


Endothelial dysfunction as a driver of microvascular injury in diabetic cardiomyopathy
 0
0 
Abstract
Diabetic cardiomyopathy (DCM) is a serious complication of diabetic mellitus that occurs independently of other known cardiac diseases and is associated with significant morbidity and mortality. Microvascular injury plays a central role in the pathogenesis of DCM, contributing to its hallmark features, such as cardiac contractile dysfunction and myocardial fibrosis. Current evidence points to endothelial dysfunction (ED) as the key contributor to the development of microvascular injury. Chronic hyperglycemia, hyperinsulinemia, and insulin resistance progressively promote ED, characterized by alterations in gene expression, shifts in endothelial cell (EC) subpopulation dynamics, and dysregulated crosstalk between EC and other cardiac cell types. Ultimately, these changes result in microvascular impairments such as chronic inflammation, EC loss leading to microvascular rarefaction, endothelial-to-mesenchymal transition (EndoMT) promoting myocardial fibrosis, and loss of vasodilatory function. If left uncorrected, these impairments will progress to contractile dysfunction and widespread myocardial fibrosis, manifesting as systolic heart failure. Over the past decade, single-cell RNA sequencing (scRNA-seq) has allowed for the transcriptional profiling of individual cells, enabling the identification of distinct subpopulations within the same cell type and providing deeper insights into cellular crosstalk under both normal and disease conditions. Although research on DCM using scRNA-seq remains an emerging field, studies have identified distinct EC subpopulations, their gene expression profiles, and their contributions to DCM pathogenesis. Moreover, scRNA-seq has revealed dysregulated interactions between ECs and other cardiac cell types in DCM. The expanding application of scRNA-seq holds significant promise for mapping EC subpopulation dynamics and communication pathways in DCM, which may ultimately support the development of novel EC-targeted therapeutic strategies against ED in this condition.
Keywords
INTRODUCTION
Diabetes mellitus (DM) is a globally prevalent disorder currently affecting over 580 million individuals, a number projected to rise to at least 850 million by 2050[1]. Among its many systemic complications, cardiovascular manifestations remain one of the most common, affecting roughly one-third of patients with DM and may lead to a variety of structural, functional, and molecular changes in both the heart and the vascular system[2]. One particularly understudied complication is diabetic cardiomyopathy (DCM), a diabetes-induced cardiac dysfunction that develops independently of other risk factors, such as coronary artery disease (CAD), hypertension, or major valvular disease[3,4]. Since its initial recognition in the late 20th century, DCM has been the subject of growing research because of its strong association with an elevated risk of heart failure (HF), contributing substantially to an increased morbidity and mortality in diabetic populations[5].
Several risk factors have been determined to contribute to the pathogenesis of DCM, including chronic hyperglycemia, insulin resistance, and oxidative stress[6]. Such factors disrupt cellular metabolism and signaling pathways, promoting inflammation, mitochondrial damage, and the accumulation of advanced glycation end products (AGEs), all of which play a role in the progression of myocardial structural and functional deterioration. Epidemiological data suggest that individuals with DM exhibit a notably elevated risk of developing HF, approximately two to five times greater than non-diabetic individuals. This is accompanied by further association with glycemic control, particularly HbA1c levels[5-8]. Over time, these risk factors promote dynamic alterations within the myocardial tissue, potentially leading to left ventricular hypertrophy (LVH), myocardial fibrosis, and abnormal diastolic function[6]. A key underlying pathological feature is microvascular injury, which involves endothelial dysfunction (ED), capillary rarefaction, and pericyte loss[6,7]. Together, these changes lead to severe complications, ultimately resulting in myocardial remodeling and HF, prompting the need for early diagnosis and intervention.
OVERVIEW OF MICROVASCULAR INJURY IN DCM
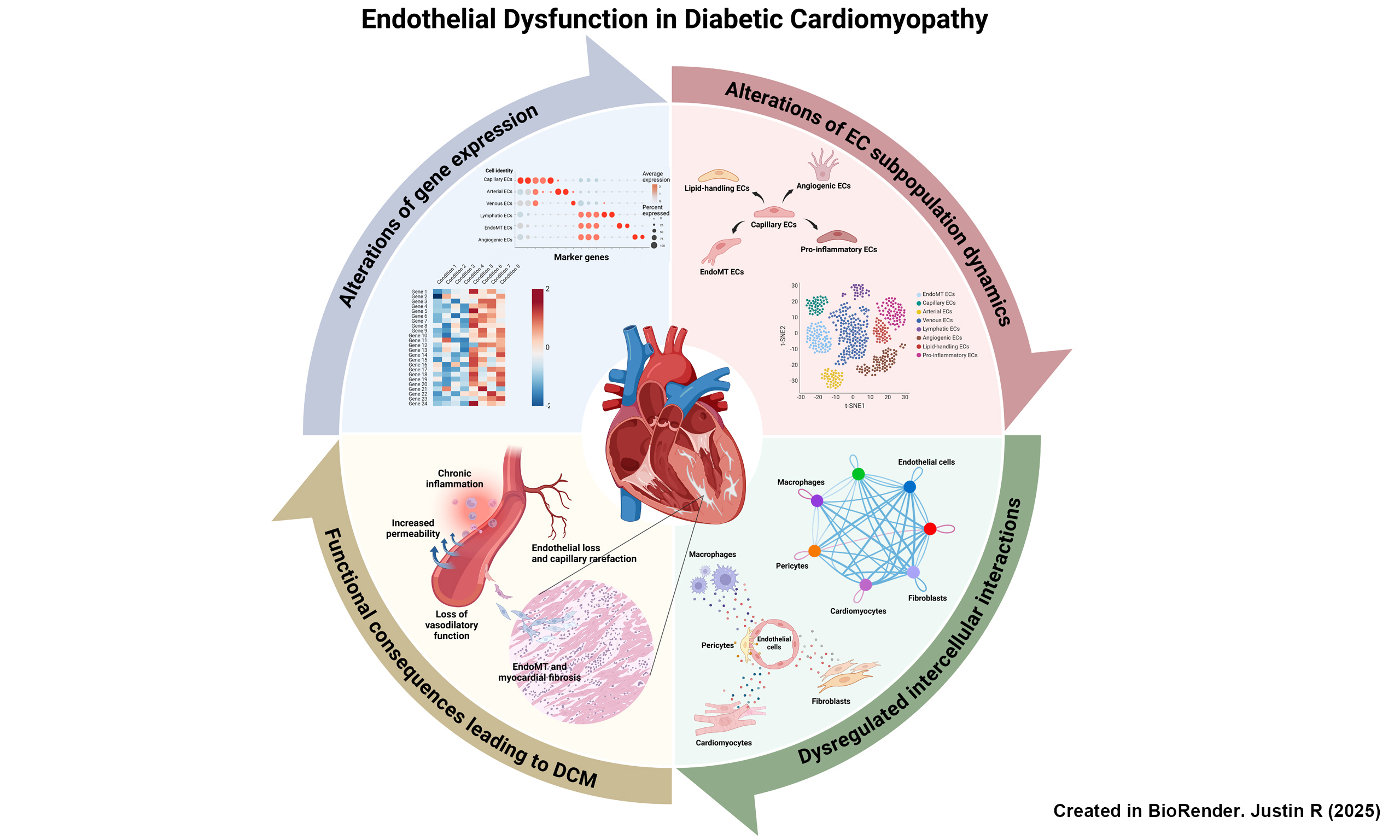
Microvascular injury is a major pathophysiological process in DCM that contributes to cardiac dysfunction and HF. This injury involves a series of complex molecular interactions between the primary components of the capillary network, endothelial cells (ECs) and pericytes, which together maintain microvascular homeostasis. Chronic hyperglycemia and insulin resistance are key drivers of microvascular injury in DCM, substantially leading to oxidative stress through the accumulation of AGEs and inflammatory components. These changes result in ED, one of the earliest manifestations in disease progression [Figure 1]. Typical characteristics of ED arise from the loss of EC markers alongside an increase in fibroblast markers causing EndoMT, enhanced vascular permeability, elevated production of inflammatory factors [interleukin (IL)-6, IL-1β, tumor necrosis factor (TNF)-α, vascular cell adhesion molecule (VCAM)-1, intercellular adhesion molecule (ICAM)-1, monocyte chemoattractant protein (MCP)-1], impaired nitric oxide (NO) bioavailability with reduced vasodilation, and abnormal angiogenesis[3,9,10]. Concurrently, pericytes, specialized cells that wrap around ECs within capillary basement membranes, also undergo changes in response to induced hyperglycemia and oxidative stress, leading progressively to cellular dysfunction and apoptosis[11,12]. More importantly, DCM disrupts EC-pericyte crosstalk, impairing vascular formation, which in turn causes capillary rarefaction and ultimately HF[12]. Together, both ECs and pericytes play central roles in the development of DCM, cardiac remodeling (including fibrosis and hypertrophy), and cardiac dysfunction.
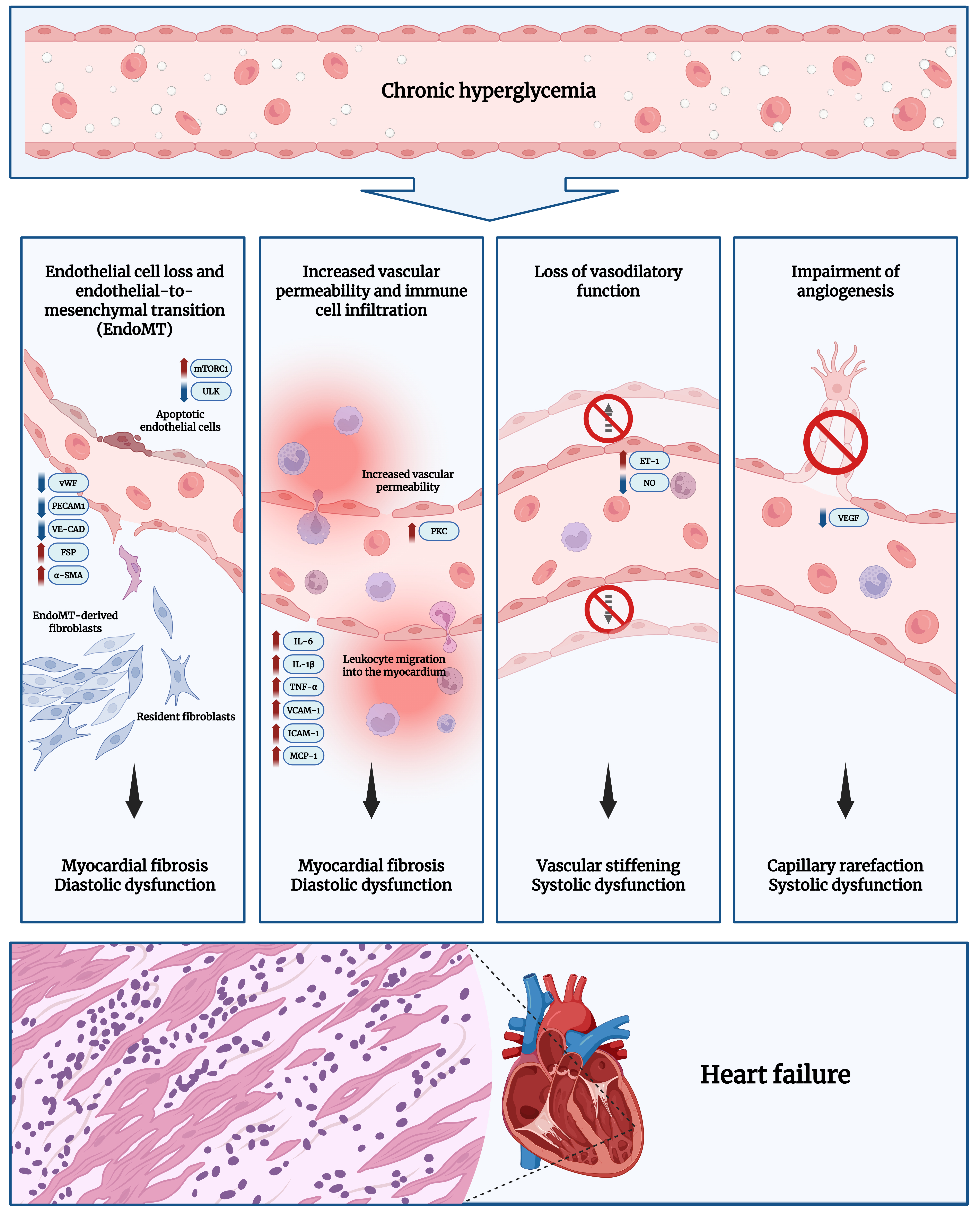
Figure 1. Pathogenesis of DCM. Prolonged hyperglycemia leads to metabolic changes in the myocardium, which converge on ED. This impairs normal endothelial function, manifesting as increased EC loss, increased transformation of ECs into fibroblasts (through EndoMT), elevated vascular permeability with immune cell infiltration into the myocardium, impaired vasodilation, and defective angiogenesis[3,4,7,10]. Excessive fibroblast transition and heightened inflammatory signaling drive progressive myocardial fibrosis, reducing ventricular relaxation and leading to diastolic dysfunction. Meanwhile, impaired endothelial vasodilation and angiogenesis causes vascular stiffening and rarefaction, reducing myocardial perfusion. The resulting nutrient and oxygen deficiency in
To enhance the understanding of cellular and molecular alterations associated with DCM, recent advances in transcriptomics, particularly single-cell RNA sequencing (scRNA-seq), have been increasingly applied. ScRNA-seq enables the identification of various cell types and their gene expression profiles at the cellular level[13]. Beyond identifying subpopulations, it allows for trajectory analyses of genomic heterogeneity and differentiation. Moreover, scRNA-seq offers transcriptomic and epigenetic mapping of specific tissues, thereby facilitating the determination of signaling pathways and intercellular communication among ECs, pericytes, and other myocardial cell types during DCM development[14]. Accordingly, this review highlights current insights into microvascular injury induced by DCM, with a focus on ED as revealed by scRNA-seq.
ENDOTHELIAL INJURY AND DYSFUNCTION IN DCM
Alterations of EC gene expression in DCM
Under normal physiological conditions, ECs are considered non-homogeneous, exhibiting organ-specific gene expression patterns that support homeostasis and tissue-specific functions. scRNA-seq enables the identification of differentially expressed genes (DEGs) and associated molecular pathways, providing insights into cellular interactions in both healthy and diseased states. Paik et al. used scRNA-seq to identify organ-specific ECs under physiological conditions, revealing DEGs across 12 different organs in mice. In cardiac ECs, the top 10 DEGs included Wt1, Slc28a2, Eepd1, Kcna5, Car8, Fbln1, Meox2, Rftn1, Lamb1, and Myadm. Identification of the most common DEGs enables the determination of molecular pathways associated with endothelial function. For instance, ErbB signaling pathways, primarily responsible for angiogenesis and the control of cellular permeability, were enriched in the brain, whereas axon guidance pathways, which modulate angiogenesis mainly through vascular endothelial growth factors (VEGFs), are specific to cardiac ECs, reflecting tissue-specific physiological role. Additionally, shared molecular pathways mediating angiogenesis and vascularization - such as canonical (β-catenin-dependent) and non-canonical (β-catenin-independent) Wingless-related integration site (WNT) pathways - are present in the brain, lung, heart, and liver[15-18].
In DCM, ECs exhibit dynamic alterations in gene expression, with DEGs upregulated or downregulated at different stages of disease progression. These gene expression changes are associated with a variety of molecular, structural, and functional alterations that contribute to ED. Application of scRNA-seq enables visualization of DEGs in multiple cell types, including ECs, cardiomyocytes (CMs), fibroblasts, and pericytes, allowing for identification of pathways, cellular subgroups, and interactions implicated in microvascular injury during DCM.
Genes related to inflammatory pathways
One of the major molecular pathways that may often be associated with ED during DCM is the inflammatory pathway. Multiple inflammatory signaling pathways, such as nuclear factor κB (NF-κB), nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) inflammasome, IL-6/signal transducer and activator of transcription 3 (STAT3), Janus kinase (JAK)/STAT, and p38/ mitogen-activated protein kinase (MAPK), have been shown to be involved[3,4,10,19-21]. scRNA-seq studies have revealed upregulation of DEGs such as Cebpb, Ctsh, Plvap, Igfbp5, and Irf7 [Figure 2][14,22-24]. These genes are crucial in modulating immune responses, particularly through the secretion of cytokines such as NF-κB and IL-6, which also contribute to cardiac fibrosis and remodeling. Li et al. reported that Irf7 is highly expressed in ECs, regulates the NF-κB pathway, and may be associated with the pathological development of cardiac hypertrophy and HF[24]. Plvap is upregulated in certain EC subgroups, facilitating the migration of leukocytes to inflammatory sites[14]. Igfbp5 is also upregulated during DCM progression and activates cardiac fibroblasts, thereby contributing to the progression of fibrosis[14]. Although several studies suggest Igfbp5 triggers inflammatory pathways in the kidney via cytokines such as IL-6, MCP-1, ICAM-1, and NF-κB, its mechanisms in the heart remain poorly understood, warranting further research[25,26]. Similarly, although Ctsh was found to be upregulated in ECs[14], studies on other cathepsins such as Ctsb and Ctsd indicate that their upregulation promotes inflammation through the NF-κB signaling pathway, highlighting the limited knowledge on Ctsh’s role in ECs and related mechanisms under DCM conditions[27,28].
