


Macrophage responses to extracellular matrix stiffening in cardiac fibrosis: mechanisms and implications
 0
0 Abstract
Cardiac fibrosis is a hallmark of various cardiovascular diseases and is characterized by excessive extracellular matrix (ECM) deposition and progressive tissue stiffening. Emerging evidence suggests that ECM stiffening is not merely a mechanical consequence of fibrosis but also an active driver of pathological cellular responses. Among the mechanosensitive cells within the myocardium, macrophages play a central role in sensing and responding to mechanical cues. Through defined mechanotransduction pathways, macrophages undergo phenotypic and functional reprogramming that shapes inflammatory signaling and the progression of fibrosis. In this review, we summarize current knowledge on ECM stiffening in cardiac fibrosis and its impact on macrophage behavior. We also highlight key mechanosensors that mediate macrophage responses to biomechanical stress. Finally, we discuss emerging therapeutic strategies targeting macrophage mechanotransduction and ECM remodeling and explore their potential as antifibrotic interventions in cardiovascular disease.
Keywords
INTRODUCTION
Cardiac fibrosis is a common pathological feature in the progression of many cardiovascular diseases, including hypertension[1], myocardial infarction (MI)[2], and cardiomyopathies[3], and it is also associated with aging[4]. It is characterized by excessive deposition and crosslinking of extracellular matrix (ECM) components within the myocardium. This process distorts cardiac tissue architecture, promotes arrhythmia and contractile dysfunction, and ultimately worsens clinical outcomes in patients with heart failure (HF)[5]. While alterations in ECM composition are recognized as key drivers of fibrotic remodeling, recent research indicates that ECM stiffening itself functions as a potent biomechanical signal regulating cellular behavior and tissue remodeling[6-8]. This concept shifts our understanding of cardiac fibrosis from a mainly biochemical process to a dynamic interplay between mechanical and molecular cues.
Cardiac-resident macrophages constitute a heterogeneous population that coordinates inflammation, tissue repair, and myocardial remodeling[9]. Beyond classical immune stimuli such as pathogen- or damage-associated signals, macrophages are increasingly recognized as mechanosensitive cells that detect and respond to changes in ECM stiffness[10]. Mechanoreceptors involved include integrins[11,12], mechanosensitive ion channel Piezo1[13,14], and cytoskeletal elements. Activation of downstream pathways such as Yes-associated protein (YAP) and transcriptional coactivator with PSD-95/Dlg/ZO-1 (PDZ)-binding motif (TAZ)[15,16] as well as Notch signaling[17,18], transduces mechanical cues into transcriptional and metabolic reprogramming. These mechano-adaptive responses modulate macrophage polarization, cytokine production, and crosstalk with fibroblasts, thereby influencing both the progression and resolution of cardiac fibrosis.
Importantly, cardiac fibrosis frequently develops and progresses with aging, even in the absence of overt ischemic injury or pressure overload[19]. Aging-associated alterations in the myocardial ECM, including increased collagen crosslinking, altered viscoelastic properties, and reduced matrix turnover[20], create a persistently stiffened and less elastic cardiac microenvironment[21]. Concurrently, aging reshapes the cardiac macrophage compartment, characterized by loss of tissue-resident macrophages and expansion of inflammatory, monocyte-derived C-C chemokine receptor type 2 (CCR2+) macrophages[9,22], which contributes to chronic low-grade inflammation. Given extensive evidence from non-cardiac tissues such as tumor[23], bone[24], and vascular environment[25] showing that infiltrating macrophages actively sense and respond to ECM mechanical cues, it is plausible that ECM mechanics play a central role in shaping macrophage function during aging-associated cardiac fibrosis.
Despite growing interest in macrophage mechanobiology, the precise mechanisms by which ECM stiffness modulates macrophage behavior in the fibrotic heart remain incompletely understood. Moreover, how these immune-mechanical interactions can be therapeutically exploited to mitigate cardiac fibrosis is still largely unexplored. In this review, we summarize current insights into extracellular matrix stiffening in cardiac fibrosis, highlight emerging molecular mechanisms of macrophage mechanosensing, and discuss potential biophysical strategies that target mechanotransduction pathways to influence fibrotic remodeling.
EXTRACELLULAR MATRIX STIFFENING IN CARDIAC FIBROSIS
Mechanical stress in the cardiac microenvironment
The cardiac microenvironment is continuously exposed to multiple forms of mechanical stress. These include dynamic stimuli such as cyclic strain and shear stress[26]. In addition, cells experience forces transmitted through cell-cell interactions as well as mechanical cues derived from the ECM, including stiffness, topography, and geometric confinement. These ECM-derived cues are generally considered static biomechanical properties[26-28]. Together, these mechanical stimuli drive functional changes in multiple cardiac cell types, including cardiomyocytes, endothelial cells, fibroblasts, and immune cells [Figure 1].
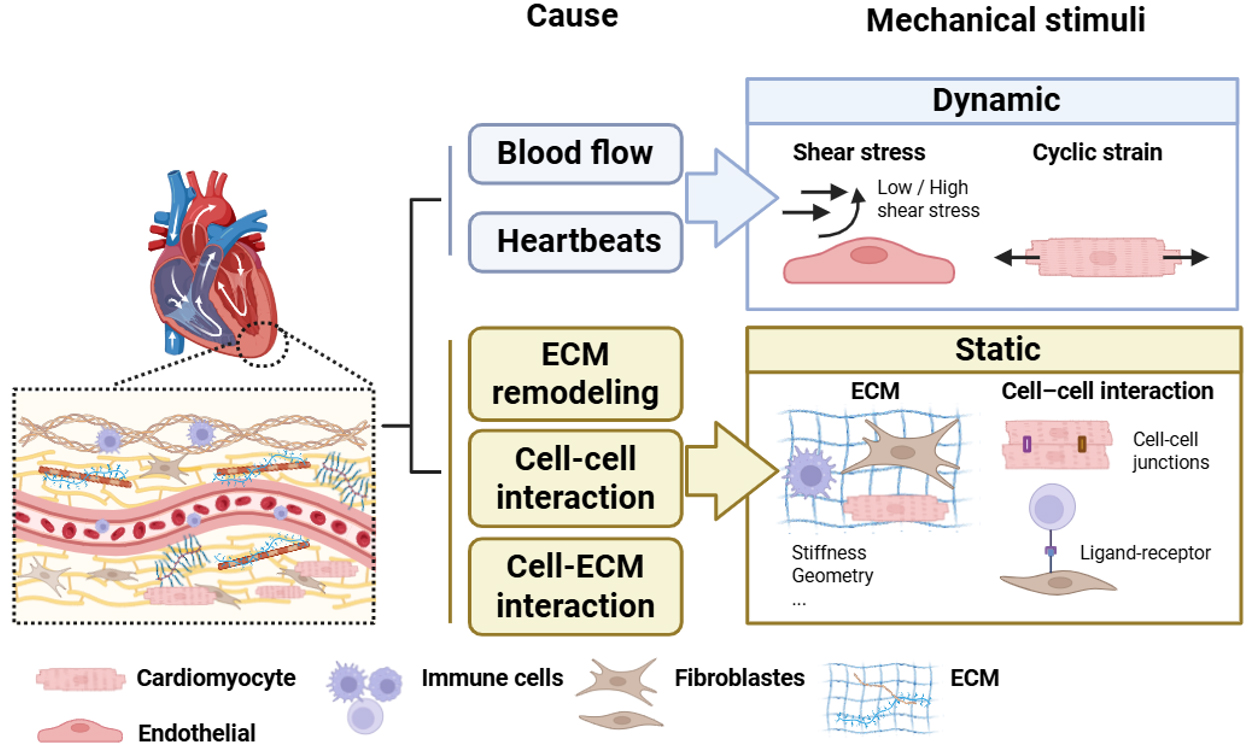
Figure 1. Mechanical stress in the cardiac microenvironment. Mechanical forces in the cardiac microenvironment arise from both hemodynamic activity and structural remodeling. Blood flow and heartbeats generate dynamic mechanical stimuli, including shear stress and cyclic strain. In contrast, ECM remodeling and cell-ECM adhesions produce relatively static mechanical cues, such as ECM stiffness, geometric constraints, and signaling mediated by cell-cell interaction. These mechanical stimuli collectively shape the heart’s biomechanical environment and influence cellular responses during cardiac homeostasis and disease.
Among the dynamic stimuli, cyclic strain is a mechanical force generated by the heartbeat. During myocardial contraction and chamber filling, cardiac tissues undergo repetitive stretching. Experimental data indicate that physiological valvular tissues typically experience approximately 10% cyclic strain, whereas pathological conditions such as valvular disease or abnormal hemodynamic loading, including hypertension, HF, and pressure overload, can increase strain levels toward ~20%[29].
Another important mechanical stimulus in the cardiovascular system is wall shear stress, which arises from blood flow acting on the vascular endothelium. In coronary arteries, shear stress varies throughout the cardiac cycle and contributes to endothelial homeostasis. Physiological shear stress promotes anti-inflammatory signaling and vascular stability. In contrast, low shear stress is associated with endothelial dysfunction and initiation of atherosclerotic plaques, while abnormally high shear stress in stenotic regions may contribute to plaque destabilization and vascular remodeling[30].
In addition to hemodynamic forces, progressive ECM remodeling during cardiac aging and fibrosis alters matrix stiffness and mechanical signaling, which in turn regulates cellular behavior. In this review, we primarily focus on ECM-derived mechanical cues and their roles in the initiation, progression, and resolution of cardiac fibrosis.
Myocardial stiffness in physiological and pathological conditions
ECM stiffening is a defining biomechanical feature of cardiac fibrosis and represents a critical contextual signal that shapes cellular behavior within the myocardium. Changes in myocardial stiffness not only reflect disease severity but also actively regulate mechanoresponsive cells, including macrophages. Under physiological conditions, the stiffness of healthy myocardium typically ranges from ~10 to 15 kPa. However, during pathological remodeling, such as that induced by pressure overload, myocardial stiffness can increase significantly. In murine models subjected to transverse aortic constriction (TAC), stiffness has been shown to rise to 30-50 kPa, as measured by atomic force microscopy (AFM)[31]. By contrast, clinical in vivo elastography and ultrasound-based assessments report increased myocardial stiffness in patients with HF, rising from approximately 4 kPa in healthy hearts to about 12 kPa in diseased myocardium[32-34]. These differences underscore the importance of reporting measurement modality and species when comparing stiffness values across studies.
Myocardial stiffness is primarily governed by dynamic remodeling of the cardiac ECM [Figure 2]. Under physiological conditions, the ECM comprises a highly organized network of fibrillar collagens (primarily types I and III), elastin, laminin, fibronectin, proteoglycans, and glycosaminoglycans[35]. Together, these components maintain myocardial structural integrity, elasticity, and essential cell-matrix interactions. In response to pathological stimuli, such as pressure overload, ischemic injury, or aging, the homeostatic balance between ECM synthesis and degradation is disrupted[36]. Following MI, ECM remodeling is temporally staged, with early matrix degradation and provisional matrix formation followed by collagen accumulation and progressive stiffening during scar maturation[37,38].
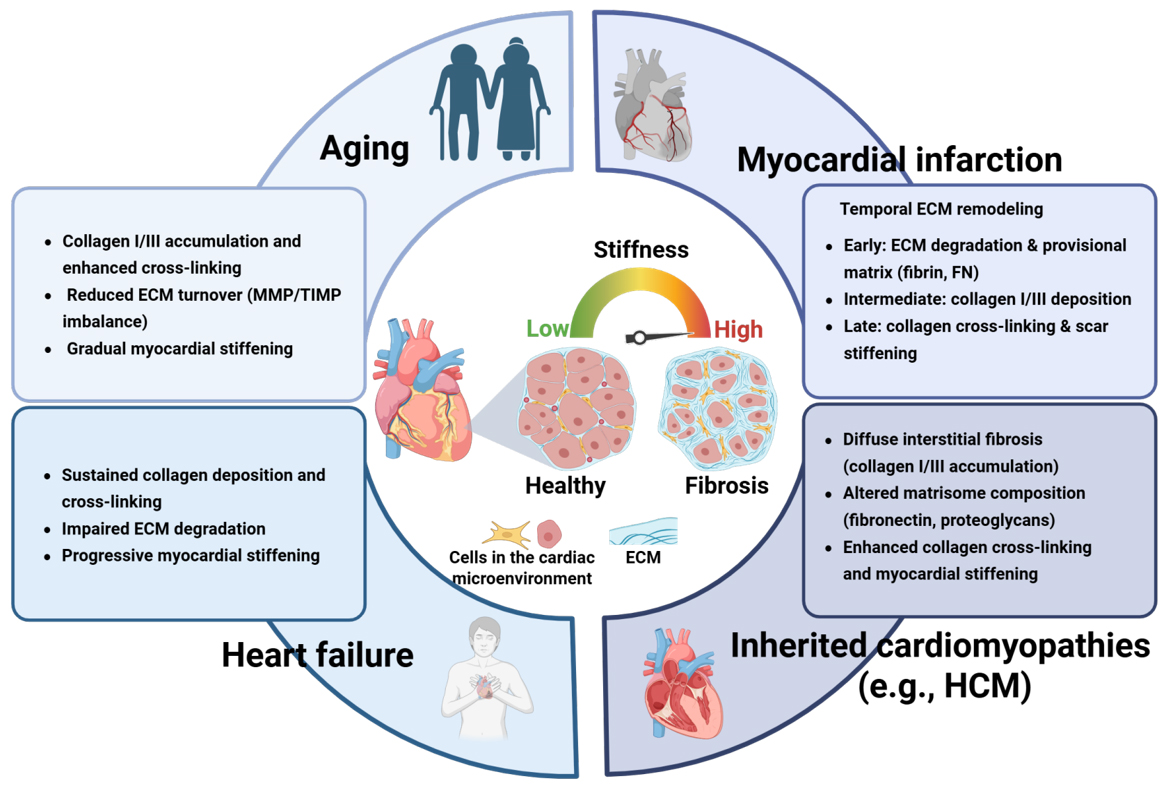
Figure 2. Extracellular matrix remodeling across physiological aging and major cardiac disease contexts. Schematic overview of dynamic extracellular matrix (ECM) remodeling and associated changes in myocardial stiffness under physiological and pathological conditions, including aging, myocardial infarction (MI), heart failure (HF), and inherited cardiomyopathies (e.g., hypertrophic cardiomyopathy, HCM).
