
CAR-T cell therapy in neuro-oncology: applications and toxicity

Abstract
A new era for cancer treatment has been ushered in with the field of cancer immunotherapy. After initial success with systemic malignancies, several of these promising treatments are being investigated for efficacy with primary and secondary brain tumors. Chimeric antigen receptor (CAR) T cells are being studied, both with systemic infusion and direct administration to the tumor and into the cerebrospinal fluid, with promising early results. Systemic CAR-T treatment can have serious systemic and neurological toxicities that are important for the practicing neurologist and neuro-oncologist to know and understand. This review aims to discuss adoptive cell therapies with a focus on CAR-T treatment. We review use of this therapy in brain cancers, particularly malignant glioma, and provide an overview of the toxicity of CAR-T treatment and its appropriate management.
Keywords
Introduction
The immune system plays many roles in cancer prevention. Not only does it protect against viral infections and the development of virus-driven tumors, it also eliminates tumor cells, and rapidly identifies and eliminates foreign pathogens, minimizing the duration of an inflammatory, tumor-promoting environment. It is also understood that the immune system can impact the immunogenicity of the tumor itself given intact immune systems impact the very nature of the tumor and contribute to its ability to progress and grow. Understanding the impact of the immune system, including this knowledge of “cancer immunoediting” has allowed for the development of additional therapies to treat cancer[1]. Various classes of immunotherapies now exist that attempt to harness and exploit different strengths of the immune system, ranging from adoptive T cell therapy and cytokine therapy to checkpoint blockade therapy and oncolytic virus therapy[2]. Our improved understanding of the central nervous system (CNS) over the last several decades has led to the growth and continuous innovative design of immunotherapeutics for different malignancies, including those in the CNS. Lymphatics have been identified in the meninges and the dura, demonstrating a communication route for lymphocytes to and from the brain and cervical lymph nodes[3]. We also now know that the CNS has processes of immunosurveillance which allow for autoimmune disease and paraneoplastic syndromes to exist within its realm. The immune sytem can thus be used to our advantage in fighting cancer even in the CNS, contrary to prior belief. It has been shown that T cells infused intravenously will make their way to the central nervous system, but there are still many challenges to resolve before we are able to harness the immune system to attack cancer without excessive toxicity[4]. The responses to antigens must be narrow and limited only to those particular epitopes; otherwise, the response can spread and damage healthy, normal brain tissue. Tumor specific mutations are also less common in brain tumors compared to other malignancies, and may be unique to each individual, requiring optimal individualized medicine which can be cost-prohibitive[5]. Another challenge is that gliomas appear to have natural mechanisms to inhibit T cell activation and actually decrease peripheral T-cell counts, resulting in the lymphopenia seen in these patients even prior to the initiation of therapy[6]. Glioma cells are even able to evade detection by the immune system by down-regulating major histocompatibility complexes (MHC) or other components necessary for antigen presentation[7]. Thus, there are many avenues and opportunities for further exploration in the field of immunotherapy as it relates to brain tumor treatments, and as we learn and understand more, we slowly improve in our ability to develop novel therapeutics for CNS tumors.
Adoptive cell therapies - introduction to chimeric antigen receptor-T
The ability to harness the body’s own immune system to create a sustained anti-tumor response has transformed conventional therapeutic strategies for patients with cancer. Cell-based therapies involve the allogeneic or autologous transfer of immune-derived cells into cancer patients to enhance the host immune system’s ability to recognize and destroy cancer cells[8,9]. T-lymphocytes are the backbone of the body’s defense system due to their critical role in orchestrating and executing an effective immune response. They have been identified as prime candidates for their robust response against foreign pathogens and persistent anti-tumor activity[10]. Although endogenous T cells are frequently unable to eradicate progressing tumor on their own, modern technology has made it possible to genetically engineer the function, specificity, and longevity of T-cell anti-tumor response. Perhaps most importantly, the adoptive transfer of these cells can elicit a high degree of efficacy and specificity, thus, minimizing off-target toxicities.
