


Immune microenvironment and immunotherapy strategies in MAFLD-related hepatocellular carcinoma
 0
0 Abstract
Driven by the global pandemic of obesity and metabolic syndrome, metabolic-associated fatty liver disease (MAFLD) has emerged as a principal driver of hepatocellular carcinoma (HCC). MAFLD-related HCC (MAFLD-HCC) exhibits distinct pathological features and a unique immune microenvironment, which collectively contribute to its poorer response to immune checkpoint inhibitors (ICIs) compared to HCC of other etiologies. This review systematically outlines the immune microenvironment in MAFLD-HCC, focusing on the dynamic changes and interactions among dysfunctional CD8+ T cells, regulatory T cells, myeloid cells, and B cells, as well as their crosstalk with metabolic reprogramming. These changes collectively result in a highly immunosuppressive tumor microenvironment, leading to resistance to ICIs. Addressing the challenges in treating MAFLD-HCC, this paper discusses the current progress in immunotherapy for MAFLD-HCC and explores potential directions for future immunotherapy research.
Keywords
INTRODUCTION
Liver cancer is a major global health burden, currently ranking as the sixth most common cancer, and the third leading cause of cancer-related deaths[1]. Hepatocellular carcinoma (HCC) is the main histological subtype of liver cancer, accounting for approximately 80% of global cases[2]. It is a highly fatal and difficult-to-treat malignancy, with a five-year survival rate generally ranging from 5% to 30%[3]. The occurrence of HCC is closely associated with chronic liver disease, with major risk factors including chronic infection with hepatitis B virus (HBV) or hepatitis C virus (HCV), alcohol-related liver disease (ALD), and metabolic-associated fatty liver disease (MAFLD)[4,5].
In recent years, the nomenclature and diagnostic framework for fatty liver disease have evolved to emphasize metabolic dysfunction as the core driver. The term MAFLD was proposed in 2020 to emphasize metabolic dysfunction as central to disease classification[6]. More recently, a multi-society Delphi process reframed the condition within the broader category of steatotic liver disease (SLD) and introduced the term metabolic dysfunction-associated SLD (MASLD) to replace “non-alcoholic fatty liver disease (NAFLD)” in that consensus taxonomy[7]. Importantly, MAFLD and MASLD largely overlap in practice: the diagnostic criteria are substantially congruent, and most patients meeting earlier NAFLD/MAFLD definitions also meet MASLD criteria, although the specific wording and the minimal metabolic requirements differ slightly between definitions (MAFLD: steatosis + either overweight/obesity, type 2 diabetes mellitus (T2DM), or metabolic dysregulation; MASLD: steatosis + ≥ 1 of five classical metabolic syndrome-related risk factors)[8-10]. The conceptual emphasis of both frameworks shifts from alcohol exclusion to metabolic dysfunction as the unifying pathophysiology. Accordingly, MAFLD is employed as the principal term herein.
Driven by the global prevalence of obesity, type 2 diabetes, and metabolic syndrome, MAFLD has become the most common form of chronic liver disease[11], affecting approximately 24% of adults. Its incidence has nearly doubled over the past decade[12]. This trend has directly fueled a rapid increase in non-viral, non-alcoholic etiologies among HCC cases[11]. The progressive form of MAFLD - metabolic-associated steatohepatitis (MASH) - is now the fastest-growing cause of HCC worldwide. Predictive models project that the proportion of liver cancer cases associated with MASH will increase from 8% in 2022 to 11% in 2050. Conversely, the proportions of HBV- and HCV-related cases are projected to decline from 39% to 37% and from 29% to 26%, respectively, over the same period[13]. Compared to viral HCC, MAFLD-related HCC (MAFLD-HCC) exhibits distinct pathological features, notably the occurrence of 20%-50% of cases without underlying cirrhosis. These patients often face a poorer prognosis due to delayed diagnosis[12].
