



Profiling the composition of resistome and bacteriome in the upper respiratory tract of domestic cats with respiratory signs in China
 0
0 
Abstract
Aim: Domestic cats, among the most popular pets globally, may harbor antimicrobial resistance genes (ARGs) and zoonotic pathogens that impact human health. This study aims to investigate the resistome and bacteriome composition in the upper respiratory tract of domestic cats with respiratory signs in China.
Methods: We performed metagenomic sequencing on 1,454 oropharyngeal-nasal swabs from cats with respiratory signs across diverse living conditions in 22 Chinese provinces. Resistome and bacteriome profiles were analyzed using these sequencing data.
Results: We characterized the resistome and bacteriome in the upper respiratory tract of cats, identifying a wide range of ARGs - including those conferring resistance to last-resort antibiotics {e.g., carbapenems (blaNDM,
Conclusion: The findings highlight the potential zoonotic risks posed by cats. Including monitoring of this companion species within the One Health approach to address public health concerns is necessary.
Keywords
INTRODUCTION
Antimicrobial resistance (AMR) is one of the most critical global public health threats[1]. Without urgent intervention, drug-resistant infections are projected to cause 10 million deaths annually worldwide by 2050, with a cumulative economic loss of 100 trillion USD[2]. A recent systematic analysis revealed that lower respiratory infections were the leading AMR-associated infectious syndrome in 2019, responsible for over 1.5 million deaths[3]. The same study identified Escherichia coli, Staphylococcus aureus, Klebsiella pneumoniae, Streptococcus pneumoniae, Acinetobacter baumannii, and Pseudomonas aeruginosa as the six deadliest AMR-related pathogens[3].
Domesticated animals, particularly pets, are recognized as potential reservoirs for transmitting antimicrobial-resistant pathogens to humans[4,5]. Among them, domestic cats (Felis catus), one of the world’s most popular household pets, have experienced a dramatic population surge over the past 40 years. By 2021, the global cat population reached 600 million, with 400 million in Asia[6]. In China, the number of household cats exceeded 96 million in 2021 and continues to rise[7], alongside a substantial stray cat population. Given their close interaction with humans, cats harbor antimicrobial resistance genes (ARGs) and zoonotic pathogens that pose public health risks. For example, studies have documented that companion cats can carry human-associated pathogens and multidrug-resistant bacteria, including MRSA and β-lactam-resistant Enterobacteriaceae[8,9]. Recent evidence also highlights the transmission of resistomes from cats to their owners and living environment[10]. While the oral microbiota of healthy cats is predominantly composed of Pasteurellaceae, Moraxellaceae, Thermomonas, and Comamonadaceae[11], an increasing number of human infections - particularly following cat bites - have been linked to bacterial species within these families[12,13]. Despite these risks, comprehensive surveillance of AMR in domestic cats remains limited.
Recently, metagenomic sequencing, combined with bioinformatic analysis, has emerged as a powerful tool for AMR and microorganism surveillance[14,15]. In this study, we employed nationwide metagenomic sequencing to profile the resistome and bacteriome in the upper respiratory tract of domestic cats across China. Our findings aim to elucidate the public health risks posed by these ubiquitous companion animals.
METHODS
Ethics statement
This study was approved by the Animal Management and Ethics Committee of Huazhong Agricultural University (Approval ID: HZAUCA-2023-0005).
Sample collection, treatment, and metagenomic sequencing
From September 1 to October 30, 2022, we collected 1,454 oropharyngeal-nasal swabs from domestic cats exhibiting respiratory signs across diverse environments in 22 Chinese provinces [Figure 1 and Supplementary Table 1]. Sampling sites include: catteries (509 samples), animal hospitals (449 samples), and stray bases (496 samples). Samples were geographically distributed as follows: Northeast China, 135 samples (39 samples from catteries, 39 samples from animal hospitals, 57 samples from stray bases); Northern China, 223 samples (96 from catteries, 75 from animal hospitals, 52 from stray bases); Eastern China, 285 samples (111 from catteries, 58 from animal hospitals, 116 from stray bases); Central China, 273 samples (96 from catteries, 77 from animal hospitals, 100 from stray bases); Southern China, 160 samples (36 from catteries, 45 from animal hospitals, 79 from stray bases); Northwest China, 138 samples (67 from catteries, 36 from animal hospitals, 35 from stray bases); Southwest China, 240 samples (64 from catteries, 119 from animal hospitals, 57 from stray bases). Within each geographical region, samples from catteries, animal hospitals, and stray bases were pooled separately, yielding 63 pools for analysis [Figure 1].
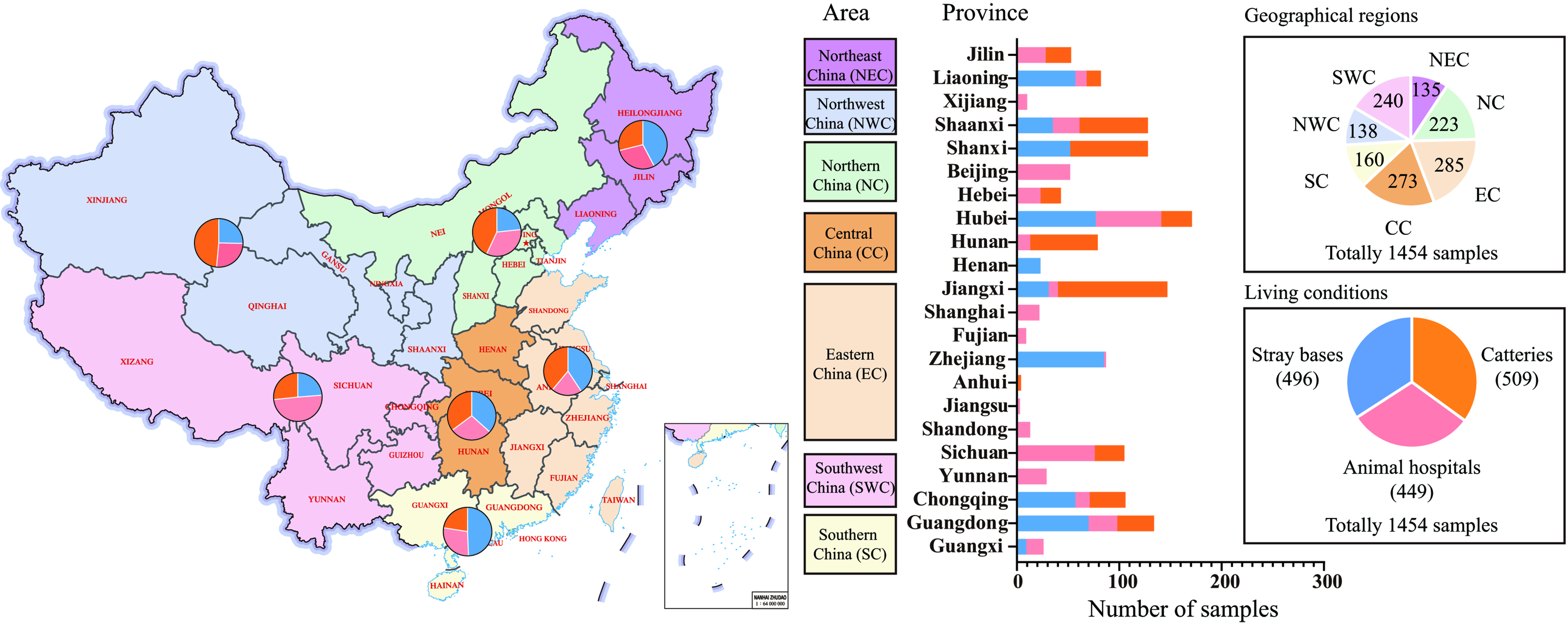
Figure 1. Distribution of oral-nasal swabs collected from domestic cats in various living conditions across different regions in China. Different Chinese regions: NEC - Northeast China, NC - Northern China, EC - Eastern China, CC - Central China, SC - Southern China, NWC - Northwest China, SWC - Southwest China.