Beyond injury-induced fibrosis, inherited cardiomyopathies also exhibit prominent ECM remodeling and myocardial stiffening. For example, hypertrophic cardiomyopathy (HCM) is frequently accompanied by abnormal ECM turnover, interstitial fibrosis, and increased left ventricular stiffness, which correlates with disease severity and clinical symptoms[39]. Notably, these fibrotic and biomechanical changes can develop in the absence of classic ischemic or pressure-overload injury, highlighting ECM remodeling as an intrinsic driver of myocardial stiffening in certain genetic heart diseases.
In ischemic cardiomyopathy (ICM), a leading cause of HF, ECM remodeling largely represents a secondary response to cardiomyocyte injury and death and is characterized by progressive accumulation of collagens, proteoglycans, glycoproteins, and regulators of ECM cross-linking, as revealed by quantitative ECM proteomics[40]. This injury-driven ECM deposition initially serves an adaptive role in maintaining structural integrity but ultimately becomes maladaptive, forming a feed-forward loop in which increased matrix stiffness further amplifies profibrotic signaling and adverse cardiac remodeling[20].
Extracellular matrix components contributing to myocardial stiffening
Aging represents a distinct and progressive driver of myocardial stiffening, characterized by cumulative collagen cross-linking and impaired matrix turnover. Alterations in the composition of specific ECM proteins are key determinants of myocardial stiffness. Among fibrillar collagens, type I and type III are the major structural proteins in the heart. Numerous studies reported an increased collagen I/III ratio across diverse cardiovascular diseases, a change that correlates with progressive myocardial stiffening and reduced tissue compliance[41,42]. In addition to collagens, other structural ECM components (for example, fibronectin, elastin) and matricellular proteins (for example, tenascin, osteopontin, secreted protein acidic and rich in cysteine (SPARC)) play crucial roles in maintaining ECM homeostasis[43]. Fibronectin assembly is essential for ECM organization because fibronectin fibrillogenesis is required for the proper deposition of collagen I and thrombospondin 1[44].
Beyond structural proteins, non-structural enzymes such as matrix metalloproteinases (MMPs) play a critical role in ECM remodeling and myocardial stiffening during cardiac fibrosis. MMPs are a large family of zinc-dependent endopeptidases that degrade various ECM components, including collagens, elastin, fibronectin, laminin, and proteoglycans[45]. To date, over 20 MMPs have been identified and are typically classified based on substrate specificity and structural domains, such as collagenases (e.g., MMP-1, MMP-8, MMP-13), gelatinases (e.g., MMP-2, MMP-9), stromelysins (e.g., MMP-3, MMP-10), and membrane-type MMPs (MT-MMPs)[46]. In the heart, MMPs are produced by cardiac fibroblasts, macrophages, neutrophils, and endothelial cells in response to injury or stress signals. These enzymes play essential roles in ECM turnover, angiogenesis, and tissue repair following injury. However, dysregulated MMP activity is a hallmark of pathological cardiac remodeling. For instance, elevated levels of circulating MMP-9 have been demonstrated in patients with coronary artery disease (CAD)[47]. A study using transgenic mice with macrophage-specific overexpression of MMP-9 demonstrated that elevated MMP-9 levels led to age-dependent cardiac inflammation and fibrosis[48]. This finding was evidenced by increased expression of pro-inflammatory genes and excessive collagen deposition, ultimately resulting in myocardial stiffening and impaired cardiac function[49].
Secreted phosphoprotein 1 (SPP1), also known as osteopontin, has emerged as a critical regulator of ECM remodeling in several cardiovascular conditions, including thrombus formation[50], vascular disease[10,51], and HF[52]. Via interactions with integrins and CD44, SPP1 modulates ECM synthesis, crosslinking, and degradation, thereby influencing tissue repair and fibrotic remodeling[53,54]. Notably, SPP1 expression is also mechanosensitive. In tumor models, elevated ECM stiffness induces SPP1 expression and secretion, thereby contributing to tumor-associated fibrosis[55,56]. Aberrant SPP1 production is also associated with vascular stiffening and pathological calcification[57,58]. Although direct evidence that SPP1 acts as an intrinsic ECM stiffness sensor in the cardiovascular system remains unclear, our recent data demonstrate that increasing matrix stiffness upregulates SPP1 expression and secretion in macrophages, and this in turn exacerbates vascular ECM remodeling[10]. As the most abundant immune cell population in the heart, macrophages are essential for tissue homeostasis and for orchestrating early inflammatory responses after cardiovascular injury[59-61]. Nonetheless, the precise mechanisms by which cardiac macrophages sense and transduce ECM-derived mechanical cues remain largely unresolved.
Cellular responses to myocardial stiffening
Progressive ECM stiffening is a hallmark of cardiac aging and fibrosis and profoundly influences the behavior of multiple cardiac cell types. For cardiomyocytes, increased ECM stiffness disrupts contractile function and calcium handling, impairs sarcomere organization, and promotes pathological hypertrophy. Cardiomyocytes cultured on stiff substrates exhibit altered mechanotransduction signaling and reduced contractile efficiency, suggesting that pathological stiffening of the myocardium can directly contribute to impaired cardiac performance and HF progression[62,63].
Endothelial cells also respond dynamically to changes in matrix stiffness. Increased substrate stiffness enhances endothelial proliferation, migration, and cytoskeletal remodeling via mechanosensitive signaling pathways. These stiffness-dependent endothelial responses may contribute to vascular remodeling and impaired endothelial barrier function, thereby influencing myocardial perfusion and inflammatory signaling during cardiac disease[64].
Cardiac fibroblasts are the primary ECM-producing cells in the heart and are highly responsive to mechanical cues. Elevated matrix stiffness promotes fibroblast activation into myofibroblasts, characterized by increased proliferation, upregulation of α-smooth muscle actin (α-SMA), and excessive collagen synthesis. This stiffness-driven fibroblast activation establishes a positive feedback loop: ECM deposition raises tissue stiffness, which in turn further exacerbates myocardial fibrosis and structural remodeling[43,65,66].
By comparison, the mechanosensitivity of immune cells, particularly macrophages, in the cardiac microenvironment is less well characterized. In multiple noncardiac tissues, macrophages and other immune cells respond to matrix stiffness by altering polarization, migration, and inflammatory signaling, but evidence from the heart remains limited[6]. Understanding how immune cells, particularly macrophages, sense and respond to ECM stiffening may provide new insights into the regulation of inflammation and fibrosis in the diseased heart[62].
Macrophage heterogeneity in the aging and fibrotic heart
Cardiac aging and age-associated fibrosis are accompanied by profound remodeling of the cardiac immune microenvironment, with macrophages forming a highly heterogeneous and functionally diverse population. Under steady-state conditions, the adult heart is predominantly populated by embryonically derived CCR2- resident macrophages that maintain tissue homeostasis. In contrast, aging and chronic cardiac stress increase the proportion of monocyte-derived CCR2+ macrophages that display pro-inflammatory and profibrotic phenotypes[67]. Single-cell transcriptomic and lineage-tracing studies reveal that aging is associated with shifts in cardiac macrophage composition, including reduced self-renewal capacity, impaired efferocytosis, and upregulation of inflammatory and ECM-related gene programs[68].
Fibrotic remodeling of the aged heart is closely linked to the expansion of distinct macrophage subsets that express profibrotic mediators, SPP1, transforming growth factor-beta (TGF-β), and matrix-remodeling enzymes, and that spatially colocalize with activated fibroblasts within fibrotic niches[67]. Collectively, these findings indicate that cardiac macrophages do not constitute a uniform population during aging. Rather, they adopt diverse, context-dependent states that integrate inflammatory signaling, fibrotic remodeling, and tissue-specific cues, thereby shaping disease progression in the cardiac microenvironment.
Disease etiology further shapes macrophage heterogeneity in the fibrotic heart. Integrated single-cell transcriptomic analysis across distinct cardiac conditions—MI, ICM, dilated cardiomyopathy (DCM), and heart failure (HF)[69,70]—has demonstrated that pathological environments differentially regulate macrophage gene programs. In DCM and MI, multiple macrophage clusters exhibit robust profibrotic signatures characterized by secretion of ECM-related mediators such as Fibronectin 1 (FN1), SPP1, and Thrombospondin-1 (THBS1)[69,71], promoting widespread macrophage-fibroblast communication. By contrast, in ICM and HF, profibrotic activity is often confined to a smaller subset of macrophage clusters, producing a more localized fibrotic phenotype[72]. Despite these disease-specific differences, all conditions converge on enhanced ECM remodeling and activation of ECM-receptor signaling pathways, suggesting a shared fibrotic microenvironment that engages common cellular sensing mechanisms.
Importantly, the relationship between ECM remodeling and macrophage activation is bidirectional. Increasing ECM stiffness can directly modulate macrophage mechanosensing and drive functional reprogramming[73,74], while activated macrophages in turn promote ECM deposition, cross-linking, and fibroblast activation through inflammatory cytokines and matricellular signals[75,76]. These reciprocal interactions form a feed-forward loop that amplifies myocardial stiffening and fibrotic remodeling
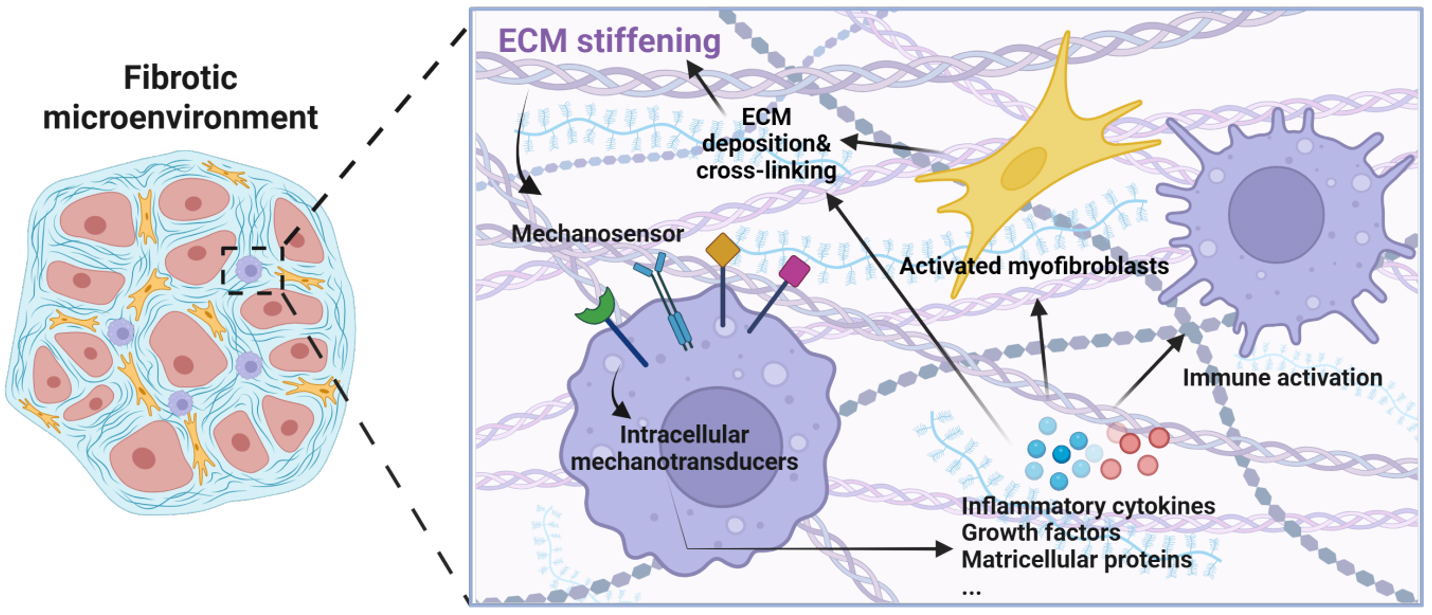
Figure 3. Feed-forward interactions between ECM remodeling and macrophage mechanotransduction in cardiac fibrosis. ECM stiffening modulates macrophage mechanosensing and functional reprogramming through mechanosensors and intracellular mechanotransduction pathways. Macrophages in turn promote ECM deposition, cross-linking, and fibroblast activation via inflammatory cytokines and matricellular signals, forming a self-amplifying loop that drives progressive myocardial fibrosis and stiffening.