MAFLD encompasses a spectrum of liver damage, progressing from simple steatosis to MASH and potentially to cirrhosis and HCC. This progression is driven by core pathological mechanisms, including lipotoxicity, insulin resistance, and oxidative stress. Specifically, disturbances in hepatic metabolism lead to the accumulation of cytotoxic lipids, which, synergistically with endoplasmic reticulum stress (ERS), promote hepatocyte death and trigger a critical driver of disease progression, necroeinflammation[14]. It is essential to recognize that MAFLD encompasses distinct pathological subtypes - the generally non-progressive metabolic-associated fatty liver (MAFL) and the progressive MASH, which is characterized by inflammation and specific histopathological hallmarks, such as hepatocyte ballooning. Emerging research further suggests that qualitative differences in steatosis, influenced by varying metabolite-lipid combinations, may impact the risk of transitioning to MASH, highlighting the complexity and heterogeneity of the disease[15]. Notably, a significant proportion of MAFLD-HCC arises without preceding cirrhosis, underscoring the distinct and potent oncogenic potential of this disease entity[16].
Immune checkpoint inhibitors (ICIs) and their combination strategies with anti-angiogenesis drugs (e.g., atezolizumab + bevacizumab) have become the first-line systemic treatment standard for unresectable or metastatic HCC, significantly improving overall survival (OS) and objective response rates (ORR), thus changing the treatment landscape of HCC. Despite the overall significant efficacy, increasing clinical subgroup analyses and real-world data suggest that HCCs with different etiologies (especially non-viral etiologies, such as MAFLD/MASH) may exhibit heterogeneity in response to ICI or ICI combination therapies[17]. Converging mechanistic and clinical evidence from crossover studies has uncovered a critical issue: the liver immune microenvironment in MAFLD/MASH may not only reduce ICI efficacy but also, in a paradoxical phenomenon observed in some preclinical models, lead to anti-programmed cell death 1 (PD-1) therapy to promote tumor progression[18]. This review aims to systematically outline the metabolic reprogramming mechanisms within the MAFLD-HCC immune microenvironment, elucidate the dynamic functional transitions of key immune cells [including T cells, B cells, myeloid cells, and natural killer T (NKT) cells], and discuss current advances in immunotherapy for this disease.
DISTINCT IMMUNE LANDSCAPE OF MAFLD-HCC
Immune cells orchestrate the progression of MAFLD and its transition to HCC. Within the complex MAFLD-HCC milieu, immune cells undergo substantial remodeling, resulting in a distinctive immunological landscape that distinguishes it from HCCs of other etiologies, such as viral hepatitis. This uniqueness is largely driven by the pervasive metabolic dysregulation - including lipotoxicity, cholesterol accumulation, and gut microbiota dysbiosis - which in turn fundamentally shapes the phenotypes and functions of immune cells. The functional states of key immune cells - including CD8+ T cells, regulatory T cells (Tregs), CD4+ T cells, natural killer (NK) cells, macrophages, dendritic cells (DCs), and neutrophils - critically influence both MAFLD-HCC pathogenesis and response to immunotherapy [Figure 1].
