


Metabolic reprogramming and the tumor microenvironment in hepatocellular carcinoma: mechanisms and therapeutic targeting
 0
0 Abstract
Metabolic reprogramming is a fundamental mechanism through which tumor cells reshape their energy metabolism to sustain rapid proliferation. It facilitates malignant growth by reprogramming key pathways, including glycolysis and amino acid metabolism. The tumor microenvironment (TME) is composed of tumor cells, stromal cells, and immune cells. The characteristics of hypoxia, acidity, and nutrient deficiency are mainly driven by the metabolic products and cytokines secreted by tumor cells. This metabolic pressure not only inhibits the functions of immune cells, but also further enhances immune evasion through nutrient competition. Targeting metabolic reprogramming can reverse immunosuppression within the TME and enhance the response to immunotherapy. This article systematically reviews the regulatory mechanisms of metabolic reprogramming in hepatocellular carcinoma and its impact on the TME, while also exploring therapeutic strategies based on metabolic interventions.
Keywords
INTRODUCTION
Liver cancer ranks as the sixth most common malignancy and the third leading cause of cancer-related mortality globally. The number of new liver cancers will nearly double, from 0.87 million in 2022 to 1.52 million in 2050, if current trends continue. Hepatocellular carcinoma (HCC), the predominant histological subtype, accounts for nearly 80% of all primary liver cancers[1-5]. The pathogenesis of HCC is complex, involving genetic, environmental, and lifestyle factors, but it is usually based on chronic liver disease. Key etiological risk factors include chronic infection with hepatitis B virus (HBV) or hepatitis C virus (HCV), excessive alcohol consumption, non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH), and exposure to dietary aflatoxins[6,7]. Due to its insidious onset and lack of specific symptoms in early stages, a substantial proportion of patients are diagnosed at intermediate or advanced stages, thereby missing the window for potentially curative interventions. This late diagnosis contributes to the notoriously poor overall prognosis associated with HCC[8].
The management of HCC is predominantly guided by the Barcelona Clinic Liver Cancer (BCLC) staging system and involves a multidisciplinary team (MDT) approach. Current therapeutic strategies for HCC include surgical resection, liver transplantation, local ablation, transarterial chemoembolization (TACE), and so on. However, the long-term therapeutic outcomes remain unsatisfactory. The advent of systemic therapies, notably molecular targeted agents (e.g., sorafenib, lenvatinib) and immune checkpoint inhibitors (ICIs) [e.g., anti-programmed cell death protein 1/programmed death-ligand 1 (anti-PD-1/PD-L1) antibodies], has revolutionized the treatment paradigm for advanced HCC[9,10]. Nevertheless, major challenges persist, including limited therapeutic efficacy, primary and acquired drug resistance, the absence of reliable predictive biomarkers, and difficulties in managing adverse effects[11,12].
Driven by these challenges, researchers continue to explore new intervention targets. In recent years, metabolic reprogramming has gained significant attention as a pivotal emerging hallmark of cancer in HCC research[13,14]. It refers to the process by which cancer cells reshape their metabolic networks through alterations in gene expression and signaling pathways to meet the bioenergetic and biosynthetic demands of rapid proliferation[15]. This phenomenon is particularly prominent in HCC, characterized by enhanced aerobic glycolysis, aberrant lipid synthesis, and dependence on glutamine metabolism. These adaptations not only promote tumor progression but also drive resistance to therapeutics[16]. Given the central role of metabolic reprogramming in HCC, targeting related metabolic pathways has emerged as a promising strategy to overcome current treatment limitations[17]. The therapeutic implications include: first, providing new targets for suppressing tumor growth; second, potentially reversing resistance to existing therapies; and third, enhancing the efficacy of immunotherapy by modulating the tumor microenvironment (TME)[18-21].
In addition to metabolic reprogramming, the TME also plays a critical role in the pathogenesis, progression, and treatment response of HCC. The TME in HCC is highly immunosuppressive, characterized by dysfunctional T cells, abundant infiltrating immunosuppressive cells, and various immunosuppressive factors[22,23]. This microenvironment significantly contributes to the limited efficacy of ICI monotherapy. Notably, a close bidirectional crosstalk exists between the metabolic reprogramming of tumor cells and the shaping of the immune microenvironment[15]. By competitively consuming nutrients and releasing metabolic waste, tumor cells create a nutrient-depleted and acidified microenvironment that directly suppresses the function of effector immune cells while promoting the activation and proliferation of immunosuppressive cells[16,17,22,24,25]. Therefore, a deeper understanding of the complex network of metabolic-immune interactions in HCC will not only help elucidate the mechanisms of tumor development but also provide new insights for combining metabolic interventions with immunotherapy to overcome current therapeutic limitations[26-28].
This review focuses on the interplay between metabolic reprogramming and the tumor immune microenvironment in HCC, and discusses potential strategies and recent advances in targeting metabolism to enhance the efficacy of immunotherapy.
METABOLIC REPROGRAMMING AND ITS REGULATORY MECHANISMS IN HCC CELLS
Metabolic reprogramming in HCC encompasses multiple hallmark alterations, including the Warburg effect, glutamine metabolism, dysregulated fatty acid synthesis and oxidation, aberrant amino acid metabolism, and mitochondrial dysfunction[29-31]. This section focuses on three representative pathways, examining them from two distinct angles: their core metabolic functions and the non-metabolic (“moonlighting”) roles of the key enzymes involved.
The Warburg effect
The Warburg effect refers to a metabolic adaptation in which tumor and other proliferating or developing cells dramatically increase glucose uptake and produce lactate even in the presence of oxygen and fully functional mitochondria[32]. In HCC, cancer cells preferentially utilize aerobic glycolysis and exhibit this classical Warburg effect. Through such metabolic reprogramming, tumor cells supply energy to promote their own survival, growth, and metastasis[22].
The Warburg effect plays a central role in the metabolic reprogramming of HCC cells, contributing to tumor progression through multiple interconnected mechanisms. Specifically, HCC cells upregulate key glycolytic enzymes including hexokinase 2 (HK2), pyruvate kinase M2 (PKM2), lactate dehydrogenase A (LDHA), and other metabolic enzymes. This leads to a markedly accelerated glycolytic flux, accompanied by enhanced glucose uptake and lactate secretion[25]. Concurrently, mitochondrial oxidative metabolism is functionally attenuated, favoring the diversion of pyruvate away from the tricarboxylic acid (TCA) cycle and reinforcing glycolytic dependence[33,34]. Beyond providing rapid energy, glycolytic intermediates also support tumor growth through various mechanisms. For instance, the intermediate glucose-6-phosphate (G6P) can enter the pentose phosphate pathway (PPP) upon activation of glucose-6-phosphate dehydrogenase (G6PD, the rate-limiting enzyme of PPP), generating nicotinamide adenine dinucleotide phosphate (NADPH) to maintain redox homeostasis[35,36]. Moreover, another intermediate, 3-phosphoglycerate (3-PG), can be channeled into anabolic processes such as serine metabolism and one-carbon metabolism, thereby supporting nucleotide biosynthesis and cellular proliferation[37].
In addition to the mechanisms described above, recent studies have further revealed the important impact of glycolysis on protein post-translational modifications. A particularly direct pathway is lactylation - a novel type of histone and non-histone modification mediated by lactate, the end product of glycolysis. Lactylation has emerged as a significant discovery in cancer research[38-40]. In HCC, lactate is no longer merely a metabolic byproduct; rather, it acts as a key signaling molecule that directly regulates tumor stemness, immune evasion, and microenvironment remodeling through lactylation. For example, tumor-derived lactate can promote the polarization of tumor-associated macrophages toward the pro-tumor M2 phenotype by upregulating nuclear protein 1 (NUPR1) expression via lactylation, thereby suppressing anti-tumor immunity. Meanwhile, a high-lactate environment impairs cluster of differentiation 8 positive (CD8+) T-cell function and induces lactylation at the K72 site of moesin protein (MOESIN), which enhances its binding to the transforming growth factor beta (TGF-β) receptor and facilitates regulatory T-cell differentiation, further undermining immune surveillance. Additionally, histone lactylation can activate the transcription of stemness-related genes, maintaining the self-renewal capacity and chemotherapy resistance of liver cancer stem cells[41,42]. Together, these findings underscore the important role of glycolysis in regulating post-translational modifications.
Moreover, this intrinsic metabolic reprogramming is synergistically enhanced by extrinsic factors. Notably, hypoxic conditions within the TME further amplify glycolytic flux through hypoxia-inducible factor 1α (HIF-1α)–mediated transcriptional activation of glycolysis-related genes, thereby establishing a metabolic landscape that favors HCC survival and progression[35,36,43,44] [Figure 1].
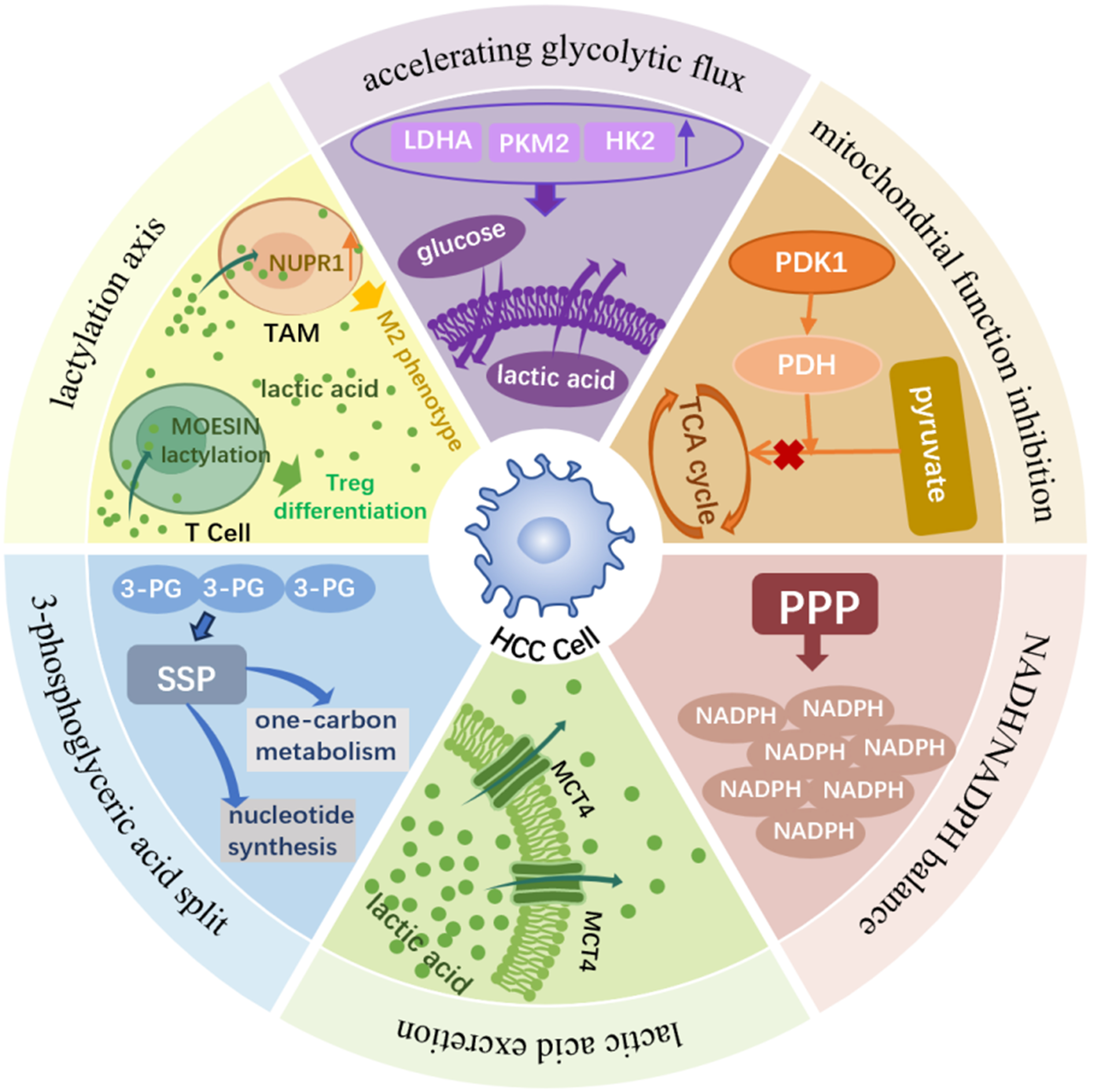
Figure 1. The Warburg effect in HCC: mechanisms and functional consequences. HCC cells preferentially rely on aerobic glycolysis - the Warburg effect - even in the presence of oxygen and functional mitochondria. Upregulation of key glycolytic enzymes (HK2, PKM2, LDHA) enhances glucose uptake, glycolytic flux, and lactate production/export. PDH inhibition diverts pyruvate away from the TCA cycle, reinforcing glycolytic dependency. Glycolytic intermediates are channeled into anabolic pathways: G6P fuels the PPP to generate NADPH for redox balance, while 3-PG supports serine and one-carbon metabolism for nucleotide biosynthesis and proliferation. Excess lactate is exported via MCTs, accumulates in the TME, and acts as a signaling molecule that induces lysine lactylation. Lactylation promotes HCC progression by activating stemness-related transcriptional programs and reshaping the immune microenvironment - driving M2 polarization of TAMs via NUPR1 upregulation and facilitating regulatory T-cell differentiation through MOESIN K72 lactylation. Hypoxia further amplifies these processes via HIF-1α–mediated transcriptional activation of glycolytic genes, collectively establishing a metabolic–epigenetic landscape that supports HCC survival, immune evasion, and progression. HCC: Hepatocellular carcinoma; HK2: hexokinase 2; PKM2: pyruvate kinase M2; LDHA: lactate dehydrogenase A; PDH: pyruvate dehydrogenase; TCA: tricarboxylic acid; G6P: glucose-6-phosphate; PPP: pentose phosphate pathway; NADPH: nicotinamide adenine dinucleotide phosphate; 3-PG: 3-phosphoglycerate; MCTs: monocarboxylate transporters; TME: tumor microenvironment; TAMs: tumor-associated macrophages; NUPR1: nuclear protein 1; HIF-1α: hypoxia-inducible factor 1α; PDK1: pyruvate dehydrogenase kinase 1; NADH: nicotinamide adenine dinucleotide; SSP: serine synthesis pathway; Treg: regulatory T cell.
