



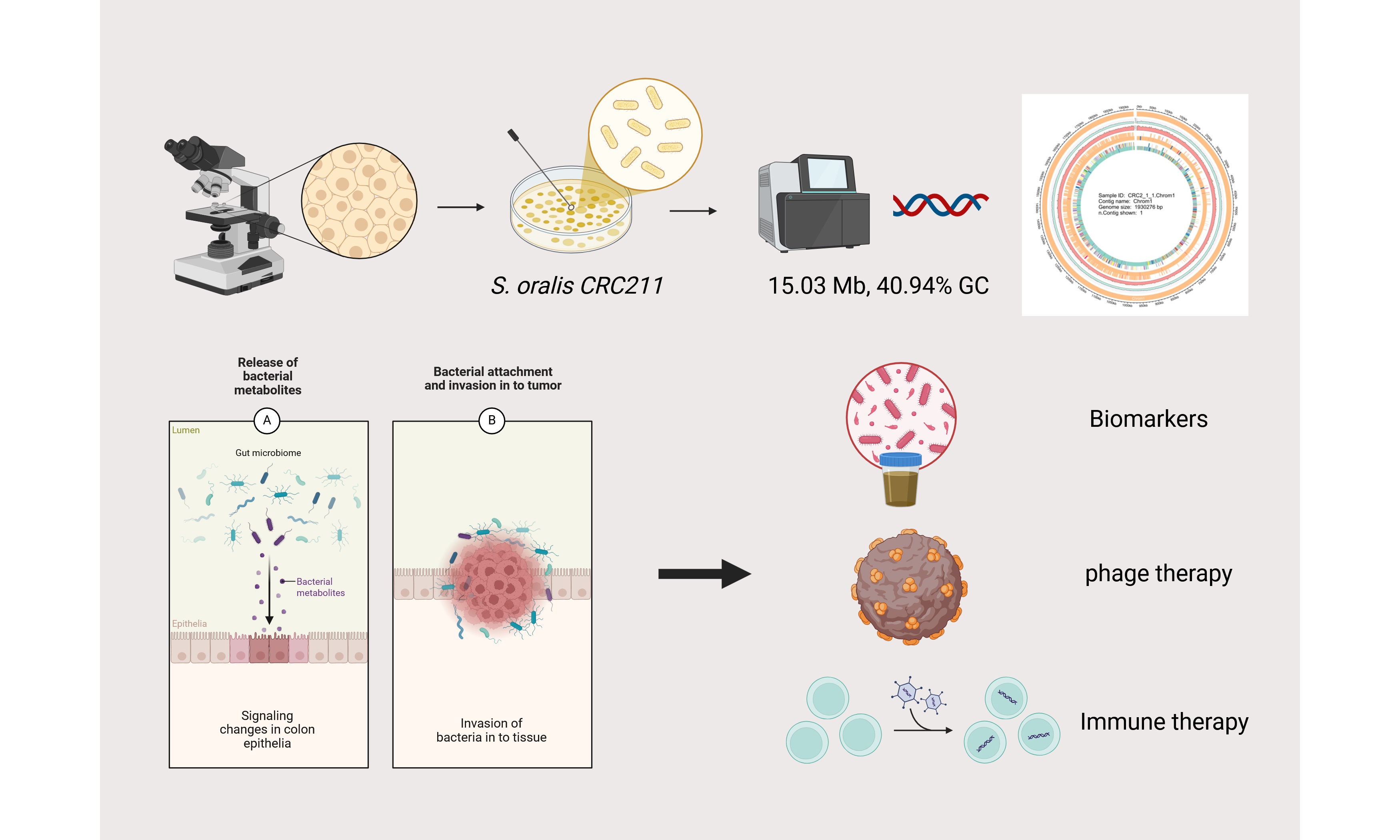
Whole-genome sequencing and analysis of a novel strain Streptococcus oralis CRC211 from colorectal tumor
 0
0 Abstract
Aim: This study provides a comprehensive genomic characterization of Streptococcus oralis CRC211, a novel bacterial strain isolated from colorectal tumor tissue.
Methods: Whole-genome sequencing and comparative genomic analyses were performed.
Results: The high-quality assembled genome (15.03 Mb, 40.94% guanine-cytosine content) contains 2 prophage regions spanning 160.5 kb, which may facilitate the horizontal transfer of virulence genes. Functional annotation identified 3,674 genes, with significant enrichment in metabolic pathways (amino acid and carbohydrate metabolism) and virulence factors (116 genes in Virulence Factors Batabase), including adhesins and biofilm-associated proteins that likely promote tumor colonization. Comparative genomic analysis revealed that CRC211 shares 92.29% average nucleotide identity with reference Streptococcus oralis strains, while pan-genome analysis demonstrated an open genome structure with 1,222 conserved core genes. In addition, the strain also carries 75 antimicrobial resistance genes, underscoring its potential clinical relevance. Notably, the genomic profile indicates adaptations for nutrient acquisition and immune evasion in the tumor microenvironment.
Conclusion: These findings establish CRC211 as a colorectal cancer (CRC)-associated strain with distinct genomic features that may contribute to tumor progression. The study provides critical insights into its possible oncogenic mechanisms and highlights potential applications in mic ases,indels - changerobiota-based diagnostics or therapeutics for colorectal cancer.
Keywords
INTRODUCTION
The intratumoral microbiota has gained significant attention in recent years as a critical component of the tumor microenvironment, with emerging evidence highlighting its impact on tumorigenesis, immune modulation, and therapeutic response. Large-scale profiling studies have identified tumor-type-specific microbial signatures across multiple cancers, suggesting potential diagnostic and prognostic applications[1,2]. Notably, the gut-tumor axis has been implicated in colorectal cancer (CRC), where Fusobacterium nucleatum promotes chemoresistance through autophagy activation[3]. In pancreatic cancer, intratumoral bacteria have been shown to metabolize chemotherapy drugs and modulate tumor-associated macrophages[4]. Recent single-cell analyses have also revealed the spatial organization of microbiota within tumors and their interactions with immune cells[5]. These findings have spurred growing interest in microbiota-targeted interventions, including phage therapy and microbial modulation strategies to enhance immunotherapy efficacy[6].
Bacteria affect the tumor microenvironment (TME) through multiple key mechanisms. Certain species, such as Fusobacterium nucleatum, can infiltrate tumors directly and activate pro-survival signaling pathways (e.g., TLR4/NF-κB) in cancer cells, thereby promoting proliferation and inflammation[7]. Additionally, bacteria can modulate local immune responses by recruiting immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs), creating an immune-tolerant TME that supports tumor progression[8]. Bacterial metabolites, including short-chain fatty acids, can also epigenetically regulate gene expression in host and immune cells, further influencing TME functionality[9]. More recently, the oral-gut axis has been proposed as a key route for bacterial translocation to colorectal tumors, with oral pathogens such as Streptococcus oralis (S. oralis) exploiting mucosal inflammation and immune dysregulation to establish intratumoral niches. Together, these interactions highlight the multifaceted role of intratumoral bacteria in shaping a permissive tumor microenvironment. However, challenges remain in distinguishing causative microbial drivers from passenger species, as well as in elucidating the mechanisms of microbial translocation and colonization within tumors.
