods can produce SACs at large scales, achieving efficient, simple, and scalable production remains a significant challenge. The future of SAC development will center on addressing challenges related to large-scale, sustainable, and green production processes. In conclusion, the design and synthesis of SACs are entering a new era of precision, where atomic-level control and scalable production are keys to realizing their industrial applications.
Industrial applications and challenges of single-atom catalysts
The transition from laboratory-scale synthesis to industrial application presents significant challenges related to the cost, stability, reproducibility, and large-scale production of catalysts. While SACs exhibit excellent performance at the laboratory scale, issues such as non-uniform distribution, high production costs, and low efficiency hinder their large-scale adoption. Industrial catalysts, particularly those used in heterogeneous catalysis, often operate in more complex environments, involving multiple phases, multiple states, and multiple scales. These catalytic systems encompass active particles, porous supports, catalyst particles, and reactor components, all of which must be carefully integrated[216,217]. In industrial catalysis, the design of the reactor and the compatibility of the catalyst are key factors for successful application[218]. Additionally, factors such as the catalyst surface structure, pore configuration, and reaction atmosphere significantly influence reaction kinetics and product selectivity[219,220].
From an industrial application standpoint, catalyst stability and space-time yield are the two most critical concerns[221]. (1) Stability: Catalysts, especially SACs, tend to lose activity under high-temperature or oxidative conditions, a common scenario in industrial applications. Despite their superior catalytic activity at lower temperatures, SACs are prone to sintering or aggregation over prolonged use, which significantly diminishes their catalytic performance[41]. Ensuring the long-term stability of the SA structure is thus a critical challenge for industrial applications. Strategies such as defect engineering, spatial confinement, and ligand design can enhance the stability of the active sites, suppress aggregation, and improve both the activity and durability of the catalysts[222-224]; (2) Space-time yield: Space-time yield, which measures the productivity of a catalyst per unit volume per unit time, is a key indicator of the economic efficiency of industrial catalysis. In large-scale applications, SACs typically exhibit lower space-time yields compared to traditional NP catalysts[225]. Increasing the density and improving the distribution of active sites in SACs, along with exploring new synthesis methods, is crucial for achieving high-yield, well-dispersed SACs that maintain excellent catalytic performance across various reactions, thereby enhancing industrial productivity[226].
SACs offer significant environmental and economic benefits due to their 100% atomic utilization and fully exposed active sites. Traditional catalysts often suffer from deactivation due to sintering or poisoning, which results in their disposal. These discarded catalysts, especially those containing noble metals, can contribute to environmental pollution, contaminating soil and water[227]. By designing recovery systems that convert used catalysts back into their SACs form, we can minimize the need for catalyst replacement, reduce production costs, and lower waste disposal expenses. This approach also reduces the environmental impact of discarded catalysts[228,229]. For instance, using nitrogen-doped carbon-based thermal atomization techniques[230], sintered noble metal particles can be redistributed into isolated atoms, recovering their catalytic activity.
As catalyst synthesis techniques continue to improve and production scales expand, SACs are expected to demonstrate significant environmental and economic benefits across various industrial fields, including automotive exhaust treatment, industrial waste gas processing, wastewater treatment, and energy conversion. This highlights the promising potential of SACs in driving both environmental sustainability and economic efficiency.
Future directions and research trends
With the continuous advancement of catalytic technology, future research on SACs will focus on innovative catalyst design, the enhancement of multifunctionality, and a deeper understanding of catalytic mechanisms[231]. SAACs, through the synergistic effects between metals, provide optimized catalytic activity across multiple reaction pathways. These catalysts not only regulate the selectivity of reactions but also exhibit excellent stability and efficiency in complex reaction systems. In addition, significant progress has been made in the study of single-atom nano-island catalysts[232-236]. Nano-islands comprise small particles distributed on the support surface, where SAs can selectively adsorb. This structural design effectively enhances the thermal stability of SAs, preventing their agglomeration under high-temperature reaction conditions, while optimizing the electronic structure to improve catalytic selectivity and efficiency.
With the continuous improvement of experimental equipment, especially the development of aberration-corrected high-resolution transmission electron microscopy (AC-HRTEM), the structure of individual atoms can now be captured with precision, providing a solid foundation for the rational design of SACs and the concept of catalysis. Future research will rely on more precise characterization techniques to reveal the microstructure and reaction processes of catalysts[237,238]. For example, AC-HRTEM can reveal the atomic-level structure of catalysts, analyzing the distribution and morphological changes of active sites[239]; synchrotron radiation technology can explore changes in the catalyst surface, oxidation state, and coordination structure[240,241]; while in situ Raman spectroscopy can capture dynamic processes during catalytic reactions and establish dynamic characterization methods at the atomic/molecular scale[242]. Additionally, in situ characterization techniques, such as in situ transmission electron microscopy (TEM), can observe the structural evolution of catalysts and the changes in reaction intermediates under reaction conditions[243]. These advancements will provide robust support for the design, optimization, and practical application of catalysts.
Theoretical calculations also provide valuable guidance for the design of SACs. Compared to traditional nano-catalysts, SACs have a more uniform atomic configuration, allowing methods based on DFT to more accurately simulate the catalytic mechanisms and changes in active sites during reactions[244,245]. Future research should strengthen the integration of theoretical calculations and experimental validation, particularly in the fine-tuning of catalytic reactions and optimization of catalytic performance, using computational results to predict the behavior and reaction mechanisms of catalysts[246]. Furthermore, theoretical studies should explore the interaction between reactants and catalyst surfaces and the stability of active centers, promoting the design and optimization of catalysts under various reaction conditions. In addition, machine learning is emerging as a powerful tool for accelerating the discovery of SACs, enabling rapid screening of active site structures, prediction of catalytic performance, and identification of optimal material combinations[247-249]. By combining theoretical calculations with dynamic characterization techniques, catalysts can be precisely designed for a wide range of reaction environments, ensuring their efficiency and stability[250].
The future development of SACs will rely on the synergy among catalyst design, advanced characterization, and theoretical calculations. The rational design of SAACs and nano-island structures enhances stability and selectivity, while cutting-edge characterization techniques provide atomic-level insights into structural evolution. Meanwhile, theoretical calculations guide experimental optimization by predicting catalytic behaviors. The integration of these approaches will deepen mechanistic understanding and accelerate the practical application of SACs.
CONCLUSION
This review summarizes the application and progress of SA catalytic systems in CO-SCR reaction. It begins by introducing various types of SACs, including conventional SACs, negatively charged SACs, DACs, singly dispersed bimetallic sites catalysts, SAACs, and single-atom-cluster/NP catalysts. The unique properties of each catalyst and their application in CO-SCR are discussed in detail.
SACs promote the adsorption and activation of NO through synergistic interactions between the SAs and the support, lowering the activation energy of NO reduction. This results in reaction pathways that differ from those of NP catalysts, exhibiting significantly higher catalytic activity. Additionally, modifications of the local structure of SAs (such as coordination environment and electronic structure) can influence the formation of intermediate species, further enhancing catalytic performance. Negatively charged SACs, due to the charge transfer between metal atoms and supports, demonstrate stronger NO adsorption ability. This electronic transfer leads to higher catalytic efficiency in NO reduction and alters the binding strength at the active sites, enhancing resistance to O2 and SO2 poisoning. DACs demonstrate important synergistic effects in CO-SCR reactions. The interactions between the two metal atoms significantly boost catalytic activity, with high metal loading typically offering better performance than SACs. Moreover, adjusting the acidity/redox properties of the dual-metal sites can effectively reduce their affinity for H2O and SO2, enhancing the resistance to H2O and SO2 poisoning. Singly dispersed bimetallic sites catalysts, by incorporating a second metal, optimize the electronic structure of the single-metal catalyst, improving catalytic properties. During the reaction, the M1An site plays a critical role in the adsorption and activation of NO, weakening the N-O bond and lowering the activation barrier for N2O decomposition, which further enhances catalytic activity. SAACs combine the high activity of SACs with the superior stability of alloy catalysts. The co-adsorption of active sites changes the energy distribution of the reaction, increasing the NO adsorption capacity and facilitating N-O bond cleavage. The presence of the second metal also enhances the oxidation performance, further improving catalytic activity and selectivity. Single-atom-cluster/NP catalysts combine the advantages of both SAs and clusters/NPs, making them particularly effective in catalytic processes involving multiple reactants or complex reaction conditions. The synergistic effect between SAs and clusters/NPs alleviates the competitive adsorption of NO and CO on the catalyst surface, regulating the electronic structure and improving catalytic efficiency. Overall, the application of SACs in CO-SCR reactions demonstrates significant advantages, optimizing reaction pathways and enhancing overall catalytic performance.
