


The cross-talk between neurodegeneration and metabolic disorders
 0
0 Abstract
Despite substantial advances extending human lifespan, the gap between healthspan and lifespan continues to widen, with neurodegenerative diseases (NDDs) and metabolic disorders representing major contributors to this disparity. Growing epidemiological and genetic evidence indicates a bidirectional relationship between NDDs and metabolic disorders, suggesting shared pathophysiological mechanisms that transcend organ-specific boundaries. In this narrative review, we sought to explore the interconnections between neurodegeneration and metabolic dysfunction through the lens of the twelve established hallmarks of aging. We conducted a comprehensive literature search across multiple databases (PubMed, Google Scholar, Scopus, ScienceDirect) from January 2013 to April 2025, focusing on studies examining aging hallmarks in both NDDs (particularly Alzheimer’s disease and Parkinson’s disease) and metabolic disorders (obesity, type 2 diabetes mellitus, and metabolic dysfunction-associated steatotic liver disease). Our analysis reveals that all twelve hallmarks - i.e., genome instability, telomere attrition, epigenetic alterations, loss of proteostasis, impaired autophagy, dysregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, chronic inflammation, and dysbiosis - may serve as convergence points linking these seemingly disparate conditions. These findings support an integrated pathophysiological model wherein aging-related processes simultaneously promote neurodegeneration and metabolic dysfunction through shared molecular pathways. Understanding these mechanistic intersections offers promising opportunities for developing therapeutic interventions that could simultaneously target both neurodegenerative and metabolic diseases, potentially helping to close the healthspan-lifespan gap.
Keywords
INTRODUCTION
Despite major advancements in modern medicine that substantially extended human lifespan - particularly evident in the increase in life expectancy throughout the latter half of the 20th century - there has been a notable lack of corresponding compression of morbidity or proportional improvement of healthspan[1]. Conversely, the observed disparity between healthspan and lifespan - currently estimated to be approximately 9 years[2] - highlights the increasing burden posed by age-related conditions, including neurodegenerative diseases (NDDs) and metabolic disorders[3]. In general, NDDs are defined by the progressive and irreversible dysfunction and loss of selectively vulnerable neuronal populations - a process that underlies their distinct clinical features and disease trajectories[4]. Among the numerous clinically recognized NDDs, Alzheimer’s disease (AD) and Parkinson’s disease (PD) emerged as leading public health concerns[4]. These entities are unified by a fundamental pathophysiological hallmark - namely, the aberrant accumulation of misfolded proteins - which constitutes the basis for their classification as neurodegenerative proteinopathies[5]. Paralleling the rising incidence of NDDs, metabolic disorders reached epidemic proportions globally, manifesting as a constellation of interrelated conditions - including obesity, type 2 diabetes mellitus (T2DM), and metabolic-dysfunction associated steatotic liver disease (MASLD)[6].
A growing body of epidemiological and genetic evidence has shown a bidirectional relationship between NDDs and metabolic disorders[7-12], suggesting the presence of complex pathophysiological interconnections that extend beyond traditional organ-specific frameworks. This evolving perspective has challenged the conventional conceptualization of these conditions as discrete entities - instead situating them along a continuum of systemic dysfunction characterized by shared molecular and cellular mechanisms. Despite increasing recognition of these complex associations, the precise mechanisms mediating the cross-talk between neurodegenerative and metabolic disorders remain incompletely understood, representing a significant gap in current knowledge. Nonetheless, a pivotal observation is that aging has consistently emerged as the predominant risk factor for both NDDs[13] and metabolic diseases[14], modulating their onset and progression through the convergence of genetic, environmental, and systemic determinants. Building upon this context, we undertook a narrative review to systematically explore the interplay between neurodegeneration and metabolic disorders through the conceptual framework of the twelve established hallmarks of aging[15,16]: genome instability, telomere attrition, epigenetic alterations, loss of proteostasis, impaired macroautophagy, dysregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, chronic inflammation, and dysbiosis. This integrative approach may provide a comprehensive basis for elucidating mechanistic connections and identifying therapeutic strategies that may simultaneously target neurodegenerative and metabolic diseases, ultimately aiming to mitigate their public health impact and bridge the gap between lifespan and healthspan.
METHODS
We conducted a literature search across four major electronic databases (Medline/PubMed, Google Scholar, Scopus, and ScienceDirect) to identify pertinent studies examining the hallmarks of aging in NDDs and metabolic disorders, with a focus on elucidating the molecular interconnections between these conditions. The search centered on publications in English between January 1, 2013, and April 13, 2025, using the following keyword combinations and Boolean operators: (“neurodegeneration” OR “Alzheimer’s disease” OR “Parkinson’s disease” OR “neurodegenerative diseases”) AND (“metabolic disorders” OR “obesity” OR “type 2 diabetes” OR “MASLD” OR “fatty liver”) AND (“aging” OR “senescence” OR “hallmarks of aging” OR “geroscience”). The initial database query resulted in 553 articles, which were screened based on their titles and abstracts [Figure 1]. We prioritized studies that examined the relationship between the twelve established hallmarks of aging and either neurodegeneration or metabolic dysfunction. Case reports, editorials, and articles not published in English were excluded from the analysis. Following the screening phase, full-text articles were retrieved for analysis. In addition, the reference lists of selected studies were manually examined to identify further relevant publications. The data synthesis was conducted using a narrative approach, in which the evidence was organized to highlight the shared molecular pathways underlying both neurodegeneration and metabolic disorders, and was consistently analyzed within the framework of the aging hallmarks [Figure 2].
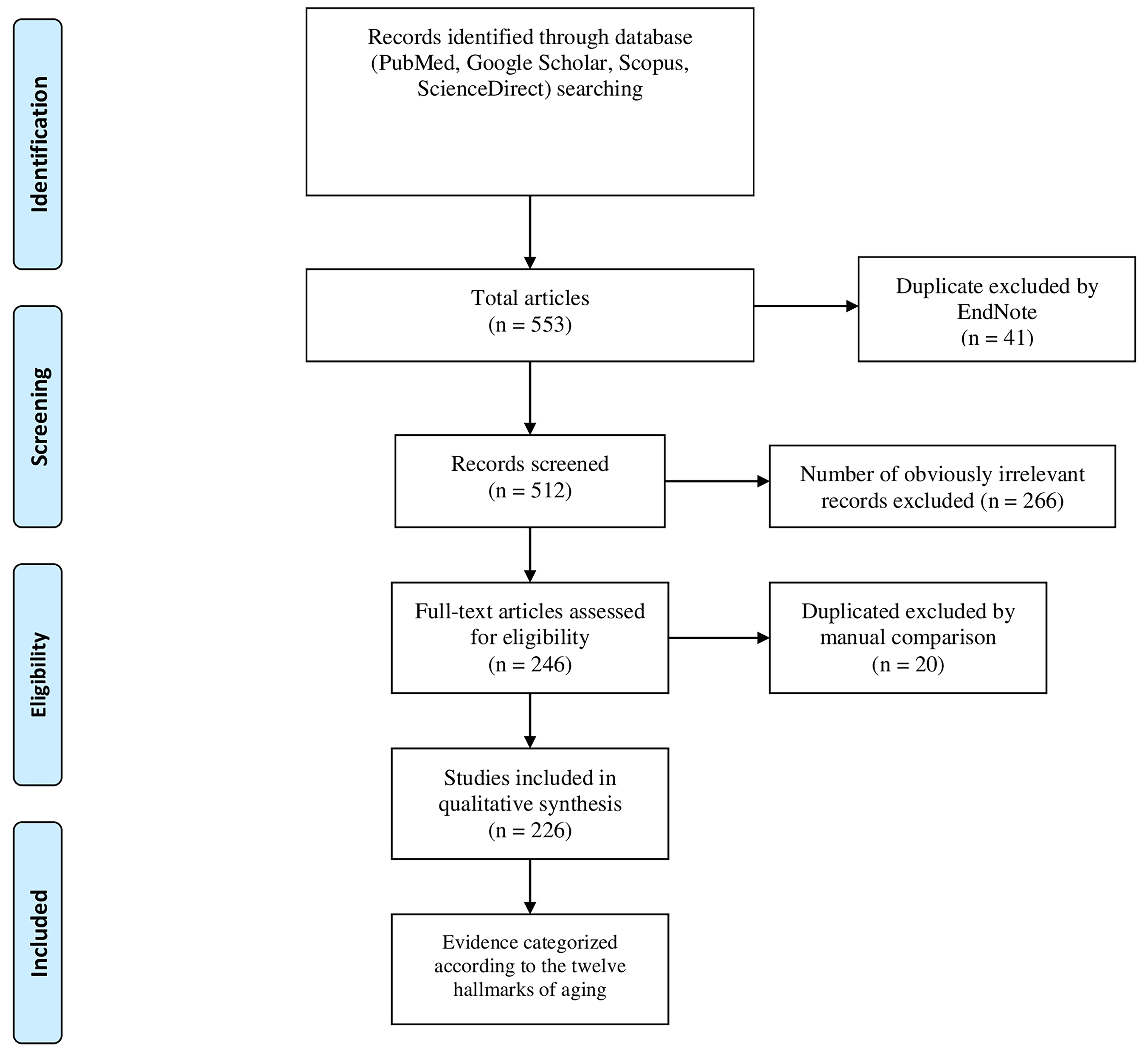
Figure 1. Flow diagram of studies’ screening and selection.
