


Glucose metabolic dysregulation and oxidative stress in cholangiocarcinoma: molecular mechanisms, oncogenic drivers, and novel therapeutic targets
 0
0 
Abstract
Cholangiocarcinoma (CCA) is an aggressive malignancy marked by profound glucose metabolic dysregulation and oxidative stress. Central to this reprogramming is the upregulation of glucose transporters such as GLUT1, driving enhanced glycolytic flux, activation of the pentose phosphate pathway (PPP), increased lactate production, and alterations in the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS). These metabolic shifts support tumor proliferation, redox balance, and stemness, and are closely linked to recurrent oncogenic mutations, including KRAS, TP53, IDH1/2, ARID1A, FGFR2, and HER2. These mutations converge on key signaling networks that promote metabolic plasticity and therapeutic resistance. Recent evidence suggests that targeting metabolic vulnerabilities offers promising avenues for intervention. Inhibitors of glycolytic enzymes (HKII, PKM2), PPP regulators (G6PD, TKT), TCA cycle components [IDH, glutaminase (GLS)], lactate metabolism (LDHA), and OXPHOS machinery (Complex I) have demonstrated potential in preclinical models. Additionally, repurposing antidiabetic drugs such as metformin and SGLT2 inhibitors may offer novel metabolic therapies. Regulatory non-coding RNAs, including microRNAs and long non-coding RNAs, further modulate key enzymes and transporters, highlighting their emerging roles as both biomarkers and therapeutic targets. However, challenges such as tumor heterogeneity, metabolic redundancy, off-target toxicity, and resistance mechanisms continue to hinder clinical translation. Integrated therapeutic approaches combining metabolic inhibitors with chemotherapy, immunotherapy, or targeted agents are likely necessary to overcome these barriers. This review synthesizes the current understanding of glucose metabolism and redox dysregulation in CCA, emphasizing the molecular drivers, therapeutic opportunities, and translational challenges, with the goal of guiding future research toward more effective and personalized treatment strategies.
Keywords
INTRODUCTION
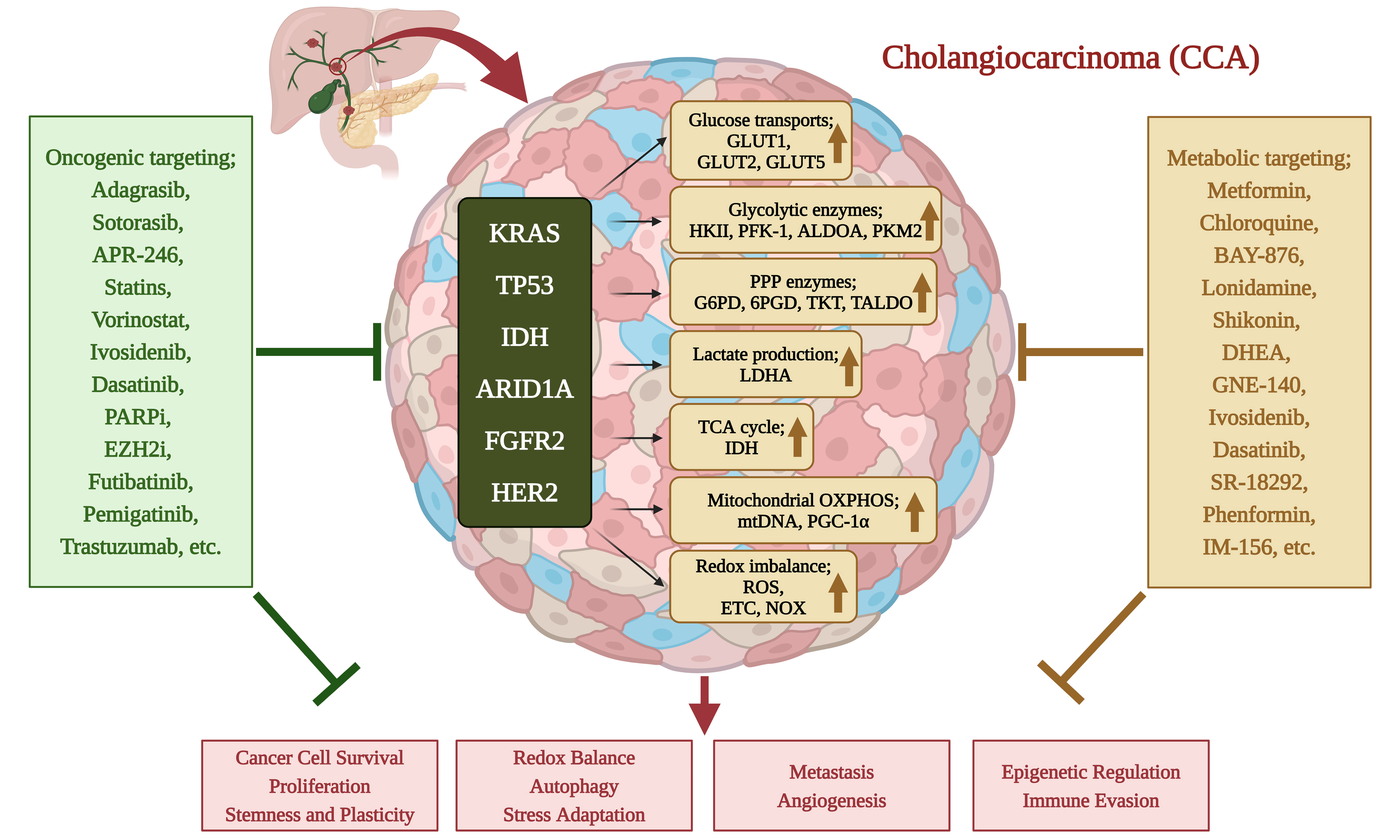
Cholangiocarcinoma (CCA) is an aggressive adenocarcinoma and the second most common primary liver cancer after hepatocellular carcinoma (HCC). It originates in the biliary tract, representing approximately 15% of primary liver tumors and 3% of gastrointestinal cancers[1]. CCA is classified into intrahepatic (iCCA), arising within the liver, and extrahepatic (eCCA), which is further subdivided into perihilar (pCCA) and distal (dCCA) subtypes[1,2]. Among these, pCCA is the most common, accounting for 50%-60% of cases, followed by dCCA (20%-30%) and iCCA (10%-20%)[1] [Figure 1]. While rare in Western countries, CCA is highly prevalent in Southeast Asia, with rising global mortality rates[1,3]. In Thailand, CCA poses a major public health concern, with Northeastern Thailand reporting the highest incidence rates globally (113/100,000 males and 50/100,000 females), making it the leading cause of cancer-related death in the region[4-6].
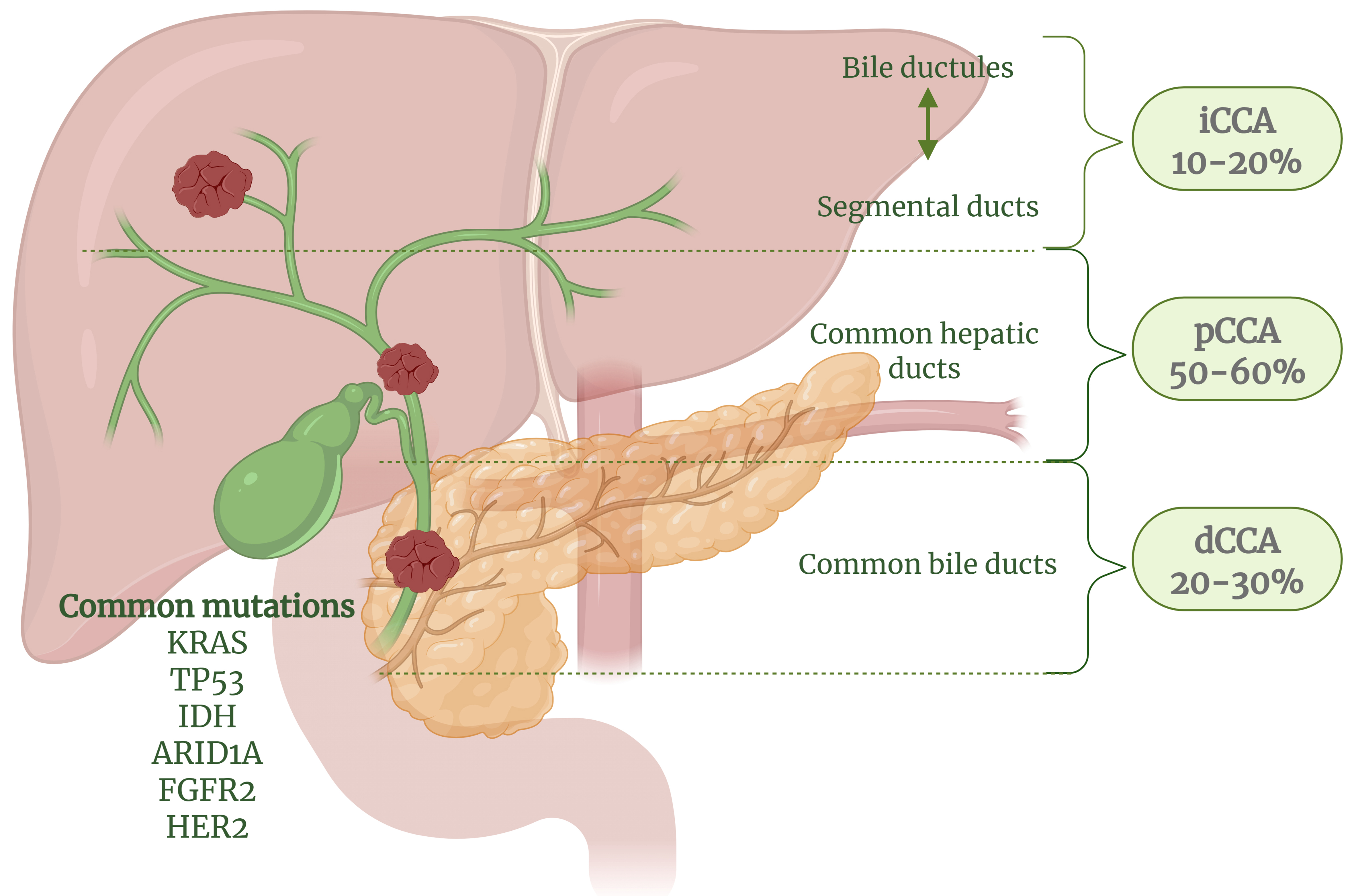
Figure 1. Classification of CCA based on its anatomical site of origin. iCCA originates in the periphery of the second-order bile ducts, pCCA arises in the right and/or left hepatic ducts and their junction, and dCCA involves the common bile duct (choledochus). CCA presents with three growth patterns: mass-forming, periductal-infiltrating, and intraductal-growing. Mass-forming CCA appears as a hepatic mass lesion, periductal-infiltrating iCCA grows along the duct wall, and intraductal-growing CCA forms a polypoid or papillary tumor extending into the duct lumen. Common oncogenic mutations contributing to CCA include: KRAS, TP53, IDH, ARID1A, FGFR2, HER2. These mutations drive tumorigenesis by altering key cellular pathways. Created in BioRender. Chanda M. (2025) https://BioRender.com/s4pk8iv. CCA: Cholangiocarcinoma; iCCA: intrahepatic cholangiocarcinoma; pCCA: perihilar cholangiocarcinoma; dCCA: distal cholangiocarcinoma; KRAS: Kirsten rat sarcoma viral oncogene homolog; TP53: tumor protein p53; IDH: isocitrate dehydrogenase; ARID1A: AT-rich interaction domain 1A; FGFR2: fibroblast growth factor receptor 2; HER2: human epidermal growth factor receptor 2.
Patients diagnosed with CCA face a poor prognosis, with a median survival time of 24 months[7] and a 5-year survival rate of less than 5%[6]. Early-stage diagnosis is challenging due to the asymptomatic nature of the disease, with most patients being diagnosed at advanced stages when surgical options are no longer viable. Only about 35% of cases are detected early, and even fewer (25%) are eligible for surgery due to delayed detection[8]. The poor prognosis is further complicated by the genetic, epigenetic, and functional heterogeneity of CCA, which includes diverse histopathological subtypes, molecular mutations [e.g., Kirsten rat sarcoma virus (KRAS), tumor protein p53 (TP53), isocitrate dehydrogenase (IDH), AT-rich interactive domain 1A (ARID1A), fibroblast growth factor receptor (FGFR), and human epidermal growth factor receptor 2 (HER2)], and alterations in oncogenic and inflammatory pathways[9-13]. This complexity underscores the need for a better understanding of the mechanisms driving resistance to treatment.
Currently, the first-line chemotherapy for CCA is gemcitabine[14], often in combination with cisplatin for advanced or unresectable cases[1,15,16]. However, the effectiveness of these treatments is limited due to chemoresistance[7,8], driven by mechanisms such as altered cellular metabolism, drug-metabolizing enzymes, efflux transporter overexpression, and disruptions in intracellular signaling[17]. These resistances underscore the need for targeted therapies and immunotherapies to improve treatment outcomes[1,15]. Understanding the underlying mechanisms of resistance to chemotherapy and targeted therapies, as well as the role of metabolic reprogramming in CCA progression, is essential for developing more effective, molecularly targeted treatments[18].
Altered glucose metabolism plays a key role in CCA and many other cancers, contributing to tumorigenesis by supporting rapid proliferation and chemoresistance[6]. This dysregulation is a fundamental characteristic of cancer, as it interacts with oncogenic signaling pathways to sustain tumor growth and various cancer phenotypes[19,20]. A prominent feature in many cancers, including CCA, is the Warburg effect, in which glucose metabolism shifts toward increased glycolysis, even in the presence of oxygen, to maintain energy balance and support rapid proliferation[20,21]. Glucose enters the cell via glucose transporters (GLUTs) and is metabolized through glycolysis by several rate-limiting enzymes, including hexokinase (HK), phosphofructokinase (PFK), pyruvate kinase (PK), and glucose-6-phosphate (G6P) dehydrogenase (G6PD) in the pentose phosphate pathway (PPP)[22]. This process generates ATP, with pyruvate converted to acetyl-CoA for oxidative phosphorylation (OXPHOS) in mitochondria, producing up to 36-38 ATP molecules per glucose molecule under normal conditions[22,23]. However, in cancer cells, pyruvate is predominantly converted to lactate by lactate dehydrogenase A (LDHA), even under aerobic conditions, driving the Warburg effect and promoting lactate production [Figure 2]. This metabolic reprogramming, which includes alterations in glycolysis, the PPP, lactate production, the tricarboxylic acid (TCA) cycle, and OXPHOS, is essential for tumorigenesis and survival[24]. Energy metabolic reprogramming (EMR) and altered redox homeostasis are increasingly recognized as hallmarks of cancer[24,25]. CCA cells exhibit significant metabolic plasticity, adapting to fluctuating environmental conditions and energy demands[8,20,21]. Elevated LDH levels are frequently observed in CCA[26], further highlighting these metabolic alterations.
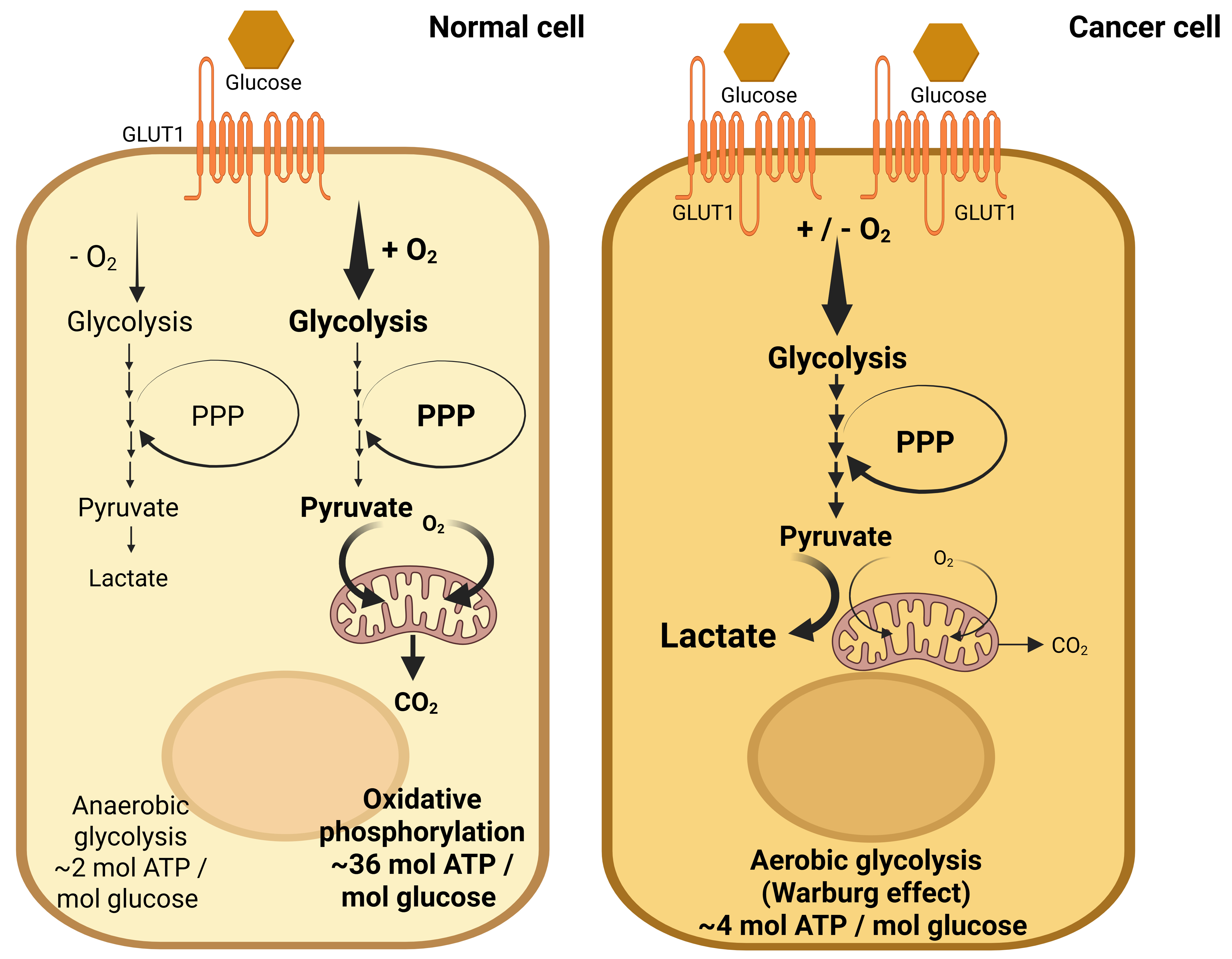
Figure 2. Glucose metabolism in normal cells vs. the Warburg effect in cancer cells. In normal cells (left), glucose is transported via GLUT1 and metabolized through glycolysis and the PPP, generating lactate and ATP, while OXPHOS produces additional ATP in the presence of oxygen. In CCA cells (right), GLUT1 expression is upregulated, leading to increased glucose uptake. This enhances glycolysis and the PPP, promoting greater lactate production. Despite the availability of oxygen, CCA cells exhibit reduced OXPHOS activity, characteristic of the Warburg effect, in which glucose metabolism shifts toward increased glycolysis to maintain energy balance and support rapid proliferation. Created in BioRender. Chanda, M. (2025) https://BioRender.com/d1db3yv. GLUT1: Glucose transporters 1; PPP: pentose phosphate pathway; ATP: adenosine triphosphate; OXPHOS: oxidative phosphorylation; CCA: cholangiocarcinoma.
Recent studies highlight the link between common genetic alterations in CCA and metabolic shifts, investigating these reprogramming pathways as promising therapeutic targets to improve treatment efficacy[8,20,21]. Genetic alterations in oncogenes and tumor suppressors, such as KRAS[27,28], TP53[29-31], IDH[32,33], ARID1A[34], FGFR[35], and HER2[36], drive this metabolic shift, supporting tumor growth by enhancing biosynthesis for rapid cell division[6]. KRAS mutations promote glycolysis to support cancer cell growth, while TP53 mutations impair mitochondrial function and favor glycolysis, both of which create opportunities for therapeutic interventions targeting these metabolic alterations[27,28,31]. IDH1 mutations in CCA disrupt TCA and lead to oncometabolite accumulation. Ivosidenib, an IDH1 inhibitor targeting a key TCA enzyme, has recently been approved for the treatment of chemoresistant metastatic CCA harboring IDH1 mutations[37]. ARID1A loss in cancers such as HCC activates the AMP-activated protein kinase (AMPK) pathway, altering glycolysis and lipid metabolism, and offering a potential target for AMPK inhibition[34]. FGFR2 activation in CCA drives aerobic glycolysis through NF-κB signaling, making FGFR2 inhibitors a viable strategy for targeting metabolic reprogramming[35]. HER2 overexpression enhances glycolysis through the Akt and mTORC1 pathways, suggesting that targeting HER2-associated glycolysis could overcome treatment resistance[36]. Moreover, increased metabolic activity in cancer cells elevates reactive oxygen species (ROS) levels, countered by nuclear factor erythroid 2-related factor 2 (NRF2)-mediated antioxidant defenses, which can be dysregulated to support tumor survival and therapy resistance, presenting further therapeutic opportunities[38]. Integrating tumor metabolism into diagnostic and treatment strategies could enable earlier detection, enhance therapeutic effectiveness, and improve overall outcomes[8]. Therapeutic interventions targeting these metabolic dysregulations, including IDH1 inhibitors, metformin, GLS inhibitors, and sphingosine kinase inhibitors, offer promising prospects for improving CCA treatment outcomes[37]. These therapies could counteract the metabolic adaptability of CCA, enhancing survival and quality of life. Continued research into tumor metabolism and cancer progression remains crucial for developing next-generation therapies that target the metabolic vulnerabilities of CCA and other cancers[37].
In this review, we explore the key metabolic alterations in CCA, focusing on glucose metabolism, including the reprogramming of glycolysis, the PPP, lactate production, the TCA cycle, and OXPHOS. Additionally, we examine how these metabolic dysregulations are intricately linked to oncogenic enzymatic pathways and disrupted redox homeostasis - common features of cancer that are central to its pathogenesis. Importantly, we will discuss the potential to exploit these metabolic vulnerabilities for novel therapeutic strategies. By mapping the metabolic networks in CCA, this review aims to identify key alterations that could serve as targets for therapy, paving the way for the discovery of effective compounds for CCA treatment - and potentially other cancers. Ultimately, our goal is to emphasize how these insights may inform treatment strategies, enhance patient outcomes, and contribute to the development of next-generation cancer therapies.
ALTERED GLUCOSE METABOLISM IN CCA
In CCA, altered glucose metabolism is supported by key pathways, including glycolysis, the PPP, lactate production, the TCA cycle, and OXPHOS[39]. CCA cells preferentially convert pyruvate to lactate over OXPHOS despite the presence of oxygen, driving metabolic reprogramming or the Warburg effect, which plays a critical role in tumor progression, survival, and therapy resistance[21]. This shift ensures a continuous supply of energy and intermediates for rapid proliferation, generating key metabolites like G6P and fructose-6-phosphate (F6P) that are essential for nucleotide and lipid biosynthesis, supporting tumor growth[21]. Genetic alterations in oncogenes and tumor suppressors, including KRAS[27,28], TP53[29-31], IDH[32,33], ARID1A[34], FGFR[35], and HER2[36], contribute to this reprogramming of glucose metabolism in CCA. Although the evidence in CCA remains limited, these findings suggest that genetic mutations in key oncogenes and tumor suppressors may drive alterations in glucose metabolism and tumor progression. These genetic mutations likely drive alterations in glucose metabolism, further fueling tumor progression. Therefore, in the context of altered glucose metabolism in CCA, we will focus on key aspects, including glucose uptake via GLUTs, rate-limiting enzymes in glycolysis and the PPP, lactate production, the TCA cycle, and OXPHOS. We will also address the genetic alterations commonly observed in CCA, which contribute to the reprogramming of these metabolic pathways [Figure 3 and Table 1].