As catalyst design and synthesis techniques progress, future research will focus on enhancing catalytic performance by tuning the spatial arrangement of metal atoms, coordination environment, and MSIs. Despite the outstanding catalytic performance of SACs under laboratory conditions, their industrial application still faces many challenges, particularly related to catalyst stability, production costs, and scalability. With the continued advancement of high-resolution microscopy and computational tools, future research will place more emphasis on the structural characterization and mechanistic elucidation of catalysts. The integration of theoretical calculations and experimental results will provide crucial guidance for optimizing catalyst design.
In conclusion, even though SACs show great potential in CO-SCR reactions, their practical industrial application still faces several challenges. Future research will focus on optimizing catalyst design and synthesis methods, improving catalyst stability and industrial scalability, and addressing key issues related to large-scale production. These efforts will drive the broader application of SACs in environmental protection and energy conversion.
DECLARATIONS
Authors’ contributions
Prepared the manuscript: Yang, L.
Designed and revised the manuscript: Li, J.; Liu, B.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the Fundamental Research Funds for the Central Universities (No. N2229002), the Basic and Applied Basic Research Foundation of Guangdong Province (No. 2023A1515010678), the Research and the Development Start-up Foundation of Foshan Graduate School of Innovation, Northeastern University (Nos. FSNEU20201016001 and FSNEU20201016003), and the Scientific Research Project of Foshan Talents (Nos. 200076622001 and 200076622004).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Richter, A.; Burrows, J. P.; Nüss, H.; Granier, C.; Niemeier, U. Increase in tropospheric nitrogen dioxide over China observed from space. Nature 2005, 437, 129-32.
2. Han, L.; Cai, S.; Gao, M.; et al. Selective catalytic reduction of NOx with NH3 by using novel catalysts: state of the art and future prospects. Chem. Rev. 2019, 119, 10916-76.
3. Wang, Y.; Xu, W.; Liu, H.; Chen, W.; Zhu, T. Catalytic removal of gaseous pollutant NO using CO: catalyst structure and reaction mechanism. Environ. Res. 2024, 246, 118037.
4. Zhang, R.; Liu, N.; Lei, Z.; Chen, B. Selective transformation of various nitrogen-containing exhaust gases toward N2 over zeolite catalysts. Chem. Rev. 2016, 116, 3658-721.
5. Beale, A. M.; Gao, F.; Lezcano-Gonzalez, I.; Peden, C. H.; Szanyi, J. Recent advances in automotive catalysis for NOx emission control by small-pore microporous materials. Chem. Soc. Rev. 2015, 44, 7371-405.
6. Li, G.; Wang, B.; Ma, Z.; et al. An anti-poisoning defective catalyst without metal active sites for NH 3 -SCR via in situ stabilization. EES. Catal. 2023, 1, 134-43.
7. Feng, S.; Li, Z.; Shen, B.; et al. An overview of the deactivation mechanism and modification methods of the SCR catalysts for denitration from marine engine exhaust. J. Environ. Manage. 2022, 317, 115457.
8. Skalska, K.; Miller, J. S.; Ledakowicz, S. Trends in NOx abatement: a review. Sci. Total. Environ. 2010, 408, 3976-89.
9. Lu, Y.; Zhang, Z.; Lin, F.; Wang, H.; Wang, Y. Single-atom automobile exhaust catalysts. ChemNanoMat 2020, 6, 1659-82.
10. Liu, Y.; Zhao, J.; Lee, J. Conventional and new materials for selective catalytic reduction (SCR) of NOx. ChemCatChem 2018, 10, 1499-511.
11. Javed, M. T.; Irfan, N.; Gibbs, B. M. Control of combustion-generated nitrogen oxides by selective non-catalytic reduction. J. Environ. Manage. 2007, 83, 251-89.
12. Heck, R. M. Catalytic abatement of nitrogen oxides-stationary applications. Catal. Today. 1999, 53, 519-23.
13. Xu, G.; Guo, X.; Cheng, X.; Yu, J.; Fang, B. A review of Mn-based catalysts for low-temperature NH3-SCR: NOx removal and H2O/SO2 resistance. Nanoscale 2021;13:7052-80.[DOI:10.1039/d1nr00248a] Caution!.
14. Xie, R.; Ma, L.; Li, Z.; Qu, Z.; Yan, N.; Li, J. Review of sulfur promotion effects on metal oxide catalysts for NOx emission control. ACS. Catal. 2021, 11, 13119-39.
15. Ye, B.; Jeong, B.; Lee, M. J.; et al. Recent trends in vanadium-based SCR catalysts for NOx reduction in industrial applications: stationary sources. Nano. Converg. 2022, 9, 51.
16. Chen, D.; Yan, Y.; Guo, A.; et al. Mechanistic insights into the promotion of low-temperature NH3-SCR catalysis by copper auto-reduction in Cu-zeolites. Appl. Catal. B. Environ. 2023, 322, 122118.
17. Zhang, N.; Wang, J.; Li, Q.; et al. Enhanced selective catalytic reduction of NO with NH3 over homoatomic dinuclear sites in defective α-Fe2O3. Chem. Eng. J. 2021, 426, 131845.
18. Liu, Q.; Yang, J.; Zhang, S.; et al. Flattened Pt clusters constructed on CeO2 for efficient selective oxidation of NH3. Appl. Catal. B. Environ. Energy. 2025, 365, 124877.
19. Zhang, N.; He, H.; Wang, D.; Li, Y. Challenges and opportunities for manganese oxides in low-temperature selective catalytic reduction of NOx with NH3: H2O resistance ability. J. Solid. State. Chem. 2020, 289, 121464.
20. Cheng, K.; Liu, J.; Zhao, Z.; Wei, Y.; Jiang, G.; Duan, A. Direct synthesis of V-W-Ti nanoparticle catalysts for selective catalytic reduction of NO with NH3. RSC. Adv. 2015, 5, 45172-83.
21. Zhang, Z.; Yang, L.; Luo, W.; An, V.; Li, J.; Liu, B. Mesh-supported V2O5-WO3/TiO2 nanosheet array catalysts for efficient removal of NOx. Tungsten 2025, 7, 100-11.
22. Hu, W.; He, J.; Liu, X.; et al. SO2- and H2O-tolerant catalytic reduction of NOx at a low temperature via engineering polymeric VOx species by CeO2. Environ. Sci. Technol. 2022, 56, 5170-8.
23. Liu, H.; Xiong, S.; Ou, H.; et al. The contradictory impact of sulfation on a CeOx/TiO2 NH3-SCR catalyst: a combined experimental and DFT study. Energy. Fuels. 2023, 37, 6674-82.
24. Bian, M.; Liu, K.; Zheng, D.; et al. Metal-free β zeolite used as an efficient NH3-SCR catalyst can achieve complete immunity to SO2: unique design strategy of sulfur-resistant catalyst. Chem. Eng. J. 2024, 481, 148563.
25. Luo, N.; Gao, F.; Liu, H.; et al. Hierarchical structured Ti-doped CeO2 stabilized CoMn2O4 for enhancing the low-temperature NH3-SCR performance within highly H2O and SO2 resistance. Appl. Catal. B. Environ. 2024, 343, 123442.
26. Chen, Y.; Liu, X.; Wang, P.; et al. Challenges and perspectives of environmental catalysis for NOx reduction. JACS. Au. 2024, 4, 2767-91.
27. Luo, W.; Yang, L.; Zhang, Z.; Cao, G.; Li, J.; Liu, B. Flexible Ti mesh-supported MnOx-CuOx/TiO2 nanosheet monolithic catalysts for low-temperature selective catalytic reduction of NOx with NH3. ACS. Appl. Nano. Mater. 2024, 7, 6262-72.
28. Chen, M.; Lian, D.; Wang, H.; et al. The catalytic mechanisms and design strategies of noble metal catalysts for selective reduction of NOx with CO. ChemCatChem 2024, 16, e202400323.
29. Chen, X.; Liu, Y.; Liu, Y.; et al. Recent advances of cu-based catalysts for NO reduction by CO under O2-containing conditions. Catalysts 2022, 12, 1402.