TCA cycle
The TCA cycle, also known as the citric acid cycle or Krebs cycle, serves as a central hub of cellular energy metabolism. Located in the mitochondrial matrix, it represents the terminal pathway of oxidative catabolism for carbohydrates, lipids, and amino acids. Beyond generating reducing equivalents for adenosine triphosphate (ATP) production, the TCA cycle supplies critical metabolic intermediates - such as citrate, α-ketoglutarate (α-KG), and oxaloacetate - that serve as precursors for the biosynthesis of lipids, nucleotides, and amino acids. Thus, the cycle functions not only as an “energy generator” but also as a fundamental “metabolic nexus” integrating anabolic and catabolic processes[45-47].
In cancer cells, however, the TCA cycle undergoes profound metabolic reprogramming. It transitions from being primarily an energy-yielding pathway into a dynamic biosynthetic factory, redirecting carbon flux to support the massive demand for macromolecular synthesis - such as fatty acids, non-essential amino acids, and nucleotides - required for rapid proliferation. HCC, widely regarded as a metabolic disorder, exhibits particularly prominent dependencies and adaptations in its TCA cycle, providing a strong rationale for novel therapeutic targeting[46,48].
The TCA cycle functions not only as a central hub of cellular energy metabolism but also as a key regulator of epigenetic dynamics. Recent studies have demonstrated that select TCA cycle metabolites and enzymes can translocate into the nucleus, where they directly modulate chromatin modifications and gene expression - a phenomenon termed metabolic–epigenetic coupling[49,50] [Figure 2].
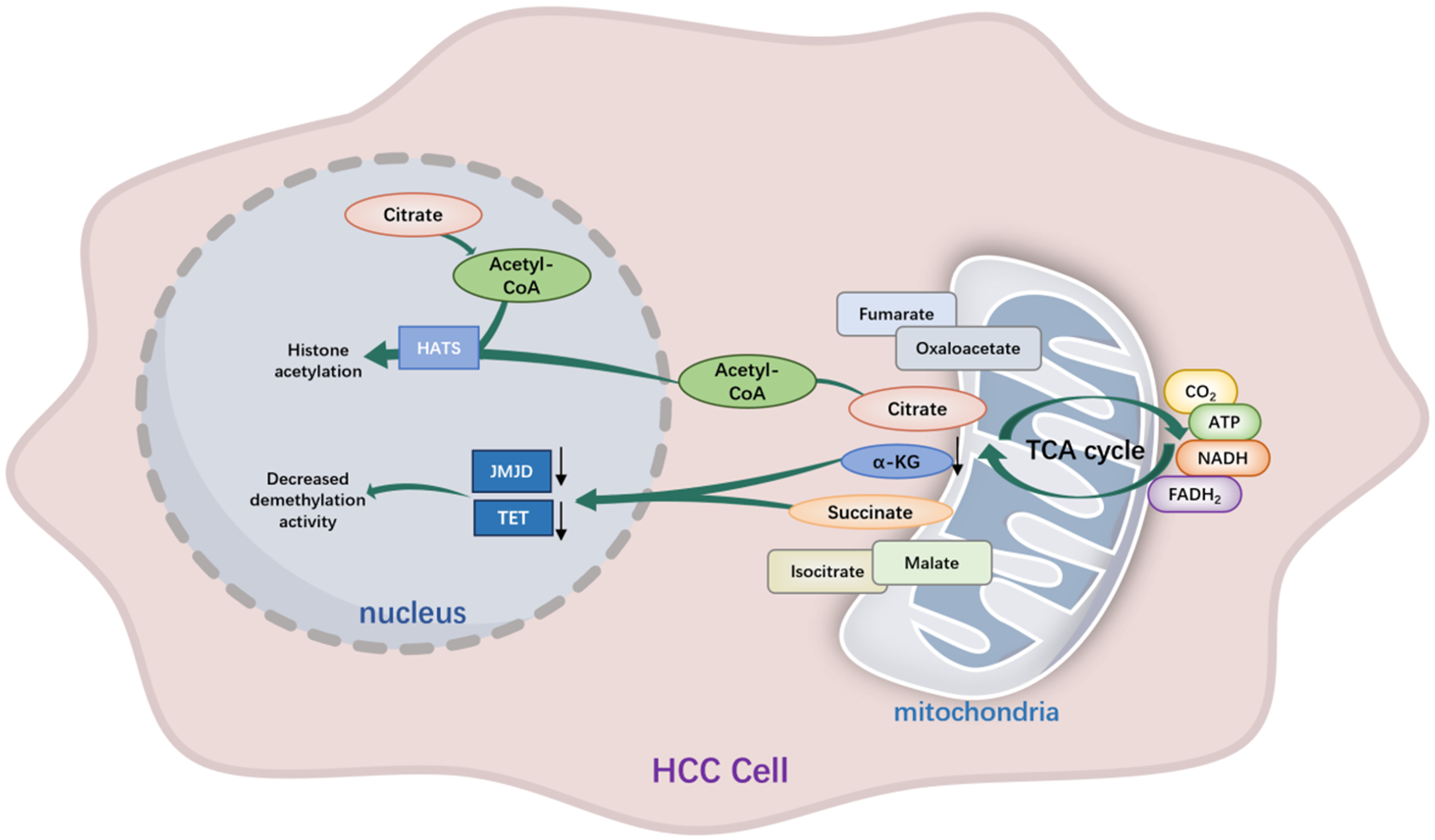
Figure 2. Metabolic–epigenetic coupling of the TCA cycle in HCC. Reprogrammed TCA cycle flux in HCC generates metabolic signals that directly regulate nuclear chromatin states. Succinate accumulation inhibits α-KG–dependent TET and JMJD demethylases, promoting DNA and histone hypermethylation, whereas reduced α-KG availability further constrains demethylation and limits enhancer plasticity. In parallel, mitochondrial-derived citrate is exported and converted to acetyl-CoA to fuel histone acetyltransferase activity, reinforcing oncogenic transcriptional programs. TCA: Tricarboxylic acid; HCC: hepatocellular carcinoma; α-KG: α-ketoglutarate; TET: ten-eleven translocation; JMJD: Jumonji C domain-containing; HATs: histone acetyltransferases; ATP: adenosine triphosphate; NADH: nicotinamide adenine dinucleotide; FADH2: flavin adenine dinucleotide.
Flux through the TCA cycle thus dynamically shapes the epigenetic landscape. For instance, accumulated succinate acts as a competitive inhibitor of α-KG–dependent dioxygenases, including the ten-eleven translocation (TET) family of DNA demethylases and Jumonji C domain-containing (JMJD) family histone demethylases, thereby promoting global DNA hypermethylation and transcriptional silencing[51]. In HCC, disruption of succinate dehydrogenase (SDH) function exacerbates this metabolite-driven epigenetic constraint by coupling succinate accumulation to sustained chromatin repression across cell division cycles, thereby impairing the replication-coupled resetting of regulatory chromatin domains. Rather than inducing transient alterations in methylation states, this metabolic imbalance stabilizes silenced regulatory regions that govern hepatocyte lineage identity, inflammatory responsiveness, and antigen-presentation pathways through mitotically heritable repression of enhancer accessibility. As a result, HCC cells adopt a transcriptionally inflexible, poorly differentiated state that favors anabolic metabolism, unchecked proliferation, and blunted immune recognition - effectively translating metabolic perturbations into durable oncogenic and immune-evasive phenotypes[52-54].
Conversely, α-KG serves as an essential cofactor for TET and JMJD enzymes, supporting active DNA and histone demethylation and facilitating transcriptional activation. In HCC, metabolic reprogramming that restricts intracellular α-KG availability compromises the dynamic responsiveness of regulatory chromatin, particularly at enhancer regions governing hepatocyte differentiation and tumor-suppressive signaling. This constraint preferentially dampens stimulus-induced chromatin remodeling rather than basal transcription, thereby locking in oncogenic transcriptional hierarchies and limiting lineage reprogramming. Consequently, reduced α-KG availability reinforces a poorly differentiated, metabolically adaptive state that supports sustained tumor growth and therapeutic resistance in HCC[55,56].
Furthermore, mitochondrial-derived citrate is exported to the cytosol and converted to acetyl coenzyme A (acetyl-CoA), which serves as the essential substrate for histone acetyltransferases, thereby promoting chromatin relaxation and gene activation[57]. In HCC, enhanced mitochondrial citrate export and increased cytosolic–nuclear carbon flux bias transcriptional control toward growth-promoting programs by preferentially reinforcing highly active regulatory regions. This spatially constrained epigenetic reinforcement selectively stabilizes oncogenic promoters and super-enhancers, lowering their activation threshold and rendering their transcription less dependent on extrinsic growth or immune signals. Consequently, citrate-driven metabolic signaling concentrates transcriptional capacity on lipid biosynthesis, cell-cycle progression, and survival pathways, establishing a transcriptionally polarized state that sustains rapid tumor expansion and adaptive resistance in HCC[58-60].
Glutamine metabolism
In tumor metabolic reprogramming, TCA cycle relies not only on glucose-derived carbons but also on various nutrient inputs, among which glutamine metabolism plays an especially critical role. Beyond serving as a substrate for protein synthesis, glutamine acts as a key precursor for energy production, nucleotide biosynthesis, and the synthesis of the antioxidant glutathione[61]. Highly proliferative cancer cells display “glutamine addiction”, consuming large amounts of glutamine through glutaminolysis: glutaminase (GLS) converts glutamine to glutamate, which is further metabolized to α-KG for entry into the TCA cycle, thereby supplying both energy and biosynthetic intermediates. This metabolic rewiring, often referred to as “glutamine addiction”, is a hallmark of cancer cell metabolism.
In parallel, glutamine-derived carbon and nitrogen atoms are utilized for purine and pyrimidine biosynthesis, meeting the elevated nucleic acid demands of rapidly dividing tumor cells[62]. Glutamine metabolism also contributes to cellular redox balance through NADPH generation mediated by malic enzyme 1, thereby limiting reactive oxygen species accumulation and supporting tumor cell survival[63].
Under glutamine-deprived conditions, tumor cells do not remain passive; instead, they activate multiple adaptive mechanisms to ensure survival. During glutamine starvation, the expression of DNA damage-inducible transcript 3 [DDIT3; also known as C/EBP homologous protein (CHOP)] is upregulated. DDIT3 promotes glycolysis by inhibiting TP53-induced glycolysis and apoptosis regulator (TIGAR) to generate ATP, while simultaneously facilitating the degradation of mitochondrial electron transport chain proteins [such as coenzyme Q9 homolog (COQ9) and cytochrome c oxidase subunit 4 (COX4)] via lon peptidase 1, mitochondrial (LONP1), thereby suppressing oxidative phosphorylation and reducing reactive oxygen species (ROS) production. This dual regulation coordinates glycolysis and the TCA cycle, supporting the survival of HCC cells under metabolic stress. Additionally, the long non-coding RNA glutamine insufficiency regulator of glutaminase lncRNA (GIRGL) is induced under glutamine deficiency. GIRGL promotes cell cycle-associated protein 1 (CAPRIN1)-mediated liquid-liquid phase separation, leading to the formation of stress granules that suppress the translation of glutaminase 1 (GLS1) messenger RNA (mRNA). This mechanism helps HCC cells survive prolonged nutrient stress. Furthermore, studies have shown that interferon-related developmental regulator 1 (IFRD1) is induced under glutamine starvation. In the absence of glutamine, loss of IFRD1 enhances nucleophagy, promotes the degradation of histone H1.0, increases chromatin accessibility, and upregulates the expression of genes related to ribosome biogenesis. The resulting hyperactive protein synthesis, faced with limited external glutamine sources, ultimately promotes autophagy-mediated death of HCC cells[64,65].
In summary, through the coordinated actions of DDIT3, GIRGL, and IFRD1, liver cancer cells establish a robust survival network to resist glutamine-deficient environments. These adaptive mechanisms highlight potential therapeutic targets for intervention, suggesting that strategies combining DDIT3, GIRGL, or IFRD1 inhibition could overcome the limitations of current glutamine-targeting therapies.
Fatty acid metabolism
Fat synthesis drives cancer progression through multiple mechanisms[66-68]. For instance, fatty acids can contribute to multiple facets of tumorigenesis by serving as building blocks for membrane phospholipid synthesis, providing energy substrates, and acting as precursors for tumor-promoting lipid signaling molecules[67,69]. Meanwhile, fatty acids also play a significant role in altering membrane dynamics and drug resistance.
In terms of metabolic functions, fatty acid metabolism in HCC can be broadly divided into two major aspects: the cancer-promoting role of fatty acid synthesis (lipogenesis) and the metabolic adaptation mediated by fatty acid oxidation (FAO). Together, these dual processes highlight the metabolic flexibility of HCC cells in sustaining growth, survival, and metastasis[66-68,70,71].
From this, we can summarize that the FAO has a dual effect.
FAO plays a context-dependent dual role within the TME, serving as a critical adaptive survival mechanism for cancer cells under metabolic stress, while often being associated with functional exhaustion in antitumor effector T cells. This fundamental dichotomy underscores the metabolic competition and distinct cellular responses within the TME[72,73].
For cancer cells, particularly in glucose-deprived regions of the tumor core, FAO via mitochondrial β-oxidation provides an essential alternative energy source, supporting their survival and proliferation. Key molecules such as carnitine palmitoyltransferase 1A (CPT1A), which facilitates fatty acid transport into mitochondria, and transcriptional regulators such as peroxisome proliferator-activated receptors (PPARs), are centrally involved in maintaining this metabolic adaptation, enabling cancer cells to withstand hypoxia and nutrient scarcity[74-76]. Notably, even perturbations in FAO, such as those caused by deficiencies in enzymes such as long-chain acyl-CoA dehydrogenase (ACADL), can lead to the accumulation of intermediate metabolites (e.g., octanoic acid) that may paradoxically enhance the metastatic potential of cancer cells[74-76].
In stark contrast, a reliance on FAO in effector T cells infiltrating the tumor is frequently linked to functional suppression[61,77]. Under conditions such as obesity or persistent antigen exposure, signaling through pathways (e.g., PD-1) or via adipokines (e.g., leptin) can activate transcriptional regulators [e.g., signal transducer and activator of transcription 3 (STAT3)][73,78]. This drives a metabolic reprogramming in T cells, shifting their primary energy metabolism from efficient glycolysis towards FAO. This metabolic shift is concomitant with a diminished capacity to produce effector cytokines such as interferon-γ (IFN-γ) and progression towards a functionally exhausted state. Furthermore, increased lipid accumulation in the TME, often mediated by receptors (e.g., CD36), can lead to lipotoxicity and oxidative stress within T cells, exacerbating their dysfunction[73,78,79].
