


Extracellular vesicles in vascular diseases: pathological mechanisms and therapeutic application progress
 0
0 Abstract
Vascular diseases-including atherosclerosis, essential hypertension, pulmonary arterial hypertension, aortic dissection, and aneurysms-share core pathological mechanisms including endothelial dysfunction, inflammation-oxidative stress cascades, and aberrant activation of vascular smooth muscle cells (VSMCs). This review focuses on these four disorders as classic case studies to elucidate the multifaceted roles of extracellular vesicles (EVs) in vascular pathophysiology. Emerging evidence highlights that autoimmune-mediated vascular wall injury exacerbates these common pathogenic pathways in a subset of patients. EVs, nanoscale mediators endogenously produced by cells, are increasingly recognized for their multifaceted roles in vascular pathophysiology. They act not only as carriers of pathological signals [e.g., proteins such as cytokines, autoantigens, and proteases, as well as nucleic acids such as microRNAs (miRNAs)], but also as regulators of immune homeostasis. Beyond their
Keywords
INTRODUCTION
Cardiovascular diseases (CVDs) remain the leading cause of death globally. According to the World Health Organization (WHO), CVDs accounted for approximately 19.8 million deaths in 2022, representing 32% of all global mortality. Notably, heart attacks and strokes were responsible for 85% of these fatalities, with over three-quarters occurring in low- and middle-income countries[1]. Beyond their immense mortality, CVDs inflict severe disability and impose substantial economic burdens on healthcare systems and societies globally. Among these conditions, vascular diseases, characterized by structural and functional abnormalities within the circulatory system, cause reduced exercise tolerance and multi-organ ischemic injury. Four major vascular conditions, including atherosclerosis (AS), essential hypertension (EH), and pulmonary arterial hypertension (PAH), together with acute and life-threatening events such as aortic dissection and aneurysm rupture, impose a substantial global health burden. The core pathological cascade typically commences with endothelial dysfunction, which elicits a synergistic amplification of inflammation and oxidative stress[2]. When coupled with dysregulated immune responses targeting vascular self-antigens under specific contexts, this cascade drives vascular smooth muscle cell (VSMC) phenotypic switching-transitioning from a quiescent, contractile state to a proliferative, synthetic phenotype characterized by enhanced migration, matrix degradation, and cytokine secretion[3]. In turn, this phenotypic transition further perpetuates endothelial dysfunction and promotes pathological vascular remodeling. These interconnected processes ultimately lead to progressive and often maladaptive vascular remodeling, manifesting as vessel occlusion, pathological dilation (aneurysm), or wall dissection, and end-organ damage[4]. In addition, a growing body of evidence also implicates autoimmune-mediated injury as a critical pathogenic driver in a subset of these vascular disorders. Current clinical interventions-including anticoagulation, antihypertensive therapy, and revascularization-are often inadequate to prevent disease onset or halt progression, particularly against autoimmune components. Limited therapeutic options leave patients at risk of severe complications, including limb amputation, organ failure, and catastrophic vascular rupture, underscoring the urgent need for novel therapeutic strategies.
Extracellular vesicles (EVs) are phospholipid bilayer-enclosed particles with a diameter ranging from 30 nanometers to several micrometers. They are generated through diverse cellular processes, including plasma membrane budding, the fusion of multivesicular bodies with the plasma membrane, and the fragmentation of the plasma membrane during apoptosis (see more details on EV biogenesis, classification, and functional mechanisms in our previous review[5]). These nanoscale particles released by cells function as critical mediators of intercellular communication by transferring bioactive cargo such as proteins, nucleic acids, and lipids. While much attention has been given to microRNA (miRNA)-mediated effects, it is crucial to emphasize that EV-associated proteins are equally critical functional effectors, directly activating signaling pathways in recipient cells and driving vascular pathogenesis and repair[6]. They actively participate in these pathological cascades. More importantly, EVs are emerging as pivotal mediators in autoimmune vascular injury, capable of shuttling autoantigens, cytokines, and other immunostimulatory molecules that break tolerance and perpetuate autoimmunity, thereby degrading the vascular extracellular matrix and compromising wall integrity. However, their inherent biocompatibility, low immunogenicity, and intrinsic tissue-penetrating properties also position EVs as promising therapeutic targets and highly efficient delivery vehicles. Moreover, analyzing EV cargo and function provides profound mechanistic insights into disease pathogenesis, offering a novel lens to identify autoimmune drivers and rupture risk biomarkers and clinical therapy.
Here, we summarize the multifaceted roles of EVs in vascular disease therapy, focusing on these shared pathways and the unique role of autoimmunity. We also explore cutting-edge engineering strategies designed to enhance their therapeutic efficacy or targeting specificity, including their potential to
OVERVIEW AND CLASSIFICATION OF VASCULAR DISEASES
AS, PAH, EH, and aortic aneurysms and dissections share fundamental pathological mechanisms initiated by vascular endothelial dysfunction and/or autoimmune-mediated vascular wall injury. This initial insult triggers a cascade involving synergistic amplification of inflammation and oxidative stress, which drives pathological VSMC activation and extracellular matrix degradation. These processes culminate in maladaptive vascular remodeling, leading to irreversible structural and functional impairment that manifests as either obstructive luminal narrowing or destructive wall weakening and rupture. Consequently, targeting endothelial protection, suppressing inflammatory and oxidative stress pathways, counteracting autoimmunity, and modulating VSMC activity and matrix homeostasis represent conserved therapeutic strategies across these vascular disorders.
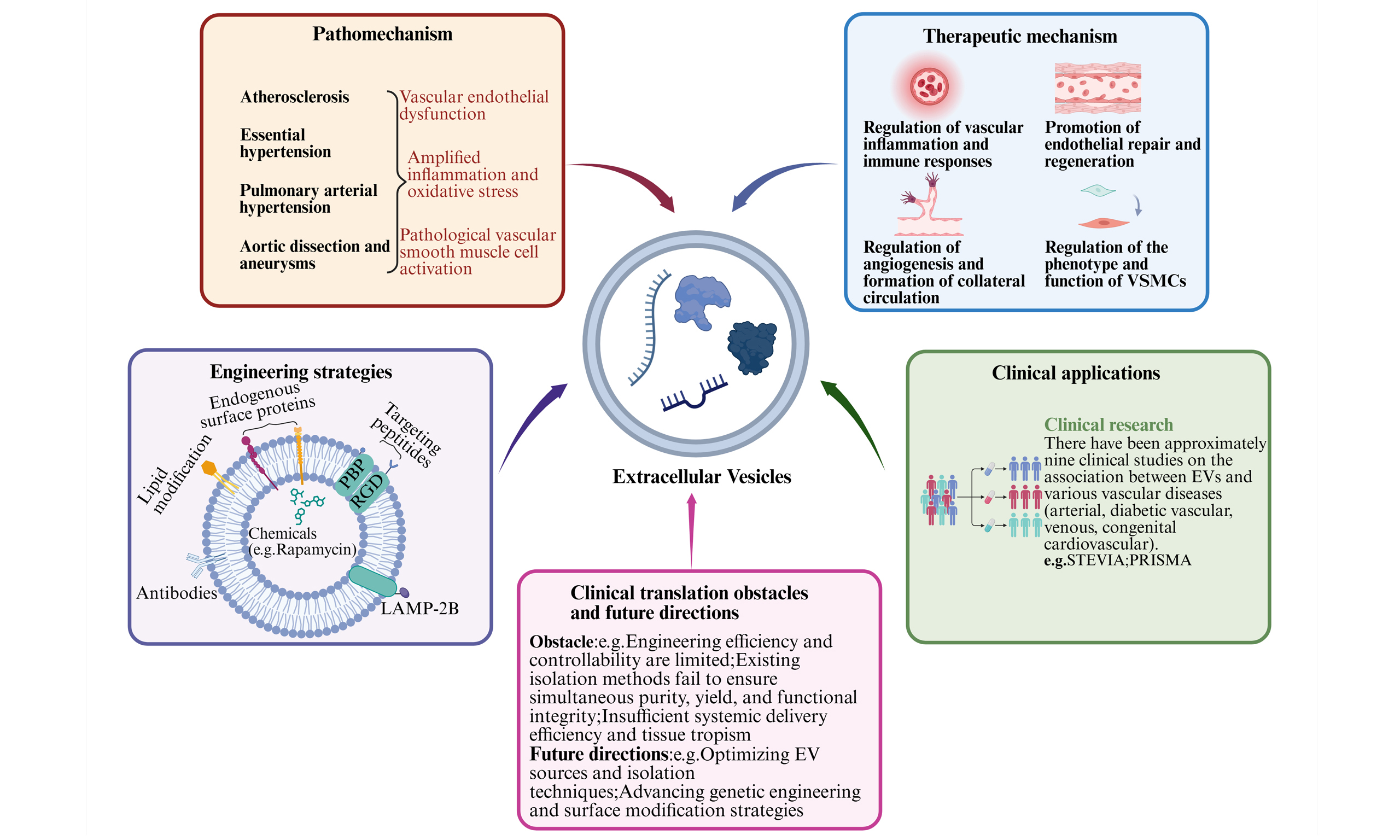
As depicted in Figure 1, EV-mediated pathological cascades in five major vascular diseases: (1) Atherosclerosis: Dual pathogenic cascades drive AS: Classical intimal pathway: Platelet-derived EVs [carrying CD40L, interleukin-1β (IL-1β)] induce endothelial activation, vascular cell adhesion molecule-1 (VCAM-1) upregulation, and monocyte adhesion, promoting lipid deposition, foam cell formation, and eventual plaque rupture[7]; Emerging adventitial pathway: adventitial fibroblast (AF)-EVs [carrying vascular endothelial growth factor (VEGF)] trigger vasa vasorum (VV) endothelial activation and pathological angiogenesis, leading to adventitial VV dysplasia and plaque vulnerability via leaky neovessels[8,9]. (2) Essential Hypertension: Fibroblast-derived EVs carrying miR-21-3p suppress SORBS2 expression, triggering VSMC proliferation. This contributes to vascular wall thickening, increased resistance/stiffness, and hemodynamic dysfunction[10]. (3) Pulmonary Arterial Hypertension: pulmonary artery smooth muscle cell (PASMC)-derived EVs with reduced bone morphogenetic protein receptor type 2 (BMPR2) expression promote endothelial apoptosis and PASMC hyperproliferation, establishing a self-amplifying cycle of vascular obstruction and right ventricular failure[11]. (4) Aortic Dissection and Aneurysm: The schematic illustrates the pathogenesis of aortic dissection and aneurysm: macrophage- and smooth muscle cell (SMC)-derived EVs deliver increased matrix metalloproteinases (MMPs), mediating extracellular matrix (ECM) degradation. This drives medial degeneration and proteolysis, culminating in vessel dilation or dissection. MMPs directly cause elastin/collagen breakdown and wall weakening[12]. Furthermore, in the context of aneurysms, dissection, and autoimmunity, EVs are particularly implicated in ferrying proteolytic enzymes, cell death signals, and immunostimulatory cargo that directly compromise vessel wall integrity.