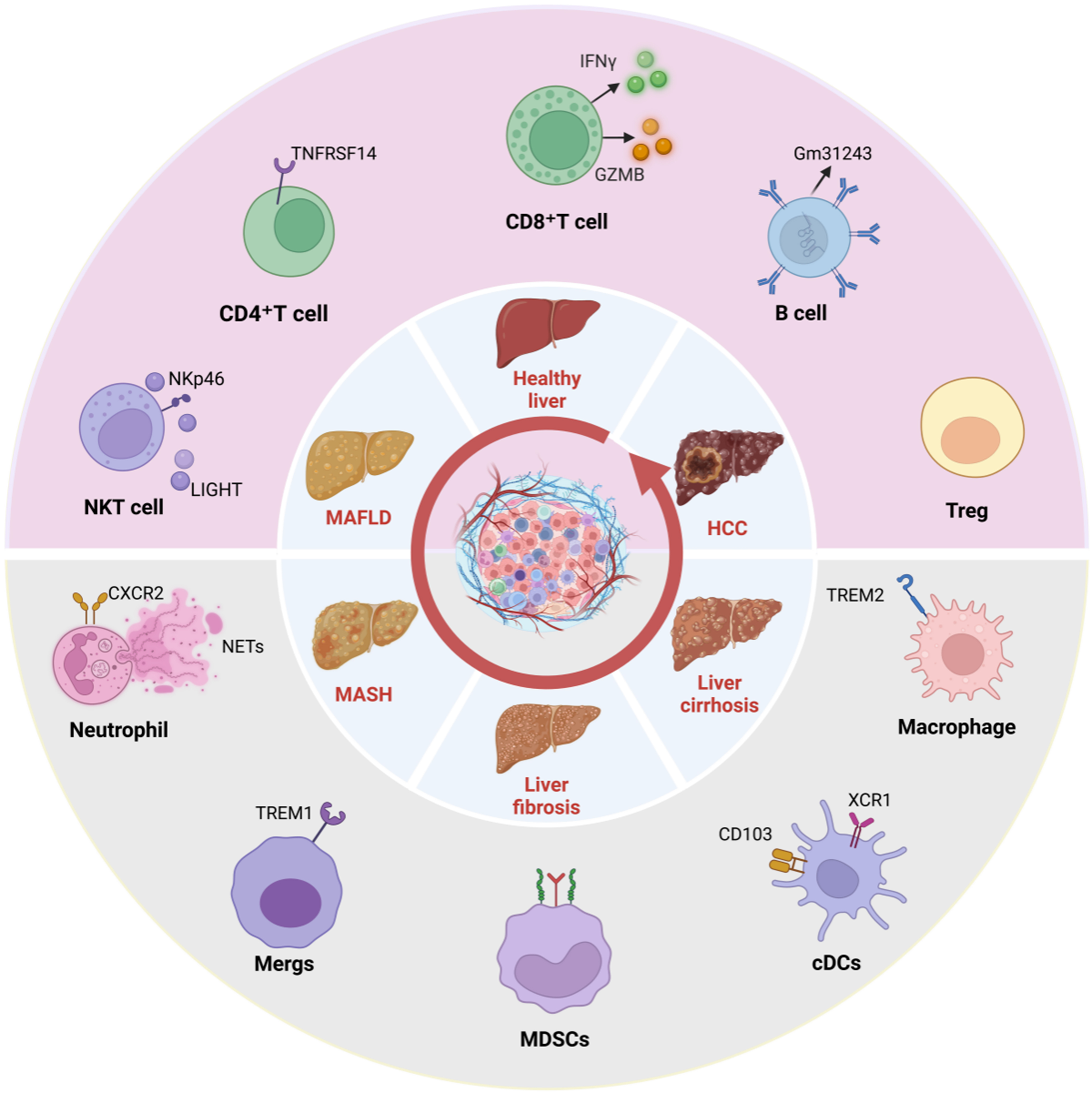
Figure 1. Disease progression of MAFLD and the immune landscape of associated HCC. Created in BioRender. Han, H. (2025) https://BioRender.com/qwm30hm. MAFLD: Metabolic-associated fatty liver disease; HCC: hepatocellular carcinoma; NKT: natural killer T; LIGHT: tumor necrosis factor superfamily member 14; CD4+: cluster of differentiation 4 positive; IFNγ: interferon gamma; GZMB: granzyme B; CD8+: cluster of differentiation 8 positive; Treg: regulatory T cell; CXCR2: C-X-C motif chemokine receptor 2; NETs: neutrophil extracellular traps; TREM1/2: triggering receptor expressed on myeloid cells 1/2; MDSCs: myeloid-derived suppressor cells; cDCs: conventional dendritic cells; XCR1: X-C motif chemokine receptor 1; MASH: metabolic-associated steatohepatitis.
