
Beyond the brain: a multisystem review of pathogenesis, comorbidity, and therapeutic progress in sporadic Parkinson’s disease
 0
0 
Abstract
Sporadic Parkinson’s disease (PD) is increasingly viewed as a multifactorial neurodegenerative syndrome driven by complex interactions between genetic predisposition, environmental exposures, aging-related vulnerability, and systemic dysfunctions beyond the central nervous system. Far from just a localized dopaminergic deficit, PD pathology encompasses the sensory–gut–midbrain axis and incorporates metabolic, immune, and peripheral nervous system abnormalities. This review summarizes recent advances in four dimensions. First, we highlight key genetic contributors, such as SNCA, NEAT1, and ATP13A2/PARK9, alongside environmental stressors and aging-related changes that collectively influence disease susceptibility. Second, using cross-species transcriptomic data from humans and mice, we pinpoint conserved gene networks shared across the midbrain, gut, and sensory systems. These networks include genes (PINK1, PRKN1, LRRK2, PARK7/DJ-1, SCN9A, FAAH and GCH1) involved in key pathways regulating mitochondrial quality control and oxidative stress, autophagy-mediated protein clearance, neural development and synaptic signaling, and immune-metabolic regulation, providing molecular evidence that PD manifests as a systemic network disorder. Third, we examine current and emerging therapeutic strategies, emphasizing the move from symptom relief to disease-modifying interventions through systemic, network-level, and precision approaches. Finally, we address the expanding role of early diagnostic biomarkers and computational subtyping in enabling precision-guided therapy. These insights point to future PD care relying on multidimensional strategies - combining targets co-expressed in central and peripheral neural systems, systemic regulatory modulation, and individualized care - to slow progression and ultimately prevent disease onset. This systemic and integrative perspective could shed new light on clinical and research paradigms for PD over the coming decade.
Keywords
INTRODUCTION
Parkinson’s disease (PD) ranks as the second most common neurodegenerative disorder, impacting over 10 million people globally[1]. Traditionally characterized by motor dysfunction resulting from dopaminergic (DA) neuron loss in the substantia nigra, PD has long been framed as a brain-centric condition rooted in dopamine deficiency. While DA therapies have demonstrated clear symptomatic benefits, they offer limited efficacy in halting or reversing disease progression, especially in later stages when non-motor symptoms such as gastrointestinal dysfunction, sleep disturbances, autonomic dysregulation, and cognitive impairment become prominent[2,3].
Over recent decades, advances in molecular biology, neuroimaging, and systems-level analysis have transformed our understanding of PD into a complex, multisystem disease. A growing body of evidence indicates that its pathogenesis is not confined to the central nervous system, but rather involves distributed dysfunction across peripheral organs, including the gastrointestinal tract, olfactory system, cardiovascular network, and immune system[4,5]. These peripheral manifestations often precede classic motor symptoms by years, underscoring the limitations of symptom-based diagnoses and revealing a broader pathophysiological spectrum.
Despite substantial progress in identifying PD-associated genes (PAGs), toxic protein species such as α-synuclein (α-syn), and mechanisms such as mitochondrial dysfunction and neuroinflammation, a unified model that connects these factors across organs and disease stages remains elusive. Increasingly, PD is regarded as a systems disorder - one in which genetic, environmental, and aging-related factors converge on a vulnerable biological network that spans both central and peripheral compartments[6,7].
In this review, we re-evaluate PD from a network-centric and cross-organ perspective. We begin by summarizing recent advances in understanding its systemic pathogenesis and comorbidity patterns. We then discuss emerging therapeutic strategies designed to modulate multiple targets and restore inter-organ homeostasis. Finally, we explore how precision medicine, early diagnosis tools, and data-driven subtyping are redefining the future of PD management. By synthesizing insights from molecular, clinical, and transcriptomic research, this review seeks to present a cohesive view of PD as a whole-body disease and chart potential pathways toward more effective, personalized interventions.
METHODOLOGY AND DATA SOURCES
To ensure reproducibility and transparency, explicit inclusion, filtering, and integration criteria were applied to all transcriptomic datasets synthesized in this review. Both developmental and adult datasets from human and mouse sources were incorporated, focusing on four PD–relevant systems: midbrain dopaminergic (mDA) neurons, olfactory bulb (OB), dorsal root ganglia (DRG), and the enteric nervous system (ENS). All datasets were obtained from public repositories, and accession numbers are provided in the Gene Expression Omnibus (GEO): GSE245310, GSE139088, GSE134355, GSE149524, GSE156905, GSE121891, GSE70896, GSE76381, and GSE178265.
Dataset-specific filtering criteria were applied to minimize low-level transcriptional noise while accounting for differences in tissue origin, species, developmental stage, and sequencing modality. For DRG datasets, human spatial transcriptomics data were filtered using a log10(M + 1) > 0.3 threshold. In adult mouse DRG single-cell RNA-seq datasets, genes were retained if their expression level relative to β-actin exceeded 0.2. Developmental DRG single-cell datasets from both human and mouse sources were filtered using a minimum detection rate of ≥ 5% across cells and an average expression level of ≥ 1 counts per million (CPM).
For OB datasets, developmental bulk RNA-seq data from mouse samples were filtered using a fragments per kilobase per million mapped reads (FPKM) threshold of ≥ 1. Mouse OB scRNA-seq datasets were filtered using a detection rate of ≥ 5% and an average CPM ≥ 1, whereas human OB bulk RNA-seq datasets adopted differentially expressed genes as defined in the original studies using a significance threshold of P < 0.05.
For mDA neuron datasets, both human and mouse developmental scRNA-seq data were filtered using a detection rate ≥ 5% and an average CPM ≥ 1, with adult mouse datasets processed using identical thresholds. Mouse ENS developmental scRNA-seq datasets were filtered using the same criteria (detection rate ≥ 5%, average CPM ≥ 1). Across all datasets, only genes passing dataset-specific quality and expression thresholds were retained for downstream cross-system and cross-species integration analyses.
Primary data preprocessing and normalization were performed in a dataset-dependent manner. For bulk RNA-seq datasets, differential expression analyses followed the pipelines reported in the original studies, most commonly based on standard tools such as DESeq2. For scRNA-seq datasets involving midbrain and ENS dopaminergic populations, raw or count-level data were reanalyzed using established workflows implemented in Seurat. This reanalysis was performed to enable consistent identification and filtering of dopaminergic neurons across datasets based on unified marker-based criteria. Standard single-cell processing steps, including normalization, dimensionality reduction, and cell-type selection, were applied as described in the respective analytical frameworks. For other single-cell or bulk datasets not requiring dopaminergic neuron–specific resolution (e.g., OB and DRG), normalized expression matrices, cell-type annotations, or gene lists reported by the original authors were adopted directly for downstream integration. This hybrid strategy - combining targeted reanalysis of single-cell datasets with reuse of published results - was implemented to ensure methodological consistency where required, while preserving the integrity of original analyses across heterogeneous transcriptomic platforms.
Cell-type resolution was adapted across tissues according to biological relevance and data structure. For midbrain and ENS single-cell datasets, analyses focused specifically on DA or catecholaminergic neurons, reflecting their established central role in PD pathogenesis. DA neurons were identified using canonical marker-based criteria, including expression of tyrosine hydroxylase (TH), dopa decarboxylase (DDC), the DAT (SLC6A3), and the vesicular monoamine transporter 2 (SLC18A2), with marker nomenclature adapted to species-specific conventions. Only cells expressing at least two canonical DA markers, as reported in the original datasets, were retained for analysis.
In contrast, for OB and DRG datasets, analyses were conducted at the level of pan-neuronal populations or bulk tissue. In these systems, dopaminergic neurons are sparse, heterogeneous, or not clearly defined as dominant pathological substrates, and restricting analyses to putative DA-like cells would risk excluding biologically relevant neuronal subtypes. Accordingly, all neuronal populations (or bulk transcriptomes, where applicable) were retained to capture system-level vulnerability patterns. This tissue-specific analytical strategy was adopted to balance cell-type specificity with biological representativeness and to ensure that cross-system comparisons reflect shared vulnerability architectures rather than forced uniformity across fundamentally distinct neural systems.
Functional enrichment and protein–protein interaction (PPI) analyses were performed using the STRING database. Gene sets identified from cross-system and cross-species transcriptomic integration were submitted to STRING for annotation of enriched biological pathways, molecular functions, and interaction networks[8,9].
PATHOGENIC MECHANISMS
Genetic factors
Research indicates that genetic factors play a pivotal role in sporadic PD. To date, over 20 high penetrance monogenic variants have been identified as potential causes of familial PD, while more than 100 genetic risk loci are associated with sporadic PD[10-13]. During neurodevelopment, specific PAGs such as OTX2[14-16], PITX3[17-24], and NURR1 (NR4A2)[25-28] are highly expressed and significantly influence the formation of the DA system[29]. In adulthood, these genes continue to maintain neuronal homeostasis[28-30]. Additionally, other genes exert sustained effects on DA neuron development and function throughout both developmental and adult stages. For instance, studies have categorized known PD genes into two modules based on their expression patterns during early and late brain development: the first module shows significantly higher expression in adulthood compared to the embryonic stage, while the second module exhibits the opposite pattern, with most PD genes belonging to the first module[12]. Interestingly, patients with PD caused by mutations in the first module (highly expressed in adulthood) tend to develop the disease at a younger age than those with mutations in the second module (highly expressed during development)[12]. This suggests that mutations affecting genes active in adulthood may be more likely to lead to early-onset disease.
During neurodevelopment, certain susceptibility genes regulate the formation and survival of mDA neurons. For instance, transcription factors such as NURR1, LMX1A/B, and EN1 mediate DA neuron differentiation during embryogenesis[28-33]. Insufficient expression of these genes may reduce congenital DA neuron reserves, thereby increasing the risk of developing PD. NURR1 is not only crucial for early DA neuron differentiation but also maintains expression in adulthood, making it a potential therapeutic target for PD[28,29]. Reduced NURR1 activity in PD patients correlates with substantia nigra neuron loss and inflammation, suggesting that enhancing NURR1 activity could provide protective benefits[28,29,34]. Mice with Engrailed-1 (En1) single-allelic deletion spontaneously develop substantia nigra DA neuron degeneration and motor dysfunction, indicating EN1’s protective role in adult brains[30]. Cohort studies also reveal associations between EN1 gene polymorphisms and susceptibility to idiopathic PD. These findings demonstrate that developmental genes continue influencing neuronal fate in adulthood, with reduced expression levels potentially contributing to PD pathogenesis. LMX1A plays a key role in mDA system development, leading to the hypothesis that its low expression in sensory nervous systems may correlate with early symptoms like olfactory dysfunction in PD patients (see Section “A Conserved genetic network links PD and olfactory dysfunction”).
In metabolic homeostasis and cellular clearance, multiple PD genes are directly involved in mitochondrial function, protein degradation, and lysosomal pathways. For instance, PINK1 and PRKN encode key proteins in the mitochondrial quality control pathway, playing crucial roles in eliminating damaged mitochondria[35,36]. Specifically, serine protein kinase PINK1 identifies damaged mitochondria, while E3 ubiquitin ligase Parkin mediates their autophagic clearance. Mutations in PINK1 or PRKN genes can lead to autosomal recessive early-onset PD, characterized by significant mitochondrial dysfunction and elevated oxidative stress. Research shows that these genetic mutations disrupt normal mitochondrial function, causing abnormal mitochondrial accumulation in neurons. This impairs energy production while generating harmful reactive oxygen species, delivering a dual blow that ultimately triggers PD. Another example is DJ-1, which plays a role in cellular antioxidant defense[37]. Its mutations can also cause autosomal recessive PD. These genetic defects make neurons more susceptible to metabolic stress damage. For instance, animals lacking Parkin exhibit heightened sensitivity to mitochondrial toxin rotenone. Additionally, heterozygous mutations in the GBA gene (encoding lysosomal enzyme glucocerebrosidase) are among the strongest risk factors for PD, leading to impaired lysosomal degradation and accumulation of toxic substrates[13]. GBA mutations also disrupt glycolipid metabolism. The disease manifests through impaired degradation of α-syn in the brain, leading to its pathological aggregation. Research indicates that GBA mutations disrupt lipid metabolism, particularly by elevating levels of critical cerebroside lipids, suggesting lipid homeostasis imbalance as a key contributor to PD[38]. The LRRK2 gene (encoding lysine-rich repeat kinase 2) is the most frequently mutated gene in autosomal dominant PD, with its protein complex regulating lysosomal function, mitochondrial dynamics, and inflammatory responses[39,40]. LRRK2 mutations not only affect DA synapses and autophagy pathways in the brain but are also highly expressed in peripheral immune cells, establishing a critical link between metabolic and immune mechanisms in PD. Collectively, genes associated with sporadic PD influence key pathways including mitochondrial function, protein homeostasis, and neuroinflammation through multiple biological mechanisms[12,13,35,36,38,39].
Notably, certain molecular biomarkers show promise as potential indicators for early PD diagnosis or disease severity assessment. For instance, α-syn, encoded by the SNCA gene, exhibits abnormal aggregation as the core pathological feature of PD[41,42]. Studies have demonstrated that abnormal α-syn conformational seeds can be detected in cerebrospinal fluid, with highly positive detection rates using the RT-QuIC amplification method[43]. This suggests the technology could identify the disease years before motor symptoms emerge[44]. Additionally, the C05-05 probe developed by Japanese scientists specifically targets α-syn fibers, detecting pathological α-syn presence through positron emission tomography (PET) imaging in both animal models and PD patients[45]. Another example is neurofilament light chain (NfL), a sensitive axonal injury marker. In PD, particularly in advanced stages or dementia-associated cases, elevated blood NfL levels correlate closely with disease severity and progression[46,47]. Furthermore, variations in blood concentrations of certain gene products like DJ-1 protein are being explored as potential biomarkers reflecting cellular oxidative stress and disease activity[37]. Although no single genetic or molecular marker can currently fully predict PD, these biomarkers demonstrate promising applications in early diagnosis and disease severity evaluation.
While many of the genes discussed above were initially identified through familial forms of PD, their biological functions extend beyond monogenic inheritance and contribute to sporadic PD pathogenesis. Classical familial PD genes - including SNCA, LRRK2, PINK1, PRKN (Parkin), and DJ-1 - harbor highly penetrant mutations that directly cause Mendelian PD through mechanisms such as protein aggregation, mitochondrial dysfunction, and oxidative stress[35,42,48-58]. Among these, SNCA duplication or triplication and LRRK2 gain-of-function mutations (e.g., G2019S) represent the most common dominant causes, whereas PINK1, PRKN, and DJ-1 loss-of-function variants lead to autosomal-recessive, early-onset forms of PD[39,52,56,58]. Importantly, the molecular pathways governed by these genes - mitophagy, lysosomal degradation, and proteostasis - are also dysregulated in apparently sporadic cases, implying shared mechanistic foundations[59,60].
In contrast, several developmental and regulatory genes - such as NURR1, LMX1A/B, EN1, OTX2, PITX3, and WNT5A - do not typically carry high-penetrance mutations but influence disease susceptibility through transcriptional or epigenetic modulation of mDA neuron differentiation and maintenance[25,30,61-63]. Variants or reduced expression of these factors may lower the “developmental reserve” of DA neuron, predisposing individuals to later degeneration under environmental or metabolic stress. Additionally, heterozygous GBA variants occupy an intermediate position between familial and sporadic PD, functioning as strong risk factors that accelerate α-syn aggregation and cognitive decline[64]. These mechanisms, which are amplified in familial PD, also apply to sporadic PD development, though the latter is typically triggered by the cumulative effects of multiple minor genetic variants and environmental factors. In-depth research into monogenic PD mechanisms has provided targeted intervention strategies for sporadic PD, including developing biologics to clear α-syn, activating mitochondrial quality control, or enhancing lysosomal activity, with the goal of slowing or halting disease progression.
Environmental factors
Environmental exposure plays a significant role in the development of PD, typically influencing disease risk at the population level while interacting with the susceptibility genes at the individual level. Epidemiological studies have demonstrated that exposure to pesticides, heavy metals, and air pollutants, particularly fine particulate matter (PM2.5) and nitrogen dioxide (NO2), is significantly associated with an increased risk of PD[65-70].
Pesticides and chemical toxins
Prolonged exposure to certain pesticides, including organophosphorus insecticides, the herbicide paraquat, and pyrethroid insecticides, is believed to significantly increase the risk of PD. For instance, paraquat induces oxidative stress, while rotenone suppresses mitochondrial complex I, both of which can trigger PD-like pathological changes in animals, such as loss of DA neuron in the substantia nigra and aggregation of α-syn[65,67,69,71]. These toxins may cause mitochondrial dysfunction, neuroinflammation, and impaired protein degradation, with mechanisms highly similar to those in hereditary PD. Notably, some environmental toxins can directly target PD-related genes. Studies show that pesticide exposure and heavy metal exposure may cause cellular damage comparable to mutations in LRRK2 and PINK1, thereby exacerbating oxidative stress and protein buildup[65]. Thus, environmental toxins can drive PD-related pathological processes independently or through convergence on genetically vulnerable molecular pathways. Additionally, exposure to organic solvents, drinking well water, and repeated head trauma have been reported as environmental risk factors associated with PD[72-75].
Heavy metal exposure
Prolonged contact with heavy metals like manganese - particularly among miners and welders - may heighten risks of neurodegenerative disorders such as PD. Manganese poisoning (common in smelting workers) can induce movement symptoms resembling PD, known as “manganese-induced Parkinsonism”, indicating that elevated manganese levels damage substantia nigra neurons and disrupt dopamine transmission[76]. While pathological differences exist between manganese poisoning and typical PD (lacking Lewy bodies), this suggests that heavy metal overload triggers mDA neuron death through oxidative stress and mitochondrial damage[77,78]. Studies show that transition metals like iron and copper promote α-syn aggregation, generating toxic free radicals. Pathological examinations reveal increased iron ion accumulation in substantia nigra of PD patients, which may exacerbate oxygen free radicals produced by the Fenton reaction, causing chronic neuronal damage[79]. Additionally, industrial air pollution containing fine particulate matter (PM2.5) with multiple metal components has been linked to higher PD incidence in heavily trafficked areas. Ultrafine particles in air pollutants may enter the bloodstream through the lungs, cross the blood-brain barrier, and activate microglia cells, accelerating neurodegeneration through systemic inflammation[70,80,81].
Gut microbiota and infection
Recent studies indicate that gut microbiota imbalance may be a critical environmental factor in PD. PD patients exhibit significantly reduced levels of short-chain fatty acids (SCFAs) produced by beneficial gut bacteria[82,83]. SCFAs (e.g., butyric acid) exert neuroprotective effects by suppressing inflammation and maintaining intestinal epithelial integrity. Their deficiency leads to intestinal immune activation, increased mucosal permeability, and distal neuropathic inflammation. Concurrently, harmful bacterial products like Gram-negative lipopolysaccharide (LPS) accumulate, triggering central nervous system inflammation via vagus nerve and blood pathways. Clinical observations reveal correlations between Helicobacter pylori infection and PD symptom fluctuations, with eradication therapy enhancing levodopa efficacy - a clear example of gut microbiota influencing drug metabolism[84-86]. Regarding viral infections, some researchers propose that certain neurotropic viruses may induce latent encephalitis before PD onset, exacerbating neuronal vulnerability. For instance, post-influenza encephalopathy patients often develop Parkinsonian symptoms years after recovery, suggesting infection-induced immune damage as a potential trigger[86]. However, specific pathogens remain undetermined. Overall, gut microbiota dysbiosis, chronic infections, and persistent intestinal inflammation interact through the “gut-brain axis” to contribute to PD development.
Lifestyle and other factors
Multiple observational studies have found that current smoking and higher coffee/caffeine intake are associated with a lower risk of PD[87-90]. While the exact mechanisms remain unclear, research suggests this may be related to nicotine’s neuroprotective effects in tobacco or caffeine’s antagonistic action on adenosine A2A receptors. Conversely, metabolic syndrome factors like diabetes and obesity have been shown to increase PD incidence[91,92]. Physical activity serves as a protective factor, with lower PD rates observed in regular exercisers, indicating that healthy exercise habits help maintain healthy dopamine circuits in the brain[93,94]. Additionally, vitamin D deficiency, rural well water use (potentially containing pesticides), and high dairy intake have been linked to PD risk in some studies[7]. However, overall, environmental and lifestyle factors primarily contribute to PD development through cumulative, long-term effects, which are often modulated by underlying genetic susceptibility. As described in the “Multiple Hit Hypothesis”, age-related neuronal fragility (see Section “Aging and Systemic Vulnerability”) combined with chronic exposure to environmental toxins ultimately leads to clinical PD.