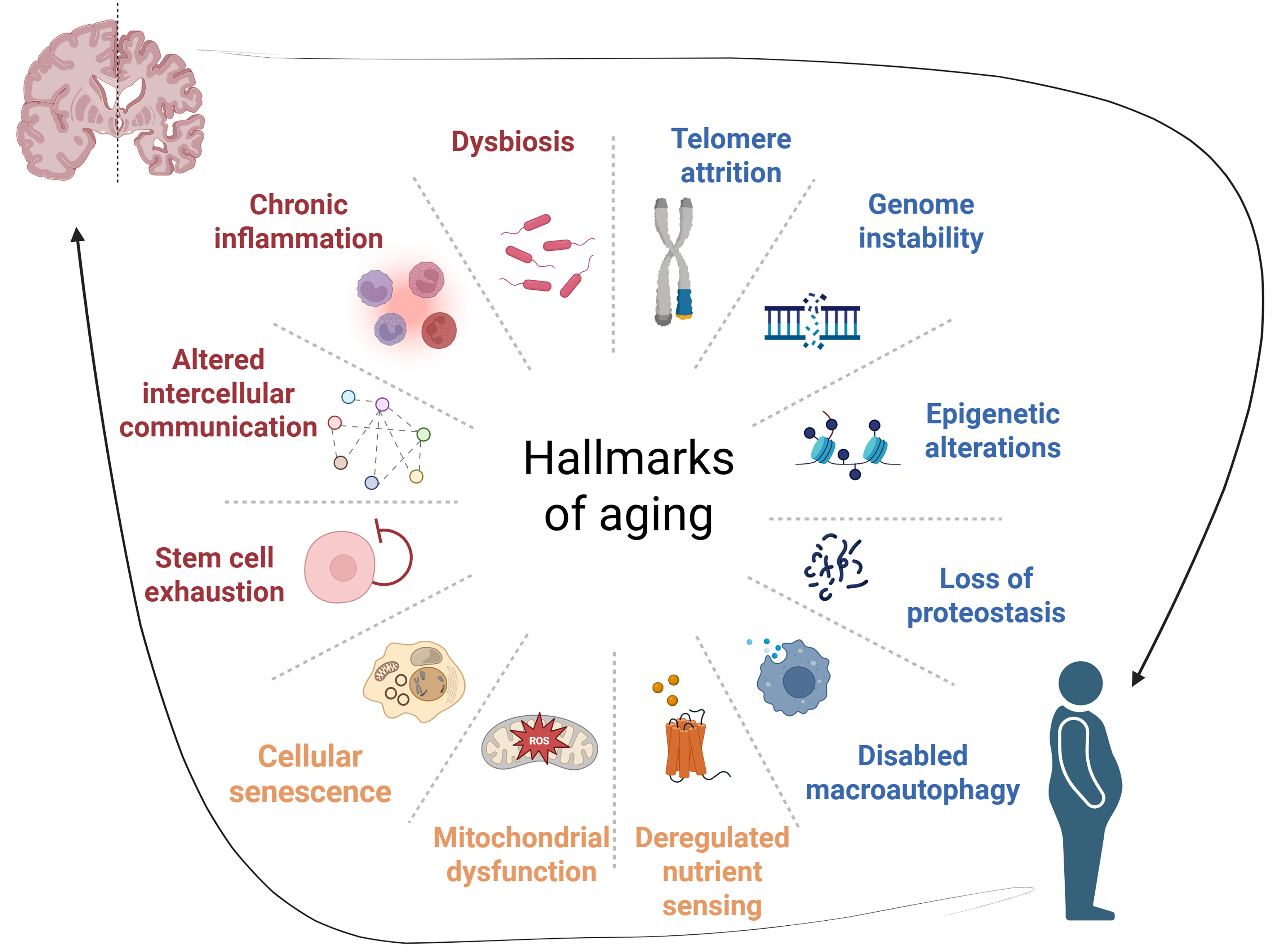
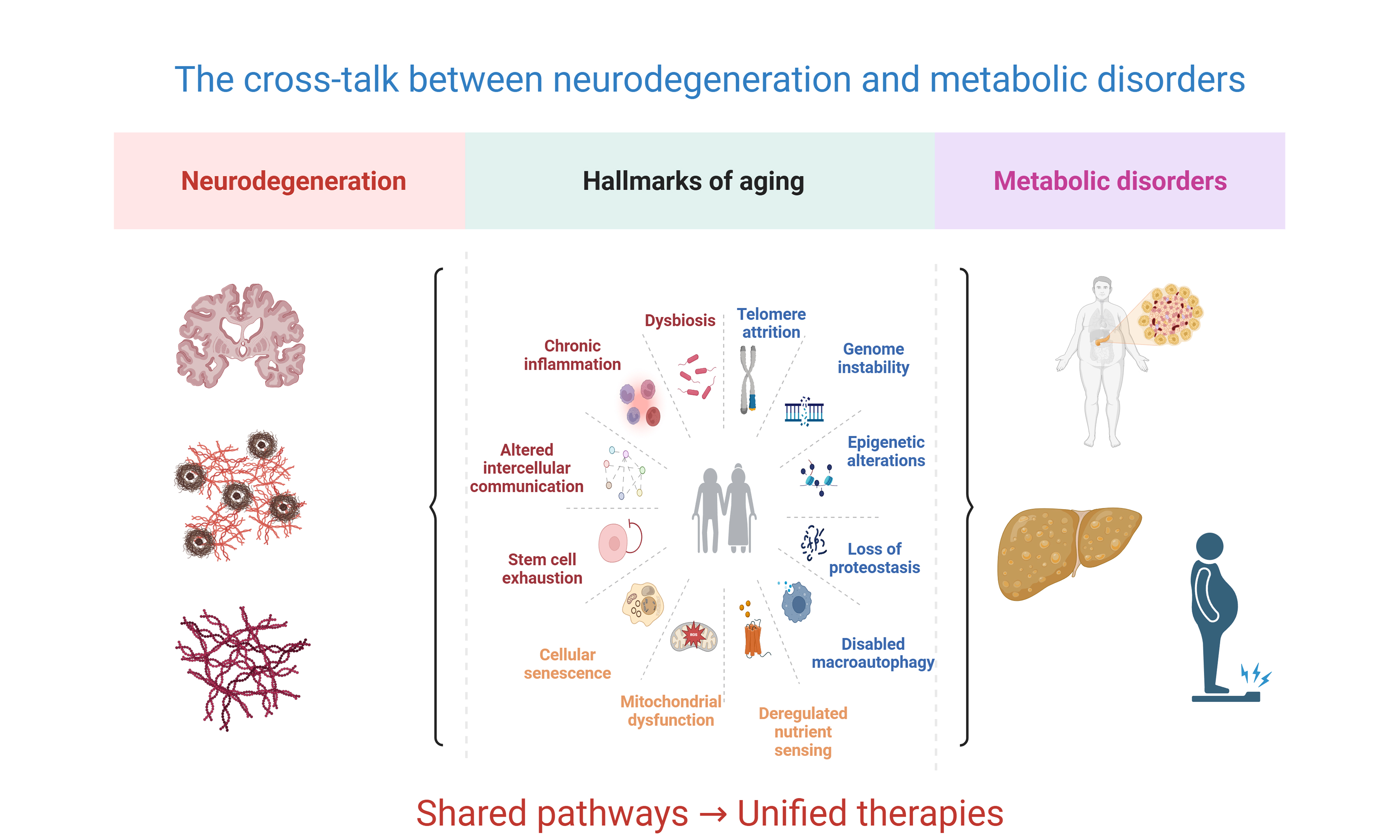
Figure 2. The twelve hallmarks of aging as convergence points linking neurodegeneration and metabolic disorders. The established hallmarks of aging are depicted as interconnected processes surrounding the central aging framework, with neurodegeneration represented by the brain illustration (top) and metabolic disorders represented by the human silhouette showing metabolic dysfunction (bottom). The hallmarks are color-coded by mechanistic category: blue indicates primary damage-accumulating hallmarks (genome instability, telomere attrition, epigenetic alterations, loss of proteostasis, disabled macroautophagy); orange represents antagonistic hallmarks that arise as compensatory responses but become detrimental over time (deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence); and red/purple denotes integrative hallmarks reflecting systemic consequences of aging (stem cell exhaustion, altered intercellular communication, chronic inflammation, dysbiosis). Dotted lines illustrate the interconnected nature of these processes, while bidirectional arrows emphasize the reciprocal relationship between neurodegeneration and metabolic dysfunction. This framework demonstrates how aging-related mechanisms simultaneously drive pathology in both domains through overlapping molecular pathways, supporting therapeutic strategies targeting shared aging processes [Created in BioRender. YILMAZ Y. (2025) https://BioRender.com/41ww4cq].
NEURODEGENERATION, METABOLIC DISORDERS, AND THE HALLMARKS OF AGING
Genome instability
Genomic stability becomes compromised in presence of an elevated propensity for genomic damage accumulation, which may originate from either impaired repair mechanisms or chronic exposure to exogenous and endogenous genotoxic sources[17]. Neural cells are known to exhibit exceptional vulnerability to DNA damage accumulation owing to their distinctive metabolic profiles and post-mitotic nature, which significantly constrains their capacity for cellular renewal[18]. The spectrum of DNA damage associated with neurodegeneration includes a wide range of lesions, such as nucleotide mismatches, complex adducts, single- and double-strand breaks, abasic sites, and frameshift alterations resulting from insertions or deletions[19]. In the pathogenesis of AD, oxidative damage to DNA appears particularly consequential. Accordingly, several studies conducted on post mortem neural tissues from patients with AD revealed elevated levels of oxidized DNA[20-22]. These observations led to the hypothesis that DNA oxidation constitutes a fundamental pathophysiological process in AD that significantly compromises neuronal functionality[23]. Similar observations have been reported for patients with PD, wherein investigations demonstrated that 8-oxoguanine - an established biomarker of oxidative DNA damage - exhibits particularly high concentrations in the substantia nigra of affected individuals[24]. Significantly, metabolic disorders increasingly emerged as a critical determinant of DNA damage and genomic instability[25]. In this regard, as observed in NDDs, a diverse spectrum of DNA lesions - including double-strand breaks, single-strand breaks, and oxidized nucleotides - has been documented in individuals with obesity [26]. Moreover, the burden of DNA damage in lymphocytes isolated from individuals with obesity has been shown to be two-fold higher compared to normal-weight counterparts, with robust statistical correlations between body mass index (BMI) and the magnitude of genomic lesions[27]. Ex vivo analyses also revealed that lymphocytes derived from individuals with obesity exhibit heightened vulnerability to exogenous genotoxic agents, as evidenced by their amplified susceptibility to mitomycin C-induced DNA damage compared to cells obtained from normal-weight individuals[28]. In addition to obesity, T2DM constitutes a major metabolic contributor to genotoxic damage[29]. Notably, individuals with T2DM display pathologically elevated levels of advanced glycation end-products (AGEs)[30], which can in turn form deleterious adducts with DNA and compromise genomic integrity[31]. Interestingly, one of the principal mediators of AGEs signaling is the receptor for AGEs (RAGE), which has been implicated as a key contributor to neurodegeneration through mechanisms that include disruption of the blood-brain barrier, induction of neuroinflammation, and remodeling of the extracellular matrix[32]. Collectively, these findings support a strong association between genomic instability, metabolic disturbances, and neural degeneration. When considered alongside the inherent susceptibility of neuronal populations to genotoxic damage, this relationship defines a critical pathophysiological axis that may underlie the epidemiological link between NDDs and metabolic dysregulation.
Telomere attrition
Telomeres are tandemly repeated nucleotide sequences (TTAGGG in humans) that cap the terminal regions of linear chromosomes, serving as critical guardians of genomic stability by preventing chromosomal degradation and aberrant end-to-end fusion[33]. The number of these repetitive elements varies considerably among different cell types and individual chromosomes. Due to the intrinsic limitations of DNA polymerase in replicating terminal DNA segments - a phenomenon known as the “end-replication problem” - telomeres undergo progressive attrition with successive cellular divisions, ultimately culminating in replicative senescence[34]. Consequently, telomere length (TL) functions as a molecular chronometer reflecting cellular senescence and represents a compelling biomarker for biological aging and age-associated diseases[35]. In NDDs, particularly AD, telomere dynamics have been extensively characterized. Significantly shorter leukocyte TL has been repeatedly reported in AD cohorts - establishing a robust correlation between cognitive deterioration, telomeric attrition, and progression from mild cognitive impairment (MCI) to clinically overt AD[36]. Notably, a systematic meta-analysis that included 13 studies - comprising 860 patients with AD and 2,022 control individuals - provided compelling evidence of telomere shortening in AD pathology, with this phenomenon being particularly pronounced in leukocytes[37]. In a 25-year longitudinal study, short leukocyte TL at baseline was also found to predict a higher incidence of AD among individuals who did not carry the apolipoprotein E (APOE) ε4 allele, even after accounting for competing risks such as vascular dementia and mortality[38]. Importantly, metabolic dysregulation - notably in obesity and T2DM - appears to accelerate telomere attrition through sustained low-grade inflammation and oxidative stress, potentially establishing a mechanistic link to AD pathogenesis. In obesity, multiple investigations demonstrated inverse correlations between TL and various anthropometric parameters including BMI[39,40], weight-adjusted-waist index[41], and total adiposity[42]. Notably, visceral adipose tissue accumulation appears particularly deleterious to telomere maintenance, resulting in accelerating cellular aging trajectories (“adipaging”)[43]. Accordingly, in a longitudinal assessment of 2,912 Chinese female individuals aged 40-70 years, telomere shortening was significantly pronounced when post-middle-age weight gain exceeded 15%[39]. Similarly, a comprehensive meta-analysis of 37 observational studies (n = 18,181 participants) demonstrated that patients with T2DM consistently exhibited shorter TL compared with normoglycemic counterparts[44]. Collectively, this evidence establishes telomere attrition as a critical mechanistic bridge linking metabolic dysfunction to accelerated neurodegeneration, wherein chronic inflammation and oxidative stress from obesity and diabetes accelerate cellular senescence pathways that compromise neuronal resilience.
Epigenetics alterations
Epigenetic mechanisms - including DNA methylation at cytosine residues, diverse histone post-translational modifications including acetylation and methylation, and the regulatory influence of non-coding RNA species - represent a sophisticated regulatory framework of reversible biochemical modifications that modulate gene expression patterns[45]. In NDDs, distinct epigenetic dysregulation patterns emerge with molecular specificity[46]. Notably, methylome-wide association studies consistently showed that aberrant DNA methylation, particularly promoter hypermethylation, is associated with AD and creates epigenetic barriers that prevent gene transcription[47]. This epigenetic imbalance may in turn affect transcriptional regulation of genes involved in tau phosphorylation[48] and neuroinflammatory cascades[49]. In addition, histone modifications - particularly aberrant acetylation and methylation patterns - may directly impact chromatin accessibility at neuroplasticity-associated genes, ultimately compromising cognitive resilience[50]. There is also evidence suggesting that methylation and other post-translational modifications may affect the formation of intracellular neurofibrillary tangles made of microtubule-associated protein tau and extracellular amyloid-β plaques[51]. Metabolic disorders similarly manifest distinctive epigenetic signatures with direct pathophysiological relevance[52,53]. Specifically, epigenetic modifications have been shown to regulate inflammatory gene expression in adipose tissue during obesity, with an increased expression of DNA methyltransferases promoting pro-inflammatory responses in both adipocytes and adipose tissue macrophages[52]. Similarly, T2DM exhibits characteristic epigenetic reprogramming across peripheral tissues, with aberrant DNA methylation particularly evident at genes regulating mitochondrial function and glucose homeostasis[53]. These methylation patterns demonstrate substantial overlap with age-associated epigenetic signatures, suggesting accelerated epigenetic aging in metabolic disorders[54]. The molecular convergence of neurodegeneration and metabolic dysregulation may therefore occur through shared epigenetic pathways. In this scenario, NOD-like receptor protein 3 (NLRP3) inflammasome regulation represents one critical nexus, with its epigenetic control affected by both metabolic[55] and neurodegenerative triggers[56]. Accordingly, the inflammasome activation cascade - regulated through specific DNA methylation patterns and histone modifications - perpetuates inflammatory signaling that simultaneously contributes to insulin resistance[57] and neuronal dysfunction[58]. Additionally, mitochondrial dysfunction emerges as another connecting mechanism, with epigenetic dysregulation of mitochondrial genes occurring in both pathological states through aberrant DNA methylation and altered histone acetylation patterns[59,60]. The shared epigenetic landscape between these conditions further manifests through dysregulated non-coding RNAs, particularly microRNAs targeting metabolic and neuronal health. Specific microRNAs - including miR-34a and miR-125b - demonstrate altered expression in both neurodegenerative[61] and metabolic[62] contexts, potentially serving as molecular mediators connecting these pathological domains. These findings underscore the potential therapeutic value of interventions targeting epigenetic mechanisms to simultaneously address both metabolic dysregulation and neurodegeneration through precise molecular reprogramming.
