

Noble and non-noble metal catalysts in methane oxidation: a comparative study under various oxidants and low temperatures
 0
0 Abstract
Selective oxidation of methane (SOM) offers a sustainable pathway for energy conversion and chemical synthesis. This review critically compares noble metal (Au, Pd, Ru, Rh) and non-noble metal (Fe, Cu, Cr, Zn, Ni) catalysts for methane activation at low temperatures, evaluating their performance under H2O2 and O2 as oxidants in environments, with CO as a promoter. Through a detailed analysis of the structure of typical systems, we have established key design principles involving active site engineering, metal-support interactions, and reactive oxygen species. Advanced characterization and density functional theory studies reveal that metal-oxygen interfaces govern methane activation mechanisms, where dynamic oxygen species, such as O*, OH*, and OOH*, dictate reaction pathways. Catalyst dimensionality, such as single-atom vs. clusters, and electronic modifications are shown to critically influence C–H bond cleavage energetics and methanol desorption. While noble metals excel in oxygen activation, modified non-noble catalysts achieve comparable efficacy by optimizing their coordination environments. This review summarizes recent advances in the SOM under mild conditions, providing a systematic qualitative and quantitative kinetic comparison of noble metal and non-noble metal catalysts across various oxidant systems. It offers valuable insights into reaction pathways and mechanisms in different catalytic environments, contributing to a deeper understanding of methane activation and functionalization. It is anticipated that this review will provide a useful guide to chemists and materials scientists attempting to design better metal catalysts for the SOM.
Keywords
INTRODUCTION
Hydrocarbons, as a core research subject in both the petrochemical industry and catalytic science, hold strategic significance for sustainable energy development[1]. Among them, CH4, the simplest hydrocarbon, constitutes more than 90% of natural gas by volume, characterized by its global abundance, high calorific value (56 MJ·kg-1), and low extraction costs[2]. The conversion of gaseous methane into liquid methanol represents a pivotal step in the “methane economy”, significantly reducing transportation costs and enabling the replacement of petroleum for the production of high-value-added chemicals. Currently, over 90% of methane is directly combusted, with its greenhouse effect being 25 times greater than that of CO2[3].
Therefore, advancing methane-directed conversion technologies not only alleviates the fossil energy crisis but also effectively mitigates greenhouse gas emissions while generating economic benefits. This strategy is particularly critical in the current context of energy transition and sustainable development. Replacing petroleum with methane to produce liquid fuels and foundational chemicals has long been a focal point in research and development across academia and industry. Concurrently, the direct conversion of CH4 into oxygenates (e.g., CH3OH, HCHO, CH3COOH) or olefins (e.g., C2H4) has emerged as a critical pathway to maximize its resource value[4,5], particularly due to the substantial advantages in transport economics of liquid-phase products (30%-40% cost reduction compared to gaseous forms)[6].
However, at the molecular scale, methane’s tetrahedral symmetry imparts exceptional thermodynamic stability. Its average C–H bond dissociation energy (BDE) amounts to 440 kJ·mol-1, coupled with a low proton affinity of 544 kJ·mol-1, rendering it an extremely weak Brønsted acid (pKa ≈ 48)[7]. These properties introduce significant kinetic barriers for both homolytic cleavage (mediated by radical pathways) and heterolytic cleavage (polar pathways) of C–H bonds. Consequently, selective conversion into high-value products is severely limited[8,9,10]. Notably, the minimum C-H BDE of target products such as CH3OH at
Conventional CH4 conversion technologies, exemplified by steam methane reforming (SMR) to syngas, predominantly rely on harsh conditions - high temperatures (1,073-1,273 K) and pressures (3-5 MPa) - with subsequent CH3OH or Fischer-Tropsch synthesis via syngas (CO/H2) intermediates[12]. Such indirect routes suffer from inherent drawbacks, including excessive energy consumption, complex multistep integration, and prohibitive capital investment[13]. Consequently, the development of novel catalytic systems to bypass syngas intermediates and achieve direct selective oxidation of CH4 under mild conditions - such as oxidative coupling of methane (OCM) and partial oxidation of methane (POM) - has become a shared objective across academia and industry.
Despite the significant challenges in scaling up conventional catalytic processes, research teams have been inspired by the binuclear iron-based active centers in nature’s methane monooxygenase (MMO) to develop a series of transition metal (Fe, Cu)-based biomimetic catalysts[14,15,16]. These catalysts emulate the chemical microenvironment of MMO, including cooperative sites and confined pore architectures, and have been designed to achieve selective methane oxidation under mild conditions, such as low temperatures or ambient temperature[17], using H2O2 as the oxidant. The catalysts exhibit catalytic efficiency comparable to enzymatic systems, with CH4-to-CH3OH selectivity exceeding 80%[16]. Building on the success of Fe/Cu-based catalysts, additional transition metals (e.g., Zn, Ni, Mo, Cr) have been incorporated into active site designs, significantly enhancing C–H bond activation efficiency and product distribution controllability through the construction of oxide/zeolite/carbon-based composite catalytic materials[18,19]. Noble metal catalysts, such as Pd, Ru, Rh, and Au, have demonstrated unique catalytic advantages in selective oxidation of methane (SOM). For instance, Pd-based catalysts exhibit exceptional hydrogen activation capability and surface electronic properties, which enhance CH4 adsorption and effectively catalyze POM in H2O2 or O2 oxidation systems[20,21,22]. Studies confirm that the distinctive electronic states in the noble metal d-band[23,24,25] (e.g., upward shift of the d-band center, hybridization of unoccupied d-orbitals with C-H σ* orbitals) play a critical role in reducing C–H bond activation energy barriers[12,26,27,28]. These attributes endow noble metal catalysts with immense potential in modulating energy barriers and optimizing selective oxidation pathways.
To balance catalytic performance and economic feasibility, researchers have proposed constructing diatomic catalysts (DACs) by integrating noble and non-noble metals, leveraging synergistic effects at heteronuclear metal sites (e.g., Cu-Pd, Ag-Ru) to overcome the limitations of single-metal systems[29]. For example, Cu sites suppress the formation of byproducts (e.g., HCOOH)[16], while Pd sites facilitate C–H bond cleavage via d-electron transfer, and bimetallic synergy elevates CH3OH selectivity beyond 90%[30]. Furthermore, efforts to address the high cost of noble metals have focused on integrating oxidant engineering (e.g., in situ generation of H2O2, molecular oxygen) with atomic-level dispersion techniques [single-atom catalysts (SACs)], aiming to maximize noble metal atom utilization efficiency and reduce noble metal loading per unit catalyst mass.
In the oxidant engineering of methane selective oxidation systems, green and safe H2O2 has been extensively explored due to its controllable reactivity. Through homolytic cleavage, H2O2 generates ·OH and ·OOH, which coordinate with metal active sites (e.g., Fe2+, Cu+) on the catalyst surface[31,32,33], forming highly reactive metal-oxo intermediates (e.g., Fe4+ = O, Cu-O-O) that drive the stepwise oxidation of methane C–H bonds[16,34]. Although SOM using commercially sourced H2O2 shows promise at the laboratory scale (e.g., CH3OH selectivity reaching 85% in Fe-ZSM-5 systems), its industrial application remains constrained by economic and safety challenges associated with H2O2 synthesis, transport, and storage[35].
To address this challenge, researchers have proposed in situ generation of H2O2 through H2/CO and O2 reactions or direct utilization of O2 as the terminal oxidant[33,36,37,38]. Experimental studies have confirmed that in situ H2O2 enables effective CH3OH synthesis. Furthermore, in photocatalytic systems such as TiO2/ZSM-5, the generation of ·OH radicals in situ facilitates CH4-to-CH3OH conversion under mild conditions (298 K, selectivity > 75%) via proton-coupled electron transfer mechanisms[39,40,41]. However, when O2 is employed as the oxidant, the spin-forbidden reaction between the triplet-state O2 and singlet-state CH4 obstructs direct hydroxylation pathways[42], thereby limiting the potential of H2O2 in industrial applications.
Overcoming this barrier requires dioxygen activation at reduced metal centers such as Pd0, Fe2+, where spin inversion generates reactive oxygen species (ROS) such as metal-superoxo (Mn+1O2·-), μ-peroxo (Mn+2O22-), and terminal oxo [Mn+4(O2-)2][13]. Notably, the O-O BDE of metal-superoxo species (~200 kJ·mol-1) is significantly lower than that of free O2 (498 kJ·mol-1), enabling heterolytic cleavage of C–H bond. However, O2 activation typically requires elevated temperatures (373-513 K) to overcome kinetic limitations, which poses a risk of overoxidation of target products, such as CH3OH to CO2[43,44].
Strategies such as atomic-level dispersion (e.g., single-atom Fe/ZSM-5) to modulate metal site electronic states[45] or zeolite confinement effects (e.g., MFI topology) to suppress radical chain reactions[46] are critical for terminating the reaction at desired oxygenates. While the regulatory effects of oxidant type (H2O2, in situ generated H2O2, O2) on methane oxidation activity and selectivity are preliminarily established, the dynamic adaptation mechanisms between oxidants and catalytic active sites - such as oxygen species migration pathways and metal valence cycling efficiency - remain underexplored. The core scientific challenge lies in achieving efficient and directional synthesis of oxygenates through synergistic innovation in active site microenvironment design, such as bimetallic cooperation and defect engineering, and optimized oxidant delivery strategies, such as pulsed feeding and membrane-enhanced separation. In summary, noble metal catalysts (e.g., Au, Pd, Rh) exhibit superior peak activity [turnover frequency (TOF) > 102 h-1] in an O2 atmosphere due to their exceptional proficiency in O2 activation[47-49]. In contrast, non-noble metal catalysts (e.g., Fe- or Cu-zeolites) demonstrate more consistent and excellent selectivity (generally > 90%) within H2O2 systems, alongside significantly enhanced cost-effectiveness[50]. Although both catalyst classes can achieve high selectivity, the performance of noble metal systems is highly sensitive to reaction conditions, whereas non-noble metal pathways offer greater robustness. Consequently, a fundamental distinction lies in their oxidant dependence: noble metals excel in the direct activation of O2, while non-noble metals currently achieve high efficiency primarily through H2O2 utilization [Table 1].
Comparison of the catalytic performance of various reported catalysts in the DSOM reaction under mild conditions
| Oxidants | Catalysts | TOF (h-1) | Productivity (mmol/gcat./h) | Selectivity of liquid oxygenates (%) | Reference |
| H2O2 | 0.15 wt% Pd1/2DT | 3.3 | 4.6 × 10-2 | 94% | [22] |
| H2O2 | 0.10 wt Pd1-ZSM-5 | 2.1 × 102 | 2.0 | ~100% | [69] |
| H2O2 | 0.01 wt% Pd/ZSM-5 +2 wt% CuO | 2.8 | 9.1 | 86.4% | [30] |
| H2O2 | Ag1Cu1/ZSM-5 | 9.2 × 104 | 40 | 81% | [72] |
| H2O2 | 0.13Ru1/UiO-66 | 1.9 × 102 | - | ~100% | [75] |
| H2O2 | FeN4/GN-2.7 | 0.50 | - | 94% | [17] |
| H2O2 | Cu2@C3N4-D2 | 83 | 4.9 | - | [81] |
| H2O2 | Cu-Pd/Anatase | - | 28 | > 99% | [110] |
| H2O2 | 0.1%Fe/ZSM-5 | 66 | 1.2 | - | [85] |
| H2O2 | Fe-silicalite-1 | 70 | 6.7 | 96% | [16] |
| H2O2 | Cu-Fe (2/0.1)/ZSM-5 | 4.3 × 102 | 7.7 | 80% | [86] |
| H2O2 | 1%Cu/TiO2 | 16 | 25 | ~100% | [87] |
| H2O2 | 1wt%Cr1/TiO2 | 2.9 | 5.5 × 10-1 | 93% | [89] |
| H2O2 | Cr1/ZSM-5 SAC | 87 | 2.1 × 10 | 99.8% | [90] |
| H2O2 | Cu-BTC-P-235 | - | 11 | 99.6% | [91] |
| H2O2 | CUS-M-P-210 | 3.3 × 102 | 83 | ~100% | [92] |
| H2O2 | Fe1/Cu1-C3Nx | 5.4 × 102 | 19 | ~100% | [93] |
| H2O2 | 0.31 wt% Fe-BN/ZSM-5 | 6.1 × 10-1 | 3.4 × 10-2 | 100% | [50] |
| H2O2 | Fe/ZSM-5 (0.25) | 2.7 × 102 | 12 | 63.2% | [152] |
| O2 | 0.5 wt% Rh-ZSM-5 | 2.9 × 102 | 14 | 70.5% | [100] |
| O2 | Pd3Au1NS | - | 1.5 × 102 | 98% | [28] |
| O2 | AuPd@ZSM-5-C16 | 14 | 4.7 | 92% | [38] |
| O2 | Pd9Au1NWs | - | 2.9 × 102 | 99% | [128] |
| O2 | Au-ZSM-5-0.25 | 7.3 | 9.2 × 10-2 | 100% | [108] |
| O2 | PdCu/Z-5 | 1.3 × 102 | 1.6 | 95% | [48] |
| O2 | Au-Fe/ZSM-5 | 1.5 × 102 | 1.9 | 92% | [49] |
| O2 | 0.10wt%Rh/ZSM-5 | 2.6 × 102 | 2.5 | 70% | [47] |
| O2 | 1.0%AuPd0.5/ZnO | - | 81 | 88.2% | [109] |
| O2 | Pd/CsPMA-H | - | 7.0 × 10-2 | 100% | [111] |
| O2 | Pd1/TS-1@CN | 7.3 | 6.5 × 10-1 | 100% | [112] |
| Air | IrO2/CuO | 16 | 6.5 × 10-1 | - | [116] |
| O2 | MoS2 | 5.7 | 1.4 × 10-3 | > 99.9% | [139] |
This review systematically summarizes recent advancements in SOM over noble metal (e.g., Rh, Pd, Ru, Pt, Au) and non-noble metal (e.g., Fe, Cu, Mo, Cr, Zn, Ni)-based catalysts under diverse oxidant conditions
REACTION TEMPERATURE AND LOW-TEMPERATURE STRATEGIES FOR SOM
The SOM is a pivotal route for natural gas valorization yet faces a fundamental challenge: the high stability of CH4 vs. the high reactivity of desired products (e.g., CH3OH, CH3COOH), dictating temperature choice and catalytic strategy. While early processes required high temperatures (973-1,353 K) for sufficient yield, research now focuses on mild-condition activation (< 473 K, often < 373 K)[52,53].
The central strategy to achieve this goal involves designing advanced catalytic systems capable of lowering the activation energy barrier. Mechanistically, C–H bond cleavage of methane at low temperatures proceeds mainly through two pathways [Figure 1A][54]: first, electrophilic activation, where active species (such as
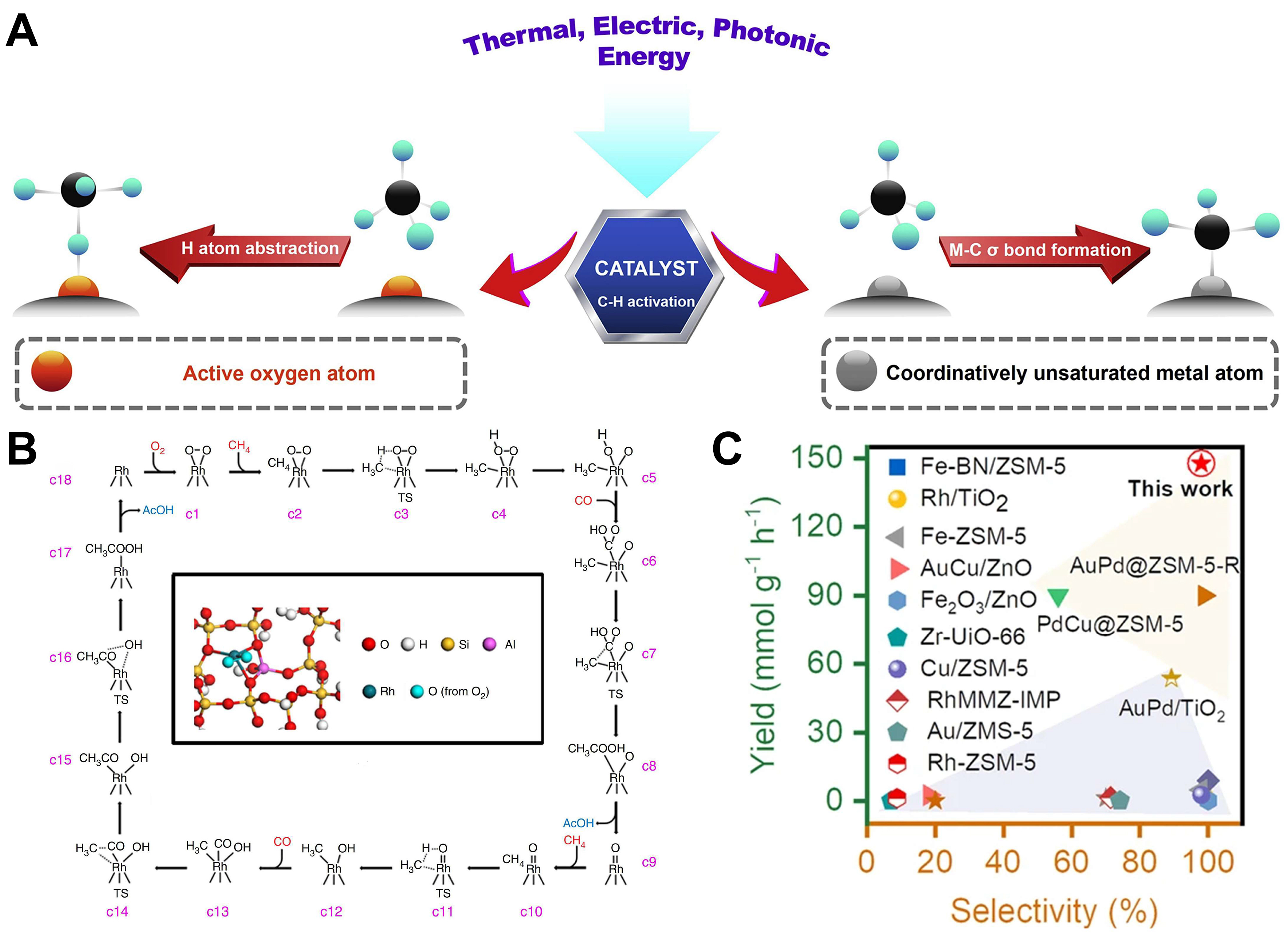
Figure 1. (A) The mechanism of C–H bond cleavage of methane at low-temperature reaction systems[54]. Copyright 2019, Elsevier; (B) Computational studies of reaction pathway. Minimum-energy paths and reaction schematic for formation of acetic acid from CH4, CO, and O2 on Rh1O5/ZSM-5. Intermediates and transition states for a complete catalytic cycle, starting with Rh1O5 (c1). Transition states are highlighted with the double dagger symbols[47]. Copyright 2018, Springer Nature; (C) Comparison of the performance and CH3OH selectivity of different catalysts for methane conversion in the methane selective oxidation process[28]. Copyright 2024, Springer Nature.