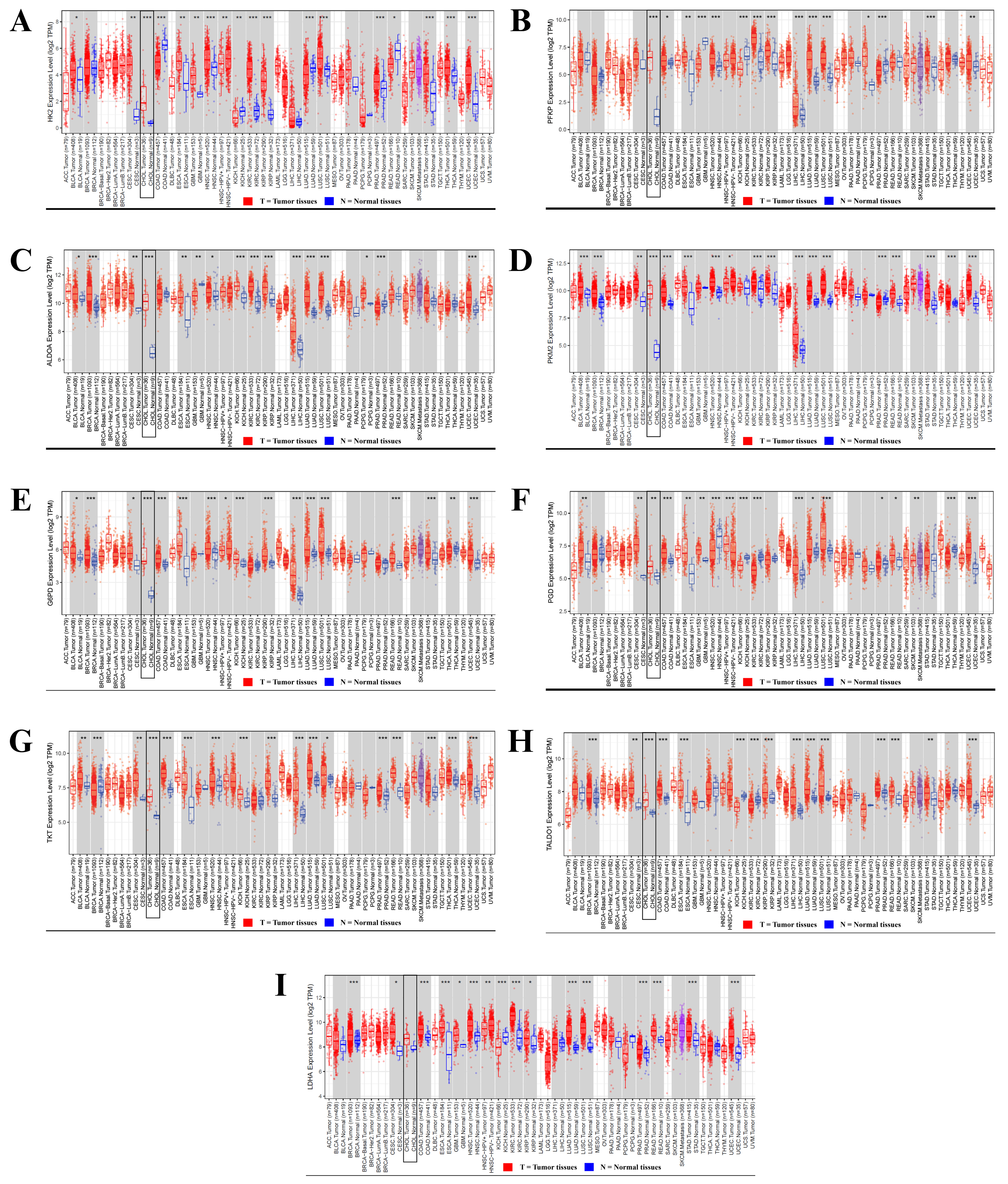
Figure 3. Differential expression of glucose metabolic enzymes in CCA. The distribution of gene expression levels is displayed using box plots (red: tumor tissues; blue: normal tissues) in panels A-I. (A) HKII; (B) PFKP; (C) ALDOA; (D) PKM2; (E) G6PD; (F) 6PGD; (G) TKT; (H) TALDO; (I) LDHA. Statistical significance was determined using the Wilcoxon test, with P-values indicated by the number of stars
Metabolic rewiring and targetable pathways in CCA
| Metabolic process | Target/Enzyme (Abbreviation) | Mechanistic role in CCA | References |
| Glucose uptake | GLUT1 | Overexpressed in CCA; correlates with poor survival, tumor aggressiveness, KRAS mutations, and FDG uptake. Knockdown reduces proliferation and invasion | [22,40-43,47,48] |
| GLUT2 | Expressed in large duct CCA and precursor lesions; involved in glucose/fructose transport | [44] | |
| GLUT5 | Upregulated in CCA; supports fructose metabolism. Knockdown impairs proliferation and migration | [45,46] | |
| GLUTs 3, 4, 6-14 | Roles in CCA unclear; implicated in glucose handling in other cancers | [45,49] | |
| Glycolysis | HKII | Catalyzes glucose to G6P; overexpressed in CCA; promotes proliferation, invasion; regulated by c-MYC. Knockdown impairs tumor growth | [43,50-52] |
| PFK1 / PFKP | Converts F6P to F1,6BP; overexpressed in CCA, especially IDH1-mutant iCCA; correlates with poor prognosis | [43,53,54] | |
| ALDOA | Cleaves F1,6BP to G3P and DHAP; elevated in CCA; enhances proliferation and migration; associated with poor survival | [43,55] | |
| PKM2 | Final glycolytic step; upregulated in CCA; promotes proliferation, recurrence, chemoresistance; c-MYC-regulated. Targetable by Shikonin | [43,51,56-60] | |
| PPP | G6PD | Rate-limiting enzyme; promotes NADPH production, redox balance, DNA repair, metastasis, and cisplatin resistance. Inhibition restores chemosensitivity | [8,61-64,78] |
| 6PGD | Supports tumor proliferation and migration via oxidative PPP flux; regulated by NRF2; linked to poor prognosis | [62,65-67] | |
| TKT | Enhances proliferation, invasion, chemoresistance; linked to AKT, NRF2, and EGFR signaling; suppressed by NRF2 knockdown | [62,72,73,77] | |
| TALDO | Contributes to metabolic rewiring and tumor progression; interacts with oncogenic proteins (e.g., TP53, HKII) | [62,68,74] | |
| Lactate metabolism | LDHA | Catalyzes pyruvate to lactate; regenerates NAD+; supports ATP production, tumor invasiveness, and redox balance; upregulated in iCCA | [26,81] |
| TCA cycle | IDH1-mut | Converts α-KG to 2-HG; promotes ROS, impairs differentiation, enhances glycolysis and tumor progression; found in ~15%-20% iCCA | [54,85-87] |
| OXPHOS | mtDNA | mtDNA depletion or mutation impairs respiratory complexes, reducing OXPHOS function in CCA cells | [89,90] |
| PGC-1α | Regulates mitochondrial biogenesis and stemness; promotes CSC self-renewal, invasion, and drug resistance in CCA | [94,95] |
Role of GLUTs in CCA
GLUTs are essential regulators of glucose uptake and glycolysis, central to tumor metabolism. Among the 14 GLUTs (GLUT1-14, encoded by SLC2A1-14), GLUT1 is the most prominently upregulated in malignancies and is strongly associated with aggressive cancer traits such as proliferation, metastasis, and therapy resistance[22]. In CCA, GLUT1 is significantly overexpressed and serves as both a diagnostic and prognostic biomarker. Elevated GLUT1 expression correlates with reduced overall survival, especially in distal CCA, and is enriched in moderately to poorly differentiated tumors[40-42]. Overexpression is linked to adverse pathological features, including larger tumor size, lymphovascular invasion, and lymph node metastasis[42], while siRNA-mediated knockdown reduces proliferation, migration, and invasion in CCA cell lines[42]. GLUT1 expression also correlates with KRAS mutations and increased FDG-PET uptake, highlighting its role in metabolic reprogramming and potential as an imaging biomarker[43]. These findings establish GLUT1 as a central driver of glycolysis and a promising therapeutic target in CCA.
Other GLUTs have also been implicated in CCA. GLUT2, a low-affinity glucose and fructose transporter, is associated with large bile duct adenocarcinoma and early lesions such as high-grade biliary intraepithelial neoplasia[44]. GLUT5, a fructose-specific transporter, is significantly overexpressed in CCA compared to normal biliary tissue; its knockdown impairs CCA cell proliferation, migration, and invasion, underscoring a metabolic reliance on fructose[45]. In liver-fluke-associated CCA models, both GLUT1 and GLUT5 expression progressively increase throughout tumorigenesis[45,46]. These data suggest that targeting fructose metabolism via GLUT5, in parallel with GLUT1-mediated glucose transport, may offer combinatorial therapeutic potential.
Beyond CCA, GLUT1 overexpression is a hallmark of various cancers, including colorectal, lung, and breast cancers, where it enhances glucose uptake and supports tumor growth under hypoxic conditions[47]. GLUT1 knockdown reduces proliferation in preclinical models, and selective inhibition by BAY-876 has demonstrated efficacy in vitro and in vivo[48]. Other GLUTs also play roles in oncogenesis: GLUT3 is regulated by TP53 and enriched in high-glucose-demand tumors such as glioblastoma; GLUT4 is involved in insulin-responsive glucose uptake via the PI3K-AKT pathway and contributes to cancer cell viability[49]. While Class II and III GLUTs such as GLUT6, GLUT8, and GLUT12 are less well characterized, some exhibit altered expression in cancers and may modulate intracellular glucose handling or metabolic signaling.
Collectively, GLUT1 is a critical metabolic regulator in CCA and other tumors, with strong potential as a therapeutic target. Additional GLUTs, particularly GLUT2 and GLUT5 in CCA, may further refine metabolic vulnerabilities for intervention.
Glycolytic enzyme dysregulation in CCA
Glycolysis plays a central role in the altered glucose metabolism observed in CCA, with several glycolytic enzymes exhibiting dysregulated expression, thereby enhancing glycolytic flux to support the energy demands of rapidly proliferating tumor cells. Key enzymes such as hexokinase II (HKII), phosphofructokinase-1 (PFK-1), aldolase A (ALDOA), and pyruvate kinase M2 (PKM2) are pivotal in driving tumor aggressiveness[22]. Upregulation of these enzymes contributes to the rapid conversion of glucose to pyruvate, facilitating biosynthesis and ATP production necessary for cancer cell proliferation. These enzymes are central to the altered metabolic network in CCA, positioning them as potential therapeutic targets for disrupting cancer cell metabolism[43].
HKII
HKII, a rate-limiting enzyme in the glycolytic pathway, catalyzes the phosphorylation of glucose to G6P. The overexpression of HKII has been observed in various gastrointestinal cancers, including CCA [Figure 3A], where it contributes to the metabolic reprogramming characteristic of tumor progression[50]. In addition, the inhibition of HKII expression through siRNA transfection results in reduced CCA cell proliferation and invasiveness, further underscoring its pivotal role in driving malignancy[50]. HKII is overexpressed in a significant proportion of CCA cases and has been linked to increased tumor aggressiveness. This enzyme plays a crucial role in tumor progression, as inhibition of HKII, along with other glycolytic enzymes, has shown potential for reducing tumor growth, positioning glycolysis as a promising therapeutic target in CCA[51]. HKII expression is regulated by myelocytomatosis oncogene (c-MYC), a key regulator of cellular metabolism and proliferation. c-MYC also influences FGFR-mediated lymphangiogenesis and lymphatic metastasis in iCCA, highlighting the possible implications of HKII regulation in tumor biology[52].
PFK-1
PFK-1 is a key rate-limiting enzyme in glycolysis, catalyzing the conversion of F6P to fructose 1,6-bisphosphate (F1,6P), a crucial regulatory step. Among its isoforms, platelet-type PFK (PFKP) plays a significant role in cancer metabolism. Overexpression of PFK-1 has been observed in various cancers, including CCA [Figure 3B], and is linked to poorer clinical outcomes[53]. Elevated PFK-1 expression is associated with enhanced glycolytic activity, driving cancer cell proliferation and metastasis[53]. In iCCA with IDH1 mutations, increased PFKP expression and PFK-1 activity were observed, suggesting that IDH1 mutations promote glycolysis through PFK-1 upregulation[54]. These findings underscore the potential of PFK-1 as a prognostic marker and therapeutic target in CCA and other cancers.
ALDOA
ALDOA, a key enzyme in the glycolytic pathway, catalyzes the conversion of F1,6P to glyceraldehyde-3-phosphate (G3P) and dihydroxyacetone phosphate (DHAP). High ALDOA expression has been observed in various cancers, including CCA [Figure 3C], and is strongly associated with tumor malignancy and poor prognosis[43,55]. In iCCA, ALDOA expression is elevated and correlates with poor prognosis[55]. By enhancing glycolysis, ALDOA promotes iCCA progression, making its enzymatic activity a promising therapeutic target[55]. ALDOA plays a pivotal role in tumor progression by promoting cell proliferation and migration. Knockdown of ALDOA significantly inhibits these processes, while overexpression enhances them. Elevated ALDOA expression is inversely correlated with overall survival and recurrence-free survival in CCA patients, highlighting its potential as a prognostic marker and therapeutic target. Inhibition of ALDOA using siRNA in iCCA cells (HuCCT1 and RBE) suppresses glycolysis, reduces tumor growth, and impairs proliferation and invasion. Studies have demonstrated that ALDOA regulates the metabolic and biological characteristics of iCCA cells, further suggesting its potential as a therapeutic target[43,55].
PKM2
PKM2, the final enzyme in glycolysis, catalyzes the conversion of phosphoenolpyruvate to pyruvate. PKM2 is upregulated in various cancers, including CCA [Figure 3D], where it plays a critical role in tumor progression. In CCA, elevated PKM2 expression is associated with aggressive clinical features such as neural invasion and tumor recurrence and is regulated by c-MYC, linking it to oncogenic pathways[56,57]. In iCCA, high PKM2 expression correlates with poor prognosis and is inversely related to overall survival and recurrence-free survival[58]. PKM2 promotes cell proliferation, migration, and angiogenesis in CCA cells, with increased levels serving as an independent predictor of tumor progression and recurrence after resection of pCCA[51,58]. In vitro knockdown of PKM2 significantly inhibits these processes and enhances the sensitivity of iCCA cells to gemcitabine treatment by inhibiting the β-catenin signaling pathway[59]. Targeting PKM2 with inhibitors like Shikonin can regulate tumor metabolism, reduce growth in CCA xenograft models, and provide antitumor effects, making PKM2 a promising therapeutic target in CCA[43,60].
The dysregulation of the glycolytic pathway in CCA underscores the essential role of key metabolic enzymes in driving tumorigenesis. These enzymes, which are crucial for the progression of CCA, represent promising targets for metabolism-based therapies. Inhibiting their activity could effectively disrupt glycolytic flux, attenuate tumor growth, and limit metastatic potential, thereby offering a novel and potentially impactful therapeutic approach. Consequently, targeting the metabolic reprogramming in CCA holds substantial promise, providing new therapeutic avenues to counteract the metabolic alterations and improve patient outcomes.
Dysregulation of the PPP in CCA
An increasing body of evidence links dysregulated redox homeostasis to cancer metabolic reprogramming[61]. The PPP, a critical branch of glycolysis, plays a central role in this reprogramming, particularly in CCA. This pathway is essential for supporting the rapid proliferation of cancer cells by generating ribose-5-phosphate (R5P) for nucleotide biosynthesis and nicotinamide adenine dinucleotide phosphate (NADPH) for maintaining redox homeostasis and mitigating oxidative stress[8]. The rate-limiting enzyme of the PPP, G6PD, directs glucose flux toward these products, which are crucial for cancer cell survival and DNA damage repair[61]. The PPP consists of two phases: oxidative and nonoxidative. The oxidative phase involves three enzymatic reactions that produce two NADPH molecules and ribulose-5-phosphate (Ru5P), with key enzymes such as G6PD and 6-phosphogluconate dehydrogenase (6PGD). The reversible nonoxidative phase begins with Ru5P, which is converted into R5P through the actions of transketolase (TKT) and transaldolase (TALDO)[62]. Together, these processes contribute to the dysregulation of redox balance, further supporting cancer progression and therapeutic resistance[61].
Irreversible oxidative PPP: G6PD and 6PGD
G6PD and 6PGD play crucial roles in tumorigenesis. G6PD has been shown to be upregulated in various cancers including CCA [Figure 3E], where it is associated with metastasis, advanced disease stages, and poor overall survival[63]. Elevated G6PD expression correlates with worse prognosis in multiple malignancies, including gastrointestinal tract cancers, as well as in breast, renal, and lung cancers[61,62]. Notably, patients with G6PD-positive tumors tend to have a lower survival rate compared to those with G6PD-deficient tumors[64]. Enhanced G6PD activity has also been reported in cancers such as thyroid, colorectal, renal, and prostate cancer. This upregulation is driven by oncogenes like platelet-derived growth factor, epidermal growth factor, phosphoinositide 3-kinase, and Ras. Conversely, TP53 acts as a negative regulator by directly binding to G6PD, preventing its activation[62].
Similar to G6PD, 6PGD plays a pivotal role in tumorigenesis and high 6PGD expression has been observed in various cancers, including CCA [Figure 3F]. Genetic silencing of 6PGD leads to the accumulation of cellular TP53 protein, induces senescence in lung cancer cells, and slows tumor growth in mouse xenograft models[62,65]. Furthermore, 6PGD has been identified as an independent prognostic factor in HCC, with higher expression levels correlating with worse prognosis and enhanced efficacy of immunotherapy[66]. In HCC, overexpression of NRF2 binds to the antioxidant response element in the 6PGD promoter, enhancing its expression. Elevated 6PGD levels, in turn, upregulate NRF2, creating a positive feedback loop that promotes cell proliferation, survival, and migration. These findings suggest that 6PGD is a key regulator of proteins involved in redox processes[67].
Both G6PD and 6PGD are highly expressed in gastrointestinal cancers, where they serve as significant carcinogenic indicators closely linked to prognosis. These enzymes show promise as potential diagnostic markers for gastrointestinal cancers, offering opportunities for innovative therapeutic strategies. G6PD is expected to emerge as a valuable target for cancer therapies in the near future[68,69]. Furthermore, preclinical evidence suggests that inhibiting 6PGD may enhance the efficacy of standard cancer treatments[70]. However, direct studies in CCA are lacking and need to be performed. Collectively, G6PD and 6PGD present compelling opportunities as therapeutic targets in cancer treatment.
Reversible oxidative PPP: TKT and TALDO
TKT and TALDO are key enzymes upregulated in various cancers[71], including CCA High ALDOA expression has been observed in various cancers, including CCA [Figure 3G and H]. TKT drives tumorigenesis, progression, and treatment resistance by enhancing cell proliferation, invasion, and metastasis, particularly in gastrointestinal cancers[72]. High TKT expression correlates with poor prognosis in colorectal cancer (CRC), where it promotes glycolysis through increased AKT phosphorylation, facilitating metastasis. This positions TKT as a potential prognostic biomarker and therapeutic target for CRC[73]. The isoform transketolase-like-1 (TKTL1) is also associated with poor prognosis and reduced chemosensitivity in gastric cancer, with its silencing inhibiting tumor growth[72]. In HCC, TKT alleviates oxidative stress via the NRF2/KEAP1 pathway and promotes tumor progression through the EGFR pathway[72]. TALDO, a member of the aldolase enzyme family, is elevated in various tumors, including squamous cell carcinomas and HCC, where it contributes to tumor progression[62]. Enrichment analysis of G6PD-related genes in gastrointestinal cancers revealed significant interactions with proteins such as TALDO, GAPDH, and TP53. STRING analysis identified 47 G6PD-interacting proteins, including HKII, 6PGD, LDH, PKM, PGLS, TALDO, GAPDH, and TP53, underscoring their role in regulating PPP activation[68]. Additionally, KRAS activation in a pancreatic cancer model stimulates the nonoxidative PPP[74], further highlighting the roles of both TKT and TALDO in cancer progression.
The PPP plays a critical role by providing R5P for nucleotide synthesis and NADPH, which protects cells from oxidative stress and supports DNA damage repair[75]. PPP activity is upregulated and regulated by the antioxidant transcription factor NRF2, reducing ROS levels and protecting against oxidative stress-induced cell death[76]. Glycolytic intermediates also fuel the serine synthesis pathway, enhancing biosynthesis and antioxidant defense.
In CCA, high levels of sirtuin 2 (SIRT2) and cellular c-Myc further promote this metabolic shift by inhibiting OXPHOS and maintaining redox balance[51,77]. In cisplatin-resistant CCA cells, PPP activity and antioxidant capacity are elevated, with increased G6PD expression and NADPH/NADP ratios compared to HCC cells. This enhanced PPP activity promotes autophagy and strengthens antioxidant defenses, contributing to chemoresistance[62,78]. These findings underscore the critical role of glucose metabolism and PPP-mediated antioxidant capacity in conferring cisplatin resistance. G6PD inhibition by dehydroepiandrosterone (DHEA), a clinical-stage inhibitor, increases ROS levels, sensitizing CCA cells to cisplatin and overcoming chemoresistance[62,78]. The combination of cisplatin and 6-Aminonicotinamide (6-AN), a G6PD inhibitor, increases platinum-DNA adducts, enhances treatment sensitivity in HCC, and reduces G6PD activity while reversing doxorubicin resistance in CRC[62]. Furthermore, in KKU-M156 and KKU-100 CCA cells, silencing NRF2 enhances cisplatin sensitivity, reduces Bcl-xL expression, and suppresses colony formation. NRF2 knockdown also reduces the expression of NRF2-regulated genes like G6PD and TKT, further supporting NRF2 inhibition as a potential strategy to target metabolic dysregulation and overcome chemoresistance[77]. Additionally, ALDOA is highly expressed in iCCA and correlates with poor overall survival and recurrence-free survival. Knocking down ALDOA expression in iCCA cells (HuCCT1 and RBE) with siRNA inhibits iCCA cell proliferation and migration in vitro and
Dysregulation of LDHA in CCA
An important feature of altered metabolic reprogramming in cancer is the conversion of pyruvate to lactate under oxygen-rich conditions. This conversion is driven by LDHA, an enzyme encoded by the LDHA gene, which catalyzes the final step of glycolysis. LDHA is primarily responsible for converting pyruvate to lactate, and its overexpression has been observed in various cancers, including CCA[26] [Figure 3I]. Elevated LDHA expression has been correlated with tumor stage, size, histological grade, and patient survival in cancers such as renal and gastric cancers[26,79]. Moreover, LDHA has a higher affinity for pyruvate, promoting its conversion to lactate, while lactate dehydrogenase B (LDHB) favors the reverse process. In cancer, LDHA expression is upregulated, and LDHB expression is downregulated, supporting aerobic glycolysis by converting pyruvate to lactate[80].
Serum LDH activity is a well-established clinical marker associated with poor prognosis in various cancers[81-83]. In gastrointestinal cancers, gene silencing of LDHA in colon cancer cells led to increased HIF1α levels in cell cultures and tumor lysates, independent of LDH activity in vivo. These findings suggested that LDHA regulates HIF1α activity, facilitating cellular adaptation to the hypoxic tumor microenvironment[81].
In CCA, increased LDHA expression leads to lactate accumulation, supporting ATP production via glycolysis and helping tumor cells maintain redox balance. This metabolic shift enhances the metabolic flexibility of CCA cells, enabling survival in hypoxic conditions. Elevated LDHA expression is also associated with accelerated tumor progression and correlates with adverse histopathological features, including tumor invasiveness and poor prognosis in iCCA tissues. Given its central role in metabolic adaptation and tumor growth, LDHA inhibition could mitigate lactate accumulation and enhance the efficacy of other treatments[26].
Disruption of the TCA cycle in CCA
An important feature of altered IDH is that it is a critical enzyme in the TCA cycle, catalyzing the conversion of isocitrate (ICT) to α-ketoglutarate (α-KG) while concurrently producing NADPH, which plays a vital role in protecting cells against oxidative stress[84]. Mutations in IDH occur in approximately 15%-20% of iCCA cases[85]. The most common mutation affects the cytoplasmic form, IDH1, leading to the conversion of α-KG to the oncometabolite 2-hydroxyglutarate (2-HG), which disrupts normal cellular differentiation and promotes tumorigenesis by altering metabolism[86].
In addition to blocking differentiation, mutated IDH1 (mIDH1) contributes to the accumulation of ROS, enhancing OXPHOS. This metabolic shift reactivates aerobic glycolysis, fueling the TCA cycle and accelerating tumor progression[87]. Experimental evidence shows that mIDH1 promotes the formation of intrahepatic biliary organoids and upregulates PFK-1, a rate-limiting enzyme in glycolysis. The upregulation of PFK-1 supports the oncogenic role of mIDH1 by enhancing glycolytic flux and linking the TCA cycle to aerobic glycolysis, further promoting cancer cell survival and growth[54].
Furthermore, tumors with IDH mutations exhibit greater heterogeneity and more complex tumor-supportive microenvironments, suggesting that these mutations not only alter cellular metabolism but also contribute to tumor progression and resistance to conventional therapies[88]. These findings highlight the importance of mIDH1 mutations in tumorigenesis and suggest that mIDH1 represents a promising therapeutic target in iCCA. Inhibiting mIDH1 could disrupt tumor metabolism and hinder disease progression, offering new opportunities for targeted therapy in iCCA.
Altered OXPHOS in CCA
According to the Warburg effect, OXPHOS is typically downregulated in cancer cells, correlating with an increase in glycolytic flux. Studies have shown that the expression of nearly all OXPHOS complexes is reduced in several CCA cell lines, suggesting compromised OXPHOS function in these cells[89]. Decreased mitochondrial DNA (mtDNA) content or mutations in mtDNA have been linked to reduced OXPHOS activity[90]. Mitochondrial membrane potential, crucial for energy storage during OXPHOS, has been targeted therapeutically in CCA. For instance, Niclosamide, a compound known to interfere with mitochondrial membrane potential, has been shown to inhibit OXPHOS in CCA cells[91].
Recent studies indicate that the Warburg effect is not the only metabolic shift in cancer cells. The “reverse” Warburg effect describes a process in which tumor cells induce oxidative stress in surrounding stromal fibroblasts, prompting them to undergo aerobic glycolysis and produce lactate. This lactate is then exported to and taken up by cancer cells, where it is converted into pyruvate to fuel OXPHOS. The “lactate shuttle” model emphasizes the role of stromal cells in modulating tumor metabolism, in contrast to the Warburg effect, which focuses on increased glycolysis within cancer cells[8]. Once inside cancer cells, lactate enters the TCA cycle to support OXPHOS, highlighting the interplay between stromal cells and cancer cells in metabolic regulation. Unlike the Warburg effect, which primarily emphasizes glycolysis in tumor cells, the reverse Warburg effect supports the high energy demands of cancer cells by enhancing OXPHOS[8,92,93].
Mitochondrial OXPHOS in cancer cells has been identified as a promising strategy to reduce tumor growth and chemotherapy resistance in CCA[94]. While mitochondrial metabolism provides new therapeutic targets, metabolic reprogramming in CCA remains underexplored. Additionally, the metabolic profiles of cancer stem cells (CSCs) may play a crucial role in maintaining stemness and contributing to the failure of anti-cancer treatments. However, limited research exists regarding this phenomenon in human CCA. These findings suggest that mitochondrial oxidative metabolism plays a pivotal role in maintaining stemness in CCA cells, thereby enhancing tumorigenic potential and drug resistance in vivo[94].
Although the mechanisms underlying the observed OXPHOS phenotype remain unclear, a connection between mitochondrial biogenesis regulators and stemness in CCA cells has been investigated. Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a key transcription factor that enhances OXPHOS and self-renewal in malignant cancer cells, including CSCs, has been implicated in this process[95]. Knockdown of PGC-1α in CCA cells resulted in reduced expression of CSC markers, impaired self-renewal, and diminished invasiveness. These findings were further confirmed using SR-18292, a specific inhibitor of PGC-1α, which led to decreased mitochondrial biogenesis and impaired sphere formation. Moreover, downregulation of malignancy-related pathways, epithelial-mesenchymal transition (EMT), and genes associated with self-renewal, pluripotency, and drug transport was observed. These results underscore the dependence of stem-like cells on mitochondrial activity and biogenesis in this metabolic dysregulation[94].
Experimental studies have demonstrated that CSCs in CCA exhibit heightened OXPHOS activity, characterized by a more respiratory phenotype, elevated mitochondrial membrane potential, increased mitochondrial mass, and upregulation of PGC-1α, a key regulator of mitochondrial biogenesis[94]. Inhibition of mitochondrial complex I or silencing of PGC-1α in CCA tumor xenografts significantly reduced tumor growth and stem-like features, further highlighting the potential of targeting OXPHOS as a therapeutic approach in CCA[94].
Additionally, several cancer signaling pathways are activated to induce OXPHOS activity, which is critical for resistance to cancer therapies[96]. Therefore, further investigation into OXPHOS is needed to identify promising therapeutic targets capable of eliminating CSCs and preventing the development of drug resistance. Taken together, these findings underscore the high dependence of CCA cells on glucose metabolism and consumption. Several key glucose metabolites and glycolytic enzymes play pivotal roles in cancer progression, presenting them as promising therapeutic targets for CCA treatment[6].
REDOX AND METABOLIC DYSREGULATION IN CCA
Redox balance, the equilibrium between ROS and antioxidants, is crucial for cellular homeostasis and plays a key role in cancer progression. ROS are generated as byproducts during metabolic processes, particularly through the mitochondrial electron transport chain (ETC) and NADPH oxidase complexes (NOX). In the ETC, electron leakage from Complex I (NADH dehydrogenase) and Complex III (ubiquinone-cytochrome c reductase) leads to the production of superoxide, a primary ROS[97]. While ROS are vital for normal cellular signaling, excessive accumulation results in oxidative stress, contributing to DNA damage, lipid peroxidation, and cellular dysfunction. In cancer cells, including CCA, there is often a dysregulation of metabolism, where glycolysis predominates over OXPHOS, a phenomenon known as the Warburg effect[98]. This metabolic shift increases ROS production, positioning cancer cells closer to the toxic threshold of oxidative stress.
To counteract oxidative damage and ensure survival, cancer cells activate antioxidant defense mechanisms. A central player in this adaptive response is NRF2, a transcription factor that regulates the expression of genes involved in ROS detoxification. NRF2 activation promotes the synthesis of glutathione (GSH) and the production of NADPH, both of which are crucial for maintaining redox balance. The PPP is a key metabolic route that generates NADPH, supporting antioxidant defenses and fueling biosynthetic processes essential for rapid tumor growth[38,99]. By upregulating enzymes such as glutathione peroxidases (GPXs), peroxiredoxins (PRXs), and catalases (CAT), NRF2 ensures that ROS levels remain at a pro-tumor level, enhancing cancer cell proliferation while protecting against excessive oxidative damage[38].