Loss of proteostasis
Proteostasis comprises the integrated cellular processes that maintain protein homeostasis, including protein synthesis, folding, trafficking, and degradation through an intricate network comprising molecular chaperones and the ubiquitin-proteasome system (UPS)[63]. Age-associated deterioration of this proteostatic network represents a central pathophysiological mechanism underlying neurodegenerative proteinopathies and establishes a critical molecular bridge to metabolic dysfunction[64]. Multiple proteostatic impairments contribute mechanistically to neurodegeneration, including UPS dysfunction and endoplasmic reticulum stress triggering maladaptive unfolded protein responses[65]. Notably, UPS dysfunction represents a particularly critical nexus in the pathogenesis of neurodegenerative proteinopathies, as the progressive decline in proteasomal efficiency observed in aging neurons leads to accumulation of ubiquitinated proteins and formation of intracellular inclusions[66]. Metabolic perturbations similarly influence proteostatic machinery function[67], establishing a mechanistic bridge to neurodegeneration through multiple converging pathways. Chronic hyperglycemia[68] and insulin resistance[69] may impair proteasomal activity via glycation of proteasomal subunits, oxidative damage to proteolytic machinery, and dysregulation of molecular chaperones, resulting in proteostatic compromise that accelerates protein misfolding and aggregation within metabolically stressed neural tissues[70]. Furthermore, the inhibition of insulin growth factor (IGF)-1 signaling has been shown to promote proteostasis in mammalian cells by inducing the aggregation and deposition of misfolded proteins into aggresomes[71]. In general, the molecular mechanisms connecting metabolic dysfunction and proteostasis failure in neurodegeneration include oxidative stress-mediated generation of structurally altered proteins resistant to normal degradation pathways[72], mitochondrial dysfunction that compromises cellular energetics required for optimal chaperone function and protein clearance mechanisms[73], and chronic inflammation that activates stress response pathways, particularly those involving mitogen-activated protein kinase (MAPK) signaling[74]. These inflammatory pathways modulate proteostasis network components, with prolonged signaling ultimately undermining effective protein quality control[75]. Therapeutic strategies targeting proteostasis are promising for addressing the metabolic-neurodegenerative intersection, as experimental evidence demonstrates that natural compounds such as carnosol can restore proteostasis by simultaneously modulating the IGF-1 pathway, regulating MAPK signaling, and activating molecular chaperones[76], thereby mitigating pathological processes common to both metabolic and NDDs. Collectively, these findings support the therapeutic potential of proteostasis-enhancing interventions to concurrently ameliorate metabolic disturbances and neurodegeneration by correcting the underlying proteome dysfunction that links these pathological processes.
Impaired autophagy
Autophagy represents a fundamental cellular quality control mechanism characterized by the sequestration of cytosolic constituents within double-membrane vesicles (autophagosomes) that subsequently fuse with lysosomes for degradation[77]. This process involves a highly coordinated molecular machinery comprising over 30 autophagy-related proteins that mediate vesicle nucleation, membrane elongation, cargo selection, and autophagosome-lysosome fusion[78]. Autophagy exhibits remarkable sensitivity to nutrient availability and energetic status, serving as a critical interface between cellular metabolism and proteostasis[79]. In NDDs, substantial evidence indicates progressive autophagy dysfunction as a central pathogenic mechanism[80,81]. In AD, amyloid beta has been shown to disrupt autophagy by blocking the fusion of autophagosomes with lysosomes and impairing lysosomal function, leading to the accumulation of autophagosomes in brain tissue from AD patients, animal models, and cell cultures[82]. Notably, a complex bidirectional relationship between autophagic-endolysosomal networks and tau protein accumulation has been reported in tauopathies, where dysfunction in cellular clearance mechanisms both contributes to and results from tau pathology[83]. In addition, autophagy dysfunction in NDDs disrupts fundamental cellular metabolic homeostasis by impairing nutrient recycling mechanisms and organellar quality control, creating a pathological feedback loop wherein metabolic stress and protein aggregation perpetuate autophagic insufficiency[84]. Metabolic disorders similarly demonstrate profound autophagy dysregulation that may disrupt homeostasis in various cell types[85,86] - including neurons and glial cells. Notably, obesity establishes a complex paradoxical relationship with autophagy, wherein the lipotoxic, proteotoxic, and oxidative stresses inherent to this condition simultaneously necessitate autophagy for cellular protection while disrupting autophagic flux through tissue-specific mechanisms, including defective autophagosomal-lysosomal fusion and compromised lysosomal acidification[87]. In addition, AGEs resulting from diabetic hyperglycemia may critically impair autophagy through specific molecular mechanisms involving mammalian target of rapamycin (mTOR) hyperactivation[88]. Notably, autophagy impairment - along with insulin signaling dysfunction - has increasingly emerged as a common pathophysiological mechanism linking obesity and T2DM to AD, PD, and other NDDs[89]. Furthermore, lipophagy - the selective autophagic degradation of lipid droplets through lysosomal pathways including macroautophagy, microautophagy, and chaperone-mediated autophagy - serves as a critical mechanism linking lipid homeostasis and energy metabolism essential for proper central nervous system function[90]. Accordingly, dysregulation of lipophagic machinery contributes to the pathogenesis of diverse NDDs by disrupting neuronal lipid balance and compromising cellular energetic capacity[91]. Further substantiating the pivotal role of autophagy in linking metabolic dysfunction with neurodegeneration, pharmacological autophagy activators exhibit therapeutic efficacy across both metabolic disorders[92] and NDDs[93].
Dysregulated nutrient sensing
Nutrient sensing pathways constitute sophisticated molecular networks that detect and respond to fluctuations in metabolic substrates, coordinating cellular processes with nutrient availability and energetic demands[94]. These evolutionarily conserved mechanisms - including the insulin/IGF-1 signaling cascade, mTOR, AMP-activated protein kinase (AMPK), and sirtuins[95] - integrate environmental cues with cellular metabolism, and their age-related decline represents a fundamental hallmark of aging[96] that contributes substantially to both neurodegenerative and metabolic diseases. Brain insulin resistance - characterized by increased insulin receptor substrate (IRS) phosphorylation markers in the hippocampal formation - represents an early and diabetes-independent feature of AD, strongly correlating with the accumulation of amyloid-beta oligomers and cognitive decline[97]. In a rat model of sporadic AD induced by intracerebroventricular streptozotocin, neurodegeneration and cognitive deficits have been closely associated with inhibition of mTOR signaling networks downstream of insulin/IGF-1 pathways, linking metabolic dysfunction to impaired brain energy metabolism, neuronal plasticity, and white matter integrity[98]. In AD, reduced sirtuin levels [particularly sirtuin 1 (SIRT1)/sirtuin 3 (SIRT3)] and diminished AMPK activity contribute to synaptic dysfunction, amyloid-beta accumulation, tau hyperphosphorylation, and neuroinflammation, while exacerbating mitochondrial impairment and tau pathology[99]. Metabolic disorders exhibit parallel disruptions in nutrient sensing machinery. Obesity is a well-known driver of insulin resistance, which is central to the development of T2DM and related metabolic disorders, through complex mechanisms involving adipocyte dysfunction and altered adipokine signaling[100]. Similarly, MASLD is primarily driven by insulin resistance, with current research focusing on the therapeutic potential of hypoglycemic agents such as glucagon-like peptide-1 (GLP-1) receptor agonists and peroxisome proliferator-activated receptor (PPAR)-γ agonists to address its underlying metabolic abnormalities[101]. Emerging evidence also indicates that obesity triggers chronic mTOR overactivation across multiple tissues[102]. In addition, obese individuals with insulin resistance exhibit reduced SIRT1/sirtuin 2 (SIRT2) gene expression and AMPK activity, which correlate with elevated BMI, waist circumference, and insulin resistance markers[103]. The molecular intersection between neurodegenerative and metabolic disorders through dysregulated nutrient sensing involves several convergent mechanisms. First, peripheral hyperinsulinemia accelerates brain insulin resistance by promoting receptor desensitization and internalization while restricting insulin transport across the blood-brain barrier[104]. Second, chronic mTOR overactivation triggers gliosis and neuroinflammation while impairing autophagy-dependent clearance of amyloid-beta/tau pathology[105]. Third, defective AMPK activation may compromise both synaptic connectivity and neuroprotection[106]. In this scenario, therapeutic strategies targeting dysregulated nutrient sensing pathways offer dual therapeutic potential for metabolic and neurodegenerative disorders. For example, caloric restriction (CR) can restore insulin sensitivity[107] and normalize mTOR activity[108], while enhancing AMPK signaling[109]. Notably, preclinical evidence has shown that CR may ameliorate both metabolic dysfunction[110] and neurodegenerative pathology[111], positioning nutrient sensing modulation as a promising unified therapeutic strategy.
Mitochondrial dysfunction
Mitochondria function as essential cellular organelles orchestrating bioenergetic homeostasis, calcium buffering, redox signaling, and apoptotic regulation through their sophisticated double-membrane architecture and autonomous circular genome[112]. Age-associated decline in mitochondrial function[113] represents a fundamental hallmark of aging that contributes substantially to both neurodegenerative processes and metabolic perturbations. Recently, Ashleigh et al. proposed that mitochondrial dysfunction - rather than amyloid beta pathology as suggested by the conventional amyloid cascade hypothesis - may be the primary driver of AD pathogenesis, as mitochondrial function is central to maintaining synaptic integrity[114]. Accordingly, patients with AD have been shown to exhibit structural and functional mitochondrial alterations - including disrupted distribution and motility, abnormal morphology, dysregulated enzymatic activity, and compromised biogenesis - suggesting mitochondrial dysfunction as a critical contributor to disease progression[115]. Metabolic disorders similarly exhibit profound mitochondrial dysfunction that establishes molecular connections to neurodegeneration. Accordingly, obesity and T2DM have been associated with impaired mitochondrial oxidative capacity through mechanisms involving altered substrate availability, inflammatory mediator production, and lipotoxicity[116]. Adipose tissue from individuals with obesity demonstrates reduced mitochondrial content, compromised respiratory chain complex activity, and mitochondrial DNA damage, triggering compensatory adaptations that ultimately prove maladaptive, including incomplete fatty acid oxidation and elevated reactive oxygen species production[117]. Furthermore, hyperglycemia has been shown to induce mitochondrial fragmentation through dysregulated fusion-fission dynamics[118]. The molecular intersection between neurodegenerative and metabolic disorders through mitochondrial dysfunction involves several convergent mechanisms. In this regard, excessive mitochondrial fission has been reported in both insulin-resistant tissues[119] and neurodegenerative models[120], leading to mitochondrial fragmentation and subsequent cellular dysfunction. Furthermore, disruption of calcium homeostasis further impairs metabolic-bioenergetic coupling and promotes activation of the mitochondrial permeability transition pore, exacerbating cellular dysfunction in both pathological states[121,122]. Mitochondria also provide a crucial link between metabolic and neurodegenerative disorders through their central role in inflammasome regulation and inflammatory signaling[123]. Following disruption, mitochondria release damage-associated molecular patterns, including mitochondrial DNA and cardiolipin, which activate NLRP3 inflammasomes and initiate inflammatory cascades implicated in both insulin resistance[124] and neurodegeneration[125]. This process establishes a self-perpetuating cycle, whereby initial mitochondrial dysfunction triggers inflammatory signaling that further impairs mitochondrial function through cytokine-mediated inhibition of the respiratory chain and increased production of reactive oxygen species[126]. Within this framework, therapeutic strategies that target mitochondrial dysfunction are showing considerable promise for both pathologies. For example, mitochondrial-targeted antioxidants have been shown to reduce oxidative damage and preserve respiratory chain function in preclinical models of metabolic dysfunction[127] and neurodegeneration[128]. Importantly, compounds that enhance mitochondrial biogenesis demonstrated the ability to improve metabolic parameters and confer neuroprotection[129,130], underscoring the potential of mitochondria-focused therapies to address these interconnected conditions simultaneously.