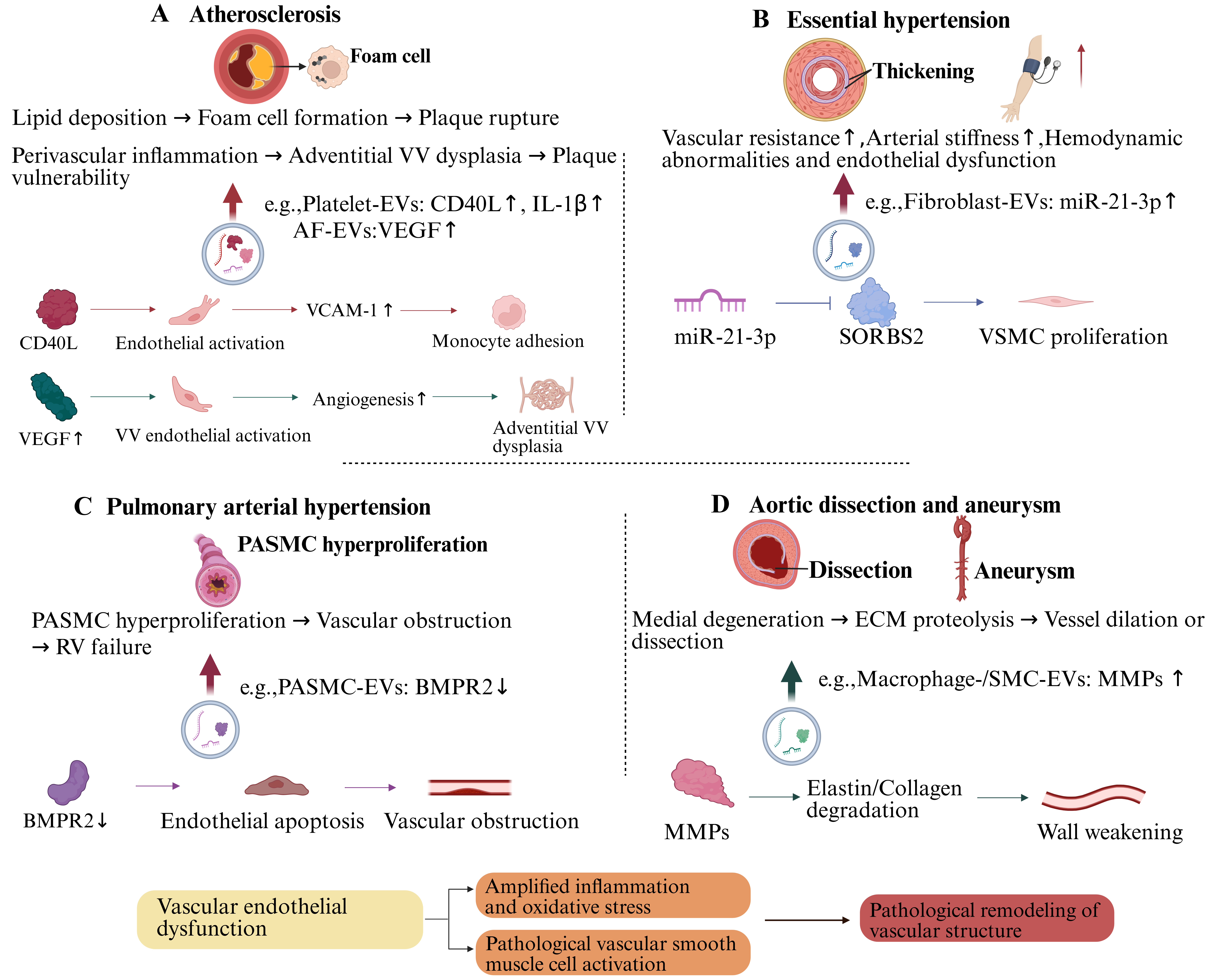
Figure 1. Mechanisms underlying the roles of disease-specific extracellular vesicles in vascular pathologies. (A) Schematic illustration of EV-mediated molecular pathological cascades in atherosclerosis; (B) Schematic illustration of EV-mediated molecular pathological cascades in essential hypertension; (C) Schematic illustration of EV-mediated molecular pathological cascades in pulmonary arterial hypertension; (D) Schematic illustration of EV-mediated molecular pathological cascades in aortic dissection and aneurysm. Created in BioRender. gN, lý. (2025) https://BioRender.com/4w1wlxx. EV: Extracellular vesicle; VV: vasa vasorum; AF-EVs: adventitial fibroblast extracellular vesicles; VEGF: vascular endothelial growth factor; IL-1β: interleukin-1β; VCAM-1: vascular cell adhesion molecule-1; PASMC: pulmonary artery smooth muscle cell; BMPR2: bone morphogenetic protein receptor type 2; VSMC: vascular smooth muscle cell; ECM: extracellular matrix; MMPs: matrix metalloproteinases; RV: right ventricle.
AS represents a chronic inflammatory vascular disorder and constitutes one of the most prevalent diseases within the cardiovascular system. Its pathophysiological essence involves the aberrant accumulation of lipids within the arterial wall, culminating in the formation of fibrous or atheromatous plaques within the intima. This process results in vascular wall stiffening, luminal narrowing, and diminished elasticity. The characteristic pathological progression encompasses endothelial injury, lipid deposition, and inflammatory cell infiltration, which may include autoimmune responses against vessel wall antigens such as oxidized low-density lipoprotein (LDL)[13], phenotypic modulation of VSMCs, foam cell formation, and ultimately, plaque development[14]. Importantly, beyond the classic “inside-out” paradigm, the adventitia is now recognized as an active participant from the early stages. Pathological angiogenesis of the VV, the microvasculature nourishing the arterial wall, and inflammation originating from perivascular adipose tissue (PVAT) facilitate the entry of inflammatory cells into the vessel wall, contributing to the “outside-in” pathway of atherosclerotic plaque development and vulnerability[15]. This pathology predominantly EVs large- and medium-sized arteries, particularly at sites experiencing elevated hemodynamic stress, such as areas of turbulence. AS serves as the fundamental pathological substrate underpinning major cardiovascular diseases, including coronary artery disease, cerebrovascular disease, and peripheral arterial disease[16].
EH is a chronic condition characterized by persistently elevated blood pressure of unknown etiology. Its development involves complex interactions between genetic predisposition, environmental factors, and lifestyle influences[17,18]. The underlying pathophysiology encompasses multiple interrelated mechanisms, including increased vascular resistance, elevated arterial stiffness, hemodynamic abnormalities, endothelial dysfunction, heightened sympathetic nervous system activity, dysregulation of the renin-angiotensin-aldosterone system (RAAS), vascular remodeling, inflammation, and in some patients, autoantibody-mediated activation (e.g., targeting angiotensin II type 1 receptors)[19], and sodium retention[20-22]. These mechanisms collectively sustain elevated blood pressure and heighten cardiovascular risk. EH is strongly associated with metabolic syndrome and diabetes mellitus, further contributing to the increased risk of cardiovascular complications[23].
PAH is driven by progressive pulmonary vascular remodeling, characterized by abnormal PASMC proliferation with impaired apoptosis, causing vascular wall thickening and luminal obstruction[24]. Concurrent endothelial dysfunction, exacerbated by BMPR2 signaling deficiency, manifests as increased apoptosis and compromised barrier integrity[11]. Enhanced inflammation and metabolic dysregulation, frequently fueled by autoimmune phenomena as seen in PAH associated with connective tissue diseases, amplify endothelial injury and oxidative stress, accelerating remodeling[25]. Critically, these processes form a positive feedback loop: endothelial damage promotes PASMC hyperproliferation, which in turn exacerbates vascular dysfunction, culminating in irreversible pulmonary vascular remodeling and right ventricular failure.
Aortic dissection and aneurysms represent life-threatening structural failures of the vascular wall, characterized by pathological dilation (aneurysm) or tearing of the intima leading to false lumen formation (dissection). Their development involves a multifactorial etiology stemming from both hereditary predispositions and acquired risk factors (such as chronic hypertension, AS)[26,27]. The underlying pathophysiology is driven by a cascade of interrelated mechanisms, including medial degeneration with smooth muscle cell apoptosis, fragmentation of elastic fibers, excessive proteolysis of the extracellular matrix, chronic inflammation, and increasingly recognized, autoimmune-mediated injury targeting vascular components[12]. These mechanisms collectively undermine the structural integrity of the aortic wall, culminating in either progressive dilation or acute dissection. This pathology is strongly associated with hypertension and AS, which exert heightened hemodynamic stress on the weakened vessel wall, further contributing to the risk of rupture or dissection.
Notably, beyond these disease-specific triggers, autoimmune-mediated injury emerges as a critical shared upstream mechanism that can instigate or exacerbate these pathological cascades across the vascular spectrum[28]. The core pathway, beginning with a rise in autoantigens, followed by T cell and B cell activation, and ultimately leading to amplified inflammation, is increasingly recognized. This process, often initiated by the exposure of vascular self-antigens (e.g., oxidized LDL in AS, vascular wall components in aortic dissection and aneurysms) and potently amplified by EV-mediated communication, drives sustained inflammation and cellular dysfunction[29-31]. It thereby constitutes a common pathogenic thread that contributes to endothelial activation in AS and EH, PASMC hyperproliferation in PAH, and medial degeneration with ECM degradation in aortic dissection and aneurysms, ultimately accelerating pathological remodeling and heightening cardiovascular risk.
THE ROLE OF EVS AS DELIVERY VEHICLES
EVs are a heterogeneous population of nanoscale, lipid bilayer-enclosed structures actively secreted by cells. Their nucleic acid content-including mRNAs, miRNAs, and small interfering RNAs (siRNAs)-can modulate gene expression in recipient cells. For instance, EV-derived miR-155 enhances macrophage inflammation[32]. Additionally, EV-delivered TGF-β1 mRNA contributes to tissue fibrosis, and epidermal growth factor receptor (EGFR) mRNA can promote tumor cell proliferation within the tumor microenvironment (TME)[33,34]. In addition to nucleic acids, EVs carry a diverse array of proteins with significant biological functions. These include cytokines, growth factors, and metalloproteinases, which play crucial roles in inflammation, angiogenesis, and extracellular matrix remodeling. Such proteins can directly activate signaling pathways upon delivery to recipient cells[35-37]. Furthermore, EVs are enriched with various lipid species, such as sphingomyelin, cholesterol, phosphatidylserine, and prostaglandins. These lipids not only form the structural foundation of EVs but also actively participate in essential biological processes including cellular signaling, membrane fusion, and immunomodulation[38-40]. Circulating via the bloodstream, EVs demonstrate significant potential as non-invasive biomarkers for disease auxiliary diagnosis[41]. Critically, their inherent biocompatibility, low cytotoxicity, capacity to traverse biological barriers, and amenability to bioengineering position EVs as promising platforms for targeted drug delivery, offering substantial theranostic potential in cardiovascular diseases and beyond[42-44].
Although the role of EV-encapsulated miRNAs has been extensively explored, the contribution of EV-associated proteins to vascular pathophysiology is equally profound and indispensable. EVs serve as mobile repositories of functional proteins, including cytokines, growth factors, receptors, and enzymes, which can be directly delivered to recipient cells to elicit rapid biological responses. A seminal example is the finding that EVs from mice subjected to pressure overload are enriched with angiotensin II type 1 receptor (AT1R). These AT1R-positive EVs can transfer the receptor to other cells, activating angiotensin II signaling pathways and directly contributing to pathological cardiovascular remodeling, independent of miRNA cargo[45]. This represents a paradigm shift, demonstrating that EVs can deliver fully functional transmembrane receptors, thereby conferring new signaling capabilities to recipient cells. Similarly, EVs carry and transfer active MMPs to degrade the extracellular matrix in aneurysms, deliver proinflammatory cytokines to exacerbate inflammation, and present autoantigens to trigger autoimmune responses[46]. Furthermore, EVs are enriched with cell-type-specific surface proteins [e.g., tetraspanins CD9, CD63, and CD81 for general EVs; P-selectin for platelet-EVs; major histocompatibility complex (MHC) class II for immune cell EVs] that dictate their cellular tropism and biological fate[47,48]. Therefore, a comprehensive understanding of EV functions necessitates equal consideration of their protein cargo.
ROLES OF EVS IN CARDIOVASCULAR DISEASES
EVs exert multifaceted therapeutic effects on vascular pathologies through precise modulation of intercellular communication. As illustrated in Figure 2, EVs orchestrate four interconnected functional axes critical for vascular homeostasis: (1) Regulation of vascular inflammation and immune response: EVs harbor molecules such as IL-1β, miR-185-3p, miR-210, miR-126, and miR-223. By regulating the activation state of immune cells (e.g., macrophages, T cells) or the secretion of inflammatory factors by vascular endothelial cells, these cargos suppress excessive vascular inflammatory responses and maintain immune homeostasis. (2) Promotion of vascular endothelial repair and regeneration: EVs carry molecules including Annexin A1 (ANXA1), interleukin-10 (IL-10), miR-126, VEGF, and MMPs. These cargos regulate endothelial cell proliferation, migration, and damage repair, directly facilitating the repair and regeneration of the vascular endothelium and preserving endothelial barrier integrity. (3) Regulation of angiogenesis and formation of collateral circulation: EVs deliver molecules such as VEGF, Ang-1, Ang-2, Cellular Communication Network Factor 2 (CCN-2 protein), and miR-210-3p. Acting on vascular endothelial cells or pericytes, these cargos modulate key steps of angiogenesis (e.g., endothelial sprouting, lumen formation) and induce the establishment of collateral circulation, thereby improving blood supply to ischemic tissues. (4) Regulation of the phenotype and function of VSMCs: EVs transmit molecules such as high-mobility group box 1 (HMGB1) and HMGB2. These cargos control the phenotypic transition of vascular smooth muscle cells (specifically, from a contractile to a synthetic phenotype) and influence their proliferation, migration, and capacity to secrete extracellular matrix, thus participating in vascular tone regulation and vascular remodeling processes. In the following sections, we provide a detailed review of these four aspects.
Figure 2. Roles of EVs in cardiovascular diseases. EVs promote vascular endothelial repair and regeneration, regulate angiogenesis and formation of collateral circulation, modulate vascular inflammation and immune response, and regulate the phenotype and function of vascular smooth muscle cells. Created in BioRender. gN, lý. (2025) https://BioRender.com/r0i0fed. EVs: Extracellular vesicles; IL-1β: interleukin-1β; VEGF: vascular endothelial growth factor; IL-10: interleukin-10; CCN-2 protein: cellular communication network factor 2; MSCs: mesenchymal stem cells; ASC: adipose-derived stem cell; hiPSC: human induced pluripotent stem cell; AnxA1: annexin A1; VEGF: vascular endothelial growth factor; MMPs: matrix metalloproteinases; HMGB1: high-mobility group box 1; NSCs: neural stem cells.