In CCA, dysregulated redox balance has significant consequences. Increased ROS levels lead to DNA damage, particularly oxidative modifications to guanine, which can result in mutations if not properly repaired by base excision repair (BER) or nucleotide excision repair (NER) systems[38]. Furthermore, ROS-induced lipid peroxidation generates reactive intermediates that propagate ROS formation, driving genomic instability and tumor progression[38]. Elevated iron levels, a common feature in CCA, further exacerbate oxidative stress through the Fenton reaction, where excess iron catalyzes the formation of highly reactive hydroxyl radicals, contributing to mutagenesis and promoting tumor growth[100]. These processes highlight the importance of redox regulation in CCA and its role in cancer progression.
Targeting redox homeostasis presents a promising therapeutic strategy in CCA. Redox-active compounds can specifically target cancer cells by increasing intracellular ROS levels or inhibiting antioxidant defenses, disrupting the cellular redox environment and inducing cell death[98]. Many chemotherapeutic agents induce ROS formation, overwhelming the cancer cell’s antioxidant defenses and pushing them beyond their threshold, ultimately leading to oxidative stress-induced cell death[38]. This strategy offers potential therapeutic benefits for CCA, as targeting redox signaling pathways such as NRF2 could enhance treatment efficacy and overcome therapeutic resistance, making ROS modulation a promising approach for combating cancer.
CORRELATION OF ONCOGENIC AND METABOLIC DYSREGULATION IN CCA
Genetic alterations commonly found in CCA are closely linked to metabolic reprogramming, making these pathways important targets for therapy. Mutations in key oncogenes and tumor suppressors - such as KRAS - promote glycolysis to support tumor growth[27,28]. TP53 mutations impair mitochondrial function and favor glycolysis[29-31]. IDH1 mutations disrupt the TCA cycle by causing oncometabolite buildup[32,33]. Loss of ARID1A affects glucose and lipid metabolism through AMPK signaling[34]. Activation of FGFR2 stimulates glycolysis via NF-κB signaling[35]. HER2 overexpression enhances glycolysis through Akt and mTOR pathways[36]. These metabolic changes contribute to cancer progression and offer promising avenues for targeted treatment in CCA [Table 2].
Genetic alterations and their associated metabolic dysregulation in CCA
| Gene/Target | Mutation frequency and role in CCA | Metabolic impact | References |
| KRAS | ~10%-20% in CCA; G12/G13/Q61 missense mutations (G12D common, G12C rare) | Enhances glycolysis via upregulation of GLUT1, HKII, PFK-1, LDHA; activates MAPK and PI3K/AKT/mTOR; promotes glucose uptake and Warburg effect | [27,43,74,110-112] |
| TP53 | ~20% in iCCA; loss-of-function mutations | Loss leads to increased glycolysis, decreased OXPHOS; upregulates GLUT3, LDHA; activates NF-κB and HIF-1α; suppresses TIGAR, SCO2, GLS2 | [114-117,121-123] |
| IDH | ~5%-36% in iCCA; R132H mutation common | Produces 2-HG, inhibits α-KG-dependent enzymes; promotes glycolysis via PFK-1 upregulation; activates AMPK | [11,33,124-126,129,130] |
| ARID1A | ~10%-30% in CCA; loss-of-function | Alters glycolysis and lipid metabolism; shifts glucose metabolism toward OXPHOS; sensitizes cells to AMPK and copper stress | [34,134-137] |
| FGFR2 | ~15% in iCCA; gene fusions/rearrangements | Promotes glycolysis via NF-κB activation; inhibition induces metabolic vulnerability and shift to FAO/autophagy | [35,138,139] |
| HER2 | ~17.4% in eCCA; overexpression | Enhances glycolysis via PI3K/AKT/mTOR activation; promotes Warburg effect, survival, and resistance | [36,140] |
KRAS
The KRAS proto-oncogene encodes a 21 kDa small GTPase that cycles between an active GTP-bound state and an inactive GDP-bound state. Oncogenic KRAS mutations lead to its constitutive activation, disrupting cellular processes such as morphology, proliferation, motility, and survival by continuously activating downstream signaling pathways, including MAPK and PI3K/AKT/mTOR[27]. These mutations are most frequently found in pancreatic ductal adenocarcinoma[101], CRC[102], non-small cell lung cancer (NSCLC)[103], and CCA[104,105]. KRAS mutations are primarily characterized by single-base missense mutations, with 98% occurring at codon 12 (G12), codon 13 (G13), or codon 61 (Q61), which significantly contribute to the oncogenic activation of KRAS in these cancers[106].
KRAS-driven cancers, comprising approximately 30% of cases, including CCA, exhibit significant resistance to therapeutic interventions. Due to its structural characteristics, KRAS has long been considered “undruggable”. However, Sotorasib (AMG 510) and Adagrasib (MRTX849) have proven effective in targeting KRAS G12C mutations in lung cancers[107,108]. These drugs are highly specific, irreversible small-molecule allosteric inhibitors that trap KRAS G12C in its inactive state. In vitro and xenograft mouse models of KRAS G12C-driven cancers, both treatments have been associated with downregulation of MAPK signaling and durable tumor regression[103]. Despite this progress, KRAS G12D mutations, common in gastrointestinal cancers such as CRC, pancreatic cancer, and CCA, still lack effective targeted therapies. KRAS G12D remains a significant challenge in CCA, and targeting this mutation could offer a promising therapeutic opportunity[109].
Oncogenic KRAS drives tumor growth by reprogramming glucose metabolism, particularly through the dysregulation of glycolysis, a hallmark of many cancers. While the precise mechanisms remain incompletely understood, evidence from various cancer types supports the role of KRAS in promoting glycolysis-dependent tumorigenesis[27].
In CRC, KRAS mutations upregulate GLUT1, enhancing glucose uptake and glycolysis, which supports survival under low-glucose conditions. While wild-type KRAS cells struggle to survive in such environments, KRAS mutations are linked to increased GLUT1 expression and lactate production, positioning GLUT1 as a potential therapeutic target. The glycolysis inhibitor 3-bromopyruvate selectively targets KRAS-mutant cells[110]. In pancreatic cancer, oncogenic KRAS drives glycolysis through GLUT1, facilitating glycosphingolipid (GSL) synthesis necessary for KRAS plasma membrane localization and signaling. Additionally, mutated KRAS enhances GLUT1 expression and key glycolytic enzymes such as HKII, PFK-1, and LDHA, driving glucose uptake and diverting glucose into metabolic pathways such as glycolysis and the PPP, which promote tumor growth and protein glycosylation. Targeting GLUT1 and these metabolic pathways presents a potential therapeutic strategy[74,111]. HKII, which catalyzes the first step of glucose metabolism, has been identified as a promising target in KRAS-driven cancers, including NSCLC. Deletion of HKII in mouse models of KRAS-driven NSCLC inhibits tumor initiation and maintenance by disrupting glucose-derived ribonucleotide synthesis and impairing the incorporation of glutamine-derived carbon into the TCA cycle[112]. In KRAS-mutant cancers such as lung, colorectal, and pancreatic cancers, KRAS upregulates GLUT and glycolytic enzymes, promoting glucose uptake and adaptation to low-glucose environments[74,110,111]. In CCA, a rare and aggressive malignancy, KRAS mutations are closely associated with GLUT1 expression and 18F-FDG-PET volumetric parameters, which modulate tumor glucose uptake and correlate with patient prognosis. This evidence underscores GLUT1 as a potential prognostic biomarker for CCA[43]. Although the detailed mechanisms of KRAS-driven metabolic reprogramming in CCA remain insufficiently understood, insights derived from other KRAS-driven cancers provide a promising framework for elucidating the metabolic pathways underlying this highly lethal disease. Targeting the GLUT1-GSL axis, along with the metabolic pathways involving GLUT1, glycolysis, the PPP, and LDH, represents a promising therapeutic strategy for pancreatic cancer and other KRAS-driven malignancies, as it has the potential to disrupt critical oncogenic pathways and enhance the efficacy of treatment in CCA [Figure 4].
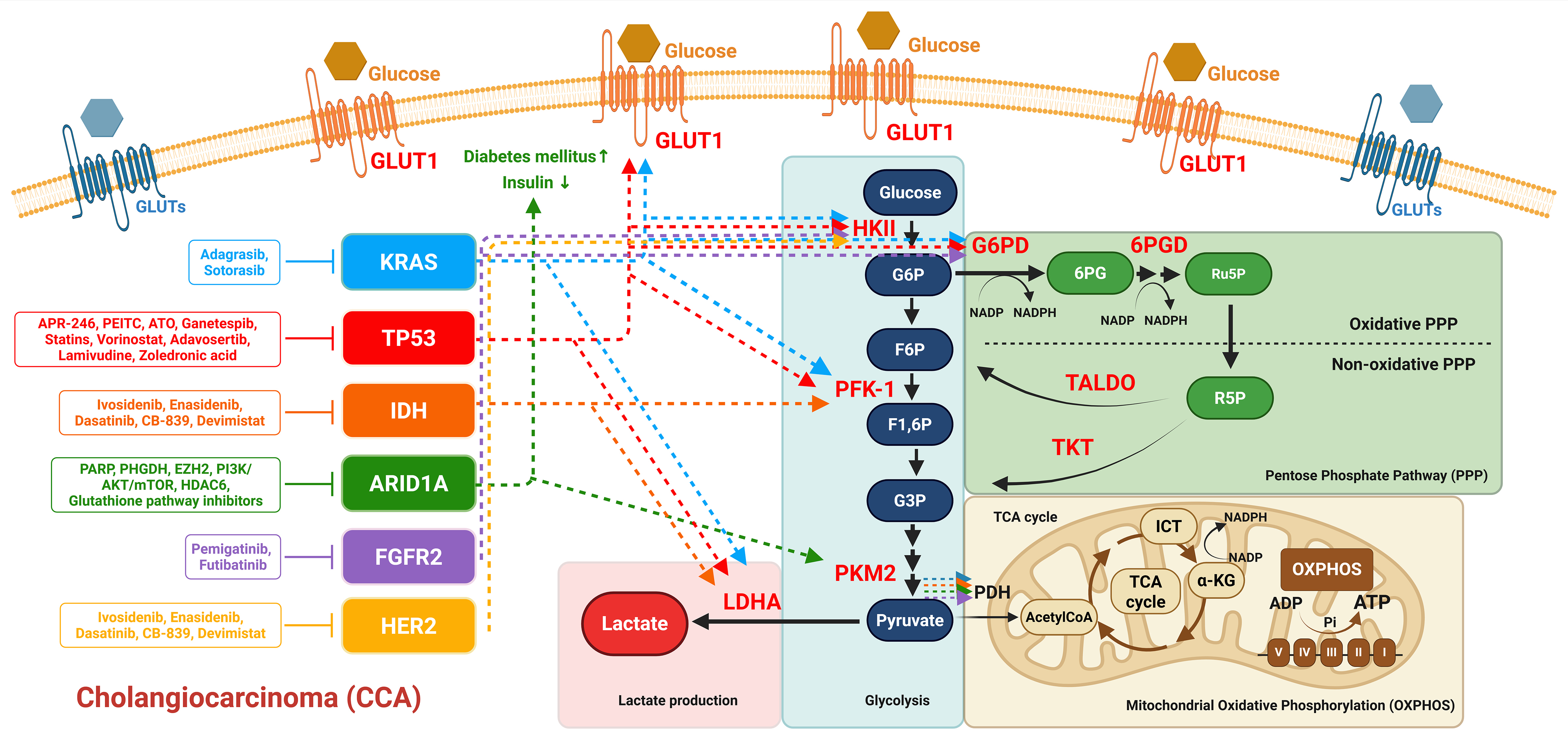
Figure 4. Oncogenic Mutations and Glucose Metabolic Dysregulation in CCA: Therapeutic Targets and Metabolic Shifts. This figure illustrates the upregulation of key glycolytic enzymes (highlighted in red) and the associated metabolites involved in the metabolic alterations observed in cholangiocarcinoma. Additionally, it depicts common oncogenic mutations that modulate glucose metabolic reprogramming in CCA. The dotted arrows represent the regulation of glycolytic enzyme alterations and glucose metabolism dysregulation driven by common oncogenic mutations in CCA. Therapeutic targets are indicated adjacent to each corresponding oncogenic mutation to highlight potential intervention points. Created in BioRender.
TP53
TP53, along with KRAS, is among the most frequently mutated genes in CCA. In iCCA, loss-of-function mutations in TP53 occur in approximately 20% of cases and are significantly associated with poor prognosis[11]. Across three independent cohorts, patients harboring concurrent mutations in both TP53 and KRAS demonstrated worse clinical outcomes, as supported by Kaplan-Meier survival curves and both univariate and multivariate analyses[113]. Often referred to as the “guardian of the genome”, TP53 plays a crucial tumor-suppressive role by maintaining genomic stability and regulating cellular metabolism[114]. In the context of CCA, TP53 mutations contribute not only to genomic instability but also to metabolic reprogramming that promotes tumor progression, aggressiveness, and therapy resistance. One of the key metabolic alterations associated with TP53 loss is the enhancement of aerobic glycolysis, commonly known as the Warburg effect, which allows cancer cells to meet the energy and biosynthetic demands required for rapid proliferation and survival under stress conditions[115].
Under normal conditions, TP53 maintains metabolic balance by inhibiting glycolysis and promoting OXPHOS[114]. It achieves this through several mechanisms, including transcriptional repression of glucose transporters such as GLUT1 and GLUT4, downregulation of rate-limiting glycolytic enzymes, inhibition of lactate transporters like monocarboxylate transporter 1 (MCT1)[116], and suppression of oncogenic signaling pathways including AKT/mTOR and NF-κB[114,117]. TP53 also upregulates key components of mitochondrial metabolism, such as cytochrome c oxidase 2 (SCO2) and glutaminase 2 (GLS2), enhancing mitochondrial respiration and supporting antioxidant defense via GSH production[118-120].
To further limit glycolytic flux, TP53 induces TP53-induced glycolysis and apoptosis regulator (TIGAR), which decreases fructose-2,6-bisphosphate levels, reducing glycolysis and ROS accumulation while redirecting glucose metabolism toward the PPP to generate NADPH[121]. TP53 also downregulates HKII by destabilizing its mRNA and inhibits phosphoglycerate mutase (PGM), further dampening glycolytic activity[121,122]. Additionally, TP53 activates Parkin, which binds to and promotes the degradation of HIF-1α, a transcription factor that normally drives expression of glycolytic genes such as GLUT1 and LDHA[122]. In the PPP, TP53 suppresses G6PD by directly binding to the enzyme, preventing its activation and reducing nucleotide biosynthesis capacity[123].
Loss of TP53 disrupts these regulatory mechanisms, resulting in enhanced glycolysis, lactate production, and increased adaptability to metabolic stress - features that contribute to the aggressiveness of CCA[115]. In TP53-deficient cells, increased IKKα/β kinase activity leads to NF-κB activation, which upregulates GLUT3 and promotes aerobic glycolysis[117]. This metabolic shift also synergizes with oncogenic Ras signaling to enhance glucose uptake and energy production. Importantly, Ras-driven glycolysis is suppressed in the absence of NF-κB subunit p65, but restored upon GLUT3 re-expression, emphasizing the importance of this axis in TP53-deficient tumors[117]. Hypoxic conditions further exacerbate this metabolic phenotype by stabilizing HIF-1α, which upregulates glycolytic enzymes such as HKII, PDK1, and LDHA, reinforcing the Warburg effect[115].
Collectively, these findings highlight the dual impact of TP53 mutations in iCCA - both through loss of genome surveillance and through the promotion of metabolic reprogramming. This shift toward glycolysis not only fuels tumor progression and metastasis but also contributes to resistance against therapeutic interventions, underscoring TP53’s central role in CCA pathogenesis[114,115,117].
IDH
IDH mutations, particularly in IDH1, are identified in approximately 5%-36% of iCCA cases, making them one of the most frequent metabolic alterations in this cancer subtype[124]. These mutations are generally mutually exclusive with FGFR2 rearrangements, though rare co-occurrence has been reported in FGFR2-fused iCCA patients[125]. IDH1 mutations in iCCA are also associated with decreased ARID1A expression, implicating a broader epigenetic disruption[125]. Functionally, mutant IDH enzymes acquire a neomorphic activity, converting α-KG into the oncometabolite 2-HG. This metabolite accumulates in cells and inhibits α-KG-dependent dioxygenases, including histone demethylases, leading to epigenetic dysregulation, impaired differentiation, and tumorigenesis[11].
In iCCA cells, mutant IDH1 drives metabolic reprogramming by upregulating PFK-1 via histone modification, promoting glycolytic flux and supporting proliferation. Silencing of PFK-1 attenuates this effect, and high PFK-1 levels correlate with IDH1-mutant CCA, reinforcing its role in metabolic adaptation[11]. Additionally, mutant IDH1 activates AMPK signaling, enabling maintenance of ATP levels under metabolic stress, further supporting tumor cell survival[11]. These findings establish a clear link between IDH mutations and glycolytic enhancement in iCCA, highlighting their role in tumor growth and potential therapeutic resistance.
While mechanistic studies in gliomas have demonstrated that mutant IDH1 can affect PI3K/Akt and HIF-1α pathways, potentially altering glycolysis and mitochondrial metabolism[126-128], these insights primarily serve to inform understanding of similar metabolic consequences in iCCA, where direct evidence is increasingly emerging. For instance, modulation of LDH isoform expression and pyruvate metabolism seen in other IDH1-mutant tumors may similarly contribute to metabolic inflexibility in CCA, though more studies are warranted[33].
Therapeutically, these metabolic vulnerabilities have clinical relevance. Ivosidenib, a targeted oral IDH1 inhibitor, has shown clinical efficacy in patients with IDH1-mutant iCCA, providing a strong rationale for metabolic-targeted therapy in this subset[129,130].
ARID1A
The ARID1A gene, an essential component of the SWI/SNF chromatin remodeling complex, is frequently mutated in various cancers, facilitating protein access to DNA. Loss-of-function mutations in ARID1A are common across several cancer types, including gastric, hepatocellular, colorectal, and pancreatic cancers[131-134]. Recent studies have also linked ARID1A mutations to clinicopathologic features of CCA, with mutations found in approximately 10%-30% of CCA cases. ARID1A is one of the most frequently mutated tumor suppressor genes in CCA[134]. ARID1A variation may serve as a critical prognostic marker for predicting mortality, metastasis, and recurrence in CCA patients. Its role in CCA pathogenesis involves mechanisms such as cell cycle disruption, chromatin remodeling, oxidative stress, DNA hypermethylation, and gene interactions. Targeting ARID1A may provide potential diagnostic and therapeutic strategies, offering opportunities for precision medicine in CCA management[134].
Loss of ARID1A in pancreatic cancer disrupts glucose metabolism, impairing islet development and insulin production, which may contribute to altered glucose homeostasis and the onset of diabetes mellitus in affected individuals[132]. Thus, ARID1A loss plays a critical role in metabolic dysregulation in pancreatic cancer. In HCC, a CRISPR-Cas9 synthetic lethality screen revealed key genes essential for the survival of ARID1A-deficient cells, highlighting a dependence on the TCA cycle. ARID1A loss downregulates the glycolysis-related gene PKM, shifting glucose metabolism from aerobic glycolysis to reliance on the TCA cycle and OXPHOS [Figure 4]. Furthermore, ARID1A-deficient HCC cells and xenograft tumors show heightened sensitivity to copper treatment, a form of cuproptosis that targets the TCA cycle. These findings suggest synthetic lethality between ARID1A deficiency and mitochondrial respiration impairment, positioning copper treatment as a promising therapeutic approach for ARID1A-deficient HCC[135]. Additionally, recent studies have identified synthetic lethality between ARID1A deficiency and AMPK inactivation in HCC. Treatment with the AMPK inhibitor Compound C suppressed HCC growth in an orthotopic mouse model, with ARID1A knockout cells exhibiting increased sensitivity and prolonged survival in tumor-bearing mice. A regulatory axis involving ARID1A, histone deacetylase 1 (HDAC1), ubiquitin-specific peptidase 9 X-linked (USP9X), and AMPK has been identified as a critical driver of HCC progression. Targeting the metabolic vulnerabilities of ARID1A-deficient HCC cells with AMPK inhibitors may offer a potential therapeutic strategy and may also identify ARID1A deficiency as a biomarker for precision treatment in HCC[136].
ARID1A’s involvement in chromatin remodeling and cell cycle regulation suggests a significant role in CCA tumorigenesis. Studies have shown that ARID1A alterations lead to DNA hypermethylation and a reduction in IDH expression in the mutant subtype of CCA. These changes imply that ARID1A may be involved in the dysregulation of glycolytic metabolism in CCA, contributing to tumor progression[137]. Knockdown of ARID1A disrupts cell cycle progression, promoting cellular proliferation and enhancing tumorigenesis. Given ARID1A’s function in these processes, further investigation into its impact on metabolism in CCA is crucial[134]. ARID1A variations have shown potential as prognostic markers for disease mortality, metastasis, and recurrence in CCA patients. However, their precise role in CCA progression remains to be fully elucidated. The lack of definitive evidence regarding the prognostic significance of ARID1A alterations and their effect on metabolism in CCA highlights the need for further studies with larger sample sizes. While several clinical studies suggest a causal relationship between ARID1A mutations and CCA, a deeper understanding of its biological functions and mechanisms in CCA development is essential. Further research is needed to validate the impact of ARID1A alterations on CCA progression and metabolism, as well as to explore their therapeutic potential[134].
FGFR2
FGFR is a crucial receptor that regulates cell growth, differentiation, and tissue homeostasis. FGFRs consist of four highly conserved transmembrane receptor tyrosine kinases (FGFR1-4). The FGF-FGFR signaling pathway is essential for various biological processes, including angiogenesis, wound healing, and tissue regeneration. FGFR2 fusions or rearrangements occur in approximately 15% of iCCA patients, making them a promising target for therapeutic interventions[138,139]. These alterations contribute to metabolic reprogramming in iCCA, particularly influencing glycolysis and glucose metabolism.
Activation of FGFR2 signaling in iCCA drives aerobic glycolysis, a process regulated by the NF-κB pathway. NF-κB activation, often observed in cancers, enables tumor cells to meet their high metabolic demands by upregulating glycolysis-related gene expression. Inhibition of either FGFR2 or NF-κB disrupts this glycolytic activity, impairing tumor cell growth and survival[35]. FGFR2 inhibition also affects glucose metabolism by blocking glucose uptake and glycolysis via key enzymes like HKII, while inducing adaptive changes such as fatty acid oxidation, increased mitochondrial fusion, and autophagy. This metabolic shift reduces metabolites in the PPP and TCA cycle, further disrupting cellular metabolism[35].
These findings highlight the crucial relationship between FGFR2 signaling and glucose metabolism in iCCA [Figure 4], demonstrating that FGFR2-mediated metabolic reprogramming depends on NF-κB activation. Inhibition of FGFR2 creates a metabolic vulnerability in iCCA cells, suggesting that targeting glucose metabolism through FGFR2 inhibition could provide a promising therapeutic strategy. Additionally, the reduction in glucose utilization caused by FGFR2 inhibition opens new avenues for treatment, including pharmacological and dietary strategies, to enhance the efficacy of FGFR inhibitors and improve patient outcomes in iCCA[35].
HER2
HER2 is a critical regulator in the pathogenesis of various cancers, particularly in breast and gastric cancers. Recent studies have also implicated HER2 in biliary tract cancers, with HER2 overexpression observed in approximately 17.4% of eCCA cases[140].
In breast cancer, HER2-positive tumors are often associated with the hyperactivation of the mTOR signaling pathway, which drives a metabolic shift from OXPHOS to glycolysis, a phenomenon characteristic of the Warburg effect. This metabolic reprogramming supports tumor cell proliferation, survival, and resistance to therapeutic interventions. Moreover, both the mTOR pathway and glycolysis contribute to tumor recurrence and chemoresistance, making them promising therapeutic targets for HER2-positive breast cancer[36]. Given its role in regulating cell survival, proliferation, and metabolic adaptation, HER2 represents a critical target for therapeutic strategies aimed at improving outcomes in HER2-driven cancers [Figure 4].
ASSOCIATION OF METABOLIC DYSREGULATION AND SIGNALING PATHWAY IN CCA
Glucose metabolic dysregulation is a hallmark of cancer and involves bidirectional interactions between oncogenic and tumor suppressor signaling pathways. Mutations in these pathways contribute to metabolic dysregulation, which, in turn, can alter the pathways, driving carcinogenesis and various cancer phenotypes[19,20]. Dysregulated glucose metabolism in CCA enhances tumor progression, invasion, and migration through mechanisms involving the activation of RAS/RAF-MAPK/ERK, PI3K/AKT/mTOR, IDH1/2, JAK-STAT, and NF-κB signaling pathways, as well as increased ROS levels[141-144]. For example, AKT activation inhibits pyruvate entry into the mitochondrial TCA cycle, thereby boosting glycolytic flux by promoting GLUT1 expression, increasing PFK-1 activity, phosphorylating HKII, and reducing PKM2 activity[144]. The PI3K/AKT pathway, often activated in CCA, supports metastasis and poor clinical outcomes, making it a potential target for treatment[145].
In a hyperglycemic environment, oncogenic molecules such as O-GlcNAcylated proteins, glucosamine-F6P amidotransferase (GFAT), and O-GlcNAc transferase are upregulated, promoting a highly metastatic tumor phenotype[146]. High glucose levels in CCA cell lines also upregulate Forkhead box protein M1 (FOXM1), a cell cycle regulator that contributes to CCA cell aggressiveness, and epidermal growth factor receptor (EGFR), which enhances migration and invasion capabilities[147]. Additionally, KRAS and mTORC1 can induce the Warburg effect and channel glycolytic intermediates toward anabolic pathways, further promoting metabolic dysregulation[148,149]. Thus, the interplay between glucose metabolism and oncogenic signaling is bidirectional.
Glucose metabolic dysregulation is primarily driven by transcription factors such as HIF-1α and c-Myc, with secondary contributions from pathways like PI3K-Akt-mTOR and the activation of oncogenes or inactivation of tumor suppressors[8]. The PI3K/AKT pathway enhances aerobic glycolysis, which not only provides energy but also increases the activity of ABC transporters, enhancing drug efflux[144]. Triptolide (TP), an anti-cancer drug that inhibits glycolysis through the AKT-mTOR pathway, has shown promise in inhibiting iCCA growth both in vitro and in vivo, though further clinical investigation is needed[150].
Several signaling pathways, including RAS/RAF-MAPK/ERK, PI3K-AKT-mTOR, IDH1/2, and JAK-STAT, contribute to CCA development and progression, with their activities closely linked to metabolic alterations. Growth factor receptors like EGFR and FGFR2 transmit signals via the PI3K-AKT and RAF-MAPK pathways, influencing glucose and lipid metabolism[1,2]. High glucose concentrations promote CCA progression by upregulating FOXM1 expression via the EGFR/STAT3 pathway, increasing migration and invasion capabilities[147]. Furthermore, the PI3K-AKT pathway supports aerobic glycolysis in CCA cells, with mTOR enhancing the activity of key glycolytic enzymes like HK and PK[150].
TP53 plays a central role in regulating glycolysis and energy metabolism by controlling various processes. It inhibits glycolysis by repressing glucose transporter transcription, downregulating key rate-limiting glycolytic enzymes, and reducing lactate transporter activity. TP53 also modulates critical signaling pathways such as AKT/mTOR and NF-κB, further suppressing glycolytic flux[114].
In IDH1 mutant tumors, mutations lead to the conversion of α-KG to D-2HG, contributing to metabolic stress by inhibiting the TCA cycle and ETC. This metabolic reprogramming is further influenced by glutamine metabolism, which provides 2-HG, a potent oncometabolite[151-153]. IDH mutations activate the PI3K/Akt pathway, suggesting that Akt regulation of glucose metabolism in IDH mutant tumors is modulated by additional oncogenic mutations and the tumor microenvironment[127]. In IDH1 mutant CCA cells, AMPK activation helps maintain ATP levels and supports cell survival under metabolic stress, while also upregulating glycolysis by stimulating PFK-1 expression[54]. These findings highlight metabolic reprogramming as a key driver of IDH-driven tumorigenesis.