Cellular senescence
Cellular senescence represents a state of permanent cell cycle arrest characterized by distinctive morphological alterations, chromatin reorganization, and metabolic reprogramming[131]. Senescent cells exhibit resistance to apoptotic signals while maintaining metabolic activity, accumulating progressively with age across diverse tissues[132]. The hallmark characteristic of cellular senescence is the development of a senescence-associated secretory phenotype (SASP) - a complex secretome containing pro-inflammatory cytokines, chemokines, growth factors, and matrix-remodeling enzymes that exerts deleterious effects on the surrounding tissue microenvironment through paracrine signaling mechanisms[133]. Senescence induction occurs through multiple molecular mechanisms - including but not limited to telomere attrition, oncogenic activation, oxidative damage, and genotoxic stress - converging on activation of p16INK4a/Rb and p53/p21 tumor suppressor pathways[134]. In AD, senescent astrocytes, microglia, and oligodendrocytes accumulate - especially in proximity to amyloid plaques - and secrete pro-inflammatory SASP factors, ultimately promoting neuroinflammation, synaptic dysfunction, and neuronal loss[135]. Notably, both hyperphosphorylated tau and amyloid-beta oligomers have been shown to induce cellular senescence in AD[136]. In addition, overexpression of mutant α-synuclein in the substantia nigra of a murine PD model can rapidly induce cellular senescence, as evidenced by elevated SASP factors, increased expression of senescence markers, mitochondrial dysfunction, and iron dysregulation - all preceding dopaminergic neuron loss and motor deficits[137]. Significantly, the selective elimination of senescent glial cells using senolytic compounds ameliorates neurodegenerative pathology in preclinical models, establishing a causal relationship between cellular senescence and neurodegeneration[138]. Metabolic disorders similarly demonstrate accelerated cellular senescence with distinctive tissue distribution[139]. Adipose tissue from individuals with obesity exhibits significant senescent cell accumulation, particularly within visceral depots, characterized by elevated senescence markers and SASP production[140]. Importantly, senescent adipocytes and pre-adipocytes have been shown to contribute to insulin resistance through paracrine effects of their inflammatory secretome, compromising insulin signaling in neighboring cells[141]. Furthermore, hyperglycemia directly induces cellular senescence through mechanisms involving oxidative stress, AGEs formation, and mitochondrial dysfunction[142]. The resulting SASP further exacerbates metabolic dysfunction through promoting macrophage recruitment, inhibiting adipogenesis, and inducing systemic insulin resistance[143]. The interplay between NDDs and metabolic disorders through cellular senescence involves several convergent mechanisms. In this regard, chronic systemic hyperinsulinemia can lead to neuronal insulin resistance, which impairs glycolysis and disrupts protein degradation pathways, ultimately resulting in aberrant cell cycle re-entry and a senescent-like state in neurons[143]. Similarly, obesity can promote neuronal senescence by driving chronic low-grade inflammation that triggers a SASP in neurons[144]. Moreover, increased oxidative stress associated with obesity contributes to neuronal mitochondrial dysfunction, DNA damage, and the activation of cell cycle arrest pathways[144]. Cellular senescence further connects these conditions through metabolic reprogramming that affects energy substrate availability for neuronal populations[145].
Therapeutic strategies targeting cellular senescence represent promising approaches for addressing both neurodegenerative and metabolic diseases. Senolytics - which can be defined as compounds that selectively eliminate senescent cells - demonstrated efficacy in ameliorating metabolic dysfunction[139,141] and neurodegeneration[138] in preclinical models. Complementarily, SASP modulators offer an alternative therapeutic approach by suppressing the pro-inflammatory secretome while preserving senescent cells[146], potentially reducing inflammation-mediated damage across both conditions. These findings position cellular senescence as a compelling therapeutic target for treating these mechanistically interconnected disorders.
Stem cell exhaustion
Adult stem cells preserve tissue homeostasis by virtue of their intrinsic self-renewal capabilities and multipotent differentiation potential, thereby orchestrating repair mechanisms and regenerative processes throughout an organism’s lifespan[147]. The phenomenon of stem cell exhaustion is characterized by a progressive decline in stem cell functional competence and regenerative efficacy associated with chronological aging[148]. This age-related senescence phenomenon precipitates the gradual deterioration of tissue architectural integrity, ultimately impairing organ-level functionality[149]. In NDDs, mitochondrial dysfunction has been shown to impair neural stem cell (NSC) activation and proliferation, leading to reduced neurogenesis and associated learning and memory deficits that can be rescued through hypoxia-mediated oxidative stress reduction[150]. In the AD mouse model, adult NSCs showed significantly reduced proliferation capacity and failed to undergo terminal differentiation into mature neurons as early as 1.5 months of age - a presymptomatic stage preceding amyloid-beta accumulation and neurodegeneration - due to the pathological accumulation of intracellular amyloid-beta oligomers within the endoplasmic reticulum, which triggered tau-mediated microtubule hyperstabilization[151]. Metabolic disorders similarly exhibit accelerated stem cell exhaustion with distinctive tissue manifestations. Obesity and insulin resistance promote adipose-derived stem cell senescence and compromise their differentiation capacity, particularly affecting brown adipogenesis and contributing to thermogenic defects[152]. Importantly, hyperglycemia has been shown to suppress NSCs differentiation by inducing oxidative stress and subsequent endoplasmic reticulum stress[153]. A separate study found that diabetes alters gene expression related to insulin production and neural function in adult NSCs derived from both the hippocampus and the olfactory bulb, with similar gene-expression changes observed in both cell types[154]. The authors also proposed that olfactory bulb NSCs may serve as a potential tool for monitoring diabetes-induced neurodegeneration and for screening drugs targeting central nervous system disorders[154].
Compromised neurotrophic factor production further connects metabolism and neurodegeneration through stem cell dysfunction. Accordingly, metabolic perturbations can reduce systemic levels of brain-derived neurotrophic factor[155,156] - a molecule that promotes NSCs proliferation and drives their differentiation into neurons and glia[157]. Therapeutic interventions aimed at rejuvenating or replacing senescent stem cells demonstrated potential to restore tissue homeostasis and improve outcomes in preclinical models of both NDDs and metabolic disorders. Inducing endogenous neurogenesis, for instance, by modulating the NSC niche with growth factors or small molecules, can reactivate quiescent NSCs and promote the generation of new neurons[158]. This approach has shown efficacy in ameliorating cognitive deficits in models of neurodegeneration, suggesting that restoring the regenerative capacity of the brain may counteract disease progression[159]. In addition, exogenous stem cell transplantation, using sources such as mesenchymal stem cells or induced pluripotent stem cells, can offer a multifaceted mechanism of action[160]. Accordingly, transplanted cells not only replace lost or dysfunctional cells but also secrete trophic factors, modulate immune responses, and stimulate endogenous repair pathways[160]. These effects may be beneficial in both neurodegenerative and metabolic contexts, as they can support neuronal survival, enhance insulin sensitivity, and reduce chronic inflammation.
Altered intercellular communication
Intercellular communication involves diverse molecular mechanisms - including direct cell-cell contact, soluble factors, and extracellular vesicles (EVs) - that enable cells to exchange signals, coordinate activities, and sustain tissue homeostasis[161]. Age-related disruptions in these networks constitute a hallmark of aging, driving pathophysiology in both neurodegenerative and metabolic disorders by impairing tissue function and amplifying pathological signaling cascades[162]. During neurodegeneration, EVs contribute to disease progression by facilitating the prion-like spread of amyloid-beta, tau, and alpha-synuclein between neurons and glial cells[163]. Notably, the cargo of EVs in patients with NDDs is distinctly altered, frequently enriched with pathological proteins, disease-associated microRNAs, and pro-inflammatory mediators that can intensify neuroinflammation and disrupt synaptic function[163]. Similarly, EVs isolated in individuals with morbid obesity and T2DM carry distinct microRNA and protein profiles that may disrupt insulin signaling in the liver, muscle, and brain, impairing gluconeogenesis, glucose uptake, and energy homeostasis[164]. Interestingly, EVs from adipocytes, macrophages, adipose-derived stem cells, and endothelial cells have the potential to contribute to the development or amelioration of obesity, type 2 diabetes, and MASLD. For example, EVs from healthy adipocytes can enhance pancreatic β-cell function and insulin release, while those from obese or inflamed adipose tissue can impair insulin signaling and promote inflammation[165]. As with other hallmarks of aging, neurodegenerative and metabolic disease may intersect through shared altered communication pathways. The gut-brain axis exemplifies this link, with dysbiosis-driven microbial metabolites and EVs compromising blood-brain barrier integrity and promoting neuroinflammation[166]. Similarly, peripherally derived EVs from metabolically impaired tissues can infiltrate the central nervous system and disrupt neuronal proteostasis and metabolism[167]. Consequently, the dynamic and context-dependent cargo of EVs - shaped by the physiological or pathological state of adipose and neural tissues - not only mediates local and systemic signaling but also bridges metabolic dysfunction and neuroinflammation. As evidence accumulates that EVs can propagate pathological signals, they emerge as both biomarkers and active participants in disease progression. Harnessing the diagnostic and therapeutic potential of EVs - by refining their isolation, targeting, and engineering - may offer a transformative strategy for early detection, monitoring, and intervention across these interconnected disorders[168].
Chronic inflammation
Chronic inflammation represents a maladaptive state characterized by persistent production of pro-inflammatory mediators, continuous immune cell activation, and self-perpetuating molecular cascades involving intricate networks of cytokines, chemokines, Toll-like receptors, pattern recognition receptors, and inflammasome complexes[169]. Within the NDDs spectrum, chronic neuroinflammation has increasingly emerged as a critical pathophysiological axis driving protein aggregation and progressive neuronal loss[170]. In this scenario, the released inflammatory mediators can directly compromise neuronal viability through different mechanisms - including excitotoxicity, oxidative stress induction, and disruption of synaptic homeostasis[171]. Notably, AD pathology is characterized by pronounced glial activation, with reactive microglia and astrocytes clustering around amyloid plaques and neurofibrillary tangles and releasing elevated pro-inflammatory molecules that are increasingly being exploited as peripheral disease markers[172]. Mechanistically, misfolded protein aggregates activate microglial pattern recognition receptors and inflammasome complexes, initiating interleukin-1β production that amplifies glial activation and accelerates pathological protein deposition[173]. This establishes a detrimental feedforward loop wherein initial proteinopathy triggers neuroinflammation, which reciprocally enhances amyloid accumulation and tau hyperphosphorylation through inflammatory signaling cascades[174]. Metabolic disorders manifest equally significant inflammatory dysregulation that fundamentally disrupts systemic homeostasis. Obesity induces chronic low-grade inflammation characterized by macrophage infiltration into expanding adipose tissue, creating crown-like structures around hypertrophic adipocytes while secreting pro-inflammatory cytokines and adipokines[175]. These inflammatory mediators, in turn, can directly impair insulin signaling through serine phosphorylation of IRSs, establishing peripheral insulin resistance[176]. Multiple metabolic stressors - including adipocyte hypertrophy-induced hypoxia[177], lipotoxicity from ectopic lipid accumulation[178], and endoplasmic reticulum stress[179] - have the potential to converge to activate inflammatory pathways. The resulting chronic inflammatory milieu can establish a self-sustaining cycle of metabolic dysfunction that impairs adipocyte endocrine activity, disrupts hepatic glucose regulation, and compromises hypothalamic control of energy balance[180]. The molecular convergence between neurodegenerative and metabolic pathologies through inflammatory mechanisms involves multiple shared pathways. In presence of neuroinflammation, systemic metabolic inflammation can compromise blood-brain barrier integrity via cytokine-mediated disruption of tight junction proteins, facilitating peripheral immune cell infiltration[181]. Platelet-activating factor and its precursor plasmalogen represent shared inflammatory mediators, with elevated platelet-activating factor levels promoting inflammatory cascades while plasmalogen depletion compromises anti-inflammatory signaling[182,183]. In addition, the NLRP3 inflammasome has been suggested to serve as a critical convergence point, demonstrating activation in adipose tissue[184], hepatocytes[185], and microglial cells[186] - potentially linking peripheral metabolic inflammation with neuroinflammatory processes. Cellular senescence can further interconnect these conditions through the SASP - which establishes chronic inflammatory microenvironments in both peripheral and central tissues[146]. In this scenario, therapeutic interventions targeting chronic inflammation are increasingly emerging as promising strategies for addressing both metabolic dysfunction[187] and neurodegeneration[188] through restoration of immune homeostasis.