Regulation of vascular inflammation and immune response
EV-mediated proinflammatory communication
EVs from specific cellular sources directly propagate proinflammatory signaling in vascular pathologies. Platelet-derived EVs deliver CD40L and IL-1β, triggering endothelial activation to induce the expression of VCAM (such as VCAM-1, ICAM-1) and leukocyte recruitment[49,50]. Activated endothelial EVs amplify local inflammation and thrombosis through coagulant factors[51,52]. M1 macrophage-derived EVs deliver proinflammatory cargo, including miRNAs (such as miR-185-3p) and proteins [such as tumor necrosis factor-α (TNF-α), IL-1β], that exacerbate AS by promoting lipid accumulation, endothelial dysfunction, and oxidative stress[53]. Atherogenic EVs further mediate miR-30a-3p-dependent suppression of cholesterol efflux, accelerating foam cell formation[54], while factor kappa-light-chain-enhancer of activated B cells
EVs mediate anti-inflammatory effects
EVs mediate anti-inflammatory responses in vascular diseases through bioactive cargo delivery.
EVs as key mediators in autoimmune vascular injury
Beyond their roles in general inflammation, EVs are increasingly recognized as pivotal mediators in the initiation and propagation of autoimmune-mediated vascular injury, a critical pathogenic mechanism shared by several vasculopathies. They function as mobile reservoirs of bioactive cargo, encompassing both proteinaceous autoantigens (e.g., oxidized LDL in AS, vascular wall components in aortic pathologies) and nucleic acids. When released from stressed or dying vascular cells, these EVs deliver cryptic self-antigens to antigen-presenting cells (APCs), thereby potentially breaking immune tolerance and activating autoreactive T and B lymphocytes[62,63]. This process can initiate an adaptive immune response against the vessel wall. Furthermore, EVs derived from activated immune cells in autoimmune settings are packaged with a potent proinflammatory cargo. They deliver specific miRNAs (such as miR-155, a key regulator of T cell and macrophage inflammation) and cytokines (such as IL-17, IFN-γ) that can exacerbate endothelial dysfunction, promote pathological VSMC phenotypic switching, and drive macrophage polarization towards a proinflammatory (M1) phenotype[32]. For instance, T cell-derived EVs have been shown to promote macrophage lipid peroxidation and migration, contributing to abdominal aortic aneurysm progression. This EV-facilitated crosstalk creates a self-amplifying loop between the vascular wall and the immune system, sustaining a chronic state of autoimmunity and inflammation that accelerates vascular remodeling[31].
However, the inherent immunomodulatory properties of certain therapeutic EVs, particularly those from MSCs, offer a promising therapeutic avenue. MSC-EVs are enriched with anti-inflammatory cytokines (e.g., IL-10, TGF-β) and tolerance-inducing molecules that can suppress effector T cell responses, promote the expansion of regulatory T cells (Tregs), and induce macrophage polarization towards a pro-repair (M2) phenotype. This protein-mediated immunomodulation is a key mechanism. This capacity to restore immune homeostasis positions engineered EVs as potential novel biologics for mitigating autoimmune attacks and promoting vascular integrity in autoimmune vasculopathies[64].
EVs act as a communication bridge within the vascular-immune microenvironment
EVs serve as pivotal mediators of bidirectional crosstalk between vascular wall cells (endothelial cells, smooth muscle cells) and immune cells (monocytes/macrophages, lymphocytes) in vascular homeostasis and disease. Under inflammatory conditions, endothelial-derived EVs recruit monocytes via adhesion molecules and chemokines[65], while activated macrophage EVs deliver proinflammatory miRNAs and cytokines that disrupt endothelial barrier function and induce pathological VSMC phenotypic switching[53]. This aberrant EV-mediated communication establishes a self-amplifying circuit that exacerbates vascular inflammation and remodeling, presenting critical targets for therapeutic intervention.
Collectively, EVs mediate critical intercellular communication networks that coordinate vascular inflammation and immune responses. This establishes novel mechanistic insights into vascular pathogenesis and provides a foundation for targeted therapeutic strategies.
Promoting vascular endothelial repair and regeneration
Mechanisms of EVs in repairing injured vascular endothelium
Vascular endothelial injury initiates cardiovascular pathologies such as AS. EVs critically promote endothelial repair and regeneration via a sequential, multistage process. Upon injury, EVs are selectively recruited to damaged sites through surface ligand-receptor interactions (e.g., P-selectin-binding EVs targeting activated endothelium)[66]. Subsequently, EVs deliver anti-inflammatory mediators such as ANXA1 and IL-10, suppressing NF-κB signaling and mitigating inflammation to protect endothelial cells[67-71]. The delivery of these functional proteins provides immediate anti-inflammatory signals. This establishes a microenvironment conducive to repair. EVs then enhance endothelial cell proliferation and migration, facilitated by cargo such as miR-126[56]. Progressing to angiogenesis, EVs promote lumen formation and vascular network assembly by delivering pro-angiogenic factors and miRNAs (e.g., miR-126 via SPRED1 inhibition)[72-74]. Ultimately, EVs contribute to vascular homeostasis restoration by modulating immune tolerance, suppressing excessive proliferation/inflammation, and enhancing endothelial barrier/anticoagulant functions, thereby preventing pathological remodeling[36]. This coordinated cascade-targeted recruitment, anti-inflammatory protection, cellular activation, angiogenesis, and homeostasis restoration-underpins EV-mediated endothelial repair.
Roles of EVs derived from different cell sources
While the multistage mechanism of EVs in promoting endothelial repair and regeneration is well-established (as summarized above), the functional heterogeneity of endothelial cells (ECs) themselves necessitates careful selection of EC sources for optimal therapeutic outcomes in vascular injury diseases. Consequently, identifying appropriate EV systems is critical. Herein, we highlight three prominent EV sources with therapeutic potential for vascular injury: Macrophage-derived EVs, Platelet-derived microparticles (PMPs), and MSC-EVs.
Macrophage-derived EVs critically promote vascular endothelial repair and regeneration. Upon vascular injury, these EVs exhibit innate tropism for chemokine-enriched sites at damaged endothelium, enabling targeted therapeutic delivery[75]. M2-polarized macrophage EVs deliver anti-inflammatory cytokines and specific miRNAs (such as miR-99a/146b/378a) that suppress pathological inflammation, reduce atherosclerotic plaque burden, and remodel the local microenvironment to facilitate vascular regeneration[76]. Furthermore, macrophage membrane-engineered nanocarriers, including rapamycin-loaded macrophage membrane-coated nanoparticles, leverage this intrinsic targeting capability to deliver therapeutics precisely to inflamed endothelium, effectively inhibiting neointimal hyperplasia with minimal systemic toxicity, as demonstrated in AS models[77]. Thus, macrophage EVs function dually as endogenous immunomodulators and engineered platforms for targeted vascular therapy.
PMPs significantly enhance vascular endothelial repair and regeneration[78]. These vesicles transfer bioactive cargo-including nucleic acids, VEGF, and other signaling molecules-to recipient cells. PMPs promote angiogenesis by augmenting endothelial cell proliferation, migration, and capillary-like structure formation, primarily through VEGF-mediated signaling pathways[79,80]. Inheriting the inherent vascular tropism of parental platelets, PMPs serve as precision nanocarriers for targeted drug delivery (such as paclitaxel, rapamycin). This capability attenuates coronary restenosis and atherosclerotic progression while modulating post-injury inflammatory responses, thereby creating a regenerative microenvironment for endothelial restoration[81,82].
MSC-EVs critically contribute to vascular repair through their multifaceted regenerative properties. Enriched in anti-inflammatory and pro-angiogenic miRNAs[83], these EVs accelerate re-endothelialization post-stent implantation[84] and mitigate cardiac injury. Specifically, miR-182-5p in MSC-EVs targets gasdermin D (GSDMD) to suppress inflammation and oxidative stress, reducing myocardial infarct size while enhancing cardiac contractility[85]. Additionally, MSC-EVs polarize macrophages toward pro-repair M2 phenotypes, attenuating vascular inflammation[86]. This synergistic modulation of inflammation, angiogenesis, and cellular repair underpins their therapeutic efficacy in cardiovascular regeneration.
EVs coordinate vascular endothelial repair through multistage mechanisms involving inflammation modulation, angiogenesis promotion, and cellular regeneration. The source-specific functions of
Regulation of angiogenesis and formation of collateral circulation
Vascular remodeling is an integrative process that entails not only the formation of new microvessels (angiogenesis) but also the phenotypic and functional adaptation of VSMCs. EVs have emerged as master coordinators of this process, enabling simultaneous communication with both ECs and VSMCs to ensure harmonious tissue repair or, conversely, to drive disease progression. This section details the mechanisms by which EVs regulate EC-driven angiogenesis and integrates how these processes are intrinsically coupled with the modulation of VSMC behavior, highlighting the sophisticated dual-cell targeting that underpins holistic vascular remodeling.
The regulatory role of EVs in the formation of collateral circulation
EVs promote collateral circulation reconstruction in ischemic tissues through synergistic multi-mechanisms. In chronic myocardial ischemia models, MSC-EVs significantly enhance endothelial cell proliferative activity and neovascularization capacity by dual activation of the VEGF signaling pathway and its downstream VEGFR2/MAPK ERK1/ERK2 phosphorylation cascade. However, it is noteworthy that high-fat diet-induced metabolic syndrome can substantially alter the biological properties of EVs, impairing their pro-angiogenic function, as evidenced by aberrantly increased FOXO1 transcriptional activity and dysregulated endothelial nitric oxide synthase (eNOS) phosphorylation, which consequently interferes with vascular endothelial growth factor A (VEGF-A)-mediated endothelial repair processes[87]. Although metabolic disorders compromise endogenous EV function, exogenous endothelial progenitor cell-derived EVs (EPC-EVs) can effectively restore damaged vascular barrier function through specific delivery of protective miRNAs[88], providing a critical theoretical basis for developing engineered EVs to overcome pathological microenvironment limitations. More importantly, EV therapy can significantly downregulate the expression levels of angiogenesis inhibitors (including angiostatin and endostatin), thereby relieving their negative regulatory effects on neovascularization and ultimately improving microcirculatory perfusion in ischemic myocardial tissue[88]. These results delineate the multi-dimensional mechanisms of EV-driven ischemic vascular reconstruction, highlighting key signaling nodes, including VEGF/extracellular
The therapeutic potential of EVs in vascular barrier repair has been well-documented in various pathological models. In glomerulonephritis models, endothelial progenitor cell (EPC)-derived EVs efficiently deliver bioactive miRNAs and protein components through L-selectin-mediated specific endocytic pathways. These effector molecules exert therapeutic effects via dual regulatory mechanisms: (1) significantly suppressing TNF-α- and IL-6-induced overexpression of VCAM-1/ICAM-1, thereby effectively reducing inflammatory leukocyte infiltration; and (2) upregulating VEGF-A and hepatocyte growth factor (HGF) expression to promote structural and functional reconstruction of the glomerular filtration barrier[89]. In intracerebral hemorrhage models, proteomic profiling of EVs reveals their dynamic changes strongly correlate with clinical outcomes: circulating EVs from patients with a favorable prognosis are significantly enriched with pro-angiogenic proteins (such as RHG01) during the subacute phase, while those from poor-prognosis patients overexpress inflammatory markers such as C-reactive protein (CRP). This finding not only confirms the temporal dynamics of EV molecular cargo but also suggests that EV proteome reprogramming directly influences collateral circulation compensation capacity[90]. Collectively, these results elucidate the molecular mechanisms by which EVs repair vascular barrier function through multi-target regulatory networks, providing important theoretical foundations for developing EV-based vascular protective strategies.
At the molecular regulation level, EVs precisely modulate angiogenic signaling pathways through the
Cumulative evidence demonstrates that EVs precisely regulate angiogenesis and collateral circulation formation through complex, multi-target signaling networks operating at multiple levels[96]. These regulatory effects exhibit remarkable microenvironment-dependent characteristics, wherein pathological conditions including hypoxia, inflammation, and metabolic disorders dynamically reshape EV biological properties. Importantly, bioengineering approaches enabling directed modification of EVs can significantly enhance their tissue tropism and therapeutic efficacy, providing innovative solutions to overcome pathological microenvironment constraints. These systematic research findings not only advance our understanding of vascular regeneration mechanisms but also establish a solid theoretical foundation for developing novel therapeutic strategies against ischemic diseases, holding significant clinical translation potential.