This metabolic competition is further intensified by the superior ability of cancer cells to scavenge and utilize available lipids. In models of obesity-associated cancer, tumor cells can significantly increase their fatty acid uptake, potentially creating a local depletion of metabolic substrates that contributes to the “fuel shortage” experienced by effector T cells, placing them at a metabolic disadvantage. This competitive consumption of nutrients, coupled with the accumulation of immunosuppressive metabolites, collaboratively fosters an immunosuppressive TME[77,80].
This critical comparison reveals a therapeutic paradox: broad inhibition of FAO might simultaneously affect both pro-tumoral and anti-tumoral cells, leading to complex and potentially unpredictable outcomes. Therefore, future therapeutic strategies targeting metabolism must be precisely designed. Rather than indiscriminately targeting the FAO pathway, interventions could focus on disrupting the specific mechanisms that underpin this detrimental metabolic crosstalk[81]. Promising approaches may include selectively blocking specific lipid uptake pathways in cancer cells or reversing the aberrant signaling that drives the dysfunctional metabolic reprogramming in T cells, thereby converting a metabolic vulnerability into a therapeutic advantage[74-76].
NON-CANONICAL REGULATORY FUNCTIONS OF METABOLIC ENZYMES IN HCC
In recent years, accumulating evidence has revealed that metabolic enzymes in HCC exhibit dual functionalities: not only do they sustain tumor growth through their canonical catalytic activities, they also exert diverse non-enzymatic functions that contribute significantly to oncogenesis[82-84]. Rather than reflecting metabolic reprogramming per se, these non-canonical activities constitute an additional regulatory layer mediated by metabolic enzymes, encompassing epigenetic modulation, transcriptional control, signal transduction, cytoskeletal organization, and immune evasion. A systematic overview of representative metabolic enzymes and their dual canonical and non-canonical roles in HCC is provided in Table 1.
Dual roles of metabolic enzymes in HCC
| Enzyme | Metabolic function | Non-enzymatic function |
| HK2 | Catalyzes glucose phosphorylation in glycolysis[30] | Binds VDAC1 to block MPTP opening, inhibits apoptosis; regulates Rheb–mTORC1 axis[30] |
| PKM2 | Catalyzes PEP → pyruvate in glycolysis[87] | Nuclear translocation → phosphorylates STAT3 (EMT, metastasis); stabilizes HIF-1α[85,86] |
| LDHA | Converts pyruvate ↔ lactate[32,43] | Promotes histone lactylation (H3K18la); lactate upregulates PD-L1 and suppresses CD8+ T cells[88] |
| PDK1 | Inhibits PDH, blocks pyruvate entry into TCA[32,43] | Activates NF-κB via IKKβ phosphorylation, promotes inflammation[89] |
| PGAM1 | Catalyzes 3-PG ↔ 2-PG in glycolysis[32,43] | Interacts with ACTA2 to stabilize F-actin, maintains cytokinesis, prevents polyploidy[90] |
| GAPDH | Catalyzes G3P → 1,3-BPG in glycolysis[32,43] | Acts as RNA-binding protein, regulates mRNA stability[91] |
| ENO1 | Catalyzes 2-PG → PEP[32,43] | Serves as plasminogen receptor, promotes invasion[92] |
| PFKFB3 | Produces F2,6BP to regulate glycolysis[32,43] | Controls cell cycle progression independent of metabolism[93] |
| GLS1 | Converts glutamine → glutamate[62] | Interacts with KEAP1 to release NRF2, enhances antioxidant defense[94] |
| GLUD1 | Converts glutamate ↔ α-KG[95] | Modulates TET/JmjC demethylases via α-KG, regulates epigenetics[96] |
| ASCT2 | Transports glutamine[104] | Interacts with EGFR, amplifies MAPK/ERK signaling[97] |
| IDH1/2 | Catalyzes isocitrate → α-KG[104] | Mutants produce 2-HG, inhibiting α-KG–dependent dioxygenases (epigenetic dysregulation)[100] |
| SLC7A5 | Exchanges glutamine with leucine[104] | Activates mTORC1 signaling indirectly[101] |
| GOT1 | Links glutamine metabolism with TCA intermediates[104] | Maintains NADPH balance, supports redox homeostasis[96] |
| GDH2 | Catalyzes glutamate → α-KG[104] | Enhances mitochondrial signaling, promotes apoptosis resistance[105] |
| FASN | Synthesizes palmitate from acetyl-CoA and malonyl-CoA[114] | Nuclear translocation → acetylates p53; interacts with HER2 to enhance EGFR signaling; stabilizes PD-L1[106-109] |
| ACC | Converts acetyl-CoA → malonyl-CoA[114] | Malonyl-CoA inhibits HDAC3, facilitating c-Myc expression[110] |
| CPT1A | Imports fatty acids into mitochondria for FAO[114] | Modulates BCL-2 family proteins, suppresses mitochondrial apoptosis[111] |
| ACLY | Converts citrate → acetyl-CoA[114] | Provides acetyl-CoA for histone acetylation (H3K27ac), activates Wnt/β-catenin signaling[112] |
| SCD1 | Synthesizes MUFAs from saturated fatty acids[114] | Regulates oncogenic lipid signaling pathways[115] |
| ACOX1 | Catalyzes first step of peroxisomal FAO[114] | Modulates ROS balance and oxidative stress[116] |
| HADHA | Catalyzes β-oxidation of fatty acids[114] | Regulates mitochondrial dynamics and apoptosis[117] |
Glycolytic enzymes associated with the Warburg effect exemplify this phenomenon. HK2 binds to mitochondrial voltage-dependent anion channel 1 (VDAC1) to prevent the opening of the mitochondrial permeability transition pore (MPTP)[30,37], thereby inhibiting apoptosis, while also regulating the Ras homolog enriched in brain (Rheb)–mechanistic target of rapamycin complex 1 (mTORC1) axis to promote proliferation. PKM2 translocates into the nucleus, where it phosphorylates STAT3 to induce epithelial–mesenchymal transition (EMT) and metastasis[85], and simultaneously stabilizes HIF-1α to enhance hypoxia-responsive transcription[86,87]. LDHA contributes to histone lactylation, linking lactate metabolism to epigenetic regulation, while lactate itself promotes PD-L1 expression and suppresses CD8+ T cells, facilitating immune escape[88]. Pyruvate dehydrogenase kinase 1 (PDK1) activates nuclear factor kappa B (NF-κB) signaling via inhibitor of nuclear factor kappa B kinase beta (IKKβ) phosphorylation, reinforcing a pro-inflammatory TME. Phosphoglycerate mutase 1 (PGAM1), beyond its glycolytic role, facilitates F-actin assembly through interaction with actin alpha 2 (ACTA2), thereby maintaining cytokinesis and genomic stability; its downregulation leads to cytokinetic failure, polyploidy, and growth arrest, underscoring its structural role in tumor cell proliferation. Additional enzymes such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (as an RNA-binding protein), Enolase 1 (ENO1) (as a plasminogen receptor), and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) (as a regulator of the cell cycle) further illustrate how glycolytic enzymes act beyond metabolism to directly control oncogenic signaling[89-93].
Similarly, glutamine metabolic enzymes also display significant non-enzymatic roles. GLS1 interacts with Kelch-like ECH-associated protein 1 (KEAP1) to release nuclear factor erythroid 2-related factor 2 (NRF2), enhancing antioxidant defenses and promoting chemotherapy resistance, while glutamine-derived glutamate activates mTORC1 via Rag GTPases[62,94]. Glutamate dehydrogenase 1 (GLUD1), through α-KG, regulates TET and Jumonji C domain-containing (JmjC) demethylases, thereby modulating epigenetic programs[95,96]. Alanine serine cysteine transporter 2 (ASCT2/SLC1A5) acts not only as a transporter but also interacts with epidermal growth factor receptor (EGFR) to amplify mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling[97]. Mutant isocitrate dehydrogenase 1/2 (IDH1/2) produces the oncometabolite 2-hydroxyglutarate (2-HG), which inhibits α-KG–dependent dioxygenases, resulting in widespread epigenetic dysregulation[98-100]. Moreover, solute carrier family 7 member 5 (SLC7A5/LAT1) couples with ASCT2 to mediate glutamine–leucine exchange, indirectly activating mTORC1[101]. Meanwhile, glutamate oxaloacetate transaminase 1 (GOT1) supports redox homeostasis by maintaining NADPH balance, and glutamate dehydrogenase 2 (GDH2) contributes to mitochondrial signaling that enhances apoptosis resistance[96,102-105].
In addition, enzymes involved in fatty acid metabolism contribute to HCC through their non-enzymatic actions[67,68]. Fatty acid synthase (FASN) translocates to the nucleus to acetylate and inactivate tumor protein p53 (p53)[106], while also enhancing EGFR signaling via human epidermal growth factor receptor 2 (HER2) interaction and stabilizing PD-L1 through glycosylation to promote immune escape[107-109]. Acetyl-CoA carboxylase (ACC) produces malonyl-CoA, which inhibits histone deacetylase 3 (HDAC3) and thereby facilitates the expression of oncogenes such as c-Myc[110]. CPT1A modulates B-cell lymphoma 2 (BCL-2) family proteins to suppress mitochondrial apoptosis, while ATP citrate lyase (ACLY) provides acetyl-CoA for histone acetylation[111], activating the Wingless/integrated (Wnt)/β-catenin signaling pathway[112]. Other enzymes, including stearoyl-CoA desaturase 1 (SCD1), acyl-CoA oxidase 1 (ACOX1), and hydroxyacyl-CoA dehydrogenase subunit alpha (HADHA), also regulate lipid signaling, oxidative stress responses, and mitochondrial dynamics independent of their catalytic roles[113-117].
In summary, these findings highlight a unifying theme: metabolic enzymes in HCC possess dual identities. They not only sustain tumor bioenergetics but also act as signaling regulators, transcriptional modulators, epigenetic remodelers, and immune checkpoints[82-84]. This dual functionality suggests that therapeutic strategies targeting metabolic enzymes must consider both enzymatic and non-enzymatic functions to effectively disrupt metabolic reprogramming and overcome drug resistance in HCC[107,108,111].
INFLUENCE OF HCC TUMOR CELL METABOLIC REPROGRAMMING ON THE TME
The TME of HCC is a highly organized and metabolically dynamic ecosystem composed of immune cells, stromal components - including cancer-associated fibroblasts (CAFs) and endothelial cells - extracellular matrix (ECM), and locally enriched signaling molecules. Rather than serving as a passive structural context, the HCC TME actively regulates tumor initiation, progression, and therapeutic response by shaping nutrient availability, metabolic constraints, and immune cell states. Consistent with Paget’s “seed and soil” hypothesis, the successful growth and dissemination of malignant hepatocytes depend on a permissive microenvironment; however, in HCC, this “soil” exhibits pronounced spatial and metabolic specialization. In this context, an immune–metabolic niche refers to a spatially confined microdomain in which localized metabolic conditions - such as hypoxia, nutrient deprivation, and metabolite accumulation - intersect with specific immune and stromal cell assemblies to coordinately regulate immune cell function and tumor behavior.
Advances in single-cell transcriptomics and spatially resolved multi-omics have revealed that the HCC TME is far from homogeneous. Integrated single-cell and spatial transcriptomic profiling across primary HCC specimens demonstrates that distinct cellular assemblies are spatially segregated across tumor cores, tumor–normal interfaces, perivascular regions, and fibrotic capsules, with corresponding differences in transcriptional programs and functional states[118,119]. Spatial transcriptomics analyses further uncovered structured immunosuppressive modules at tumor boundaries composed of secreted phosphoprotein 1 (SPP1, also known as osteopontin)–positive macrophages and CAFs that correlate with immune exclusion and checkpoint blockade resistance[120,121]. High-resolution spatial platforms such as CosMx Spatial Molecular Imaging platform spatial molecular imaging have enabled the dissection of cell–cell interactions and ligand–receptor signaling within defined anatomical contexts, identifying metabolically adapted myeloid subsets in hypoxic niches and lymphoid populations in more nutrient-permissive regions[122].
Importantly, these spatially defined architectures are tightly coupled to local metabolic conditions within the TME. Recent single-cell studies have identified immunosuppressive populations - including SPP1+ tumor-associated macrophages (TAMs) and transcriptionally exhausted T-cell states - that preferentially localize to regions marked by hypoxia, elevated lactate, and altered nutrient availability, features that are associated with disease progression and poor prognosis in HCC patients[123,124]. Collectively, these high-resolution observations demonstrate that immune dysfunction in HCC arises within discrete immune–metabolic niches, rather than uniformly across the tumor.
Together, these findings underscore that metabolic reprogramming in HCC operates within a spatially heterogeneous TME, where localized metabolic constraints and cellular interactions cooperatively shape immune suppression and tumor evolution. On this basis, the following sections systematically examine how HCC metabolic reprogramming influences four core components of the TME - immunosuppressive cells, immune effector cells, stromal cells, and non-cellular elements - in a context- and niche-dependent manner [Figure 3].