MECHANOTRANSDUCTION IN MACROPHAGES
Mechanotransduction is the process by which cells convert mechanical signals into biochemical responses via membrane receptors, the cytoskeleton, and nuclear signaling pathways. Through mechanotransduction, macrophages dynamically adapt by altering gene expression, inflammatory state, metabolism, and interactions with neighboring cells, including fibroblasts and endothelial cells. Here, we focus on mechanotransduction mechanisms relevant to macrophages in the fibrotic cardiac microenvironment [Figure 4].
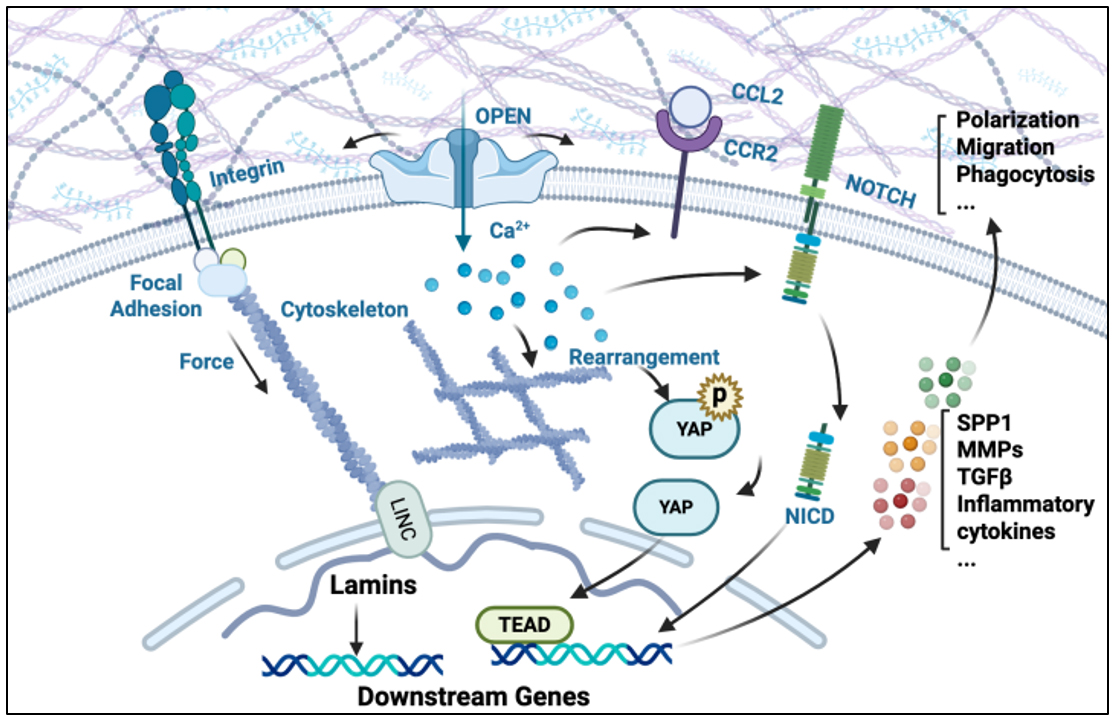
Figure 4. Schematic illustration of macrophage mechanotransduction in response to elevated extracellular matrix (ECM) stiffness during cardiac fibrosis. Macrophages sense elevated ECM stiffness through diverse mechanosensitive elements, including integrins, Piezo1 mechanosensitive ion channels, Notch receptors, and the YAP/TAZ transcription module. These mechanical inputs are transduced via cytoskeletal remodeling and downstream signaling including YAP/TAZ activation, leading to transcriptional reprogramming of pro-fibrotic and inflammatory genes. Piezo1-mediated Ca2+ influx further amplifies CCR2 and Notch signaling, coordinating macrophage recruitment, polarization, and effector functions. Together, these mechano-adaptive responses contribute to cardiac fibrosis by promoting cytokine production, migration, and fibroblast activation.
Accumulating evidence identifies macrophages as highly mechanosensitive immune cells that respond to changes in their extracellular mechanical microenvironment[77-79]. In fibrotic tissues such as the heart[80], blood vessel[81], lung[82] and liver[83], ECM stiffening emerges as a critical biophysical cue that regulates macrophage behavior beyond classical immunological stimuli. Notably, many mechanistic insights into macrophage mechanotransduction derive from noncardiac tissues or from in vitro stiffness-controlled systems, whereas direct evidence in the aging or fibrotic heart remains comparatively limited. Nevertheless, a growing body of studies demonstrates that the fibrotic microenvironment of the diseased heart profoundly reshapes macrophage phenotypes and functions, suggesting that cardiac macrophages may engage mechanosensing mechanisms shared across fibrotic tissues. These shared mechanobiological principles support the relevance of non-cardiac studies for interpreting macrophage responses to ECM stiffening in the aging heart. Below, we summarize well-established mechanosensitive pathways in macrophages from various fibrotic tissues and discuss the current evidence for their roles in aging-associated cardiac fibrosis.
Integrin-mediated mechanotransduction
Integrins in cardiac macrophage-mediated heart disease
Integrins are heterodimeric transmembrane receptors formed by 18 α and 8 β subunits that combine to generate 24 distinct functional αβ heterodimers in mammals[84,85]. They serve as critical mechanosensors across diverse cell types, linking the ECM to intracellular cytoskeletal structures and signaling pathways. Upon sensing increased ECM stiffness, integrins undergo conformational change and clustering, leading to the formation of focal adhesions and the activation of downstream effectors such as focal adhesion kinase (FAK), Src family kinases, and Rho GTPases[86,87]. These signaling cascades coordinate cytoskeletal remodeling, nuclear mechanotransduction, and transcriptional reprogramming, ultimately modulating macrophage phenotype, polarization, and effector functions.
Accumulating evidence suggests that integrin signaling is actively involved in cardiac aging and fibrotic remodeling, largely in response to age- and disease-associated alterations in ECM composition. Aging promotes progressive modifications of fibronectin, which can acquire isoAspartate-glycine-arginine (isoDGR) motifs that enhance binding to integrins expressed on monocytes and macrophages, thereby promoting integrin-dependent macrophage activation in aging-associated cardiovascular pathology[88]. Beyond cardiovascular aging, alpha V (αV)-containing integrins have also been implicated in cardiac injury and fibrotic remodeling, as disruption of αV signaling attenuates adverse remodeling following MI[89]. In addition, leukocyte-specific β2 integrins, particularly CD11b/CD18 (Macrophage-1 antigen (Mac-1)), regulate macrophage recruitment and inflammatory activation in MI and angiotensin II-induced cardiac fibrosis, and genetic or antibody-mediated blockade mitigates fibrotic remodeling[90]. Notably, the pro-fibrotic effects of αV integrin appear, in some contexts, to be independent of classical fibronectin binding, suggesting that integrins can respond broadly to pathological ECM alterations rather than specific chemical ligands[91]. Collectively, these findings indicate that integrin signaling in cardiac macrophages is sensitive to ECM abnormalities characteristic of aging and fibrosis, supporting a potential link between myocardial matrix remodeling and macrophage activation in the aging heart.
Mechanism of integrin-mediated macrophage mechanotransduction
Much of the mechanistic evidence for integrin-mediated macrophage mechanotransduction has been derived from non-cardiac disease models or in vitro systems that control matrix stiffness. For instance,
Studies directly addressing this pathway in cardiac disease are still limited. Cardiovascular research, however, provides supporting evidence that integrins mediate macrophage-ECM interactions in fibrosis. For example, Shimojo et al.[94] reported that integrin αVβ3 senses the ECM protein Tenascin-C and activates the Nuclear factor kappa B (NF-κB)/Interleukin-6 (IL-6) axis, thereby exacerbating cardiac fibrosis. Together, these findings suggest that integrin-dependent mechanotransduction helps shape macrophage behavior within the fibrotic heart microenvironment.
YAP/TAZ-mediated mechanotransduction
YAP in cardiac macrophage-mediated heart disease
YAP and its paralog TAZ are transcriptional coactivators that act as key effectors of mechanical signaling in many cell types, including macrophages[95,96]. In a mouse model of MI, macrophage-specific deletion of YAP/TAZ reduced the expression of pro-inflammatory cytokines (such as TNF-α and IL-6) and increased Arginase-1 (Arg1) expression, changes that improved infarct healing and cardiac function[97]. Mechanistically, YAP/TAZ can promote inflammatory programs (for example, raising IL-6) and repress reparative signatures such as Arg1 via interaction with the Histone deacetylase 3 (HDAC3)-Nuclear receptor corepressor 1 (NCoR1) complex, thereby exacerbating MI-induced fibrosis and remodeling[98].
YAP activity responds to multiple mechanical cues present in the cardiovascular system, including tissue stiffness[99], shear stress[100], cell shape[101] and cyclic stretch[102]. In cardiac macrophages, YAP phosphorylation has been linked to regulation of NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome after MI, which influences cytokine sensing[103]. Phosphorylation at Y357 enhances YAP transcriptional activity[104,105], whereas phosphorylation at S127 promotes its cytoplasmic retention and inhibits nuclear translocation[106]. Moreover, K63-linked ubiquitination of YAP promotes nuclear localization and has been implicated in exacerbating vascular pathology such as atherosclerosis[107].
Mechanism of YAP-mediated macrophage mechanotransduction
Although studies specifically addressing YAP-mediated mechanotransduction in cardiac macrophages remain limited, the mechanisms by which YAP responds to mechanical cues have been well characterized in vitro. In cultured macrophages, YAP activity is dynamically regulated by substrate stiffness, with dephosphorylation and enhanced nuclear translocation observed on stiffer matrices[102]. Activation of the YAP pathway modulates macrophage activation programs: it increases expression of pro-inflammatory cytokines (such as IL-6, IL-1β, and IL-12β) and can regulate anti-inflammatory markers (such as Arg178), thereby reprogramming macrophage functional states[25,108-110]. On rigid substrates, including glass or fibrotic ECM, macrophages exhibit enhanced F-actin polymerization and increased traction forces, which promote YAP nuclear localization and transcriptional activation. Consistently, Meli et al.[111] demonstrated that macrophages cultured on soft fibrin hydrogels display reduced YAP nuclear translocation and attenuated pro-inflammatory cytokine expression upon lipopolysaccharide (LPS) stimulation, whereas stiffer matrices enhanced YAP-dependent transcription and amplified inflammatory responses. These effects were abolished by cytochalasin D, an inhibitor of actin polymerization[111] , highlighting the critical role of cytoskeletal remodeling in YAP-mediated mechanotransduction.
To date, most studies of YAP-mediated mechanotransduction in cardiac macrophages have examined inflammatory activation. However, other fundamental macrophage functions—such as migration and phagocytosis, which are essential for responding to cardiac injury and driving fibrotic progression—remain poorly understood in relation to YAP signaling. While direct evidence from the heart is limited, findings from other ECM-rich environments provide indirect mechanistic insights. For example, in the bone microenvironment, YAP-dependent mechanosensing regulates actin cytoskeletal integrity and is required for the formation of podosomes and sealing zones in osteoclasts, structures that are essential for osteoclastogenesis[112]. By analogy, similar cytoskeletal control mechanisms may operate in macrophages within fibrotic cardiac tissue. Taken together, available evidence supports the concept that YAP/TAZ-mediated mechanotransduction links increased ECM stiffness to sustained inflammation and fibrosis in the heart.
Piezo-mediated mechanotransduction
Piezo1 in cardiac macrophage-mediated heart disease
The Piezo family of mechanosensitive ion channels consists of two members: Piezo1 and Piezo2, both of which serve as primary force sensors that transduce mechanical stimuli into biochemical signals[113]. Piezo1 assembles into an approximately 900 kDa homotrimeric complex, forming a propeller-shaped ion channel with a central pore module embedded in the plasma membrane[83]. Piezo1 opens in response to various mechanical cues such as membrane stretch[114], shear stress[115], and ECM rigidity[116], permitting rapid Ca2+ influx and activation of downstream signaling cascades. While both Piezo subtypes contribute to mechanotransduction, Piezo2 is predominantly expressed in sensory neurons and is essential for touch sensation and proprioception[113,117], whereas Piezo1 is more broadly expressed in non-neuronal cells, including endothelial cells, osteoblasts, and various immune cells such as macrophages[13,118]. Here, we focus on Piezo1-mediated mechanotransduction in cardiac-infiltrating macrophages and its potential role linking ECM stiffness to immune activation and fibrotic remodeling.
