


On the potential of group-specific component (GC) inhibition for treating metabolic disease
 0
0 
Abstract
Recent advances in obesity and type 2 diabetes treatment using glucagon-like peptide-1 receptor agonist (GLP1RA)-based therapeutics have enabled major improvements in the management of these metabolic disorders. However, many patients cannot tolerate side effects associated with appetite suppression, and around 10% of those adhering to treatment do not lose weight. Thus, there is a need for alternative and/or additional treatment. Here, we examine the therapeutic potential of targeting a gene called Group-specific component (GC) to treat metabolic diseases. First, we review GC’s established roles in vitamin D transport, metabolism, and inflammation, including its structure-activity relationships as well as the phenotypes associated with genetic variation in GC. Next, we summarize studies of GC-null humans and mice, which were both generally healthy and viable, demonstrating the safety of inhibiting GC. Finally, we discuss novel evidence that among mice fed a high fat diet, Gc ablation confers protection against the development of obesity and type 2 diabetes. Notably, these benefits came without reductions in food intake or lean mass, the primary drivers of GLP1RA-associated adverse effects. GC therefore represents a promising novel therapeutic target for metabolic diseases.
Keywords
INTRODUCTION
Background
Obesity and type 2 diabetes are driven by inadequate exercise, poor dietary habits, and genetic predisposition, and are major causes of morbidity and mortality worldwide. Defined as having excess adipose tissue resulting in a disproportionate ratio of body weight relative to height [body mass index (BMI) ≥
Prior to overt hyperglycemia and a formal type 2 diabetes diagnosis is prediabetes or intermediate hyperglycemia[4], defined as a high-risk state characterized by one of the following criteria: (1) elevated fasting glucose levels (100-125 mg/dL) due to the liver’s resistance to insulin’s suppression of hepatic glucose production; (2) impaired glucose tolerance (based on an oral glucose tolerance test) due to insulin resistance in skeletal muscle, resulting in reduced postprandial glucose uptake and consequent elevated blood glucose (140 to 199 mg/dL); and (3) increased levels of glycated hemoglobin A1c (HbA1c; 5.7%-6.4%) representing the proportion of hemoglobin beta-chains with non-enzymatically added glucose.
Nutrient-stimulated hormones such as glucagon-like peptide-1 (GLP-1) and gastric inhibitory peptide (GIP) promote insulin secretion in metabolically healthy individuals, and their receptors have been targeted by GLP-1 receptor agonist (GLP1RA)-based therapeutics that have revolutionized the management and treatment of type 2 diabetes and obesity. Remarkably, when combined with diet and exercise, these treatments can result in good glycemic control and up to 20% body weight loss[5]; for the first time in over
GC (vitamin D binding protein)
GC is a multifunctional circulating protein and a member of the albumin superfamily of binding proteins which includes albumin, alpha-albumin, and alpha-fetoprotein[9]. The mature form of human GC protein is made up of 458 amino acids (AAs), and is primarily expressed in the liver, where it is synthesized into a
The three major electrophoretic variants of GC protein
| GC form | rs7041 | AA 416 | rs4588 | AA 420 |
| GC1F | T | Asp (D) | C | Thr (T) |
| GC1S | G | Glu (E) | C | Thr (T) |
| GC2 | T | Asp (D) | A | Lys (K) |
BIOLOGICAL BASIS AND PLEIOTROPIC FUNCTIONS OF GC
Molecular structure of GC: structure-activity relationships
GC likely arose from a gene triplication event[15] and possesses three domains, one of which binds to vitamin D. Based on its crystal structure[16-19] [Figure 1A], GC has an all alpha-helical structure with 10 helices in domain I (residues 1-191) and 9 helices in domain II (residues 192-378), while the third domain is truncated at the C-terminus and only contains 4 alpha helices (residues 379-458)[19]. The N-terminal part of domain I contains a cleft structure formed by helices 1-6 enabling high-affinity binding to vitamin D[20] [Figure 1B]. Interestingly although rs7041 and rs4588 affect AAs in domain III, these single nucleotide polymorphisms (SNPs) are associated with vitamin D binding affinity[21]. Domain I also contains residues at AAs 130-149 that are essential for enhancing the chemotactic function of complement component 5-derived peptides (C5a/C5adesArg)[22] [Figure 1C], although this activity does not involve direct binding to C5[23]. Residues spanning both domains I and III (AAs 150-172 and 379-402) enable binding to fatty acids in the cell membrane[24] [Figure 1D]; mono- and polyunsaturated fatty acids interfere with vitamin D binding to GC, while saturated fatty acids do not[25]. This suggests competition for a shared binding pocket or allosteric modulation. Lastly, GC can bind to monomeric globular actin (G-actin)[18,26] with high avidity (Kd = 2 × 109 M-1) via a large binding surface of
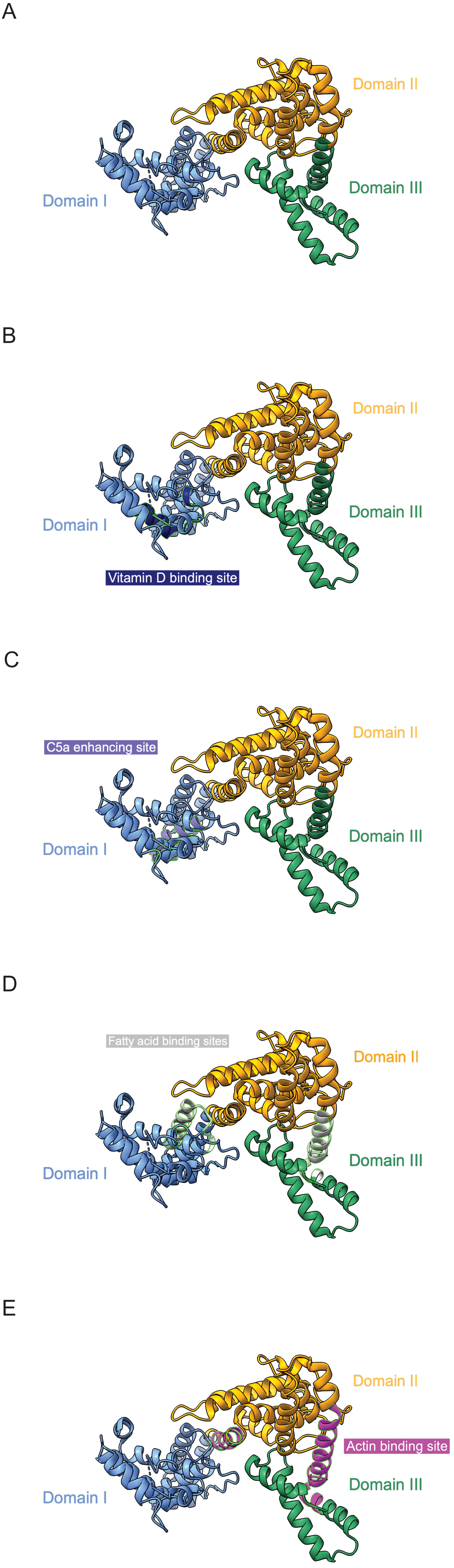
Figure 1. Three-dimensional structural diagram of GC protein and its domains (A). Subsequent panels highlight GC subdomains containing its vitamin D binding site (B), C5a enhancing site (C), fatty acid binding sites (D), and actin binding site (E). GC: Group-specific component; C5a: complement component 5a.
Human GC structure-activity relationships
| Protein domain | AA | Helices | Function | Notes | Ref. |
| I | 1-191 | 1-10 | [19] | ||
| II | 192-378 | 1-6,8,9,10 | |||
| III | 379-458 | 1-4 | |||
| I | 35-49 | 1-6 | Vitamin D binding 25(OH)D: Kd = 5 × 108 M-1-1 × 1011 M-1 1,25(OH)2D: Kd = 4 × 107 M-1 | Cleft, in contrast with vitamin D receptor (closed pocket) | [20] |
| I | 130-149 | 7,8 | Enhance C5 chemotactic function | Not a C5 binding site | [22] |
| I | 150-172 | 8,9 | Fatty acid (cell membrane) binding | [24] | |
| III | 379-402 | 1,2 | |||
| I | 194-207 | 10 | G-actin binding: Kd = 2 × 109 M-1 | Does not interfere with vitamin D binding | [16,17,27-30] |
| II | 373-403 | 6 | |||
| III | 3 | ||||
| III | 418, 420 | Threonine residues required for conversion to GCMAF via O-glycosylation | Primarily in GC1F/S, vs. GC2 carriers | [53] |
Classical functions and mechanisms of GC: the core transport protein in the vitamin D system
Vitamin D is a sterol needed for maintaining homeostatic serum levels of calcium in addition to bone development. GC is alternatively named for its role in binding vitamin D metabolites, and transports and sequesters the majority (85%-88%)[31,32] of vitamin D in the body. According to the free hormone hypothesis[33], lipophilic sterols such as vitamin D require their respective carrier proteins to travel through the circulatory system, but must be unbound and therefore free to enter cell membranes in order to act on their target tissues. In the liver, vitamin D from either sun exposure or dietary supplementation is converted into 25-hydroxy vitamin D [25(OH)D], which is the intermediate prohormone form in which the majority of vitamin D is transported within the organism. GC-bound 25(OH)D is taken up in the kidney via the megalin/cubilin transport system[34,35], where it is further metabolized to its active form 1,25-dihydroxyvitamin D3 [1,25(OH)2D] in the proximal tubules[34]. GC binds to 25(OH)D with at least 10× higher affinity (Kd between 5 × 108 M-1 and 1 × 1011 M-1)[36-38] compared to 1,25(OH)2D (Kd = 4 × 107 M-1)[38]. Once GC-bound 1,25(OH)2D reaches its target tissues, it dissociates from GC to enter cell membranes and govern transcription of vitamin D-responsive genes. Vitamin D binds to the nuclear transcription factor vitamin D receptor (VDR), which then forms a heterodimeric complex with retinoic acid X receptor (RXR). GC-bound vitamin D thus serves as a circulatory reservoir for local delivery of vitamin D[39]. Notably, only 1%-2% of sterol-binding sites on GC are occupied at any given time, suggesting GC’s primary biological role may not be limited to binding and transporting vitamin D[36].