EVs drive angiogenesis by EV-mediated transference of pro-angiogenic factors
EVs drive angiogenesis through the coordinated delivery of both pro-angiogenic proteins and regulatory miRNAs[97]. EVs, serving as pivotal mediators of intercellular communication, exert dual regulatory effects on both physiological and pathological angiogenesis. Accumulating evidence demonstrates that tumor-derived EVs can potently drive pathological angiogenesis through their specific molecular cargo. For instance, EVs secreted by lung adenocarcinoma H1975 cells efficiently deliver the transcriptional coactivator Yes - associated protein (YAP) protein, which dose-dependently enhances the pro-angiogenic phenotype of human umbilical vein endothelial cells (HUVECs). Notably, siRNA-mediated knockdown of YAP completely abolishes EV-induced vascular formation capacity, definitively establishing YAP as an indispensable effector molecule in EV-mediated vascular remodeling[98]. Further clinical investigations reveal significantly elevated levels of EV-associated pro-angiogenic factors angiopoietin-2 (ANGPT2) and cell migration-inducing protein (CEMIP) in the plasma of lung cancer patients with brain metastases. These molecules cooperatively promote endothelial cell migration and tube formation, thereby facilitating the establishment of metastatic niches. Notably, postoperative dynamic reduction of these factors shows significant correlation with improved patient outcomes. Mechanistically, such EVs aberrantly activate the YAP/HIF-1α signaling pathway to induce pathological vascular proliferation, exhibiting striking similarities to the neovascularization process observed in atherosclerotic plaques[99]. These findings systematically elucidate the conserved molecular mechanisms by which EVs drive aberrant angiogenesis across pathologies such as AS, but also provide a rationale for developing combined therapeutic strategies targeting ANGPT2/YAP, with their potential as non-invasive biomarkers further expanding clinical translation prospects.
In the field of therapeutic angiogenesis, EVs derived from various stem cell sources have demonstrated remarkable potential in promoting vascular regeneration. Compelling evidence shows that EVs secreted by human induced pluripotent stem cell-derived cardiomyocyte (hiPSC)-CMs significantly enhance endothelial cell function through paracrine mechanisms, as evidenced by their ability to promote tube formation, accelerate wound healing, and augment cellular proliferation. Notably, these EVs can upregulate the expression of multiple pro-angiogenic growth factors in endothelial cells, thereby optimizing the angiogenic microenvironment through multi-target synergistic effects[100]. Further investigations reveal that cardiac mesenchymal stromal cell (CMC)-derived EVs are selectively enriched with Ang-1 and Ang-2, which specifically activate the Tie2 receptor and its downstream STAT3 and PI3K-activate the protein kinase B (Akt) signaling pathways, leading to significantly improved endothelial cell migration and survival rates. Of particular interest, pharmacological inhibition of Tie2 kinase completely abolishes Ang-1-mediated pro-migratory effects, a finding that not only confirms the molecular specificity of ligand-receptor interactions but also provides a theoretical basis for targeted therapy[101]. In diabetic vascular complication models, adipose-derived stem cell EVs (ADSC-EVs) effectively activate the PI3K/AKT signaling cascade through CCN2 protein delivery, thereby improving endothelial cell function. Additionally, hypoxia-preconditioned olfactory mucosa mesenchymal stem cell (OM-MSC) EVs are enriched with miR-612, which potently suppresses TP53 gene expression, consequently relieving its negative regulation on HIF-1α and ultimately activating the HIF-1α-VEGF signaling axis to significantly enhance the angiogenic capacity of brain microvascular endothelial cells[102]. These findings systematically elucidate the distinct molecular mechanisms through which stem cell-derived EVs promote vascular regeneration, offering novel strategic options for the treatment of ischemic diseases.
Cell-type-specific effects of EVs in vascular remodeling
EVs orchestrate vascular remodeling through sophisticated cell-type-specific regulatory mechanisms. As master coordinators of intercellular communication, EVs demonstrate remarkable functional plasticity in modulating endothelial cell (EC) biology and VSMC phenotype, with their effects being critically determined by cellular origin and microenvironmental context.
EVs precisely regulate endothelial cell function through multifaceted molecular mechanisms governing proliferation, migration, and differentiation - fundamental processes in angiogenesis. The impact on EC proliferation exhibits significant context-dependency: while mesenchymal stem cell-derived EVs generally promote proliferation through mechanisms such as lysyl oxidase homolog 2 (LOXL2)-mediated extracellular matrix remodeling in ischemic myocardium[103], EVs from specialized sources such as human corneal endothelial cells paradoxically inhibit proliferation via unique miRNA profiles[104]. This functional heterogeneity extends to migratory regulation, where hypoxia-preconditioned olfactory mucosa MSC-EVs enhance migration through the miR-612/TP53/HIF-1α/VEGF axis[102], and ultrasound-stimulated adipose-derived stem cell EVs promote wound healing via miR-148a-3p enrichment[105]. Conversely, pathological EVs from preeclamptic trophoblasts impair migration through miR-150-3p-mediated chondroitin polymerizing factor (CHPF) targeting[106]. In differentiation control, EVs demonstrate bidirectional regulation-JAG1-enriched EVs suppress vasculogenic potential by inhibiting Notch1 activation[106], while cardiac MSC-EVs promote functional vascular differentiation through miR-210-mediated Ephrin-A3 suppression and VEGF pathway activation[107]. Notch-activated EVs further enhance neovessel stability through biglycan-mediated structural maturation, illustrating the sophisticated multi-level control of vascular development. In addition, MSC-derived EVs transfer protective miRNAs such as miR-146a-5p to promote the contractile phenotype, while EC-derived EVs deliver miR-126-5p to suppress aberrant proliferation, thereby collectively maintaining vascular homeostasis[62,89].
However, pathological microenvironments reprogram EV function toward disease promotion: Lipopolysaccharide (LPS)-activated EC-EVs deliver miR-92a-3p to drive atherosclerotic progression through human aortic smooth muscle cell (HASMC) proliferation[108], while activated cell-derived EVs in hypertensive conditions induce synthetic phenotypes via mediators such as miR-221-3p or IL-1β[109]. Crucially, EV-mediated effects on VSMCs are intrinsically coupled with EC-driven angiogenesis, enabling coordinated vessel maturation through dual-cell targeting that ensures mechanistic integration of luminal formation and wall remodeling processes[110].
The role of EVs in adventitial angiogenesis and vasa vasorum remodeling during atherosclerosis
Beyond the intima, emerging evidence suggests that the adventitia may play an active role in the early stages of AS, potentially contributing to an “outside-in” pathological pathway[111]. This process is thought to be largely driven by pathological angiogenesis of the VV and perivascular inflammation. Extracellular vesicles are hypothesized to be key mediators in this cascade[15].
In the hypoxic and inflammatory microenvironment of a developing atherosclerotic plaque, activated adventitial fibroblasts, macrophages, and other cells may release EVs loaded with pro-angiogenic cargo (e.g., VEGF, MMPs, and miRNAs such as miR-210 and miR-132)[112]. These EVs could then facilitate communication with the existing VV endothelium, potentially stimulating excessive and aberrant neovessel formation[113]. This expanded microvascular network is believed to facilitate the delivery of inflammatory cells and lipids into the vessel wall, thereby exacerbating plaque growth. Furthermore, the leaky nature of these neovessels might contribute to intraplaque hemorrhage, which deposits erythrocyte-derived cholesterol and fuels local inflammation, potentially increasing plaque vulnerability[114].
Thus, EV-driven adventitial angiogenesis is increasingly recognized as a potential “outside-in” pathway in AS, and targeting this specific EV-mediated communication could represent a novel therapeutic avenue for stabilizing vulnerable plaques.
Regulating the phenotype and function of VSMCs
Regulation of phenotypic transformation of VSMCs by EVs
As crucial mediators of intercellular communication, EVs play a decisive role in regulating VSMC phenotypic plasticity. Emerging evidence indicates that endothelial cell-derived EVs (EC-EVs) dynamically modulate VSMC phenotypic switching via their specific molecular cargo. Under physiological homeostasis, EC-EVs are significantly enriched with high-mobility group box 1/2 (HMGB1/2)-damage-associated molecular pattern proteins (DAMPs) that dose-dependently upregulate VCAM-1 expression in VSMCs, thereby promoting leukocyte-endothelial adhesion and accelerating cellular senescence. Many studies show that pharmacological inhibition (e.g., glycyrrhizin treatment) or clustered regularly interspaced short palindromic repeats (CRISPR)/associated protein 9 (Cas9)-mediated knockout of HMGB1/2 signaling completely abolishes EC-EV-induced VSMC phenotypic modulation, unequivocally confirming HMGB1/2’s central role in this regulatory network. In-depth proteomic profiling further identifies multiple effector proteins carried by EC-EVs-including S100 calcium-binding protein family members and MMPs-which may cooperatively trigger VSMC inflammatory responses and drive phenotypic switching, providing novel insights into the molecular mechanisms of vascular remodeling[115].
Beyond DAMPs, EVs act as vehicles for intercellular transfer of fully functional transmembrane receptors and enzymes, directly endowing recipient VSMCs with new pathological capabilities. A notable example is the transfer of MMPs such as MMP-2 and MMP-14 (MT1-MMP). EVs from activated macrophages or endothelial cells carry these proteases on their surface in an active form; upon fusion with VSMCs,
Besides lipid metabolic disorders, EC-EVs also play a critical role in mediating VSMC damage under diabetic conditions. Studies show that diabetic EC-EVs carry specific cargo that promotes VSMC transition to a proinflammatory, synthetic state, accelerating diabetic vascular remodeling and injury. For example, under diabetic hyperglycemic conditions, EC-EVs are specifically enriched with circRNA hsa_circ_0008362. Upon uptake by VSMCs, this circRNA facilitates osteogenic/calcified phenotypic transition via two synergistic mechanisms: (1) acting as a molecular sponge for miR-1251-5p to relieve repression of Runx2, and (2) directly binding Runx2 to enhance its stability. This significantly exacerbates diabetic arterial calcification[116]. This evidence highlights diabetes as a key pathological context for EC-EV-mediated VSMC damage, broadening the scope of EC-EV mechanisms in metabolic vasculopathies.
The aforementioned proinflammatory and phenotypic-switching effects of EVs on VSMCs are central to the pathogenesis of aortic aneurysms and dissections. EVs from activated endothelium or macrophages deliver pro-apoptotic signals (e.g., HMGB1/2), matrix-degrading proteins, and proinflammatory cytokines, directly promoting VSMC apoptosis, acquisition of a synthetic phenotype, and extracellular matrix degradation-key processes driving medial layer degeneration and aortic wall weakening[115]. Conversely, as previously detailed, the therapeutic potential of MSC-EVs to suppress VSMC apoptosis and maintain the contractile phenotype offers a promising strategy to stabilize the aortic wall in these life-threatening conditions.
The role of EVs on the proliferation, migration and contraction functions of VSMCs
EVs exhibit a highly hierarchical molecular regulatory architecture in regulating VSMC functional activation. Regarding pro-activation regulation, EVs from various pathological sources have been demonstrated to promote VSMC proliferation and migration through distinct molecular mechanisms. Angiotensin II (Ang II)-stimulated human umbilical vein endothelial cell (Ang II)-derived EVs significantly enhance VSMC proliferation rate, migratory capacity, and proinflammatory factor secretion in a
In the inhibitory regulation of aberrant VSMC activation, therapeutic EVs demonstrate unique therapeutic advantages and functional specificity. MSC-derived EVs significantly attenuate neointimal hyperplasia by 60%-70% in vein graft models through multi-target inhibition of Akt/ERK/p38 pro-proliferative signaling pathway phosphorylation[118]. Genetically modified Notch1-overexpressing cardiac mesenchymal stem cell (C-MSC)-secreted EVs exhibit distinct molecular cargo characteristics, being specifically enriched with extracellular matrix remodeling proteins such as LOXL2, which not only effectively promote vascular network formation but also significantly improve contractile function in ischemic myocardium[119]. More importantly, neural stem cell-derived EVs multidimensionally suppress PASMC aberrant proliferation and phenotypic switching in pulmonary hypertension models through synergistic delivery of inhibitory miRNA networks including let-7b-5p, miR-92b-3p and miR-100-5p, demonstrating superior therapeutic efficacy compared to single miRNA intervention[120]. These findings systematically elucidate the molecular basis by which therapeutic EVs inhibit VSMC abnormal activation through multiple mechanisms, including signaling pathway suppression, matrix remodeling, and epigenetic regulation, providing novel interventional strategies for the treatment of vascular proliferative disorders.
EVs demonstrate unique molecular mechanisms in the precise regulation of VSMC contractile function. Research reveals that synthetic phenotype VSMC-derived EVs significantly influence target cell function through dual delivery mechanisms: (1) directly altering intracellular calcium homeostasis via bioactive calcium ion transport; and (2) specifically increasing intracellular reactive oxygen species (ROS) generation through nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 5 (NOX5) transfer, establishing a persistent positive feedback loop that ultimately accelerates VSMC calcification and progressively impairs contractile function[121]. More intriguingly, during obesity-associated pathological processes, endothelial cell-derived EVs mediate endothelial-to-mesenchymal transition (EndoMT) by transferring specific functional signatures and metabolic reprogramming signals to VSMCs. This intercellular communication not only modifies VSMC phenotypic characteristics but also further impacts contractile function maintenance through metabolic pathway remodeling[122]. These findings dissect the synergistic network of EV-mediated VSMC contractility regulation via ion homeostasis, oxidative stress, and metabolic reprogramming, identifying critical targets for precision interventions to reverse vascular dysfunction.