Taken together, environmental and lifestyle factors should be viewed as the primary external pressures acting on vulnerable biological systems, rather than as isolated triggers or mere phenocopies of genetic defects. At the population level, cumulative exposures shape the overall disease burden and risk. Yet exposure alone rarely determines clinical outcome: individuals with broadly similar environmental histories often follow markedly different trajectories. This heterogeneity arises from differences in intrinsic resilience - shaped by genetic architecture, developmental reserve, and progressively eroded by aging. Thus, environmental pressures and intrinsic susceptibility interact to determine whether, and when, pathological thresholds are crossed, bridging the environmental factors discussed here with the genetic and aging-related mechanisms in Section “Genetic Factors” and “Aging and Systemic Vulnerability”.
Aging and systemic vulnerability
Ageing is the primary risk factor for PD. The prevalence of PD rises exponentially with age: approximately 1% in individuals over 60, and reaching 3% to 5% in those over 85. Population ageing is recognized as the driving force behind the rapid increase in PD cases this century[1,95]. Aging increases the vulnerability of the central nervous system through multiple changes, vividly described as the background amplifier of PD[96].
Mitochondrial dysfunction
Advancing age is accompanied by progressive declines in mitochondrial number, ultrastructural integrity, and respiratory‐chain activity, resulting in insufficient ATP generation and excess reactive oxygen species. In DA neurons of the substantia nigra pars compacta - cells with vast axonal arbors, pacemaking activity, and large Ca2+ fluxes - this bioenergetic frailty is magnified. Post-mortem PD tissue shows complex I deficits and age-related mtDNA deletions that accumulate in substantia nigra neurons, consistent with a lifelong trajectory toward bioenergetic failure and oxidative dopamine quinone formation[97-99]. These processes heighten intrinsic DA neuron toxicity and help explain the age dependency of PD onset.
Loss of proteostasis
Aging compromises protein quality control by diminishing chaperone capacity, attenuating ubiquitin–proteasome performance, and weakening autophagy–lysosome flux. As a result, damaged proteins and organelles accumulate, including α-syn species prone to misfolding and aggregation[100]. Proteostasis decline is a core molecular hallmark of aging and neurodegeneration; in PD, reduced proteasome biogenesis (e.g., via age-related NFE2L1/Nrf1 dysregulation) and impaired autophagy synergize to promote Lewy pathology and neuron loss[101-103]. The aged brain therefore tolerates misfolded protein stress poorly, converting modest burdens into toxic, self-propagating aggregates.
Inflammaging and immune dysregulation
Aging reshapes systemic and brain immunity toward a chronic, low-grade pro-inflammatory state (“inflammaging”), with persistent elevations of interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor α (TNF-α). Microglia become “primed”, responding disproportionately to α-syn and neuronal debris, while immunosenescence reduces lymphocyte diversity and self-tolerance, enabling autoreactive T cells to breach tolerance checkpoints[100]. This milieu establishes a feed-forward loop in which misfolded α-syn activates innate cells that, in turn, amplify oxidative and cytokine stress, sustaining chronic neuroinflammation typical of aged and PD brains[104,105].
Vascular and blood–brain barrier dysfunction
Age-related cerebrovascular remodeling - loss of pericytes and astrocytic endfeet coverage, basement-membrane thickening, and reduced tight-junction proteins - weakens blood–brain barrier (BBB) selectivity and increases permeability[106]. In vivo imaging demonstrates measurable BBB leak in cognitively normal older adults; in PD, vascular fragility coincides with α-syn pathology and heightened neuroinflammation, facilitating entry of peripheral cytokines, immune cells, and xenobiotics into vulnerable midbrain circuits[107,108]. This vascular gateway effect likely amplifies DA neuron injury from systemic inflammatory or toxic exposures in late life.
Reduced DA neuronal reserve and sex differences
Humans are born with a finite pool of nigral DA neurons, of which ~30%-50% are lost by age 80 even without PD[109]. Classic stereology and modern reconstructions indicate a decades-long decline that reduces functional reserve; surviving neurons also show age-dependent alterations in pacemaker fidelity and calcium handling, further curtailing dopamine output[110]. Individuals with a lower congenital “DA neuronal reserve” (shaped by developmental programs) cross the clinical threshold earlier as aging proceeds - consistent with a “brain-reserve” model of PD risk. Male sex confers ~1.5-fold higher prevalence than female sex; estrogen’s mitochondrial and antioxidant effects, and its modulation of DA signaling, may partly underlie this difference and the rise in risk after menopause[111].
In this section, we have outlined PD’s core pathogenic mechanisms, which span a continuum from genetic architecture to environmental triggers, amplified by aging. Despite diverse triggers, these inputs converge on a limited set of vulnerable cellular programs in DA circuits. Developmental determinants shape a finite DA neuronal reserve, helping explain inter-individual susceptibility and the timing of clinical conversion. Crucially, these mechanisms are not confined to the brain. They are modulated by signals and stressors originating in peripheral organ that communicate via cytokines, lipids, metabolites, extracellular vesicles, and autonomic or vagal pathways. Thus, PD pathogenesis is better framed as a systems problem, where brain-intrinsic vulnerabilities intersect with cross-tissue regulatory networks to produce prodromal features and comorbid trajectories. And these will be elaborated in detail in the next section.
Environmental pressure and genetically constrained resilience
Sporadic PD arises from the interplay of sustained environmental pressure, genetically constrained resilience, and age-related erosion of compensatory capacity. As detailed in Section “Genetic Factors”, “Environmental Factors” and “Aging and Systemic Vulnerability”, genetic architecture sets the baseline robustness of molecular and cellular networks, environmental exposures impose chronic, cumulative stress, and aging progressively diminishes the capacity to buffer these insults. Disease onset thus reflects the point at which environmental load exceeds the tolerance limits defined by genetic and developmental resilience.
At the population level, environmental and lifestyle factors act as the primary external risk factors of PD risk. The exposome concept underscores that lifelong, cumulative non-genetic influences - chemical exposures, microbial shifts, metabolic stress, infections, and behavioral patterns - shape overall disease burden more than isolated events[112]. Epidemiological evidence consistently links PD to pesticides, air pollution, and gut-related factors, reinforcing the role of environmental pressure in initiating stress across peripheral and central systems.
Yet exposure alone cannot account for the striking inter-individual heterogeneity in sporadic PD. Individuals with comparable environmental histories often exhibit divergent outcomes, from lifelong resilience to early neurodegeneration. This variability stems from differences in intrinsic resilience - shaped by genetic architecture, developmental reserve, and eroded over time by aging. Population studies integrating polygenic risk scores (PRS) with exposure data confirm that genetic susceptibility modulates environmental risk magnitude, with high PRS and high environmental burden conferring substantially greater risk than either factor alone[113,114].
Aging serves as a critical temporal modifier, progressively narrowing resilience windows through declines in mitochondrial function, proteostasis, immune regulation, and vascular integrity. In this view, aging does not initiate PD but lowers the threshold at which cumulative environmental pressure destabilizes vulnerable networks.
Epigenetic mechanisms provide a key interface, enabling environmental exposures to induce durable changes in DNA methylation, histone modification, and non-coding RNA regulation - thereby encoding exposure history into gene expression programs without altering DNA sequence[115,116]. Genetic background and aging further shape this epigenetic responsiveness, helping explain divergent outcomes from similar exposures.
Collectively, environmental pressure supplies the primary external load, genetic architecture defines tolerance thresholds, and aging erodes these thresholds. This dynamic framework accounts for disease heterogeneity, incomplete penetrance, and multisystem involvement in sporadic PD, while laying the groundwork for the network analyses in subsequent sections.
Immune dysregulation and autoimmunity
Immune dysregulation, particularly chronic inflammation, markedly exacerbates neurodegeneration in PD. From the adaptive immunity perspective, genome-wide association studies (GWAS) have consistently linked variants within the major histocompatibility complex (MHC) class II region - including HLA-DQB1 and HLA-DRB5 - to PD susceptibility, implicating antigen presentation and T-cell–mediated immune mechanisms in disease risk[117].
α-Syn spreads in a prion-like manner through neuron-to-neuron transmission, with microglia playing a key facilitating role. Consistent with these genetic associations, α-syn-specific T-cell reactivity has been detected in the peripheral blood of individuals with preclinical and early PD, supporting a role for autoreactive adaptive immune responses in the disease process[118,119].
In parallel, innate immune activation within the central nervous system plays a pivotal role in PD pathogenesis. Extracellular α-syn aggregates are efficiently sensed by microglia through pattern-recognition receptors, particularly TLR2 and TLR4, triggering robust microglial activation and inflammatory signaling[120-123]. While microglia internalize α-syn species, this process simultaneously promotes neuroinflammatory responses that contribute to DA neuronal vulnerability. Downstream of Toll-like receptor (TLR) signaling, α-syn-induced activation of the NLRP3 inflammasome leads to the maturation and release of pro-inflammatory cytokines such as IL-1β, thereby amplifying neuroinflammation and DA neurodegeneration[124,125]. As highlighted by Wang et al. (2015), sustained neuroinflammation and microglial activation constitute a central pathogenic mechanism in PD, providing a permissive immune environment in which α-syn–driven pathology can be amplified[126]. Collectively, these findings support a self-reinforcing immune–α-syn axis in PD, in which adaptive immune susceptibility, innate immune activation, and microglia-mediated neuroinflammation converge to drive disease progression - a framework that highlights autoimmune-like features in sporadic PD.
To weave together the roles of genetics, environment, and aging in sporadic PD, we suggest classifying cases into three subtypes based on dominant drivers. First, environment-driven PD arises when intense exposures - like heavy pesticides or pollution - overpower even resilient genetic networks, leading to disease in people who might otherwise stay healthy[65,69,127]; this echoes Ascherio et al.’s (2006) emphasis on environmental dominance[128]. Second, gene network-driven PD - our review’s core focus - occurs when vulnerable conserved modules (e.g., SNCA, PINK1, PRKN) make individuals susceptible even to everyday or mild stressors. Third, mixed-driver PD involves two factors exerting roughly equal influence, such as genetics paired with environment or aging. This model recognizes the limits of our gene-focused lens for purely exposure-based cases, yet stresses genetics as the primary internal force in most sporadic PD, shaped by external elements.
MULTISYSTEM COMORBIDITY MECHANISMS AT THE MOLECULAR AND GENETIC NETWORK LEVEL
PD has traditionally been recognized as a movement disorder primarily affecting the central nervous system[10]. However, growing evidences indicate that sporadic PD is a multisystem disorder, with molecular pathological changes involving both central and peripheral systems[129,130]. Common non-motor symptoms in PD patients (such as reduced sense of smell, constipation, depression, and sleep behavior disorders) serve as clear evidence[3,131,132]. These symptoms suggest that PD pathology extends beyond the substantia nigra-striatum pathway, involving sensory, autonomic, digestive, and immune systems[3,130,133]. In other words, PD susceptibility genes and molecular networks overlap with certain sensory disorders, gastrointestinal diseases, metabolic disturbances, and autoimmune diseases[119,134,135]. Since PD pathology involves multiple interconnected systems and the molecular mechanisms behind are unveiled, we performed a cross-species integrative analysis to elucidate the molecular network underlying comorbidity across the midbrain-sensory-intestinal axis. Specifically, both developmental and adult transcriptomic datasets from human and mouse sources were incorporated, focusing on four PD-relevant regions: mDA neurons[136,137], OB[138-140], DRG[141,142], and the ENS[143-145]. All data were sourced from public databases, with genes from single-cell sequencing datasets required to have a detection rate ≥ 5% and an average CPM ≥ 1, genes from spatial transcriptomics datasets filtered using log10(M + 1) > 0.3 to exclude low-level noise, and differentially expressed genes (DEGs) from human bulk RNA-seq defined using a threshold of P < 0.05.Building on this concept, we systematically map cross-tissue transcriptomic and network connections between PD and its major comorbid systems in this chapter. While this cross-species transcriptomic approach offers a powerful systems-level perspective on conserved molecular architectures, we acknowledge that it remains inherently gene-centric. Rather than being limited to evolutionarily conserved programs, our approach identifies both conserved and human-specific vulnerability genes, which together assemble a coherent vulnerability framework. Thus, the resulting networks are best interpreted as scaffolds that organize shared and human-specific susceptibility pathways, instead of direct causal drivers of sporadic PD.
A conserved genetic network links PD and olfactory dysfunction
The sensory system in PD becomes affected earlier than typical motor symptoms, with olfactory dysfunction being particularly prominent. Epidemiological and longitudinal studies show that 70%-90% of patients develop hyposmia or anosmia, often 4-10 years before motor onset, supporting an “olfactory-first” or bottom-up subtype in at least a subset of cases[129,130,146-148]. We speculated a core genetic network are shared between mDA neurons and OB neurons. To validate this hypothesis, we analyzed single-cell RNA sequencing (scRNA-seq)-derived DEGs through a comparison between PD and control mDA neurons[136], and bulk RNA-seq DEGs in OB between PD patients and controls[140]. We identified that 712 DEGs are shared in PD mDA and OB tissues. These genes showed enrichment in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways related to PD, Non-alcoholic fatty liver disease, Prion disease, Alzheimer disease, Oxidative phosphorylation and Huntington disease [Figure 1A]. By integrating human developmental mDA neuron scRNA-seq data[137] (no human developmental OB transcriptome data being available), we found that 528 of the 712 genes are expressed from development through adulthood in mDA neurons. Among these, the top enriched KEGG pathway remained the PD pathway [Figure 1A]. These results suggest the existence of a core genetic network that may drive the co-occurrence of PD and olfactory dysfunction along aging. Since mouse models are commonly used to investigate PD mechanism, we also analyzed the transcriptome data from mouse models[137-139]. We identified 4,468 and 8,570 genes shared between mDA and OB neurons in adult and developmental-stage mouse, respectively. Similar to findings in human, these genes were enriched in KEGG pathways such as Ribosome, PD, Huntington disease and Spliceosome, among others [Figure 1B]. Among them, 3,918 genes are co-expressed in mouse mDA and OB throughout development and adulthood, and are also enriched in KEGG pathways such as Ribosome, PD and Huntington disease [Figure 1B]. This recurrent enrichment across independent datasets and stages indicates that mouse mDA and OB neurons share a PD-related molecular program, suggesting that olfactory structures are embedded within a common vulnerability network underlying progressive nigrostriatal degeneration.
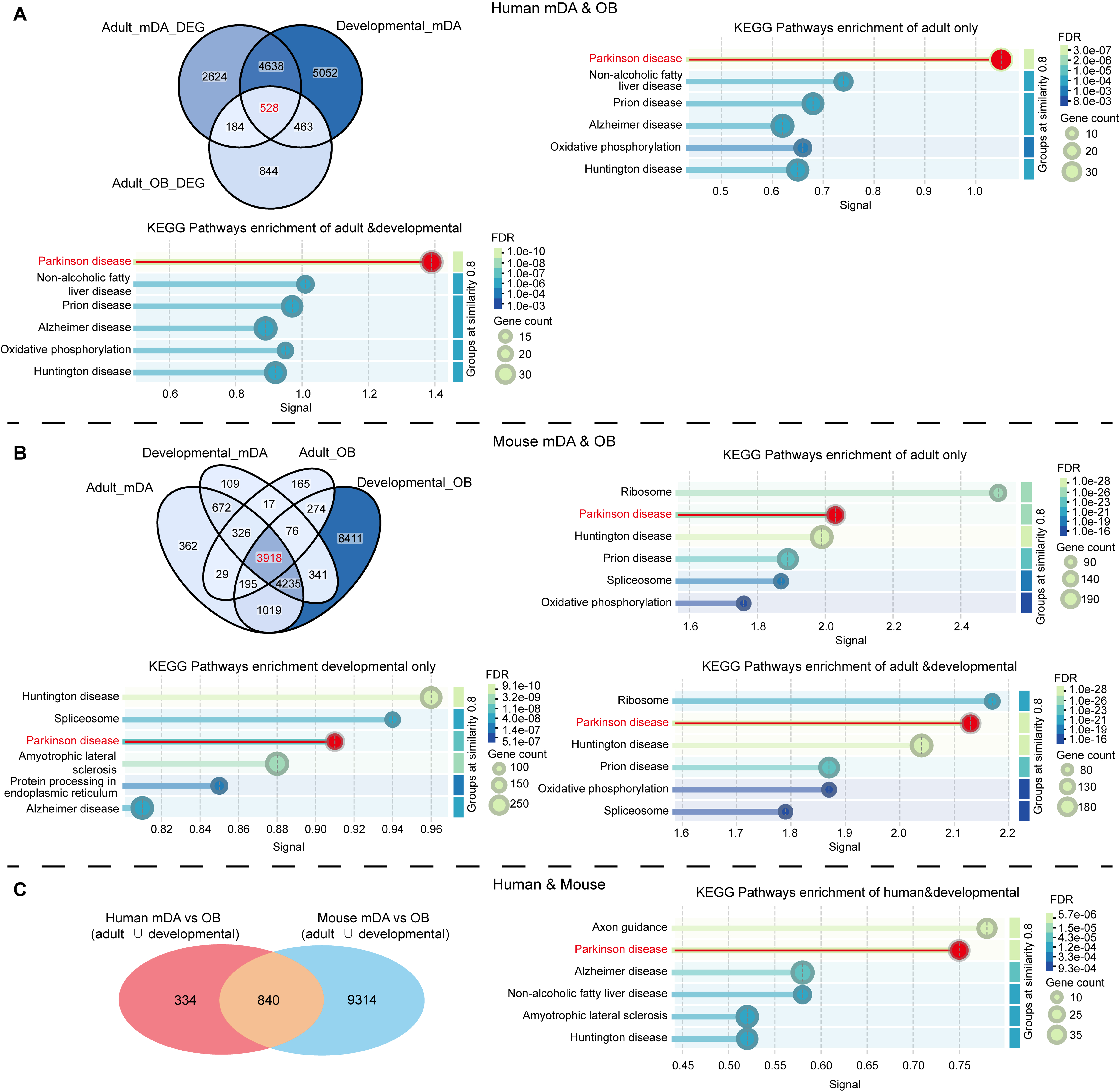
Figure 1. Integrated analysis of shared molecular programs between mDA neurons and the OB. (A) Venn diagram illustrates the overlap of gene sets derived from adult human DEG from mDA neurons (PD vs. control), developing human mDA neurons, and DEGs from OB samples (PD vs. control). KEGG pathway enrichment was conducted separately for genes shared only in the adult stage, only in the developmental stage, and those shared across both stages; (B) Venn diagram illustrates the overlap of gene sets derived from adult mouse mDA neurons, developing mouse mDA neurons, adult mouse OB and developing mouse OB. KEGG pathway enrichment was conducted separately for genes shared only in the adult stage, only in the developmental stage, and those shared across both stages; (C) Venn diagram illustrates the overlap of gene sets derived from human mDA vs. OB and mouse mDA vs. OB. KEGG pathway enrichment was also conducted for common gene set. The top6 enriched pathways are visualized based on signal strength, gene count, and FDR. Human adult OB transcriptomic data with sufficient quality were not available and are therefore not included in this comparison. mDA: Midbrain dopaminergic; OB: olfactory bulb; DEG: differentially expressed gene; PD: Parkinson’s disease; KEGG: Kyoto Encyclopedia of Genes and Genomes; FDR: false discovery rate.
To further test whether this mDA–OB vulnerability module is evolutionarily conserved rather than dataset-specific, we integrated human and mouse gene sets derived from shared signatures. By intersecting human developmental and adult mDA- and OB-related gene sets (with no developmental OB data available in human) with corresponding mouse developmental/adult mDA and OB datasets, we found that a conserved cross-species module of 840 genes co-expressed in mDA and OB neurons in both species [Figure 1C]. KEGG enrichment analysis of this conserved gene set again revealed significant over-representation of the PD pathway [Figure 1C], indicating that the shared mDA–OB network constitutes a PD-relevant, evolutionarily conserved vulnerability scaffold. These findings strongly support for the use of mouse models as biologically meaningful surrogates for investigating human susceptibility to PD and olfactory dysfunction.
Within the conserved mDA-OB module, several well-established PD genes provide concrete anchors supporting the biological relevance of this shared network. SNCA is central: α-syn aggregation in the olfactory epithelium and OB, together with its capacity for prion-like propagation along olfactory tracts, provides a direct molecular substrate for prodromal hyposmia and for rostrocaudal spread of pathology[146,147]. UCHL1 is highly expressed in olfactory receptor neurons and their axons and is widely used as a pan-neuronal or sensory marker[129,149,150]. Impaired UCHL1 function compromises ubiquitin–proteasome–mediated protein turnover and can facilitate α-syn accumulation, reducing proteostatic reserve in olfactory neurons exposed to environmental toxins[151]. Dopamine D2 receptor (DRD2) is abundant in the OB DA interneurons and plays a role in adjusting synaptic input and odor discrimination. Modulation of DRD2 signaling within OB circuits can modify olfactory performance and has the potential to improve olfactory deficits in parkinsonian experimental models. This functionally links DA signaling to early sensory impairments associated with PD[152-154]. ATP13A2, a lysosomal P-type ATPase whose loss-of-function mutations cause early-onset parkinsonism[155,156], and whose deficiency induces lysosomal dysfunction and α-syn accumulation[157], is expected to be equally important for protein clearance in OB neurons; subclinical ATP12A2 insufficiency could thus contribute to early olfactory α-syn vulnerability. The convergence of these canonical PD genes within the cross-species mDA-OB overlap strongly argues that this 840-gene set captures a genuine, PD-relevant vulnerability architecture, rather than a generic developmental or sensory transcriptome. Environmental exposures interact with this conserved mDA–OB network, particularly through inhalational routes that preferentially engage olfactory structures. Airborne pollutants, including PM2.5 and NO2, as well as certain pesticides, have been associated with oxidative stress and mitochondrial dysfunction in olfactory and DA systems. Consistent with this, epidemiological studies by Ritz et al. (2016) demonstrated that long-term exposure to traffic-related air pollution is associated with increased PD risk, while mechanistic studies have suggested that such exposures may impair oxidative phosphorylation and related mitochondrial pathways, thereby amplifying vulnerability in the OB and connected networks[158]. These conserved vulnerability networks likely define intrinsic limits of resilience, which may be preferentially engaged - and thresholds lowered - by environmental pressures entering via the nasal interface, potentially contributing to the early and prominent olfactory involvement observed in sporadic PD.