30. Lian, D.; Chen, M.; Wang, H.; et al. Recent advancements in Fe-based catalysts for the efficient reduction of NOx by CO. Chem. Asian. J. 2024, 19, e202400802.
31. Lian, D.; Chen, M.; Wang, H.; et al. Promising selective catalytic reduction of NOx by CO: status, challenges, and perspective. Chem. Eng. J. 2024, 496, 154242.
32. Liu, S.; Gao, J.; Xu, W.; et al. Transition metal-based catalysts for selective catalytic reduction of NO by CO: a state-of-the-art review. Chem. Eng. J. 2024, 486, 150285.
33. Wang, H.; Lian, D.; Chen, M.; et al. Enhancing the resistance of single-atom and cluster catalysts in CO-SCR to water, sulfur, and oxygen via structural engineering. Chem. Eng. J. 2024, 500, 157326.
34. Du, Y.; Gao, F.; Zhou, Y.; Yi, H.; Tang, X.; Qi, Z. Recent advance of CuO-CeO2 catalysts for catalytic elimination of CO and NO. J. Environ. Chem. Eng. 2021, 9, 106372.
35. Wang, J.; Gao, F.; Dang, P.; et al. Recent advances in NO reduction with CO over copper-based catalysts: reaction mechanisms, optimization strategies, and anti-inactivation measures. Chem. Eng. J. 2022, 450, 137374.
36. Yang, Y.; Yang, L.; Cao, G.; et al. Monolithic CuMnO2 -nanosheet-based catalysts in situ grown on stainless steel mesh for selective catalytic reduction of NO with CO. ACS. Appl. Nano. Mater. 2023, 6, 4803-11.
37. Dong, C.; Li, Y.; Cheng, D.; et al. Supported metal clusters: fabrication and application in heterogeneous catalysis. ACS. Catal. 2020, 10, 11011-45.
38. Li, X.; Yang, X.; Huang, Y.; Zhang, T.; Liu, B. Supported noble-metal single atoms for heterogeneous catalysis. Adv. Mater. 2019, 31, e1902031.
39. Yan, H.; Zhang, N.; Wang, D. Highly efficient CeO2-supported noble-metal catalysts: from single atoms to nanoclusters. Chem. Catal. 2022, 2, 1594-623.
40. Leybo, D.; Etim, U. J.; Monai, M.; Bare, S. R.; Zhong, Z.; Vogt, C. Metal-support interactions in metal oxide-supported atomic, cluster, and nanoparticle catalysis. Chem. Soc. Rev. 2024, 53, 10450-90.
41. Zeng, L.; Cheng, K.; Sun, F.; et al. Stable anchoring of single rhodium atoms by indium in zeolite alkane dehydrogenation catalysts. Science 2024, 383, 998-1004.
42. Qiao, B.; Wang, A.; Yang, X.; et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634-41.
43. Yang, X. F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740-8.
44. Liu, L.; Corma, A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem. Rev. 2018, 118, 4981-5079.
45. Hannagan, R. T.; Giannakakis, G.; Réocreux, R.; et al. First-principles design of a single-atom-alloy propane dehydrogenation catalyst. Science 2021, 372, 1444-7.
46. Tao, F.; Li, Y. A new type of catalysts: catalysts of singly dispersed bimetallic sites. Trends. Chem. 2023, 5, 486-99.
47. Li, Y.; Li, Y.; Sun, H.; et al. Current status and perspectives of dual-atom catalysts towards sustainable energy utilization. Nanomicro. Lett. 2024, 16, 139.
48. Liang, X.; Fu, N.; Yao, S.; Li, Z.; Li, Y. The progress and outlook of metal single-atom-site catalysis. J. Am. Chem. Soc. 2022, 144, 18155-74.
49. Liang, X.; Yao, S.; Li, Z.; Li, Y. Challenge and chance of single atom catalysis: the development and application of the single atom site catalysts toolbox. Acc. Chem. Res. 2025, 58, 1607-19.
50. Lang, R.; Xi, W.; Liu, J. C.; et al. Non defect-stabilized thermally stable single-atom catalyst. Nat. Commun. 2019, 10, 234.
51. Fonseca, J.; Lu, J. Single-atom catalysts designed and prepared by the atomic layer deposition technique. ACS. Catal. 2021, 11, 7018-59.
52. Chang, J.; Jing, W.; Yong, X.; et al. Synthesis of ultrahigh-metal-density single-atom catalysts via metal sulfide-mediated atomic trapping. Nat. Synth. 2024, 3, 1427-38.
53. Ji, S.; Chen, Y.; Wang, X.; Zhang, Z.; Wang, D.; Li, Y. Chemical synthesis of single atomic site catalysts. Chem. Rev. 2020, 120, 11900-55.
54. Lv, H.; Guo, W.; Chen, M.; Zhou, H.; Wu, Y. Rational construction of thermally stable single atom catalysts: From atomic structure to practical applications. Chin. J. Catal. 2022, 43, 71-91.
55. Eck NJ, Waltman L. Software survey: VOSviewer, a computer program for bibliometric mapping. Scientometrics 2010, 84, 523-38.
56. Web of science all databases. Available from: https://www.webofscience.com (accessed 2025-8-7).
57. Zhang, N.; He, C.; Jing, Y.; et al. Enhanced nitrous oxide decomposition on zirconium-supported rhodium catalysts by iridium augmentation. Environ. Sci. Technol. 2025, 59, 1598-607.
58. Yang, L.; Li, X.; He, H.; et al. Enhanced low-temperature activity of Rh-Pt alloy clusters supported on TiO2/Ti nanosheets for selective catalytic reduction of NO by CO. Appl. Surf. Sci. 2025, 695, 162824.
59. Yang, L.; Li, X.; Li, S.; Li, J.; Liu, B. Cooperative Rh-Ov-Co sites boosting the wide reaction temperature window for NO reduction with CO. J. Environ. Chem. Eng. 2025, 13, 115994.
60. Cheng, X.; Zhang, X.; Su, D.; Wang, Z.; Chang, J.; Ma, C. NO reduction by CO over copper catalyst supported on mixed CeO2 and Fe2O3: catalyst design and activity test. Appl. Catal. B. Environ. 2018, 239, 485-501.
61. He, Y.; Liu, J.; Zhang, G.; et al. Interfacial effects promote the catalytic performance of CuCoO2-CeO2 metal oxides for the selective reduction of NO by CO. Chem. Eng. J. 2023, 465, 142856.
62. Zhang, L.; Yao, X.; Lu, Y.; et al. Effect of precursors on the structure and activity of CuO-CoOx/γ-Al2O3 catalysts for NO reduction by CO. J. Colloid. Interface. Sci. 2018, 509, 334-45.
63. Xu, Z.; Li, Y.; Lin, Y.; Zhu, T. A review of the catalysts used in the reduction of NO by CO for gas purification. Environ. Sci. Pollut. Res. Int. 2020, 27, 6723-48.
64. Gholami, Z.; Luo, G.; Gholami, F.; Yang, F. Recent advances in selective catalytic reduction of NOx by carbon monoxide for flue gas cleaning process: a review. Catal. Rev. 2021, 63, 68-119.
65. Chen, K.; Han, X.; Wang, Q.; Liu, K.; Yang, X.; Zhang, Y. Enhanced CO-SCR denitration on supported Rh-Mn/CoAlOx catalysts through Rh-Mn interaction. Appl. Surf. Sci. 2024, 665, 160357.
66. Fernández, E.; Liu, L.; Boronat, M.; Arenal, R.; Concepcion, P.; Corma, A. Low-temperature catalytic NO reduction with CO by subnanometric Pt clusters. ACS. Catal. 2019, 9, 11530-41.
67. Pan, Y.; Li, N.; Li, K.; et al. Enhanced low-temperature behavior of selective catalytic reduction of NOx by CO on Fe-based catalyst with looping oxygen vacancy. Chem. Eng. J. 2023, 461, 141814.
68. Zang, P.; Liu, J.; Zhang, G.; et al. Insights into the highly activity CuMgFe oxides for the selective catalytic reduction of NO by CO: structure-activity relationships and K/SO2 poisoning mechanism. Fuel 2023, 331, 125800.