The immune landscape of MAFLD-HCC is not merely a collection of altered cell populations, but a coherent ecosystem shaped by pervasive metabolic stress, including lipotoxicity, cholesterol accumulation, and gut-derived signals. Unlike the antigen-driven immunity characteristic of viral hepatitis, the MAFLD-HCC microenvironment is fundamentally organized around a paradoxical coexistence of chronic inflammation and profound immunosuppression. This duality creates a “false-hot” or immune-resistant tumor phenotype: while pro-inflammatory, often auto-aggressive immune cells operate in parallel with a powerfully orchestrated immunosuppressive network that actively restrains anti-tumor immunity, underlining the marked resistance to immunotherapy[19,20]. The following sections detail how individual lymphoid and myeloid cell types are metabolically reprogrammed and functionally polarized to contribute to this overarching immunopathological theme.
Lymphoid cells
CD8+ T cells
In the MAFLD-HCC tumor microenvironment, CD8+ T cells exhibit distinct functional characteristics and are pivotal drivers of tumor progression and immune escape. In mouse models of MAFLD, liver PD-1+CD8+ T cells are significantly increased and aberrantly co-express effector function markers [such as granzyme K (Gzmk) and C-C motif chemokine ligand 3 (Ccl3)] and exhaustion markers [such as programmed cell death 1 (Pdcd1) and thymocyte selection-associated high mobility group box (Tox)][19]. This contrasts with the more “classically exhausted” T cell phenotype often observed in chronic viral infections[21]. Among these cells in a mixed activation-exhaustion state, a specific CXCR6+PD-1+CD8+ (CXCR6: C-X-C motif chemokine receptor 6) subset drives the MASH-to-HCC transition by eliminating parenchymal cells in a major histocompatibility complex (MHC) class I-independent manner via the P2X purinoceptor 7 (P2X7) receptor signaling pathway[15,22]. In mice, the depletion of CD8+ T cells significantly reduces liver injury and HCC incidence[19,22]. Spatially, CD8+ T cell density progressively decreases from the tumor periphery to the core, with a sharp drop at the marginal zone (MZ), indicating progressive T cell exclusion from the tumor core as a key mechanism of immune escape[23]. CD8+ T cell dysfunction is further shaped by metabolic regulation, unique to the MAFLD milieu. Methyltransferase-like 3 (METTL3) promotes sterol regulatory element-binding protein cleavage-activating protein (SCAP)-mediated cholesterol biosynthesis via an N6-methyladenosine (m6A)-dependent mechanism, thereby suppressing granzyme B (GZMB) and interferon gamma (IFN-γ) production in intra-tumoral CD8+ T cells and facilitating immune escape[24]. Notably, despite the significant oncogenic role of the PD-1+CD8+ T cell subset in MAFLD-HCC transformation, anti-PD-1 therapy has poor efficacy in MAFLD-HCC and may even exacerbate tissue damage and fibrosis[18,23]. Mechanistically, PD-1/programmed death-ligand 1 (PD-L1) blockade overactivates the pathogenic CD8+PD-1+CXCR6+ population, providing an explanation for the clinical observation of therapy resistance despite substantial intra-tumoral T-cell infiltration[20,22].