DNA extraction was performed using the cetyltrimethylammonium bromide protocol[16]. DNA quality/quantity was assessed through 1% agarose gel electrophoresis, NanoDrop2000 (Thermo Fisher Scientific, Waltham, US), and Agilent 2100 Bioanalyzer (Agilent, Santa Clara, US), respectively. Libraries were prepared with the NEBNext® UltraTM DNA Library Prep Kit (NEB, Ipswich, US; 350 bp insert size) and sequenced on an Illumina Nova600 PE150 platform (Illumina, San Diego, China) employing the paired-end 150 bp strategy. All data were deposited into the NCBI Sequence Read Archive (SRA) database (Bioproject: PRJNA998709; Accessions: SRR33674915-SRR33674924, SRR33696099-SRR33696115, SRR34261881-SRR34261890).
Sequence assembly and identification of ARGs
Metagenomic sequencing generated ~10.84 gigabytes of (Gb) reads per sample [Supplementary Table 2]. The quality of the raw reads was assessed using FastQC (v 0.12.1) (https://github.com/s-andrews/FastQC), and reads containing low-quality and adaptors were eliminated using Trimmomatic (quality score ≥ 20)[17]. Clean reads were then assembled using MEGAHIT (v1.1.2; parameters: -k-min 61 -min- contig-len 1000)[18], with assembly quality evaluated via QUAST 5.2.0[19]. Contigs carrying ARGs were identified following a protocol outlined in a previously published article[20]. Briefly, contigs derived from sequence assembly were utilized to predict open reading frames (ORFs) using Prodigal[21]. ARG-like ORFs were identified by comparing the amino acid sequences of the predicted ORFs against the Structured ARG (SARG) database (v 2.0)[22] using Diamond v0.8.35[23], with the following BLASTP parameters: e-value ≤ 10-10, an identity ≥ 80%, and query coverage ≥ 70%[20].
Identification of bacterial species
For bacterial species characterization, predicted ORFs were matched against the NCBI NR database using Diamond (version 0.8.35; E-value ≤ 1e-5)[23]. The alignment outcomes were subsequently assessed utilizing the Lowest Common Ancestor (LCA) algorithm implemented in MEGAN[24]. Taxonomic lineage details were retrieved for the top BLAST hit of each contig. DNA abundances were depicted visually using Krona[25]. The bacterial taxonomic contributors to the ARGs were identified employing FishTaco[26] with default parameters.
Statistical analysis
Transcripts per million (TPM) per predicted gene was calculated as a proxy for gene abundance. TPM was defined as the number of metagenomic reads mapped to a given gene (× 106), divided by the gene’s length, and normalized by the sum of all reads mapped to all genes, also adjusted by their respective lengths, as previously described[27,28]. Analysis of similarities (ANOSIM) was used to evaluate whether the grouping makes sense (0 < R < 1)[29]. For comparing the abundances between two groups, Mann-Whitney U test (known as the Wilcoxon rank-sum test) was performed using pairwise multiple comparison adjustments according to the Benjamini-Hochberg procedure, as previously described[30,31]. P < 0.05 (*) indicates statistically significant.
RESULTS
Composition of resistome in the upper respiratory tract of cats in China
Following assembly, an average of 107,726 contigs with an average length of 136,712,231 bp (average N50: 2,085 bp) were generated for each pool [Supplementary Figure 1 and Supplementary Table 2]. ARG-like ORFs identified from the assembled contigs showed an average sequence identity of 93.39% (ranged: 80.00%-100%) and an average coverage of 98.35% (ranged: 70.24%-100%) [Supplementary Table 3]. ANOSIM analysis confirmed the validity of sample grouping by both geographic regions (R = 0.04) and living conditions (R = 0.056) [Figure 2A and B]. Subsequent comparison of resistome abundances using the Mann-Whitney U test revealed significant regional variations [Figure 2C]. Specifically, cats from Northeast China showed lower resistome abundances compared to those from Southern and Southwest China, while Southern China cats exhibited higher resistome levels than those from Northwest and Northern China (P < 0.05). No significant differences in resistome abundance were detected among different living conditions [Figure 2D].