Non-classical functions and mechanisms of GC
GC plays pro-inflammatory roles based on its structure-activity relationships and post-translational modifications. First, GC behaves as an adjuvant, promoting inflammation by binding peptides derived from complement 5 (C5), one of the most potent activators of the inflammatory response[40]. Specifically, GC binds and enhances the chemoattractant functions of C5a and its stable degradation product C5adesArg on phagocytic cells, including neutrophils[41], monocytes/macrophages[42], and fibroblasts[42,43]. GC itself does not have chemotactic functions[23,40,44-46]. Second, independent of its functional domains, GC can be converted into GCMAF (GC Macrophage Activating Factor)[47,48] via O-glycosylation by B-cells and T- cells during inflammation[49]. Specifically, this occurs on threonine residues at positions 418 or 420 and GC1 carriers therefore have a higher capacity for GCMAF formation than GC2 carriers[50]. As its name suggests, GCMAF rapidly stimulates macrophage phagocytic activity[51], and can also trigger macrophage death when these cells’ presence is no longer needed[52]. Interestingly, cancer cells can cause immunosuppression via loss of macrophage activation by releasing the enzyme endoglycosidase alpha-N-acetylgalactosaminidase (alpha-NaGalase), which can deglycosylate GC and thereby remove the substrate needed for its conversion to GCMAF[53]. Third, GCMAF is involved in bone resorption, as its exogenous administration can rescue skeletal defects in rat models of osteoporosis caused by defects in the pathway involved in the conversion of GC to GCMAF[54]. Notably, this effect was found to be independent of vitamin D binding to GCMAF[55], and subsequent studies implicated the glycosylation site in domain III of GC[56].
During tissue injury-induced cell lysis, filamentous (F-) actin is released and severed by gelsolin into globular (G-) actin. According to the actin scavenger hypothesis[57], GC binds and sequesters G-actin to prevent its spontaneous re-polymerization into F-actin that would otherwise damage and obstruct the microvasculature[58], thereby exerting an anti-inflammatory effect. Depletion of GC would consequently be expected to damage small blood vessels and increase inflammation by reducing extracellular actin binding capacity. However, based on more recent experimental evidence, intravenous injection of G-actin results in less severe acute lung inflammation in Gc knockout (GcKO) mice compared to wild-type (WT) mice[59]. This result suggests that GC promotes inflammation in the presence of actin. Furthermore, consistent with this notion, the addition of GC-actin complexes to human endothelial cells causes inflammation and injury. These data indicate that the classical actin scavenger hypothesis is incomplete, and that GC promotes inflammation by forming GC-actin complexes. GC ablation is therefore expected to help reduce inflammation by preventing GC-actin complex formation.
EVIDENCE LINKING GC TO METABOLIC DISEASES
GC genetic variation and metabolic diseases in humans
To corroborate the above in vitro and in vivo studies of GC, we next searched for human genetic associations between GC and human health and disease. Availability of human genetic evidence linking drug targets to diseases suggests causal relationships, and more than doubles the success rate of drug development[60]. Given the highly polymorphic nature of GC protein[61], GC genotypes have long been investigated for associations with numerous human diseases, including cancer, chronic disease, infectious diseases, and neurodegenerative diseases, and the results of these studies have been systematically reviewed and summarized by others[14,62-64]. Many of these early studies generally featured small cross-sectional designs focused on specific variants such as rs7041 and rs4588 and were therefore less robust than larger and more comprehensive genome-wide scans for disease associations.
We therefore surveyed genome-wide association studies (GWAS) published and entered into the National Human Genome Research Institute - European Bioinformatics Institute (NHGRI-EBI) catalog as of August 25th, 2025, and found GC-associated phenotypes consistent with GC’s structure-activity relationships [Figure 2 and Supplementary Table 1]. Specifically, GC variants are associated with serum levels of GC (vitamin D binding protein)[21,65-80] as well as vitamin D and its metabolites[21,67,70,74,79,81-95], in addition to immune cell-related traits such as white blood cell counts[65,79,96-101] and modulators of the immune response[102,103]. GC variants also have genome-wide significant associations with metabolic traits including blood lipid levels[104-107], blood pressure[108,109], bone mineral density[110], and non-alcoholic fatty liver disease (NAFLD)[111], neurological[112-114] and other traits[115-120], as well as serum levels of albumin[121] and other proteins[65,73,122-125].
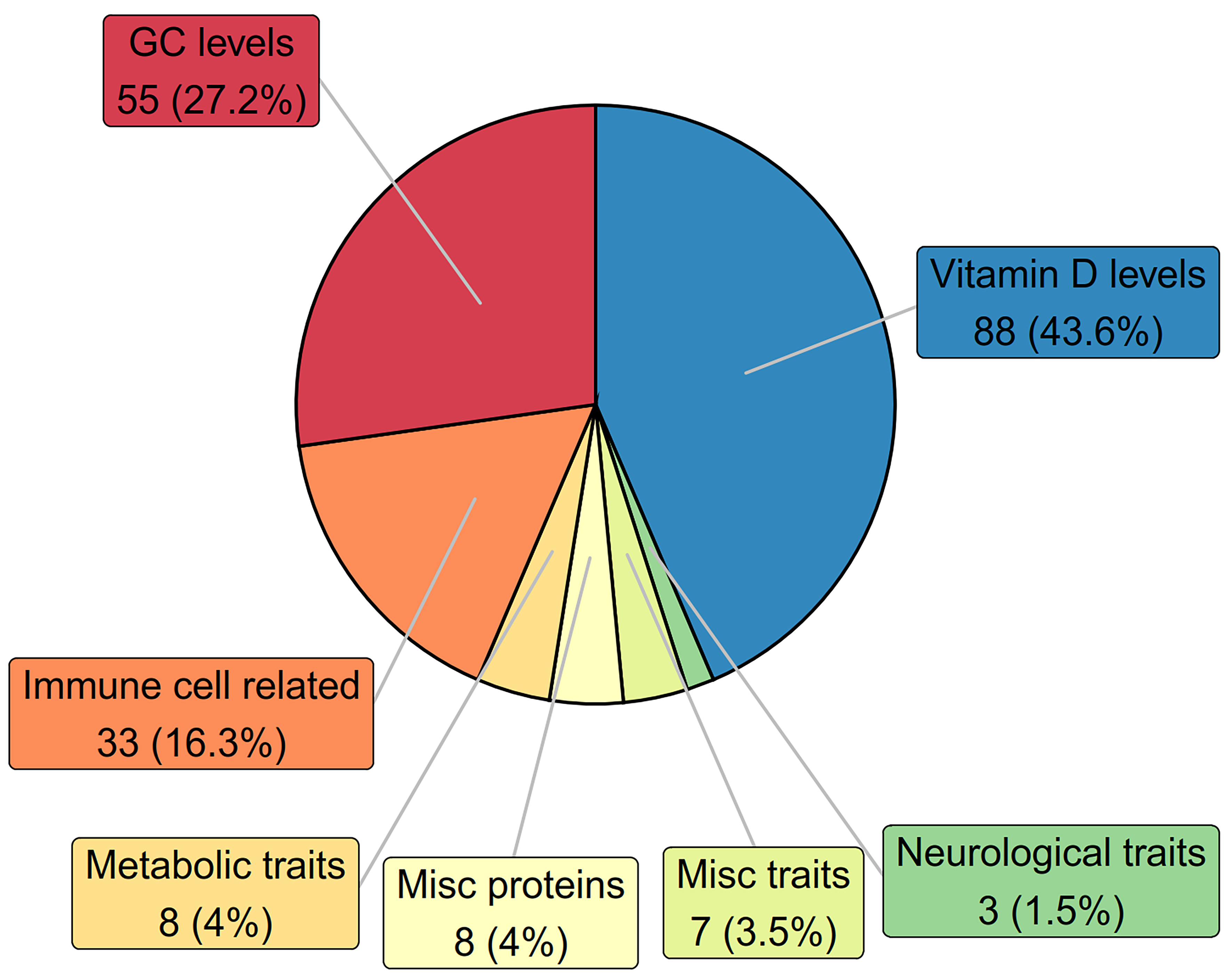
Figure 2. Number and proportion of GC variants associated with distinct groups of phenotypes in the NHGRI-EBI GWAS Catalog as of August 25th, 2025. https://www.ebi.ac.uk/gwas/genes/GC. GC: Group-specific component; NHGRI: National Human Genome Research Institute; EBI: European Bioinformatics Institute; GWAS: genome-wide association study.