CD4+ T cells
CD4+ T cells play a crucial role in both the development of MASH and its progression to HCC. In murine MASH models, type 1 T helper (Th1), cytotoxic CD4+ T cells, and Tregs are significantly enriched[25]. These cells actively participate in the inflammation and fibrosis processes associated with MAFLD, with different subpopulations having distinct roles in disease progression. The pro-MASH role of CD4+ T cells has been experimentally confirmed; the depletion of CD4+ T cells alleviates liver injury, inflammation, and fibrosis induced by a fructose-fat-cholesterol (FFC) diet without affecting systemic metabolic function. Functional experiments further reveal that pro-inflammatory cytokines, IFN-γ and tumor necrosis factor-alpha
Tregs
Tregs, a key subset of CD4+ T lymphocytes, maintain immune tolerance and homeostasis primarily through the secretion of immunosuppressive cytokines[29]. They exhibit a critical, stage-dependent duality throughout the MAFLD-HCC continuum. In the early stages of MAFLD, intrahepatic Treg numbers decrease, and their depletion exacerbates liver injury and steatosis, underscoring their initial anti-inflammatory role[30,31]. The inflammatory milieu, which includes signals such as Toll-like receptor 7 (TLR7) activation, can promote Treg apoptosis, thereby contributing to their numerical decline[31]. As disease progresses to fibrosis, Tregs exert complex and context-dependent effects, influenced by the balance of cytokines, such as the anti-fibrotic interleukin (IL)-10 and the pro-fibrotic transforming growth factor-β (TGF-β)[32-34]. In the late stages of MASH and HCC, a profound shift occurs: Tregs undergo a marked expansion within the liver[35]. This increase is driven by factors such as neutrophil extracellular traps (NETs) and the gut microbiota, which promote the differentiation and function of immunosuppressive Tregs (e.g., IL-10+), thereby fostering a pro-tumor microenvironment and reducing CD8+ T cell infiltration[35,36]. Consequently, elevated Treg levels in HCC patients correlate with tumor progression, primarily by suppressing CD8+ T cell cytotoxicity[36,37]. Collectively, this NET-driven expansion of Tregs constitutes a signature immunosuppressive mechanism in MAFLD-HCC, starkly contrasting with the immunobiology of viral- or alcohol-related HCC and underpinning its pronounced resistance to immunotherapy[35].
NKT
NKT cells in MAFLD-HCC exhibit a distinct immunometabolic profile that fundamentally differs from viral-related HCC. Unlike the virus-specific, antigen-driven T cell responses characteristic of hepatitis-associated HCC, NKT cell activation in MAFLD is primarily mediated by metabolic cues and occurs in an antigen-independent manner[15,38]. This metabolite-driven activation paradigm represents a hallmark feature of MAFLD-HCC immunopathology.
The role of NKT cells in MAFLD and HCC remains not fully understood, with their function potentially exhibiting duality due to different pathological states and immune microenvironments. Studies have found that NKT cells, by secreting the tumor necrosis factor superfamily member 14 (LIGHT) molecule, synergistically activate the lymphotoxin-β receptor (LTβR)/nuclear factor−kappaB (NF-κB) signaling axis in hepatocytes in cooperation with CD8+ T cells, promoting HCC transformation[39]. However, the activation receptor natural killer cell p46 (NKp46)/natural cytotoxicity triggering receptor 1 (NCR1) on NK cells mediates direct cytotoxicity against hepatic stellate cells (HSCs), inhibiting the progression of liver fibrosis[40]. On the other hand, cholesterol accumulation driven by the mechanistic target of rapamycin complex 1 (mTORC1)/sterol regulatory element-binding protein 2 (SREBP2) pathway can induce lipid peroxidation in NKT cells, thereby impairing their cytotoxicity and disrupting anti-tumor surveillance[41]. Furthermore, specific gut microbiota (e.g., Bacteroides vulgatus) can restore NKT cell function by inhibiting transforming growth factor-β1 (TGF-β1)/IL-10, thus slowing the progression of HCC[42]. Therefore, NKT cells may have both protective and detrimental effects on MAFLD, depending on the stage of the disease and the microenvironment. In the context of obesity, NKT cells may also activate the ERS pathway in hepatocytes by producing factors such as osteopontin (OPN), thereby promoting inflammation and metabolic abnormalities[43]. In summary, various T cell subsets regulate the progression of MAFLD-HCC through unique and interacting mechanisms, and their net effects and targeting potential still require further investigation.