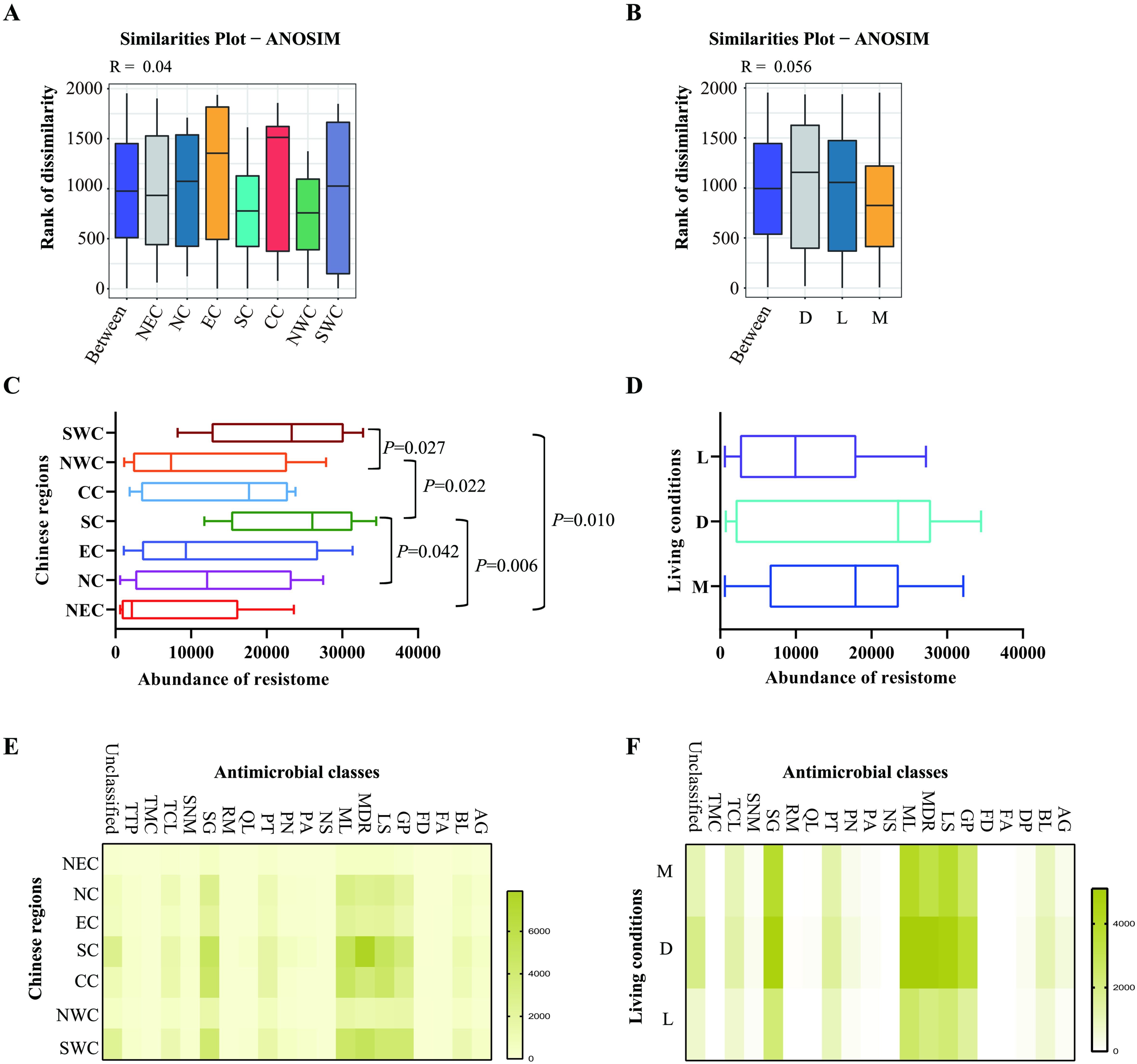
Figure 2. Composition of the resistome in the upper respiratory tract of domestic cats determined by metagenomic sequencing. (A and B) ANOSIM evaluating the resistomes in the upper respiratory tract of domestic cats from different Chinese regions (A) and/or different living conditions (B); (C and D) Abundances of the resistomes in the upper respiratory tract of domestic cats from different Chinese regions (C) and/or different living conditions (D); (E and F) Heatmaps showing the abundances of genes conferring resistance to different antimicrobial classes in the upper respiratory tract of domestic cats from different Chinese regions (E) and/or different living conditions (F). Mann-Whitney U test was performed to compare the abundances between two groups, using pairwise multiple comparison adjustments according to the Benjamini-Hochberg procedure. The significance level was set at a P value of < 0.05 (*) and P > 0.05 is not marked. Different Chinese regions: NEC - Northeast China, NC - Northern China, EC - Eastern China, CC - Central China, SC - Southern China, NWC - Northwest China, SWC - Southwest China. Different living conditions: M - catteries, D - animal hospitals,
After removing the duplicate genes, we identified 444 unique ARGs conferring resistance to 16 antimicrobial classes: aminoglycosides, β-lactams, diaminopyrimidines, glycopeptides, lincosamides, macrolides, nucleosides, peptides, phenicols, phosphonic acids, quinolones, rifamycin, streptogramins, sulfonamides, tetracenomycins, and tetracyclines [Supplementary Figure 2]. Notably prevalent were ARGs conferring multidrug resistance [Figure 2E]. High detection rates were also observed for resistance genes targeting aminoglycosides, β-lactams, glycopeptides, macrolides, lincosamides, peptides, phenicols, streptogramins, and tetracyclines [Figure 2E]. Regional variations in ARG prevalence across antimicrobial classes were evident [Figure 2E], while differences in the distribution of multidrug resistance genes were observed among cats from different living conditions [Figure 2F].
Distribution of public health concern ARGs in the upper respiratory tract of cats
We also identified several ARGs conferring resistance to critically important agents listed by the World Health Organization (WHO) as the highest priority for human medicine[32]. These included genes responsible for resistance to 3rd/4th/5th-generation cephalosporins (e.g., blaCTX-M, blaTEM-1, blaSHV-1, blaVEB-5, blaGES-13, blaOXA-9/59/209), vancomycin (e.g., vanA, vanB, vanC, vanD, vanE, vanG, vanH, vanM, vanN, vanR, vanS, vanT, vanU, vanV, vanX, vanY, vanZ), macrolides and ketolides (e.g., ermA, ermB, ermF), polymyxins (e.g., mcr-1.2, mcr-1.4, mcr-1.5, mcr-2.1, mcr-2.2, mcr-3, mcr-4, mcr-5), and quinolones (e.g., qnrA, qnrB, qnrS) [Figure 3A and Supplementary Figure 2]. Of particular concern were ARGs conferring resistance to the last-resort antimicrobials, including carbapenems (blaNDM-1, blaNDM-10, blaOXA-244, blaVIM-13, blaVIM-33), colistin (mcr-1.2, mcr-1.4, mcr-1.5, mcr-2.1, mcr-2.2, mcr-3, mcr-4, mcr-5), high-level tigecycline [MIC ≥ 4 µg/mL;
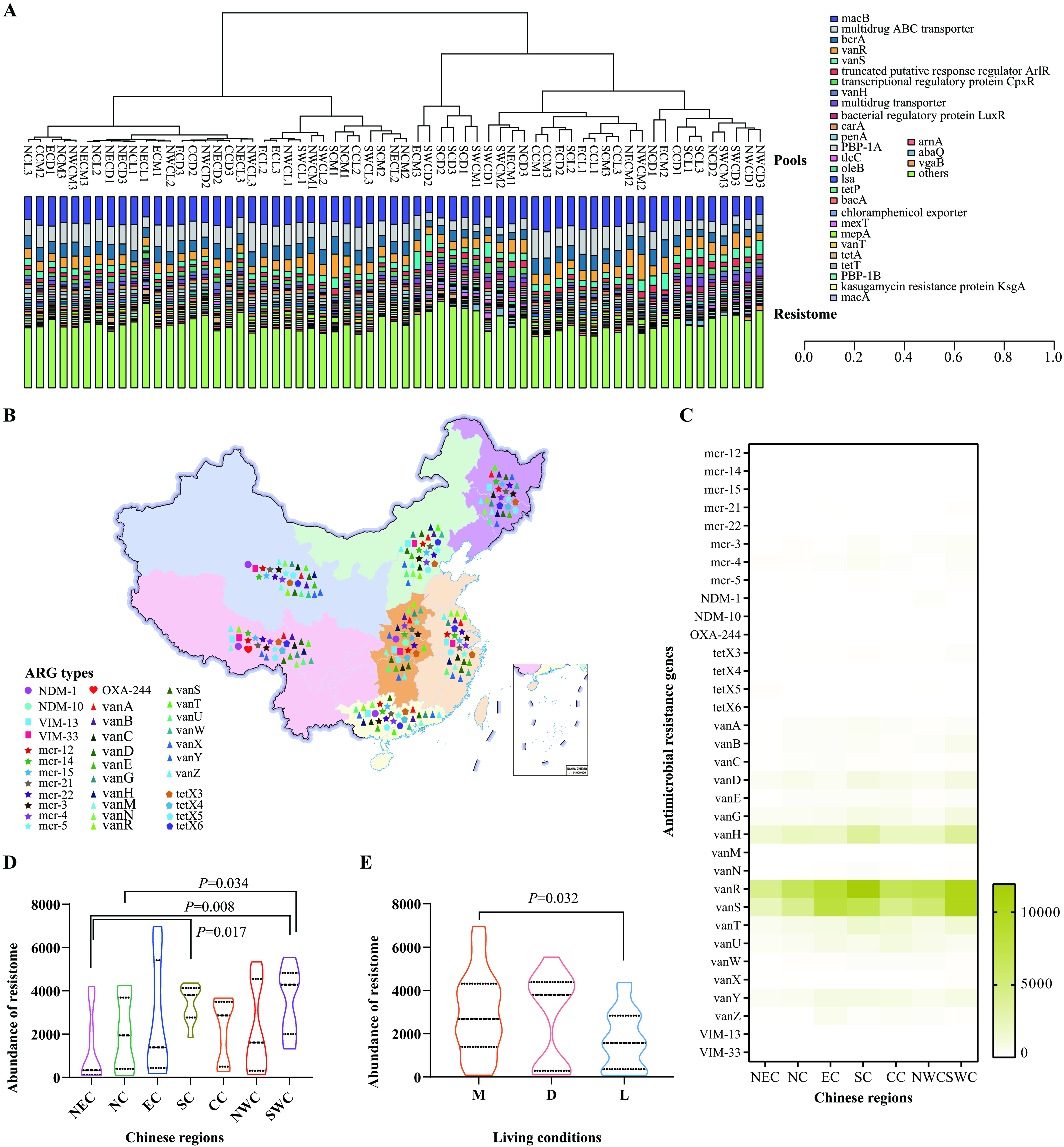
