


Novel insights into the purinergic P2X7 receptor signaling in obesity and cardiovascular-kidney-metabolic syndrome: a brief overview
 0
0 
Abstract
It has long been debated whether targeting systemic inflammation can lessen the burden of obesity-related cardiometabolic complications. In the cardiovascular field, various pharmacological approaches using anti-inflammatory agents as preventive strategies have been conducted, but results have been mixed. In this context, understanding the pathophysiology of meta-inflammation remains incomplete, as the problem must be approached from multiple perspectives. The molecular pattern regulated by the purinergic receptor P2X7 and the subsequent activation of the inflammasome play a crucial role in inflammatory responses and could serve as a target. Specifically, the P2X7 receptor pathway appears to be involved in the development of cardiovascular, hepatic, and renal abnormalities associated with metabolic syndrome. In this review, we briefly outline the current state of knowledge and our perspective on the role of the P2X7 receptor in obesity and its related complications, as highlighted in the new definition of cardiovascular-kidney-metabolic syndrome. Since P2X7 receptor antagonists are currently under development, particularly for rheumatological diseases, this approach merits investigation in future translational studies, especially in combination with incretin-based therapies.
Keywords
INTRODUCTION
Beyond its well-established roles in energy storage and mechanical protection, adipose tissue - particularly white adipose tissue (WAT) - is increasingly recognized as a key regulator of endocrine and immune functions[1]. WAT can modulate hormonal responses, as demonstrated by abnormalities of the reproductive system observed both in conditions of excess adiposity, such as obesity and metabolic syndrome[2], and in conditions of adiposity scarcity, such as anorexia nervosa or lipodystrophy syndromes. WAT also contributes to immune responses, as both very high and very low body mass index (BMI) are associated with reduced immune system efficacy. These properties relate to the biochemical machinery of adipocytes and other resident cells in WAT, such as fibroblasts, macrophages, and endothelial cells, which can synthesize molecules including chemokines, cytokines, and specific adipose-derived factors - collectively called adipokines - that act as endocrine hormones[3]. When adipose depots expand abnormally in obesity, WAT becomes hyperactive, and local inflammatory reactions can spread systemically, affecting the function and even the structure of distant organs and tissues. In this context, the alterations in WAT lead to abnormalities responsible for the onset of obesity-related clinical (and sub-clinical) complications. Obesity is the most significant risk factor for developing cardiometabolic diseases such as type 2 diabetes (T2DM)[4], coronary artery disease (CAD), heart failure (HF)[5], and a trigger for chronic kidney disease (CKD) progression[6]. In this regard, the expression of meta-inflammation (inflammation triggered by metabolic abnormalities) has been coined. Therefore, a better understanding of the inflammatory responses activated during WAT dysfunction and their potential pharmacological modulation may have a significant impact on clinical practice.
In addition to well-established pathways, other inflammatory signaling routes are involved in adipose tissue biology and pathophysiology. One such pathway is the purinergic system, which includes extracellular nucleosides and nucleotides - such as adenosine (A) and adenosine triphosphate (ATP) - that function as regulatory molecules by interacting with specific surface receptors. Multiple purinergic receptor subtypes exist, each with unique properties. Identifying purinergic receptors on the surface of adipocytes has paved the way for characterizing a purinergic system also within adipose tissue. Consequently, a new avenue in obesity pathophysiology research has opened. Since WAT dysfunction is characterized by the development and progression of inflammatory reactions, ranging from local cytokine release and inflammatory cell recruitment to fibrogenesis and abnormalities in extracellular matrix (ECM) deposition[7], with subsequent systemic complications, particular attention has been paid to defining the features of the purinergic system within adipose tissue. To date, the receptor subtype P2X7 (P2X7R) has been recognized as playing a key role in WAT inflammation as it is specifically involved in the inflammatory process, and is a central component of the complex cellular machinery called “inflammasome”.
Obesity is a complex, systemic disease. The local and systemic activation of the P2X7 pathway, resulting from WAT enlargement and dysfunction and resembling interorgan crosstalk, may contribute to the development of obesity-associated comorbidities, including cardiovascular, renal, and hepatic complications, now collectively referred to as cardiovascular-kidney-metabolic syndrome. In this review, we briefly present the current state of the art and our perspective on the evidence regarding the P2X7R-inflammasome axis in obesity pathophysiology and associated cardiovascular-kidney-metabolic complications, as well as its potential implications for future pharmacological interventions.
THE INFLAMMASOME, THE INTERLEUKIN-1β, AND THE P2X7 RECEPTOR
Interleukin-1β (IL-1β) is one of the major effectors of the inflammatory cascade, first identified in 1980, but only after decades, its pathway of secretion was described. IL-1β is produced as an inactive cytoplasmic precursor cleaved to the mature active form by the enzyme caspase-1, a component of the inflammasome arrangement[8]. Inflammasomes are constituted by cytosolic multiprotein complexes that activate in response to noxa patogena, such as microbes or other inflammatory stimuli, such as extracellular molecules related to pathogen activity defined as pathogen-associated molecular patterns (PAMPs). The lipopolysaccharide (LPS), mannose, and other microbial components, such as portions of nucleic acids, represent classic examples of PAMPs. However, inflammasomes are also responsive to self-derived molecules released from stress-induced cell damage, such as uric acid, nucleotides, and nucleosides, termed danger-associated molecular patterns (DAMPs). Different inflammasome complexes can be distinguished; among them, (NOD-, LRR- and pyrin domain-containing protein 3) NLRP3 is the most studied since its first identification in 2004[9].
In tissue damage areas, purine nucleotides are continuously released. Acting as DAMPs[10], ATP constitutes a significant mediator of inflammation, whose effects are mediated by interaction with purinergic receptors. The purinergic receptor subtypes include P1 receptors, which are selective for adenosine, and P2 receptors (P2Rs), which are preferentially selective for ATP and adenosine diphosphate (ADP) but are also sensitive to other ATP-related nucleotides; this is particularly true for the P2Y subtypes [Table 1]. All cells potentially express P2Rs, although it is generally considered that P2Rs are primarily expressed by components of the immune system. Among them, the P2X7R is the main purinergic receptor involved in inflammation and immunity[11].