Adoptive T-cell therapy (ACT) consists of isolating, expanding and reintroducing tumor-specific T lymphocytes into patients with cancer. Currently, there are four forms of ACT actively being investigated for cancer treatment: (1) tumor-infiltrating lymphocytes (TILs); (2) cytotoxic T lymphocytes (CTLs); (3) T-cell receptors (TCRs); and (4) chimeric antigen receptors (CARs) [Table 1][9,11]. TILs have unique anti-tumor properties as they are extracted from tumor biopsies where they have been exposed and conditioned to the tumor microenvironment. These cells can be isolated from tumor tissue and expanded in IL-2 cytokine prior to re-introduction and have shown to be an effective approach for patients with metastatic malignant melanoma[12]. CTLs are naturally circulating tumor-specific T cells taken from a patient’s peripheral blood and can be expanded using antigen presenting cells (APCs). Both TILs and CTLs, once infused back into the patient, recognize tumor associated antigens (TAA) via their TCR as antigeneic peptides presented by MHC, and subsequently execute T cell activation[13]. T cells can also be engineered to express TCRs and the ability to genetically clone, and affinity optimize TCRs can substantially increase their potential in recognizing tumor-specific antigens[14,15]. Lastly, CARs are genetically engineered surface receptors composed of extracellular antigen-binding domains fused to intracellular T cell signaling domains of the T-cell receptor. These modified T-cells expressing tumor-targeted CARs redirect antineoplastic specificity towards cancer cells without MHC restriction. CARs have advanced the furthest in clinical development with their recent FDA approval and their use showing remarkable clinical outcomes for patients with hematological malignancies[14-19].
Types of adoptive T cell therapies. Summary of T cell therapeutic strategies, mechanistic action, benefits and limitations
| Modified T-cell product | Description | Mechanism | Benefits | Limitations |
|---|---|---|---|---|
| TILs | Natural T lymphocytes extracted from autologous tumor biopsy and ex vivo cytokine fortification | Recognition of TAA via MHC-I complex by conditioned TCRs | Extensive intracellular and extracellular TAA specificity and recognition | Restricted to MHC-I complex Isolation and expansion complexities Low frequency of antigenic peptides expressed by specialized cells |
| CTLs | Enhanced and expanded in ex vivo isolated circulating tumor-specific T cells extracted from the patients’ peripheral blood using APCs | Via MHC-I complex, enriched TCRs recognize processed TAAs | Highly advantageous of helper T cells and APC augmentation of persistent anti-tumor activity and prolonged survival in vivo. Specific to viral or non-viral antigenic peptides | Restricted to MHC-I complex. Limited efficacy in non-viral tumor-specific antigenic peptides Low affinity of TAA TCR. Low frequency of TAA specific cells |
| TCRs | T lymphocytes modified to express a tgTCR with optimal specificity towards TAAs | Recognition of tumor antigenic peptides by tgTCR presented on MHC-I molecules | Augmented specificity in targeting extracellular or intracellular TAAs | Restricted to MHC-I complex and HLA-A2 patients tgTCR genetic mis-match with native TCR |
| CARs | Engineered T cells expressing an antibody-binding scFv exodomain fused with a CD3z chain intracellular domain via a transmembrane domain | CAR extracellular domain recognizes and binds to specific TAAs in a MCH-independent manner | Highly specific modified T cells with reliable production and unrestricted to any MHC complexes | Cytokine release syndrome largely due to persistent T cell proliferation and subsequent cytokine secretion |
CARs consist of an extracellular recognition domain that can bind specifically to a target molecule expressed on the surface of tumor cells, and an intracellular signaling domain that provides an activation signal upon target binding, linked via a transmembrane spacer/hinge domain. The extracellular domain is usually comprised of an antibody single-chain variable fragment (scFv)[20,21]. Interestingly, ligands of cell-surface receptors are also alternative molecules being used at multiple institutions[22,23]. The transmembrane spacer domain is important in conferring stability, flexibility and spacial orientation for CAR-antigen immunological synapse formation. Once the extracellular domain binds to a tumor-specific antigen, it will communicate and activate the intracellular domain, which directly initiates CAR-T cell cytolytic activity, cytokine production and proliferation[24]. The intracellular domain usually incorporates a region of the TCR CD3z chain to provide the primary activating signal. Most CAR designs also incorporate one or more domains from co-stimulatory receptors, such as CD28, CD134 (OX40), and/or CD137 (4-1BB) to provide the secondary signal for the optimal activation of downstream signaling cascades[25,26].
