





Cancer stem cells, plasticity, and drug resistance

Abstract
Melanoma is a highly aggressive tumor and almost always fatal when metastatic. Herein, we discuss recent findings on the mechanisms of resistance of human cutaneous melanoma. To achieve a precision medicine approach, the heterogeneity and plasticity of tumor cells are two crucial aspects to be investigated in depth. In fact, to understand the mechanisms that cells use to acquire a resistant phenotype after chemotherapy or how resistant cells inside the tumor are selected, it is the most important issue for a successful therapy. Since new therapeutic strategies are trying to go in this direction, we discuss here the state of the art of the research and the clinical impact of these strategies. We also discuss and suggest future research directions to develop approaches able to define the best concentration and time of exposure of the drug or the cocktails of drugs for each specific patient based on his/her biological features.
Keywords
Current therapeutic strategies for the treatment of human cutaneous melanoma
Melanoma arises from mutated melanocytes, the pigment producing cells. Although melanoma is a rare tumor, occurring in about 1% of all skin malignant tumors, it represents almost 2% of all cancer deaths worldwide[1]; the survival rate is strictly related to the stage of the tumor and thus to the ability to perform an early diagnosis[2,3][Figure 1]. Furthermore, the age-adjusted rate of new cases reported in the USA between 1999 and 2016 shows an important increase of new cases of melanoma per year with respect to others kinds of cancer such as lung, breast, and colon cancers [Figure 1]. The overall survival is higher in the case of localized disease, but patients with metastatic melanoma show a very poor prognosis, with a median survival rate ranging 3-6 months[2-4]. While low-grade primary tumors are usually successfully treated by surgical excision, systemic treatment of advanced metastatic disease treated with chemotherapy shows a low response rate and generally no overall survival rate improvement[5].
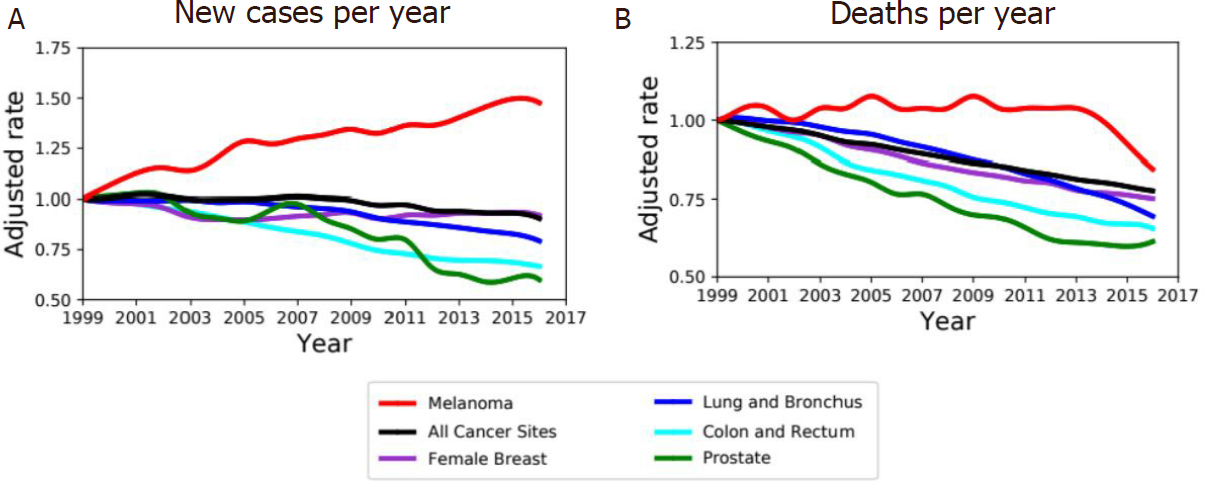
Figure 1. Melanoma incidence and death rate. A: age-adjusted rate of new cancer cases diagnosed in USA between 1999 and 2016 normalized over corresponding rate in 1999 for both sexes. Incidence is increasing in melanoma (red) compared to other type of tumors (colors as in legend). B: Age-adjusted death rate, for both sexes, between 1999 and 2016 in USA normalized over corresponding rate in 1999 for melanoma and top-rated cancers by rates of cancer deaths
Cutaneous melanoma is characterized by a series of peculiar somatic genetic alterations, many of which are common in others tumors such as genes responsible for the control of cell cycle and proliferation, metabolism, growth, and apoptosis that typically lead to the deregulation of mitogen-activated protein kinase (MAPK) and the phosphoinositol-3-kinase (PI3K)/AKT pathways[6,7]. The most frequently mutated gene is BRAF, and in particular the missense mutation V600E is typical of this kind of tumor since it is the most frequent mutation occurring in melanoma[8-10] while NRAS activating mutations have been detected in a small percentage of these tumor cases[8,11]. BRAF is a serine-threonine kinase involved in the RAF/MEK/MAPK pathway controlling through ERK1/2 cellular proliferation, survival, and differentiation[9]. Notably, NRAS and BRAF mutations are generally mutually exclusive; only in a minor proportion of patients the coexistence of both genetic alterations is reported[9,12]. According to Genomic Data Commons portal data, mutations on BRAF and NRAS genes taken together affect about 10% of all the considered cancers but are detected in about 52% and 27%, respectively, of the melanoma patients in this cohort. Additionally, genetic alterations of telomerase reverse transcriptase promoter (TERT) and cyclin dependent kinase inhibitor 2A (CDK2A) or phosphatase and tensin homolog (PTEN) loss-of-function have been frequently observed in advanced melanoma[13-18].
