


MASH as a network disease: therapeutic modulation of interorgan immunometabolic architecture
 0
0 
MASH AS A NETWORK DISEASE
The study by Jara et al. represents a mechanistic contribution of particular significance to the field of metabolic dysfunction-associated steatohepatitis (MASH), integrating histological, proteomic and preclinical data to delineate the coordinated modulation of metabolic, inflammatory and fibrogenic pathways under semaglutide treatment[1]. Beyond documenting histological resolution, the study provides consistent evidence of a coordinated reorganization of interconnected regulatory nodes, reinforcing the interpretation of MASH as a tissue-level manifestation of a systemic immunometabolic network.
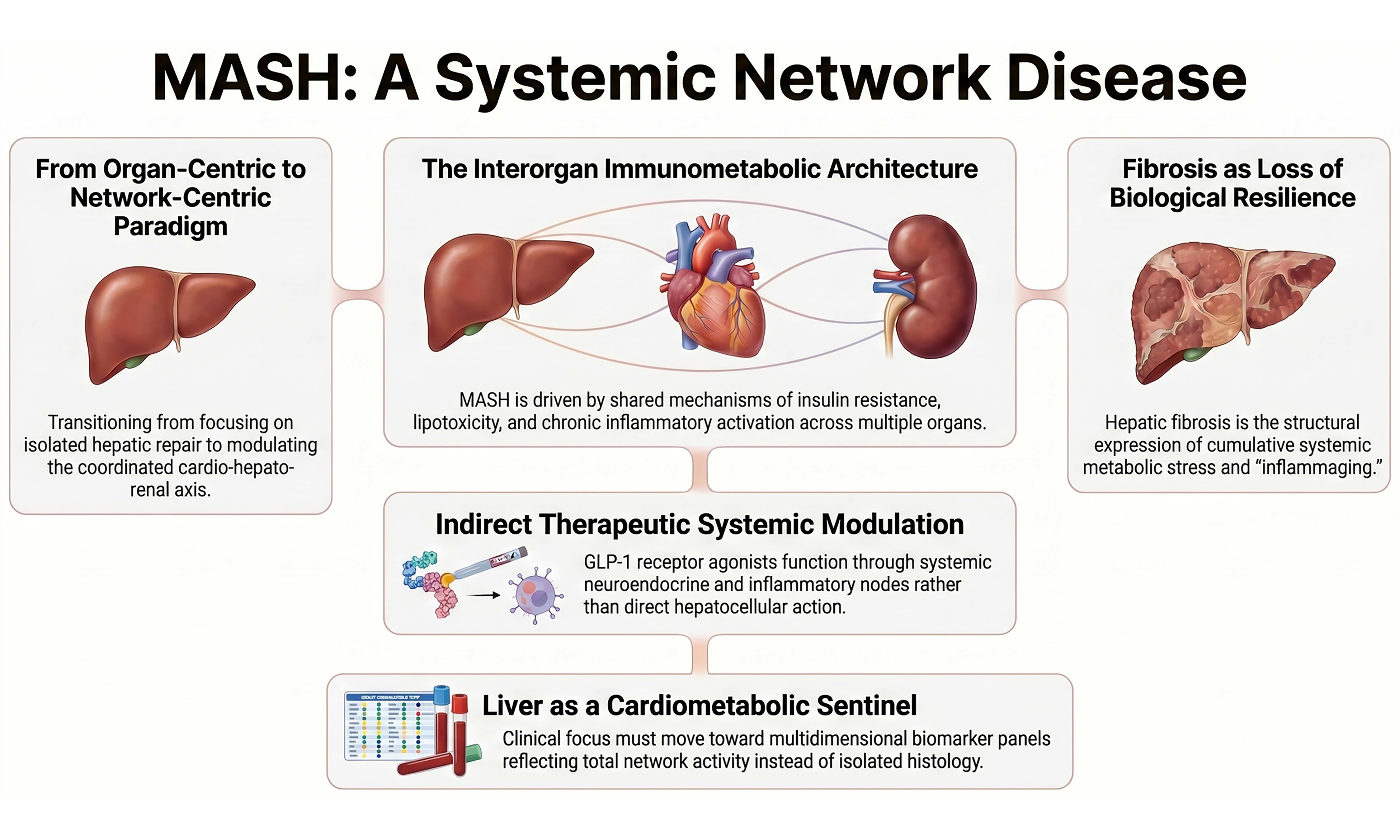
MASH constitutes the hepatic expression of a systemic metabolic derangement whose population burden has reached global proportions[2]. Its bidirectional association with type 2 diabetes is not merely epidemiological but pathophysiological, reflecting shared mechanisms of insulin resistance, lipotoxicity and chronic inflammatory activation[3]. Convergently, its integration within the cardio-hepato-renal axis does not represent additive comorbidity but rather parallel manifestations of a common disrupted metabolic architecture[4,5], as shown in Figure 1. The joint European Association for the Study of the Liver (EASL)-European Association for the Study of Diabetes (EASD)-European Association for the Study of Obesity (EASO) guidelines have formalized this conceptual shift by embedding hepatic, cardiovascular and metabolic risk stratification within a unified clinical framework[6].