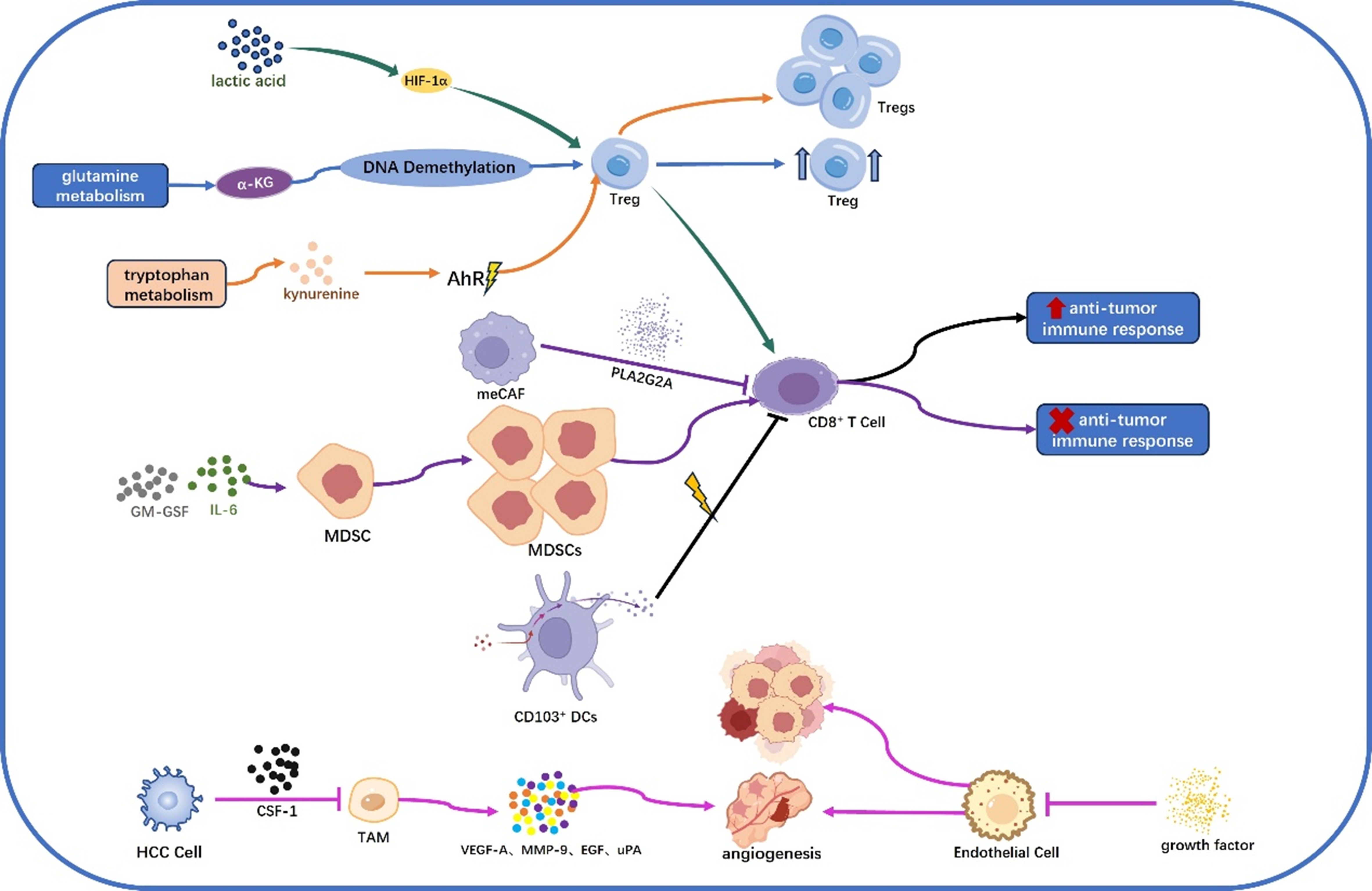
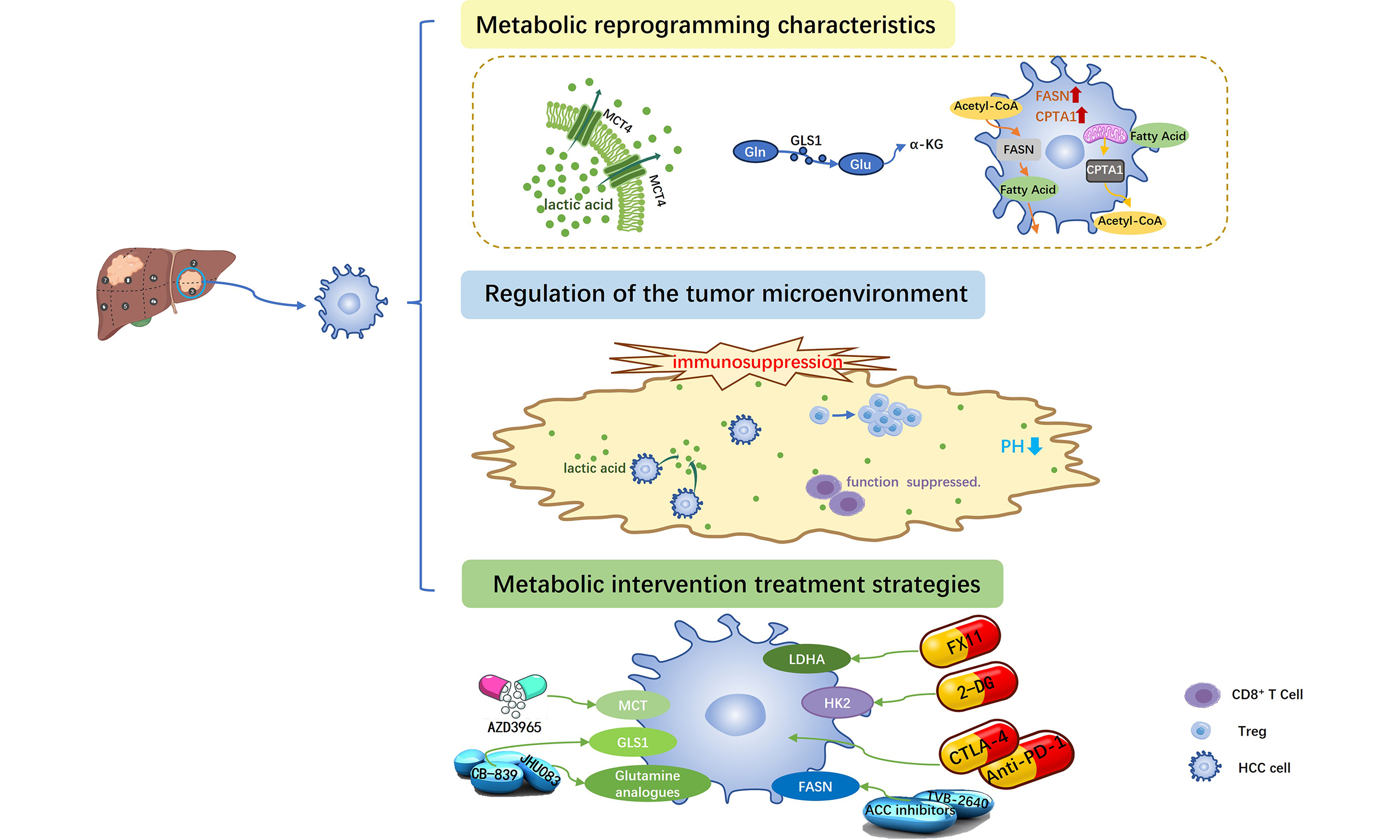
Figure 3. Metabolic reprogramming in HCC modulates a multi-cellular immune microenvironment. HCC metabolic reprogramming actively shapes an immune microenvironment by modulating the function of both immune and stromal cells. Key metabolic pathways, such as tryptophan, glutamine, and lactate metabolism, regulate immune cell functions. Tregs: Metabolites such as lactate, α-KG, and kynurenine promote Treg differentiation and function, supporting immune suppression in the TME. MDSCs: Tumor-derived factors such as GM-CSF and IL-6 drive MDSC expansion, which inhibits anti-tumor immunity. TAMs M2: Tumor cells and metabolic reprogramming influence TAM polarization, with factors such as VEGF-A and MMP-9 promoting tumor growth and metastasis. CD8+ T Cells: PLA2G2A+ CAFs and metabolic changes in HCC inhibit CD8+ T cell anti-tumor function via signaling pathways such as MAPK/Erk and NF-κB. DCs: Metabolic reprogramming affects the function of CD103+ DCs, essential for CD8+ T cell activation and immune surveillance. Metabolic changes in CAFs and Endothelial Cells promote tumor growth, angiogenesis, and immune evasion, contributing to a pro-TME. Additionally, metabolites such as lactate and exosomes from tumor cells further shape the immunosuppressive environment, promoting tumor progression and immune resistance. HCC: Hepatocellular carcinoma; α-KG: α-ketoglutarate; Treg: regulatory T cell; TME: tumor microenvironment; MDSCs: myeloid-derived suppressor cells; GM-CSF: granulocyte–macrophage colony-stimulating factor; IL-6: interleukin-6; TAMs: tumor-associated macrophages; VEGF-A: vascular endothelial growth factor A; MMP-9: matrix metalloproteinase-9; CD8+: cluster of differentiation 8 positive; PLA2G2A+: phospholipase A2 group IIA positive; CAFs: cancer-associated fibroblasts; MAPK: mitogen-activated protein kinase; Erk: extracellular signal-regulated kinase; NF-κB: nuclear factor kappa B; DCs: dendritic cells; HIF-1α: hypoxia-inducible factor 1α; AhR: aryl hydrocarbon receptor; EGF: epidermal growth factor; uPA: urokinase-type plasminogen activator.
The effect of HCC metabolic reprogramming on immunosuppressive cells
Regulatory T cells
Regulatory T cells (Tregs) serve as indispensable “peacekeeping forces” within the immune system. Rather than targeting pathogens, their primary role is to suppress excessive activation of other immune cells, thereby maintaining immune homeostasis and preventing autoimmune attacks against self-tissues. Dysregulation of Treg function is closely linked to various pathological conditions, including tumor immune evasion. In HCC, a dynamic interplay exists between tumor cell metabolic reprogramming and the enrichment and functional enhancement of Tregs[125,126].
HCC cells undergo metabolic rewiring - most notably the Warburg effect - leading to intense glucose consumption and substantial lactate production. This reshapes TME into an immunosuppressive niche, which selectively favors Treg survival, expansion, and suppressive activity. Specifically, lactate accumulated in the TME activates the HIF-1α pathway in Tregs, promoting their differentiation, stability, and functional maturation. Moreover, kynurenine - a downstream metabolite of tryptophan catabolism - activates the aryl hydrocarbon receptor (AhR) signaling axis, further driving Treg proliferation and immunosuppressive capacity[126]. Conversely, Tregs themselves exhibit remarkable metabolic plasticity and adaptive reprogramming. For instance, in HBV-associated HCC, a stem-like C-C chemokine receptor type 4 (CCR4)-positive Treg subset within the tumor acquires enhanced suppressive potency and longevity through epigenetic remodeling, thereby dominating the immunosuppressive landscape[127,128].
In summary, metabolic reprogramming in HCC not only creates a permissive milieu for Treg accumulation and function, but the metabolic and epigenetic adaptations of Tregs reciprocally reinforce immunosuppression - establishing a self-perpetuating, vicious cycle that accelerates HCC progression.
Myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSCs) represent a heterogeneous population of immature myeloid progenitor cells that are present at extremely low frequencies under physiological conditions. They expand dramatically and become activated in pathological states such as chronic inflammation, infection, and malignancies - including HCC[129]. Based on phenotypic and morphological features, MDSCs are primarily classified into two subsets: monocytic MDSCs [M-MDSCs; CD14+HLA-DRlow/-CD15- (HLA-DR: human leukocyte antigen–DR isotype)] and polymorphonuclear MDSCs (PMN-MDSCs; CD14-CD15+ or CD66b+)[129,130]. Both subsets exhibit potent immunosuppressive capacities, effectively inhibiting effector immune cells - including T lymphocytes and natural killer (NK) cells - thereby fostering tumor immune evasion and disease progression[130,131].
In HCC, tumor cells undergo extensive metabolic reprogramming across glucose, lipid, and amino acid metabolism. This metabolic rewiring not only fulfills the bioenergetic and biosynthetic demands of rapid tumor proliferation but also dominantly orchestrates the recruitment, expansion, and functional polarization of MDSCs within the TME[132].
Glucose metabolism reprogramming (i.e., the Warburg effect) leads to lactate accumulation and extracellular acidification; lactate, acting via the G-protein-coupled receptor GPR81, drives MDSC differentiation toward an immunosuppressive phenotype and upregulates PD-L1 expression. Dysregulated lipid metabolism - characterized by enhanced de novo fatty acid synthesis and β-oxidation - provides MDSCs with energetic substrates and signaling intermediates, promoting the secretion of immunosuppressive cytokines such as interleukin (IL)-10 and transforming growth factor β (TGF-β)[133]. Amino acid metabolic alterations, particularly upregulation of indoleamine 2,3-dioxygenase 1 (IDO1) and arginase-1 (ARG1), result in kynurenine accumulation and L-arginine depletion, synergistically suppressing T cell function while reinforcing MDSC activation[131]. Moreover, aberrant glycogen metabolism supports sustained high glycolytic flux in MDSCs, thereby enhancing their survival and functional stability[132].
Notably, MDSCs exhibit close developmental and functional links with TAMs. In particular, M-MDSCs can further differentiate into M2-like TAMs, which likewise exert critical immunosuppressive functions[134]. TAMs originate from distinct lineages - including yolk sac-derived erythro-myeloid progenitors, fetal liver monocytes, and bone marrow monocytes. The latter are recruited into tumor sites by inflammatory signals such as colony-stimulating factor 1 (CSF-1). In HCC patients, canonical TAM subsets (e.g., SPP1+ TAMs) promote tumor progression through the secretion of factors including macrophage migration inhibitory factor (MIF) and secreted phosphoprotein 1 (SPP1/osteopontin). Of particular interest, immunosuppressive TAMs derived from M-MDSCs persistently express S100A9, a protein that sustains their T cell–suppressive activity and is associated with poor response to immunotherapy in multiple cancers[133]. These TAMs further support tumor angiogenesis and progression by secreting vascular endothelial growth factor A (VEGF-A), matrix metalloproteinase-9 (MMP-9), and urokinase-type plasminogen activator (uPA).
In summary, MDSCs serve as central cellular mediators of immune evasion in HCC. Their biology is exquisitely regulated by tumor-driven metabolic reprogramming, positioning them as pivotal architects of the immunosuppressive TME. Targeting key metabolic pathways that drive MDSC immunosuppression - such as the lactate–GPR81 axis, FAO, and the IDO1/ARG1 cascade - holds promise for reversing immune suppression and enhancing the efficacy of immune checkpoint blockade. This provides a compelling rationale for integrating metabolic modulation with immunotherapy in the development of novel combination strategies for HCC treatment[135].
The influence of HCC metabolic reprogramming on immune effector cells
CD8+ T cells
Intratumoral glucose deprivation caused by dominant tumor glycolysis constrains glycolytic flux in activated CD8+ T cells. Under these conditions, the glycolytic enzyme GAPDH can bind AU-rich elements in the 3′ untranslated region (3′UTR) of IFN-γ mRNA and repress its translation, directly lowering IFN-γ production and cytotoxic function[136]. Tumor-derived lactate accumulates and acidifies the TME, which suppresses CD8+ T cell proliferation, reduces granzyme B and cytokine expression, and perturbs pyruvate/TCA routing, thereby promoting a metabolically exhausted phenotype[137]. In addition, extracellular ATP hydrolysis by CD39/CD73 produces adenosine that signals through adenosine A2A and A2B receptors (A2AR/A2BR) on T cells to raise intracellular cyclic adenosine monophosphate (cAMP) and inhibit proliferation and effector cytokine release, a pathway increasingly validated as a major immunosuppressive circuit in tumors[138].