Shared genetic architecture underlying PD and somatosensory deficits
Beyond olfaction, the somatosensory system is frequently affected in patients with PD, with pain representing one of the most prevalent non-motor symptoms. Systematic reviews estimate that approximately 40%-85% of PD patients experience pain, most commonly musculoskeletal, dystonic, radicular or central in origin[159-161]. Neuropathological evidence shows that Lewy-related α-syn pathology is not confined to the brain: Lewy bodies and Lewy neurites have been reported in DRG, spinal dorsal horn, and other peripheral sensory structures in PD and Lewy body spectrum disease. Population-based mapping studies confirm the presence of peripheral and spinal Lewy pathology in defined disease subtypes[162-164].
Inspired by the conserved molecular architecture common to mDA and OB neurons, we subsequently inquired whether similar genetic vulnerability modules extend to peripheral sensory neurons, particularly those in DRG. To explore this, we intersected PD-related genes from mDA signatures with transcriptomic profiles of DRG neurons from human and mouse datasets, encompassing developmental and adult stages. Utilizing gene sets derived from developing and adult mDA neurons, including PD-relevant DA modules[136,137], we compared their expression with curated DRG datasets enriched for sensory neurons[141,165]. Cross-tissue intersections analysis was then performed to identify conserved and disease-relevant molecular modules. We identified 3,651 shared genes between PD mDA DEGs and adult DRG neurons. These genes showed enrichment in KEGG pathways including Ribosome, PD, Oxidative phosphorylation, Prion disease, Huntington disease and amyotrophic lateral sclerosis (ALS) [Figure 2A]. During development, 8,595 genes are co-expressed in both human mDA and DRG neurons. Among these, the PD pathway was significantly enriched, ranking among the top five KEGG pathways. By integrating human developmental and adult data, we identified a robust shared gene set of 3,089 genes co-expressed in both tissues and significantly associated with PD-related alterations, with the PD pathway ranking among the top two enriched KEGG pathways [Figure 2A]. A similar pattern was observed in mouse model[137,141], where 3,936 genes were shared between mDA and DRG from development throughout adulthood, and the PD pathway ranked among the top three significantly enriched KEGG pathways [Figure 2B].
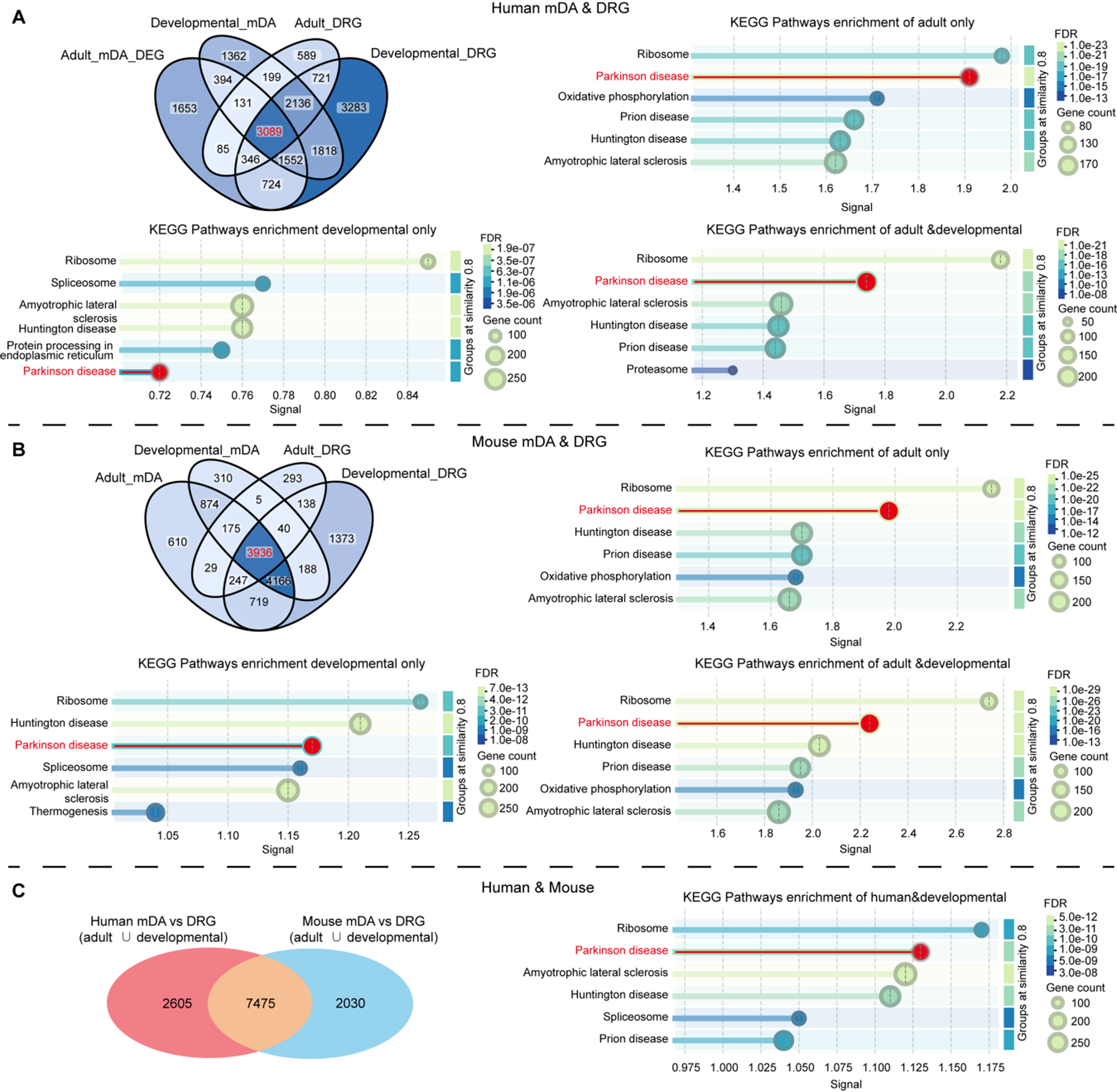
Figure 2. Overlapping gene networks between mDA neurons and dorsal root ganglion sensory neurons (mDA and DRG). (A) Venn diagram illustrates the overlap of gene sets derived from adult human DEG from mDA neurons (PD vs. control), developing human mDA neurons, adult human DRG and developing human DRG. KEGG pathway enrichment was conducted separately for genes shared only in the adult stage, only in the developmental stage, and those shared across both stages; (B) Venn diagram illustrates the overlap of gene sets derived from adult mouse mDA neurons, developing mouse mDA neurons, adult mouse DRG and developing mouse DRG. KEGG pathway enrichment was conducted separately for genes shared only in the adult stage, only in the developmental stage, and those shared across both stages; (C) Venn diagram illustrates the overlap of gene sets derived from human mDA vs. DRG and mouse mDA vs. DRG. KEGG pathway enrichment was also conducted for common gene set. The top6 enriched pathways are visualized based on signal strength, gene count, and FDR. mDA: Midbrain dopaminergic; DRG: dorsal root ganglia; DEG: differentially expressed gene; PD: Parkinson’s disease; KEGG: Kyoto Encyclopedia of Genes and Genomes; FDR: false discovery rate.
To evaluate whether the mDA–DRG vulnerability module represents a conserved biological program rather than dataset-specific artifacts, we performed a cross-species integration of the overlapping signatures. Human developmental and adult mDA- and DRG-associated gene sets were systematically compared with matched mouse developmental and adult mDA/DRG transcriptomes. This analysis revealed a reproducible conserved module of 7,475 genes consistently co-expressed in both mDA and DRG neurons across species [Figure 2C]. KEGG pathway analysis of this conserved subset again showed significant enrichment for the PD pathway [Figure 2C], along with Ribosome, ALS, Huntington disease, Spliceosome and Prion disease, confirming that the mDA–DRG overlap captures a PD-relevant, evolutionarily conserved vulnerability network. Together, these findings support the use of mouse midbrain and DRG models as mechanistically relevant systems for investigating shared molecular mechanisms underlying nigrostriatal degeneration and somatosensory deficits in PD.
Within the conserved mDA-DRG module, several well-established PD genes offer concrete anchors that support the biological relevance of this shared network. SCN9A, FAAH and GCH1 define a peripheral “sensory gain” module within DRG neurons that interfaces with central DA vulnerability in PD. Nav1.7 encoded by SCN9A is a key determinant of nociceptor excitability, and PD cohorts show SCN9A variants enriched in patients with refractory or severe pain, supporting a genetic lowering of pain threshold in the parkinsonian state[166,167]. Loss-of-function or hypomorphic variation in FAAH, a major endocannabinoid-degrading enzyme, can prolong endogenous analgesic signaling and conversely, risk alleles have been associated with heightened pain sensitivity, including in PD, indicating that endocannabinoid tone shapes individual susceptibility to PD-related pain[168,169]. GCH1, via control of tetrahydrobiopterin (BH4) and thus dopamine and nociceptive monoamine synthesis, acts as both a PD risk/modifier gene and a pain gene, with variants influencing PD risk, nigrostriatal integrity and inflammatory–nociceptive signaling, positioning BH4 metabolism at the intersection of mDA degeneration and sensory hyperexcitability[170-172]. In parallel, pathogenic mutations in core monogenic PD genes, such as SNCA, PINK1, PARK7, ATP13A2, VPS35, DNAJC6, FBXO7, SYNJ1, RAB39B, CHCHD2, and PLA2G6, converge on mitochondrial quality control, lysosomal-autophagic flux, and synaptic vesicle/endocytic pathways in mDA neurons[173-176]. These mutations converge on pathways governing mitochondrial quality control, lysosomal–autophagic function, and synaptic vesicle/endocytic trafficking, and are well-established contributors to nigrostriatal vulnerability in PD. When mapped together with peripheral sensory channel and neuromodulator genes such as SCN9A, FAAH, and GCH1, they delineate a partially shared gene network between PD-linked molecular pathology and DRG-based nociceptive regulation. This overlapping mDA-DRG 7475-gene module suggests a genetic interface between PD biology and pain/somatosensory pathways, rather than demonstrates a simple causal mechanism. For example, epidemiological and experimental studies have shown that exposure to pesticides and other environmental toxicants is associated with increased PD risk, with oxidative stress and mitochondrial dysfunction, overlapping with mitochondrial quality-control pathways implicated in PD, such as those involving PINK1[65]. In parallel, airborne pollutants have been shown to elicit inflammatory responses in exposed peripheral tissues, including sensory systems, by directly activating peripheral sensory neurons and associated inflammatory signaling, thereby potentially modulating DRG-mediated vulnerability relevant to PD[177]. These shared molecular architectures likely constrain neuronal resilience under sustained stress, providing a plausible mechanistic substrate through which environmental load contributes to somatosensory dysfunction - such as heightened pain sensitivity - and overall vulnerability in sporadic PD.
A conserved genetic network bridging central and peripheral neuronal vulnerability
The ENS embedded in the gastrointestinal tract is increasingly recognized as an early site of PD pathology. We hypothesize that the ENS shares a developmental and molecular homology with central DA structures, rendering it susceptible to parallel pathological processes under parkinsonian conditions. To test this hypothesis, we integrated and compared recent high-resolution transcriptomic datasets from the adult or developmental human enteric and mDA neurons[136,137,143,145], performing overlap analysis to identify conserved gene modules that may underlie their shared vulnerability in PD. We identified 736 DEGs that are shared in PD mDA and enteric neurons. These genes showed enrichment in KEGG pathways related to Ribosome, PD, Oxidative phosphorylation, Non-alcoholic fatty liver disease, Thermogenesis and Cardiac muscle contraction [Figure 3A]. During development, 1,594 genes are co-expressed in both human mDA and enteric neurons. Among these, the PD pathway was significantly enriched, ranking among the top two KEGG pathways. By integrating human developmental and adult data, we identified 153 genes co-expressed in both tissues and significantly associated with PD-related alterations, and the PD pathway was significantly enriched, ranking among the top two KEGG pathways [Figure 3A]. A similar pattern was observed in mouse model[145,178], where 8,873 and 8,817 genes were shared between mDA and enteric neurons in adult and developmental stages, respectively, and the PD pathway ranked among the top four significantly enriched KEGG pathways [Figure 3B]. Among them, 7,778 genes are co-expressed from development throughout adulthood and enriched in PD pathway [Figure 3B].
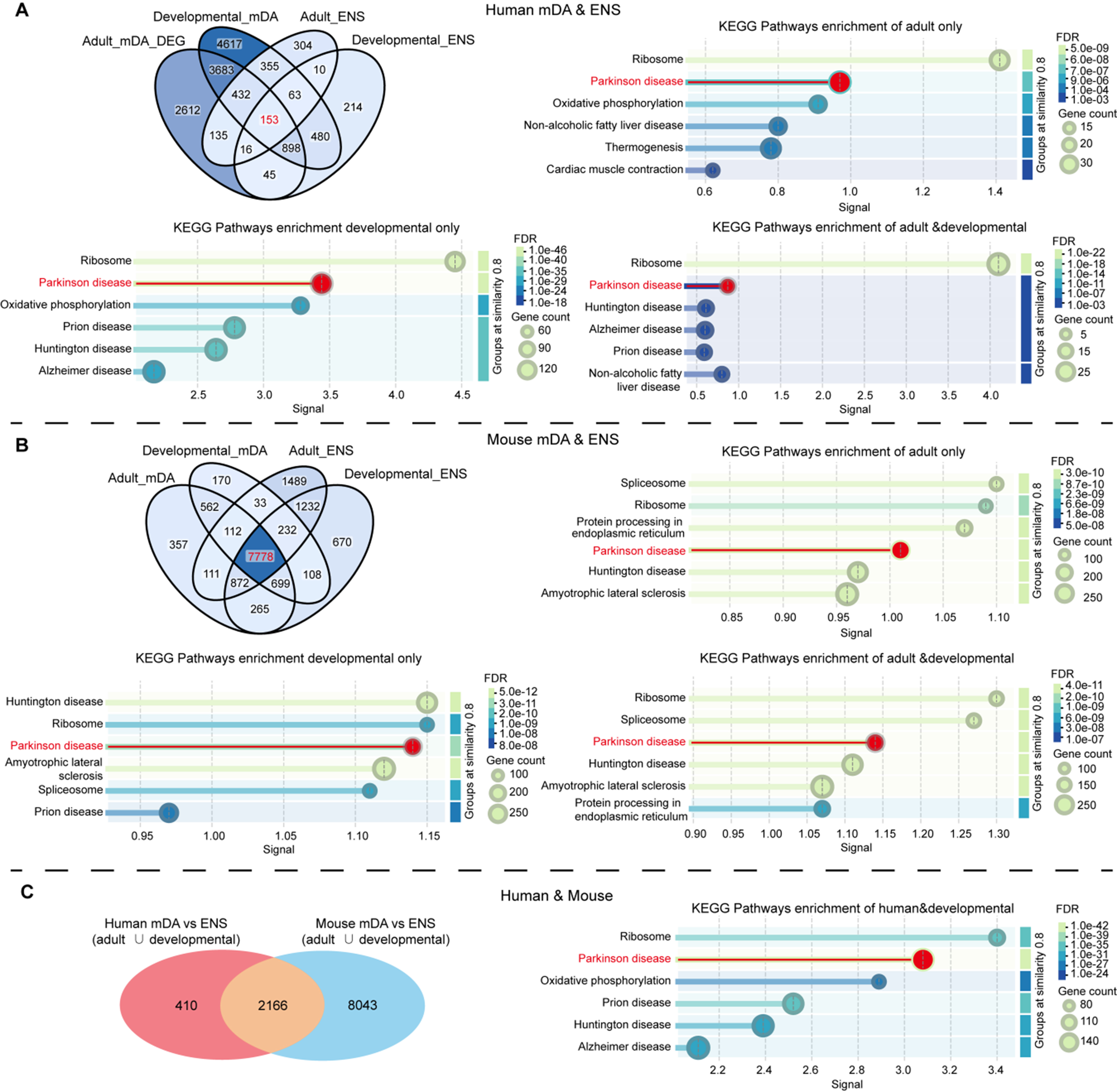
Figure 3. Shared transcriptional modules between mDA neurons and the ENS. (A) Venn diagram illustrates the overlap of gene sets derived from adult human DEG from mDA neurons (PD vs. control), developing human mDA neurons, adult human ENS and developing human ENS. KEGG pathway enrichment was conducted separately for genes shared only in the adult stage, only in the developmental stage, and those shared across both stages; (B) Venn diagram illustrates the overlap of gene sets derived from adult mouse mDA neurons, developing mouse mDA neurons, adult mouse ENS and developing mouse ENS. KEGG pathway enrichment was conducted separately for genes shared only in the adult stage, only in the developmental stage, and those shared across both stages; (C) Venn diagram illustrates the overlap of gene sets derived from human mDA vs. ENS and mouse mDA vs. ENS. KEGG pathway enrichment was also conducted for common gene set. The top6 enriched pathways are visualized based on signal strength, gene count, and FDR. mDA: Midbrain dopaminergic; ENS: enteric nervous system; DEG: differentially expressed gene; PD: Parkinson’s disease; KEGG: Kyoto Encyclopedia of Genes and Genomes; FDR: false discovery rate.
To determine whether the mDA–ENS vulnerability module represents an evolutionarily conserved architecture rather than a dataset-specific artifact, we next integrated shared gene signatures derived from both human and mouse. Specifically, we intersected gene sets co-expressed in human mDA and enteric neurons at developmental and/or adult with corresponding mouse developmental and/or adult mDA and enteric neurons gene sets. This cross-species analysis identified a robust conserved module of 2,166 genes jointly represented in mDA and enteric neurons across species [Figure 3C]. KEGG pathway analysis of this conserved set again revealed significant enrichment of the PD pathway [Figure 3C], indicating that the mDA–ENS network reflects not merely a generic developmental or peripheral neuronal program, but a PD-relevant, evolutionarily conserved vulnerability scaffold.
Within the conserved mDA-ENS module, several well-established PD genes offer concrete anchors that support the biological relevance of this shared network. SNCA sits at the core: α-syn pathology is repeatedly detected in the ENS and other peripheral autonomic sites in PD-findings that helped inspire “gut-first” hypotheses of disease spread[130,179,180]. PINK1-PRKN encode the mitophagy axis that safeguards neuronal and enteric mitochondrial quality. In the intestine, infection can trigger mitochondrial-antigen presentation and PD-like phenotypes in Pink1-/- mice[181], while human PD patients harbor T-cell responses to PINK1-derived mitochondrial peptides, linking this pathway directly to gut immune mechanisms relevant to PD[182]. LRRK2 mechanistically links PD genetics to mucosal immunity: it modulates myeloid responses, and its overexpression can exacerbate experimental colitis, supporting a role for LRRK2-driven immune signaling at the gut interface, with potential implications for gut-brain communication in PD[183]. PARK7 (DJ-1) buffers oxidative and inflammatory stress; loss of DJ-1 heightens gut mucosal inflammation and worsens colitis in vivo, consistent with a model in which impaired cytoprotection in the intestinal milieu can feed forward onto PD-relevant ENS vulnerability[184]. This overlapping mDA-ENS 2166-gene module suggests a genetic interface brain-gut axis in PD biology, rather than demonstrates a simple hypothesis.
Convergent modules along the sensory-gut-midbrain axis
To refine the gene sets most relevant to human PD pathology, we performed intersection analysis between mDA and each of the three peripheral tissues (OB, DRG, ENS). This approach identified two distinct groups: 265 genes co-expressed exclusively in humans and 244 genes shared between humans and mice [Figure 4A]. Each gene was manually classified into one of three categories - established PD relevance, potential PD relevance, or no current evidence of PD association. Based on these results, a comprehensive PPI network was constructed [Figure 4B and C], identifying several central “hub” regulators and potential regulatory clusters.
