




ROS1 mutations promote an immunosuppressive tumor microenvironment via MYC to confer immune evasion in head and neck cancer
 0
0 Abstract
Aim: Immune checkpoint inhibitors (ICIs) have transformed cancer therapy; however, their efficacy in head and neck cancer (HNC) remains limited, with only a minority of patients achieving durable responses. Understanding the molecular mechanisms underlying ICI resistance in HNC is therefore crucial.
Methods: We conducted an integrative analysis of genomic, transcriptomic, and clinical data from 139 ICI-treated HNC patients (MSKCC cohort) and 502 treatment-naïve HNC cases (TCGA cohort). ROS1 mutation status, tumor mutational burden (TMB), neoantigen load, immune cell infiltration (via CIBERSORT), and immune-related gene expression were evaluated. Gene set enrichment analysis (GSEA) was performed to identify dysregulated pathways. Survival outcomes were assessed using Kaplan-Meier analysis and Cox regression, with statistical significance defined as P < 0.05.
Results: Patients harboring ROS1 mutations exhibited significantly poorer outcomes following ICI therapy, with shorter median overall survival [OS: 5.0 vs. 11.0 months, hazard ratio (HR) = 3.22, 95%CI: 1.26-8.19, P = 0.011] compared to ROS1 wild-type counterparts. Multivariate analysis confirmed ROS1 mutation as an independent predictor of poor OS in ICI-treated patients (HR = 4.78, 95%CI: 1.70-13.43, P = 0.003). In contrast, ROS1 mutations showed no prognostic significance in the treatment-naïve TCGA-HNC cohort (P = 0.26), confirming their role as a predictive (not prognostic) biomarker for ICI response. Interestingly, despite exhibiting higher TMB and neoantigen levels, ROS1-mutant patients showed inferior survival, underscoring the context-dependent limitations of TMB as a predictive biomarker. Mechanistically, ROS1-mutant tumors displayed an immunosuppressive tumor microenvironment characterized by diminished CD8+ T cell infiltration, attenuated interferon-γ signaling, and downregulation of immune-related genes (CXCL9, CXCL10, IFNG, PD-L1). GSEA revealed enrichment of MYC pathway activity in ROS1-mutant tumors, which suppressed antigen presentation and T cell activation pathways.
Conclusion: ROS1 mutations drive ICI resistance in HNC by promoting an immunosuppressive TME via MYC-mediated transcriptional reprogramming, impairing antigen presentation and T cell function. Incorporating ROS1 status into biomarker panels may improve patient stratification and guide combinatorial therapies targeting both immune evasion and oncogenic pathways.
Keywords
INTRODUCTION
Head and neck cancer (HNC), encompassing malignancies of the oral cavity, pharynx, and larynx, is the sixth most common cancer globally, with approximately 890,000 new cases and 450,000 deaths reported in 2022[1]. Despite advances in multimodal therapies, the prognosis for advanced HNC remains poor, with a 5-year survival rate below 50% for recurrent or metastatic disease[1,2].
Treatment options for recurrent or metastatic HNC have evolved significantly over the past decade, improving survival outcomes[3,4]. The immune checkpoint inhibitors (ICIs) pembrolizumab and nivolumab are FDA-approved for cisplatin-refractory recurrent or metastatic HNC. Current national and regional guidelines recommend first-line therapy based primarily on programmed cell death ligand 1 (PD-L1) expression levels. Options include pembrolizumab (with or without chemotherapy) or cetuximab-based regimens (e.g., cetuximab combined with platinum/5-FU chemotherapy)[5-7]. For second-line treatment, nivolumab or pembrolizumab is advised, with alternatives including cetuximab (with or without chemotherapy) or biomarker-directed therapy. Despite these advances, sustained clinical benefits are observed in only 10%-20% of patients, highlighting the urgent need for predictive biomarkers to optimize patient stratification[8,9].
Recent studies have identified tumor mutational burden (TMB), microsatellite instability (MSI), and PD-L1 expression as key determinants of ICI response; however, their predictive power in HNC remains suboptimal. For instance, PD-L1 positivity is associated with improved ICI response in HNC, yet approximately 80% of PD-L1-positive patients fail to achieve objective responses[7], highlighting the complexity of tumor-immune interactions[10]. Similarly, while high TMB correlates with enhanced neoantigen presentation and ICI efficacy in melanoma and lung cancer[11,12], its utility in HNC is limited by molecular heterogeneity[5,8]. These observations emphasize the need for comprehensive molecular characterization and immune profiling to integrate prognostic and predictive biomarkers into clinical practice.
ROS1, a receptor tyrosine kinase, is implicated in the carcinogenesis of multiple cancers. ROS1 fusions are established therapeutic targets in lung adenocarcinoma, with inhibitors such as entrectinib and taletrectinib demonstrating efficacy[13-15]. However, the role of somatic ROS1 mutations (ROS1-Mut) in HNC remains unexplored. Emerging evidence suggests that somatic mutations in driver genes can modulate tumor immunogenicity; for example, ALK rearrangements, EGFR mutations, and KRAS mutations correlate with immunosuppressive tumor microenvironments (TMEs)[16,17]. Paradoxically, colorectal cancers with an ultramutated phenotype exhibit significantly higher objective response rates and more favorable outcomes following ICI treatment compared to dMMR/MSI-H tumors[18]. These findings raise the intriguing possibility that ROS1-Mut may similarly influence immune response in HNC.
Given the critical need to overcome ICI resistance in HNC, we hypothesize that ROS1-Mut might drive immunosuppressive mechanisms similar to those induced by oncogenic drivers such as ALK or EGFR. To investigate this, we integrated multi-omics analyses of 139 ICI-treated HNC patients (MSKCC cohort) and 502 treatment-naïve cases (TCGA cohort) with three specific aims: (1) determine whether ROS1-Mut predict poor ICI response independently of TMB/PD-L1; (2) characterize the immunogenomic landscape of ROS1-Mut tumors; and (3) elucidate mechanistic links between ROS1-Mut and MYC-driven immune evasion. This study identifies ROS1-Mut as candidate mediators of ICI resistance and proposes potential therapeutic strategies.