S. oralis, a commensal bacterium of the human oral cavity, has recently emerged as a potential oncogenic contributor in intratumoral microbiota studies, particularly in CRC. Research indicates that S. oralis is enriched in CRC tissues compared to adjacent normal mucosa, suggesting its possible role in tumorigenesis[10]. Mechanistically, S. oralis may promote CRC progression by inducing chronic inflammation, activating oncogenic pathways such as NF-κB, and producing genotoxic metabolites including reactive oxygen species[11,12]. Recent studies have shown that streptococcal species can enhance the invasiveness of CRC cells in vitro and promote tumor development in xenograft models through modulation of host signaling pathways[13]. Similar to other oncogenic bacteria such as Fusobacterium nucleatum, S. oralis may also contribute to tumor progression by modulating the tumor immune microenvironment, as bacterial presence within tumors has been associated with altered patient prognosis[14]. The ability of S. oralis to form biofilms on colorectal mucosa may further facilitate its persistence and chronic inflammatory effects[15]. Additionally, S. oralis has been reported to enhance the invasiveness of CRC cells in vitro by modulating the TME[16]. Further studies are therefore needed to clarify the precise mechanisms through which S. oralis influences CRC development and to assess its potential as a diagnostic or therapeutic target.
In this study, we isolated an S. oralis strain, CRC211, from the tumor tissue of a patient with CRC, and performed sequencing and comparative genomic analyses with other S. oralis strains to better understand its pathogenicity.
METHODS
Bacterial strain and DNA preparation
The study protocol was reviewed and approved by the Ethics Committee of Changhai Hospital, Naval Medical University (Ethics No. CHEC2023-307). Written informed consent was obtained from all participants. Tumor tissue samples were collected from ten treatment-naïve CRC patients and processed for bacterial isolation. Following anaerobic culture on Columbia blood agar, multiple colonies were selected based on morphology and identified by 16S rRNA Sanger sequencing. Streptococcus species were predominant among the isolates (detected in 8/10 patients), with S. oralis being the most frequent (6/10 patients). Three S. oralis strains with high sequencing quality were selected for preliminary genomic comparison. From these, strain CRC211 was chosen for comprehensive whole-genome sequencing because it displayed representative genetic features (e.g., virulence genes and prophage regions) and was isolated from a mid-stage CRC patient who had not received antimicrobial pretreatment, minimizing potential therapy-induced microbial bias.
S. oralis CRC211 was isolated from tumor tissue obtained from a patient with early-to-mid-stage colon cancer at the Changhai Hospital, Shanghai, China. The patient had not undergone neoadjuvant chemoradiotherapy prior to surgical treatment. The tissue sample was homogenized in sterile phosphate-buffered saline and plated on Columbia blood agar plates (BioMérieux, France). After anaerobic incubation at 37 °C for 48 h, single colonies were selected and identified by 16S rRNA Sanger sequencing. Genomic DNA was extracted using the NucleoBond® HMW DNA kit (MN NucleoBond, Germany, 740160.20). DNA concentration and purity were determined via Qubit4.0 (Thermo, Q33226) and Nanodrop (SMA4000, Taiwan, China). DNA integrity was assessed by 0.75% agarose gel electrophoresis.
Genome sequencing, assembly, and gene annotation
Genomic DNA (gDNA) was processed in parallel: one portion was randomly fragmented to construct a 300-bp insert library for paired-end 150-bp sequencing on the Illumina NovaSeq 6000 (Illumina, USA), while the other was sheared using g-TUBEs (Covaris, USA) to generate ~ 20-kb fragments. The Illumina library was prepared using the NEBNext Ultra II DNA Library Prep Kit (NEB, USA) according to the manufacturer’s instructions. The PacBio library was constructed using the SMRTbell Express Template Prep Kit 2.0 (PacBio, USA). After purification with AMPure PB beads (Pacific Biosciences, USA) and quality assessment by 0.7% agarose gel electrophoresis, qualified samples were sequenced on the PacBio Sequel II platform using the Sequel II Binding Kit 2.0 and Sequel II SMRT Cells 8M, with a 10-hour movie time. Assembly of PacBio/Nanopore long-read data was performed using Canu (v2.2) and Unicycler (v0.5.0) with default parameters. Next-generation sequencing (NGS) data were incorporated to close assembly gaps using GapFiller (v1.10). GapFiller utilizes paired-end read information to iteratively extend contigs into gap regions, applying a minimum overlap of 30 bp and base quality of 20. This approach effectively resolved 92% of gaps in the preliminary assembly. Sequence polishing was conducted with Pilon (v1.24) to correct base-calling errors and small indels. Pilon was run for 2 iterative cycles using the Illumina reads aligned to the draft assembly with BWA-MEM (v0.7.17). The following parameters were applied: - fix bases,indels - changes - vcf - tracks. Assembly completeness and contamination were assessed using CheckM (v1.2.2).
Gene prediction was performed with Prokka (v1.14.6) using default parameters. Functional annotation was conducted by aligning predicted protein sequences against multiple databases using BLAST+ (v2.13.0) with an e-value cutoff of 1e-5, percent identity > 30%, and query coverage > 50%. Specifically, COG annotation was performed using rpsBLAST against the COG database; KEGG pathways were assigned using KAAS with the bidirectional best-hit method; Gene Ontology (GO) terms were annotated using InterProScan (v5.56-89.0); virulence factors were identified using BLASTp (e-value < 1e-10) against the Virulence Factors Database (VFDB); and antimicrobial resistance genes were annotated with RGI (Resistance Gene Identifier) against the CARD database using strict cutoff criteria.
Comparative genomic analysis
The assembled genome was aligned against the NCBI nt database using BLAST+ (v2.13.0) to identify homologous bacterial strains. Thirty representative reference strains with complete genome annotations were selected from the NCBI RefSeq database to ensure diversity, based on the following criteria: (1) geographical distribution (strains from Asia, Europe, and North America); (2) varied clinical sources (oral, blood, and colorectal isolates); and (3) representation of both commensal and pathogenic lifestyles. For phylogenetic analysis, 16S rRNA sequences were aligned with MUSCLE (v3.8.31) using default parameters, and a maximum-likelihood tree was constructed in MEGA (v11.0.13) with the Tamura-Nei model and 1,000 bootstrap replicates. Whole-genome average nucleotide identity (ANI) was assessed using FastANI (v1.33) with default settings (minimum fragment length: 1,000 bp; identity threshold ≥ 95% for species delineation). Pan-genome analysis and orthologous gene cluster identification were performed with Roary (v3.13.0) using a minimum BLASTp identity of 95% and a minimum coverage of 80%. Core genes were defined as those present in ≥ 99% of strains, and accessory genes as those present in < 99%. A phylogenetic tree was reconstructed from the core gene set using RAxML (v8.2.12) under the GTRGAMMA model.