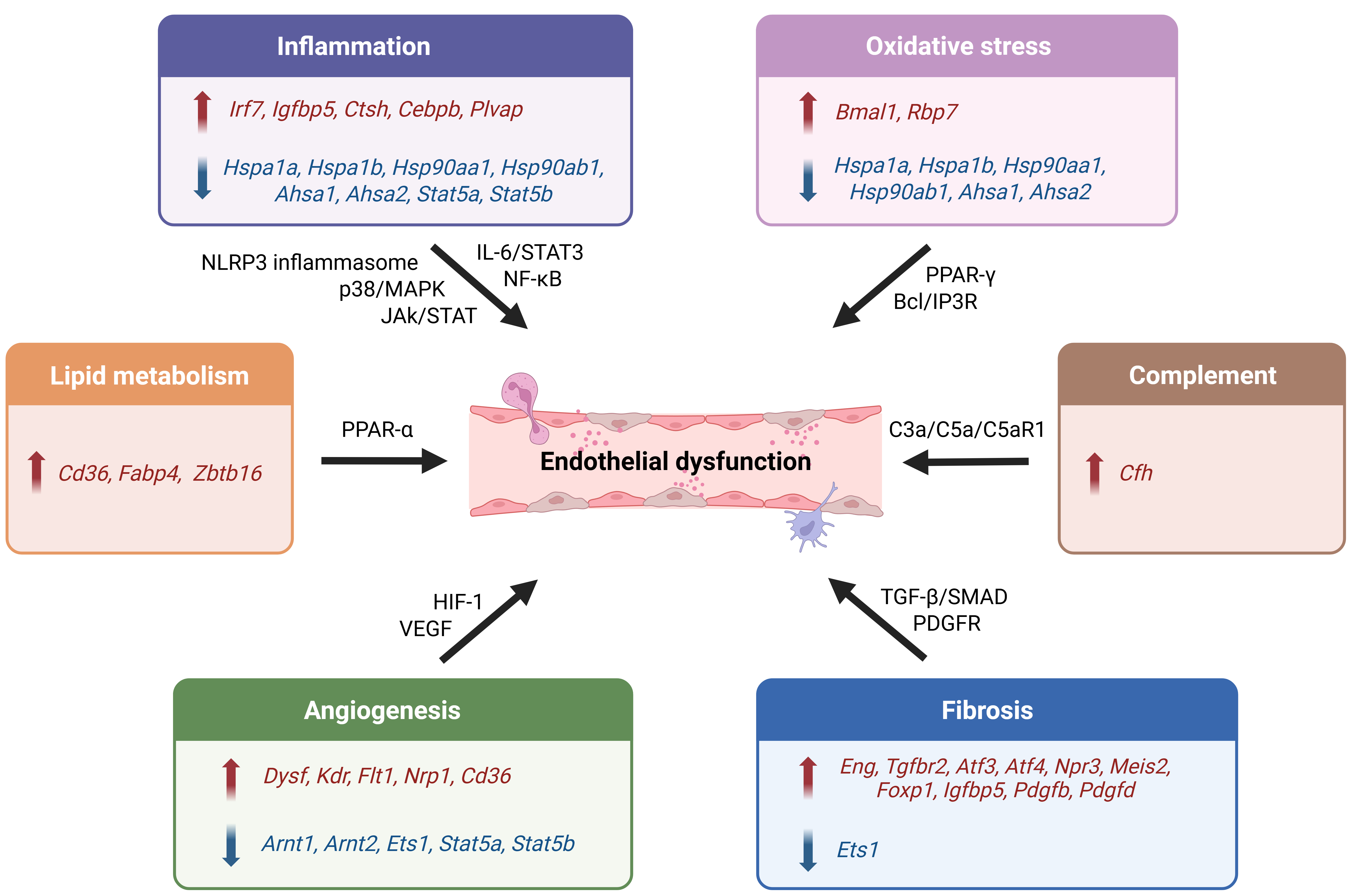
Figure 2. DEGs in ECs during DCM pathogenesis. Prolonged hyperglycemia in diabetes results in altered gene expression, preceding ED and subsequent microvascular injury. In general, these genes can be classified into six functional categories based on vascular pathology, including inflammation, oxidative stress response, complement activation, fibrosis, angiogenesis, and lipid metabolism[14,22,23]. Overlapping gene pathway assignments are supported by GO/KEGG analyses [Supplementary Table 1], with additional functional validation from experimental literature. DEG: Differentially expressed gene; DCM: diabetic cardiomyopathy; EC: endothelial cell; ED: endothelial dysfunction; GO: gene ontology; KEGG: kyoto encyclopedia of genes and genomes; MAPK: mitogen-activated protein kinase; JAK: Janus kinase. STAT: signal transducers and activators of transcription; NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; PPAR-γ: peroxisome proliferator-activated receptor γ; BCL2: B-cell lymphoma 2; HIF-1: hypoxia-inducible factor 1; VEGF: vascular endothelial growth factor; IP3R: inositol 1,4,5-trisphosphate receptor; TGF: transforming growth factor; PDGFR: Platelet-derived growth factor receptor; SMAD: Sma- and Mothers against decapentaplegic-related proteins [Created in BioRender. Justin R (2025)].
Conversely, several DEGs related to inflammatory pathways are downregulated, including molecular chaperones from the heat shock proteins (HSP) family (Hspa1a, Hspa1b, Hsp99aa/b1), Ahsa1, Ahsa2, Stat5a, and Stat5b [Figure 2][22,29]. Decreased HSP expression impairs NF-κB and p38/MAPK pathways, compromising cellular protective functions such as protein folding. This dysfunction results in the accumulation of reactive oxygen species (ROS), thereby promoting inflammation[30]. In addition, downregulation of Stat5a and Stat5b, key components of the JAK/STAT pathway, further exacerbates inflammation in DCM. Although gene ontology (GO)/Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses demonstrate that these pathways are primarily inflammatory [JAK/STAT, AGE/receptor for advanced glycation end products (RAGEs)], studies show that they also participate in the regulation of angiogenesis. Therefore, loss of these factors may lead to microvascular rarefaction[22].
Genes related to fibrosis and EndoMT
EndoMT and subsequent myocardial fibrosis are hallmarks of DCM, involving pathways such as transforming growth factor (TGF)-β/Sma- and Mothers against decapentaplegic-related proteins (SMAD), IL-6/STAT3, and peroxisome proliferator-activated receptor γ (PPAR-γ). Several upregulated DEGs , including Pdgfb, Pdgfd, Tgfbr2, Eng, Npr3, Igfbp5, and transcription factors such as Meis2, Tcf21, Atf3, Atf4, and Foxp1 have been identified [Figure 2][14,23,29,31]. According to Li et al., both Pdgfb and Pdgfd are expressed in ECs and contribute to myocardial fibrosis through Pdgf receptors on other cell types, while Pdgfc and Pdgfa are expressed in macrophages and fibroblasts, respectively, suggesting potential crosstalk between these two cell types in promoting fibrotic changes[24]. Notably, Tgfbr2 and Eng upregulation during DCM is directly associated with the TGF-β pathway, promoting vascular remodeling and fibrosis[29,32,33]. Npr3 expression increases in specific EC clusters[14], and its deletion attenuates cardiac fibrosis in diabetic mice by decreasing TGF-β/SMAD signaling, indicating its role in disease progression[34].
Upregulation of DEGs encoding transcription factors has also been implicated in the development of DCM. Zhang et al. reported that the upregulation of Meis2 and Foxp1 is associated with the additional upregulation of 73 and 50 genes, respectively. Although the upregulation of these transcription factors may suppress the TGF-β/SMAD pathway and alleviate cardiac dysfunction, complete loss of the pathway promotes vascular and chamber dilation and adverse cardiac remodeling[14,35]. Conversely, reduced Foxp1 expression can lead to the overactivation of genes targeting the TGF-β/SMAD and NLRP3 inflammasome pathways (e.g., Casp1 and Il1b), resulting in inflammation and fibrosis, which suggests that Foxp1 may have complex molecular roles in remodeling during DCM[36]. Moreover, Tcf21 upregulation facilitates mesenchymal transition; its loss prevents fibroblast differentiation, indicating that its upregulation may drive the transition of ECs into fibroblasts[22,37]. Additionally, Atf3 and Atf4 are elevated in ECs during DCM, targeting the TGF-β/SMAD3 pathways, thus causing EndoMT[31]. Atf4 also contributes to the upregulation of Smurf2, which further promotes oxidative stress and cardiac fibrosis[38]. In contrast, downregulated Ets1 normally protects ECs and regulates over 154 genes involved in angiogenesis, including Alk1, Cldn5, Sox18, Robo14, Esm1, Notch14, Kdr, and Ppp1r16b. GO/KEGG analyses indicate that loss of Ets1 expression in ECs during EndoMT leads to reduced expression of genes involved in extracellular matrix (ECM) remodeling and cell migration. This loss also impairs vascular angiogenesis, as direct target genes-including Alk1, Cldn5, Sox18, Robo4, Esm1, and Kdr-are downregulated following Ets1 knockout. These changes primarily contribute to coronary vascular defects and altered ECM composition in the trabecular layer, while also facilitating EndoMT[14,39].
Genes related to oxidative stress and mitochondrial dysfunction
In addition to fibrosis and inflammation, ECs also exhibit oxidative stress, endoplasmic reticulum (ER) stress, and mitochondrial dysfunction under diabetic conditions. Notable DEGs include Rbp7 and Bmal1
Genes related to insulin signaling and lipid metabolism
Insulin signaling is essential for maintaining vascular homeostasis. Early DCM is characterized by disruptions in this pathway, leading to structural and functional changes. PPAR-α pathway dysfunction promotes lipotoxicity via increased fatty acid uptake, mediated by upregulation of genes such as Cd36 and Fabp4 in ECs [Figure 2][22]. This enhances lipid accumulation and oxidative stress, which are crucial hallmarks of DCM. Cd36 is central to long-chain fatty acid uptake; its overexpression exacerbates metabolic disturbances and endothelial dysfunction. Additionally, Cd36 negatively regulates angiogenesis in ECs by suppressing VEGF signaling, impairing capillary formation, and contributing to myocardial remodeling[43]. Insulin resistance promotes Cd36 translocation and PPAR-α activation[43], driving abnormal Cd36 expression and accelerating endothelial damage and DCM progression. The transcription factor Zbtb16 regulates Cd36 and Fabp4; its upregulation in lipid metabolism-related EC clusters further elevates these DEGs, exacerbating lipid accumulation and disease progression[22].
Genes related to angiogenesis
Angiogenesis plays a crucial role in maintaining endothelial function and vascular integrity. It is well established that this process becomes dysregulated in diabetic hearts, contributing to ED and microvascular damage. Key pathways such as VEGF, Wnt/β-catenin, and hypoxia-inducible factor 1 (HIF-1) are disrupted during DCM, leading to impaired vascular regeneration and EC apoptosis. Major DEGs with increased expression include Kdr, Flt1, Nrp1, Dysf, and Cd36 [Figure 2][23,29]. These alterations initially promote angiogenic signaling through the VEGF and Wnt pathways, triggering a compensatory response in early disease stages. However, in later stages, a decompensatory response occurs, characterized by reduced angiogenic signaling. Thus, disease progression involves the downregulation of angiogenesis-related genes, which in turn suppresses vessel formation by impairing these signaling pathways. As noted earlier, Ets1 is downregulated in DCM, leading to impaired angiogenesis through interactions with VEGF and TGF-β pathways. This downregulation further suppresses direct angiogenic target genes, thereby impairing vascular development[44]. Similarly, Stat5a and Stat5b regulate angiogenesis through a positive feedback loop of prolactin secretion, which upregulates VEGF signaling in ECs. Their downregulation, therefore, diminishes angiogenesis [Figure 2][22,45]. Disruption of VEGF signaling also decreases NO bioavailability, increases ROS production, and activates inflammatory cascades, collectively driving disease progression[46]. Furthermore, decreased expression of Arnt1 and Arnt2 under hyperglycemic conditions disrupts the HIF-1 pathway, impairing cellular signaling and limiting EC responsiveness to hypoxia[47-49]. These alterations contribute to EC loss and microvascular rarefaction in DCM. Targeting the HIF-1 pathway has been proposed as a promising therapeutic strategy to correct angiogenesis dysregulation and mitigate microvascular complications.
Genes related to complement pathways
The complement pathway, though less studied, also contributes to ED in DCM. Upregulation of the Cfh gene [Figure 2][14] excessively activates the C3a/C5a/C5aR1 pathway, disrupting the endothelial barrier and triggering inflammation. Several studies have reported that Cfh is associated with an increased risk of type 2 DM, identifying it as an early marker of disease progression[50,51]. Zhang et al. reported that Cfh was among the most distinctively expressed genes in endothelial clusters, with expression levels increasing during DCM progression, indicating its role as a biomarker for microvascular damage[14]. However, additional research is needed to clarify the direct effects of this pathway on ECs in DCM, its role in vascular injury, and its therapeutic potential.
Genes targeting multiple pathways
Comparative analyses of scRNA-seq data indicate that ECs express several genes that act across multiple pathways, influencing diverse processes during disease progression. Notable upregulated DEGs include Igfbp5, Cebpb, and Cd36 [Figure 2][24,14,23]. Zhang et al. reported increased expression of the transcription factor Cebpb[14], which regulates more than 37 genes and is implicated in multiple inflammatory pathways. Li et al. showed that the upregulation of Cebpb promotes NLRP3 inflammasome binding, driving cytokine production, inflammation, and ED[52]. Similarly, Manea et al. found that under hyperglycemic conditions, the upregulation of Cebpb increases endothelin-1 (ET-1) transcription in ECs, promoting vasoconstriction and inflammation through the MAPK signaling pathway[53]. Although GO/KEGG pathway annotations classify Igfbp5 under growth factor binding and ECM organization, experimental studies demonstrate that its upregulation contributes to cardiac fibrosis by activating fibroblasts via mixed-lineage kinase 1 (MLK-1). It also enhances inflammatory responses through the activation of cytokines, although its precise role in DCM pathogenesis remains unclear, warranting further exploration[14,25,54]. Cd36 is validated by GO/KEGG as a key regulator of lipid metabolism via the PPAR-α pathway in ECs. However, experimental studies indicate that Cd36 also targets angiogenesis via the VEGF pathway, underscoring its significance in DCM pathogenesis. Consequently, Cd36 may serve as both a biomarker for diagnosis and a potential therapeutic target[43,55].
In addition, downregulation of HSPs and their co-chaperones (Ahsa1 and Ahsa2) has been observed in DCM. GO and KEGG analyses show that these genes are primarily responsible for cellular stress responses and protein folding. Their reduced expression leads to protein misfolding and subsequent EC apoptosis. Moreover, HSP dysfunction indirectly activates inflammatory and oxidative stress pathways[56]. A summary of key upregulated and downregulated signaling pathways in DCM is provided in Table 1. Because
DEGs in ECs identified by scRNA-seq in murine models of DCM
| Pathway | Upregulated DEGs | Downregulated DEGs | Citation |
| Angiogenesis | Dysf, Kdr, Flt1, Nrp1, Cd36 | Ets1, Arnt1, Arnt2, Stat5a, Stat5b | Cohen et al.[29] Su et al.[22] Zhang et al.[14] |
| Fibrosis | Eng, Tgfbr2, Atf4, Tcf21, Pdgfb, Pdgfd, Npr3, Meis2, Foxp1, Atf3, Igfbp5 | Ets1 | Cohen et al.[29] Guo et al.[31] Li et al.[24] Zhang et al.[14] |
| Lipid metabolism | Cd36, Fabp4, Zbtb16 | Cohen et al.[29] Su et al.[22] Wen and Wang[23] | |
| Oxidative stress | Bmal1, Rbp7 | Hspa1a, Hspa1b, Hsp99aa/b1, Ahsa 1, Ahsa 2 | Cohen et al.[29] Su et al.[22] Wen and Wang[23] Zhang et al.[14] |
| Inflammation | Irf7, Igfbp5, Ctsh, Cebpb, Plvap | Hspa1a, Hspa1b, Hsp99aa/b1, Ahsa 1, Ahsa 2, Stat5a, Stat5B | Cohen et al.[29] Li et al.[24] Su et al.[22] Zhang et al.[14] |
| Complement | Cfh | Zhang et al.[14] |
Subpopulations of ECs and their dynamics in DCM
ECs are important mediators of microvascular injury in DCM, driving pathological processes such as myocardial fibrosis, inflammation, and impaired angiogenesis. Although the role of ECs in DCM pathogenesis is well recognized, they were long considered a relatively homogeneous population. Advances in scRNA-seq and spatial transcriptomics have challenged this idea, revealing significant heterogeneity among cardiac ECs, which comprise multiple subpopulations marked by distinct gene expression profiles and functions. This section reviews the current understanding of EC subpopulations in normal heart tissue and their dynamic changes in the diabetic heart that contribute to the pathogenesis of DCM.