Comparison of CH4 activation energy barrier data among different oxidant systems
In summary, the key to efficient SOM under mild conditions lies in the rational design of catalysts that modulate reaction mechanisms and oxidant activation pathways.
H2O2-BASED OXIDATION SYSTEM
In a typical low-temperature methane oxidation system, the liquid-phase reaction is conventionally conducted in pressurized autoclave reactors employing aqueous media as the solvent and H2O2 as an environmentally benign oxidant[33]. The reaction proceeds via a radical-mediated selective oxidation mechanism: At transition metal active sites, H2O2 undergoes homolytic cleavage to generate ·OH and ·OOH, while the activated ·CH3 derived from C–H bond scission of methane reacts with these ROS to yield CH3OH and CH3OOH. Notably, CH3OOH serves as a critical intermediate governing the subsequent formation of oxygenated products. The decomposition of H2O2 ultimately produces water[43], endowing this system with inherent environmental compatibility and alignment with green chemistry principles.
Recent studies highlight the efficacy of supported noble metal catalysts (e.g., Au-Pd/TiO2) and zeolite-encapsulated transition metal catalysts (e.g., Fe-ZSM-5, Cu-ZSM-5) in H2O2-driven methane oxidation. These catalysts leverage synergistic interactions between metal active sites and acidic supports to modulate reaction pathways, enabling selective conversion of CH4 to CH3OH, HCOOH, and other oxygenates[16]. However, excess H2O2 over-oxidizes CH3OH to HOCH2OOH and HCOOH, reducing selectivity. Incorporating cocatalysts such as copper addresses this by scavenging excess H2O2 to generate ·OH, promoting *CH3 coupling to boost CH3OH yield and selectivity. Concurrently, sodium additives modify the Brønsted acidity of zeolitic frameworks, promoting CH4 protonation and C–H bond activation[61]. Such acid-base cooperativity not only elevates CH4 conversion efficiency but also suppresses undesired side reactions, offering a mechanistic foundation for designing high-performance catalysts for SOM.
Noble metal catalysts for H2O2-driven SOM
Atomic-scale two-dimensional (2D) catalysts have emerged as a paradigm-shifting platform in heterogeneous catalysis, leveraging maximally exposed active sites and quantum confinement-induced electronic structure modulation to transcend the performance limitations of conventional bulk counterparts. The synergistic integration of atomically thin 2D semiconductor matrices with atomic-level metal cocatalysts enables precise control over charge carrier dynamics and surface reaction pathways, offering unprecedented catalytic efficiency.
A study by Wu et al. synthesized atomically dispersed Pd on 2D TiO2 nanosheets (NSs) (Pd1/2DT), achieving 94% CH3OH selectivity in visible-light-driven methane photooxidation[22]. H2O2 undergoes heterolytic cleavage at electron-rich Pd sites, generating ·OH/·OOH radicals promoted by Pd–O bonds and oxygen vacancies (OVs), which stabilize Pd1 and improve charge separation. The 2D confinement limits radical diffusion, suppressing overoxidation and highlighting H2O2’s dual role as oxidant and radical precursor.
The catalytic performance of Pd in methane combustion and selective oxidation is intrinsically governed by its particle size[62,63], oxidation state[64,65], and interfacial electronic modulation with support matrices[66,67]. Recent advances demonstrate that atomically dispersed Pd species on oxide substrates exhibit distinct electronic configurations compared to nanoparticle analogues, enabling exceptional activity/selectivity in SOM[12,68].
For example, Xu et al. fabricated a CO-coordinated Pd1/ZSM-5 SAC via wet impregnation [Figure 2A], achieving near-100% oxygenate selectivity (TOF = 207 h-1) under ambient conditions[69]. CO-induced electron transfer lowers the C-H activation barrier from 1.27 to 0.48 eV and weakens Pd–O bonding, promoting H2O2 heterolysis to form critical ·OH species. The stabilized L-Pd1-O structure further directs hydroxyl coupling and methyl insertion toward CH3COOH, while suppressing H2O2 overconsumption. This electronic modulation and controlled radical pathway underscore the dual role of H2O2 as both oxidant and radical precursor in steering selectivity[70]. Supported AuPd nanoparticles enable methane oxidation under extremely mild conditions using H2O2 as the oxidant. Studies by Ab Rahim et al. revealed that methyl hydroperoxide is observed as the primary oxidation product, which subsequently undergoes gradual conversion to methanol, with no formic acid detected[71]. Notably, the current utilization of H2O2 as an oxidant in Au-Pd catalytic systems may incur prohibitive costs. In bimetallic catalysts, the yield and selectivity of CH3OH are modulated by the surface Pd0/Pd2+ ratio, while H2O2 concentration exerts pronounced and profound influences on catalytic activity.
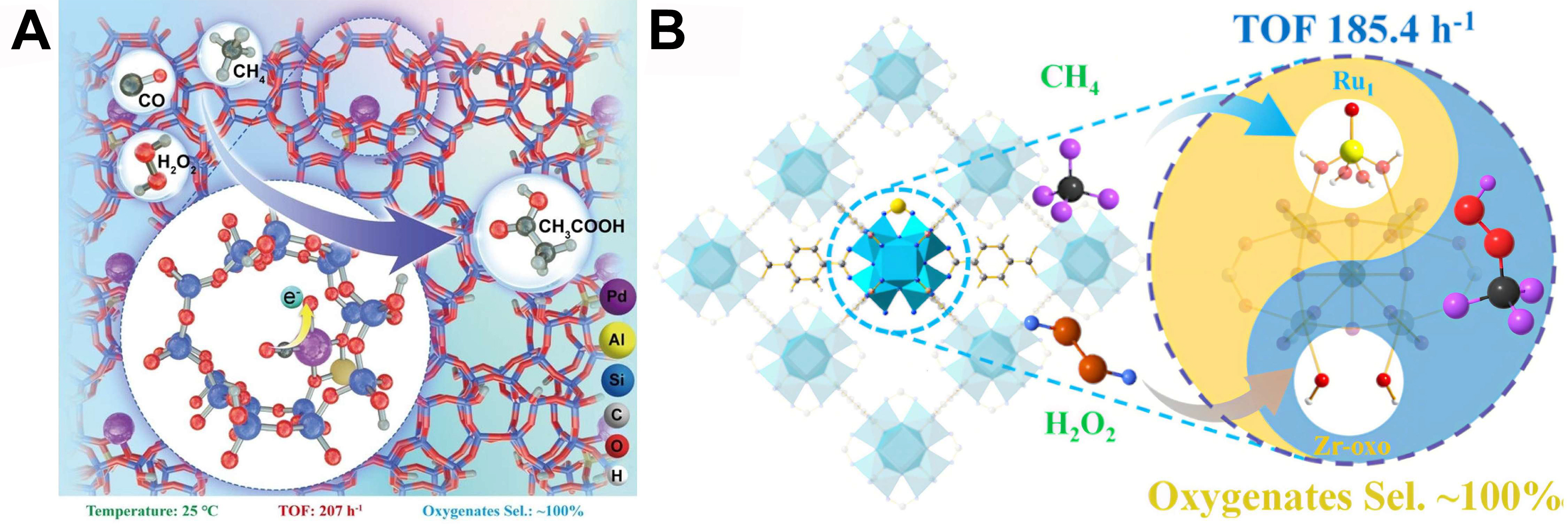
However, over stoichiometric H2O2 in methane oxidation systems induces cascade oxidation of CH3OH to acidic byproducts (e.g., formic acid), compromising oxygenate selectivity. Strategic integration of H2O2-scavenging cocatalysts emerges as an effective mitigation strategy. Huang et al. achieved 86% CH3OH selectivity in liquid-phase CH4 oxidation using microporous silicate-anchored Pd1O4 single sites, where confined pores suppress overoxidation[30]. In contrast, conventional 0.01 wt% Pd/ZSM-5 primarily yields HCOOH due to CH3OH overoxidation by residual H2O2. Introducing 2 wt% CuO as a sacrificial cocatalyst enhances CH3OH selectivity by 9% via catalytic H2O2 decomposition (H2O + O2), effectively quenching secondary oxidation[30]. While microscopy confirms atomically dispersed Pd as active centers, mass transport limitations in ZSM-5 micropores hinder CH4 diffusion and product egress. Optimizing pore dimensions or hierarchical architectures could improve accessibility to Pd1O4 sites, amplifying the cocatalytic efficacy of CuO.
Similarly, Yu et al. engineered a synergistic dual SAC (Ag1-Cu1/ZSM-5 hetero-SAC) by coupling Ag and Cu single atoms[72]. Spectroscopic analyses and computational modeling showed that the electronic interaction between Ag and Cu single atoms led to charge redistribution, facilitating the generation of reactive ·OH* for C–H bond activation. Cu species-modulated Ag sites endow the Ag-OH moieties with robust C–H bond cleavage capability for methane while suppressing over-oxidation of the products[73,74]. This electronic synergy was the key to catalytic enhancement. In methane partial oxidation, the hetero-SAC achieved a CH3OH productivity of 20 mmol·gcat-1 with 81% selectivity under mild conditions, outperforming single-metal SACs. The work offers mechanistic insights into heteronuclear dual-atom interactions and sets a design model for future cooperative catalysis systems.
Regarding support selection, metal-organic frameworks (MOFs) emerge as outstanding catalytic supports by virtue of their programmable architectures, exceptional surface areas (> 1,000 m2/g), and precisely tunable porosity (0.5-3.8 nm). Fang et al. developed a Ru1/UiO-66 SAC that achieves near-100% oxygenate selectivity and a TOF of 185 h-1 in H2O2-driven methane oxidation[75] [Figure 2B]. The system features dual active centers: Ru1 sites form Ru1 = O* to activate CH4, while Zr-oxo nodes generate oxygen radicals. Modified nodes also decompose excess H2O2 to O2, inhibiting overoxidation. This synergy between Ru1 and Zr-oxo sites regulates ·OH concentration, enabling optimal balance between activity and selectivity.
Non-noble metal catalysts for H2O2-driven SOM
Methane monooxygenases - soluble (sMMO) and particulate (pMMO) variants - employ distinct metalloactive sites for ambient CH4-to-CH3OH conversion: sMMO features a binuclear iron cluster with crystallographically resolved geometry, while copper coordination motifs in pMMO remain structurally ambiguous[76-78].
Iron and copper centers demonstrate dual functionality as catalytic motifs for H2O2 synthesis and methane oxidation. Cu(II) incorporation suppresses CH3OH overoxidation by modulating ·OH concentrations through radical scavenging mechanisms[79,80]. Biomimetic catalyst design leveraging these principles has spurred development of Fe/Cu-based systems for homogeneous and heterogeneous applications. Engineered copper-zinc oxides and iron-porphyrin complexes exemplify this approach, achieving methane activation through controlled radical generation while mitigating cascade oxidation pathways.
Moreover, Cui et al. synthesized a graphene-confined FeN4 SAC via ball-milling, achieving 94% C1 oxygenate selectivity with 10 h stability at room temperature[17] [Figure 3A]. Performance declined at higher Fe loadings due to active-site aggregation. Combined in situ and density functional theory (DFT) studies identified an O-FeN4-O center that promotes methane conversion via a radical pathway, with enhanced density of states near the Fermi level boosting activity [Figure 3B and C]. This work offers a strategy for mild methane functionalization using non-noble Fe catalysts and underscores the role of electronic structure in low-temperature catalytic design.
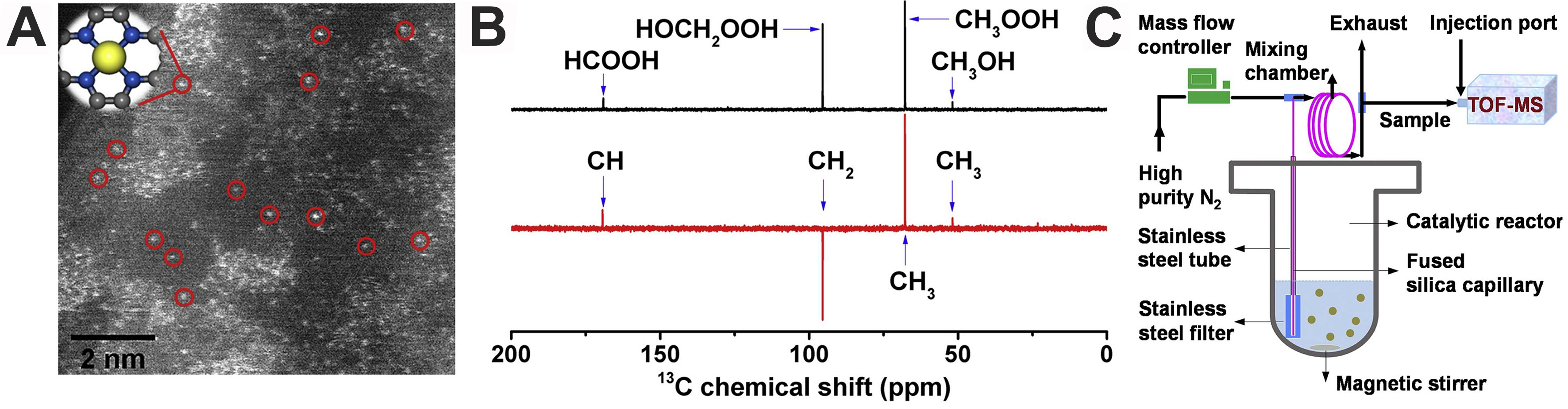
Figure 3. (A) An HAADF-STEM image of FeN4/GN-2.7. The red circles show some single iron atoms in the matrix of graphene nanosheets. The inset presents the model of FeN4/GN; (B) 13C NMR spectra obtained from typical reaction products of methane oxidation; (C) A scheme of the designed in operando TOF-MS[17]. Copyright 2018, Elsevier. HAADF-STEM: High-angle annular dark-field scanning transmission electron microscopy; FeN4: iron coordinated with four nitrogen atoms; GN: graphene nanosheets; NMR: nuclear magnetic resonance; TOF-MS: time-of-flight mass spectrometry.