Dysbiosis
Dysbiosis - characterized by pathological alterations in gut microbiota composition and function - has emerged as a critical hallmark of aging[189] that may bridge neurodegenerative and metabolic disorders. The gastrointestinal microbiota - comprising trillions of microorganisms functioning as an integrated ecosystem - has been shown to profoundly influence host physiology through metabolite production, immune modulation, and maintenance of intestinal barrier integrity[190]. Age-related disruptions in this delicate microbial equilibrium manifest as reduced diversity, depletion of beneficial commensals, and expansion of potentially pathogenic species[191,192], establishing dysbiosis as a fundamental driver of both neurological and metabolic dysfunction. In NDDs, distinctive dysbiotic signatures emerge with significant functional consequences[193]. For example, patients with AD exhibit marked reductions in short-chain fatty acid (SCFA)-producing bacteria, particularly Bifidobacterium and Akkermansia species, concurrent with expansions of pro-inflammatory taxa[194]. These microbial shifts compromise intestinal permeability, facilitating bacterial endotoxin translocation that amplifies neuroinflammatory cascades[194]. Critically, gut-derived metabolites directly influence amyloid aggregation dynamics, while microbially-produced neurotransmitters modulate neuronal vulnerability[195]. In patients with PD, a consistent enrichment of the genera Lactobacillus, Akkermansia, and Bifidobacterium, alongside a marked depletion of SCFA-producing bacteria from the Lachnospiraceae family and the Faecalibacterium genus, has been observed[196]. Metabolic disorders similarly manifest profound dysbiotic alterations with mechanistic implications[197]. Notably, reproducible gut microbial patterns in obesity have been shown to include reduced microbial diversity and depletion of SCFA-producing bacteria (Alistipes spp., Odoribacter splanchnicus) and gut barrier promoters
DISCUSSION
While NDDs and metabolic disorders are traditionally conceptualized as distinct, organ-specific conditions, the convergence of evidence across epidemiology, genetics, and molecular biology currently points to a deeply intertwined pathophysiology[8-12,209,210], unified under the framework of the twelve hallmarks of aging. In this review, we discussed how the biological processes driving neurodegeneration and metabolic dysfunction might be not only parallel but also mutually reinforcing[11], with a plethora of shared molecular mechanisms. Accordingly, we showed that all twelve hallmarks of aging can serve as critical nodes of intersection between NDDs and metabolic disorders. Importantly, the relationship between neurodegeneration and metabolic dysfunction appears to be profoundly bidirectional. In this regard, insulin resistance not only accelerates amyloid and tau pathology in AD[211] but may be itself exacerbated by neuroinflammation, immunosenescence, inflamm-aging, and metainflammation [Figure 3][212,213]. Similarly, telomere attrition[214] and epigenetic alterations[215] actively participate in disease progression by modulating gene expression networks that govern both neuronal survival and metabolic homeostasis. In this scenario, interventions targeting fundamental age-related processes - including CR, senolytics, or mitochondrial-targeted antioxidants - can display pleiotropic benefits across both neurodegenerative and metabolic domains [Table 1]. For instance, CR not only improves insulin sensitivity[107] but also upregulates the autophagic flux[216] and attenuates neuroinflammation[217], supporting the notion of unified geroscience-based interventions. Similarly, metformin - a canonical metabolic drug - modulates nutrient-sensing pathways implicated in both metabolic and neurodegenerative disorders[218]. Nonetheless, the translation of these insights into clinical practice is fraught with complexity. The effects of interventions can indeed be highly context-dependent, varying across disease stages and individual genetic backgrounds. For example, while autophagy enhancement is generally neuroprotective, excessive activation may be deleterious in certain contexts[219], underscoring the need for precision medicine approaches. Moreover, the temporal dynamics of hallmark interactions remain poorly characterized, complicating the identification of optimal intervention windows and the prediction of long-term outcomes.
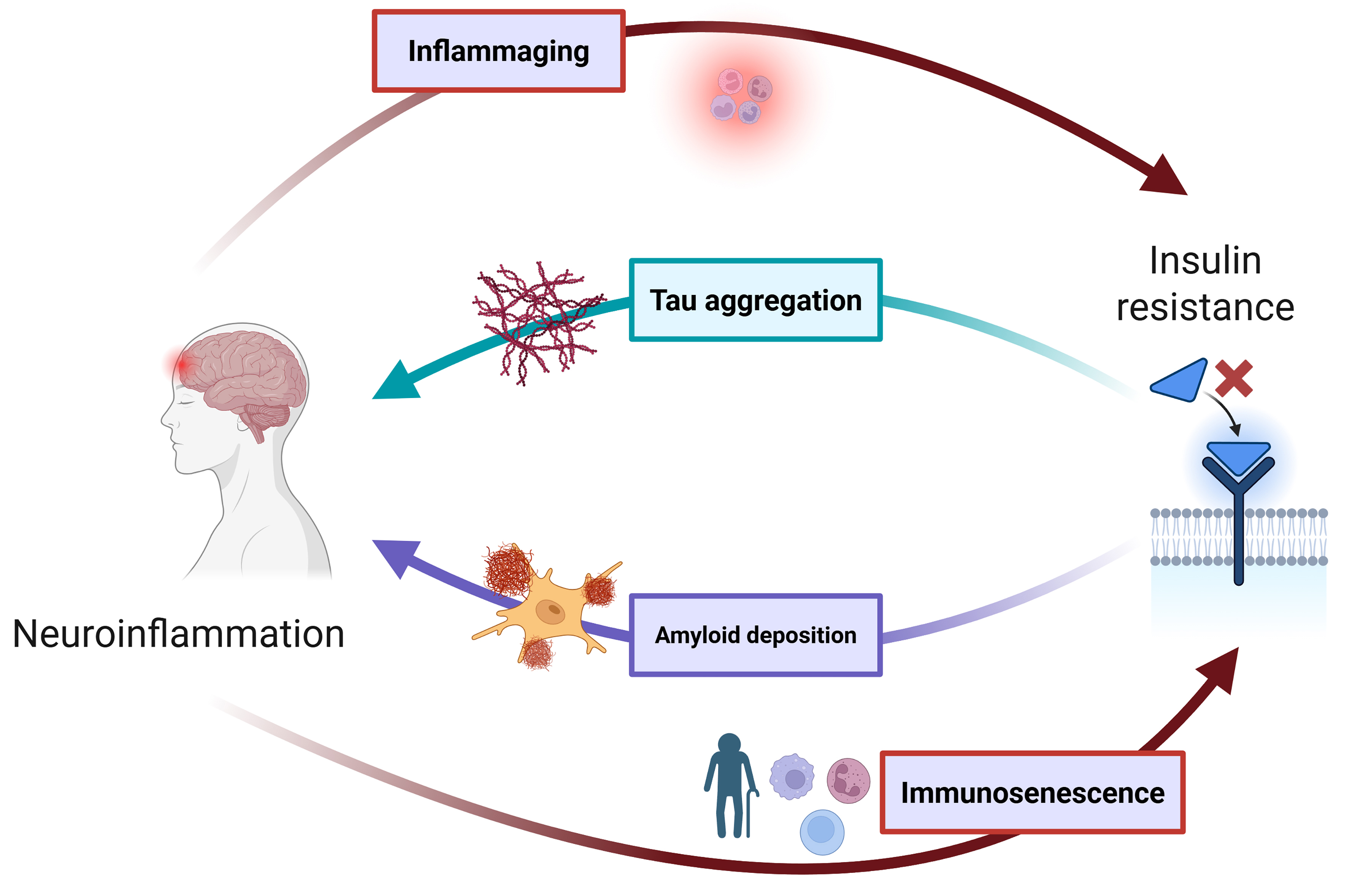
Figure 3. Bidirectional relationship between insulin resistance and neurodegeneration. Impaired insulin signaling amplifies amyloid-β deposition and tau aggregation, both of which trigger microglial-mediated neuroinflammation. The resulting neuroinflammatory environment - further intensified by age-associated immunosenescence and inflammaging - feeds back to aggravate neuronal and systemic insulin resistance, creating a self-sustaining cycle of metabolic and neurodegenerative damage[213] [Created in BioRender. YILMAZ Y. (2025) https://BioRender.com/er2vljj].
Potential interventions targeting the twelve hallmarks of aging in neurodegeneration and metabolic disorders
| Hallmark | Example interventions | Key outcomes in neurodegeneration | Key outcomes in metabolic disorders |
| Genome instability | DNA repair enhancers; antioxidants; RAGE antagonists | Less oxidative DNA damage; improved neuronal survival | Lower AGE-DNA adducts; improved genomic stability |
| Telomere attrition | Telomerase activators; lifestyle/anti-inflammatory measures | Slower cognitive decline; improved resilience | Slower “adipaging”; improved cellular longevity |
| Epigenetic alterations | DNMT modulators; HDAC inhibitors; microRNA therapeutics | Restored neuroplasticity genes; less Tau phosphorylation/inflammation | Normalized inflammatory genes; improved insulin sensitivity/mitochondrial function |
| Loss of proteostasis | Chaperone enhancers; proteasome activators; carnosol | Reduced aggregates; better amyloid β clearance; improved synapses | Restored protein QC; reduced ER stress; enhanced insulin signaling |
| Impaired autophagy | Autophagy activators; mTOR inhibitors; lipophagy enhancers | Better amyloid β/Tau clearance; restored synaptic homeostasis | Improved lipid handling; energy homeostasis; reduced lipotoxicity |
| Dysregulated nutrient sensing | Caloric restriction; metformin; AMPK/sirtuin modulators | Improved brain insulin sensitivity; less amyloid β; enhanced plasticity | Better insulin sensitivity; normalized mTOR; greater metabolic flexibility |
| Mitochondrial dysfunction | Mitochondria-targeted antioxidants; biogenesis enhancers; fusion-fission modulators | Reduced oxidative stress; improved bioenergetics/synapses | Restored oxidative capacity; better FA oxidation; reduced ROS |
| Cellular senescence | Senolytics; SASP modulators; p16/p21 pathway inhibitors | Reduced neuroinflammation; better cognition/synaptic connectivity | Improved insulin signaling; less adipose inflammation; better metabolism |
| Stem cell exhaustion | Growth factors; stem cell transplantation; niche rejuvenation | Enhanced neurogenesis; improved cognition/plasticity | Improved adipogenesis/regeneration; restored homeostasis |
| Altered intercellular communication | EV targeting; BBB stabilizers; gut-brain axis modulators | Less pathological protein spread; improved synaptic function | Better insulin signaling; reduced systemic inflammation |
| Chronic inflammation | NLRP3 inhibitors; anti-inflammatories; cytokine modulators | Lower microglial activation; improved BBB integrity | Reduced adipose inflammation; enhanced insulin sensitivity |
| Dysbiosis | Pre-/probiotics; FMT; SCFA supplementation | Better BBB function; less neuroinflammation; improved cognition | Improved gut barrier; lower endotoxemia; better insulin sensitivity |
Several critical gaps also persist in the literature. First, most studies examined individual hallmarks in isolation, potentially overlooking synergistic or antagonistic interactions that may be mechanistically relevant. Second, the temporal sequence and causality of hallmark interactions are not fully elucidated. Consequently, it remains unclear whether targeting one hallmark can induce compensatory changes in others, or whether combination therapies will be required for durable disease modification. Third, current biomarker panels are insufficiently sensitive or specific to capture the multidimensional nature of aging hallmarks, limiting their utility for patient stratification and treatment monitoring[220]. To address these challenges, future research should prioritize systems biology approaches that integrate multi-omics data (e.g., transcriptomics, proteomics, metabolomics, and epigenomics)[221] to map the hierarchical organization and temporal evolution of hallmark interactions. The development of composite biomarkers that reflect multiple hallmarks simultaneously will be essential for advancing personalized medicine. Furthermore, longitudinal studies employing comprehensive biomarker panels are required to disentangle causality and inform the design of rational, multi-targeted interventions.
The concept of hormetic interventions - defined as therapies that activate adaptive stress responses across multiple biological systems - holds particular promise in this context[222]. For example, intermittent fasting/CR[223], exercise[224], and NAD+ boosters[225] may simultaneously modulate mitochondrial function, autophagy, and inflammatory signaling, offering broad-spectrum benefits. In addition, the promising efficacy of senolytic therapies[226] and mitochondrial-targeted compounds[227] in preclinical models provides proof-of-concept for hallmark-directed interventions, but combination strategies that address multiple hallmarks are likely to yield superior outcomes. The convergence of neurodegenerative and metabolic diseases through shared aging hallmarks represents more than a theoretical construct and has profound implications for public health and clinical practice. As global populations age, the dual burden of NDDs and metabolic disorders threatens to overwhelm healthcare systems. In this scenario, targeting the fundamental processes that drive both conditions may make it possible to compress morbidity, extend healthspan, and reduce the societal and economic costs of age-related disease.
In summary, the reviewed evidence supports a paradigm in which neurodegeneration and metabolic dysfunction are manifestations of a common biological substrate - i.e., biological aging - operating through interconnected hallmarks that drive systemic decline. Recognizing and therapeutically targeting these shared mechanisms offers a rational path toward disease modification. In this regard, geroscience-guided strategies - including CR mimetics[228], senolytics[229], and mitochondrial-targeted antioxidants[230] - are prime examples of interventions with the potential to transform the prevention and treatment of multiple age-related disorders by addressing their fundamental biological drivers. Future efforts must focus on translating these mechanistic insights into clinically actionable strategies, leveraging advances in systems biology, biomarker discovery, and precision medicine to realize the promise of integrated geroscience for human health.
DECLARATIONS
Authors’ contributions
Made substantial contributions to conception and design of the study, wrote the first draft of the manuscript and performed data analysis and interpretation: Emanuele E, Minoretti P, Yılmaz Y, Lista S
Performed study revision and editing and provided administrative, technical, and material support: Khoramipour K, Crespo-Escobar P, Merino País M, López-Ortiz S, Santos-Lozano A
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Emanuele E is the sole owner of 2E Science, a for-profit scientific consulting company, and Minoretti P is the sole owner of Studio Minoretti, a for-profit outpatient facility. Both authors confirm that neither they nor their respective companies have any commercial or financial interests in connection with this article. Emanuele E is an Editorial Board Member of the journal Metabolism and Target Organ Damage. Emanuele E was not involved in any steps of editorial processing, notably including reviewers’ selection, manuscript handling and decision making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
2. Garmany A, Terzic A. Global healthspan-lifespan gaps among 183 World Health Organization member states. JAMA Netw Open. 2024;7:e2450241.