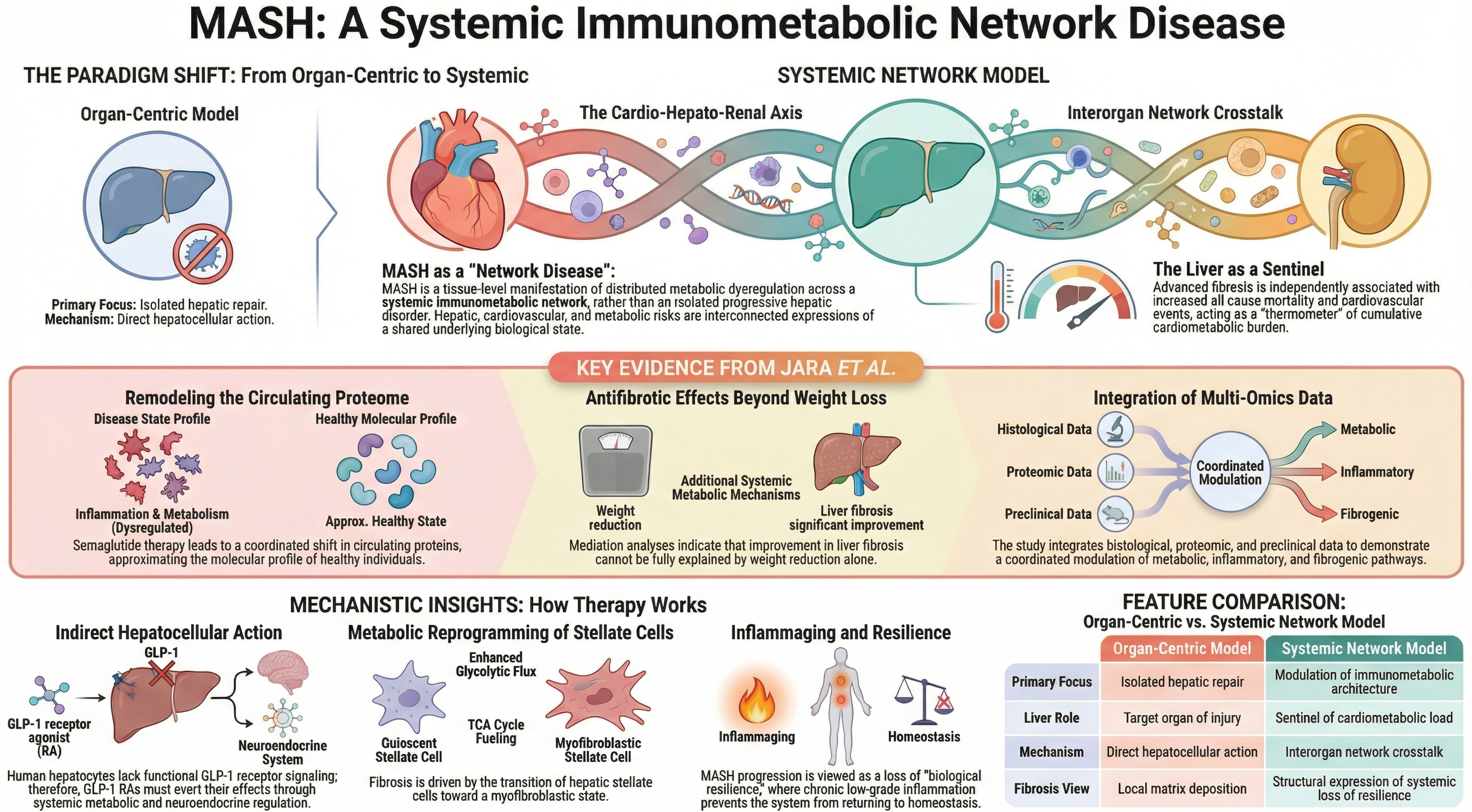
Figure 1. MASH as a systemic immunometabolic network disease. Schematic representation of the paradigm shift from an organ-centric model - focused on isolated hepatic repair and direct hepatocellular mechanisms - to a systemic network framework integrating the cardio-hepato-renal axis. Key evidence from Jara et al. demonstrates coordinated remodeling of the circulating proteome toward a non-disease molecular profile and antifibrotic effects not fully explained by weight reduction alone[1]. Mechanistically, GLP-1 receptor agonism acts indirectly through systemic metabolic and neuroendocrine pathways, modulates stellate cell metabolic reprogramming, and attenuates fibrogenic activation. Advanced fibrosis is conceptualized as a structural expression of systemic immunometabolic dysregulation and loss of biological resilience, positioning the liver as a sentinel of cumulative cardiometabolic burden rather than an isolated target organ. MASH: Metabolic dysfunction-associated steatohepatitis; GLP-1: glucagon-like peptide-1; GLP-1 RA: glucagon-like peptide-1 receptor agonist; TCA: tricarboxylic acid.
International recognition of metabolic dysfunction-associated steatotic liver disease (MASLD)/MASH as a systemic entity further consolidates this paradigm[7]. Within this framework, the liver may be interpreted as a sensor of cumulative cardiometabolic burden, given that advanced fibrosis is independently associated with increased all-cause mortality and cardiovascular events[4,8]. This association suggests that hepatic stage reflects the temporal integration of systemic metabolic exposures rather than purely local injury.
INTERORGAN IMMUNOMETABOLIC ARCHITECTURE