Over the past two decades, CAR-T cell designs have dramatically improved in their ability to mediate anti-tumor activity. The first-generation CARs only included a CD3z signaling domain, and although these first-generation CARs were sufficient to redirect T cell cytotoxic activity, they exhibited suboptimal persistence and in vivo killing potential. By contrast, second generation CARs include the addition of a costimulatory signaling domain (i.e., CD28, CD134, CD137) to enhance the signal function of the CD3z signaling domain. Indeed, second generation CARs have shown robust antitumor activity in patients in the setting of hematological malignancies. In third generation CARs, the main difference compared to second-generation CARs is the inclusion of two costimulatory signaling domains. The costimulatory additions aim to amplify the anti-tumor effect of second generation CARs, however, it remains to be determined whether the inclusion of additional co-stimulatory domains improves CAR function. The fourth-generation CARs are referred as T-cell redirected for universal cytokine killing (TRUCK) - and a variety of cytokine genes have been added to this structure[13]. There have also been recent creative designs that involve infusion of nanoparticles aimed to enhance T-cells anti-tumor response[27] and synthetic Notch (synNotch) receptors to increase tumor specificity[28]. Inducible CAR systems are also being developed by many researchers centered around the idea that the T cells would only turn “on” and become cytotoxic in the presence of certain other drugs - for example, a tetracycline regulation system that only turns on with doxycycline introduction[12]. Dual targeting CARs have also been developed that can target different receptors and induce signaling that will results in different outcomes - for example, one pathway elicits cytoxicity while another promotes the proliferation of T cells[14].
CAR-T cells in systemic cancer
The first clinical application of CD19 CAR-T therapy allowing for the products to be studied for their efficacy in humans was initiated in 2005 (NCT00182650; PMID: 20304086). Multiple clinical trials since then have shown impressive outcomes in patients with relapsed, refractory B-cell hematologic malignancies. This includes pediatric lymphoblastic leukemia (ALL), aggressive B cell non-Hodgkin’s lymphoma, and chronic lymphocytic leukemia (CLL)[15-21]. The response has not only been dramatic in terms of overall response rate and complete remission rate progression-free survival, but it has also been very durable in the majority of cases. Consequently, on August 30th, 2017, the first anti-CD19 CAR-T cell product, tisagenlecleucel, was approved for the treatment of children and young adults with relapsed or refractory B-cell precursor ALL and subsequently also for the treatment of aggressive B cell lymphoma[21,22]. This was followed by the approval of axicabtagene ciloleucel, another autologous anti-CD19 CAR-T cell therapy for the treatment of relapsed large B-cell lymphoma, in October of 2017[23].
CAR-T cells in brain cancer
CAR-T cell therapy has shown tremendous clinical success in the treatment of hematological malignancies and also solid tumors extending to the central nervous system (CNS). Within the JCAR017 clinical trial, Abramson reported a case study of a 68-year-old patient with a systemic refractory diffuse large-B-cell lymphoma who had developed a new brain lesion in the right temporal lobe. The patient was given CD19-targeted CAR-T cells and exhibited complete remission for 12 months in the absence of any neurotoxicity[24]. Interestingly, CAR-T cells were identified in the cerebrospinal fluid (CSF) after systemic administration, suggesting an alternative delivery route and their ability to cross the blood brain barrier, and trigger a durable anti-tumor response[25,26].
Several clinical trials across the nation are evaluating the efficacy of CAR-T cell therapy for patients with primary neuro-oncological malignancies. Multiple institutions are looking into several tumor targets as well as alternative delivery routes for optimal anti-tumor response. Brown et al.[27] described three patients with recurrent glioblastoma treated with first-generation interleukin-13 receptor alpha 2 (IL13Rα2) redirected CAR-T cells. Using an Ommaya reservoir, patients were given up to 12 intracavity infusions with two patients showing transient antitumor response. CNS inflammation was observed in all three patients and the degree of inflammation appeared to correlate with IL13Rα2 antigen expression. This indicated a persistent anti-tumor response through the activation of CAR-T cells once they recognized and were bound to the TAA[27]. Additionally, Brown et al.[28] reported the use of second-generation CAR-T cells targeting IL13Rα2 in a patient with recurrent multifocal leptomeningeal glioblastoma (GBM). The patient showed no disease progression at the tumor site receiving CAR-T cell infusions via an Ommaya catheter; however this intratumoral treatment failed to prevent the development of additional tumors over time. Due to metastatic progression, the patient subsequently received 10 intrathecal infusions into the right lateral ventricle. This was the first time intraventricular administration of CAR-T cells was performed and surprisingly showed remarkable tumor regression at the 6th infusion (out of 10). Complete response was observed for 7.5 months with increased production of cytokines and immune cells but without any major systemic associated side effects. The study suggested the ability to induce transient anti-tumor activity against multifocal glioblastoma with leptomeningeal seeding through multiple delivery routes with very manageable therapy-related toxicity [Figure 1][28].