Genomic variation analysis was conducted with Snippy (v4.6.0) using a minimum base quality of 20 and a minimum mapping quality of 30. SNP profiles were used to construct a maximum-likelihood phylogenetic tree with IQ-TREE (v2.2.0), employing 1,000 ultrafast bootstrap replicates. All scripts and parameter files used in the bioinformatic analyses are available from the corresponding authors upon reasonable request.
Taxonomic validation using dDDH and GTDB-Tk
To confirm the taxonomic assignment of strain CRC211, digital DNA-DNA hybridization (dDDH) and genome-based taxonomy analyses were performed. dDDH was conducted using the Type Strain Genome Server (TYGS)[17], an online platform for prokaryotic species delineation. In addition, the Genome Taxonomy Database Toolkit (GTDB-Tk v2.3.0)[18] was applied with the Genome Taxonomy Database (GTDB R214) to classify the strain within a standardized bacterial taxonomy framework.
RESULTS
Phylogenetic analysis identified the isolate as a novel species related to CRC211
The whole genome of strain CRC211 was sequenced and assembled, yielding a final sequence of 15,026,974 bp. The coding region spanned 423,942 bp, with an average read length of 149.75 bp. The guanine-cytosine (GC) content was 40.94%. The estimated haploid length ranged from 1,948,637 to 1,950,904 bp, with repeat regions spanning 67,691-67,769 bp and unique regions covering 1,880,947-1,883,135 bp. The model fit showed a goodness-of-fit between 91.664% and 92.850%. Strain CRC211 harbored 61 tRNA genes, 12 rRNA genes, and 3 ncRNA genes, as well as 2 prophages totaling 160,506 bp. In-depth analysis of the prophage regions (designated ΦCRC211-1 and ΦCRC211-2) was performed using PHASTER. ΦCRC211-1 (~ 89.2 kb) was identified as intact (score: 110) and was integrated into a gene encoding a putative helicase. It encoded several potential virulence factors, including a pyocin lipase family protein (associated with host cell damage) and a mitomycin-induced bacteriocin. ΦCRC211-2 (~ 71.3 kb) was classified as questionable (score: 70) and integrated adjacent to a tRNA-Arg gene. Its cargo genes included a lysogeny regulation protein and an extracellular binding protein. Both prophages contained genes associated with lysogeny (integrase, excisionase) as well as lytic functions (capsid, tail fiber proteins). The presence of toxin-related and host interaction genes suggested that these prophages may contribute to the fitness and pathogenicity of S. oralis CRC211 within the tumor niche. Analysis against the NR database showed that S. oralis was the most frequently annotated species, accounting for 70.21% of hits [Figure 1A]. To further verify the nucleotide BLAST results, a phylogenetic tree was constructed [Figure 1B]. The 16S rRNA sequence of strain CRC211 clustered within the S. oralis branch and shared 99% homology with other strains. These findings indicated that strain CRC211 belonged to the genus Streptococcus and was closely related to S. oralis. However, its ANI value (92.29%) fell below the conventional species threshold (95%-96%), suggesting possible genomic divergence. To resolve its taxonomic status, we performed dDDH and GTDB-Tk analyses. The dDDH value between CRC211 and the S. oralis type strain ATCC 35037 was 70.2%, exceeding the recommended 70% species boundary. Moreover, GTDB-Tk confidently classified CRC211 as S. oralis. Collectively, these results confirmed that strain CRC211 represents a genomically divergent strain of S. oralis.
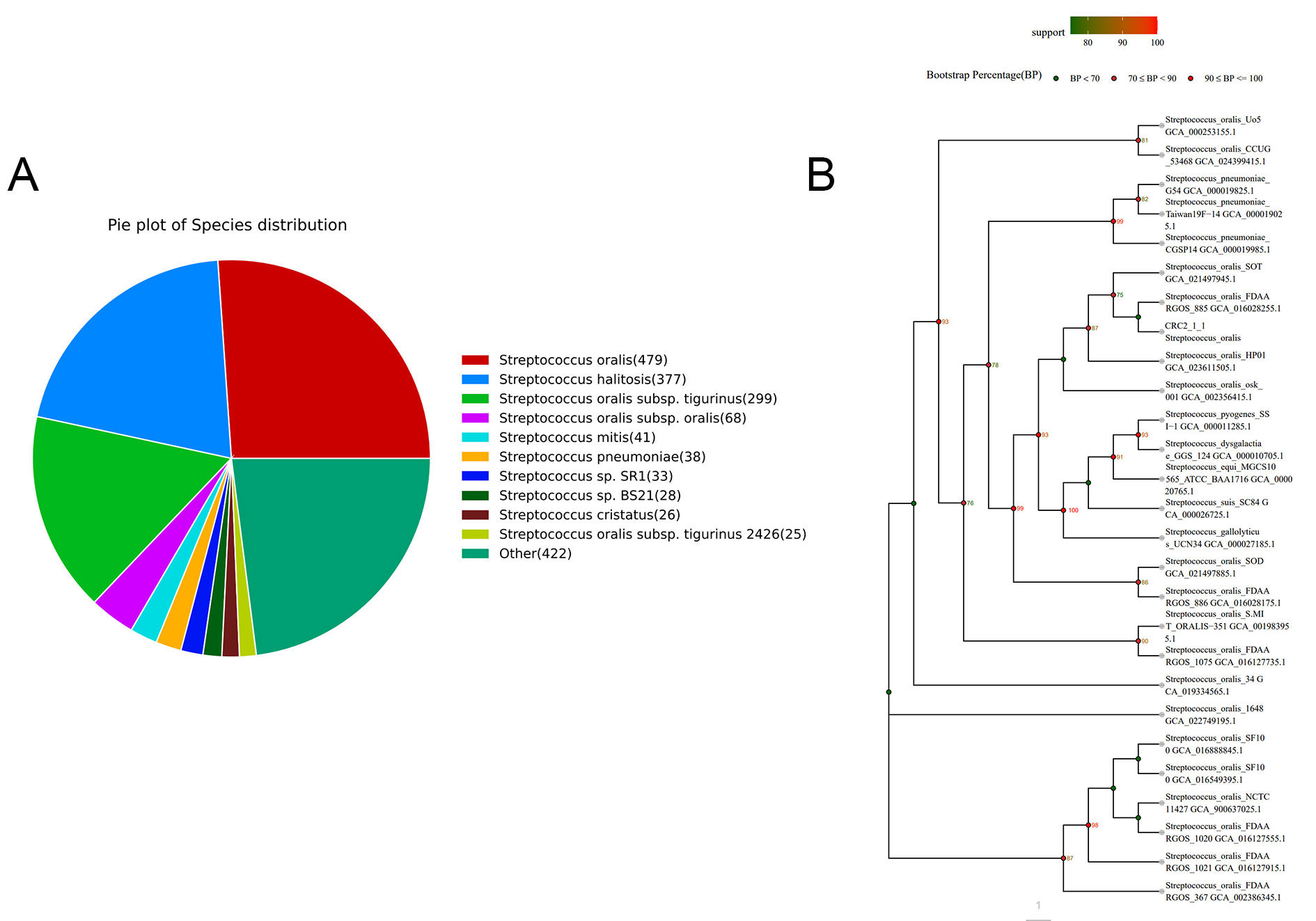
Figure 1. Phylogenetic identification and taxonomic classification of strain CRC211. (A) Species distribution of the top BLAST hits against the NR database, showing S. oralis as the predominant species (70.21%); (B) Maximum-likelihood phylogenetic tree based on 16S rRNA gene sequences, demonstrating the clustering of CRC211 with other S. oralis strains (99% homology), confirming its taxonomic placement within this species. S. oralis: Streptococcus oralis.