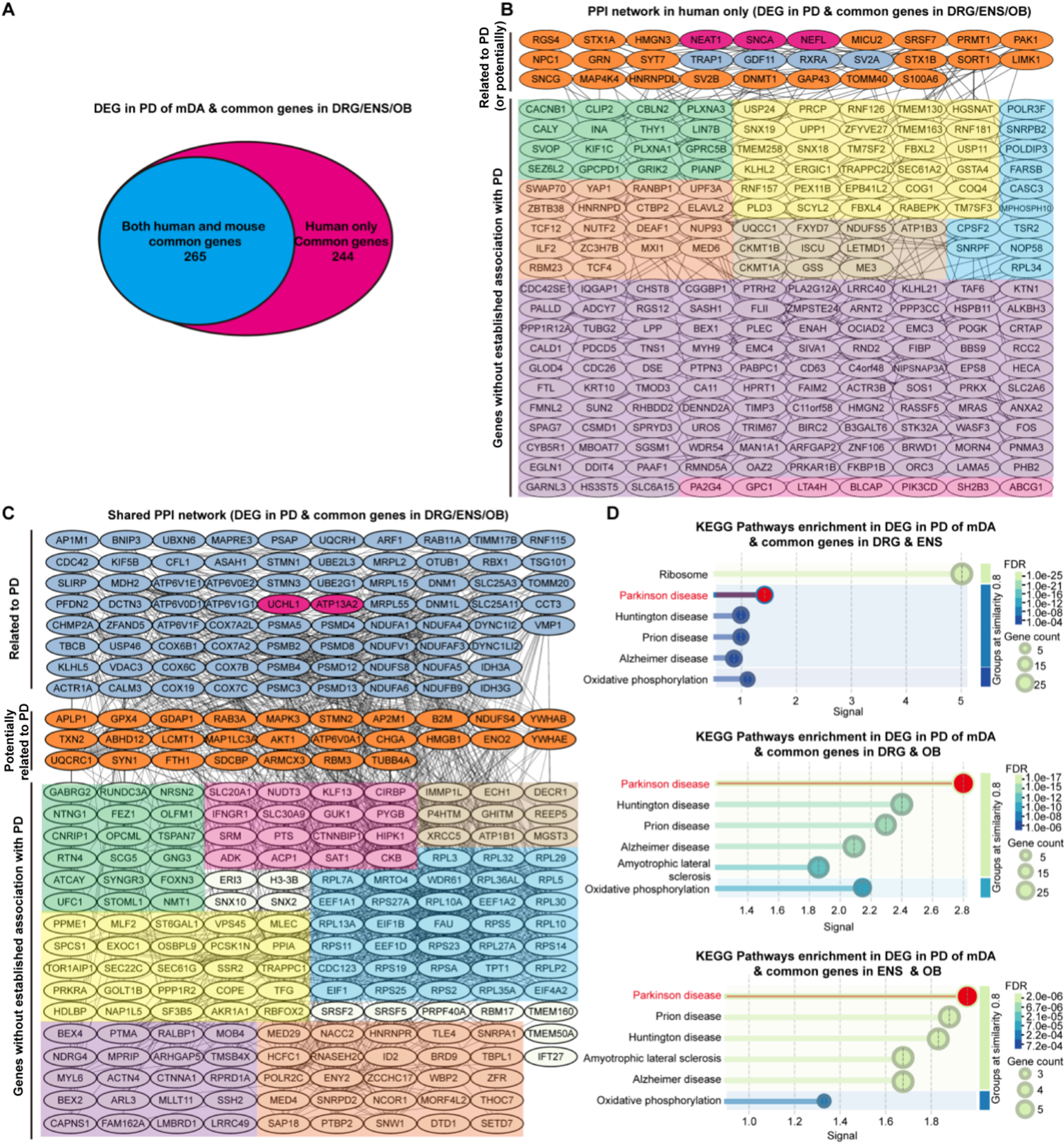
Figure 4. Cross-species comorbidity gene overlap and functional enrichment analysis. (A) Venn diagram showing the overlap between comorbidity-associated genes identified in humans only (244 genes) and those conserved across both human and mouse datasets (265 genes); (B) PPI network of comorbidity genes identified only in humans; (C) PPI network of comorbidity genes shared between human and mouse. Divided into three clusters: Related to PD, Potentially related to PD and Genes without established association with PD. Note: This classification is based on the literature and serves as a hypothesis-generating framework rather than a definitive annotation. “Genes without established association with PD” cluster are further grouped into seven functional submodules based on biological relevance: neural development and synapses (green), autophagy and protein clearance transport (yellow), mitochondrial function and oxidative stress (brown), inflammation and immune metabolism (pink), translation and ribosome/protein synthesis (blue), basic cellular functions and cytoskeleton (purple), and RNA processing/splicing/nuclear export and transcriptional regulation(orange); (D) KEGG pathway enrichment analysis of the conserved comorbidity genes expressed in both species. Related to PD: established PD relevance; Potentially related to PD: potential PD relevance; Genes without established association with PD. PPI: Protein–protein interaction; PD: Parkinson’s disease; KEGG: Kyoto Encyclopedia of Genes and Genomes; DEG: differentially expressed gene; mDA: midbrain dopaminergic; DRG: dorsal root ganglia; ENS: enteric nervous system; OB: olfactory bulb; FDR: false discovery rate.
Among these intersection sets, several genes are well-established PD-associated molecules, including SNCA, NEAT1, NEFL, UCHL1, and ATP13A2. SNCA, encoding α-syn, remains the most validated causative gene in familial and sporadic PD. Point mutations and multiplications of SNCA drive α-syn aggregation and inter-neuronal propagation, forming the molecular basis for the “prion-like” spreading mode[49,185]. NEAT1, a long noncoding RNA, regulates SNCA expression and neuroinflammatory responses. Elevated NEAT1 in PD patient brains and in vitro models enhances α-syn accumulation via PINK1- and NLRP3-dependent pathways, amplifying oxidative stress and inflammation[163,186,187]. NfL, encoded by NEFL, is a key structural component of axons; knockout and mutant mouse studies demonstrate that loss or disruption of NEFL induces axonal degeneration and altered neuronal integrity[188,189], while clinical cohorts consistently show elevated CSF and blood NfL in PD and other parkinsonian syndromes, with higher levels associated with more severe disease and faster progression[190-193], establishing NEFL/NfL as a robust marker of axonal damage and prognosis. Ubiquitin C-terminal hydrolase L1 (UCHL1), is a neuron-enriched deubiquitinase essential for maintaining the free ubiquitin pool and proteasomal function; rare PD-associated mutations (such as I93M) and functional studies implicate UCHL1 dysfunction in impaired protein clearance, α-syn aggregation, and axonal vulnerability, positioning it as a proteostasis-sensitive node within PD-relevant circuits[194-196]. ATP13A2 (PARK9) was discovered as the gene mutated in Kufor–Rakeb syndrome, an autosomal recessive juvenile-onset parkinsonism[155], and Atp13a2-deficient models develop lysosomal storage pathology, impaired autophagy, and sensorimotor deficits[157,197,198]. Cell and animal studies further demonstrate that ATP13A2 loss-of-function causes generalized lysosomal dysfunction and promotes α-syn accumulation and neurodegeneration[199,200], firmly linking this lysosomal P-type ATPase to PD pathogenesis.
Genes without established association with PD were further grouped into seven functional categories - neural development and synapses, autophagy and protein clearance transport, mitochondrial function and oxidative stress, inflammation and immune metabolism, translation and ribosome/protein synthesis, basic cellular functions and cytoskeleton, and RNA processing/splicing/nuclear export and transcriptional regulation - to serve as a reference resource for hypothesis-driven exploration [Figure 4B and C]. Within the “Genes without established association with PD” subgroup, several genes nevertheless suggest plausible PD-related mechanisms. In the human-only set, SYNGR3 encodes a synaptic vesicle membrane protein that interacts with the dopamine transporter (DAT) and shapes presynaptic dopamine handling; overexpression of Synaptogyrin-3 enhances striatal dopamine uptake and partially rescues DA dysfunction in LRRK2R1441G mutant mice, pointing to a potential role as a synaptic modifier of PD vulnerability[201]. RTN4 (Nogo-A) is an ER membrane protein that shapes tubular ER and axons. RTN4 levels in CSF are elevated in neurodegenerative diseases including PD, and Nogo-A-targeting strategies improve DA neuron survival in PD models, suggesting a link between ER/axon integrity and PD pathology[202]. Among human-mouse common genes, TMEM163 encodes a zinc-transporting membrane protein. GWAS and follow-up studies implicate TMEM163 as a PD risk locus and associate rare variants with early-onset PD, supporting a role for disturbed synaptic Zn2+ homeostasis in PD[203]. Finally, SNCG encodes γ-synuclein, a synuclein family member capable of aggregation. Genetic and pathological data indicate that SNCG variants and
Rather than proposing a definitive mechanistic model, this integrative cross-species analysis provides a structured reference framework for future studies. By mapping conserved and tissue-specific molecular overlaps across sensory, gut, midbrain, and peripheral systems, our findings highlight candidate genes and pathways that warrant targeted experimental validation. The inclusion of both well-established PD-related genes (e.g., SNCA, NEAT1, NEFL, UCHL1, and ATP13A2) and previously uncharacterized “Genes without established association with PD” genes offers a prioritized landscape of potential molecular nodes within this multisystem network [Figure 4]. Building on this framework, the subsequent sections of this chapter dissect major clinical and pathological domains, sensory dysfunction, gut–brain axis alterations, and systemic metabolic and immune changes, to assess how existing evidence aligns with and reinforces the cross-system modules identified above. Environmental factors influence this conserved mDA–ENS network, particularly through chronic metabolic, microbial, and inflammatory pressures that selectively challenge gut vulnerability. For instance, pesticides and heavy metals ingested via water or food impair autophagy and mitochondrial pathways in genes such as LRRK2 and PINK1-PRKN, as evidenced by Tanner et al. (2011) and Ascherio et al. (2006)[128]. Similarly, gut dysbiosis and infection-driven inflammation induce mitochondrial stress and disrupt mitophagy and proteostasis - processes that overlap with the PD-enriched mitochondrial and lysosomal modules identified here. These shared vulnerability networks position the gut as a key interface where environmental load intersects with genetically constrained resilience, potentially exacerbating gut-brain axis vulnerability and contributing to early non-motor manifestations in sporadic PD.
Systemic amplifiers: metabolic and immune dysfunction
In the pathogenesis of PD, systemic metabolic status and immune system function act as “amplifiers” that can accelerate or exacerbate central nervous system pathology. First, metabolic disorders and PD share a bidirectional relationship. Epidemiological studies indicate that metabolic syndromes such as type 2 diabetes increase both the risk and progression rate of PD. For instance, a 2021 meta-analysis showed that type 2 diabetes is associated with a 27% higher risk of developing PD[65,205,206]. Additionally, medications that improve insulin resistance, such as glucagon-like peptide-1 receptor agonists (GLP-1RA), not only help lower blood sugar but may also enhance DA neuron function, thereby reducing PD risk or improving symptoms; observational cohorts link GLP-1RA or DPP-4 inhibitor use to lower incident PD in diabetes[207-209]. A typical example is GLP-1RA: randomized trials in PD show signal for disease-modifying benefit with lixisenatide[210], while exenatide had earlier phase-2 benefit but a later phase-3 did not meet its endpoints, underscoring ongoing uncertainty[211,212]. Meanwhile, preclinical/early translational work supports GLP-1RA neuroprotection and anti-inflammatory effects[213]. Its mechanisms involve multiple aspects, including enhancing systemic and brain-level insulin signaling, mitochondrial support, and suppression of inflammatory pathways; AMPK activation with downstream mTOR inhibition promotes autophagy and mitophagy, a pathway engaged by agents like metformin in DA models[214]. The AMPK-mTOR pathway, serving as a core mechanism for cellular energy sensing, plays a pivotal role in PD pathology. Under conditions of nutritional excess or abnormal insulin signaling, excessive mTOR activation inhibits autophagy, fostering toxic protein accumulation; conversely, AMPK activation (e.g., by metformin) suppresses mTOR, promotes autophagy/mitochondrial renewal, and protects mDA neuron in vivo[214]. Parkinson’s patients also show characteristic brain glucose-metabolism patterns on FDG-PET (the PD-related metabolic network), which correlate with disease stage and can aid differential diagnosis[215]. Some researchers therefore frame a shared “metabolic-neurodegenerative” axis between diabetes and PD.
Secondly, immune dysregulation, particularly chronic inflammatory states, is considered to significantly exacerbate the neurodegenerative process in PD. Evidence of immune system activation is commonly observed in PD patients, including elevated levels of pro-inflammatory cytokines (such as TNF-α and IL-1β) in peripheral blood compared to healthy individuals, as well as reactive proliferation and phagocytic phenotype of microglia in the central nervous system. Genetic studies further validate immune involvement in PD: Large-scale GWAS have identified significant associations between genetic variants encoding Class II major histocompatibility complex (MHC II) molecules (e.g., HLA-DQB1, HLA-DRB5) and PD susceptibility[117]. This suggests that antigen-presenting and T-cell-mediated autoimmune processes may be implicated in PD. Recent research has revealed the presence of autoreactive T cells targeting neuroantigens such as α-syn in PD patients, with increased numbers of these cells in peripheral blood that may cross the blood-brain barrier to infiltrate the central nervous system and participate in aggressive attacks on DA neuron[216]. These adaptive immune abnormalities add a hidden dimension to PD as an autoimmune-like condition, leading some scholars to propose that autoimmune mechanisms may contribute to disease progression, rather than classifying PD as a classical chronic autoimmune disorder.
More specifically, the neuroinflammatory response observed in PD brains amplifies neuronal damage through a feedback loop. Aggregated α-syn itself serves as a potent stimulus for immune responses - studies have shown that α-syn oligomers can activate TLRs, particularly TLR2 and TLR4, on microglia surfaces[122,123]. Activation of these receptors triggers the downstream NF-κB signaling pathway, inducing extensive gene transcription that leads to the release of pro-inflammatory cytokines such as IL-1β and TNF-α[217]. These cytokines can directly impact DA neurons, contributing to mitochondrial dysfunction and apoptotic signaling. Simultaneously, they induce neighboring astrocytes to adopt an A1 harmful phenotype, resulting in loss of neurotrophic support and increased production of toxic substances. Additionally, NF-κB upregulates and activates the NLRP3 inflammasome, leading to the maturation and amplification of inflammatory cascades like IL-1β[218,219]. Importantly, this feedback loop primarily functions to amplify ongoing neuronal stress and proteinopathy, rather than acting as an independent initiating cascade. In summary, during PD neurodegeneration, immune activation and inflammatory factors form a positive feedback loop: initial neuronal damage (potentially caused by toxic proteins or environmental toxins) triggers glial immune responses[220]. The released inflammatory mediators further damage neurons, expose more autoantigens, and recruit additional immune cells, perpetuating this vicious cycle that progressively worsens the disease[124].
Recent studies indicate that PD may result from the interaction of multiple factors including genetics, environment, metabolism, and immunity. Abnormalities in the immune system, such as associations between specific immune genes and PD, suggest immune factors play a role in disease progression. Therefore, the amplifying effects of metabolic and immune factors on PD further emphasize the necessity of viewing PD as a multifactorial outcome. Some researchers propose adding a third “hit” - metabolic/immune abnormalities - as a modulatory layer to Braaks “double hit” hypothesis, forming the “triple hit” hypothesis to comprehensively explain PDs pathogenesis. In summary, while the pathological pathology of α-syn protein and the vulnerability of DA neuron are central to PD, their progression rate and severity are significantly influenced by systemic metabolic status and immune environment. These “amplifiers” not only exacerbate the pathological burden of PD but also represent critical factors that require focused consideration in future comprehensive prevention and treatment strategies.
Taken together, these findings position immune dysregulation and α-syn–associated inflammatory responses not as independent initiators of PD, but as potent systemic amplifiers that exacerbate neuronal vulnerability and accelerate disease progression in the context of genetic susceptibility and environmental pressure.
A systematic model of multisystem vulnerability in PD
Based on the evidence reviewed above regarding PD-associated olfactory dysfunction, somatosensory deficits, central–peripheral neuronal vulnerability, and the sensory–gut–midbrain axis, PD can be understood as a cross-system disorder shaped by sustained environmental pressures acting upon genetically constrained vulnerability networks across sensory, intestinal, and midbrain systems[221,222]. Rather than reflecting a single-site lesion or a unidirectional propagation process, the cross-system networks identified here delineate shared architectures of susceptibility that define where pathological stress is most likely to accumulate under sustained challenge. Peripheral sensory circuits, such as olfactory and somatosensory pathways, and the ENS may represent the earliest sites of pathological stress, where environmental or endogenous pressures impose cumulative stress that promotes protein misfolding, mitochondrial impairment, and chronic inflammation[223]. These local disturbances gradually converge on the mDA system through neural, immune, and metabolic routes, amplifying neurodegenerative cascades via systemic feedback loops[224]. Importantly, such pressures do not act as isolated triggers but interact with pre-existing network constraints that shape individual tolerance and adaptive capacity. Within this framework, mDA degeneration emerges not as the inevitable endpoint of peripheral pathology, but as part of a coupled, multisystem process in which vulnerable networks across peripheral and central domains undergo parallel destabilization. Neural, immune, and metabolic connectivity enables stress signals to integrate across organs, allowing local failures to amplify one another through feedback loops rather than simple anatomical spread.
Genetic architecture defines the structure and robustness of these vulnerability networks, while aging progressively erodes compensatory mechanisms and lowers activation thresholds across interconnected systems[225]. Age-related declines in mitochondrial bioenergetics, proteostasis, immune resolution, and vascular integrity narrow the margin between physiological stress and pathological failure, providing a mechanistic basis for the delayed onset, incomplete penetrance, and heterogeneous clinical trajectories characteristic of sporadic PD.
Critically, the interaction between genetic architecture and environmental exposure is likely multiplicative rather than additive[226]. Genetic networks establish baseline resilience and compensatory capacity across tissues, whereas cumulative environmental exposures - toxicants, metabolic stress, microbial perturbations, and inflammatory burden - determine the magnitude and duration of imposed stress. Gene–environment interactions (G×E) thus shape individual disease trajectories by modulating vulnerability thresholds rather than acting independently. This model also accounts for incomplete penetrance and heterogeneity in sporadic PD, where many genetically at-risk individuals remain disease-free in the absence of sufficient environmental load. Consequently, conserved genetic programs primarily delineate susceptibility architecture, while human-specific exposures dictate whether, when, and where this vulnerability manifests clinically.
In this environment-influenced, genetically constrained framework, multisystem involvement reflects coordinated loss of resilience across sensory, intestinal, metabolic, and immune domains, with systemic amplifiers intensifying the impact of cumulative environmental load. Rather than proposing a definitive mechanistic pathway, this integrative model serves as a conceptual reference for understanding prodromal features, identifying early biomarkers, and guiding therapeutic strategies that extend beyond DA replacement toward restoring molecular homeostasis across the sensory–gut–midbrain axis.
Building upon these insights, the next section focuses on current and emerging therapeutic strategies, tracing the evolution from symptomatic relief to disease modification and exploring how systemic mechanisms can inform multi-target, cross-organ interventions for PD.
CURRENT AND EMERGING THERAPEUTIC STRATEGIES
Building on the molecular insights from the sensory–gut–midbrain axis and the systemic regulatory networks described in Chapter 2, it becomes evident that PD cannot be effectively addressed through DA therapy alone. The cross-organ propagation of pathological signals, encompassing olfactory and somatosensory dysfunction, gut-brain communication, metabolic disruption, and immune dysregulation, calls for a paradigm shift in treatment strategies. Therapeutic development must therefore move beyond focal interventions to encompass multi-target, cross-system approaches capable of restoring disrupted molecular homeostasis across central and peripheral tissues. The following section explores this evolution - charting the progression from traditional symptomatic control to disease-modifying therapies - and evaluates emerging strategies that aim to intervene along diverse mechanistic axes of PD pathology.
Existing drugs and standard therapies
DA replacement
Levodopa remains the gold-standard symptomatic therapy for PD, administered with a peripheral decarboxylase inhibitor (carbidopa or benserazide) to increase central delivery[227,228]. It effectively improves bradykinesia, rigidity, and overall motor function across disease stages, markedly enhancing quality of life[229]. However, chronic use leads to motor fluctuations (“wearing-off”) and levodopa-induced dyskinesias, arising from pulsatile DA stimulation and progressive neuronal sensitization[230,231]. To minimize these complications, levodopa is started at the lowest effective dose and dosing schedules are carefully optimized[227]. Despite its limitations, levodopa has profoundly extended lifespan and functional independence for PD patients[228].