We further queried GWAS data from population-scale biobanks consisting of electronic medical record phenotypes linked to genotypes [Supplementary Table 1]. In FinnGen Release 12, GC variants are significantly associated with phenotypes in the “Endocrine, nutritional and metabolic diseases” category including lipidemias, cholesterolemias, and metabolic disorders, in addition to statin medication prescription, height, cystic kidney disease, and diseases of the respiratory system[126]. In analyses of UK Biobank data by AstraZeneca, GC SNPs are significantly associated with vitamin D levels among Europeans and with vitamin D, reduced inflammation, whole-body impedance (fat mass), as well as reduced fat-free mass among Africans[127]. In summary, there is consistent human genetic evidence from large-scale studies linking GC with vitamin D metabolism, inflammation, and metabolic diseases.
Characterization of GC deficiency in humans and mice
Two homozygous GC-null individuals have been identified[128,129]. The first patient (Patient 1) was a 58-year-old Lebanese woman who carried two 139-kb deletions on chromosome 4q13.3 eliminating the entirety of GC, as well as nearby 144-kb deletions eliminating part of the neuropeptide FF receptor 2 (NPFFR2) gene[129]. The second patient (Patient 2) was a 60-year-old Pakistani man who had two copies of a c.702-1G>A variant, resulting in exon 7 skipping and the consequent loss of GC protein due to nonsense-mediated decay[128].
Both patients had consanguineous parents (first and second cousins, respectively) and exhibited undetectable GC protein and very low 25(OH)D and 1,25(OH)D levels. In contrast, serum measures of albumin, calcium, phosphate, parathyroid hormone, alkaline phosphatase, as well as magnesium and calcitonin in Patient 1, were within normal clinical ranges. Neither patient exhibited bone metabolism or skeletal abnormalities associated with vitamin D deficiency, such as rickets, osteomalacia, or osteoporosis. Only Patient 1 had progressive ankylosing spondylitis as well as osteopenia and fragility fractures in the fifth decade of life, suggesting her low bone density and fractures were attributable to her rheumatic disease, rather than the apparent vitamin D deficiency[128]. The observed clinical differences between the two GC-null patients may therefore be attributable to genetic differences beyond the absence of GC, such as disruption of NPFFR2 or nearby regulatory elements, and/or other genetic differences.
Gc-null mouse phenotypes generally mirror those of GC-null humans [Table 3], with plasma 25(OH)D and 1,25(OH)2D levels decreasing based on the dosage of inactive Gc and GC alleles while the remaining measurements available in both species are stable across genotypes. Mice with one (Gc+/-) or zero (Gc-/-) copies of functional Gc are viable, fertile, and do not exhibit any obvious phenotype compared to WT mice when placed on a normal (non-vitamin D deficient) mouse chow diet, despite a 95% reduction in serum 25(OH)D and 1,25(OH)2D[38]. Compared to WT mice, Gc-null mice only develop hyperparathyroidism and osteopathy (bone mineralization defects) secondary to vitamin D deficiency when placed on vitamin D-deficient diets. These mice also develop vitamin D deficiency symptoms more rapidly, likely due to accelerated 25(OH)D clearance in the absence of a reservoir of Gc-bound vitamin D[38]. Together, the evidence demonstrating that Gc-null mice phenocopy GC-null humans suggests that murine models of Gc deficiency are representative of GC deficiency in humans.
Laboratory measurements in GC-null humans and Gc-null mice, compared to wild-type
| Clinical and biochemical data from studies of human GC-null cases | Phenotypic data from studies of Gc-null mouse models | |||||||
| Study | Henderson et al. 2019[129] | Banerjee et al. 2021[128] | Safadi et al. 1999[38] | |||||
| Lab result | Normal clinical ranges | +/+ Sibling | +/- Sibling | GC-null Patient 1 | GC-null Patient 2 | WT mouse | +/- mouse | Gc-null mouse |
| Total plasma 25(OH)D ng/mL | 20-100 | 19.655 | 13.139 | 0.079 | 1.07 | 34 | 21 | 3 |
| Total plasma 1,25(OH)2D pg/mL | 21.2-73.1 | 55.7 | 19.5 | 5.6 | 14.2 | 24 | 17 | 4 |
| Free 25(OH)D pg/mL | 3.2 | 3.4 | 1.8 | 3.45 | ||||
| GC µg/mL | 104-477 | 224.5 | 95.3 | < 3.0 | < 1 | |||
| Albumin (g/dL) | 3.3-4.8 | 3.8-4.2 | 4.5 | |||||
| Serum calcium (mg/dL) | 8.4-10.2 | 9.9 | 9.66 | 9.58 | 8.9-10.2 | 8.0 ± 0.3 | 7.9 ± 0.8 | |
| Phosphate (mg/dL) | 2.4-5.0 | 2.76 | 2.23 | 2.45 | 2.5-3.7 | 11.4 ± 1 | 10.3 ± 1.5 | |
| Parathyroid hormone (pg/mL) | 12-90 | 43 | 42 | 57 | 54.1-94.2 | 566 | 633 | |
| Alkaline phosphatase (U/L) | 37-117 | 30-115 | 52-68 | 74 ± 28 | 94 ± 26 | |||
| Magnesium (mg/dL) | 1.6-2.6 | 1.7-2.1 | ||||||
| Calcitonin (ng/L) | < 12 | < 2 | ||||||
| Urinary Ca excretion (mg/d) | 100.2-300.6 | 94-414 | 177.0 @55y 671.0 @59y | |||||
| Urinary Mg excretion (mg/d) | 388.9-1,166.6 | 608 | ||||||
| Calculated urinary Mg (mg/d) | 72-103 | 91 | ||||||
| Calculated urinary creatinine (mg/d) | 1,000-2,000 | 2,000 @55y 2,024 @59y | ||||||
The function of Gc in mouse metabolic tissues
Gc in the pancreatic beta cell
With respect to metabolic diseases, Gc was initially identified as an effector gene in beta cell failure[130]. In healthy pancreatic beta cells, the expression of Gc is repressed at the transcript level by key beta cell transcription factors such as Foxo1. In contrast, the expression of Gc protein is increased during the progression of beta cell dedifferentiation and dysfunction. We next subjected mice to a metabolic challenge, in the form of a high-fat diet (HFD), to further investigate the role of Gc in beta cell function under obesogenic and diabetogenic conditions. Since we did not observe a significant deterioration of glucose tolerance in female mice, which are known to be more resistant to HFD challenges[131-133], we focused our experiments on mice with diet-induced obesity. In hyperglycemic clamp experiments, which assess beta cells’ ability to secrete insulin by varying the rate of glucose infusion necessary to maintain (i.e., clamp) a high blood glucose concentration, male whole body GcKO mice displayed improved insulin secretory capacity compared to WT [Figure 3A][130]. Furthermore, acute Gc knockdown in WT male mice on a HFD preserved circulating insulin levels[134]. Together, these data indicate that reducing Gc expression protects beta cell function and insulin secretion.