Functional analysis of strain CRC211
Gene annotation revealed that 649, 400, 1,354, and 1,836 genes were successfully mapped to the KEGG, GO, COG, and NR databases, respectively [Table 1]. In total, 3,674 genes were functionally annotated in the GO database. Within the biological process category, cellular processes (279 genes) and metabolic processes (260 genes) exhibited the highest gene enrichment. In the cellular component category, protein-containing complex (69 genes) and membrane (55 genes) were the most represented. For molecular function, binding (95 genes) and catalytic activity (201 genes) were the dominant categories [Figure 2A]. A total of 1283 orthologous protein-coding genes were assigned to 25 KEGG metabolic pathways. The pathways most enriched were amino acid metabolism, carbohydrate metabolism, and overview, all of which are essential for maintaining bacterial metabolism [Figure 2B]. Similarly, 1,355 genes were annotated in the COG (Clusters of Orthologous Groups) database and classified into 20 functional categories (C-V) [Figure 2C]. The predominant categories included cell wall/membrane/envelope biosynthesis, metabolic pathways, amino acid transport, and transcription, reflecting fundamental cellular processes in bacteria. These findings aligned well with the KEGG pathway analysis, further confirming the enrichment of genes involved in core metabolic functions critical for sustaining bacterial life.
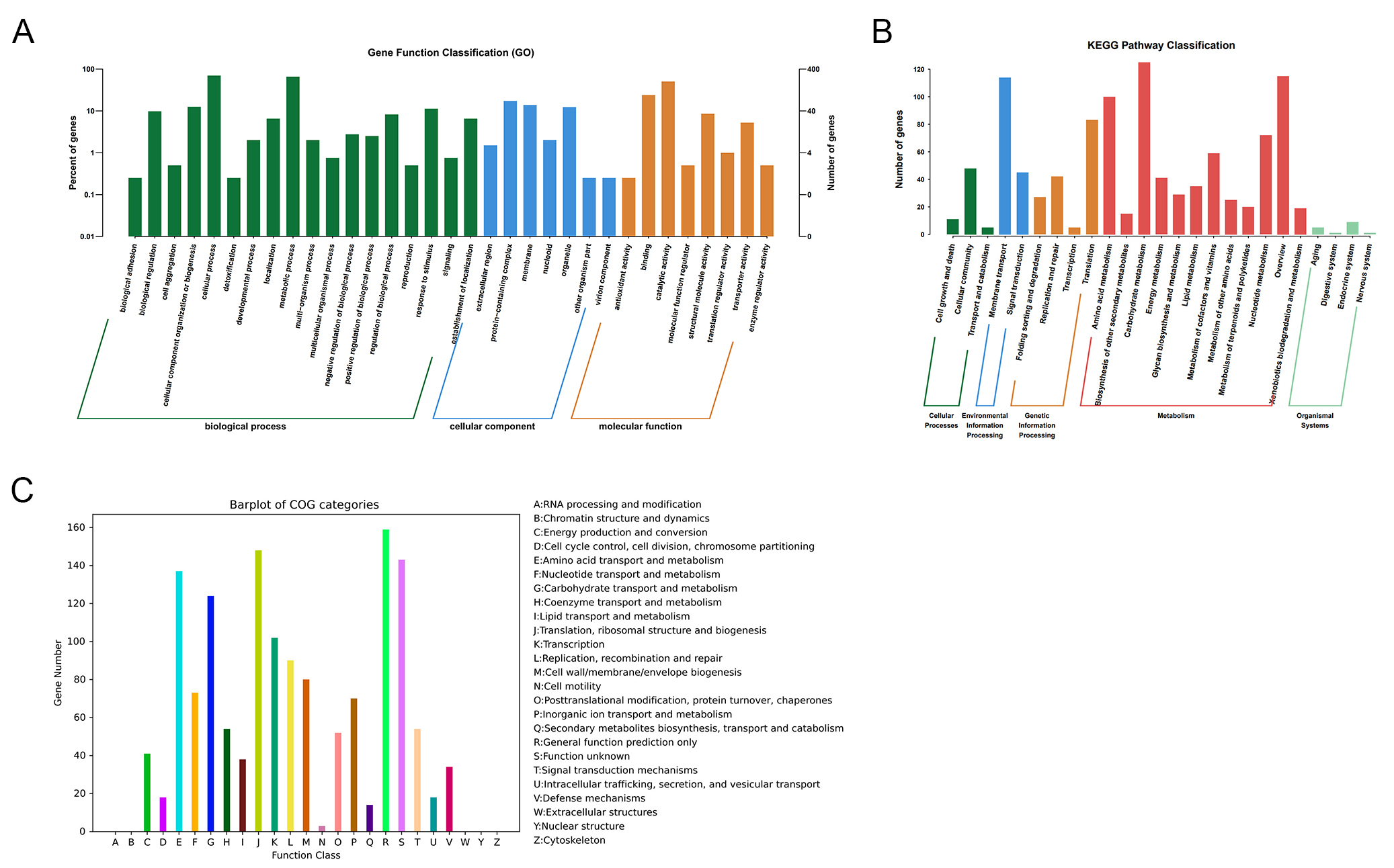
Figure 2. Functional annotation of the CRC211 genome. (A) GO classification of annotated genes across three main categories: Biological Process, Cellular Component, and Molecular Function; (B) KEGG pathway enrichment analysis showing the distribution of genes across metabolic pathways. The most enriched pathways were amino acid metabolism, carbohydrate metabolism, and overview; (C) COG functional categorization of predicted proteins. Major categories include: Translation, ribosomal structure, and biogenesis (J): protein synthesis; Amino acid transport and metabolism (E): nutrient uptake and utilization; Cell wall/membrane/envelope biogenesis (M): bacterial integrity and host interaction; Transcription (K), and Replication, recombination, and repair (L): genetic information processing. GO: Gene Ontology; COG: Clusters of Orthologous Groups.