ARID1A, a tumor suppressor gene frequently downregulated in CCA[134], can undergo methylation by 2-HG, linking it to metabolic dysregulation[137,154]. This methylation event disrupts its tumor-suppressive functions, impairing chromatin remodeling and facilitating oncogenic processes that promote CCA tumorigenesis and progression. Additionally, a novel ARID1A/HDAC1/USP9X/AMPK axis has been identified as crucial for HCC progression. Targeting metabolic vulnerabilities in ARID1A-deficient HCC cells through AMPK inhibition may offer a potential therapeutic strategy, with ARID1A deficiency serving as a biomarker for precision treatment in HCC[136]. Thus, the intricate interplay between glucose metabolic dysregulation and oncogenic signaling pathways in CCA underscores the critical role of metabolic reprogramming in driving tumor progression. Targeting these pathways could present novel therapeutic strategies for CCA and other malignancies with analogous metabolic and signaling alterations.
GLUCOSE METABOLISM AS A POTENTIAL THERAPEUTIC TARGET IN CCA
Metabolic reprogramming is an established hallmark of cancer, yet direct therapeutic strategies targeting altered glucose metabolism in CCA remain limited. Most current treatments affect glucose metabolism indirectly, with few agents specifically tailored to the metabolic profile of CCA. Therapeutic efforts have focused on inhibiting glucose transporters (e.g., GLUT1 via WZB117 and BAY-876), glycolytic enzymes (HKII, PKM2), components of the PPP (G6PD), lactate metabolism (LDHA), and mIDH in the TCA cycle. While direct OXPHOS inhibition is uncommon, mitochondrial factors such as mtDNA content and PGC-1α are emerging as markers of metabolic adaptation and potential therapeutic targets.
Due to the scarcity of CCA-specific data, many of these targets have been investigated primarily in other cancers. Nevertheless, their established roles in tumor progression, metabolic plasticity, and resistance support their translational relevance in CCA [Table 3]. Further elucidation of these metabolic pathways may lead to more effective and precise therapies for this aggressive malignancy.
Potential therapeutic targets and agents in metabolic pathways in CCA
| Metabolic process | Target/Enzyme | Therapeutic agent(s) | Targeted in CCA therapeutics? | References |
| Antidiabetic repurposing | AMPK/mTORC1/NAD+/SIRT1 | Metformin; chloroquine | Yes - preclinical and phase I/II | [155-157] |
| Glucose uptake | GLUT1 | BAY-876; WZB117 | Yes - preclinical promising | [46,165-167] |
| GLUT2 | Selective inhibitors (preclinical) | No - not yet explored in CCA | [168] | |
| GLUT5 | Selective inhibitors (preclinical) | No - not yet explored in CCA | [168] | |
| Glycolysis | HKII | Lonidamine | Yes - preclinical; trials in other cancers | [50,169-172,174,175] |
| PFK-1 | TLAM + mitochondrial inhibitor (synthetic lethality) | No - not yet explored in CCA | [176] | |
| ALDOA | Dimethyl itaconate | No - not yet explored in CCA | [177,178] | |
| PKM2 | Shikonin; lapachol; vitamin K derivatives (VK3/VK5); compound 3k; TEPP 46; DASA 58; TLN232; micheliolide; metformin; benserazide; MS 001 | Yes - Shikonin in CCA; others preclinical | [179-186] | |
| PPP | G6PD | DHEA (inhibitor; suppresses MYC, oxidative stress reduction), polydatin, 6-AN | Yes - DHEA in CCA models; others not yet tested | [194-196,197-199,62] |
| 6PGD | Physcion (inhibits 6PGD, activates AMPK) | No - not yet explored in CCA | [200-202] | |
| TKT | OT, genistein (natural isoflavonoid) | No - not yet explored in CCA | [203-212,62] | |
| TALDO | F1, 6BP (glycolytic intermediate), sugar phosphate analogues | No - not yet explored in CCA | [62,213,214] | |
| Lactate metabolism | LDHA | GNE-140 (selective LDHA inhibitor), FX11, oxamate | Not studied in CCA | [215-228] |
| TCA cycle | IDH1 mutant | Ivosidenib (FDA approved, improves PFS), enasidenib (limited activity in CCA) | Yes - clinical use | [37,229-233,234] |
| Src kinase (IDH1 mutant related resistance) | Dasatinib | Phase II trial in IDH-mutant iCCA | [235] | |
| GLS1 | CB-839 (telaglenastat) | Not yet specific CCA data | [236,237] | |
| PDH / α-KG dehydrogenase | Devimistat (CPI-613), synergizes with gemcitabine/cisplatin | Phase I trial, preclinical synergy | [94,238] | |
| OXPHOS/mitochondrial metabolism | Complex I and PGC-1α | Metformin (indirect Complex I inhibitor); SR-18292 (PGC-1α inhibitor) | Yes (preclinical and clinical trials) | [94,155,238] |
| Rotenone, deguelin (direct Complex I inhibitors) | No (preclinical; toxicity limits use) | [239,240] | ||
| Phenformin, IM156 (potent Complex I inhibitors) | Yes (clinical evaluation ongoing) | [241] | ||
| BAY-87-2243, IACS-010759 (Complex I inhibitors) | No (clinical trials terminated due to toxicity) | [242-244] | ||
| Mubritinib (dual HER2/Complex I inhibitor) | No (preclinical; untested in CCA) | [245] | ||
| AG311, Mdivi-1 (emerging agents) | No (preclinical; unclear specificity) | [246,247] | ||
| Biological relevance: CSCs enriched by sphere culture show increased mitochondrial mass, membrane potential, and PGC-1α; high PGC-1α or Complex II correlates with poor prognosis | - | [94] |
Repurposing antidiabetic agents as metabolic therapies in CCA
Metformin
Metformin, a widely prescribed antidiabetic drug, exhibits antitumor effects in CCA by modulating energy metabolism. It activates AMPK, thereby inhibiting mTORC1 signaling and reducing cell proliferation, protein synthesis, and lipogenesis[155,156]. Preclinical studies demonstrate its efficacy in suppressing tumor growth in both in vitro and in vivo CCA models. Its activity is particularly notable in tumors harboring IDH1/2 mutations, which are prevalent in intrahepatic CCA and associated with altered mitochondrial metabolism and OXPHOS dependence.
A phase I/II clinical trial NCT02496741 evaluated the combination of metformin and chloroquine in patients with IDH1/2-mutated solid tumors, including CCA, assessing pharmacokinetics, tumor response, and suppression of IDH-derived metabolites[157]. Chloroquine, which inhibits autophagy and glutaminolysis, was used to augment metabolic stress. While the trial demonstrated potential pathway modulation, it did not yield significant tumor responses, highlighting the need for further studies with larger cohorts and optimized combinations.
SGLT2 Inhibitors
Sodium-glucose cotransporter 2 (SGLT2) inhibitors, approved for type 2 diabetes, have shown clinical benefit in heart failure and renal disease[158]. Among them, canagliflozin (CANA) exhibits anti-cancer effects across multiple malignancies, including liver and pancreatic cancers[159-161]. In CCA, CANA inhibits cell proliferation and migration by inducing cell cycle arrest, independent of SGLT2 expression[162]. However, CANA also activates the NAD+ salvage pathway and upregulates sirtuin 1 (SIRT1), potentially promoting EMT and tumor growth[162-164].
Interestingly, combining CANA with nicotinamide phosphoribosyltransferase inhibitors, which block the NAD+ salvage pathway, enhances its antitumor activity in CCA models[162]. These findings underscore CANA’s dual role in CCA and the need for further in vivo studies to evaluate efficacy and safety.
Inhibition of glucose transport in CCA: GLUT inhibitors
Inhibition of glucose transport in CCA: GLUT inhibitors
GLUT1 inhibitors
GLUT1 is significantly overexpressed in CCA and is associated with carcinogenesis, progression, and poor prognosis[46]. Silencing GLUT1 in CCA cells reduces tumor aggressiveness, identifying it as a promising metabolic target[46]. BAY-876, a potent and selective GLUT1 inhibitor, has shown strong antitumor activity in colorectal and head and neck squamous cell carcinoma (HNSCC) models. In CRC, it suppresses proliferation by reducing GLUT1 expression, enhancing mitochondrial respiration and ROS production, and inducing apoptosis[165]. In HNSCC, BAY-876 reduces glucose uptake, metabolism, and IL-8 production, with enhanced apoptotic effects when combined with bitter taste receptor (T2R) agonists, suggesting potential for combination therapies[48].
WZB117, another GLUT1 inhibitor, reduces cancer cell growth, migration, and invasion in vitro and
Other GLUT inhibitors
Recent in silico and experimental research has identified several inhibitors selective for GLUT2, GLUT5, and pan-Class I GLUTs (GLUT1-4) that impede glucose transport by binding near the substrate-binding site[168]. While these compounds show potential to reduce glucose uptake in cancer cells, their efficacy and safety in CCA remain uninvestigated. Given the complex and context-dependent role of GLUT2 in metabolism, further studies are essential to evaluate these inhibitors before clinical translation in CCA.
Therapeutic targeting of glycolysis in CCA: HKII, PFK-1, ALDOA and PKM2 inhibitors
HKII inhibitors
Lonidamine (LND), a selective HKII inhibitor, demonstrates potent anti-proliferative effects against both drug-sensitive and resistant CCA cell lines in a dose- and time-dependent manner[50]. CCA cells exhibit higher sensitivity to LND compared to other cancers, including nasopharyngeal carcinoma and leukemia[169]. Its efficacy correlates with HKII expression, and sub-cytotoxic doses effectively inhibit migration and invasion, indicating anti-metastatic potential consistent with observations in breast and colon cancers[170,171]. Although siRNA-mediated HKII knockdown reduces CCA cell growth and invasiveness, LND at higher concentrations may exert off-target effects on mitochondrial complex II and the mitochondrial pyruvate carrier[172,173]. LND has progressed to phase II clinical trials for various cancers, supporting its translational potential[174,175]. Given HKII’s selective overexpression in CCA compared to normal liver tissue, targeting HKII with LND represents a promising therapeutic strategy, warranting further in vivo and clinical validation[50]. Further in vivo and clinical investigations are warranted to validate LND’s efficacy and safety in CCA treatment.
PFK-1 inhibitors
PFK-1 plays a central role in sustaining glycolysis in cancer. Although tryptolinamide (TLAM), a PFK-1 inhibitor, has limited efficacy alone, it dramatically sensitizes cancer cells to mitochondrial ATP synthase inhibitors such as oligomycin A, increasing cytotoxicity by over 13,000-fold. This synthetic lethality selectively targets cancer cells over normal fibroblasts, likely due to differential metabolic demands. While not yet studied in CCA, this dual-targeting approach highlights the potential of PFK-1 inhibition as a therapeutic strategy[176].
ALDOA inhibitors
Synthetic itaconate derivatives, including dimethyl itaconate, show anti-cancer potential by modulating cytokine production and tumor metabolism[177]. Itaconate inhibits ALDOA enzymatic activity via covalent modification without affecting its expression, leading to glycolytic disruption and impaired tumor cell energy metabolism[178]. In retinoblastoma models, itaconate reduces tumor viability and progression with minimal toxicity compared to standard chemotherapy, supporting ALDOA as a viable metabolic target. Further investigation is warranted to explore its dual immunometabolic effects in CCA[177,178].
PKM2 inhibitors
Shikonin, a selective PKM2 inhibitor, exhibits significant antitumor effects in CCA by reducing proliferation, migration, and invasion, and promoting apoptosis via ROS accumulation and caspase activation[179,180]. It enhances caspase-3 and -8 expression while downregulating MMP-9 and EGFR. Shikonin’s efficacy is further amplified in combination with tumor necrosis factor-related apoptosis-inducing ligand, which triggers JNK signaling and DR5 expression[181]. PKM2 inhibition also reverses chemoresistance in cisplatin- and gemcitabine-resistant cancers; in bladder cancer, shikonin induces necroptosis and suppresses resistance[182,183]. Several other PKM2 inhibitors have demonstrated preclinical potential: lapachol (ferroptosis in melanoma), vitamin K derivatives (chemosensitization in leukemia), compound 3k (broad-spectrum PKM2 inhibition), and metabolic activators such as TEPP-46 and DASA-58 (lung cancer)[184-186].
Additional candidates include peptide TLN232 (phase II melanoma trials)[187], micheliolide (targeting leukemia and glioma)[188,189], and repurposed drugs like metformin[190], benserazide[191], and MS-001, which targets CSC metabolism[192]. While these remain largely untested in CCA, they represent promising avenues for future investigation. Notably, benserazide also inhibits HKII, reducing glycolytic activity and tumor growth in vivo with low toxicity, suggesting its value as a dual-action metabolic therapy[193].
Targeting the PPP: G6PD, 6PGD, TKT, and TALDO inhibitors
G6PD inhibitors
G6PD, the rate-limiting enzyme of the oxidative PPP, is increasingly recognized as a metabolic vulnerability in cancer. DHEA, a known G6PD inhibitor[194], protects cholangiocytes by promoting proliferation and reducing bile acid-induced apoptosis via ERα/ERβ and G-protein-coupled estrogen receptor pathways, which reduce oxidative stress[195]. In CCA, DHEA also suppresses MYC expression, thereby inhibiting cell viability, proliferation, and migration. In silico modeling confirmed stable MYC-DHEA binding, supporting its role as a low-toxicity metabolic and oncogenic modulator[196].
Other G6PD inhibitors studied in non-CCA cancers also show promise. Polydatin, a natural resveratrol glucoside, inhibits G6PD activity, disrupting NADPH production and inducing oxidative stress, apoptosis, and tumor suppression in HNSCC and CRC models[197,198]. It demonstrates high tolerability in phase II clinical trials[199]. Similarly, 6-AN, a niacin analog, inhibits both G6PD and 6PGD, enhancing ROS generation and sensitizing tumors to chemotherapy and radiotherapy in breast, colorectal, and hepatocellular models[62]. While neither polydatin nor 6-AN has been tested in CCA, their mechanisms suggest potential for translational research.
6PGD inhibitors
6PGD maintains redox homeostasis and anabolic metabolism via NADPH production. In HCC models, the selective 6PGD inhibitor physcion disrupts this pathway, triggering AMPK activation, impairing cell viability, and altering the NADPH/NAD+ ratio[200,201]. The dependence of physcion’s efficacy on AMPK signaling supports prior findings that 6PGD-AMPK crosstalk regulates metabolic reprogramming in cancer[202]. Though not yet studied in CCA, the close metabolic parallels with HCC suggest 6PGD inhibition may offer therapeutic benefit.
TKT inhibitors
TKT, the key enzyme of the non-oxidative PPP, facilitates NADPH and ribose production critical for tumor progression[203]. In HCC, TKT is the most highly expressed PPP enzyme and correlates with aggressive features such as large tumor size and vascular invasion, while TKTL1/2 remain undetectable[204]. Oxythiamine (OT), a thiamine antimetabolite and irreversible TKT inhibitor, competitively binds the active site and enhances sorafenib efficacy in HCC models[204,205]. OT induces G0/G1 arrest[206], inhibits MAPK signaling[207], and suppresses migration and MMP-2/9 activity[208]. Co-treatment with agents like lovastatin or 6-AN amplifies ROS-mediated apoptosis[209,210].
Genistein, a natural isoflavonoid, also inhibits TKT, disrupting the non-oxidative PPP, reducing nucleotide synthesis, and restoring sorafenib sensitivity in resistant HCC cells[211,212]. It downregulates HIF-1α, HK2, and GLUT1, and inhibits NF-κB and AKT signaling, showing synergy with agents like tamoxifen and cisplatin[62]. While direct studies in CCA are lacking, TKT’s role in hepatobiliary cancer metabolism warrants further investigation.
TALDO inhibitors
TALDO, another enzyme in the non-oxidative PPP, supports redox balance and mitochondrial function[213]. TALDO inhibition disrupts mitochondrial respiration, membrane potential, and induces oxidative stress in both animal and patient-derived models[62].
F1,6BP, a glycolytic intermediate, competitively inhibits TALDO by binding its active site[214]. Additional sugar phosphates like arabinose-5-phosphate and D-tagatose 6-phosphate irreversibly inhibit TALDO via Schiff base formation with catalytic lysines[62]. Although untested in CCA, TALDO’s role in maintaining oxidative metabolism positions it as a candidate for future targeting.
Modulation of LDHA in CCA: LDH inhibitors
GNE-140
GNE-140, a selective LDHA inhibitor, reduces glucose uptake and downregulates GLUT1 and LDH expression in tumor cells, while enhancing glucose availability and GLUT1 expression in tumor-infiltrating T cells - preserving immune function while impairing tumor glycolysis[215,216]. This tumor-selective metabolic disruption provides a therapeutic window for combining LDHi with immune checkpoint blockade (ICB). However, LDH inhibitors have not yet reached clinical trials, and optimal combination strategies with ICB are under investigation due to the shared glycolytic demands of immune and tumor cells[217-221].
FX11
FX11 selectively inhibits LDHA, increasing mitochondrial oxygen consumption and ROS generation, thereby inducing apoptosis in glycolysis-dependent lymphoma and pancreatic cancer cells. In vivo, FX11 significantly reduces tumor burden[222,223]. Other LDHA-targeting compounds, including galloflavin and N-hydroxyindole derivatives, demonstrate similar activity, though clinical validation is pending[224,225].
Oxamate and combination approaches
Inhibition of LDHA has been shown to enhance chemosensitivity. Oxamate, a well-characterized LDH inhibitor, sensitizes resistant breast cancer and erythroblastic oncogene B2 (ERBB2/HER2)-positive cancer cells to paclitaxel and trastuzumab, respectively, both in vitro and in vivo[226,227]. LDHA silencing via shRNA also increases paclitaxel-induced apoptosis in A549 lung cancer cells[228]. Additionally, co-administration of FX11 with FK866, a NAD+ biosynthesis inhibitor, induces synergistic tumor regression in lymphoma models[222]. While data in CCA remain lacking, LDHA’s metabolic and immunologic roles support further investigation of these agents.
Targeting TCA cycle enzymes in CCA: IDH, GLS, PDH, and α-KG dehydrogenase inhibitors
IDH inhibitors
Ivosidenib, a selective IDH1 inhibitor, is approved for metastatic mIDH1 CCA[37]. In the phase III ClarIDHy trial NCT02496741, it significantly improved progression-free survival (PFS) versus placebo (HR = 0.37; 95%CI: 0.25-0.54), indicating a 63% reduction in progression or death risk[229]. While overall survival (OS) showed a numerical increase (10.3 months vs. 7.5 months), it was not statistically significant (HR = 0.79; 95%CI: 0.56-1.12), likely due to ~70% crossover from placebo to ivosidenib. After adjustment, the OS benefit reached statistical significance, with the placebo arm’s median OS reduced to 5.1 months (P < 0.001)[229]. Ivosidenib’s efficacy compares favorably with second-line regimens such as FOLFOX and liposomal irinotecan plus fluorouracil[230,231], and it is recommended in ESMO guidelines for IDH1-mutant biliary cancers post-first-line chemoimmunotherapy[232]. Current trials are evaluating its combinations with nivolumab registered as NCT04056910, gemcitabine-cisplatin registered as NCT04088188[85], and other IDH-targeted agents including enasidenib registered as NCT02273739 and LY3410738 registered as NCT04521686, supported by references[85,233].
Enasidenib, a selective IDH2 inhibitor approved for acute myeloid leukemia, was assessed in the NCT02273739 phase I/II trial for solid tumors, including CCA. Among 10 evaluable patients, no objective responses were observed[234]. Its limited activity, along with the low prevalence of IDH2 mutations in CCA, restricts its clinical relevance.
Dasatinib, a Src/BCR-Abl kinase inhibitor, retains activity in IDH-mutant CCA cell lines resistant to IDH inhibitors. SRC, but not other dasatinib targets, mediates this sensitivity[235]. A phase II trial (NCT02428855) in IDH-mutant intrahepatic CCA reported a median PFS of 8.7 months and OS of 37.9 months, supporting its potential as monotherapy or in combination with IDH inhibitors[235].
GLS inhibitors
In mIDH1-expressing cancers, indirect metabolic therapies such as GLS inhibitors have been investigated[236]. Glutamine is a major 2-HG source in mIDH1 CCA and is essential for tumor growth. Glutaminase 1 (GLS1), often upregulated in CCA, is associated with enhanced invasiveness and migration[237]. Therefore, GLS inhibitors may be effective in CCA regardless of IDH mutation status. CB-839 (telaglenastat), a GLS1 inhibitor, was evaluated in a phase I trial NCT02071862 involving IDH-mutant solid tumors, though mIDH1-specific outcomes remain unpublished[237].
PDH and α-KG dehydrogenase inhibitor
Devimistat (CPI-613), an inhibitor of PDH and α-KG dehydrogenase - critical enzymes in the TCA cycle and CCA stemness maintenance[94,238] - was studied in trial NCT0176629. Among tested doses, 2,300 mg/m2 yielded the longest OS (4.7 months), while PFS was highest (2.5 months) at 600/3,000 mg/m2. Preclinical studies revealed synergy with gemcitabine and cisplatin in CCA models[238], suggesting combination potential for future clinical development.
Therapeutic targeting of OXPHOS in CCA: complex I and associated inhibitors
Metformin and SR-18292
Metformin, an indirect Complex I inhibitor, impairs mitochondrial respiration and PGC-1α-associated OXPHOS, reducing sphere formation, downregulating stemness and EMT markers, and suppressing in vivo tumor growth in CCA models[94]. SR-18292, a selective PGC-1α inhibitor, similarly inhibits mitochondrial biogenesis and stem-like phenotypes, reinforcing the therapeutic relevance of targeting PGC-1α in CCA[94].
Rotenone and deguelin
Rotenone and its derivative deguelin directly inhibit NADH oxidation and electron transfer at Complex I, inducing apoptosis and tumor growth suppression in preclinical settings[239]. However, rotenone’s clinical application is limited by systemic toxicity affecting the nervous system, liver, and bone marrow[240].
Phenformin and IM156
Phenformin and IM156, more potent Complex I inhibitors than metformin, exhibit enhanced mitochondrial targeting. IM156 is currently under clinical evaluation due to its improved efficacy profile, although higher doses are typically required to achieve antitumor effects[241].
BAY-87-2243
Initially identified as a HIF-1α inhibitor, BAY-87-2243 was later shown to directly inhibit Complex I, decreasing oxygen consumption and mitigating tumor hypoxia. Despite encouraging preclinical efficacy, its development was halted due to dose-limiting toxicities in a phase I trial NCT01297530[242].
IACS-010759
A derivative of BAY-87-2243, IACS-010759 is a potent Complex I inhibitor that blocks ubiquinone binding to the ND1 subunit, demonstrating nanomolar IC50 values and significant antitumor effects in preclinical models. However, Phase I trials in hematologic and solid malignancies (NCT02882321 and NCT03291938) were terminated due to toxicity and limited clinical benefit[243,244].
Mubritinib
Originally developed as a HER2 kinase inhibitor, mubritinib has been reclassified as a Complex I inhibitor targeting the ubiquinone-binding site. Its dual mechanism of action offers a novel therapeutic angle, although its application in CCA remains untested clinically[245].
Emerging inhibitors: AG311 and mdivi-1
AG311 is an experimental agent affecting mitochondrial respiration; however, its mechanism and Complex I specificity remain unclear. Mdivi-1, initially proposed as a Complex I inhibitor, is now understood to target mitochondrial fission via dynamin-related protein 1 (Drp1), rather than directly inhibiting Complex I[246,247]. Both require further preclinical validation.
OXPHOS is essential for energy metabolism in CCA, particularly in CSCs enriched via sphere culture, which show elevated mitochondrial mass, membrane potential, and PGC-1α expression. High levels of PGC-1α or Complex II correlate with poor prognosis[94]. While OXPHOS inhibition is a promising strategy, current agents are limited by toxicity, low efficacy, or poor specificity, highlighting the need for improved mitochondrial-targeted therapies.
THERAPEUTIC TARGETING OF ONCOGENIC MUTATIONS IN CCA
CCA is a molecularly heterogeneous malignancy increasingly addressed through precision oncology. KRAS mutations are targeted by inhibitors such as adagrasib and sotorasib. TP53-directed strategies involve reactivation, degradation, and synthetic lethality. IDH mutations, associated with metabolic reprogramming, are targeted via mutant IDH and related enzyme inhibition. ARID1A-deficient tumors present opportunities for synthetic lethality and epigenetic therapies. FGFR2 fusions, common in intrahepatic CCA, are treated with agents such as futibatinib and pemigatinib, alongside resistance management. HER2 alterations are targeted by monoclonal antibodies, ADCs, TKIs, and immune conjugates [Table 4]. Although MET alterations have been reported, their therapeutic targeting requires further investigation. These advances highlight the shift toward mutation-driven therapies to improve CCA outcomes.