Current therapeutic approaches for cutaneous melanoma include surgical resection, chemotherapy, photodynamic therapy, immunotherapy, biochemotherapy, and targeted therapy, depending on the features of the tumor such as its localization, stage, and genetic profile. Chemotherapy combinations have been shown to improve the clinical response, but the overall survival does not change significantly[20]. Dacarbazine, approved in 1974 by Federal and Drug Administration (FDA), is the standard drug used for metastatic melanoma. Temozolomide, which is an oral pro-drug of the active metabolite of dacarbazine, is used in advanced melanoma and it seems to improve the median progression-free survival but not the overall survival[21,22]. Electrochemotherapy is a technique that combines the use of cytotoxic drugs such as bleomycin and cisplatin with high-intensity electric pulses, which facilitate the drug delivery inside the cells[23,24]. Light-based therapy is a promising adjuvant therapy useful for palliative treatment in advanced metastatic melanomas[25].
Immunotherapy is mainly based on the frequent presence of chronic inflammation and of immune cells inside the tumor[26]. The possibility to target the immunogenic tumor microenvironment is nowadays one of the more promising strategies for a successful cancer treatment. Regarding cutaneous melanoma, there are immunotherapies approved by FDA (e.g., nivolumab, pembrolizumab, and gp100 vaccine). Nivolumab and pembrolizumab, approved for the treatment of metastatic melanoma, re anti-Programmed cell Death protein (PD1) antibodies that block the interaction between PD-1, which is a membrane antigen, and its ligand programmed death-ligand 1 (PD-L1)/PD-L2. The blockade of the interaction between this ligand and its receptor induces antitumor activity, showing a reduction of tumor progression through the modulation of the immune system[27]. Another interesting drug is ipilimumab, which is an anti-cytotoxic T-lymphocyte-associated protein 4 antibody that acts as a receptor antagonist, enhancing pro-inflammatory T-cell cytokine production and promoting clonal T-cell expansion[28,29]. In Figure 2, we report a scheme of the pathways on which these drugs work.

Figure 2. A: tumor target therapy. Simplified schematic of the key molecular component of MAPK and PI3K/Akt signaling pathway related to melanoma tumorigenesis and targeted inhibitors of representative drugs and therapies with the main resistant mechanisms. B: immunotherapy. The target of the tumor cell environment, the most important drugs, and the main resistance mechanisms. HLA: human leukocyte antigen
Gp100 is a glycoprotein expressed by melanoma cells with few exceptions (healthy epidermal melanocytes and retina) and it is recognized by cytotoxic T cells (CTL). The administration of gp100 epitopes enhances CTLs activity; however, it is reported to have limited clinical benefits and it is used as adjuvant therapy only[30].
Biochemotherapy is a combination of chemotherapy and immunotherapy. In fact, some conventional chemotherapies may act partially through immune-stimulatory mechanisms[31]. The most common use of biochemotherapy is the combination of dacarbazin, cisplatin, and vinblastine with interleukine-2 (IL-2) and interferon (IFN)α-2b as immunoregulators.