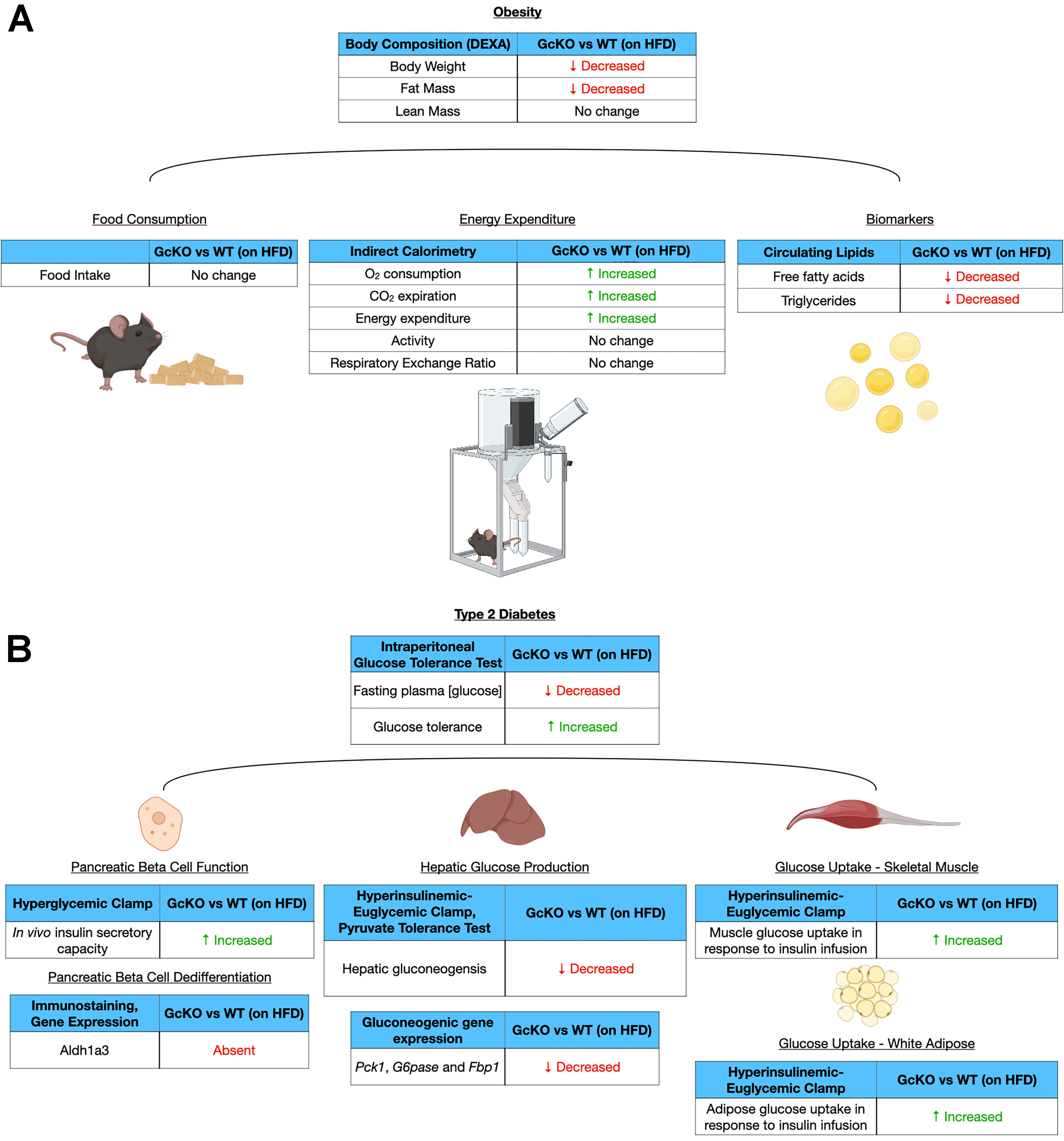
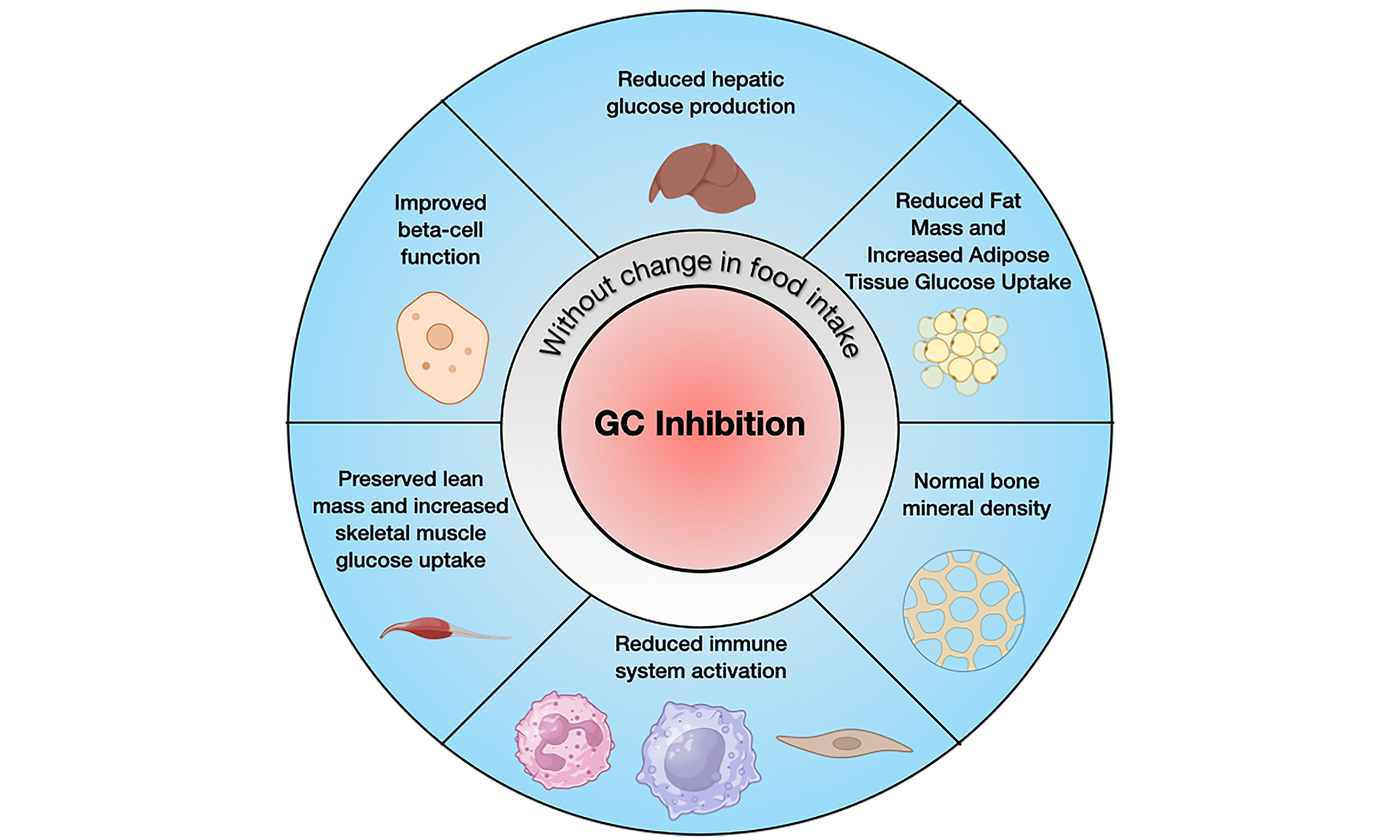
Figure 3. Effects of Gc inhibition on hallmarks of (A) type 2 diabetes and (B) obesity in high fat diet-fed mice, based on previous studies[130,134]. GcKO: Gc knockout mice. WT: Wild-type mice. HFD: High-fat diet. DEXA: dual-energy X-ray absorptiometry. Created in BioRender. Gill, R. (2026) https://BioRender.com/buhul0t. Gc: Group-specific component; GcKO: Gc knockout mice; WT: wild-type mice; HFD: high-fat diet; DEXA: dual-energy X-ray absorptiometry; Aldh1a3: aldehyde dehydrogenase 1 family member A3; Pck1: phosphoenolpyruvate carboxykinase 1; G6pase: glucose-6-phosphatase; Fbp1: fructose-1,6-bisphosphatase 1.
Gc in metabolic tissues
When challenged with a HFD, male GcKO mice showed a ~22% decrease in fasting glycemia compared to WT. These mice also demonstrated improvements in glucose tolerance based on intraperitoneal injection of a bolus of glucose, improvements in insulin sensitivity based on insulin tolerance tests, and reduced gluconeogenesis after a meal based on pyruvate tolerance tests [Figure 3A]. Moreover, compared to WT mice on HFD, GcKO mice displayed (i) ~15% reduced body weight; (ii) ~27% decreased fat mass; (iii) preserved muscle mass; (iv) ~17% reduced circulating non-esterified fatty acids; (v) ~35% reduced triglycerides; (vi) improved glucose uptake in skeletal muscle (~22%); (vii) improved glucose uptake in adipose (~30%); and (viii) suppression of hepatic glucose production by ~3 fold after refeeding [Figure 3A and B]. Notably, the weight loss in GcKO mice did not result from a reduction in food intake. Furthermore, acute ~70% knockdown of Gc in the liver was sufficient to recapitulate the phenotypes of chronic GcKO mice, as evidenced by improved glucose tolerance, preserved hepatic insulin signaling, and protected circulating insulin levels[134].