Therapeutic targeting of oncogenic mutations in CCA
| Gene/Target | Treatment regimen | Therapeutic target | Combination | Phase of clinical trial | NCT number(s) | Results | References |
| KRAS | Adagrasib (KRYSTAL-1 trial) | KRAS G12C mutant (inactive GDP-bound form) | Monotherapy | Phase I/II | NCT03785249 | 41% ORR, 100% DCR in GI tumors incl. CCA; well tolerated; reduces GLUT1 and lipid uptake | [248,249] |
| Sotorasib | Selective KRAS G12C inhibitor | Monotherapy | FDA-approved in NSCLC | NCT03600883 | FDA-approved for NSCLC; inhibits glycolysis via TXNIP upregulation; limited CCA data | [250-254] | |
| TP53 | APR‑246 (eprenetapopt) | Reactivates mutant TP53 via cysteine binding | ± Azacitidine | Phase III | NCT03745716 | ~73% ORR, 60% 1‑year relapse-free survival in MDS/ALL; no CCA data | [256-259] |
| PEITC | Refolds R175H/L mutants, induces oxidative stress | Monotherapy | Phase II | NCT01790204 | Ongoing; data pending | [260] | |
| ATO | Degrades/refolds mutant TP53; disrupts antioxidant defense | Mono/combo | Early-phase | NCT03855371, NCT04869475, NCT04489706, NCT04695223 | Ongoing; no CCA-specific results | [261-265] | |
| Ganetespib (HSP90 inhibitor) | Destabilizes mutant TP53 via CHIP/MDM2 activation | + Paclitaxel | Phase I/II | NCT02012192 | Safe, but no added survival in ovarian cancer | [267-271] | |
| Statins (e.g., atorvastatin) | Promotes TP53 degradation via mevalonate/CHIP pathway | Monotherapy | Phase II | NCT04767984, NCT03560882 | Promising preclinical; CCA data lacking | [272-277] | |
| Vorinostat (HDAC inhibitor) | Induces TP53 degradation via ubiquitination machinery | ± 17AAG/chemo | Phase II | NCT02042989, NCT01339871 | Modest benefit; mutation specificity unclear | [278-280] | |
| Adavosertib (WEE1 inhibitor) | Synthetic lethality in TP53-deficient cells | Mono or combo | Phase I/II | NCT01164995, NCT01357161, NCT02272790, NCT03668340 | Modest PFS/RR in TP53-mutated tumors | [284-289] | |
| Lamivudine (LINE‑1 RTi) | Inhibits retrotransposon activity in TP53-deficient cells | Monotherapy | Phase II | NCT03144804 | Disease stabilization in TP53-mutant CRC | [290-293] | |
| Zoledronic acid + statins | Mevalonate pathway inhibition; reduces TP53-YAP/TAZ axis | + Chemotherapy | Phase II | NCT03358017 | Ongoing in TNBC; TP53-stratified | [294-298] | |
| IDH1/2 | Ivosidenib | Selective IDH1 inhibition | Monotherapy | Phase III (ClarIDHy) | NCT02496741, NCT04056910, NCT04088188 | Improved PFS vs. placebo (HR 0.37); adjusted OS significant; FDA approved | [37,229-232] |
| Enasidenib | Selective IDH2 inhibition | Monotherapy | Phase I/II | NCT02273739 | No objective response in CCA; limited activity | [233,234] | |
| Dasatinib | SRC inhibition (overcomes IDHi resistance) | Mono/combo | Phase II | NCT02428855 | PFS 8.7 mo; OS 37.9 mo in mIDH1 CCA | [235] | |
| CB-839 (Telaglenastat) | Glutaminase 1 (GLS1) inhibition | Monotherapy | Phase I | NCT02071862 | Study completed; no published results in mIDH1 CCA | [237,238] | |
| Devimistat (CPI-613) | Mitochondrial metabolism inhibition (PDH/α-KGDH) | Combo with gemcitabine + cisplatin | Phase I/II | NCT01766297 | Synergistic with chemo; max OS 4.7 mo at 2,300 mg/m2 | [97,239] | |
| ARID1A | PARP inhibitors (olaparib, rucaparib, niraparib, talazoparib) | Synthetic lethality via defective homologous recombination DNA repair | Preclinical/early clinical | Various ongoing and exploratory | - | Sensitizes ARID1A-deficient tumors to PARP inhibition; increased PARP-DNA trapping and replication stress | [299,301] |
| PHGDH inhibitor (CBR-5884) + olaparib | Metabolic disruption enhances PARP inhibitor efficacy | Preclinical | - | - | Increases ROS and activates ROS/Wnt/β-catenin pathway; synergistic antitumor effect with olaparib | [301] | |
| EZH2 inhibitors (tazemetostat, GSK2816126, CPI-1205) | Reverses polycomb-mediated gene silencing, induces apoptosis | Early clinical/exploratory | Phase II | NCT05023655 (solid tumors with ARID1A mutations), NCT03348631 (ovarian, peritoneal, endometrial cancers) | Selective apoptosis in ARID1A-mutant cells; no CCA-specific data yet | [299,300,302,303] | |
| PI3K/AKT/mTOR inhibitors (idelalisib, copanlisib, MK2206, everolimus, temsirolimus) | Blocks dysregulated survival signaling in ARID1A-deficient cells | Preclinical/exploratory | - | - | Heightened sensitivity in ARID1A-mutant models; potential monotherapy or radiosensitizer | [299] | |
| HDAC6 inhibitors (tubacin, NQN-1) | Epigenetic modulation, induces apoptosis, enhances immune checkpoint sensitivity | Preclinical | - | - | Upregulates PD-L1, synergizes with PD-L1 blockade in ARID1A-deficient tumors | [299] | |
| Glutathione pathway inhibitors (buthionine sulfoximine, APR-246) | Exploits redox imbalance by reducing glutathione synthesis | Preclinical | - | - | Induces oxidative stress and apoptosis selectively in ARID1A-mutant cells | [299] | |
| FGFR2 | Pemigatinib | FGFR2 fusion/rearrangement | Monotherapy | Phase II | NCT02924376 | ORR 35.5% in FIGHT-202 trial; FDA approved for FGFR2-altered CCA | [35,141,142,296,304,306] |
| Futibatinib (TAS-120) | Pan-FGFR inhibitor (irreversible) | Monotherapy | Phase II/III | NCT02052778 | Overcomes resistance to ATP-competitive FGFR inhibitors; FDA approved in 2022 | [294,295,304,305] | |
| HER2 | Pertuzumab + trastuzumab | Dual HER2 antibodies (domains II and IV) | Dual antibody combo | Phase IIa (MyPathway) | NCT02091141 | Synergistic inhibition; clinical activity in HER2+ CCA; ongoing | [36,143,297,307] |
| Trastuzumab deruxtecan (T-DXd) | HER2-targeted antibody-drug conjugate | Monotherapy | Phase II | NCT04482309 | ORR ~37%; median PFS 4.4-11.9 mo; OS 7.1-21.1 mo; pulmonary toxicity requires monitoring | [298,299,308-310] | |
| Zanidatamab (bispecific antibody) | Bispecific antibody targeting HER2 extracellular domains | Monotherapy | Phase I/II | NCT02892123, NCT04466891 | ORR 41.3% in HER2-amplified CCA; manageable toxicity; promising | [300,311,312] | |
| Neratinib | Irreversible HER2/4 TKI | Monotherapy | Phase II | NCT01953926 | ORR 16% in HER2-mutant, refractory CCA; S310F and V777L mutations common; further study needed | [301,313] | |
| Tucatinib + trastuzumab | Selective HER2 TKI + antibody | Combo | Phase II | NCT04579380 | ORR 46.7%, DCR 76.7%; median PFS 5.5 mo; well tolerated; promising combo | [302,314] | |
| Trastuzumab + chemotherapy | HER2 + chemo (FOLFOX or Gem + Cis) | Combo | Phase II | NCT04722133, CTRI/2019/11/021955 | ORR 29.4% (FOLFOX), 55.5% (Gem-Cis); good disease control; ongoing trials | [303,304,315,316] |
Targeting KRAS mutations in CCA: adagrasib, the KRYSTAL-1 trial, and sotorasib
KRAS is a key oncogenic driver in CCA, with activating mutations predominantly at codons 12 (G12), 13 (G13), and 61 (Q61)[106], among which G12D is most prevalent[109]. Although KRAS G12C mutations are relatively rare in CCA, selective inhibitors targeting this variant have shown clinical potential, including modulation of glucose metabolism.
Adagrasib and the KRYSTAL-1 trial
Adagrasib is a covalent KRAS G12C inhibitor that traps the mutant protein in its inactive GDP-bound state. It exhibits favorable pharmacokinetics, including a ~24-h half-life, broad tissue distribution, and central nervous system penetration. In the KRYSTAL-1 Phase I/II trial NCT03785249, adagrasib monotherapy demonstrated an objective response rate of 41% and a disease control rate of 100% in patients with KRAS G12C-mutant gastrointestinal tumors, including biliary tract cancers. The treatment was well tolerated, underscoring the value of genotype-driven therapy despite the low frequency of KRAS G12C in CCA[248].
Beyond direct pathway inhibition, adagrasib induces metabolic reprogramming in KRAS G12C-mutant tumors. Transcriptomic analyses from murine xenografts and patient biopsies revealed downregulation of GLUT1 and low-density lipoprotein receptor, indicating reduced glucose and cholesterol uptake. Simultaneously, genes involved in cholesterol transport were upregulated, suggesting enhanced lipid efflux. These coordinated changes reflect a shift in tumor metabolism and may reveal targetable vulnerabilities for combination strategies[249].
Sotorasib
The recent FDA approval of sotorasib, a selective KRAS G12C inhibitor for NSCLC, was supported by sensitive detection of KRAS mutations in circulating cell-free DNA via droplet digital PCR, highlighting the expanding role of liquid biopsies in mutation profiling[250-252]. However, the concordance and clinical significance of cfDNA-detected KRAS mutations in CCA remain unclear[253].
Metabolically, while KRAS G12D is the predominant mutation in CCA and known to promote glycolysis, KRAS G12C mutations - found in bladder cancer - also drive altered glucose metabolism. Sotorasib inhibits glucose uptake and glycolysis in KRAS G12C mutant bladder cancer cells by stabilizing thioredoxin-interacting protein (TXNIP), a negative regulator of glucose metabolism, via suppression of the RAS/RAF/ERK pathway. This metabolic inhibition is selective for mutant cells, revealing a targeted therapeutic mechanism[254].
Targeting TP53 mutations in cancer: reactivation, degradation, and synthetic lethality
Restoring wild-type TP53 function in missense TP53 mutants: APR-246, phenethyl isothiocyanate, and arsenic trioxide
Missense TP53 mutations abrogate tumor suppressor function by destabilizing the protein and impairing transcriptional activity. Agents such as APR-246 (also known as eprenetapopt/PRIMA-1MET), phenethyl isothiocyanate (PEITC), and arsenic trioxide (ATO) restore wild-type TP53 function[255].
APR-246 covalently modifies cysteine residues in mutant TP53, reactivating its transcriptional function and synergizing with DNA-damaging agents[256]. Mechanistically, APR-246 induces oxidative stress by inhibiting thioredoxin reductase 1, converting it into a pro-oxidant NADPH oxidase that disrupts redox homeostasis and triggers endoplasmic reticulum stress in TP53-mutant cells. It further depletes GSH, inducing lipid peroxidation and ferroptosis, while upregulating HMOX1 and affecting metabolic pathways such as the PPP and OXPHOS[257]. In TP53-mutant myelodysplastic syndrome and acute lymphoblastic leukemia, APR-246 combined with azacitidine achieved up to 73% response rates and 60% one-year relapse-free survival[258,259]. A phase 3 trial (NCT03745716) is ongoing to evaluate its efficacy in TP53-mutated malignancies, although data in CCA are not available.
PEITC, derived from cruciferous vegetables, refolds TP53R175H/L mutants, induces oxidative stress via GSH disruption[260], and is currently under investigation in a phase 2 study (NCT01790204) of PEITC-rich watercress juice in oral TP53mutant lesions; its role in CCA remains unaddressed.
ATO, approved for the treatment of acute promyelocytic leukemia, promotes degradation of mutant TP53 through ubiquitin-mediated mechanisms and facilitates the refolding of structural TP53 mutants via covalent cysteine binding[261-264]. This reactivation of TP53 function restores expression of downstream genes involved in cell cycle regulation and apoptosis, leading to suppressed tumor growth in models harboring certain structural TP53 mutations but not DNA contact mutants[263]. Additionally, ATO disrupts the Nrf2-mediated antioxidant defense system, reducing ROS detoxification capacity and promoting oxidative stress-associated metabolic dysfunction, particularly in hepatic tissue[265]. Several clinical trials are currently investigating the therapeutic potential of ATO in TP53-mutant malignancies, including NCT03855371, NCT04869475, NCT04489706, and NCT04695223; however, clinical data specific to CCA have yet to be reported.
Targeted removal and degradation of mutant TP53: HSP90 inhibitors, statins, ATO, and vorinostat
Mutant TP53 is stabilized by oncogenic stress and contributes to gain-of-function oncogenic phenotypes[266]. Its depletion suppresses tumorigenesis, demonstrating tumor dependence[267]. Agents such as HSP90 inhibitors, statins, ATO, and vorinostat selectively degrade mutant TP53[267,268].
Heat shock protein 90 (HSP90) prevents mutant TP53 degradation by inhibiting murine double minute 2 (MDM2) and c-terminus of hsc70-interacting protein (CHIP), key E3 ubiquitin ligases. Ganetespib, an HSP90 inhibitor, selectively depletes mutant TP53 in vitro and in vivo with minimal effects on wild-type TP53[267,269]. Although a phase 1/2 ovarian cancer trial of ganetespib combined with paclitaxel demonstrated good tolerability, it showed limited efficacy[270,271]. The therapeutic relevance of this approach in CCA remains unexplored.
Statins, competitive inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase), lower plasma cholesterol by suppressing de novo biosynthesis and altering LDL receptor expression[272]. In the tumor microenvironment, simvastatin induces metabolic reprogramming by reducing lactate production and enhancing sensitivity to MCT1 inhibition, suppressing growth in HNSCC[273]. Notably, atorvastatin inhibits the mevalonate pathway and promotes CHIP-mediated degradation of conformational TP53 mutants by disrupting the interaction between mutant TP53 and DnaJ homolog subfamily A member 1, a heat shock co-chaperone[274]. Statins have shown antitumor activity in preclinical models[274,275] and clinical benefit in TP53-mutant lung adenocarcinoma, though not in colon cancer[276,277]. Ongoing trials (NCT04767984 and NCT03560882) may clarify their therapeutic potential; however, no data specific to CCA are currently available.
ATO promotes degradation of conformational and DNA contact TP53 mutants by enhancing ubiquitin-mediated proteasomal turnover and restores partial protein function through covalent cysteine binding[263,264]. It also shows synergy with the HSP90 inhibitor tanespimycin and the histone deacetylase inhibitor vorinostat. Clinical trials (NCT03381781 and NCT03377725) are ongoing, though none currently include CCA.
Vorinostat (SAHA), an HDAC inhibitor, promotes degradation of mutant TP53 proteins via ubiquitin ligases such as MDM2/CHIP, disrupts the HSP90-HDAC6 complex, represses transcription, and induces autophagy[278-280]. It enhances sensitivity to chemotherapy and acts synergistically with 17-N-allylamino-17-demethoxygeldanamycin (17AAG). Clinical trials (NCT02042989 and NCT01339871) demonstrated modest benefits; however, TP53 mutation-specific responses and CCA data remain limited.
Leveraging synthetic lethality to target TP53 mutations and deletions: adavosertib, LINE-1 RT inhibitors, and mevalonate pathway blockade
Synthetic lethality, where simultaneous disruption of two genes causes cell death but loss of either alone is tolerated, enables selective targeting of TP53-deficient cancers by exploiting essential compensatory pathways[281]. Key synthetic lethal partners such as WEE1, PLK1, and PARP regulate the G2/M checkpoint; inhibition of these is especially lethal to TP53-deficient cells due to TP53’s primary role in the G1 checkpoint[281-283].
Adavosertib (AZD1775), a selective WEE1 inhibitor, induces synthetic lethality in TP53-mutant tumors by disrupting the G2/M checkpoint and increasing DNA damage sensitivity[284]. Clinical trials in TP53-mutated ovarian cancer (NCT01164995, NCT01357161, NCT02272790), CRC (FOCUS4-C)[285-288], and uterine serous carcinoma (NCT03668340)[289] demonstrated modest improvements in PFS and response rates, often requiring co-occurring alterations such as CCNE1 or MYC amplification for optimal efficacy. Other G2/M regulators (e.g., UCN-01, BI-2536) have not undergone TP53-stratified evaluation.
LINE-1 retrotransposons, normally silenced in somatic tissues, are derepressed in TP53-deficient cancers, promoting genomic instability and tumorigenesis[290-292]. Reverse transcriptase inhibitors like efavirenz and lamivudine suppress LINE-1 activity, with lamivudine preferentially inhibiting the growth of mutant TP53 CRC cells and xenografts[293]. A phase 2 trial (NCT03144804) reported disease stabilization in mutant TP53 metastatic CRC, supporting LINE-1 RT inhibition as a therapeutic strategy.
Gain-of-function TP53 mutants enhance YAP/TAZ signaling via the mevalonate pathway, driving cancer progression[294,295]. Statins and zoledronic acid inhibit this pathway, reducing mutant TP53 and YAP/TAZ activity and showing synergistic cytotoxicity[296-298]. A phase 2 trial (NCT03358017) is evaluating their combination with chemotherapy in triple-negative breast cancer stratified by TP53 status.
TP53 mutations are frequent in cancer and offer therapeutic opportunities via reactivation, degradation, or synthetic lethality. While agents such as APR-246, statins, and WEE1 inhibitors show preclinical and early clinical promise, their application to CCA remains limited. Most studies lack TP53-specific stratification, and the heterogeneity of TP53 mutations underscores the need for tailored, mutation-guided approaches.
Targeting ARID1A-deficient tumors in CCA: synthetic lethality and epigenetic vulnerabilities
ARID1A mutations, common in CCA, impair chromatin remodeling, cell cycle control, and DNA repair, creating therapeutic vulnerabilities exploitable through synthetic lethality. Targeted strategies - such as PARP, enhancer of zeste homolog 2 (EZH2), PI3K/AKT/mTOR, HDAC6, and GSH pathway inhibition - have shown preclinical or early clinical promise in ARID1A-deficient cancers[299,300].
PARP inhibitors: targeting defective DNA repair pathways
ARID1A loss impairs homologous recombination, sensitizing cancer cells to PARP inhibitors that block single-strand DNA repair and induce synthetic lethality. FDA-approved agents - olaparib, rucaparib, niraparib, and talazoparib - initially developed for BRCA-mutant cancers, are now under evaluation in ARID1A-deficient models, which exhibit increased PARP-DNA trapping and replication stress[299].
Recent evidence shows that metabolic disruption via phosphoglycerate dehydrogenase (PHGDH) inhibition can enhance PARP inhibitor efficacy. PHGDH maintains mitochondrial redox balance; its inhibition by CBR-5884 elevates ROS and activates the ROS/Wnt/β-catenin pathway, suppressing epithelial ovarian cancer growth. Co-treatment with olaparib amplifies these effects, indicating a synergistic antitumor strategy linking metabolic stress to DNA repair vulnerability[301].
EZH2 inhibitors: reversing polycomb-mediated gene silencing
Loss of SWI/SNF (SWItch/Sucrose Non-Fermentable) complex antagonism in ARID1A-deficient tumors enhances EZH2-mediated gene silencing, promoting tumor progression. EZH2 inhibitors such as tazemetostat, GSK2816126, and CPI-1205 reactivate silenced tumor suppressors and selectively induce apoptosis in ARID1A-mutated cells[300]. While no clinical trials specifically target ARID1A-mutated cancers, a phase II study (NCT05023655) is evaluating tazemetostat in solid tumors with ARID1A mutations, and NCT03348631 is assessing it in recurrent ovarian, peritoneal, and endometrial cancers regardless of ARID1A status.
Beyond epigenetics, EZH2 regulates metabolism, affecting therapy response. In ovarian cancer, EZH2 noncatalytically upregulates IDH2, enhancing TCA cycle activity and tumor growth[302]. In CRC, EZH2 loss induces GLS, increasing glutamate and GSH synthesis, reducing ROS, and conferring resistance to nutrient deprivation[303]. This metabolic plasticity contributes to EZH2 inhibitor resistance, supporting combined targeting of EZH2 and metabolic pathways.
PI3K/AKT/mTOR inhibitors: targeting dysregulated growth signaling
ARID1A loss activates PI3K/AKT/mTOR signaling, enhancing cell survival and resistance to apoptosis. This pathway becomes a therapeutic dependency, with ARID1A-deficient models showing heightened sensitivity to PI3K (idelalisib, copanlisib), AKT (MK2206), and mTOR (everolimus, temsirolimus) inhibitors[299]. Mechanistically, ARID1A deficiency increases AKT Ser473 phosphorylation and downstream mTOR signaling, supporting pathway inhibition as a monotherapy or radiosensitizing strategy in ARID1A-mutant CCA.
HDAC6 inhibitors: epigenetic modulation and immunotherapy synergy
HDAC6 regulates protein degradation, stress responses, and immune signaling. In ARID1A-deficient tumors, selective HDAC6 inhibitors (e.g., tubacin, NQN-1) trigger apoptosis and suppress tumor growth[299]. HDAC6 inhibition also elevates PD-L1 expression, enhancing immune recognition and synergizing with PD-L1 checkpoint blockade in preclinical ARID1A-deficient models. These findings support an epigenetic-immunotherapy combination approach in CCA.
GSH pathway inhibitors: exploiting redox imbalance
ARID1A mutations suppress SLC7A11, impairing GSH synthesis and sensitizing cells to oxidative stress. Buthionine sulfoximine and APR-246, which target glutamate-cysteine ligase catalytic subunit, enhance ROS and induce apoptosis in ARID1A-deficient cells[299]. This redox imbalance offers a promising therapeutic strategy, particularly in combination with chemo- or radiotherapy.
Collectively, these approaches highlight the growing potential of targeting synthetic lethal and epigenetic vulnerabilities in ARID1A-mutated CCA. Further validation of ARID1A as a biomarker will be essential for advancing precision oncology in this setting.
Targeting FGFR2 alterations in CCA: futibatinib, pemigatinib, and overcoming resistance
Futibatinib
Futibatinib (TAS-120), a pyrazolopyrimidine derivative, is a potent and selective pan-FGFR inhibitor, exhibiting IC50 values of 3.9, 1.3, 1.6, and 8.3 nM against FGFR1-4, respectively. It demonstrates high specificity with no reported off-target kinase inhibition. In 2022, futibatinib and pemigatinib received FDA approval for CCA patients harboring FGFR2 fusions or rearrangements, representing a significant advance in targeted therapy[304]. Unlike ATP-competitive FGFR inhibitors (e.g., infigratinib, BGJ398, Debio 1347), which often encounter resistance due to FGFR2 kinase domain mutations, futibatinib irreversibly inhibits FGFRs and has shown clinical efficacy in overcoming resistance in iCCA patients with such mutations[305].
Pemigatinib
Pemigatinib was evaluated in the phase II FIGHT-202 trial, an open-label, single-arm study enrolling CCA patients with FGFR2 fusions or rearrangements who had progressed on prior therapy. Patients received
Therapeutic targeting of HER2 in CCA: monoclonal antibodies, ADCs, TKIs, and immune-conjugates
HER2 overexpression or gene amplification defines a therapeutically actionable subset of CCA, with ongoing trials evaluating various HER2-directed strategies.
Pertuzumab and trastuzumab (Dual HER2 monoclonal antibodies)
The phase IIa MyPathway trial (NCT02091141) demonstrated synergistic HER2 blockade using pertuzumab (targeting domain II) and trastuzumab (targeting domain IV), resulting in clinical activity in HER2-positive CCA[307].
Trastuzumab deruxtecan
In phase II trials, trastuzumab deruxtecan (T-DXd) demonstrated notable efficacy in HER2-positive CCA. The NCCH1805 HERB trial[308] and the JMA-IIA00423 HERB trial reported a 36.4% objective response rate in patients refractory to gemcitabine, with median PFS of 4.4 months and overall survival of 7.1 months, though 25% experienced interstitial lung disease necessitating close monitoring[309]. The phase II DESTINY-PanTumor02 trial (NCT04482309) showed a 37.1% overall response rate in HER2-positive advanced tumors - including CCA - rising to 61.3% in HER2 IHC 3+ cases, with median progression-free and overall survival of 11.9 and 21.1 months, respectively; grade ≥ 3 adverse events occurred in 40.8%, and serious lung toxicity in 10.5%[310]. Collectively, these data support T-DXd as a promising therapeutic option in HER2-expressing CCA, warranting vigilant management of pulmonary toxicity.
Zanidatamab (Bispecific monoclonal antibody)
Zanidatamab is a bispecific monoclonal antibody targeting two distinct extracellular domains of HER2, designed to enhance receptor clustering and degradation. In the phase IIb HERIZON-BTC-01 trial (NCT04466891), zanidatamab demonstrated a 41.3% objective response rate with manageable toxicity in patients with treatment-refractory, HER2-amplified CCA, supporting its potential as a targeted therapy in this setting[311]. It also showed antitumor activity in HER2-positive malignancies in a phase I trial (NCT02892123)[312].
Neratinib (HER2/4 tyrosine kinase inhibitor)
In the SUMMIT phase II trial NCT01953926, neratinib - an irreversible tyrosine kinase inhibitor of HER2 and HER4 - was evaluated in patients with HER2-mutant, treatment-refractory CCA. Among 25 patients, including 11 with CCA, the overall response rate was 16%, with S310F and V777L identified as the most frequent HER2 mutations. Although the primary endpoint was not met, neratinib demonstrated clinical activity in select HER2-mutant CCA cases, supporting further investigation in combination strategies[313].
Tucatinib plus trastuzumab (Selective HER2 TKI combination)
In the phase II SGNTUC-019 trial NCT04579380, tucatinib - a selective HER2 tyrosine kinase inhibitor - combined with trastuzumab demonstrated encouraging antitumor activity in patients with previously treated HER2-positive metastatic CCA[314]. Among 30 enrolled patients, the confirmed objective response rate was 46.7%, with a disease control rate of 76.7%. Median PFS was 5.5 months, and overall survival at 12 months was 53.6%. The regimen was generally well tolerated; most adverse events were low-grade, and no treatment-related deaths were reported. These findings support the combination as a promising HER2-directed strategy in this setting.
Trastuzumab plus chemotherapy (FOLFOX or gemcitabine-cisplatin)
Trastuzumab combined with chemotherapy has demonstrated clinical activity in HER2-positive CCA. In the phase II KCSG-HB19-14 trial (NCT04722133), trastuzumab plus FOLFOX achieved an objective response rate of 29.4% and a disease control rate of 79.4% in patients who had progressed after gemcitabine-based therapy, with an acceptable safety profile[315]. Similarly, in the Indian TAB phase II trial (CTRI/2019/11/021955), trastuzumab combined with first-line gemcitabine-cisplatin resulted in a 6-month PFS rate of 75.6%, with an overall response rate of 55.5% and disease control in 80% of patients[316]. These data support the utility of trastuzumab-based chemotherapy in HER2-positive CCA.
HER2-targeted therapies show promise in CCA but benefit only select patients. Variability in response highlights the need for standardized diagnostics, predictive biomarkers, and optimized combination strategies. Emerging trials aim to refine patient selection and expand HER2-directed approaches into earlier treatment lines.
REGULATORY NON-CODING RNAS IN CCA: ROLES IN METABOLIC DYSREGULATION
MicroRNAs (miRNAs) and long non-coding RNAs (lncRNAs) are critical non-coding RNAs involved in gene regulation. miRNAs (~18-23 nucleotides) repress gene expression by binding to the 3’ untranslated regions (3’-UTRs) of target mRNAs, promoting degradation or translational inhibition[317]. lncRNAs, defined as transcripts longer than 200 nucleotides, form complex structures that interact with DNA, RNA, and proteins to regulate gene expression at both transcriptional and post-transcriptional levels[318]. Both classes play essential roles in carcinogenesis[317,318].
In CCA, dysregulated expression of specific miRNAs and lncRNAs contributes to tumor proliferation, invasion, metastasis, and metabolic reprogramming, acting as oncogenes or tumor suppressors[317-319]. High-throughput analyses of CCA tissues, cell lines, and circulating samples using RNA sequencing, microarrays, and NanoString have identified numerous deregulated non-coding RNAs[318,320-322]. These findings support their potential as prognostic biomarkers and therapeutic targets, particularly in modulating glucose metabolism, a key driver of CCA progression.
Non-coding RNA-mediated regulation of glucose transporters in CCA
miR-148a and GLUT1 regulation in CCA
GLUT1 is significantly overexpressed in iCCA and correlates with poor overall and disease-free survival. Functionally, GLUT1 enhances tumor growth, invasion, and gemcitabine resistance, while its inhibition reduces these malignant traits. miR-148a, which directly targets GLUT1, is downregulated in iCCA, leading to GLUT1 overexpression and tumor progression. Restoration of miR-148a suppresses GLUT1 expression and impairs tumor aggressiveness. Additionally, pharmacologic inhibition of GLUT1 with WZB117 markedly reduced tumor growth in patient-derived xenograft models, highlighting the miR-148a-GLUT1 axis as a promising therapeutic target in iCCA[323].