69. Li, R.; Li, A.; Li, J.; Wang, Y.; Liu, Z. Unveiling the significant promoting effect of SO2 on Ir/SiO2 catalyst for the CO-SCR of NOx in the presence of O2. J. Catal. 2024, 430, 115336.
70. Bai, Y.; Miao, C.; Deng, S.; Wang, H.; Wu, Z. Unveiling the effect of SO2 on CO selective catalytic reduction of NO in the presence of O2 over IrRb@SBA-15 catalyst. Sep. Purif. Technol. 2025, 352, 128039.
71. Zhang, N.; Ye, C.; Yan, H.; et al. Single-atom site catalysts for environmental catalysis. Nano. Res. 2020, 13, 3165-82.
72. Liu, H.; Wang, Y.; Xu, W.; et al. Unraveling the synergistic mechanism of Ir species with various electron densities over an Ir/ZSM-5 catalyst enables high-efficiency NO reduction by CO. Environ. Sci. Technol. 2024, 58, 12082-90.
73. Sun, Y.; Wu, Y.; Bai, Y.; Wu, X.; Wang, H.; Wu, Z. High performance iridium loaded on natural halloysite nanotubes for CO-SCR reaction. Fuel 2024, 357, 129938.
74. Jiang, R.; Liu, S.; Li, L.; et al. Single Ir atoms anchored on ordered mesoporous WO3 are highly efficient for the selective catalytic reduction of NO with CO under oxygen-rich conditions. ChemCatChem 2021, 13, 1834-46.
75. Inomata, H.; Shimokawabe, M.; Kuwana, A.; Arai, M. Selective reduction of NO with CO in the presence of O2 with Ir/WO3 catalysts: influence of preparation variables on the catalytic performance. Appl. Catal. B. Environ. 2008, 84, 783-9.
76. Nanba, T.; Shinohara, S.; Masukawa, S.; Uchisawa, J.; Ohi, A.; Obuchi, A. Formation of active sites on Ir/WO3-SiO2 for selective catalytic reduction of NO by CO. Appl. Catal. B. Environ. 2008, 84, 420-5.
77. Wang, J.; Gao, F.; Yi, H.; et al. Strong Ir-W interaction boosts CO-SCR denitration over supported Ir-based catalysts and influential mechanism of oxygen. Sep. Purif. Technol. 2023, 325, 124684.
78. Jiang, D.; Yao, Y.; Li, T.; et al. Tailoring the local environment of platinum in single-atom Pt1/CeO2 catalysts for robust low-temperature CO oxidation. Angew. Chem. Int. Ed. Engl. 2021, 60, 26054-62.
79. Ji, Y.; Chen, X.; Liu, S.; et al. Tailoring the electronic structure of single Ag atoms in Ag/WO3 for efficient NO reduction by CO in the presence of O2. ACS. Catal. 2023, 13, 1230-9.
80. Roy, S.; Hegde, M. Pd ion substituted CeO2: a superior de-NO catalyst to Pt or Rh metal ion doped ceria. Catal. Commun. 2008, 9, 811-5.
81. Roy, S.; Marimuthu, A.; Hegde, M.; Madras, G. High rates of CO and hydrocarbon oxidation and NO reduction by CO over
82. Roy, S.; Marimuthu, A.; Hegde, M.; Madras, G. High rates of NO and N2O reduction by CO, CO and hydrocarbon oxidation by O2 over nano crystalline Ce0.98Pd0.02O2-δ: catalytic and kinetic studies. Appl. Catal. B. Environ. 2007, 71, 23-31.
83. Xu, Q.; Cheng, X.; Zhang, N.; et al. Unraveling the advantages of Pd/CeO2 single-atom catalysts in the NO + CO reaction by model catalysts. Nano. Res. 2023, 16, 8882-92.
84. Ikemoto, S.; Muratsugu, S.; Koitaya, T.; Tada, M. Chromium oxides as structural modulators of Rhodium dispersion on ceria to generate active sites for NO reduction. ACS. Catal. 2022, 12, 431-41.
85. Cai, D.; Zhang, J.; Kong, Z.; Li, Z. Synergistic effect of single-atom catalysts and vacancies of support for versatile catalytic applications. ChemCatChem 2024, 16, e202301414.
86. Luo, L.; Fu, L.; Liu, H.; et al. Synergy of Pd atoms and oxygen vacancies on In2O3 for methane conversion under visible light. Nat. Commun. 2022, 13, 2930.
87. Bai, Y.; Zong, X.; Jin, C.; Wang, S.; Wang, S. Synergy of single-atom Fe1 and Ce-Ov sites on mesoporous CeO2-Al2O3 for efficient selective catalytic reduction of NO with CO. ACS. Catal. 2024, 14, 827-36.
88. Song, I.; Wang, Y.; Szanyi, J.; Khivantsev, K. Co-existence of atomically dispersed Ru and Ce3+ sites is responsible for excellent low temperature N2O reduction activity of Ru/CeO2. Appl. Cataly. B. Environ. 2024, 343, 123487.
89. Song, I.; Kovarik, L.; Engelhard, M. H.; Szanyi, J.; Wang, Y.; Khivantsev, K. Developing robust ceria-supported catalysts for catalytic NO reduction and CO/hydrocarbon oxidation. ACS. Catal. 2024, 14, 18247-55.
90. Wu, J.; Li, Y.; Yang, Y.; et al. A heterogeneous single Cu catalyst of Cu atoms confined in the spinel lattice of MgAl2O4 with good catalytic activity and stability for NO reduction by CO. J. Mater. Chem. A. 2019, 7, 7202-12.
91. Asokan, C.; Yang, Y.; Dang, A.; Getsoian, A.; Christopher, P. Low-temperature ammonia production during NO reduction by CO Is due to atomically dispersed rhodium active sites. ACS. Catal. 2020, 10, 5217-22.
92. Yang, C.; Garl, C. W. Infrared studies of carbon monoxide chemisorbed on Rhodium. J. Phys. Chem. 1957, 61, 1504-12.
93. Matsubu, J. C.; Yang, V. N.; Christopher, P. Isolated metal active site concentration and stability control catalytic CO2 reduction selectivity. J. Am. Chem. Soc. 2015, 137, 3076-84.
94. Wu, D.; Liu, S.; Zhong, M.; et al. Nature and dynamic evolution of Rh single atoms trapped by CeO2 in CO hydrogenation. ACS. Catal. 2022, 12, 12253-67.
95. Khivantsev, K.; Vargas, C. G.; Tian, J.; et al. Economizing on precious metals in three-way catalysts: thermally stable and highly active single-atom Rhodium on ceria for NO abatement under dry and industrially relevant conditions*. Angew. Chem. Int. Ed. Engl. 2021, 60, 391-8.
96. Khivantsev, K.; Jaegers, N. R.; Aleksandrov, H. A.; et al. Single Ru(II) ions on ceria as a highly active catalyst for abatement of NO. J. Am. Chem. Soc. 2023, 145, 5029-40.
97. Tian, J.; Khivantsev, K.; Lu, Y.; et al. NO reduction with CO on low-loaded platinum-group metals (Rh, Ru, Pd, Pt, and Ir) atomically dispersed on ceria. ChemCatChem 2024, 16, e202301227.
98. Wang, T.; Hu, J.; Ouyang, R.; et al. Nature of metal-support interaction for metal catalysts on oxide supports. Science 2024, 386, 915-20.
99. S.; Fung, S.; Garten, R. L. Strong metal-support interactions. Group 8 noble metals supported on titanium dioxide. J. Am. Chem. Soc. 1978, 100, 170-5.
100. Tauster, S. Strong metal-support interactions: occurrence among the binary oxides of groups IIA? J. Catal. 1978, 55, 29-35.
101. S. J.; Fung, S. C.; Baker, R. T. K.; Horsley, J. A. Strong interactions in supported-metal catalysts. Science 1981, 211, 1121-5.
102. Tang, Y.; Zhao, S.; Long, B.; Liu, J.; Li, J. On the Nature of support effects of metal dioxides MO2 (M = Ti, Zr, Hf, Ce, Th) in single-atom gold catalysts: importance of quantum primogenic effect. J. Phys. Chem. C. 2016, 120, 17514-26.
103. Adachi,
104. Wan, Q.; Fung, V.; Lin, S.; Wu, Z.; Jiang, D. Perovskite-supported Pt single atoms for methane activation. J. Mater. Chem. A. 2020, 8, 4362-8.