METHODS
Study cohorts and data acquisition
We analyzed two independent HNC cohorts: 139 advanced HNC patients treated with ICIs [anti-programmed death 1 (PD-1)/PD-L1 ± anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)] from the MSKCC cohort, and 502 treatment-naïve HNC cases from The Cancer Genome Atlas (TCGA) cohort. Clinical and genomic data were retrieved from the following sources: (1) MSKCC cohort from Samstein et al.[9]; (2) whole-exome sequencing (WES) and TMB data from Hoadley et al.[19]; (3) RNA-seq data from the Genomic Data Commons (GDC; https://portal.gdc.cancer.gov/); and (4) survival data from the UCSC Xena Browser (https://xenabrowser.net).
Genomic profiling
ROS1 mutation was defined as non-synonymous somatic mutations, including missense, nonsense, splice-site mutations, and in-frame indels in the coding region of the ROS1 gene. TMB was defined as the total count of non-synonymous mutations per megabase (mut/Mb), with a TMB-high (TMB-H) threshold set at > 10 mut/Mb. Neoantigen prediction was performed as previously described[20]. Expressed somatic variants and patient-specific HLA alleles (predicted using POLYSOLVER) were used as inputs for the NetMHCpan 4.0 algorithm[21]. Strong-binding peptides (IC50 < 500 nM) were counted as neoantigens to calculate tumor neoantigen burden (TNB).
Transcriptomic and immune analyses
Immune cell composition was estimated using CIBERSORT, which quantified 22 immune cell subsets from TCGA RNA-seq data based on the LM22 signature matrix (1,000 permutations)[22]. Differential gene expression was analyzed using the R package DESeq2[23], with thresholds of FDR < 0.05 and log2 fold change > 0.5. Immune-related genes were obtained from Danaher et al. to compare expression between ROS1-Mut and ROS1-wild-type (ROS1-WT) HNC cases in TCGA[24]. Gene set enrichment analysis (GSEA) was performed using the R Package ClusterProfiler v3.18.1[25]. Gene sets were considered significantly enriched at an adjusted P value < 0.05 (Benjamini-Hochberg correction).
Statistical analysis
All statistical analyses were performed in R version 4.0.3 (http://www.r-project.org). Categorical variables were compared using Fisher’s exact test, and continuous variables were assessed with the Wilcoxon rank-sum test. Overall survival (OS) differences were evaluated using Kaplan-Meier curves (log-rank test) and Cox proportional hazards models. Multivariate analyses were adjusted for age, gender, metastatic status, TMB, and treatment regimen. PD-L1 expression across ROS1 subgroups was compared using Fisher’s exact test, and the association between ROS1 status and TMB was assessed using the Mann-Whitney U-test. Statistical significance was defined as two-sided P < 0.05.
RESULTS
Clinicopathologic characteristics of ICI-treated HNC patients
A total of 139 patients with advanced HNC were enrolled (median age 62 years; range, 17-81 years; 78.4% male). Patients were classified according to primary tumor site: oropharynx squamous cell carcinoma (37, 26.6%), oral cavity squamous cell carcinoma (25, 18.0%), larynx squamous cell carcinoma (9, 6.5%), nasopharyngeal carcinoma (9, 6.5%), sinonasal squamous cell carcinoma (6, 4.3%), hypopharynx squamous cell carcinoma (5, 3.6%), and unspecified head and neck squamous carcinoma (48, 34.5%).
Regarding ICI treatment, 131 patients (94.2%) received PD-1/PD-L1 inhibitor monotherapy, while 8 patients (5.8%) received combination therapy with CTLA-4 inhibitors. Tumor samples included 42 primary tumors (30.2%) and 97 metastatic lesions (69.8%), all analyzed by DNA-based next-generation sequencing (NGS). Detailed clinicopathologic characteristics are listed in Supplementary Table 1.
ROS1 mutations in MSKCC and TCGA HNC cohorts
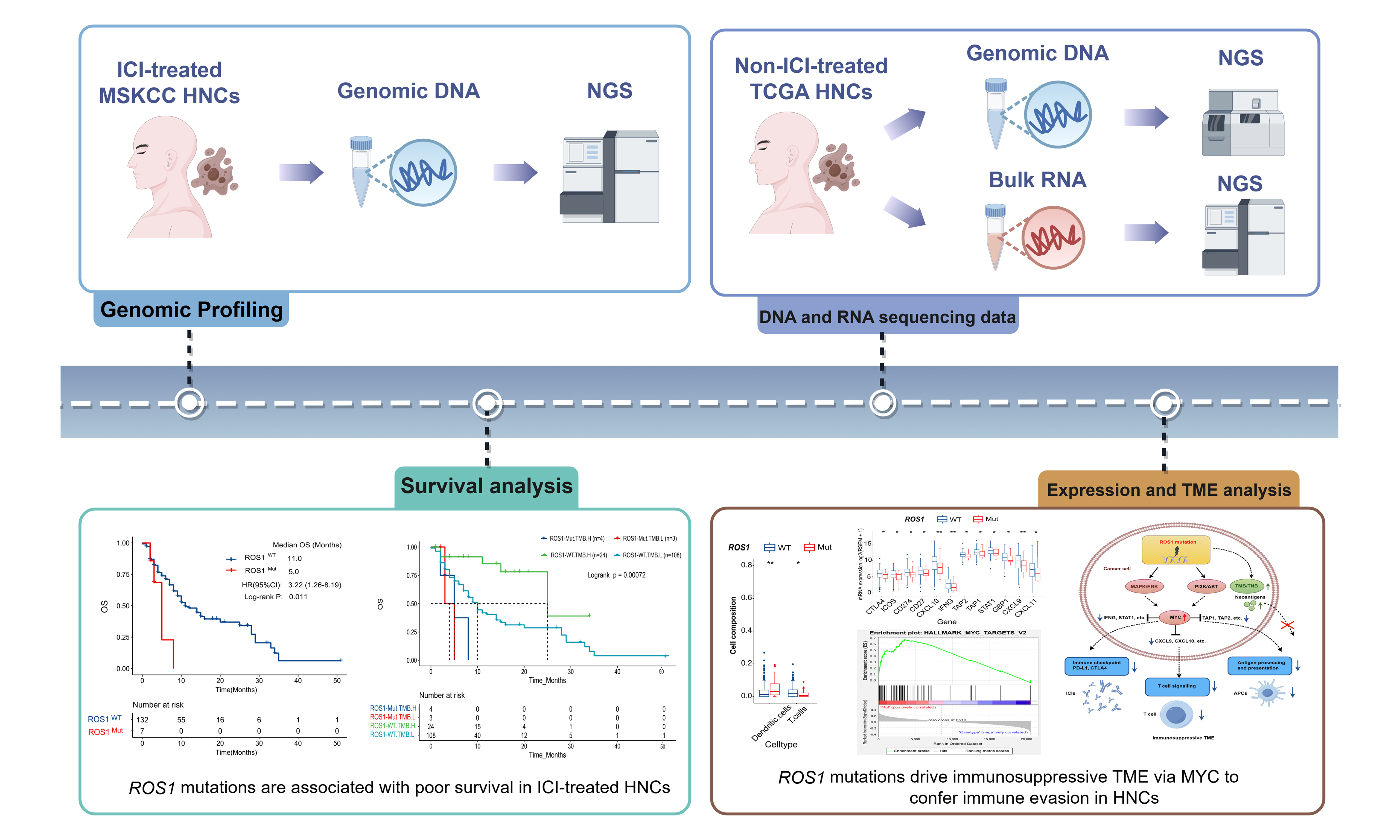
ROS1-Mut were detected in 5.0% (7/139) of the MSKCC cohort and 7.2% (36/502) of the TCGA-HNC cohort. Unlike ROS1 kinase domain mutations that drive tyrosine kinase inhibitor (TKI) resistance in fusion-positive cancers[26], somatic ROS1-Mut in both the MSKCC [Figure 1A] and TCGA [Figure 1B] cohorts were distributed broadly across the protein (e.g., extracellular, transmembrane, cytoplasmic, and kinase domains). Most variants were classified as variants of unknown significance (VUS) according to current CAP/AMP guidelines, and their clinical relevance requires functional validation.