Other miRNAs targeting GLUTs in human cancers: lessons for CCA
Several miRNAs regulate glucose transporters across cancer types, offering mechanistic insights applicable to CCA. In breast cancer, miR-22 targets GLUT1 and suppresses tumor proliferation and invasion, with levels inversely correlated to tumor stage and prognosis[324]. miR-143 downregulates GLUT1 in immune cells, limiting glycolytic flux and affecting T cell differentiation[325]. In bladder cancer, miR-340 inhibits GLUT1 and the PI3K/AKT pathway, promoting apoptosis and reducing proliferation[326]. In oral squamous cell carcinoma (OSCC), miR-340, miR-10a, miR-150-5p, and miR-200c regulate GLUT1 and GLUT3, contributing to tumor progression and metastasis[327]. Furthermore, let-7a-5p suppresses GLUT12 in triple-negative breast cancer, reducing cell proliferation and migration[328]. These findings suggest a conserved role of GLUT-targeting miRNAs in tumor metabolism and underscore the need to investigate similar regulatory networks in CCA.
lncRNA-mediated pathways potentially involving GLUT regulation in CCA
Although direct links between lncRNAs and glucose transporters in CCA remain limited, indirect evidence suggests metabolic involvement. HOX transcript antisense intergenic RNA (HOTAIR) facilitates CCA cell proliferation and suppresses apoptosis by modulating the miR-204-5p/high mobility group box 1 (HMGB1) signaling axis[329], and in macrophages, it enhances GLUT1 expression through NF-κB activation, increasing glucose uptake[330]. Motor neuron and pancreas homeobox 1 antisense RNA 1 (MNX1-AS1) promotes iCCA progression by activating cellular c-Myc and Myc-associated zinc finger protein (MAZ), which upregulate MNX1 and Ajuba LIM protein (Ajuba), resulting in inhibition of the Hippo signaling pathway and activation of Yes-associated protein (YAP), both involved in metabolic regulation[331]. These findings point to potential roles for HOTAIR and MNX1-AS1 in modulating glucose metabolism in CCA, warranting further exploration.
Non-coding RNAs regulating glycolytic enzymes in CCA
Non-coding RNA-mediated regulation of HKII in CCA and other cancers
In CCA, the lncRNA PVT1 is upregulated and functions as a competing endogenous RNA (ceRNA), sequestering tumor-suppressive miR-143 to prevent inhibition of HKII. This enhances glycolysis, increasing glucose uptake and lactate production, thereby promoting tumor proliferation, invasion, and poor prognosis. PVT1 knockdown or miR-143 overexpression decreases HKII expression and impairs tumor progression in vitro and in vivo, highlighting the PVT1/miR-143/HKII axis as a promising therapeutic target[332].
Although studies on other miRNAs targeting HKII in CCA are limited, miR-143 and others are well characterized in diverse cancers. For example, miR-143 suppresses HKII protein in lung cancer, reducing glycolysis under mTOR regulation[333]. Similarly, miR-216b downregulates HKII and mTOR signaling in breast cancer, inhibiting proliferation and enhancing apoptosis[334]. In HCC, hypoxia-mediated downregulation of miR-125a permits HKII overexpression, promoting the Warburg effect; restoring miR-125a reduces lactate, glucose uptake, ATP, and ROS, impairing tumor growth[335]. Additional miRNAs, including miR-199a-5p, miR-155, and miR-138, modulate HKII and related isoforms across various cancers, emphasizing conserved miRNA-HKII regulatory mechanisms with translational potential in CCA[336].
Non-coding RNA-mediated regulation of PFK-1 in CCA and other cancers
PFK-1, a rate-limiting glycolytic enzyme catalyzing F6P to F1,6P, is negatively regulated by miR-135. This miRNA directly targets PFK-1, inhibiting aerobic glycolysis and tumor growth, underscoring post-transcriptional regulation as a key node in cancer metabolic reprogramming with therapeutic implications[336,337].
Non-coding RNA-mediated regulation of ALDOA in CCA and other cancers
In CCA, ALDOA is suppressed by miR-122-5p, inhibiting proliferation, invasion, and tumor growth[338]. Conversely, the lncRNA urothelial cancer associated 1 promotes metastasis via the miR-122/ chloride intracellular channel 1 axis and ERK/MAPK pathway activation[339]. Hepatocyte nuclear factor 6, studied in HCC, upregulates miR-122, reinforcing tumor suppression[340].
In other cancers, miR-122 acts as a tumor suppressor by targeting ALDOA, and its inhibition by the upregulated lncRNA DIO3OS promotes pancreatic cancer progression via the miR-122/ALDOA axis[341]. The miR-15a/16-1 cluster also modulates ALDOA-linked metabolic reprogramming[342]. In esophageal squamous cell carcinoma (ESCC), miR-378a-3p targets ALDOA, GLUT1, and PKM2, reducing ATP production and inducing apoptosis[343]. In CRC, lncRNA NONHSAG028908.3 acts as a ceRNA for miR-34, derepressing ALDOA to promote glycolysis and proliferation[344]. Lung cancer studies reveal ALDOA enhances stemness and drug resistance through a non-enzymatic pathway by suppressing miR-145, independent of its enzymatic role; miR-378 may regulate this axis[345]. These findings position ALDOA as a critical metabolic regulator modulated by diverse non-coding RNAs, presenting therapeutic targets in CCA.
Non-coding RNA-mediated regulation of PKM2 in CCA and other cancers
MiRNAs such as miR-122, miR-1286, miR-21, and miR-520a-3p regulate PKM2 and LDHA, key glycolytic enzymes in cancer metabolism. In CCA and ESCC, miR-122 suppresses PKM2, inhibiting glycolysis, EMT, and tumor progression[346,347]. Downregulation of miR-122 in gallbladder and CRCs enhances PKM2 activity, promoting tumor growth and therapy resistance; restoring miR-122 reverses these effects[347]. In NSCLC, miR-1286 targets PKM2 to reduce lactate production and proliferation, suggesting potential applicability in CCA[348]. Additionally, miR-21 and miR-520a-3p inhibit PKM2 and LDHA in cisplatin-resistant NSCLC and gastric cancer, disrupting glycolysis and PI3K/AKT/mTOR/HIF-1α signaling[349,350]. Given miR-122’s dual regulation of ALDOA and PKM2 in CCA, it emerges as a pivotal modulator of glycolytic enzymes driving tumor metabolism and progression. These insights emphasize miRNA-enzyme interactions as critical targets for metabolic intervention in CCA.
Non-coding RNA control of the PPP in CCA
Non-coding RNA-mediated regulation of oxidative PPP enzymes: G6PD and 6PGD in CCA and other cancers
The oxidative PPP, essential for NADPH production and redox homeostasis, is tightly regulated by non-coding RNAs. G6PD, the rate-limiting enzyme, is directly suppressed by miR-1, which inhibits tumor metabolism[351]. Liver-specific miR-122 also downregulates G6PD; its loss disrupts redox balance and drives metabolic reprogramming in liver cancers[352]. Another key enzyme, 6PGD, overexpressed in cancers including leukemia[353], is post-transcriptionally repressed by miR-206 and miR-613. These miRNAs attenuate 6PGD-driven metabolic rewiring and cisplatin resistance in ovarian and lung cancers[354,355]. Although direct evidence in CCA is sparse, these conserved mechanisms emphasize the therapeutic promise of targeting oxidative PPP enzymes via non-coding RNAs.
Non-coding RNA-mediated regulation of non-oxidative PPP enzymes: TKT and TALDO in CCA and beyond
TKT, a key non-oxidative PPP enzyme, is increasingly recognized as a miRNA-regulated metabolic node. In pancreatic cancer, prolactin receptor (PRLR) signaling upregulates miR-4763-3p and miR-3663-5p, which suppress TKT and G6PD, reducing nucleotide biosynthesis and enhancing gemcitabine sensitivity[356]. Similarly, in CRC, miR-124 targets PRPS1 and RPIA, impairing ribose production and tumor proliferation[357]. In cervical cancer, miR-497 directly downregulates TKT, inducing GSH depletion and ROS accumulation, thereby increasing cisplatin sensitivity[358]. Although these miRNA-mediated controls remain unconfirmed in CCA, their metabolic significance suggests that targeting TKT via miRNAs like miR-124, miR-497, or miR-4763-3p may offer novel therapeutic avenues to modulate nucleotide synthesis and chemoresistance.
Non-coding RNA regulation of LDHA in CCA
miRNAs control of LDHA and export
Though specific CCA data are limited, numerous miRNAs regulate LDHA in other cancers. LDHA, critical for lactate production, is targeted by miR-30d-5p (gallbladder cancer), miR-30a-5p and miR-34a (breast cancer), miR-383 (ovarian cancer), miR-449a (NSCLC), miR-142-3p (HCC), miR-204-3p (bladder cancer), miR-329-3p and miR-323a-3p (osteosarcoma), and miR-489-3p (pancreatic cancer)[359]. Similarly, lactate transporters MCT1 and MCT4 are regulated by miRNAs including miR-124 (medulloblastoma, breast cancer), miR-342-3p (HCC, triple-negative breast cancer), miR-449a (glioma), and miR-145-5p (HCC). Although unexplored in CCA, these miRNA-mediated pathways present promising targets for disrupting tumor LDHA.
Non-coding RNAs modulating the TCA cycle in CCA
Regulation of the IDH by miRNAs
IDH mutations characterize a CCA subtype with mitochondrial activation, DNA hypermethylation, and altered chromatin modifier gene expression. miR-194-5p is significantly upregulated in this subtype, inversely correlating with chromatin modifier signatures, implicating it in epigenetic and metabolic reprogramming linked to IDH-driven tumorigenesis[137]. Evidence from IDH-mutant gliomas further highlights miRNA involvement: accumulation of the oncometabolite 2-HG induces promoter hypermethylation, suppressing tumor-suppressive miRNAs such as miR-148a. Additional miRNAs influenced by IDH status - miR-106b, miR-130b, miR-98-3p, miR-185 - modulate proliferation and progression, while the IDH1 R132H mutation downregulates miR-141-3p, miR-7-5p, and miR-223-3p, upregulating IGF1R and promoting aggressiveness. Moreover, 2-HG-mediated EMT involves suppression of miR-200b/c and induction of ZEB1[360]. Although these pathways remain unconfirmed in CCA, they underscore miRNAs as critical regulators and potential therapeutic targets in IDH-mutant cancers.
Non-coding RNA influence on OXPHOS and mitochondrial biogenesis in CCA
Direct evidence in CCA is lacking; however, miRNAs modulating PGC-1α, a master regulator of mitochondrial biogenesis and OXPHOS, have been identified in other cancers. In breast cancer, miR-217 suppresses PGC-1α; its downregulation enhances PGC-1α expression and reduces proliferation, suggesting a tumor-suppressive function[361]. In HCC, miR-23a binds PGC-1α 3’-UTR, decreasing mitochondrial function and gluconeogenesis[362]. Likewise, in osteosarcoma, miR-23b-3p targets PGC-1α, suppressing OXPHOS and promoting aerobic glycolysis consistent with the Warburg effect[363]. These conserved miRNA-PGC-1α interactions offer promising targets for modulating metabolic reprogramming in CCA.
LIMITATIONS AND CHALLENGES IN METABOLIC TARGETING OF CCA: DRUG RESISTANCE, TOXICITY, AND TUMOR SPECIFICITY
Despite promising preclinical data supporting the repurposing of antidiabetic agents such as metformin and SGLT2 inhibitors in CCA, important limitations persist. Tumor cells exhibit substantial metabolic flexibility, often bypassing inhibited pathways and leading to therapeutic resistance over time[155,157]. Furthermore, systemic agents like SGLT2 inhibitors may activate unintended pro-tumorigenic processes, including the NAD+ salvage pathway, thereby complicating their safety profile[162-164]. Targeting GLUTs, particularly GLUT1, faces similar challenges. Although overexpressed in CCA, GLUT1 is also essential for glucose uptake in normal tissues, and its inhibition risks off-target effects and metabolic disruption in non-malignant cells. In addition, the heterogeneous expression of glucose transporters across tumor subtypes undermines therapeutic specificity[46,166-168].
Therapeutic targeting of key glycolytic enzymes - HKII, PFK-1, ALDOA, and PKM2 - offers a direct approach to disrupt tumor metabolism, yet resistance mechanisms remain a significant hurdle. Metabolic reprogramming may allow tumor cells to compensate via alternate pathways, reducing the efficacy of single-agent inhibitors[50,176,179]. Similarly, compounds such as LND and shikonin show potential in preclinical models but may exert off-target or dose-limiting toxicities[172,173,180-183]. Parallel strategies targeting the PPP, including inhibition of G6PD, 6PGD, TKT, and TALDO, are hampered by the ubiquitous expression of these enzymes in both malignant and healthy cells. This raises concerns over systemic toxicity and restricts therapeutic windows, especially given their vital roles in maintaining redox balance and nucleotide biosynthesis[194,200,203].
Targeting LDHA via LDHA inhibitors like FX11 and oxamate holds promise in reshaping the tumor microenvironment and enhancing antitumor immunity. However, because both cancer and immune cells rely on glycolysis, these approaches risk collateral effects on immune function and potential tumor adaptation[191,215-219,222]. Inhibition of TCA cycle enzymes, including IDH1/2, GLS, and α-KG dehydrogenase, has shown early efficacy, particularly with agents like ivosidenib and devimistat. Nevertheless, acquired resistance - such as activation of compensatory pathways like SRC - and limited efficacy in IDH2-mutant or wild-type cases remain critical concerns[229,235,237,238].
Finally, mitochondrial OXPHOS inhibition, especially targeting Complex I, has demonstrated robust antitumor effects in CCA stem-like cells. Yet, toxicity has impeded clinical translation. Agents such as rotenone, BAY-87-2243, and IACS-010759 were discontinued due to neurotoxicity, hepatotoxicity, and hematologic side effects[240,242-244]. Even clinically relevant agents like metformin and phenformin exert systemic mitochondrial effects that may impact high-energy-demand normal tissues[94,241]. While selective inhibitors of PGC-1α, such as SR-18292, offer a more targeted approach by exploiting CSC vulnerabilities, tumor heterogeneity in mitochondrial dependency and variable expression of metabolic targets like PGC-1α or IDH1/2 further complicate personalized application[94,229].
Overall, major limitations across all metabolic targeting strategies in CCA include metabolic plasticity and redundancy, allowing tumor cells to adapt by switching among glycolysis, PPP, LDHA, TCA cycle, and OXPHOS pathways to evade single-target inhibition. Tumor heterogeneity adds complexity, with diverse metabolic phenotypes and genetic backgrounds complicating the broad application of metabolic inhibitors. Off-target toxicity poses significant risks due to the essential roles of many metabolic enzymes in normal proliferative and immune cells. Furthermore, the lack of CCA-specific clinical data means that most metabolic inhibitors have been tested primarily in other cancer types, limiting direct translational evidence. The complex effects of metabolic interventions on immune modulation may also inadvertently impair antitumor immunity or activate compensatory tumor-promoting pathways. Addressing these challenges will require comprehensive, integrated approaches combining metabolic inhibitors with chemotherapy, immunotherapy, and molecularly targeted agents, guided by detailed tumor metabolic profiling. Continued preclinical and clinical research is essential to optimize dosing, minimize toxicity, and develop effective combination regimens that exploit the unique metabolic vulnerabilities of CCA.
CONCLUSION
Metabolic dysregulation, particularly in glucose metabolism, plays a critical role in the progression and carcinogenesis of CCA. Elevated glucose levels in CCA cells are driven by increased GLUT1 expression, resulting in enhanced glycolytic flux. While the Warburg effect predominates, OXPHOS also contributes to CCA cell stemness and proliferation. Dysregulation of key enzymes involved in glycolysis, the PPP, LDHA, and the TCA cycle further supports tumor growth and survival by maintaining redox balance. These metabolic alterations, driven by common oncogenic mutations in CCA, shape critical cell signaling pathways that promote tumorigenesis.
The disruption of glucose metabolism, including glycolysis and OXPHOS, and the regulation of redox homeostasis, present valuable targets for therapeutic intervention in CCA. Current strategies, such as IDH1 inhibitors like Ivosidenib and GLS1 inhibitors, show potential in targeting metabolic pathways. However, the full extent of metabolic dysregulation in CCA remains inadequately explored, and additional research is needed to elucidate the underlying mechanisms and their role in tumor progression.
Moreover, repurposing existing drugs, such as those used in diabetes management, may offer new opportunities for targeting upregulated metabolic pathways in CCA. Future studies should prioritize investigating glucose metabolic reprogramming in chemoresistant CCA, as this could lead to novel therapeutic combinations of chemotherapy and targeted metabolic therapies. Advancing our understanding of these metabolic targets is essential for developing more effective treatment strategies for patients with advanced CCA.
DECLARATIONS
Acknowledgments
The authors would like to thank Siriraj SiCORE-M Unit for the support.
Authors’ contributions
Conceptualization, writing of the original draft, and writing - review and editing of the manuscript: Makamas C
Conceptualization and writing of the original draft: Mekasuwandumrong C, Wilasrusmee K
Critical revision of the manuscript: Panich U
Conceptualization, study design and coordination, and critical revision of the manuscript: Jirawatnotai S
All authors read and approved the final version of the manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
The project was funded by the National Research Council of Thailand (NRCT) (N41A640162 and N42A670195) [HOME - NATIONAL RESEARCH COUNCIL OF THAILAND], the Foundation for Cancer Care Siriraj Hospital, and Siriraj Foundation (D003421). Jirawatnotai S is supported by the Siriraj Hospital’s Research Excellence Development Program (RED). The funding bodies had no role in experiment design, collection, analysis and interpretation of data, and writing of the manuscript.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Banales JM, Marin JJG, Lamarca A, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17:557-88.
3. Vithayathil M, Khan SA. Current epidemiology of cholangiocarcinoma in Western countries. J Hepatol. 2022;77:1690-8.
4. Sripa B, Pairojkul C. Cholangiocarcinoma: lessons from Thailand. Curr Opin Gastroenterol. 2008;24:349-56.
5. Treeprasertsuk S, Poovorawan K, Soonthornworasiri N, et al. A significant cancer burden and high mortality of intrahepatic cholangiocarcinoma in Thailand: a nationwide database study. BMC Gastroenterol. 2017;17:3.
6. Pant K, Richard S, Peixoto E, Gradilone SA. Role of glucose metabolism reprogramming in the pathogenesis of cholangiocarcinoma. Front Med. 2020;7:113.
7. Ilyas SI, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology. 2013;145:1215-29.
8. Pastore M, Lori G, Gentilini A, et al. Multifaceted aspects of metabolic plasticity in human cholangiocarcinoma: an overview of current perspectives. Cells. 2020;9:596.
9. Stenzinger A, Vogel A, Lehmann U, et al. Molecular profiling in cholangiocarcinoma: a practical guide to next-generation sequencing. Cancer Treat Rev. 2024;122:102649.
10. Chen W, Xu D, Liu Q, Wu Y, Wang Y, Yang J. Unraveling the heterogeneity of cholangiocarcinoma and identifying biomarkers and therapeutic strategies with single-cell sequencing technology. Biomed Pharmacother. 2023;162:114697.
11. Carotenuto M, Sacco A, Forgione L, Normanno N. Genomic alterations in cholangiocarcinoma: clinical significance and relevance to therapy. Explor Target Antitumor Ther. 2022;3:200-23.
12. Andraus W, Tustumi F, de Meira Junior JD, et al. Molecular profile of intrahepatic cholangiocarcinoma. Int J Mol Sci. 2023;25:461.
13. Porreca V, Barbagallo C, Corbella E, et al. Unveil intrahepatic cholangiocarcinoma heterogeneity through the lens of omics and multi-omics approaches. Cancers. 2024;16:2889.
14. Lu M, Qin X, Zhou Y, et al. Long non-coding RNA LINC00665 promotes gemcitabine resistance of cholangiocarcinoma cells via regulating EMT and stemness properties through miR-424-5p/BCL9L axis. Cell Death Dis. 2021;12:72.
15. Elvevi A, Laffusa A, Scaravaglio M, et al. Clinical treatment of cholangiocarcinoma: an updated comprehensive review. Ann Hepatol. 2022;27:100737.
16. Valle J, Wasan H, Palmer DH, et al; ABC-02 Trial Investigators. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362:1273-81.
17. Srijiwangsa P, Ponnikorn S, Na-Bangchang K. Effect of β-Eudesmol on NQO1 suppression-enhanced sensitivity of cholangiocarcinoma cells to chemotherapeutic agents. BMC Pharmacol Toxicol. 2018;19:32.
18. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714-26.
20. Park JH, Pyun WY, Park HW. Cancer metabolism: phenotype, signaling and therapeutic targets. Cells. 2020;9:2308.
21. Vaupel P, Multhoff G. Revisiting the Warburg effect: historical dogma versus current understanding. J Physiol. 2021;599:1745-57.
22. Zhang Y, Li Q, Huang Z, et al. Targeting glucose metabolism enzymes in cancer treatment: current and emerging strategies. Cancers. 2022;14:4568.
23. Zheng J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol Lett. 2012;4:1151-7.
24. Xing F, Hu Q, Qin Y, et al. The relationship of redox with hallmarks of cancer: the importance of homeostasis and context. Front Oncol. 2022;12:862743.
25. De Santis MC, Porporato PE, Martini M, Morandi A. Signaling pathways regulating redox balance in cancer metabolism. Front Oncol. 2018;8:126.
26. Thonsri U, Seubwai W, Waraasawapati S, et al. Overexpression of lactate dehydrogenase A in cholangiocarcinoma is correlated with poor prognosis. Histol Histopathol. 2017;32:503-10.
27. Kawada K, Toda K, Sakai Y. Targeting metabolic reprogramming in KRAS-driven cancers. Int J Clin Oncol. 2017;22:651-9.
28. Ma Q, Zhang W, Wu K, Shi L. The roles of KRAS in cancer metabolism, tumor microenvironment and clinical therapy. Mol Cancer. 2025;24:14.
29. Zhang C, Liu J, Liang Y, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935.
30. Gomes AS, Ramos H, Soares J, Saraiva L. p53 and glucose metabolism: an orchestra to be directed in cancer therapy. Pharmacol Res. 2018;131:75-86.
31. Abukwaik R, Vera-Siguenza E, Tennant D, Spill F. p53 orchestrates cancer metabolism: unveiling strategies to reverse the Warburg effect. Bull Math Biol. 2024;86:124.
32. Li J, He Y, Tan Z, et al. Wild-type IDH2 promotes the Warburg effect and tumor growth through HIF1α in lung cancer. Theranostics. 2018;8:4050-61.
33. Parker SJ, Metallo CM. Metabolic consequences of oncogenic IDH mutations. Pharmacol Ther. 2015;152:54-62.
34. Chan FF, Wong CM. Targeting the metabolic vulnerability of ARID1A-deficient hepatocellular carcinoma. Cell Mol Gastroenterol Hepatol. 2022;14:241-2.
35. Zhen Y, Liu K, Shi L, et al. FGFR inhibition blocks NF-ĸB-dependent glucose metabolism and confers metabolic vulnerabilities in cholangiocarcinoma. Nat Commun. 2024;15:3805.
36. Holloway RW, Marignani PA. Targeting mTOR and glycolysis in HER2-positive breast cancer. Cancers. 2021;13:2922.
37. Casak SJ, Pradhan S, Fashoyin-Aje LA, et al. FDA approval summary: ivosidenib for the treatment of patients with advanced unresectable or metastatic, chemotherapy refractory cholangiocarcinoma with an IDH1 mutation. Clin Cancer Res. 2022;28:2733-7.
38. Caligiuri A, Becatti M, Porro N, et al. Oxidative stress and redox-dependent pathways in cholangiocarcinoma. Antioxidants. 2023;13:28.
39. Shestov AA, Liu X, Ser Z, et al. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. Elife. 2014;3:e03342.
40. Kinjo Y, Naito Y, Akiba J, et al. SUOX and GLUT1 are biomarkers for the prognosis in large duct type intrahepatic cholangiocarcinoma. Hum Pathol. 2022;128:11-9.
41. Kozaka K, Kobayashi S, Takamura H, et al. Differences in 18F-FDG uptake and expression of glucose transporter between 2 distinct subtypes of mass-forming intrahepatic cholangiocarcinomas. Clin Nucl Med. 2020;45:e267-73.
42. Kubo Y, Aishima S, Tanaka Y, et al. Different expression of glucose transporters in the progression of intrahepatic cholangiocarcinoma. Hum Pathol. 2014;45:1610-7.
43. Hao L, Li S, Peng Q, Zhang J, Deng J, Hu X. Targeting glycolytic reprogramming in cholangiocarcinoma: a novel approach for metabolic therapy. J Inflamm Res. 2024;17:9665-81.
44. Szablewski L. Glucose transporters as markers of diagnosis and prognosis in cancer diseases. Oncol Rev. 2022;16:561.
45. Suwannakul N, Armartmuntree N, Thanan R, et al. Targeting fructose metabolism by glucose transporter 5 regulation in human cholangiocarcinoma. Genes Dis. 2022;9:1727-41.
46. Thamrongwaranggoon U, Sangkhamanon S, Seubwai W, Saranaruk P, Cha’on U, Wongkham S. Aberrant GLUT1 expression is associated with carcinogenesis and progression of liver fluke-associated cholangiocarcinoma. In Vivo. 2021;35:267-74.
47. Luo XM, Zhou SH, Fan J. Glucose transporter-1 as a new therapeutic target in laryngeal carcinoma. J Int Med Res. 2010;38:1885-92.
48. Miller ZA, Muthuswami S, Mueller A, et al. GLUT1 inhibitor BAY-876 induces apoptosis and enhances anti-cancer effects of bitter receptor agonists in head and neck squamous carcinoma cells. Cell Death Discov. 2024;10:339.
49. Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol. 2012;24:650-4.
50. Thamrongwaranggoon U, Seubwai W, Phoomak C, et al. Targeting hexokinase II as a possible therapy for cholangiocarcinoma. Biochem Biophys Res Commun. 2017;484:409-15.
51. Raggi C, Taddei ML, Rae C, Braconi C, Marra F. Metabolic reprogramming in cholangiocarcinoma. J Hepatol. 2022;77:849-64.
52. Peng M, Li H, Cao H, et al. Dual FGFR and VEGFR inhibition synergistically restrain hexokinase 2-dependent lymphangiogenesis and immune escape in intrahepatic cholangiocarcinoma. J Gastroenterol. 2023;58:908-24.
53. Peng J, Li P, Li Y, et al. PFKP is a prospective prognostic, diagnostic, immunological and drug sensitivity predictor across pan-cancer. Sci Rep. 2023;13:17399.
54. Fujiwara H, Tateishi K, Misumi K, et al. Mutant IDH1 confers resistance to energy stress in normal biliary cells through PFKP-induced aerobic glycolysis and AMPK activation. Sci Rep. 2019;9:18859.
55. Li X, Yu C, Luo Y, et al. Aldolase A enhances intrahepatic cholangiocarcinoma proliferation and invasion through promoting glycolysis. Int J Biol Sci. 2021;17:1782-94.
56. Yu G, Yu W, Jin G, et al. PKM2 regulates neural invasion of and predicts poor prognosis for human hilar cholangiocarcinoma. Mol Cancer. 2015;14:193.
57. Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15:6479-83.
58. Qian Z, Hu W, Lv Z, et al. PKM2 upregulation promotes malignancy and indicates poor prognosis for intrahepatic cholangiocarcinoma. Clin Res Hepatol Gastroenterol. 2020;44:162-73.
59. Yu W, Zeng F, Xiao Y, et al. Targeting PKM2 improves the gemcitabine sensitivity of intrahepatic cholangiocarcinoma cells via inhibiting β-catenin signaling pathway. Chem Biol Interact. 2024;387:110816.
60. Zhou Q, Yin Y, Yu M, et al. GTPBP4 promotes hepatocellular carcinoma progression and metastasis via the PKM2 dependent glucose metabolism. Redox Biol. 2022;56:102458.
61. Chanda M, Anuntasomboon P, Ruangritchankul K, Cheepsunthorn P, Cheepsunthorn CL. Inhibition of non-small cell lung cancer (NSCLC) proliferation through targeting G6PD. PeerJ. 2023;11:e16503.