The data generated by Jara et al. demonstrate that histological resolution is accompanied by coordinated modulation of circulating proteins implicated in inflammation, metabolism and fibrogenesis, approximating the molecular profile observed in individuals without disease[1]. Such a pattern is consistent with an intervention acting on interorgan regulatory networks rather than on an isolated anatomical compartment. The functional absence of glucagon-like peptide-1 (GLP-1) receptor expression in human hepatocytes[9-11] further excludes a dominant direct hepatocellular mechanism, although more complex intrahepatic effects cannot be fully excluded. Emerging evidence suggests potential contributions from paracrine signaling, modulation of non-parenchymal cells, and hepatic microenvironment remodeling. MASH progression depends on sustained activation of the hepatic microenvironment, wherein lipotoxic hepatocytes trigger pro-inflammatory signaling and activation of hepatic stellate cells[12,13]. Metabolic reprogramming of these cells constitutes a critical axis of fibrogenesis. Loss of phosphoenolpyruvate carboxykinase 1 (PCK1) in hepatic stellate cells drives enhanced glycolytic flux and tricarboxylic acid cycle fueling, promoting fibrogenic activation[14]. This myofibroblastic transition entails mitochondrial remodeling and redox alterations, reflecting integrated metabolic adaptation rather than an exclusively local phenomenon.
Recent single-cell transcriptomic analyses further demonstrate that hepatic stellate cell activation in MASLD is characterized by coordinated shifts in metabolic gene expression programs, including enhanced glycolysis, altered oxidative phosphorylation, and pro-fibrogenic cytokine signaling, reinforcing that fibrogenesis represents a metabolically rewired cellular state rather than a passive scarring process[15]. Parallel experimental evidence indicates that adipose tissue inflammation and dysregulated adipokine secretion amplify hepatic stellate cell activation and extracellular matrix deposition, thereby functionally linking visceral adiposity to hepatic fibrosis progression[16].