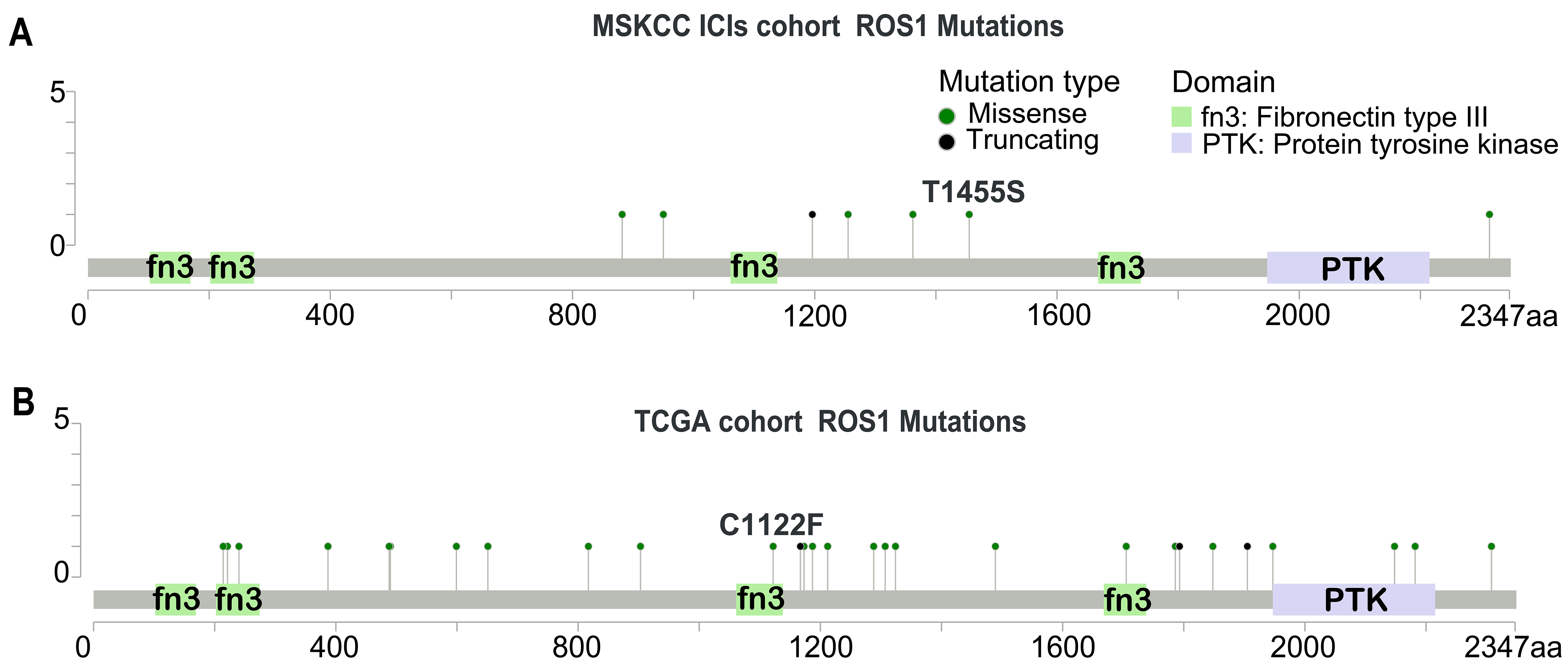
Figure 1. Somatic mutations of ROS1 in patients with HNC. Lollipop plots show somatic mutation resulting in amino acid changes in (A) MSKCC HNC cohort and (B) TCGA HNC cohort. HNC: Head and neck cancer; TCGA: The Cancer Genome Atlas.
ROS1 mutations are associated with poor survival in the ICI-treated HNC cohort
To assess the impact of ROS1-Mut on treatment outcomes, we examined their association with response to ICI therapy. Patients with ROS1-WT mutations exhibited a median OS of 11.0 months, whereas patients with ROS1-Mut had a significantly shorter median OS of 5.0 months [Figure 2A]. The hazard ratio (HR) for OS comparing ROS1-Mut to ROS1-WT was 3.22 (95%CI: 1.26-8.19; P = 0.011). Multivariate analysis confirmed ROS1 mutations as an independent predictor of poor OS in ICI-treated HNC patients (HR = 4.78; 95%CI: 1.70-13.43; P = 0.003), after adjusting for age, gender, metastatic status, TMB, and treatment regimen [Figure 2B].