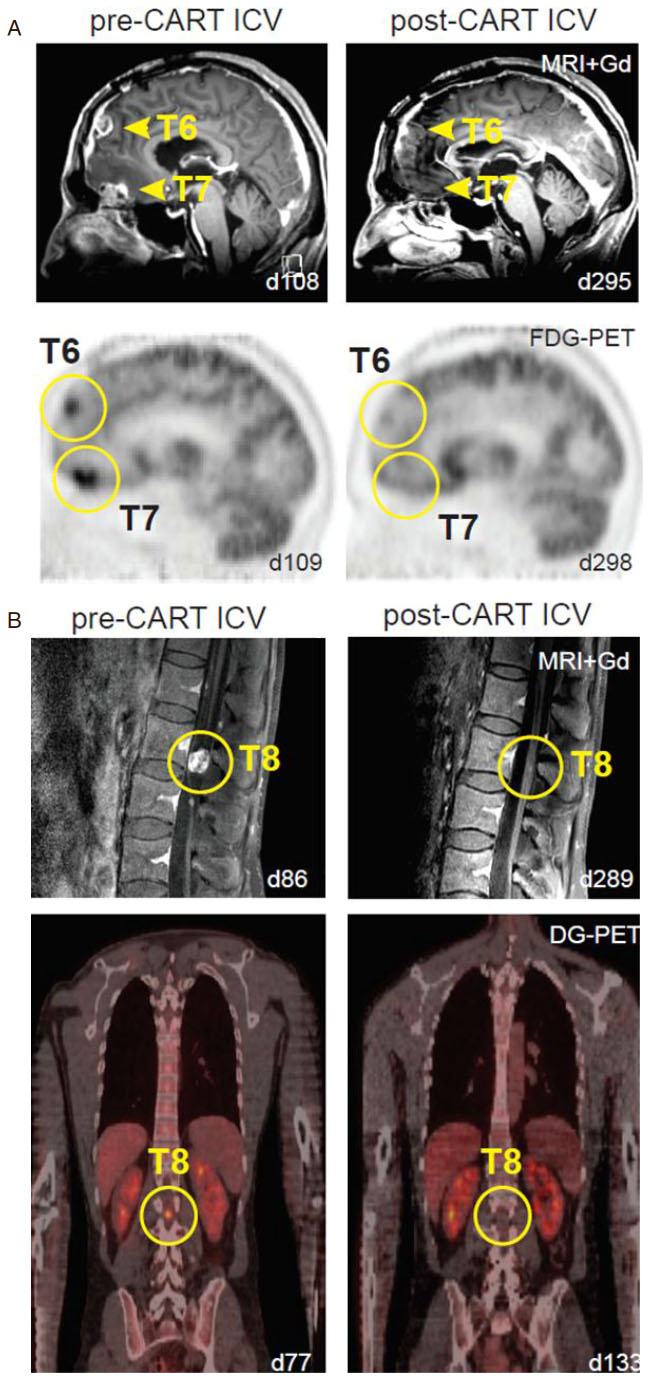
Figure 1. Multifocal glioblastoma responding to intraventricular delivery of IL13Ra2-redirected CAR T-cell therapy (modified from Brown et al.[28], permission for use granted by NEJM). A: sagittal view of gadolinium-enhanced MRI (top row) and FDG-PET (bottom row) images showing tumor regression of tumors 6 and 7 in the brain (yellow arrowheads and circles); B: sagittal gadolinium-enhanced MRI (top row) and coronal DG-PET (bottom row) images exhibiting tumor regression of tumor 8 in the spine (yellow circles)
O’Rourke et al.[29] reported their experience with 10 patients with recurrent GBM who received a single dose of peripherally infused EGFRvIII directed CAR-Ts. In their phase-I open label study, all 10 patients had an unmethylated MGMT promoter, known to be a poor prognostic factor. The median level of EGFRvIII expression was 71% (6%-96%). Importantly, there were no dose-limiting toxicities, and CAR-T therapy was not associated with EGFR-directed toxicity, systemic toxicity (CRS - see below) or neurotoxicity - signs and symptoms observed with CD-19- CAR-T cells. Median overall survival (OS) was 251 days (8 months) with one subject alive without further therapy 18 months after a single infusion of CART-EGFRvIII[29].