Classification of the purinergic receptors
| Purinergic receptor type | Purinergic receptor subtype | Endogenous agonist |
| P1 | A1 A2a A2b A3 | Adenosine Adenosine Adenosine Adenosine |
| P2X | P2X1 P2X2 P2X3 P2X4 P2X5 P2X6 P2X7 | ATP ATP ATP ATP ATP ATP ATP |
| P2Y | P2Y1 P2Y2 P2Y4 P2Y6 P2Y11 P2Y12 P2Y13 P2Y14 | ADP ATP, UTP ATP, UTP UDP ATP ADP ADP UDP |
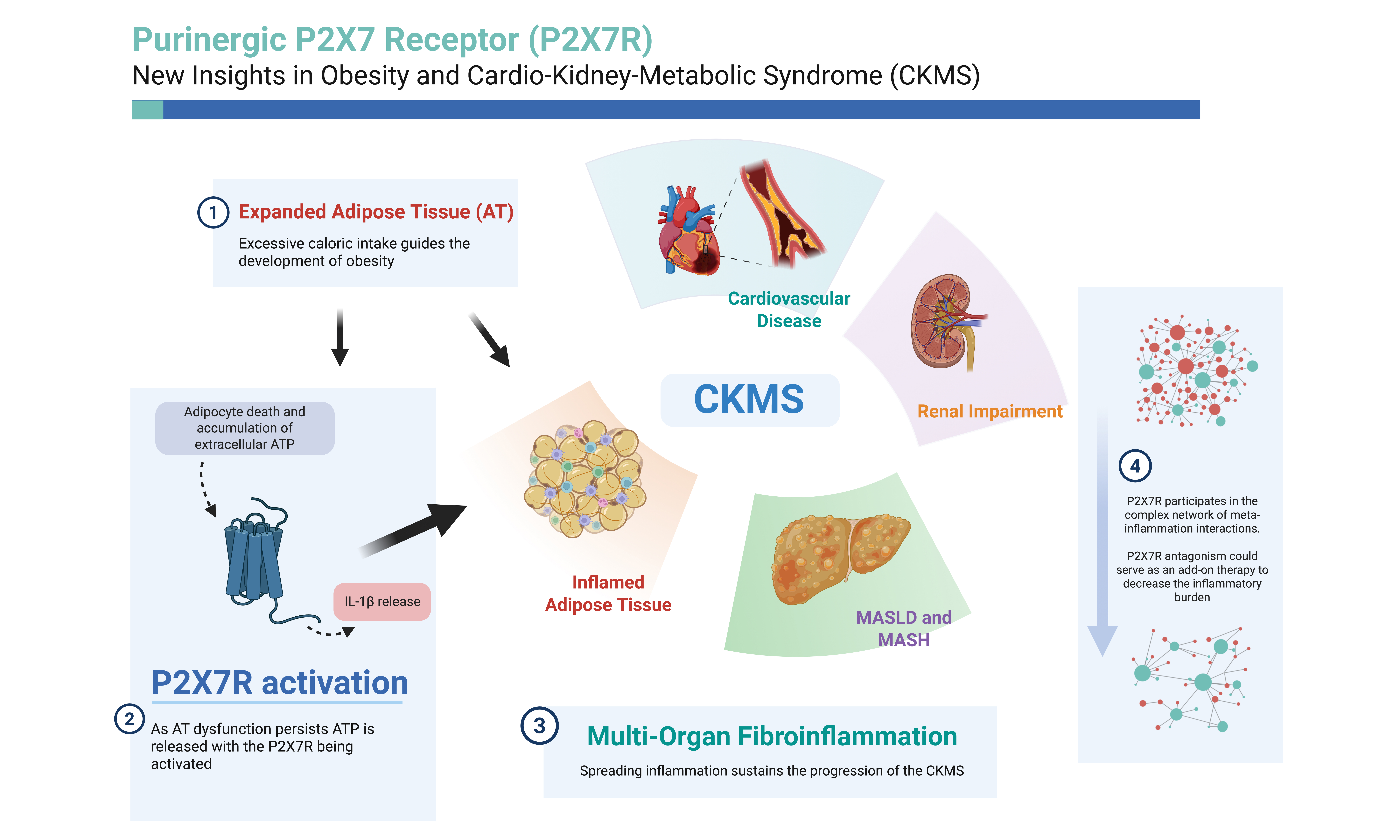
The P2X7R is expressed on cell types such as blood cells, endothelial cells, muscle, renal, and skin cells[12]. It is the main stimulus for the NLRP3 inflammasome activation, gaining attention as a potential target for anti-inflammatory therapy[13,14]. P2X7R-mediated assembly of the NLRP3 inflammasome complex promotes IL-1β[15] as part of a “two-signal” model. In this model, the first signal involves stimulation of Toll-like receptors, leading to accumulation of cytoplasmic pro-IL-1β, followed by a second, ATP-dependent phase in which P2X7R stimulation triggers inflammasome-related caspase-1 activation[16]. Consequently, P2X7R represents the rate-limiting step for IL-1β release and the subsequent downstream inflammatory cascade. A summary of the potential downstream effects of P2X7R activation in cardiovascular-kidney-metabolic syndrome is shown in Figure 1, and tissue expression of P2X7R is reported in Table 2.
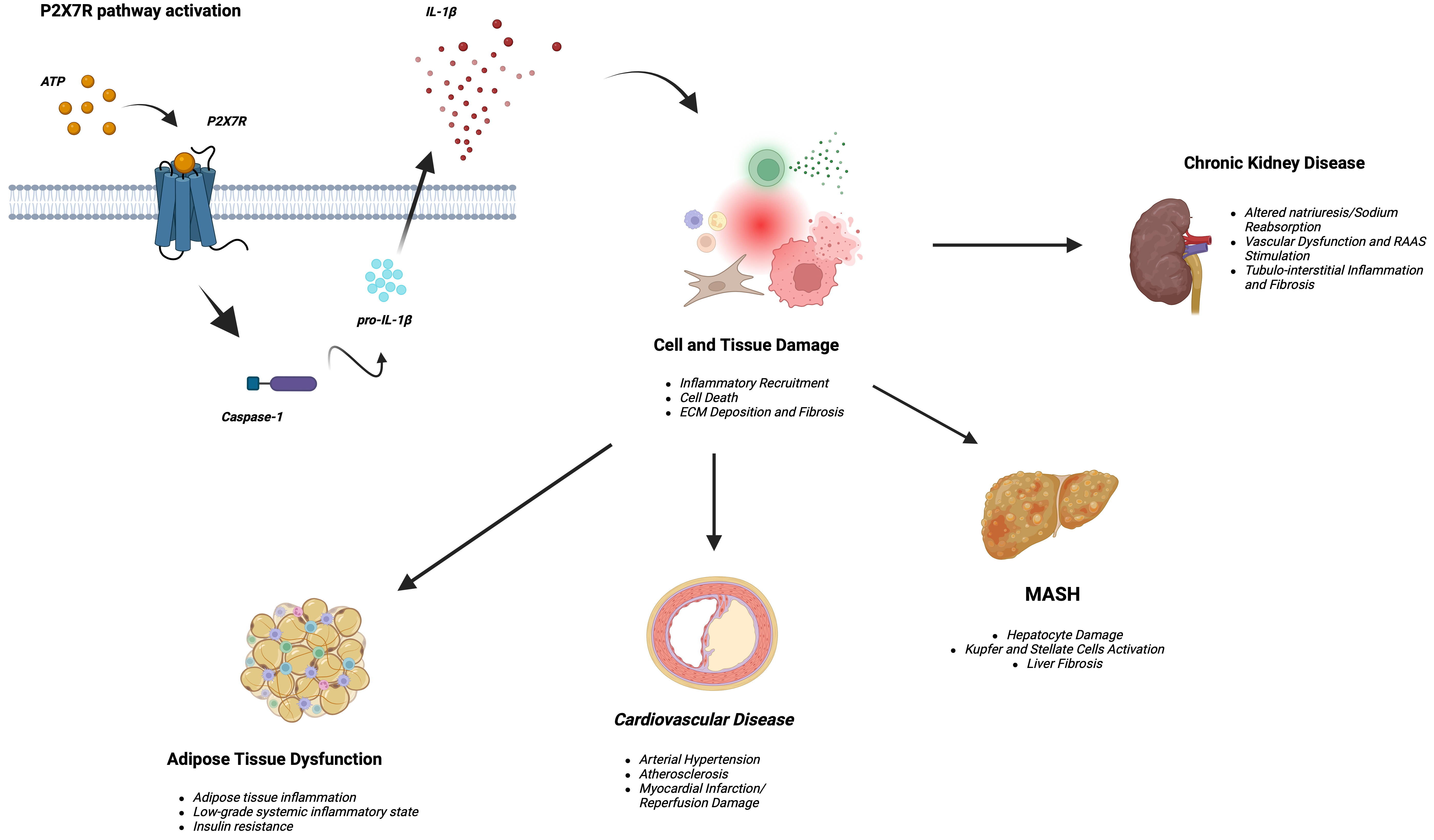
Figure 1. P2X7R-caspase 1-IL-1β axis activation, inflammatory cascade, and resulting clinical scenarios in cardiovascular-kidney-metabolic syndrome. (Created in BioRender; Di Vincenzo A, 2025. https://BioRender.com/frz4vwz). MASH: Metabolic-associated steatohepatitis; IL-1β: interleukin-1β.