The therapeutic rationale for intervention “beyond the liver” therefore gains mechanistic coherence[17]. Incretin biology modulates energy intake, energy expenditure, systemic inflammation and central neuroendocrine regulation[18,19]. Cardiovascular benefits demonstrated in clinical trials[20], together with the efficacy of dual GLP-1/glucose-dependent insulinotropic polypeptide (GIP) agonism in MASH[19], confirm that therapeutic effects operate at shared regulatory nodes. Mediation analyses reported by Jara et al. further indicate that improvement in fibrosis cannot be explained solely by weight reduction, implying the contribution of additional metabolic mechanisms[1]. Large cardiovascular outcome trials with GLP-1 receptor agonists have consistently shown reductions in major adverse cardiovascular events independent of baseline body mass index, supporting the concept that incretin-based therapies act on systemic inflammatory and metabolic pathways beyond weight loss alone[21]. In addition, the AMPK (adenosine monophosphate-activated protein kinase)-centric hypothesis further supports this systemic view, as AMPK acts as a central metabolic regulator linking energy homeostasis, inflammation and fibrogenesis, reinforcing the notion that therapeutic efficacy in MASH extends beyond the liver[22].
In summary, these findings support the view that fibrogenesis in MASH reflects coordinated immunometabolic dysregulation across interconnected organ systems rather than an isolated hepatic process.
TEMPORAL BIOLOGY, INFLAMMAGING AND FIBROSIS TRAJECTORIES
Histological reversal following substantial weight loss is well established[23,24]. However, equivalent weight reductions may be associated with divergent metabolic signatures[25], and inflammatory changes may precede maximal weight loss under incretin agonism[26]. These findings suggest that systemic inflammatory modulation may exert an independent influence on reversal trajectories.
From a temporal perspective, MASH progression can be interpreted as the tissue crystallization of a deviated biological trajectory shaped by chronic low-grade inflammation and persistent metabolic dysregulation. This framework converges with the concept of inflammaging, which defines sustained immunometabolic activation as a transversal substrate of biological aging and multiple chronic diseases[27,28]. The updated hallmarks of aging further integrate metabolic dysfunction and loss of adaptive resilience as core components of the aging process[29].
Chronic low-grade inflammation has been mechanistically linked to progressive organ fibrosis through sustained activation of innate immune pathways, redox imbalance, and epigenetic remodeling, thereby connecting metabolic stress to durable structural tissue change[27].
Epigenetic alterations induced by metabolic overload - including DNA methylation changes and histone modifications - have been described in MASLD and may contribute to persistent fibrogenic programming even after partial metabolic improvement[30].
Interaction with the intestinal microbiome introduces an eco-biological dimension to metabolic susceptibility[31]. Within this context, fibrosis may be understood as the cumulative structural expression of impaired biological resilience, defined as the capacity of the system to absorb metabolic and inflammatory perturbations and return to homeostasis[32]. Gut-derived microbial metabolites, including short-chain fatty acids and endotoxin-related signals, modulate hepatic inflammation and stellate cell activation, providing a mechanistic bridge between intestinal ecology and hepatic fibrogenesis[33]. A neuroimmunometabolic dimension further broadens this interpretation. Obesity-induced inflammation alters hypothalamic circuits and peripheral-central communication[31], and GLP-1 receptor agonists are being explored as modulators in neurodegenerative disease[34]. Digestive chronobiology introduces an additional regulatory layer linking circadian rhythms, metabolism and inflammation[35].
Circadian misalignment has been shown to exacerbate insulin resistance and hepatic steatosis through disruption of clock-controlled metabolic genes, indicating that temporal regulation constitutes an additional layer of vulnerability in MASLD progression[36].