EC subpopulations in the normal and diabetic hearts
The heart consists of a heterogeneous mixture of cell types whose interactions influence both normal function and disease progression. In addition to CMs, the heart comprises diverse cell types, including fibroblasts, smooth muscle cells (SMCs), immune cells, pericytes, and ECs[14]. ECs, which are identified by expression of pan-endothelial markers such as Pecam1, Cdh5, and Vwf, form the inner lining of the microvasculature and outnumber CMs by approximately 3:1[15,57,58]. While currently there is no consensus on the exact number of EC subpopulations in the healthy heart, cardiac ECs are traditionally divided into several subtypes based on their characteristic markers: arterial (Notch1, Efnb2, Dll4, Hey1), capillary (Ca4), venous (Nr2f2, Ackr1), lymphatic (Lyve1, Prox1, Pdpn), and endocardial (Npr3) cells[58-60]. Among these, capillary ECs are the predominant subtype, accounting for roughly half of the total EC population[58,59,61].
With the advent of scRNA-seq, previously unrecognized EC subpopulations have begun to emerge. Recent studies on human and murine cardiac tissue using scRNA-seq have refined the traditional vascular EC subtypes, identifying additional subpopulations within each category. Notably, capillary ECs, which are the most abundant subtype, have been shown in several studies to contain multiple functional subclusters. Depending on the study, researchers reported three[62], four[58], five[60], or seven[14] distinct capillary EC subpopulations based on differential gene expression, while other studies did not identify clear subdivisions within this group[16,10,42,45]. These specialized subpopulations are primarily associated with fatty acid metabolism, angiogenesis, EC proliferation, and NO production[14,22,23,61]. Some are sufficiently distinct that certain studies have classified them as independent EC subclusters[61]. While most evidence highlights capillary EC heterogeneity, finer subclassification has also been observed within venous[31] and arterial[60] EC clusters, although less extensively studied. One study discerned five venous EC subclusters[31], while another described two[61]. Similarly, one study demonstrated three separate subclusters of arterial ECs[60]. This lack of consensus underscores the need for further investigation, particularly for venous ECs, which play an important role in DCM-related microvascular injury.
Beyond these established categories, novel EC subpopulations with specialized functions have been identified, such as those involved in angiogenesis[23,61,62], inflammation[14,58,62], and lipid metabolism[23]. Angiogenic ECs, characterized by expression of genes such as Egfl7 and Apln, regulate vascular development and maintain vascular integrity[23,62]. Pro-inflammatory ECs represent another unique subtype strongly implicated in DCM. These cells express genes involved in immune cell recruitment (Cx3cl1, Ccl2, Il6), leukocyte adhesion and migration (Vcam1, Icam1, Plvap, Igfbp5), and interferon response (Ifit1, Ifit3)[14,23,58,61,62]. While most studies report a single pro-inflammatory subcluster[58], others describe two[23] or even three[20,61]. Their origins are debated: some evidence suggests derivation from capillary ECs[58], whereas other studies noted that they are of all three vascular EC lineages[61]. ECs specialized in lipid metabolism, hereafter referred to as lipid-handling ECs, have also been observed across several DCM studies. Expressing genes such as Cd36, Lpl, and Gpihbp1, these ECs are responsible for lipid localization, uptake, and metabolism[23,61]. Marker analysis suggests that this subpopulation is most likely derived from capillary ECs[61], and their expansion is especially pronounced in DCM [Figure 3].
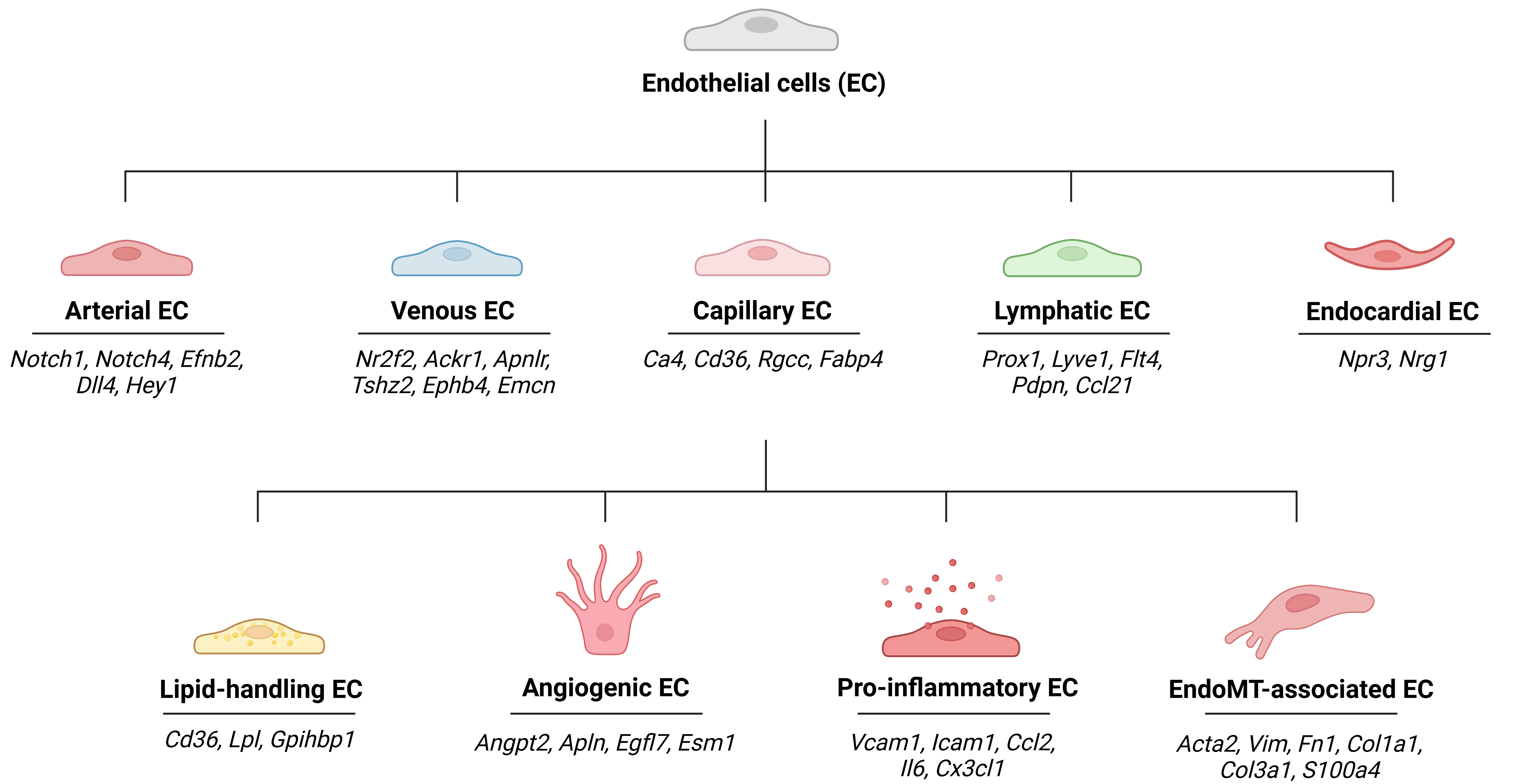
Figure 3. Cardiac EC subpopulations identified by scRNA-seq. Cardiac ECs represent a heterogeneous population that can be subdivided into several functionally distinct groups using scRNA-seq based on their specific cell markers. Traditional vascular EC subtypes include arterial, venous, capillary, and lymphatic ECs[58-60]. Recent studies in DCM have also revealed novel EC subtypes that play important roles in disease progression, such as lipid metabolism-associated ECs, angiogenic ECs, and pro-inflammatory ECs[14,23,62].
Interestingly, some ECs displayed intermediate transcriptional profiles, expressing markers characteristic of more than one traditional subtype. For example, one study identified one such EC subcluster that exhibits both capillary and arterial markers, likely reflecting transitional states between EC phenotypes[60]. Additionally, some studies have reported EC subclusters composed of cells traditionally classified into different EC subtypes, yet sharing similar gene expression profiles, suggesting a potential functional overlap among canonical EC subtypes[14,23]. Despite the growing consensus on EC heterogeneity, some studies have yielded conflicting results. For example, Skelly et al. did not detect transcriptionally distinct EC subpopulations in their analysis, instead reporting a relatively homogeneous EC population in mature murine cardiac tissue. Such discrepancies may be attributed to differences in cell isolation protocols, sequencing platforms, or sequencing depth[63]. A summary of all published scRNA-seq studies on EC subpopulations in DCM is provided in Table 2.
EC subpopulations identified across different scRNA-seq studies in DCM
| Species | Subclusters | Cell markers | Citation |
| Mice | Ec-0 (upregulated in DCM) | Fabp4, Zbtb16, Cd36 | Su et al.[22] |
| Ec-1 (predominant in normal) | Robo2, Plxnd1, Dlg2 | ||
| Mice | Cluster 3 (capillary ECs) | Car4, Timp4, Aqp1 | Zhang et al.[14] |
| Cluster 5 (capillary ECs) | Cxcl12, Btnl9, Rbp7 | ||
| Cluster 10 (capillary ECs) | mt-Cytb, mt-Co3, mt-Nd2 | ||
| Cluster 12 (diverse/unspecified vascular ECs) | H19, Npr3, Cfh | ||
| Cluster 15 (diverse/unspecified vascular ECs) | Sema3g, Gsn, Fbln5 | ||
| Cluster 20 (capillary ECs) | Rsad2, Ifit1, Isg15 | ||
| Cluster 21 (venous ECs) | Slfn2, Fmo2, Odc1 | ||
| Cluster 22 (capillary ECs) | Prnd, Tmsb10, Apln | ||
| Cluster 26 (diverse/unspecified vascular ECs) | Fgl2, Mmrn1, Ccl21a | ||
| Cluster 31 (capillary ECs) | Hba-a2, Hba-a1, Hbb-bs | ||
| Cluster 34 (capillary ECs) | Stmn1, Hmgb2, Lgals1 | ||
| Mice | EC_1 (lipid metabolism) | Cd36, Lpl, Gpihbp1 | Zhao et al.[61] |
| EC_2 (atheroprotection) | Klf2, Klf4 | ||
| EC_3 (maintenance of endothelial barrier) | Cdh5, Gpihbp5, Vim | ||
| EC_4 (endocardial-lineage ECs) | Fabp3, Myl2, Myl3 | ||
| EC_5 (EndoMT, inflammation) | Abca1, Vcam1, Vim | ||
| EC_6 (EndoMT) | Vwf, Eln, Vim | ||
| EC_7 (maintenance of endothelial functions) | Abcg1, Scarb1, Cd36 | ||
| EC_8 (EndoMT, cell adhesion, EC activation) | Abca1, Fgl2, Ccl21a, Il7, Vim | ||
| Mice | Cluster 1 (lipid homeostasis, interferon response) | Rbm3, Fabp4, S100a1, Angptl4 | Wen and Wang[23] |
| Cluster 2 (vasculature development) | Ifit1, Zfp36, Fos, Atf3, Hspa1a, Hspa1b | ||
| Cluster 3 (interferon response) | Lpl, Egfl7 | ||
| Cluster 4 (ATP synthesis, lipid localization) | Pth1r, Hrct1, Cyp1a1, Lars2, Cd36 | ||
| Cluster 5 (protein synthesis) | Rflnb, Anp32a, Rps3a1, Gm424 | ||
| Cluster 6 (vasculature development) | Fosb, Fus, Malat1 |
Changes in EC subpopulations in the pathogenesis of DCM: a biphasic model
Microvascular injury in DCM can be attributed to several interrelated molecular processes, including EndoMT, chronic inflammation, impaired angiogenesis, and capillary rarefaction. ECs and their dysfunction play a central role in driving these pathological processes. Under normal conditions, capillary ECs constitute the major EC subpopulation in the heart, supporting myocardial homeostasis and normal vascular function[59]. Recent single-cell and in vitro studies suggest a biphasic alteration in EC subpopulations during diabetes, reflecting their remarkable plasticity. In the early stages of DCM, capillary ECs mount a compensatory response to hyperglycemia-induced metabolic stress by generating angiogenic and lipid-handling EC subpopulations[2,21,27,37]. These adaptive changes help preserve endothelial integrity and maintain vascular homeostasis. However, as the disease progresses, this compensatory mechanism becomes maladaptive. Pro-inflammatory and EndoMT-associated EC subclusters expand and eventually predominate in late-stage DCM, promoting chronic inflammation, myocardial fibrosis, and contractile dysfunction, ultimately culminating in HF [Figure 4][21,27].
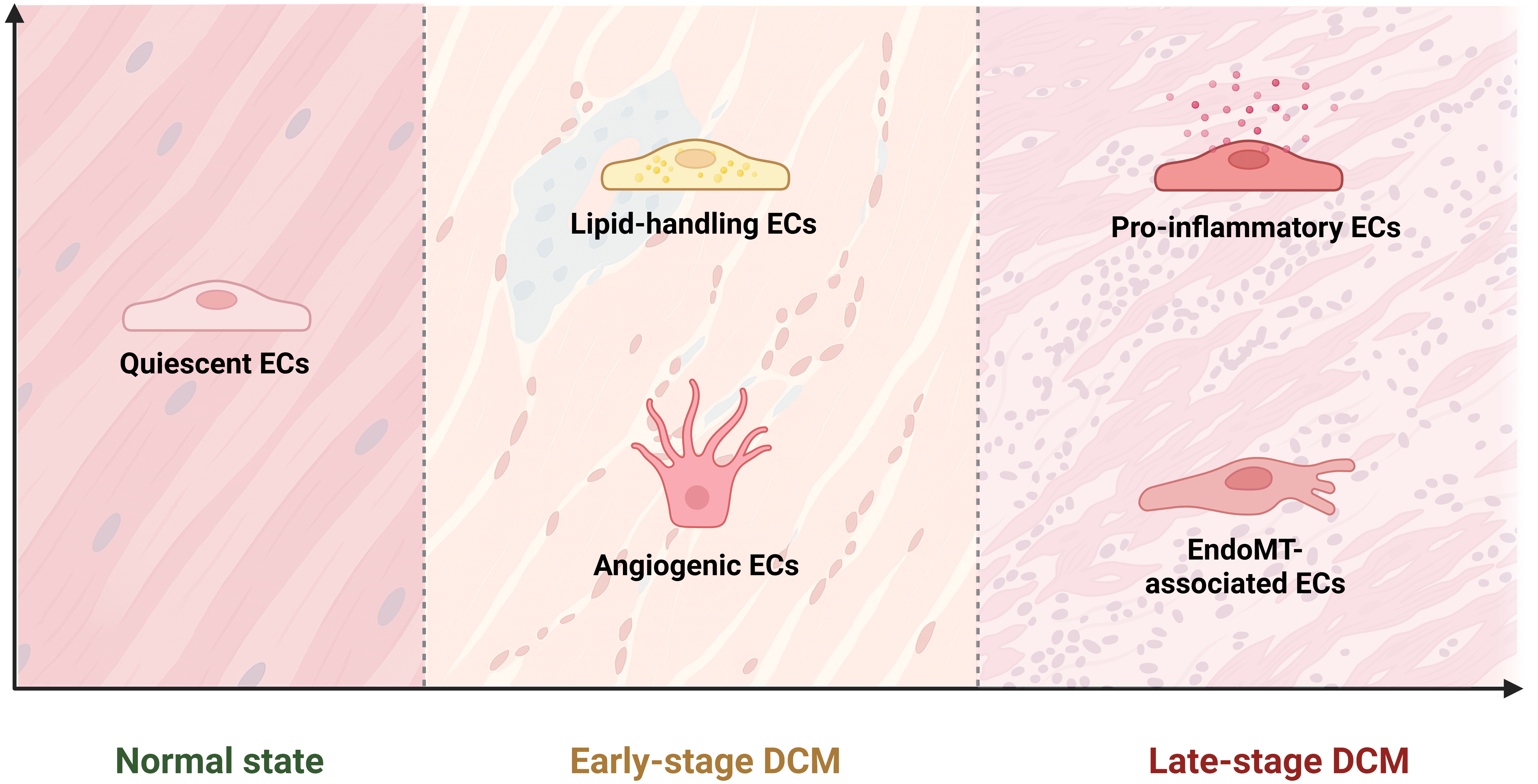
Figure 4. Dynamic changes in EC subpopulations during DCM progression. EC dynamics in DCM progression follow a biphasic pattern: an initial adaptive response is followed by a progressive maladaptive response at later stages. Under normal physiological conditions, quiescent ECs predominate. In the early stage of DCM, lipid-handling and angiogenic ECs emerge in response to hyperglycemia-induced metabolic stress[14,23,61]. During the late stage, there is a shift toward pro-inflammatory and EndoMT-associated ECs, representing a decompensatory response that disrupts vascular homeostasis[14,31,61]. This transition promotes inflammation, myocardial fibrosis, and ultimately HF. EC: Endothelial cell; DCM: diabetic cardiomyopathy; EndoMT: endothelial-to-mesenchymal transition; HF: heart failure. [(Created in BioRender. Justin R (2025)].