P2X7 receptor distribution and potential effects across the different tissues and organs
| Tissue/Organ | Action | Potential effects |
| White adipose tissue | Activation/Agonism/Overexpression | Adipogenesis and lipogenesis modulation[24,29,33] Triggering inflammation[34] Immune Cells Recruitment and Cytokine Release[34] |
| Inhibition/Antagonism/Genetic KO | Reduction in whole-body energy expenditure (? limited evidence)[26-28] | |
| Brown adipose tissue | Activation/Agonism/Overexpression | Reduction/Null effect in UCP-1 activity (? limited evidence)[41-44] |
| Inhibition/Antagonism/Genetic KO | Increased UCP-1 expression and reduction in whole-body energy expenditure[45] | |
| Vessels and endothelium | Activation/Agonism/Overexpression | Arterial hypertension[49] Atherosclerotic Plaque Progression[51,52] Aggravating ischemia/Reperfusion damage[53] |
| Inhibition/Antagonism/Genetic KO | Blood pressure reduction[49,50] Cardiovascular risk reduction (indirect effects)[50] | |
| Myocardium | Activation/Agonism/Overexpression | Cell hypertrophy[53,54] Increased extracellular matrix deposition and fibrosis[53,54] |
| Inhibition/Antagonism/Genetic KO | Improved cardiomyocyte function[55] Anti-arrhythmic effect[56,57] Amelioration of cardiac dysautonomia[58,59] | |
| Kidney | Activation/Agonism/Overexpression | Increased sodium reabsorption [64] RAAS Modulation [68] Fibrotic Progression [66] |
| Inhibition/Antagonism/Genetic KO | Anti-inflammatory effect [67, 70,71,72] Slowing proteinuria progression [73] | |
| Liver | Activation/Agonism/Overexpression | Enhancement of sepsis-related and drug-associated toxicity[86,87] Activation of Kupfer cells[89] |
| Inhibition/Antagonism/Genetic KO | Blunting inflammatory and fibrotic responses[91,92,96,97,100] |
P2X7R IN WHITE ADIPOSE TISSUE FUNCTION AND DYSFUNCTION
Obesity complications arise when adipocytes in WAT progressively expand, reaching a threshold beyond which a series of detrimental biochemical abnormalities occur. Among the two distinct phenotypic patterns of WAT deposition - subcutaneous adipose tissue (SAT) and visceral adipose tissue (VAT) - VAT is more frequently implicated in the systemic consequences of obesity[17]. The adipocytes of VAT become dysfunctional, leading to the synthesis of hypoxia-induced mediators and pro-inflammatory molecules[18]. When reaching the circulation, the high concentrations of these adipokines establish a chronic, low-grade inflammation, the meta-inflammation responsible for the development of systemic comorbidities[19,20]. VAT inflammation may also account for local effects. The biochemical abnormalities arising in dysfunctional adipocytes contribute to reduced insulin sensitivity, increasing the risk of developing T2DM. Beyond this autocrine effect, both abdominal and ectopic VAT depots also act in a paracrine manner, influencing the function and structure of neighboring tissues and cells. This is the case of the epicardial fat, whose abnormalities are associated with the development of CAD and HF[21], but also of perirenal adipose tissue, whose dysfunction has been hypothesized to contribute to CKD[22].
P2X7 signaling contributes to VAT dysfunction in obesity and metabolic syndrome. Accumulating evidence shows that adipocytes express purinergic components[23,24]; see also Figure 2, with A and P2Rs involved in lipolysis, lipogenesis, and glucose intake[25]. In bone-derived mesenchymal progenitor cells from P2X7R knockout (KO) mice, higher expression of adipogenic markers has been observed compared with cultures from wild-type mice[26]. Furthermore, some reports indicate that P2X7R is involved in the regulation of energy homeostasis in animal models. However, results have often been conflicting, with some studies reporting benefits of P2X7R antagonism on whole-body energy expenditure[27], while others do not[28].
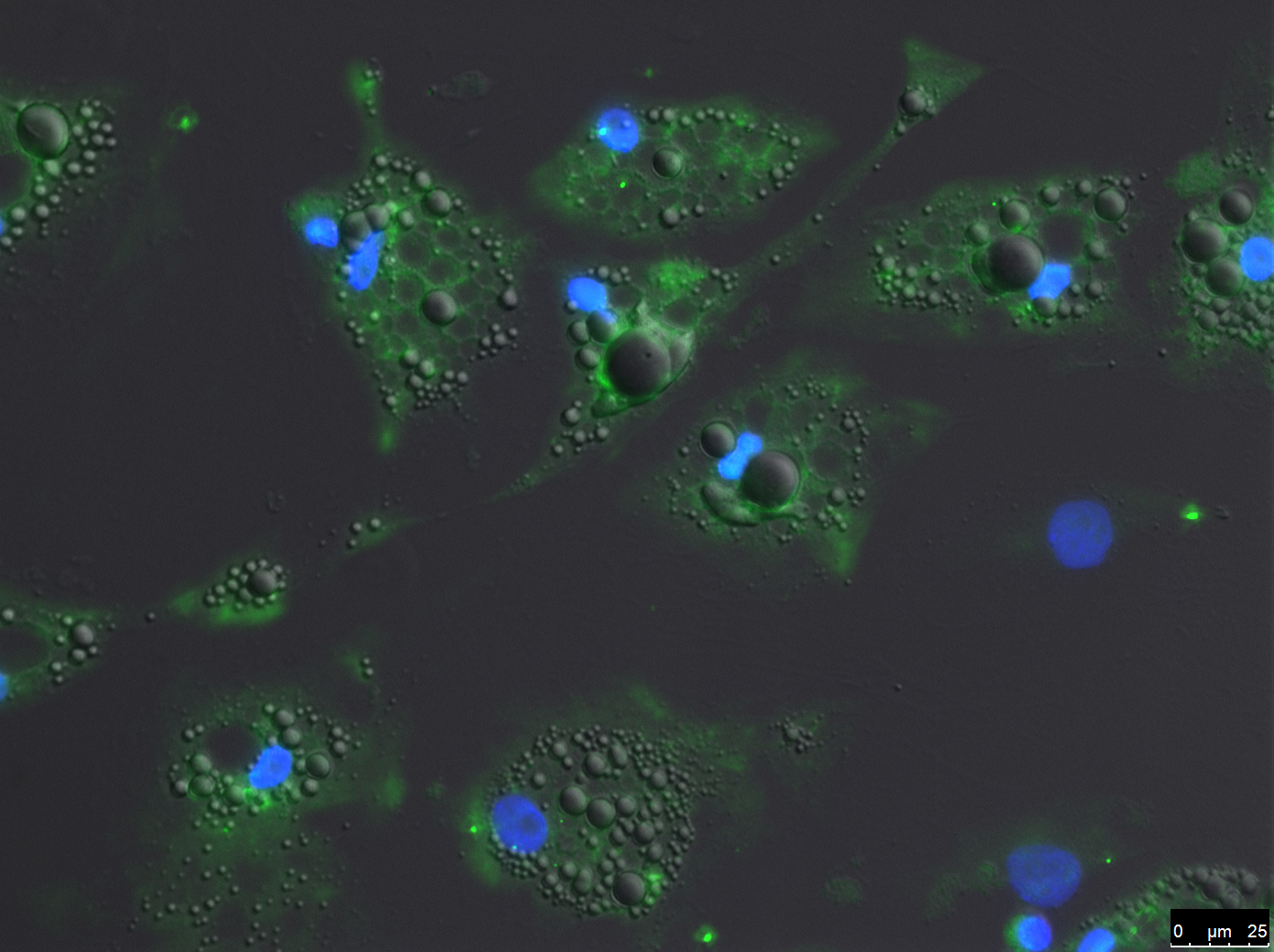
Figure 2. Evaluation of P2X7R expression (green; antibody APR004, 0.8 mg/mL, overnight at 4 °C) on human mature adipocytes by immunofluorescence assay (IFA). Nuclei were stained with DAPI. The image shows a merge of the fluorescent channels with the bright-field image. The scale bar is indicated in the image. P2X7R: Receptor subtype P2X7.