Other tumor biomarkers expressed by GBM are being tested in several institutions across the world. Yang and colleagues at Hefei Binhu Hospital are testing a modified CAR-T cell redirected to target MUC1 positive tumors in patients with recurrent/relapsed malignant glioma (NCT02617134). Lin and colleagues at Xuanwu Hospital in China are recruiting patients with recurrent malignant gliomas to test a spectrum of tumor specific/associated antigens including EGFRvIII, IL12Ra2, HER2, CD133, EphA2 and GD2 (NCT03423992). Rosenberg’s group from the National Cancer Institute is investigating an anti-EGFRvIII CAR in patients with malignant glioma who are positive for EGFRvIII (NCT01454596). In a phase I clinical trial, Vitanza from Seattle Children’s Hospital is testing a HER2-specific CAR in recurrent/refractory pediatric patients diagnosed with CNS tumors (NCT03500991). At Baylor College of Medicine, Ahmed and colleagues developed an autologous second-generation Cytomegalovirus (CMV)-specific CAR targeting HER2-positive GBM cells. It is currently undergoing evaluation in a clinical trial for patients diagnosed with GBM (NCT01109095). Chinese PLA General Hospital has deployed a phase I clinical trial investigating an anti-CD133 CAR in relapsed patients with advanced malignancies including brain tumors (NCT02541370). Brown et al.[28] at the City of Hope Medical Center have developed and deployed a phase I clinical trial testing a second-generation IL13Rα2-specific CAR in recurrent patients with malignant glioma (NCT02208362). Her group is also testing single and combined delivery routes including intracavity, intratumoral and intraventricular in order to identify the optimal route for T-cell infusion that will maximize antitumor response. These studies are further summarized in Table 2.
List of clinical trials investigating the efficacy of chimeric antigen receptor T-cell therapy across multiple antigens expressed in brain cancer
| Antigen | Disease | Intervention/treatment | Identifier |
|---|---|---|---|
| MUC1 | Malignant glioma | Anti-MUC1 CAR-T cells in patients with malignant glioma | NCT02617134 |
| EGFRvIII, IL12Ra2, HER2, CD133, EphA2, GD2 | Recurrent malignant glioma | CAR T cells redirected to target tumor specific/associated antigens in patients with recurrent malignant gliomas | NCT03423992 |
| HER2 | Pediatric CNS tumors | HER2-specific CAR T cell for recurrent/refractory pediatric CNS tumors | NCT03500991 |
| EGFRvIII | Malignant gliomas | Anti-EGFRvIII CAR T cells in patients with malignant glioma | NCT01454596 |
| IL13Ra2 | Recurrent malignant glioma | IL13Ra2-specific, hinge-optimized, 41BB-costimulatory CAR/truncated CD19-expressing autologous T lymphocytes | NCT02208362 |
| CD133 | Glioma | Anti-CD133 modified CAR T cells in relapsed patients with malignant glioma | NCT02541370 |
| HER2 | GBM | CMV-specific CTLs expressing CAR targeting HER2 positive recurrent GBM patients | NCT01109095 |
Systemic and neurological toxicity of CAR-T cells
CAR-T treatment is not without toxicities, which have been well-studied over the years for all of the different products created and investigated in clinical trials. One of these products (JCAR015) had to be taken off the market after the death of five patients from severe cerebral edema and herniation[30].
The primary toxicity that has been seen across trials and is now well-described is cytokine release syndrome or cytokine release syndrome (CRS). This is characterized by fever, malaise, anorexia, myalgias, hypotension, and can include multi-organ dysfunction. The mechanism continues to be explored but the release of cytokines by infused T-cells plays a significant role in this syndrome. A variety of inflammatory cytokines have been found to be elevated in the serum of patients experiencing CRS, including interleukin(IL)-6, interferon-gamma, IL-15, IL-8, IL-10, and IL-2. Higher disease burden has been predicted to lead to more toxicity, but other factors continue to be explored. Various organ systems have been noted to be impacted by CRS to varying degrees[23,31][Figure 2]. CRS is generally seen within the first week after infusion of CAR-T cell therapy and the peak risk is within the first two weeks of administration - thus, patients are typically monitored in the acute inpatient setting (with access to an intensive care unit), and with frequent monitoring of vital signs and laboratory parameters including blood counts, coagulation factors, measures of organ function, and inflammatory markers[23,31].