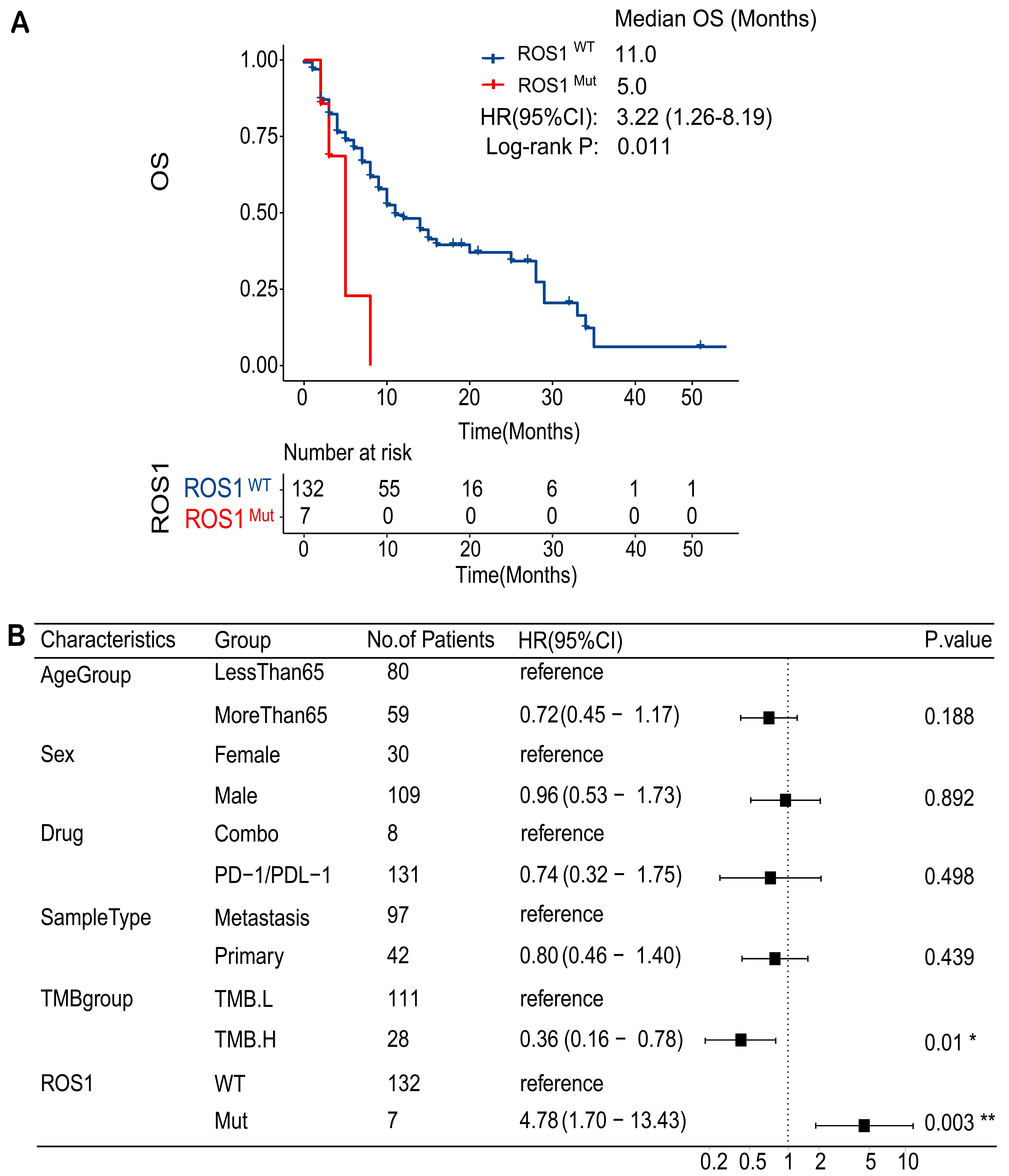
Figure 2. ROS1 mutation as a predictor of ICI resistance in patients with HNC. (A) Kaplan-Meier survival curves comparing OS between ROS1-Mut and ROS1-WT groups in the ICI-treated HNC cohort; (B) Multivariate analysis of OS in the ICI-treated HNC cohort. ICI: Immune checkpoint inhibitor; HNC: head and neck cancer; OS: overall survival; ROS1-Mut: ROS1 mutations; ROS1-WT: ROS1-wild-type.
ROS1-Mut tumors exhibited significantly higher TMB compared to ROS1-WT tumors (P = 0.043) [Figure 3A]. This pattern was more pronounced in the TCGA HNC cohort [Figure 3B], where ROS1-Mut tumors showed markedly elevated TMB (P < 0.001). Similarly, analysis of TCGA data [Figure 3C] demonstrated that ROS1-Mut tumors were associated with higher neoantigen levels (P < 0.001), suggesting enhanced immunogenicity in ROS1-mutated cases. Despite the presumed predictive value of high TMB for ICI response, survival analysis demonstrated that ROS1-Mut cases with TMB-H had shorter OS compared to ROS1-WT cases [Figure 3D]. These findings highlight a critical interaction between ROS1 mutations and TMB, where mutational burden fails to mitigate the aggressive biology of ROS1-mutant tumors in the context of ICI therapy.
Figure 3. Association of ROS1 mutations with TMB, neoantigen levels, and patient survival in HNC. Boxplots comparing log2-transformed TMB between ROS1-Mut and ROS1-WT tumors in (A) MSKCC HNC cohort and (B) TCGA HNC cohort; (C) Boxplot comparing log2-transformed neoantigen levels in TCGA HNC tumors by ROS1 mutation status; (D) Kaplan-Meier analysis of OS stratified by ROS1 mutation status and TMB levels. TMB: Tumor mutational burden; HNC: head and neck cancer; ROS1-Mut: ROS1 mutations; ROS1-WT: ROS1-wild-type; TCGA: The Cancer Genome Atlas; OS: overall survival.
ROS1 mutation is not a prognostic factor in HNC
To evaluate the prognostic value of ROS1 mutation in HNC, survival analyses were conducted using TCGA data. No significant differences in progression-free survival (PFS) or OS were observed between ROS1-Mut and ROS1-WT patients (log-rank P = 0.26 for OS; Supplementary Figure 1), indicating that ROS1 mutations do not serve as a prognostic factor in untreated HNC.