In the cardiovascular system, Piezo1 plays a central role in sensing ECM rigidity. Multiple studies report enriched Piezo1 expression in CCR2+ monocyte-derived macrophages (MoMFs). These cells infiltrate solid tissues from the periphery[119-121] and represent a major subtype of cardiac-infiltrating macrophages. While the mechanisms driving CCR2+ monocyte recruitment into fibrotic cardiac tissue remain poorly understood, several lines of evidence in fibrotic diseases suggest that Piezo1 activation promotes C-C motif chemokine ligand 2 (CCL2)-CCR2 axis and Notch signaling, facilitating macrophage recruitment[122]. Additionally, in vascular studies, Piezo1 activation was shown to enhance oxidized low-density lipoprotein (oxLDL) uptake, promoting foam cell formation and contributing to atherosclerotic plaque progression[123]. Genetic deletion of Piezo1 in myeloid cells reduced Ca2+ influx, suppressed the expression of scavenger receptors CD36 and Class A1 scavenger receptor (SRA1), and ultimately mitigated plaque formation in vivo. In the context of cardiac disease, the fibrotic myocardium provides a highly stiffened microenvironment likely to activate Piezo. Peng et al.[124] reported that Piezo1 is expressed in MoMFs and contributes to tissue inflammation following MI. Mechanistically, Piezo1 activation (via Yoda1) increased ferroptosis and impaired efferocytosis by MoMFs, thereby exacerbating inflammation[124].
Mechanism of piezo1-mediated macrophage mechanotransduction
Emerging in vivo studies suggest that macrophage-specific deletion or knockdown of Piezo1 can attenuate cardiac inflammation and ameliorate myocardial injury in inflammatory cardiac conditions[125], highlighting that suppressing Piezo1 signaling is protective in the heart. However, whether these cardioprotective effects are directly mediated by altered mechanical sensing by cardiac macrophages remains insufficiently explored. Furthermore, mechanistic insights linking Piezo1 to macrophage mechanotransduction within the cardiac microenvironment remain limited.
Accumulating in vitro studies have extensively characterized Piezo1 as a central mechanosensor governing macrophage responses to extracellular mechanical cues. Among the classical immune functions of macrophages, Piezo1 has been widely recognized as a key regulator of polarization in response to matrix stiffness. Atcha et al.[126] demonstrated that ECM stiffening promotes Piezo1 activation, evidenced by increased Piezo1 expression and intracellular Ca2+ influx. Activation of Piezo1 augmented the proinflammatory M1 polarization of macrophages on stiff hydrogels, marked by increased inducible nitric oxide synthase (iNOS) expression and NF-κB signaling, while simultaneously suppressing M2 polarization, as indicated by downregulated Arg1 levels. Moreover, Piezo1 has been shown to be essential for durotaxis—the migration of cells along stiffness gradients-in various non-immune cell types[127-129]. Whether Piezo1-mediated mechanical sensing also guides the transition from circulating monocytes to CCR2+ cardiac-infiltrating macrophages under fibrotic conditions represents an important area for future investigation.
Notch-mediated mechanotransduction
Notch in cardiac macrophage-mediated heart disease
It is well known that the activation of Notch signaling depends on mechanical stimuli[130]. In mammals, four single-pass transmembrane Notch receptors (NOTCH1-4) interact with five canonical ligands expressed on adjacent cells: Jagged1 and Jagged2, and Delta-like ligand 1 (DLL1), Delta-like ligand 3 (DLL3), Delta-like ligand 4 (DLL4)[131]. Upon ligand binding, mechanical forces induce conformational changes in the Notch receptor that permit proteolytic cleavage first by a disintegrin and metalloproteinase (ADAM)-family metalloproteases at the S2 site and then by the γ-secretase complex at the S3 site[131]. This process releases the Notch intracellular domain (NICD), which translocates into the nucleus and binds to CBF1/suppressor of hairless/LAG-1 (CSL/RBPJ) to drive target gene transcription[132]. In the cardiovascular system, particularly in endothelial cells, Notch signaling has been shown to respond to various biomechanical cues, including shear stress[133] and cyclic stretch[134].
Functionally, in the cardiac context, studies have demonstrated that inhibition of Notch signaling in a mouse model of MI suppresses M2 macrophage polarization[135], a phenotype typically associated with fibrotic, stiffened ECM. This suggests that Notch activation may mimic cellular responses to increased ECM stiffness. Beyond macrophage polarization, Notch signaling also mediates intercellular communication. One study reported that during cardiac transplant rejection, endothelial DLL4 interacts with Notch receptors on macrophages, inducing IL-6 production and promoting antibody-mediated rejection[136]. Collectively, these findings highlight the dual roles of Notch in sensing mechanical and inflammatory signals through both cell-ECM and cell-cell interactions and underscore its therapeutic potential as a mechanosensitive target in cardiac fibrosis and inflammation.
Mechanism of notch-mediated macrophage mechanotransduction
In myeloid lineages, Notch2 is the predominant receptor and plays a crucial role in regulating monocyte fate and tissue-resident macrophage composition[137]. In a mouse model of lung injury and fibrosis, antibody-mediated blockade of Notch2 attenuated fibrotic progression by modulating the population of MoMFs characterized by high expression of fibrotic markers such as Spp1, cluster of differentiation 9 (Cd9), glycoprotein non-metastatic melanoma protein B (Gpnmb), and fatty acid binding protein 5 (Fabp5)[138]. These findings suggest that Notch signaling may serve as an upstream regulator of profibrotic pathways in infiltrating macrophages. Although Notch can respond to fibrotic environmental cues surrounding macrophages, it remains unclear whether it directly senses ECM stiffness. Nevertheless, current evidence in vitro indicates that Notch interacts with classical mechanosensors in macrophages, such as Piezo1[122] and YAP[139], supporting its role in mechanotransduction.
Limitations and emerging experimental models for cardiac macrophage mechanotransduction
Current experimental models investigating macrophage mechanosensing in fibrosis predominantly rely on stiffness-tunable hydrogels, such as polyacrylamide gel[24], alginate[140], or gelatin-based matrices[141], to mimic ECM mechanics. While these reductionist systems have provided important mechanistic insights, several limitations become apparent when they are applied to the cardiac context. First, most mechanistic insights into macrophage responses to matrix stiffness are derived from tumor models or non-cardiac fibrotic tissues, whereas studies focusing specifically on cardiac macrophages remain relatively limited. Given that different organs exhibit distinct stiffness profiles[142], macrophage mechanotransduction is likely shaped by tissue-specific microenvironments.
Indeed, hydrogel-based studies have shown that macrophages exposed to stiffened substrates resembling the aging cardiac matrix adopt pro-inflammatory phenotypes, underscoring the relevance of mechanical cues in the aging heart[74]. In addition to matrix stiffness, recent studies have shown that cardiac macrophages can directly sense mechanical stretch through stretch-activated ion channels, such as Piezo1, highlighting stretch as another physiologically relevant mechanical cue in the myocardium[143]. Nevertheless, current studies largely examine individual mechanical parameters separately. Although composite mechanical models integrating multiple cues, such as cyclic stretch and matrix stiffness, have been developed to study cardiomyocyte-fibroblast interactions[144], their application to cardiac macrophages remains limited. This highlights the need for more physiologically relevant experimental models. Such models are required to better recapitulate the complex mechanical landscape of the cardiac microenvironment.
Looking ahead, AFM provides a powerful approach to quantify the mechanical properties of the cardiac microenvironment under distinct physiological and pathological conditions[145]. AFM-based measurements have enabled high-resolution mapping of myocardial stiffness across different disease contexts and anatomical regions, revealing pronounced mechanical heterogeneity within the fibrotic heart[146]. For example, AFM analyses in MI models have demonstrated region-specific increases in tissue stiffness spanning several hundred kilopascals[147], underscoring the spatial complexity of myocardial mechanics under pathological conditions. Such AFM-derived stiffness profiles may serve as an important reference for calibrating in vitro mechanical models, thereby improving their physiological relevance.
In parallel, organoid-based platforms represent a promising in vitro strategy to recapitulate the complex cellular and mechanical features of the cardiac microenvironment. Emerging studies in non-cardiac organoid systems, including liver and intestinal organoids, have demonstrated that hydrogel stiffness and viscoelasticity function as instructive biomechanical cues regulating tissue organization, cell fate decisions, and inflammatory responses[148,149]. By contrast, despite rapid advances in cardiac organoid technologies, systematic incorporation of tunable mechanical properties into human cardiac organoids remains limited. Notably, recent human heart-macrophage assembloid models have begun to recapitulate immune-cardiac interactions in a three-dimensional context, and single-cell transcriptomic analyses have revealed that macrophages within these assembloids exhibit profibrotic transcriptional signatures, including high expression of SPP1, suggesting their potential role in ECM remodeling[150]. Together, the integration of AFM-guided mechanical calibration with advanced cardiac organoid and assembloid systems may offer a powerful framework to investigate macrophage mechanotransduction in a cardiac-specific and physiologically relevant manner.
POTENTIAL THERAPEUTIC STRATEGIES TARGETING MACROPHAGES MECHANOBIOLOGY
Targeting mechanosensors in macrophages
Two main therapeutic strategies have been proposed to modulate cell mechanotransduction: one targets intracellular mechanosensitive molecules, and the other focuses on remodeling the ECM [Table 1].
Current therapeutic strategies targeting cellular mechanotransduction
| Target type | Target | Drug name | Mechanism of action | Disease context | Clinical stage | Reference |
| Intracellular mechanosensor | Integrin α4 | Natalizumab (monoclonal antibody) | Inhibiting integrin α4 mediated immune cell adhesion | Multiple sclerosis, AIDS-induced cardiac fibrosis (animal model) | Approved (multiple sclerosis); preclinical in CVD | [151,152] |
| IntegrinαIIbβ3 | Abciximab (monoclonal antibody) | Inhibiting platelet aggregation via blocking αIIbβ3 | Coronary artery disease | Approved | [153] | |
| Integrin αL | Efalizumab (monoclonal antibody) | Inhibiting αL-mediated T cell adhesion and activation | Plaque psoriasis | Approved (2003), withdrawn (2009) | [154,155] | |
| Integrinα4β7 | Vedolizumab (monoclonal antibody) | Blocking α4β7-mediated lymphocyte trafficking | Inflammatory bowel | Approved | [156] | |
| Integrin αLβ2 | Lifitegrast (small molecule) | Blocking LFA-1/ICAM-1 interaction via αLβ2 | Dry eye disease | Approved | [157] | |
| Integrin α4 | Carotegrast (AJM300, small molecule) | Orally inhibiting α4-integrin to block leukocyte trafficking | Ulcerative colitis | Approved | [158] | |
| Integrin αvβ6 | BG00011 (monoclonal antibody) | Targeting the non-internalized, bent-closed αvβ6 conformation | Idiopathic pulmonary fibrosis | Phase II | [159,160] | |
| YAP | Verteporfin (photosensitizer) | Inducing sequestration of YAP in cytoplasm through increasing levels of 14-3-3σ | Macular degeneration, atherosclerosis (animal model) | Approved (macular degeneration), preclinical in atherosclerosis | [161,162] | |
| FAK | Defactinib (small molecule) | FAK inhibitor, inhibiting non-angiogenic vascularization via reduced cell migration | Cancer | Phase II | NCT02758587, NCT02943317[163] | |
| ROCK2 | KD025 (small molecule) | Selective ROCK2 inhibitor that modulates cytoskeletal tension by reducing myosin light chain phosphorylation | Lung fibrosis | Phase II | NCT02688647 | |
| Notch | CB-103 (small molecule) | Pan-notch pathway inhibitor selectively blocks the CSL-NICD interaction | Cancer | Phase I/II | NCT03422679[164] | |
| Piezo1 | GsMTx4 (small molecule) | Inhibitor that can turn off Piezo1 by modulating the local membrane tension | Heart failure (animal models) | Preclinical | [165] | |
| Extracellular matrix component | LOX | PXS-5505 (small molecule) | Pan-LOX inhibitor, reduces ECM stiffness | Thrombocythemia myelofibrosis | Phase I/II | NCT04676529 |
| LOXL2 | Simtuzumab (monoclonal antibody) | Monoclonal antibody against LOXL2, inhibits collagen cross-linking | Liver fibrosis | Phase II | NCT01707472, NCT01672853 | |
| LOXL2 | SNT-5382 (small molecule) | LOXL2 inhibitor, inhibits collagen cross-linking | myocardial infarction (animal model) | Phase I | [166] | |
| LOX | BAPN (small molecule) | Irreversible inhibitor of LOX | Cardiac fibrosis (animal model) | Preclinical | [167] | |
| P4H | EDHB (small molecule) | Inhibiting hyaluronan synthase | Breast cancer | Preclinical | [168] | |
| MMP9 | Andecaliximab (monoclonal antibody) | Specifically blocks matrix metalloproteinase-9 (MMP9) | COPD Gastric cancer | Phase III/Phase I | NCT02077465, NCT02545504 |
Most current approaches are based on cancer studies, where immune mechanosensing is modified to enhance immune cell function. For example, Mout et al.[130] designed multivalent soluble agonists targeting Notch, a well-established mechanosensor involved in T cell development and activation[130,131,169-171], to deliver mechanical force and promote Notch signaling in scalable T cell differentiation platforms. Another team developed an antibody targeting discoidin domain receptor 1 (DDR1), a mechanosensor that we[172,173] and others[174] have found to sense ECM stiffness independently of its classical ligand in the vascular wall. In breast cancer models, the DDR1 antibody blocked the interaction between DDR1 and its canonical ligand, collagen, thereby altering collagen fiber alignment and enhancing immune cell infiltration[175]. Another strategy involves targeting the intracellular cytoskeleton, a core component of mechanotransduction. For example, inhibiting ras homolog family member A (RhoA), a small Rho GTPase that regulates cytoskeleton dynamics[176], has been shown to improve the regenerative capacity of aged hematopoietic stem cells and to correct lympho/myeloid skewing in vivo[177].