105. Ji, Y.; Liu, S.; Song, S.; et al. Negatively charged single-atom Pt catalyst shows superior SO2 tolerance in NOx reduction by CO. ACS. Catal. 2023, 13, 224-36.
106. Gloag, L.; Somerville, S. V.; Gooding, J. J.; Tilley, R. D. Co-catalytic metal-support interactions in single-atom electrocatalysts. Nat. Rev. Mater. 2024, 9, 173-89.
107. Ji, Y.; Liu, S.; Zhu, H.; et al. Isolating contiguous Ir atoms and forming Ir-W intermetallics with negatively charged Ir for efficient NO reduction by CO. Adv. Mater. 2022, 34, e2205703.
108. Jia, C.; Wang, Q.; Yang, J.; et al. Toward rational design of dual-metal-site catalysts: catalytic descriptor exploration. ACS. Catal. 2022, 12, 3420-9.
109. Hu, Y.; Li, Z.; Li, B.; Yu, C. Recent Progress of diatomic catalysts: general design fundamentals and diversified catalytic applications. Small 2022, 18, e2203589.
110. Liu, W.; Long, G.; Xiang, Z.; et al. Extremely active and robust Ir-Mn dual-atom electrocatalyst for oxygen evolution reaction by oxygen-oxygen radical coupling mechanism. Angew. Chem. Int. Ed. Engl. 2024, 63, e202411014.
111. Wang, B.; Yang, X.; Xie, C.; et al. A General metal ion recognition strategy to mediate dual-atomic-site catalysts. J. Am. Chem. Soc. 2024, 146, 24945-55.
112. Zhang, Y. X.; Zhang, S.; Huang, H.; et al. General synthesis of a diatomic catalyst library via a macrocyclic precursor-mediated approach. J. Am. Chem. Soc. 2023, 145, 4819-27.
113. Sun, X.; Qiu, Y.; Jiang, B.; et al. Isolated Fe-Co heteronuclear diatomic sites as efficient bifunctional catalysts for high-performance lithium-sulfur batteries. Nat. Commun. 2023, 14, 291.
114. Hao, Q.; Zhong, H.; Wang, J.; et al. Nickel dual-atom sites for electrochemical carbon dioxide reduction. Nat. Synth. 2022, 1, 719-28.
115. Jiang, S.; Xue, J.; Liu, T.; et al. Visualization of the distance-dependent synergistic interaction in heterogeneous dual-site catalysis. J. Am. Chem. Soc. 2024, 146, 29084-93.
116. Li, L.; Yuan, K.; Chen, Y. Breaking the scaling relationship limit: from single-atom to dual-atom catalysts. Acc. Mater. Res. 2022, 3, 584-96.
117. Chen, Y.; Lin, J.; Pan, Q.; Liu, X.; Ma, T.; Wang, X. Inter-metal interaction of dual-atom catalysts in heterogeneous catalysis. Angew. Chem. Int. Ed. Engl. 2023, 62, e202306469.
118. Pan, Y.; Zhang, C.; Liu, Z.; Chen, C.; Li, Y. Structural regulation with atomic-level precision: from single-atomic site to diatomic and atomic interface catalysis. Matter 2020, 2, 78-110.
119. Wang, H.; Bootharaju, M. S.; Kim, J. H.; et al. Synergistic interactions of neighboring platinum and iron atoms enhance reverse water-gas shift reaction performance. J. Am. Chem. Soc. 2023, 145, 2264-70.
120. Fu, J.; Dong, J.; Si, R.; et al. Synergistic effects for enhanced catalysis in a dual single-atom catalyst. ACS. Catal. 2021, 11, 1952-61.
121. Huang, F.; Peng, M.; Chen, Y.; et al. Low-temperature acetylene semi-hydrogenation over the Pd1-Cu1 dual-atom catalyst. J. Am. Chem. Soc. 2022, 144, 18485-93.
122. Tan, Z.; Haneda, M.; Kitagawa, H.; Huang, B. Slow synthesis methodology-directed immiscible octahedral Pdx Rh1-x dual-atom-site catalysts for superior three-way catalytic activities over Rh. Angew. Chem. Int. Ed. Engl. 2022, 61, e202202588.
123. Zhao, X.; Wang, F.; Kong, X. P.; Fang, R.; Li, Y. Dual-metal hetero-single-atoms with different coordination for efficient synergistic catalysis. J. Am. Chem. Soc. 2021, 143, 16068-77.
124. Liu, Z.; Wen, Y.; Wang, Z.; et al. Synergistic dual-atom catalysts on ceria for enhanced CO preferential oxidation: insights from high-throughput first-principles microkinetics. ACS. Catal. 2025, 15, 664-75.
125. Tang, X.; Zhang, Y.; Tang, S.; Lützenkirchen-hecht, D.; Yuan, K.; Chen, Y. Structural and chemical origin of dual-atom sites for enhanced oxygen electroreduction. ACS. Catal. 2024, 14, 13065-80.
126. Han, A.; Wang, X.; Tang, K.; et al. An adjacent atomic platinum site enables single-atom iron with high oxygen reduction reaction performance. Angew. Chem. Int. Ed. Engl. 2021, 60, 19262-71.
127. Gao, Y.; Liu, B.; Wang, D. Microenvironment engineering of single/dual-atom catalysts for electrocatalytic application. Adv. Mater. 2023, 35, e2209654.
128. Kim, J.; Choi, S.; Cho, J.; Kim, S. Y.; Jang, H. W. Toward multicomponent single-atom catalysis for efficient electrochemical energy conversion. ACS. Mater. Au. 2022, 2, 1-20.
129. Ding, Y.; Shi, Y.; Xiong, W.; et al. Insights into N-coordinated bimetallic site synergy during no selective catalytic reduction by CO. ACS. Appl. Mater. Interfaces. 2021, 13, 57182-92.
130. Zhou, X.; Han, K.; Li, K.; et al. Dual-site single-atom catalysts with high performance for three-way catalysis. Adv. Mater. 2022, 34, e2201859.
131. Vogt, C.; Weckhuysen, B. M. The concept of active site in heterogeneous catalysis. Nat. Rev. Chem. 2022, 6, 89-111.
132. Hannagan, R. T.; Giannakakis, G.; Flytzani-Stephanopoulos, M.; Sykes, E. C. H. Single-atom alloy catalysis. Chem. Rev. 2020, 120, 12044-88.
133. Liu, L.; Corma, A. Bimetallic sites for catalysis: from binuclear metal sites to bimetallic nanoclusters and nanoparticles. Chem. Rev. 2023, 123, 4855-933.
134. Jiang, T.; Li, Y.; Tang, Y.; et al. Breaking continuously packed bimetallic sites to singly dispersed on nonmetallic support for efficient hydrogen production. ACS. Appl. Mater. Interfaces. 2024, 16, 21757-70.
135. Zhang, S.; Nguyen, L.; Liang, J. X.; et al. Catalysis on singly dispersed bimetallic sites. Nat. Commun. 2015, 6, 7938.
136. Liu, N.; Ma, X.; Li, J.; Xiao, H. Singly Dispersed bimetallic sites as stable and efficient single-cluster catalysts for activating N2 and CO2. J. Phys. Chem. C. 2021, 125, 27192-8.
137. Nguyen, L.; Zhang, S.; Wang, L.; et al. Reduction of nitric oxide with hydrogen on catalysts of singly dispersed bimetallic sites
138. Ma, X.; Yang, Y.; Xu, L.; Xiao, H.; Yao, W.; Li, J. Theoretical investigation on hydrogenation of dinitrogen triggered by singly dispersed bimetallic sites. J. Mater. Chem. A. 2022, 10, 6146-52.
139. Zhang, S.; Shan, J. J.; Zhu, Y.; et al. WGS catalysis and in situ studies of CoO1-x, PtCon/Co3O4, and PtmCom’/CoO1-x nanorod catalysts. J. Am. Chem. Soc. 2013, 135, 8283-93.
140. Nguyen, L.; Zhang, S.; Tan, L.; Tang, Y.; Liu, J.; Tao, F. F. Ir1Znn bimetallic site for efficient production of hydrogen from methanol. ACS. Sustainable. Chem. Eng. 2019, 7, 18793-800.
141. Yang, H.; Yu, R.; Fang, Y.; et al. Singly dispersed Ir1Ti3 bimetallic site for partial oxidation of methane at high temperature. Appl. Surf. Sci. 2022, 599, 153863.