As WAT inflammation persists and the concentration of extracellular ATP increases, the P2X7R pathway remains activated[29]. P2X7R is overexpressed in the adipocytes of patients with obesity compared to healthy subjects, enhancing the burden of WAT dysfunction[30]. This increased expression may also be quantified by detecting in the blood portions of the P2X7R shed from the cell surface, identified as soluble P2X7R (sP2X7R)[31]. Our lab showed increased levels of sP2X7R in the blood plasma of patients with severe obesity, and the reduction following weight loss by bariatric surgery as a result of the loss of dysfunctional fat mass[32]. However, it is still not clear if sP2X7R could represent a reliable biomarker of systemic inflammation, and further studies are required to justify its application in clinical practice.
The P2X7R expression in adipocytes is related to the severity of tissue dysfunction, suggesting a potential pharmacological target: in models of diet-induced obesity, the antagonism of the P2X7R pathway through the inhibition of caspase-1 seems to be protective[33]. However, the overall biological role of the P2X7R in obesity is far from being completely elucidated. The P2X7R activation may be more relevant when WAT dysfunction has occurred, as it promotes a pro-inflammatory secretory profile in activated M1 macrophages; however, it also appears to exhibit anti-inflammatory activity in the context of inflammation resolution[34]. Furthermore, P2X7R activity is complex and modulated by multiple factors. For example, a sex dimorphism has been observed: in male P2X7R-KO mice, increased ectopic lipid deposition in the kidney and pancreas has been reported, whereas this effect is not seen in female animals, suggesting that P2X7R regulates adipogenesis and lipid metabolism in a sex-dependent manner[35]. The relationship between P2X7R and sex hormones seems dichotomous, as the exposure of human-derived adipocytes to androgens enhances the P2X7R expression when incubated with an inflammatory agent[36]. These reports merit further investigation, as in obesity sex hormone abnormalities are common[37].
P2X7R IN BROWN ADIPOSE TISSUE ACTIVATION
On the opposite of the dysfunctional WAT, brown adipose tissue (BAT) has protective effects for cardiovascular health[38]. Several attempts have been made so far to enhance BAT activation in vivo. Both physical exercise and cold exposure are known to activate BAT through sympathetic stimulation, so pharmacological adrenergic stimulation, mainly through β3-receptor agonism, has been proposed; however, to date, it has been limited in its clinical applicability. Mirabegron, the only β3-adrenergic receptor agonist approved for human use, has shown promising results[39]; however, there are still some perplexities regarding the safety profile due to the risk of tachycardia and other adverse cardiovascular effects. Therefore, the identification of other approaches is awaited.
BAT is a highly vascularized tissue rich in mitochondria, capable of burning energy from fat (“fat-burning” tissue) and generating heat through the activation of uncoupling protein 1 (UCP1), which uncouples mitochondrial respiration and the electron transport chain from ATP generation, leading to non-shivering thermogenesis[40]. Considering P2X7R as a sensor of extracellular ATP, a possible role in BAT activity has been suggested. To date, evidence regarding the ability of P2X7R to modulate energy expenditure remains limited and conflicting. P2X7R genetic depletion - and, to a lesser extent, pharmacological inhibition - appears to reduce energy expenditure and metabolic rate in mice; in contrast, receptor hyperactivation shows no effect[41,42]. Other reports described a non-relevant, or even negative, effect of the P2X7R on thermogenesis[43,44]. In primary adipocytes, inflammasome activation attenuates UCP-1 function[45], whereas extracellular ATP activates purinergic receptors on BAT-resident myeloid cells, impairing the thermogenic profile[46].
P2X7R IN CARDIOVASCULAR DISEASE
It is widely accepted that atherosclerosis and cardiovascular diseases (CVDs) involve inflammatory reactions in their pathophysiology, with meta-inflammation representing the underlying condition that places patients with metabolic syndrome at higher atherosclerotic risk. In this context, it should be emphasized that obesity is a risk factor per se, independent of other conditions such as arterial hypertension or T2DM, and that careful attention is warranted when defining metabolically healthy obesity, as inflammatory processes may begin well before overt clinical manifestations.
Different inflammatory pathways contribute to the distinct abnormalities in various vascular beds[47], yet all are characterized by local recruitment of inflammatory cells and the release of inflammatory cytokines. Among these, IL-1β plays a pivotal role; accordingly, the involvement of P2X7R in CVD has been investigated. It is plausible that P2X7R activation within the visceral adipose depot, leading to increased circulating IL-1β levels, promotes peripheral vascular damage. In addition, the paracrine role of perivascular adipose tissue must be considered, as local IL-1β release from depots surrounding the vasculature may contribute significantly. Adipose P2X7R expression may also be influenced bidirectionally by classical cardiovascular risk factors. Notably, one study reported higher P2X7R expression in perivascular adipocytes from smokers compared with healthy non-smokers[48].
P2X7R has a defined localization and role in the cardiovascular system, as it is expressed on endothelial cells, contributing to vascular tone regulation. In animal models, hyperactivation of the NLRP3 inflammasome promotes arterial hypertension, and in humans, both elevated IL-1β levels and P2X7R polymorphisms have been associated with an increased risk of developing hypertension[49]. However, whereas IL-1β antagonism effectively reduces blood pressure in mice, treatment with the anti-IL-1β agent canakinumab does not control hypertension in humans, despite reducing major cardiovascular events in high-risk patients[50].
P2X7R appears to be involved in both the development and stability of atherosclerotic plaques. Basic studies have demonstrated that P2X7R activation increases plaque size. Furthermore, oxidized low-density lipoproteins that cross the endothelial layer stimulate NLRP3 and P2X7R, enhancing the recruitment of monocytes and leukocytes into the plaque and creating a self-sustaining inflammatory cycle. P2X7R may also promote plaque rupture by modulating ECM deposition and metalloproteinase activation[51]. In this context, differential expression of P2X7R has been observed in vascular regions near atherosclerotic plaques compared with healthy vessels[52], suggesting a role for P2X7R in the progression from simple fatty deposition to unstable plaque. Following plaque rupture and consequent myocardial ischemia, the release of large amounts of intracellular ATP further amplifies P2X7R activation, exacerbating ischemia/reperfusion injury.
P2X7R is also involved in the pathological remodeling of the myocardium characteristic of CVD. In animal models, P2X7R promotes structural changes such as cardiac hypertrophy, increased ECM deposition, fibrosis, and ventricular dilation, all of which predispose to the development of HF[53]. P2X7R is expressed in cardiac fibroblasts, where its activation contributes to the transforming growth factor β (TGF-β)-mediated myocardial fibrotic process. Consequently, antagonism of the P2X7R-IL-1β axis may reduce the risk of HF[54,55]. Furthermore, aberrant P2X7R signaling has pro-arrhythmogenic potential through mechanisms involving both ion channel function and intramyocardial fibrosis. In animal models, inhibition of the purinergic receptor preserves normal atrial physiology[56,57], suggesting a role in the genesis of atrial fibrillation and other cardiac rhythm disturbances. In addition, P2X7R inhibition has been shown to protect against sympathetic hyperinnervation and the resulting pro-arrhythmic milieu following acute myocardial
P2X7R IN CHRONIC RENAL DISEASE
Obesity and metabolic syndrome are well-established risk factors for CKD. As metabolic abnormalities progress, worsening insulin resistance and the development of T2DM are major contributors; however, this model does not account for other mechanisms that may also play a role in this relationship.
In central obesity, the kidneys undergo structural and functional changes due to increased abdominal pressure. In addition, fat accumulation around the kidneys in the para-renal and peri-renal regions, combined with mechanical compression, promotes deleterious paracrine signaling through the release of inflammatory mediators[60,61]. In this context, a role for P2X7 in metabolic-related CKD has been proposed. However, it is important to note that, similar to vascular beds, the purinergic system is expressed in various renal structures, where it may exert distinct biological functions.