In the context of fibrosis, targeting macrophage-specific mechanotransduction is an emerging direction. Chen et al.[178] reported that inhibition of FAK-mediated signaling in macrophages attenuated scar formation in multiple fibrotic models. Although cardiac fibrosis represents the terminal stage of many heart diseases, limited studies have directly explored macrophage-specific mechanical signaling in this process. Cho et al.[31] found that targeting Src, an upstream mechanosensitive kinase regulating YAP/TAZ signaling in cardiac fibroblasts, reduced cardiac fibrosis. Given the pivotal macrophage-fibroblast crosstalk in fibrosis progression, this study highlights the feasibility of modulating macrophage mechanotransduction as a therapeutic strategy. Furthermore, since macrophages are essential players in inflammation and immune activation in infectious diseases, strategies targeting macrophage-ECM interactions have shown promise in mitigating infection-related cardiac fibrosis, such as human immunodeficiency virus (HIV)-associated cardiovascular events[179,180]. For instance, the anti-α4 integrin antibody natalizumab was found to reduce macrophage infiltration and fibrosis in the left ventricle of simian immunodeficiency virus macaque 251 strain (SIVmac251)-infected rhesus macaques, a model of HIV-induced heart disease[151].
Among the mechanosensors, integrins are the most widely targeted, likely due to their accessibility on the cell surface and responsiveness to molecular blockade[181]. The first integrin-targeting drug approved for clinical use was Abciximab, an αIIbβ3 antagonist approved in 1994 for coronary artery disease, highlighting the cardiovascular relevance of integrin inhibition[153]. Efalizumab, an αL integrin inhibitor approved in 2003 for treating plaque psoriasis, was withdrawn in 2009 because of serious adverse effects[154,155]. In 2004, natalizumab, a broad-spectrum α4 integrin inhibitor, was approved for multiple sclerosis[152]. Since then, several subtype-specific integrin inhibitors have reached the clinic: vedolizumab (α4β7) for inflammatory bowel disease (approved in 2014) and lifitegrast (αLβ2) for dry eye disease (approved in 2016)[156,157]. Carotegrast (AJM300), the first oral integrin inhibitor, was approved in 2022 for the treatment of active ulcerative colitis[158]. Together, these advances reflect renewed momentum in integrin-targeted therapy development.
In addition to integrins, other mechanosensitive pathways have been explored. Verteporfin, an inhibitor of YAP signaling, is an approved small-molecule photosensitizer for neovascular macular degeneration[182]. Several other agents are under clinical investigation. For example, Defactinib (VS-6063), a FAK inhibitor from Verastem Oncology, is in phase II trials for pancreatic cancer due to its anti-fibrotic properties (NCT02758587). Rho-associated coiled-coil containing protein kinase (ROCK) is a key mechanotransducer that controls cytoskeletal tension via myosin activation[183]. Among its isoforms, ROCK2 has attracted particular attention; with the selective inhibitor, KD025 has completed phase II clinical trials for lung fibrosis (NCT02688647). Notch, functioning both as a mechanosensitive receptor and a transcriptional regulator, represents a particularly promising therapeutic target. The pan-Notch pathway inhibitor CB-103 is currently under clinical evaluation for the treatment of solid tumors[164].
Despite these developments, no drugs have yet entered clinical trials specifically targeting macrophage mechanotransduction in cardiovascular disease. Although the peptide inhibitor of Piezo1, GsMTx4, has demonstrated cardioprotective effects in preclinical HF models[165], its clinical utility remains to be validated. Importantly, recent studies suggest potential in this direction: Cho et al.[31] reported that suppressing stromal mechanosensing reduced cardiac fibrosis, while Chen et al.[178] demonstrated that targeting circulating mechanoresponsive monocytes and macrophages alleviated fibrosis in a humanized scarring model. Together, these findings support further exploration of macrophage-targeted mechanotherapies in cardiac fibrosis.
Modifying the ECM to regulate macrophage function
In addition to cellular targets, normalizing the ECM has emerged as a promising therapeutic strategy, particularly in the context of tumor immunotherapy. For instance, studies have shown that chimeric antigen receptor T (CAR-T) cells expanded on softened Matrigel exhibited enhanced stemness and increased cytotoxicity against solid tumors compared with cells cultured on conventional plastic surfaces[184]. This supports the notion that ECM softening can enhance immune cell function and suggests a broader role for matrix modulation in regulating macrophage behavior.
Among the contributors to ECM stiffening, members of the lysyl oxidase (LOX) family, including LOX, LOXL1, and LOXL2, are key drivers of collagen crosslinking during fibrosis[185-187]. β-Aminopropionitrile (BAPN), a well-known pan-LOX inhibitor, disrupts collagen crosslinking and thus softens the ECM. In animal studies, BAPN has been shown to reduce skin fibrosis and improve scar appearance[188]. It also appears to modulate macrophage polarization, shifting the balance between M1 and M2 phenotypes[189]. In cardiovascular studies, BAPN reduced myocardial fibrosis and altered macrophage populations in models of age-related cardiac fibrosis, suggesting potential utility in cardiac fibrosis therapy[190]. However, BAPN has not progressed to current clinical studies. Only two early studies, conducted before the 1980s, evaluated its ability to inhibit collagen crosslinking in patients with scleroderma and urethral strictures[191,192]. The lack of clinical development is primarily due to its in vivo toxicity[193]. In animal models, BAPN induces lathyrism—characterized by extensive connective tissue damage and development of abdominal aortic aneurysm (AAA)[194,195]. Another pan-LOX inhibitor, PXS-5505, has shown promising effects in treating myelofibrosis and thrombocythemia, which are associated with elevated cardiovascular risk due to chronic inflammation, abnormal platelet activity, and a prothrombotic state[196].
Among LOX family members, LOXL2 has been the most frequently targeted in cardiovascular research. Elevated levels of circulating LOXL2 have been detected in patients with HF[197]. A small-molecule LOXL2 inhibitor, SNT-5382, has been shown to reduce fibrosis and improve cardiac function in a mouse model of MI, with phase I trials demonstrating good safety[166]. Another LOXL2 inhibitor, the monoclonal antibody Simtuzumab, has been tested in clinical trials for fibrotic diseases such as liver and lung fibrosis (e.g., NCT01707472, NCT01672853), but its efficacy in cardiac fibrosis remains unclear.
Beyond the LOX family, other ECM-modulating strategies are being explored. For example, targeting hyaluronan synthesis using the natural inhibitor ethyl-3,4-dihydroxybenzoate has shown efficacy in preclinical models of breast cancer[168]. MMP-9, a MMP that degrades ECM components such as collagen and gelatin, has also been investigated. Monoclonal antibodies targeting MMP-9 have entered clinical trials for fibrotic diseases, chronic obstructive pulmonary disease (COPD), and solid tumors (e.g., NCT02077465, NCT02545504). Although numerous studies have shown that plasma MMP-9 levels correlate with the extent of myocardial remodeling and the progression of HF[198], therapeutic strategies targeting MMP-9 remain largely restricted to animal models rather than clinical studies in cardiac disease. The difficulty in clinically translating MMP-9-targeted therapies may be partially attributed to the dual, stage-dependent roles of MMP-9 in cardiac disease. In early stages of tissue injury, MMP-9 contributes to ECM degradation, immune cell infiltration, and clearance of damaged matrix, facilitating tissue repair. Specifically, elevated MMP-9 expression in macrophages has been shown to improve ventricular function following MI by modulating inflammatory and fibrotic responses during the early stages of tissue repair[199]. In this context, MMP-9 facilitates ECM degradation, promotes immune cell infiltration, and clearance of damaged tissue, collectively supporting tissue recovery. However, sustained or excessive MMP-9 activity may exert detrimental effects by promoting the release and activation of profibrotic cytokines, particularly TGF-β[200]. Elevated TGF-β levels enhance crosstalk between macrophages and fibroblasts, stimulating fibroblast activation and excessive collagen production[201]. This profibrotic cascade contributes to maladaptive ECM remodeling, progressive fibrosis, and ultimately HF. These contrasting roles highlight the stage-dependent duality of MMPs in cardiac disease. While transient activation may be beneficial during early repair phases, chronic overexpression can drive pathological remodeling, suggesting that selective or temporally controlled modulation, rather than broad-spectrum inhibition, may offer a more effective and safer approach for treating fibrosis-related cardiac conditions.
In summary, although many ECM-targeting approaches were initially developed for cancer and fibrotic disorders, the shared mechanisms of ECM remodeling across organ systems suggest strong translational potential for treating cardiac fibrosis and HF. Continued preclinical validation and the development of targeted delivery strategies will be essential to overcome systemic toxicity and to optimize therapeutic efficacy in cardiovascular settings.
CONCLUSION AND FUTURE PERSPECTIVES
Collectively, recent advances underscore the pivotal role of macrophage mechanotransduction in modulating immune responses and fibrosis under conditions of mechanical stress. Mechanosensitive pathways such as those mediated by integrins, YAP/TAZ, and Piezo1 have been shown to influence macrophage polarization, cytokine production, and crosstalk with cardiac fibroblasts, especially under stiffened fibrotic ECM. These findings highlight promising therapeutic avenues involving the targeting of cellular mechanotransduction.
Despite significant advances in the field, our current understanding of macrophage mechanotransduction largely stems from in vitro models or studies conducted in non-cardiac contexts, such as tumors or fibrosis in other organs. The specific roles and mechanisms of macrophage mechanosensing within the pathological cardiac microenvironment remain poorly characterized. In addition, although cardiac macrophages comprise multiple subsets as revealed by single-cell RNA sequencing, direct comparisons of matrix stiffness sensing between monocyte-derived and resident macrophages remain limited. Recent studies have demonstrated that mechanical stretch activates distinct ion channels in macrophages, with differential involvement of Piezo1 and TRPV4 depending on macrophage origin[143]. Whether analogous subset-specific mechanisms govern macrophage sensing of ECM stiffness has yet to be systematically investigated. Moreover, translational progress has been limited, as most mechanobiology-targeted therapies are explored predominantly in oncology. Decades of research have established mechanotransduction and ECM stiffness as critical regulators of immune responses. Building on this foundation, the emerging field of mechanomedicine offers new opportunities for the treatment of cardiovascular disease. Furthermore, the most promising therapeutic strategies will likely involve combination approaches that target multiple mechanosensitive pathways in parallel to overcome cellular redundancy and heterogeneity.
Looking ahead, four key aspects should be considered in the development of novel strategies targeting macrophage mechanotransduction. First, emerging technologies-such as single-cell transcriptomics and spatial omics-should be leveraged to identify novel mechanosensors or mechanosensitive macrophage subsets, particularly given the heterogeneity of macrophage populations within the cardiac microenvironment. Second, mechanical heterogeneity across different cardiac regions and disease stages must be carefully evaluated, as the mechanical microenvironment evolves dynamically during disease progression. Third, the selection of appropriate animal models that closely recapitulate human cardiac pathophysiology is essential to bridge the frequent translational gap between preclinical efficacy and clinical outcomes. Finally, translating mechanotherapeutic insights into safe and effective clinical applications will require precision-targeted delivery systems to minimize off-target effects and systemic toxicity. Promising approaches include nanoparticle-based carriers[202], antibody-drug conjugates[203], and hydrogel-mediated local delivery[204], which may enhance therapeutic specificity and reduce adverse effects.
DECLARATIONS
Acknowledgments
Figures 1-4 were created in BioRender. Wang, J. (2026) https://BioRender.com/a877sp0.
Authors’ contributions
Conceptualized the review theme, conducted the literature search, and drafted the manuscript: Wang J
Performed literature retrieval and contributed to the preparation of figures and tables: Xu W
Provided clinical insights and perspectives, identified unmet clinical needs in the field, and provided professional guidance on clinical translation: Guan H
Supervised the project, provided funding support, supervised the study, and critically revised the manuscript for important intellectual content: Wang X
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (OpenAI, version based on GPT-5.3) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by the grants from the National Natural Science Foundation of China (32270635, 32300658), Young Elite Scientist Sponsorship Program by Beijing Association For Science and Technology, Excellent Youth Training Programme of Beijing Ditan Hospital (ZZ202401), and the Special Program for Clinical Research and Scientific Innovation Transformation of Beijing Ditan Hospital, Xuzhou Hospital, Capital Medical University (KCZL202505).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026
REFERENCES
1. González A, López B, Ravassa S, et al. Myocardial interstitial fibrosis in hypertensive heart disease: from mechanisms to clinical management. Hypertension. 2024;81:218-28.