Adaptive endothelial responses in early DCM
The early compensatory response of ECs to hyperglycemia is characterized by enhanced lipid handling and angiogenic activation. Across multiple studies, capillary ECs and their functionally specialized derivatives have been observed to expand during the initial stages of diabetes[14,22,23,61]. Zhang et al. reported that three of seven capillary ECs were expanded in early diabetes[14], enriched in genes associated with Fatty acid oxidation (FAO) (Fabp4, Cd36), angiogenesis (Flt1, Kdr), and NO production[14,22,59,61]. Similarly, Wen and Wang identified a capillary EC subcluster with specialized function in lipid handling and metabolism[23], whereas Zhao et al. reported two metabolically activated subclusters likely originating from capillary ECs[61]. These findings suggest that ECs initially activate transcriptional programs to buffer lipid and glucose overload. However, as diabetes progresses, ECs eventually undergo decompensation, marked by downregulation of FAO-related genes, leading to intracellular lipid accumulation, lipotoxic stress, and eventual transition to EndoMT. In parallel, proangiogenic responses characterized by the upregulation of tip cell-associated genes (such as Apln, Adm, Lxn, Fscn1, Col4a1) are also noted in early DCM or following anti-angiogenic interventions[14,64,65]. Over time, however, angiogenic capacity declines, resulting in progressive capillary rarefaction[65]. Studies suggest that this decompensation can be attributed to specific EC subpopulations that lose angiogenic potential and acquire pro-inflammatory phenotypes, thereby further driving DCM progression.
Endothelial decompensation in late-stage DCM
As DCM progresses, the compensatory endothelial response wanes, concurrent with expansion of EC subpopulations with pro-inflammatory and EndoMT phenotypes. Multiple studies have consistently reported a progressive increase in pro-inflammatory EC subpopulations during disease progression[14,31,61]. Two studies identified two such clusters, which gradually expand as diabetes progresses[14,31]. These
EndoMT and myocardial fibrosis become prominent in late-stage DCM. Traditionally, cardiac fibrosis was attributed solely to the activation and proliferation of resident fibroblasts. However, recent advances in scRNA-seq have revealed that a substantial proportion of fibroblasts in DCM also express endothelial markers, supporting the role of EndoMT in fibrotic remodeling[61,63]. Approximately one-third of fibroblasts in cardiac fibrosis are estimated to arise from ECs undergoing EndoMT, characterized by downregulation of EC marker genes (Pecam1, Cdh5, Vwf) and gain of mesenchymal markers (Vim, Cdh2)[66]. In diabetic murine models, three studies identified EndoMT-associated EC clusters: clusters 12 and 26 (Zhang et al.[14]), EC-5, EC-6, and EC-8 (Zhao et al.[61]), and C1 and C3 (Guo et al.[31]). All three studies demonstrated consistent expansion of these EndoMT-associated EC subsets with increasing disease severity. While most evidence points to capillary ECs as the primary source, some studies suggest that arterial and venous ECs also contribute[14,67]. Moreover, pro-inflammatory ECs were also suggested as an intermediate state, as a subset of these cells eventually transition to EndoMT phenotypes[14,31,61]. In Guo et al.’s study, the two venous EC subclusters undergoing EndoMT exhibit increased expression of Cxcl1 and Vcam1, which promotes immune cell recruitment and vascular inflammation[31,68]. Similarly, Zhao et al. reported that one EndoMT EC subcluster also shows high expression of Vcam1[61].
Meanwhile, the early proangiogenic response of capillary ECs diminishes over time, leading to leaky vessels and eventually capillary rarefaction. As DCM progresses, angiogenic genes are markedly downregulated, concurrent with the upregulation of pro-inflammatory and EndoMT genes[14,61]. Only a limited pool of ECs retains angiogenic capacity, potentially counteracting vascular loss. Zhang et al. reported that two
Lipid-handling ECs may persist into late stages, though their functional capacity progressively declines. Notably, changes in EC subpopulations are gradual, representing a continuum in which cells often display transitional phenotypes.
Metabolic reprogramming as a driver of endothelial decompensation
The shift toward pro-inflammatory and EndoMT phenotypes in DCM is driven by metabolic reprogramming. ECs primarily rely on glycolysis for adenosine triphosphate (ATP) production, with only a small portion of their energy derived from FAO[69]. Nevertheless, FAO and lipid signaling pathways are important in maintaining endothelial identity and function, supporting pathways that preserve EC quiescence and suppress EndoMT[61]. In the diabetic heart, these pathways become dysregulated, following a biphasic pattern of changes in lipid metabolism consistent with the compensatory and decompensatory phases of EC function. Persistent hyperglycemia, insulin resistance, and metabolic dysregulation lead to the accumulation of metabolites such as methylglyoxal and AGEs, promoting oxidative stress and inflammatory signaling that ultimately triggers metabolic reprogramming in ECs[70,71]. This reprogramming, in turn, induces transcriptional alterations, such as upregulation of Igfbp5, reinforcing the EndoMT phenotype[14]. Consistent with this, studies have shown that ECs undergoing EndoMT exhibit significantly reduced FAO activity, both in vivo or in vitro, when stimulated by TGF-β1 and IL-1β[61,72]. Acetate supplementation can suppress TGF-β1-induced EndoMT by increasing acetyl-CoA levels, underscoring the role of FAO in maintaining endothelial identity[72].
Biphasic endothelial dynamics in other cardiovascular diseases
A similar biphasic pattern of EC dynamics has been observed in other cardiovascular pathologies, including atherosclerosis[73,74], myocardial infarction (MI)[75-77], and HF[65,78]. In these conditions, the early compensatory phase is characterized by expansion of EC subpopulations involved in angiogenesis and endothelial repair. However, with persistent pathological stimuli, such as disturbed blood flow, this compensatory response transitions into a maladaptive phase dominated by pro-inflammatory and EndoMT EC phenotypes[74,79,80]. Interestingly, partial and reversible EndoMT has been observed following MI, where it may facilitate regeneration[75]. Such reversibility has not been demonstrated in DCM, presenting a promising area for further investigation.
Intercellular interactions between ECs and other cardiac cell types in DCM
As diabetes progresses, cellular interactions within the heart become increasingly dysregulated and complex[31]. ECs play a central role in this process, promoting the development of DCM through a series of crosstalk with other cardiac cells such as fibroblasts, CMs, and immune cells. In recent years, scRNA-seq has provided valuable insights into intercellular communication networks by mapping ligand-receptor interactions and their associated pathways, which enabled the discovery of previously unknown communication axes in DCM [Figure 5]. Two main modes of EC-mediated signaling have been described, reflecting the biphasic dynamics of EC subpopulations described previously[14]. The first mode involves signals related to cellular growth and development, such as VEGF signaling. In contrast, the second mode is associated with leukocyte adhesion and inflammation, exemplified by VCAM-1 signaling. During DCM progression, the growth-related signaling pathways decline, while pro-inflammatory pathways increase, indicating a shift from proliferative to inflammatory signaling[14].
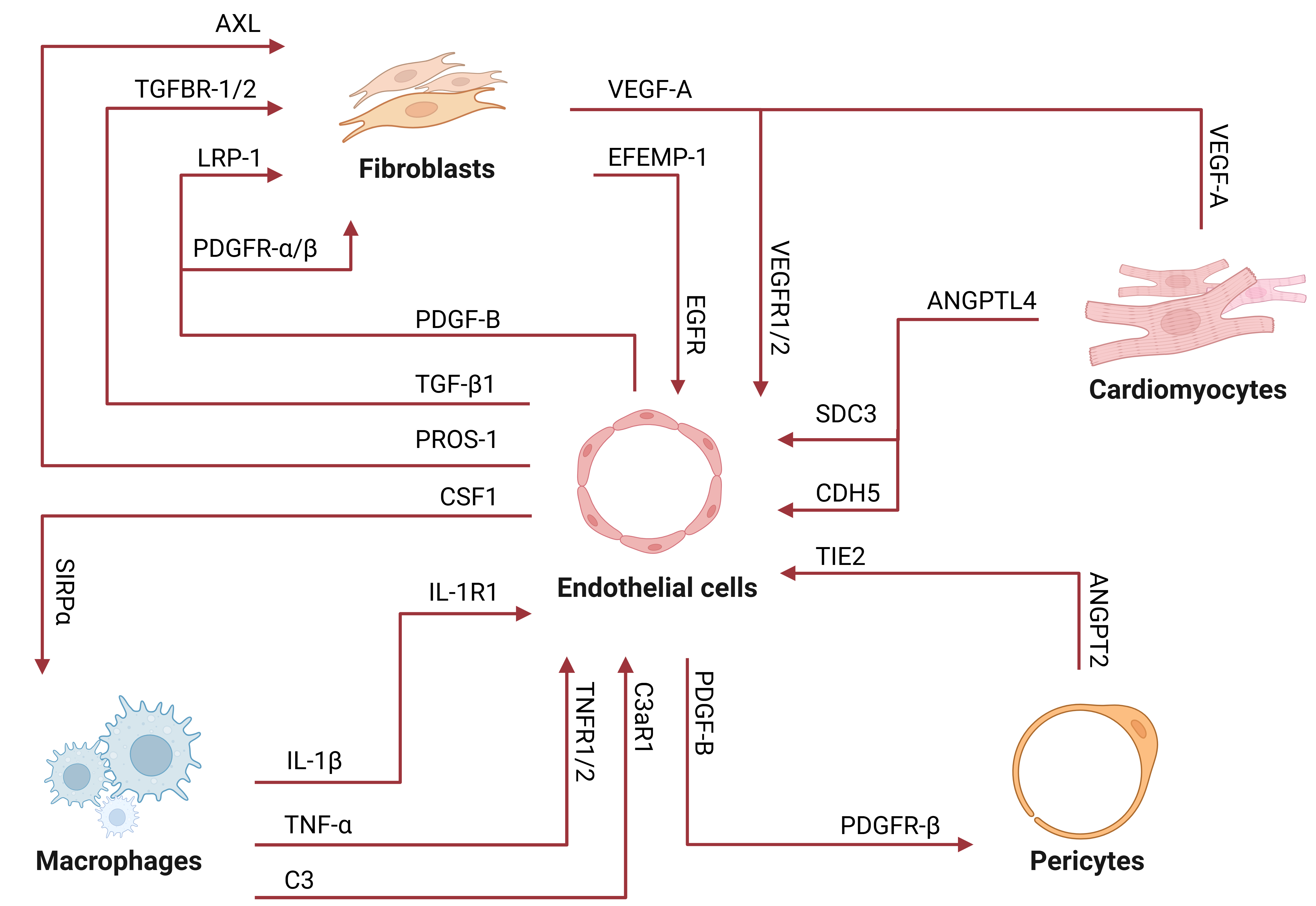
Figure 5. Cell-cell interactions and their associated ligand-receptor pairs in DCM identified using scRNA-seq. ECs serve as central hubs for the increased cellular signaling observed in DCM. Arrows indicate signaling direction, with the protein at the arrow tail denoting the ligand and the protein at the arrowhead representing the receptor. DCM: Diabetic cardiomyopathy; scRNA-seq: single-cell RNA sequencing; EC: endothelial cell; TGFBR: transforming growth factor β receptor; LRP1: low-density lipoprotein receptor-related protein 1; PDGF: platelet-derived growth factor. PDGFR: platelet-derived growth factor receptor; TGF: transforming growth factor; CSF1:
EC-fibroblast interactions
ECs and fibroblasts are central mediators of autocrine and paracrine signaling in DCM[24]. In the healthy heart, approximately 90 ligand-receptor pairs have been identified that mediate EC-fibroblast interactions, many of which regulate ECM turnover and maintain fibroblast quiescence[63]. Conversely, fibroblasts support vascular integrity by secreting pro-survival factors such as insulin-like growth factor 1 (IGF1) and nerve growth factor (NGF), which sustain EC and pericyte viability[24,63]. Additionally, fibroblasts constitute the principal cell type in the tunica adventitia, the outermost layer of blood vessels, where they play a critical role in maintaining vascular wall integrity[81]. Under diabetic conditions, adventitial fibroblasts become pathologically activated, leading to increased ROS production, secretion of pro-inflammatory mediators, and ECM remodeling[81-83]. These changes collectively contribute to fibrotic remodeling of the vascular wall. In DCM, ECs progressively decrease in number, whereas fibroblasts expand, particularly during the later stages of the disease[22]. ScRNA-seq has identified expansion of specific fibroblast subpopulations with high expression of Hrc, Postn, Lox, and Fbln, which are strongly implicated in the development of myocardial fibrosis[24]. Several ligand-receptor pairs mediating EC-fibroblast crosstalk are dysregulated in DCM, including TGF-β1-TGFBR1/2, platelet-derived growth factor B/platelet-derived growth factor receptor α (PDGFB-PDGFRα)/PDGFRβ/Low-density lipoprotein receptor-related protein 1 (LRP1), PROS1-AXL, EFEMP1-EGFR, and VEGFA-FLT1/KDR[22].
TGF-β1, a growth factor primarily produced by ECs and immune cells, binds to transforming growth factor β receptor (TGFBR) 1/2 on fibroblasts to activate Smad-dependent and non-canonical signaling pathways [extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), p38]. TGF-β signaling is markedly upregulated under hyperglycemia, leading to cascades that induce fibroblast transformation into myofibroblasts and ECM deposition, ultimately resulting in cardiac fibrosis[84-87]. In ECs, increased TGF-β signaling also promotes EndoMT through activation of transcription factors such as Snail, Slug, and Twist[85,86]. Similarly, PDGF-B, secreted by ECs, binds PDGFRα on fibroblasts to stimulate their proliferation and ECM deposition. Elevated PDGF-B expression in ECs, along with the upregulation of PDGFRα and PDGFRβ in fibroblasts, has been observed in DCM[22,24]. Additionally, the PDGFB-LRP1 interaction between ECs and both fibroblasts and myocardial cells is enhanced in DCM, further amplifying fibrotic signaling[22]. By contrast, upregulation of the PROS1-AXL axis, another endothelial-fibroblast/myocardial communication pathway, promotes EC survival and suppresses inflammation, suggesting the presence of compensatory mechanisms within dysregulated crosstalk[22].
Fibroblast-to-EC signaling also plays a critical role. EGF-containing fibulin-like extracellular matrix protein 1 (EFEMP1), a growth factor predominantly produced by fibroblasts, activates epidermal growth factor receptor (EGFR) expressed on ECs, stimulating the downstream MAPK/ERK, phosphoinositide 3-kinase (PI3K)/AKT, and JAK/STAT signaling pathways. These signals collectively promote EC proliferation and angiogenic remodeling. In DCM, however, overactivation of EFEMP1-EGFR signaling leads to maladaptive angiogenesis and excessive fibroblast expansion[24]. Fibroblasts, along with hypoxic cardiomyocytes and inflammatory cells, also secrete VEGF-A, which acts on ECs by binding to the vascular endothelial growth factor receptor-1 (VEGFR-1) (encoded by the gene Flt1) and VEGFR-2 (Kdr), with VEGFR-2 being the primary mediator of angiogenic sprouting, EC survival, and vessel permeability. In DCM, despite upregulation of Vegfa, Flt1, and Kdr, angiogenic responses are impaired, likely due to reduced EC responsiveness to VEGF-A[22]. It is thought that chronic factors, such as inflammation and oxidative stress, may have prevented ECs from properly activating the downstream signaling pathways required for angiogenesis[24]. Consequently, angiogenesis becomes inefficient, leading to the formation of disorganized, immature, and leaky vessels, which ultimately contribute to vascular rarefaction and worsened fibrotic remodeling.