The EV-mediated dysregulation of VSMC contractility, as detailed above, involving calcium mishandling, ROS generation, and metabolic reprogramming, represents a key mechanism contributing to the progression of aortic dissection and aneurysm. The impairment of VSMC contraction and the promotion of a synthetic phenotype by pathological EVs can critically compromise aortic wall integrity and tone regulation[121]. Furthermore, the capacity of engineered therapeutic EVs, such as those from MSCs, to improve VSMC contractile function and promote a contractile phenotype, as previously discussed, highlights their potential therapeutic value for these aortic pathologies[123].
The regulatory role of EVs in VSMCs during vascular calcification and inflammatory responses
EVs establish a highly sophisticated regulatory network in vascular calcification and inflammatory responses. During vascular calcification progression, pathologically stimulated VSMC-derived EVs are specifically enriched with mineralization-related effector molecules, including tissue-nonspecific alkaline phosphatase (TNAP) and calcium-binding proteins, which promote ectopic hydroxyapatite crystal deposition through both autocrine and paracrine mechanisms, playing a decisive role in atherosclerotic plaque microcalcification formation[124-126]. Notably, EVs from different anatomical locations exhibit distinct tissue-specific regulatory patterns: carotid plaque-derived EVs primarily mediate their pro-calcific effects through Notch signaling, while those from aortic valve calcification preferentially activate the canonical Wnt/β-catenin pathway, providing a molecular basis for understanding vascular calcification heterogeneity[127]. More importantly, pro-calcific factors such as lipoprotein(a) {Lp[a]} dynamically remodel EV molecular composition, significantly promoting CD29+ microvesicle release. These EVs demonstrate enhanced calcification propensity and mineralization activity within collagen matrices, potentially serving as key mediators in early arterial calcification initiation events[126,128]. These findings systematically reveal the precise mechanisms by which EVs participate in vascular calcification regulation through tissue-specific signaling pathways and dynamic molecular reprogramming, providing novel theoretical foundations for developing targeted anti-calcification therapeutic strategies.
Inflammatory responses and extracellular vesicle (EV)-mediated vascular calcification processes form a mutually reinforcing pathological cycle. LPS-activated macrophages release EVs enriched with proinflammatory cytokines and oxidative stress-related proteins, which dose-dependently activate the
Targeted therapeutic strategies leveraging the heterogeneous characteristics of EVs demonstrate remarkable translational potential in vascular disease intervention. State-of-the-art engineered biomimetic EVs, surface-modified with hydroxyapatite-binding peptide (HABP), constitute a precision delivery system for sodium thiosulfate (STS) with dual therapeutic advantages: (1) achieving local drug enrichment through specific targeting of calcified regions to effectively inhibit osteogenic-like differentiation of VSMCs; and (2) modulating macrophage polarization toward the anti-inflammatory (M2) phenotype, thereby simultaneously ameliorating vascular calcification and inflammatory responses[130,132]. Furthermore, exogenous supplementation of γ-carboxylated Gla-rich protein (GRP) can specifically inhibit the nucleation and deposition of calcifying crystals, providing innovative molecular targets for developing EV-based targeted delivery systems[130,131]. These breakthroughs decode EV heterogeneity and tissue-specific regulation patterns, translating the targeted therapeutic potential of engineered EVs into clinical solutions against the vicious cycle of vascular calcification-inflammation.
The EV-mediated crosstalk between inflammation and calcification described above is highly relevant to the pathogenesis of aortic aneurysm and dissection. The capacity of inflammatory EVs to promote VSMC osteogenic transition and matrix degradation, coupled with their role in sustaining a proinflammatory microenvironment through macrophage polarization, can significantly contribute to medial layer degeneration and aortic wall weakening[121,133]. The emerging strategies using engineered EVs to target calcification and inflammation simultaneously, as detailed in this section, may therefore hold particular promise for mitigating the progression of these aortic diseases.
EVs exhibit complex dual regulatory properties in vascular pathologies such as AS and hypertension: while promoting aberrant VSMC activation and phenotypic switching through delivery of pro-proliferative and pro-calcific molecules, they simultaneously exert vasculoprotective effects by maintaining endothelial homeostasis and suppressing inflammatory responses. This functional duality suggests that developing specific EV-based intervention strategies-either inhibiting the detrimental effects of pathological EVs or enhancing the beneficial actions of therapeutic EVs-based on the molecular signatures of distinct vascular lesions holds significant clinical translational value. Current research has demonstrated that precise modulation of EV biogenesis or engineered modification of their molecular cargo enables targeted intervention in vascular disease progression, providing innovative approaches for developing
EVS AS A PROMISING THERAPEUTIC TOOL
Unique advantages of EVs as a novel therapeutic tool
EVs exhibit distinct therapeutic advantages over conventional therapies for vascular pathologies, underpinned by unique biological properties that accelerate clinical translation. A primary advantage lies in their generally favorable biocompatibility profile. EVs derived from autologous or human-derived sources (e.g., mesenchymal stem cells) typically demonstrate low immunogenicity, a feature attributed to their endogenous origin and the preservation of “self” surface markers, which reduces the risk of rapid clearance and adverse immune reactions[83,134]. However, it is critical to contextualize this advantage; immunogenicity is not absolute and is highly dependent on the EV source (xenogeneic vs. allogeneic vs. autologous), dose, administration route, and the recipient's immune status. EVs from non-human sources or allogeneic donors may still elicit immune responses, and concerns regarding the potential transfer of immunogenic cargo or the provocation of proinflammatory effects in certain microenvironments warrant careful evaluation. Their capacity to traverse physiological barriers (e.g., blood-brain barrier, vascular endothelium) enables targeted delivery to ischemic tissues, overcoming a fundamental limitation of traditional therapeutics. Functioning as versatile molecular shuttles for bioactive cargoes (nucleic acids, proteins, lipids), EVs facilitate
The therapeutic potential of EVs is highly dependent on their cellular origin, and a comprehensive evaluation of major sources is essential. Among stem cell-derived EVs, MSCs remain the most widely investigated, with evidence supporting the efficacy of EVs from diverse MSC populations. Adipose-derived stem cell extracellular vesicles (ASC-EVs) are particularly promising due to their high yield, minimally invasive acquisition, and enrichment with pro-angiogenic factors such as VEGF, basic fibroblast growth factor (bFGF), and miR-31, as demonstrated in preclinical models of peripheral artery disease and myocardial ischemia[136]. Bone marrow MSC-EVs (BMSC-EVs) are well characterized for their immunomodulatory functions mediated via TGF-β and PGE2 pathways, as well as their efficacy in vascular repair in hindlimb ischemia models. Umbilical cord MSC-EVs (UC-MSC-EVs), characterized by primitive marker expression, potent pro-angiogenic and tissue-reparative capacities, and low immunogenicity, hold promise for allogeneic applications in vascular repair and broader tissue injuries[137], while placental-derived MSC-EVs represent an abundant source with promising angiogenic effects in diabetic wound healing[138]. Furthermore, EVs from other stem cell sources are gaining increasing attention, such as induced pluripotent stem cell-derived extracellular vesicles (iPSC-EVs), which are increasingly recognized as a breakthrough in regenerative medicine, offering virtually unlimited scalability, potent pro-angiogenic capacity comparable to embryonic stem cells, and validated therapeutic efficacy in myocardial infarction models, while circumventing ethical limitations[139]. Additionally, dental pulp stem cell-derived EVs (DPSC-EVs), originating from neural crest-derived cells, have shown remarkable angiogenic and neuro-regenerative capabilities, making them particularly promising for the treatment of peripheral neuropathy and ischemic tissue repair[140].
Notably, beyond stem cell sources, serum-derived EVs have also emerged as clinically significant therapeutic agents, with a growing body of evidence from multiple independent studies confirming their potent pro-angiogenic capacity. These circulating vesicles are enriched in a diverse repertoire of bioactive signaling molecules-including functional microRNAs such as miR-126-3p, miR-143-3p, and miR-939, as well as key proteins such as HIF-1α and VEGF-A. By efficiently delivering this cargo to recipient cells, serum EVs robustly promote endothelial cell migration, proliferation, and tube formation, orchestrating a pro-regenerative response.
This therapeutic potential has been consistently demonstrated across various preclinical models of vascular injury. For instance, it has been demonstrated that peripheral serum exosomes from patients with acute myocardial infarction (AMI) significantly enhanced angiogenesis both in vitro and in vivo by delivering miR-126-3p, which targeted TSC1 to activate the mTORC1/HIF-1α signaling axis[141]. Similarly, exosomes derived from the coronary serum of AMI patients were reported to promote endothelial network formation through the miR-143/IGF-IR pathway[142]. Furthermore, coronary serum exosomes from patients with myocardial ischemia were shown to regulate angiogenesis via a miR-939-mediated nitric oxide signaling mechanism[143]. The role of miR-126-3p is further underscored by work that highlighted its critical function in exosome-mediated repair of ischemic hindlimbs[144]. It is important to note that the protein cargo (e.g., VEGF-A, IGF-1) within these serum EVs likely acts in concert with miRNAs to produce the observed
The expanding therapeutic landscape of EVs now includes not only naturally derived EVs but also engineered EVs tailored through surface modification or cargo loading, offering enhanced targeting and potency for specific vascular pathologies. These translational attributes are actively being validated in human studies. Notably, the SER-VES-HEAL trial (NCT04652531) represents a significant step in this direction, evaluating the efficacy of serum-derived EVs in patients with venous trophic lesions. Published findings from this study have demonstrated that topical application of EV-based therapy promoted healing of refractory venous ulcers, likely through mechanisms involving enhanced angiogenesis and modulation of the local inflammatory microenvironment. This clinical evidence provides crucial proof-of-concept for the therapeutic application of native EVs in vascular repair, bridging the gap between robust preclinical data and human efficacy.
However, a critical evaluation of the SER-VES-HEAL trial reveals several important considerations. As summarized in Table 1, this trial, together with several other ongoing clinical studies, is investigating
Clinical research on the association between EVs and vascular diseases
| Classification | Name | Institute | Related vascular disease | Stage | NCT number |
| Arterial diseases | AFFECT EV | Medical University of Warsaw | Acute myocardial infarction | Phase 4 | NCT02931045 |
| STEVIA | S&Ebio Co. Ltd. | Acute ischemic stroke | Phase 1 | NCT06995625 | |
| PRISMA | Fondazione Don Carlo Gnocchi Onlus | Stroke and brain injury | / | NCT06871800 | |
| EXO4STROKE | Fondazione Don Carlo Gnocchi Onlus | Stroke | / | NCT05370105 | |
| EXOPLT | Centro Cardiologico Monzino | Vascular disease | / | NCT06298682 | |
| Diabetic vascular complications | EVDFUUJCTC | University of Jordan | Diabetic foot ulcers | Phase 1 | NCT06825884 |
| EVs, Insulin Action, and Exercise | Rutgers, The State University of New Jersey | Type 2 diabetes related cardiovascular disease | Not Applicable | NCT06546085 | |
| Venous diseases | SER-VES-HEAL | University of Turin, Italy | Venous trophic lesions | Not Applicable | NCT04652531 |
| Congenital cardiovascular diseases | EVmiRNA | Boston Children’s Hospital | Congenital heart disease | / | NCT06434207 |
Optimization strategies for engineering EVs
Engineering strategies enhance the therapeutic efficacy of EVs for vascular diseases through multifaceted optimization. Genetic modification of donor cells - for instance, overexpression of CCND2 - augments EV bioactivity, thereby promoting cardiomyocyte proliferation and angiogenesis[145]. Post-isolation surface engineering, such as conjugation of Lamp2b-fused targeting peptides (e.g., CSTSMLKAC), improves
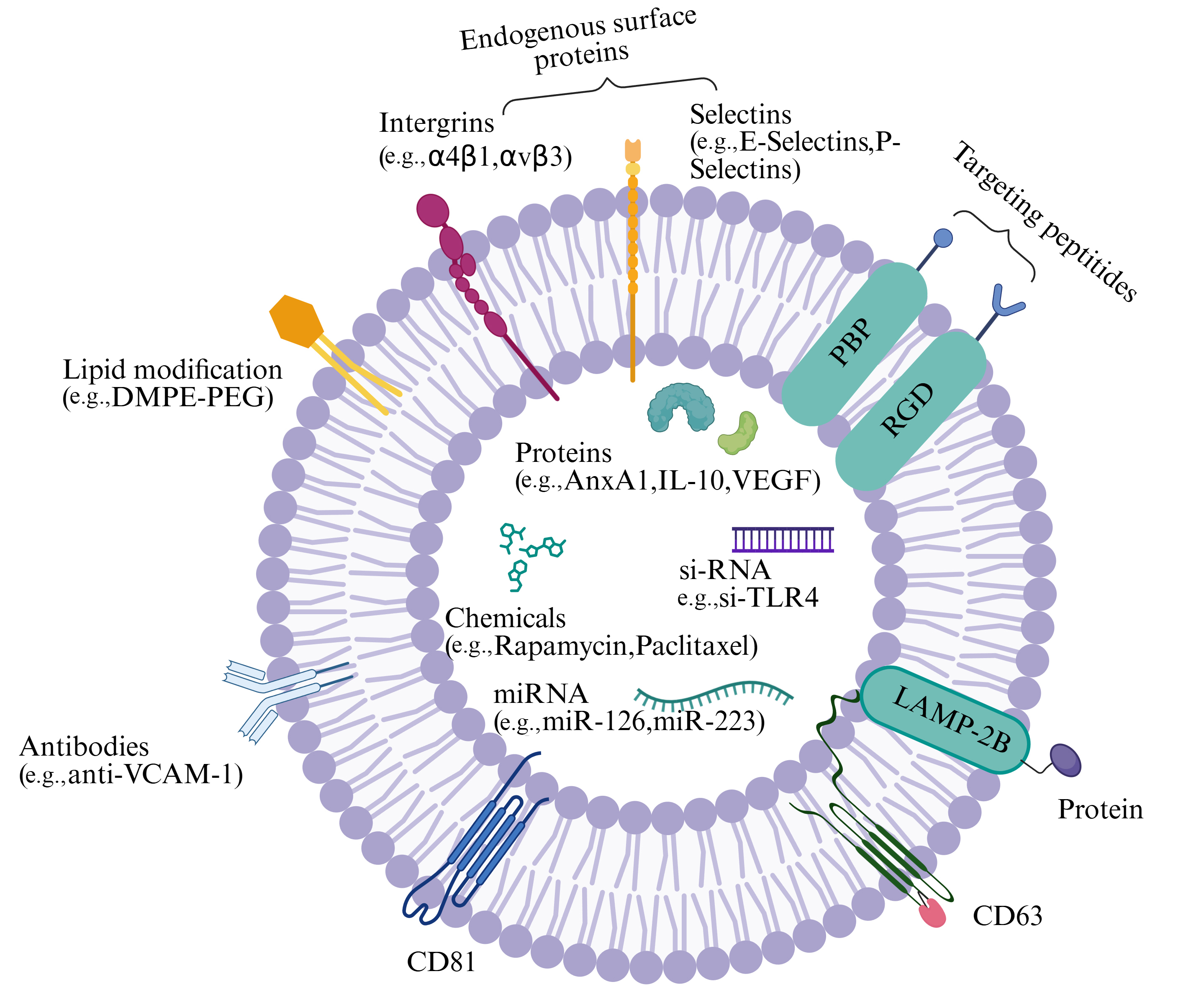
Figure 3. Engineering EVs for vascular disease therapy. EVs can be engineered to incorporate diverse bioactive agents, including chemicals, nucleic acids, peptides, and proteins. Created in BioRender. gN, lý. (2025) https://BioRender.com/ltqoohy. EVs: Extracellular vesicles; VCAM-1: vascular cell adhesion molecule-1; AnxA1: annexin A1; VEGF: vascular endothelial growth factor; si-TLR4: small interfering RNA targeting Toll-like receptor 4; DMPE-PEG: 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-polyethylene glycol; PBP: proline-rich bactericidal peptide; RGD: arginine-glycine-aspartic acid; LAMP-2B: lysosome-associated membrane protein 2B.