Figure 3. Detection of ARGs in the upper respiratory tract of cats in various Chinese regions. (A) A bar graph displaying the distribution of identified ARGs in the upper respiratory tract of cats across different Chinese regions (Only those with abundances ranking among the top thirty are shown); (B) A geographic map illustrating the detection of ARGs conferring resistance to last-resort antimicrobials (carbapenems, colistin, high-level tigecycline, vancomycin) in samples collected from cats in diverse living conditions in China; (C) A heatmap presenting the abundances of ARGs conferring resistance to last-resort antimicrobials (carbapenems, colistin, high-level tigecycline, vancomycin) in the upper respiratory tract of domestic cats from various Chinese regions; (D) A violin plot demonstrating the abundances of ARGs conferring resistance to last-resort antimicrobials (carbapenems, colistin, high-level tigecycline, vancomycin) in the upper respiratory tract of domestic cats across different Chinese regions; (E) A violin plot showing the abundances of ARGs conferring resistance to last-resort antimicrobials (carbapenems, colistin, high-level tigecycline, vancomycin) in the upper respiratory tract of domestic cats in different living conditions. Mann-Whitney U test was performed to compare the abundances between two groups, using pairwise multiple comparison adjustments according to the Benjamini-Hochberg procedure. The significance level was set at a P value of < 0.05 (*) and P > 0.05 is not marked. Different Chinese regions: NEC - Northeast China, NC - Northern China, EC - Eastern China, CC - Central China, SC - Southern China, NWC - Northwest China, SWC - Southwest China. Different living conditions:
Distribution of main bacterial species associated with AMR that cause global burdens
Antimicrobial-resistant bacteria were identified through metagenomic sequencing-based bacteriome analysis, revealing a total of 8,190 known bacterial species belonging to families such as Enterobacteriaceae, Pasteurellaceae, Moraxellaceae, and Comamonadaceae [Supplementary Figure 3 and Supplementary Table 4]. Notably, the six major pathogens linked to global AMR-related fatalities - E. coli, S. aureus, K. pneumoniae, S. pneumoniae, A. baumannii, and P. aeruginosa[3] - were widely detected in cats across different regions and living conditions [Figure 4A and B]. While no significant difference was observed in the overall abundance of these bacteria among regions (P > 0.05), variations in the abundances of individual species were noted [Figure 4C]. Additionally, the combined abundance of these six pathogens in the upper respiratory tract of cats from animal hospitals was significantly higher than in those from stray bases [Figure 4D]. Specifically, A. baumannii, K. pneumoniae, S. aureus, and E. coli exhibited statistically significant differences in abundance depending on living conditions [Figure 4E].
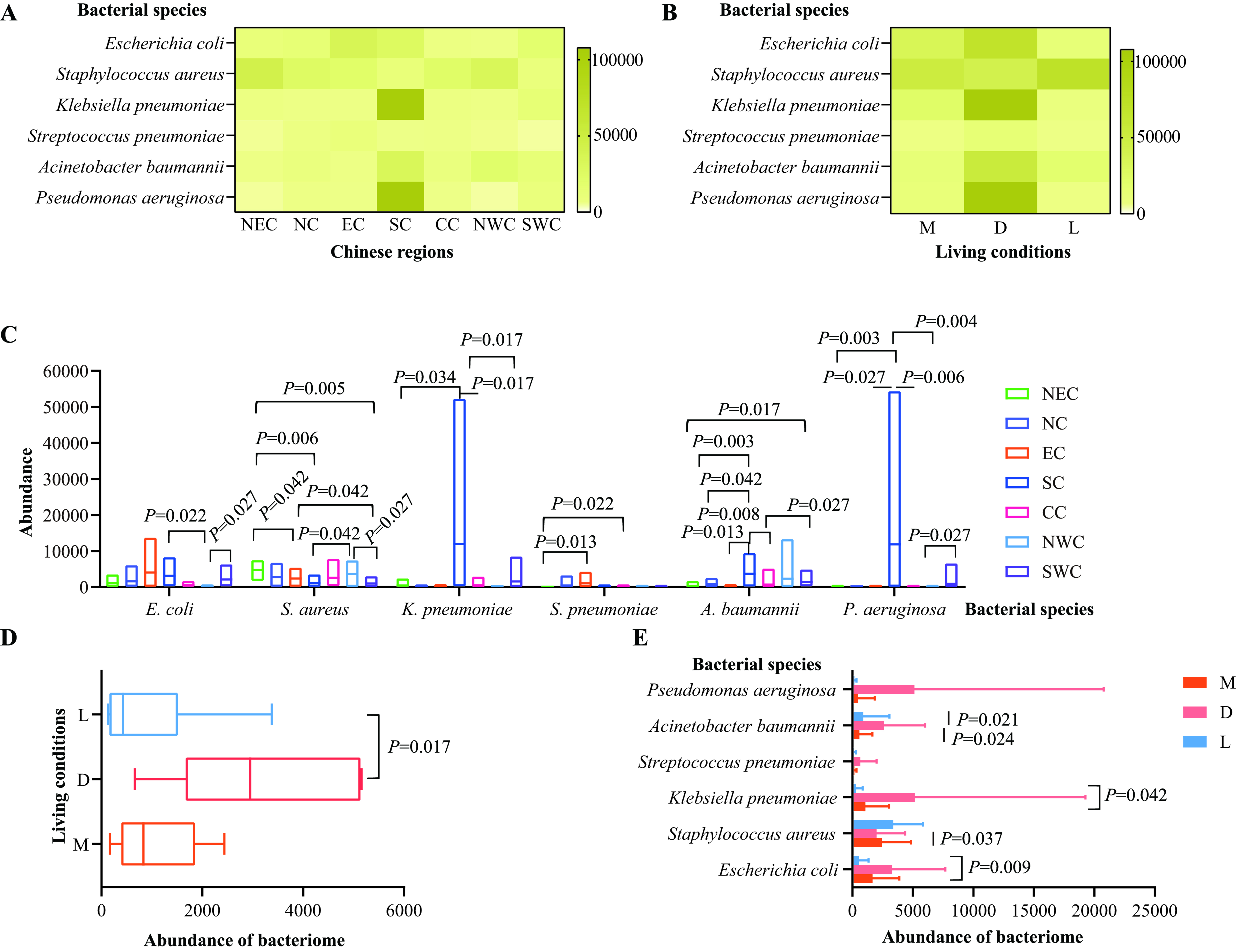