Overview of genome function analysis of strain CRC211
| Database | Number of genes | Percentage (%) |
| NR | 1,836 | 100 |
| COG | 1,354 | 73.75 |
| CDD | 826 | 44.99 |
| PFAM | 1,260 | 68.63 |
| GO | 400 | 21.79 |
| KEGG | 649 | 35.35 |
| CAzy | 36 | 1.96 |
| TCDB | 202 | 11 |
| PHI | 331 | 18.03 |
| VFDB A | 116 | 6.32 |
| VFDB B | 191 | 10.4 |
| CARD | 75 | 4.08 |
| BactMet exp | 83 | 4.52 |
| BactMet pre | 98 | 5.34 |
| SARG | 27 | 1.47 |
Importantly, several virulence-related genes were identified. Genes encoding pili (pilA, pilB) and biofilm formation factors (gtf, ftf) were enriched, suggesting roles in adhesion to host epithelial cells and persistence within the tumor microenvironment. Genes involved in oxidative stress response (sodA, ahpC) were also present, potentially enhancing bacterial survival under host immune pressure. Moreover, 75 antimicrobial resistance (AMR) genes were annotated in the CARD database, including genes conferring resistance to tetracyclines (tetM) and macrolides (ermB), indicating that CRC211 may persist in the tumor microenvironment despite antibiotic exposure. In addition, 116 virulence factors were identified in VFDB A, including adhesins and biofilm-associated genes. These factors likely promote colonization of the colorectal mucosa, disruption of epithelial integrity, and maintenance of pro-tumorigenic inflammation, thereby contributing to CRC progression.
Genomic features of the CRC211 strain
The complete genome of the newly isolated strain S. oralis CRC211 was sequenced due to its potential clinical significance in CRC patients. The genomic characteristics are presented in Figure 3.
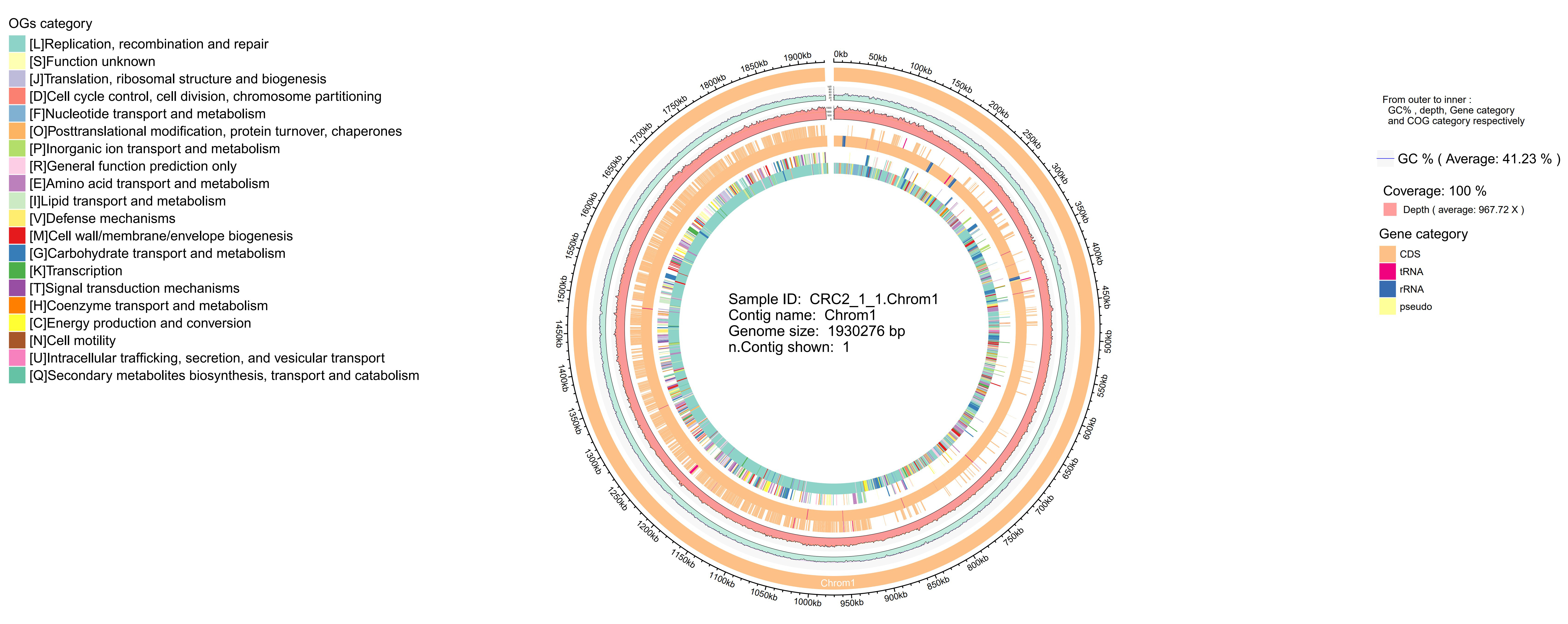
Figure 3. Circular genome map of S. oralis CRC211. From outer to inner rings: (1) GC skew (purple/green); (2) GC content (black); (3) protein-coding genes on the forward strand (colored by COG categories); (4) protein-coding genes on the reverse strand (colored by COG categories); (5) rRNA genes (red); (6) tRNA genes (green); (7) prophage regions (blue). The map illustrates the overall genomic architecture and key features, including the two prophage regions. S. oralis: Streptococcus oralis; GC: guanine-cytosine; COG: Clusters of Orthologous Groups.
Whole-genome similarity
Strain CRC211, along with 29 other phylogenetically related S. oralis strains with publicly available whole-genome sequences, was analyzed using ANI. The ANI value between CRC211 and S. oralis _ GCA_016028255.1 was 92.29%, which falls below the conventional species cutoff but was supported by complementary taxonomic methods (dDDH and GTDB-Tk), as detailed in Section 3.1. The ANI heatmap
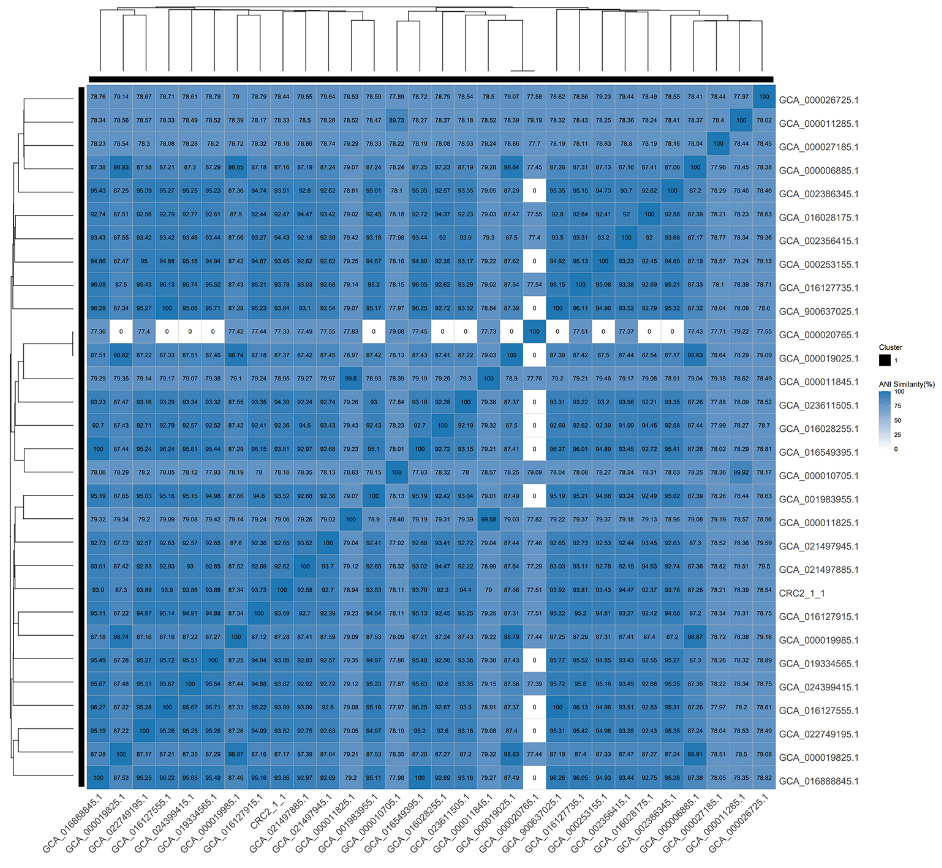
Figure 4. Heatmap of ANI between CRC211 and 29 closely related S. oralis strains. The ANI value of 92.29% between CRC211 and S. oralis GCA_016028255.1 (arrow) confirms classification within the species while suggesting potential genomic divergence. Warmer colors represent higher sequence similarity. ANI: Average nucleotide identity; S. oralis: Streptococcus oralis.