Dopamine agonists and enzyme inhibitors
Adjunct agents enhance or prolong DA signaling. Dopamine receptor agonists (e.g., pramipexole, ropinirole, rotigotine) directly stimulate striatal receptors, offering moderate benefit and delayed dyskinesia onset vs. initial levodopa in trials, though with trade-offs in motor improvement[230-232]. These agents carry higher risks of somnolence, hallucinations, and impulse-control disorders, particularly at higher doses and with chronic exposure[233]. MAO-B inhibitors (selegiline, rasagiline, safinamide) and COMT inhibitors (entacapone, opicapone) reduce dopamine breakdown and extend levodopa effect duration, decreasing “off” time in fluctuating disease[234]. These agents enable more stable DA tone and allow levodopa dose reduction, forming the backbone of individualized pharmacotherapy.
Surgical and infusion therapies
For advanced PD with refractory motor fluctuations or tremor, deep brain stimulation (DBS) of the subthalamic nucleus or globus pallidus interna provides durable improvement in motor symptoms and allows medication reduction[235-237]. Though surgical risks exist, DBS is safe and effective in expert centers and can maintain benefit for years. Lesioning procedures (thalamotomy, pallidotomy) and continuous DA infusions (levodopa/carbidopa intestinal gel or subcutaneous apomorphine) serve as alternatives when oral therapy is insufficient[236,238,239].
Management of non-motor symptoms
Non-motor symptoms - depression, anxiety, cognitive decline, sleep and autonomic disturbances, pain - often impair quality of life as much as motor dysfunction. SSRIs/SNRIs (paroxetine, venlafaxine) and cognitive-behavioral therapy improve depressive symptoms in RCTs[240-242]; cholinesterase inhibitors (rivastigmine) aid cognition in PD dementia[243]; pimavanserin treats PD psychosis without worsening motor function[244]; and droxidopa benefits neurogenic orthostatic hypotension[245]. Targeted treatments for orthostatic hypotension, constipation, urinary issues, sleep disorders, and pain, combined with physical, occupational, and speech therapy, form the basis of comprehensive multidisciplinary care[246].
Limitations of current therapies
All current treatments are symptomatic; none halt neurodegeneration. DA drugs restore function but not neuronal integrity, and complications accumulate with disease progression. Surgical and infusion therapies similarly fail to address non-DA and cognitive decline. Thus, despite remarkable symptomatic advances, PD remains incurable - highlighting the urgent need for disease-modifying interventions aimed at slowing or preventing neurodegeneration[2,232].
Emerging disease-modifying therapies
Given the limitations of symptomatic treatments, current research in PD is shifting toward interventions that directly target the molecular and cellular drivers of neurodegeneration. These emerging approaches reflect PD’s multifactorial nature - encompassing protein aggregation, genetic mutations, mitochondrial dysfunction, and loss of neuronal integrity - and aim to alter disease trajectory rather than simply alleviate symptoms. The following sections summarize major categories of disease-modifying strategies under active investigation.
Protein aggregation and clearance
A central pathological hallmark of PD is α-syn misfolding and aggregation into toxic oligomers and Lewy bodies. Therapeutic efforts therefore focus on reducing α-syn burden through immunotherapy, gene silencing, or small-molecule inhibition. Passive immunotherapies such as prasinezumab (PRX002/RG7935) and cinpanemab (BIIB054) have completed Phase 2 trials; although neither met their primary endpoints, prasinezumab showed target engagement and a possible trend toward slower motor decline, prompting ongoing Phase 2b/3 evaluation[247]. Active vaccination (e.g., AFFITOPE PD01A/PD03A) has also proven immunogenic and well tolerated in early trials[248,249]. Parallel gene-targeted approaches, including antisense oligonucleotides against SNCA (BIIB101), seek to lower α-syn synthesis, while small molecules such as Anle138b aim to block oligomer formation. Despite mixed clinical outcomes so far, α-syn–directed therapies remain one of the most intensively pursued routes toward upstream disease modification[250,251].
Genetic and molecular modulators
Progress in PD genetics has catalyzed precision strategies that correct specific molecular defects. The best-validated target is LRRK2, whose pathogenic mutations increase kinase activity and disrupt lysosomal function. Brain-penetrant inhibitors (DNL151/BIIB122 and DNL201) and antisense oligonucleotides (BIIB094) have shown safety and biological target engagement, though recent trials have yielded inconclusive efficacy results. Another major focus is GBA, encoding the lysosomal enzyme glucocerebrosidase (GCase). Reduced GCase activity, whether due to mutation or secondary impairment, contributes to α-syn buildup. Chaperone therapies such as ambroxol have raised CSF GCase levels and hinted at clinical benefit, while gene therapy (AAV-GBA, PR001) is under evaluation to restore enzyme production[252,253]. Other gene-based interventions, including AAV-mediated delivery of aromatic L-amino acid decarboxylase (AADC) to enhance dopamine synthesis, illustrate the promise of targeted molecular restoration[254,255]. Collectively, these approaches herald a precision-medicine era in PD, where treatments are tailored to a patient’s genetic and biochemical profile.
Mitochondrial and metabolic protection
Mitochondrial impairment and oxidative stress are core contributors to PD pathogenesis, driving efforts to boost neuronal energy metabolism and reduce cellular stress. GLP-1RA, originally developed for diabetes, have demonstrated neuroprotective properties in preclinical models. Although the Phase 3 Exenatide-PD3 trial found no significant clinical benefit, other agents with improved CNS penetration (liraglutide, semaglutide) remain in testing[210,213,256]. Complementary metabolic interventions aim to reinforce mitochondrial resilience. NAD+ precursors (nicotinamide riboside, nicotinamide mononucleotide) have shown increased brain NAD+ levels and reduced inflammatory markers, while compounds such as mitoquinone (MitoQ), idebenone, urolithin A, and spermidine target oxidative stress and mitophagy enhancement[211,257]. Emerging NOX2 inhibitors further promise to blunt reactive oxygen species generation in microglia and neurons[258-260]. Together, these agents strive to maintain cellular homeostasis and protect DA neuron from chronic metabolic injury.
Neurorestoration and regeneration
Beyond neuroprotection, regenerative therapies aim to rebuild DA circuits lost to PD. Gene-based delivery of neurotrophic factors such as glial cell line-derived neurotrophic factor (GDNF) has shown encouraging long-term safety and biological activity in early AAV2-GDNF (AB-1005) trials, with evidence of motor stabilization[261,262]. In parallel, Japan has granted conditional approval for an iPSC-derived dopaminergic progenitor therapy (Amchepry/raguneprocel) for PD, showing safety and efficacy without graft-induced dyskinesia in a Phase I/II trial. Similar candidate (bemdaneprocel/MSK-DA01) continues clinical development[263], demonstrating safety and early functional improvement without graft-induced dyskinesia. These advances revive the vision of reconstructing the nigrostriatal pathway, possibly in combination with neurotrophic support or gene editing to enhance integration and survival. Though still experimental, regenerative strategies represent a paradigm shift - from slowing degeneration to restoring function.
Together, these emerging modalities embody a multidimensional push toward disease modification. By tackling complementary mechanisms - protein misfolding, lysosomal dysfunction, mitochondrial stress, and neuronal loss - they mirror the complex network nature of PD. Future therapies will likely combine modalities (e.g., anti-α-syn antibodies with metabolic enhancers or trophic gene delivery) to achieve synergistic benefit. The coming decade will determine whether these innovative strategies can move PD treatment beyond symptomatic relief toward true prevention and restoration of neural integrity.
Future trends of PD therapy
Systemic and population-scalable prevention strategies
Importantly, as the majority of PD cases are sporadic and driven by cumulative environmental exposures, aging-related decline, and systemic physiological states, future therapeutic progress will likely depend not only on molecular precision strategies but also on population-scalable interventions that target upstream disease drivers. Growing evidence supports the benefits of physical exercise, metabolic optimization (e.g., through diet and glucose control), dietary modulation, sleep regulation, and environmental risk reduction (e.g., minimizing exposure to pollutants and toxins). These approaches enhance mitochondrial function, bolster proteostasis, mitigate neuroinflammation, and strengthen organism-level resilience, thereby reducing the cumulative stress on vulnerable neuronal networks. From a public health perspective, such systemic and scalable strategies may deliver broader population-level impact than highly individualized gene-targeted therapies alone, particularly in preventing or delaying onset in at-risk groups.
Network-level and multi-system therapeutic strategies
Building upon these systemic perspectives, another clear trend is a move from single-target therapies to network-level, multi-system interventions. PD is now understood as a multi-organ, systemic disorder involving central and peripheral nervous systems, immune pathways, and the gut; treating one facet alone often yields partial benefit[130,264]. Future strategies will likely use combinations that act on interconnected pathways - an approach already explored in α-syn immunotherapy and lysosomal/immune modulation programs. For example, pairing an anti-α-syn therapy (to reduce toxic aggregates) with a lysosomal enhancer (e.g., GCase/GLP-1–linked approaches) and an anti-inflammatory or metabolic modulator is conceptually plausible and increasingly tested across trials and mechanistic studies[247,265]. Importantly, multi-organ involvement is being recognized: targeting peripheral pathology such as the gut microbiome and systemic inflammation is entering clinical investigation (probiotics/FMT and diet-based trials), reflecting PD’s whole-body nature[252,264,266,267]. The goal is to interrupt disease processes at central and peripheral levels for greater impact on progression.
Representative molecular targets within vulnerability networks
Research is increasingly identifying key molecular targets spanning the PD network. These targets illustrate how future therapies may intervene on several fronts in parallel:
SNCA (α-syn): The principal protein in Lewy bodies and a central driver of PD pathology. Reducing α-syn aggregation is a prime therapeutic focus[48]. Immunotherapies (monoclonal antibodies or vaccines) are in trials to neutralize toxic α-syn species, aiming to slow neurodegeneration as a disease-modifying strategy. Indeed, substantial evidence implicates α-syn in PD pathogenesis, making it an attractive target[247,268].
Nuclear enriched abundant transcript 1 (NEAT1): A long non-coding RNA emerging as a regulatory hub in PD. NEAT1 is upregulated in PD and can modulate the expression of PD-related genes (including SNCA) and interact with pathways like PINK1/Parkin-mediated mitophagy. Intriguingly, some studies suggest NEAT1 might exacerbate PD by promoting protein aggregation and inflammation, whereas others indicate it could play a protective role (e.g. acting as a natural inhibitor of LRRK2 kinase). This dual nature makes NEAT1 a potential biomarker of PD activity and a novel therapeutic target, pending further research to clarify its role. In the future, drugs or gene therapies might aim to dial NEAT1 activity up or down to restore cellular balance in PD[163,269,270].
Ubiquitin C-terminal hydrolase L1 (UCH-L1): A neuron-specific enzyme involved in the protein degradation (ubiquitin-proteasome) system, also known as Park5 in genetic forms of PD. Impaired UCH-L1 function has been implicated in PD pathogenesis, as it can lead to reduced clearance of misfolded proteins like α-syn. Notably, certain variants of the UCHL1 gene have been associated with altered PD risk, and genetic studies identify UCH-L1 as a potential target for disease-modifying therapy. Future approaches might seek to modulate UCH-L1 activity: for instance, inhibiting a pathological form of UCH-L1 that contributes to α-syn aggregation, or enhancing its normal protease activity to boost protein clearance[271,272]. By targeting UCH-L1, therapies could impact the broader proteostasis network, complementing other treatments like autophagy enhancers or lysosomal activators in a combined regimen.
ATP13A2 (PARK9): A lysosomal ATPase whose mutations cause a familial form of PD (Kufor-Rakeb syndrome) characterized by early-onset parkinsonism and dementia. ATP13A2 is crucial for lysosomal and mitochondrial function, helping cells handle toxic metals and oxidative stress. Loss of ATP13A2 activity leads to lysosomal dysfunction, impaired degradation of proteins, and neuronal death. This makes it an appealing target for neuroprotective therapy[273]. Researchers are now developing small-molecule activators of ATP13A2 to restore lysosomal health. The hypothesis is that agonists of ATP13A2 will bolster the clearing of cellular waste and reduce oxidative damage, thereby slowing or halting PD progression[157,274,275]. Such a drug would address one of the root problems in PD (protein accumulation and organelle stress), and could be used broadly - not only in patients with ATP13A2 mutations, but in sporadic PD as well, to enhance the cell’s natural housekeeping functions.
These examples underscore the multi-faceted strategy emerging in PD therapy. Rather than a one-size-fits-all drug, the future likely involves a cocktail of therapies, each targeting a different node in the disease network (protein aggregation, RNA regulation, axonal health, proteolysis, lysosomal function, etc.). By combining these approaches, there is hope for synergistic effects - for example, an α-syn antibody could reduce ongoing toxicity, while an ATP13A2 agonist improves cleanup of existing aggregates, and an anti-inflammatory agent or lncRNA modulator (like a NEAT1-targeting therapy) calms the harmful immune response[273]. This network-level thinking also means that non-neuronal aspects of PD will be addressed. For instance, the role of the gut-brain axis in PD is drawing intense interest: misfolded α-syn protein is often found in the ENS and may propagate from the gut to the brain. Therapies targeting the gut microbiome or peripheral α-syn are being explored as part of a systemic treatment vision. Early clinical trials are already testing interventions like high-fiber diets, probiotics, or even fecal microbiota transplants to restore a healthy gut microbiome in PD. The immune system is another facet - immune cells can contribute to neuroinflammation, so immunomodulatory treatments (for example, targeting overactive microglia or peripheral immune triggers) might complement direct neuronal therapies. In summary, the future of PD management looks holistic: multiple organs and pathological processes will be targeted together to achieve a more complete therapeutic effect.
Precision medicine tailored to patient subtypes
Hand-in-hand with network-based therapy comes the realization that “one size will not fit all” in PD. PD is a remarkably heterogeneous disorder - patients vary in their symptom profiles, rates of progression, and underlying molecular drivers. Consequently, there is a growing push toward precision medicine in PD, meaning treatments customized to an individual’s specific disease characteristics (genetic, biochemical, and clinical). In the future, patients may be stratified into subgroups (or “endophenotypes”) and receive personalized combinations of therapies most likely to benefit their form of PD[130,264].
Several factors could define these subgroups:
Genetics: A proportion of PD patients carry gene mutations (such as LRRK2, GBA, SNCA, VPS35, UCHL1, ATP13A2, etc.) that influence disease pathways[252,276]. Knowing a patient’s genetic makeup allows targeted treatment - for example, LRRK2 inhibitors are being developed for LRRK2 mutation carriers to normalize their kinase activity and lysosomal function[277,278]. Likewise, if a patient has a duplication of the SNCA gene (leading to excess α-syn), future gene therapies or antisense oligonucleotides might be used to reduce SNCA expression[279,280]. As mentioned, ATP13A2 agonists could be especially relevant for those with ATP13A2 loss-of-function, and so on. Genetics can also predict who might respond to certain drugs[156,200]; for instance, GBA mutation carriers (who have glucocerebrosidase enzyme deficiency) might benefit from therapies that boost lysosomal enzyme activity[252]. In short, clinicians envision genotype-driven treatment regimens, where a PD patient’s DNA guides the selection of disease-modifying strategies.
Molecular Biomarkers: Beyond DNA, each patient’s disease may have different dominant molecular features. Some individuals exhibit higher α-syn pathology, while others have more tau/amyloid co-pathology or more pronounced neuroinflammation. Emerging biomarkers - including α-syn seed amplification assays in CSF, blood/CSF NfL, inflammatory cytokine panels, and even lncRNA profiles (e.g., NEAT1 levels) - can help map the active pathways in a given patient[163,281-283]. For example, a patient with biomarker evidence of high inflammation might receive an immunosuppressive or glial-modulating therapy[264]. Another patient with signs of impaired protein clearance [e.g., high CSF α-syn oligomers/positive seed amplification analysis (SAA), low lysosomal activity] might be funneled toward autophagy/lysosomal enhancers; UCH-L1 remains mechanistically linked to proteostasis, though human genetic causality is limited compared with major PD genes[284,285]. NEAT1 has context-dependent roles (reports of protective and deleterious effects), so any NEAT1-targeting approach would likely be subtype-specific pending further validation[163,281].
Clinical Phenotype: Patients vary in their clinical presentation - for example, tremor-dominant vs. posture/gait-dominant PD, or those with rapid progression and cognitive decline vs. those with milder courses. These clinical subtypes likely reflect underlying differences in pathology distribution and progression[286,287]. In the future, clinical profiling will be used alongside biomarkers to tailor therapy. A patient with severe gait difficulty and falls (suggesting more widespread neuronal loss and perhaps cholinergic deficits) might benefit from an early addition of falls-prevention strategies or cognitive enhancers, whereas a tremor-dominant patient might be managed with a different emphasis. Even the non-motor symptoms (like autonomic dysfunction or mood symptoms) could guide adjunct therapies (e.g. targeting peripheral autonomic nerves or serotonin pathways). Precision medicine means moving away from treating every PD patient the same way, and instead developing an individualized treatment plan that targets the most troublesome and fundamental aspects of their disease.
Taken together, the future of PD therapy will likely integrate two complementary dimensions: broad systemic interventions that reduce cumulative environmental and metabolic stress across populations, and precision molecular strategies that directly reinforce genetically constrained vulnerability networks. Rather than competing, these approaches should operate synergistically to enhance resilience at both organismal and cellular levels, ultimately delaying onset, slowing progression, and improving long-term outcomes in sporadic PD.
Realizing such an integrated framework requires not only therapeutic innovation but also the ability to identify which patients are most likely to benefit from specific interventions. Stratification based on molecular, genetic, and clinical profiles - particularly biomarkers reflecting the network-level dysfunctions described above - will therefore be essential for translating both systemic and precision strategies into effective practice. In this context, integrative biomarker platforms and early-stage diagnostic models become critical tools for determining who to treat, when to intervene, and how best to match therapies to disease subtypes. The following section thus focuses on emerging diagnostic and predictive frameworks that enable personalized and timely intervention.
EARLY DIAGNOSIS AND PREDICTIVE FRAMEWORKS
Existing modalities and progress
A major challenge in PD is diagnostic delay: clinical diagnosis still relies largely on the emergence of cardinal motor symptoms, which occur only after substantial degeneration of the nigrostriatal system, with estimates suggesting about 30%-50% loss of substantia nigra DA neurons and ~50%-80% depletion of striatal dopamine terminals at motor onset[109,130,288-290]. Therefore, developing early diagnostic techniques to intervene when neural damage is still minimal is crucial for improving prognosis. Recent advancements across multiple fields have brought us closer to the goal of advancing the time of diagnosis by 5 to 10 years:
Imaging biomarkers: Functional imaging can detect changes in the dopamine system before clinical symptoms emerge. Dopamine transporter imaging (DAT-SPECT) utilizes radioactive tracers such as [123I]-ioflupane binding to striatal DAT enables quantification of DA neuron terminal density[291-293]. Early-stage PD can be detected through reduced uptake in one side of the striatum via PET, which aids in differentiating atypical tremor symptoms. PET imaging using tracers like [18F]-DOPA reflects decreased dopamine synthesis function, playing a crucial role in clinical diagnosis[294]. As an emerging MRI technique, neuro-melanin-sensitive magnetic resonance imaging (NM-MRI) holds significant diagnostic value for PD. In normal midbrain substantia nigra, abundant neuro-melanin produces high signal intensity on T1-weighted sequences. However, in PD patients, this high signal band markedly diminishes or disappears. Studies demonstrate that NM-MRI achieves high accuracy in distinguishing PD patients from healthy elderly individuals using 7T ultra-high field MRI technology, making it a promising non-invasive early diagnostic tool[295,296]. Additionally, MRIs iron load imaging (SWI or Quantitative Susceptibility Mapping) detects iron deposition in substantia nigra, which increases in early PD patients and shows negative correlation with reduced NM signals[297-300]. MRI techniques such as subcortical nucleus volume measurement and brain network connectivity analysis also provide robust support for related research. Overall, the imaging field is progressively advancing. The diagnostic approach is evolving from exclusionary methods (such as ruling out stroke or tumors) to quantitative detection of early PD pathology. With advancements in high-resolution MRI technology and specialized sequences, it is anticipated that a single MRI scan could screen high-risk PD candidates [e.g., individuals with anosmia combined with resting-state brain dysfunction (RBD) symptoms][297,300]. Integrating imaging techniques with AI analysis further enhances diagnostic sensitivity and specificity[301].