ROS1-Mut tumors exhibit a cold immune microenvironment in HNC
To investigate the molecular mechanisms underlying ROS1-mediated resistance to ICI therapy in HNC, RNA-seq and WES data from the TCGA-HNC cohort were analyzed. Assessment of immune cell composition revealed minimal differences between ROS1-Mut and ROS1-WT tumors [Figure 4A]. Among 22 immune cell types evaluated, only activated dendritic cells exhibited significantly higher infiltration in ROS1-Mut tumors (P < 0.01), whereas regulatory T cells (Tregs) were reduced in ROS1-Mut tumors (P < 0.05). No other immune cell types, including CD8+ T cells or NK cells, showed statistically significant differences. Differential expression analysis showed broad downregulation of immune-related genes in ROS1-Mut tumors. Notably, immune checkpoint genes (CTLA4, ICOS, CD274/PD-L1), tumor necrosis factor receptor family member CD27, IFN-γ mediating signaling pathway genes (CXCL9, CXCL10, CXCL11, GBP1, IFNG, STAT1), and antigen-processing genes (TAP1, TAP2) were significantly reduced in ROS1-Mut tumors [Figure 4B]. These results suggest that ROS1 mutations in HNC are associated with an immunosuppressive microenvironment characterized by downregulation of immune-related genes and reduced Treg infiltration, which may contribute to resistance to ICI therapy.
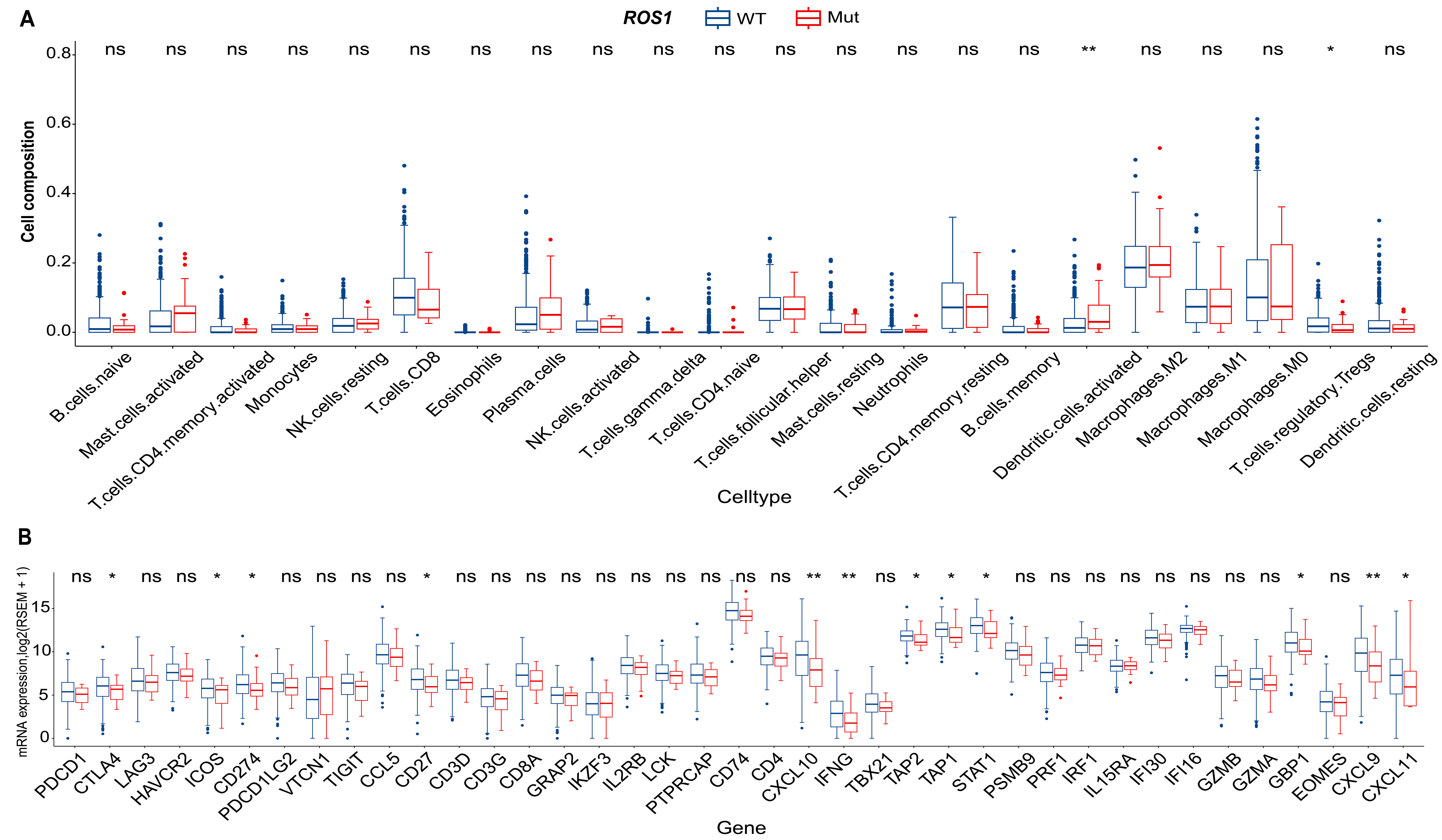
Figure 4. Molecular differences in immune cell composition and immune-related gene expression between ROS1-WT and ROS1-Mut tumors in the TCGA HNC cohort. (A) Comparison of 22 predefined immune cell types; (B) expression of 39 immune-related genes. *P < 0.05; **P < 0.01; ns: not significant (P ≥ 0.05). ROS1-WT: ROS1-wild-type; ROS1-Mut: ROS1 mutations; TCGA: The Cancer Genome Atlas; HNC: head and neck cancer.
Pathway analysis links ROS1 mutation to immune evasion mechanisms
GSEA revealed distinct pathway activation patterns between ROS1-Mut and ROS1-WT tumors, elucidating potential mechanisms of ICI resistance. In ROS1-Mut tumors, genes associated with the MYC signaling pathway were significantly upregulated [Figure 5A], mediating tumor proliferation, metabolic adaptation, and immune evasion. In contrast, immune-activating pathways including antigen receptor-mediated signaling, T cell receptor (TCR) signaling, and cell adhesion molecules were downregulated in ROS1-Mut tumors [Figure 5B-D]. These findings collectively highlight that ROS1 mutations drive transcriptional reprogramming toward an immunosuppressive phenotype.