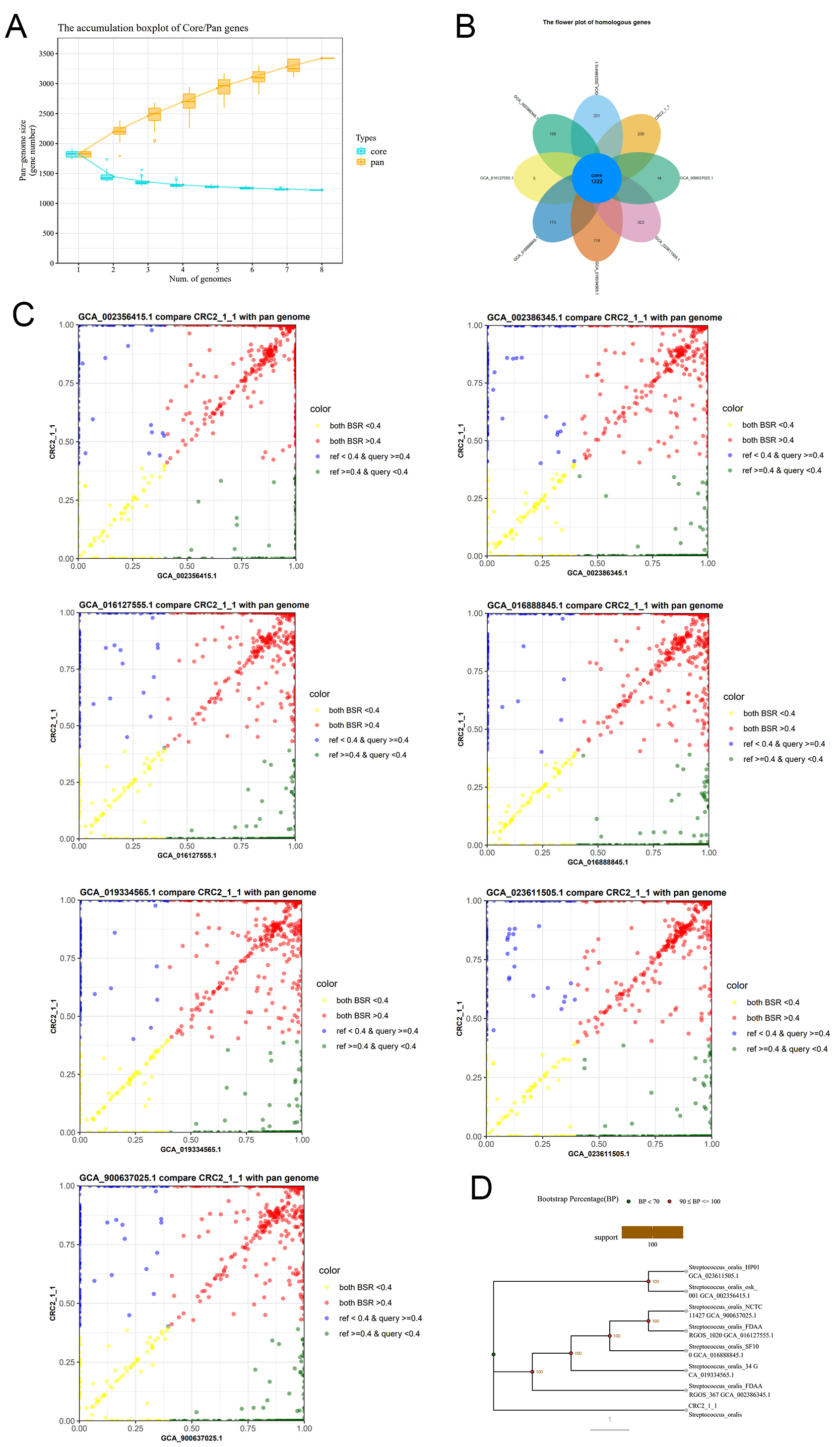
Figure 5. Pan-genome analysis of CRC211 and seven other S. oralis strains (A) Core- and pan-genome curves showing an open pan-genome (increasing with additional strains) and a decreasing core genome, indicative of high genomic plasticity; (B) Flower plot showing the distribution of gene families. The center represents the 1,222 core genes shared by all strains, while the petals represent strain-specific genes; (C) BSR scatter plots comparing CRC211 against each of the other strains. Points above the diagonal indicate higher gene similarity; (D) Phylogenetic tree based on single-copy orthologous genes, illustrating the evolutionary relationships among the eight S. oralis strains. S. oralis: Streptococcus oralis; BSR: Blast Score Ratio.
Functions of homologous gene clusters
COG annotation of the Orthologs Clusters provided a summary of functional categories for Core, Dispensable, and Strain-Specific Clusters. The predominant functions of the core genes were associated with amino acid transport and metabolism, as well as translation, ribosomal structure, and biogenesis [Table 2]. The enrichment of core genes in amino acid transport and metabolism (COG category E) and in translation (COG category J) highlights their essential metabolic roles conserved across strains. In contrast, accessory genes were enriched in carbohydrate metabolism (COG category G) and defense mechanisms (COG category V), indicating adaptive traits that may facilitate niche-specific survival, such as in the nutrient-rich yet immune-active tumor microenvironment.