Body fluid biomarkers: An ideal PD biomarker should originate from easily accessible samples while demonstrating high sensitivity and specificity[302]. The most promising research direction currently is detecting abnormal conformations of α-syn. Leveraging the self-propagating characteristic of pathological α-syn, scientists have developed SAA techniques encompassing RT-QuIC and PMCA methods[283,303]. The procedure involves adding patient samples (cerebrospinal fluid or peripheral tissue homogenates) to normal α-syn protein solution. If trace amounts of abnormal α-syn seeds are present, they induce aggregation of normal α-syn, which can be detected by fluorescent probes (e.g., ThT). Multicenter studies show that RT-QuIC demonstrates ~85%-95% sensitivity and ~90%-96% specificity in cerebrospinal fluid PD detection, particularly exhibiting exceptionally high positivity rates for patients with RBD[43,304,305]. To reduce invasiveness, skin and nasal/olfactory mucosa samples have been explored: skin RT-QuIC and skin phospho-α-syn IHC show high specificity and high positivity in PD/synucleinopathies in prospective multicenter studies[304,305]. Another highly promising biomarker is NfL, a protein found in the axonal skeleton that leaks into cerebrospinal fluid and bloodstream when brain neurons are damaged. While early changes in NfL levels during PD may not be as pronounced as in other neurodegenerative diseases like ALS, recent studies show that elevated plasma NfL levels are closely linked to rapid disease progression and cognitive decline risks in PD patients. This makes NfL a potential key biomarker for monitoring PD’s progression[46,306]. Other biomarkers such as DJ-1 protein, tau protein, and inflammatory factors have been reported, but they lack specificity and consistency. Currently, multiple institutions are collaborating to improve diagnostic accuracy through biomarker combinations. For instance, the PPMI study suggests combining α-syn RT-QuIC results, saliva-based miRNAs regulating DJ-1 gene expression, and urine NfL testing for better predictive power. In summary, the field of biomarkers is advancing rapidly and holds promise for clinical application in the near future.
Peripheral tissue biopsies: As previously mentioned, skin biopsies serve as a window to obtain central pathological information. Similarly, colon mucosal biopsies and salivary gland biopsies are also under investigation. Results indicate that approximately 70% to 80% of PD patients show immunohistochemical detection of phosphorylated α-syn inclusions in intestinal mucosal or lower lip salivary gland biopsy specimens, while controls rarely exhibit positive findings. While these biopsies combined with highly specific antibodies demonstrate diagnostic value, their sensitivity remains limited and sampling errors may occur[306,307,308]. A more minimally invasive approach involves nasal endoscopic scraping of olfactory mucosa cells, which has been applied in RT-QuIC testing. Peripheral tissue samples also hold promise for transcriptomic/metabolomic analysis to identify diagnostic biomarkers through pattern recognition. However, these methods are currently in the research phase and remain distant from widespread clinical application.
Digital phenotyping: Leveraging wearable devices, smartphone sensors, and AI algorithms, this technology captures subtle motor and behavioral changes in PD patients. For instance, wrist-mounted accelerometers track gait patterns and tremor frequency, while smartphone touchscreen tapping rhythms reflect finger dexterity. Typing error rates indicate fine motor dysfunction. Voice analysis detects weak vocalizations and slowed speech rates to identify speech disorders early. Sleep monitors can detect abnormal nocturnal movements and heart rate variations that may help screen for REM sleep behavior disorder[309]. The advantages of digital phenotyping - continuity, objectivity, and non-invasiveness - have been validated in multiple PD studies as correlating with traditional clinical assessments. Particularly for individuals with family history or mild symptoms, digital monitoring establishes baseline data and tracks trend changes[310]. Some companies are developing mobile Apps for self-monitoring, enabling patients to perform home-based finger tapping and voice tests, then upload data to cloud platforms for analysis. While these tools cannot yet serve as standalone diagnoses, they show great promise as clinical assessment aids and high-risk screening tools. A study published in Science Advances demonstrated that integrating keystroke dynamics from intelligent keyboard systems (IKS) with gait acceleration patterns reveals behavioral biomarkers[311]. By analyzing degree and speech spectrum data, it can effectively distinguish patients with mild PD from healthy individuals with over 80% accuracy. In the future, digital phenotypes are expected to integrate with biomarkers to enable remote, early, and multidimensional detection of PD.
Future directions
Although multiple methods are now available for early diagnosis of PD, new approaches and tools are required to achieve more advanced prediction and personalized risk assessment. The following are some future directions:
Evaluation of Developmental Risk Factors: Although multiple methods are now available for early diagnosis of PD, new approaches and tools are required to achieve more advanced prediction and personalized risk assessment. The following directions emphasize risk identification and early-life susceptibility profiling rather than therapeutic intervention. Developmental factors may influence lifelong vulnerability to PD. For instance, variations in the number of DA neuron in the substantia nigra at birth mean individuals with lower neural “reserves” may be more prone to functional deficits in middle age[312]. Another example: Could developmental injuries like brain inflammation or trauma during childhood potentially create hidden risks? Current cohort studies are tracking premature infants, low birth weight babies, and those exposed to maternal infections during pregnancy to observe whether their PD incidence increases in old age. Research also suggests that hormone levels around puberty and high-intensity exercise habits may shape adult brains resistance to PD[313]. Future developments might include scoring systems based on developmental indicators - such as motor coordination in childhood, cognitive development, and family history - to predict lifelong PD risk. While long-term data validation remains necessary, this highlights the concept that “PD origins may trace back to childhood”. These findings underscore the importance of incorporating developmental indicators into long-term risk models and early surveillance frameworks.
Multi-omics integrated Risk Stratification: Currently, PRS provide modest predictive value when used alone, as they primarily reflect inherited DNA variation rather than downstream biological states[314,315]. Because PD risk is shaped not only by genetics but also by epigenetic regulation, immune-metabolic status, and environmental exposures, next-generation models are expected to integrate multiple layers - genome, epigenome, transcriptome, metabolome, and exposome - for more comprehensive risk assessment[316]. For example, an individual carrying high-risk variants (e.g., LRRK2 or a high PRS) who also shows lipidomic changes such as elevated sphingolipids and pro-inflammatory cytokine profiles may have a substantially higher integrated risk estimate, whereas some high-risk gene carriers may remain unaffected when metabolic and immune indicators are unremarkable[316-318]. Machine-learning methods can identify informative combinations across these modalities, but models require external validation and careful assessment of generalizability[316]. In microbiome-based work, some single-cohort studies report high internal discrimination; however, a multi-study meta-analysis across thousands of samples found an average AUC of ~0.72, underscoring both promise and current limitations[319]. Large longitudinal resources such as PPMI 2.0 are now assembling and sharing multimodal clinical, imaging, genetic, and biospecimen data to enable such integrative prediction efforts. Early multimodal-fusion studies using PPMI already show that combining imaging with clinical features improves discrimination of early/prodromal PD, illustrating a realistic near-term path for integrative models. As these tools move toward earlier-life risk estimation, ethical communication and counseling protocols for long-horizon risk prediction will be essential.
Artificial Intelligence Assisted Predictive Analytics: With the accumulation of multi-modal datasets, artificial intelligence (AI) is expected to play a pivotal role in early detection and disease trajectory modeling. Deep learning algorithms can extract features from complex genomic, microbiome, imaging, and behavioral data that may not be apparent through conventional analyses. Rather than directing treatment decisions, these tools primarily enable individualized risk estimation, progression forecasting, and dynamic monitoring, helping identify individuals who may benefit from closer clinical surveillance or additional testing. Such predictive models may facilitate earlier enrollment into observational studies or biomarker-validation cohorts, thereby improving understanding of disease evolution. Of course, achieving this vision requires collaboration among neurologists, data scientists, geneticists, and other interdisciplinary experts to jointly build credible models. Fortunately, several international teams have launched prototype projects to apply AI technology to early screening and comprehensive data analysis of PD.
In the future, PD diagnosis is expected to evolve toward early and more accurate detection-identifying subtle biological and behavioral changes years before motor symptoms emerge and providing quantitative risk profiles for individuals. This predictive framework aims to enable timely surveillance and improved staging rather than late recognition of established disease. Similar to cardiovascular risk scores or dementia prediction models, PD research is progressing toward scientifically grounded early risk estimation systems that support proactive clinical monitoring.
CONCLUSION
Sporadic PD should no longer be viewed as a focal nigrostriatal disorder, but as a heterogeneous systemic syndrome emerging from the interplay of genetic susceptibility, environmental exposures, aging, and network-level physiological disturbances across the gut, sensory systems, immune signaling, and metabolism. Our cross-species integrative analyses support this framework by identifying hub genes such as SNCA, NEAT1, ATP13A2, NEFL, and UCHL1 that connect peripheral tissues with mDA circuits. These findings suggest that DA degeneration reflects the cumulative failure of interconnected systems rather than a single initiating lesion.
This perspective holds direct therapeutic implications. Future strategies should transcend isolated DA replacement and progress towards coordinated, multi - target interventions that tackle α-syn pathology, mitochondrial and lysosomal dysfunction, systemic inflammation, and gut-brain dysregulation across organs. Concurrently, network- aware precision medicine, which integrates genetics, transcriptomics, and fluid biomarkers, will facilitate molecular subtyping, biomarker-guided trials, and customized combination regimens. The early identification of high-risk or prodromal individuals (e.g., those with RBD, hyposmia, or mutation carriers) provides a crucial window for disease interception via gene-, immune-, and microbiome- based approaches. Collectively, these advancements advocate for a transition from reactive symptom control to proactive, system-level modification, paving a feasible path for delaying and ultimately preventing PD.
DECLARATIONS
Authors’ contributions
Literature search, writing, and original draft preparation: Li Y, Guo L
Conceptualization, review, revision, and editing: Peng C, Li Y, Yu X
Availability of data and materials
All original data referenced and analyzed in this review are publicly available from existing repositories. The transcriptomic datasets used for cross-system analysis of midbrain dopaminergic neurons (mDA), olfactory bulb (OB), enteric nervous system (ENS), and dorsal root ganglia (DRG) can be accessed through the Gene Expression Omnibus (GEO) under the following accession numbers: GSE245310, GSE139088, GSE134355, GSE149524, GSE156905, GSE121891, GSE70896, GSE76381 and GSE178265.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by grants from the National Natural Science Foundation of China (32070977, 51971236, 31871063 and 31741057 to Peng C), from the Basic Research Project of the Discipline Promotion Program of Shanghai Fourth People’s Hospital (SY-XKZT-2025-1003 to Peng C), and from the Major National Science and Technology Projects of China (2018ZX09733001-006-005 to Peng C).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Dorsey ER, Sherer T, Okun MS, Bloem BR. The emerging evidence of the Parkinson pandemic. J Parkinsons Dis. 2018;8:S3-8.
2. Athauda D, Foltynie T. The ongoing pursuit of neuroprotective therapies in Parkinson disease. Nat Rev Neurol. 2014;11:25-40.
3. Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol. 2006;5:235-45.
4. Parkkinen L, Pirttilä T, Alafuzoff I. Applicability of current staging/categorization of α-synuclein pathology and their clinical relevance. Acta Neuropathol. 2008;115:399–407.
5. Gelpi E, Navarro-Otano J, Tolosa E, et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov Disord. 2014;29:1010–8.
6. Obeso J, Stamelou M, Goetz C, et al. Past, present, and future of Parkinson’s disease: a special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord. 2017;32:1264-310.
7. Ascherio A, Schwarzschild MA. The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol. 2016;15:1257-72.
8. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498-504.
9. Szklarczyk D, Kirsch R, Koutrouli M, et al. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51:D638-46.
11. Kim JJ, Vitale D, Véliz Otani D, et al. Multi-ancestry genome-wide association meta-analysis of Parkinson’s disease. Nat Genet. 2024;56:27–36.
12. Li B, Zhao G, Li K, et al. Characterizing the expression patterns of Parkinson’s disease associated genes. Front Neurosci. 2021;15:629156.
13. Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651-61.
14. Vernay B, Koch M, Vaccarino F, et al. Otx2 regulates subtype specification and neurogenesis in the midbrain. J Neurosci. 2005;25:4856-67.
15. Di Salvio M, Di Giovannantonio LG, Acampora D, et al. Otx2 controls neuron subtype identity in ventral tegmental area and antagonizes vulnerability to MPTP. Nat Neurosci. 2010;13:1481-8.
16. Frantz G, Weimann J, Levin M, Mcconnell S. Otx1 and Otx2 define layers and regions in developing cerebral cortex and cerebellum. J Neurosci. 1994;14:5725-40.
17. Nunes I, Tovmasian LT, Silva RM, Burke RE, Goff SP. Pitx3 is required for development of substantia nigra dopaminergic neurons. Proc Natl Acad Sci U S A. 2003;100:4245-50.
18. Peng C, Fan S, Li X, et al. Overexpression of pitx3 upregulates expression of BDNF and GDNF in SH‐SY5Y cells and primary ventral mesencephalic cultures. FEBS Lett. 2007;581:1357-61.
19. Peng C, Aron L, Klein R, et al. Pitx3 is a critical mediator of GDNF-induced BDNF expression in nigrostriatal dopaminergic neurons. J Neurosci. 2011;31:12802-15.
20. Van Den Munckhof P, Luk KC, Ste-Marie L, et al. Pitx3 is required for motor activity and for survival of a subset of midbrain dopaminergic neurons. Development. 2003;130:2535-42.
21. Martinat C, Bacci J, Leete T, et al. Cooperative transcription activation by Nurr1 and Pitx3 induces embryonic stem cell maturation to the midbrain dopamine neuron phenotype. Proc Natl Acad Sci U S A. 2006;103:2874-9.
22. Semina EV, Ferrell RE, Mintz-Hittner HA, et al. A novel homeobox gene PITX3 is mutated in families with autosomal-dominant cataracts and ASMD. Nat Genet. 1998;19:167-70.
23. Semina EV. Deletion in the promoter region and altered expression of Pitx3 homeobox gene in aphakia mice. Hum Mol Genet. 2000;9:1575-85.
24. Kouwenhoven WM, Von Oerthel L, Smidt MP. Pitx3 and En1 determine the size and molecular programming of the dopaminergic neuronal pool. PLoS ONE. 2017;12:e0182421.
25. Le W, Xu P, Jankovic J, et al. Mutations in NR4A2 associated with familial Parkinson disease. Nat Genet. 2002;33:85-9.
26. Saucedo-Cardenas O, Quintana-Hau JD, Le W, et al. Nurr1 is essential for the induction of the dopaminergic phenotype and the survival of ventral mesencephalic late dopaminergic precursor neurons. Proc Natl Acad Sci U S A. 1998;95:4013-8.
27. Zetterström RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276:248-50.
28. Kadkhodaei B, Ito T, Joodmardi E, et al. Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J Neurosci. 2009;29:15923-32.
29. Laguna A, Schintu N, Nobre A, et al. Dopaminergic control of autophagic-lysosomal function implicates Lmx1b in Parkinson’s disease. Nat Neurosci. 2015;18:826-35.
30. Sgadò P, Albéri L, Gherbassi D, et al. Slow progressive degeneration of nigral dopaminergic neurons in postnatal Engrailed mutant mice. Proc Natl Acad Sci U S A. 2006;103:15242-7.
31. Wurst W, Auerbach AB, Joyner AL. Multiple developmental defects in Engrailed-1 mutant mice: an early mid-hindbrain deletion and patterning defects in forelimbs and sternum. Development. 1994;120:2065-75.
32. Wurst W, Prakash N. Wnt1-regulated genetic networks in midbrain dopaminergic neuron development. J Mol Cell Biol. 2013;6:34-41.
33. Zhang J, Götz S, Vogt Weisenhorn DM, Simeone A, Wurst W, Prakash N. A WNT1-regulated developmental gene cascade prevents dopaminergic neurodegeneration in adult En1+/- mice. Neurobiol Dis. 2015;82:32-45.
34. Goodier JL, Kazazian HH. Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008;135:23-35.
35. Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605-8.
36. Valente EM, Salvi S, Ialongo T, et al. PINK1 mutations are associated with sporadic early‐onset parkinsonism. Ann Neurol. 2004;56:336-41.
37. Lin X, Cook TJ, Zabetian CP, et al. DJ-1 isoforms in whole blood as potential biomarkers of Parkinson disease. Sci Rep. 2012;2:954.
38. Kedaigle AJ, Fraenkel E, Atwal RS, et al.; The HD iPSC Consortium. Bioenergetic deficits in Huntington’s disease iPSC-derived neural cells and rescue with glycolytic metabolites. Hum Mol Genet. 2020;29:1757-71.
39. Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601-7.
40. Hu Y, Yang H, Song C, et al. LRRK2G2019S gene mutation causes skeletal muscle impairment in animal model of Parkinson’s disease. J Cachexia Sarcopenia Muscle. 2024;15:2595-607.
41. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469-73.
42. Singleton AB, Farrer M, Johnson J, et al. α-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841.
43. Zhuang D, Ren Z, Sheng J, et al. A dynamic nomogram for predicting unfavorable prognosis after aneurysmal subarachnoid hemorrhage. Ann Clin Transl Neurol. 2023;10:1058-71.
44. Iranzo A, Fairfoul G, Ayudhaya ACN, et al. Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: a longitudinal observational study. Lancet Neurol. 2021;20:203-12.
45. Goto R, Matsuoka K, Kimura Y, et al. Human biodistribution and radiation dosimetry of two novel α-synuclein PET tracers, 18F-SPAL-T-06 and 18F-C05-05. Sci Rep. 2025;15:8640.
46. Hällqvist J, Bartl M, Dakna M, et al. Plasma proteomics identify biomarkers predicting Parkinson’s disease up to 7 years before symptom onset. Nat Commun. 2024;15:4759.
47. Buhmann C, Magnus T, Choe C. Blood neurofilament light chain in Parkinson’s disease. J Neural Transm. 2023;130:755-62.
48. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839-40.
49. Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045-7.
50. Shimura H, Hattori N, Kubo S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302-5.
51. Patek CE. Murine Denys-Drash syndrome: evidence of podocyte de-differentiation and systemic mediation of glomerulosclerosis. Hum Mol Genet. 2003;12:2379-94.
52. Bonifati V, Rizzu P, Van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256-9.
53. Paisán-Ruı́z C, Jain S, Evans E, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595-600.
54. Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158-60.
55. Morimoto M, Takahashi Y, Endo M, Saga Y. The Mesp2 transcription factor establishes segmental borders by suppressing Notch activity. Nature. 2005;435:354-9.
56. Dell’agnello C, Leo S, Agostino A, et al. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet. 2007;16:431-44.
57. Healy DG, Falchi M, O’Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7:583-90.
58. Nörenberg A, Hu H, Vida I, Bartos M, Jonas P. Distinct nonuniform cable properties optimize rapid and efficient activation of fast-spiking GABAergic interneurons. Proc Natl Acad Sci U S A. 2009;107:894-9.
59. Pickrell AM, Youle RJ. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257-73.
60. Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020;19:170-8.
61. Andersson ER, Saltó C, Villaescusa JC, et al. Wnt5a cooperates with canonical Wnts to generate midbrain dopaminergic neurons in vivo and in stem cells. Proc Natl Acad Sci U S A. 2013;110:E602-10.
62. Nouri P, Götz S, Rauser B, et al. Dose-dependent and subset-specific regulation of midbrain dopaminergic neuron differentiation by LEF1-mediated WNT1/b-catenin signaling. Front Cell Dev Biol. 2020;8:587778.
63. Yang D, Peng C, Li X, et al. Pitx3‐transfected astrocytes secrete brain‐derived neurotrophic factor and glial cell line‐derived neurotrophic factor and protect dopamine neurons in mesencephalon cultures. J Neurosci Res. 2008;86:3393-400.
64. Smith L, Schapira AHV. GBA variants and Parkinson disease: mechanisms and treatments. Cells. 2022;11:1261.
65. Tanner CM, Kamel F, Ross GW, et al. Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect. 2011;119:866-72.
66. Firestone JA, Smith-Weller T, Franklin G, Swanson P, Longstreth WT, Checkoway H. Pesticides and risk of Parkinson disease: a population-based case-control study. Arch Neurol. 2005;62:91.
67. Costello S, Cockburn M, Bronstein J, Zhang X, Ritz B. Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am J Epidemiol. 2009;169:919-26.
68. Pouchieu C, Piel C, Carles C, et al. Pesticide use in agriculture and Parkinson’s disease in the AGRICAN cohort study. Int J Epidemiol. 2018;47:299-310.
69. Hancock DB, Martin ER, Mayhew GM, et al. Pesticide exposure and risk of Parkinson’s disease: a family-based case-control study. BMC Neurol. 2008;8:6.
70. Liu R, Young MT, Chen J, Kaufman JD, Chen H. Ambient air pollution exposures and risk of Parkinson disease. Environ Health Perspect. 2016;124:1759-65.
71. Betarbet R, Sherer TB, Mackenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301-6.
72. Goldman SM, Tanner CM, Oakes D, Bhudhihanok GSS, Gupta A, Langston JW. Head injury and Parkinson’s disease risk in twins. Ann Neurol. 2006;60:65-72.
73. Van Maele-Fabry G, Hoet P, Vilain F, Lison D. Occupational exposure to pesticides and Parkinson’s disease: a systematic review and meta-analysis of cohort studies. Environ Int. 2012;46:30-43.
74. Lock EA, Zhang J, Checkoway H. Solvents and Parkinson disease: a systematic review of toxicological and epidemiological evidence. Toxicol Appl Pharmacol. 2013;266:345-55.
75. Sallmén M, Burstyn I, Uuksulainen S, Koskinen A, Hublin C, Sainio M. Parkinson’s disease and occupational exposure to organic solvents in Finland: a nationwide case-control study. Scand J Work Environ Health. 2024;50:39-48.