Lipid dysregulation within the TME - notably uptake of oxidized lipids via scavenger receptors such as CD36 - causes lipid peroxidation, mitochondrial stress, and ROS accumulation in tumor-infiltrating CD8+ T cells, leading to functional impairment. Conversely, controlled enhancement of fatty-acid oxidation [e.g., via peroxisome proliferator-activated receptor alpha (PPARα) signaling] can sometimes rescue T cell energetic capacity in low-glucose settings, highlighting a context-dependent, bidirectional role of lipid metabolism[139]. Amino-acid pathways further modulate cytotoxic responses: tryptophan catabolism by indoleamine 2,3-dioxygenase/tryptophan 2,3-dioxygenase (IDO/TDO) yields kynurenine, which activates the AhR and skews local immunity towards regulatory/tolerogenic phenotypes. Similarly, tumor consumption of glutamine limits its availability for effector T cells and for type 1 conventional dendritic cells (cDC1s), impairing mitochondrial metabolism and persistence of effectors[140,141]. Finally, hypoxia, impaired mitochondrial oxidative phosphorylation, and chronic oxidative stress in the TME converge to undermine CD8+ T cell memory formation and responsiveness to immunotherapies[135,142].
Dendritic cells
Tumor metabolic remodeling likewise disrupts dendritic cell (DC) biology at multiple levels. Tumor-intrinsic Wnt/β-catenin signaling reduces C–C motif chemokine ligand 4 (CCL4) secretion and thereby limits recruitment of basic leucine zipper ATF-like transcription factor 3 (Batf3)-dependent CD103+ (cDC1) cross-presenting DCs, which is associated with immune exclusion and poor CD8+ priming[143]. Lactate and acidification impair monocyte-to-DC differentiation and decrease IL-12 production, compromising DC cross-priming capacity.
Aberrant lipid accumulation and oxidatively truncated lipids in tumor-associated DCs trigger endoplasmic reticulum (ER) stress and X-box binding protein 1 (XBP1) activation, which interferes with antigen processing/trafficking and blocks effective cross-presentation. Lipid body accumulation thereby converts DCs toward tolerogenic states and reduces IL-12 output. IDO/TDO–kynurenine–AhR signaling similarly conditions DCs toward immune-suppressive phenotypes and enhances Treg induction. In parallel, glutamine competition between tumor cells and cDC1s [via solute carrier family 38 member 2 (SLC38A2)] limits cDC1 metabolic fitness and cross-priming - phenomena that can be reversed in preclinical models by modulating glutamine availability or uptake[140]. Moreover, oncogenic and inflammatory programs [e.g., STAT3 activation, elevated IL-6/IL-10/vascular endothelial growth factor (VEGF)] both directly inhibit DC maturation and shift DC metabolism (toward FAO and ER-stress signatures), reinforcing tolerogenic differentiation[144].
The influence of HCC metabolic reprogramming on stromal cells
CAFs
The metabolic reprogramming of HCC has a significant impact on stromal cells, especially CAFs. Some literature has discovered a novel metabolically active cancer-associated metabolically active cancer-associated fibroblasts[145], which is characterized by high expression of phospholipase A2 group IIA (PLA2G2A) and active glycolytic activity. These PLA2G2A+ meCAFs significantly inhibit the anti-tumor function of CD8+ T cells by secreting PLA2G2A protein, thereby forming an immunosuppressive microenvironment. There is also a view suggesting that tumor cells recruit and activate CAFs by secreting specific cytokines [such as CSF-1 and C–C motif chemokine ligand 2 (CCL2)], and these activated CAFs further secrete factors such as VEGF and MMP-9 to promote tumor angiogenesis and invasion[144].
Endothelial cells
Single-cell sequencing analyses revealing the high heterogeneity of endothelial cells in HCC demonstrate that metabolic reprogramming drives endothelial cell proliferation and angiogenesis by activating growth factor–related pathways. Gene regulatory network (GRN) analysis indicates that the metabolic activity of endothelial cells is regulated by key transcription factors such as forkhead box A1 (FOXA1) and nuclear receptor subfamily 2 group E member 3 (NR2E3). Pseudo-temporal analysis further reveals that endothelial cell subpopulation En_RAC2 (defined based on RAC2 expression) is in the initial stage of development, whereas tumor endothelial cell (TEC)_glutathione peroxidase 1 (GPX1) is in the terminal differentiation state, suggesting that the highly metabolically active TEC subpopulation may maintain tumor vascular stability through antioxidant stress (such as GPX1 expression). In addition, endothelial cells establish metabolic crosstalk with tumor cells through the ErbB receptor tyrosine kinase signaling pathway (ErbB pathway)[146].
The influence of HCC metabolic reprogramming on non-cellular components
The metabolic reprogramming of HCC profoundly affects non-cellular components in the TME, particularly by altering the levels of key metabolites such as lactate, ketone bodies, and extracellular vesicles[46,147]. Tumor cells not only affect the function of immune cells by secreting factors such as IL-6, IL-10 and VEGF, but also markedly alter the composition and physical properties of the ECM in the microenvironment[147]. Under conditions of nutrient deprivation, HCC cells activate ketolysis, leading to the accumulation of ketone bodies such as β-hydroxybutyric acid (β-HB) and acetoacetic acid in the microenvironment. These metabolites affect the energy perception of surrounding cells by inhibiting the AMP-activated protein kinase (AMPK) signaling pathway.
Lactate plays a central role in shaping the immunosuppressive microenvironment. Research has found that the high glycolytic activity of PLA2G2A+ meCAFs leads to excessive lactate secretion, forming an acidic microenvironment[148]. This acidic environment not only directly suppresses the function of CD8+ T cells, but also acts through the following mechanisms: activation of the MAPK/Erk and NF-κB signaling pathways in CAFs, promotion of ECM-remodeling proteins (e.g., MMP-9), and stabilization of HIF-1α expression, thereby sustaining a tumor-promoting microenvironment.
In addition, tumor-derived exosomes also play a significant role in metabolic reprogramming. These nano-sized vesicles can deliver metabolic enzymes (e.g., PKM2), non-coding RNAs (e.g., miR-122), and metabolic intermediates (e.g., succinate), which influence the metabolic state of recipient cells via paracrine signaling, thereby establishing a metabolic “field effect” that promotes tumor progression[149].
THERAPEUTIC STRATEGIES TARGETING HCC METABOLIC REPROGRAMMING: RESHAPING THE TME AND INFLUENCING TREATMENT OUTCOMES
Based on an integrated analysis of the current literature, it is evident that the limited clinical success of earlier metabolic interventions in cancer - particularly in HCC - can be broadly attributed to three interrelated factors: the complexity of the TME, the redundancy and plasticity of metabolic networks, and pronounced inter-tumoral heterogeneity[146,150].
First, TME of HCC is characterized by hypoxia, acidity, and nutrient deprivation, conditions that are actively reinforced by tumor-intrinsic metabolic reprogramming. Excessive glycolysis and metabolite accumulation not only support tumor growth but also induce metabolic dysfunction in immune cells, including CD8+ T cells and macrophages, thereby limiting effective antitumor immunity[15,151]. Under such conditions, inhibition of a single metabolic node is often insufficient to overcome the global immunosuppressive milieu of the TME[150].
Second, both tumor cells and immune cells exhibit substantial metabolic redundancy and adaptability. Compensatory activation of alternative pathways - such as lipid or amino acid metabolism following glycolytic blockade - frequently undermines the durability of single-pathway interventions[61,77]. Moreover, metabolic crosstalk within the TME, mediated by metabolites such as lactate or kynurenine, further reinforces immune suppression and therapeutic resistance[152,153].
Third, marked inter- and intra-tumoral heterogeneity in HCC - including distinct molecular subtypes and context-dependent metabolic dependencies - poses a major challenge to unselected metabolic therapies. In the absence of robust biomarker stratification, metabolic inhibitors may exhibit limited efficacy or unacceptable systemic toxicity, thereby restricting their clinical applicability[99,154].
Nevertheless, these limitations do not diminish the central role of metabolic reprogramming in HCC pathogenesis; rather, they highlight the need for more refined and context-aware therapeutic strategies. Building upon these metabolic dependencies, HCC cells exploit multiple rewired pathways - including glycolysis, glutamine metabolism, lipid synthesis, and amino acid metabolism - to sustain growth and evade immune surveillance[152]. Accordingly, therapeutic strategies targeting key metabolic nodes have emerged as promising approaches for disrupting tumor metabolism and reshaping the immunosuppressive TME[80,154].
Targeting key metabolic pathways in HCC
Building on the metabolic reprogramming characteristic of HCC, multiple therapeutic strategies have been explored to target key metabolic dependencies of HCC cells and the TME, with varying degrees of translational maturity. An overview of these approaches is summarized in Table 2.
Therapeutic strategies targeting metabolic reprogramming in HCC
| Targeted pathway | Representative drug/inhibitor | Mechanism of action | Impact on TME | Clinical progress/notes |
| Glycolysis (Warburg effect) | 2-DG[156,157] | Inhibits HK2, reduces glycolytic flux and lactate production | Enhances glucose availability for CD8+ T cells, alleviates acidosis | Assessed in early phase I studies in non-HCC solid tumors, mainly for safety, with no ICI combination or HCC-specific clinical data |
| FX11[151,160] | Inhibits LDHA, blocking pyruvate → lactate conversion | Reduces lactate accumulation, limits M2 macrophage polarization | Limited to preclinical models and no clinical development | |
| Galloc acid[151,158,159] | Inhibits LDH activity, reduces lactate generation | Similar effects to FX11, suppressing immunosuppressive TME | Demonstrates antitumor effects in preclinical liver-related models, but lacks clinical validation | |
| AZD3965[151,161,162] | Inhibits MCTs MCT1/2, blocks lactate export | Improves TME pH, decreases MDSC recruitment | Evaluated in phase I trials in advanced solid tumors and lymphomas, with no HCC-specific efficacy data reported | |
| PKM2 inhibitors[136] | Block PKM2 activity, reduce aerobic glycolysis | Decrease lactate secretion, enhance T cell function | Preclinical stage | |
| Glutamine metabolism | CB-839[77,99,163,164] | Inhibits GLS, blocks glutamine → α-KG conversion and TCA cycle entry | Reduces CAF-derived IL-6 secretion, weakens inflammatory signaling | Advanced through early-phase and biomarker-driven phase II trials in selected solid tumors, but not specifically in HCC |
| BPTES[165,166] | Selective GLS inhibitor, suppresses glutamine catabolism | Promotes pro-inflammatory TAM phenotype | First-generation GLS inhibitor used as a preclinical tool compound only | |
| DON[77,167] | Glutamine analogue, competitively inhibits multiple glutamine-utilizing enzymes | Suppresses MDSC generation and function | Investigated in early clinical trials but discontinued due to toxicity and limited efficacy, with no benefit shown in HCC | |
| JHU083[77,168] | Improves DON delivery, targets glutamine metabolism in tumors | Reduces MDSCs, enhances T cell immunity | Orally bioavailable DON prodrug currently restricted to preclinical evaluation | |
| Lipid metabolism | TVB-2640[15,169] | Inhibits FASN, reduces lipid raft formation and PD-L1 localization | Decreases lipid accumulation in TAMs, enhances T cell cytotoxicity | Completed phase I and entered phase II studies in non-HCC malignancies, with no clinical evaluation in HCC |
| Tryptophan metabolism | Epacadostat[152,153,170] | Inhibits IDO1, blocks tryptophan → kynurenine pathway | Prevents kynurenine-driven T cell exhaustion, reduces Treg induction | Failed to demonstrate clinical benefit in a pivotal phase III trial, highlighting translational limitations |
| Arginine metabolism | CB-1158[152,171] | Inhibits arginase, restores arginine availability for T cells | Enhances T cell proliferation and effector function | Limited to early phase I/II studies in advanced solid tumors, reporting mainly safety and pharmacodynamic data without HCC-specific efficacy |
Targeting glycolysis: inhibiting the Warburg effect
Mode of action: Inhibiting HK2 or PKM2 can reduce lactate secretion, alleviate TME acidification, and reverse the functional inhibition of CD8+ T cells (such as through PD-L1 down-regulation)[155].
The glycolysis inhibitor 2-deoxy-D-glucose (2-DG), combined with ICIs, has been shown to enhance T-cell infiltration and antitumor immune responses in preclinical models. In the clinical setting, 2-DG has thus far been evaluated only in early-phase trials focused on safety and dose escalation, either as monotherapy or in combination with chemotherapy or radiotherapy, including NCT00096707/NCT00196707, NCT00633087, and related studies, which enrolled patients with various advanced solid tumors or high-grade gliomas. These trials reported manageable metabolic and cardiac toxicities but did not investigate combinations with ICIs or provide definitive efficacy endpoints. Importantly, no clinical trials to date have assessed 2-DG in combination with ICIs, nor have any studies specifically enrolled patients with HCC[156,157].
Lactate dehydrogenase (LDH) inhibitors, such as 2,3-dihydroxy-6-methyl-7-(phenylmethyl)-4-propyl-1-naphthalenecarboxylic acid (FX11) and Gallic acid, inhibit the activity of LDH and block the conversion of pyruvate to lactate[151]. Among these inhibitors, a notable distinction exists in their translational progress. While studies suggest Gallic acid possesses potential in combating hepatic precancerous lesions and inhibiting liver metastasis in animal models, its clinical efficacy remains to be established in humans[158,159]. In contrast, FX-11 is confined to the preclinical stage, as no clinical trial has yet been conducted to evaluate its activity in humans[160].