CLINICAL TRANSLATION: FROM ORGAN-CENTRIC TREATMENT TO NETWORK MODULATION
Taken together, the work of Jara et al. compels a definitive departure from an organ-centric reading of MASH[1]. Their findings not only support a systemic interpretation of the disease but suggest that incretin-based interventions act upon shared regulatory nodes within an interorgan immunometabolic network. The conceptual implication is unambiguous: MASH can no longer be regarded as an isolated progressive hepatic disorder, but rather as the tissue-level manifestation of distributed metabolic dysregulation. In this light, the liver ceases to be merely a target organ and emerges as a sentinel of cumulative cardiometabolic load[8]. Fibrosis is not simply extracellular matrix deposition but the structural expression of progressive loss of biological resilience within a context of chronic low-grade inflammation and immunometabolic aging. Under this framework, GLP-1 receptor agonists do not act exclusively upon hepatic parenchyma; they modulate neuroendocrine, inflammatory and metabolic axes that organize systemic homeostasis. The shift is neither rhetorical nor terminological - it redefines the clinical objective. Treating MASH entails intervening upon the integrated metabolic architecture of the system, not upon an isolated anatomical compartment. Liver, heart and kidney are not independent entities, but coordinated expressions of a shared underlying biological state. The identification of dynamic biomarker panels reflecting immunometabolic network activity - rather than single-organ injury - represents a critical step toward validating the network disease model and translating it into clinical practice. Importantly, the immunometabolic network underlying MASH is unlikely to be uniform across all patient populations. Variability related to phenotypes such as lean MASH, sex differences, and pediatric disease may reflect distinct configurations of interorgan signaling and metabolic regulation, highlighting the need for more personalized approaches within this framework[25,27,37].
This systemic reframing has implications for endpoint selection in clinical trials, favoring composite cardio-hepato-metabolic outcomes and multidimensional biomarker panels over isolated histological improvement as sole markers of therapeutic success[38]. Regulatory science is progressively incorporating such multidimensional risk constructs, recognizing that MASLD/MASH therapies may confer benefits across cardiovascular, renal and metabolic domains beyond hepatic histology alone[19].
DECLARATIONS
Authors’ contributions
Responsible for the ideation, supervision and review: Crespo J, Iruzubieta P
Performed the literature search, original draft preparation and editing: Jiménez-González C
All authors have read and agreed to the published version of the manuscript.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tools Claude Opu (Claude 4.8, released 2026-05-28) and ChatGPT Instant (ChatGPT 5.5, released 2026-04-26) were used solely for language editing without unsupervised automatic generation. In addition, the AI tool NotebookLM (NotebookLM IA Google Pro, released 2025-02-10) was used solely to generate the Figure 1 and the Graphical Abstract. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This project has received funding from the European Horizon’s research and innovation program HORIZON-HLTH-2022-STAYHLTH-02 (Agreement No. 101095679), Spanish Instituto de Salud Carlos III-FEDER Grant (FIS-PI22/01853) and European Union-NextGenerationEU (PMP21/00112). The funding sources were not involved in the research design or preparation of the article.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Jara M, Norlin J, Kjær MS, et al. Modulation of metabolic, inflammatory and fibrotic pathways by semaglutide in metabolic dysfunction-associated steatohepatitis. Nat Med. 2025;31:3128-40.
2. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73-84.
3. Mantovani A, Byrne CD, Bonora E, Targher G. Nonalcoholic fatty liver disease and risk of incident type 2 diabetes: a meta-analysis. Diabetes Care. 2018;41:372-82.
4. Targher G, Tilg H, Byrne CD. Non-alcoholic fatty liver disease: a multisystem disease requiring a multidisciplinary and holistic approach. Lancet Gastroenterol Hepatol. 2021;6:578-88.
5. Targher G, Byrne CD, Tilg H. NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut. 2020;69:1691-705.
6. Tacke F, Horn P, Wong VW-S, et al. Publisher correction: EASL–EASD–EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD): executive summary. Diabetologia. 2024;67:2608.
7. Lazarus JV, Pessoa MG, Shawcross DL, Schwarz P, Su GL, Barquera S. Name MASLD/MASH - and act on it. JHEP Rep. 2026;8:101674.
8. Crespo J, Iruzubieta P, Fernández Rodríguez CM. The liver as a thermometer of cardiometabolic health: time to prioritize MASLD in global health policy. Rev Esp Enferm Dig. 2025;117:709-11.