Beyond carbon-supported Fe single-atom systems, Yang et al. fabricated diatomic Cu catalysts
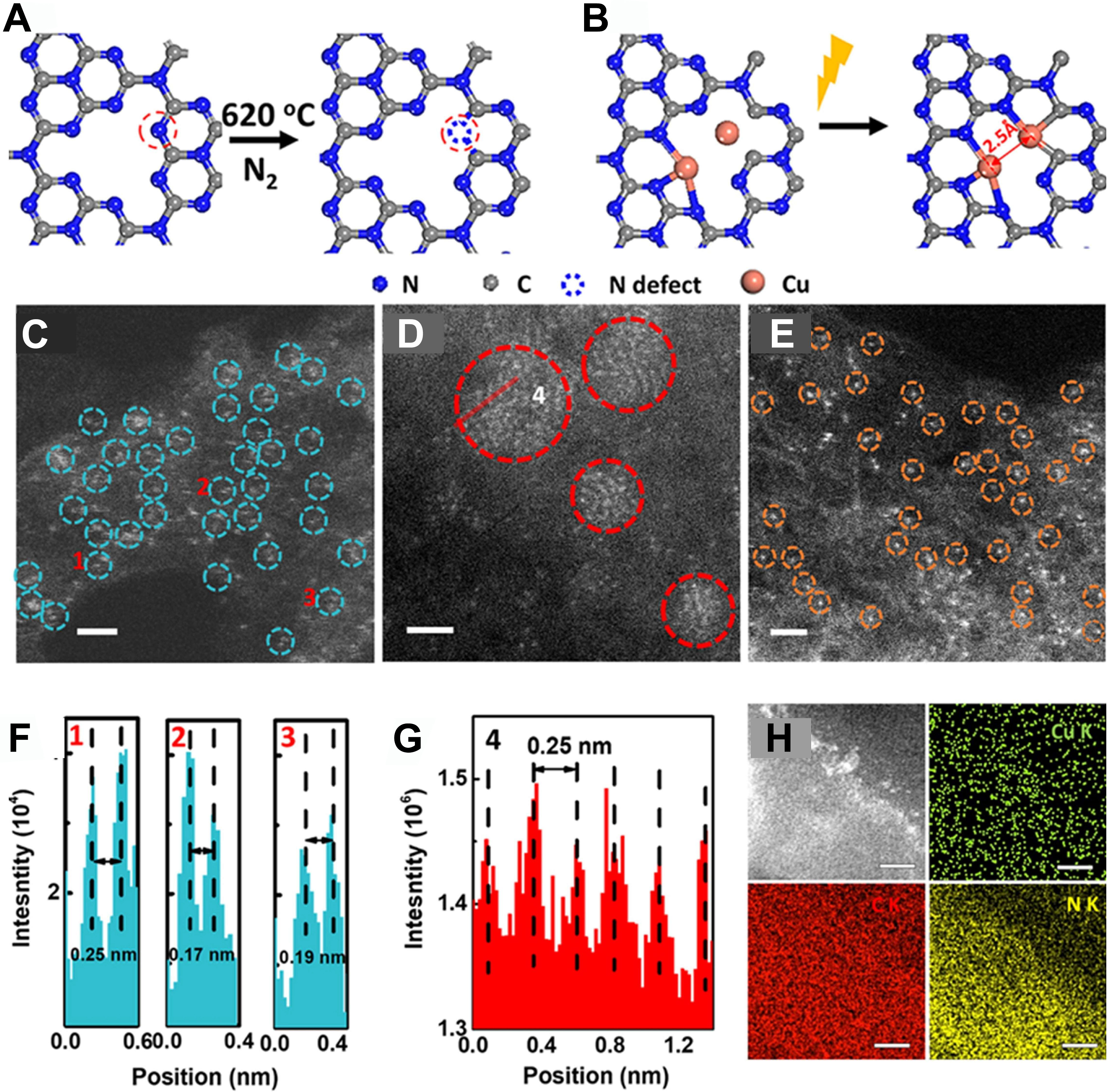
Figure 4. Schematics of the preparation of (A) C3N4-D and (B) Cu2@C3N4-D. AC-HAADF STEM images of (C) Cu2@C3N4-D1, (D) Cu2@C3N4-D2, and (E) Cu1@C3N4-D1. (F) and (G) Atomic distance from the selected areas in (C) and (D), respectively. (H) Elemental distribution of C, N and Cu on Cu2@C3N4-D1. Scale bars are 1 nm for c-e and 10 nm for h, respectively[81]. Copyright 2021, Elsevier. AC-HAADF-STEM: Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy; STEM: scanning transmission electron microscopy; nm: nanometer.
Comparative analysis shows that Cu2@C3N4-D has better C–H bond scission efficiency, with a CH4 activation barrier of 1.14 eV, 0.26 eV lower than Cu1@C3N4-D. The electronic synergy results from interatomic charge redistribution in diatomic configurations, enhancing electron density at active sites and maintaining atomic dispersion at high Cu loadings, highlighting the superiority of paired metal sites over single atoms in methane activation.
The level of activity ZSM-5 exhibits as a catalyst support has so far proven to be unique, other zeolites with similar compositions but different pore structures are an order of magnitude less active. Therefore, many bionic catalysts choose zeolite molecular sieves as carriers. Adding iron to inactive silica-1 can significantly increase activity, while adding copper to silica-1 has a limited contribution to activity. This suggests that the key active ingredient for methane activation under low-temperature liquid phase conditions is iron species rather than copper species, an observation consistent with previously reported results[17]. Likewise,
However, due to the inability to effectively inhibit the peroxidation of CH3OH to form formic acid, the monomer 0.1Fe/ZSM-5 catalyst system is difficult to achieve high selectivity control of CH3OH[85].
In order to improve CH3OH selectivity, adding Cu as a cocatalyst is an effective method. Hammond et al. introduced Fe into an inactive silica-1 matrix via hydrothermal synthesis, increasing activity significantly[16]. H2O2 reacts at Fe centers to form CH3OOH, and Cu promotes CH3OH formation by suppressing over-oxidation. Similarly, Yu et al. prepared Cu-Fe(2/0.1)/ZSM-5 via co-impregnation[86]. Using H2O2 as oxidant at 323 K, it directly converts CH4 to CH3OH with high yield and selectivity. Cu facilitates the formation of ·OH radicals, while adjacent Brønsted acid sites enable low-energy generation of ·CH3 radicals. In Cu-Fe(2/0.1)/ZSM-5, the Cu-promoted ·OH radicals rapidly combine with ·CH3 to form CH3OH [Figure 5A and B]. Hydroxyl groups thermodynamically favor CH3OH formation. Despite thorough characterization of Fe- and Cu-based zeolites, Cu-zeolite catalysts still require further research to develop superior catalytic systems.
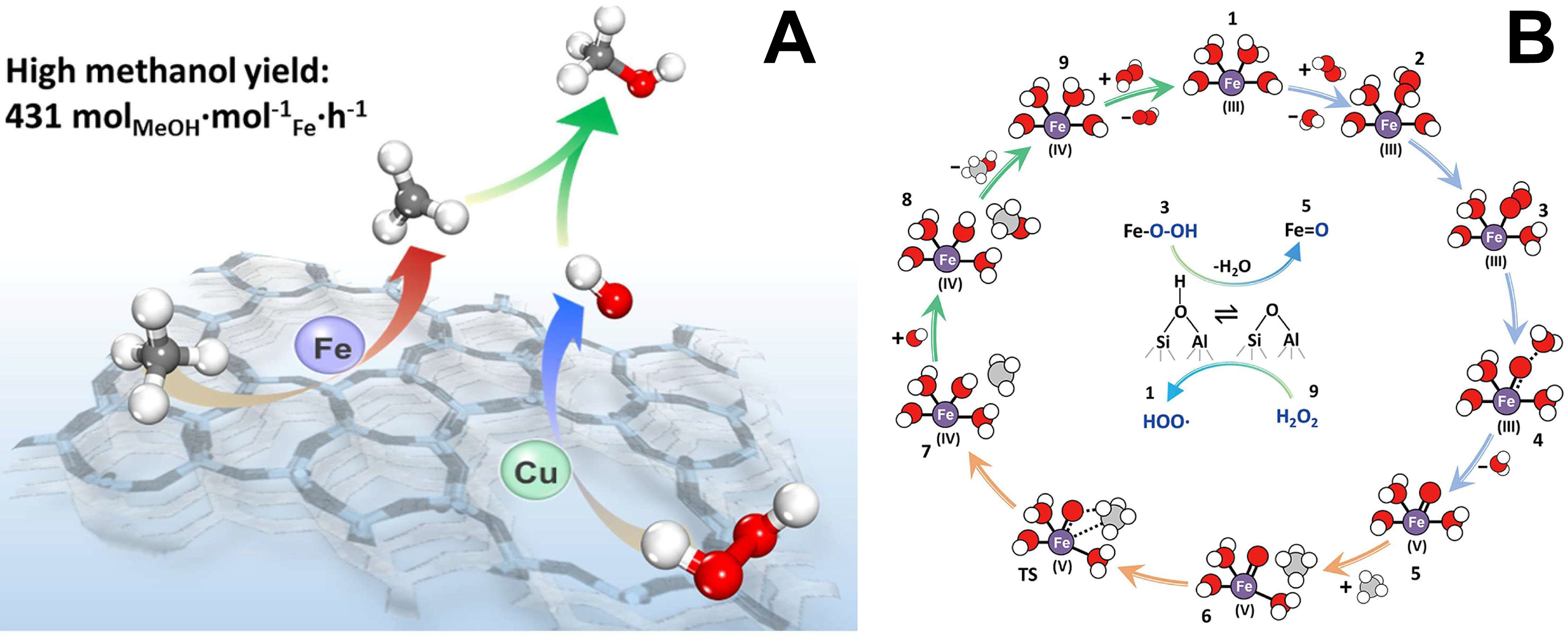
Figure 5. (A) Conceptual diagram of Cu-Fe(2/0.1)/ZSM-5 catalyst for selective oxidation of methane; (B) Proposed reaction scheme of reaction pathway for direct methane oxidation to methanol over Cu-Fe(2/0.1)/ZSM-5 using H2O2 as the oxidant. Red, purple, gray, and white balls represent O, Fe, C, and H atoms, respectively[86]. Copyright 2021, American Chemical Society.
Li et al. constructed Cu/TiO2 nanofiber composites through photodeposition, achieving a record C1 oxygenate yield of 2,511 mmol·gCu-1·h-1 with 100% selectivity at 353 K within 5 min[87]. Rutile-anatase TiO2 heterojunctions boost activity by elevating the Cu+/Cu0 ratio. DFT confirms that Cu+ sites on Cu2O(111) promote H2O2 heterolysis to form ROS, triggering C-H activation and a radical cascade pathway. The synergy between TiO2 phase junctions and Cu oxidation state modulation offers a generalized design principle for selective photocatalytic methane conversion.
In addition to the immobilization of Cu species on TiO2, Cr with low loading has also been demonstrated to exhibit superior performance in the SOM, where high-valent Cr species (particularly hexavalent chromium, Cr6+) display enhanced oxidative activity. Prior studies suggest that Cr6+ may serve as a highly active site[88], endowing it with significant potential for methane-selective oxidation. For instance, Shen et al. developed a 1 wt% Cr1/TiO2 catalyst that achieved a C1 oxygenate yield of 58 mol·molCr-1 with 93% selectivity at 323 K[89]. Spectroscopic studies revealed Cr-TiO2 electronic interactions synchronously cleave CH4 C–H and H2O2 bonds, generating ·CH3 and ·OH radicals that couple to form CH3OOH. Electron transfer from Cr to TiO2 lowered the Ti 2p binding energy and enhanced redox activity, while electron paramagnetic resonance (EPR) confirmed dominant radical pathways[89]. The single-atom Cr structure maximizes atomic efficiency, offering a design model for selective methane oxidation.
Inspired by the M1-O4 anchoring sites within ZSM-5 micropores, Zeng et al. developed a Cr1/ZSM-5 SAC, where the Cr1-O4 active center drives the formation of adsorbed ·OH* species via Cr3+ → Cr6+ oxidation, achieving a record C1 oxygenate productivity of 21, 100 μmol·gcat-1·h-1 (> 99% selectivity) at 323 K, outperforming nanoparticle counterparts by threefold[90]. Extended X-ray absorption fine structure (EXAFS) confirms Cr1 atoms anchored in ZSM-5 micropores as Cr1-O4. This tetrahedral coordination stabilizes Cr3+ and promotes Cr6+ formation. DFT shows Cr1-O4 generates OH* via low-barrier heterolytic O-O cleavage (0.16 eV with H2O aid), while higher-coordination Cr (e.g., nanoparticles) exhibits barriers > 1.21 eV. The atomically dispersed Cr1-O4 configuration suppresses over-oxidation, achieving 99.8% C1 oxygenate selectivity, vs. < 96.8% for nanoparticulate Cr. Reducing coordination from 6 to 4 enhanced C1 yield 50-fold (to 21,100 μmol·gcat-1·h-1), identifying Cr1-O4 as an efficient SOM active center. To address the hydrothermal instability of MOFs, Li et al. engineered hydrophobic Cu-BTC-P-235 (11 mmol·g-1·h-1)[91] and CUS-M-P-210 (83 mmol·g-1·h-1)[92] [Figure 6A and B] catalysts via polydimethylsiloxane (PDMS) modification, where Cu(I) and Fe(II) sites heterolytically cleave H2O2 and form Fe(IV)=O species [Figure 6C], respectively, for distinct methane activation pathways. Leveraging the complementary roles of Fe and Cu in SOM, the team further designed a nitrogen vacancy-enriched Fe/Cu heteronuclear DAC (Fe1/Cu1-C3Nx) [Figure 7A], which delivered a C1 oxygenate productivity of 9 mol·g-1·h-1 (TOF = 543 h-1) at
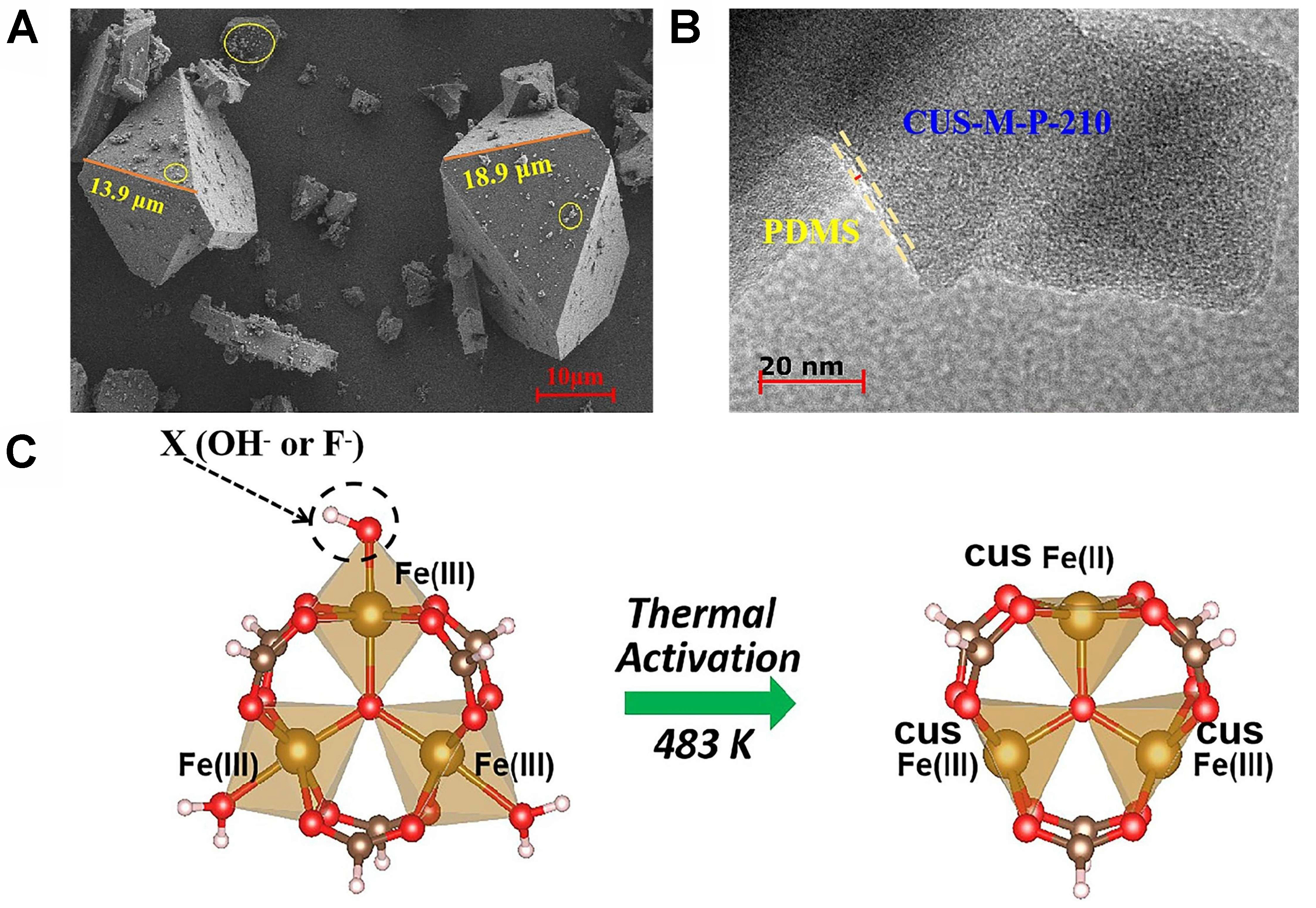
Figure 6. To address the hydrothermal instability of MOFs, Li et al. engineered hydrophobic Cu-BTC-P-235 (11 mmol·g-1·h-1)[87] and CUS-M-P-210 (83 mmol·g-1·h-1)[88] (A and B) catalysts via polydimethylsiloxane (PDMS) modification, where Cu(I) and Fe(II) sites heterolytically cleave H2O2 and form Fe(IV)=O species (C), respectively, for distinct methane activation pathways.