Con-Pan comparative analysis of gene sets across functional categories
| #COG Abbr. | COG Category | Core genes | Shell genes | Cloud genes | Full pan genes |
| A | RNA processing and modification | 0 | 0 | 0 | 0 |
| B | Chromatin structure and dynamics | 0 | 0 | 0 | 0 |
| C | Energy production and conversion | 29 | 16 | 4 | 49 |
| D | Cell cycle control, cell division, chromosome partitioning | 12 | 4 | 3 | 19 |
| E | Amino acid transport and metabolism | 112 | 28 | 6 | 146 |
| F | Nucleotide transport and metabolism | 64 | 8 | 8 | 80 |
| G | Carbohydrate transport and metabolism | 86 | 19 | 27 | 132 |
| H | Coenzyme transport and metabolism | 39 | 19 | 7 | 65 |
| I | Lipid transport and metabolism | 26 | 10 | 3 | 39 |
| J | Translation, ribosomal structure and biogenesis | 135 | 5 | 5 | 145 |
| K | Transcription | 68 | 27 | 20 | 115 |
| L | Replication, recombination and repair | 70 | 12 | 40 | 122 |
| M | Cell wall/membrane/envelope biogenesis | 57 | 15 | 8 | 80 |
| N | Cell motility | 3 | 0 | 0 | 3 |
| O | Posttranslational modification, protein turnover, chaperones | 39 | 6 | 7 | 52 |
| P | Inorganic ion transport and metabolism | 50 | 21 | 7 | 78 |
| Q | Secondary metabolites biosynthesis, transport and catabolism | 10 | 0 | 3 | 13 |
| R | General function prediction only | 113 | 30 | 35 | 178 |
| S | Function unknown | 101 | 51 | 39 | 191 |
| T | Signal transduction mechanisms | 37 | 10 | 10 | 57 |
| U | Intracellular trafficking, secretion, and vesicular transport | 15 | 4 | 3 | 22 |
| V | Defense mechanisms | 20 | 14 | 28 | 62 |
| W | Extracellular structures | 0 | 0 | 0 | 0 |
| Y | Nuclear structure | 0 | 0 | 0 | 0 |
| Z | Cytoskeleton | 0 | 0 | 0 | 0 |
Genomic variation analysis
Comparative SNP analysis identified numerous non-synonymous mutations (nsSNPs) in CRC211 relative to the reference strain. Notably, nsSNPs were found in genes encoding virulence factors (e.g., adhesins, biofilm-related proteins) and antimicrobial resistance determinants (e.g., ermB, tetM). These variations suggest potential functional changes that may enhance CRC211’s adaptation, persistence, and pathogenicity within the colorectal tumor microenvironment.
DISCUSSION
The intratumoral microbiota has emerged as a key modulator of CRC progression, with specific bacterial species implicated in tumorigenesis, immune evasion, and therapeutic resistance[19]. In this study, we sequenced and characterized the genome of S. oralis strain CRC211, isolated from CRC tumor tissue, and identified genomic features that may contribute to its potential oncogenic role. Our findings are consistent with previous evidence showing that S. oralis is enriched in CRC tissues compared to adjacent normal mucosa, suggesting its active involvement in the tumor microenvironment[20].
The assembled genome of S. oralis CRC211 (15.03 Mb) exhibited typical features of Streptococcus species, including a GC content of 40.94% and a functional repertoire of genes for core metabolic processes (e.g., amino acid and carbohydrate metabolism). Notably, CRC211 harbored 2 prophages (160.5 kb total), which may facilitate horizontal gene transfer of virulence or antibiotic resistance genes, as observed in other oncobacteria such as Fusobacterium nucleatum[21]. Since prophages often encode toxins or immune evasion factors[22,23], their presence in CRC211 warrants further investigation into their role in CRC progression. Comparative genomics revealed that CRC211 shares 92.29% ANI with S. oralis reference strains, confirming its taxonomic classification. The open pan-genome of S. oralis and the 1,222 core genes conserved across strains highlight its genomic plasticity, which may enable niche adaptation within tumors[24]. The enrichment of COG categories related to amino acid transport (112 genes) and cell wall biogenesis (57 genes) suggests mechanisms that CRC211 may use to exploit nutrient-rich tumor environments and evade host immune responses[25].
Functional annotations linked CRC211 to pathways associated with bacterial persistence and host interaction: (1) KEGG: The dominance of amino acid and carbohydrate metabolism genes aligns with S. oralis’ reliance on host-derived nutrients in the tumor microenvironment[26,27]; (2) Virulence factors: The enrichment of adhesins and biofilm-forming genes suggests a mechanism for CRC211 to establish and maintain colonization within tumors, potentially inducing chronic inflammation and activating oncogenic pathways such as NF-κB[28,29]; (3) Antimicrobial resistance (AMR): The repertoire of 75 AMR genes could enable CRC211 to withstand antibiotic therapy, promoting persistence and the formation of a therapy-resistant dysbiotic niche that compromises treatment efficacy and contributes to poor patient outcomes[30,31]. The 2 intact prophage regions identified in CRC211 (160.5 kb total) are of particular interest. Prophages often carry virulence genes that can be horizontally transferred, potentially enhancing bacterial pathogenicity. For instance, prophage-encoded cytolysins or superantigens could exacerbate tissue damage and chronic inflammation, fostering a tumor-permissive microenvironment. Furthermore, the enrichment of adhesins (e.g., pilus genes) and biofilm-associated proteins suggests that CRC211 may form microbial communities within tumors, shielding itself from immune clearance and antibiotics. The presence of 75 AMR genes, including those conferring resistance to tetracycline (tetM) and macrolides (ermB), highlights the potential role of CRC211 in therapy failure among CRC patients. Metabolically, the dominance of amino acid and carbohydrate metabolism pathways is consistent with CRC211’s adaptation to utilize tumor-derived nutrient sources, such as lactate and amino acids from necrotic tumor cells. This metabolic flexibility may not only support bacterial survival but also influence tumor cell metabolism through cross-talk, potentially accelerating cancer progression.
Overall, the genomic features of CRC211 suggest multiple mechanisms through which it may directly influence tumor progression, including persistent colonization that induces chronic inflammation, host DNA damage mediated by genotoxic metabolites, and modulation of local immune responses. Experimental validation is essential to establish causality.
Our findings also point to several potential avenues for clinical translation: (1) Diagnostic Biomarker: The enrichment of S. oralis in CRC tissues, along with strain-specific genes or prophage regions in CRC211, may aid the development of microbial biomarkers for non-invasive CRC detection using stool or blood-based microbial DNA assays; Phage-Mediated Therapy: (2) The prophage regions identified in CRC211 could be leveraged for engineered phage therapies that selectively target and lyse CRC-associated S. oralis, thereby reducing its pro-tumorigenic effects; (3) Microbiota Modulation: Given its antimicrobial resistance profile and metabolic adaptability, CRC211 may represent a target for microbiota-modulating interventions, such as probiotics or antibiotics, to restore microbial balance and enhance chemotherapeutic efficacy; (4) Immunotherapy Synergy: Further research could explore whether targeting CRC211 alters the immune landscape of CRC tumors, potentially synergizing with immune checkpoint inhibitors by reducing immunosuppressive bacterial influences.