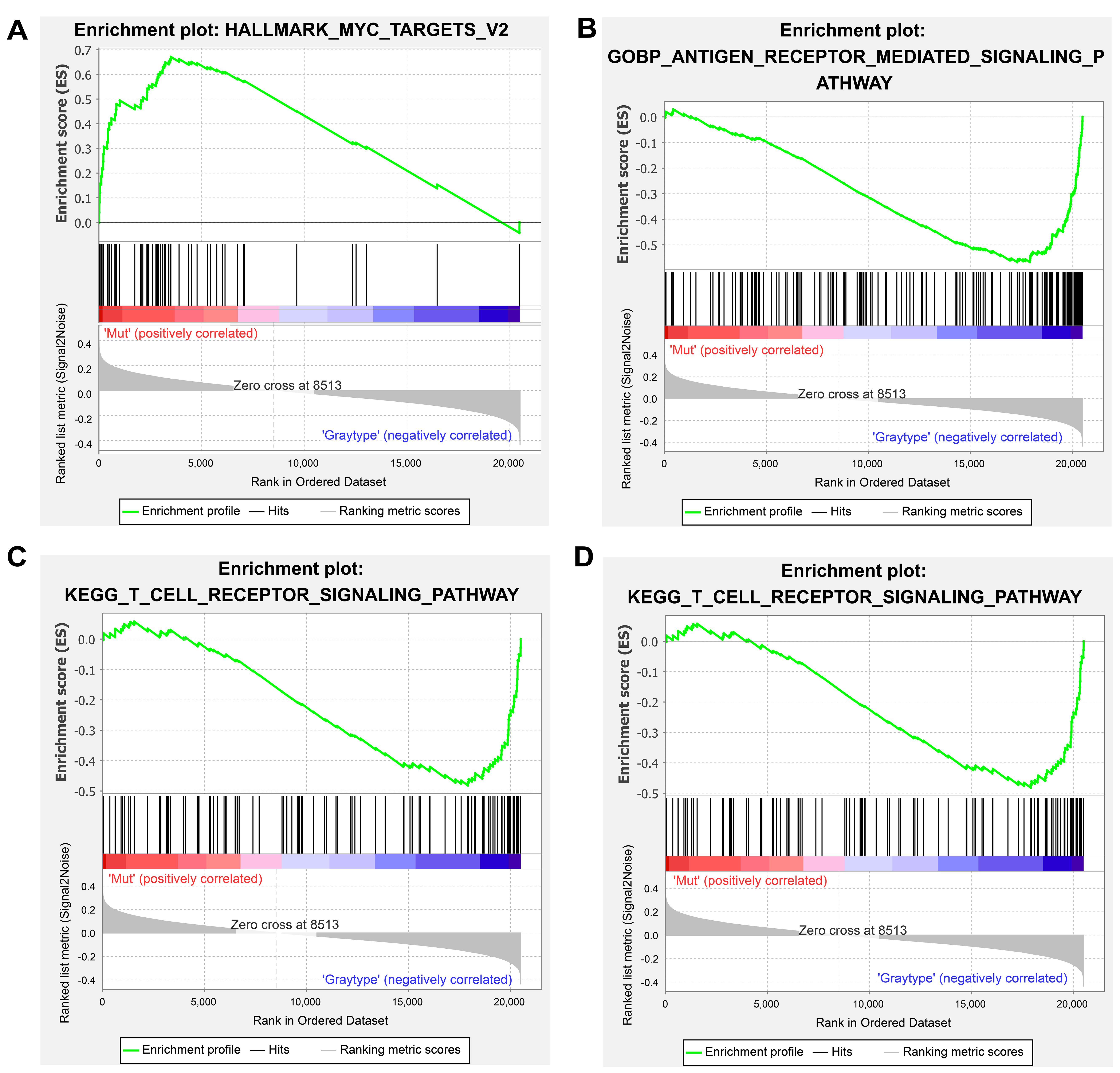
Figure 5. GSEA results for hallmark gene sets (A) and KEGG pathways (B-D). A t-value > 0 (red) indicates higher scores in ROS1-Mut tumors; t-value < 0 (blue) indicates higher scores in ROS1-WT tumors. GSEA: Gene set enrichment analysis; ROS1-Mut: ROS1 mutations; ROS1-WT: ROS1-wild-type.
Integrative multi-omics analyses further revealed that ROS1-mutant tumors exhibit hyperactivation of the MYC pathway, which transcriptionally represses IFN-γ signature genes (e.g., IFNG, STAT1), T cell chemokines (e.g., CXCL9, CXCL10), and components of the antigen presentation machinery (TAP1 and TAP2), collectively fostering an immunosuppressive TME. Despite elevated TMB and neoantigen load, these tumors show impaired CD8+ T cell infiltration and reduced responsiveness to ICI therapy. Figure 6 provides a schematic overview of the proposed mechanistic cascade by which ROS1 mutations drive immune evasion and ICI resistance in HNC.