Figure 4. Characterization of the six primary bacterial pathogens associated with deaths due to AMR worldwide. (A) A heatmap illustrating the distribution and abundance of these six bacterial species in the upper respiratory tract of cats across various Chinese regions; (B) A heatmap displaying the distribution and abundance of these six bacterial species in the upper respiratory tract of cats in different living conditions; (C) Comparisons of the abundances of the bacteriome of these six bacterial species in the upper respiratory tract of cats from different Chinese regions; (D) Comparisons of the total abundances of the bacteriome of these six bacterial species in the upper respiratory tract of cats in different living conditions; (E) Comparisons of the abundances of the bacteriome of each of these six bacterial species in the upper respiratory tract of cats across different living conditions. Mann-Whitney U test was performed to compare the abundances between two groups, using pairwise multiple comparison adjustments according to the Benjamini-Hochberg procedure. The significance level was set at a P value of < 0.05 (*) and P > 0.05 is not marked. Different Chinese regions: NEC - Northeast China, NC - Northern China, EC - Eastern China, CC - Central China, SC - Southern China, NWC - Northwest China, SWC - Southwest China. Different living conditions: M - catteries, D - animal hospitals, L - stray bases. AMR: Antimicrobial resistance.
Further analysis clarified the contributions of different bacterial species to AMR, revealing distinct associations with specific ARGs [Figure 5]. The six aforementioned pathogens played critical roles in conferring resistance to key antimicrobials, including those classified by the WHO as highest-priority critically important agents [Figure 5]. Among them, E. coli was primarily associated with resistance to aminoglycosides and tetracyclines, mainly linked to aph3-I and tetA, respectively [Figure 5A and B].
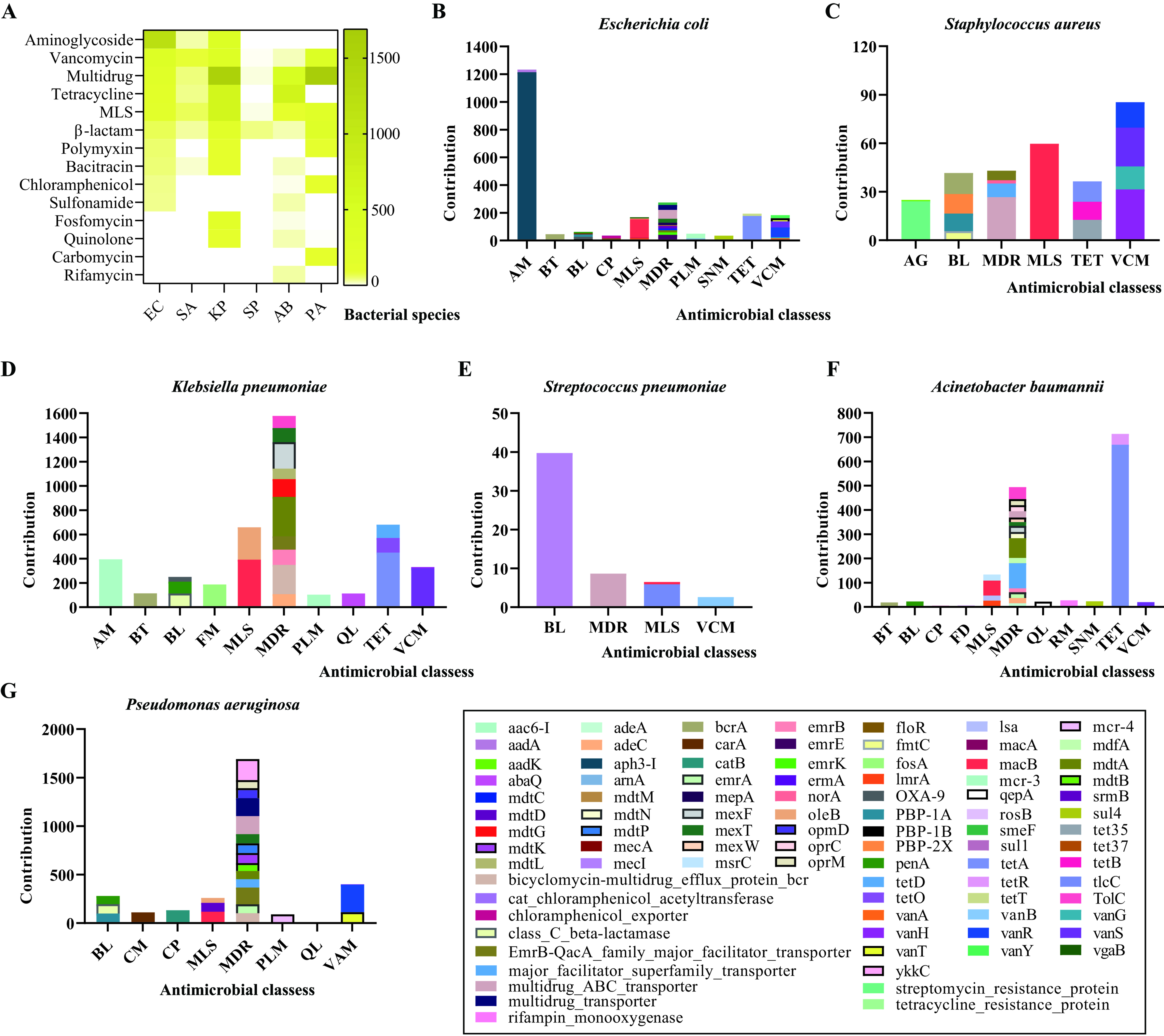
Figure 5. Contributions of the six primary bacterial pathogens to the abundance of genes mediating resistance against various antimicrobial classes. (A) A heatmap illustrating the contributions of the six leading bacterial pathogens to the resistant phenotypes against different antimicrobial classes; (B) A column chart showing the contributions of Escherichia coli to the abundance of genes mediating resistance against various antimicrobial classes; (C) A column chart showing the contributions of Staphylococcus aureus to the abundance of genes mediating resistance against various antimicrobial classes; (D) A column chart showing the contributions of Klebsiella pneumoniae to the abundance of genes mediating resistance against various antimicrobial classes; (E) A column chart showing the contributions of Streptococcus pneumoniae to the abundance of genes mediating resistance against various antimicrobial classes; (F) A column chart showing the contributions of Acinetobacter baumannii to the abundance of genes mediating resistance against various antimicrobial classes; (G) A column chart showing the contributions of Pseudomonas aeruginosa to the abundance of genes mediating resistance against various antimicrobial classes. Bacterial species: EC - E. coli, SA - S. aureus, KP - K. pneumoniae, SP - S. pneumoniae, AB - A. baumannii, PA - P. aeruginosa.