PROSPECT
EVs possess unprecedented potential to transform the diagnosis, treatment, and management of cardiovascular diseases; however, substantial challenges must be addressed to fully capitalize on this promise. A core hurdle lies in inherent EV heterogeneity, which complicates functional characterization and undermines the standardization of clinical-grade production-current isolation methodologies struggle to concurrently achieve high purity, yield, and preservation of functional integrity[151]. Additionally, the efficiency and controllability of EV engineering remain limited, whether through donor cell modification or direct manipulation of EVs for cargo loading or targeted delivery. Compounding this, immature scalable manufacturing processes drive up costs, hindering translational progress[152]. Systemic delivery also faces critical limitations: EVs are rapidly cleared by the mononuclear phagocyte system (MPS) and exhibit poor specific accumulation in target tissues (such as myocardium, vasculature), leading to off-target effects that compromise efficacy and safety[153]. Safety concerns are further amplified by the lack of unified protocols for EV characterization, quality control, and standardization, including risks associated with xenogeneic EV immunogenicity and unintended delivery of harmful cargo. Finally, an incomplete understanding of EV biogenesis, cellular uptake mechanisms, and cargo function within the cardiovascular microenvironment remains a major barrier to rational engineering optimization[154]. Overcoming these obstacles will require robust interdisciplinary collaboration, which is essential to unlocking EVs’ clinical potential in vascular therapy.
The future advancement of engineered EVs for vascular disease applications depends on integrating multidisciplinary expertise to surmount current limitations and unleash their therapeutic promise. Key strategic priorities include: (1) optimizing EV sources and isolation techniques to enhance biocompatibility and tissue tropism, enabling targeted delivery of therapeutic cargo to diseased vasculature[155,156]; (2) advancing genetic engineering and surface modification approaches to precisely load regulatory molecules that modulate endothelial function, suppress inflammation, and promote angiogenesis/repair[157,158]; (3) developing sustained-release delivery systems using biocompatible biomaterials to improve long-term efficacy, particularly for chronic vascular conditions[158]; and (4) innovating scalable production technologies (e.g., to enhance drug loading efficiency and batch uniformity) alongside rigorous efficacy and mechanistic validation in complex, clinically relevant vascular disease models[159]. Systematically exploring and integrating these strategies will be critical to boosting therapeutic efficacy, resolving translational bottlenecks, and advancing precision medicine for vascular disorders.
CONCLUSION
As natural nanoscale signaling mediators, EVs exhibit dual potential in vascular pathobiology: they serve as highly specific non-invasive diagnostic biomarkers and promising targeted therapeutic vehicles. By delivering bioactive molecules-including proteins, nucleic acids, and lipids-EVs precisely modulate vascular inflammatory-immune responses, promote endothelial repair and regeneration, facilitate angiogenesis and collateral vessel formation, and regulate VSMC phenotypic switching and function.
In this paper, we emphasize that the protein cargo of EVs-such as cytokines, receptors, enzymes, and structural proteins-is not merely ancillary but often plays a dominant, direct role in these processes, as illustrated by the transfer of functional AT1R, MMPs, and HMGB1/2. This multifaceted activity enables EVs to intervene in the shared pathological mechanisms underlying major vascular diseases, including AS, hypertension, and PAH. Engineering strategies-such as targeting peptide conjugation and functional cargo loading-further enhance EV enrichment at lesion sites and therapeutic efficacy, offering novel approaches to overcome the limitations of conventional therapies. While clinical translation faces hurdles related to EV heterogeneity, scalable production, and in vivo delivery efficiency, ongoing innovations in isolation technologies, multi-omics analyses, and biomaterial integration hold substantial promise for establishing engineered EVs as transformative tools for the precision diagnosis and treatment of vascular disorders.
DECLARATIONS
Acknowledgments
We thank all members of the Han laboratory for their critical comments and helpful suggestions. We apologize for being unable to cite many important papers in this field due to space limitations. The graphical abstract was created with BioRender.com (Created in BioRender. gN, lý. (2025) https://BioRender.com/qrqe1ga).
Authors’ contributions
Drafted the manuscript: Wang Y, Zhou Y
Constructed a framework: Wang H
Provided important instructions: Li K
Drew diagrams: Hao Q
Collected materials: Li P,
Revised the paper: Guan L, Zhou X
Supervised the project and revised the paper: Gao B
Provided critical revisions for important intellectual content, supervised the review, and is responsible for correspondence regarding the work: Han T
All authors read and approved the final manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported in part by the International Science and Technology Cooperation Projects in Henan Province (252102521001 to Han T), the National Natural Science Foundation of China (82172891 to Han T, and 82273098 to Zhou X), the Talent Support Plan of Xinxiang Medical University (505559 to Han T), and the Doctoral Foundation of Xinxiang Medical University (XYBSKYZZ202001 to Han T).
Conflicts of interest
Gao B from Umibio Co. Ltd., Shanghai, supervised the project and revised the manuscript. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Bashatah A, Syed W, Al-Rawi MBA. Knowledge of cardiovascular disease risk factors and its primary prevention practices among the saudi public - a questionnaire-based cross-sectional study. Int J Gen Med. 2023;16:4745-4756.
2. Higashi Y. Roles of Oxidative Stress and inflammation in vascular endothelial dysfunction-related disease. Antioxidants. 2022;11:1958.
3. Cao G, Lu Z, Gu R, et al. Deciphering the intercellular communication between immune cells and altered vascular smooth muscle cell phenotypes in aortic aneurysm from single-cell transcriptome data. Front Cardiovasc Med. 2022;9:936287.
4. Tang HY, Chen AQ, Zhang H, Gao XF, Kong XQ, Zhang JJ. Vascular smooth muscle cells phenotypic switching in cardiovascular diseases. Cells. 2022;11:4060.
5. Buzas EI. The roles of extracellular vesicles in the immune system. Nat Rev Immunol. 2023;23:236-50.
6. Boyer MJ, Kimura Y, Akiyama T, et al. Endothelial cell-derived extracellular vesicles alter vascular smooth muscle cell phenotype through high-mobility group box proteins. J Extracell Vesicles. 2020;9:1781427.
7. Galindo CL, Khan S, Zhang X, Yeh YS, Liu Z, Razani B. Lipid-laden foam cells in the pathology of atherosclerosis: shedding light on new therapeutic targets. Expert Opin Ther Targets. 2023;27:1231-45.
8. Li XD, Hong MN, Chen J, et al. Adventitial fibroblast-derived vascular endothelial growth factor promotes vasa vasorum-associated neointima formation and macrophage recruitment. Cardiovasc Res. 2020;116:708-20.
9. Hu CL, Xiang JZ, Hu FF, Huang CX. Adventitial inflammation: a possible pathogenic link to the instability of atherosclerotic plaque. Med Hypotheses. 2007;68:1262-4.
10. Bang C, Batkai S, Dangwal S, et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124:2136-46.
11. Long L, Ormiston ML, Yang X, et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med. 2015;21:777-85.
12. Baumann F, Makaloski V, Diehm N. Aortic aneurysms and aortic dissection: epidemiology, pathophysiology and diagnostics. Internist. 2013;54:535-42.
13. Mandal K, Jahangiri M, Xu Q. Autoimmune mechanisms of atherosclerosis. In: von Eckardstein A, editor. Atherosclerosis: diet and drugs. Berlin: Springer Berlin Heidelberg; 2005. pp. 723-43.
14. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317-25.
15. Sedding DG, Boyle EC, Demandt JAF, et al. Vasa vasorum angiogenesis: key player in the initiation and progression of atherosclerosis and potential target for the treatment of cardiovascular disease. Front Immunol. 2018;9:706.
16. Fernández M, Calligaris SD. Circulating microparticles in cardiovascular disease: going on stage! Biomarkers. 2019;24:423-8.
19. Li H, Kem DC, Zhang L, et al. Novel retro-inverso peptide inhibitor reverses angiotensin receptor autoantibody-induced hypertension in the rabbit. Hypertension. 2015;65:793-9.
20. Safar ME. Arterial stiffness as a risk factor for clinical hypertension. Nat Rev Cardiol. 2018;15:97-105.
21. Brod J, Fencl V, Hejl Z, Jirka J. Haemodynamics in essential hypertension. Nature. 1959;184(Suppl 21):1643-4.
22. Wilson C. East-west symposium on pathogenesis of essential hypertension. Lancet. 1960;2:1077-80.
23. Monticone S, D’Ascenzo F, Moretti C, et al. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: a systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2018;6:41-50.
24. Paulin R, Dromparis P, Sutendra G, et al. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab. 2014;20:827-39.
25. Harvey LD, Alotaibi M, Tai YY, et al. Lysosomal dysfunction and inflammatory sterol metabolism in pulmonary arterial hypertension. Science. 2025;387:eadn7277.
26. Silaschi M, Byrne J, Wendler O. Aortic dissection: medical, interventional and surgical management. Heart. 2017;103:78-87.
27. Bossone E, Eagle KA. Epidemiology and management of aortic disease: aortic aneurysms and acute aortic syndromes. Nat Rev Cardiol. 2021;18:331-48.
28. Porsch F, Binder CJ. Autoimmune diseases and atherosclerotic cardiovascular disease. Nat Rev Cardiol. 2024;21:780-807.
29. Virella G, Lopes-Virella MF. The pathogenic role of the adaptive immune response to modified LDL in diabetes. Front Endocrinol. 2012;3:76.
30. Chakravarti R, Gupta K, Swain M, et al. 14-3-3 in thoracic aortic aneurysms: identification of a novel autoantigen in large vessel vasculitis. Arthritis Rheumatol. 2015;67:1913-21.
31. Dang G, Li T, Yang D, et al. T lymphocyte-derived extracellular vesicles aggravate abdominal aortic aneurysm by promoting macrophage lipid peroxidation and migration via pyruvate kinase muscle isozyme 2. Redox Biol. 2022;50:102257.
32. Giri BR, Li S, Cheng G. Exogenous modification of EL-4 T cell extracellular vesicles with miR-155 induce macrophage into M1-type polarization. Drug Deliv Transl Res. 2024;14:934-44.
33. Clancy JW, D'Souza-Schorey C. Tumor-derived extracellular vesicles: multifunctional entities in the tumor microenvironment. Annu Rev Pathol. 2023;18:205-29.