Multiple P2R subtypes are expressed in the vasculature and glomerulus. Studies on human renal biopsies reported increased P2X7R expression in patients with diabetic and non-diabetic kidney disease[62]. Notably, in a healthy kidney, P2X7R expression is usually low and confined to the microvasculature, but inflammatory responses upregulate this expression. This is highly relevant because repeated kidney injuries increase local ATP concentrations, promoting renal P2R activation and glomerular and tubular stress[63].
P2X7R polymorphisms are linked to a higher risk of arterial hypertension, due to its modulatory role in natriuresis and sodium reabsorption along the proximal tubule[64]. P2X7R also impairs the effects of angiotensin II and endothelin-1 on the renal vasculature[65,66]. Activation of P2X7R promotes tubular fibrosis by enhancing the TGF-β pathway[67] and participates in a multi-level renal response to injury, contributing to hypertension-related damage. In P2X7R KO mice, a high-fat diet used to induce metabolic-associated kidney disease results in reduced renal inflammation[68]. As for CAD, P2X7R also appears involved in acute ischemia-reperfusion syndrome in the kidney, with early inhibition showing protective effects[69].
Therefore, antagonism of P2X7R represents a potential therapeutic strategy for renal protection, despite mixed results from clinical trials in rheumatologic disorders. Brilliant Blue G, a P2X7R antagonist, has been shown to reduce kidney inflammation in mice[70] and also exhibits protective effects against proteinuria and interstitial fibrosis in salt-sensitive hypertensive rats[49]. Additionally, translational studies suggest benefits from messenger RNA-based therapies targeting P2X7R[71], although other reports present conflicting findings[72]. Recently, a preclinical study introduced a novel, orally administered, long-acting P2X7R antagonist that demonstrated promising results in slowing CKD progression[73].
P2X7R IN METABOLIC DYSFUNCTION-ASSOCIATED STEATOTIC LIVER DISEASE
Currently, metabolic dysfunction-associated steatotic liver disease (MASLD), formerly known as non-alcoholic fatty liver disease (NAFLD), is the leading cause of liver disease worldwide and is expected to become the primary indication for liver transplantation starting from 2023[74].
The role of adipose tissue expansion in its development is well established, as MASLD is characterized by abnormal fat accumulation in the liver, typically defined as at least 5% steatosis, in the absence of pre-existing liver disease. Under the influence of genetic predisposition and environmental factors, this condition often progresses to hepatocyte damage, inflammation, and metabolic-associated steatohepatitis (MASH), and further advances to liver fibrosis and ultimately cirrhosis[75]. MASLD carries an increased risk of hepatocellular carcinoma, even before the development of overt cirrhosis. In addition, it has been identified as an independent risk factor for cardiovascular morbidity and mortality, with cardiovascular complications being the leading cause of death among affected patients[76]. Early-stage MASLD is associated with manifestations of sub-clinical atherosclerosis, such as impaired flow-mediated dilation and increased carotid intima-media thickness[77,78], with liver disease acting both as a driver of inflammatory cascade amplification and as a consequence of metabolic derangement. These epidemiological data underscore the importance of MASLD screening in all at-risk subjects, as the treatment of MASLD may significantly impact clinical outcomes.
The primary treatment for MASLD remains weight-loss interventions, which have demonstrated progressively greater histological improvement with increasing weight reduction[79]. Data from phase III clinical trials also indicate significant benefits from incretin-based therapies, particularly in the early stages of the disease[80], although the histological improvements achieved with bariatric surgery are more pronounced[81]. Currently, fibrosis regression remains an unmet goal, with the exception of the recently Food and Drug Administration (FDA)-approved drug Resmetirom, which may represent a breakthrough for the treatment of liver cirrhosis[82]. Therefore, early and effective treatment strategies remain essential.
The triggers driving the progression from simple steatosis to MASH and fibrosis are not yet fully understood. Lipotoxicity plays a central role, as lipid-derived products induce hepatocyte damage and initiate local inflammatory responses[83]. Following hepatocellular injury, increased extracellular ATP promotes pathological responses that sustain cell death and fibrogenesis through the purinergic pathway[84,85]. Moreover, P2X7R contributes to acute liver injury, including drug-induced hepatotoxicity[86] and sepsis-related liver damage[87]. P2X7R also plays a key role in MASH development by activating IL-1β, which stimulates hepatic Kupffer cells and stellate cells, leading to inflammation and ECM deposition, respectively[88,89]. IL-1β antagonism has shown protective effects in alcohol-related steatohepatitis[90]. Human liver samples indicate higher inflammasome activity in steatoinflammatory conditions compared to simple fatty liver[91,92]. P2X7R-mediated inflammasome activation is required for abnormal ECM synthesis by stellate cells, and P2X7R antagonists inhibit liver fibrosis, as demonstrated in carbon tetrachloride-induced models, where they reduced the expression of α-smooth muscle actin, TGF-β, and other ECM components[93]. The inflammasome is integral to liver damage mechanisms[94,95], with studies in NLRP3, caspase-1, or IL-1β KO mice showing less susceptibility to high-fat diet-induced hepatitis and fibrosis[96,97]. Nutritional stress triggers fatty liver infiltration and inflammation via inflammasome activation in mice, with P2X7R deletion blunting this inflammatory response[98]. Interestingly, some reports suggest that P2X7R activation might support autophagy and lipid droplet degradation[99]. A recent hypothesis proposes that loss of P2X7R function in the intestinal mucosa, more than its overexpression, contributes to hepatic steatosis through increased glucose transport across the gut barrier[100]. Furthermore, leptin levels seem to influence the effect of P2X7R on the liver[101].
Therefore, targeting the inflammasome may be a promising therapeutic approach for MASLD and MASH. Animal studies using antagonists have yielded promising results[102]. As treatment strategies are shifting from single drugs to combination therapies[103], reflecting the complex MASLD pathogenesis[104], future translational studies should adopt a multi-targeted approach, including the P2X7R antagonism.
P2X7R-ADIPOSE TISSUE INTERACTION IN CANCER
As epidemiological data certified the influence of metabolic abnormalities and obesity on the risk of certain types of cancer[105], pathophysiological hypotheses for this relationship are accumulating, mainly focusing on the role of meta-inflammation.
Adipose dysfunction and chronic inflammation are related to the development of some gastrointestinal tract and reproductive organ cancers[106]. Obesity disrupts adipokines profile and insulin signaling, which in turn may foster cancer development[107]. The interactions between adipose tissue and cancer lie in both direct contact-dependent signaling and the paracrine communication between adipocytes and the stromal-vascular fraction[108]. Inadequate oxygenation and hypoxia result in a vicious cycle of cell death and further recruitment of immune cells and release of cytokines, chemokines, growth factors, and matrix-degrading enzymes such as matrix metalloproteases (MMPs)[109]. As a consequence of this tissue debridement, extracellular purine concentrations increase, supporting a role for P2X7R in cancer development.
P2X7R splice variants with gain-of-function contribute to tumor microenvironment (TME) characteristics, enhancing cell proliferation, angiogenesis, ECM degradation, and metastatic spreading[110]. In addition, ATP is actively accumulated near TME, with a chronic stimulation of the purinergic-mediated inflammation[111]. Animal models have evaluated the potential role of P2X7R antagonism, reporting promising results in pancreatic and mammary cancers[112,113]. Thus, it is plausible that P2X7R contributes to the adipose tissue-cancer crosstalk, suggesting a new area for pathophysiological and pharmacological research.
PAST ATTEMPTS AND FUTURE DIRECTIONS FOR P2X7R AS PHARMACOLOGICAL TARGET
Accumulating evidence is demonstrating the potential of immune-modulating therapy in improving clinical outcomes in cardiometabolic and atherosclerotic diseases. However, to date, their application remains limited due to the potential harmful side effects.