Bacterial species with public health concerns characterized in the upper respiratory tract of cats
Metagenomic sequencing revealed the presence of bacterial species with significant public health implications due to their association with human infections following cat exposure [Supplementary Figure 3]. Of particular concern were Enterococcus faecium, Enterobacter spp., Pasteurella multocida, Streptococcus spp., Staphylococcus spp., Neisseria spp., Moraxella spp., Bordetella bronchiseptica, Clostridioides difficile, Salmonella spp., Listeria monocytogenes, Shigella spp., Campylobacter spp., and Haemophilus influenzae. These pathogens were detected in the upper respiratory tract of cats across various Chinese regions [Figure 6A] and under different living conditions [Figure 6B], with many showing high detection abundances
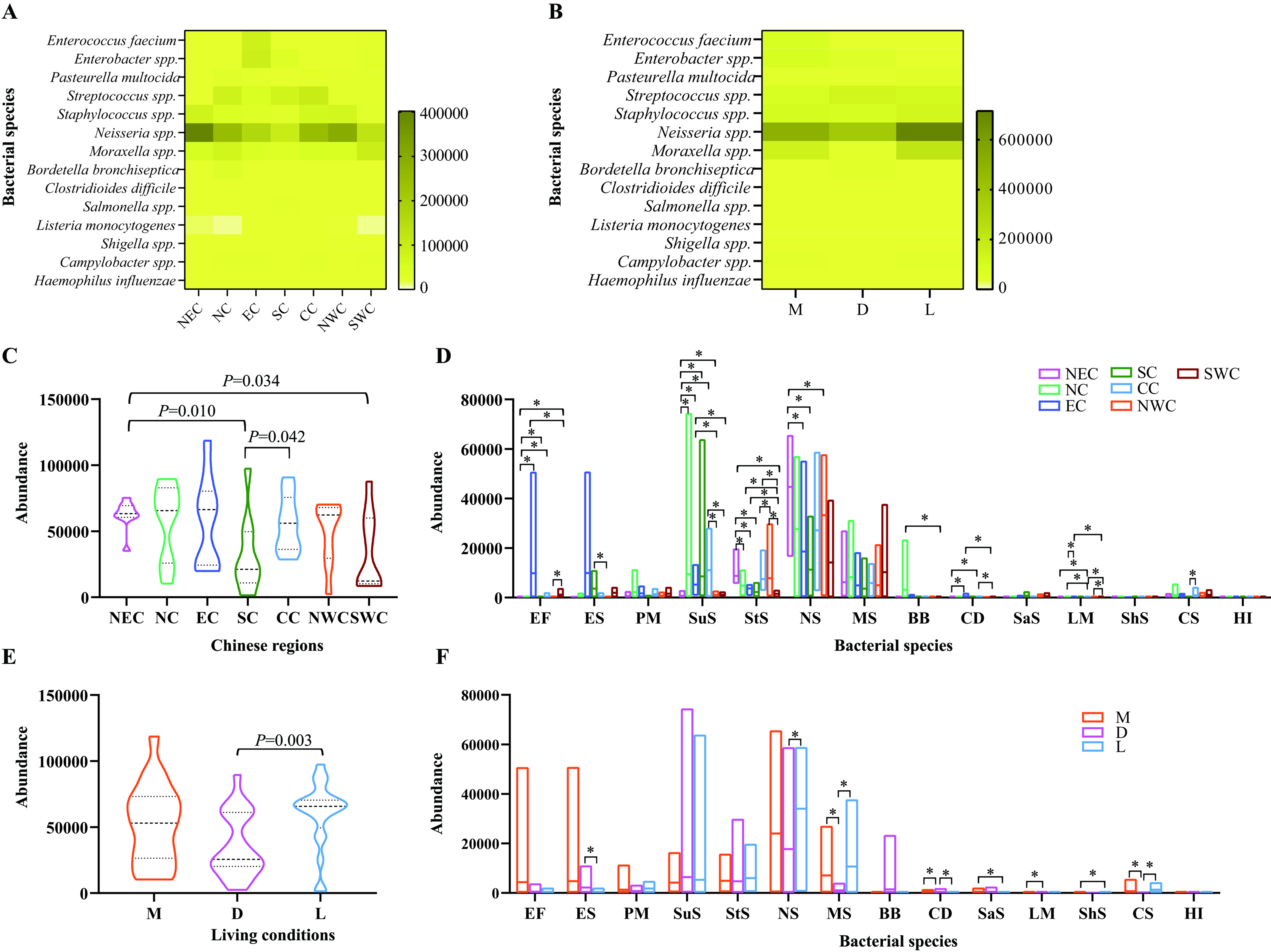
Figure 6. Characterization of bacterial species associated with public health concerns. (A) A heatmap displaying the distribution and detection abundance of specific bacterial species linked to public health concerns in the upper respiratory tract of cats from various Chinese regions; (B) A heatmap illustrating the distribution and detection abundance of specific bacterial species associated with public health concerns in the upper respiratory tract of cats across different living conditions; (C) Comparisons of the total abundances of the bacteriome of these bacterial species in the upper respiratory tract of cats from different Chinese regions (P < 0.05); (D) Comparisons of the abundances of bacteriome of different bacterial species in the upper respiratory tract of cats across various Chinese regions; (E) Comparisons of the total abundances of the bacteriome of these bacterial species in the upper respiratory tract of cats in different living conditions; (F) Comparisons of the abundances of the bacteriome of different bacterial species in the upper respiratory tract of cats across diverse living conditions. Mann-Whitney U test was performed to compare the abundances between two groups, using pairwise multiple comparison adjustments according to the Benjamini-Hochberg procedure. The significance level was set at a P value of < 0.05 (*) and P > 0.05 is not marked. Different Chinese regions: NEC - Northeast China, NC - Northern China, EC - Eastern China, CC - Central China, SC - Southern China, NWC - Northwest China, SWC - Southwest China. Different living conditions: M - catteries, D - animal hospitals, L - stray bases. Bacterial species: EF - Enterococcus faecium, ES - Enterobacter spp., PM - Pasteurella multocida, SuS - Streptococcus spp., StS - Staphylococcus spp., NS - Neisseria spp., MS - Moraxella spp., BB - Bordetella bronchiseptica, CD - Clostridioides difficile, SaS - Salmonella spp., LM - Listeria monocytogenes, ShS - Shigella spp., CS - Campylobacter spp., HI - Haemophilus influenzae.