Most cutaneous melanomas are treated with targeted therapy, since about 70% of these tumors express specific mutations related to key signaling pathways (e.g., BRAF V600E) [Figure 2]. Targeted therapy by using small molecule inhibitors or antibodies affecting those mutated proteins which play a critical role in the progression of the tumor [Figure 2] are discussed in the next section.
Genetic and epigenetic mechanisms of resistance
Melanoma is a highly resistant tumor. The appearance of resistance after chemotherapy or the presence of intrinsic resistance leads to great difficulties in devising an effective and durable therapy and, finally, to a poor survival of the patients, in particular when they are already metastatic. In recent years, many studies were designed with the aim to understand the molecular basis of resistance. We herein discuss the main biological mechanisms displayed by melanoma to become resistant to current therapies.
Treatment of advanced BRAF V600E mutant melanoma using a BRAF inhibitor or its combination with a MEK inhibitor typically elicits only partial response. It has been reported that new genetic alterations arise in patients carrying BRAF mutation when treated with anti-BRAF antibody as well as in patients displaying both BRAF and MEK mutations and treated with inhibitors for both factors[32,33]. In particular, it has been reported that one of the main mechanisms of resistance is the reactivation of MAPK signaling[33]. Moreover, in a recent paper, a comparison between the transcriptomes of melanoma patient-derived tumors regressing after MAPK inhibitor (MAPKi) treatment with respect to MAPKi-induced temporal transcriptomic states showed that residual melanoma on MAPKi therapy displays adaptive transcriptomic, epigenomic, and immune-regulomic alterations[34].
Hannan and coworkers observed that an incorrect analysis of the melanoma polyclonal population affects the choice of the therapy[35]. This aspect is relevant for melanoma since drug therapy is usually applied when the disease is in advanced state[2]. The deletion or loss of function of PTEN is also quite common in drug-resistant melanoma, reactivating PI3K/ATK pathway in a MAPK-independent manner[36-38]. On the other hand, transient resistance can be induced by compensatory changes in gene expression such as the upregulation of the receptor of tyrosine kinases, the overexpression of CRAF, or the amplification or truncation of BRAF gene[37,39-41].
The high heterogeneity of the tumor cells and their plasticity lead to the possibility that the same drug might induce the switch to slow-cycling resistant phenotype associated to high melanocyte inducing transcription factor levels and to a mesenchymal-like phenotype[42-46]. Early adaptation involving transcriptome reprogramming seems to be particularly relevant even at long time scale, allowing the tumor to survive until a genetic mutation and permanent resistance mechanism is acquired[43,47]. Interestingly, melanoma cells can display profound transcriptional variability at the level of single cell that can involve the transcription of a number of resistance markers at high level in a very small percentage of cells[48]. The presence of a drug can, therefore, induce an epigenetic reprogramming in these cells, converting the transient transcriptional state into a stable one[48].
Other important actors of drug resistance in melanoma are non-coding RNAs[49-51]. In this context, the use of combined and coadjuvant therapies has been proposed to avoid successive treatment failures due to the acquisition of a cross-resistance or changes in tumor environment[52-57]. Moreover, the tumor niche can play an important role and a long-term success of targeted therapies seems to be strictly related to a favorable microenvironment and immunologic signature[54,58,59]. In this connection, a recent paper shows that the development of drug resistance to anti-BRAF treatment is dominated by a dynamic deregulation of a large population of microRNA (miRNA)[60]. The latter leads to the alteration of the intrinsic proliferation and survival pathways, enhancing proinflammatory and proangiogenic cues[60].