Monocarboxylate transporter (MCT) inhibitors, such as (S)-5-(4-hydroxy-4-methylisoxazolidine-2-carbonyl)-1-isopropyl-3-methyl-6-((5-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)methyl)thieno[2,3-d]pyrimidine-2,4(1H,3H)-dione (AZD3965), inhibit the lactate transporter monocarboxylate transporter 1 and 2 (MCT1/2) and block lactate excretion[151]. AZD3965 has advanced into a Cancer Research UK–sponsored phase I clinical trial (NCT01791595) involving patients with advanced solid tumors and lymphomas; however, the study has primarily reported safety, tolerability, and dose-escalation data, and formal efficacy outcomes such as objective response rate or disease control rate have not yet been disclosed. Importantly, these early-phase clinical investigations have not been restricted to HCC. Preclinical and mechanistic studies indicate that AZD3965 is particularly effective in tumors characterized by high MCT1 and low MCT4 expression, especially in certain lymphoma subtypes, where inhibition of lactate transport induces intracellular acidification and metabolic stress. These findings suggest that AZD3965 may hold therapeutic potential in metabolically defined tumor subsets, supporting its further exploration in HCC and other malignancies with similar metabolic dependencies[161,162] [Figure 4].
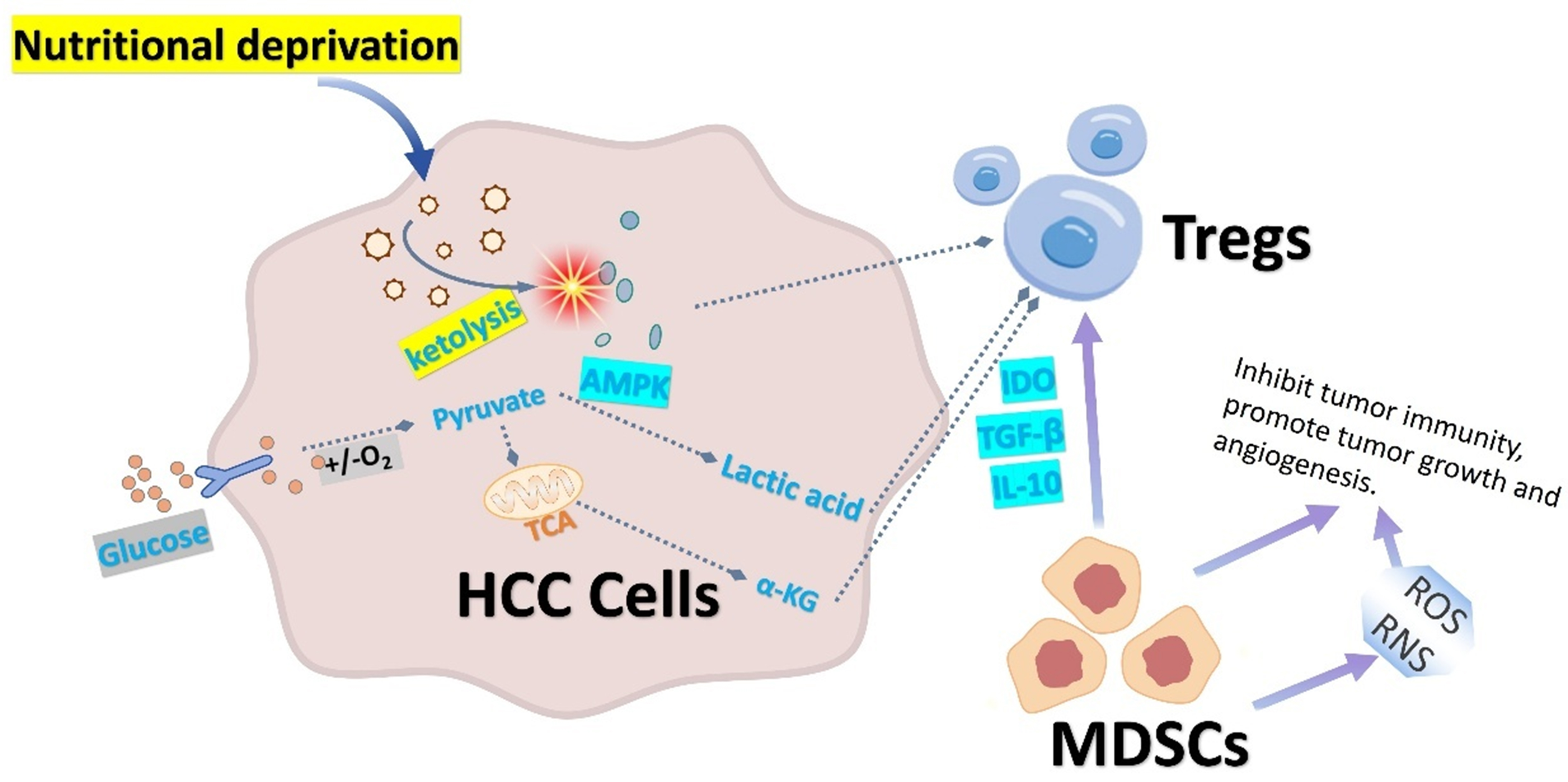
Figure 4. Metabolic reprogramming in HCC shapes an immunosuppressive microenvironment by modulating Tregs and MDSCs. Under nutrient deprivation, HCC cells activate ketolysis to maintain energy supply, suppressing AMPK activity and inhibiting Tregs for rapid energy acquisition, HCC cells produce lactate via the Warburg effect; this lactate promotes Treg differentiation by activating HIF-1α. α-KG from glutamine metabolism enhances Treg function. HCC-derived factors (e.g., GM-CSF, IL-6) expand bone marrow myeloid precursors, increasing MDSCs. MDSCs infiltrating the TME suppress antitumor immunity through IDO, TGF-β, and IL-10, thereby modulating Treg activity. HCC: Hepatocellular carcinoma; Tregs: regulatory T cells; MDSCs: myeloid-derived suppressor cells; AMPK: AMP-activated protein kinase; HIF-1α: hypoxia-inducible factor 1α; α-KG: α-ketoglutarate; GM-CSF: granulocyte–macrophage colony-stimulating factor; IL-6: interleukin-6; TME: tumor microenvironment; IDO: indoleamine 2,3-dioxygenase; TGF-β: transforming growth factor β; IL-10: interleukin-10; ROS: reactive oxygen species; RNS: reactive nitrogen species.
Impact on the TME: Reducing lactate levels can decrease the polarization of M2-type macrophages and the recruitment of MDSCs. These findings provide a strong rationale for combining glycolytic inhibition with immune checkpoint blockade, as reducing lactate accumulation and TME acidification may restore CD8+ T-cell effector function and enhance responsiveness to immunotherapy, despite the current lack of HCC-specific clinical validation.
Targeting glutamine metabolism: blocking biosynthetic precursors
Mode of action: GLS inhibitors: such as Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide (BPTES) and telaglenastat (CB-839)[77,99]. To date, CB-839 has completed phase I and phase I/II dose-escalation studies as monotherapy and advanced into phase Ib/II combination trials and phase II biomarker-driven basket studies, primarily enrolling patients with KEAP1/NRF2-, NF1 (neurofibromin 1)-altered, or LKB1 (liver kinase B1)-altered solid tumors (e.g., NCT03047993, NCT03875313, NCT03872427). Among currently explored biomarkers, KEAP1/NRF2 alterations represent the most clinically substantiated genetic context for CB-839 sensitivity. In the phase II BeGIN basket trial (Biomarker-driven Evaluation of Glutaminase Inhibition in Neoplasms; NCT03872427), KEAP1/NRF2 mutations were prospectively defined as key inclusion criteria, and pharmacodynamic analyses demonstrated elevated circulating lactate levels, consistent with in vivo GLS target engagement. A phase I/II trial combining CB-839 with the PD-1 antibody nivolumab in advanced melanoma, clear cell renal cell carcinoma, and non-small cell lung cancer showed acceptable tolerability but modest overall response rates, with no HCC cohort included[163]. Importantly, no registered clinical trial to date has enrolled HCC patients as an independent cohort, and HCC-specific efficacy or liver-metastasis–focused analyses have not been reported. Thus, while early-phase studies support biological activity and clinical feasibility, direct clinical validation of CB-839 in HCC remains lacking[163,164].
BPTES, a first-generation GLS inhibitor, has primarily been used as a preclinical tool compound, with evidence limited to experimental models. Clinical investigation of GLS inhibition has instead focused on optimized agents such as CB-839[165,166].
Glutamine analogs, such as 6-diazo-5-oxo-L-norleucine (DON), interfere with glutamine metabolism[77]. JHU083 (DON prodrug) can effectively inhibit the generation and recruitment of MDSCs and promote the transformation of TAMs to pro-inflammatory phenotypes. By 2020, DON had advanced through phase I–II clinical testing in more than 700 patients, but dose-limiting gastrointestinal toxicity and limited efficacy curtailed further development[167]. Subsequent efforts to improve the therapeutic index led to the development of JHU083, an orally bioavailable DON prodrug, which has thus far been evaluated only in preclinical models[168].
Impact on TME: Glutamine deprivation can lead to a reduction in the secretion of IL-6 by CAFs, thereby weakening tumor-promoting inflammatory signals. Given the central role of glutamine metabolism in both tumor growth and immune regulation, GLS or glutamine-antagonist–based therapies are particularly well suited for combination with ICIs, as they can simultaneously restrict tumor bioenergetics and relieve immunosuppressive constraints within the TME [Figure 4].
Targeting lipid metabolism: inhibiting fatty acid synthesis
Mode of action: FASN inhibitors [such as (R)-N-(3-acetylphenyl)-7-(3-fluorophenyl)-6-isopropyl-3,4-dihydro-1,8-naphthyridine-2(1H)-carboxamide (TVB-2640)] inhibit the PD-L1 membrane localization of HCC cells by reducing lipid raft formation, enhance T cell killing, suppress fatty acid synthase, and block lipid synthesis[15]. However, clinical translation of FASN inhibition remains at an early stage. TVB-2640 has completed a phase I dose-escalation trial in advanced solid tumors (NCT02223247), establishing safety and maximum tolerated dose, and has subsequently advanced into phase II studies in selected non-HCC malignancies, including Kirsten rat sarcoma viral oncogene homolog (KRAS)-mutant non-small cell lung cancer (NCT03808558) and astrocytoma (NCT03032484). To date, no phase I or II clinical trials have evaluated TVB-2640 in patients with HCC, and evidence in HCC is therefore limited to preclinical models[169].
Impact on TME: Reduced lipid accumulation suppresses pro-tumor polarization of TAMs (e.g., by inhibiting ARG1 expression). These findings suggest that inhibiting lipid synthesis may sensitize HCC to immunotherapy by modulating immune checkpoint localization and macrophage polarization, supporting the exploration of FASN-targeted strategies in combination with immune-based treatments.
Targeting amino acid metabolism: modulating immune cell function
Tryptophan Metabolism: IDO1 inhibitors, such as Epacadostat, can block the conversion of tryptophan to kynurenine[152], prevent the accumulation of kynuurine, and reverse T cell exhaustion. In the pivotal Phase III ECHO-301/KEYNOTE-252 trial (NCT02752074), the combination of Epacadostat and the PD-1 inhibitor pembrolizumab in patients with advanced melanoma failed to improve progression-free survival (PFS) or overall survival (OS) compared with pembrolizumab monotherapy. This outcome starkly contradicted the promising antitumor activity observed in early Phase I/II studies, exposing the discrepancy between preclinical/early-phase signals and large-scale clinical efficacy. The failure of ECHO-301/KEYNOTE-252 underscores key limitations, including insufficient biomarker-driven patient selection, inadequate pharmacokinetic–pharmacodynamic correlation, and underestimation of metabolic pathway redundancy[153]. This experience provides a critical lesson for metabolism–immunotherapy combinations: successful clinical translation requires biomarker-guided enrollment, adaptive trial design, and early verification of true target engagement[153,170].
Arginine Metabolism: Arginase inhibitors (such as CB-1158) block arginine metabolism and restore T cell function[152]. CB-1158 is currently in early clinical development, with evaluation limited to a first-in-human phase I dose-escalation study as monotherapy (NCT03361228; “CB-1158-101”) and a phase I/II exploratory study in combination with PD-1 blockade (INCB001158-202, internal study number). Both studies have been conducted in patients with advanced or metastatic solid tumors. Consistent with this early developmental stage, published clinical data - including a first-in-human phase I trial of INCB001158 alone or combined with pembrolizumab (NCT02903914) and a phase Ib study assessing INCB001158 with or without retifanlimab in Japanese patients (NCT03361228) - primarily report safety, pharmacokinetics/pharmacodynamics, and only limited antitumor activity in small cohorts[171]. To date, the overall level of evidence remains at level III, and no phase II or phase III efficacy data are available.
Although clinical outcomes have been heterogeneous, targeting amino acid metabolic pathways - particularly tryptophan and arginine metabolism - remains conceptually attractive for immunotherapy combinations, as these pathways directly regulate T-cell exhaustion, regulatory T-cell expansion, and immune checkpoint responsiveness.
Reshaping the TME through metabolic therapy
Metabolic therapy can reshape the TME through multiple mechanisms. It can increase apoptosis of DSCs and suppress their immunosuppressive function[77]. It improves glucose availability and enhances the effector function of CD8+ T cells[151]. IDO inhibitors reduce the generation and function of Tregs. Moreover, metabolic therapy enhances the cytotoxicity and cytokine production of NK cells while promoting their metabolic activity and overall function[15]. In addition, targeted glycolysis reduces lactate accumulation in the TME and alleviates acidic stress[15,151]. MCT inhibitors reduce lactate excretion and normalize the pH of the TME. Metabolic therapy reduces the competition between tumor cells and immune cells for nutrients such as glucose and glutamine[77]. Some articles point out that KEAP1-mutated cells have an increased dependence on glutamine, and inhibiting GLS can selectively kill these cells. Targeting the NRF2/KEAP1 pathway also regulates oxidative stress responses. NRF2 activation upregulates the expression of antioxidant genes, while reducing ROS can further enhance therapeutic efficacy[99].
Antitumor effects of different therapeutic strategies
According to the literature, different therapeutic strategies lead to distinct antitumor effects. Monotherapies include GLS inhibitors such as CB-839, which significantly suppress the growth of KEAP1-mutated tumors and markedly reduce the volume of both subcutaneous and orthotopic tumor grafts[99]. Glycolysis inhibitors such as 2-DG and MCT inhibitors also demonstrate broad antitumor activity across multiple tumor models[151]. Additionally, IDO inhibitors such as epacadostat exhibit objective single-agent responses in certain tumor types[152].