Despite providing a genomic foundation for understanding CRC211’s role in CRC, several key questions remain: (1) Mechanistic and in vivo validation: The role of CRC211 in tumorigenesis should be evaluated in vivo using gnotobiotic or conventional mouse models of CRC (e.g., AOM/DSS). Colonization with wild-type CRC211 versus isogenic mutants (e.g., lacking adhesion or biofilm genes) would enable direct assessment of its effects on tumor burden and immune infiltration, moving from association to causality; (2) Host-microbe interactions: Single-cell spatial transcriptomics could reveal CRC211's localization within tumors and its immune-modulatory effects; (3) Therapeutic targeting: Prophage-encoded virulence factors may represent novel targets for microbiota-directed therapies.
In conclusion, the genomic characterization of S. oralis CRC211 underscores its potential as a CRC-associated bacterium with adaptive traits that promote tumor colonization. These insights not only enhance our understanding of S. oralis CRC211’s role in CRC but also provide a foundation for developing microbiota-based diagnostic tools and targeted therapies, contributing to personalized cancer medicine. Future work should integrate in vitro and in vivo models to validate its oncogenic mechanisms and explore its utility as both a diagnostic and therapeutic target.
DECLARATIONS
Acknowledgments
The graphical abstract was created with BioRender.com/ (Created in BioRender. Shi, Y. (2025) https://BioRender.com/mvhax6a/).
Authors’ contributions
Conceptualization: Shi Y, Hou W, Wang H
Formal analysis: Li X, Liu L
Investigation: Shi Y, Gao M
Writing - original draft preparation: Shi Y, Wu J
Writing - review and editing: Sheng K
Supervision: Wang H
Funding acquisition: Wang H
All authors have read and agreed to the published version of the manuscript.
Availability of data and materials
Raw sequencing data have been uploaded to the National Center for Biotechnology Information (NCBI) (Accession GEO number: GSE294724). All data presented in this study are available from the corresponding authors upon reasonable request.
Financial support and sponsorship
This research was funded by the National Natural Science Foundation of China (Grant No. 82173005).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethics approval and consent to participate
The study protocol was reviewed and approved by the Ethics Committee of Changhai Hospital, Naval Medical University (Ethics No. CHEC2023-307). Written informed consent was obtained from all participants.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Sears CL, Salzberg SL. Microbial diagnostics for cancer: a step forward but not prime time yet. Cancer Cell. 2020;37:625-7.
2. Livyatan I, Nejman D, Shental N, Straussman R. Characterization of the human tumor microbiome reveals tumor-type specific intra-cellular bacteria. Oncoimmunology. 2020;9:1800957.
3. Li N, Yu Y, Chen Q, et al. A gene delivery system with autophagy blockade for enhanced anti-angiogenic therapy against Fusobacterium nucleatum-associated colorectal cancer. Acta Biomater. 2024;183:278-91.
4. Tavano F, Napoli A, Gioffreda D, et al. Could the microbial profiling of normal pancreatic tissue from healthy organ donors contribute to understanding the intratumoral microbiota signature in pancreatic ductal adenocarcinoma? Microorganisms. 2025;13:452.
5. Uribe-herranz M, Rafail S, Beghi S, et al. Gut microbiota modulate dendritic cell antigen presentation and radiotherapy-induced antitumor immune response. J Clin Invest. 2019;130:466-79.
6. Yang Q, Wang B, Zheng Q, et al. A review of gut microbiota-derived metabolites in tumor progression and cancer therapy. Adv Sci (Weinh). 2023;10:2207366.
7. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14:195-206.
8. Galeano Niño JL, Wu H, Lacourse KD, et al. Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature. 2022;611:810-7.
9. Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. 2014;12:661-72.
10. Castellarin M, Warren RL, Freeman JD, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299-306.
11. Kostic AD, Chun E, Robertson L, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207-15.
12. Gur C, Ibrahim Y, Isaacson B, et al. Binding of the Fap2 Protein of fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015;42:344-55.
13. Kumar R, Herold JL, Schady D, et al. Streptococcus gallolyticus subsp. gallolyticus promotes colorectal tumor development. PLoS Pathog. 2017;13:e1006440.
14. Yamamura K, Baba Y, Nakagawa S, et al. Human microbiome in esophageal cancer tissue is associated with prognosis. Clin Cancer Res. 2016;22:5574-81.
15. Abed J, Maalouf N, Manson AL, et al. Colon cancer-associated fusobacterium nucleatum may originate from the oral cavity and reach colon tumors via the circulatory system. Front Cell Infect Microbiol. 2020;10:400.
16. Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012;10:575-82.
17. Meier-kolthoff JP, Auch AF, Klenk HP, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14:60.
18. Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH, Hancock J. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2020;36:1925-7.
19. Ding T, Liu C, Li Z. The mycobiome in human cancer: analytical challenges, molecular mechanisms, and therapeutic implications. Mol Cancer. 2025;24:18.
20. Camañes-gonzalvo S, Montiel-company JM, Lobo-de-mena M, et al. Relationship between oral microbiota and colorectal cancer: a systematic review. J Periodontal Res. 2024;59:1071-82.
21. Shigematsu Y, Saito R, Amori G, et al. Fusobacterium nucleatum, immune responses, and metastatic organ diversity in colorectal cancer liver metastasis. Cancer Sci. 2024;115:3248-55.
22. Ribeiro HG, Nilsson A, Melo LDR, Oliveira A. Analysis of intact prophages in genomes of Paenibacillus larvae: an important pathogen for bees. Front Microbiol. 2022;13:903861.
23. Bucher MJ, Czyż DM. Phage against the machine: the SIE-ence of superinfection exclusion. Viruses. 2024;16:1348.
24. Joyce LR, Youngblom MA, Cormaty H, et al. Comparative genomics of streptococcus oralis identifies large scale homologous recombination and a genetic variant associated with infection. mSphere. 2022;7:e00509-22.
25. Liu X, Sun X, Mu W, et al. Autophagic flux-lipid droplet biogenesis cascade sustains mitochondrial fitness in colorectal cancer cells adapted to acidosis. Cell Death Discov. 2025;11:21.
26. Wang L, Wang Q, Zhou Y. Oral microbial translocation genes in gastrointestinal cancers: insights from metagenomic analysis. Microorganisms. 2024;12:2086.
27. Sun J, Tang Q, Yu S, et al. F. nucleatum facilitates oral squamous cell carcinoma progression via GLUT1-driven lactate production. EBioMedicine. 2023;88:104444.
28. Marongiu GL, Fink U, Schöpf F, Oder A, Von Kries JP, Roderer D. Structural basis for immune cell binding of via the trimeric autotransporter adhesin CbpF. Proc Natl Acad Sci U S A. 2025;122:e2418155122.
29. Duizer C, Salomons M, Van Gogh M, et al. Fusobacterium nucleatum upregulates the immune inhibitory receptor in colorectal cancer cells via the activation of ALPK1. Gut Microbes. 2025;17:2458203.
30. Afordoanyi DM, Akosah YA, Shnakhova L, Saparmyradov K, Diabankana RGC, Validov S. Biotechnological key genes of the rhodococcus erythropolis MGMM8 genome: genes for bioremediation, antibiotics, plant protection, and growth stimulation. Microorganisms. 2023;12:88.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].