EC-CM interactions
ECs play an important role in maintaining myocardial homeostasis by regulating CM contractility and nutrient supply. Under hyperglycemic conditions, intercellular communication between ECs and CMs is disrupted, contributing to cardiac remodeling and contractile dysfunction. While EC numbers decline in DCM, there is an observed expansion of CMs and fibroblasts[22,23]. However, scRNA-seq studies have identified an increase in EC-CM molecular signaling throughout diabetes progression despite the reduced EC number[31]. Among the upregulated ligand-receptor pairs, most are involved in lipid trafficking and nutrient delivery, processes that are impaired in CMs under hyperglycemia. Prolonged hyperglycemia reduces glucose uptake by CMs, shifting their energy metabolism from glucose to fatty acids[23]. Simultaneously, hyperglycemia impairs the vasodilatory function of ECs, reducing oxygen delivery to CMs and exacerbating oxidative stress[23,31].
One upregulated EC-CM crosstalk pathway in DCM is the angiopoietin-like protein (ANGPTL) signaling pathway[23,31]. Specifically, ANGPTL4-SDC3 and ANGPTL4-CDH5 interactions are markedly upregulated, resulting in enhanced endothelial permeability, impaired lipid trafficking, and vascular inflammation[23]. Wen and Wang reported that Angptl4, along with several genes related to lipid homeostasis (Rbm3, Fabp4, S100a1), is upregulated in one of six EC subclusters[23]. Angptl4 expression is also elevated in CMs under diabetic conditions, suggesting bidirectional signaling under hyperglycemic stress. Angptl4 is a known inhibitor of lipoprotein lipase (LPL), an enzyme located at the vascular lumen that hydrolyzes circulating triglycerides into free fatty acids (FFAs), which can be utilized as energy by CMs. Under normal conditions, Angptl4 causes inactivation of LPL, thereby limiting FFA uptake and protecting against lipid overload. However, in DCM, paradoxical upregulation of Angptl4 increases LPL activity, promoting FFA influx, lipid accumulation, and lipotoxicity, culminating in contractile dysfunction of CMs[69].
ECs also contribute to CM survival under hypoxic conditions via the heparanase-VEGF signaling axis.
Exosomes, a subtype of extracellular vesicles, are increasingly recognized as important mediators of intercellular communication. In the heart, particularly between ECs and CMs, bidirectional signaling via exosomes carrying microRNAs (miRNAs), proteins, transcription factors, and lipids has been shown to regulate both physiological and pathological processes[93]. In DCM, dysregulation of exosome-mediated miRNA signaling has been consistently reported. Notably, exosomes derived from ECs exhibit reductions in miR-30d-5p, miR-126a-5p, miR-320-3p, and miR-378-3p, coupled with an upregulation of miR-15-5p, miR-16-5p, miR-34a-5p, and miR-532[94-97]. These alterations promote CM apoptosis and are associated with heart failure phenotypes in the diabetic heart. Conversely, the upregulation of CM-derived exosomal
EC-macrophage interactions
The cardiac microenvironment in DCM is characterized by persistent, low-grade inflammation. Among the contributors are ECs and macrophages, which play central roles in vascular integrity and innate immunity. In DCM, both ECs and macrophages are decreased in number, leading to vascular rarefaction, impaired immune function, and chronic inflammation. Under prolonged hyperglycemia, ECs and macrophages engage in aberrant intercellular signaling, with both shifting toward pro-inflammatory and pro-fibrotic phenotypes. Several molecular ligand-receptor pairs have been implicated in this process, such as IL-1β, TGF-β1, colony-stimulating factor 1 (CSF1)-signal regulatory protein α (SIRPA), C3-C3AR1, and triggering receptor expressed on myeloid cells 2 (TREM2).
During hyperglycemia, there is an increased polarization of macrophages toward the pro-inflammatory M1 phenotype, accompanied by upregulation of genes encoding inflammatory mediators, including Il1b, Tgfb1, Lgals3, Anxa1, and Itgam[31]. These ligands interact with ECs to promote EndoMT, suppress EC proliferation, and enhance EC secretion of pro-inflammatory cytokines. IL-1β, in particular, induces EC expression of Vcam1 and Cxcl1, thereby facilitating leukocyte adhesion and recruitment[31]. Moreover, IL-1β activates cardiac fibroblasts, contributing to both immune activation and fibrotic remodeling in the diabetic heart[31]. Beyond phenotypic switching, hyperglycemia also alters macrophage metabolism. Under prolonged diabetic conditions, macrophages increase glucose uptake and glycolytic activity, which further enhances the secretion of IL-1β and TNF-α[99,100].
Recent studies have identified additional ligand-receptor pairs that mediate EC-macrophage interactions in DCM. For example, one EC subpopulation enriched in DCM exhibited altered crosstalk with macrophages compared to ECs in normal hearts. Specifically, downregulation of the CSF1-SIRPA axis and upregulation of the C3-C3AR1 pathway were observed[22]. CSF1 signaling is essential for macrophage survival and immune tolerance, whereas C3-C3AR1 promotes inflammatory signaling. These findings suggest a loss of immunoregulation and an increase in complement-mediated inflammation in DCM. Other aberrant ligand-receptor interactions have also been reported. High expression of leukocyte immunoglobulin-like receptor A5 (LILRA5) is normally observed in both ECs and macrophages, but is significantly reduced in DCM and other nonischemic cardiomyopathies. Although its precise role remains unclear, this downregulation is thought to contribute to immune dysregulation and ED[101]. Additionally, TREM2 signaling between macrophages and ECs is upregulated in the peripheral vasculature during diabetes. Hyperglycemia increases TREM2 expression on macrophages along with the expression of its ligand in ECs. Enhanced TREM2 signaling impairs EC migration and promotes inflammatory gene expression, although its relevance in the cardiac microvasculature remains to be determined[102].
EC-pericyte interactions
Cardiac pericytes are specialized mural cells embedded within the EC basement membrane, primarily derived from epicardial mesothelium[103]. These cells envelop the microvasculature, where they play a critical role in maintaining vascular integrity and regulating angiogenesis[104]. In DCM, pericyte loss occurs as a consequence of prolonged hyperglycemia. This process is driven by multiple factors, including AGE formation, ROS, oxidative stress, and inflammatory responses, which collectively promote pericyte apoptosis[11,12]. Pericyte depletion further activates inflammatory signaling cascades, ultimately resulting in diastolic dysfunction and HF[105-107].
In DCM, signaling pathways crucial for EC-pericyte crosstalk are disrupted. PDGFB-PDGFRβ signaling plays a vital role in pericyte recruitment during angiogenesis. Newly formed blood vessels are inherently unstable and are therefore covered by pericytes to ensure stabilization. Under physiological conditions, PDGF-B ligand secreted by ECs during sprouting angiogenesis binds to PDGFRβ on pericytes and activates a signaling pathway that promotes their proliferation and migration during vessel maturation[108]. However, hyperglycemia triggers the inactivation of PDGFRβ, leading to impaired pericyte recruitment. This causes endothelial hyperplasia and abnormal vascular morphogenesis, resulting in ED[6,43]. Another key pathway in EC-pericyte communication is angiopoietin (ANGPT)-Tyrosine kinase with immunoglobulin-like and EGF-like domains 2 (TIE2) signaling, which facilitates pericyte-EC attachment, vessel stabilization, and maintenance of vascular permeability. During DCM progression, ANGPT2 is upregulated and antagonizes the effects of ANGPT1, resulting in vascular destabilization, pericyte detachment, and eventual cellular loss[6]. Collectively, the disruption of EC-pericyte interactions in DCM promotes microvascular instability, which in turn impairs cardiac function.
Functional consequences of ED
ED in DCM has a series of functional consequences that could potentially lead to further complications such as systolic and diastolic dysfunction. These consequences arise from various signaling pathways, molecular interactions, and genetic factors, making ED a critical area of research. ED may result from prolonged exposure to hyperglycemia, which is regarded as an initial alteration in DCM progression. This generates a cascade of cellular events such as disruptions in signaling pathways and cellular interactions, eventually contributing to functional outcomes of ED, including inflammation, EC loss, increased vascular permeability, EndoMT, and vasodilator dysfunction.
Chronic inflammatory response
One of the earliest responses to hyperglycemia is inflammation. ED is driven initially by increased intracellular ROS, which induces oxidative stress and mitochondrial dysfunction. This redox imbalance activates pro-inflammatory mediators, including IL-6, IL-1β, TNF-α, IFN-γ, and NF-κB[3,26]. Dysregulation of the PI3K and MAPK signaling pathways further elevates mediators such as VCAM-1, ICAM-1, and plasminogen activator inhibitor-1 (PAI-1), which impair myocardial perfusion and promote a prothrombotic state[4,10]. In addition, a study has shown that VEGF is activated during inflammation, promoting leukocyte infiltration into white adipose tissue in individuals with type 2 DM[10]. This inflammatory cascade is often exacerbated by the activation of the renin-angiotensin-aldosterone system (RAAS), which not only enhances leukocyte infiltration but also triggers local inflammation in myocardial tissue. Together, these molecular pathways result in both structural and functional remodeling[21]. Another study reported that type 2 DM is associated with VEGF activation, elevated inflammatory markers, and impaired glycemic control, indicating that these factors may act synergistically in the development of ED in DCM and related complications[109]. Moreover, inflammatory cytokine expression is mainly regulated by NF-κB. In DM, insulin resistance promotes the upregulation of IL-6, IL-1β, TNF-α, ICAM-1, VCAM-1, and MCP-1. Activation of these cytokines leads to leukocyte migration into myocardial tissue, contributing to adverse structural and functional cardiac remodeling, including myocardial fibrosis and both systolic and diastolic dysfunction[4,19,10]. Importantly, the IL-6 and TGF-β signaling pathways mediate EC-fibroblast crosstalk. Under high glucose conditions, this interaction is disrupted, leading to increased tissue stiffness, myocardial fibrosis, and diastolic dysfunction[110]. Additionally, previous studies have established a clear association between levels of inflammation and an increased risk of HF especially in DM patients, highlighting the pivotal role of ECs as inflammatory mediators in both ED and the cardiac remodeling that characterizes DCM[21].
Subsequent loss of endothelial cell
EC loss arises from multiple interconnected dysregulated pathways, including autophagy impairment, chronic inflammation, and mitochondrial dysfunction. During the progression of DCM, prolonged hyperglycemia is one of the earliest triggers that initiates a series of molecular interactions, ultimately leading to EC loss. Autophagy, a cellular degradation mechanism, plays a crucial role in regulating EC function through pathways related to inflammation, mitochondrial homeostasis, and angiogenesis. Salemkour and Lenoir noted that high glucose concentrations induce autophagic flux impairment by increasing the activation of mechanistic target of rapamycin complex 1 (mTORC1), which inhibits the Unc-51-like autophagy activating kinase 1 (ULK1) complex, a molecular complex required for autophagy initiation[111]. This results in reduced autophagic flux, accumulation of intracellular waste, and EC loss due to increased apoptosis. Chronic inflammation also contributes to EC depletion. The accumulation of oxygen-derived radicals stimulates the PI3K/AKT signaling pathway in ECs, promoting caspase activation through NF-κB-mediated upregulation of cyclooxygenase (COX)-2, which subsequently induces apoptosis[112,113]. Interestingly, autophagy has also been shown to regulate inflammatory signaling, further highlighting the association between these pathways[111]. Another mechanism contributing to EC reduction is mitochondrial dysfunction. This dysfunction is often triggered by the exposure of AGEs and oxidized low-density lipoprotein (ox-LDL), which increase mitochondrial ROS levels, causing cellular damage and death. Moreover, AGEs and ox-LDL are known to block autophagic flux, further reducing EC viability[111].
The functional consequences of EC loss are diverse. One such outcome is microvascular rarefaction, driven by ROS accumulation in mitochondrial ECs, which is shown to cause endothelial nitric oxide synthase (eNOS) uncoupling, impairing autophagy and vasomotor responses. Alongside apoptosis-induced reductions in EC population, capillary density decreases, promoting capillary rarefaction, which clinically contributes to both systolic and diastolic HF[111,114]. Cardiac fibrosis is another consequence of EC loss, frequently initiated by EndoMT. Hyperglycemia-induced chronic inflammation via NF-κB-mediated pathways promotes EndoMT by upregulating FN expression, facilitating the transition of ECs into myofibroblasts. This process increases myocardial stiffness, resulting in cardiac fibrosis[111]. Finally, EC loss disrupts angiogenetic function. Angiogenesis, primarily mediated by VEGF signaling pathways, drives new vessel formation. Hyperglycemia impairs this process by reducing VEGF signaling. Salemkour and Lenoir further noted that VEGF signaling is intrinsically linked to autophagy, as both pathways induce ULK1 phosphorylation[111]. Since ULK1 complex activity is suppressed by impaired autophagic flux, VEGF-mediated angiogenesis is also reduced, preventing new vessel formation and thereby worsening cardiac function.
Increased permeability and barrier dysfunction
ED leads to increased cellular permeability and barrier dysfunction, which are often associated with oxidative stress as an initial change[20]. A common pathway linked to cellular permeability is the diacylglycerol (DAG)-protein kinase C (PKC) pathway, whose activity is elevated under diabetic conditions[115]. Activation of this pathway is thought to contribute to ED by promoting leukocyte adhesion and increasing cellular permeability[4]. In addition, the DAG-PKC signaling pathway is associated with intracellular Ca2+ levels, as Ca2+ acts as a second messenger for eNOS activation and modulates cellular trafficking. In DCM, oxidative stress induces Ca2+ influx into endothelial cells and disrupts tight junctions, leading to neutrophil migration and endothelial barrier disruption[3]. Increased cellular permeability and endothelial barrier disruption have multiple consequences during disease progression. Greater permeability facilitates leukocyte migration into myocardial tissue, allowing immune cells to infiltrate more freely. This stimulates persistent inflammation and ROS accumulation and results in cardiac fibrosis, myocardial wall stiffness, and diastolic dysfunction[3,6,103]. A compromised cellular barrier also permits the leakage of other proteins, causing cellular edema[103]. Concurrently, NO signaling becomes impaired, resulting in dysfunctional vascular relaxation and, in turn, cardiac fibrosis, hypertrophy, and myocardial dysfunction[7,103,116]. Collectively, these changes underscore the pivotal role of ECs in maintaining myocardial structure and function and highlight the need to develop therapeutic strategies targeting these mechanisms.
EndoMT and fibrotic remodeling
One of the key functional alterations in response to ED is EndoMT. This process is closely associated with inflammation and oxidative stress, which are common early changes. In particular, ECs undergo phenotypic transformation in response to abnormal homeostatic disturbances such as hyperglycemia, observed in the early stages of DCM[117,118]. EndoMT can be triggered through multiple pathways, including the TGF-β/SMAD signaling cascade, as well as non-TGF-mediated pathways such as Wnt, Notch, ET-1, and cytokine-driven inflammatory signaling. Among these, the TGF-β-mediated pathway is the best characterized and is considered a major driver of EndoMT in DCM. A typical cascade begins with TGF-β1 activation following its binding to TGF-β2 and the recruitment of anaplastic lymphoma kinase 5 (ALK5). ALK5 subsequently associates with SMAD proteins to form transcriptional complexes that translocate into the nucleus and induce the expression of certain genes such as Notch1, Twist1, and Snai1/2[117,119]. Although TGF-β-mediated is regarded as the primary mechanism, evidence also supports contributions from the Wnt and Notch pathways. These pathways can modulate cell signaling by upregulating molecules such as carbonic anhydrase (CA) and NOTCH2, thereby promoting fibrosis and microvascular dysfunction. These processes may act synergistically with, or independently of, TGF-β signaling[10,117,118,120,121]. During EndoMT, ECs acquire fibroblast-like characteristics. This transition involves the loss of cell-cell adhesion due to depletion of adhesion proteins, cytoskeletal reorganization with actin filament disruption, and a morphological shift from a cobblestone appearance to a spindle-shaped morphology. There is also reduced expression of key EC markers, including von Willebrand factor (vWF), CD31/platelet endothelial cell adhesion molecule 1 (PECAM1), and vascular endothelial cadherin (VE-CAD), alongside increased expression of mesenchymal markers such as α-smooth muscle actin (α-SMA), fibroblast-specific protein (FSP), fibronectin (FN), and collagen, leading to altered cellular identity[110]. These alterations are accompanied by increased ECM production, collectively contributing to enhanced cellular migration[3,117-119,122]. Ultimately, EndoMT contributes significantly to cardiac remodeling in DCM by expanding fibroblast populations within the myocardium. This expansion increases ECM deposition, such as collagen, leading to myocardial stiffness and reduced ventricular compliance[4,7,116]. Interstitial fibrosis and hypertrophy may further contribute to diastolic dysfunction, which manifests clinically as both systolic and diastolic HF[4,7]. These changes co-occur with microvascular rarefaction due to impaired myocardial perfusion, further accelerating HF progression in DCM[116,123].