Combination therapies appear to enhance efficacy significantly. For instance, the glutamine antagonist JHU083 combined with ICIs [anti–PD-1/anti–cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)] strongly enhances antitumor immunity, even eliciting responses in immunotherapy-resistant models[77]. Similarly, the FASN inhibitor TVB-2640 shows synergistic effects when co-administered with paclitaxel[15]. There is also growing consensus that simultaneously targeting multiple metabolic pathways - such as both glycolysis and glutamine metabolism - may yield greater therapeutic benefits[77].
Biomarkers in predicting and evaluating metabolic therapy response
Biomarkers play an essential role in predicting and evaluating treatment responses. KEAP1/NRF2 mutation status may serve as a predictive biomarker for GLS inhibitor efficacy[99]. Expression levels of metabolic enzymes such as IDO and GLS can also help identify patients likely to respond to corresponding inhibitors[99,154]. Furthermore, radiographic evaluations such as fluorodeoxyglucose positron emission tomography (FDG-PET) provide a non-invasive means of monitoring the response to glycolysis-targeted therapies[32].
CONCLUSION
This review outlines the key mechanisms of metabolic reprogramming in HCC and their impact on the TME, highlighting therapeutic opportunities based on metabolic targeting. Metabolic rewiring - characterized by enhanced aerobic glycolysis, glutamine dependency, and dysregulated lipid metabolism - not only supports tumor proliferation but also establishes an immunosuppressive TME through hypoxia, acidosis, and nutrient competition. In addition to their metabolic roles, key enzymes exert non-metabolic regulatory functions that further promote tumor progression. These alterations collectively drive metabolic–immune crosstalk, impairing antitumor immunity and facilitating disease progression.
Despite strong preclinical evidence, clinical translation remains limited due to metabolic heterogeneity, adaptive rewiring, systemic toxicity, and the lack of robust predictive biomarkers. Future efforts should focus on precision-based strategies, including biomarker-guided patient stratification, multi-omics–driven metabolic profiling, rational combination therapies, and advanced drug delivery systems to enhance efficacy and specificity. Overall, integrating metabolic insights with translational innovation may provide new avenues to improve therapeutic outcomes in HCC.
DECLARATIONS
Authors’ contributions
Conceived and designed the review: Ruan Z, Sun X
Drafted the manuscript: Ruan Z
Revised the manuscript: Sun X
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This study was supported by the National Natural Science Foundation of China (Grant Nos. 32541002 and 32570636).
Conflicts of interest
Both authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Chan YT, Zhang C, Wu J, et al. Biomarkers for diagnosis and therapeutic options in hepatocellular carcinoma. Mol Cancer. 2024;23:189.
2. Allemani C, Matsuda T, Di Carlo V, et al.; CONCORD Working Group. Global surveillance of trends in cancer survival 2000-14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet. 2018;391:1023-75.
3. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229-63.
4. Chan SL, Sun HC, Xu Y, et al. The Lancet Commission on addressing the global hepatocellular carcinoma burden: comprehensive strategies from prevention to treatment. Lancet. 2025;406:731-78.
5. Rumgay H, Ferlay J, de Martel C, et al. Global, regional and national burden of primary liver cancer by subtype. Eur J Cancer. 2022;161:108-18.
6. Al Noshokaty T, Mesbah N, Abo-Elmatty D, Abulsoud A, Abdel-Hamed A. Hepatocellular carcinoma pathogenesis: epigenetics and relationship with cancer hallmarks. Rec Pharm Biomed Sci. 2022;6:136-57.
7. Hytiroglou P, Bioulac-Sage P, Theise ND, Sempoux C. Etiology, pathogenesis, diagnosis, and practical implications of hepatocellular neoplasms. Cancers. 2022;14:3670.
8. Mao JJ, Pillai GG, Andrade CJ, et al. Integrative oncology: addressing the global challenges of cancer prevention and treatment. CA Cancer J Clin. 2022;72:144-64.
9. Federico P, Petrillo A, Giordano P, et al. Immune checkpoint inhibitors in hepatocellular carcinoma: current status and novel perspectives. Cancers. 2020;12:3025.
10. Leowattana W, Leowattana T, Leowattana P. Systemic treatment for unresectable hepatocellular carcinoma. World J Gastroenterol. 2023;29:1551-68.
11. Sankar K, Gong J, Osipov A, et al. Recent advances in the management of hepatocellular carcinoma. Clin Mol Hepatol. 2024;30:1-15.
12. Keenan BP, Fong L, Kelley RK. Immunotherapy in hepatocellular carcinoma: the complex interface between inflammation, fibrosis, and the immune response. J Immunother Cancer. 2019;7:267.
13. Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2:881-98.
14. Han J, Li Q, Chen Y, Yang Y. Recent metabolomics analysis in tumor metabolism reprogramming. Front Mol Biosci. 2021;8:763902.
15. Lin J, Qin J, Jiang L. Progress in metabolism of the immune cells in tumor microenvironment. J Shanghai Jiaotong Univ. 2022;42:1122-30.
16. Foglia B, Beltrà M, Sutti S, Cannito S. Metabolic reprogramming of HCC: a new microenvironment for immune responses. Int J Mol Sci. 2023;24:7463.
17. Hu N, Li H, Tao C, Xiao T, Rong W. The role of metabolic reprogramming in the tumor immune microenvironment: mechanisms and opportunities for immunotherapy in hepatocellular carcinoma. Int J Mol Sci. 2024;25:5584.
18. Yang F, Hilakivi-Clarke L, Shaha A, et al. Metabolic reprogramming and its clinical implication for liver cancer. Hepatology. 2023;78:1602-24.
19. Liu Q, Zhang X, Qi J, et al. Comprehensive profiling of lipid metabolic reprogramming expands precision medicine for HCC. Hepatology. 2025;81:1164-80.
20. Gao B, Lu Y, Lai X, et al. Metabolic reprogramming in hepatocellular carcinoma: mechanisms of immune evasion and therapeutic implications. Front Immunol. 2025;16:1592837.
21. Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10:267-77.
22. Guerra L, Bonetti L, Brenner D. Metabolic modulation of immunity: a new concept in cancer immunotherapy. Cell Rep. 2020;32:107848.
23. Shen KY, Zhu Y, Xie SZ, Qin LX. Immunosuppressive tumor microenvironment and immunotherapy of hepatocellular carcinoma: current status and prospectives. J Hematol Oncol. 2024;17:25.
24. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11-20.
25. Elia I, Haigis MC. Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat Metab. 2021;3:21-32.
26. Oura K, Morishita A, Tani J, Masaki T. Tumor immune microenvironment and immunosuppressive therapy in hepatocellular carcinoma: a review. Int J Mol Sci. 2021;22:5801.
27. Lv B, Wang Y, Ma D, et al. Immunotherapy: reshape the tumor immune microenvironment. Front Immunol. 2022;13:844142.
28. Fu T, Dai LJ, Wu SY, et al. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J Hematol Oncol. 2021;14:98.
30. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297-308.
31. Chang CH, Qiu J, O’Sullivan D, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229-41.
32. Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211-8.
33. Vaupel P, Multhoff G. Revisiting the Warburg effect: historical dogma versus current understanding. J Physiol. 2021;599:1745-57.
34. Deng X, Wang Q, Cheng M, et al. Pyruvate dehydrogenase kinase 1 interferes with glucose metabolism reprogramming and mitochondrial quality control to aggravate stress damage in cancer. J Cancer. 2020;11:962-73.
35. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325-37.
36. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441-64.
37. Bouillaud F, Hammad N, Schwartz L. Warburg effect, glutamine, succinate, alanine, when oxygen matters. Biology. 2021;10:1000.
38. Zhang D, Tang Z, Huang H, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575-80.
39. Zhang D, Gao J, Zhu Z, et al. Lysine L-lactylation is the dominant lactylation isomer induced by glycolysis. Nat Chem Biol. 2025;21:91-9.
40. Rho H, Hay N. Protein lactylation in cancer: mechanisms and potential therapeutic implications. Exp Mol Med. 2025;57:545-53.
41. Feng F, Wu J, Chi Q, et al. Lactylome analysis unveils lactylation-dependent mechanisms of stemness remodeling in the liver cancer stem cells. Adv Sci. 2024;11:e2405975.
42. Cai J, Zhang P, Cai Y, et al. Lactylation-driven NUPR1 promotes immunosuppression of tumor-infiltrating macrophages in hepatocellular carcinoma. Adv Sci. 2025;12:e2413095.
43. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029-33.
44. Epstein T, Xu L, Gillies RJ, Gatenby RA. Separation of metabolic supply and demand: aerobic glycolysis as a normal physiological response to fluctuating energetic demands in the membrane. Cancer Metab. 2014;2:7.
45. Inigo M, Deja S, Burgess SC. Ins and outs of the TCA cycle: the central role of anaplerosis. Annu Rev Nutr. 2021;41:19-47.
46. Yang H, Li J, Niu Y, et al. Interactions between the metabolic reprogramming of liver cancer and tumor microenvironment. Front Immunol. 2025;16:1494788.
47. DeBerardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345-50.
48. Eniafe J, Jiang S. The functional roles of TCA cycle metabolites in cancer. Oncogene. 2021;40:3351-63.
49. Ghisletti S, Russo M. TCA-cycle metabolites in the nucleus: drivers of chromatin and epigenetic control. BMC Biol. 2025;23:316.
50. Li W, Long Q, Wu H, et al. Nuclear localization of mitochondrial TCA cycle enzymes modulates pluripotency via histone acetylation. Nat Commun. 2022;13:7414.
51. Morin A, Goncalves J, Moog S, et al. TET-mediated hypermethylation primes SDH-deficient cells for HIF2α-driven mesenchymal transition. Cell Rep. 2020;30:4551-66.e7.
52. Wang Z, Liu Z, Lv M, Luan Z, Li T, Hu J. Novel histone modifications and liver cancer: emerging frontiers in epigenetic regulation. Clin Epigenetics. 2025;17:30.
53. Yuan T, Zhou T, Qian M, et al. SDHA/B reduction promotes hepatocellular carcinoma by facilitating the deNEDDylation of cullin1 and stabilizing YAP/TAZ. Hepatology. 2023;78:103-19.
54. Verma A, Lindroth AM. The emerging intertwined activities of metabolism and epigenetics unveils culprits and prospects in cancer. Exp Mol Med. 2025;57:1928-39.
55. Fernández-Barrena MG, Arechederra M, Colyn L, Berasain C, Avila MA. Epigenetics in hepatocellular carcinoma development and therapy: the tip of the iceberg. JHEP Rep. 2020;2:100167.
56. Wang P, Chen LL, Xiong Y, Ye D. Metabolite regulation of epigenetics in cancer. Cell Rep. 2024;43:114815.
57. Zhao S, Torres A, Henry RA, et al. ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep. 2016;17:1037-52.
58. Cai Q, Zhu H, Dai Y, Zhou Q, Zhang Q, Zhu Q. ATP citrate lyase promotes the progression of hepatocellular carcinoma by activating the REGγ-proteasome pathway. Mol Carcinog. 2024;63:1874-91.
59. Petillo A, Abruzzese V, Koshal P, Ostuni A, Bisaccia F. Extracellular citrate is a Trojan Horse for cancer cells. Front Mol Biosci. 2020;7:593866.
60. Wang G, Han JJ. Connections between metabolism and epigenetic modifications in cancer. Med Rev. 2021;1:199-221.
61. Yang C, Ko B, Hensley CT, et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell. 2014;56:414-24.
62. Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. 2002;277:30409-12.
63. Ying M, You D, Zhu X, Cai L, Zeng S, Hu X. Lactate and glutamine support NADPH generation in cancer cells under glucose deprived conditions. Redox Biol. 2021;46:102065.
64. Wang R, Cao L, Thorne RF, et al. LncRNA GIRGL drives CAPRIN1-mediated phase separation to suppress glutaminase-1 translation under glutamine deprivation. Sci Adv. 2021;7:eabe5708.
65. Huang Y, Meng F, Zeng T, et al. IFRD1 promotes tumor cells “low-cost” survival under glutamine starvation via inhibiting histone H1.0 nucleophagy. Cell Discov. 2024;10:57.
66. Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res. 2013;52:585-9.
67. Ray U, Roy SS. Aberrant lipid metabolism in cancer cells - the role of oncolipid-activated signaling. FEBS J. 2018;285:432-43.
68. Kuhajda FP, Pizer ES, Li JN, Mani NS, Frehywot GL, Townsend CA. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc Natl Acad Sci U S A. 2000;97:3450-4.
69. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153-61.
70. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763-77.
71. Rysman E, Brusselmans K, Scheys K, et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010;70:8117-26.
72. Liu Z, Liu W, Wang W, et al. CPT1A-mediated fatty acid oxidation confers cancer cell resistance to immune-mediated cytolytic killing. Proc Natl Acad Sci U S A. 2023;120:e2302878120.
73. Patsoukis N, Bardhan K, Chatterjee P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692.
74. Yu TCY, Tsui YM, Zhang VX, Ma H, Ng IOL. Lipid metabolism and immune response in hepatocellular carcinoma: interplay driving tumor progression. Cancer Heterog Plast. 2025;2:0006.
75. Ren M, Xu H, Xia H, Tang Q, Bi F. Simultaneously targeting SOAT1 and CPT1A ameliorates hepatocellular carcinoma by disrupting lipid homeostasis. Cell Death Discov. 2021;7:125.
76. Sangineto M, Villani R, Cavallone F, Romano A, Loizzi D, Serviddio G. Lipid metabolism in development and progression of hepatocellular carcinoma. Cancers. 2020;12:1419.
77. Oh MH, Sun IH, Zhao L, et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest. 2020;130:3865-84.
78. Zhang C, Yue C, Herrmann A, et al. STAT3 activation-induced fatty acid oxidation in CD8+ T effector cells is critical for obesity-promoted breast tumor growth. Cell Metab. 2020;31:148-61.e5.
79. Ma X, Xiao L, Liu L, et al. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33:1001-12.e5.
80. Brown TP, Ganapathy V. Lactate/GPR81 signaling and proton motive force in cancer: role in angiogenesis, immune escape, nutrition, and Warburg phenomenon. Pharmacol Ther. 2020;206:107451.
81. Hossain F, Al-Khami AA, Wyczechowska D, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. 2015;3:1236-47.
82. Wu J, Xue R, Jiang RT, Meng QH. Characterization of metabolic landscape in hepatocellular carcinoma. World J Gastrointest Oncol. 2021;13:1144-56.
83. Yang M, McKay D, Pollard JW, Lewis CE. Diverse functions of macrophages in different tumor microenvironments. Cancer Res. 2018;78:5492-503.