3. Su B, Luo Y, Zhong P, Zheng X. Long-term trends in healthy and unhealthy life expectancy among adults aged 60 - a global perspective, 1990-2019. China CDC Wkly. 2023;5:877-83.
4. Wang S, Jiang Y, Yang A, Meng F, Zhang J. The expanding burden of neurodegenerative diseases: an unmet medical and social need. Aging Dis. 2024;16:2937-52.
5. Noor A, Zafar S, Zerr I. Neurodegenerative proteinopathies in the proteoform spectrum-tools and challenges. Int J Mol Sci. 2021;22:1085.
7. Stranahan AM, Mattson MP. Bidirectional metabolic regulation of neurocognitive function. Neurobiol Learn Mem. 2011;96:507-16.
8. Procaccini C, Santopaolo M, Faicchia D, et al. Role of metabolism in neurodegenerative disorders. Metabolism. 2016;65:1376-90.
9. Shinohara M, Sato N. Bidirectional interactions between diabetes and Alzheimer’ disease. Neurochem Int. 2017;108:296-302.
10. Muddapu VR, Dharshini SAP, Chakravarthy VS, Gromiha MM. Neurodegenerative diseases - is metabolic deficiency the root cause? Front Neurosci. 2020;14:213.
11. Song D, Li Y, Yang LL, Luo YX, Yao XQ. Bridging systemic metabolic dysfunction and Alzheimer’s disease: the liver interface. Mol Neurodegener. 2025;20:61.
12. Kaya E, Yılmaz Y. Association of metabolic dysfunction-associated fatty liver disease with cognitive impairment and all-cause dementia: a comprehensive review. Turk J Gastroenterol. 2024;35:76-82.
13. Hou Y, Dan X, Babbar M, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15:565-81.
14. Palmer AK, Jensen MD. Metabolic changes in aging humans: current evidence and therapeutic strategies. J Clin Invest. 2022;132:e158451.
15. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186:243-78.
16. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194-217.
17. Niedernhofer LJ, Gurkar AU, Wang Y, Vijg J, Hoeijmakers JHJ, Robbins PD. Nuclear genomic instability and aging. Annu Rev Biochem. 2018;87:295-322.
18. Shadfar S, Brocardo M, Atkin JD. The complex mechanisms by which neurons die following DNA damage in neurodegenerative diseases. Int J Mol Sci. 2022;23:2484.
19. Welch G, Tsai LH. Mechanisms of DNA damage-mediated neurotoxicity in neurodegenerative disease. EMBO Rep. 2022;23:e54217.
20. Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J Neurochem. 2005;93:953-62.
21. Markesbery WR, Lovell MA. DNA oxidation in Alzheimer’s disease. Antioxid Redox Signal. 2006;8:2039-45.
22. Bradley-Whitman MA, Timmons MD, Beckett TL, Murphy MP, Lynn BC, Lovell MA. Nucleic acid oxidation: an early feature of Alzheimer’s disease. J Neurochem. 2014;128:294-304.
23. Santos RX, Correia SC, Zhu X, et al. Nuclear and mitochondrial DNA oxidation in Alzheimer’s disease. Free Radic Res. 2012;46:565-76.
24. Fukae J, Takanashi M, Kubo S, et al. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson’s disease and related neurodegenerative disorders. Acta Neuropathol. 2005;109:256-62.
25. Shimizu I, Yoshida Y, Suda M, Minamino T. DNA damage response and metabolic disease. Cell Metab. 2014;20:967-77.
26. Włodarczyk M, Nowicka G. Obesity, DNA damage, and development of obesity-related diseases. Int J Mol Sci. 2019;20:1146.
27. Włodarczyk M, Jabłonowska-Lietz B, Olejarz W, Nowicka G. Anthropometric and dietary factors as predictors of DNA damage in obese women. Nutrients. 2018;10:578.
28. Scarpato R, Verola C, Fabiani B, Bianchi V, Saggese G, Federico G. Nuclear damage in peripheral lymphocytes of obese and overweight Italian children as evaluated by the gamma-H2AX focus assay and micronucleus test. FASEB J. 2011;25:685-93.
29. Zhong A, Chang M, Yu T, et al. Aberrant DNA damage response and DNA repair pathway in high glucose conditions. J Can Res Updates. 2018;7:64-74.
30. Khalid M, Petroianu G, Adem A. Advanced glycation end products and diabetes mellitus: mechanisms and perspectives. Biomolecules. 2022;12:542.
31. Stopper H, Schinzel R, Sebekova K, Heidland A. Genotoxicity of advanced glycation end products in mammalian cells. Cancer Lett. 2003;190:151-6.
32. Bhattacharya R, Alam MR, Kamal MA, Seo KJ, Singh LR. AGE-RAGE axis culminates into multiple pathogenic processes: a central road to neurodegeneration. Front Mol Neurosci. 2023;16:1155175.
33. Alanazi AFR, Parkinson GN, Haider S. Structural motifs at the telomeres and their role in regulatory pathways. Biochemistry. 2024;63:827-42.
34. Harman A, Bryan TM. Telomere maintenance and the DNA damage response: a paradoxical alliance. Front Cell Dev Biol. 2024;12:1472906.
35. Vaiserman A, Krasnienkov D. Telomere length as a marker of biological age: state-of-the-art, open issues, and future perspectives. Front Genet. 2020;11:630186.
36. Levstek T, Kozjek E, Dolžan V, Trebušak Podkrajšek K. Telomere attrition in neurodegenerative disorders. Front Cell Neurosci. 2020;14:219.
37. Forero DA, González-Giraldo Y, López-Quintero C, Castro-Vega LJ, Barreto GE, Perry G. Meta-analysis of telomere length in Alzheimer’s disease. J Gerontol A Biol Sci Med Sci. 2016;71:1069-73.
38. Hackenhaar FS, Josefsson M, Adolfsson AN, et al. Short leukocyte telomeres predict 25-year Alzheimer’s disease incidence in non-APOE ε4-carriers. Alzheimers Res Ther. 2021;13:130.
39. Cui Y, Gao YT, Cai Q, et al. Associations of leukocyte telomere length with body anthropometric indices and weight change in Chinese women. Obesity. 2013;21:2582-8.
40. Gielen M, Hageman GJ, Antoniou EE, et al; TELOMAAS group. Body mass index is negatively associated with telomere length: a collaborative cross-sectional meta-analysis of 87 observational studies. Am J Clin Nutr. 2018;108:453-75.
41. Xia J, Xu L, Yu Y, et al. Associations between weight-adjusted-waist index and telomere length: results from NHANES: an observational study. Medicine. 2024;103:e37905.
42. Batsis JA, Mackenzie TA, Vasquez E, et al. Association of adiposity, telomere length and mortality: data from the NHANES 1999-2002. Int J Obes. 2018;42:198-204.
43. Tzanetakou IP, Katsilambros NL, Benetos A, Mikhailidis DP, Perrea DN. “Is obesity linked to aging? Ageing Res Rev. 2012;11:220-9.
44. He X, Cao L, Fu X, et al. The association between telomere length and diabetes mellitus: accumulated evidence from observational studies. J Clin Endocrinol Metab. 2024;110:e177-85.
46. Słowikowski B, Owecki W, Jeske J, et al. Epigenetics and the neurodegenerative process. Epigenomics. 2024;16:473-91.
47. Liu Y, Wang M, Marcora EM, Zhang B, Goate AM. Promoter DNA hypermethylation - implications for Alzheimer’s disease. Neurosci Lett. 2019;711:134403.
48. Yu CC, Jiang T, Yang AF, Du YJ, Wu M, Kong LH. Epigenetic modulation on Tau phosphorylation in Alzheimer’s disease. Neural Plast. 2019;2019:6856327.
49. Ma Y, Wang W, Liu S, et al. Epigenetic regulation of neuroinflammation in Alzheimer’s disease. Cells. 2023;13:79.
50. Park J, Lee K, Kim K, Yi SJ. The role of histone modifications: from neurodevelopment to neurodiseases. Signal Transduct Target Ther. 2022;7:217.
51. Balmik AA, Chinnathambi S. Methylation as a key regulator of Tau aggregation and neuronal health in Alzheimer’s disease. Cell Commun Signal. 2021;19:51.
52. Jung BC, Kang S. Epigenetic regulation of inflammatory factors in adipose tissue. Biochim Biophys Acta Mol Cell Biol Lipids. 2021;1866:159019.
53. Ling C, Bacos K, Rönn T. Epigenetics of type 2 diabetes mellitus and weight change - a tool for precision medicine? Nat Rev Endocrinol. 2022;18:433-48.
54. Franzago M, Pilenzi L, Di Rado S, Vitacolonna E, Stuppia L. The epigenetic aging, obesity, and lifestyle. Front Cell Dev Biol. 2022;10:985274.
55. Meyers AK, Zhu X. The NLRP3 inflammasome: metabolic regulation and contribution to inflammaging. Cells. 2020;9:1808.
56. La Rosa F, Mancuso R, Agostini S, et al. Pharmacological and epigenetic regulators of NLRP3 inflammasome activation in Alzheimer’s disease. Pharmaceuticals. 2021;14:1187.
57. Lu S, Li Y, Qian Z, et al. Role of the inflammasome in insulin resistance and type 2 diabetes mellitus. Front Immunol. 2023;14:1052756.
58. Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci. 2018;19:610-21.
59. Coppedè F. Mitochondrial DNA methylation and mitochondria-related epigenetics in neurodegeneration. Neural Regen Res. 2024;19:405-6.
60. Low HC, Chilian WM, Ratnam W, et al. Changes in mitochondrial epigenome in type 2 diabetes mellitus. Br J Biomed Sci. 2023;80:10884.
61. Maldonado-Lasuncion I, Atienza M, Sanchez-Espinosa MP, Cantero JL. Aging-related changes in cognition and cortical integrity are associated with serum expression of candidate microRNAs for Alzheimer disease. Cereb Cortex. 2019;29:4426-37.
62. Shen Y, Xu H, Pan X, et al. miR-34a and miR-125b are upregulated in peripheral blood mononuclear cells from patients with type 2 diabetes mellitus. Exp Ther Med. 2017;14:5589-96.
63. Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. 2019;20:421-35.
64. Klaips CL, Jayaraj GG, Hartl FU. Pathways of cellular proteostasis in aging and disease. J Cell Biol. 2018;217:51-63.
65. Kurtishi A, Rosen B, Patil KS, Alves GW, Møller SG. Cellular proteostasis in neurodegeneration. Mol Neurobiol. 2019;56:3676-89.
66. Rao G, Croft B, Teng C, Awasthi V. Ubiquitin-proteasome system in neurodegenerative disorders. J Drug Metab Toxicol. 2015;6:187.
67. Ottens F, Franz A, Hoppe T. Build-UPS and break-downs: metabolism impacts on proteostasis and aging. Cell Death Differ. 2021;28:505-21.
68. Queisser MA, Yao D, Geisler S, et al. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes. 2010;59:670-8.
69. James HA, O’Neill BT, Nair KS. Insulin regulation of proteostasis and clinical implications. Cell Metab. 2017;26:310-23.
70. Santiago-Lopez AJ, Berglund K, Gross RE, Gutekunst CN. Kinetic monitoring of neuronal stress response to proteostasis dysfunction. Mol Cell Neurosci. 2022;118:103682.
71. Moll L, Ben-Gedalya T, Reuveni H, Cohen E. The inhibition of IGF-1 signaling promotes proteostasis by enhancing protein aggregation and deposition. FASEB J. 2016;30:1656-69.
72. Höhn A, Tramutola A, Cascella R. Proteostasis failure in neurodegenerative diseases: focus on oxidative stress. Oxid Med Cell Longev. 2020;2020:5497046.
73. Ross JM, Olson L, Coppotelli G. Mitochondrial and ubiquitin proteasome system dysfunction in ageing and disease: two sides of the same coin? Int J Mol Sci. 2015;16:19458-76.
74. Ahmed T, Zulfiqar A, Arguelles S, et al. Map kinase signaling as therapeutic target for neurodegeneration. Pharmacol Res. 2020;160:105090.
75. Sonninen TM, Goldsteins G, Laham-Karam N, Koistinaho J, Lehtonen Š. Proteostasis disturbances and inflammation in neurodegenerative diseases. Cells. 2020;9:2183.
76. Chen Y, Qin Q, Zhao W, et al. Carnosol reduced pathogenic protein aggregation and cognitive impairment in neurodegenerative diseases models via improving proteostasis and ameliorating mitochondrial disorders. J Agric Food Chem. 2022;70:10490-505.
77. Saha S, Panigrahi DP, Patil S, Bhutia SK. Autophagy in health and disease: a comprehensive review. Biomed Pharmacother. 2018;104:485-95.
78. Mokarram P, Ghavami S. Autophagy unveiled: new horizons in health and disease. Biochim Biophys Acta Mol Basis Dis. 2024;1870:167289.