Figure 2. Toxicity from chimeric antigen receptor-T by organ system. Chimeric antigen receptor-T cell treatment has been found to result in a number of toxicities throughout the body. Professional illustration by Ryan Stemen
Various grading systems to evaluate CRS have been developed including one by Lee et al.[32] which has been used at various centers where the therapy is offered[32]. The group modified a National Cancer Institute Common Terminology Criteria for Adverse Events or CTCAE to define mild, moderate, severe and life-threatening degrees of CRS. Grade 1 symptoms are mild, requiring supportive treatment, and include fever, nausea, headache, etc. The grading is subsequently raised based on the degree of organ toxicity noted and the level of intervention required to maintain a stable hemodynamic status, with grade 5 toxicity translating to death [Table 3][32].
Cytokine release syndrome and chimeric antigen receptor-T cell related encephalopathy syndrome*
| Grade | CRS | CRES |
|---|---|---|
| Grade 1 | Non-life threatening symptoms (includes fever, nausea, fatigue, headache, myalgias, grade 1 organ toxicity) | CARTOX-10 score 7-9 (mild impairment) |
| Grade 2 | Symptoms require moderate intervention (Oxygen requirement < 40%, hypotensive but responsive to fluids or low dose pressors, grade 2 organ toxicity) | CARTOX-10 score 3-6 (moderate impairment) |
| Grade 3 | Symptoms require aggressive intervention (Oxygen requirement ≥ 40%, hypotensive requiring high dose or multiple pressors, grade 2 organ toxicity or grade 4 transaminitis) | CARTOX-10 score 0-1 (severe impairment), intracranial pressure as noted by papilledema or CSF opening pressure < 20 mmHg, partial seizures or non-convulsive seizures on EEG responsive to benzodiazepines. |
| Grade 4 | Life-threatening symptoms (ventilator support, grade 4 organ toxicity) | Obtunded, high grade papilledema or CSF opening pressure ≥ 20 mmHg or cerebral edema on imaging, generalized seizures or status epilepticus, new motor weakness |
| Grade 5 | Death |
Neurological toxicity is the other well-described and important phenomenon seen in these patients and has been noted from the very onset of CAR-T trials. A wide spectrum of symptoms have been reported including headaches, global encephalopathy, seizures, tremors, ataxia, hemiparesis, aphasia, ataxia, apraxia, dysmetria, and cranial nerve palsies. In very rare cases, diffuse cerebral edema has been noted, which has in some cases been fatal. Elevated cytokine levels are implicated although it is not yet completely clear what is the mechanism of neurological dysfunction observed across patients. The role of endothelial activation and increased blood-brain barrier permeability as well as role of IL-1 have been recently described in association with neurotoxicity[33]. Evaluation of several cases has demonstrated anti-CD19 CAR T cells may be found at a higher level in the CSF of patients who experience neurotoxicity, and these patients may also have higher levels of IL-6 in the CSF[32,34]. While the initial neurotoxicity is seen early in the course with concurrent CRS, delayed neurological toxicity up to several weeks after infusion has also been noted, though the mechanism for this remains unclear.
Neelapu et al.[23] have described the neurological toxicity seen with CAR-T infusions and developed the term CAR-T related encephalopathy syndrome (CRES), and have provided a helpful grading system for this entity[23]. Similar to CRS, Grade 1-4 are assigned based on a combination of a neurological assessment score, raised intracranial pressure and seizures or motor weakness. The neurological status is assessed with CARTOX-10, a 10-point neurological assessment tool developed by the authors that evaluates attention, speech and writing ability [Table 3].
It should be noted that direct CNS infusions of CAR-T have not resulted in the same toxicity (CRS or CRES), though only a few patients have had this treatment at this point. As previously discussed, Brown et al.[28] described a patient with glioblastoma who was treated with an intrathecal delivery of CAR-T cells. They reported only grade 1-2 toxicity, the majority of which lasted less than 1 day. Grade 1 fatigue was the most lasting adverse event (4 days). Headache, myalgias and lymphopenia were the worst higher grade toxicities (Grade 2)[28].