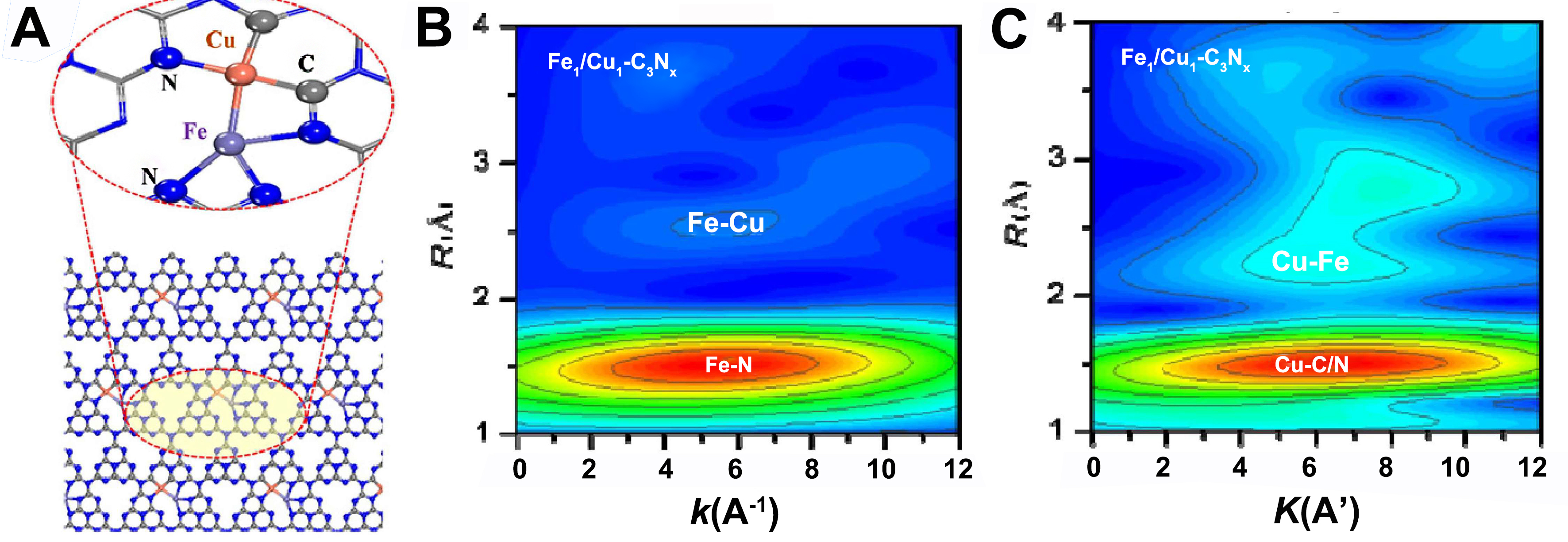
Figure 7. (A) Structure of Fe1/Cu1-C3Nx; (B and C) Wavelet transform for the k2-weighted EXAFS signals of Fe1/Cu1-C3Nx K-edge[93]. Copyright 2024, American Chemical Society. WT: Wavelet transform; EXAFS: extended X-ray absorption fine structure.
Both Cr1/ZSM-5 and Fe1/Cu1-C3Nx rely on dynamic metal valence transitions [Cr3+/Cr6+, Fe(II)/Fe(IV)] to generate critical reactive oxygen intermediates (·OH*, Fe=O), while localized hydrophobic microenvironments (PDMS-modified) suppress competitive H2O adsorption, synergistically enhancing selectivity and stability. Comparative studies demonstrate that atomic dispersion elevates Cr1/ZSM-5 SAC’s productivity by threefold over nanoparticles, and heteronuclear synergy doubles the TOF of Fe/Cu catalysts compared to monometallic systems, underscoring the multiplicative effects of atomic precision design. By integrating atomic-level active site dispersion, support coordination modulation, and microenvironment engineering, this work[93] establishes a multidimensional synergistic framework, offering systematic Fe/Cu-based SAC and diatomic solutions for low-temperature methane valorization.
Future efforts should prioritize understanding hydrophobic interfacial mass-transfer kinetics and multistep cascade mechanisms at active sites, combined with machine learning-guided support-metal screening, to surpass current activity/selectivity limits. Despite their potential, scalable DAC synthesis is challenged by metal aggregation. Advanced strategies - such as defect-mediated anchoring (e.g., N vacancies, OVs) and confined precursor decomposition - are essential to stabilize diatomic pairs. Selecting chelating precursors (e.g., metalloporphyrins) with trapping supports (e.g., pyrrolic N, zeolitic defects) enables high metal dispersion, leveraging support-metal electronic interactions to inhibit aggregation while preserving open coordination sites.
O2-BASED OXIDATION SYSTEM
Although H2O2 demonstrates excellent catalytic activity, its industrial application is constrained by economic limitations, prompting researchers to explore more cost-effective O2 as an alternative oxidant. Despite the industrial promise of O2 as a terminal oxidant under mild conditions, its inherent low activation efficiency typically requires elevated temperatures or multistep chemical looping to generate reactive metal-oxo intermediates (e.g., Fe-O, Cu-O)[94,95].
The direct oxidative conversion of CH4 to CH3OH (CH4 + O2 → CH3OH) is regarded as the “holy grail” of catalysis due to the high inertness of C–H bonds and kinetic challenges of overoxidation, with current yields constrained to millimolar scales (mmol·gcat-1). Extending this to C2+ oxygenates such as CH3COOH, (global demand: 18 Mt/a) necessitates concurrent C-C coupling and selective oxidation - traditionally reliant on methanol carbonylation, whereas direct routes offer sustainable potential[14,96] For instance, high-temperature O2 activation (~773 K) combined with low-temperature CH4 functionalization (~398 K) achieves measurable conversion but suffers from poor energy efficiency and prohibitive scaling costs[97-99]. In contrast, noble metal catalysts (e.g., Rh/ZSM-5) enable CH3COOH synthesis from CH4/CO/O2 mixtures at 423 K, yet face selectivity trade-offs[100]. Reductive promoters (e.g., CO) enhance conversion efficiency but struggle to balance C1/C2 oxygenate yields[101].
Recent research has pivoted toward bioinspired catalytic strategies, encapsulating synthetic analogs of di/tri-nuclear Fe/Cu clusters within zeolitic matrices to mimic the active sites of pMMO[102,103]. These systems leverage confinement effects and tailored acidity (Brønsted/Lewis) to stabilize intermediates (e.g., ·CH3,
Mechanistically, ·OH radicals - the pivotal ROS for C-H activation - originate from two pathways[104]: (1) photooxidation of H2O by photogenerated holes (h+) or (2) stepwise O2 reduction via *OOH and H2O2 intermediates (O2 → *OOH → H2O2 → ·OH)[105]. The latter dominates due to favorable redox potentials [O2/H2O2: 0.695 V; H2O2/·OH: 1.14 V vs. normal hydrogen electrode (NHE)]. Aqueous-phase systems mitigate overoxidation (e.g., CH3OH → HCHO) by accelerating product desorption, yet face mass transfer limitations and competitive adsorption that hinder H2O2 enrichment[106].
Future breakthroughs require hierarchical catalyst design to balance O2 activation with CH4 functionalization. Key strategies involve tuning active sites to modulate ·OOH/·OH balance, optimizing electronic (e.g., d-orbital hybridization) and geometric properties (e.g., mesoporosity) to stabilize intermediates and control ROS lifetimes. Combined with operando spectroscopy and modeling, these efforts can resolve the “activity-selectivity-stability” trade-off, advancing methane valorization toward industrial application.
Noble metal catalysts for O2-driven SOM
Utilizing cheap O2 as an oxidant to convert CH4 into value-added chemicals at low temperatures is expected to enable commercial applications of methane hydroxylation. However, this approach faces challenges due to the chemical stability of CH4 and the low reactivity of O2[107]. A series of heterogeneous catalysts have been developed with O2 as the terminal oxidant, typically requiring relatively high temperatures (373-513 K). For example, atomically dispersed Rh species have demonstrated high efficiency when using CO as a co-reducing agent. Recent studies show that loading Au nanoparticles achieves good performance in the absence of co-reducing agents. Qi et al. employed Au nanoparticles on ZSM-5 to catalyze methane oxidation with O2 between 393-513 K, yielding CH3OH and CH3COOH[108]. Electron microscopy confirmed that Au nanoparticles - not atoms or clusters - constituted the active sites. Although CH3OH and CH3COOH were proposed to form via parallel surface-mediated pathways, significant over-oxidation to byproducts such as HCOOH remained unavoidable.
Most reports indicate that Au and Pd are effective catalysts for O2 activation and subsequent oxidation reactions in the SOM, but precisely regulating the O2 evolution pathway using only single-metal catalysts remains challenging. For example, Au, as an excellent electron conductor, can promote the separation of photogenerated carriers that reduce O2 to *OOH and H2O2; however, its ability to cleave O–O bonds into *O and *OH is limited. In contrast, Pd exhibits strong adsorption toward O2, leading to efficient cleavage of
Palladium-based catalysts excel in methane activation. Recent progress has demonstrated bimetallic synergies to boost CH3OH selectivity; for instance, Wang et al. reported a CuPd/Anatase catalyst that achieves a CH3OH yield of 32 mmol·gcat-1·h-1 through bimetallic synergy[110]. Cu promotes H2O2 formation while Pd activates CH4 via heterolytic cleavage. DFT and in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) revealed that Cu-Pd interfacial electron redistribution lowers the *CH3 desorption barrier. Cu’s α-spin d-orbitals facilitate CH4 adsorption, with adjacent Pd aiding hydrogen abstraction. The stable Pd-O-Cu linkages suppress metal leaching, and under chloride-free conditions, the system preferentially directs *CH3-·OH coupling over deep oxidation, balancing high activity and selectivity.
Compared with pure O2, introducing H2 simultaneously can generate more ROS in a short time. Wang et al. synthesized a palladium-doped cesium phosphomolybdate catalyst (Pd/CsPMA) via precipitation[111]. This method leverages Pd’s hydrogen activation and spillover effects to reduce Mo sites in the CsPMA framework. Notably, the reduced catalyst (Pd/CsPMA-H) achieves sequential activation of O2 and CH4 under a H2/CH4/O2 mixed atmosphere, enabling methane-to-methanol conversion with 100% selectivity and a productivity of 67 μmol·g-1·h-1 at ambient conditions without external energy input, thus markedly surpassing conventional systems reliant on H2O2/·OH radicals [Figure 8A]. Mechanistic investigations reveal that O2 activation follows a “vacancy-refilling mechanism” at bridge-site OVs on partially reduced Mo centers, where O2 dissociates to generate ROS that directly abstract hydrogen atoms from CH4. Notably, Pd solely facilitates H2 activation and support reduction, playing no role in methane activation pathways
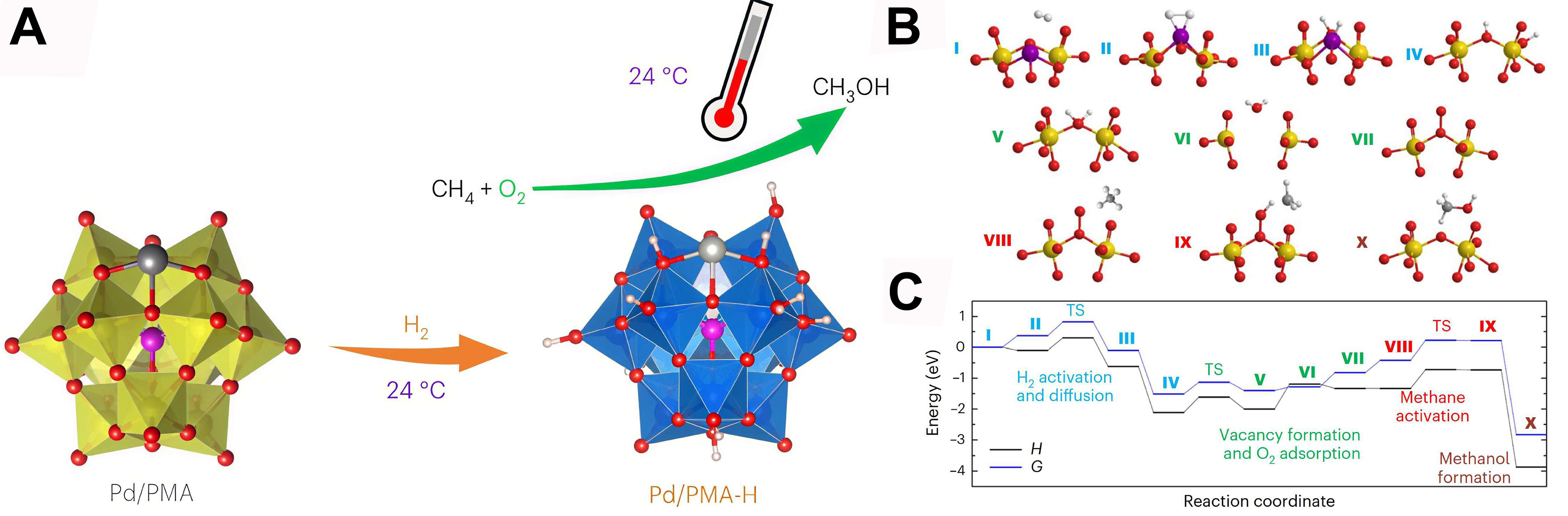
Figure 8. (A) Conceptual diagram of the preparation of Pd/PMA-H and its conversion of methane to methanol under oxygen conditions. The reaction intermediates (B) and the reaction profiles for both the enthalpy (H) and the Gibbs free energy (G) (C). The reaction proceeds initially via H2 activation and diffusion (intermediates I-IV), followed by vacancy formation and O2 adsorption (intermediates V-VII), then methane activation (intermediates VIII and IX) and methanol formation (intermediate X). Atom color code: Mo: yellow; Pd: purple; O: red; C: dark grey; H: light grey. TS: transition state[111]. Copyright 2023, Springer Nature.
Advanced characterization and theory reveal that efficient ambient O2 utilization stems from synergy between Mo5+/Mo6+ mixed valence and OVs, uncovering a dynamic O2-to-ROS pathway via vacancy engineering. This insight enables optimized O2 activation through valence modulation, establishing an OV-driven paradigm for direct CH4-O2 conversion that avoids energy-intensive oxidants.
Traditionally, the co-feeding of O2 and H2 has been assumed to preferentially generate H2O2, which subsequently produces free ·OH radicals for downstream methane oxidation, thereby overlooking the mechanistic role of adsorbed ·OH species during catalytic cycles. However, Yu et al. developed a single-atom Pd catalyst on N-doped carbon-coated TS-1 (Pd1/TS-1@CN), achieving 100% selective methane conversion at 288 K using O2 and H2[112]. O2 activation on Pd sites followed by H2 incorporation via a non-Horiuti-Polanyi mechanism generates surface-bound ·OH* species, which enable efficient C-H activation while minimizing overoxidation[113]. This adsorbed hydroxyl intermediate dictates selectivity and offers a new design strategy for mild O2-driven catalysis. The role of Pd varies with support: it activates H2 on metal oxides but directly mediates O2 dissociation and ·OH formation in zeolitic frameworks.
IrO2 exhibits exceptional C–H bond activation in methane due to the strong electrophilicity of Ir4+, where low-coordinate Ir sites cleave C–H bonds via Ir-C σ–bond formation, while O2 acts as the terminal oxidant to replenish OVs and sustain catalytic cycles[114,115]. Building on this, Yang et al. engineered a bifunctional IrO2/CuO catalyst via a bottom-up strategy[116]. Under mild conditions (423 K), O2 synergizes with H2O to drive methane oxidation: IrO2 first activates CH4 to generate ·CH3 radicals. Concurrently, CuO provides surface oxygen species (-O) to transform ·CH3 into -OCH3, with H2O subsequently facilitating desorption to form CH3OH [Figure 9A and B]. Critically, O2 dynamically refills OVs to close the catalytic loop. Notably, increasing CH4 pressure to 20 bar enhances O2 mass transfer, boosting methanol productivity to
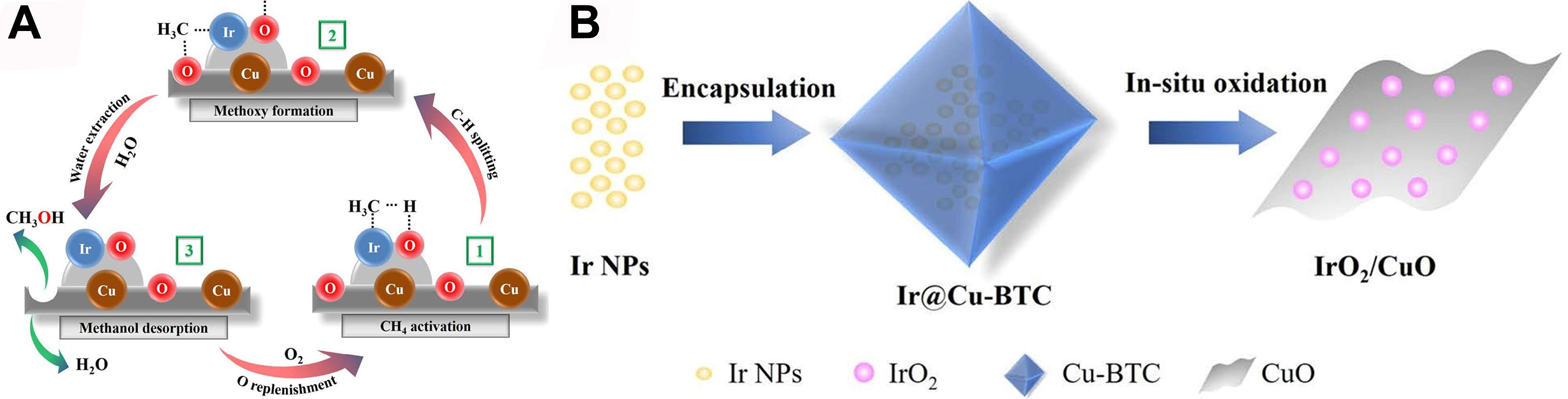
Figure 9. (A) Proposed Catalytic Principle of Methane Oxidation to Methanol on IrO2/CuO and (B) Schematic Illustration of the IrO2/CuO Synthesis Process[116]. Copyright 2019, American Chemical Society.
Compared to conventional impregnation, the bottom-up approach utilizes the confinement effect of Cu-BTC to achieve uniform IrO2 dispersion on CuO, strengthening IrO2-CuO interfaces and enhancing O2 activation into ROS (Cu-O-Ir)[118]. X-ray photoelectron spectroscopy (XPS) confirms higher Cu-O-Ir oxygen content in such systems than in impregnated counterparts, due to O2-induced strong metal-support interaction (SMSI) and lattice distortion improving oxygen mobility. This system mimics pMMO’s O2 activation: IrO2 mimics Fe/Cu sites for CH4 activation, while CuO reduces O2 to reactive oxygen intermediates, enabling biomimetic oxidation. O2 serves dual roles: refilling OVs and modulating Cu-O-Ir interfacial species, enabling a cross-scale synergy for efficient methane oxidation[119,120].