34. Zhang Y, Zhu Z, Wang T, Dong Y, Fan Y, Sun D. TGF-β1-containing exosomes from cardiac microvascular endothelial cells mediate cardiac fibroblast activation under high glucose conditions. Biochem Cell Biol. 2021;99:693-9.
35. Yang Y, Boza-Serrano A, Dunning CJR, Clausen BH, Lambertsen KL, Deierborg T. Inflammation leads to distinct populations of extracellular vesicles from microglia. J Neuroinflammation. 2018;15:168.
36. Cavallari C, Ranghino A, Tapparo M, et al. Serum-derived extracellular vesicles (EVs) impact on vascular remodeling and prevent muscle damage in acute hind limb ischemia. Sci Rep. 2017;7:8180.
37. Garcia-Hernandez A, Leal-Orta E, Ramirez-Ricardo J, Cortes-Reynosa P, Thompson-Bonilla R, Salazar EP. Linoleic acid induces secretion of extracellular vesicles from MDA-MB-231 breast cancer cells that mediate cellular processes involved with angiogenesis in HUVECs. Prostaglandins Other Lipid Mediat. 2021;153:106519.
38. Pfrieger FW, Vitale N. Cholesterol and the journey of extracellular vesicles. J Lipid Res. 2018;59:2255-61.
39. Groß R, Reßin H, von Maltitz P, et al. Phosphatidylserine-exposing extracellular vesicles in body fluids are an innate defence against apoptotic mimicry viral pathogens. Nat Microbiol. 2024;9:905-21.
40. Lacy SH, Woeller CF, Thatcher TH, et al. Activated human lung fibroblasts produce extracellular vesicles with antifibrotic prostaglandins. Am J Respir Cell Mol Biol. 2019;60:269-78.
41. Iannotta D, Amruta A, Kijas AW, Rowan AE, Wolfram J. Entry and exit of extracellular vesicles to and from the blood circulation. Nat Nanotechnol. 2024;19:13-20.
42. der Meel R, Fens MH, Vader P, van Solinge WW, Eniola-Adefeso O, Schiffelers RM. Extracellular vesicles as drug delivery systems: lessons from the liposome field. J Control Release. 2014;195:72-85.
43. Abreu RC, Fernandes H, da Costa Martins PA, Sahoo S, Emanueli C, Ferreira L. Native and bioengineered extracellular vesicles for cardiovascular therapeutics. Nat Rev Cardiol. 2020;17:685-97.
44. Kang Y, Meng L, Bai S, Li S. Extracellular vesicles: the “Trojan Horse” within breast cancer host microenvironments. Mol Cancer. 2025;24:183.
45. Pironti G, Strachan RT, Abraham D, et al. Circulating exosomes induced by cardiac pressure overload contain functional angiotensin II Type 1 receptors. Circulation. 2015;131:2120-30.
46. Bravo-Miana RDC, Soler MF, Ceschin DG, et al. Extracellular vesicles from thyroid cancer harbor a functional machinery involved in extracellular matrix remodeling. Eur J Cell Biol. 2022;101:151254.
47. Zuppone S, Zarovni N, Vago R. The cell type dependent sorting of CD9- and CD81 to extracellular vesicles can be exploited to convey tumor sensitive cargo to target cells. Drug Deliv. 2023;30:2162161.
48. Hu H, Li N, Yngen M, Ostenson CG, Wallén NH, Hjemdahl P. Enhanced leukocyte-platelet cross-talk in type 1 diabetes mellitus: relationship to microangiopathy. J Thromb Haemost. 2004;2:58-64.
49. Henn V, Slupsky JR, Gräfe M, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391:591-4.
50. Nhek S, Clancy R, Lee KA, et al. Activated platelets induce endothelial cell activation via an interleukin-1β pathway in systemic lupus erythematosus. Arterioscler Thromb Vasc Biol. 2017;37:707-16.
51. Mesri M, Altieri DC. Endothelial cell activation by leukocyte microparticles. J Immunol. 1998;161:4382-7.
52. Hezel MEV, Nieuwland R, Bruggen RV, Juffermans NP. The Ability of extracellular vesicles to induce a Pro-inflammatory host response. Int J Mol Sci. 2017;18:1285.
53. Li K, Cui M, Zhang K, Wang G, Zhai S. M1 macrophages-derived extracellular vesicles elevate microRNA-185-3p to aggravate the development of atherosclerosis in ApoE-/- mice by inhibiting small mothers against decapentaplegic 7. Int Immunopharmacol. 2021;90:107138.
54. Chen X, Chen S, Pang J, et al. Hepatic steatosis aggravates atherosclerosis via small extracellular vesicle-mediated inhibition of cellular cholesterol efflux. J Hepatol. 2023;79:1491-501.
55. Jiang F, Chen Q, Wang W, Ling Y, Yan Y, Xia P. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J Hepatol. 2020;72:156-66.
56. Jansen F, Yang X, Hoelscher M, et al. Endothelial microparticle-mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation. 2013;128:2026-38.
57. Chen Q, Wang H, Liu Y, et al. Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1β production in macrophages by targeting STAT3. PLoS One. 2012;7:e42971.
58. Siasos G, Bletsa E, Stampouloglou PK, et al. MicroRNAs in cardiovascular disease. Hellenic J Cardiol. 2020;61:165-73.
59. Buntsma N, van der Pol E, Nieuwland R, Gąsecka A. Extracellular vesicles in coronary artery disease. In: Xiao J, editor. Extracellular vesicles in cardiovascular and metabolic diseases. Singapore: Springer Nature; 2023. pp. 81-103.
60. Dang XTT, Phung CD, Lim CMH, et al. Dendritic cell-targeted delivery of antigens using extracellular vesicles for anti-cancer immunotherapy. Cell Prolif. 2024;57:e13622.
61. Leoni G, Neumann PA, Kamaly N, et al. Annexin A1-containing extracellular vesicles and polymeric nanoparticles promote epithelial wound repair. J Clin Invest. 2015;125:1215-27.
62. Dieudé M, Turgeon J, Rimbaud AK, et al. Extracellular vesicles derived from injured vascular tissue promote the formation of tertiary lymphoid structures in vascular allografts. Am J Transplant. 2020;20:726-38.
63. Wu WC, Song SJ, Zhang Y, Li X. Role of extracellular vesicles in autoimmune pathogenesis. Front Immunol. 2020;11:579043.
64. Chen L, Liu Y, Wang Z, et al. Mesenchymal stem cell-derived extracellular vesicles protect against abdominal aortic aneurysm formation by inhibiting NET-induced ferroptosis. Exp Mol Med. 2023;55:939-51.
65. Hosseinkhani B, van den Akker NMS, Molin DGM, Michiels L. (Sub)populations of extracellular vesicles released by TNF-α -triggered human endothelial cells promote vascular inflammation and monocyte migration. J Extracell Vesicles. 2020;9:1801153.
66. Zhang K, Li R, Chen X, et al. Renal endothelial cell-targeted extracellular vesicles protect the kidney from ischemic injury. Adv Sci. 2023;10:e2204626.
67. Fujita Y, Kadota T, Kaneko R, et al. Mitigation of acute lung injury by human bronchial epithelial cell-derived extracellular vesicles via ANXA1-mediated FPR signaling. Commun Biol. 2024;7:514.
68. Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240-6.
69. York AG, Skadow MH, Oh J, et al. IL-10 constrains sphingolipid metabolism to limit inflammation. Nature. 2024;627:628-35.
70. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356:513-9.
71. Novizio N, Belvedere R, Pessolano E, et al. ANXA1 contained in EVs regulates macrophage polarization in tumor microenvironment and promotes pancreatic cancer progression and metastasis. Int J Mol Sci. 2021;22:11018.
72. Wang S, Aurora AB, Johnson BA, et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261-71.
73. Al-Nedawi K, Meehan B, Kerbel RS, Allison AC, Rak J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc Natl Acad Sci U S A. 2009;106:3794-9.
74. Taraboletti G, D'Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol. 2002;160:673-80.
75. Holtzman J, Lee H. Emerging role of extracellular vesicles in the respiratory system. Exp Mol Med. 2020;52:887-95.
76. Bouchareychas L, Duong P, Covarrubias S, et al. Macrophage exosomes resolve atherosclerosis by regulating hematopoiesis and inflammation via microRNA cargo. Cell Rep. 2020;32:107881.
77. Wang Y, Zhang K, Li T, et al. Macrophage membrane functionalized biomimetic nanoparticles for targeted anti-atherosclerosis applications. Theranostics. 2021;11:164-80.
78. Mause SF, Ritzel E, Liehn EA, et al. Platelet microparticles enhance the vasoregenerative potential of angiogenic early outgrowth cells after vascular injury. Circulation. 2010;122:495-506.
79. Tan KT, Lip GY. The potential role of platelet microparticles in atherosclerosis. Thromb Haemost. 2005;94:488-92.
80. Brill A, Dashevsky O, Rivo J, Gozal Y, Varon D. Platelet-derived microparticles induce angiogenesis and stimulate post-ischemic revascularization. Cardiovasc Res. 2005;67:30-8.
81. Hu CM, Fang RH, Wang KC, et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526:118-21.
82. Song Y, Huang Z, Liu X, et al. Platelet membrane-coated nanoparticle-mediated targeting delivery of Rapamycin blocks atherosclerotic plaque development and stabilizes plaque in apolipoprotein E-deficient (ApoE-/-) mice. Nanomedicine. 2019;15:13-24.
83. Yu B, Kim HW, Gong M, et al. Exosomes secreted from GATA-4 overexpressing mesenchymal stem cells serve as a reservoir of anti-apoptotic microRNAs for cardioprotection. Int J Cardiol. 2015;182:349-60.
84. Hu S, Li Z, Shen D, et al. Exosome-eluting stents for vascular healing after ischaemic injury. Nat Biomed Eng. 2021;5:1174-88.
85. Yue R, Lu S, Luo Y, et al. Mesenchymal stem cell-derived exosomal microRNA-182-5p alleviates myocardial ischemia/reperfusion injury by targeting GSDMD in mice. Cell Death Discov. 2022;8:202.
86. Chuah SJ, Yong CW, Teo KYW, et al. Mesenchymal stromal cell-derived small extracellular vesicles modulate macrophage polarization and enhance angio-osteogenesis to promote bone healing. Genes Dis. 2022;9:841-4.
87. Aboulgheit A, Potz BA, Scrimgeour LA, et al. Effects of high fat versus normal diet on extracellular vesicle-induced angiogenesis in a swine model of chronic myocardial ischemia. J Am Heart Assoc. 2021;10:e017437.
88. Sabe SA, Scrimgeour LA, Xu CM, et al. Extracellular vesicle therapy attenuates antiangiogenic signaling in ischemic myocardium of swine with metabolic syndrome. J Thorac Cardiovasc Surg. 2023;166:e5-e14.
89. Medica D, Franzin R, Stasi A, et al. Extracellular vesicles derived from endothelial progenitor cells protect human glomerular endothelial cells and podocytes from complement- and cytokine-mediated injury. Cells. 2021;10:1675.
90. Casado-Fernández L, Laso-García F, Piniella D, et al. The proteomic signature of circulating extracellular vesicles following intracerebral hemorrhage: novel insights into mechanisms underlying recovery. Neurobiol Dis. 2024;201:106665.
91. Hosen MR, Li Q, Liu Y, et al. CAD increases the long noncoding RNA PUNISHER in small extracellular vesicles and regulates endothelial cell function via vesicular shuttling. Mol Ther Nucleic Acids. 2021;25:388-405.
92. Xie C, Ji N, Tang Z, Li J, Chen Q. The role of extracellular vesicles from different origin in the microenvironment of head and neck cancers. Mol Cancer. 2019;18:83.
93. Zhou H, Li X, Wu RX, et al. Periodontitis-compromised dental pulp stem cells secrete extracellular vesicles carrying miRNA-378a promote local angiogenesis by targeting Sufu to activate the Hedgehog/Gli1 signalling. Cell Prolif. 2021;54:e13026.
94. Gu T, Just J, Stenz KT, et al. The role of plasma extracellular vesicles in remote ischemic conditioning and exercise-induced ischemic tolerance. Int J Mol Sci. 2022;23:3334.
95. Femminò S, D’Ascenzo F, Ravera F, et al. Percutaneous coronary intervention (PCI) reprograms circulating extracellular vesicles from ACS patients impairing their cardio-protective properties. Int J Mol Sci. 2021;22:10270.
96. Todorova D, Simoncini S, Lacroix R, Sabatier F, Dignat-George F. Extracellular vesicles in angiogenesis. Circ Res. 2017;120:1658-73.
97. Lombardo G, Dentelli P, Togliatto G, et al. Activated stat5 trafficking via endothelial cell-derived extracellular vesicles controls IL-3 pro-angiogenic paracrine action. Sci Rep. 2016;6:25689.
98. Wang Y, Dong L, Zhong H, et al. Extracellular vesicles (EVs) from lung adenocarcinoma cells promote human umbilical vein endothelial cell (HUVEC) angiogenesis through yes kinase-associated protein (YAP) transport. Int J Biol Sci. 2019;15:2110-8.