We observed several phenomena that may underlie the insulin sensitizing effects of Gc inhibition. First, compared to WT, indirect calorimetry studies found that GcKO mice had increased energy expenditure, which may be partially explained by a modest but significant increase in mitochondrial brown fat uncoupling protein 1 (Ucp1) expression in white adipose tissue. Ucp1 is involved in generating heat in brown adipose tissue (non-shivering thermogenesis)[135]. Second, Gc may also protect against oxidative stress in the liver based on RNA-seq followed by Gene Ontology analysis[134]. Moreover, Gc ablation increases Ceramide kinase expression. Cerk upregulates Nuclear factor erythroid 2-related factor 2 (NRF2), which is essential to maintain redox homeostasis[136]. Third, several changes in hepatic gene expression with known metabolic effects provide further mechanistic explanation. GcKO liver showed reduced Cytochrome P450 family 8 subfamily B member 1 (Cyp8b1) expression, whose ablation leads to resistance to hepatic steatosis and insulin resistance[137]. GcKO liver also displayed lower levels of 11β-hydroxysteroid dehydrogenase type 1 (Hsd11b1), and its upregulation is associated with metabolic disorders and obesity in humans[138]. GcKO mice had decreased liver expression of Krüppel-like factor 15 (Klf15), which encodes a transcription factor that promotes gluconeogenesis[139], and Cytochrome P450 2E1 (Cyp2e1), which encodes an enzyme that is elevated in NAFLD and nonalcoholic steatohepatitis (NASH)[140]. Lastly, we found improved insulin signaling in the liver, based on increased levels of Ak strain transforming (Akt)/Protein Kinase B (PKB) phosphorylated at serine 473, a mediator of insulin signaling.
As GC’s primary role has been recognized as a vitamin D transporter, does the absence of GC lower tissue-level vitamin D? GC deletion could eliminate the barrier preventing vitamin D from entering target tissues, as only the kidney can uptake vitamin D-bound GC via the megalin-cubulin complex, but at the same time, shorten the half-life of vitamin D by depleting its reservoir. Based on experiments conducted by Safadi et al., although GcKO mice have low levels of vitamin D metabolites in the bloodstream, they are normal in general[38]. Only when GcKO mice are intentionally placed on a vitamin D-deficient diet do they develop bone defects due to vitamin D deficiency. Therefore, Gc ablation most likely does not affect tissue-level vitamin D with sufficient vitamin D intake. Moreover, clinical trial evidence does not support a role for vitamin D in the development of diabetes, as randomized control trials of vitamin D supplementation in individuals at risk of developing diabetes found no significant association with diabetes incidence[141-143].
The question of how GC directly promotes insulin sensitivity remains open. Beyond transporting vitamin D, GC could carry biochemical entities that confer insulin resistance; these entities would be eliminated by GC ablation. Alternatively, GC could promote insulin receptor signaling through an unknown intracellular mechanism, although only kidney cells possess the molecular machinery needed for GC uptake. Considering low-grade inflammation is associated with insulin resistance[144], it is plausible that GC deficiency improves insulin sensitivity by blunting inflammatory cascades driven by GC-actin and GC-fatty acid complexes, to reduce hepatic oxidative stress and fatty liver disease. More work is necessary to test these hypotheses.
EVALUATION AND PROSPECTS OF GC AS A POTENTIAL THERAPEUTIC TARGET FOR METABOLIC DISEASES
While GLP1RAs have dramatically improved management of metabolic diseases, treatment-associated side effects limit tolerability and dosing, indicating an unmet need for a drug that promotes insulin secretion and sensitivity as well as fat mass reduction, without reducing food intake or muscle mass. The effects of GLP-1 receptor (GLP1R) agonism and GC inhibition on type 2 diabetes and obesity have not been compared head-to-head, but the improved metabolic phenotypes observed in acute and incomplete knockdown of Gc in mice demonstrate the possible utility of targeting GC to treat metabolic diseases via a novel mechanism of action that avoids appetite suppression and preserves lean mass, and could potentially improve outcomes in GLP1RA non-responders.
To our knowledge, there are no publicly disclosed therapeutic agents that inhibit GC. GC is a circulating protein that has an in vivo half-life of 2.5-3 days[38] and is primarily expressed in the liver by hepatocytes[134,145,146]. Considering the evidence above demonstrating the insulin-sensitizing effects of GC inhibition in the liver, and to minimize any off-target effects given GC’s many functions, it would be prudent to use a liver-specific GC inhibitor to treat metabolic diseases. While Gc-null mice and GC-null humans were viable and fertile, and acute knockdown achieved metabolic improvements observed in knockout mice, potential long-term safety considerations for pharmacological inhibition of GC revolve around its roles in vitamin D homeostasis, bone resorption, and immunity. The design, dosing, and clinical testing of such agents and subsequent monitoring will need to be done with these safety concerns in mind and are areas of ongoing investigation.
Conclusions and future directions
Based on the available data, GC represents a promising novel therapeutic target to treat metabolic disorders such as obesity and diabetes. Gc inhibition in mice preserves beta cell function with improved insulin secretion[130], and increases insulin sensitivity in the liver, skeletal muscle, and adipose[134]. Moreover, these mouse studies show that chronic Gc ablation leads to reduced body weight due to reduced fat mass, while maintaining muscle mass and bone mass[38,134]. Although GC has long been recognized for its role in vitamin D and metabolite transport, recent findings strongly suggest additional functions in metabolism. GcKO mice only showed bone defects associated with vitamin D deficiency or phenotypes associated with vitamin D toxicity when deliberately placed on diets with deficient or excessive vitamin D, respectively[38]. Similar observations were made in the case reports of GC-null humans[128].
The metabolic health benefits of GC inhibition are now clear, and studies to illuminate the mechanisms underlying these improvements are underway. Future directions include identifying the effectors of Gc in metabolic tissues, and signaling pathways involved in mediating its role in metabolism. These findings will provide insights important for developing next-generation therapeutics for metabolic diseases.
DECLARATIONS
Acknowledgment
The Graphical Abstract was created in BioRender [Gill, R. (2026) https://BioRender.com/buhul0t].
Authors’ contributions
Wrote the manuscript, analyzed data, made figures, and revised the manuscript: Gill R
Wrote the manuscript, analyzed data, and revised the manuscript: Kuo T
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
University of California, Davis (UC Davis) start-up fund (to Kuo T); National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) pilot and feasibility grants P30DK98722, P30DK63608, P30DK116074, and P30DK135103 (to Kuo T); National Institutes of Health (NIH) grant R01DK132227 (to Kuo T).
Conflicts of interest
Both authors declared there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Emmerich SD, Fryar CD, Stierman B, Ogden CL. Obesity and Severe Obesity Prevalence in Adults: United States, August 2021-August 2023. NCHS Data Brief. 2024.
2. WHO. Obesity and Overweight. Available from: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. [Last accessed on 27 Feb 2026].
3. Solis-Herrera C, Triplitt C, Cersosimo E, DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. South Dartmouth (MA): MDText.com, Inc.; 2000. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279115/. [Last accessed on 27 Feb 2026].
5. Jastreboff AM, Aronne LJ, Ahmad NN, et al; SURMOUNT-1 Investigators. Tirzepatide once weekly for the treatment of obesity. N Engl J Med. 2022;387:205-16.
6. Rader B, Hazan R, Brownstein JS. Changes in adult obesity trends in the US. JAMA Health Forum. 2024;5:e243685.
7. Jensen SBK, Sørensen V, Sandsdal RM, et al. Bone health after exercise alone, GLP-1 receptor agonist treatment, or combination treatment: a secondary analysis of a randomized clinical trial. JAMA Netw Open. 2024;7:e2416775.
8. Wilding JPH, Batterham RL, Calanna S, et al; STEP 1 Study Group. Once-weekly semaglutide in adults with overweight or obesity. N Engl J Med. 2021;384:989-1002.
9. Cooke NE. Rat vitamin D binding protein. Determination of the full-length primary structure from cloned cDNA. J Biol Chem. ;261:3441-50.
10. Haddad JG Jr., Walgate J. 25-Hydroxyvitamin D transport in human plasma. Isolation and partial characterization of calcifidiol-binding protein. J Biol Chem. 1976;251:4803-9.
11. Hirschfeld J. Immune-electrophoretic demonstration of qualitative differences in human sera and their relation to the haptoglobins. Acta Pathol Microbiol Scand. 1959;47:160-8.
12. Hirschfeld J, Jonsson B, Rasmuson M. Inheritance of a new group-specific system demonstrated in normal human sera by means of an immuno-electrophoretic technique. Nature. 1960;185:931-2.
13. Hirschfeld J, Soderberg U. Immuno-electrophoretic demonstration of precipitating components in sera from pregnant women. Nature. 1960;187:332-3.
14. Malik S, Fu L, Juras DJ, et al. Common variants of the vitamin D binding protein gene and adverse health outcomes. Crit Rev Clin Lab Sci. 2013;50:1-22.