Loss of vasodilatory function
ED may present as a loss of vasodilatory function and is considered a major contributor to myocardial dysfunction in DCM[3]. This impairment primarily results from an imbalance between vasoconstrictors and vasodilators. NO acts as a major vasodilator in the body. In DCM, NO bioavailability is reduced due to elevated levels of asymmetric dimethylarginine (ADMA) and decreased levels of tetrahydrobiopterin (BH4), respectively[9]. High glucose increases ADMA levels by impairing dimethylarginine dimethylaminohydrolase (DDAH), the enzyme responsible for ADMA degradation and thus for supporting NO production. The accumulation of ADMA during hyperglycemia, in turn, leads to a loss of endothelial vasodilatory function[115]. Conversely, BH4 is deficient in diabetic conditions[124]. As an important cofactor for eNOS coupling, reduced BH4 promotes eNOS uncoupling, resulting in enzyme dysfunction, which is characterized by decreased NO production along with increased superoxide generation[9,124]. With reduced NO and an imbalance between vasodilation and vasoconstriction, it is inferred that vasoconstrictors such as ET-1 are increased in the heart[3]. These changes lead to harmful cardiovascular consequences. One of the most well-known outcomes is microvascular rarefaction, which refers to reduced coronary blood flow. This process is driven by multiple factors, including impaired angiogenesis, decreased NO bioavailability, and imbalances between vasoconstrictors and vasodilators. These changes diminish new vessel formation and impair vasodilatory function. Together, these defects further worsen capillary rarefaction, thereby promoting cardiac dysfunction. The resulting dysfunction increases the risk of myocardial ischemia and hypoxia during periods of increased blood supply demand in the myocardium, thus promoting the development of HF[116]. Additionally, diminished NO signaling contributes to myocardial stiffness, a hallmark of diastolic dysfunction. By promoting vasoconstriction, vascular compliance is reduced, which stimulates fibroblast-to-myofibroblast differentiation through the TGF-β pathway[3,6,7,116]. Furthermore, NO has anti-inflammatory properties, and NO deficiency in ECs exacerbates the inflammatory response, leading to chronic inflammation and ultimately cardiac remodeling[116]. Studies have also established that reduced NO bioavailability disrupts signaling pathways that regulate myocardial hypertrophy, indicating that NO loss promotes hypertrophy - one of the key features of DCM[7,116]. Collectively, these observations illustrate how NO deficiency in ECs drives a cascade of events, including dysregulation of vascular tone and compliance, alterations in cell populations, chronic inflammation, and cardiac remodeling, eventually contributing to HF.
ENDOTHELIAL CELL-CENTERED THERAPEUTIC STRATEGIES FOR DCM
Given the increasing recognition of EC involvement in the pathogenesis of DCM, strategies targeting ED present a fresh perspective and a valuable addition to current treatment approaches. Multiple studies have described a biphasic pattern in EC subpopulation dynamics, beginning with compensatory expansion of capillary ECs followed by a decompensatory phase characterized by EndoMT, inflammation, and fibrosis. Since endothelial decompensation plays a pivotal role in DCM progression, it is crucial to initiate endothelial-protective and antifibrotic interventions early, alongside standard glycemic management.
In the early stages of DCM, treatment strategies should extend beyond glycemic control to include restoration of endothelial homeostasis and mitigation of metabolic stress. Sodium-glucose cotransporter 2 (SGLT2) inhibitors (empagliflozin, dapagliflozin) and glucagon-like peptide-1 (GLP-1) receptor agonists (exenatide), which are recommended as first-line glycemic control agents for diabetic patients with cardiac involvement, also provide vascular benefits by reducing oxidative stress and inflammation[71,125-127]. Inhibition of EndoMT is another key strategy in DCM treatment. EndoMT is primarily driven by maladaptive EC subpopulations and activation of pathways such as NF-κB and TGF-β, along with metabolic reprogramming characterized by reduced reliance on FAO. Targeted interventions that disrupt these pathways or suppress glycolytic conversion may therefore mitigate fibrosis and inflammation in DCM[26]. Finerenone, a mineralocorticoid receptor antagonist currently recommended for DCM, has shown efficacy in modulating ED and reducing cardiac fibrosis, partly by suppressing persistent NF-κB and TGF-β signaling[125]. Similarly, SGLT2 inhibitors such as empagliflozin and dapagliflozin have also been shown to repress EndoMT in preclinical studies[126,128]. In non-DCM fibrotic models, agents including losartan[129], dipeptidyl peptidase-4 (DPP-4) inhibitors (linagliptin)[130,131], cinacalcet[132], kallistatin[133], and macitentan[134] have also shown EndoMT-inhibitory and antifibrotic effects, warranting investigation of their applicability in DCM. Moreover, compounds such as high-density lipoprotein (HDL), apolipoprotein A1 (ApoA1), and icariin have been reported to modulate EndoMT in vitro[135]. Future efforts should further explore therapeutic targeting of key EndoMT mediators, including calpain[136] and ET-1[6,137].
A key challenge to early intervention is the concept of “hyperglycemic memory”, whereby EC dysfunction and maladaptive gene expression induced during prolonged hyperglycemia persist even after blood glucose is normalized[70]. This suggests that glycemic control alone during early DCM may not be sufficient to completely reverse ED or prevent progression to EndoMT. The mechanism underlying metabolic memory is thought to involve a self-sustaining signaling loop between calcium/calmodulin-dependent protein kinase II α (CaMKIIα) and its O-linked attachment of N-acetylglucosamine (O-GlcNAc)[138]. Chronic hyperglycemia increases O-GlcNAcylation of CaMKIIα, which in turn activates CaMKIIα that promotes further O-GlcNAcylation, establishing a positive feedback loop that perpetuates inflammatory and profibrotic signaling despite restored normoglycemia. Strategies targeting O-GlcNAcylation with O-GlcNAcase activators or O-GlcNAc transferase inhibitors have shown promise in breaking this loop and reducing persistent pathogenic signaling[139]. Agents such as SGLT2 inhibitors, nuclear factor erythroid 2-related factor 2 (NRF2) activators, and miR-27a-3p inhibitors have also exhibited several benefits in targeting hyperglycemic memory, although further investigation is needed[126,140].
Despite advances in understanding DCM, diagnosis and staging remain difficult, particularly in asymptomatic early stages[125]. Currently, no sensitive and specific tools exist to detect subclinical changes in DCM, such as dysregulated signaling, altered EC subpopulations, or microvascular dysfunction. Improved methods are needed to capture molecular and microvascular hallmarks of early DCM and its progression. Circulating biomarkers represent one avenue, but their use remains limited by a lack of reliable candidates. Current promising biomarkers include fibrinogen-like protein 1 (FGL-1), lysyl oxidase-like 2 (LOXL2), and electron transfer flavoprotein β subunit (ETFβ), all of which have been found to be elevated in early DCM[141,142]. Complement factor H (CFH), the product of the Cfh gene previously linked to EC phase transitions in DCM, has also emerged as a potential serum biomarker for type 2 DM and warrants evaluation for DCM staging[14,50]. Additionally, decreased serum D-glutamine has demonstrated a strong association with DCM[143]. Circulating miRNAs offer another promising biomarker class due to their stability and involvement in pathogenesis. Among these, miR-126, miR-146a, miR-221-3p, and miR-320a are the most consistently validated in human studies[144-146]. Reduced miR-146a appears particularly specific to DCM, given its implication in enhancing NF-κB signaling in ECs and promoting pro-inflammatory cytokine release and cardiac fibrosis[146,145]. While miR-126 is less cardiac-specific, it is extensively studied in diabetes, and its reduction is linked to ED and impaired angiogenesis[95,144-147]. Elevated miR-221-3p and miR-320a have also been widely validated for correlation with DCM, with strong evidence on cardiovascular risk prediction and disease progression[145,146]. Additional promising but less extensively validated miRNAs, including miR-1, miR-19b-3p, miR-30d-5p, miR-181b-5p, miR-29a-3p, miR-199a-3p, miR-132, miR-133a, and miR-483-3p, have been linked to early microvascular dysfunction and metabolic memory and warrant further investigation as potential biomarkers[95,97,147-154]. Therapeutically, miRNA modulation provides a novel EC-centered approach for DCM treatment. Upregulation of miR-146a and miR-30, as well as inhibition of miR-21 and miR-125b, has been shown in preclinical studies to improve endothelial function and attenuate cardiac fibrosis[97,146,155]. Although the preclinical data are encouraging, further studies are required to validate diagnostic specificity, establish therapeutic efficacy, and standardize quantification methods before clinical application[156].
In addition to biomarkers, imaging technologies play a key role in DCM diagnosis by enabling assessment of cardiac microvascular changes. Conventional modalities such as ultrasound, cardiovascular magnetic resonance imaging (CMRI), and positron emission tomography (PET), while useful for evaluating late-stage myocardial fibrosis, have limited value in detecting early microvascular alterations. Emerging techniques, such as multiphoton microscopy (MPM), provide higher resolution for assessing endothelial and microvascular changes but require further refinement for clinical implementation[70]. Future studies should integrate biomarker-based approaches with advanced imaging to develop composite diagnostic strategies that enable more accurate staging and timely intervention.
Beyond diagnostic advances, EC-targeted gene therapies, particularly small interfering RNAs (siRNAs), offer a promising therapeutic avenue in DCM due to their ability to silence genes involved in inflammation, fibrosis, and angiogenesis. For instance, siRNA against ANGPT2 has shown efficacy in diabetic retinopathy by counteracting HIF-1 and increasing VEGF expression, thus promoting angiogenesis and reducing vascular damage[157]. Similarly, siRNA inhibition of pathways such as Toll-like receptor 4 (TLR4), chemokine-like receptor 1 (CMKLR1), and NLRP3 has demonstrated the suppression of inflammation and fibrosis by reducing markers such as VCAM-1, ICAM-1, IL-1, ROS, and even TGFB1 mRNA expression[158]. These findings support further development of EC-centered siRNA delivery to restore vascular function and limit cardiac remodeling in DCM.
Within this context, Igfbp5 has emerged as a potential gene target, as its elevation induces EC inflammation in diabetic kidney disease and may affect other tissues such as the heart and lungs[25]. Evidence indicates that Igfbp5 knockout attenuates glucose-mediated fibroblast activation by downregulating proangiogenic genes and reducing apoptosis, cellular proliferation, and collagen synthesis, highlighting its functional relevance in diabetic conditions[159,160]. While hyperglycemia-induced Igfbp5 upregulation is thought to be indirect, EC-targeted siRNA delivery represents a promising therapeutic strategy. At the transcriptional level, Igfbp5 is believed to act through MLK-1-mediated inflammatory responses, and MLK-1 deletion has been shown to attenuate cardiac fibrosis in both fibroblasts and ECs. Moreover, microfibrillar-associated protein 5 (MFAP5), regulated by the insulin-like growth factor-binding protein 5 (IGFBP5)-nuclear factor of activated T cells 4 (NFAT4) axis, promotes increased expression of myofibroblast markers, ECM remodeling, and fibrosis[160]. Collectively, these findings indicate the therapeutic potential of targeting the axis, inhibiting proteins such as MLK-1, and developing EC-specific siRNA delivery strategies. However, further studies are needed to validate target specificity, efficacy, and off-target effects prior to clinical translation.
FUTURE PERSPECTIVES
The scRNA-seq technique has enabled the identification of DEGs, along with cellular subpopulations and intercellular interactions during DCM pathogenesis. However, continued research in this area remains necessary. Because scRNA-seq cannot preserve the spatial information of the RNA transcriptome, spatial transcriptomics presents a valuable tool for future studies in DCM[161,162]. In addition, gene expression during pathological states is greatly influenced by transcription factors, underscoring the need for regulatory factor profiling, which may pave the way for future therapeutic strategies that target entire gene networks. Despite the growing understanding of EC heterogenicity, inconsistencies in the classification of EC subpopulations have made it difficult to compare findings across studies, highlighting the need to standardize and establish EC markers and align key markers to facilitate EC cluster separation under both physiological and pathological conditions. Moreover, dysregulation of ligand-receptor interactions between cellular subtypes, such as EC-fibroblast and EC-CM, is common in DCM, emphasizing the need to investigate both new and existing receptor pairs, as they may provide therapeutic targets for disrupted signaling pathways. Importantly, most transcriptomic experiments have been conducted in murine models, which differ from humans, particularly in immune system regulation. This creates a need for further comparative studies of DCM pathogenesis and drug responses across species to support the development of future treatment strategies.
Looking forward, integrating scRNA-seq with technologies such as single-cell ATAQ-seq, spatial transcriptomics, and proteomics will enable a more comprehensive understanding of cellular and molecular mechanisms promoting ED in DCM. These approaches can determine cell-specific epigenetic features and metabolic network patterns that are not captured by scRNA-seq alone, thereby improving the identification of novel biomarkers. Current biomarkers identified using scRNA-seq include transcription factors such as ETS1, transcription factor 21 (TCF21), STAT5A, and STAT5B; metabolic enzymes including pyruvate dehydrogenase kinase isozyme 4 (PDK4), PPARγ, and tropomyosin 1 (TPM1); and major glycoproteins like CFH, which is often associated with vascular damage in diabetes[14,22]. These biomarkers can be visualized and validated in blood or tissue in mice, enabling earlier diagnosis and treatment. Moreover, integrating multi-omic datasets may support patient-specific risk stratification and guide personalized therapies, advancing precision medicine. Although research remains limited, the use of multiple transcriptomic approaches represents a promising direction.
From a clinical perspective, scRNA-seq holds strong potential for translation into diagnostic and therapeutic applications. For example, the identification of transcription factors such as ETS1, signaling pathways involved in fibrosis (e.g., TGF-β, AGE, and HIF), and inflammatory cytokines such as IL-1β, TNF-α, and IL-6 could provide new therapeutic targets. ETS1 was identified via scRNA-seq as a protective factor in ECs in diabetic hearts, and its knockdown has been shown to promote fibrosis in DCM, potentially through the ETS1-thrombospondin-1 (THBS1) axis[163]. This highlights the need for therapies targeting ETS1 and the ETS1-THBS1 pathway, which may lead to novel strategies to prevent fibrosis in DCM patients[14,163]. Preclinical studies have shown that inhibition of the TGF-β pathway can alleviate myocardial dysfunction and fibrosis. Compounds such as cinnamoyl anthranilate and its derivatives (FT23 and FT011) have been shown to block this signaling cascade and mitigate structural and functional damage in animal models of DCM[7]. Notably, commonly used DM drugs such as pioglitazone, rosiglitazone, and empagliflozin have also been reported to inhibit TGF-β signaling and may be considered in future therapeutic regimens[164,165]. The AGE/RAGE axis is also deeply involved in cardiac fibrosis and inflammation in DCM. Aminoguanidine, a widely used AGE inhibitor, has been shown in mice to reduce vascular protein crosslinks and limit fibrosis in diabetic hearts[165]. HIF-induced upregulation of VEGF plays a significant role in glucose uptake via glucose transporter (GLUT) expression. Dysregulation of the HIF pathway causes downregulation of VEGF-mediated GLUT interactions; thus, therapies targeting HIF-1 could potentially alleviate ED in diabetic hearts[48]. Additionally, IL-1β antibodies such as canakinumab have been shown to reduce cardiovascular events and improve glycemic control, although additional evidence is still required[165].
Several challenges in translating current therapeutic strategies to clinical use have been identified. Pathways such as TGF-β are pleiotropic, meaning individuals may show different phenotypes due to genetic variation. Therefore, systemic inhibition of these pathways may cause off-target toxicity and disrupt multiple physiological processes, including angiogenesis, vascular repair, and immune responses[166]. Addressing these issues will require the development of cell-specific modulation strategies that reduce toxicity and improve outcomes in different patient groups. Additionally, biomarker validation in human cohorts remains understudied. Without standardized validation platforms across species, these markers cannot be incorporated into clinical practice. Because most studies are conducted in murine models, they fail to capture the full complexity of human cellular and metabolic interactions in DCM. Developing animal models with more human-like characteristics would improve understanding of pharmacological responses and facilitate the functional validation of biomarkers for clinical diagnostic use[165,167,168]. Overcoming these barriers will help advance the clinical translation of scRNA-seq-guided therapies within precision medicine for DCM patients.