76. Gorell JM, Johnson CC, Rybicki BA, et al. Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology. 1997;48:650-8.
77. Racette BA, Tabbal SD, Jennings D, Good L, Perlmutter JS, Evanoff B. Prevalence of parkinsonism and relationship to exposure in a large sample of Alabama welders. Neurology. 2005;64:230–35.
78. Kwakye G, Paoliello M, Mukhopadhyay S, Bowman A, Aschner M. Manganese-induced parkinsonism and Parkinson’s disease: shared and distinguishable features. Int J Environ Res Public Health. 2015;12:7519-40.
79. Zeng W, Cai J, Zhang L, Peng Q. Iron deposition in Parkinson’s disease: a mini-review. Cell Mol Neurobiol. 2024;44:26.
80. Krzyzanowski B, Mullan AF, Turcano P, Camerucci E, Bower JH, Savica R. Air pollution and Parkinson disease in a population-based study. JAMA Netw Open. 2024;7:e2433602.
81. Wang VA, Delaney S, Flynn LE, et al. The effect of air pollution on hospitalizations with Parkinson’s disease among medicare beneficiaries nationwide. NPJ Parkinsons Dis. 2024;10:196.
82. Nishiwaki H, Hamaguchi T, Ito M, et al. Short-chain fatty acid-producing gut microbiota is decreased in Parkinson’s disease but not in rapid-eye-movement sleep behavior disorder. mSystems. 2020;5:e00797-20.
83. Chen S, Chen C, Liao H, et al. Association of fecal and plasma levels of short-chain fatty acids with gut microbiota and clinical severity in patients with Parkinson disease. Neurology. 2022;98:e848-58.
84. Goldman JG, Holden S, Ouyang B, Bernard B, Goetz CG, Stebbins GT. Diagnosing PD‐MCI by MDS task force criteria: How many and which neuropsychological tests? Mov Disord. 2014;30:402-6.
85. Pierantozzi M, Pietroiusti A, Brusa L, et al. Helicobacter pylori eradication and l-dopa absorption in patients with PD and motor fluctuations. Neurology. 2006;66:1824-9.
86. Hashim H, Azmin S, Razlan H, et al. Eradication of Helicobacter pylori infection improves levodopa action, clinical symptoms and quality of life in patients with Parkinson’s disease. PLoS ONE. 2014;9:e112330.
87. Chen H, Huang X, Guo X, et al. Smoking duration, intensity, and risk of Parkinson disease. Neurology. 2010;74:878–84.
88. Leentjens AF, Dujardin K, Marsh L, Richard IH, Starkstein SE, Martinez‐Martin P. Anxiety rating scales in Parkinson’s disease: a validation study of the Hamilton anxiety rating scale, the Beck anxiety inventory, and the hospital anxiety and depression scale. Mov Disord. 2011;26:407-15.
89. Jafari S, Etminan M, Aminzadeh F, Samii A. Head injury and risk of Parkinson disease: a systematic review and meta‐analysis. Mov Disord. 2013;28:1222-9.
90. Hernán MA, Takkouche B, Caamaño‐Isorna F, Gestal‐Otero JJ. A meta‐analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann Neurol. 2002;52:276-84.
91. Yue X, Li H, Yan H, Zhang P, Chang L, Li T. Risk of Parkinson disease in diabetes mellitus: an updated meta-analysis of population-based cohort studies. Medicine. 2016;95:e3549.
92. Roth NM, Saidha S, Zimmermann H, et al. Photoreceptor layer thinning in idiopathic Parkinson’s disease. Mov Disord. 2014;29:1163-70.
93. Petzinger GM, Fisher BE, McEwen S, Beeler JA, Walsh JP, Jakowec MW. Exercise-enhanced neuroplasticity targeting motor and cognitive circuitry in Parkinson’s disease. Lancet Neurol. 2013;12:716–26.
94. Fang X, Han D, Cheng Q, et al. Association of levels of physical activity with risk of Parkinson disease: a systematic review and meta-analysis. JAMA Netw Open. 2018;1:e182421.
96. GBD 2016 Parkinson’s Disease Collaborators Global, regional, and national burden of Parkinson’s disease, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018;17:939-53.
97. Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;333:1269.
98. Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515-7.
99. Monzani E, Nicolis S, Dell’Acqua S, et al. Dopamine, oxidative stress and protein–quinone modifications in Parkinson’s and other neurodegenerative diseases. Angew Chem Int Ed. 2019;58:6512-27.
100. Song D, Li J. Revisiting the advance of age-dependent α-synuclein propagation and aggregation. Ageing Neur Dis. 2025;5:4.
101. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194-217.
102. Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, Deshaies RJ. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell. 2010;38:17-28.
104. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69 Suppl:S4-9.
105. Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10:217-24.
106. Bendig J, Frank A, Reichmann H. Aging and Parkinson’s disease: a complex interplay of vulnerable neurons, the immune system and the blood-brain barrier. Ageing Neur Dis. 2024;4:5.
107. Montagne A, Barnes SR, Sweeney MD, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296-302.
108. Al-Bachari S, Naish JH, Parker GJM, Emsley HCA, Parkes LM. Blood–brain barrier leakage is increased in Parkinson’s disease. Front Physiol. 2020;11:593026.
109. Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114:2283-301.
110. Branch SY, Sharma R, Beckstead MJ. Aging decreases L-type calcium channel currents and pacemaker firing fidelity in substantia nigra dopamine neurons. J Neurosci. 2014;34:9310-8.
111. Gillies GE, Pienaar IS, Vohra S, Qamhawi Z. Sex differences in Parkinson’s disease. Front Neuroendocrinol. 2014;35:370–84.
113. Jacobs BM, Belete D, Bestwick J, et al. Parkinson’s disease determinants, prediction and gene–environment interactions in the UK Biobank. J Neurol Neurosurg Psychiatry. 2020;91:1046-54.
114. Kwon D, Paul KC, Kusters C, et al. Interaction between traffic-related air pollution and Parkinson disease polygenic risk score. JAMA Netw Open. 2025;8:e250854.
115. Tsalenchuk M, Gentleman SM, Marzi SJ. Linking environmental risk factors with epigenetic mechanisms in Parkinson’s disease. NPJ Parkinsons Dis. 2023;9:123.
116. Angelopoulou E, Piperi C. Epigenetics: the missing link between environmental exposures and Parkinson’s disease? Epigenomics. 2024;16:921-7.
117. Hamza TH, Zabetian CP, Tenesa A, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010;42:781-5.
118. Lindestam Arlehamn CS, Dhanwani R, Pham J, et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun. 2020;11:1875.
119. Yu E, Ambati A, Andersen MS, et al. Fine mapping of the HLA locus in Parkinson’s disease in Europeans. NPJ Parkinsons Dis. 2021;7:84.
120. Noelker C, Morel L, Lescot T, et al. Toll like receptor 4 mediates cell death in a mouse MPTP model of Parkinson disease. Sci Rep. 2013;3:1393.
121. Hughes CD, Choi ML, Ryten M, et al. Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson’s disease pathogenesis. Acta Neuropathol. 2018;137:103-20.
122. Fellner L, Irschick R, Schanda K, et al. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia. 2013;61:349–60.
123. Kim C, Ho D, Suk J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562.
124. Pike AF, Varanita T, Herrebout MAC, et al. α‐Synuclein evokes NLRP3 inflammasome‐mediated IL‐1β secretion from primary human microglia. Glia. 2021;69:1413-28.
125. Lee E, Hwang I, Park S, et al. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2018;26:213-28.
126. Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener. 2015;4:19.
127. Pezzoli G, Cereda E. Exposure to pesticides or solvents and risk of Parkinson disease. Neurology. 2013;80:2035-41.
128. Ascherio A, Chen H, Weisskopf MG, et al. Pesticide exposure and risk for Parkinson’s disease. Ann Neurol. 2006;60:197-203.
129. Rietdijk CD, Perez-Pardo P, Garssen J, Van Wezel RJA, Kraneveld AD. Exploring Braak’s hypothesis of Parkinson’s disease. Front Neurol. 2017;8:37.
130. Braak H, Tredici KD, Rüb U, De Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197-211.
131. Zis P, Erro R, Walton CC, Sauerbier A, Chaudhuri KR. The range and nature of non-motor symptoms in drug-naive Parkinson’s disease patients: a state-of-the-art systematic review. NPJ Parkinsons Dis. 2015;1:15013.
132. Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 2009;8:464-74.
133. Braak H, Del Tredici K. Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff. J Parkinsons Dis. 2017;7:S71-85.
134. Kang X, Ploner A, Wang Y, et al. Genetic overlap between Parkinson disease and inflammatory bowel disease. medRxiv. 2022.
135. Witoelar A, Jansen IE, Wang Y, et al.; for the International Parkinson’s Disease Genomics Consortium (IPDGC), North American Brain Expression Consortium (NABEC), and United Kingdom Brain Expression Consortium (UKBEC) Investigators. Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol. 2017;74:780-92.
136. Kamath T, Abdulraouf A, Burris SJ, et al. Single-cell genomic profiling of human dopamine neurons identifies a population that selectively degenerates in Parkinson’s disease. Nat Neurosci. 2022;25:588-95.
137. La Manno G, Gyllborg D, Codeluppi S, et al. Molecular diversity of midbrain development in mouse, human, and stem cells. Cell. 2016;167:566-80.e19.
138. Tepe B, Hill MC, Pekarek BT, et al. Single-cell RNA-seq of mouse olfactory bulb reveals cellular heterogeneity and activity-dependent molecular census of adult-born neurons. Cell Rep. 2018;25:2689-703.e3.
139. Kawasawa YI, Salzberg AC, Li M, Sestan N, Greer CA, Imamura F. RNA-seq analysis of developing olfactory bulb projection neurons. Mol Cell Neurosci. 2016;74:78-86.
140. Tremblay C, Aslam S, Walker JE, et al. RNA sequencing of olfactory bulb in Parkinson’s disease reveals gene alterations associated with olfactory dysfunction. Neurobiol Dis. 2024;196:106514.
141. Lu T, Wang M, Zhou W, et al. Decoding transcriptional identity in developing human sensory neurons and organoid modeling. Cell. 2024;187:7374-93.e28.
142. Sharma N, Flaherty K, Lezgiyeva K, Wagner DE, Klein AM, Ginty DD. The emergence of transcriptional identity in somatosensory neurons. Nature. 2020;577:392-8.
143. Han X, Zhou Z, Fei L, et al. Construction of a human cell landscape at single-cell level. Nature. 2020;581:303-9.
144. Rossi LF, Harris KD, Carandini M. Spatial connectivity matches direction selectivity in visual cortex. Nature. 2020;588:648-52.
145. Wright CM, Schneider S, Smith-Edwards KM, et al. scRNA-Seq reveals new enteric nervous system roles for GDNF, NRTN, and TBX3. Cell Mol Gastroenterol Hepatol. 2021;11:1548-92.e1.
146. Beach TG, White CL, Hladik CL, et al.; The Arizona Parkinson’s Disease Consortium. Olfactory bulb α-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol. 2008;117:169.
147. Duda JE. Olfactory system pathology as a model of Lewy neurodegenerative disease. J Neurol Sci. 2010;289:49-54.
148. Puig J, Pedraza S, Blasco G, et al. Acute damage to the posterior limb of the internal capsule on diffusion tensor tractography as an early imaging predictor of motor outcome after stroke. AJNR Am J Neuroradiol. 2011;32:857-63.
149. Day IN, Thompson RJ. UCHL1 (PGP 9.5): neuronal biomarker and ubiquitin system protein. Prog Neurobiol. 2010;90:327-62.
150. Genç B, Lagrimas AKB, Kuru P, et al. Visualization of sensory neurons and their projections in an upper motor neuron reporter line. PLoS ONE. 2015;10:e0132815.
151. Taniguchi K, Harumi S, Okamura M, Ogawa K. Immunohistochemical demonstration of protein gene product 9.5 (PGP 9.5) in the primary olfactory system of the rat. Neurosci Lett. 1993;156:24-6.
152. Kruzich PJ, Grandy DK. Dopamine D2 receptors mediate two-odor discrimination and reversal learning in C57BL/6 mice. BMC Neurosci. 2004;5:12.
153. Zhou H, Zhuang L, Bao H, et al. Olfactory regulation by dopamine and DRD2 receptor in the nose. Proc Natl Acad Sci U S A. 2022;119:e2118570119.
154. Medeiros D, Masini D, Plewnia C, et al. Dopamine D2 receptor activation counteracts olfactory dysfunction and related cellular abnormalities in experimental parkinsonism. Heliyon. 2024;10:e35948.
155. Ramirez A, Heimbach A, Gründemann J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184-91.
156. Yang X, Xu Y. Mutations in the ATP13A2 gene and parkinsonism: a preliminary review. Biomed Res Int. 2014;2014:371256.
157. Dehay B, Ramirez A, Martinez-Vicente M, et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. 2012;109:9611-6.
158. Ritz B, Lee P, Hansen J, et al. Traffic-related air pollution and Parkinson’s disease in denmark: a case–control study. Environ Health Perspect. 2016;124:351-6.
159. Broen MP, Braaksma MM, Patijn J, Weber WE. Prevalence of pain in Parkinson’s disease: a systematic review using the modified QUADAS tool. Mov Disord. 2012;27:480-4.
161. Fil A, Cano-de-la-Cuerda R, Muñoz-Hellín E, Vela L, Ramiro-González M, Fernández-de-las-Peñas C. Pain in Parkinson disease: a review of the literature. Parkinsonism Relat Disord. 2013;19:285-94.
162. Taly AB, Muthane UB. Involvement of peripheral nervous system in juvenile Parkinson’s disease. Acta Neurol Scand. 2009;85:272-5.
163. Simchovitz A, Hanan M, Niederhoffer N, et al. NEAT1 is overexpressed in Parkinson’s disease substantia nigra and confers drug‐inducible neuroprotection from oxidative stress. FASEB J. 2019;33:11223-34.
164. Raunio A, Kivistö V, Kero M, et al. Distribution of Lewy-related pathology in the brain, spinal cord, and periphery: the population-based Vantaa 85 + study. Acta Neuropathol Commun. 2022;10:178.
165. Tavares-Ferreira D, Shiers S, Ray PR, et al. Spatial transcriptomics of dorsal root ganglia identifies molecular signatures of human nociceptors. Sci Transl Med. 2022;14:eabj8186.
166. Greenbaum L, Tegeder I, Barhum Y, Melamed E, Roditi Y, Djaldetti. Contribution of genetic variants to pain susceptibility in Parkinson disease. Eur J Pain. 2012;16:1243-50.
167. Reimann F, Cox JJ, Belfer I, et al. Pain perception is altered by a nucleotide polymorphism in SCN9A. Proc Natl Acad Sci U S A. 2010;107:5148-53.
168. Habib AM, Okorokov AL, Hill MN, et al. Microdeletion in a FAAH pseudogene identified in a patient with high anandamide concentrations and pain insensitivity. Br J Anaesth. 2019;123:e249-53.
169. Cajanus K, Holmström EJ, Wessman M, Anttila V, Kaunisto MA, Kalso E. Effect of endocannabinoid degradation on pain: role of FAAH polymorphisms in experimental and postoperative pain in women treated for breast cancer. Pain. 2016;157:361-9.
170. Latremoliere A, Latini A, Andrews N, et al. Reduction of neuropathic and inflammatory pain through inhibition of the tetrahydrobiopterin pathway. Neuron. 2015;86:1393-406.
171. Pan H, Zhao Y, Mei J, et al. GCH1 variants contribute to the risk and earlier age-at-onset of Parkinson’s disease: a two-cohort case-control study. Transl Neurodegener. 2020;9:31.
172. Larbalestier H, Keatinge M, Watson L, et al. GCH1 deficiency activates brain innate immune response and impairs tyrosine hydroxylase homeostasis. J Neurosci. 2022;42:702-16.
173. Franco R, Rivas-Santisteban R, Navarro G, Pinna A, Reyes-Resina I. Genes implicated in familial Parkinson’s disease provide a dual picture of nigral dopaminergic neurodegeneration with mitochondria taking center stage. Int J Mol Sci. 2021;22:4643.
174. Nguyen M, Wong YC, Ysselstein D, Severino A, Krainc D. Synaptic, mitochondrial, and lysosomal dysfunction in Parkinson’s disease. Trends Neurosci. 2019;42:140-9.
175. Smolders S, Van Broeckhoven C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson’s disease pathogenesis. Acta Neuropathol Commun. 2020;8:63.
176. Zhang F, Wu Z, Long F, et al. The roles of ATP13A2 gene mutations leading to abnormal aggregation of α-synuclein in Parkinson’s disease. Front Cell Neurosci. 2022;16:927682.
177. Veronesi B, Oortgiesen M. Neurogenic inflammation and particulate matter (PM) air pollutants. Neurotoxicology. 2001;22:795-810.
178. Morarach K, Mikhailova A, Knoflach V, et al. Diversification of molecularly defined myenteric neuron classes revealed by single-cell RNA sequencing. Nat Neurosci. 2020;24:34-46.
179. Beach TG, Adler CH, Sue LI, et al.; Arizona Parkinson’s Disease Consortium. Multi-organ distribution of phosphorylated α-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010;119:689-702.
180. Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: a dual‐hit hypothesis. Neuropathol Appl Neurobiol. 2007;33:599-614.
181. Matheoud D, Cannon T, Voisin A, et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1-/- mice. Nature. 2019;571:565-9.
182. Matheoud D, Sugiura A, Bellemare-Pelletier A, et al. Parkinson’s disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell. 2016;166:314-27.
183. Wallings RL, Tansey MG. LRRK2 regulation of immune-pathways and inflammatory disease. Biochem Soc Trans. 2019;47:1581-95.
184. Singh Y, Trautwein C, Dhariwal A, et al. DJ-1 (Park7) affects the gut microbiome, metabolites and the development of innate lymphoid cells (ILCs). Sci Rep. 2020;10:16131.
185. Masliah E, Rockenstein E, Veinbergs I, et al. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265-9.
186. Clemson CM, Hutchinson JN, Sara SA, et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell. 2009;33:717-26.
187. Boros FA, Maszlag-Török R, Vécsei L, Klivényi P. Increased level of NEAT1 long non-coding RNA is detectable in peripheral blood cells of patients with Parkinson’s disease. Brain Res. 2020;1730:146672.
188. Walker KL, Yoo HK, Undamatla J, Szaro BG. Loss of neurofilaments alters axonal growth dynamics. J Neurosci. 2001;21:9655-66.
189. Zhu Q, Couillard-Després S, Julien J. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol. 1997;148:299-316.
190. Hansson O, Janelidze S, Hall S, et al.; For the Swedish BioFINDER study. Blood-based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology. 2017;88:930-7.
191. Ashton NJ, Hye A, Rajkumar AP, et al. An update on blood-based biomarkers for non-Alzheimer neurodegenerative disorders. Nat Rev Neurol. 2020;16:265-84.
192. Liu Y, Dou K, Xue L, Li X, Xie A. Neurofilament light as a biomarker for motor decline in Parkinson’s disease. Front Neurosci. 2022;16:959261.
193. Hall S, Öhrfelt A, Constantinescu R, et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch Neurol. 2012;69:1445-52.
194. Saigoh K, Wang YL, Suh JG, et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet. 1999;23:47-51.
195. Leroy E, Boyer R, Auburger G, et al. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395:451-2.
196. Mayer RJ, Tipler C, Arnold J, et al. Endosome-lysosomes, ubiquitin and neurodegeneration. Adv Exp Med Biol. 1996;389:261-9.
197. Schultheis PJ, Fleming SM, Clippinger AK, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited α-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. 2013;22:2067-82.
198. Vrijsen S, Besora-Casals L, Van Veen S, et al. ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress. Proc Natl Acad Sci U S A. 2020;117:31198-207.
199. Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32:4240-6.
200. Fujii T, Nagamori S, Wiriyasermkul P, et al. Parkinson’s disease-associated ATP13A2/PARK9 functions as a lysosomal H+,K+-ATPase. Nat Commun. 2023;14:2174.
201. Ho PWL, Li L, Liu HF, et al. In vivo overexpression of synaptogyrin‐3 promotes striatal synaptic dopamine uptake in LRRK2R1441G mutant mouse model of Parkinson’s disease. Brain Behav. 2023;13:e2886.
202. Kulczyńska-Przybik A, Dulewicz M, Słowik A, et al. The clinical significance of cerebrospinal fluid reticulon 4 (RTN4) levels in the differential diagnosis of neurodegenerative diseases. J Clin Med. 2021;10:5281.
203. Styrpejko DJ, Cuajungco MP. Transmembrane 163 (TMEM163) protein: a new member of the zinc efflux transporter family. Biomedicines. 2021;9:220.
204. Nishioka K, Wider C, Vilariño-Güell C, et al. Association of α-, β-, and γ-synuclein with diffuse Lewy body disease. Arch Neurol. 2010;67:970-5.
205. Aune D, Schlesinger S, Mahamat-Saleh Y, Zheng B, Udeh-Momoh CT, Middleton LT. Diabetes mellitus, prediabetes and the risk of Parkinson’s disease: a systematic review and meta-analysis of 15 cohort studies with 29.9 million participants and 86,345 cases. Eur J Epidemiol. 2023;38:591-604.