Ir-based catalysts excel in C-H activation: cationic Ir(III) complexes are among the most active C-H activators, while metallic Ir surfaces show high alkane C-H cleavage activity, driven by low-coordination sites[121]. The key mechanism involves coordinatively unsaturated Ir centers that strongly adsorb and activate alkanes, leveraging synergistic electronic (e.g., electron-rich d-orbitals) and geometric (e.g., defect sites) features to facilitate C–H bond scission[96].
Metal oxides feature defect-rich surfaces with hydroxyl groups, enabling selective oxidation. Using oxides that promote methoxy formation and exhibit low M–O bond strength (e.g., IrO2) as cocatalysts can enhance methanol productivity and selectivity. Such materials may broaden the applicability of dual-component catalysts for selective methane conversion under oxygen-rich conditions.
Recent studies have shown that AuPd bimetallic alloy catalysts exhibit excellent performance in the low-temperature conversion of methane to methanol. The Hutchings team discovered that in this catalytic system, H2O2 serves as the oxidant, activating methane through ·OH radicals to generate ·CH3 intermediates (verified by EPR spectroscopy). Moreover, when H2O2 and CO coexist, ·CH3 is more likely to couple with CO and ·OH to form CH3COOH[50,122]. The introduction of O2 can promote the generation of CH3OOH, accelerating the termination step of methanol formation. In addition, the excellent catalytic performance of AuPd bimetallic nanostructures stems from the synergy between the ligand effect and the geometric effect. These two effects optimize the electronic structure and the accessibility of active sites, and the electron transfer and structural changes are triggered by the electronegativity differences of atoms[123].
In the SOM, the in situ generation of H2O2 from O2 and H2 (instead of direct addition) is crucial for optimizing the efficiency[124]. Currently, PdAu alloy catalysts have set a performance record with a production rate of 92 mmol·g-1·h-1 and a selectivity of 92%. However, existing studies focus on 0D PdAu nanoparticles[38]. Their structural heterogeneity and disordered atomic arrangements hinder the precise analysis of the structure-activity relationship and the functions of Pd/Au sites[33,43,125]. In addition, although the C–H bond activation mediated by ·OH radicals dominates the H2O2-driven SOM, the formation and activation mechanisms of ·OH remain unclear, limiting the rational design of 0D PdAu nanoparticle catalysts[126].
Furthermore, one-dimensional nanowires (NWs) exhibit superior activity and durability over 0D counterparts in methane conversion due to anisotropy and rapid mass transfer[127]. For instance, Xu et al. synthesized zigzag-shaped Au-Pd NWs via self-assembly and galvanic replacement, where Pd9Au1 NWs feature Pd-rich cores and Au-enriched surfaces[128] [Figure 10A and B]. Notably, under in situ H2O2 generation at 343 K, this catalyst achieved a C1 oxygenate productivity of 2,890 μmol·g-1·h-1 (99% selectivity), outperforming pre-added H2O2 systems by nearly ninefold[123]. Mechanistic insights reveal that Au electronically modulates Pd via ligand effects, weakening intermediate (·OH/·OOH/·CH3) binding strength to balance C-H activation and product desorption [Figure 10C].
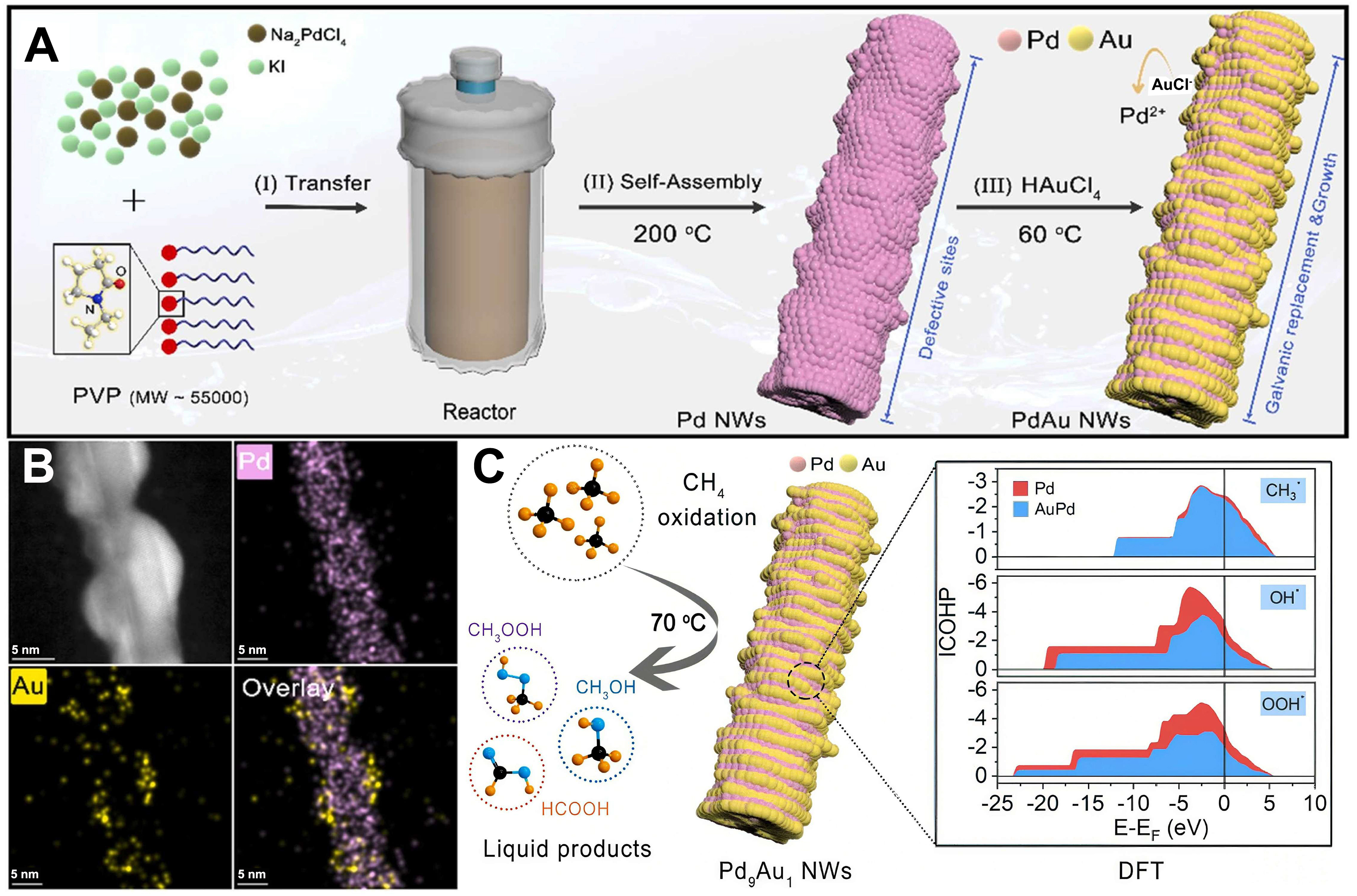
Figure 10. (A) Schematic illustrations for the synthesis of PdxAuy NWs; (B) High-angle annular dark-field scanning transmission electron microscopy (HADDF-STEM) images and the corresponding STEM-EDS element mapping of Pd9Au1 NWs; (C) Conceptual diagram of methane oxidation by Pd9Au1 NMs at 343 K[128]. Copyright 2022, Elsevier. PdxAuy NWs: Palladium-gold nanowires; HAADF-STEM: high-angle annular dark-field scanning transmission electron microscopy; STEM-EDS: scanning transmission electron microscopy-energy dispersive X-ray spectroscopy; Pd9Au1 NMs: palladium-gold nanomaterials; DFT: density functional theory.
In contrast, ultrathin 2D NSs, with uniformly exposed facets and high surface atom ratios, enable precise structure-activity correlation[129,130]. As demonstrated by Xu et al., tailoring Au coverage on PdxAuy NSs via galvanic displacement yielded a volcano-shaped activity trend, with Pd3Au1 NSs delivering optimal performance: 148 mmol·g-1·h-1 CH3OH yield (98% selectivity) at 343 K[28]. The enhanced activity stems from electronic regulation of Pd by adjacent inert Au atoms, highlighting the pivotal role of ligand effects in fine-tuning active-site microenvironments in bimetallic systems.
Unlike the aforementioned AuPd system, the Cu-Pd/anatase catalyst developed by the Hutchings team exhibits a unique product regulation mechanism in methane oxidation: in the presence of CO, this system directs the reaction towards a highly selective CH3OH pathway (selectivity > 90%) by suppressing the formation of CH3OOH[108]. The Cu-Pd synergy selectively decomposes H2O2 into ·OH over ·OOH and stabilizes ·CH3 via electronic effects, promoting ·OH coupling to form CH3OH. Competitive CO adsorption destabilizes peroxo-intermediates, while low in situ H2O2 concentration (< 0.1 mM) further suppresses ·OOH formation, collectively avoiding CH3OOH production. This complements AuPd systems: Cu-Pd regulates radical speciation (·OH/·OOH), whereas AuPd enhances reaction kinetics, together highlighting the potential of multimetal designs in directing methane oxidation[16,131].
Similarly, Wu et al. engineered a PdCu/ZSM-5 catalyst that achieves efficient methane oxidation via in situ H2O2 generation and activation: PdO nanoparticles catalyze H2 and O2 to produce H2O2, while atomically dispersed Cu sites activate H2O2 into ·OH radicals, driving C–H bond cleavage in CH4[48]. The high selectivity (95%) and productivity (1,178 mmol·gPd-1·h-1) arise from rapid recombination of ·CH3 intermediates with ·OH to form CH3OH, circumventing the ·OOH pathway triggered by excessive H2O2 decomposition [Figure 11A]. This highlights the critical synergy between H2O2-generating (PdO) and radical-activating (Cu) sites in zeolitic frameworks for selective C1 oxygenation.
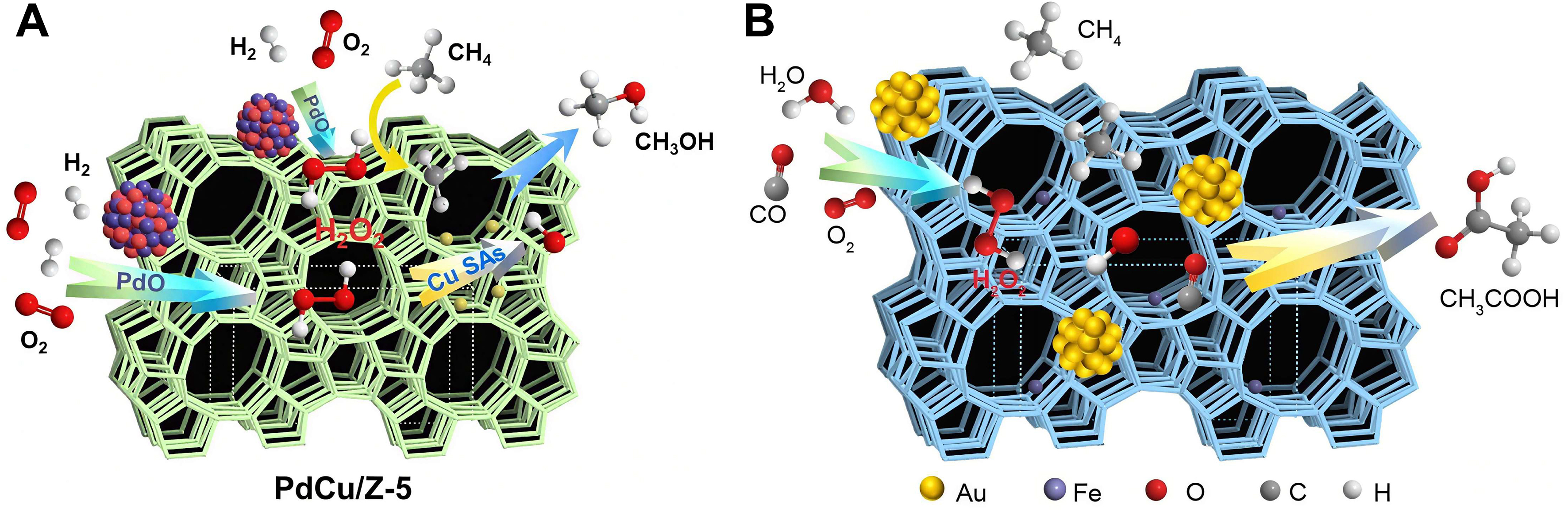
Beyond in situ oxidant synthesis, Wu et al. revealed CO’s dual role: as both a reductant and a ligand, CO modulates oxidation pathways[49,132-134]. In Au-Fe/ZSM-5, Au nanoparticles catalyze hydroxyl species formation via CO/O2/H2O reactions, while atomically dispersed Fe sites mediate coupling of CH4-derived methyl species with hydroxyl groups to yield CH3COOH (92% selectivity, 6 mmol·gcat-1). CO indirectly enhances oxidant supply via the water-gas shift reaction (CO + H2O → CO2 + H2) and subsequent H2O2 synthesis (H2 + O2 → H2O2), whose decomposition generates ·OH to activate CH4 [Figure 11B].
Although the 3D pore structure of ZSM-5 facilitates reactant diffusion and product desorption, the hydrophilic nature of in situ-generated H2O2 causes its diffusion into bulk solution, reducing local oxidant concentration and limiting activity[31]. Jin et al. hydrophobically modified AuPd@ZSM-5 with C16 organosilane, creating a molecular fence that confines 78%-86% of H2O2 within the pores[38]. This local enrichment sustains high ·OH flux, suppresses overoxidation, and achieves 92 mmol·gAuPd-1·h-1 productivity with 92% selectivity at 343 K. Future work should employ operando techniques to dynamically track oxidant dynamics and their roles in complex reaction networks.
In addition to ZSM-5-supported bimetallic systems, single-metal Rh catalysts also exhibit superior performance. Studies on ZSM-5-supported single-atom Rh catalysts demonstrated that CH4 can be converted into C1-C2 products at 423 K with CO assistance[47]. CO serves dual roles: as a ligand to promote C–H bond activation (e.g., Rh single sites activate CH4 into Rh-CH3, followed by CO insertion to form CH3COOH) and as a reductant that enhances O2 activation via surface reduction, albeit with partial oxidation to CO2 that inhibits the main reaction. Furthermore, CO stabilizes metal dispersion, e.g., suppressing Au nanoparticle agglomeration, while isolated cations or subnanometer clusters remain critical for methane activation[135].
Given CO’s indispensability, C-H activation likely occurs at cooperative metal sites (CO as a ligand) or via CO-derived ROS (e.g., dissociated O atoms, M=O). In either case, CO must first coordinate with metal sites. Tang et al. proposed that Rh1O5 sites mediate CH4/CO/O2 coupling to CH3COOH: Rh single centers activate CH4 into Rh-CH3 and O2 into Rh-OH; CO inserts into Rh-OH to form Rh-COOH, which couples with Rh-CH3 to yield CH3COOH[47]. Beyond Rh, other platinum group metals (e.g., Ru, Ir) show catalytic potential[136]. Moteki et al. demonstrated that ZSM-5-supported transition and platinum group metals (e.g., Fe, Cu, Ru, Ir) enable CO-assisted oxidative carbonylation of CH4 to C1/C2 oxygenates[137]. Noble metals, particularly Ru and Ir, outperform others - Ru favors CH3OH, while Ir selectively produces HCOOH - suggesting a universal CO-facilitated mechanism rooted in their distinct electronic and coordination properties. CO enhances reaction kinetics and methanol yield by stabilizing the catalyst in a reduced state via competitive adsorption and electronic modulation, thereby suppressing overoxidation and promoting selective product formation.
Considering the impact of zeolite framework structure on product selectivity, Moteki et al. experimentally studied CH4 conversion over zeolite-supported single-atom Rh catalysts with varying Si/Al ratios[136]. They found that Rh-SSZ-13 exhibits higher CH3OH selectivity, with small-pore zeolites such as SSZ-13 favoring C1 oxygenates due to pore window constraints on larger products. Adjusting the Si/Al ratio and proton exchange with basic metal cations confirmed the critical role of acidic sites in C2 product (e.g., CH3COOH) formation[138]. To further enhance Rh catalyst selectivity, Gu et al. introduced Cu2+ as a cocatalyst in a Rh/TiO2/Cu2+ system, achieving > 99% CH3OH selectivity and millimolar-level yields at 423 K and 31 bar[101]. Key to this performance is Cu2+’s dual role: stabilizing Rh in a low oxidation state via in situ H2 reduction and suppressing methyl peroxide formation. This underscores the synergy between promoter ions (Cu2+) and atomically dispersed noble metals for efficient CH3OH production under mild conditions.
However, current noble metal systems (Rh-, Ir-, Pd-based) for CH4-to-CH3COOH conversion face persistent activity-selectivity trade-offs, with performance metrics lagging industrial benchmarks[50]. Challenges include balancing C-H activation energetics with C-C coupling selectivity and suppressing thermodynamically favored overoxidized products (e.g., CO2).
This performance gap highlights the urgent need for catalysts that reconcile: (1) precise control of radical speciation (·CH3, ·OH) to suppress side reactions; (2) active sites synergistically driving C-H cleavage and
Non-noble metal catalysts for O2-driven SOM
As non-noble metal catalysts for SOM, zeolites with iron and copper as active sites are a very promising class of materials. They are able to selectively oxidize CH4 to surface-bound methoxy species through an active site similar to that of MMO. In addition, the exploration of other non-noble metal catalysts (such as Mo[139], Ni[140], etc.) is also ongoing and has achieved good results. Although the large-scale production of such catalysts that react efficiently under O2 conditions requires long-term research, the properties of their active sites provide inspiration for the development of synthetic oxidation catalysts.