84. Lu M, Zhu WW, Wang X, et al. ACOT12-dependent alteration of acetyl-CoA drives hepatocellular carcinoma metastasis by epigenetic induction of epithelial-mesenchymal transition. Cell Metab. 2019;29:886-900.e5.
85. Hamabe A, Konno M, Tanuma N, et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc Natl Acad Sci U S A. 2014;111:15526-31.
86. Lee JW, Bae SH, Jeong JW, Kim SH, Kim KW. Hypoxia-inducible factor (HIF-1)alpha: its protein stability and biological functions. Exp Mol Med. 2004;36:1-12.
87. Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230-3.
88. Colegio OR, Chu NQ, Szabo AL, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559-63.
89. Lee KY, D’Acquisto F, Hayden MS, Shim JH, Ghosh S. PDK1 nucleates T cell receptor-induced signaling complex for NF-kappaB activation. Science. 2005;308:114-8.
90. Sun X, He L, Liu H, et al. The diapause-like colorectal cancer cells induced by SMC4 attenuation are characterized by low proliferation and chemotherapy insensitivity. Cell Metab. 2023;35:1563-79.e8.
91. Sen N, Hara MR, Kornberg MD, et al. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat Cell Biol. 2008;10:866-73.
92. Hon KW, Naidu R. Unveiling metabolic signatures as potential biomarkers in common cancers: insights from lung, breast, colorectal, liver, and gastric tumours. Biomolecules. 2025;15:1376.
93. Yalcin A, Clem BF, Imbert-Fernandez Y, et al. 6-Phosphofructo-2-kinase (PFKFB3) promotes cell cycle progression and suppresses apoptosis via Cdk1-mediated phosphorylation of p27. Cell Death Dis. 2014;5:e1337.
94. De Los Santos-Jiménez J, Campos-Sandoval JA, Alonso FJ, Márquez J, Matés JM. GLS and GLS2 glutaminase isoenzymes in the antioxidant system of cancer cells. Antioxidants. 2024;13:745.
95. Soro-Arnáiz I, Fitzgerald G, Cherkaoui S, et al. GLUD1 determines murine muscle stem cell fate by controlling mitochondrial glutamate levels. Dev Cell. 2024;59:2850-65.e8.
96. Hao Y, Yi Q, XiaoWu X, WeiBo C, GuangChen Z, XueMin C. Acetyl-CoA: an interplay between metabolism and epigenetics in cancer. Front Mol Med. 2022;2:1044585.
97. Lopes C, Pereira C, Medeiros R. ASCT2 and LAT1 contribution to the hallmarks of cancer: from a molecular perspective to clinical translation. Cancers. 2021;13:203.
98. Zhao L, Kang M, Liu X, et al. UBR7 inhibits HCC tumorigenesis by targeting Keap1/Nrf2/Bach1/HK2 and glycolysis. J Exp Clin Cancer Res. 2022;41:330.
99. Romero R, Sayin VI, Davidson SM, et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med. 2017;23:1362-8.
100. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739-44.
101. Nicklin P, Bergman P, Zhang B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521-34.
102. Wahi K, Holst J. ASCT2: a potential cancer drug target. Expert Opin Ther Targets. 2019;23:555-8.
103. Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016;27:599-608.
104. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619-34.
105. Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, Haigis MC. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science. 2017;358:941-46.
106. Liu J, Shen Y, Liu J, et al. Lipogenic enzyme FASN promotes mutant p53 accumulation and gain-of-function through palmitoylation. Nat Commun. 2025;16:1762.
107. Jin Q, Yuan LX, Boulbes D, et al. Fatty acid synthase phosphorylation: a novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 2010;12:R96.
108. Menendez JA, Vellon L, Mehmi I, et al. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc Natl Acad Sci U S A. 2004;101:10715-20.
109. Zhang H, Chen H, Yin S, et al. Docosahexaenoic acid reverses PD-L1-mediated immune suppression by accelerating its ubiquitin-proteasome degradation. J Nutr Biochem. 2023;112:109186.
110. Chen G, Bao B, Cheng Y, et al. Acetyl-CoA metabolism as a therapeutic target for cancer. Biomed Pharmacother. 2023;168:115741.
111. Gu L, Surolia R, Larson-Casey JL, et al. Targeting Cpt1a-Bcl-2 interaction modulates apoptosis resistance and fibrotic remodeling. Cell Death Differ. 2022;29:118-32.
112. Yu X, Wu H, Su J, et al. Acetyl-CoA metabolism maintains histone acetylation for syncytialization of human placental trophoblast stem cells. Cell Stem Cell. 2024;31:1280-97.e7.
113. Icard P, Wu Z, Fournel L, Coquerel A, Lincet H, Alifano M. ATP citrate lyase: a central metabolic enzyme in cancer. Cancer Lett. 2020;471:125-34.
114. Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. 2020;31:62-76.
115. Bian X, Liu R, Meng Y, Xing D, Xu D, Lu Z. Lipid metabolism and cancer. J Exp Med. 2021;218:e20201606.
116. Chen XF, Tian MX, Sun RQ, et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018;19:e45124.
117. Ding X, Shao L, Wang J, Jin Y, Chen H, Li B. HADHA promotes esophageal cancer progression by activating mTOR signaling and the SP1/MDM2 axis. Acta Biochim Biophys Sin. 2024;57:378-88.
118. Ye J, Lin Y, Liao Z, et al. Single cell-spatial transcriptomics and bulk multi-omics analysis of heterogeneity and ecosystems in hepatocellular carcinoma. NPJ Precis Oncol. 2024;8:262.
119. Li Y, Huan C, Sun H, et al. Spatial transcriptomics and snRNA-seq expose CAF niches orchestrating dual stromal-immune barriers in hepatocellular carcinoma. Adv Sci. 2025;12:e14661.
120. Tong W, Wang T, Bai Y, et al. Spatial transcriptomics reveals tumor-derived SPP1 induces fibroblast chemotaxis and activation in the hepatocellular carcinoma microenvironment. J Transl Med. 2024;22:840.
121. Liu Y, Xun Z, Ma K, et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J Hepatol. 2023;78:770-82.
122. Fan G, Xie T, Li L, Tang L, Han X, Shi Y. Single-cell and spatial analyses revealed the co-location of cancer stem cells and SPP1+ macrophage in hypoxic region that determines the poor prognosis in hepatocellular carcinoma. NPJ Precis Oncol. 2024;8:75.
123. Su R, Du Y, Tian P, Ma W, Hui Y, Yang S. Single-cell and spatial transcriptomics reveal correlation between RNA methylation-related miRNA risk model and immune infiltration in hepatocellular carcinoma. Front Oncol. 2025;15:1553239.
124. Xu WX, Ye YM, Chen JL, et al. SPP1+ tumor-associated macrophages define a high-risk subgroup and inform personalized therapy in hepatocellular carcinoma. Front Oncol. 2025;15:1606195.
125. Wang Y, Liu T, Tang W, et al. Hepatocellular carcinoma cells induce regulatory T cells and lead to poor prognosis via production of transforming growth factor-β1. Cell Physiol Biochem. 2016;38:306-18.
126. Gu J, Zhou J, Chen Q, et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-β signaling in regulatory T cells. Cell Rep. 2022;39:110986.
127. Zhou Y, Lin F, Wan T, et al. ZEB1 enhances Warburg effect to facilitate tumorigenesis and metastasis of HCC by transcriptionally activating PFKM. Theranostics. 2021;11:5926-38.
128. Kumagai S, Koyama S, Itahashi K, et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. 2022;40:201-18.e9.
129. Chivite-Lacaba M, Justo I, Utrero-Rico A, et al. Delineation of monocytic and early-stage myeloid-derived suppressor cells in the peripheral blood of patients with hepatocarcinoma. Int J Cancer. 2025;156:2416-28.
130. Iwata T, Kondo Y, Kimura O, et al. PD-L1+MDSCs are increased in HCC patients and induced by soluble factor in the tumor microenvironment. Sci Rep. 2016;6:39296.
131. Hoechst B, Ormandy LA, Ballmaier M, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4+CD25+Foxp3+ T cells. Gastroenterology. 2008;135:234-43.
132. Yang X, Lu Y, Hang J, et al. Lactate-modulated immunosuppression of myeloid-derived suppressor cells contributes to the radioresistance of pancreatic cancer. Cancer Immunol Res. 2020;8:1440-51.
133. Wang H, Zhou F, Qin W, Yang Y, Li X, Liu R. Metabolic regulation of myeloid-derived suppressor cells in tumor immune microenvironment: targets and therapeutic strategies. Theranostics. 2025;15:2159-84.
134. Yang Y, Li S, To KKW, Zhu S, Wang F, Fu L. Tumor-associated macrophages remodel the suppressive tumor immune microenvironment and targeted therapy for immunotherapy. J Exp Clin Cancer Res. 2025;44:145.
135. Huang D, Li T, Wang L, et al. Hepatocellular carcinoma redirects to ketolysis for progression under nutrition deprivation stress. Cell Res. 2016;26:1112-30.
136. Chang CH, Curtis JD, Maggi LB Jr, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239-51.
137. Elia I, Rowe JH, Johnson S, et al. Tumor cells dictate anti-tumor immune responses by altering pyruvate utilization and succinate signaling in CD8+ T cells. Cell Metab. 2022;34:1137-50.e6.
138. Xia C, Yin S, To KKW, Fu L. CD39/CD73/A2AR pathway and cancer immunotherapy. Mol Cancer. 2023;22:44.
139. Xu S, Chaudhary O, Rodríguez-Morales P, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity. 2021;54:1561-77.e7.
140. Guo C, You Z, Shi H, et al. SLC38A2 and glutamine signalling in cDC1s dictate anti-tumour immunity. Nature. 2023;620:200-8.
141. Pallotta MT, Rossini S, Suvieri C, et al. Indoleamine 2,3-dioxygenase 1 (IDO1): an up-to-date overview of an eclectic immunoregulatory enzyme. FEBS J. 2022;289:6099-118.
142. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161:1527-38.
143. Veglia F, Tyurin VA, Mohammadyani D, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun. 2017;8:2122.
144. Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541-50.
145. Xiang H, Yang R, Tu J, et al. Metabolic reprogramming of immune cells in pancreatic cancer progression. Biomed Pharmacother. 2023;157:113992.
146. Xie Z, Huang J, Li Y, et al. Single-cell RNA sequencing revealed potential targets for immunotherapy studies in hepatocellular carcinoma. Sci Rep. 2023;13:18799.
147. Wang K, Li X, Guo S, et al. Metabolic reprogramming of glucose: the metabolic basis for the occurrence and development of hepatocellular carcinoma. Front Oncol. 2025;15:1545086.
148. Wang JX, Choi SYC, Niu X, et al. Lactic acid and an acidic tumor microenvironment suppress anticancer immunity. Int J Mol Sci. 2020;21:8363.
149. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238-42.
150. Quail DF, Joyce JA. Molecular pathways: deciphering mechanisms of resistance to macrophage-targeted therapies. Clin Cancer Res. 2017;23:876-84.
151. Kouidhi S, Ben Ayed F, Benammar Elgaaied A. Targeting tumor metabolism: a new challenge to improve immunotherapy. Front Immunol. 2018;9:353.
152. Yang L, Chu Z, Liu M, et al. Amino acid metabolism in immune cells: essential regulators of the effector functions, and promising opportunities to enhance cancer immunotherapy. J Hematol Oncol. 2023;16:59.
153. Morchón-Araujo D, Catani G, Mirallas O, et al. Emerging immunotherapy targets in early drug development. Int J Mol Sci. 2025;26:5394.
154. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27-47.
155. Jin H, Wu P, Lv C, et al. Mannose inhibits PKM2 lactylation to induce pyroptosis in bladder cancer and activate antitumor immune responses. Commun Biol. 2025;8:689.
156. Stein M, Lin H, Jeyamohan C, et al. Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate. 2010;70:1388-94.
157. Wang R, Zhuang J, Zhang Q, et al. Decoding the metabolic dialogue in the tumor microenvironment: from immune suppression to precision cancer therapies. Exp Hematol Oncol. 2025;14:99.
158. Escutia-Gutiérrez R, Reyes-Esparza J, Rodriguez-Fragoso L. Effect of gallic acid on preneoplastic liver lesions in vivo. FASEB J. 2016;30:1268.7.
159. Li P, Song Y, Lv L, et al. The role of gallic acid in liver disease: a review of its phytochemistry, pharmacology, and safety. Front Pharmacol. 2025;16:1595508.
160. Woodford MR, Chen VZ, Backe SJ, Bratslavsky G, Mollapour M. Structural and functional regulation of lactate dehydrogenase-A in cancer. Future Med Chem. 2020;12:439-55.
161. Benyahia Z, Blackman MCNM, Hamelin L, et al. In vitro and in vivo characterization of MCT1 inhibitor AZD3965 confirms preclinical safety compatible with breast cancer treatment. Cancers. 2021;13:569.
162. Noble RA, Bell N, Blair H, et al. Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica. 2017;102:1247-57.
163. Gouda MA, Voss MH, Tawbi H, et al. A phase I/II study of the safety and efficacy of telaglenastat (CB-839) in combination with nivolumab in patients with metastatic melanoma, renal cell carcinoma, and non-small-cell lung cancer. ESMO Open. 2025;10:104536.
164. Chen Y, Zhao Y, Bajor DL, Wang Z, Selfridge JE. A facile and sensitive method of quantifying glutaminase binding to its inhibitor CB-839 in tissues. J Genet Genomics. 2020;47:389-95.
165. Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer. 2017;3:169-80.
166. Zhang Y, Kumata K, Xie L, et al. The glutaminase-1 inhibitor [11C-carbony]BPTES: synthesis and positron emission tomography study in mice. Pharmaceuticals. 2023;16:963.
167. Cervantes-Madrid D, Romero Y, Dueñas-González A. Reviving lonidamine and 6-diazo-5-oxo-L-norleucine to be used in combination for metabolic cancer therapy. Biomed Res Int. 2015;2015:690492.
168. Bell BJ, Hollinger KR, Deme P, et al. Glutamine antagonist JHU083 improves psychosocial behavior and sleep deficits in EcoHIV-infected mice. Brain Behav Immun Health. 2022;23:100478.
169. Falchook G, Infante J, Arkenau HT, et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine. 2021;34:100797.
170. Chen S, Tan J, Zhang A. The ups, downs and new trends of IDO1 inhibitors. Bioorg Chem. 2021;110:104815.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].