CONCLUSION
DM is a global health concern, and DCM is one of its significant complications. ED is a major pathological event that contributes to microvascular injury in DCM. Prolonged hyperglycemia induces various changes in cardiac tissue, including chronic inflammation, EndoMT, oxidative stress, and impaired angiogenesis, which ultimately culminate in ED. The scRNA-seq technique has revolutionized this research field by enabling transcriptional profiling of individual cardiac cells and providing deeper insight into DCM pathogenesis. Data from scRNA-seq studies have shown that ECs undergo dynamic alterations in gene expression as DCM progresses, leading to pathological functional changes. These studies have also confirmed the heterogeneity of cardiac EC populations and the asymmetrical functional changes among EC subpopulations during diabetic progression. Furthermore, scRNA-seq data have revealed dysregulated intercellular interactions between ECs and other cardiac cell types, including CMs, fibroblasts, macrophages, and pericytes. Collectively, these insights open new avenues for the development of EC-targeted therapies aimed at preserving vascular function and preventing disease progression.
DECLARATIONS
Acknowledgments
All figures were created with BioRender.com. The graphic abstract was created in BioRender. Justin R (2025) https://BioRender.com/vj8mclr.
Authors’ contributions
Conceptualization, data analysis and interpretation: Siriwattanawong P, Justin R
Methodology: Siriwattanawong P, Justin R, Chen T
Writing-original draft preparation: Siriwattanawong P, Justin R, Gamachchige PR
Writing-review and editing: Siriwattanawong P, Justin R, Zhou D, Chen T
Project administration, funding acquisition: Chen T
All authors have read and agreed to the published version of the manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by a grant from the National Key Research and Development Program of China (2023YFC3606500), two National Natural Science Foundation of China (No.82470428, 81873484), a grant from the Natural Science Foundation of Zhejiang Province (No. LMS25H020003), and a grant from the Project of Medical Science Research Foundation from the Health Department of Zhejiang Province (No. WKJ-ZJ-2312), and supported by Key Laboratory of Precision Medicine for Atherosclerotic Diseases of Zhejiang Province, China (Grant No. 2022E10026).
Conflicts of interest
Chen T serves as the Guest Editor of the Special Issue “Decoding Vascular Remodelling: Stem Cell Dynamics and Single-Cell Omics Insights” and as a Youth Editorial Board member of Vessel Plus. She was not involved in any stage of the editorial process for this manuscript, including reviewer selection, manuscript handling, or decision making. Other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
Supplementary Materials
REFERENCES
1. International Diabetes Federation. Over 250 million people worldwide unaware they have diabetes, according to new IDF research. 2025. Available from: https://idf.org/news/idf-diabetes-atlas-11th-edition/ [Last accessed on 13 Oct 2025].
2. Einarson TR, Acs A, Ludwig C, Panton UH. Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007-2017. Cardiovasc Diabetol. 2018;17:83.
3. Wang M, Li Y, Li S, Lv J. Endothelial dysfunction and diabetic cardiomyopathy. Front Endocrinol. 2022;13:851941.
4. Salvatore T, Pafundi PC, Galiero R, et al. The diabetic cardiomyopathy: the contributing pathophysiological mechanisms. Front Med. 2021;8:695792.
5. Radzioch E, Dąbek B, Balcerczyk-Lis M, et al. Diabetic cardiomyopathy-from basics through diagnosis to treatment. Biomedicines. 2024;12:765.
6. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. 2018;122:624-38.
7. Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. 2020;17:585-607.
8. Nakamura K, Miyoshi T, Yoshida M, et al. Pathophysiology and treatment of diabetic cardiomyopathy and heart failure in patients with diabetes mellitus. Int J Mol Sci. 2022;23:3587.
9. Shi Y, Vanhoutte PM. Macro- and microvascular endothelial dysfunction in diabetes. J Diabetes. 2017;9:434-49.
10. Yang DR, Wang MY, Zhang CL, Wang Y. Endothelial dysfunction in vascular complications of diabetes: a comprehensive review of mechanisms and implications. Front Endocrinol. 2024;15:1359255.
12. Chilton R, Iranpour EI, Bloomgarden Z. Deciphering the connection: diabetes, pericyte dysfunction, and their impact on cardiovascular health. J Diabetes. 2024;16:e13539.
13. Paik DT, Cho S, Tian L, Chang HY, Wu JC. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat Rev Cardiol. 2020;17:457-73.
14. Zhang Y, Cao Y, Zhang X, et al. Single-cell RNA sequencing uncovers pathological processes and crucial targets for vascular endothelial injury in diabetic hearts. Adv Sci. 2024;11:e2405543.
15. Paik DT, Tian L, Williams IM, et al. Single-cell RNA sequencing unveils unique transcriptomic signatures of organ-specific endothelial cells. Circulation. 2020;142:1848-62.
16. Wu L, Islam MR, Lee J, et al. ErbB3 is a critical regulator of cytoskeletal dynamics in brain microvascular endothelial cells: implications for vascular remodeling and blood brain barrier modulation. J Cereb Blood Flow Metab. 2021;41:2242-55.
17. Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. 2011;12:551-64.
18. Dejana E. The role of wnt signaling in physiological and pathological angiogenesis. Circ Res. 2010;107:943-52.
19. Kaur N, Guan Y, Raja R, Ruiz-Velasco A, Liu W. Mechanisms and therapeutic prospects of diabetic cardiomyopathy through the inflammatory response. Front Physiol. 2021;12:694864.
20. Bellemare M, Bourcier L, Iglesies-Grau J, Boulet J, O’Meara E, Bouabdallaoui N. Mechanisms of diabetic cardiomyopathy: Focus on inflammation. Diabetes Obes Metab. 2025;27:2326-38.
21. Frati G, Schirone L, Chimenti I, et al. An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc Res. 2017;113:378-88.
22. Su Q, Huang W, Huang Y, et al. Single-cell insights: pioneering an integrated atlas of chromatin accessibility and transcriptomic landscapes in diabetic cardiomyopathy. Cardiovasc Diabetol. 2024;23:139.
23. Wen Y, Wang Q. Cardiac endothelial cells and cardiomyocytes alter their communication properties in diabetic mice. Biol Res. 2025;58:23.
24. Li W, Lou X, Zha Y, et al. Single-cell RNA-seq of heart reveals intercellular communication drivers of myocardial fibrosis in diabetic cardiomyopathy. eLife. 2023:12.
25. Song C, Wang S, Fu Z, et al. IGFBP5 promotes diabetic kidney disease progression by enhancing PFKFB3-mediated endothelial glycolysis. Cell Death Dis. 2022;13:340.
26. Chatterjee A, Tumarin J, Prabhakar S. Role of inflammation in the progression of diabetic kidney disease. Vessel Plus. 2024;8:28.
27. Sayed S, Faruq O, Preya UH, Kim JT. Cathepsin S knockdown suppresses endothelial inflammation, angiogenesis, and complement protein activity under hyperglycemic conditions in vitro by inhibiting NF-κB signaling. Int J Mol Sci. 2023;24:5428.
28. Kobayashi S, Zhao F, Kobayashi T, et al. Hyperglycemia-induced cardiomyocyte death is mediated by lysosomal membrane injury and aberrant expression of cathepsin D. Biochem Biophys Res Commun. 2020;523:239-45.
29. Cohen CD, De Blasio MJ, Farrugia GE, et al. Mapping the cellular and molecular landscape of cardiac non-myocytes in murine diabetic cardiomyopathy. iScience. 2023;26:107759.
30. Zygmunciak P, Stróżna K, Błażowska O, Mrozikiewicz-Rakowska B. Extracellular vesicles in diabetic cardiomyopathy-state of the art and future perspectives. Int J Mol Sci. 2024;25:6117.
31. Guo W, Yang C, Zou J, et al. Interleukin-1β polarization in M1 macrophage mediates myocardial fibrosis in diabetes. Int Immunopharmacol. 2024;131:111858.
32. Li C, Meng X, Wang L, Dai X. Mechanism of action of non-coding RNAs and traditional Chinese medicine in myocardial fibrosis: Focus on the TGF-β/Smad signaling pathway. Front Pharmacol. 2023;14:1092148.
33. Rossi E, Bernabeu C, Smadja DM. Endoglin as an adhesion molecule in mature and progenitor endothelial cells: a function beyond TGF-β. Front Med. 2019;6:10.
34. Meng L, Lu Y, Wang X, et al. NPRC deletion attenuates cardiac fibrosis in diabetic mice by activating PKA/PKG and inhibiting TGF-β1/Smad pathways. Sci Adv. 2023;9:eadd4222.
35. Biernacka A, Cavalera M, Wang J, et al. Smad3 signaling promotes fibrosis while preserving cardiac and aortic geometry in obese diabetic mice. Circ Heart Fail. 2015;8:788-98.
37. Kanisicak O, Khalil H, Ivey MJ, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260.
38. Li Y, He Q, He CY, Cai C, Chen Z, Duan JZ. Activating transcription factor 4 drives the progression of diabetic cardiac fibrosis. ESC Heart Fail. 2023;10:2510-23.
39. Wang L, Lin L, Qi H, Chen J, Grossfeld P. Endothelial loss of ETS1 impairs coronary vascular development and leads to ventricular non-compaction. Circ Res. 2022;131:371-87.
40. Peng ML, Fu Y, Wu CW, Zhang Y, Ren H, Zhou SS. Signaling pathways related to oxidative stress in diabetic cardiomyopathy. Front Endocrinol. 2022;13:907757.
41. Zheng ZQ, Cai DH, Song YF. Identification of immune feature genes and intercellular profiles in diabetic cardiomyopathy. World J Diabetes. 2024;15:2093-110.
42. Woll AW, Quelle FW, Sigmund CD. PPARγ and retinol binding protein 7 form a regulatory hub promoting antioxidant properties of the endothelium. Physiol Genomics. 2017;49:653-8.
43. Zhang X, Fan J, Li H, Chen C, Wang Y. CD36 signaling in diabetic cardiomyopathy. Aging Dis. 2021;12:826-40.
44. Chen J, Fu Y, Day DS, et al. VEGF amplifies transcription through ETS1 acetylation to enable angiogenesis. Nat Commun. 2017;8:383.
45. Yang X, Meyer K, Friedl A. STAT5 and prolactin participate in a positive autocrine feedback loop that promotes angiogenesis. J Biol Chem. 2013;288:21184-96.
46. Park CW, Kim HW, Lim JH, et al. Vascular endothelial growth factor inhibition by dRK6 causes endothelial apoptosis, fibrosis, and inflammation in the heart via the Akt/eNOS axis in db/db mice. Diabetes. 2009;58:2666-76.
47. Cerychova R, Pavlinkova G. HIF-1, metabolism, and diabetes in the embryonic and adult heart. Front Endocrinol. 2018;9:460.
48. Ullah K, Wu R. Hypoxia-inducible factor regulates endothelial metabolism in cardiovascular disease. Front Physiol. 2021;12:670653.
49. Sabolová G, Kočan L, Rabajdová M, Rapčanová S, Vašková J. Association of inflammation, oxidative stress, and deteriorated cognitive functions in patients after cardiac surgery. Vessel Plus. 2024;8:27.
50. Gou W, Yue L, Tang XY, et al. Circulating proteome and progression of type 2 diabetes. J Clin Endocrinol Metab. 2022;107:1616-25.
51. Ngo D, Benson MD, Long JZ, et al. Proteomic profiling reveals biomarkers and pathways in type 2 diabetes risk. JCI Insight. 2021;6:144392.
52. Li JP, Qiu S, Tai GJ, et al. NLRP3 inflammasome-modulated angiogenic function of EPC via PI3K/ Akt/mTOR pathway in diabetic myocardial infarction. Cardiovasc Diabetol. 2025;24:6.
53. Manea SA, Todirita A, Manea A. High glucose-induced increased expression of endothelin-1 in human endothelial cells is mediated by activated CCAAT/enhancer-binding proteins. PLoS One. 2013;8:e84170.
54. Hu X, Chen W, Yang M, Li M, Li X, Ouyang S. IGFBP5 promotes EndoMT and renal fibrosis through H3K18 lactylation in diabetic nephropathy. Cell Mol Life Sci. 2025;82:215.
55. Glatz JFC, Heather LC, Luiken JJFP. CD36 as a gatekeeper of myocardial lipid metabolism and therapeutic target for metabolic disease. Physiol Rev. 2024;104:727-64.
56. Bellini S, Barutta F, Mastrocola R, Imperatore L, Bruno G, Gruden G. Heat shock proteins in vascular diabetic complications: review and future perspective. Int J Mol Sci. 2017;18:2709.
57. Horton WB, Barrett EJ. Microvascular dysfunction in diabetes mellitus and cardiometabolic disease. Endocr Rev. 2021;42:29-55.
58. Litviňuková M, Talavera-López C, Maatz H, et al. Cells of the adult human heart. Nature. 2020;588:466-72.
59. Koenig AL, Shchukina I, Amrute J, et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat Cardiovasc Res. 2022;1:263-80.
60. McCracken IR, Dobie R, Bennett M, et al. Mapping the developing human cardiac endothelium at single-cell resolution identifies MECOM as a regulator of arteriovenous gene expression. Cardiovasc Res. 2022;118:2960-72.
61. Zhao G, Lu H, Liu Y, et al. Single-cell transcriptomics reveals endothelial plasticity during diabetic atherogenesis. Front Cell Dev Biol. 2021;9:689469.
62. Kalucka J, de Rooij LPMH, Goveia J, et al. Single-cell transcriptome atlas of murine endothelial cells. Cell. 2020;180:764-779.e20.
63. Skelly DA, Squiers GT, McLellan MA, et al. Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep. 2018;22:600-10.
64. Zhao Q, Eichten A, Parveen A, et al. Single-cell transcriptome analyses reveal endothelial cell heterogeneity in tumors and changes following antiangiogenic treatment. Cancer Res. 2018;78:2370-82.
65. Peisker F, Halder M, Nagai J, et al. Mapping the cardiac vascular niche in heart failure. Nat Commun. 2022;13:3027.
66. Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. 2016;8:a021873.
67. Alvandi Z, Bischoff J. Endothelial-mesenchymal transition in cardiovascular disease. Arterioscler Thromb Vasc Biol. 2021;41:2357-69.
68. Liu Y, Zhang J, Han Q, Li Y, Xue Y, Liu X. Identification of biomarkers associated with macrophage polarization in diabetic cardiomyopathy based on bioinformatics and machine learning approaches. Life Sci. 2025;364:123443.
69. Wan A, Rodrigues B. Endothelial cell-cardiomyocyte crosstalk in diabetic cardiomyopathy. Cardiovasc Res. 2016;111:172-83.
70. Guan H, Zhao S, Fang X, et al. Frontier technologies for investigating endothelial heterogeneity and function in diabetic vascular disease: an updated review. Biomed Pharmacother. 2025;191:118445.
71. Tate M, Grieve DJ, Ritchie RH. Are targeted therapies for diabetic cardiomyopathy on the horizon? Clin Sci. 2017;131:897-915.
72. Lovisa S, Kalluri R. Fatty acid oxidation regulates the activation of endothelial-to-mesenchymal transition. Trends Mol Med. 2018;24:432-4.
73. Botros M, Fadah K, Mukherjee D. The role of inflammatory response in the development of atherosclerosis, myocardial infarction, and remodeling. Vessel Plus. 2024;8:31.
74. Zhang Z, Fang Z, Ge J, Li H. Endothelial-to-mesenchymal transition in cardiovascular diseases. Trends Mol Med. ;2025:S1471-4914(25)00113.
75. Tombor LS, John D, Glaser SF, et al. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat Commun. 2021;12:681.
76. Martín-Bórnez M, Falcón D, Morrugares R, et al. New insights into the reparative angiogenesis after myocardial infarction. Int J Mol Sci. 2023;24:12298.
77. Dubé KN, Thomas TM, Munshaw S, Rohling M, Riley PR, Smart N. Recapitulation of developmental mechanisms to revascularize the ischemic heart. JCI Insight. 2017;2:96800.
78. Gogiraju R, Bochenek ML, Schäfer K. Angiogenic endothelial cell signaling in cardiac hypertrophy and heart failure. Front Cardiovasc Med. 2019;6:20.
79. Peng Z, Shu B, Zhang Y, Wang M. Endothelial response to pathophysiological stress. Arterioscler Thromb Vasc Biol. 2019;39:e233-43.
80. Islam S, Boström KI, Di Carlo D, et al. The mechanobiology of endothelial-to-mesenchymal transition in cardiovascular disease. Front Physiol. 2021;12:734215.
81. Csányi G, Taylor WR, Pagano PJ. NOX and inflammation in the vascular adventitia. Free Radic Biol Med. 2009;47:1254-66.
82. Lee SJ, Bae SS, Kim KH, et al. High glucose enhances MMP-2 production in adventitial fibroblasts via Akt1-dependent NF-κB pathway. FEBS Lett. 2007;581:4189-94.
83. Titus AS, Ushakumary MG, Venugopal H, Wang M, Lakatta EG, Kailasam S. Metformin attenuates hyperglycaemia-stimulated pro-fibrotic gene expression in adventitial fibroblasts via inhibition of discoidin domain receptor 2. Int J Mol Sci. 2022;24:585.