Role of immunity in resistance
Chronic inflammation is a hallmark of cancer[61-63]. Innate and adaptive immune responses contribute to select aggressive clones, stimulating cancer cell proliferation and migration[64]. Natural killers and CTL can recognize and eliminate the immunogenic cancer cells and in this way less immunogenic cells are selected[65]. Tumor associated macrophages and neutrophils can also promote angiogenesis and lymphangiogenesis as well as cancer cell proliferation and epithelial-mesenchymal transition (EMT) by secreting a set of stimulating cytokines[66-68]. However, the same tumor cells can secrete immunosuppressive factors, controlling the immune response[68-70]. Tumor-associated endothelial cells also contribute to make cancer physically inaccessible to the immune system by increasing deposition of factors that, on the one hand, confer a higher stiffness of the extracellular matrix and, on the other hand, prevent immune infiltration in the tumor tissue and favor tumor cell proliferation[69,71]. In light of these findings, several different immunotherapeutic strategies have been developed. Cytokines with immunomodulatory, antiangiogenic, anti-proliferative, and antitumor activities, such as IFNs and IL-2, have been combined with chemotherapy but with less satisfying results[72]. Immune-checkpoint inhibitors, a class of target-specific drugs that interfere with critical inhibitory signaling pathways promoting immune-mediated target of tumor cells (e.g., ipilimumab and nivolumab), gave more successful results[73].
Adoptive T-cells transfer therapy is, at the moment, one of the personalized and effective treatment methods available for the management of metastatic melanoma. In this case, tumor-infiltrating lymphocytes directly derived from the patients or genetically engineered melanoma-specific T-cells are expanded ex-vivo and then injected into the patient[74]. Although the complex anti-tumor mechanism triggered by this therapeutic approach has not yet been fully elucidated, the obtained results are very promising. Adoptive T-cells transfer therapy has been reported to be associated with complete and durable responses also in metastatic melanomas[75]. This approach results effective not only alone but also in combination with other standard therapies for melanoma management[76].
Phenotypic plasticity and drug resistance
Cancer is highly heterogeneous. This fact brings many important consequences: there is profound variation between different individuals with the same type of cancer as well as a high grade of genetic and phenotypical variability in the cancer cell population of a specific subject. The different phenotypes of tumor cells are due not only to genetic and epigenetic intratumor heterogeneity[77] but also to epigenetic changes due to the impact of the environment. It has been reported that genetically homogeneous tumor cells show a remarkable diversity in their response to therapy or other environmental stimuli[78,79]. Epigenetic gene regulation at the molecular level from DNA methylation, post-translational modification of histones, non-coding RNAs, and chromatin remodeling are the most common mechanisms contributing to cellular epigenetic heterogeneity. For a cancer, the robustness of the system comes from the ability of the cells to adapt to another environment, having the capacity to evolve into new cellular ecosystems. The ability of cancer cells to adapt to critical changes in the environment leads to the difficulty in finding a successful strategy. The main genetic mechanism that contributes to the ability of cancer to adapt to different microenvironments is genetic destabilization[80,81]. Furthermore, differences in tumor cell metabolism, due to genetic mutations and/or altered microenvironment, have a direct impact on epigenetic changes[82]. In this connection, our group recently demonstrated that human melanoma cells can change their phenotype, expressing EMT markers dynamically, thanks to a complex network of miRNAs[83]. The direct and more important consequence of these findings is that the cells show an intrinsic ability to dynamically change the phenotype, depending on the environment[83]. Similar results were published recently for breast cancer[84].
The impact of plasticity of tumor on drug resistance is a key factor and is crucial to develop new therapeutic strategies. In this connection, a recent interesting paper describes the dynamics of single melanoma cells after the treatment with a drug and shows that the cells reprogram to a stable resistant state[48]. The reprogramming involves the loss of SOX-10, which mediates differentiation, and the activation of Jun-Activator protein 1 and TEAD[48].
Conclusion and perspectives
Plasticity of tumor cells including melanoma is a critical issue for a successful therapeutic strategy. The ability of tumor cells to change their status using epigenetic mechanisms in dependence of the environment, such as the tumor niche, has been shown to play a critical role in the acquisition of a resistant phenotype in response to a specific drug. In light of these findings, in our opinion, the following will be crucial for precision medicine treatments: (1) knowing the epigenetic profile of each specific tumor of each specific patient before the treatment to start the best therapy; and (2) avoiding the ability of tumor cells to change their phenotype during the treatment, thus acquiring a resistant phenotype, by acting both at the level of the tumor cells and at the level of the tumor niche.
Declarations
Authors’ contributionsDesign and write the paper: Lionetti MC, Fumagalli MR, La Porta CAM
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe project was supported by funded from the Center for Complexity and Biosystems of UniMI. The research leading to these results has received funding from AIRC under IG 2019 - ID. 23141 project - P.I. La Porta Caterina.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. World Health Organization. Cancer mortality database. Available from: http://www-dep.iarc.fr/WHOdb/WHOdb.htm. [Last accessed on 18 Feb 2020].