DISCUSSION
In this study, we conducted a nationwide genomic survey to characterize the resistome and bacteriome in the upper respiratory tract of domestic cats in China. As one of the world’s fastest-growing economies, China has experienced a dramatic increase in pet ownership, particularly cats, in recent years. By 2022, an estimated 65.4 million cats were kept as pets in urban households in China[33]. Given this close human-animal interaction, understanding the antimicrobial-resistant bacterial profile in domestic cats carries significant public health implications. While several epidemiological studies have monitored the prevalence of zoonotic antimicrobial-resistant bacteria in Chinese cats[34,35], many remain limited in scope. To our knowledge, this study represents the first large-scale metagenomic sequencing-based surveillance of antimicrobial-resistome and bacteriome in the upper respiratory tract of domestic cats across China.
By analyzing 1,454 samples, we comprehensively characterized AMR bacteria in feline respiratory tracts. Compared to other anatomical sites, the upper respiratory tract poses a higher transmission risk to humans due to frequent close contact. Notably, respiratory infections caused by antimicrobial-resistant bacteria contribute substantially to global AMR-related deaths[3]. Additionally, increasing reports of human infections following cat bites or scratches further justify our focus on oropharyngeal/nasal swabs[36-38]. These factors collectively highlight the importance of this study.
Our results indicate that domestic cats may serve as reservoirs for ARGs, which were abundantly detected in their upper respiratory tracts. Many of these ARGs confer resistance to antibiotics critical for human medicine, as classified by the WHO[32], including: 3rd/4th/5th-generation cephalosporins (blaCTX-M, blaTEM-1, blaSHV-1, blaVEB-5, blaGES-13, blaOXA-9/59/209), vancomycin (vanABCDEGHMNRSTUVXYZ), polymyxins (mcr-1.2, mcr-1.4, mcr-1.5, mcr-2.1, mcr-2.2, mcr-3, mcr-4, mcr-5), and quinolones (qnrA, qnrB, qnrS). Of particular concern were mobile ARGs conferring resistance to last-resort antibiotics such as carbapenems (blaNDM,
Beyond ARGs, we identified numerous bacterial species capable of causing severe infections in humans, including ESKAPE pathogens (E. faecium, Enterobacter spp., S. aureus, K. pneumoniae, A. baumannii, and P. aeruginosa), which pose major challenges in global healthcare[42,43]. Among them, E. coli, S. aureus,
Several identified bacteria are also causative agents of respiratory disease in cats. For example,
This study has several limitations that should be acknowledged. To improve dataset comprehensiveness, future research should include samples from a wider variety of cat breeds and geographic regions. Additionally, while stray base cats were included, many had been in captivity, potentially limiting their representativeness of truly feral populations. However, sampling fully free-roaming cats is logistically challenging due to their elusive behavior. Due to the high cost of individual sequencing, pooled samples were used - a common and acceptable approach for resistome, bacteriome, and virome studies[30,51,52], despite potential limitations in tracking ARGs and bacterial species at the individual level. Furthermore, antibiotic usage data were unavailable, which may influence resistome differences. Future work should incorporate detailed drug administration records to distinguish environmental effects from antibiotic-driven selection.
Despite these limitations, our findings still provide valuable insights into the AMR landscape of domestic cats across China and highlight their potential zoonotic risks. Regular health monitoring of cats under the One Health framework is essential to address global public health challenges.
DECLARATIONS
Acknowledgments
The authors thank their colleagues for their contribution to sample collection. The authors also acknowledge the Ministry of Natural Resources of the People’s Republic of China for providing standard maps for data presentation.
Authors’ contributions
Conceptualization: Peng Z, Ye J, Cao S
Data curation: Li Q, Zhou D, Cao L, Li Y, Li J
Formal analysis: Li Q, Zhou D, Cao L, Li Y, Li J
Funding acquisition: Peng Z, Ye J, Cao S, Li Q
Investigation: Li Q, Zhou D, Cao L, Li Y, Li J
Methodology: Li Q, Zhou D, Peng Z
Project administration: Peng Z, Ye J, Cao S
Supervision: Peng Z, Ye J, Cao S, Chen H
Writing - original draft: Li Q, Peng Z
Writing - review and editing: Peng Z, Ye J, Cao S, Zhao J, Chen H
Availability of data and materials
All sequencing data were deposited into the NCBI Sequence Read Archive (SRA) database under the BioProject accession number PRJNA998709. SRA accession numbers are SRR33674915-SRR33674924, SRR33696099-SRR33696115, and SRR34261881-SRR34261890.
Financial support and sponsorship
This work was supported by National Key Research and Development Program of China (2023YFD1800402, 2022YFD1800105, 2022YFD1801500, 2021YFC2600204), National Natural Science Foundation of China (32022082, 31972721), Fundamental Research Funds for the Central Universities (Project 2662023PY005), Natural Science Foundation of Hubei Province (2021CFA056, 2023AFA094), and Hubei Hongshan Laboratory & Huazhong Agricultural University Start up fund. The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Sample collection was approved by the Animal Management and Ethics Committee of Huazhong Agricultural University (Approval ID: HZAUCA-2023-0005). All samples were collected only after informed consent had been obtained from the pet owners by the head of the sampling institution.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
Supplementary Materials
REFERENCES
1. Walsh TR, Gales AC, Laxminarayan R, Dodd PC. Antimicrobial resistance: addressing a global threat to humanity. PLoS Med. 2023;20:e1004264.
2. O’Neill J. Tackling drug-resistant infections globally: final report and recommendations. 2016. Available from: https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf. [Last accessed on 24 Jul 2025].
3. Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet. 2022;399:629-55.
4. Graham DW, Bergeron G, Bourassa MW, et al. Complexities in understanding antimicrobial resistance across domesticated animal, human, and environmental systems. Ann N Y Acad Sci. 2019;1441:17-30.
5. Jin M, Osman M, Green BA, et al. Evidence for the transmission of antimicrobial resistant bacteria between humans and companion animals: a scoping review. One Health. 2023;17:100593.
6. Schubiger V. How many cats are in the world? Available from: https://a-z-animals.com/blog/how-many-cats-are-in-the-world/. 2023. [Last accessed on 24 Jul 2025].
7. Deloitte. Chinese Pet Food Industry White Paper. Available from: https://www.199it.com/archives/1527220.html. [Last accessed on 24 Jul 2025].
8. Jung WK, Shin S, Park YK, et al. Distribution and antimicrobial resistance profiles of bacterial species in stray cats, hospital-admitted cats, and veterinary staff in South Korea. BMC Vet Res. 2020;16:109.
9. Faires MC, Gard S, Aucoin D, Weese JS. Inducible clindamycin-resistance in methicillin-resistant Staphylococcus aureus and methicillin-resistant Staphylococcus pseudintermedius isolates from dogs and cats. Vet Microbiol. 2009;139:419-20.