Although it is thermodynamically and kinetically feasible to oxidize the C–H bond of CH4 at low temperature, the high dissociation energy of this molecule (435 kJ·mol-1) hinders the C-H cleavage reaction through homolytic or heterolytic pathways. Therefore, few catalysts can prevent over-oxidation to CO2. Incorporation of copper ions is an effective strategy to improve CH3OH selectivity. Hammond et al. reported that the addition of Cu2+ reduced the peroxidation process of CH3OH because the concentration of ·OH decreased[16]. Shan et al. proposed that Cu ions improve CH3OH selectivity by inhibiting the formation of formic acid[100]. However, since these strategies are indirectly involved in the reaction, their ability to improve the selective conversion of methane is limited. Therefore, developing strategies that directly involve the methane activation and key intermediate desorption steps is expected to significantly improve the yield and selectivity of CH3OH. Notably, zeolite-supported transition metal catalysts represent a compelling class of materials for methane oxidation, combining the structural rigidity, tunable acidity, and confinement effects of zeolitic frameworks with the redox versatility of transition metals. Over the past two decades, such systems - particularly metal-exchanged zeolites (e.g., Fe-, Cu-ZSM-5) - have demonstrated exceptional selectivity toward CH3OH and other oxygenates under mild conditions[42]. The effectiveness of these catalysts relies on confining and stabilizing key radicals (e.g., ·CH3, ·OH) within zeolite pores while inhibiting over-oxidation. For example, Fe-zeolites activate methane via radical pathways, whereas Cu species enable oxygen insertion into C–H bonds, mimicking enzymatic systems. This biomimetic strategy enhances both efficiency and sustainability by avoiding harsh conditions. Recent work underscores how zeolite topology - such as pore geometry and acid site distribution - modulates metal redox behavior and intermediate stability, providing a basis for rational catalyst design.
The binuclear [Cu2O]2+ site first discovered by Woertink et al. is the oxo-bridged binuclear Cu(II) site[103]. In Cu-exchanged ZSM-5 zeolites, dinuclear CuII-O-CuII species are critical intermediates for single-electron transfer mediated CH3OH generation, requiring a Cu/Al molar ratio > 0.2 to stabilize the dinuclear configuration[141]. Narsimhan et al. engineered a Cu-exchanged mordenite (Cu-MOR) catalyst that converts CH4 to CH3COOH via oxidation-carbonylation coupling, achieving a significantly higher yield
Despite the ability of Cu-zeolites to catalytically oxidize CH4 to CH3OH selectively at low temperatures using only O2 and H2O, the precise nature and origin of active species remain unresolved. Beyond Cu-based systems, explorations of other metals have yielded breakthroughs. Mao et al. achieved direct CH4-to-C1 oxygenate conversion with O2 on edge-rich MoS2 catalysts at 298 K, attaining a CH4 conversion of 4% and C1 oxygenate selectivity > 99% while suppressing CO2 formation[139] [Figure 12A and B]. Sulfur vacancies in edge-rich MoS2 directly dissociate O2 to form O=Mo=O active species, enabling high-selectivity CH4 activation [Figure 12C and D]. The binuclear Mo (bi-Mo) sites in MoS2 mirror the binuclear Fe (bi-Fe) sites in MMO, utilizing O2-derived Fe-O species to activate CH4, thus enabling one-step catalytic conversion of CH4 and O2.
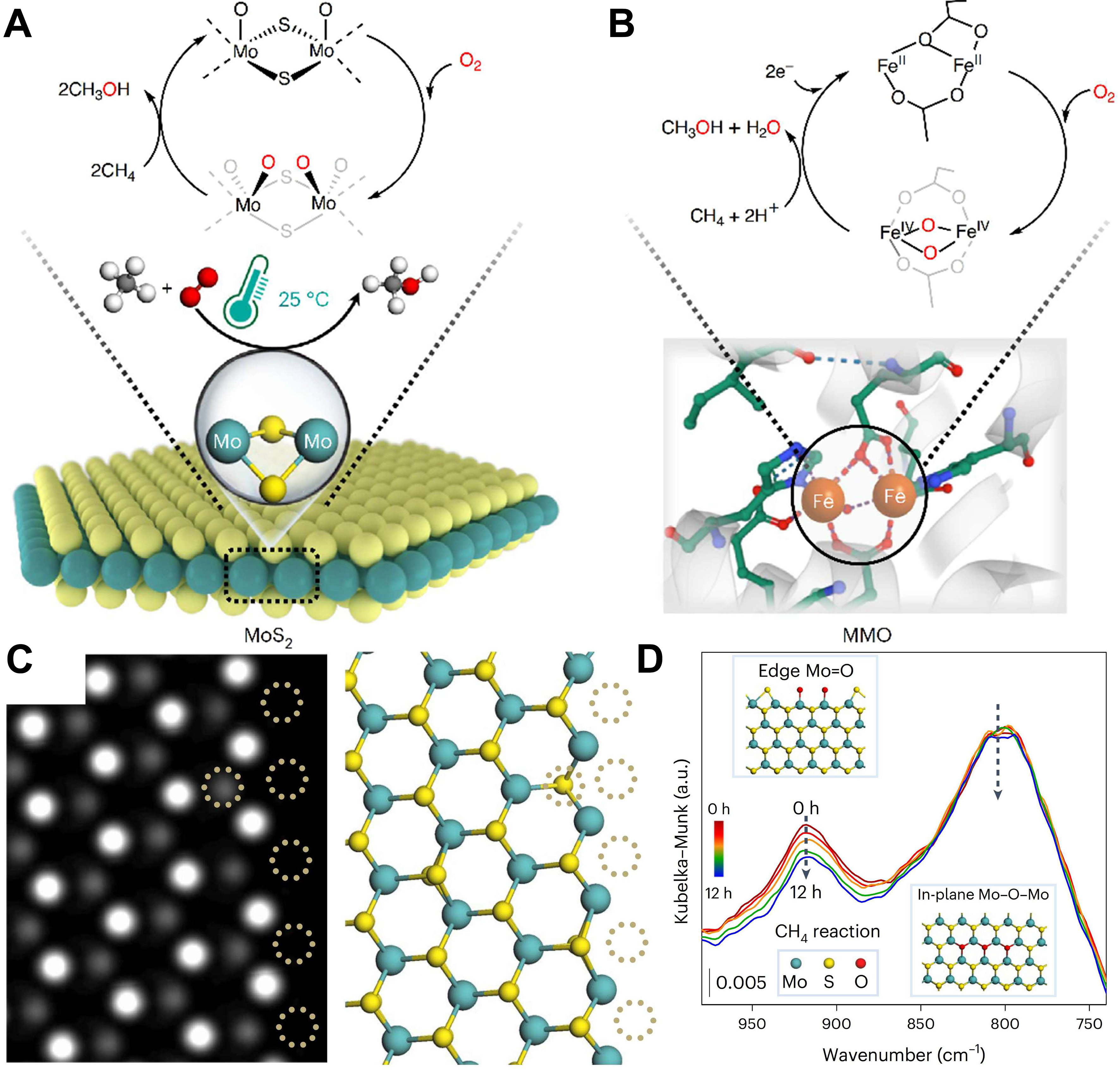
Figure 12. Room-temperature CH4 conversion by O2 over the MoS2 and MMO catalysts. Schematic illustration for the reaction cycle over the bi-Mo sites of SVs at the MoS2 edge (A) and the bi-Fe sites in the hydroxylase of MMO (Protein Data Bank 1FYZ) (B). Mo: cyan; S: yellow; O: red; C: grey (A) or green (B); H: white; Fe: orange; N: blue; (C) Simulated image and the corresponding atomic configurations of MoS2 showing the quintuple SVs at the edge denoted by dotted circles; (D) In situ DRIFTS spectra showing the variation of relative intensities of the Mo = O and Mo-O-Mo peaks when CH4 passes through the O2-pretreated and H2O-pretreated MoS2 catalyst at
Additionally, Lustemberg et al. demonstrated that water critically regulates SOM on Ni/CeO2[140]. SMSI and water confinement enhance CH4-to-CH3OH conversion. While bulk Ni surfaces remain inert, the Ni/CeO2(111) interface activates CH4 at 300 K and utilizes water to inhibit over-oxidation, enabling low-temperature (450 K) methanol production. This system exhibits superior CH3OH selectivity attributable to the reducible CeO2 support tuning the electronic structure and oxidation state of interfacial Ni sites.
H2O dissociation and CH4 activation are central to O2-assisted CH4-to-CH3OH conversion. At the Ni-CeO2 interface, SMSI promotes heterolytic H2O cleavage into H/OH, stabilizing OCH3 and inhibiting CO/CO2 formation - a mechanism distinct from Co-zeolites or CeO2/CuOx systems lacking similar water-mediated pathways. Selectivity and stability depend on balancing surface hydration, optimizing interfacial charge transfer, and tailoring OVs/hydroxyl groups. These insights highlight the potential of SMSI and solvent effects to direct methane partial oxidation, guiding the design of next-generation oxide-supported catalysts beyond conventional high-temperature or radical-based systems. Despite progress, industrial scalability challenges remain, particularly in stability and cost-effective synthesis. Advances in atomically precise catalysts (e.g., SACs) and operando studies are essential to decipher active-site dynamics and improve catalytic cycles. Integrating computational modeling (e.g., DFT, microkinetics) with experiment will accelerate the development of non-noble catalysts that combine enzymatic precision with industrial scalability. Recent studies also indicate that Zn-modified zeolites enable low-temperature co-activation of CH4 and CO2 for acetic acid synthesis[150]. Wu et al. engineered Zn/H-ZSM-5 to promote CH4-CO2 co-conversion at moderate temperatures (523-773 K), enabling selective CH3COOH formation via well-defined surface intermediates[151]. Advanced solid-state NMR identified Zn-methyl (-Zn-CH3) and carbonate species as key chemisorbed intermediates, highlighting Zn2+ Lewis acid sites in stabilizing reactive fragments while suppressing over-functionalization. This work advanced methods for probing transient active sites under operando conditions. Building on these insights, Wu et al. synthesized Fe-BN/ZSM-5 via calcination-impregnation, embedding binuclear Fe centers that achieved 89% total oxidation rate and 66% CH3COOH selectivity at 303 K[50]. The performance stems from unique [Fe(III)-(μ-O)2-Fe(III)-(OH)2] active sites
Figure 13. (A) Proposed reaction network for the CH3OH/CH3COOH formation, the calculated Gibbs reaction energy as well as the free-energy barriers; (B) Initial [Fe(III)-(-O)2-Fe(III)-(OH)2] model placed in the ZSM-5 zeolite pore[50]. Copyright 2022, Elsevier.
Wang et al. synthesized Fe/ZSM-5 via vacuum impregnation, with Fe/ZSM-5(0.25) achieving a record space-time yield of 12 mmol·gcat-1·h-1 and 63% CH3COOH selectivity at 323 K, attributed to enzyme-mimetic mononuclear Fe3+ sites[152]. The catalyst shows reduced Brønsted acidity vs. H-ZSM-5, indicating Fe anchored near Si-O-Al bridges forming Lewis acid centers. While questions remain regarding methyl radical dynamics and zeolite topology’s role in selectivity, oxygen’s function as the key oxidant is clear.
CONCLUSION AND OUTLOOK
This review systematically evaluates noble and non-noble metal catalysts for methane oxidation across diverse oxidant systems (H2O2, O2), correlating active site architectures with catalytic performance through advanced characterization and theoretical modeling. Critical insights into C–H bond activation mechanisms and product selectivity regulation are established, revealing that optimal catalytic efficiency hinges on balancing two fundamental challenges: (1) overcoming the kinetic inertia of methane C–H bond cleavage and (2) preventing overoxidation of oxygenated intermediates.
However, significant challenges remain. Scalability is limited by poor reproducibility and high costs in synthesizing atomically dispersed non-noble catalysts. While H2O2 enables low-temperature oxidation, its expense and handling risks reduce practicality; O2 faces mass-transfer constraints due to low CH4 solubility. Selectivity at high conversion is often overestimated, as over-oxidation intensifies under industrial conditions - exacerbated by dynamic active sites (e.g., Fe(II)/Fe(III) cycling generating nonselective ·OH). Stability issues include leaching, sintering, and support degradation (e.g., zeolite dealumination) for non-noble metals, and over-oxidation/poisoning for noble metals. Critically, mechanistic ambiguities persist regarding the true active sites in Fe/Cu-zeolites and the competition between H2O2 homolysis/heterolysis, hindering rational design.
Therefore, future efforts should focus on:
(1) Mechanistic elucidation
Advanced operando characterization techniques (synchronized in situ Raman, XAS, and DRIFTS) coupled with multiscale simulations are imperative to resolve dynamic active site evolution and transient intermediate stabilization. Emphasis should be placed on deciphering oxidant-specific activation pathways, particularly the divergent roles of O2-derived (O*, OOH*) vs. H2O2-generated (·OH) oxygen species in governing reaction selectivity.
(2) Catalyst design paradigms
Multinuclear Synergy: Engineered di-/polyatomic catalysts (e.g., Fe-Co, Cu-Mo) leveraging metal-metal electronic interactions could enhance C-H activation while suppressing CH3OH overoxidation. Confinement Engineering: Zeolitic frameworks and defect-rich supports (MXenes, doped MOFs) offer spatial control to stabilize reactive intermediates and modulate oxygen mobility. DAC: Photothermal/electrocatalytic systems integrating multiple activation modes (light, heat, voltage) may bypass thermodynamic limitations of purely thermal processes. Photocatalysis utilizes photogenerated electron-hole pairs to activate reactants: under light irradiation, semiconductor catalysts produce holes for CH4 activation, while electrons reduce O2 to ROS, facilitating reactions near ambient temperature. In contrast, electrochemical oxidation applies potential to precisely modulate catalyst valence states and surface adsorption, enabling on-demand generation of active oxygen species. Furthermore, photothermal/electro-thermal synergistic catalysis integrates multiple energy modes, often yielding a “1 + 1 > 2” effect. For instance, photothermal synergy simultaneously leverages photon energy to lower reaction barriers and thermal energy to enhance mass transfer, markedly increasing reaction rates - as demonstrated in O2 cleavage over Au/TiO2 via photothermal catalysis.
(3) Process innovation
Transition from H2O2-dependent systems to sustainable O2/H2O-based protocols requires bifunctional catalysts capable of in situ H2O2 generation and selective CH4 oxidation. Gas-phase tandem reactors with CO co-feeding could enable C-C coupling for C2+ products, contingent on precise control of CO oxidation kinetics.
(4) Industrial translation
The scalable synthesis of atomically dispersed catalysts requires breakthroughs in precursor engineering (e.g., sintering-resistant ligands) and support functionalization to address the reproducibility and economic challenges in the mass production of single-atom or bimetallic catalysts. Industrial feasibility hinges on achieving > 10% CH4 conversion with > 80% oxygenate selectivity under mild conditions (T < 473 K, P < 20 bar), necessitating reactor designs that enhance CH4 solubility and gas-liquid mass transfer. Conventional fixed-bed reactors are limited by mass transfer efficiency despite their operational simplicity. In contrast, continuously flowing slurry-bed or microchannel reactors, with superior mass and thermal management, represent a more promising industrial alternative. While noble metals (Pd, Rh) currently dominate low-temperature performance metrics, emerging Fe-/Cu-based systems exhibit comparable efficacy through optimized coordination environments. Bridging the gap between mechanistic understanding and practical implementation will require concerted efforts in catalyst informatics, machine learning-guided optimization, and cross-scale simulations. The ultimate goal remains the development of earth-abundant catalytic systems that reconcile activity, stability, and economic viability for decentralized methane valorization.
(5) Long-term stability and deactivation mechanisms
Catalyst durability remains a major obstacle for industrial methane oxidation. Noble metals (e.g., Pd, Pt) deactivate mainly through sintering, over-oxidation, and coking, while non-noble catalysts (e.g., Fe/ZSM-5) face leaching, aggregation, support degradation, and valence instability. Stabilization requires tailored strategies: confined supports and atmosphere control for noble metals, and enhanced metal-support interaction (e.g., M-O4 coordination), reduced acidity, hydrophobic coatings, and chemical looping for non-noble systems. Standardized stability testing is essential for assessing industrial viability.
DECLARATIONS
Acknowledgements
We acknowledge that the Graphic Abstract icons used in this paper are copyrighted by the following references. TOC-1, Copyright 2024, John Wiley and Sons, Reproduced with permission[130]; TOC-2, Copyright 2023, Springer Nature, Reproduced with permission[147]; TOC-3, Copyright 2018, Elsevier, Reproduced with permission[17]; TOC-4, Copyright 2024, American Chemical Society, Reproduced with permission[89]; TOC-5, Copyright 2023, Royal Society of Chemistry, Reproduced with permission[86]; TOC-6, Copyright 2022, Elsevier, Reproduced with permission[118]; TOC-7, Copyright 2025, American Chemical Society, Reproduced with permission.
Authors’ contributions
Topic proposal: Zhao, Z.; Li, W.
Manuscript preparation: Shen, H.
Collective discussion and revision: Li, W.; Shen, H.; Xie, Z.; Cai, J.; Sun, Y.; Wang, W.