2. Gershenwald JE, Scolyer RA, Hess KR, Sondak VK, Long GV, et al. Melanoma staging: evidence-based changes in the american joint committee on cancer eighth edition cancer staging manual. CA Cancer J Clin 2017;67:472-92.
3. Isaksson K, Katsarelias D, Mikiver R, Carneiro A, Ny L, et al. A population-based comparison of the AJCC 7th and AJCC 8th editions for patients diagnosed with stage III cutaneous malignant melanoma in Sweden. Ann Surg Oncol 2019;26:2839-45.
4. Tas F. Metastatic behavior in melanoma: timing, pattern, survival, and influencing factors. J Oncol 2012:647684.
5. Ives N J, Stowe RL, Lorigan P, Wheatley K. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2,621 patients. J Clin Oncol 2007;25:5426-34.
6. Palmieri G, Ombra M, Colombino M, Casula M, Sini M, et al. Multiple molecular pathways in melanomagenesis: characterization of therapeutic targets. Front Oncol 2015;5:183.
7. Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, et al. RAS/RAF/MEK/ERK and PI3K/PTEN/AKT/MTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget 2011;2:135-64.
8. NCI National Cancer Institute. The cancer genome atlas (TCGA). Available from: https://www.cancer.gov/tcga. [Last accessed on 18 Feb 2020].
9. Curtin J A, Fridlyand J, Kageshita T, Patel HN, Busam KJ, et al. Distinct sets of genetic alterations in melanoma. New Engl J Med 2005;353:2135-47.
10. Rubinstein JC, Sznol M, Pavlick AC, Ariyan S, Cheng E, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010;8:67.
11. Jakob JA, Bassett RL Jr, Ng CS, Curry JL, Joseph RW, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2012;118:4014-23.
12. Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, et al. The genetic evolution of melanoma from precursor lesions. New Engl J Med 2015;373:1926-36.
13. Fedorenko IV, Gibney GT, Smalley KSM. NRAS mutant melanoma: biological behavior and future strategies for therapeutic management. Oncogene 2013;32:3009-18.
14. MacKie R, Hauschild A, Eggermont A. Epidemiology of invasive cutaneous melanoma. Ann Oncol 2009;20:vi1-7.
16. Nogueira C, Kim KH, Sung H, Paraiso KH, Dannenberg JH, et al. Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Oncogene 2010;29:6222-32.
17. Mirmohammadsadegh A, Marini A, Nambiar S, Hassan M, Tannapfel A, et al. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res 2006;66:6546-52.
19. Murphy EA, Shields DJ, Stoletov K, Dneprovskaia E, McElroy M, et al. Disruption of angiogenesis and tumor growth with an orally active drug that stabilizes the inactive state of PDGFRbeta/B-RAF. Proc Natl Acad Sci U S A 2010;107:4299-304.
21. Kim C, Lee CW, Kovacic L, Shah A, Klasa R, et al. Long-term survival in patients with metastatic melanoma treated with DTIC or temozolomide. Oncologist 2010;15:765-71.
22. Middleton MR, Lorigan P, Owen J, Ashcroft L, Lee SM, Harper P, et al. A randomized phase III study comparing dacarbazine, BCNU, cisplatin and tamoxifen with dacarbazine and interferon in advanced melanoma. Br J Cancer 2000;82:1158-62.
23. Testori A, Ribero S, Bataille V. Diagnosis and treatment of in-transit melanoma metastases. Eur J Surg Oncol 2017;43:544-60.
24. Miklavčič D, Serša G, Brecelj E, Gehl J, Soden D, et al. Electrochemotherapy: technological advancements for efficient electroporation-based treatment of internal tumors. Med Biol Eng Comput 2012;50:1213-25.
25. Baldea I, Giurgiu L, Teacoe ID, Olteanu DE, Olteanu FC, et al. Photodynamic therapy in melanoma - where do we stand? Curr Med Chem 2018;25:5540-63.
28. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, et al. A new member of the immunoglobulin superfamily - CTLA-4. Nature 1987;328:267-70.
29. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, et al. Lymphoproliferative disorders with early lethality in mice deficient in CTLA-4. Science 1995;270:985-8.
30. Panelli MC, Wunderlich J, Jeffries J, Wang E, Mixon A, et al. Phase 1 study in patients with metastatic melanoma of immunization with dendritic cells presenting epitopes derived from the melanoma-associated antigens MART-1 and gp100. J Immunother 2000;23:487-98.
31. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 2015;28:690-714.
32. Moriceau G, Hugo W, Hong A, Shi H, Kong X, et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015;27:240-56.
33. Shi H, Hugo W, Kong X, Hong A, Koya RC, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80-93.
34. Song C, Piva M, Sun L, Hong A, Moriceau G, et al. Recurrent tumor cell-intrinsic and -extrinsic alterations during MAPKi-induced melanoma regression and early adaptation. Cancer Discov 2017;7:1248-65.
35. Hannan E, O’Leary DP, MacNally SP, Kay EW, Farrell MA, et al. The significance of BRAF V600E mutation status discordance between primary cutaneous melanoma and brain metastases: the implications for BRAF inhibitor therapy. Medicine (Baltimore) 2017;96:e8404.
36. Roesch A, Paschen A, Landsberg J, Helfrich I, Becker JC, et al. Phenotypic tumour cell plasticity as a resistance mechanism and therapeutic target in melanoma. Eur J Cancer 2016;59:109-12.
37. Johnson D, Menzies AM, Zimmer L, Eroglu Z, Ye F, et al. Acquired BRAF inhibitor resistance: a multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer 2015;51:2792-9.
38. Shi H, Hugo W, Kong X, Hong A, Koya RC, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2013;4:80-93.
39. Brighton HE, Angus SP, Bo T, Roques J, Tagliatela AC, et al. New mechanisms of resistance to MEK inhibitors in melanoma revealed by intravital imaging. Cancer Res 2018;78:542-57.
40. Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res 2008;68:4853-61.
41. Doudican N, Orlow S. Inhibition of the CRAF/prohibitin interaction reverses CRAF-dependent resistance to vemurafenib. Oncogene 2016;36:423-8.
42. Müller J, Krijgsman O, Tsoi J, Robert L, Hugo W, et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 2014;5:5712.
43. Ramsdale R, Jorissen RN, Li FZ, Al-Obaidi S, Ward T, et al. The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci Signal 2015;8:ra82.
44. Wouters J, Kalender-Atak Z, Minnoye L, Spanier KI, De Waegeneer M, et al. Single-cell gene regulatory network analysis reveals new melanoma cell states and transition trajectories during phenotype switching. BiorXiv 2019; doi: 10.1101/715995.
45. Su Y, Bintz M, Yang Y, Robert L, Ng AHC, et al. Phenotypic heterogeneity and evolution of melanoma cells associated with targeted therapy resistance. PLOS Comput Biol 2019;15:e1007034.
46. Perego M, Maurer M, Wang JX, Shaffer S, Müller AC, et al. A slow-cycling subpopulation of melanoma cells with highly invasive properties. Oncogene 2018;37:302-12.
47. Emmons MF, Faião-Flores F, Sharma R, Thapa R, Messina JL, et al. HDAC8 regulates a stress response pathway in melanoma to mediate escape from BRAF inhibitor therapy. Cancer Res 2019;79:2947-61.
48. Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017;546:431-5.
49. Kim JH, Ahn JH, Lee M. Upregulation of microRNA-1246 is associated with BRAF inhibitor resistance in melanoma cells with mutant BRAF. Cancer Res Treat 2017;49:947-59.
50. Hwang SH, Ahn JH, Lee M. Upregulation of S100A9 contributes to the acquired resistance to BRAF inhibitors. Genes Genomics 2019;41:1273-80.
51. Caporali S, Amaro A, Levati L, Alvino E, Lacal PM, et al. mir-126-3p down-regulation contributes to dabrafenib acquired resistance in melanoma by up-regulating ADAM9 and VEGF-a. J Exp Clin Cancer Res 2019;38:272.
52. Erdmann S, Seidel D, Jahnke HG, Eichler M, Simon JC, et al. Induced cross-resistance of BRAF V600E melanoma cells to standard chemotherapeutic dacarbazine after chronic PLX4032 treatment. Sci Rep 2019;9:30.