10. Yang Y, Hu X, Cai S, et al. Pet cats may shape the antibiotic resistome of their owner’s gut and living environment. Microbiome. 2023;11:235.
11. Sturgeon A, Pinder SL, Costa MC, Weese JS. Characterization of the oral microbiota of healthy cats using next-generation sequencing. Vet J. 2014;201:223-9.
12. Rottmann FA, Schorle P, Giesen R, Jäger C. Prosthetic valve endocarditis caused by Pasteurella dagmatis, Germany. Emerg Infect Dis. 2024;30:2202-4.
13. Westling K, Farra A, Cars B, et al. Cat bite wound infections: a prospective clinical and microbiological study at three emergency wards in Stockholm, Sweden. J Infect. 2006;53:403-7.
14. Hendriksen RS, Munk P, Njage P, et al; Global Sewage Surveillance project consortium. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat Commun. 2019;10:1124.
15. Huang X, Yao X, Song W, et al. Discovery of viruses and bacteria associated with swine respiratory disease on farms at a nationwide scale in China using metatranscriptomic and metagenomic sequencing. mSystems. 2025;10:e0002525.
16. Allen GC, Flores-Vergara MA, Krasynanski S, Kumar S, Thompson WF. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat Protoc. 2006;1:2320-5.
17. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-20.
18. Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674-6.
19. Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072-5.
20. Zhao R, Yu K, Zhang J, et al. Deciphering the mobility and bacterial hosts of antibiotic resistance genes under antibiotic selection pressure by metagenomic assembly and binning approaches. Water Res. 2020;186:116318.
21. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119.
22. Yin X, Jiang XT, Chai B, et al. ARGs-OAP v2.0 with an expanded SARG database and Hidden Markov Models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics. 2018;34:2263-70.
23. Buchfink B, Reuter K, Drost HG. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. 2021;18:366-8.
24. Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377-86.
25. Ondov BD, Bergman NH, Phillippy AM. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics. 2011;12:385.
26. Manor O, Borenstein E. Systematic characterization and analysis of the taxonomic drivers of functional shifts in the human microbiome. Cell Host Microbe. 2017;21:254-67.
27. Ayala-Muñoz D, Burgos WD, Sánchez-España J, Couradeau E, Falagán C, Macalady JL. Metagenomic and metatranscriptomic study of microbial metal resistance in an acidic pit lake. Microorganisms. 2020;8:1350.
28. Huang X, Wu W, Tian X, et al. A total infectome approach to understand the etiology of infectious disease in pigs. Microbiome. 2022;10:73.
29. Somerfield PJ, Clarke KR, Gorley RN. A generalised analysis of similarities (ANOSIM) statistic for designs with ordered factors. Austral Ecol. 2021;46:901-10.
30. Hu Y, Cheng M, Liu B, et al. Metagenomic analysis of the lung microbiome in pulmonary tuberculosis - a pilot study. Emerg Microbes Infect. 2020;9:1444-52.
31. Nearing JT, Douglas GM, Hayes MG, et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat Commun. 2022;13:342.
32. WHO. WHO list of medically important antimicrobials. 2024. Available from: https://cdn.who.int/media/docs/default-source/gcp/who-mia-list-2024-lv.pdf. [Last accessed on 24 Jul 2025].
33. Ou X. Number of domestic cats and dogs in urban China 2020-2024. Available from: https://www.statista.com/statistics/1196715/china-number-of-domestic-dogs-and-cats-kept-as-pets/. [Last accessed on 24 Jul 2025].
34. Ma S, Chen S, Lyu Y, et al. China antimicrobial resistance surveillance network for pets (CARPet), 2018 to 2021. One Health Adv. 2023;1:7.
35. Teng L, Liao S, Zhou X, et al. Prevalence and genomic investigation of multidrug-resistant Salmonella isolates from companion animals in Hangzhou, China. Antibiotics. 2022;11:625.
36. Shinohara K, Tsuchido Y, Suzuki M, et al. Putative novel species of genus Capnocytophaga, Capnocytophaga stomatis bacteremia in a patient with multiple myeloma after direct contact with a cat. Intern Med. 2022;61:2233-7.
37. Vecilla DF, Matheus MPR, Hidalgo GI, de Tuesta Del Arco JLD. Osteomyelitis caused by Pasteurella multocida and Bacteroides pyogenes after cat bite. Eur J Clin Microbiol Infect Dis. 2023;42:125-8.
38. Fennell AG, Wilson KS, Caja KR, Parikh PM.
39. Peng Z, Hu Z, Li Z, et al. Antimicrobial resistance and population genomics of multidrug-resistant Escherichia coli in pig farms in mainland China. Nat Commun. 2022;13:1116.
40. McEwen SA, Collignon PJ. Antimicrobial resistance: a one health perspective. Microbiol Spectr. 2018;6:10.1128/microbiolspec.arba-0009-2017.
41. Velazquez-Meza ME, Galarde-López M, Carrillo-Quiróz B, Alpuche-Aranda CM. Antimicrobial resistance: one health approach. Vet World. 2022;15:743-9.
42. Roch M, Sierra R, Andrey DO. Antibiotic heteroresistance in ESKAPE pathogens, from bench to bedside. Clin Microbiol Infect. 2023;29:320-5.
43. Zhen X, Lundborg CS, Sun X, Hu X, Dong H. Economic burden of antibiotic resistance in ESKAPE organisms: a systematic review. Antimicrob Resist Infect Control. 2019;8:137.
44. Peng Z, Lin L, Wang X, Chen H, Wu B. The public health concern of Pasteurella multocida should not be ignored. Lancet Microbe. 2022;3:e560.
45. Goldstein EJC, Abrahamian FM. Diseases transmitted by cats. Microbiol Spectr. 2015;3:10.1128/microbiolspec.iol5-0013-2015.
46. Abrahamian FM, Goldstein EJ. Microbiology of animal bite wound infections. Clin Microbiol Rev. 2011;24:231-46.
47. CDC. 2019 Antibiotic Resistance Threats Report. Available from: https://www.cdc.gov/antimicrobial-resistance/data-research/threats/index.html. [Last accessed on 24 Jul 2025].
48. Lappin MR, Blondeau J, Boothe D, et al. Antimicrobial use guidelines for treatment of respiratory tract disease in dogs and cats: Antimicrobial Guidelines Working Group of the International Society for Companion Animal Infectious Diseases. J Vet Intern Med. 2017;31:279-94.
49. Maboni G, Che S, Tallmadge R, et al. Feline respiratory disease complex: insights into the role of viral and bacterial co-infections. Front Microbiol. 2024;15:1455453.
50. Kadlec K, Schwarz S. Antimicrobial resistance in Bordetella bronchiseptica. Microbiol Spectr. 2018;6:10.1128/microbiolspec.arba-0024-2017.
51. He T, Xie J, Jin L, et al. Seasonal dynamics of the phage-bacterium linkage and associated antibiotic resistome in airborne PM2.5 of urban areas. Environ Int. 2024;194:109155.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].