62. Ghanem N, El-Baba C, Araji K, El-Khoury R, Usta J, Darwiche N. The pentose phosphate pathway in cancer: regulation and therapeutic opportunities. Chemotherapy. 2021;66:179-91.
63. Yang HC, Stern A, Chiu DT. G6PD: A hub for metabolic reprogramming and redox signaling in cancer. Biomed J. 2021;44:285-92.
64. Nagashio R, Oikawa S, Yanagita K, et al. Prognostic significance of G6PD expression and localization in lung adenocarcinoma. Biochim Biophys Acta Proteins Proteom. 2019;1867:38-46.
65. Liu R, Li W, Tao B, et al. Tyrosine phosphorylation activates 6-phosphogluconate dehydrogenase and promotes tumor growth and radiation resistance. Nat Commun. 2019;10:991.
66. Liu T, Qi J, Wu H, et al. Phosphogluconate dehydrogenase is a predictive biomarker for immunotherapy in hepatocellular carcinoma. Front Oncol. 2022;12:993503.
67. Ong AJ, Saeidi S, Chi NHK, et al. The positive feedback loop between Nrf2 and phosphogluconate dehydrogenase stimulates proliferation and clonogenicity of human hepatoma cells. Free Radic Res. 2020;54:906-17.
68. Zheng P, Pan HH, Zhou XH, Qiu YY, Hu J, et al. Glucose 6 phosphatase dehydrogenase (G6PD): a novel diagnosis marker related to gastrointestinal cancers. Am J Transl Res. 2023;15:4:2304-28.
69. Li R, Wang W, Yang Y, Gu C. Exploring the role of glucose6phosphate dehydrogenase in cancer (Review). Oncol Rep. 2020;44:2325-36.
70. Cao J, Sun X, Zhang X, Chen D. 6PGD upregulation is associated with chemo- and immuno-resistance of renal cell carcinoma via AMPK signaling-dependent NADPH-mediated metabolic reprograming. Am J Med Sci. 2020;360:279-86.
71. Jin L, Zhou Y. Crucial role of the pentose phosphate pathway in malignant tumors. Oncol Lett. 2019;17:4213-21.
72. Qiao J, Yu Z, Zhou H, Wang W, Wu H, Ye J. The pentose phosphate pathway: from mechanisms to implications for gastrointestinal cancers. Int J Mol Sci. 2025;26:610.
73. Li M, Zhao X, Yong H, et al. Transketolase promotes colorectal cancer metastasis through regulating AKT phosphorylation. Cell Death Dis. 2022;13:99.
74. Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656-70.
75. Marbaniang C, Kma L. Dysregulation of glucose metabolism by oncogenes and tumor suppressors in cancer cells. Asian Pac J Cancer Prev. 2018;19:2377-90.
76. He F, Ru X, Wen T. NRF2, a transcription factor for stress response and beyond. Int J Mol Sci. 2020;21:4777.
77. Samatiwat P, Prawan A, Senggunprai L, Kukongviriyapan U, Kukongviriyapan V. Nrf2 inhibition sensitizes cholangiocarcinoma cells to cytotoxic and antiproliferative activities of chemotherapeutic agents. Tumour Biol. 2016;37:11495-507.
78. Qu X, Sheng J, Shen L, et al. Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PLoS One. 2017;12:e0173712.
79. Yu Y, Liao M, Liu R, Chen J, Feng H, Fu Z. Overexpression of lactate dehydrogenase-A in human intrahepatic cholangiocarcinoma: its implication for treatment. World J Surg Oncol. 2014;12:78.
80. Mathew M, Nguyen NT, Bhutia YD, Sivaprakasam S, Ganapathy V. Metabolic signature of Warburg effect in cancer: an effective and obligatory interplay between nutrient transporters and catabolic/anabolic pathways to promote tumor growth. Cancers. 2024;16:504.
81. Langhammer S, Najjar M, Hess-Stumpp H, Thierauch KH.
82. Liu D, Wang D, Wu C, et al. Prognostic significance of serum lactate dehydrogenase in patients with breast cancer: a meta-analysis. Cancer Manag Res. 2019;11:3611-9.
83. Malhotra G, Gattani RG, Shinde RK, Gianchandani SG, Nayak K, Salwan A. Significance of serum lactate dehydrogenase as a prognostic marker and outcome predictor in patients with breast cancer. Cureus. 2024;16:e55932.
84. Al-Khallaf H. Isocitrate dehydrogenases in physiology and cancer: biochemical and molecular insight. Cell Biosci. 2017;7:37.
85. Rizzo A, Ricci AD, Brandi G. IDH inhibitors in advanced cholangiocarcinoma: another arrow in the quiver? Cancer Treat Res Commun. 2021;27:100356.
86. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21:117:373-80.
87. Gu Y, Yang R, Yang Y, et al. IDH1 mutation contributes to myeloid dysplasia in mice by disturbing heme biosynthesis and erythropoiesis. Blood. 2021;137:945-58.
88. Xiang X, Liu Z, Zhang C, et al. IDH mutation subgroup status associates with intratumor heterogeneity and the tumor microenvironment in intrahepatic cholangiocarcinoma. Adv Sci. 2021;8:e2101230.
89. Feichtinger RG, Neureiter D, Kemmerling R, Mayr JA, Kiesslich T, Kofler B. Low VDAC1 expression is associated with an aggressive phenotype and reduced overall patient survival in cholangiocellular carcinoma. Cells. 2019;8:539.
90. Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res. 2018;24:2482-90.
91. Kulthawatsiri T, Kittirat Y, Phetcharaburanin J, et al. Metabolomic analyses uncover an inhibitory effect of niclosamide on mitochondrial membrane potential in cholangiocarcinoma cells. PeerJ. 2023;11:e16512.
92. Bonuccelli G, Tsirigos A, Whitaker-Menezes D, et al. Ketones and lactate “fuel” tumor growth and metastasis: evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010;9:3506-14.
93. Pavlides S, Whitaker-Menezes D, Castello-Cros R, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984-4001.
94. Raggi C, Taddei ML, Sacco E, et al. Mitochondrial oxidative metabolism contributes to a cancer stem cell phenotype in cholangiocarcinoma. J Hepatol. 2021;74:1373-85.
95. Sancho P, Burgos-Ramos E, Tavera A, et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015;22:590-605.
96. Zhao Z, Mei Y, Wang Z, He W. The effect of oxidative phosphorylation on cancer drug resistance. Cancers. 2022;15:62.
97. Ghoneum A, Abdulfattah AY, Warren BO, Shu J, Said N. Redox homeostasis and metabolism in cancer: a complex mechanism and potential targeted therapeutics. Int J Mol Sci. 2020;21:3100.
98. Mcbeth C. Redox in cancer metabolism: manipulation of cancer metabolism through exploitation of redox environments. ChemElectroChem. 2023;10:e202300117.
100. Yang M, Li M, Lyu Z, Yang Z. Implication of ferroptosis in cholangiocarcinoma: a potential future target? Cancer Manag Res. 2023;15:335-42.
102. Zhu G, Pei L, Xia H, Tang Q, Bi F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol Cancer. 2021;20:143.
103. Luo J, Ostrem J, Pellini B, et al. Overcoming KRAS-mutant lung cancer. Am Soc Clin Oncol Educ Book. 2022;42:1-11.
104. Moffat GT, Hu ZI, Meric-Bernstam F, et al. KRAS allelic variants in biliary tract cancers. JAMA Netw Open. 2024;7:e249840.
105. Zhou SL, Xin HY, Sun RQ, et al. Association of KRAS variant subtypes with survival and recurrence in patients with surgically treated intrahepatic cholangiocarcinoma. JAMA Surg. 2022;157:59-65.
106. Huang L, Guo Z, Wang F, Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther. 2021;6:386.
107. Fell JB, Fischer JP, Baer BR, et al. Identification of the clinical development candidate MRTX849, a covalent KRASG12C inhibitor for the treatment of cancer. J Med Chem. 2020;63:6679-93.
108. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548-51.
109. Mukhopadhyay S, Vander Heiden MG, McCormick F. The metabolic landscape of RAS-driven cancers from biology to therapy. Nat Cancer. 2021;2:271-83.
110. Yun J, Rago C, Cheong I, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555-9.
111. Liu J, van der Hoeven R, Kattan WE, et al. Glycolysis regulates KRAS plasma membrane localization and function through defined glycosphingolipids. Nat Commun. 2023;14:465.
112. Patra KC, Wang Q, Bhaskar PT, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013;24:213-28.
113. Guo C, Liu Z, Yu Y, et al. TP53 /KRAS co-mutations create divergent prognosis signatures in intrahepatic cholangiocarcinoma. Front Genet. 2022;13:844800.
114. Harami-Papp H, Pongor LS, Munkácsy G, et al. TP53 mutation hits energy metabolism and increases glycolysis in breast cancer. Oncotarget. 2016;7:67183-95.
115. Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget. 2011;2:948-57.
116. Boidot R, Végran F, Meulle A, et al. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res. 2012;72:939-48.
117. Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008;10:611-8.
118. Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012;72:560-7.
119. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010;107:7455-60.
120. Suzuki S, Tanaka T, Poyurovsky MV, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A. 2010;107:7461-6.
122. Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and metabolism. J Mol Cell Biol. 2019;11:284-92.
123. Jiang P, Du W, Wang X, et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011;13:310-6.
124. Salati M, Caputo F, Baldessari C, et al. IDH signalling pathway in cholangiocarcinoma: from biological rationale to therapeutic targeting. Cancers. 2020;12:3310.
125. Ntanasis-Stathopoulos I, Tsilimigras DI, Gavriatopoulou M, Schizas D, Pawlik TM. Cholangiocarcinoma: investigations into pathway-targeted therapies. Expert Rev Anticancer Ther. 2020;20:765-73.
126. Bralten LB, Kloosterhof NK, Balvers R, et al. IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Ann Neurol. 2011;69:455-63.
127. Birner P, Pusch S, Christov C, et al. Mutant IDH1 inhibits PI3K/Akt signaling in human glioma. Cancer. 2014;120:2440-7.
128. Cheng T, Sudderth J, Yang C, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A. 2011;108:8674-9.
129. Du D, Liu C, Qin M, et al. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm Sin B. 2022;12:558-80.
130. Abou-Alfa GK, Macarulla T, Javle MM, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020;21:796-807.
131. Jiang Z, Gu Z, Lu X, Wen W. The role of dysregulated metabolism and associated genes in gastric cancer initiation and development. Transl Cancer Res. 2024;13:3854-68.
132. Kuo TL, Cheng KH, Chen LT, Hung WC.
133. Guo B, Friedland SC, Alexander W, et al. Arid1a mutation suppresses TGF-β signaling and induces cholangiocarcinoma. Cell Rep. 2022;40:111253.
134. Zhao S, Xu Y, Wu W, et al.
135. Xing T, Li L, Chen Y, et al. Targeting the TCA cycle through cuproptosis confers synthetic lethality on ARID1A-deficient hepatocellular carcinoma. Cell Rep Med. 2023;4:101264.
136. Zhang FK, Ni QZ, Wang K, et al. Targeting USP9X-AMPK axis in ARID1A-deficient hepatocellular carcinoma. Cell Mol Gastroenterol Hepatol. 2022;14:101-27.
137. Farshidfar F, Zheng S, Gingras MC, et al; Cancer Genome Atlas Network. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 2017;18:2780-94.
138. Goyal L, Meric-Bernstam F, Hollebecque A, et al; FOENIX-CCA2 Study Investigators. Futibatinib for FGFR2-rearranged intrahepatic cholangiocarcinoma. N Engl J Med. 2023;388:228-39.
139. Goyal L, Kongpetch S, Crolley VE, Bridgewater J. Targeting FGFR inhibition in cholangiocarcinoma. Cancer Treat Rev. 2021;95:102170.
140. Ayasun R, Ozer M, Sahin I. The role of HER2 status in the biliary tract cancers. Cancers. 2023;15:2628.
141. Saengboonmee C, Seubwai W, Pairojkul C, Wongkham S. High glucose enhances progression of cholangiocarcinoma cells via STAT3 activation. Sci Rep. 2016;6:18995.
142. Thonsri U, Wongkham S, Wongkham C, et al. High glucose-ROS conditions enhance the progression in cholangiocarcinoma via upregulation of MAN2A2 and CHD8. Cancer Sci. 2021;112:254-64.
143. Saengboonmee C, Phoomak C, Supabphol S, et al. NF-κB and STAT3 co-operation enhances high glucose induced aggressiveness of cholangiocarcinoma cells. Life Sci. 2020;262:118548.
144. Liu C, Jin Y, Fan Z. The mechanism of warburg effect-induced chemoresistance in cancer. Front Oncol. 2021;11:698023.
145. Yothaisong S, Dokduang H, Techasen A, et al. Increased activation of PI3K/AKT signaling pathway is associated with cholangiocarcinoma metastasis and PI3K/mTOR inhibition presents a possible therapeutic strategy. Tumour Biol. 2013;34:3637-48.
146. Phoomak C, Vaeteewoottacharn K, Silsirivanit A, et al. High glucose levels boost the aggressiveness of highly metastatic cholangiocarcinoma cells via O-GlcNAcylation. Sci Rep. 2017;7:43842.
147. Detarya M, Thaenkaew S, Seubwai W, et al. High glucose upregulates FOXM1 expression via EGFR/STAT3 dependent activation to promote progression of cholangiocarcinoma. Life Sci. 2021;271:119114.
148. Colyn L, Alvarez-Sola G, Latasa MU, et al. New molecular mechanisms in cholangiocarcinoma: signals triggering interleukin-6 production in tumor cells and KRAS co-opted epigenetic mediators driving metabolic reprogramming. J Exp Clin Cancer Res. 2022;41:183.
149. Fujinaga A, Hirashita T, Hirashita Y, et al. Glucose metabolic upregulation via phosphorylation of S6 ribosomal protein affects tumor progression in distal cholangiocarcinoma. BMC Gastroenterol. 2023;23:157.
150. Li L, Wang C, Qiu Z, et al. Triptolide inhibits intrahepatic cholangiocarcinoma growth by suppressing glycolysis via the AKT/mTOR pathway. Phytomedicine. 2023;109:154575.
151. Salamanca-Cardona L, Shah H, Poot AJ, et al.
152. Gonsalves WI, Ramakrishnan V, Hitosugi T, et al. Glutamine-derived 2-hydroxyglutarate is associated with disease progression in plasma cell malignancies. JCI Insight. 2018;3:94543.
153. Thomas T, Thakur S, Young R. Imaging 2-hydroxyglutarate and other brain oncometabolites pertinent to critical genomic alterations in brain tumors. BJR Open. 2023;5:20210070.
154. Rimini M, Loi E, Fabregat-Franco C, et al. Next-generation sequencing analysis of cholangiocarcinoma identifies distinct IDH1-mutated clusters. Eur J Cancer. 2022;175:299-310.
155. Kaewpitoon SJ, Loyd RA, Rujirakul R, et al. Benefits of metformin use for cholangiocarcinoma. Asian Pac J Cancer Prev. 2015;16:8079-83.
156. Jiang X, Ma N, Wang D, et al. Metformin inhibits tumor growth by regulating multiple miRNAs in human cholangiocarcinoma. Oncotarget. 2015;6:3178-94.
157. Molenaar RJ, Coelen RJS, Khurshed M, et al. Study protocol of a phase IB/II clinical trial of metformin and chloroquine in patients with IDH1-mutated or IDH2-mutated solid tumours. BMJ Open. 2017;7:e014961.
158. Dutka M, Bobiński R, Francuz T, et al. SGLT-2 inhibitors in cancer treatment-mechanisms of action and emerging new perspectives. Cancers. 2022;14:5811.
159. Kaji K, Nishimura N, Seki K, et al. Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int J Cancer. 2018;142:1712-22.
160. Scheen AJ. Pharmacodynamics, efficacy and safety of sodium-glucose co-transporter type 2 (SGLT2) inhibitors for the treatment of type 2 diabetes mellitus. Drugs. 2015;75:33-59.
161. Xu D, Zhou Y, Xie X, et al. Inhibitory effects of canagliflozin on pancreatic cancer are mediated via the downregulation of glucose transporter1 and lactate dehydrogenase A. Int J Oncol. 2020;57:1223-33.
162. Taguchi D, Shirakami Y, Sakai H, et al. Dual roles of canagliflozin on cholangiocarcinoma cell growth and enhanced growth suppression in combination with FK866. Int J Mol Sci. 2025;26:978.
163. Lee YH, Kim SJ, Fang X, et al. JNK-mediated Ser27 phosphorylation and stabilization of SIRT1 promote growth and progression of colon cancer through deacetylation-dependent activation of Snail. Mol Oncol. 2022;16:1555-71.
164. Yu XJ, Guo XZ, Li C, et al. SIRT1-ZEB1-positive feedback promotes epithelial-mesenchymal transition process and metastasis of osteosarcoma. J Cell Biochem. 2019;120:3727-35.
165. Hayashi M, Nakamura K, Harada S, et al. GLUT1 inhibition by BAY-876 induces metabolic changes and cell death in human colorectal cancer cells. BMC Cancer. 2025;25:716.
166. Liu Y, Cao Y, Zhang W, et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther. 2012;11:1672-82.
167. Li YL, Weng HC, Hsu JL, Lin SW, Guh JH, Hsu LC. The combination of MK-2206 and WZB117 exerts a synergistic cytotoxic effect against breast cancer cells. Front Pharmacol. 2019;10:1311.
168. Schmidl S, Ursu O, Iancu CV, Oreb M, Oprea TI, Choe JY. Identification of new GLUT2-selective inhibitors through in silico ligand screening and validation in eukaryotic expression systems. Sci Rep. 2021;11:13751.
169. Silvestrini B, Hahn GM, Cioli V, De Martino C. Effects of lonidamine alone or combined with hyperthermia in some experimental cell and tumour systems. Br J Cancer. 1983;47:221-31.
170. Morais-Santos F, Miranda-Gonçalves V, Pinheiro S, et al. Differential sensitivities to lactate transport inhibitors of breast cancer cell lines. Endocr Relat Cancer. 2014;21:27-38.
171. Lee GH, Chae HJ, Kim HR. Monoamine carboxylate transporters are involved in BI-1-associated cancer metastasis in HT1080 colon fibrosarcoma cells. Int J Oncol. 2011;39:209-16.
172. Guo L, Shestov AA, Worth AJ, et al. Inhibition of mitochondrial complex II by the anticancer agent lonidamine. J Biol Chem. 2016;291:42-57.
173. Nancolas B, Guo L, Zhou R, et al. The anti-tumour agent lonidamine is a potent inhibitor of the mitochondrial pyruvate carrier and plasma membrane monocarboxylate transporters. Biochem J. 2016;473:929-36.
174. Carapella CM, Paggi MG, Cattani F, et al. The potential role of lonidamine (LND) in the treatment of malignant glioma. Phase II study. J Neurooncol. 1989;7:103-8.
175. De Lena M, Lorusso V, Latorre A, et al. Paclitaxel, cisplatin and lonidamine in advanced ovarian cancer. A phase II study. Eur J Cancer. 2001;37:364-8.
176. Kobayashi H, Takase S, Nishimura H, Matsumoto K, Harada H, Yoshida M. RNAi screening reveals a synthetic chemical-genetic interaction between ATP synthase and PFK1 in cancer cells. Cancer Sci. 2023;114:1663-71.
177. Chen F, Dowerg B, Cordes T. The yin and yang of itaconate metabolism and its impact on the tumor microenvironment. Curr Opin Biotechnol. 2023;84:102996.
178. Wang Y, Tang J, Liu Y, et al. Targeting ALDOA to modulate tumorigenesis and energy metabolism in retinoblastoma. iScience. 2024;27:110725.
179. Thonsri U, Seubwai W, Waraasawapati S, et al. Antitumor effect of shikonin, a PKM2 inhibitor, in cholangiocarcinoma cell lines. Anticancer Res. 2020;40:5115-24.
180. Liu C, Xuan LQ, Li K, et al. Shikonin inhibits cholangiocarcinoma cell line QBC939 by regulating apoptosis, proliferation, and invasion. Cell Transplant. 2021;30:963689720979162.
181. Zhou G, Yang Z, Wang X, Tao R, Zhou Y. TRAIL enhances shikonin induced apoptosis through ROS/JNK signaling in cholangiocarcinoma cells. Cell Physiol Biochem. 2017;42:1073-86.
182. Kim DJ, Park YS, Kang MG, et al. Pyruvate kinase isoenzyme M2 is a therapeutic target of gemcitabine-resistant pancreatic cancer cells. Exp Cell Res. 2015;336:119-29.
183. Wang Y, Hao F, Nan Y, et al. PKM2 inhibitor shikonin overcomes the cisplatin resistance in bladder cancer by inducing necroptosis. Int J Biol Sci. 2018;14:1883-91.
184. Su Q, Luo S, Tan Q, et al. The role of pyruvate kinase M2 in anticancer therapeutic treatments. Oncol Lett. 2019;18:5663-72.
185. Ning X, Qi H, Li R, Jin Y, McNutt MA, Yin Y. Synthesis and antitumor activity of novel 2, 3-didithiocarbamate substituted naphthoquinones as inhibitors of pyruvate kinase M2 isoform. J Enzyme Inhib Med Chem. 2018;33:126-9.
186. Lu W, Cao Y, Zhang Y, et al. Up-regulation of PKM2 promote malignancy and related to adverse prognostic risk factor in human gallbladder cancer. Sci Rep. 2016;6:26351.
187. Ning X, Qi H, Li R, et al. Discovery of novel naphthoquinone derivatives as inhibitors of the tumor cell specific M2 isoform of pyruvate kinase. Eur J Med Chem. 2017;138:343-52.
188. An Y, Guo W, Li L, et al. Micheliolide derivative DMAMCL inhibits glioma cell growth in vitro and in vivo. PLoS One. 2015;10:e0116202.
189. Ji Q, Ding YH, Sun Y, et al. Antineoplastic effects and mechanisms of micheliolide in acute myelogenous leukemia stem cells. Oncotarget. 2016;7:65012-23.
190. Shang D, Wu J, Guo L, Xu Y, Liu L, Lu J. Metformin increases sensitivity of osteosarcoma stem cells to cisplatin by inhibiting expression of PKM2. Int J Oncol. 2017;50:1848-56.
191. Zhou Y, Huang Z, Su J, et al. Benserazide is a novel inhibitor targeting PKM2 for melanoma treatment. Int J Cancer. 2020;147:139-51.
192. Francesco EM, Sotgia F, Lisanti MP. Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. Biochem J. 2018;475:1611-34.
193. Li W, Zheng M, Wu S, et al. Benserazide, a dopadecarboxylase inhibitor, suppresses tumor growth by targeting hexokinase 2. J Exp Clin Cancer Res. 2017;36:58.
195. Kilanczyk E, Ruminkiewicz D, Banales JM, Milkiewicz P, Milkiewicz M. DHEA protects human cholangiocytes and hepatocytes against apoptosis and oxidative stress. Cells. 2022;11:1038.
196. Qin L, Kuai J, Yang F, et al. Selected by bioinformatics and molecular docking analysis, Dhea and 2-14,15-Eg are effective against cholangiocarcinoma. PLoS One. 2022;17:e0260180.
197. Mele L, Paino F, Papaccio F, et al. A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell Death Dis. 2018;9:572.
198. De Maria S, Scognamiglio I, Lombardi A, et al. Polydatin, a natural precursor of resveratrol, induces cell cycle arrest and differentiation of human colorectal Caco-2 cell. J Transl Med. 2013;11:264.
199. Indraccolo U, Indraccolo SR, Mignini F. Micronized palmitoylethanolamide/trans-polydatin treatment of endometriosis-related pain: a meta-analysis. Ann Ist Super Sanita. 2017;53:125-34.
200. Chen H, Wu D, Bao L, et al. 6PGD inhibition sensitizes hepatocellular carcinoma to chemotherapy via AMPK activation and metabolic reprogramming. Biomed Pharmacother. 2019;111:1353-8.
201. Lin R, Elf S, Shan C, et al. 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1-AMPK signalling. Nat Cell Biol. 2015;17:1484-96.
202. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016-23.
203. Xu IM, Lai RK, Lin SH, et al. Transketolase counteracts oxidative stress to drive cancer development. Proc Natl Acad Sci U S A. 2016;113:E725-34.
204. Kowalik MA, Columbano A, Perra A. Emerging role of the pentose phosphate pathway in hepatocellular carcinoma. Front Oncol. 2017;7:87.
205. Mariadasse R, Biswal J, Jayaprakash P, et al. Mechanical insights of oxythiamine compound as potent inhibitor for human transketolase-like protein 1 (TKTL1 protein). J Recept Signal Transduct Res. 2016;36:233-42.
206. Raïs B, Comin B, Puigjaner J, et al. Oxythiamine and dehydroepiandrosterone induce a G1 phase cycle arrest in Ehrlich’s tumor cells through inhibition of the pentose cycle. FEBS Lett. 1999;456:113-8.
207. Zhang H, Cao R, Lee WN, et al. Inhibition of protein phosphorylation in MIA pancreatic cancer cells: confluence of metabolic and signaling pathways. J Proteome Res. 2010;9:980-9.
208. Yang CM, Liu YZ, Liao JW, Hu ML. The in vitro and in vivo anti-metastatic efficacy of oxythiamine and the possible mechanisms of action. Clin Exp Metastasis. 2010;27:341-9.
209. Wang CY, Shui HA, Chang TC. Dual effects for lovastatin in anaplastic thyroid cancer: the pivotal effect of transketolase (TKT) on lovastatin and tumor proliferation. J Investig Med. 2018;66:1-9.
210. Liu CL, Hsu YC, Lee JJ, et al. Targeting the pentose phosphate pathway increases reactive oxygen species and induces apoptosis in thyroid cancer cells. Mol Cell Endocrinol. 2020;499:110595.
211. Boros LG, Bassilian S, Lim S, Lee WN. Genistein inhibits nonoxidative ribose synthesis in MIA pancreatic adenocarcinoma cells: a new mechanism of controlling tumor growth. Pancreas. 2001;22:1-7.
212. Li S, Li J, Dai W, et al. Genistein suppresses aerobic glycolysis and induces hepatocellular carcinoma cell death. Br J Cancer. 2017;117:1518-28.
213. Lachaise F, Martin G, Drougard C, et al. Relationship between posttranslational modification of transaldolase and catalase deficiency in UV-sensitive repair-deficient xeroderma pigmentosum fibroblasts and SV40-transformed human cells. Free Radic Biol Med. 2001;30:1365-73.
214. Ogawa T, Murakami K, Yoshino M. Inhibition by fructose 1,6-bisphosphate of transaldolase from Escherichia coli. FEMS Microbiol Lett. 2016;363:fnw183.
215. Verma S, Budhu S, Serganova I, et al. Pharmacologic LDH inhibition redirects intratumoral glucose uptake and improves antitumor immunity in solid tumor models. J Clin Invest. 2024;134:e177606.
216. Boudreau A, Purkey HE, Hitz A, et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat Chem Biol. 2016;12:779-86.
217. Xiao Y, Yu TJ, Xu Y, et al. Emerging therapies in cancer metabolism. Cell Metab. 2023;35:1283-303.
218. Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123:3685-92.
219. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell. 2017;169:570-86.
220. O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16:553-65.
221. Xu K, Yin N, Peng M, et al. Glycolysis fuels phosphoinositide 3-kinase signaling to bolster T cell immunity. Science. 2021;371:405-10.
222. Le A, Cooper CR, Gouw AM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107:2037-42.
223. Ooi AT, Gomperts BN. Molecular pathways: targeting cellular energy metabolism in cancer via inhibition of SLC2A1 and LDHA. Clin Cancer Res. 2015;21:2440-4.