142. Jin, X.; Wen, H.; Liu, J. Insight into the reaction mechanism of the reduction of NO by H2 on the singly dispersed bimetallic Pt(Rh)Co4/Co3O4 catalysts: a first-principles study. J. Phys. Chem. C. 2020, 124, 9908-16.
143. Wang, P.; Yang, M.; Liao, H.; et al. Restructured zeolites anchoring singly dispersed bimetallic platinum and zinc catalysts for propane dehydrogenation. Cell. Reports. Physical. Science. 2023, 4, 101311.
144. Ma, X. L.; Liu, J. C.; Xiao, H.; Li, J. Surface single-cluster catalyst for N2-to-NH3 thermal conversion. J. Am. Chem. Soc. 2018, 140, 46-9.
145. Zhang, T.; Walsh, A. G.; Yu, J.; Zhang, P. Single-atom alloy catalysts: structural analysis, electronic properties and catalytic activities. Chem. Soc. Rev. 2021, 50, 569-88.
146. Jin, Z.; Xu, Y.; Chhetri, M.; et al. Recent developments of single atom alloy catalysts for electrocatalytic hydrogenation reactions. Chem. Eng. J. 2024, 491, 152072.
147. Papanikolaou, K. G.; Darby, M. T.; Stamatakis, M. Engineering the surface architecture of highly dilute alloys: an ab initio monte Carlo approach. ACS. Catal. 2020, 10, 1224-36.
148. Papanikolaou, K. G.; Darby, M. T.; Stamatakis, M. CO-induced aggregation and segregation of highly dilute alloys: a density functional theory study. J. Phys. Chem. C. 2019, 123, 9128-38.
149. Ouyang, M.; Papanikolaou, K. G.; Boubnov, A.; et al. Directing reaction pathways via in situ control of active site geometries in PdAu single-atom alloy catalysts. Nat. Commun. 2021, 12, 1549.
150. Pei, G. X.; Liu, X. Y.; Yang, X.; et al. Performance of Cu-alloyed Pd single-atom catalyst for semihydrogenation of acetylene under simulated front-end conditions. ACS. Catal. 2017, 7, 1491-500.
151. Zhang, S.; Wang, R.; Zhang, X.; Zhao, H. Recent advances in single-atom alloys: preparation methods and applications in heterogeneous catalysis. RSC. Adv. 2024, 14, 3936-51.
152. Schumann, J.; Stamatakis, M.; Michaelides, A.; Réocreux, R. Ten-electron count rule for the binding of adsorbates on single-atom alloy catalysts. Nat. Chem. 2024, 16, 749-54.
153. Liu, J.; Lucci, F. R.; Yang, M.; et al. Tackling CO poisoning with single-atom alloy catalysts. J. Am. Chem. Soc. 2016, 138, 6396-9.
154. Mao, J.; Yin, J.; Pei, J.; Wang, D.; Li, Y. Single atom alloy: an emerging atomic site material for catalytic applications. Nano. Today. 2020, 34, 100917.
155. Bunting, R. J.; Wodaczek, F.; Torabi, T.; Cheng, B. Reactivity of single-atom alloy nanoparticles: modeling the dehydrogenation of propane. J. Am. Chem. Soc. 2023, 145, 14894-902.
156. Berger, F.; Schumann, J.; Réocreux, R.; Stamatakis, M.; Michaelides, A. Bringing molecules together: synergistic coadsorption at dopant sites of single atom alloys. J. Am. Chem. Soc. 2024, 146, 28119-30.
157. Bai, Y.; Gao, S.; Sun, Y.; et al. Insight into the mechanism of selective catalytic reduction of NO by CO over a bimetallic IrRu/ZSM-5 catalyst in the absence/presence of O2 by Isotopic C13O tracing methods. Environ. Sci. Technol. 2023, 57, 9105-14.
158. Song, J. H.; Park, D. C.; You, Y.; et al. Kinetic and DRIFTS studies of IrRu/Al2O3 catalysts for lean NOx reduction by CO at low temperature. Catal. Sci. Technol. 2020, 10, 8182-95.
159. Arshad, M. W.; Kim, D. H.; You, Y.; Kim, S. M.; Heo, I.; Kim, S. K. A first-principles understanding of the CO-assisted NO reduction on the IrRu/Al2O3 catalyst under O2-rich conditions. Catal. Sci. Technol. 2021, 11, 4353-66.
160. Song, J. H.; Park, D. C.; You, Y.; Chang, T. S.; Heo, I.; Kim, D. H. Lean NOx reduction by CO at low temperature over bimetallic IrRu/Al2O3 catalysts with different Ir : Ru ratios. Catal. Sci. Technol. 2020, 10, 2120-36.
161. Papanikolaou, K. G.; Stamatakis, M. On the behaviour of structure-sensitive reactions on single atom and dilute alloy surfaces. Catal. Sci. Technol. 2020, 10, 5815-28.
162. Beniya, A.; Ikuta, Y.; Isomura, N.; Hirata, H.; Watanabe, Y. Synergistic promotion of NO-CO reaction cycle by gold and nickel elucidated using a well-defined model bimetallic catalyst surface. ACS. Catal. 2017, 7, 1369-77.
163. Wen, H.; Huai, L.; Jin, X.; Liu, J. Mechanism of nitric oxide reduction by hydrogen on Ni(110) and Ir/Ni(110): first principles and microkinetic modeling. J. Phys. Chem. C. 2019, 123, 4825-36.
164. Giannakakis, G.; Flytzani-Stephanopoulos, M.; Sykes, E. C. H. Single-atom alloys as a reductionist approach to the rational design of heterogeneous catalysts. Acc. Chem. Res. 2019, 52, 237-47.
165. Xing, F.; Jeon, J.; Toyao, T.; Shimizu, K. I.; Furukawa, S. A Cu-Pd single-atom alloy catalyst for highly efficient NO reduction. Chem. Sci. 2019, 10, 8292-8.
166. Darby, M. T.; Stamatakis, M.; Michaelides, A.; Sykes, E. C. H. Lonely atoms with special gifts: breaking linear scaling relationships in heterogeneous catalysis with single-atom alloys. J. Phys. Chem. Lett. 2018, 9, 5636-46.
167. Jeon, J.; Kon, K. I.; Toyao, T.; Shimizu, K. I.; Furukawa, S. Design of Pd-based pseudo-binary alloy catalysts for highly active and selective NO reduction. Chem. Sci. 2019, 10, 4148-62.
168. Jeon, J.; Ham, H.; Xing, F.; Nakaya, Y.; Shimizu, K.; Furukawa, S. PdIn-based pseudo-binary alloy as a catalyst for NOx removal under lean conditions. ACS. Catal. 2020, 10, 11380-4.
169. Zhang, J.; Wang, M.; Gao, Z.; et al. Importance of species heterogeneity in supported metal catalysts. J. Am. Chem. Soc. 2022, 144, 5108-15.
170. Luo, G.; Song, M.; Zhang, Q.; et al. Advances of synergistic electrocatalysis between single atoms and nanoparticles/clusters. Nanomicro. Lett. 2024, 16, 241.
171. Su, D.; Wang, Y.; Sheng, H.; et al. Efficient amine-assisted CO2 hydrogenation to methanol co-catalyzed by metallic and oxidized sites within ruthenium clusters. Nat. Commun. 2025, 16, 590.
172. Mo, F.; Song, C.; Zhou, Q.; et al. The optimized Fenton-like activity of Fe single-atom sites by Fe atomic clusters-mediated electronic configuration modulation. Proc. Natl. Acad. Sci. 2023, 120, e2300281120.
173. Lan, G.; Ye, Q.; Zhu, Y.; Tang, H.; Han, W.; Li, Y. Single-site Au/carbon catalysts with single-atom and Au nanoparticles for acetylene hydrochlorination. ACS. Appl. Nano. Mater. 2020, 3, 3004-10.
174. Jiang, R.; Li, Q.; Zheng, X.; et al. Metal-organic framework-derived Co nanoparticles and single atoms as efficient electrocatalyst for pH universal hydrogen evolution reaction. Nano. Res. 2022, 15, 7917-24.
175. Gao, M.; Tian, F.; Guo, Z.; et al. Mutual-modification effect in adjacent Pt nanoparticles and single atoms with sub-nanometer inter-site distances to boost photocatalytic hydrogen evolution. Chem. Eng. J. 2022, 446, 137127.