53. Hugo W, Shi H, Sun L, Piva M, Song C, et al. Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell 2015;162:1271-85.
54. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2016;165:35-44.
55. Jarkowski A 3rd, Norris L, Trinh VA. Controversies in the management of advanced melanoma: “gray” areas amid the “black and blue”. Ann Pharmacother 2014;48:1456-68.
56. Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015;372:30-9.
57. Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer cell 2010;18:683-95.
58. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in keynote-001. Ann Oncol 2019;30:582-8.
59. Ascierto PA, Dummer R. Immunological effects of BRAF + MEK inhibition. Oncoimmunology 2018;7:e1468955.
60. Fattore L, Ruggiero CF, Pisanu ME, Liguoro D, Cerri A, et al. Reprogramming mirnas global expression orchestrates development of drug resistance in BRAF mutated melanoma. Cell Death Differ 2019;26:1267-82.
61. Hussain SP, Amstad P, Raja K, Ambs S, Nagashima M, et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res 2000;60:3333-7.
63. Beaugerie L, Svrcek M, Seksik P, Bouvier AM, Simon T, et al. Risk of colorectal high-grade dysplasia and cancer in a prospective observational cohort of patients with inflammatory bowel disease. Gastroenterology 2013;145:166-75.
64. di Carlo E, Iezzi M, Pannellini T, Zaccardi F, Modesti A, et al. Neutrophils in anti-cancer immunological strategies: old players in new games. J Hematother Stem Cell Res 2001;10:739-48.
65. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev 2018;32:1267-84.
66. Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest 2015;125:3338-46.
68. Costanza B, Umelo IA, Bellier J, Castronovo V, Turtoi A. Stromal modulators of TGF-b in cancer. J Clin Med 2017;6.
69. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature 2004;432:332-7.
70. Hasmim M, Messai Y, Ziani L, Thiery J, Bouhris JH, et al. Critical role of tumor microenvironment in shaping NK cell functions: Implication of hypoxic stress. Front Immunol 2015;6:482.
71. Hida K, Maishi N, Annan DA, Hida Y. Contribution of tumor endothelial cells in cancer progression. Int J Mol Sci 2018;19.
72. Eggermont AM, Suciu S, Rutkowski P, Kruit WH, Punt CJ, et al. Long term follow up of the EORTC 18952 trial of adjuvant therapy in resected stage IIB-III cutaneous melanoma patients comparing intermediate doses of interferon-alpha-2b (IFN) with observation: ulceration of primary is key determinant for IFN-sensitivity. Eur J Cancer 2016;55:111-21.
73. Brahmer JR, Pardoll DM. Immune checkpoint inhibitors: making immunotherapy a reality for the treatment of lung cancer. Cancer Immunol Res 2013;1:85-91.
74. Labarrière N, PandGervois N, Khammari A, Tessier MH, et al. Therapeutic efficacy of melanoma-reactive TIL injected in stage III melanoma patients. Cancer Immunol Immunother 2002;51:532-8.
75. Klemen ND, Feingold PL, Goff SL, Hughes MS, Kammula US, et al. Metastasectomy following immunotherapy with adoptive cell transfer for patients with advanced melanoma. Ann sur oncol 2017;24:135-41.
76. Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, et al. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin Cancer Res 2016;22:3734-45.
77. Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 2014;54.
78. Kreso A, O’Brien CA, van Galen P, Gan OI, Notta F, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013;339:543-8.
79. Kreso A, van Galen P, Pedley NM, Lima-Fernandes E, Frelin C, et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat Med 2014;20:29-36.
80. Luoto KR, Kumareswaran R, Bristow RG. Tumor hypoxia as a driving force in genetic instability. Genome Integr 2013;4:5.
81. Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer 2012;12:487-93.
82. Kinnaird A, Zhao S, Wellen KE, Michelakis ED. Metabolic control of epigenetics in cancer. Nat Rev Cancer 2016;16:694-707.
83. Sellerio AL, Ciusani E, Ben-Moshe NB, Coco S, Piccinini A, et al. Overshoot during phenotypic switching of cancer cell populations. Sci Rep 2015;5:15464.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright















Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].