15. Roger B, Rene FC, Frans S. Chapter 7 - vitamin D-binding protein. In: Hewison M, Bouillon R, Giovannucci E, Goltzman D, Meyer M, Welsh JE, Editors. Feldman and Pike’s Vitamin D. New York: Elsevier; 2024. pp. 111-138.
16. Head JF, Swamy N, Ray R. Crystal structure of the complex between actin and human vitamin D-binding protein at 2.5 A resolution. Biochemistry. 2002;41:9015-20.
17. Otterbein LR, Cosio C, Graceffa P, Dominguez R. Crystal structures of the vitamin D-binding protein and its complex with actin: structural basis of the actin-scavenger system. Proc Natl Acad Sci U S A. 2002;99:8003-8.
18. Verboven C, Bogaerts I, Waelkens E, et al. Actin-DBP: the perfect structural fit? Acta Crystallogr D Biol Crystallogr. 2003;59:263-73.
19. Verboven C, Rabijns A, De Maeyer M, Van Baelen H, Bouillon R, De Ranter C. A structural basis for the unique binding features of the human vitamin D-binding protein. Nat Struct Biol. 2002;9:131-6.
20. Daiger SP, Schanfield MS, Cavalli-Sforza LL. Group-specific component (Gc) proteins bind vitamin D and 25-hydroxyvitamin D. Proc Natl Acad Sci U S A. 1975;72:2076-80.
21. Zhang J, Kew RR. Identification of a region in the vitamin D-binding protein that mediates its C5a chemotactic cofactor function. J Biol Chem. 2004;279:53282-7.
22. Kew RR, Mollison KW, Webster RO. Binding of Gc globulin (vitamin D binding protein) to C5a or C5a des Arg is not necessary for co-chemotactic activity. J Leukoc Biol. 1995;58:55-8.
23. Zhang J, Habiel DM, Ramadass M, Kew RR. Identification of two distinct cell binding sequences in the vitamin D binding protein. Biochim Biophys Acta. 2010;1803:623-9.
24. Bouillon R, Xiang DZ, Convents R, Van Baelen H. Polyunsaturated fatty acids decrease the apparent affinity of vitamin D metabolites for human vitamin D-binding protein. J Steroid Biochem Mol Biol. 1992;42:855-61.
25. Van Baelen H, Bouillon R, De Moor P. Vitamin D-binding protein (Gc-globulin) binds actin. J Biol Chem. 1980;255:2270-2.
26. Buch S, Gremm D, Wegner A, Mannherz HG. Binding of a C-terminal fragment (residues 369 to 435) of vitamin D-binding protein to actin. Biol Chem. 2002;383:1621-31.
27. Swamy N, Head JF, Weitz D, Ray R. Biochemical and preliminary crystallographic characterization of the vitamin D sterol- and actin-binding by human vitamin D-binding protein. Arch Biochem Biophys. 2002;402:14-23.
28. Haddad JG, Hu YZ, Kowalski MA, et al. Identification of the sterol- and actin-binding domains of plasma vitamin D binding protein (Gc-globulin). Biochemistry. 1992;31:7174-81.
29. Mc Leod JF, Kowalski MA, Haddad JG Jr. Interactions among serum vitamin D binding protein, monomeric actin, profilin, and profilactin. J Biol Chem. 1989;264:1260-7.
30. Bikle DD, Gee E, Halloran B, Kowalski MA, Ryzen E, Haddad JG. Assessment of the free fraction of 25-hydroxyvitamin D in serum and its regulation by albumin and the vitamin D-binding protein. J Clin Endocrinol Metab. 1986;63:954-9.
31. Bikle DD, Siiteri PK, Ryzen E, Haddad JG. Serum protein binding of 1,25-dihydroxyvitamin D: a reevaluation by direct measurement of free metabolite levels. J Clin Endocrinol Metab. 1985;61:969-75.
32. Mendel CM. The free hormone hypothesis: a physiologically based mathematical model. Endocr Rev. 1989;10:232-74.
33. Nykjaer A, Dragun D, Walther D, et al. An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D3. Cell. 1999;96:507-15.
34. Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol. 2002;3:256-66.
35. Ray R. Molecular recognition in vitamin D-binding protein. Proc Soc Exp Biol Med. 1996;212:305-12.
36. Swamy N, Dutta A, Ray R. Roles of the structure and orientation of ligands and ligand mimics inside the ligand-binding pocket of the vitamin D-binding protein. Biochemistry. 1997;36:7432-6.
37. Safadi FF, Thornton P, Magiera H, et al. Osteopathy and resistance to vitamin D toxicity in mice null for vitamin D binding protein. J Clin Invest. 1999;103:239-51.
38. Rosner W. The functions of corticosteroid-binding globulin and sex hormone-binding globulin: recent advances. Endocr Rev. 1990;11:80-91.
39. Binder R, Kress A, Kan G, Herrmann K, Kirschfink M. Neutrophil priming by cytokines and vitamin D binding protein (Gc-globulin): impact on C5a-mediated chemotaxis, degranulation and respiratory burst. Mol Immunol. 1999;36:885-92.
40. Kew RR, Webster RO. Gc-globulin (vitamin D-binding protein) enhances the neutrophil chemotactic activity of C5a and C5a des Arg. J Clin Invest. 1988;82:364-9.
41. Piquette CA, Robinson-Hill R, Webster RO. Human monocyte chemotaxis to complement-derived chemotaxins is enhanced by Gc-globulin. J Leukoc Biol. 1994;55:349-54.
42. Senior RM, Griffin GL, Perez HD, Webster RO. Human C5a and C5a des Arg exhibit chemotactic activity for fibroblasts. J Immunol. 1988;141:3570-4.
43. DiMartino SJ, Shah AB, Trujillo G, Kew RR. Elastase controls the binding of the vitamin D-binding protein (Gc-globulin) to neutrophils: a potential role in the regulation of C5a co-chemotactic activity. J Immunol. 2001;166:2688-94.
44. Kew RR, Fisher JA, Webster RO. Co-chemotactic effect of Gc-globulin (vitamin D binding protein) for C5a. Transient conversion into an active co-chemotaxin by neutrophils. J Immunol. 1995;155:5369-74.
45. Metcalf JP, Thompson AB, Gossman GL, et al. Gcglobulin functions as a cochemotaxin in the lower respiratory tract. A potential mechanism for lung neutrophil recruitment in cigarette smokers. Am Rev Respir Dis. 1991;143:844-9.
46. Yamamoto N, Naraparaju VR. Role of vitamin D3-binding protein in activation of mouse macrophages. J Immunol. 1996;157:1744-9.
47. Mohamad SB, Nagasawa H, Uto Y, Hori H. Preparation of Gc protein-derived macrophage activating factor (GcMAF) and its structural characterization and biological activities. Anticancer Res. 2002;22:4297-300.
48. Nagasawa H, Uto Y, Sasaki H, et al. Gc protein (vitamin D-binding protein): Gc genotyping and GcMAF precursor activity. Anticancer Res. 2005;25:3689-95.
49. Debruyne E, Speeckaert M, Weygaerde YV, Delanghe J. Phenotype of Gc-globulin influences the macrophage activating factor (MAF) levels in serum. Clin Chem Lab Med. 2011;49:1855-60.
50. Yamamoto N, Homma S, Millman I. Identification of the serum factor required for in vitro activation of macrophages. Role of vitamin D3-binding protein (group specific component, Gc) in lysophospholipid activation of mouse peritoneal macrophages. J Immunol. 1991;147:273-80.
51. Gumireddy K, Reddy CD, Swamy N. Mitogen-activated protein kinase pathway mediates DBP-maf-induced apoptosis in RAW 264.7 macrophages. J Cell Biochem. 2003;90:87-96.
52. Kisker O, Onizuka S, Becker CM, et al. Vitamin D binding protein-macrophage activating factor (DBP-maf) inhibits angiogenesis and tumor growth in mice. Neoplasia. 2003;5:32-40.
53. Schneider GB, Benis KA, Flay NW, Ireland RA, Popoff SN. Effects of vitamin D binding protein-macrophage activating factor (DBP-MAF) infusion on bone resorption in two osteopetrotic mutations. Bone. 1995;16:657-62.
54. Swamy N, Ghosh S, Schneider GB, Ray R. Baculovirus-expressed vitamin D-binding protein-macrophage activating factor (DBP-maf) activates osteoclasts and binding of 25-hydroxyvitamin D(3) does not influence this activity. J Cell Biochem. 2001;81:535-46.
55. Schneider GB, Grecco KJ, Safadi FF, Popoff SN. The anabolic effects of vitamin D-binding protein-macrophage activating factor (DBP-MAF) and a novel small peptide on bone. Crit Rev Eukaryot Gene Expr. 2003;13:277-84.