2. Zhang Q, He Y, Li Y, et al. Matricellular protein Cilp1 promotes myocardial fibrosis in response to myocardial infarction. Circ Res. 2021;129:1021-35.
3. Eijgenraam TR, Silljé HH, De Boer RA. Current understanding of fibrosis in genetic cardiomyopathies. Trends Cardiovasc Med. 2020;30:353-61.
4. Liu K, Zheng L, Huang Q, et al. Natural product library screening identifies Darutigenol for the treatment of myocardial infarction and ischemia/reperfusion injury. Chin Med. 2025;20:90.
5. Li L, Zhao Q, Kong W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018;68-9:490-506.
6. Sutherland TE, Dyer DP, Allen JE. The extracellular matrix and the immune system: a mutually dependent relationship. Science. 2023;379:eabp8964.
7. Pathak A, Kumar S. Independent regulation of tumor cell migration by matrix stiffness and confinement. Proc Natl Acad Sci USA. 2012;109:10334-9.
8. Wang T, Abolghasemzade S, Mckee BP, et al. Matrix stiffness drives drop like nuclear deformation and lamin A/C tension-dependent YAP nuclear localization. Nat Commun. 2024;15:10151.
9. Holt M, Lin J, Cicka M, Wong A, Epelman S, Lavine KJ. Dissecting and visualizing the functional diversity of cardiac macrophages. Circ Res. 2024;134:1791-807.
10. Wang J, Wang J, Zhang Y, et al. Identifying vascular stiffening-sensitive macrophages through integration of single-cell transcriptomics and imaging flow cytometry. Biophys Rep. 2025;11:393.
11. Khalsa P, Ge W, Zia Uddin M, Hadjiargyrou M. Integrin α2β1 affects mechano-transduction in slowly and rapidly adapting cutaneous mechanoreceptors in rat hairy skin. Neuroscience. 2004;129:447-59.
12. Ingber DE. Mechanosensation through integrins: cells act locally but think globally. Proc Natl Acad Sci USA. 2003;100:1472-4.
13. Liu J, Wang X, He F, Chen X, Yi X. Mechanosensitive calcium ion channels Piezo1: a therapeutic target in liver disease. Mol Ther Nucl Acids. 2025;36:102695.
14. Zhang X, Hou L, Li F, et al. Piezo1-mediated mechanosensation in bone marrow macrophages promotes vascular niche regeneration after irradiation injury. Theranostics. 2022;12:1621-38.
15. Meli VS, Veerasubramanian PK, Downing TL, Wang W, Liu WF. Mechanosensation to inflammation: roles for YAP/TAZ in innate immune cells. Sci Signal. 2023;16:eadc9656.
16. Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol. 2017;18:758-70.
17. Souilhol C, Tardajos Ayllon B, Li X, et al. JAG1-NOTCH4 mechanosensing drives atherosclerosis. Sci Adv. 2022;8:eabo7958.
18. Foldi J, Chung AY, Xu H, et al. Autoamplification of notch signaling in macrophages by TLR-induced and RBP-J-dependent induction of Jagged1. J Immunol. 2010;185:5023-31.
19. Ghazal R, Wang M, Liu D, Tschumperlin DJ, Pereira NL. Cardiac fibrosis in the multi-omics era: implications for heart failure. Circ Res. 2025;136:773-802.
20. Frangogiannis NG. The extracellular matrix in ischemic and nonischemic heart failure. Circ Res. 2019;125:117-46.
21. Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial J, Entman ML. Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. J Mol Cell Cardiol. 2011;50:248-56.
22. Esfahani NS, Wu Q, Kumar N, Ganesan LP, Lafuse WP, Rajaram MVS. Aging influences the cardiac macrophage phenotype and function during steady state and during inflammation. Aging Cell. 2021;20:e13438.
23. Yu K, Yuan W, Wang H, Li Y. Extracellular matrix stiffness and tumor-associated macrophage polarization: new fields affecting immune exclusion. Cancer Immunol Immunother. 2024;73:115.
24. Meli VS, Rowley AT, Veerasubramanian PK, Heedy SE, Liu WF, Wang S. Modulation of stiffness-dependent macrophage inflammatory responses by collagen deposition. ACS Biomater Sci Eng. 2024;10:2212-23.
25. Wang Y, Shi R, Zhai R, et al. Matrix stiffness regulates macrophage polarization in atherosclerosis. Pharmacol Res. 2022;179:106236.
26. Liu H, Fan P, Jin F, Huang G, Guo X, Xu F. Dynamic and static biomechanical traits of cardiac fibrosis. Front Bioeng Biotechnol. 2022;10:1042030.
27. Curtis MW, Russell B. Micromechanical regulation in cardiac myocytes and fibroblasts: implications for tissue remodeling. Pflugers Arch Eur J Physiol. 2011;462:105-17.
28. Happe CL, Engler AJ. Mechanical forces reshape differentiation cues that guide cardiomyogenesis. Circ Res. 2016;118:296-310.
29. Balachandran K, Alford PW, Wylie-Sears J, et al. Cyclic strain induces dual-mode endothelial-mesenchymal transformation of the cardiac valve. Proc Natl Acad Sci USA. 2011;108:19943-8.
30. Samady H, Eshtehardi P, Mcdaniel MC, et al. Coronary artery wall shear stress is associated with progression and transformation of atherosclerotic plaque and arterial remodeling in patients with coronary artery disease. Circulation. 2011;124:779-88.
31. Cho S, Rhee S, Madl CM, et al. Selective inhibition of stromal mechanosensing suppresses cardiac fibrosis. Nature. 2025;642:766-75.
32. Omar AMS, Bansal M, Sengupta PP. Advances in echocardiographic imaging in heart failure with reduced and preserved ejection fraction. Circ Res. 2016;119:357-74.
33. Petrescu A, Cvijic M, Wouters L, et al. Ultrasound shear wave elastography for detection of myocardial fibrosis. Eur Heart J. 2025;26:1537-45.
34. Villemain O, Correia M, Mousseaux E, et al. Myocardial stiffness evaluation using noninvasive shear wave imaging in healthy and hypertrophic cardiomyopathic adults. JACC Cardiovasc Imaging. 2019;12:1135-45.
35. Miao L, Lu Y, Nusrat A, et al. β1 integrins regulate cellular behaviour and cardiomyocyte organization during ventricular wall formation. Cardiovasc Res. 2024;120:1279-94.
36. Prakash A, Smith PO, Iskratsch T. Role of the extracellular matrix in cardiac ageing. Nat Mater. 2025;24:1337-9.
37. Cleutjens JP, Creemers EE. Integration of concepts: cardiac extracellular matrix remodeling after myocardial infarction. J Card Fail. 2002;8:S344-8.
38. Weil BR, Neelamegham S. Selectins and immune cells in acute myocardial infarction and post-infarction ventricular remodeling: pathophysiology and novel treatments. Front Immunol. 2019;10:300.
39. Ibrahim AM, Elfawy HA, Terracciano CM, Yacoub M. Matrisome remodeling in the myocardium of hypertrophic cardiomyopathy; novel targets for molecular diagnostics. Front Cell Dev Biol. 2025;13:1641584.
40. Buck KM, Rogers HT, Gregorich ZR, et al. Extracellular matrix alterations in chronic ischemic cardiomyopathy revealed by quantitative proteomics. JCI Insight. 2025;10:e196933.
41. Wittig C, Szulcek R. Extracellular matrix protein ratios in the human heart and vessels: how to distinguish pathological from physiological changes? Front Physiol. 2021;12:708656.
42. Wei S, Chow LT, Shum IO, Qin L, Sanderson JE. Left and right ventricular collagen type I/III ratios and remodeling post-myocardial infarction. J Card Fail. 1999;5:117-26.
43. Sridhar KC, Mehl J, Klingel K, et al. Loss of fibronectin fiber tension is inherent to ECM remodeling in human myocarditis and post-inflammatory fibrosis. Matrix Biology Plus. 2025;28:100182.
44. Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol Biol Cell. 2002;13:3546-59.
45. Liu P, Sun M, Sader S. Matrix metalloproteinases in cardiovascular disease. Can J Cardiol. 2006;22:25B-30B.
46. Klein T, Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids. 2010;41:271-90.
47. Garvin P, Nilsson L, Carstensen J, Jonasson L, Kristenson M. Circulating matrix metalloproteinase-9 is associated with cardiovascular risk factors in a middle-aged normal population. PLoS ONE. 2008;3:e1774.
48. Toba H, Cannon PL, Yabluchanskiy A, Iyer RP, D’armiento J, Lindsey ML. Transgenic overexpression of macrophage matrix metalloproteinase-9 exacerbates age-related cardiac hypertrophy, vessel rarefaction, inflammation, and fibrosis. Am J Physiol Heart Circ Physiol. 2017;312:H375-83.
49. Voorhees AP, Deleon-pennell KY, Ma Y, et al. Building a better infarct: modulation of collagen cross-linking to increase infarct stiffness and reduce left ventricular dilation post-myocardial infarction. J Mol Cell Cardiol. 2015;85:229-39.
50. Kitano T, Sasaki T, Matsui T, et al. Transcriptome analysis identified SPP1‐positive monocytes as a key in extracellular matrix formation in thrombi. J Am Heart Assoc. 2025;14:e044299.
51. Du X, Liu T, Shen C, et al. RETRACTED ARTICLE: anti-fibrotic mechanism of SPP1 knockdown in atrial fibrosis associates with inhibited mitochondrial DNA damage and TGF-β/SREBP2/PCSK9 signaling. Cell Death Discov. 2022;8:246.
52. Hoeft K, Schaefer GJ, Kim H, et al. Platelet-instructed SPP1+ macrophages drive myofibroblast activation in fibrosis in a CXCL4-dependent manner. Cell Rep. 2023;42:112131.
53. Lee J, Wang M, Sudhir P, Chen G, Chi C, Chen J. Osteopontin promotes integrin activation through outside-in and inside-out mechanisms: OPN-CD44V interaction enhances survival in gastrointestinal cancer cells. Cancer Res. 2007;67:2089-97.
54. Weber GF, Ashkar S, Glimcher MJ, Cantor H. Receptor-ligand interaction between CD44 and osteopontin (Eta-1). Science. 1996;271:509-12.
55. Zhang J, Song J, Tang S, et al. Multi-omics analysis reveals the chemoresistance mechanism of proliferating tissue-resident macrophages in PDAC via metabolic adaptation. Cell Rep. 2023;42:112620.
56. Ghasemi H, Mousavibahar SH, Hashemnia M, Karimi J, Khodadadi I, Tavilani H. Transitional cell carcinoma matrix stiffness regulates the osteopontin and YAP expression in recurrent patients. Mol Biol Rep. 2021;48:4253-62.
57. Lok ZSY, Lyle AN. Osteopontin in vascular disease: friend or Foe? Arterioscler Thromb Vasc Biol. 2019;39:613-22.
58. Speer MY, Mckee MD, Guldberg RE, et al. Inactivation of the osteopontin gene enhances vascular calcification of matrix gla protein-deficient mice. J Exp Med. 2002;196:1047-55.
59. Mabatha KC, Letuka P, Aremu O, Zulu MZ. Macrophages of the heart: homeostasis and disease. Biomed J. 2026;49:100867.
60. Chen R, Zhang H, Tang B, et al. Macrophages in cardiovascular diseases: molecular mechanisms and therapeutic targets. Sig Transduct Target Ther. 2024;9:130.
61. Yap J, Irei J, Lozano-gerona J, Vanapruks S, Bishop T, Boisvert WA. Macrophages in cardiac remodelling after myocardial infarction. Nat Rev Cardiol. 2023;20:373-85.
62. Querceto S, Santoro R, Gowran A, et al. The harder the climb the better the view: the impact of substrate stiffness on cardiomyocyte fate. J Mol Cell Cardiol. 2022;166:36-49.
63. Park I, Marquardt RR. Effects of niacin deficiency on pyridine nucleotide levels and enzyme activities in various organs of young growing quail. J Nut. 1982;112:863-73.
64. Xu Z, Yu J, Chen D, et al. Extracellular matrix stiffness regulates the proliferation and migration capacities of lymphatic endothelial cells via FAT1. Front Cell Dev Biol. 2025;13:1667154.
65. Li M, Wu J, Hu G, et al. Pathological matrix stiffness promotes cardiac fibroblast differentiation through the POU2F1 signaling pathway. Sci China Life Sci. 2020;64:242-54.
66. Felisbino MB, Rubino M, Travers JG, et al. Substrate stiffness modulates cardiac fibroblast activation, senescence and proinflammatory secretory phenotype. Am J Physiol Heart Circ Physiol. 2023;326:H61-73.