84. Tian J, Zhang M, Suo M, et al. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J Cell Mol Med. 2021;25:7642-59.
85. Yue Y, Meng K, Pu Y, Zhang X. Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res Clin Pract. 2017;133:124-30.
86. Xiao L, Dudley AC. Fine-tuning vascular fate during endothelial-mesenchymal transition. J Pathol. 2017;241:25-35.
87. Li H, Zhu X, Cao X, Lu Y, Zhou J, Zhang X. Single-cell analysis reveals lysyl oxidase (Lox)+ fibroblast subset involved in cardiac fibrosis of diabetic mice. J Adv Res. 2023;54:223-37.
88. Zhang S, Tian W, Duan X, et al. Melatonin attenuates diabetic cardiomyopathy by increasing autophagy of cardiomyocytes via regulation of VEGF-B/GRP78/PERK signaling pathway. Cardiovasc Diabetol. 2024;23:19.
89. Lal N, Chiu AP, Wang F, et al. Loss of VEGFB and its signaling in the diabetic heart is associated with increased cell death signaling. Am J Physiol Heart Circ Physiol. 2017;312:H1163-75.
90. Xia M, Jiao L, Wang XH, et al. Single-cell RNA sequencing reveals a unique pericyte type associated with capillary dysfunction. Theranostics. 2023;13:2515-30.
91. Lee C, Chen R, Sun G, et al. VEGF-B prevents excessive angiogenesis by inhibiting FGF2/FGFR1 pathway. Signal Transduct Target Ther. 2023;8:305.
92. Lai J, Chen F, Chen J, et al. Overexpression of decorin promoted angiogenesis in diabetic cardiomyopathy via IGF1R-AKT-VEGF signaling. Sci Rep. 2017;7:44473.
93. Phang RJ, Ritchie RH, Hausenloy DJ, Lees JG, Lim SY. Cellular interplay between cardiomyocytes and non-myocytes in diabetic cardiomyopathy. Cardiovasc Res. 2023;119:668-90.
94. Ding M, Shi R, Du Y, et al. O-GlcNAcylation-mediated endothelial metabolic memory contributes to cardiac damage via small extracellular vesicles. Cell Metab. 2025;37:1344-1363.e6.
95. Huang JP, Chang CC, Kuo CY, et al. Exosomal microRNAs miR-30d-5p and miR-126a-5p are associated with heart failure with preserved ejection fraction in STZ-induced type 1 diabetic rats. Int J Mol Sci. 2022;23:7514.
96. Chandrasekera DNK, Neale JPH, van Hout I, et al. Upregulation of microRNA-532 enhances cardiomyocyte apoptosis in the diabetic heart. Apoptosis. 2020;25:388-99.
97. Veitch S, Njock MS, Chandy M, et al. MiR-30 promotes fatty acid beta-oxidation and endothelial cell dysfunction and is a circulating biomarker of coronary microvascular dysfunction in pre-clinical models of diabetes. Cardiovasc Diabetol. 2022;21:31.
98. Wang X, Huang W, Liu G, et al. Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J Mol Cell Cardiol. 2014;74:139-50.
99. Lafuse WP, Wozniak DJ, Rajaram MVS. Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair. Cells. 2020;10:51.
100. Watanabe R, Hilhorst M, Zhang H, et al. Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI Insight. 2018;3:123047.
101. Shi K, Chen X, Zhao Y, et al. Identification of potential therapeutic targets for nonischemic cardiomyopathy in European ancestry: an integrated multiomics analysis. Cardiovasc Diabetol. 2024;23:338.
102. Malhi NK, Luo Y, tang X, et al. Abstract 2005: interrogating the cross-talk: endothelial-macrophage interactions in diabetic vascular dysfunction. Arterioscler Thromb Vasc Biol. 2024;44:A2005.
103. Shi X, Liu C, Chen J, et al. Endothelial MICU1 alleviates diabetic cardiomyopathy by attenuating nitrative stress-mediated cardiac microvascular injury. Cardiovasc Diabetol. 2023;22:216.
104. Su H, Cantrell AC, Zeng H, Zhu SH, Chen JX. Emerging role of pericytes and their secretome in the heart. Cells. 2021;10:548.
105. Liu Z. Cardiac microvascular dysfunction and cardiomyopathy in diabetes: is ferroptosis a therapeutic target? Diabetes. 2023;72:313-5.
106. Simmonds SJ, Grootaert MOJ, Cuijpers I, et al. Pericyte loss initiates microvascular dysfunction in the development of diastolic dysfunction. Eur Heart J Open. 2024;4:oead129.
107. Grootaert MOJ, Pasut A, Raman J, et al. Mural cell dysfunction contributes to diastolic heart failure by promoting endothelial dysfunction and vessel remodelling. Cardiovasc Diabetol. 2025;24:62.
108. Caporali A, Martello A, Miscianinov V, Maselli D, Vono R, Spinetti G. Contribution of pericyte paracrine regulation of the endothelium to angiogenesis. Pharmacol Ther. 2017;171:56-64.
109. Zhang Q, Fang W, Ma L, Wang ZD, Yang YM, Lu YQ. VEGF levels in plasma in relation to metabolic control, inflammation, and microvascular complications in type-2 diabetes: a cohort study. Medicine. 2018;97:e0415.
110. Zhang T, Jiang D, Zhang X, et al. The role of nonmyocardial cells in the development of diabetic cardiomyopathy and the protective effects of FGF21: a current understanding. Cell Commun Signal. 2024;22:446.
112. Sheu ML, Ho FM, Yang RS, et al. High glucose induces human endothelial cell apoptosis through a phosphoinositide 3-kinase-regulated cyclooxygenase-2 pathway. Arterioscler Thromb Vasc Biol. 2005;25:539-45.
113. Paone S, Baxter AA, Hulett MD, Poon IKH. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell Mol Life Sci. 2019;76:1093-106.
114. Seferović PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J. 2015;36:1718-27.
115. Knapp M, Tu X, Wu R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharmacol Sin. 2019;40:1-8.
116. Camici PG, Tschöpe C, Di Carli MF, Rimoldi O, Van Linthout S. Coronary microvascular dysfunction in hypertrophy and heart failure. Cardiovasc Res. 2020;116:806-16.
117. Man S, Sanchez Duffhues G, Ten Dijke P, Baker D. The therapeutic potential of targeting the endothelial-to-mesenchymal transition. Angiogenesis. 2019;22:3-13.
118. Wang E, Wang H, Chakrabarti S. Endothelial-to-mesenchymal transition: an underappreciated mediator of diabetic complications. Front Endocrinol. 2023;14:1050540.
119. Cho JG, Lee A, Chang W, Lee MS, Kim J. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front Immunol. 2018;9:294.
120. Li C, Cai D, Yuan W, et al. The canonical Wnt/β-catenin signaling pathway upregulates carbonic anhydrase 2 via transcription factor 7-like 2 to promote cardiomyopathy in type 2 diabetic mice. Life Sci. 2025;368:123506.
121. Geng H, Guan J. MiR-18a-5p inhibits endothelial-mesenchymal transition and cardiac fibrosis through the Notch2 pathway. Biochem Biophys Res Commun. 2017;491:329-36.
122. Sánchez-Duffhues G, García de Vinuesa A, Ten Dijke P. Endothelial-to-mesenchymal transition in cardiovascular diseases: developmental signaling pathways gone awry. Dev Dyn. 2018;247:492-508.
123. Gamrat A, Surdacki MA, Chyrchel B, Surdacki A. Endothelial dysfunction: a contributor to adverse cardiovascular remodeling and heart failure development in type 2 diabetes beyond accelerated atherogenesis. J Clin Med. 2020;9:2090.
124. Kolluru GK, Bir SC, Kevil CG. Endothelial dysfunction and diabetes: effects on angiogenesis, vascular remodeling, and wound healing. Int J Vasc Med. 2012;2012:918267.
125. Chen D, Sindone A, Huang MLH, Peter K, Jenkins AJ. Diabetic cardiomyopathy: insights into pathophysiology, diagnosis and clinical management. J Mol Cell Cardiol. 2025;206:55-69.
126. Schmidt K, Schmidt A, Groß S, et al. SGLT2 inhibitors attenuate endothelial to mesenchymal transition and cardiac fibroblast activation. Sci Rep. 2024;14:16459.
127. Wang D, Luo P, Wang Y, et al. Glucagon-like peptide-1 protects against cardiac microvascular injury in diabetes via a cAMP/PKA/Rho-dependent mechanism. Diabetes. 2013;62:1697-708.
128. Li N, Zhou H. SGLT2 inhibitors: a novel player in the treatment and prevention of diabetic cardiomyopathy. Drug Des Devel Ther. 2020;14:4775-88.
129. Wu M, Peng Z, Zu C, et al. Losartan attenuates myocardial endothelial-to-mesenchymal transition in spontaneous hypertensive rats via inhibiting TGF-β/Smad signaling. PLoS One. 2016;11:e0155730.
130. Kanasaki K, Shi S, Kanasaki M, et al. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes. 2014;63:2120-31.
131. Sabe SA, Harris DD, Broadwin M, et al. Comparative effects of canagliflozin and sitagliptin in chronically ischemic myocardium. Vessel Plus. 2024;8:2.
132. Wu M, Tang RN, Liu H, Xu M, Pan MM, Liu BC. Cinacalcet attenuates the renal endothelial-to-mesenchymal transition in rats with adenine-induced renal failure. Am J Physiol Renal Physiol. 2014;306:F138-46.
133. Guo Y, Li P, Bledsoe G, Yang ZR, Chao L, Chao J. Kallistatin inhibits TGF-β-induced endothelial-mesenchymal transition by differential regulation of microRNA-21 and eNOS expression. Exp Cell Res. 2015;337:103-10.
134. Cipriani P, Di Benedetto P, Ruscitti P, et al. The endothelial-mesenchymal transition in systemic sclerosis is induced by endothelin-1 and transforming growth factor-β and may be blocked by macitentan, a dual endothelin-1 receptor antagonist. J Rheumatol. 2015;42:1808-16.
135. Hall IF, Kishta F, Xu Y, Baker AH, Kovacic JC. Endothelial to mesenchymal transition: at the axis of cardiovascular health and disease. Cardiovasc Res. 2024;120:223-36.
136. Teng X, Ji C, Zhong H, et al. Selective deletion of endothelial cell calpain in mice reduces diabetic cardiomyopathy by improving angiogenesis. Diabetologia. 2019;62:860-72.
137. Widyantoro B, Emoto N, Nakayama K, et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 2010;121:2407-18.
138. Lu S, Liao Z, Lu X, et al. Hyperglycemia acutely increases cytosolic reactive oxygen species via O-linked GlcNAcylation and CaMKII activation in mouse ventricular myocytes. Circ Res. 2020;126:e80-96.
139. Prakoso D, Lim SY, Erickson JR, et al. Fine-tuning the cardiac O-GlcNAcylation regulatory enzymes governs the functional and structural phenotype of the diabetic heart. Cardiovasc Res. 2022;118:212-25.
140. Yao Y, Song Q, Hu C, et al. Endothelial cell metabolic memory causes cardiovascular dysfunction in diabetes. Cardiovasc Res. 2022;118:196-211.
141. Liu Y, Wang M, Su JB, et al. Potential clinical value of fibrinogen-like protein 1 as a serum biomarker for the identification of diabetic cardiomyopathy. Sci Rep. 2024;14:10311.
142. Johnson R, Nxele X, Cour M, et al. Identification of potential biomarkers for predicting the early onset of diabetic cardiomyopathy in a mouse model. Sci Rep. 2020;10:12352.
143. Li J, Qiu H, Wu Y, Su L. Identification of metabolic pathways and serum biomarkers in diabetic cardiomyopathy using untargeted metabolomics. Sci Rep. 2025;15:18718.
144. Zampetaki A, Kiechl S, Drozdov I, et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res. 2010;107:810-7.
145. Prattichizzo F, De Nigris V, Sabbatinelli J, et al. CD31+ extracellular vesicles from patients with type 2 diabetes shuttle a miRNA signature associated with cardiovascular complications. Diabetes. 2021;70:240-54.
146. Prattichizzo F, Giuliani A, De Nigris V, et al. Extracellular microRNAs and endothelial hyperglycaemic memory: a therapeutic opportunity? Diabetes Obes Metab. 2016;18:855-67.
147. Rawal S, Munasinghe PE, Shindikar A, et al. Down-regulation of proangiogenic microRNA-126 and microRNA-132 are early modulators of diabetic cardiac microangiopathy. Cardiovasc Res. 2017;113:90-101.
148. Kura B, Kindernay L, Singla D, Dulova U, Bartekova M. Mechanistic insight into the role of cardiac-enriched microRNAs in diabetic heart injury. Am J Physiol Heart Circ Physiol. 2025;328:H865-84.
149. Copier CU, León L, Fernández M, Contador D, Calligaris SD. Circulating miR-19b and miR-181b are potential biomarkers for diabetic cardiomyopathy. Sci Rep. 2017;7:13514.
150. Zhu H, Leung SW. MicroRNA biomarkers of type 2 diabetes: evidence synthesis from meta-analyses and pathway modelling. Diabetologia. 2023;66:288-99.
151. Bielska A, Niemira M, Kretowski A. Recent highlights of research on miRNAs as early potential biomarkers for cardiovascular complications of type 2 diabetes mellitus. Int J Mol Sci. 2021;22:3153.
152. de Gonzalo-Calvo D, van der Meer RW, Rijzewijk LJ, et al. Serum microRNA-1 and microRNA-133a levels reflect myocardial steatosis in uncomplicated type 2 diabetes. Sci Rep. 2017;7:47.
153. Kuschnerus K, Straessler ET, Müller MF, Lüscher TF, Landmesser U, Kränkel N. Increased expression of miR-483-3p impairs the vascular response to injury in type 2 diabetes. Diabetes. 2019;68:349-60.
154. Jin ZQ. MicroRNA targets and biomarker validation for diabetes-associated cardiac fibrosis. Pharmacol Res. 2021;174:105941.
155. Liu S, Li W, Xu M, Huang H, Wang J, Chen X. Micro-RNA 21Targets dual specific phosphatase 8 to promote collagen synthesis in high glucose-treated primary cardiac fibroblasts. Can J Cardiol. 2014;30:1689-99.
156. Maiocchi S, Collins EN, Peterson AR, et al. Plasma microrna quantification protocol. Vessel Plus. 2023;7:27.
157. Cabrera-Becerra SE, Vera-Juárez G, García-Rubio VG, et al. siRNA knockdown of angiopoietin 2 significantly reduces neovascularization in diabetic rats. J Drug Target. 2022;30:673-86.
158. Waghode P, Quadir SS, Choudhary D, Sharma S, Joshi G. Small interfering RNA (siRNA) as a potential gene silencing strategy for diabetes and associated complications: challenges and future perspectives. J Diabetes Metab Disord. 2024;23:365-83.
159. Zhao Q, Shao T, Huang S, et al. The insulin-like growth factor binding protein-microfibrillar associated protein-sterol regulatory element binding protein axis regulates fibroblast-myofibroblast transition and cardiac fibrosis. Br J Pharmacol. 2024;181:2492-508.
160. Song SE, Kim YW, Kim JY, Lee DH, Kim JR, Park SY. IGFBP5 mediates high glucose-induced cardiac fibroblast activation. J Mol Endocrinol. 2013;50:291-303.
161. Cao L, Chang R, Wang X, et al. Integrative Single-cell and spatial transcriptomics reveal functional and spatial heterogeneity of atrial and ventricular cardiomyocytes in the heart. Mol Biotechnol. 2025.
162. Han S, Xu Q, Du Y, et al. Single-cell spatial transcriptomics in cardiovascular development, disease, and medicine. Genes Dis. 2024;11:101163.
163. Wang X, Sawuer G, Liang C, Lu L, Wu G. ETS1-THBS1 axis regulates macrophage polarization and exacerbates myocardial injury in diabetic cardiomyopathy. J Cardiovasc Pharmacol. 2025.
164. Sun J, Zhou R, Liu M, Zhang D. The role of myocardial fibrosis in the diabetic cardiomyopathy. Diabetol Metab Syndr. 2025;17:242.
165. Tuleta I, Frangogiannis NG. Fibrosis of the diabetic heart: clinical significance, molecular mechanisms, and therapeutic opportunities. Adv Drug Deliv Rev. 2021;176:113904.
166. Vistnes M. Hitting the Target! Challenges and opportunities for TGF-β inhibition for the treatment of cardiac fibrosis. Pharmaceuticals. 2024;17:267.
167. Lezoualc’h F, Badimon L, Baker H, et al. Diabetic cardiomyopathy: the need for adjusting experimental models to meet clinical reality. Cardiovasc Res. 2023;119:1130-45.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].