79. Ryter SW, Cloonan SM, Choi AM. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells. 2013;36:7-16.
80. Guo F, Liu X, Cai H, Le W. Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol. 2018;28:3-13.
81. Park H, Kang JH, Lee S. Autophagy in neurodegenerative diseases: a hunter for aggregates. Int J Mol Sci. 2020;21:3369.
82. Long Z, Chen J, Zhao Y, et al. Dynamic changes of autophagic flux induced by Abeta in the brain of postmortem Alzheimer’s disease patients, animal models and cell models. Aging. 2020;12:10912-30.
83. Jiang S, Bhaskar K. Degradation and transmission of tau by autophagic-endolysosomal networks and potential therapeutic targets for tauopathy. Front Mol Neurosci. 2020;13:586731.
84. Palmer JE, Wilson N, Son SM, et al. Autophagy, aging, and age-related neurodegeneration. Neuron. 2025;113:29-48.
86. Menikdiwela KR, Ramalingam L, Rasha F, et al. Autophagy in metabolic syndrome: breaking the wheel by targeting the renin-angiotensin system. Cell Death Dis. 2020;11:87.
87. Namkoong S, Cho CS, Semple I, Lee JH. Autophagy dysregulation and obesity-associated pathologies. Mol Cells. 2018;41:3-10.
88. Zhao X, Chen Y, Tan X, et al. Advanced glycation end-products suppress autophagic flux in podocytes by activating mammalian target of rapamycin and inhibiting nuclear translocation of transcription factor EB. J Pathol. 2018;245:235-48.
89. de Mello NP, Orellana AM, Mazucanti CH, de Morais Lima G, Scavone C, Kawamoto EM. Insulin and autophagy in neurodegeneration. Front Neurosci. 2019;13:491.
90. Lan ZQ, Ge ZY, Lv SK, Zhao B, Li CX. The regulatory role of lipophagy in central nervous system diseases. Cell Death Discov. 2023;9:229.
91. Haidar M, Loix M, Bogie JFJ, Hendriks JJA. Lipophagy: a new player in CNS disorders. Trends Endocrinol Metab. 2021;32:941-51.
92. Park K, Lee MS. Current status of autophagy enhancers in metabolic disorders and other diseases. Front Cell Dev Biol. 2022;10:811701.
93. Rahman MA, Rhim H. Therapeutic implication of autophagy in neurodegenerative diseases. BMB Rep. 2017;50:345-54.
97. Talbot K, Wang HY, Kazi H, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316-38.
98. de la Monte SM, Tong M. Dysregulated mTOR networks in experimental sporadic Alzheimer’s disease. Front Cell Neurosci. 2024;18:1432359.
99. Fernandez F, Griffiths LR, Sutherland HG, et al. Sirtuin proteins and memory: a promising target in Alzheimer’s disease therapy? Nutrients. 2024;16:4088.
101. Maldonado-Rojas ADC, Zuarth-Vázquez JM, Uribe M, Barbero-Becerra VJ. Insulin resistance and metabolic dysfunction-associated steatotic liver disease (MASLD): pathways of action of hypoglycemic agents. Ann Hepatol. 2024;29:101182.
103. Arab Sadeghabadi Z, Nourbakhsh M, Pasalar P, et al. Reduced gene expression of sirtuins and active AMPK levels in children and adolescents with obesity and insulin resistance. Obes Res Clin Pract. 2018;12:167-73.
104. Folch J, Olloquequi J, Ettcheto M, et al. The involvement of peripheral and brain insulin resistance in late onset Alzheimer’s dementia. Front Aging Neurosci. 2019;11:236.
105. Davoody S, Asgari Taei A, Khodabakhsh P, Dargahi L. mTOR signaling and Alzheimer’s disease: what we know and where we are? CNS Neurosci Ther. 2024;30:e14463.
106. Marinangeli C, Didier S, Vingtdeux V. AMPK in neurodegenerative diseases: implications and therapeutic perspectives. Curr Drug Targets. 2016;17:890-907.
107. Johnson ML, Distelmaier K, Lanza IR, et al. Mechanism by which caloric restriction improves insulin sensitivity in sedentary obese adults. Diabetes. 2016;65:74-84.
108. Ma L, Dong W, Wang R, et al. Effect of caloric restriction on the SIRT1/mTOR signaling pathways in senile mice. Brain Res Bull. 2015;116:67-72.
109. Cantó C, Auwerx J. Calorie restriction: is AMPK a key sensor and effector? Physiology. 2011;26:214-24.
110. Dos Santos C, Cambraia A, Shrestha S, et al. Calorie restriction increases insulin sensitivity to promote beta cell homeostasis and longevity in mice. Nat Commun. 2024;15:9063.
111. Hansen B, Roomp K, Ebid H, Schneider JG. Perspective: the impact of fasting and caloric restriction on neurodegenerative diseases in humans. Adv Nutr. 2024;15:100197.
114. Ashleigh T, Swerdlow RH, Beal MF. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimers Dement. 2023;19:333-42.
115. Bhatia S, Rawal R, Sharma P, Singh T, Singh M, Singh V. Mitochondrial dysfunction in Alzheimer’s disease: opportunities for drug development. Curr Neuropharmacol. 2022;20:675-92.
116. Højlund K, Mogensen M, Sahlin K, Beck-Nielsen H. Mitochondrial dysfunction in type 2 diabetes and obesity. Endocrinol Metab Clin North Am. 2008;37:713-31, x.
117. Bournat JC, Brown CW. Mitochondrial dysfunction in obesity. Curr Opin Endocrinol Diabetes Obes. 2010;17:446-52.
118. Yoon Y, Galloway CA, Jhun BS, Yu T. Mitochondrial dynamics in diabetes. Antioxid Redox Signal. 2011;14:439-57.
119. Lin J, Zhang X, Sun Y, et al. Exercise ameliorates muscular excessive mitochondrial fission, insulin resistance and inflammation in diabetic rats via irisin/AMPK activation. Sci Rep. 2024;14:10658.
120. Sridharan PS, Koh Y, Miller E, et al. Acutely blocking excessive mitochondrial fission prevents chronic neurodegeneration after traumatic brain injury. Cell Rep Med. 2024;5:101715.
121. Belosludtsev KN, Belosludtseva NV, Dubinin MV. Diabetes mellitus, mitochondrial dysfunction and Ca2+-dependent permeability transition pore. Int J Mol Sci. 2020;21:6559.
122. Kalani K, Yan SF, Yan SS. Mitochondrial permeability transition pore: a potential drug target for neurodegeneration. Drug Discov Today. 2018;23:1983-9.
123. Gurung P, Lukens JR, Kanneganti TD. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med. 2015;21:193-201.
124. Rovira-Llopis S, Apostolova N, Bañuls C, Muntané J, Rocha M, Victor VM. Mitochondria, the NLRP3 inflammasome, and sirtuins in type 2 diabetes: new therapeutic targets. Antioxid Redox Signal. 2018;29:749-91.
125. Litwiniuk A, Baranowska-Bik A, Domańska A, Kalisz M, Bik W. Contribution of mitochondrial dysfunction combined with NLRP3 inflammasome activation in selected neurodegenerative diseases. Pharmaceuticals. 2021;14:1221.
126. Zong Y, Li H, Liao P, et al. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther. 2024;9:124.
127. Cortés-Rojo C, Vargas-Vargas MA. Don’t give up on mitochondria as a target for the treatment of diabetes and its complications. World J Diabetes. 2024;15:2015-21.
128. Fields M, Marcuzzi A, Gonelli A, Celeghini C, Maximova N, Rimondi E. Mitochondria-targeted antioxidants, an innovative class of antioxidant compounds for neurodegenerative diseases: perspectives and limitations. Int J Mol Sci. 2023;24:3739.
129. Ding W, Yang X, Lai K, Jiang Y, Liu Y. The potential of therapeutic strategies targeting mitochondrial biogenesis for the treatment of insulin resistance and type 2 diabetes mellitus. Arch Pharm Res. 2024;47:219-48.
130. Uittenbogaard M, Chiaramello A. Mitochondrial biogenesis: a therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr Pharm Des. 2014;20:5574-93.
131. Gorgoulis V, Adams PD, Alimonti A, et al. Cellular senescence: defining a path forward. Cell. 2019;179:813-27.
132. Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22:75-95.
133. Wang B, Han J, Elisseeff JH, Demaria M. The senescence-associated secretory phenotype and its physiological and pathological implications. Nat Rev Mol Cell Biol. 2024;25:958-78.
134. Mijit M, Caracciolo V, Melillo A, Amicarelli F, Giordano A. Role of p53 in the regulation of cellular senescence. Biomolecules. 2020;10:420.
135. Alshaebi F, Sciortino A, Kayed R. The role of glial cell senescence in Alzheimer’s disease. J Neurochem. 2025;169:e70051.
136. Zhu J, Wu C, Yang L. Cellular senescence in Alzheimer’s disease: from physiology to pathology. Transl Neurodegener. 2024;13:55.
137. Shen QQ, Jv XH, Ma XZ, et al. Cell senescence induced by toxic interaction between α-synuclein and iron precedes nigral dopaminergic neuron loss in a mouse model of Parkinson’s disease. Acta Pharmacol Sin. 2024;45:268-81.
138. Lee S, Wang EY, Steinberg AB, Walton CC, Chinta SJ, Andersen JK. A guide to senolytic intervention in neurodegenerative disease. Mech Ageing Dev. 2021;200:111585.
139. Zhang H, Zhou H, Shen X, et al. The role of cellular senescence in metabolic diseases and the potential for senotherapeutic interventions. Front Cell Dev Biol. 2023;11:1276707.
140. Arias C, Álvarez-Indo J, Cifuentes M, Morselli E, Kerr B, Burgos PV. Enhancing adipose tissue functionality in obesity: senotherapeutics, autophagy and cellular senescence as a target. Biol Res. 2024;57:51.
141. Palmer AK, Xu M, Zhu Y, et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell. 2019;18:e12950.
142. Narasimhan A, Flores RR, Robbins PD, Niedernhofer LJ. Role of cellular senescence in type II diabetes. Endocrinology. 2021;162:bqab136.
143. Murakami T, Inagaki N, Kondoh H. Cellular senescence in diabetes mellitus: distinct senotherapeutic strategies for adipose tissue and pancreatic β cells. Front Endocrinol. 2022;13:869414.
144. Ghosh P, Fontanella RA, Scisciola L, et al. Obesity-induced neuronal senescence: unraveling the pathophysiological links. Ageing Res Rev. 2024;101:102533.
145. Cleland NRW, Al-Juboori SI, Dobrinskikh E, Bruce KD. Altered substrate metabolism in neurodegenerative disease: new insights from metabolic imaging. J Neuroinflammation. 2021;18:248.
146. Han X, Lei Q, Xie J, et al. Potential regulators of the senescence-associated secretory phenotype during senescence and aging. J Gerontol A Biol Sci Med Sci. 2022;77:2207-18.
147. Rando TA, Jones DL. Regeneration, rejuvenation, and replacement: turning back the clock on tissue aging. Cold Spring Harb Perspect Biol. 2021;13:a040907.
148. Rezazadeh S, Ellison-Hughes GM. Editorial: stem cell exhaustion in aging. Front Aging. 2024;5:1433702.
149. Brunet A, Goodell MA, Rando TA. Ageing and rejuvenation of tissue stem cells and their niches. Nat Rev Mol Cell Biol. 2023;24:45-62.
150. Iqbal MA, Bilen M, Liu Y, et al. The integrated stress response promotes neural stem cell survival under conditions of mitochondrial dysfunction in neurodegeneration. Aging Cell. 2024;23:e14165.
151. Scopa C, Marrocco F, Latina V, et al. Impaired adult neurogenesis is an early event in Alzheimer’s disease neurodegeneration, mediated by intracellular Aβ oligomers. Cell Death Differ. 2020;27:934-48.
152. Al-Regaiey K. Crosstalk between adipogenesis and aging: role of polyphenols in combating adipogenic-associated aging. Immun Ageing. 2024;21:76.
153. Chen X, Shen WB, Yang P, Dong D, Sun W, Yang P. High glucose inhibits neural stem cell differentiation through oxidative stress and endoplasmic reticulum stress. Stem Cells Dev. 2018;27:745-55.
154. Hidaka R, Machida M, Fujimaki S, Terashima K, Asashima M, Kuwabara T. Monitoring neurodegeneration in diabetes using adult neural stem cells derived from the olfactory bulb. Stem Cell Res Ther. 2013;4:51.
155. Geroldi D, Minoretti P, Emanuele E. Brain-derived neurotrophic factor and the metabolic syndrome: more than just a hypothesis. Med Hypotheses. 2006;67:195-6.
156. Motamedi S, Karimi I, Jafari F. The interrelationship of metabolic syndrome and neurodegenerative diseases with focus on brain-derived neurotrophic factor (BDNF): kill two birds with one stone. Metab Brain Dis. 2017;32:651-65.
157. Islam O, Loo TX, Heese K. Brain-derived neurotrophic factor (BDNF) has proliferative effects on neural stem cells through the truncated TRK-B receptor, MAP kinase, AKT, and STAT-3 signaling pathways. Curr Neurovasc Res. 2009;6:42-53.