Due to its broad spectrum of biological involvement, the P2X7R has been considered in a different continuum of pathological conditions ranging from chronic pain to depression and bipolar disorder, but often with unsatisfactory results[114]. Although animal models targeting P2X7 in CVD have shown some benefits, to date, there have been no clinical trials investigating P2X7 antagonism in human CVD.
We have paid particular attention to the development of molecules such as SGM-1019, an oral P2X7R inhibitor. In healthy volunteers, administration of SGM-1019 up to twice daily for two weeks was shown to be safe and fully inhibited P2X7R in whole blood; however, in MASH patients, an unfavorable risk-benefit profile led to study discontinuation[115]. In this context, we offer several suggestions for future study design. There remains considerable debate regarding the endpoints used in clinical trials for MASH/fibrosis treatment, as several promising drugs have subsequently proven ineffective. We also highlight the need to shift from the classical single-molecule approach toward combination therapy (multi-agonism/antagonism)[116]. It would be reasonable to test P2X7R antagonism in combination with other molecules of proven efficacy, such as Semaglutide, to enhance protective effects and potentially allow dose reduction, thereby attenuating side effects; ongoing phase II studies on inflammasome inhibitors are now being conducted from this perspective[117]. Additionally, a recent study revealed structural differences between rodents and humans that may account for inconsistencies in translational outcomes [118]. All these considerations should be taken into account in the design of future studies, offering a promising avenue for P2X7R antagonism.
CONCLUSIONS
The cardiovascular-kidney-metabolic syndrome encompasses the full spectrum of clinical complications that can arise during the course of obesity and metabolic syndrome. In the context of the global obesity epidemic, this syndrome presents a major challenge to health systems worldwide, underscoring the importance of both clinical and translational research. Recent clinical trials with incretin-based therapies offer new hope; however, effective management of the syndrome requires a multi-level approach. Evidence from the scientific literature highlights the central role of the P2X7 receptor in mediating cell damage and inflammation that drive cardiometabolic complications, suggesting that receptor inhibition - at least as an add-on therapy - merits further investigation. As our understanding of the pathophysiology advances, integrating these insights into a comprehensive evaluation and treatment strategy will be critical.
DECLARATIONS
Acknowledgments
We thank Sonia Leandri for her invaluable support. The Graphical Abstract was created using BioRender (Di Vincenzo A, 2026; https://BioRender.com/9tci9qc).
Authors’ contributions
Wrote the first draft of the manuscript: Di Vincenzo A, Rossato M
Performed article revision and editing: Di Vincenzo A, Granzotto M, Crescenzi M, Capone F, Fioretto P, Rossato M
Prepared figures and images: Di Vincenzo A, Granzotto M, Crescenzi M
All authors contributed equally to the conception and design of the article.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Di Vincenzo A and Rossato M serve as the Guest Editors of the Special Issue "Adipose Tissue and Metabolism in Chronic Diseases" of Metabolism and Target Organ Damage. Di Vincenzo A is a Youth Editorial Board Member of Metabolism and Target Organ Damage. They were not involved in any stage of the editorial process for this manuscript, including reviewer selection, manuscript handling, or decision-making. The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
2. Dandona P, Dhindsa S. Update: hypogonadotropic hypogonadism in type 2 diabetes and obesity. J Clin Endocrinol Metab. 2011;96:2643-51.
3. Fuster JJ, Ouchi N, Gokce N, Walsh K. Obesity-induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ Res. 2016;118:1786-807.
4. Sbraccia P, D’Adamo M, Guglielmi V. Is type 2 diabetes an adiposity-based metabolic disease? From the origin of insulin resistance to the concept of dysfunctional adipose tissue. Eat Weight Disord. 2021;26:2429-41.
5. Powell-Wiley TM, Poirier P, Burke LE, et al. ; American Heart Association Council on Lifestyle and Cardiometabolic Health; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Epidemiology and Prevention; and Stroke Council. Obesity and cardiovascular disease: a scientific statement from the American heart association. Circulation. 2021;143:e984-e1010.
6. Jiang Z, Wang Y, Zhao X, et al. Obesity and chronic kidney disease. Am J Physiol Endocrinol Metab. 2023;324:E24-41.
8. Thornberry NA, Bull HG, Calaycay JR, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768-74.
9. Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in muckle-wells autoinflammatory disorder. Immunity. 2004;20:319-25.
10. Burnstock G, Verkhratsky A. Evolutionary origins of the purinergic signalling system. Acta Physiol (Oxf). 2009;195:415-47.
11. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity. 2017;47:15-31.
13. Di Virgilio F, Vuerich M. Purinergic signaling in the immune system. Auton Neurosci. 2015;191:117-23.
14. Di Virgilio F, Sarti AC, Coutinho-Silva R. Purinergic signaling, DAMPs, and inflammation. Am J Physiol Cell Physiol. 2020;318:C832-5.
15. Ferrari D, Pizzirani C, Adinolfi E, et al. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877-83.
17. Tchernof A, Després JP. Pathophysiology of human visceral obesity: an update. Physiol Rev. 2013;93:359-404.
18. Chouchani ET, Kajimura S. Metabolic adaptation and maladaptation in adipose tissue. Nat Metab. 2019;1:189-200.
19. Rohm TV, Meier DT, Olefsky JM, Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity. 2022;55:31-55.
20. Wu H, Ballantyne CM. Metabolic Inflammation and insulin resistance in obesity. Circ Res. 2020;126:1549-64.
21. Iacobellis G. Epicardial adipose tissue in contemporary cardiology. Nat Rev Cardiol. 2022;19:593-606.
22. D'Agati VD, Chagnac A, de Vries AP, et al. Obesity-related glomerulopathy: clinical and pathologic characteristics and pathogenesis. Nat Rev Nephrol. 2016;12:453-71.
23. Rossato M, Favaretto F, Granzotto M, et al. Molecular and pharmacological evidence for the expression of multiple functional P2 purinergic receptors in human adipocytes. Molecules. 2022;27:1913.
24. Wang D, Zhou J. Purinergic receptor: a crucial regulator of adipose tissue functions. Purinerg Signal. 2023;19:273-81.
25. Caruso V, Zuccarini M, Di Iorio P, Muhammad I, Ronci M. Metabolic changes induced by purinergic signaling: role in food intake. Front Pharmacol. 2021;12:655989.
26. Panupinthu N, Rogers JT, Zhao L, et al. P2X7 receptors on osteoblasts couple to production of lysophosphatidic acid: a signaling axis promoting osteogenesis. J Cell Biol. 2008;181:859-71.
27. Giacovazzo G, Apolloni S, Coccurello R. Loss of P2X7 receptor function dampens whole body energy expenditure and fatty acid oxidation. Purinerg Signal. 2018;14:299-305.
28. Tian T, Heine M, Evangelakos I, et al. The P2X7 ion channel is dispensable for energy and metabolic homeostasis of white and brown adipose tissues. Purinerg Signal. 2020;16:529-42.
29. Burnstock G, Gentile D. The involvement of purinergic signalling in obesity. Purinerg Signal. 2018;14:97-108.
30. Madec S, Rossi C, Chiarugi M, et al. Adipocyte P2X7 receptors expression: a role in modulating inflammatory response in subjects with metabolic syndrome? Atherosclerosis. 2011;219:552-8.
31. Giuliani AL, Berchan M, Sanz JM, et al. The P2X7 receptor is shed into circulation: correlation with c-reactive protein levels. Front Immunol. 2019;10:793.
32. Di Vincenzo A, Granzotto M, Graziani A, et al. Soluble P2X7 receptor plasma levels in obese subjects before and after weight loss via bariatric surgery. Int J Mol Sci. 2023;24:16741.