67. Yang S, Penna V, Lavine KJ. Functional diversity of cardiac macrophages in health and disease. Nat Rev Cardiol. 2025;22:431-42.
68. Jin J, Wang Y, Liu Y, Chakrabarti S, Su Z. Cardiac resident macrophages: spatiotemporal distribution, development, physiological functions, and their translational potential on cardiac diseases. Acta Pharm Sin B. 2024;14:1483-93.
69. Zhang Y, Wu Y, Li M, Mijiti A, Cheng L. Identification of macrophage driver genes in fibrosis caused by different heart diseases based on omics integration. J Transl Med. 2024;22:839.
70. Li S, Ge T, Xu X, et al. Integrating scRNA-seq to explore novel macrophage infiltration-associated biomarkers for diagnosis of heart failure. BMC Cardiovasc Disord. 2023;23:560.
71. Uhlig M, Billig S, Wienhold J, Schumacher D. Pro-fibrotic macrophage subtypes: SPP1+ Macrophages as a key player and therapeutic target in cardiac fibrosis? Cells. 2025;14:345.
72. Li R, Hanna A, Huang S, et al. Macrophages in the infarcted heart acquire a fibrogenic phenotype, expressing matricellular proteins, but do not undergo fibroblast conversion. J Mol Cell Cardiol. 2024;196:152-67.
73. Ni Y, Qi H, Zhang F, et al. Macrophages modulate stiffness-related foreign body responses through plasma membrane deformation. Proc Natl Acad Sci USA. 2023;120:e2213837120.
74. Haschak M, Lopresti S, Stahl E, Dash S, Popovich B, Brown BN. Macrophage phenotype and function are dependent upon the composition and biomechanics of the local cardiac tissue microenvironment. Aging. 2021;13:16938-56.
75. Wang Y, Chaffee TS, Larue RS, et al. Tissue-resident macrophages promote extracellular matrix homeostasis in the mammary gland stroma of nulliparous mice. eLife. 2020;9:e57438.
76. Simões FC, Cahill TJ, Kenyon A, et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat Commun. 2020;11:600.
77. Su Y, Yin X. The Molecular mechanism of macrophages in response to mechanical stress. Ann Biomed Eng. 2024;53:318-30.
78. Yin Z, Wen T, Cao X, et al. The transcription factor RBPJ is required for inflammatory macrophage activation in thoracic aortic dissection by mediating mechanotransduction-induced glycolysis. Cell Mol Life Sci. 2025;82:370.
79. Tao D, Wang H, Chang S, et al. Matrix viscoelasticity orchestrates osteogenesis via mechanotransduction mediated metabolic switch in macrophages. Adv Healthc Mater. 2025;14:2405097.
80. Zhang S, Chen R, Chakrabarti S, Su Z. Resident macrophages as potential therapeutic targets for cardiac ageing and injury. Clin Trans Imm. 2020;9:e1167.
81. Lin PK, Davis GE. Extracellular matrix remodeling in vascular disease: defining its regulators and pathological influence. Arterioscler Thromb Vasc Biol. 2023;43:1599-616.
82. Xu Y, Ying L, Lang JK, Hinz B, Zhao R. Modeling mechanical activation of macrophages during pulmonary fibrogenesis for targeted anti-fibrosis therapy. Sci Adv. 2024;10:eadj9559.
83. Wang Y, Wang J, Zhang J, et al. Stiffness sensing via Piezo1 enhances macrophage efferocytosis and promotes the resolution of liver fibrosis. Sci Adv. 2024;10:eadj3289.
85. Campbell ID, Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol. 2011;3:a004994.
87. Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10:21-33.
88. Park JE, Jebamercy G, Pazhanchamy K, et al. Aging-induced isoDGR-modified fibronectin activates monocytic and endothelial cells to promote atherosclerosis. Atherosclerosis. 2021;324:58-68.
89. Li R, Chen B, Kubota A, et al. Protective effects of macrophage-specific integrin α5 in myocardial infarction are associated with accentuated angiogenesis. Nat Commun. 2023;14:7555.
90. Chen C, Li R, Ross RS, Manso AM. Integrins and integrin-related proteins in cardiac fibrosis. J Mol Cell Cardiol. 2016;93:162-74.
91. Li R, Huang S, Hanna A, et al. Macrophage ITGAV is dispensable for post-infarction remodeling in mice and does not mediate fibronectin responses. Commun Biol. 2025;9:29.
92. Chen M, Zhang Y, Zhou P, et al. Substrate stiffness modulates bone marrow-derived macrophage polarization through NF-κB signaling pathway. Bioact Mater. 2020;5:880-90.
93. Xing X, Wang Y, Zhang X, et al. Matrix stiffness‐mediated effects on macrophages polarization and their LOXL2 expression. FEBS J. 2020;288:3465-77.
94. Shimojo N, Hashizume R, Kanayama K, et al. Tenascin-C May accelerate cardiac fibrosis by activating macrophages via the integrin αVβ3/nuclear factor-κB/interleukin-6 axis. Hypertension. 2015;66:757-66.
95. Kim H, Nam J. The multifaceted role of YAP in the tumor microenvironment and its therapeutic implications in cancer. Exp Mol Med. 2025;57:2201-13.
96. Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179-83.
97. Feng H, Hu W, Liu Y, et al. Macrophages in ventricular remodeling and heart failure: orchestrators of inflammation and repair. Front Immunol. 2025;16:1682294.
98. Li R, Huang W. Yes-associated protein and transcriptional coactivator with PDZ-binding motif in cardiovascular diseases. Int J Mol Sci. 2023;24:1666.
99. Mani A, Hwa J, Martin KA. Sugar, fat, and YAP: a recipe for vascular stiffness. Circ Res. 2022;130:868-70.
100. Wang K, Yeh Y, Nguyen P, et al. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci USA. 2016;113:11525-30.
101. Azad T, Ghahremani M, Yang X. The role of YAP and TAZ in angiogenesis and vascular mimicry. Cells. 2019;8:407.
102. Sanchez-lozano M, Chirikian O, Lane KV, Dow LP, Castillo EA, Pruitt B. Stretch-activated nuclear YAP localization in cardiomyocytes. Biophys J. 2023;122:264a.
103. Wang Y, Zhang J, Wu B, et al. IL-37 improves mice myocardial infarction via inhibiting YAP-NLRP3 signaling mediated macrophage programming. Eur J Pharmacol. 2022;934:175293.
104. Li B, He J, Lv H, et al. c-Abl regulates YAPY357 phosphorylation to activate endothelial atherogenic responses to disturbed flow. J Clin Investig. 2019;129:1167-79.
105. Sugihara T, Werneburg NW, Hernandez MC, et al. YAP tyrosine phosphorylation and nuclear localization in cholangiocarcinoma cells are regulated by LCK and independent of LATS activity. Mol Cancer Res. 2018;16:1556-67.
106. Zhao B, Li L, Tumaneng K, Wang C, Guan K. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCFβ-TRCP. Genes Dev. 2010;24:72-85.
107. Liu M, Yan M, Lv H, et al. Macrophage K63-linked ubiquitination of YAP Promotes its nuclear localization and exacerbates atherosclerosis. Cell Rep. 2020;32:107990.
108. Mei F, Guo Y, Wang Y, et al. Matrix stiffness regulates macrophage polarisation via the Piezo1‐YAP signalling axis. Cell Prolif. 2024;57:e13640.
109. Lee J, Park JH, Lee JH, Lee H, Knowles JC, Kim H. Matrix-enabled mechanobiological modulation of osteoimmunology. Matter. 2022;5:3194-224.
110. Mia MM, Cibi DM, Abdul Ghani SAB, et al. YAP/TAZ deficiency reprograms macrophage phenotype and improves infarct healing and cardiac function after myocardial infarction. PLoS Biol. 2020;18:e3000941.
111. Meli VS, Atcha H, Veerasubramanian PK, et al. YAP-mediated mechanotransduction tunes the macrophage inflammatory response. Sci Adv. 2020;6:eabb8471.
112. Zhao L, Guan H, Song C, et al. YAP1 is essential for osteoclastogenesis through a TEADs-dependent mechanism. Bone. 2018;110:177-86.
113. Xiao B. Mechanisms of mechanotransduction and physiological roles of PIEZO channels. Nat Rev Mol Cell Biol. 2024;25:886-903.
114. Pathak MM, Nourse JL, Tran T, et al. Stretch-activated ion channel Piezo1 directs lineage choice in human neural stem cells. Proc Natl Acad Sci USA. 2014;111:16148-53.
115. Lai A, Thurgood P, Cox CD, et al. Piezo1 response to shear stress is controlled by the components of the extracellular matrix. ACS Appl Mater Interfaces. 2022;14:40559-68.
116. Coste B, Delmas P. PIEZO ion channels in cardiovascular functions and diseases. Circ Res. 2024;134:572-91.
117. Chesler AT, Szczot M, Bharucha-Goebel D, et al. The role of PIEZO2 in human mechanosensation. N Engl J Med. 2016;375:1355-64.
118. Liu X, Niu W, Zhao S, Zhang W, Zhao Y, Li J. Piezo1: the potential new therapeutic target for fibrotic diseases. Prog Biophys Mol Biol. 2023;184:42-9.
119. Wen J, Guan Y, Niu H, Dang Y, Guan J. Targeting cardiac resident CCR2+ macrophage-secreted MCP-1 to attenuate inflammation after myocardial infarction. Acta Biomater. 2026;211:18-32.
120. Chen B, Frangogiannis NG. The role of macrophages in nonischemic heart failure. JACC: Basic Transl Sci. 2018;3:245-8.
121. Chen J, Zhu Z, Wang Y, Yu J, Zhang X, Xu Y. Cardiac resident macrophages in cardiovascular disease: from physiology to pathology. Heart. 2025;111:391-400.
122. He Y, Deng B, Liu S, et al. Myeloid Piezo1 deletion protects renal fibrosis by restraining macrophage infiltration and activation. Hypertension. 2022;79:918-31.
123. Atcha H, Kulkarni D, Meli VS, et al. Piezo1-mediated mechanotransduction enhances macrophage oxidized low-density lipoprotein uptake and atherogenesis. PNAS Nexus. 2024;3:pgae436.
124. Peng L, Xia Y, Zhao H, et al. Piezo1 upregulation in monocyte‐derived macrophages impairs post‐myocardial infarction cardiac repair via defective efferocytosis and enhanced ferroptosis. Adv Sci. 2025;13:e10991.
125. Zhang Y, Zhang Y, Song J, et al. Piezo1 knockdown activates PI3K/AKT and enhances SPP1 to drive M2 macrophage polarization and reduce cardiac inflammation. Sci Rep. 2026;16:4879.
126. Atcha H, Jairaman A, Holt JR, et al. Mechanically activated ion channel Piezo1 modulates macrophage polarization and stiffness sensing. Nat Commun. 2021;12:3256.
127. Budde I, Ing D, Schwab A, Denes Petho Z. Mechanosensitive ion channels are essential for the durotaxis of pancreatic stellate cells. Biophys J. 2022;121:314a.
128. Espina JA, Marchant CL, Barriga EH. Durotaxis: the mechanical control of directed cell migration. FEBS J. 2021;289:2736-54.
129. Yang Y, Xie K, Jiang H. Durotaxis index of 3T3 fibroblast cells scales with stiff-to-soft membrane tension polarity. Biophys J. 2020;119:1427-38.
130. Mout R, Jing R, Tanaka-yano M, et al. Design of soluble Notch agonists that drive T cell development and boost immunity. Cell. 2025;188:5980-94.e28.
131. Rodriguez F, Sanlidag S, Sahlgren C. Mechanical regulation of the Notch signaling pathway. Curr Opin Cell Biol. 2023;85:102244.
132. Friedrich T, Ferrante F, Pioger L, et al. Notch-dependent and -independent functions of transcription factor RBPJ. Nucleic Acids Res. 2022;50:7925-37.
133. Min E, Schwartz MA. Translocating transcription factors in fluid shear stress-mediated vascular remodeling and disease. Exp Cell Res. 2019;376:92-7.
134. Karakaya C, Van Asten JGM, Ristori T, Sahlgren CM, Loerakker S. Mechano-regulated cell-cell signaling in the context of cardiovascular tissue engineering. Biomech Model Mechanobiol. 2021;21:5-54.
135. Li Z, Nie M, Yu L, et al. Blockade of the notch signaling pathway promotes M2 macrophage polarization to suppress cardiac fibrosis remodeling in mice with myocardial infarction. Front Cardiovasc Med. 2022;8:639476.
136. Pabois A, Pagie S, Gérard N, et al. Notch signaling mediates crosstalk between endothelial cells and macrophages via Dll4 and IL6 in cardiac microvascular inflammation. Biochem Pharmacol. 2016;104:95-107.
137. Bugeon L, Taylor HB, Progatzky F, et al. The NOTCH pathway contributes to cell fate decision in myelopoiesis. Haematologica. 2011;96:1753-60.
138. Cruz Tleugabulova M, Melo SP, Wong A, et al. Induction of a distinct macrophage population and protection from lung injury and fibrosis by Notch2 blockade. Nat Commun. 2024;15:9575.