206. Chohan H, Senkevich K, Patel RK, et al. Type 2 diabetes as a determinant of Parkinson’s disease risk and progression. Mov Disord. 2021;36:1420-9.
207. Guo M, Wang J, Zhao Y, et al. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain. 2020;143:1476-97.
208. Svenningsson P, Wirdefeldt K, Yin L, et al. Reduced incidence of Parkinson’s disease after dipeptidyl peptidase‐4 inhibitors - a nationwide case‐control study. Mov Disord. 2016;31:1422-3.
209. Tang H, Lu Y, Okun MS, et al. Glucagon‐like peptide‐1 receptor agonists and risk of Parkinson’s disease in patients with type 2 diabetes: a population‐based cohort study. Mov Disord. 2024;39:1960-70.
210. Meissner WG, Remy P, Giordana C, et al. Trial of lixisenatide in early Parkinson’s disease. N Engl J Med. 2024;390:1176-85.
211. Athauda D, Maclagan K, Skene SS, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1664-75.
212. Vijiaratnam N, Girges C, Auld G, et al. Exenatide once a week versus placebo as a potential disease-modifying treatment for people with Parkinson’s disease in the UK: a phase 3, multicentre, double-blind, parallel-group, randomised, placebo-controlled trial. Lancet. 2025;405:627-36.
213. Kalinderi K, Papaliagkas V, Fidani L. GLP-1 receptor agonists: a new treatment in Parkinson’s disease. Int J Mol Sci. 2024;25:3812.
214. Lu M, Su C, Qiao C, Bian Y, Ding J, Hu G. Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS clearance. Int J Neuropsychopharmacol. 2016;19:pyw047.
215. Matthews DC, Lerman H, Lukic A, et al. FDG PET Parkinson’s disease-related pattern as a biomarker for clinical trials in early stage disease. NeuroImage Clin. 2018;20:572-9.
216. Sulzer D, Alcalay RN, Garretti F, et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature. 2017;546:656–61.
218. Paik S, Kim JK, Silwal P, Sasakawa C, Jo E. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol. 2021;18:1141-60.
219. Qiao Y, Wang P, Qi J, Zhang L, Gao C. TLR‐induced NF‐κB activation regulates NLRP3 expression in murine macrophages. FEBS Lett. 2012;586:1022-6.
220. Holm CK, Rahbek SH, Gad HH, et al. Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat Commun. 2016;7:10680.
221. Nakajima M, Schmitt L, Halassa MM. Prefrontal cortex regulates sensory filtering through a basal ganglia-to-thalamus pathway. Neuron. 2019;103:445-58.e10.
222. Fullard ME, Morley JF, Duda JE. Olfactory dysfunction as an early biomarker in Parkinson’s disease. Neurosci Bull. 2017;33:515-25.
223. Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem. 2016;139 Suppl:216-31.
224. Liu B, Fang F, Pedersen NL, et al. Vagotomy and Parkinson disease: a Swedish register–based matched-cohort study. Neurology. 2017;88:1996-2002.
225. Reeve A, Simcox E, Turnbull D. Ageing and Parkinson’s disease: why is advancing age the biggest risk factor? Ageing Res Rev. 2014;14:19-30.
226. Reynoso A, Torricelli R, Jacobs BM, et al. Gene–environment interactions for Parkinson’s disease. Ann Neurol. 2024;95:677-87.
227. Fox SH, Katzenschlager R, Lim SY, et al.; on behalf of the Movement Disorder Society Evidence‐Based Medicine Committee. International Parkinson and movement disorder society evidence‐based medicine review: update on treatments for the motor symptoms of Parkinson’s disease. Mov Disord. 2018;33:1248-66.
228. Verschuur CVM, Suwijn SR, Boel JA, et al. Randomized delayed-start trial of levodopa in Parkinson’s disease. N Engl J Med. 2019;380:315-24.
229. PD Med Collaborative Group. Long-term effectiveness of dopamine agonists and monoamine oxidase B inhibitors compared with levodopa as initial treatment for Parkinson’s disease (PD MED): a large, open-label, pragmatic randomised trial. Lancet. 2014;384:1196-205.
230. Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N Engl J Med. 2000;342:1484-91.
231. Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. JAMA. 2000;284:1931-8.
233. Weintraub D, Mamikonyan E. Impulse control disorders in Parkinson’s disease. Am J Psychiatry. 2019;176:5-11.
234. Lees AJ, Ferreira J, Rascol O, et al.; for the BIPARK-2 Study Investigators. Opicapone as adjunct to levodopa therapy in patients with Parkinson disease and motor fluctuations: a randomized clinical trial. JAMA Neurol. 2017;74:197-206.
235. Deuschl G, Schade-Brittinger C, Krack P, et al. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med. 2006;355:896-908.
236. Olanow CW, Kieburtz K, Odin P, et al. Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson’s disease: a randomised, controlled, double-blind, double-dummy study. Lancet Neurol. 2014;13:141-9.
237. Limousin P, Foltynie T. Long-term outcomes of deep brain stimulation in Parkinson disease. Nat Rev Neurol. 2019;15:234-42.
238. Katzenschlager R, Poewe W, Rascol O, et al. Apomorphine subcutaneous infusion in patients with Parkinson’s disease with persistent motor fluctuations (TOLEDO): a multicentre, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2018;17:749-59.
239. Nemade D, Subramanian T, Shivkumar V. An update on medical and surgical treatments of Parkinson’s disease. Aging Dis. 2021;12:1021–35.
240. Richard IH, Mcdermott MP, Kurlan R, et al. A randomized, double-blind, placebo-controlled trial of antidepressants in Parkinson disease. Neurology. 2012;78:1229-36.
241. Dobkin RD, Menza M, Allen LA, et al. Cognitive-behavioral therapy for depression in Parkinson’s disease: a randomized, controlled trial. Am J Psychiatry. 2011;168:1066-74.
242. Dobkin RD, Mann SL, Gara MA, Interian A, Rodriguez KM, Menza M. Telephone-based cognitive behavioral therapy for depression in Parkinson disease: a randomized controlled trial. Neurology. 2020;94:e1764-73.
243. Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med. 2004;351:2509-18.
244. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. Lancet. 2014;383:533-40.
245. Kaufmann H, Freeman R, Biaggioni I, et al. Droxidopa for neurogenic orthostatic hypotension: a randomized, placebo-controlled, phase 3 trial. Neurology. 2014;83:328-35.
246. Seppi K, Ray Chaudhuri K, Coelho M, et al.; the collaborators of the Parkinson’s Disease Update on Non‐Motor Symptoms Study Group on behalf of the Movement Disorders Society Evidence‐Based Medicine Committee. Update on treatments for nonmotor symptoms of Parkinson’s disease - an evidence‐based medicine review. Mov Disord. 2019;34:180-98.
247. Pagano G, Taylor KI, Anzures-Cabrera J, et al. Trial of prasinezumab in early-stage Parkinson’s disease. N Engl J Med. 2022;387:421-32.
248. Volc D, Poewe W, Kutzelnigg A, et al. Safety and immunogenicity of the α-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: a randomised, single-blinded, phase 1 trial. Lancet Neurol. 2020;19:591-600.
249. Poewe W, Volc D, Seppi K, et al.; AFF011 study investigators. Safety and tolerability of active immunotherapy targeting α-synuclein with PD03A in patients with early Parkinson’s disease: a randomized, placebo-controlled, phase 1 study. J Parkinsons Dis. 2021;11:1079-89.
250. Heras‐Garvin A, Weckbecker D, Ryazanov S, et al. Anle138b modulates α‐synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy. Mov Disord. 2018;34:255-63.
251. Meglio M. Continued progress in phase 1 study of antisense oligonucleotide ION464 in MSA. 2024. Available from: https://www.neurologylive.com/view/continued-progress-phase-1-study-antisense-oligonucleotide-ion464-msa. [Last accessed on 29 Apr 2026].
252. Mullin S, Smith L, Lee K, et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol. 2020;77:427.
253. Siemeling O, Slingerland S, Van Der Zee S, Van Laar T. Study protocol of the GRoningen early-PD Ambroxol treatment (GREAT) trial: a randomized, double-blind, placebo-controlled, single center trial with ambroxol in Parkinson patients with a GBA mutation. BMC Neurol. 2024;24:146.
254. Nutt JG, Curtze C, Hiller A, et al. Aromatic L‐amino acid decarboxylase gene therapy enhances levodopa response in Parkinson’s disease. Mov Disord. 2020;35:851-8.
255. Mittermeyer G, Christine CW, Rosenbluth KH, et al. Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson’s disease. Hum Gene Ther. 2012;23:377-81.
256. Bloem BR, Macklin EA, Schwarzschild MA. GLP-1 agonists to slow down Parkinson’s progression? The quest continues. Med. 2025;6:100645.
257. Brakedal B, Dölle C, Riemer F, et al. The NADPARK study: a randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab. 2022;34:396-407.e6.
258. Yamamoto K, Ito H, Fukutake S, Kamei T, Yamaguchi T, Taira T. Ventralis intermedius thalamotomy with focused ultrasound for patients with low skull density ratio. Mov Disord. 2019;34:1239-40.
259. Surace MJ, Block ML. Targeting microglia-mediated neurotoxicity: the potential of NOX2 inhibitors. Cell Mol Life Sci. 2012;69:2409-27.
260. Geng L, Fan LM, Liu F, Smith C, Li JM. Nox2 dependent redox-regulation of microglial response to amyloid-β stimulation and microgliosis in aging. Sci Rep. 2020;10:1582.
261. ClinicalTrials.gov. A study of AAV2-GDNF in adults with moderate Parkinson’s disease (REGENERATE-PD). Available from: https://clinicaltrials.ucsf.edu/trial/NCT06285643. [Last accessed on 29 Apr 2026].
262. Van Laar AD, Christine CW, Phielipp N, et al. Intraputaminal delivery of adeno-associated virus serotype 2–glial cell line–derived neurotrophic factor in mild or moderate Parkinson’s disease. Mov Disord. 2025;40:1297–306.
263. Tabar V, Sarva H, Lozano AM, et al. Phase I trial of hES cell-derived dopaminergic neurons for Parkinson’s disease. Nature. 2025;641:978-83.
264. Tansey MG, Wallings RL, Houser MC, Herrick MK, Keating CE, Joers V. Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol. 2022;22:657-73.
265. Taymans J, Fell M, Greenamyre T, et al. Perspective on the current state of the LRRK2 field. NPJ Parkinsons Dis. 2023;9:104.
266. Zhang X, Tang B, Guo J. Parkinson’s disease and gut microbiota: from clinical to mechanistic and therapeutic studies. Transl Neurodegener. 2023;12:59.
267. De Sciscio M, Bryant RV, Haylock-Jacobs S, et al. Faecal microbiota transplant in Parkinson’s disease: pilot study to establish safety & tolerability. NPJ Parkinsons Dis. 2025;11:203.
268. Hutchison RM, Fraser K, Yang M, et al. Cinpanemab in early Parkinson disease: evaluation of biomarker results from the phase 2 SPARK clinical trial. Neurology. 2024;102:e209137.
269. Wang Y, Li Z, Li J, Sun C. LncRNA NEAT1 promotes MPP+ induced injury of PC12 cells and accelerates the progression of Parkinson’s disease in mice through FUS mediated inhibition of PI3K/AKT/mTOR signalling pathway. Exp Gerontol. 2024;191:112436.
270. Yan W, Chen Z, Chen J, Chen H. LncRNA NEAT1 promotes autophagy in MPTP-induced Parkinson’s disease through stabilizing PINK1 protein. Biochem Biophys Res Commun. 2018;496:1019-24.
271. Setsuie R, Wada K. The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem Int. 2007;51:105-11.
272. Osaka H. Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum Mol Genet. 2003;12:1945-58.
273. Erb ML, Sipple K, Levine N, Chen X, Moore DJ. Adult-onset deletion of ATP13A2 in mice induces progressive nigrostriatal pathway dopaminergic degeneration and lysosomal abnormalities. NPJ Parkinsons Dis. 2024;10:133.
274. Sikora J, Dovero S, Kinet R, et al. Nigral ATP13A2 depletion induces Parkinson’s disease-related neurodegeneration in a pilot study in non-human primates. NPJ Parkinsons Dis. 2024;10:141.
275. Dehay B, Martinez-Vicente M, Ramirez A, et al. Lysosomal dysfunction in Parkinson disease: ATP13A2 gets into the groove. Autophagy. 2014;8:1389-91.
276. Jennings D, Huntwork‐Rodriguez S, Vissers MF, et al. LRRK2 inhibition by BIIB122 in healthy participants and patients with Parkinson’s disease. Mov Disord. 2023;38:386-98.
277. Ysselstein D, Nguyen M, Young TJ, et al. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat Commun. 2019;10:5570.
278. Jennings D, Huntwork-Rodriguez S, Henry AG, et al. Preclinical and clinical evaluation of the LRRK2 inhibitor DNL201 for Parkinson’s disease. Sci Transl Med. 2022;14:eabj2658.
279. Cole TA, Zhao H, Collier TJ, et al. α-Synuclein antisense oligonucleotides as a disease-modifying therapy for Parkinson’s disease. JCI Insight. 2021;6:e135633.
280. Nakamori M, Junn E, Mochizuki H, Mouradian MM. Nucleic acid–based therapeutics for Parkinson’s disease. Neurotherapeutics. 2019;16:287-98.
281. Gu L, Zhang P, Gao R, Shu H, Wang P. Predictive value of serum neurofilament light chain for cognitive impairment in Parkinson’s disease. Front Aging Neurosci. 2024;16:1465016.
282. Tönges L, Buhmann C, Klebe S, et al. Blood-based biomarker in Parkinson’s disease: potential for future applications in clinical research and practice. J Neural Transm. 2022;129:1201-17.
283. Rissardo JP, Fornari Caprara AL. Alpha-synuclein seed amplification assays in Parkinson’s disease: a systematic review and network meta-analysis. Clin Pract. 2025;15:107.
284. Brockmann K, Quadalti C, Lerche S, et al. Association between CSF alpha-synuclein seeding activity and genetic status in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol Commun. 2021;9:175.
285. Agin-Liebes J, Lodge A, Reddy H, et al. α-Synuclein biomarker assays: bridging research and patient care. Lancet Neurol. 2025;24:681-97.
286. Wilson J, Yarnall AJ, Craig CE, et al. Cholinergic basal forebrain volumes predict gait decline in Parkinson’s disease. Mov Disord. 2020;36:611-21.
287. Prime M, Mckay JL, Bay AA, et al. Differentiating Parkinson disease subtypes using clinical balance measures. J Neurol Phys Ther. 2020;44:34-41.
288. Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol. 2010;67:715-25.
289. Grosch J, Winkler J, Kohl Z. Early degeneration of both dopaminergic and serotonergic axons - a common mechanism in Parkinson’s disease. Front Cell Neurosci. 2016;10:293.
290. Heng N, Malek N, Lawton MA, et al. Striatal dopamine loss in early Parkinson’s disease: systematic review and novel analysis of dopamine transporter imaging. Movement Disord Clin Pract. 2023;10:539-46.
291. Bajaj N, Hauser RA, Grachev ID. Clinical utility of dopamine transporter single photon emission CT (DaT-SPECT) with (123I) ioflupane in diagnosis of parkinsonian syndromes. J Neurol Neurosurg Psychiatry. 2013;84:1288-95.
292. Mercer MK, Revels JW, Blacklock LC, et al. Practical overview of 123I-ioflupane imaging in parkinsonian syndromes. Radiographics. 2024;44:e230133.
293. Seckin ZI, Whitwell JL, Utianski RL, et al. Ioflupane 123I (DAT scan) SPECT identifies dopamine receptor dysfunction early in the disease course in progressive apraxia of speech. J Neurol. 2020;267:2603–11.
294. Jokinen P, Helenius H, Rauhala E, Brück A, Eskola O, Rinne JO. Simple ratio analysis of 18F-fluorodopa uptake in striatal subregions separates patients with early Parkinson disease from healthy controls. J Nucl Med. 2009;50:893-9.
295. Madelung CF, Løkkegaard A, Fuglsang SA, et al. High-resolution mapping of substantia nigra in Parkinson’s disease using 7 tesla magnetic resonance imaging. NPJ Parkinsons Dis. 2025;11:113.
296. Lakhani DA, Zhou X, Tao S, et al. Diagnostic utility of 7T neuromelanin imaging of the substantia nigra in Parkinson’s disease. NPJ Parkinsons Dis. 2024;10:13.
297. Pyatigorskaya N, Sanz-Morère CB, Gaurav R, et al. Iron imaging as a diagnostic tool for Parkinson’s disease: a systematic review and meta-analysis. Front Neurol. 2020;11:366.
298. Wang Z, Luo X, Gao C. Utility of susceptibility-weighted imaging in Parkinson’s disease and atypical Parkinsonian disorders. Transl Neurodegener. 2016;5:17.
299. Langkammer C, Pirpamer L, Seiler S, et al. Quantitative susceptibility mapping in Parkinson’s disease. PLoS ONE. 2016;11:e0162460.
300. Depierreux F, Parmentier E, Mackels L, et al. Parkinson’s disease multimodal imaging: F-DOPA PET, neuromelanin-sensitive and quantitative iron-sensitive MRI. NPJ Parkinsons Dis. 2021;7:57.
301. Sun YM, Wang ZY, Liang YY, Hao CW, Shi CH. Digital biomarkers for precision diagnosis and monitoring in Parkinson’s disease. NPJ Digit Med. 2024;7:218.
302. Weirath NA, Hurben AK, Chao C, et al. Small molecule inhibitors of TET dioxygenases: Bobcat339 activity is mediated by contaminating copper(II). ACS Med Chem Lett. 2022;13:792-8.
303. Wiseman JA, Turner CP, Faull RLM, Halliday GM, Dieriks BV. Refining α-synuclein seed amplification assays to distinguish Parkinson’s disease from multiple system atrophy. Transl Neurodegener. 2025;14:7.
304. Kuzkina A, Bargar C, Schmitt D, et al. Diagnostic value of skin RT-QuIC in Parkinson’s disease: a two-laboratory study. NPJ Parkinsons Dis. 2021;7:99.
305. Gibbons CH, Levine T, Adler C, et al. Skin biopsy detection of phosphorylated α-synuclein in patients with synucleinopathies. JAMA. 2024;331:1298-306.
306. Pedersen CC, Ushakova A, Alves G, et al. Serum neurofilament light at diagnosis: a prognostic indicator for accelerated disease progression in Parkinson’s disease. NPJ Parkinsons Dis. 2024;10:162.
307. Donadio V, Wang Z, Incensi A, et al. In vivo diagnosis of synucleinopathies: a comparative study of skin biopsy and RT-QuIC. Neurology. 2021;96:e2513-24.
308. Che N, Huang J, Wang S, et al. The potential use of plasma NfL as a diagnostic and prognostic biomarker of fatigue in early Parkinson’s disease. Ther Adv Neurol Disord. 2025;18:17562864251324406.
309. Iakovakis D, Hadjidimitriou S, Charisis V, Bostantzopoulou S, Katsarou Z, Hadjileontiadis LJ. Touchscreen typing-pattern analysis for detecting fine motor skills decline in early-stage Parkinson’s disease. Sci Rep. 2018;8:7663.
310. Bhidayasiri R, Mari Z. Digital phenotyping in Parkinson’s disease: empowering neurologists for measurement-based care. Parkinsonism Relat Disord. 2020;80:35-40.
311. Tat T, Chen G, Xu J, Zhao X, Fang Y, Chen J. Diagnosing Parkinson’s disease via behavioral biometrics of keystroke dynamics. Sci Adv. 2025;11:eadt6631.
312. Gardener H, Gao X, Chen H, Schwarzschild MA, Spiegelman D, Ascherio A. Prenatal and early life factors and risk of Parkinson’s disease. Mov Disord. 2010;25:1560-7.
313. Gardner RC, Byers AL, Barnes DE, Li Y, Boscardin J, Yaffe K. Mild TBI and risk of Parkinson disease. Neurology. 2018;90:e1771-9.
314. Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18:1091-102.
315. Saffie-Awad P, Grant SM, Makarious MB, et al.; Global Parkinson’s Genetics Program (GP2). Insights into ancestral diversity in Parkinson’s disease risk: a comparative assessment of polygenic risk scores. NPJ Parkinsons Dis. 2025;11:201.
316. Pereira PAB, Trivedi DK, Silverman J, et al. Multiomics implicate gut microbiota in altered lipid and energy metabolism in Parkinson’s disease. NPJ Parkinsons Dis. 2022;8:39.
317. Hu L, Dong M, Huang Y, et al. Integrated metabolomics and proteomics analysis reveals plasma lipid metabolic disturbance in patients with Parkinson’s disease. Front Mol Neurosci. 2020;13:80.
318. Mapstone M, Cheema AK, Fiandaca MS, et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med. 2014;20:415-8.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].