Management of CAR-T toxicity
Management of CAR-T associated toxicity is based on severity of symptoms. It begins with symptomatic management and supportive care for milder symptoms - fever is treated with acetaminophen or cooling blankets, acetaminophen is used for myalgias, anti-emetics for nausea, etc. Intravenous (IV) fluids may be started for hydration in grade 1 toxicity. Organ systems are closely monitored with frequent laboratory assessments and organ toxicities treated according to what is seen (for example, acute kidney injury or AKI may be treated with hydration). Higher grades of CRS require greater intervention. Oxygen requirements may need to be met with supplemental oxygen and/or even ventilation. Vasopressors may need to be started and titrated to maintain a stable hemodynamic status. Ultrasound may be needed to assess hemodynamic status, and frequency of vitals and assessments may need to be increased. For these reasons, patients who are not yet in the intensive care unit (ICU) are often transferred when CRS or neurotoxicity is grade 2 or higher[23,31,32,35].
Similar to CRS, management of CRES is also escalated depending on the grade of the toxicity seen. Careful and frequent neurological assessment by experienced providers is extremely important. In addition, it is also important to use a scale such as the CARTOX-10 or a similarly sensitive tool that can pick up on subtle, early neurological deficits. With even minimal signs of neurological impairment, the patient is generally placed on aspiration precautions. The patient must be closely and frequently monitored from this point onwards with careful neurological examinations by a neurologist, and regular funduscopic exams. CT head of the brain, followed by MRI of the brain (and/or spine if appropriate and if deficits are present) is usually obtained along with a lumbar puncture to assess opening pressure and rule out other conditions that these patients may be at risk for (such as leukemic or infectious meningitis). If the patient is too unstable, a CT of the brain can be obtained instead of MRI in this early stage. If patient is noted to be encephalopathic, an EEG should be considered to rule out non-convulsive status epilepticus[23].
If the patient does develop increased intracranial pressure or has seizures (convulsive or non-convulsive), management should be guided per the protocols for each of these conditions. An experienced neurology team is thus quite necessary. Increased intracranial pressure may be managed with dexamethasone, mannitol or acetazolamide, bed elevation, hyperventilation, or hyperosmolar therapy, or even drainage of CSF. Seizures, and possible status epilepticus, should be managed per nationally established guidelines that include lorazepam as a starting point with IV boluses of anti-epileptics such as levetiracetam and escalating to additional medications or a drip of midazolam or phenobarbital if seizures continue despite early efforts. Drugs such as fosphenytoin and lacosamide are typically avoided due to their potential for additional cardiotoxicity. Some centers are using prophylactic levetiracetam (at 500 mg BID) prior to starting the treatment process, though at this time no clear evidence supports the use of prophylactic anti-epileptics[23].
Tocilizumab is an IL-6 antagonistic monoclonal antibody that has been available on the market to treat rheumatological disorders. Given the high levels of IL-6 seen with the toxicity of CAR-T, it was tried and found to effectively treat CRS-related complications in clinical trials. Eventually, it was approved by the FDA for this specific purpose in 2017. It is recommended as the first course of treatment when CRS and neurological toxicity is confirmed as per the grading criteria discussed previously. Tocilizumab can be dosed at 8mg/kg IV for CRS or CRES, starting as early as grade 1 toxicity. Siltuximab, an anti-IL-6 chimeric monoclonal antibody, can also be used instead of tocilizumab in these cases, and is dosed at 11mg/kg IV. If the patient fails to respond to these drugs and proceeds to grade 2 or higher CRS/CRES, then corticosteroids may need to be used. Corticosteroids appear to be more effective for patients with neurotoxicity only without concomitant CRS. Dexamethasone is typically dosed at 10mg IV every 6 h, and continued toxicity without response to dexamethasone may need to be treated with methylprednisolone. There has been concern that steroids may impact the inflammatory response that is important for the efficacy of CAR-T cell treatment and may suppress the function of the tumor-directed T cell therapy since they can suppress T cells and induce apoptosis of these cells. However, preliminary data at this point demonstrates no clear objective difference in response rates[36]. Most management protocols advise considering steroids if patients are demonstrating grade 2 toxicity without response to tocilizumab or siltuximab[23,31].