33. Stienstra R, van Diepen JA, Tack CJ, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A. 2011;108:15324-9.
34. de Torre-Minguela C, Barberà-Cremades M, Gómez AI, Martín-Sánchez F, Pelegrín P. Macrophage activation and polarization modify P2X7 receptor secretome influencing the inflammatory process. Sci Rep. 2016;6:22586.
35. Beaucage KL, Xiao A, Pollmann SI, et al. Loss of P2X7 nucleotide receptor function leads to abnormal fat distribution in mice. Purinerg Signal. 2014;10:291-304.
36. Di Vincenzo A, Granzotto M, Crescenzi M, Vettor R, Rossato M. Non-aromatizable androgens modulate the lipopolysaccharide induced expression of the P2X7 receptor in human adipocytes. Front Pharmacol. 2023;14:1251035.
37. Escobar-Morreale HF, Santacruz E, Luque-Ramírez M, Botella Carretero JI. Prevalence of 'obesity-associated gonadal dysfunction’ in severely obese men and women and its resolution after bariatric surgery: a systematic review and meta-analysis. Hum Reprod Update. 2017;23:390-408.
38. Becher T, Palanisamy S, Kramer DJ, et al. Brown adipose tissue is associated with cardiometabolic health. Nat Med. 2021;27:58-65.
39. O’Mara AE, Johnson JW, Linderman JD, et al. Chronic mirabegron treatment increases human brown fat, HDL cholesterol, and insulin sensitivity. J Clin Invest. 2020;130:2209-19.
40. Coccurello R, Volonté C. P2X7 Receptor in the management of energy homeostasis: implications for obesity, dyslipidemia, and insulin resistance. Front Endocrinol (Lausanne). 2020;11:199.
41. Adinolfi E, Callegari MG, Ferrari D, et al. Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol Biol Cell. 2005;16:3260-72.
42. Giacovazzo G, Fabbrizio P, Apolloni S, Coccurello R, Volonté C. Stimulation of P2X7 enhances whole body energy metabolism in mice. Front Cell Neurosci. 2019;13:390.
43. Orioli E, De Marchi E, Giuliani AL, Adinolfi E. P2X7 Receptor orchestrates multiple signalling pathways triggering inflammation, autophagy and metabolic/trophic responses. Curr Med Chem. 2017;24:2261-75.
44. Chiang CH, Cheng CY, Lien YT, Huang KC, Lin WW. P2X7 Activation enhances lipid accumulation during adipocytes differentiation through suppressing the expression of sirtuin-3, sirtuin-5, and browning genes. Front Pharmacol. 2022;13:852858.
45. Okla M, Zaher W, Alfayez M, Chung S. Inhibitory Effects of toll-like receptor 4, NLRP3 inflammasome, and interleukin-1β on white adipocyte browning. Inflammation. 2018;41:626-42.
46. Jaeckstein MY, Fischer AW, Rissiek B, et al. Purinergic adipocyte-macrophage crosstalk promotes degeneration of thermogenic brown adipose tissue. EMBO Rep. 2025;26:6460-93.
47. Soehnlein O, Lutgens E, Döring Y. Distinct inflammatory pathways shape atherosclerosis in different vascular beds. Eur Heart J. 2025;46:3261-72.
48. Rossi C, Santini E, Chiarugi M, et al. The complex P2X7 receptor/inflammasome in perivascular fat tissue of heavy smokers. Eur J Clin Investig. 2014;44:295-302.
49. Ji X, Naito Y, Hirokawa G, et al. P2X(7) receptor antagonism attenuates the hypertension and renal injury in Dahl salt-sensitive rats. Hypertens Res. 2012;35:173-9.
50. Ridker PM, Everett BM, Thuren T, et al. ; CANTOS Trial Group. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119-31.
51. Lombardi M, Mantione ME, Baccellieri D, et al. P2X7 receptor antagonism modulates IL-1β and MMP9 in human atherosclerotic vessels. Sci Rep. 2017;7:4872.
52. Piscopiello M, Sessa M, Anzalone N, et al. P2X7 receptor is expressed in human vessels and might play a role in atherosclerosis. Int J Cardiol. 2013;168:2863-6.
53. Bracey NA, Gershkovich B, Chun J, et al. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J Biol Chem. 2014;289:19571-84.
54. Bracey NA, Beck PL, Muruve DA, et al. The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Exp Physiol. 2013;98:462-72.
55. Luo B, Li B, Wang W, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE. 2014;9:e104771.
56. Ye T, Zhou Y, Yang J, et al. P2X7 receptor inhibition prevents atrial fibrillation in rodent models of depression. Europace. 2024;26:euae022.
57. Ye T, Yang J, Liu Z, et al. Inhibition of the P2X7 receptor prevents atrial proarrhythmic remodeling in experimental post-operative atrial fibrillation. Int Immunopharmacol. 2024;129:111536.
58. Yin J, Wang Y, Hu H, et al. P2X(7) receptor inhibition attenuated sympathetic nerve sprouting after myocardial infarction via the NLRP3/IL-1β pathway. J Cell Mol Med. 2017;21:2695-710.
59. Sun Y, Ma X, Gong Y, et al. Inhibition of P2X7R by hypericin improves diabetic cardiac autonomic neuropathy through the proteasome- Nrf2 - GPX4 signaling axis. Neurotoxicology. 2025;109:1-10.
60. Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res. 2015;116:991-1006.
61. Choi JW, Lee CM, Kang BK, Kim M. Perirenal fat thickness is an independent predictor for metabolic syndrome in steatotic liver disease. Sci Rep. 2024;14:26548.
62. Menzies RI, Booth JWR, Mullins JJ, et al. Hyperglycemia-induced Renal P2X7 receptor activation enhances diabetes-related injury. EBioMedicine. 2017;19:73-83.
63. Menzies RI, Tam FW, Unwin RJ, Bailey MA. Purinergic signaling in kidney disease. Kidney Int. 2017;91:315-23.
64. Franco M, Bautista-Pérez R, Cano-Martínez A, et al. Physiopathological implications of P2X1 and P2X7 receptors in regulation of glomerular hemodynamics in angiotensin II-induced hypertension. Am J Physiol Renal Physiol. 2017;313:F9-F19.
65. Pandit MM, Inscho EW, Zhang S, et al. Flow regulation of endothelin-1 production in the inner medullary collecting duct. Am J Physiol Renal Physiol. 2015;308:F541-52.
66. Mounieb F, Abdel-Sattar SA, Balah A, Akool ES. P2 X 7 receptor is a critical regulator of extracellular ATP-induced profibrotic genes expression in rat kidney: implication of transforming growth factor-β/Smad signaling pathway. Purinerg Signal. 2024;20:421-30.
67. Menzies RI, Howarth AR, Unwin RJ, Tam FW, Mullins JJ, Bailey MA. Inhibition of the purinergic P2X7 receptor improves renal perfusion in angiotensin-II-infused rats. Kidney Int. 2015;88:1079-87.
68. Solini A, Menini S, Rossi C, et al. The purinergic 2X7 receptor participates in renal inflammation and injury induced by high-fat diet: possible role of NLRP3 inflammasome activation. J Pathol. 2013;231:342-53.
69. Yan Y, Bai J, Zhou X, et al. P2X7 receptor inhibition protects against ischemic acute kidney injury in mice. Am J Physiol Cell Physiol. 2015;308:C463-72.
70. Pereira JMS, Barreira AL, Gomes CR, et al. Brilliant blue G, a P2X7 receptor antagonist, attenuates early phase of renal inflammation, interstitial fibrosis and is associated with renal cell proliferation in ureteral obstruction in rats. BMC Nephrol. 2020;21:206.
71. Rodrigues AM, Serralha RS, Lima DY, et al. P2X7 siRNA targeted to the kidneys increases klotho and delays the progression of experimental diabetic nephropathy. Purinerg Signal. 2020;16:175-85.
72. Nespoux J, Monaghan MT, Jones NK, et al. P2X7 receptor knockout does not alter renal function or prevent angiotensin II-induced kidney injury in F344 rats. Sci Rep. 2024;14:9573.