99. Tamas F, Tamas CI, Suciu BA, Balasa AF. Extracellular vesicle-associated angiopoietin-2 and cell migration-inducing protein in lung cancer progression and brain metastases. Cureus. 2025;17:e80200.
100. Dougherty JA, Kumar N, Noor M, et al. Extracellular vesicles released by human induced-pluripotent stem cell-derived cardiomyocytes promote angiogenesis. Front Physiol. 2018;9:1794.
101. Wysoczynski M, Pathan A, Moore JB 4th, et al. Pro-angiogenic actions of CMC-derived extracellular vesicles rely on selective packaging of angiopoietin 1 and 2, but Not FGF-2 and VEGF. Stem Cell Rev Rep. 2019;15:530-42.
102. Ge L, Xun C, Li W, et al. Extracellular vesicles derived from hypoxia-preconditioned olfactory mucosa mesenchymal stem cells enhance angiogenesis via miR-612. J Nanobiotechnology. 2021;19:380.
103. Jong OG, van der Waals LM, Kools FRW, Verhaar MC, van Balkom BWM. Lysyl oxidase-like 2 is a regulator of angiogenesis through modulation of endothelial-to-mesenchymal transition. J Cell Physiol. 2019;234:10260-9.
104. Parekh M, Rhys H, Ramos T, Ferrari S, Ahmad S. Extracellular Vesicles derived from human corneal endothelial cells inhibit proliferation of human corneal endothelial cells. Front Med. 2021;8:753555.
105. Zheng Y, Xu P, Pan C, et al. Production and biological effects of extracellular vesicles from adipose-derived stem cells were markedly increased by low-intensity ultrasound stimulation for promoting diabetic wound healing. Stem Cell Rev Rep. 2023;19:784-806.
106. Zeng Y, Wei L, Lali MS, Chen Y, Yu J, Feng L. miR-150-5p mediates extravillous trophoblast cell migration and angiogenesis functions by regulating VEGF and MMP9. Placenta. 2020;93:94-100.
107. Song BW, Lee CY, Kim R, et al. Multiplexed targeting of miRNA-210 in stem cell-derived extracellular vesicles promotes selective regeneration in ischemic hearts. Exp Mol Med. 2021;53:695-708.
108. Wang C, Wu H, Xing Y, et al. Endothelial-derived extracellular microRNA-92a promotes arterial stiffness by regulating phenotype changes of vascular smooth muscle cells. Sci Rep. 2022;12:344.
109. Li X, Ballantyne LL, Yu Y, Funk CD. Perivascular adipose tissue-derived extracellular vesicle miR-221-3p mediates vascular remodeling. FASEB J. 2019;33:12704-22.
110. Zhang L, Wei W, Ai X, et al. Extracellular vesicles from hypoxia-preconditioned microglia promote angiogenesis and repress apoptosis in stroke mice via the TGF-β/Smad2/3 pathway. Cell Death Dis. 2021;12:1068.
111. Xu J, Lu X, Shi GP. Vasa vasorum in atherosclerosis and clinical significance. Int J Mol Sci. 2015;16:11574-608.
112. Wu R, Gao W, Yao K, et al. Roles of exosomes derived from immune cells in cardiovascular diseases. Front Immunol. 2019;10:648.
113. Akhmerov A, Parimon T. Extracellular vesicles, inflammation, and cardiovascular disease. Cells. 2022;11:2229.
114. Deng W, Tang T, Hou Y, et al. Extracellular vesicles in atherosclerosis. Clin Chim Acta. 2019;495:109-17.
115. Yuan X, Bhat OM, Samidurai A, Das A, Zhang Y, Li PL. Reversal of endothelial extracellular vesicle-induced smooth muscle phenotype transition by hypercholesterolemia stimulation: role of NLRP3 inflammasome activation. Front Cell Dev Biol. 2020;8:597423.
116. Lin X, He SQ, Shan SK, et al. Endothelial cells derived extracellular vesicles promote diabetic arterial calcification via circ_0008362/miR-1251-5p/Runx2 axial. Cardiovasc Diabetol. 2024;23:369.
117. Song T, Lv M, Sun B, Zheng L, Zhao M. Tripeptides val-pro-pro (VPP) and ile-pro-pro (IPP) regulate the proliferation and migration of vascular smooth muscle cells by interfering ang II-induced human umbilical vein endothelial cells derived evs delivering RNAs to VSMCs in the Co-culture model. J Agric Food Chem. 2020;68:6628-37.
118. Liu R, Shen H, Ma J, Sun L, Wei M. Extracellular vesicles derived from adipose mesenchymal stem cells regulate the phenotype of smooth muscle cells to limit intimal hyperplasia. Cardiovasc Drugs Ther. 2016;30:111-8.
119. Xuan W, Khan M, Ashraf M. Extracellular vesicles from notch activated cardiac mesenchymal stem cells promote myocyte proliferation and neovasculogenesis. Front Cell Dev Biol. 2020;8:11.
120. Wang J, Hu L, Huang H, et al. CAR (CARSKNKDC) peptide modified rencell-derived extracellular vesicles as a novel therapeutic agent for targeted pulmonary hypertension therapy. Hypertension. 2020;76:1147-60.
121. Furmanik M, Chatrou M, van Gorp R, et al. Reactive oxygen-forming nox5 links vascular smooth muscle cell phenotypic switching and extracellular vesicle-mediated vascular calcification. Circ Res. 2020;127:911-27.
122. Haynes BA, Yang LF, Huyck RW, et al. Endothelial-to-mesenchymal transition in human adipose tissue vasculature alters the particulate secretome and induces endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2019;39:2168-91.
123. Sajeesh S, Camardo A, Dahal S, Ramamurthi A. Surface-functionalized stem cell-derived extracellular vesicles for vascular elastic matrix regenerative repair. Mol Pharm. 2023;20:2801-13.
124. Li T, Yu H, Zhang D, et al. Matrix vesicles as a therapeutic target for vascular calcification. Front Cell Dev Biol. 2022;10:825622.
126. Rogers MA, Atkins SK, Zheng KH, et al. Lipoprotein(a) induces vesicular cardiovascular calcification revealed with single-extracellular vesicle analysis. Front Cardiovasc Med. 2022;9:778919.
127. Blaser MC, Buffolo F, Halu A, et al. Multiomics of tissue extracellular vesicles identifies unique modulators of atherosclerosis and calcific aortic valve stenosis. Circulation. 2023;148:661-78.
128. Buffolo F, Monticone S, Camussi G, Aikawa E. Role of extracellular vesicles in the pathogenesis of vascular damage. Hypertension. 2022;79:863-73.
129. Yaker L, Tebani A, Lesueur C, et al. Extracellular vesicles from LPS-treated macrophages aggravate smooth muscle cell calcification by propagating inflammation and oxidative stress. Front Cell Dev Biol. 2022;10:823450.
130. Viegas C, Carreira J, Maia TM, et al. Gla rich protein (GRP) mediates vascular smooth muscle cell (VSMC) osteogenic differentiation, extracellular vesicle (EV) calcification propensity, and immunomodulatory properties. Int J Mol Sci. 2024;25:12406.
131. Viegas CSB, Santos L, Macedo AL, et al. Chronic kidney disease circulating calciprotein particles and extracellular vesicles promote vascular calcification: a role for GRP (gla-rich protein). Arterioscler Thromb Vasc Biol. 2018;38:575-87.
132. Feng W, Teng Y, Zhong Q, et al. Biomimetic grapefruit-derived extracellular vesicles for safe and targeted delivery of sodium thiosulfate against vascular calcification. ACS Nano. 2023;17:24773-89.
133. Andreata F, Syvannarath V, Clement M, et al. Macrophage CD31 signaling in dissecting aortic aneurysm. J Am Coll Cardiol. 2018;72:45-57.
134. Chen P, Wang F, Ling B, et al. Mesenchymal stem cell-derived extracellular vesicles: emerging therapies for neurodegenerative diseases. Int J Nanomedicine. 2025;20:8547-65.
135. Wang S, Wang X, Lv Y, et al. M2 macrophage-derived exosomes inhibit atherosclerosis progression by regulating the proliferation, migration, and phenotypic transformation of smooth muscle cells. Front Biosci. 2024;29:288.
136. Kang T, Jones TM, Naddell C, et al. Adipose-derived stem cells induce angiogenesis via microvesicle transport of miRNA-31. Stem Cells Transl Med. 2016;5:440-50.
137. Yin S, Ji C, Wu P, et al. Human umbilical cord mesenchymal stem cells and exosomes: bioactive ways of tissue injury repair. Am J Transl Res. 2019;11:1230-40.
138. Su J, Wei Q, Ma K, et al. P-MSC-derived extracellular vesicles facilitate diabetic wound healing via miR-145-5p/ CDKN1A-mediated functional improvements of high glucose-induced senescent fibroblasts. Burns Trauma. 2023;11:tkad010.
139. Adamiak M, Cheng G, Bobis-Wozowicz S, et al. Induced pluripotent stem cell (iPSC)-derived extracellular vesicles are safer and more effective for cardiac repair than iPSCs. Circ Res. 2018;122:296-309.
140. Shi X, Yang G, Liu MY, Yuan MT, Wang D, Wang XF. Exosomes derived from human dental pulp stem cells increase flap survival with ischemia-reperfusion injuries. Regen Med. 2023;18:313-27.
141. Duan S, Wang C, Xu X, et al. Peripheral serum exosomes isolated from patients with acute myocardial infarction promote endothelial cell angiogenesis via the miR-126-3p/TSC1/mTORC1/HIF-1α pathway. Int J Nanomedicine. 2022;17:1577-92.
142. Geng T, Song ZY, Xing JX, Wang BX, Dai SP, Xu ZS. Exosome derived from coronary serum of patients with myocardial infarction promotes angiogenesis through the miRNA-143/IGF-IR pathway. Int J Nanomedicine. 2020;15:2647-58.
143. Li H, Liao Y, Gao L, et al. Coronary serum exosomes derived from patients with myocardial ischemia regulate angiogenesis through the miR-939-mediated nitric oxide signaling pathway. Theranostics. 2018;8:2079-93.
144. Mathiyalagan P, Liang Y, Kim D, et al. Angiogenic mechanisms of human CD34+ stem cell exosomes in the repair of ischemic hindlimb. Circ Res. 2017;120:1466-76.
145. Zhao M, Nakada Y, Wei Y, et al. Cyclin D2 overexpression enhances the efficacy of human induced pluripotent stem cell-derived cardiomyocytes for myocardial repair in a swine model of myocardial infarction. Circulation. 2021;144:210-28.
146. Wang X, Chen Y, Zhao Z, et al. Engineered exosomes with ischemic myocardium-targeting peptide for targeted therapy in myocardial infarction. J Am Heart Assoc. 2018;7:e008737.
147. Lu Q, Kou D, Lou S, et al. Nanoparticles in tumor microenvironment remodeling and cancer immunotherapy. J Hematol Oncol. 2024;17:16.
148. Lai J, Huang C, Guo Y, Rao L. Engineered extracellular vesicles and their mimics in cardiovascular diseases. J Control Release. 2022;347:27-43.
149. Carney RP, Mizenko RR, Bozkurt BT, et al. Harnessing extracellular vesicle heterogeneity for diagnostic and therapeutic applications. Nat Nanotechnol. 2025;20:14-25.
150. Ramasubramanian L, Du S, Gidda S, et al. Bioengineering extracellular vesicles for the treatment of cardiovascular diseases. Adv Biol. 2022;6:e2200087.
151. Willms E, Cabañas C, Mäger I, Wood MJA, Vader P. Extracellular vesicle heterogeneity: subpopulations, isolation techniques, and diverse functions in cancer progression. Front Immunol. 2018;9:738.
152. Roerig J, Schulz-Siegmund M. Standardization approaches for extracellular vesicle loading with oligonucleotides and biologics. Small. 2023;19:e2301763.
153. Li G, Chen T, Dahlman J, et al. Current challenges and future directions for engineering extracellular vesicles for heart, lung, blood and sleep diseases. J Extracell Vesicles. 2023;12:e12305.
154. Xiang H, Bao C, Chen Q, et al. Extracellular vesicles (EVs)’ journey in recipient cells: from recognition to cargo release. J Zhejiang Univ Sci B. 2024;25:633-55.
155. Zhang M, Hu S, Liu L, et al. Engineered exosomes from different sources for cancer-targeted therapy. Signal Transduct Target Ther. 2023;8:124.
156. Si C, Gao J, Ma X. Natural killer cell-derived exosome-based cancer therapy: from biological roles to clinical significance and implications. Mol Cancer. 2024;23:134.
157. Huang L, Rong Y, Tang X, et al. Engineered exosomes as an in situ DC-primed vaccine to boost antitumor immunity in breast cancer. Mol Cancer. 2022;21:45.
158. Li Q, Hu W, Huang Q, et al. MiR146a-loaded engineered exosomes released from silk fibroin patch promote diabetic wound healing by targeting IRAK1. Signal Transduct Target Ther. 2023;8:62.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].