Conclusion
CAR-T therapy has had promising results in hematological malignancies and has revolutionized the immunotherapy field in the last several years. There is a great hope that with the identification of the right antigens, and with improvements in delivery and safety, CNS malignancies might be successfully treated with this therapy. Early studies are demonstrating some encouraging results , but larger trials are needed to fully evaluate this modality in treatment of brain cancer (both primary and secondary). It is critically important for the providers to understand the principles of this therapy, its mechanism of action and particularly its neurologic toxicity. While the understanding of neurotoxicity continues to evolve, early integration of the neurology team in the care of patients receiving CAR-T cell therapy clearly provides benefit. Immunotherapy is an emerging area in oncology and its use, including the indications for CAR-T, are likely to expand.
Declarations
AcknowledgementsThe authors would like to thank Mr. Ryan Stemen for designing the graphics for this paper.
Authors’ contributionsResearched literature, wrote the manuscript: Sharma A, De Leon G
Provided content, reviewed the manuscript: Porter A, Grill MF, Rosenthal A, Swanson K
Provided content and figures, reviewed the manuscript: Brown CE
Article concept, literature research, writing and reviewing the manuscript: Mrugala MM
Availability of data and materialsData available on PubMed and www.clinicaltrials.gov.
Financial support and sponsorshipNone.
Conflicts of interestC. E. Brown receives patent royalties and consulting support from Mustang Bio.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011;331:1565-70.
3. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015;523:337-41.
4. Walsh JT, Watson N, Kipnis J. T cells in the central nervous system: messengers of destruction or purveyors of protection? Immunology 2014;141:340-4.
5. Mohme M, Neidert MC, Regli L, Weller M, Martin R. Immunological challenges for peptide-based immunotherapy in glioblastoma. Cancer Treat Rev 2014;40:248-58.
6. Albesiano E, Han JE, Lim M. Mechanisms of local immunoresistance in glioma. Neurosurg Clin N Am 2010;21:17-29.
7. Facoetti A, Nano R, Zelini P, Morbini P, Benericetti E, et al. Human leukocyte antigen and antigen processing machinery component defects in astrocytic tumors. Clin Cancer Res 2005;11:8304-11.
8. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62-8.
9. June CH, Riddell SR, Schumacher TN. Adoptive cellular therapy: a race to the finish line. Sci Transl Med 2015;7:280ps7.
10. Nagorsen D, Scheibenbogen C, Marincola FM, Letsch A, Keilholz U. Natural T cell immunity against cancer. Clin Cancer Res 2003;9:4296-303.
11. Fousek K, Ahmed N. The evolution of T-cell therapies for solid malignancies. Clin Cancer Res 2015;21:3384-92.
12. Sakemura R, Terakura S, Watanabe K, Julamanee J, Takagi E, et al. A Tet-On inducible system for controlling CD19-chimeric antigen receptor expression upon drug administration. Cancer Immunol Res 2016;4:658-68.
14. Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol 2012;32:1059-70.
15. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010;116:4099-102.
16. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 2011;365:725-33.
17. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 2013;5:177ra38.
18. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013;368:1509-18.
19. Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol 2018;36:2267-80.
20. Maude SL, Shpall EJ, Grupp SA. Chimeric antigen receptor T-cell therapy for ALL. Hematology Am Soc Hematol Educ Program 2014;2014:559-64.
21. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, et al. Primary analysis of Juliet: a global, pivotal, phase 2 trial of CTL019 in adult patients with relapsed or refractory diffuse large B-cell lymphoma. Blood 2017;130:577.
22. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 2018;378:439-48.
23. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol 2018;15:47-62.
24. Abramson JS, McGree B, Noyes S, Plummer S, Wong C, et al. Anti-CD19 CAR T cells in CNS diffuse large-B-cell lymphoma. N Engl J Med 2017;377:783-4.
25. Wang Z, Han W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark Res 2018;6:4.
26. Yoon DH, Osborn MJ, Tolar J, Kim CJ. Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-Ts): combination or built-in CAR-T. Int J Mol Sci 2018;19:E340.
27. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res 2015;21:4062-72.
28. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 2016;375:2561-9.
29. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017;9:eaaa0984.
30. Nelson R. Juno permanently halts CAR T-cell study. In: Medscape Medical News. Online: Medscape; 2017.
31. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 2016;127:3321-30.
32. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014;124:188-95.
33. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med 2018;24:739-48.
34. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015;385:517-28.
35. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics 2016;3:16011.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].