224. Granchi C, Roy S, Giacomelli C, et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J Med Chem. 2011;54:1599-612.
225. Manerba M, Vettraino M, Fiume L, et al. Galloflavin (CAS 568-80-9): a novel inhibitor of lactate dehydrogenase. ChemMedChem. 2012;7:311-7.
226. Zhao Y, Liu H, Liu Z, et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011;71:4585-97.
227. Zhou M, Zhao Y, Ding Y, et al. Warburg effect in chemosensitivity: targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer. 2010;9:33.
228. Seth P, Grant A, Tang J, et al. On-target inhibition of tumor fermentative glycolysis as visualized by hyperpolarized pyruvate. Neoplasia. 2011;13:60-71.
229. Zhu AX, Macarulla T, Javle MM, et al. Final overall survival efficacy results of ivosidenib for patients with advanced cholangiocarcinoma with IDH1 mutation: the phase 3 randomized clinical ClarIDHy trial. JAMA Oncol. 2021;7:1669-77.
230. Lamarca A, Palmer DH, Wasan HS, et al; Advanced Biliary Cancer Working Group. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): a phase 3, open-label, randomised, controlled trial. Lancet Oncol. 2021;22:690-701.
231. Yoo C, Kim KP, Jeong JH, et al. Liposomal irinotecan plus fluorouracil and leucovorin versus fluorouracil and leucovorin for metastatic biliary tract cancer after progression on gemcitabine plus cisplatin (NIFTY): a multicentre, open-label, randomised, phase 2b study. Lancet Oncol. 2021;22:1560-72.
232. Vogel A, Bridgewater J, Edeline J, et al; ESMO Guidelines Committee. Electronic address: [email protected]. Biliary tract cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34:127-40.
233. Rodon J, Goyal L, Mercade TM, et al. Abstract CT098: a first-in-human phase 1 study of LY3410738, a covalent inhibitor of mutant IDH, in advanced IDH-mutant cholangiocarcinoma and other solid tumors. Cancer Research. 2023;83:CT098-CT098.
234. Li Y, Yu J, Zhang Y, Peng C, Song Y, Liu S. Advances in targeted therapy of cholangiocarcinoma. Ann Med. 2024;56:2310196.
235. Saha SK, Gordan JD, Kleinstiver BP, et al. Isocitrate dehydrogenase mutations confer dasatinib hypersensitivity and SRC dependence in intrahepatic cholangiocarcinoma. Cancer Discov. 2016;6:727-39.
236. Fathi AT, Wander SA, Faramand R, Emadi A. Biochemical, epigenetic, and metabolic approaches to target IDH mutations in acute myeloid leukemia. Semin Hematol. 2015;52:165-71.
237. Yang SM, Kim J, Lee JY, Lee JS, Lee JM. Regulation of glucose and glutamine metabolism to overcome cisplatin resistance in intrahepatic cholangiocarcinoma. BMB Rep. 2023;56:600-5.
238. Mohan A, Griffith KA, Wuchu F, et al. Devimistat in combination with gemcitabine and cisplatin in biliary tract cancer: preclinical evaluation and phase Ib multicenter clinical trial (BilT-04). Clin Cancer Res. 2023;29:2394-400.
239. Russell DA, Bridges HR, Serreli R, et al. Hydroxylated rotenoids selectively inhibit the proliferation of prostate cancer cells. J Nat Prod. 2020;83:1829-45.
240. Cadassou O, Jordheim LP. OXPHOS inhibitors, metabolism and targeted therapies in cancer. Biochem Pharmacol. 2023;211:115531.
241. Rha SY, Beom S, Shin YG, et al. Phase I study of IM156, a novel potent biguanide oxidative phosphorylation (OXPHOS) inhibitor, in patients with advanced solid tumors. JCO. 2020;38:3590.
242. Ellinghaus P, Heisler I, Unterschemmann K, et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013;2:611-24.
243. Molina JR, Sun Y, Protopopova M, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24:1036-46.
244. Yap TA, Daver N, Mahendra M, et al. Complex I inhibitor of oxidative phosphorylation in advanced solid tumors and acute myeloid leukemia: phase I trials. Nat Med. 2023;29:115-26.
245. Bordt EA, Clerc P, Roelofs BA, et al. The putative drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell. 2017;40:583-94.e6.
246. Bastian A, Matsuzaki S, Humphries KM, et al. AG311, a small molecule inhibitor of complex I and hypoxia-induced HIF-1α stabilization. Cancer Lett. 2017;388:149-57.
247. Bastian A, Thorpe JE, Disch BC, et al. A small molecule with anticancer and antimetastatic activities induces rapid mitochondrial-associated necrosis in breast cancer. J Pharmacol Exp Ther. 2015;353:392-404.
248. Bekaii-saab TS, Spira AI, Yaeger R, et al. KRYSTAL-1: updated activity and safety of adagrasib (MRTX849) in patients (Pts) with unresectable or metastatic pancreatic cancer (PDAC) and other gastrointestinal (GI) tumors harboring a KRASG12C mutation. JCO. 2022;40:519.
249. Shelton F, Hoffman N, Trinh D, et al. Abstract 4448: effects of adagrasib on cholesterol, lipid and glucose gene expression regulation in tumor xenograft models and patient samples. Cancer Research. 2024;84:4448.
251. Hong DS, Fakih MG, Strickler JH, et al. KRASG12C inhibition with sotorasib in advanced solid tumors. N Engl J Med. 2020;383:1207-17.
252. Janku F, Huang HJ, Fujii T, et al. Multiplex KRASG12/G13 mutation testing of unamplified cell-free DNA from the plasma of patients with advanced cancers using droplet digital polymerase chain reaction. Ann Oncol. 2017;28:642-50.
253. Thongyoo P, Chindaprasirt J, Aphivatanasiri C, et al.
254. Zhang ZR, Liu MQ, Ji Y, et al. Sotorasib inhibits ubiquitination degradation of TXNIP and suppresses glucose metabolism in KRASG12C mutant bladder cancer. Am J Cancer Res. 2024;14:5251-68.
255. Nishikawa S, Iwakuma T. Drugs targeting p53 mutations with FDA approval and in clinical trials. Cancers. 2023;15:429.
256. Bykov VJ, Zhang Q, Zhang M, Ceder S, Abrahmsen L, Wiman KG. Targeting of mutant p53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front Oncol. 2016;6:21.
257. Liu M, Liu W, Qin Y, et al. Regulation of metabolic reprogramming by tumor suppressor genes in pancreatic cancer. Exp Hematol Oncol. 2020;9:179.
258. Mishra A, Tamari R, DeZern AE, et al. Eprenetapopt plus azacitidine after allogeneic hematopoietic stem-cell transplantation for TP53-mutant acute myeloid leukemia and myelodysplastic syndromes. J Clin Oncol. 2022;40:3985-93.
259. Sallman DA, DeZern AE, Garcia-Manero G, et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant myelodysplastic syndromes. J Clin Oncol. 2021;39:1584-94.
260. Aggarwal M, Saxena R, Sinclair E, et al. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. 2016;23:1615-27.
261. Davison K, Mann KK, Miller WH Jr. Arsenic trioxide: mechanisms of action. Semin Hematol. 2002;39:3-7.
262. Yedjou C, Tchounwou P, Jenkins J, McMurray R. Basic mechanisms of arsenic trioxide (ATO)-induced apoptosis in human leukemia (HL-60) cells. J Hematol Oncol. 2010;3:28.
263. Chen S, Wu JL, Liang Y, et al. Arsenic trioxide rescues structural p53 mutations through a cryptic allosteric site. Cancer Cell. 2021;39:225-39.e8.
264. Yan W, Jung YS, Zhang Y, Chen X. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PLoS One. 2014;9:e103497.
265. Wen J, Li A, Wang Z, et al. Hepatotoxicity induced by arsenic trioxide: clinical features, mechanisms, preventive and potential therapeutic strategies. Front Pharmacol. 2025;16:1536388.
266. Terzian T, Suh YA, Iwakuma T, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337-44.
267. Alexandrova EM, Yallowitz AR, Li D, et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature. 2015;523:352-6.
268. Lukashchuk N, Vousden KH. Ubiquitination and degradation of mutant p53. Mol Cell Biol. 2007;27:8284-95.
269. Kudryavtsev VA, Khokhlova AV, Mosina VA, Selivanova EI, Kabakov AE. Induction of Hsp70 in tumor cells treated with inhibitors of the Hsp90 activity: a predictive marker and promising target for radiosensitization. PLoS One. 2017;12:e0173640.
270. Concin N, Braicu I, Combe P, et al. Phase II results of GANNET53: a European multicenter phase I/randomized II trial of the Hsp90 inhibitor Ganetespib (G) combined with weekly Paclitaxel (P) in women with high-grade serous, high-grade endometrioid, or undifferentiated, platinum-resistant epithelial ovarian, fallopian tube or primary peritoneal cancer. JCO. 2018;36:5567.
271. Ray-Coquard I, Braicu I, Berger R, et al. Part I of GANNET53: a European multicenter phase I/II trial of the Hsp90 inhibitor ganetespib combined with weekly paclitaxel in women with high-grade, platinum-resistant epithelial ovarian cancer-a study of the GANNET53 consortium. Front Oncol. 2019;9:832.
272. Jiang W, Hu JW, He XR, Jin WL, He XY. Statins: a repurposed drug to fight cancer. J Exp Clin Cancer Res. 2021;40:241.
273. Mehibel M, Ortiz-Martinez F, Voelxen N, et al. Statin-induced metabolic reprogramming in head and neck cancer: a biomarker for targeting monocarboxylate transporters. Sci Rep. 2018;8:16804.
274. Parrales A, Ranjan A, Iyer SV, et al. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat Cell Biol. 2016;18:1233-43.
275. Ingallina E, Sorrentino G, Bertolio R, et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat Cell Biol. 2018;20:28-35.
276. Gray RT, Loughrey MB, Bankhead P, et al. Statin use, candidate mevalonate pathway biomarkers, and colon cancer survival in a population-based cohort study. Br J Cancer. 2017;116:1652-9.
277. Nishikawa S, Menju T, Takahashi K, et al. Statins may have double-edged effects in patients with lung adenocarcinoma after lung resection. Cancer Manag Res. 2019;11:3419-32.
278. Foggetti G, Ottaggio L, Russo D, et al. Autophagy induced by SAHA affects mutant P53 degradation and cancer cell survival. Biosci Rep. 2019;39:BSR20181345.
279. Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011;18:1904-13.
280. Li D, Marchenko ND, Schulz R, et al. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol Cancer Res. 2011;9:577-88.
281. Topatana W, Juengpanich S, Li S, et al. Advances in synthetic lethality for cancer therapy: cellular mechanism and clinical translation. J Hematol Oncol. 2020;13:118.
282. Wang X, Simon R. Identification of potential synthetic lethal genes to p53 using a computational biology approach. BMC Med Genomics. 2013;6:30.
283. Wu CE, Pan YR, Yeh CN, Lunec J. Targeting P53 as a future strategy to overcome gemcitabine resistance in biliary tract cancers. Biomolecules. 2020;10:1474.
284. Hirai H, Arai T, Okada M, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9:514-22.
285. Leijen S, van Geel RM, Sonke GS, et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J Clin Oncol. 2016;34:4354-61.
286. Moore KN, Chambers SK, Hamilton EP, et al. Adavosertib with chemotherapy in patients with primary platinum-resistant ovarian, fallopian tube, or peritoneal cancer: an open-label, four-arm, phase II study. Clin Cancer Res. 2022;28:36-44.
287. Oza AM, Estevez-Diz M, Grischke EM, et al. A biomarker-enriched, randomized phase II trial of adavosertib (AZD1775) plus paclitaxel and carboplatin for women with platinum-sensitive TP53-mutant ovarian cancer. Clin Cancer Res. 2020;26:4767-76.
288. Seligmann JF, Fisher DJ, Brown LC, et al; FOCUS4 Trial Investigators. Inhibition of WEE1 is effective in TP53- and RAS-mutant metastatic colorectal cancer: a randomized trial (FOCUS4-C) comparing adavosertib (AZD1775) with active monitoring. J Clin Oncol. 2021;39:3705-15.
289. Liu JF, Xiong N, Campos SM, et al. Phase II study of the WEE1 inhibitor adavosertib in recurrent uterine serous carcinoma. J Clin Oncol. 2021;39:1531-9.
290. McKerrow W, Wang X, Mendez-Dorantes C, et al. LINE-1 expression in cancer correlates with p53 mutation, copy number alteration, and S phase checkpoint. Proc Natl Acad Sci U S A. 2022;119:e2115999119.
291. Tiwari B, Jones AE, Caillet CJ, Das S, Royer SK, Abrams JM. p53 directly represses human LINE1 transposons. Genes Dev. 2020;34:1439-51.
292. Wylie A, Jones AE, D’Brot A, et al. p53 genes function to restrain mobile elements. Genes Dev. 2016;30:64-77.
293. Rajurkar M, Parikh AR, Solovyov A, et al. Reverse transcriptase inhibition disrupts repeat element life cycle in colorectal cancer. Cancer Discov. 2022;12:1462-81.
294. Di Agostino S, Sorrentino G, Ingallina E, et al. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2016;17:188-201.
295. Mo JS, Yu FX, Gong R, Brown JH, Guan KL. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev. 2012;26:2138-43.
296. Göbel A, Thiele S, Browne AJ, et al. Combined inhibition of the mevalonate pathway with statins and zoledronic acid potentiates their anti-tumor effects in human breast cancer cells. Cancer Lett. 2016;375:162-71.
297. Schmidmaier R, Simsek M, Baumann P, Emmerich B, Meinhardt G. Synergistic antimyeloma effects of zoledronate and simvastatin. Anticancer Drugs. 2006;17:621-9.
298. Sorrentino G, Ruggeri N, Specchia V, et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol. 2014;16:357-66.
299. Xu S, Tang C. The role of ARID1A in tumors: tumor initiation or tumor suppression? Front Oncol. 2021;11:745187.
300. Pant K, Peixoto E, Richard S, Gradilone SA. Role of histone deacetylases in carcinogenesis: potential role in cholangiocarcinoma. Cells. 2020;9:780.
301. Zhang X, Sun M, Jiao Y, Lin B, Yang Q. PHGDH Inhibitor CBR-5884 inhibits epithelial ovarian cancer progression via ROS/Wnt/β-catenin pathway and plays a synergistic role with PARP inhibitor olaparib. Oxid Med Cell Longev. 2022;2022:9029544.
302. Chen J, Hong JH, Huang Y, et al. EZH2 mediated metabolic rewiring promotes tumor growth independently of histone methyltransferase activity in ovarian cancer. Mol Cancer. 2023;22:85.
303. Liu Y, Tu CE, Guo X, et al. Tumor-suppressive function of EZH2 is through inhibiting glutaminase. Cell Death Dis. 2021;12:975.
304. Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors: a 2023 update. Pharmacol Res. 2023;187:106552.
305. Goyal L, Shi L, Liu LY, et al. TAS-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma. Cancer Discov. 2019;9:1064-79.
306. Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21:671-84.
307. Javle M, Borad MJ, Azad NS, et al. Pertuzumab and trastuzumab for HER2-positive, metastatic biliary tract cancer (MyPathway): a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2021;22:1290-300.
308. Ohba A, Morizane C, Kawamoto Y, et al. Trastuzumab deruxtecan in human epidermal growth factor receptor 2-expressing biliary tract cancer (HERB; NCCH1805): a multicenter, single-arm, phase II trial. J Clin Oncol. 2024;42:3207-17.
309. Ohba A, Morizane C, Kawamoto Y, et al. Trastuzumab deruxtecan (T-DXd; DS-8201) in patients (pts) with HER2-expressing unresectable or recurrent biliary tract cancer (BTC): an investigator-initiated multicenter phase 2 study (HERB trial). JCO. 2022;40:4006.
310. Meric-bernstam F, Makker V, Oaknin A, et al. Efficacy and safety of trastuzumab deruxtecan (T-DXd) in patients (pts) with HER2-expressing solid tumors: DESTINY-PanTumor02 (DP-02) interim results. JCO. 2023;41:LBA3000.
311. Harding JJ, Fan J, Oh DY, et al; HERIZON-BTC-01 study group. Zanidatamab for HER2-amplified, unresectable, locally advanced or metastatic biliary tract cancer (HERIZON-BTC-01): a multicentre, single-arm, phase 2b study. Lancet Oncol. 2023;24:772-82.
312. Meric-Bernstam F, Beeram M, Hamilton E, et al. Zanidatamab, a novel bispecific antibody, for the treatment of locally advanced or metastatic HER2-expressing or HER2-amplified cancers: a phase 1, dose-escalation and expansion study. Lancet Oncol. 2022;23:1558-70.
313. Harding JJ, Piha-Paul SA, Shah RH, et al. Antitumour activity of neratinib in patients with HER2-mutant advanced biliary tract cancers. Nat Commun. 2023;14:630.
314. Nakamura Y, Mizuno N, Sunakawa Y, et al. Tucatinib and trastuzumab for previously treated HER2-positive metastatic biliary tract cancer (SGNTUC-019): a phase 2 basket study. JCO. 2023;41:4007.
315. Lee CK, Chon HJ, Cheon J, et al. Trastuzumab plus FOLFOX for HER2-positive biliary tract cancer refractory to gemcitabine and cisplatin: a multi-institutional phase 2 trial of the Korean Cancer Study Group (KCSG-HB19-14). Lancet Gastroenterol Hepatol. 2023;8:56-65.
316. Ostwal V, Mandavkar S, Bhargava P, et al. Trastuzumab plus gemcitabine-cisplatin for treatment-naïve human epidermal growth factor receptor 2-positive biliary tract adenocarcinoma: a multicenter, open-label, phase II study (TAB). J Clin Oncol. 2024;42:800-7.
317. Li Z, Shen J, Chan MT, Wu WK. The role of microRNAs in intrahepatic cholangiocarcinoma. J Cell Mol Med. 2017;21:177-84.
318. Zheng B, Jeong S, Zhu Y, Chen L, Xia Q. miRNA and lncRNA as biomarkers in cholangiocarcinoma(CCA). Oncotarget. 2017;8:100819-30.
319. Jalil AT, Abdulhadi MA, Al-Ameer LR, et al. Small but mighty: how microRNAs drive the deadly progression of cholangiocarcinoma. Pathol Res Pract. 2023;247:154565.
320. Chen L, Yan HX, Yang W, et al. The role of microRNA expression pattern in human intrahepatic cholangiocarcinoma. J Hepatol. 2009;50:358-69.
321. Oishi N, Kumar MR, Roessler S, et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology. 2012;56:1792-803.
322. Zhang MY, Li SH, Huang GL, et al. Identification of a novel microRNA signature associated with intrahepatic cholangiocarcinoma (ICC) patient prognosis. BMC Cancer. 2015;15:64.
323. Tiemin P, Peng X, Qingfu L, et al. Dysregulation of the miR-148a-GLUT1 axis promotes the progression and chemoresistance of human intrahepatic cholangiocarcinoma. Oncogenesis. 2020;9:19.
324. Chen B, Tang H, Liu X, et al. miR-22 as a prognostic factor targets glucose transporter protein type 1 in breast cancer. Cancer Lett. 2015;356:410-7.
325. Zhang T, Zhang Z, Li F, et al. miR-143 regulates memory T cell differentiation by reprogramming T cell metabolism. J Immunol. 2018;201:2165-75.
326. Xu G, Pan S, Zhu Z, Li J. Overexpression of miR-340 inhibits cell proliferation and induces apoptosis of human bladder cancer via targeting Glut-1. BMC Urol. 2021;21:168.
327. R P Jr, Yuwanati M, Sekaran S, M S. miRNA associated with glucose transporters in oral squamous cell carcinoma: a systematic review. Cureus. 2023;15:e46057.
328. Shi Y, Zhang Y, Ran F, et al. Let-7a-5p inhibits triple-negative breast tumor growth and metastasis through GLUT12-mediated warburg effect. Cancer Lett. 2020;495:53-65.
329. Lu M, Qin X, Zhou Y, et al. LncRNA HOTAIR suppresses cell apoptosis, autophagy and induces cell proliferation in cholangiocarcinoma by modulating the miR-204-5p/HMGB1 axis. Biomed Pharmacother. 2020;130:110566.
330. Obaid M, Udden SMN, Alluri P, Mandal SS. LncRNA HOTAIR regulates glucose transporter Glut1 expression and glucose uptake in macrophages during inflammation. Sci Rep. 2021;11:232.
331. Li F, Chen Q, Xue H, Zhang L, Wang K, Shen F. LncRNA MNX1-AS1 promotes progression of intrahepatic cholangiocarcinoma through the MNX1/Hippo axis. Cell Death Dis. 2020;11:894.
332. Chen J, Yu Y, Li H, et al. Long non-coding RNA PVT1 promotes tumor progression by regulating the miR-143/HK2 axis in gallbladder cancer. Mol Cancer. 2019;18:33.
333. Fang R, Xiao T, Fang Z, et al. MicroRNA-143 (miR-143) regulates cancer glycolysis via targeting hexokinase 2 gene. J Biol Chem. 2012;287:23227-35.
334. Liu T, Ye P, Ye Y, Han B. MicroRNA-216b targets HK2 to potentiate autophagy and apoptosis of breast cancer cells via the mTOR signaling pathway. Int J Biol Sci. 2021;17:2970-83.
335. Jin F, Wang Y, Zhu Y, et al. The miR-125a/HK2 axis regulates cancer cell energy metabolism reprogramming in hepatocellular carcinoma. Sci Rep. 2017;7:3089.
336. Muthukumaran N, Velusamy P, Akino Mercy CS, Langford D, Natarajaseenivasan K, Shanmughapriya S. MicroRNAs as regulators of cancer cell energy metabolism. J Pers Med. 2022;12:1329.
337. Yang Y, Ishak Gabra MB, Hanse EA, et al. MiR-135 suppresses glycolysis and promotes pancreatic cancer cell adaptation to metabolic stress by targeting phosphofructokinase-1. Nat Commun. 2019;10:809.
338. Xu Z, Liu G, Zhang M, et al. miR-122-5p inhibits the proliferation, invasion and growth of bile duct carcinoma cells by targeting ALDOA. Cell Physiol Biochem. 2018;48:2596-606.
339. Kong L, Wu Q, Zhao L, Ye J, Li N, Yang H. Upregulated lncRNA-UCA1 contributes to metastasis of bile duct carcinoma through regulation of miR-122/CLIC1 and activation of the ERK/MAPK signaling pathway. Cell Cycle. 2019;18:1212-28.
340. Zhu H, Mi Y, Jiang X, et al. Hepatocyte nuclear factor 6 inhibits the growth and metastasis of cholangiocarcinoma cells by regulating miR-122. J Cancer Res Clin Oncol. 2016;142:969-80.
341. Cui K, Jin S, Du Y, et al. Long noncoding RNA DIO3OS interacts with miR-122 to promote proliferation and invasion of pancreatic cancer cells through upregulating ALDOA. Cancer Cell Int. 2019;19:202.
342. Chen B, Li H, Zeng X, et al. Roles of microRNA on cancer cell metabolism. J Transl Med. 2012;10:228.
343. Qu Y, Xue S, Zheng Y, et al. Upregulated miR378a3p expression suppresses energy metabolism and promotes apoptosis by targeting a GLUT1/ALDOA/PKM2 axis in esophageal carcinoma. Oncol Lett. 2023;26:421.
344. Fu J, Imani S, Wu MY, Wu RC. MicroRNA-34 family in cancers: role, mechanism, and therapeutic potential. Cancers. 2023;15:4723.
345. Chang YC, Yang YF, Chiou J, et al. Nonenzymatic function of aldolase A downregulates miR-145 to promote the Oct4/DUSP4/TRAF4 axis and the acquisition of lung cancer stemness. Cell Death Dis. 2020;11:195.
346. Peng C, Sun Z, Li O, et al. Leptin stimulates the epithelialmesenchymal transition and proangiogenic capability of cholangiocarcinoma cells through the miR122/PKM2 axis. Int J Oncol. 2019;55:298-308.
347. Faramin Lashkarian M, Hashemipour N, Niaraki N, et al. MicroRNA-122 in human cancers: from mechanistic to clinical perspectives. Cancer Cell Int. 2023;23:29.
348. Li H, Lin X, Li C, et al. MiR-1286 inhibits lung cancer growth through aerobic glycolysis by targeting PKM2. Arch Med Sci. 2023;19:151-9.
349. Sun Y, Liu W, Zhao Q, et al. Down-regulating the expression of miRNA-21 inhibits the glucose metabolism of A549/DDP cells and promotes cell death through the PI3K/AKT/mTOR/HIF-1α pathway. Front Oncol. 2021;11:653596.
350. Pan C, Liu Q, Wu X. HIF1α/miR-520a-3p/AKT1/mTOR feedback promotes the proliferation and glycolysis of gastric cancer cells. Cancer Manag Res. 2019;11:10145-56.
351. Guo Z, Maki M, Ding R, Yang Y, Zhang B, Xiong L. Genome-wide survey of tissue-specific microRNA and transcription factor regulatory networks in 12 tissues. Sci Rep. 2014;4:5150.
352. Wu HQ, Cheng ML, Lai JM, et al. Flux balance analysis predicts Warburg-like effects of mouse hepatocyte deficient in miR-122a. PLoS Comput Biol. 2017;13:e1005618.
353. Bhanot H, Weisberg EL, Reddy MM, et al. Acute myeloid leukemia cells require 6-phosphogluconate dehydrogenase for cell growth and NADPH-dependent metabolic reprogramming. Oncotarget. 2017;8:67639-50.
354. Chan B, VanderLaan PA, Sukhatme VP. 6-Phosphogluconate dehydrogenase regulates tumor cell migration in vitro by regulating receptor tyrosine kinase c-Met. Biochem Biophys Res Commun. 2013;439:247-51.
355. Zheng W, Feng Q, Liu J, et al. Inhibition of 6-phosphogluconate dehydrogenase reverses cisplatin resistance in ovarian and lung cancer. Front Pharmacol. 2017;8:421.
356. Liao YN, Huang PQ, Pan H, et al. Prolactin receptor potentiates chemotherapy through miRNAs-induced G6PD/TKT inhibition in pancreatic cancer. FASEB J. 2024;38:e23705.
357. Qiu Z, Guo W, Wang Q, et al. MicroRNA-124 reduces the pentose phosphate pathway and proliferation by targeting PRPS1 and RPIA mRNAs in human colorectal cancer cells. Gastroenterology. 2015;149:1587-98.e11.
358. Yang H, Wu XL, Wu KH, Zhang R, Ju LL, et al. MicroRNA-497 regulates cisplatin chemosensitivity of cervical cancer by targeting transketolase. Am J Cancer Res. 2016;6:11:2690-99.
359. Ding J, Wen Y, Yuan X, He X. MicroRNA-mediated reprogramming of glucose, fatty acid and amino acid metabolism in cancer. GENOME INSTAB DIS. 2023;4:47-69.
360. Nikolova E, Laleva L, Milev M, et al. miRNAs and related genetic biomarkers according to the WHO glioma classification: From diagnosis to future therapeutic targets. Noncoding RNA Res. 2024;9:141-52.
361. Zhang S, Liu X, Liu J, Guo H, Xu H, Zhang G. PGC-1 alpha interacts with microRNA-217 to functionally regulate breast cancer cell proliferation. Biomed Pharmacother. 2017;85:541-8.
362. Qian L, Zhu Y, Deng C, et al. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases. Signal Transduct Target Ther. 2024;9:50.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].