176. Zeng, Y.; Liang, J.; Li, C.; et al. Regulating catalytic properties and thermal stability of Pt and PtCo intermetallic fuel-cell catalysts via strong coupling effects between single-metal site-rich carbon and Pt. J. Am. Chem. Soc. 2023, 145, 17643-55.
177. Zhang, B.; Wang, J.; Liu, G.; et al. A strongly coupled Ru-CrOx cluste-cluster heterostructure for efficient alkaline hydrogen electrocatalysis. Nat. Catal. 2024, 7, 441-51.
178. Liu, S.; Ji, Y.; Liu, B.; et al. Co single atoms and CoOx nanoclusters anchored on Ce0.75Zr0.25O2 synergistically boosts the no reduction by CO. Adv. Funct. Mater. 2023, 33, 2303297.
179. Kuai, L.; Chen, Z.; Liu, S.; et al. Titania supported synergistic palladium single atoms and nanoparticles for room temperature ketone and aldehydes hydrogenation. Nat. Commun. 2020, 11, 48.
180. Yang, L.; Wang, J.; Liu, T.; et al. Synergistic catalysis of Rh single-atom and clusters supported on TiO2 nanosheet array for highly efficient removal of CO and NOx. Small. Struct. 2024, 5, 2400230.
181. Ren, W.; Tan, X.; Jia, C.; et al. Electronic regulation of nickel single atoms by confined nickel nanoparticles for energy-efficient CO2 electroreduction. Angew. Chem. Int. Ed. Engl. 2022, 61, e202203335.
182. He, Q.; Zhou, Y.; Shou, H.; et al. Synergic reaction kinetics over adjacent ruthenium sites for superb hydrogen generation in alkaline media. Adv. Mater. 2022, 34, e2110604.
183. Liu, A.; Liu, L.; Cao, Y.; et al. Controlling dynamic structural transformation of atomically dispersed CuOx species and influence on their catalytic performances. ACS. Catal. 2019, 9, 9840-51.
184. Mitchell, S.; Pérez-ramírez, J. Atomically precise control in the design of low-nuclearity supported metal catalysts. Nat. Rev. Mater. 2021, 6, 969-85.
185. Hsu, C. S.; Wang, J.; Chu, Y. C.; et al. Activating dynamic atomic-configuration for single-site electrocatalyst in electrochemical CO2 reduction. Nat. Commun. 2023, 14, 5245.
186. Ye, C.; Shan, J.; Zhu, C.; et al. Spatial structure engineering of interactive single platinum sites toward enhanced electrocatalytic hydrogen evolution. Adv. Energy. Mater. 2023, 13, 2302190.
187. Li, L.; Zhang, N. Atomic dispersion of bulk/nano metals to atomic-sites catalysts and their application in thermal catalysis. Nano. Res. 2023, 16, 6380-401.
188. Li, R.; Zhao, J.; Liu, B.; Wang, D. Atomic distance engineering in metal catalysts to regulate catalytic performance. Adv. Mater. 2024, 36, e2308653.
189. Feng, C.; Zhang, Z.; Wang, D.; et al. Tuning the electronic and steric interaction at the atomic interface for enhanced oxygen evolution. J. Am. Chem. Soc. 2022, 144, 9271-9.
190. Zhang, Z.; Jia, C.; Ma, P.; et al. Distance effect of single atoms on stability of cobalt oxide catalysts for acidic oxygen evolution. Nat. Commun. 2024, 15, 1767.
191. Yan, Y.; Yu, R.; Liu, M.; et al. General synthesis of neighboring dual-atomic sites with a specific pre-designed distance via an interfacial-fixing strategy. Nat. Commun. 2025, 16, 334.
192. Jiang, D.; Wan, G.; Halldin, Stenlid. J.; et al. Dynamic and reversible transformations of subnanometre-sized palladium on ceria for efficient methane removal. Nat. Catal. 2023, 6, 618-27.
193. Jiang, D.; Wan, G.; García-vargas, C. E.; et al. Elucidation of the active sites in single-atom Pd1/CeO2 catalysts for low-temperature CO oxidation. ACS. Catal. 2020, 10, 11356-64.
194. Tan, W.; Xie, S.; Le, D.; et al. Fine-tuned local coordination environment of Pt single atoms on ceria controls catalytic reactivity. Nat. Commun. 2022, 13, 7070.
195. Zhang, L.; Yang, X.; Lin, J.; et al. On the coordination environment of single-atom catalysts. Acc. Chem. Res. 2025, 58, 1878-92.
196. DeRita, L.; Dai, S.; Lopez-Zepeda, K.; et al. Catalyst architecture for stable single atom dispersion enables site-specific spectroscopic and reactivity measurements of CO adsorbed to Pt atoms, oxidized Pt clusters, and metallic Pt clusters on TiO2. J. Am. Chem. Soc. 2017, 139, 14150-65.
197. Lin, L.; Chen, Z.; Chen, W. Single atom catalysts by atomic diffusion strategy. Nano. Res. 2021, 14, 4398-416.
198. Han, L.; Cheng, H.; Liu, W.; et al. A single-atom library for guided monometallic and concentration-complex multimetallic designs. Nat. Mater. 2022, 21, 681-8.
199. Zhou, H.; Zhao, Y.; Gan, J.; et al. Cation-exchange induced precise regulation of single copper site triggers room-temperature oxidation of benzene. J. Am. Chem. Soc. 2020, 142, 12643-50.
200. Kaiser, S. K.; Chen, Z.; Faust, Akl. D.; Mitchell, S.; Pérez-Ramírez, J. Single-atom catalysts across the periodic table. Chem. Rev. 2020, 120, 11703-809.
201. Xing, L.; Jin, Y.; Weng, Y.; et al. Top-down synthetic strategies toward single atoms on the rise. Matter 2022, 5, 788-807.
202. Jones, J.; Xiong, H.; DeLaRiva, A. T.; et al. Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science 2016, 353, 150-4.
203. Hu, Y.; Li, B.; Yu, C.; Fang, H.; Li, Z. Mechanochemical preparation of single atom catalysts for versatile catalytic applications: a perspective review. Materials. Today. 2023, 63, 288-312.
204. Gan, T.; Liu, Y.; He, Q.; Zhang, H.; He, X.; Ji, H. Facile synthesis of kilogram-scale Co-alloyed Pt single-atom catalysts via ball milling for hydrodeoxygenation of 5-hydroxymethylfurfural. ACS. Sustainable. Chem. Eng. 2020, 8, 8692-9.
205. Gan, T.; He, Q.; Zhang, H.; et al. Unveiling the kilogram-scale gold single-atom catalysts via ball milling for preferential oxidation of CO in excess hydrogen. Chem. Eng. J. 2020, 389, 124490.
206. Zhang, H.; Zhang, X.; Shi, S.; et al. Highly efficient fabrication of kilogram-scale palladium single-atom catalysts for the Suzuki-Miyaura cross-coupling reaction. ACS. Appl. Mater. Interfaces. 2022, 14, 53755-60.
207. He, X.; Deng, Y.; Zhang, Y.; et al. Mechanochemical kilogram-scale synthesis of noble metal single-atom catalysts. Cell. Rep. Phys. Sci. 2020, 1, 100004.
208. Qu, Y.; Li, Z.; Chen, W.; et al. Direct transformation of bulk copper into copper single sites via emitting and trapping of atoms. Nat. Catal. 2018, 1, 781-6.
209. Tang, B.; Zhou, Y.; Ji, Q.; et al. A Janus dual-atom catalyst for electrocatalytic oxygen reduction and evolution. Nat. Synth. 2024, 3, 878-90.
210. Hai, X.; Xi, S.; Mitchell, S.; et al. Scalable two-step annealing method for preparing ultra-high-density single-atom catalyst libraries. Nat. Nanotechnol. 2022, 17, 174-81.
211. Wang, Y.; Li, C.; Han, X.; et al. General negative pressure annealing approach for creating ultra-high-loading single atom catalyst libraries. Nat. Commun. 2024, 15, 5675.
212. Al-Hilfi, S. H.; Jiang, X.; Heuer, J.; et al. Single-atom catalysts through pressure-controlled metal diffusion. J. Am. Chem. Soc. 2024, 146, 19886-95.