56. Lee WM, Galbraith RM. The extracellular actin-scavenger system and actin toxicity. N Engl J Med. 1992;326:1335-41.
57. Meier U, Gressner O, Lammert F, Gressner AM. Gc-globulin: roles in response to injury. Clin Chem. 2006;52:1247-53.
58. Ge L, Trujillo G, Miller EJ, Kew RR. Circulating complexes of the vitamin D binding protein with G-actin induce lung inflammation by targeting endothelial cells. Immunobiology. 2014;219:198-207.
59. Minikel EV, Painter JL, Dong CC, Nelson MR. Refining the impact of genetic evidence on clinical success. Nature. 2024;629:624-9.
60. Cleve H, Constans J. The mutants of the vitamin-D-binding protein: more than 120 variants of the GC/DBP system. Vox Sang. 1988;54:215-25.
61. Alathari BE, Sabta AA, Kalpana CA, Vimaleswaran KS. Vitamin D pathway-related gene polymorphisms and their association with metabolic diseases: a literature review. J Diabetes Metab Disord. 2020;19:1701-29.
62. Trefilio LM, Bottino L, de Carvalho Cardoso R, Montes GC, Fontes-Dantas FL. The impact of genetic variants related to vitamin D and autoimmunity: a systematic review. Heliyon. 2024;10:e27700.
63. Rozmus D, Płomiński J, Augustyn K, Cieślińska A. rs7041 and rs4588 polymorphisms in vitamin D binding protein gene (VDBP) and the risk of diseases. Int J Mol Sci. 2022;23:933.
64. Loya H, Kalantzis G, Cooper F, Palamara PF. A scalable variational inference approach for increased mixed-model association power. Nat Genet. 2025;57:461-8.
65. Moy KA, Mondul AM, Zhang H, et al. Genome-wide association study of circulating vitamin D-binding protein. Am J Clin Nutr. 2014;99:1424-31.
66. Palmer ND, Lu L, Register TC, et al. Genome-wide association study of vitamin D concentrations and bone mineral density in the African American-Diabetes Heart Study. PLoS One. 2021;16:e0251423.
67. Caron B, Patin E, Rotival M, et al; Milieu Intérieur Consortium. Integrative genetic and immune cell analysis of plasma proteins in healthy donors identifies novel associations involving primary immune deficiency genes. Genome Med. 2022;14:28.
68. Albiñana C, Zhu Z, Borbye-Lorenzen N, et al. Genetic correlates of vitamin D-binding protein and 25-hydroxyvitamin D in neonatal dried blood spots. Nat Commun. 2023;14:852.
69. Surapaneni A, Schlosser P, Zhou L, et al. Identification of 969 protein quantitative trait loci in an African American population with kidney disease attributed to hypertension. Kidney Int. 2022;102:1167-77.
70. Parlato LA, Welch R, Ong IM, et al. Genome-wide association study (GWAS) of circulating vitamin D outcomes among individuals of African ancestry. Am J Clin Nutr. 2023;117:308-16.
71. Pietzner M, Wheeler E, Carrasco-Zanini J, et al. Mapping the proteo-genomic convergence of human diseases. Science. 2021;374:eabj1541.
72. Ahn J, Yu K, Stolzenberg-Solomon R, et al. Genome-wide association study of circulating vitamin D levels. Hum Mol Genet. 2010;19:2739-45.
73. Choe EK, Shivakumar M, Verma A, et al. Leveraging deep phenotyping from health check-up cohort with 10,000 Korean individuals for phenome-wide association study of 136 traits. Sci Rep. 2022;12:1930.
74. Jacobs BM, Stow D, Hodgson S, et al; Genes & Health Research Team. Genetic architecture of routinely acquired blood tests in a British South Asian cohort. Nat Commun. 2024;15:8929.
75. Jiang X, O’Reilly PF, Aschard H, et al. Genome-wide association study in 79,366 European-ancestry individuals informs the genetic architecture of 25-hydroxyvitamin D levels. Nat Commun. 2018;9:260.
76. Lasky-Su J, Lange N, Brehm JM, et al. Genome-wide association analysis of circulating vitamin D levels in children with asthma. Hum Genet. 2012;131:1495-505.
77. Qiu S, Zheng K, Hu Y, Liu G. Genetic correlation, causal relationship, and shared loci between vitamin D and COVID-19: a genome-wide cross-trait analysis. J Med Virol. 2023;95:e28780.
78. Sinnott-Armstrong N, Tanigawa Y, Amar D, et al; FinnGen. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat Genet. 2021;53:185-94.
79. Verma A, Huffman JE, Rodriguez A, et al. Diversity and scale: genetic architecture of 2068 traits in the VA million veteran program. Science. 2024;385:eadj1182.
80. Sun W, Kechris K, Jacobson S, et al; SPIROMICS Research Group, COPDGene Investigators. Common genetic polymorphisms influence blood biomarker measurements in COPD. PLoS Genet. 2016;12:e1006011.
81. Amin HA, Drenos F. No evidence that vitamin D is able to prevent or affect the severity of COVID-19 in individuals with European ancestry: a Mendelian randomisation study of open data. BMJ Nutr Prev Health. 2021;4:42-8.
82. Anderson D, Holt BJ, Pennell CE, Holt PG, Hart PH, Blackwell JM. Genome-wide association study of vitamin D levels in children: replication in the Western Australian Pregnancy Cohort (Raine) study. Genes Immun. 2014;15:578-83.
83. Hong J, Hatchell KE, Bradfield JP, et al. Transethnic evaluation identifies low-frequency loci associated with 25-hydroxyvitamin D concentrations. J Clin Endocrinol Metab. 2018;103:1380-92.
84. Kämpe A, Enlund-Cerullo M, Valkama S, et al. Genetic variation in GC and CYP2R1 affects 25-hydroxyvitamin D concentration and skeletal parameters: a genome-wide association study in 24-month-old Finnish children. PLoS Genet. 2019;15:e1008530.
85. Kim YA, Yoon JW, Lee Y, et al. Unveiling genetic variants underlying vitamin D deficiency in multiple korean cohorts by a genome-wide association study. Endocrinol Metab. 2021;36:1189-200.
86. Manousaki D, Dudding T, Haworth S, et al. Low-frequency synonymous coding variation in CYP2R1 has large effects on vitamin D levels and risk of multiple sclerosis. Am J Hum Genet. 2017;101:227-38.
87. Manousaki D, Mitchell R, Dudding T, et al. Genome-wide association study for vitamin D levels reveals 69 independent loci. Am J Hum Genet. 2020;106:327-37.
88. O’Brien KM, Sandler DP, Shi M, Harmon QE, Taylor JA, Weinberg CR. Genome-wide association study of serum 25-hydroxyvitamin D in US women. Front Genet. 2018;9:67.
89. Revez JA, Lin T, Qiao Z, et al. Genome-wide association study identifies 143 loci associated with 25 hydroxyvitamin D concentration. Nat Commun. 2020;11:1647.
90. Sallinen RJ, Dethlefsen O, Ruotsalainen S, et al. Genetic risk score for serum 25-hydroxyvitamin D concentration helps to guide personalized vitamin D supplementation in healthy finnish adults. J Nutr. 2021;151:281-92.
91. Sampathkumar A, Tan KM, Chen L, et al. Genetic link determining the maternal-fetal circulation of vitamin D. Front Genet. 2021;12:721488.
92. Seo J, Gaddis NC, Patchen BK, et al. Exploiting meta-analysis of genome-wide interaction with serum 25-hydroxyvitamin D to identify novel genetic loci associated with pulmonary function. Am J Clin Nutr. 2024;119:1227-37.
93. Traglia M, Windham GC, Pearl M, et al. Genetic contributions to maternal and neonatal vitamin D levels. Genetics. 2020;214:1091-102.
94. Wang TJ, Zhang F, Richards JB, et al. Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet. 2010;376:180-8.
95. Wang X, Hivert V, Groot S, et al. Cross-ancestry analyses identify new genetic loci associated with 25-hydroxyvitamin D. PLoS Genet. 2023;19:e1011033.
96. Vuckovic D, Bao EL, Akbari P, et al; VA Million Veteran Program. The polygenic and monogenic basis of blood traits and diseases. Cell. 2020;182:1214-31.e11.
97. Sakaue S, Kanai M, Tanigawa Y, et al; FinnGen. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet. 2021;53:1415-24.
98. Astle WJ, Elding H, Jiang T, et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell. 2016;167:1415-29.e19.
99. Chen MH, Raffield LM, Mousas A, et al; VA Million Veteran Program. Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell. 2020;182:1198-213.e14.
100. Kachuri L, Jeon S, DeWan AT, et al. Genetic determinants of blood-cell traits influence susceptibility to childhood acute lymphoblastic leukemia. Am J Hum Genet. 2021;108:1823-35.
101. Kichaev G, Bhatia G, Loh PR, et al. Leveraging polygenic functional enrichment to improve GWAS power. Am J Hum Genet. 2019;104:65-75.