Review and editing, methodology, supervision, project administration, funding acquisition: Zhao, Z.; Li, W.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (Nos. 22402131 and 92145301) and the Fundamental Research Funds for the Liaoning Universities (42400502105); Doctoral Research Initiation Project of Liaoning Province (2025-BS-0970); Liaoning Province international science and technology cooperation program project (2024JH2/102100004).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Xu, Z.; Park, E. D. Gas-phase selective oxidation of methane into methane oxygenates. Catalysts 2022, 12, 314.
2. Schwach, P.; Pan, X.; Bao, X. Direct conversion of methane to value-added chemicals over heterogeneous catalysts: challenges and prospects. Chem. Rev. 2017, 117, 8497-520.
3. Yu, X.; De, Waele. V.; Löfberg, A.; Ordomsky, V.; Khodakov, A. Y. Selective photocatalytic conversion of methane into carbon monoxide over zinc-heteropolyacid-titania nanocomposites. Nat. Commun. 2019, 10, 700.
4. Taifan, W.; Baltrusaitis, J. CH4 conversion to value added products: potential, limitations and extensions of a single step heterogeneous catalysis. Appl. Catal. B. Environ. 2016, 198, 525-47.
5. Horn, R.; Schlögl, R. Methane activation by heterogeneous catalysis. Catal. Lett. 2015, 145, 23-39.
6. Lunsford, J. H. Catalytic conversion of methane to more useful chemicals and fuels: a challenge for the 21st century. Catal. Today. 2000, 63, 165-74.
7. Hunter, E. P. L.; Lias, S. G. Evaluated gas phase basicities and proton affinities of molecules: an update. J. Phys. Chem. Ref. Data. 1998, 27, 413-656.
8. Hu, D.; Ordomsky, V. V.; Khodakov, A. Y. Major routes in the photocatalytic methane conversion into chemicals and fuels under mild conditions. Appl. Catal. B. Environ. 2021, 286, 119913.
9. Wu, S.; Tan, X.; Lei, J.; Chen, H.; Wang, L.; Zhang, J. Ga-doped and Pt-loaded porous TiO2-SiO2 for photocatalytic nonoxidative coupling of methane. J. Am. Chem. Soc. 2019, 141, 6592-600.
10. Tian, Y.; Piao, L.; Chen, X. Research progress on the photocatalytic activation of methane to methanol. Green. Chem. 2021, 23, 3526-41.
11. Shen, H.; Li, W.; Cai, J.; Qin, H.; Zhang, H.; Zhao, Z. Research progress in single-atom catalysts for the selective oxidation of methane. Sci. Sin. -Chim. 2024, 54, 309-37.
12. Guo, X.; Fang, G.; Li, G.; et al. Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. Science 2014, 344, 616-9.
13. Olivos-suarez, A. I.; Szécsényi, À.; Hensen, E. J. M.; Ruiz-martinez, J.; Pidko, E. A.; Gascon, J. Strategies for the direct catalytic valorization of methane using heterogeneous catalysis: challenges and opportunities. ACS. Catal. 2016, 6, 2965-81.
14. Snyder, B. E.; Vanelderen, P.; Bols, M. L.; et al. The active site of low-temperature methane hydroxylation in iron-containing zeolites. Nature 2016, 536, 317-21.
15. Ravi, M.; Sushkevich, V. L.; Knorpp, A. J.; et al. Misconceptions and challenges in methane-to-methanol over transition-metal-exchanged zeolites. Nat. Catal. 2019, 2, 485-94.
16. Hammond, C.; Forde, M. M.; Ab rahim, M. H.; et al. Direct catalytic conversion of methane to methanol in an aqueous medium by using copper-promoted Fe-ZSM-5. Angew. Chem. 2012, 124, 5219-23.
17. Cui, X.; Li, H.; Wang, Y.; et al. Room-temperature methane conversion by graphene-confined single iron atoms. Chem 2018, 4, 1902-10.
18. Gao, F.; Gao, S.; Meng, S. Screening single-atom catalysts for methane activation: α-Al2O3(0001) -supported Ni. Phys. Rev. Mater. 2017, 1, 035801.
19. Kiani, D.; Sourav, S.; Tang, Y.; Baltrusaitis, J.; Wachs, I. E. Methane activation by ZSM-5-supported transition metal centers. Chem. Soc. Rev. 2021, 50, 1251-68.
20. Kuai, L.; Chen, Z.; Liu, S.; et al. Titania supported synergistic palladium single atoms and nanoparticles for room temperature ketone and aldehydes hydrogenation. Nat. Commun. 2020, 11, 48.
21. Zhang, R.; Wang, H.; Tang, S.; et al. Photocatalytic oxidative dehydrogenation of ethane using CO2 as a soft oxidant over Pd/TiO2 catalysts to C2H4 and syngas. ACS. Catal. 2018, 8, 9280-6.
22. Wu, X.; Zhang, Q.; Li, W.; Qiao, B.; Ma, D.; Wang, S. L. Atomic-scale Pd on 2D Titania sheets for selective oxidation of methane to methanol. ACS. Catal. 2021, 11, 14038-46.
23. Hammer, B.; Nørskov, J. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211-20.
24. Vojvodic, A.; Medford, A. J.; Studt, F.; et al. Exploring the limits: a low-pressure, low-temperature Haber-Bosch process. Chem. Phys. Lett. 2014, 598, 108-12.
25. Kong, L.; Liang, X.; Wang, M.; Lawrence Wu, C. M. Role of transition metal d-orbitals in single-atom catalysts for nitric oxide electroreduction to ammonia. J. Colloid. Interface. Sci. 2023, 647, 375-83.
26. Chen, X.; Qin, X.; Jiao, Y.; et al. Structure-dependence and metal-dependence on atomically dispersed Ir catalysts for efficient n-butane dehydrogenation. Nat. Commun. 2023, 14, 2588.
27. Chan, S. I.; Lu, Y. J.; Nagababu, P.; et al. Efficient oxidation of methane to methanol by dioxygen mediated by tricopper clusters. Angew. Chem. Int. Ed. Engl. 2013, 52, 3731-5.
28. Xu, Y.; Wu, D.; Zhang, Q.; et al. Regulating Au coverage for the direct oxidation of methane to methanol. Nat. Commun. 2024, 15, 564.
29. Zhao, G.; Yang, F.; Chen, Z.; et al. Metal/oxide interfacial effects on the selective oxidation of primary alcohols. Nat. Commun. 2017, 8, 14039.
30. Huang, W.; Zhang, S.; Tang, Y.; et al. Low-temperature transformation of methane to methanol on Pd1O4 single sites anchored on the internal surface of microporous silicate. Angew. Chem. Int. Ed. Engl. 2016, 55, 13441-5.
31. Xie, J.; Jin, R.; Li, A.; et al. Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide-supported iron species. Nat. Catal. 2018, 1, 889-96.
32. Wu, B.; Yang, R.; Shi, L.; et al. Cu single-atoms embedded in porous carbon nitride for selective oxidation of methane to oxygenates. Chem. Commun. 2020, 56, 14677-80.
33. Ab Rahim, M. H.; Forde, M. M.; Jenkins, R. L.; et al. Oxidation of methane to methanol with hydrogen peroxide using supported gold-palladium alloy nanoparticles. Angew. Chem. Int. Ed. Engl. 2013, 52, 1280-4.
34. Hammond, C.; Jenkins, R. L.; Dimitratos, N.; et al. Catalytic and mechanistic insights of the low-temperature selective oxidation of methane over Cu-promoted Fe-ZSM-5. Chem. Eur. J. 2012, 18, 15735-45.
35. Mcvicker, R.; Agarwal, N.; Freakley, S. J.; et al. Low temperature selective oxidation of methane using gold-palladium colloids. Catal. Today. 2020, 342, 32-8.
36. He, Y.; Luan, C.; Fang, Y.; et al. Low-temperature direct conversion of methane to methanol over carbon materials supported Pd-Au nanoparticles. Catal. Today. 2020, 339, 48-53.
37. Kang, J.; Puthiaraj, P.; Ahn, W.; Park, E. D. Direct synthesis of oxygenates via partial oxidation of methane in the presence of O2 and H2 over a combination of Fe-ZSM-5 and Pd supported on an acid-functionalized porous polymer. Appl. Catal. A. Gen. 2020, 602, 117711.
38. Jin, Z.; Wang, L.; Zuidema, E.; et al. Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol. Science 2020, 367, 193-7.
39. Fan, Y.; Zhou, W.; Qiu, X.; et al. Selective photocatalytic oxidation of methane by quantum-sized bismuth vanadate. Nat. Sustain. 2021, 4, 509-15.
40. An, B.; Li, Z.; Wang, Z.; et al. Direct photo-oxidation of methane to methanol over a mono-iron hydroxyl site. Nat. Mater. 2022, 21, 932-8.
41. Luo, L.; Fu, L.; Liu, H.; et al. Synergy of Pd atoms and oxygen vacancies on In2O3 for methane conversion under visible light. Nat. Commun. 2022, 13, 2930.
42. Mahyuddin, M. H.; Shiota, Y.; Yoshizawa, K. Methane selective oxidation to methanol by metal-exchanged zeolites: a review of active sites and their reactivity. Catal. Sci. Technol. 2019, 9, 1744-68.
43. Agarwal, N.; Freakley, S. J.; McVicker, R. U.; et al. Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 2017, 358, 223-7.
44. Chen, J.; Wang, S.; Peres, L.; et al. Oxidation of methane to methanol over Pd@Pt nanoparticles under mild conditions in water. Catal. Sci. Technol. 2021, 11, 3493-500.
45. Zhu, K.; Liang, S.; Cui, X.; et al. Highly efficient conversion of methane to formic acid under mild conditions at ZSM-5-confined Fe-sites. Nano. Energy. 2021, 82, 105718.
46. Yang, N.; Ren, Z.; Yang, C.; Wu, P.; Zeng, G. Direct oxidation of CH4 to HCOOH over extra-framework stabilized Fe@MFI catalyst at low temperature. Fuel 2021, 305, 121624.
47. Tang, Y.; Li, Y.; Fung, V.; et al. Single rhodium atoms anchored in micropores for efficient transformation of methane under mild conditions. Nat. Commun. 2018, 9, 1231.
48. Wu, B.; Lin, T.; Huang, M.; et al. Tandem catalysis for selective oxidation of methane to oxygenates using oxygen over PdCu/zeolite. Angew. Chem. Int. Ed. Engl. 2022, 61, e202204116.
49. Wu, B.; Yin, H.; Ma, X.; et al. Highly selective synthesis of acetic acid from hydroxyl-mediated oxidation of methane at low temperatures. Angew. Chem. Int. Ed. Engl. 2025, 64, e202412995.
50. Wu, B.; Lin, T.; Lu, Z.; et al. Fe binuclear sites convert methane to acetic acid with ultrahigh selectivity. Chem 2022, 8, 1658-72.
51. Vadodaria, D. M.; Al-Fatesh, A. S.; Alrashed, M. M.; et al. A comprehensive review on catalytic oxidation of methane in the presence of molecular oxygen: total oxidation, partial oxidation and selective oxidation. Catal Rev. 2025:1-47.
52. Hargreaves, J. S. J.; Hutchings, G. J.; Joyner, R. W. Control of product selectivity in the partial oxidation of methane. Nature 1990, 348, 428-9.
53. Cui, X.; Huang, R.; Deng, D. Catalytic conversion of C1 molecules under mild conditions. EnergyChem 2021, 3, 100050.
54. Meng, X.; Cui, X.; Rajan, N. P.; Yu, L.; Deng, D.; Bao, X. Direct methane conversion under mild condition by thermo-, electro-, or photocatalysis. Chem 2019, 5, 2296-325.
55. Su, Y. Q.; Liu, J. X.; Filot, I. A. W.; Zhang, L.; Hensen, E. J. M. Highly active and stable CH4 oxidation by substitution of Ce4+ by two Pd2+ ions in CeO2(111). ACS. Catal. 2018, 8, 6552-9.
56. Conley, B. L.; Tenn, W. J.; Young, K. J.; et al. Design and study of homogeneous catalysts for the selective, low temperature oxidation of hydrocarbons. J. Mol. Catal. A. Chem. 2006, 251, 8-23.
57. Li, J.; Staykov, A.; Ishihara, T.; Yoshizawa, K. Theoretical study of the decomposition and hydrogenation of H2O2 on Pd and Au@Pd surfaces: understanding toward high selectivity of H2O2 synthesis. J. Phys. Chem. C. 2011, 115, 7392-8.
58. Jouny, M.; Hutchings, G. S.; Jiao, F. Carbon monoxide electroreduction as an emerging platform for carbon utilization. Nat. Catal. 2019, 2, 1062-70.
59. Liu, Z.; Huang, E.; Orozco, I.; et al. Water-promoted interfacial pathways in methane oxidation to methanol on a CeO2-Cu2O catalyst. Science 2020, 368, 513-7.
60. Narsimhan, K.; Iyoki, K.; Dinh, K.; Román-Leshkov, Y. Catalytic oxidation of methane into methanol over copper-exchanged zeolites with oxygen at low temperature. ACS. Cent. Sci. 2016, 2, 424-9.
61. Zhang, H.; Ji, Y.; Xu, Y.; et al. Recent advance of atomically dispersed catalysts for direct methane oxidation under mild aqueous conditions. Mater. Today. Sustain. 2023, 22, 100351.
62. Stakheev, A. Y.; Batkin, A. M.; Teleguina, N. S.; et al. Particle size effect on CH4 oxidation over noble metals: comparison of Pt and Pd catalysts. Top. Catal. 2013, 56, 306-10.
63. Murata, K.; Kosuge, D.; Ohyama, J.; et al. Exploiting metal-support interactions to tune the redox properties of supported Pd catalysts for methane combustion. ACS. Catal. 2020, 10, 1381-7.
64. Senftle, T. P.; van Duin, A. C. T.; Janik, M. J. Role of site stability in methane activation on PdxCe1-xOδ surfaces. ACS. Catal. 2015, 5, 6187-99.
65. Chen, S.; Li, S.; You, R.; et al. Elucidation of active sites for CH4 catalytic oxidation over Pd/CeO2 via tailoring metal-support interactions. ACS. Catal. 2021, 11, 5666-77.
66. Duan, H.; You, R.; Xu, S.; et al. Pentacoordinated Al3+ -stabilized active Pd structures on Al2O3 -coated palladium catalysts for methane combustion. Angew. Chem. Int. Ed. Engl. 2019, 58, 12043-8.
67. Xiong, H.; Kunwar, D.; Jiang, D.; et al. Engineering catalyst supports to stabilize PdOx two-dimensional rafts for water-tolerant methane oxidation. Nat. Catal. 2021, 4, 830-9.
68. Li, C.; Tang, B.; Ogunbiyi, A. T.; et al. The effects of facet-dependent palladium-titania interactions on the activity of Pd/Rutile catalysts for lean methane oxidation. Mol. Catal. 2022, 528, 112475.
69. Xu, W.; Liu, H. X.; Hu, Y.; et al. Metal-Oxo electronic tuning via in situ CO decoration for promoting methane conversion to oxygenates over single-atom catalysts. Angew. Chem. Int. Ed. Engl. 2024, 63, e202315343.
70. Serra-maia, R.; Michel, F. M.; Kang, Y.; Stach, E. A. Decomposition of hydrogen peroxide catalyzed by AuPd nanocatalysts during methane oxidation to methanol. ACS. Catal. 2020, 10, 5115-23.
71. Ab Rahim, M. H.; Forde, M. M.; Hammond, C.; et al. Systematic study of the oxidation of methane using supported gold palladium nanoparticles under mild aqueous conditions. Top. Catal. 2013, 56, 1843-57.
72. Yu, B.; Cheng, L.; Dai, S.; et al. Silver and copper dual single atoms boosting direct oxidation of methane to methanol via synergistic catalysis. Adv. Sci. 2023, 10, e2302143.
73. Sushkevich, V. L.; Verel, R.; van, Bokhoven. J. A. Pathways of methane transformation over copper‐exchanged mordenite as revealed by in situ NMR and IR spectroscopy. Angew. Chem. Int. Ed. Engl. 2020, 59, 910-8.
74. Meyet, J.; Searles, K.; Newton, M. A.; et al. Monomeric copper(II) sites supported on alumina selectively convert methane to methanol. Angew. Chem. Int. Ed. Engl. 2019, 58, 9841-5.
75. Fang, G.; Wei, F.; Lin, J.; et al. Retrofitting Zr-Oxo nodes of UiO-66 by Ru single atoms to boost methane hydroxylation with nearly total selectivity. J. Am. Chem. Soc. 2023, 145, 13169-80.
76. Jacobs, A. B.; Banerjee, R.; Deweese, D. E.; et al. Nuclear resonance vibrational spectroscopic definition of the Fe(IV)2 intermediate Q in methane monooxygenase and its reactivity. J. Am. Chem. Soc. 2021, 143, 16007-29.
77. Schulz, C. E.; Castillo, R. G.; Pantazis, D. A.; DeBeer, S.; Neese, F. Structure-spectroscopy correlations for intermediate Q of soluble methane monooxygenase: insights from QM/MM calculations. J. Am. Chem. Soc. 2021, 143, 6560-77.
78. Koo, C. W.; Tucci, F. J.; He, Y.; Rosenzweig, A. C. Recovery of particulate methane monooxygenase structure and activity in a lipid bilayer. Science 2022, 375, 1287-91.
79. Xie, P.; Ding, J.; Yao, Z.; et al. Oxo dicopper anchored on carbon nitride for selective oxidation of methane. Nat. Commun. 2022, 13, 1375.