73. Zhang R, Su K, Yang L, et al. Discovery of a potent, orally active, and long-lasting P2X7 receptor antagonist as a preclinical candidate for delaying the progression of chronic kidney disease. J Med Chem. 2024;67:17472-96.
74. Eslam M, Newsome PN, Sarin SK, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73:202-9.
75. Stefan N, Yki-Järvinen H, Neuschwander-Tetri BA. Metabolic dysfunction-associated steatotic liver disease: heterogeneous pathomechanisms and effectiveness of metabolism-based treatment. Lancet Diabetes Endocrinol. 2025;13:134-48.
76. Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med. 2010;363:1341-50.
77. Al-Hamoudi W, Alsadoon A, Hassanian M, et al. Endothelial dysfunction in nonalcoholic steatohepatitis with low cardiac disease risk. Sci Rep. 2020;10:8825.
78. Tarantino G, Costantini S, Finelli C, et al. Carotid intima-media thickness is predicted by combined eotaxin levels and severity of hepatic steatosis at ultrasonography in obese patients with nonalcoholic fatty liver disease. PLoS ONE. 2014;9:e105610.
79. Romero-Gómez M, Zelber-Sagi S, Trenell M. Treatment of NAFLD with diet, physical activity and exercise. J Hepatol. 2017;67:829-46.
80. Sanyal AJ, Newsome PN, Kliers I, et al. ; ESSENCE Study Group. Phase 3 trial of semaglutide in metabolic dysfunction-associated steatohepatitis. N Engl J Med. 2025;392:2089-99.
81. Verrastro O, Panunzi S, Castagneto-Gissey L, et al. Bariatric-metabolic surgery versus lifestyle intervention plus best medical care in non-alcoholic steatohepatitis (BRAVES): a multicentre, open-label, randomised trial. Lancet. 2023;401:1786-97.
82. Harrison SA, Bedossa P, Guy CD, et al. A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. N Engl J Med. 2024;390:497-509.
83. Mladenić K, Lenartić M, Marinović S, Polić B, Wensveen FM. The “Domino effect” in MASLD: the inflammatory cascade of steatohepatitis. Eur J Immunol. 2024;54:e2149641.
84. Velázquez-Miranda E, Díaz-Muñoz M, Vázquez-Cuevas FG. Purinergic signaling in hepatic disease. Purinerg Signal. 2019;15:477-89.
85. Lu D, Insel PA. Cellular mechanisms of tissue fibrosis. 6. Purinergic signaling and response in fibroblasts and tissue fibrosis. Am J Physiol Cell Physiol. 2014;306:C779-88.
86. Hoque R, Sohail MA, Salhanick S, et al. P2X7 receptor-mediated purinergic signaling promotes liver injury in acetaminophen hepatotoxicity in mice. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1171-9.
87. Savio LEB, de Andrade Mello P, Figliuolo VR, et al. CD39 limits P2X7 receptor inflammatory signaling and attenuates sepsis-induced liver injury. J Hepatol. 2017;67:716-26.
88. Rossato M, Di Vincenzo A, Pagano C, El Hadi H, Vettor R. The P2X7 receptor and NLRP3 axis in non-alcoholic fatty liver disease: a brief review. Cells. 2020;9:1047.
89. Gaul S, Leszczynska A, Alegre F, et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol. 2021;74:156-67.
90. Petrasek J, Bala S, Csak T, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122:3476-89.
91. He K, Zhu X, Liu Y, et al. Inhibition of NLRP3 inflammasome by thioredoxin-interacting protein in mouse Kupffer cells as a regulatory mechanism for non-alcoholic fatty liver disease development. Oncotarget. 2017;8:37657-72.
92. Jiang S, Zhang Y, Zheng JH, et al. Potentiation of hepatic stellate cell activation by extracellular ATP is dependent on P2X7R-mediated NLRP3 inflammasome activation. Pharmacol Res. 2017;117:82-93.
93. Huang C, Yu W, Cui H, et al. P2X7 blockade attenuates mouse liver fibrosis. Mol Med Rep. 2014;9:57-62.
94. Huang H, Chen HW, Evankovich J, et al. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J Immunol. 2013;191:2665-79.
95. Wu T, Zhang C, Shao T, Chen J, Chen D. The role of NLRP3 inflammasome activation pathway of hepatic macrophages in liver ischemia-reperfusion injury. Front Immunol. 2022;13:905423.
96. Mridha AR, Wree A, Robertson AAB, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. 2017;66:1037-46.
97. Baeza-Raja B, Goodyear A, Liu X, et al. Pharmacological inhibition of P2RX7 ameliorates liver injury by reducing inflammation and fibrosis. PLoS ONE. 2020;15:e0234038.
98. Blasetti Fantauzzi C, Menini S, Iacobini C, et al. Deficiency of the purinergic receptor 2X(7) attenuates nonalcoholic steatohepatitis induced by high-fat diet: possible role of the NLRP3 inflammasome. Oxid Med Cell Longev. 2017;2017:8962458.
99. Dong Z, Wei Y, Tao M, Zhang L. Activation of the purinergic receptor P2X7 improves hepatosteatosis by promoting lipophagy. FEBS Lett. 2021;595:2768-80.
100. Arguin G, Bourzac JF, Placet M, et al. The loss of P2X7 receptor expression leads to increase intestinal glucose transit and hepatic steatosis. Sci Rep. 2017;7:12917.
101. Chandrashekaran V, Das S, Seth RK, et al. Purinergic receptor X7 mediates leptin induced GLUT4 function in stellate cells in nonalcoholic steatohepatitis. Biochim Biophys Acta. 2016;1862:32-45.
102. Povero D, Lazic M, McBride C, et al. Pharmacology of a potent and novel inhibitor of the NOD-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome that attenuates development of nonalcoholic steatohepatitis and liver fibrosis. J Pharmacol Exp Ther. 2023;386:242-58.
103. Xiang L, Wang G, Zhuang Y, et al. Safety and efficacy of GLP-1/FGF21 dual agonist HEC88473 in MASLD and T2DM: a randomized, double-blind, placebo-controlled study. J Hepatol. 2025;82:967-78.
104. Chatterjee S, Das S. P2X7 receptor as a key player in oxidative stress-driven cell fate in nonalcoholic steatohepatitis. Oxid Med Cell Longev. 2015;2015:172493.
105. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625-38.
107. Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nat Rev Cancer. 2011;11:886-95.
108. Lengyel E, Makowski L, DiGiovanni J, Kolonin MG. Cancer as a matter of fat: the crosstalk between adipose tissue and tumors. Trends Cancer. 2018;4:374-84.
109. Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094-101.
111. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 Purinerg Signal in the tumour microenvironment. Nat Rev Cancer. 2018;18:601-18.
112. Giannuzzo A, Saccomano M, Napp J, Ellegaard M, Alves F, Novak I. Targeting of the P2X7 receptor in pancreatic cancer and stellate cells. Int J Cancer. 2016;139:2540-52.
113. Brisson L, Chadet S, Lopez-Charcas O, et al. P2X7 receptor promotes mouse mammary cancer cell invasiveness and tumour progression, and is a target for anticancer treatment. Cancers (Basel). 2020;12:2342.
114. Shokoples BG, Paradis P, Schiffrin EL. P2X7 receptors: an untapped target for the management of cardiovascular disease. Arterioscler, Thromb, Vasc Biol. 2021;41:186-99.
115. Clinical Trial Arena Home Page. Available from: https://www.clinicaltrialsarena.com. (accessed 21 January 2026).
116. Muskiet MHA, Smits MM. Beyond a singular focus on GLP-1: why we need a new nomenclature now. Lancet Diabetes Endocrinol. 2025;13:730-3.
117. Ventrx bioscience Home Page. Available from: https://ir.ventyxbio.com. (accessed 21 January 2026).
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].