9. Smits MM, Holst JJ. Endogenous glucagon-like peptide (GLP)-1 as alternative for GLP-1 receptor agonists: could this work and how? Diabetes Metab Res Rev 2023;39:e3699.
10. Kirk RK, Pyke C, von Herrath MG, et al. Immunohistochemical assessment of glucagon-like peptide 1 receptor (GLP-1R) expression in the pancreas of patients with type 2 diabetes. Diabetes Obes Metab. 2017;19:705-12.
11. da Silva Lima N, Cabaleiro A, Novoa E, et al. GLP-1 and GIP agonism has no direct actions in human hepatocytes or hepatic stellate cells. Cell Mol Life Sci. 2024;81:468.
12. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836-46.
13. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908-22.
14. Novoa E, Parracho T, Inderhees J, et al. Lack of PCK1 in hepatic stellate cells causes liver fibrosis by fueling tricarboxylic acid cycle and increasing glycolysis. Cell Metab. 2026;38:729-45.e9.
15. Ramachandran P, Dobie R, Wilson-Kanamori JR, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019;575:512-8.
16. Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest. 2017;127:1-4.
17. Vuppalanchi R, Noureddin M, Alkhouri N, Sanyal AJ. Therapeutic pipeline in nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. 2021;18:373-92.
18. Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013;17:819-37.
19. Nauck MA, Tuttle KR, Tschöp MH, Blüher M. Glucagon-like receptor agonists and next-generation incretin-based medications: metabolic, cardiovascular, and renal benefits. Lancet. 2026;407:892-908.
20. Loomba R, Hartman ML, Lawitz EJ, et al. ; SYNERGY-NASH Investigators. Tirzepatide for metabolic dysfunction-associated steatohepatitis with liver fibrosis. N Engl J Med. 2024;391:299-310.
21. Marso SP, Daniels GH, Brown-Frandsen K, et al. ; LEADER Steering Committee, LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-22.
22. Zhao P, Saltiel AR. From overnutrition to liver injury: AMP-activated protein kinase in nonalcoholic fatty liver diseases. J Biol Chem. 2020;295:12279-89.
23. Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 2005;54:603-8.
24. Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015;149:367-78.e5.
25. Liu X, Zhu L, Liu J, Nie Z, Qiu W. Effect of weight loss interventions on metabolomic signatures in obese children with insulin resistance. Amino Acids. 2024;56:54.
26. Balcázar-Valencia CM, García-Ramos AF, Osorio-Toro LM, et al. Semaglutide effects on metabolic outcomes in diabetes mellitus patients - real world study. Diabetes Metab Syndr Obes. 2024;17:1667-73.
27. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14:576-90.
28. Furman D, Campisi J, Verdin E, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25:1822-32.
29. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186:243-78.
30. Pirola CJ, Sookoian S. Epigenetics factors in nonalcoholic fatty liver disease. Expert Rev Gastroenterol Hepatol. 2022;16:521-36.
31. Tilg H, Adolph TE, Dudek M, Knolle P. Non-alcoholic fatty liver disease: the interplay between metabolism, microbes and immunity. Nat Metab. 2021;3:1596-607.
32. Amjad AI, Sheikh BS, Aslam S. Biological resilience as a crucial determinant in preventing age-associated chronic diseases. Front Physiol. 2025;16:1741426.
33. Le Thuc O, García-Cáceres C. Obesity-induced inflammation: connecting the periphery to the brain. Nat Metab. 2024;6:1237-52.
34. Sabbagh MN, Cummings JL, Ballard C, et al. Repurposing glucagon-like peptide-1 receptor agonists for the treatment of neurodegenerative disorders. Nat Aging. 2026;6:56-67.
35. Bishehsari F, Post Z, Swanson GR, Keshavarzian A. Circadian rhythms in gastroenterology: the biological clock’s impact on gut health. Gastroenterology. 2025;169:1380-96.
36. Tahara Y, Shibata S. Circadian rhythms of liver physiology and disease: experimental and clinical evidence. Nat Rev Gastroenterol Hepatol. 2016;13:217-26.
37. Babu AF. Metabolic signatures in lean MASLD: current insights and future directions. Metabolites. 2025;15:583.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].