213. Xiong, Y.; Sun, W.; Xin, P.; et al. Gram-scale synthesis of high-loading single-atomic-site Fe catalysts for effective epoxidation of styrene. Adv. Mater. 2020, 32, e2000896.
214. He, X.; Zhang, H.; Zhang, X.; et al. Building up libraries and production line for single atom catalysts with precursor-atomization strategy. Nat. Commun. 2022, 13, 5721.
215. Liu, P.; Zhao, Y.; Qin, R.; et al. Photochemical route for synthesizing atomically dispersed palladium catalysts. Science 2016, 352, 797-801.
216. Keil, F. J. Molecular modelling for reactor design. Annu. Rev. Chem. Biomol. Eng. 2018, 9, 201-27.
217. Yu, Z.; Ji, N.; Li, X.; et al. Kinetics driven by hollow nanoreactors: an opportunity for controllable catalysis. Angew. Chem. Int. Ed. Engl. 2023, 62, e202213612.
218. Sarvestani, M.; Norouzi, O.; Di Maria, F.; Dutta, A. From catalyst development to reactor design: a comprehensive review of methanol synthesis techniques. Energy. Convers. Manag. 2024, 302, 118070.
219. Zheng, R.; Liu, Z.; Wang, Y.; Xie, Z. Industrial catalysis: strategies to enhance selectivity. Chin. J. Catal. 2020, 41, 1032-8.
220. Guo, Y.; Wang, M.; Zhu, Q.; Xiao, D.; Ma, D. Ensemble effect for single-atom, small cluster and nanoparticle catalysts. Nat. Catal. 2022, 5, 766-76.
221. Wang, C.; Wang, Y.; Ge, J.; Xie, Z. Reaction: industrial perspective on single-atom catalysis. Chem 2019, 5, 2736-7.
222. Hülsey, M. J.; Zhang, J.; Yan, N. Harnessing the wisdom in colloidal chemistry to make stable single-atom catalysts. Adv. Mater. 2018, 30, e1802304.
223. Hu, Y.; Li, H.; Li, Z.; et al. Progress in batch preparation of single-atom catalysts and application in sustainable synthesis of fine chemicals. Green. Chem. 2021, 23, 8754-94.
224. Xiong, H.; Lin, S.; Goetze, J.; et al. Thermally stable and regenerable platinum-tin clusters for propane dehydrogenation prepared by atom trapping on ceria. Angew. Chem. Int. Ed. Engl. 2017, 56, 8986-91.
225. Guo, W.; Wang, Z.; Wang, X.; Wu, Y. General design concept for single-atom catalysts toward heterogeneous catalysis. Adv. Mater. 2021, 33, e2004287.
226. Jin, H.; Song, W.; Cao, C. An overview of metal density effects in single-atom catalysts for thermal catalysis. ACS. Catal. 2023, 13, 15126-42.
227. Garole, D. J.; Hossain, R.; Garole, V. J.; Sahajwalla, V.; Nerkar, J.; Dubal, D. P. Recycle, recover and repurpose strategy of spent Li-ion batteries and catalysts: current status and future opportunities. ChemSusChem 2020, 13, 3079-100.
228. Zhou, H.; Liu, T.; Zhao, X.; et al. A supported nickel catalyst stabilized by a surface digging effect for efficient methane oxidation. Angew. Chem. Int. Ed. Engl. 2019, 58, 18388-93.
229. Fu, N.; Liang, X.; Wang, X.; et al. Controllable conversion of platinum nanoparticles to single atoms in Pt/CeO2 by laser ablation for efficient CO oxidation. J. Am. Chem. Soc. 2023, 145, 9540-7.
230. Zhou, H.; Zhao, Y.; Xu, J.; et al. Recover the activity of sintered supported catalysts by nitrogen-doped carbon atomization. Nat. Commun. 2020, 11, 335.
231. Loy, A. C. M.; Teng, S. Y.; How, B. S.; et al. Elucidation of single atom catalysts for energy and sustainable chemical production: synthesis, characterization and frontier science. Prog. Energy. Combust. Sci. 2023, 96, 101074.
232. Gashnikova, D.; Maurer, F.; Sauter, E.; et al. Highly active oxidation catalysts through confining Pd clusters on CeO2 nano-islands. Angew. Chem. Int. Ed. Engl. 2024, 63, e202408511.
233. Li, X.; Pereira-Hernández, X. I.; Chen, Y.; et al. Functional CeOx nanoglues for robust atomically dispersed catalysts. Nature 2022, 611, 284-8.
234. Li, Z.; Li, B.; Li, Q. Single-atom nano-islands (SANIs): a robust atomic-nano system for versatile heterogeneous catalysis applications. Adv. Mater. 2023, 35, e2211103.
235. Liu, X.; Zhou, Y.; Lin, J.; et al. Directional growth and density modulation of single-atom platinum for efficient electrocatalytic hydrogen evolution. Angew. Chem. Int. Ed. Engl. 2024, 63, e202406650.
236. Zhang, X.; Liu, W.; Li, J.; et al. Dehydrogenation of n -butane on metal cobalt sites confined within ceria nanoislands. ACS. Catal. 2024, 14, 15123-32.
237. Zhang, F.; Zhu, Y.; Lin, Q.; Zhang, L.; Zhang, X.; Wang, H. Noble-metal single-atoms in thermocatalysis, electrocatalysis, and photocatalysis. Energy. Environ. Sci. 2021, 14, 2954-3009.
238. Agrachev, M.; Giulimondi, V.; Surin, I.; Mitchell, S.; Jeschke, G.; Pérez-ramírez, J. Electron paramagnetic resonance spectroscopy for the analysis of single-atom catalysts. Chem. Catal. 2024, 4, 101136.
239. Tang, M.; Yuan, W.; Ou, Y.; et al. Recent progresses on structural reconstruction of nanosized metal catalysts via controlled-atmosphere transmission electron microscopy: a review. ACS. Catal. 2020, 10, 14419-50.
240. Liu, Y.; Su, X.; Ding, J.; et al. Progress and challenges in structural, in situ and operando characterization of single-atom catalysts by X-ray based synchrotron radiation techniques. Chem. Soc. Rev. 2024, 53, 11850-87.
241. Liu, Q.; Zhang, Z. Platinum single-atom catalysts: a comparative review towards effective characterization. Catal. Sci. Technol. 2019, 9, 4821-34.
242. Zhao, Y.; Ling, T.; Chen, S.; et al. Non-metal single-iodine-atom electrocatalysts for the hydrogen evolution reaction. Angew. Chem. Int. Ed. Engl. 2019, 58, 12252-7.
243. Yuan, W.; Fang, K.; You, R.; Zhang, Z.; Wang, Y. Toward in situ atomistic design of catalytic active sites via controlled atmosphere transmission electron microscopy. Acc. Mater. Res. 2023, 4, 275-86.
244. Kraushofer, F.; Parkinson, G. S. Single-atom catalysis: insights from model systems. Chem. Rev. 2022, 122, 14911-39.
245. Liberto G, Tosoni S, Cipriano LA, Pacchioni G. A few questions about single-atom catalysts: when modeling helps. Acc. Mater. Res. 2022, 3, 986-95.
246. Zhang, W.; Fu, Q.; Luo, Q.; Sheng, L.; Yang, J. Understanding single-atom catalysis in view of theory. JACS. Au. 2021, 1, 2130-45.
247. Sun, J.; Tu, R.; Xu, Y.; et al. Machine learning aided design of single-atom alloy catalysts for methane cracking. Nat. Commun. 2024, 15, 6036.
248. Zheng, J.; Wang, S.; Deng, S.; Yao, Z.; Hu, J.; Wang, J. Accelerating the screening of modified MA2Z4 catalysts for hydrogen evolution reaction by deep learning-based local geometric analysis. Energy. &. Environ. Mater. 2024, 7, e12743.
249. Xu, H.; Cheng, D.; Cao, D.; Zeng, X. C. Revisiting the universal principle for the rational design of single-atom electrocatalysts. Nat. Catal. 2024, 7, 207-18.
250. Hai, X.; Zheng, Y.; Yu, Q.; et al. Geminal-atom catalysis for cross-coupling. Nature 2023, 622, 754-60.
251. Zhang, S.; Tang, Y.; Nguyen, L.; et al. Catalysis on singly dispersed Rh atoms anchored on an inert support. ACS. Catal. 2018, 8, 110-21.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].