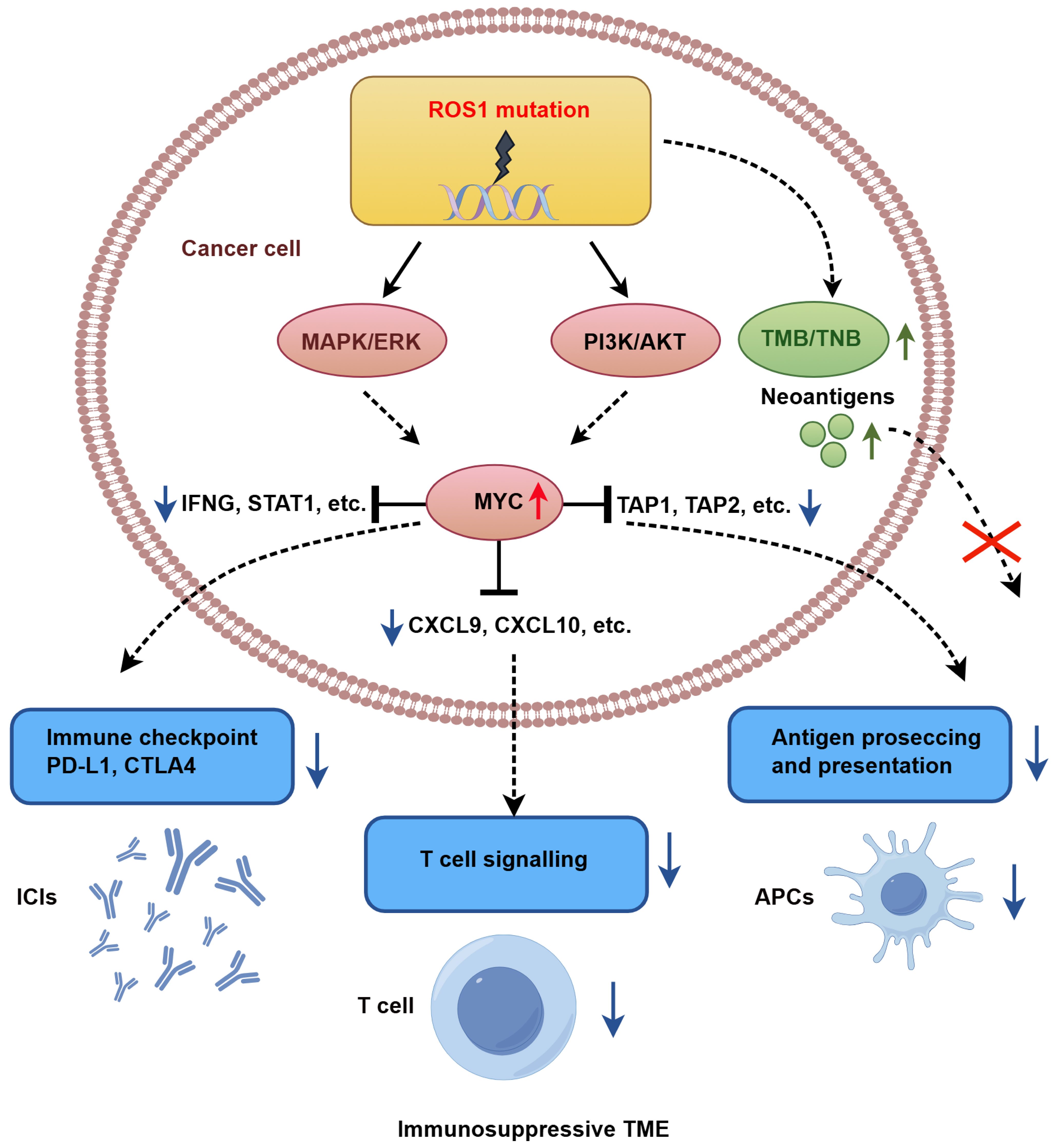
Figure 6. Proposed mechanism of ROS1 mutation-driven immune evasion in HNC. HNC: Head and neck cancer; APCs: antigen-presenting cells; ICIs: immune checkpoint inhibitors; TMB: tumor mutational burden; TME: tumor microenvironment; TNB: tumor neoantigen burden.
DISCUSSION
This study demonstrates that ROS1-Mut tumors develop an immunosuppressive TME, characterized by diminished CD8+ T cell infiltration, downregulation of key immune-related genes (e.g., CXCL9/10, IFNG, PD-L1), and impaired interferon-γ (IFN-γ) signaling. These findings identify ROS1 mutation as a potential predictive marker of resistance to ICIs in HNC, although clinical validation in larger cohorts is needed.
Despite elevated TMB and neoantigen load in ROS1-Mut tumors - features typically associated with enhanced ICI response - these patients exhibited significantly shorter OS (median OS: 5.0 vs. 11.0 months, HR = 3.22, P = 0.011). The paradoxical coexistence of high TMB with poor ICI response highlights the limitations of mutational burden as a universal biomarker. While TMB generally correlates with neoantigen visibility, ROS1-Mut tumors may override this immunogenic advantage through MYC-mediated transcriptional reprogramming. This is supported by significant downregulation of antigen presentation machinery (TAP1/TAP2) and T cell chemotaxis genes (CXCL9/CXCL10) in our transcriptomic data [Figure 4B], consistent with established MYC-immune suppression mechanism [27-32].
Our GSEA results [Figure 5A], together with the observed downregulation of MHC-I pathway genes [Figure 4B], suggest that MYC activation contributes to an immunosuppressive TME where a high neoantigen load becomes biologically irrelevant, mirroring MYC-driven immune evasion reported in pancreatic ductal adenocarcinoma (PDAC)[31] and hepatocellular carcinoma (HCC)[32]. Similar phenomena have been observed in other oncogenic alterations, such as MDM2/4 amplification, where ICI-induced hyperprogression correlates with suppressed T cell infiltration[27]. JAK1/2 mutations have also been associated with accelerated tumor growth post-ICI due to defective MHC-I expression and resistance to T cell cytotoxicity in melanoma[28].
The upregulation of MYC signaling in ROS1-Mut tumors aligns with prior studies linking MYC to immune suppression via downregulation of MHC class I molecules and recruitment of immunosuppressive myeloid cells[29-32]. These defects in antigen presentation represent a rate-limiting step that impedes neoantigen visibility[33], explaining the discordance between high TMB and poor ICI response in ROS1-mutant tumors. Concurrently, suppression of IFN-γ response genes (CXCL9, CXCL10, STAT1) mirrors observations in melanoma and lung cancer, where similar transcriptional silencing correlates with T cell exclusion and ICI resistance[34,35]. Additionally, the reduced expression of PD-L1 (CD274) and CTLA4 in ROS1-Mut tumors might further impair ICI efficacy. Collectively, these findings highlight that checkpoint molecule expression or TMB alone may be insufficient to predict ICI response, emphasizing the need for composite biomarkers that integrate genomic and immune profiling.
Clinically, ROS1 mutation status may serve as a stratification tool to identify HNC patients unlikely to benefit from ICIs. This context-dependent predictive role parallels findings for TGFBR2 mutations in NSCLC[36] and TP53 mutations in melanoma[37], but contrasts with DYNC2H1 mutations, which enhance immunogenicity[38]. The variable effects of kinase mutations highlight the need for biomarker-driven therapeutic strategies.
Therapeutically, ROS1-Mut HNC may benefit from combinatorial strategies targeting both immune evasion pathways and oncogenic drivers. Preclinical studies show that ROS1 inhibitors (e.g., entrectinib) can reverse oncogenic signaling[13,26], while MYC inhibition can restore antigen presentation in other models[39]. Thus, combinatorial strategies represent a biologically grounded approach warranting further evaluation in ROS1-mutant systems.
This study has limitations. ROS1 mutations occur at relatively low incidence in HNC (5%-7%), though this subgroup exhibits significant clinical detriment (median OS: 5 months). Our biomarker claims are constrained by the small sample size of ROS1-mutant cases (n = 7 in the ICI-treated cohort), heterogeneity of variants (predominantly VUS), and lack of functional validation. While statistically significant, these findings require prospective validation in larger cohorts and mechanistic studies to confirm ROS1’s role in immune evasion. Additionally, the absence of prognostic significance in TCGA (non-ICI-treated cohort) underscores its context-specific predictive role, akin to KRAS mutations in colorectal cancer[40].