80. Palagin, D.; Sushkevich, V. L.; van, Bokhoven. J. A. Water molecules facilitate hydrogen release in anaerobic oxidation of methane to methanol over Cu/Mordenite. ACS. Catal. 2019, 9, 10365-74.
81. Yang, N.; Zhao, Y.; Wu, P.; et al. Defective C3N4 frameworks coordinated diatomic copper catalyst: towards mild oxidation of methane to C1 oxygenates. Appl. Catal. B. Environ. 2021, 299, 120682.
82. Cheng, Q.; Li, G.; Yao, X.; et al. Maximizing active Fe species in ZSM-5 zeolite using organic-template-free synthesis for efficient selective methane oxidation. J. Am. Chem. Soc. 2023, 145, 5888-98.
83. Xie, B.; Zhang, H.; Yang, C.; et al. Seed-directed synthesis of zeolites with enhanced performance in the absence of organic templates. Chem. Commun. 2011, 47, 3945-7.
84. Hammond, C.; Dimitratos, N.; Lopez-sanchez, J. A.; et al. Aqueous-phase methane oxidation over Fe-MFI zeolites; promotion through isomorphous framework substitution. ACS. Catal. 2013, 3, 1835-44.
85. Yu, T.; Li, Z.; Jones, W.; et al. Identifying key mononuclear Fe species for low-temperature methane oxidation. Chem. Sci. 2021, 12, 3152-60.
86. Yu, T.; Li, Z.; Lin, L.; et al. Highly selective oxidation of methane into methanol over Cu-promoted monomeric Fe/ZSM-5. ACS. Catal. 2021, 11, 6684-91.
87. Li, W.; Ren, Y.; Xie, Z.; et al. TiO2 nanofiber-supported copper nanoparticle catalysts for highly efficient methane conversion to C1 oxygenates under mild conditions. Nano. Res. 2024, 17, 3844-52.
88. Gao, J.; Zheng, Y.; Tang, Y.; et al. Spectroscopic and computational study of Cr oxide structures and their anchoring sites on ZSM-5 zeolites. ACS. Catal. 2015, 5, 3078-92.
89. Shen, Q.; Cao, C.; Huang, R.; et al. Single chromium atoms supported on Titanium dioxide nanoparticles for synergic catalytic methane conversion under mild conditions. Angew. Chem. Int. Ed. Engl. 2020, 59, 1216-9.
90. Zeng, M.; Cheng, L.; Gu, Q.; et al. ZSM-5-confined Cr1-O4 active sites boost methane direct oxidation to C1 oxygenates under mild conditions. EES. Catal. 2023, 1, 153-61.
91. Li, W.; Li, Z.; Zhang, H.; et al. Efficient catalysts of surface hydrophobic Cu-BTC with coordinatively unsaturated Cu(I) sites for the direct oxidation of methane. Proc. Natl. Acad. Sci. U. S. A. 2023, 120, e2206619120.
92. Li, W.; Li, Z.; Xie, Z.; et al. Surface hydrophobic MIL-100(Fe) MOFs to boost methane oxidation with nearly total selectivity to C1 oxygenates under mild conditions. J. Catal. 2024, 429, 115243.
93. Li, W.; Li, Z.; Shen, H.; et al. Nitrogen vacancy-rich C3Nx-confined Fe-Cu diatomic catalysts for the direct selective oxidation of methane at low temperature. ACS. Catal. 2024, 14, 10689-700.
94. Grundner, S.; Markovits, M. A.; Li, G.; et al. Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat. Commun. 2015, 6, 7546.
95. Hutchings, G. J.; Scurrell, M. S.; Woodhouse, J. R. Oxidative coupling of methane using oxide catalysts. Chem. Soc. Rev. 1989, 18, 251.
96. Liang, Z.; Li, T.; Kim, M.; Asthagiri, A.; Weaver, J. F. Low-temperature activation of methane on the IrO2(110) surface. Science 2017, 356, 299-303.
97. Tomkins, P.; Ranocchiari, M.; van, Bokhoven. J. A. Direct conversion of methane to methanol under mild conditions over Cu-zeolites and beyond. Acc. Chem. Res. 2017, 50, 418-25.
98. Latimer, A. A.; Kulkarni, A. R.; Aljama, H.; et al. Understanding trends in C–H bond activation in heterogeneous catalysis. Nat. Mater. 2017, 16, 225-9.
99. Senanayake, S. D.; Rodriguez, J. A.; Weaver, J. F. Low temperature activation of methane on metal-oxides and complex interfaces: insights from surface science. Acc. Chem. Res. 2020, 53, 1488-97.
100. Shan, J.; Li, M.; Allard, L. F.; Lee, S.; Flytzani-Stephanopoulos, M. Mild oxidation of methane to methanol or acetic acid on supported isolated rhodium catalysts. Nature 2017, 551, 605-8.
101. Gu, F.; Qin, X.; Li, M.; et al. Selective catalytic oxidation of methane to methanol in aqueous medium over copper cations promoted by atomically dispersed rhodium on TiO2. Angew. Chem. Int. Ed. Engl. 2022, 61, e202201540.
102. Groothaert, M. H.; Smeets, P. J.; Sels, B. F.; Jacobs, P. A.; Schoonheydt, R. A. Selective oxidation of methane by the bis(mu-oxo)dicopper core stabilized on ZSM-5 and mordenite zeolites. J. Am. Chem. Soc. 2005, 127, 1394-5.
103. Woertink, J. S.; Smeets, P. J.; Groothaert, M. H.; et al. A [Cu2O]2+ core in Cu-ZSM-5, the active site in the oxidation of methane to methanol. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18908-13.
104. Zhou, W.; Qiu, X.; Jiang, Y.; et al. Highly selective aerobic oxidation of methane to methanol over gold decorated zinc oxide via photocatalysis. J. Mater. Chem. A. 2020, 8, 13277-84.
105. An, B.; Zhang, Q. H.; Zheng, B. S.; et al. Sulfone-decorated conjugated organic polymers activate oxygen for photocatalytic methane conversion. Angew. Chem. Int. Ed. Engl. 2022, 61, e202204661.
106. Song, H.; Huang, H.; Meng, X.; et al. Atomically dispersed nickel anchored on a nitrogen-doped carbon/TiO2 composite for efficient and selective photocatalytic CH4 oxidation to oxygenates. Angew. Chem. Int. Ed. Engl. 2023, 62, e202215057.
107. Fang, G.; Yu, W.; Wang, X.; Lin, J. Heterogeneous catalysis of methane hydroxylation with nearly total selectivity under mild conditions. Chem. Commun. 2024, 60, 11034-51.
108. Qi, G.; Davies, T. E.; Nasrallah, A.; et al. Au-ZSM-5 catalyses the selective oxidation of CH4 to CH3OH and CH3COOH using O2. Nat. Catal. 2022, 5, 45-54.
109. Zhou, Q.; Tan, X.; Wang, X.; et al. Selective photocatalytic oxidation of methane to methanol by constructing a rapid O2 conversion pathway over Au-Pd/ZnO. ACS. Catal. 2024, 14, 955-64.
110. Wang, L.; Jin, J.; Li, W.; et al. Highly selective catalytic oxidation of methane to methanol using Cu-Pd/anatase. Energy. Environ. Sci. 2024, 17, 9122-33.
111. Wang, S.; Fung, V.; Hülsey, M. J.; et al. H2-reduced phosphomolybdate promotes room-temperature aerobic oxidation of methane to methanol. Nat. Catal. 2023, 6, 895-905.
112. Yu, B.; Cheng, L.; Wu, J.; et al. Surface hydroxyl group dominating aerobic oxidation of methane below room temperature. Energy. Environ. Sci. 2024, 17, 8127-39.
113. Yang, B.; Gong, X. Q.; Wang, H. F.; Cao, X. M.; Rooney, J. J.; Hu, P. Evidence to challenge the universality of the Horiuti-Polanyi mechanism for hydrogenation in heterogeneous catalysis: origin and trend of the preference of a non-Horiuti-Polanyi mechanism. J. Am. Chem. Soc. 2013, 135, 15244-50.
114. Srivastava, R. K.; Sarangi, P. K.; Bhatia, L.; Singh, A. K.; Shadangi, K. P. Conversion of methane to methanol: technologies and future challenges. Biomass. Conv. Bioref. 2022, 12, 1851-75.
115. Wang, V. C.; Maji, S.; Chen, P. P.; Lee, H. K.; Yu, S. S.; Chan, S. I. Alkane oxidation: methane monooxygenases, related enzymes, and their biomimetics. Chem. Rev. 2017, 117, 8574-621.
116. Yang, L.; Huang, J.; Ma, R.; You, R.; Zeng, H.; Rui, Z. Metal-organic framework-derived IrO2/CuO catalyst for selective oxidation of methane to methanol. ACS. Energy. Lett. 2019, 4, 2945-51.
117. Sushkevich, V. L.; Palagin, D.; Ranocchiari, M.; van, Bokhoven. J. A. Selective anaerobic oxidation of methane enables direct synthesis of methanol. Science 2017, 356, 523-7.
118. Lin, J.; Wang, A.; Qiao, B.; et al. Remarkable performance of Ir1/FeO(x) single-atom catalyst in water gas shift reaction. J. Am. Chem. Soc. 2013, 135, 15314-7.
119. Hwang, D. Y.; Suh, D. H. Evolution of a high local strain in rolling up MoS2 sheets decorated with Ag and Au nanoparticles for surface-enhanced Raman scattering. Nanotechnology 2017, 28, 025603.
120. Wang, F.; Kusada, K.; Wu, D.; et al. Solid-solution alloy nanoparticles of the immiscible iridium-copper system with a wide composition range for enhanced electrocatalytic applications. Angew. Chem. Int. Ed. Engl. 2018, 57, 4505-9.
121. Arndtsen, B. A.; Bergman, R. G. Unusually mild and selective hydrocarbon C–H bond activation with positively charged Iridium(III) complexes. Science 1995, 270, 1970-3.
122. Zhao, E.; Chen, Y.; Xu, J.; Ma, J.; Liu, D.; Xiong, Y. Efficient photocatalytic methane conversion to oxygenates over TiO2 and Pd co-modified titanium silicalite zeolite. Chem. Synth. 2025, 5, 63.
123. Zhong, M.; Xu, Y.; Li, J.; et al. Engineering PdAu nanowires for highly efficient direct methane conversion to methanol under mild conditions. J. Phys. Chem. C. 2021, 125, 12713-20.
124. Ab, Rahim. M. H.; Armstrong, R. D.; Hammond, C.; et al. Low temperature selective oxidation of methane to methanol using titania supported gold palladium copper catalysts. Catal. Sci. Technol. 2016, 6, 3410-8.
125. Edwards, J. K.; Ntainjua, N. E.; Carley, A. F.; Herzing, A. A.; Kiely, C. J.; Hutchings, G. J. Direct synthesis of H2O2 from H2 and O2 over gold, palladium, and gold-palladium catalysts supported on acid-pretreated TiO2. Angew. Chem. Int. Ed. 2009, 48, 8512-5.
126. Luo, L.; Luo, J.; Li, H.; et al. Water enables mild oxidation of methane to methanol on gold single-atom catalysts. Nat. Commun. 2021, 12, 1218.
127. Li, M.; Zhao, Z.; Cheng, T.; et al. Ultrafine jagged platinum nanowires enable ultrahigh mass activity for the oxygen reduction reaction. Science 2016, 354, 1414-9.
128. Xu, Y.; Wu, D.; Deng, P.; et al. Au decorated Pd nanowires for methane oxidation to liquid C1 products. Appl. Catal. B. Environ. 2022, 308, 121223.
129. Wang, L.; Zeng, Z.; Gao, W.; et al. Tunable intrinsic strain in two-dimensional transition metal electrocatalysts. Science 2019, 363, 870-4.
130. Luo, M.; Zhao, Z.; Zhang, Y.; et al. PdMo bimetallene for oxygen reduction catalysis. Nature 2019, 574, 81-5.
131. Bai, S.; Liu, F.; Huang, B.; et al. High-efficiency direct methane conversion to oxygenates on a cerium dioxide nanowires supported rhodium single-atom catalyst. Nat. Commun. 2020, 11, 954.
132. Wang, W.; Zhou, W.; Tang, Y.; et al. Selective oxidation of methane to methanol over Au/H-MOR. J. Am. Chem. Soc. 2023, 145, 12928-34.
133. Bongiorno, A.; Landman, U. Water-enhanced catalysis of CO oxidation on free and supported gold nanoclusters. Phys. Rev. Lett. 2005, 95, 106102.
134. Ketchie, W. C.; Murayama, M.; Davis, R. J. Promotional effect of hydroxyl on the aqueous phase oxidation of carbon monoxide and glycerol over supported Au catalysts. Top. Catal. 2007, 44, 307-17.
135. Jin, R.; Peng, M.; Li, A.; et al. Low temperature oxidation of ethane to oxygenates by oxygen over iridium-cluster catalysts. J. Am. Chem. Soc. 2019, 141, 18921-5.
136. Moteki, T.; Tominaga, N.; Ogura, M. Mechanism investigation and product selectivity control on CO-assisted direct conversion of methane into C1 and C2 oxygenates catalyzed by zeolite-supported Rh. Appl. Catal. B. Environ. 2022, 300, 120742.
137. Moteki, T.; Tominaga, N.; Ogura, M. CO-assisted direct methane conversion into C1 and C2 oxygenates over ZSM-5 supported transition and platinum group metal catalysts using oxygen as an oxidant. ChemCatChem 2020, 12, 2957-61.
138. Cheung, P.; Bhan, A.; Sunley, G.; Law, D.; Iglesia, E. Site requirements and elementary steps in dimethyl ether carbonylation catalyzed by acidic zeolites. J. Catal. 2007, 245, 110-23.
139. Mao, J.; Liu, H.; Cui, X.; et al. Direct conversion of methane with O2 at room temperature over edge-rich MoS2. Nat. Catal. 2023, 6, 1052-61.
140. Lustemberg, P. G.; Palomino, R. M.; Gutiérrez, R. A.; et al. Direct conversion of methane to methanol on Ni-Ceria surfaces: metal-support interactions and water-enabled catalytic conversion by site blocking. J. Am. Chem. Soc. 2018, 140, 7681-7.
141. Vanelderen, P.; Vancauwenbergh, J.; Sels, B. F.; Schoonheydt, R. A. Coordination chemistry and reactivity of copper in zeolites. Coord. Chem. Rev. 2013, 257, 483-94.
142. Narsimhan, K.; Michaelis, V. K.; Mathies, G.; Gunther, W. R.; Griffin, R. G.; Román-Leshkov, Y. Methane to acetic acid over Cu-exchanged zeolites: mechanistic insights from a site-specific carbonylation reaction. J. Am. Chem. Soc. 2015, 137, 1825-32.
143. Blasco, T.; Boronat, M.; Concepción, P.; Corma, A.; Law, D.; Vidal-Moya, J. A. Carbonylation of methanol on metal-acid zeolites: evidence for a mechanism involving a multisite active center. Angew. Chem. Int. Ed. Engl. 2007, 46, 3938-41.
144. Li, B.; Xu, J.; Han, B.; et al. Insight into dimethyl ether carbonylation reaction over mordenite zeolite from in-situ solid-state NMR spectroscopy. J. Phys. Chem. C. 2013, 117, 5840-7.
145. Heyer, A. J.; Plessers, D.; Braun, A.; et al. Methane activation by a mononuclear copper active site in the zeolite mordenite: effect of metal nuclearity on reactivity. J. Am. Chem. Soc. 2022, 144, 19305-16.
146. Brezicki, G.; Kammert, J. D.; Gunnoe, T. B.; Paolucci, C.; Davis, R. J. Insights into the speciation of Cu in the Cu-H-mordenite catalyst for the oxidation of methane to methanol. ACS. Catal. 2019, 9, 5308-19.
147. Rhoda, H. M.; Plessers, D.; Heyer, A. J.; et al. Spectroscopic definition of a highly reactive site in Cu-CHA for selective methane oxidation: tuning a Mono-μ-Oxo dicopper(II) active site for reactivity. J. Am. Chem. Soc. 2021, 143, 7531-40.
148. Dinh, K. T.; Sullivan, M. M.; Narsimhan, K.; et al. Continuous partial oxidation of methane to methanol catalyzed by diffusion-paired copper dimers in copper-exchanged zeolites. J. Am. Chem. Soc. 2019, 141, 11641-50.
149. Tomkins, P.; Mansouri, A.; Bozbag, S. E.; et al. Isothermal cyclic conversion of methane into methanol over copper-exchanged zeolite at low temperature. Angew. Chem. Int. Ed. Engl. 2016, 55, 5467-71.
150. Snyder, B. E. R.; Bols, M. L.; Schoonheydt, R. A.; Sels, B. F.; Solomon, E. I. Iron and copper active sites in zeolites and their correlation to metalloenzymes. Chem. Rev. 2018, 118, 2718-68.
151. Wu, J. F.; Yu, S. M.; Wang, W. D.; et al. Mechanistic insight into the formation of acetic acid from the direct conversion of methane and carbon dioxide on zinc-modified H-ZSM-5 zeolite. J. Am. Chem. Soc. 2013, 135, 13567-73.
152. Wang, C.; Sun, Y.; Wang, L.; et al. Oxidative carbonylation of methane to acetic acid on an Fe-modified ZSM-5 zeolite. Appl. Catal. B. Environ. 2023, 329, 122549.
153. Balasubramanian, R.; Smith, S. M.; Rawat, S.; Yatsunyk, L. A.; Stemmler, T. L.; Rosenzweig, A. C. Oxidation of methane by a biological dicopper centre. Nature 2010, 465, 115-9.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].