158. Navarro Negredo P, Yeo RW, Brunet A. Aging and rejuvenation of neural stem cells and their niches. Cell Stem Cell. 2020;27:202-23.
159. Marsh SE, Blurton-Jones M. Neural stem cell therapy for neurodegenerative disorders: the role of neurotrophic support. Neurochem Int. 2017;106:94-100.
160. Zomer HD, Vidane AS, Gonçalves NN, Ambrósio CE. Mesenchymal and induced pluripotent stem cells: general insights and clinical perspectives. Stem Cells Cloning. 2015;8:125-34.
161. Sun H, Xia T, Ma S, Lv T, Li Y. Intercellular communication is crucial in the regulation of healthy aging via exosomes. Pharmacol Res. 2025;212:107591.
162. Fafián-Labora JA, O’Loghlen A. Classical and nonclassical intercellular communication in senescence and ageing. Trends Cell Biol. 2020;30:628-39.
163. Sigdel S, Swenson S, Wang J. Extracellular vesicles in neurodegenerative diseases: an update. Int J Mol Sci. 2023;24:13161.
164. Aas V, Øvstebø R, Brusletto BS, et al. Distinct microRNA and protein profiles of extracellular vesicles secreted from myotubes from morbidly obese donors with type 2 diabetes in response to electrical pulse stimulation. Front Physiol. 2023;14:1143966.
165. Liu W, Liu T, Zhao Q, Ma J, Jiang J, Shi H. Adipose tissue-derived extracellular vesicles: a promising biomarker and therapeutic strategy for metabolic disorders. Stem Cells Int. 2023;2023:9517826.
166. Park KJ, Gao Y. Gut-brain axis and neurodegeneration: mechanisms and therapeutic potentials. Front Neurosci. 2024;18:1481390.
167. Liu X, Shen L, Wan M, Xie H, Wang Z. Peripheral extracellular vesicles in neurodegeneration: pathogenic influencers and therapeutic vehicles. J Nanobiotechnology. 2024;22:170.
168. Kumar MA, Baba SK, Sadida HQ, et al. Extracellular vesicles as tools and targets in therapy for diseases. Signal Transduct Target Ther. 2024;9:27.
169. Shive C, Pandiyan P. Inflammation, immune senescence, and dysregulated immune regulation in the elderly. Front Aging. 2022;3:840827.
170. Zhang W, Xiao D, Mao Q, Xia H. Role of neuroinflammation in neurodegeneration development. Signal Transduct Target Ther. 2023;8:267.
171. Adamu A, Li S, Gao F, Xue G. The role of neuroinflammation in neurodegenerative diseases: current understanding and future therapeutic targets. Front Aging Neurosci. 2024;16:1347987.
172. Lista S, Imbimbo BP, Grasso M, et al. Tracking neuroinflammatory biomarkers in Alzheimer’s disease: a strategy for individualized therapeutic approaches? J Neuroinflammation. 2024;21:187.
173. Singh J, Habean ML, Panicker N. Inflammasome assembly in neurodegenerative diseases. Trends Neurosci. 2023;46:814-31.
174. Welikovitch LA, Do Carmo S, Maglóczky Z, et al. Early intraneuronal amyloid triggers neuron-derived inflammatory signaling in APP transgenic rats and human brain. Proc Natl Acad Sci U S A. 2020;117:6844-54.
175. Kawai T, Autieri MV, Scalia R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol. 2021;320:C375-91.
176. Li H, Meng Y, He S, et al. Macrophages, chronic inflammation, and insulin resistance. Cells. 2022;11:3001.
177. Mirabelli M, Misiti R, Sicilia L, et al. Hypoxia in human obesity: new insights from inflammation towards insulin resistance-a narrative review. Int J Mol Sci. 2024;25:9802.
178. Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res. 2016;57:2099-114.
179. Cao SS, Luo KL, Shi L. Endoplasmic reticulum stress interacts with inflammation in human diseases. J Cell Physiol. 2016;231:288-94.
180. Gkrinia EMM, Belančić A. The mechanisms of chronic inflammation in obesity and potential therapeutic strategies: a narrative review. Curr Issues Mol Biol. 2025;47:357.
181. Elwood E, Lim Z, Naveed H, Galea I. The effect of systemic inflammation on human brain barrier function. Brain Behav Immun. 2017;62:35-40.
182. Dukhinova M, Kuznetsova I, Kopeikina E, et al. Platelets mediate protective neuroinflammation and promote neuronal plasticity at the site of neuronal injury. Brain Behav Immun. 2018;74:7-27.
183. Sagar RC, Ajjan RA, Naseem KM. Non-traditional pathways for platelet pathophysiology in diabetes: implications for future therapeutic targets. Int J Mol Sci. 2022;23:4973.
184. Wu KK, Cheung SW, Cheng KK. NLRP3 inflammasome activation in adipose tissues and its implications on metabolic diseases. Int J Mol Sci. 2020;21:4184.
185. Wree A, Eguchi A, McGeough MD, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59:898-910.
186. Hanslik KL, Ulland TK. The role of microglia and the Nlrp3 inflammasome in Alzheimer’s disease. Front Neurol. 2020;11:570711.
187. Domingo E, Marques P, Francisco V, Piqueras L, Sanz MJ. Targeting systemic inflammation in metabolic disorders. A therapeutic candidate for the prevention of cardiovascular diseases? Pharmacol Res. 2024;200:107058.
188. Mohamed W, Kumar J, Alghamdi BS, Soliman AH, Toshihide Y. Neurodegeneration and inflammation crosstalk: Therapeutic targets and perspectives. IBRO Neurosci Rep. 2023;14:95-110.
189. Haran JP, McCormick BA. Aging, frailty, and the microbiome-how dysbiosis influences human aging and disease. Gastroenterology. 2021;160:507-23.
190. Marchesi JR, Adams DH, Fava F, et al. The gut microbiota and host health: a new clinical frontier. Gut. 2016;65:330-9.
191. Wu YL, Xu J, Rong XY, Wang F, Wang HJ, Zhao C. Gut microbiota alterations and health status in aging adults: from correlation to causation. Aging Med. 2021;4:206-13.
193. Intili G, Paladino L, Rappa F, et al. From dysbiosis to neurodegenerative diseases through different communication pathways: an overview. Biology. 2023;12:195.
194. Lista S, Munafò A, Caraci F, et al. Gut microbiota in Alzheimer’s disease: understanding molecular pathways and potential therapeutic perspectives. Ageing Res Rev. 2025;104:102659.
195. Ahmed H, Leyrolle Q, Koistinen V, et al. Microbiota-derived metabolites as drivers of gut-brain communication. Gut Microbes. 2022;14:2102878.
196. Romano S, Savva GM, Bedarf JR, Charles IG, Hildebrand F, Narbad A. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis. 2021;7:27.
197. Bandopadhyay P, Ganguly D. Gut dysbiosis and metabolic diseases. Prog Mol Biol Transl Sci. 2022;191:153-74.
198. Chanda D, De D. Meta-analysis reveals obesity associated gut microbial alteration patterns and reproducible contributors of functional shift. Gut Microbes. 2024;16:2304900.
199. Fan S, Chen S, Lin L. Research progress of gut microbiota and obesity caused by high-fat diet. Front Cell Infect Microbiol. 2023;13:1139800.
200. Clemente-Postigo M, Oliva-Olivera W, Coin-Aragüez L, et al. Metabolic endotoxemia promotes adipose dysfunction and inflammation in human obesity. Am J Physiol Endocrinol Metab. 2019;316:E319-32.
201. Tang W, Zhu H, Feng Y, Guo R, Wan D. The impact of gut microbiota disorders on the blood-brain barrier. Infect Drug Resist. 2020;13:3351-63.
202. Zhang D, Jian YP, Zhang YN, et al. Short-chain fatty acids in diseases. Cell Commun Signal. 2023;21:212.
203. Zhang M, Zhou C, Li X, et al. Interactions between gut microbiota, host circadian rhythms, and metabolic diseases. Adv Nutr. 2025;16:100416.
204. Hablitz LM, Plá V, Giannetto M, et al. Circadian control of brain glymphatic and lymphatic fluid flow. Nat Commun. 2020;11:4411.
205. Yoo JY, Kim SS. Probiotics and prebiotics: present status and future perspectives on metabolic disorders. Nutrients. 2016;8:173.
206. Ojha S, Patil N, Jain M, Kole C, Kaushik P. Probiotics for neurodegenerative diseases: a systemic review. Microorganisms. 2023;11:1083.
207. Zheng L, Ji YY, Wen XL, Duan SL. Fecal microbiota transplantation in the metabolic diseases: current status and perspectives. World J Gastroenterol. 2022;28:2546-60.
208. Matheson JT, Holsinger RMD. The role of fecal microbiota transplantation in the treatment of neurodegenerative diseases: a review. Int J Mol Sci. 2023;24:1001.
209. Freude KK, Moreno-Gonzalez I, Rodriguez-Ortiz CJ, Baglietto-Vargas D. Editorial: metabolic alterations in neurodegenerative disorders. Front Aging Neurosci. 2022;14:833109.
210. de Araújo Boleti AP, de Oliveira Flores TM, Moreno SE, Anjos LD, Mortari MR, Migliolo L. Neuroinflammation: an overview of neurodegenerative and metabolic diseases and of biotechnological studies. Neurochem Int. 2020;136:104714.
211. Mullins RJ, Diehl TC, Chia CW, Kapogiannis D. Insulin resistance as a link between amyloid-beta and tau pathologies in Alzheimer’s disease. Front Aging Neurosci. 2017;9:118.
212. Komleva Y, Chernykh A, Lopatina O, et al. Inflamm-aging and brain insulin resistance: new insights and role of life-style strategies on cognitive and social determinants in aging and neurodegeneration. Front Neurosci. 2020;14:618395.
213. Surguchov A. Caveolin: a new link between diabetes and AD. Cell Mol Neurobiol. 2020;40:1059-66.
214. Jinesh S, Özüpek B, Aditi P. Premature aging and metabolic diseases: the impact of telomere attrition. Front Aging. 2025;6:1541127.
215. Silberman DM. Metabolism, neurodegeneration and epigenetics: emerging role of Sirtuins. Neural Regen Res. 2018;13:417-8.
216. Bagherniya M, Butler AE, Barreto GE, Sahebkar A. The effect of fasting or calorie restriction on autophagy induction: a review of the literature. Ageing Res Rev. 2018;47:183-97.
217. Fontana L, Ghezzi L, Cross AH, Piccio L. Effects of dietary restriction on neuroinflammation in neurodegenerative diseases. J Exp Med. 2021;218:e20190086.
218. Kruczkowska W, Gałęziewska J, Buczek P, Płuciennik E, Kciuk M, Śliwińska A. Overview of metformin and neurodegeneration: a comprehensive review. Pharmaceuticals. 2025;18:486.
219. Nah J, Yuan J, Jung YK. Autophagy in neurodegenerative diseases: from mechanism to therapeutic approach. Mol Cells. 2015;38:381-9.
220. Guerville F, De Souto Barreto P, Ader I, et al. Revisiting the hallmarks of aging to identify markers of biological age. J Prev Alzheimers Dis. 2020;7:56-64.
221. Hampel H, Nisticò R, Seyfried NT, et al; Alzheimer Precision Medicine Initiative (APMI). Omics sciences for systems biology in Alzheimer’s disease: state-of-the-art of the evidence. Ageing Res Rev. 2021;69:101346.
222. Santoro A, Martucci M, Conte M, Capri M, Franceschi C, Salvioli S. Inflammaging, hormesis and the rationale for anti-aging strategies. Ageing Res Rev. 2020;64:101142.
223. James DL, Hawley NA, Mohr AE, et al. Impact of intermittent fasting and/or caloric restriction on aging-related outcomes in adults: a scoping review of randomized controlled trials. Nutrients. 2024;16:316.
224. Garatachea N, Pareja-Galeano H, Sanchis-Gomar F, et al. Exercise attenuates the major hallmarks of aging. Rejuvenation Res. 2015;18:57-89.
225. Poljšak B, Kovač V, Špalj S, Milisav I. The central role of the NAD+ molecule in the development of aging and the prevention of chronic age-related diseases: strategies for NAD+ modulation. Int J Mol Sci. 2023;24:2959.
226. Imb M, Véghelyi Z, Maurer M, Kühnel H. Exploring senolytic and senomorphic properties of medicinal plants for anti-aging therapies. Int J Mol Sci. 2024;25:10419.
227. Liu SZ, Chiao YA, Rabinovitch PS, Marcinek DJ. Mitochondrial targeted interventions for aging. Cold Spring Harb Perspect Med. 2024;14:a041199.
228. Trisal A, Singh AK. Clinical insights on caloric restriction mimetics for mitigating brain aging and related neurodegeneration. Cell Mol Neurobiol. 2024;44:67.
229. Barthet VJA, Lowe SW. Killing wisely: precision senolytics in the age of frailty. Genes Dev. 2025;39:910-3.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].