102. Ahsan M, Ek WE, Rask-Andersen M, et al. The relative contribution of DNA methylation and genetic variants on protein biomarkers for human diseases. PLoS Genet. 2017;13:e1007005.
103. Konieczny MJ, Omarov M, Zhang L, et al. The genomic architecture of circulating cytokine levels points to drug targets for immune-related diseases. Commun Biol. 2025;8:34.
104. Graham SE, Clarke SL, Wu KH, et al; VA Million Veteran Program, Global Lipids Genetics Consortium*. The power of genetic diversity in genome-wide association studies of lipids. Nature. 2021;600:675-9.
105. Karjalainen MK, Karthikeyan S, Oliver-Williams C, et al; China Kadoorie Biobank Collaborative Group, Estonian Biobank Research Team, FinnGen. Genome-wide characterization of circulating metabolic biomarkers. Nature. 2024;628:130-8.
106. Inouye M, Ripatti S, Kettunen J, et al. Novel Loci for metabolic networks and multi-tissue expression studies reveal genes for atherosclerosis. PLoS Genet. 2012;8:e1002907.
107. Kiiskinen T, Helkkula P, Krebs K, et al; FinnGen. Genetic predictors of lifelong medication-use patterns in cardiometabolic diseases. Nat Med. 2023;29:209-18.
108. Pozarickij A, Gan W, Lin K, et al; China Kadoorie Biobank Collaborative Group. Causal relevance of different blood pressure traits on risk of cardiovascular diseases: GWAS and Mendelian randomisation in 100,000 Chinese adults. Nat Commun. 2024;15:6265.
109. Asiimwe IG, Blockman M, Cohen K, et al. A genome-wide association study of plasma concentrations of warfarin enantiomers and metabolites in sub-Saharan black-African patients. Front Pharmacol. 2022;13:967082.
110. He D, Liu H, Wei W, et al. A longitudinal genome-wide association study of bone mineral density mean and variability in the UK Biobank. Osteoporos Int. 2023;34:1907-16.
111. Adams LA, White SW, Marsh JA, et al. Association between liver-specific gene polymorphisms and their expression levels with nonalcoholic fatty liver disease. Hepatology. 2013;57:590-600.
112. Arpawong TE, Pendleton N, Mekli K, et al. Genetic variants specific to aging-related verbal memory: insights from GWASs in a population-based cohort. PLoS One. 2017;12:e0182448.
113. Foss-Skiftesvik J, Li S, Rosenbaum A, et al. Multi-ancestry genome-wide association study of 4069 children with glioma identifies 9p21.3 risk locus. Neuro Oncol. 2023;25:1709-20.
114. Hall LS, Adams MJ, Arnau-Soler A, et al; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Genome-wide meta-analyses of stratified depression in Generation Scotland and UK Biobank. Transl Psychiatry. 2018;8:9.
115. Fan W, Kan H, Liu HY, et al. Association between human genetic variants and the vaginal bacteriome of pregnant women. mSystems. 2021;6:e0015821.
116. Hysi PG, Choquet H, Khawaja AP, et al; Consortium for Refractive Error and Myopia, UK Eye and Vision Consortium, 23andMe Inc. Meta-analysis of 542,934 subjects of European ancestry identifies new genes and mechanisms predisposing to refractive error and myopia. Nat Genet. 2020;52:401-7.
117. Liu J, Zhou Y, Liu S, et al; DISCO (Deciphering disorders Involving Scoliosis and COmorbidities) Study. The coexistence of copy number variations (CNVs) and single nucleotide polymorphisms (SNPs) at a locus can result in distorted calculations of the significance in associating SNPs to disease. Hum Genet. 2018;137:553-67.
118. Pasanen A, Karjalainen MK, Zhang G, et al; FinnGen. Meta-analysis of genome-wide association studies of gestational duration and spontaneous preterm birth identifies new maternal risk loci. PLoS Genet. 2023;19:e1010982.
119. Tian C, Hromatka BS, Kiefer AK, et al. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat Commun. 2017;8:599.
120. Yengo L, Vedantam S, Marouli E, et al; 23andMe Research Team, VA Million Veteran Program, DiscovEHR (DiscovEHR and MyCode Community Health Initiative), eMERGE (Electronic Medical Records and Genomics Network), Lifelines Cohort Study, PRACTICAL Consortium, Understanding Society Scientific Group. A saturated map of common genetic variants associated with human height. Nature. 2022;610:704-12.
121. Gallois A, Mefford J, Ko A, et al. A comprehensive study of metabolite genetics reveals strong pleiotropy and heterogeneity across time and context. Nat Commun. 2019;10:4788.
122. Gudjonsson A, Gudmundsdottir V, Axelsson GT, et al. A genome-wide association study of serum proteins reveals shared loci with common diseases. Nat Commun. 2022;13:480.
123. He B, Shi J, Wang X, Jiang H, Zhu HJ. Genome-wide pQTL analysis of protein expression regulatory networks in the human liver. BMC Biol. 2020;18:97.
124. Sun BB, Maranville JC, Peters JE, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73-9.
125. Tabassum R, Ruotsalainen S, Ottensmann L, et al. Lipidome- and genome-wide study to understand sex differences in circulatory lipids. J Am Heart Assoc. 2022;11:e027103.
126. Kurki MI, Karjalainen J, Palta P, et al; FinnGen. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613:508-18.
127. Wang Q, Dhindsa RS, Carss K, et al; AstraZeneca Genomics Initiative. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature. 2021;597:527-32.
128. Banerjee RR, Spence T, Frank SJ, et al. Very low vitamin D in a patient with a novel pathogenic variant in the GC gene that encodes vitamin D-binding protein. J Endocr Soc. 2021;5:bvab104.
129. Henderson CM, Fink SL, Bassyouni H, et al. Vitamin D-binding protein deficiency and homozygous deletion of the GC gene. N Engl J Med. 2019;380:1150-7.
130. Kuo T, Damle M, González BJ, Egli D, Lazar MA, Accili D. Induction of α cell-restricted Gc in dedifferentiating β cells contributes to stress-induced β-cell dysfunction. JCI Insight. 2019;5:e128351.
131. de Souza GO, Wasinski F, Donato J Jr. Characterization of the metabolic differences between male and female C57BL/6 mice. Life Sci. 2022;301:120636.
132. Pettersson US, Waldén TB, Carlsson PO, Jansson L, Phillipson M. Female mice are protected against high-fat diet induced metabolic syndrome and increase the regulatory T cell population in adipose tissue. PLoS One. 2012;7:e46057.
133. Stapleton S, Welch G, DiBerardo L, Freeman LR. Sex differences in a mouse model of diet-induced obesity: the role of the gut microbiome. Biol Sex Differ. 2024;15:5.
134. Gill R, Kuo T. Gc inhibition preserves insulin sensitivity and reduces body weight without loss of muscle mass. JCI Insight. 2025;10:e195341.
135. Ikeda K, Yamada T. UCP1 dependent and independent thermogenesis in brown and beige adipocytes. Front Endocrinol. 2020;11:498.
136. Dong W, Li Q, Lu X, et al. Ceramide kinase-mediated C1P metabolism attenuates acute liver injury by inhibiting the interaction between KEAP1 and NRF2. Exp Mol Med. 2024;56:946-58.
137. Kaur A, Patankar JV, de Haan W, et al. Loss of Cyp8b1 improves glucose homeostasis by increasing GLP-1. Diabetes. 2015;64:1168-79.
138. Baudrand R, Carvajal CA, Riquelme A, et al. Overexpression of 11beta-hydroxysteroid dehydrogenase type 1 in hepatic and visceral adipose tissue is associated with metabolic disorders in morbidly obese patients. Obes Surg. 2010;20:77-83.
139. Takashima M, Ogawa W, Hayashi K, et al. Role of KLF15 in regulation of hepatic gluconeogenesis and metformin action. Diabetes. 2010;59:1608-15.
140. Jamwal R, Barlock BJ. Nonalcoholic fatty liver disease (NAFLD) and hepatic cytochrome P450 (CYP) enzymes. Pharmaceuticals. 2020;13:222.
141. Jorde R, Schirmer H, Wilsgaard T, et al. The DBP phenotype Gc-1f/Gc-1f is associated with reduced risk of cancer. The Tromsø study. PLoS One. 2015;10:e0126359.
142. Pittas AG, Dawson-Hughes B, Sheehan P, et al; D2d Research Group. Vitamin D supplementation and prevention of type 2 diabetes. N Engl J Med. 2019;381:520-30.
143. Kawahara T, Suzuki G, Mizuno S, et al. Effect of active vitamin D treatment on development of type 2 diabetes: DPVD randomised controlled trial in Japanese population. BMJ. 2022;377:e066222.
144. Wu H, Ballantyne CM. Metabolic inflammation and insulin resistance in obesity. Circ Res. 2020;126:1549-64.
145. Guilliams M, Bonnardel J, Haest B, et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell. 2022;185:379-96.e38.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].