In conclusion, ROS1-mutant HNC defines a molecular subset with intrinsic ICI resistance driven by MYC-mediated immunosuppression. If validated prospectively, integrating ROS1 status with TMB and PD-L1 expression could optimize patient stratification for ICI therapy, though further studies are essential to establish clinical utility.
DECLARATIONS
Acknowledgments
The authors kindly thank Dr. Xing Zhang (Jiangsu Simcere Diagnostics Co., Ltd.) for providing professional technical support for this research.
Authors’ contributions
Writing - original draft, validation: Fang C
Writing - original draft, methodology, software, visualization: Zhang Q
Writing - original draft, visualization: Fang R
Writing - review and editing, formal analysis: Li Y
Visualization: Bai J
Data curation: Huang X
Project administration, data curation: Lu J
Investigation, resources: Chen D
Conceptualization, writing - review and editing: Zhang Y
Writing – reviewing and editing, supervision: Chen Z
The work reported in the paper has been performed by the authors, unless clearly specified in the text.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding authors upon reasonable request.
Financial support and sponsorship
None.
Conflicts of interest
Chen D, Zhang Q, and Zhang Y are employees of Jiangsu Simcere Diagnostics Co., Ltd. Other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
This study was approved by the Medical Ethics Committee of Nanjing Simcere Medical Laboratory (NSML-IRB-202401-MS08; January 12, 2024). Written informed consent was obtained from all patients.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
Supplementary Materials
REFERENCES
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229-63.
2. Petrelli F, Lorini L, Paderno A, et al. Treatment of primary tumor in metastatic head and neck carcinoma: a systematic review and meta-analysis. Oral Oncol. 2025;163:107248.
3. Szturz P, Fuereder T, Guo Y, et al. Treatment decision-making factors and sequencing in recurrent and/or metastatic squamous cell carcinoma of the head and neck. Cancer Treat Rev. 2025;135:102910.
4. Liu X, Harbison RA, Varvares MA, Puram SV, Peng G. Immunotherapeutic strategies in head and neck cancer: challenges and opportunities. J Clin Invest. 2025;135:e188128.
5. Ferris RL, Blumenschein G Jr, Fayette J, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856-67.
6. Seiwert TY, Burtness B, Mehra R, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956-65.
7. Burtness B, Harrington KJ, Greil R, et al; KEYNOTE-048 Investigators. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019;394:1915-28.
8. Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. 2020;6:224.
9. Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202-6.
11. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124-8.
12. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189-99.
13. Drilon A, Siena S, Dziadziuszko R, et al; trial investigators. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020;21:261-70.
14. Waliany S, Lin JJ. Taletrectinib: TRUST in the continued evolution of treatments for ROS1 fusion-positive lung cancer. J Clin Oncol. 2024;42:2622-7.
15. Li W, Xiong A, Yang N, et al. Efficacy and safety of taletrectinib in Chinese patients with ROS1+ non–small cell lung cancer: the Phase II TRUST-I study. J Clin Oncol. 2024;42:2660-70.
16. Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217-23.
17. Gainor JF, Shaw AT, Sequist LV, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin Cancer Res. 2016;22:4585-93.
18. Ambrosini M, Rousseau B, Manca P, et al. Immune checkpoint inhibitors for POLE or POLD1 proofreading-deficient metastatic colorectal cancer. Ann Oncol. 2024;35:643-55.
19. Hoadley KA, Yau C, Hinoue T, et al; Cancer Genome Atlas Network. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173:291-304.e6.
20. Angelova M, Charoentong P, Hackl H, et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015;16:64.
21. Nielsen M, Lundegaard C, Blicher T, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One. 2007;2:e796.
22. Thorsson V, Gibbs DL, Brown SD, et al; Cancer Genome Atlas Research Network. The immune landscape of cancer. Immunity. 2018;48:812-30.e14.
23. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
24. Danaher P, Warren S, Dennis L, et al. Gene expression markers of tumor infiltrating leukocytes. J Immunother Cancer. 2017;5:18.
25. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284-7.
26. Drilon A, Jenkins C, Iyer S, Schoenfeld A, Keddy C, Davare MA. ROS1-dependent cancers - biology, diagnostics and therapeutics. Nat Rev Clin Oncol. 2021;18:35-55.
27. Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017;23:4242-50.
28. Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819-29.
29. Casey SC, Tong L, Li Y, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227-31.
30. Fan Y, Song S, Li Y, et al. Galectin-3 cooperates with CD47 to suppress phagocytosis and T-cell immunity in gastric cancer peritoneal metastases. Cancer Res. 2023;83:3726-38.
31. Krenz B, Gebhardt-Wolf A, Ade CP, et al. MYC- and MIZ1-dependent vesicular transport of double-strand RNA controls immune evasion in pancreatic ductal adenocarcinoma. Cancer Res. 2021;81:4242-56.
32. Dhanasekaran R, Hansen AS, Park J, et al. MYC overexpression drives immune evasion in hepatocellular carcinoma that is reversible through restoration of proinflammatory macrophages. Cancer Res. 2023;83:626-40.
33. Dou Y, Katsnelson L, Gritsenko MA, et al; Clinical Proteomic Tumor Analysis Consortium. Proteogenomic insights suggest druggable pathways in endometrial carcinoma. Cancer Cell. 2023;41:1586-605.e15.
34. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231-5.
35. Gao J, Shi LZ, Zhao H, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167:397-404.e9.
36. Li T, Wang H, Xu J, et al. TGFBR2 mutation predicts resistance to immune checkpoint inhibitors in patients with non-small cell lung cancer. Ther Adv Med Oncol. 2021;13:17588359211038477.
37. Xiao W, Du N, Huang T, et al. TP53 mutation as potential negative predictor for response of anti-CTLA-4 therapy in metastatic melanoma. EBioMedicine. 2018;32:119-24.
38. Yang L, Feng Y, Liu X, et al. DYNC2H1 mutation as a potential predictive biomarker for immune checkpoint inhibitor efficacy in NSCLC and melanoma. Invest New Drugs. 2025;43:199-213.
39. Kortlever RM, Sodir NM, Wilson CH, et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell. 2017;171:1301-15.e14.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].