139. Yang Y, Ni M, Zong R, et al. Targeting Notch1-YAP circuit reprograms macrophage polarization and alleviates acute liver injury in mice. Cell Mol Gastroenterol Hepatol. 2023;15:1085-104.
140. Alonzo M, Kumar SA, Allen S, et al. Hydrogel scaffolds with elasticity-mimicking embryonic substrates promote cardiac cellular network formation. Prog Biomater. 2020;9:125-37.
141. Cao Y, Lee BH, Peled HB, Venkatraman SS. Synthesis of stiffness‐tunable and cell‐responsive Gelatin-poly(ethylene glycol) hydrogel for three‐dimensional cell encapsulation. J Biomed Mater Res. 2016;104:2401-11.
142. Martinez-vidal L, Murdica V, Venegoni C, et al. Causal contributors to tissue stiffness and clinical relevance in urology. Commun Biol. 2021;4:1011.
143. Simon‐chica A, Klesen A, Emig R, et al. Piezo1 stretch‐activated channel activity differs between murine bone marrow‐derived and cardiac tissue‐resident macrophages. J Physiol. 2024;602:4437-56.
144. Herum KM, Choppe J, Kumar A, Engler AJ, Mcculloch AD. Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol Biol Cell. 2017;28:1871-82.
145. Hiesinger W, Brukman MJ, Mccormick RC, et al. Myocardial tissue elastic properties determined by atomic force microscopy after stromal cell-derived factor 1α angiogenic therapy for acute myocardial infarction in a murine model. J Thorac Cardiov Sur. 2012;143:962-6.
146. Wang X, Senyo S. Microenvironment stiffness requires extracellular matrix proteins to modulate heart regeneration. FASEB J. 2021;35:03575.
147. Peña B, Adbel-hafiz M, Cavasin M, Mestroni L, Sbaizero O. Atomic force microscopy (AFM) applications in arrhythmogenic cardiomyopathy. Int J Mol Sci. 2022;23:3700.
148. Qiao Q, Sun Y, Wang J, et al. Independently tunable viscoelasticity in hydrogels as a mechanical cue for tissue engineering. Tissue Eng Part B Rev. 2026:19373341251377696.
149. Lai W, Geliang H, Bin X, Wang W. Effects of hydrogel stiffness and viscoelasticity on organoid culture: a comprehensive review. Mol Med. 2025;31:83.
150. O’hern C, Caywood S, Aminova S, et al. Human heart-macrophage assembloids mimic immune-cardiac interactions and enable arrhythmia disease modeling. Cell Stem Cell. 2025;32:1671-90.e13.
151. Walker JA, Beck GA, Campbell JH, Miller AD, Burdo TH, Williams KC. Anti‐α4 integrin antibody blocks monocyte/macrophage traffic to the heart and decreases cardiac pathology in a SIV infection model of AIDS. J Am Heart Assoc. 2015;4:e001932.
152. Craiu D, Dica AD, Pomeran C, Pescaru G, Menascu S, Simu M. Disease-modifying treatment options in very early onset multiple sclerosis—what choices are there for onset under 5 years of age? A systematic review. J Clin Med. 2025;14:8133.
153. Bledzka K, Smyth SS, Plow EF. Integrin αIIbβ3: from discovery to efficacious therapeutic target. Circ Res. 2013;112:1189-200.
154. Pugashetti R, Koo J. Efalizumab discontinuation: a practical strategy. J Dermatolog Treat. 2009;20:132-6.
155. Gordon KB, Papp KA, Hamilton TK, et al. Efalizumab for patients with moderate to severe plaque psoriasis: a randomized controlled trial. JAMA. 2003;290:3073.
156. Bouri S, Johnston E. Vedolizumab: what is the benefit from increasing the dose frequency? Drug Ther Bull. 2022;60:183-7.
157. Holland EJ, Luchs J, Karpecki PM, et al. Lifitegrast for the treatment of dry eye disease. Ophthalmology. 2017;124:53-60.
158. Matsuoka K, Watanabe M, Ohmori T, et al. AJM300 (carotegrast methyl), an oral antagonist of α4-integrin, as induction therapy for patients with moderately active ulcerative colitis: a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Gastroenterol Hepatol. 2022;7:648-57.
159. Weinreb PH, Simon KJ, Rayhorn P, et al. Function-blocking integrin αvβ6 monoclonal antibodies. J Biol Chem. 2004;279:17875-87.
160. Raghu G, Mouded M, Prasse A, et al. Randomized phase IIa clinical study of an anti-αvβ6 monoclonal antibody in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2022;206:1166-8.
161. Wang C, Zhu X, Feng W, et al. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am J Cancer Res. 2016;6:27-37.
162. Huang H, Wang T, Rousseau J, et al. Biomimetic nanodrug targets inflammation and suppresses YAP/TAZ to ameliorate atherosclerosis. Biomaterials. 2024;306:122505.
163. Masuyama M, Shimoda M, Seto I, et al. Targeting focal adhesion kinase inhibits cell migration and non-angiogenic vascularization in malignant breast cancer. Breast Cancer. 2025;33:188-99.
164. Hanna GJ, Stathis A, Lopez-Miranda E, et al. A phase I study of the pan-notch inhibitor CB-103 for patients with advanced adenoid cystic carcinoma and other tumors. Cancer Res Commun. 2023;3:1853-61.
165. Yuan W, Zhang X, Fan X. The role of the Piezo1 mechanosensitive channel in heart failure. Curr Issues Mol Biol. 2023;45:5830-48.
166. Perryman L, Findlay A, Baskar J, et al. The small molecule LOXL2 inhibitor SNT-5382 reduces cardiac fibrosis and achieves strong clinical target engagement. Sci Rep. 2025;15:22653.
167. Li H, Zhu X, Cao X, Lu Y, Zhou J, Zhang X. Single-cell analysis reveals lysyl oxidase (Lox)+ fibroblast subset involved in cardiac fibrosis of diabetic mice. J Adv Res. 2023;54:223-37.
168. Vasta JD, Andersen KA, Deck KM, Nizzi CP, Eisenstein RS, Raines RT. Selective inhibition of collagen Prolyl 4-hydroxylase in human cells. ACS Chem Biol. 2015;11:193-9.
169. Brandstadter JD, Maillard I. Notch signalling in T cell homeostasis and differentiation. Open Biol. 2019;9:190187.
171. Villanueva MT. Soluble Notch agonists drive T cell production. Nat Rev Drug Discovery. 2025;24:741.
172. Wang J, Xie S, Li N, et al. Matrix stiffness exacerbates the proinflammatory responses of vascular smooth muscle cell through the DDR1-DNMT1 mechanotransduction axis. Bioact Mater. 2022;17:406-24.
173. Liu J, Wang J, Liu Y, et al. Liquid-liquid phase separation of DDR1 counteracts the hippo pathway to orchestrate arterial stiffening. Circ Res. 2023;132:87-105.
174. Ngai D, Lino M, Rothenberg KE, Simmons CA, Fernandez-Gonzalez R, Bendeck MP. DDR1 (discoidin domain receptor-1)-RhoA (ras homolog family member a) axis senses matrix stiffness to promote vascular calcification. Arterioscler Thromb Vasc Biol. 2020;40:1763-76.
175. Sun X, Wu B, Chiang H, et al. Tumour DDR1 promotes collagen fibre alignment to instigate immune exclusion. Nature. 2021;599:673-8.
176. Zhang Y, Duan X, Cao R, et al. Small GTPase RhoA regulates cytoskeleton dynamics during porcine oocyte maturation and early embryo development. Cell Cycle. 2014;13:3390-403.
177. Mejía-ramírez E, Picazo PI, Walter B, et al. Targeting RhoA nuclear mechanoactivity rejuvenates aged hematopoietic stem cells. Nat Aging. 2025;6:68-87.
178. Chen K, Griffin M, Henn D, et al. Targeting circulating mechanoresponsive monocytes and macrophages to reduce fibrosis. Nat Biomed Eng. 2025:1479.
179. Kovacs L, Kress TC, Belin De Chantemèle EJ. HIV, Combination Antiretroviral Therapy, and Vascular Diseases in Men and Women. JACC Basic Transl Sci. 2022;7:410-21.
180. Sattentau QJ, Stevenson M. Macrophages and HIV-1: an unhealthy constellation. Cell Host Microbe. 2016;19:304-10.
181. Pang X, He X, Qiu Z, et al. Targeting integrin pathways: mechanisms and advances in therapy. Sig Transduct Target Ther. 2023;8:1.
182. Larsen M, Schmidt-erfurth U, Lanzetta P, et al. Verteporfin plus ranibizumab for choroidal neovascularization in age-related macular degeneration. Ophthalmology. 2012;119:992-1000.
183. Amano M, Nakayama M, Kaibuchi K. Rho‐kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton. 2010;67:545-54.
184. Lv J, Si T, Wang D, et al. Mechanical signaling via β2 integrin decouples T cell proliferation and differentiation for generating stem cell-like CAR T cells. Immunity. 2025;58:2289-304.e10.
185. Yang J, Savvatis K, Kang JS, et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat Commun. 2016;7:13710.
186. Poe A, Martinez Yus M, Wang H, Santhanam L. Lysyl oxidase like-2 in fibrosis and cardiovascular disease. Am J Physiol Cell Physiol. 2023;325:C694-707.
187. Wang H, Poe A, Martinez Yus M, Pak L, Nandakumar K, Santhanam L. Lysyl oxidase-like 2 processing by factor Xa modulates its activity and substrate preference. Commun Biol. 2023;6:375.
188. Chaudhari N, Findlay AD, Stevenson AW, et al. Topical application of an irreversible small molecule inhibitor of lysyl oxidases ameliorates skin scarring and fibrosis. Nat Commun. 2022;13:5555.
189. Taufalele PV, Wang W, Simmons AJ, et al. Matrix stiffness enhances cancer-macrophage interactions and M2-like macrophage accumulation in the breast tumor microenvironment. Acta Biomater. 2023;163:365-77.
190. Rosin NL, Sopel MJ, Falkenham A, Lee TD, Légaré J. Disruption of collagen homeostasis can reverse established age-related myocardial fibrosis. Am J Pathol. 2015;185:631-42.
191. Peacock EE, Madden JW. Administration of beta-aminopropionitrile to human beings with urethral strictures: A preliminary report. Am J Surg. 1978;136:600-5.
192. Keiser HR, Sjoerdsma A. Studies on beta‐aminopropionitrile in patients with scleroderma. Clin Pharmacol Ther. 2016;8:593-602.
193. González A, López B, Ravassa S, San José G, Díez J. The complex dynamics of myocardial interstitial fibrosis in heart failure. Focus on collagen cross-linking. Biochim Biophys Acta Mol Cell Res. 2019;1866:1421-32.
194. Franklin MK, Sawada H, Ito S, et al. β-aminopropionitrile induces distinct pathologies in the ascending and descending thoracic aortic regions of mice. Arterioscler Thromb Vasc Biol. 2024;44:1555-69.
195. Cullen JM, Lu G, Shannon AH, et al. A novel swine model of abdominal aortic aneurysm. J Vasc Surg. 2019;70:252-60.e2.
196. Vachhani P, Baskar J, Charlton B, et al. PXS5505-MF-101: a phase 1/2a study to evaluate safety, pharmacokinetics and pharmacodynamics of Pxs-5505 in patients with primary, post-polycythemia vera or post-essential thrombocythemia myelofibrosis. Blood. 2023;142:625.
197. Zhu Z. Serum LOXL2 is elevated and an independent biomarker in patients with coronary artery disease. Int J Gen Med. 2024;17:4071-80.
198. Frantz S, Störk S, Michels K, et al. Tissue inhibitor of metalloproteinases levels in patients with chronic heart failure: an independent predictor of mortality. Eur J Heart Fail. 2008;10:388-95.
199. Zamilpa R, Ramirez TA, Ibarra J, et al. Abstract 15699: matrix metalloproteinase-9 overexpression in macrophages improves ventricular function by regulating the inflammatory and fibrotic responses post-myocardial infarction. Circulation. 2011;124:A15699.
200. Kobayashi T, Kim H, Liu X, et al. Matrix metalloproteinase-9 activates TGF-β and stimulates fibroblast contraction of collagen gels. Am J Physiol Lung Cell Mol Physiol. 2014;306:L1006-15.
201. Froom ZS, Callaghan NI, Davenport Huyer L. Cellular crosstalk in fibrosis: insights into macrophage and fibroblast dynamics. J Biol Chem. 2025;301:110203.
202. Jha G, Sharma RB, Sridhar S, et al. Nanoparticle-based therapies for cardiovascular diseases: a literature review of recent advances and clinical potential. Cureus. 2024;16:e72808.
203. Seth L, Bhave A, Kollapaneni S, et al. Cardiotoxic effects of antibody drug conjugates vs standard chemotherapy in ERBB2-positive advanced breast cancer: a systematic review and meta-analysis. JAMA Netw Open. 2025;8:e2540336.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].