Other T-cells
Gamma delta (γδ) T cells, as a bridge between innate and adaptive immunity, also play an important role in MAFLD and MAFLD-HCC. Studies have shown that in the livers of obese individuals, γδ T cells drive pathogenic inflammation through the sustained secretion of pro-inflammatory cytokines, including IL-17A, IFN-γ, and TNF-α[44,45]. This represents a fundamental shift from their role in viral hepatitis, where they mount antigen-specific responses; in MAFLD, γδT cell activation is driven by metabolic and sterile inflammatory signals[46]. Specifically, the expansion of the interleukin-17-producing T (γδT17) cell subset and its signature cytokine, IL-17A, can recruit granulocytes and induce ROS bursts, thereby exacerbating liver injury[47]. Furthermore, studies have demonstrated that liver natural killer group 2 member D (NKG2D) ligand expression induced by MASH diets further activates γδT cells to secrete IL-17A, thereby promoting fibrosis[48]. Collectively, these findings establish γδT17 cells as key promoters of MAFLD progression.
Tissue-resident memory T (TRM) cells are markedly expanded in the livers and adipose tissue of obese individuals. By secreting factors such as IL-1β and IL-2, TRM cells form self-perpetuating inflammatory circuits that accelerate the progression of MAFLD[49]. This antigen-independent, “bystander” activation mode, driven by the sterile inflammatory milieu of MAFLD, contrasts with the antigen-specific TRM responses typical of viral hepatitis[49]. Interestingly, CD69+CD103- CD8+ TRM cells have been found to potentially play an inhibitory role in liver fibrosis during MASH[50], revealing a unique functional duality of TRM cells in a metabolic context. The heterogeneity of TRM subpopulations (e.g., CD103+ vs. CD103-) and their crosstalk with the metabolic microenvironment (e.g., lipotoxicity, hypoxia) remain areas for future investigation.
Mucosal-associated invariant T (MAIT) cells constitute a highly abundant innate-like T cell population in the human liver, playing a pivotal role in the MAFLD immune microenvironment. Studies have shown that in MAFLD patients, MAIT cells exhibit a dysfunctional state, particularly with high expression of CD39[51]. The MR1-dependent activation pathway of MAIT cells can induce alternatively activated (M2) macrophage polarization, which helps improve the disease course[52]. On the other hand, by inducing classically activated (M1) macrophage polarization, MAIT cells can promote disease progression[53]. Recent studies have also found that the accumulation of polyunsaturated fatty acids (PUFAs) in the liver significantly impairs MAIT cell function by inhibiting T cell activation genes, downregulating IFN-γ/GZMB expression, and activating ferroptosis pathways[54]. These observations highlight the dual role of the liver metabolic environment in regulating MAIT cell function.
B cell
During the progression of MAFLD to HCC, B cells exhibit complex and context-dependent dual functions, influenced by subset heterogeneity and gut-liver axis interactions[55]. Notably, the B cell response in MAFLD-HCC is distinct from that in other etiologies such as viral hepatitis, being uniquely shaped by metabolic stress and gut dysbiosis[28]. In MASH-driven HCC, certain B cell subsets promote immunosuppression and tumor progression. For instance, immunosuppressive immunoglobulin A (IgA)+ B cells, induced via the IL-21R-signal transducer and activator of transcription 1 (STAT1) signaling axis, suppress the function of cytotoxic T cells[56]. Another subset, Gm31243+ B cells, expands during disease progression; the long non-coding RNA (lncRNA) Gm31243 is suggested to enhance their immunosuppressive function through metabolic reprogramming[57]. Furthermore, regulatory B cells (Bregs), including specific subsets identified by surface markers (e.g., CD19-B220+CD5+CD1d-), accumulate in the liver. They suppress immunity via PD-L1 expression and IL-10 secretion, and their depletion improves survival in HCC mice[58]. The gut-liver axis has a significant influence on B-cell activity. Gut B cells can be activated by metabolic factors, exacerbating T cell-driven inflammation and fibrosis in MASH; B cell depletion alleviates these effects[59]. Additionally, gut dysbiosis can synergistically activate Toll-like receptor–myeloid differentiation primary response 88 (TLR-MyD88) signaling and B cell receptor (BCR) signaling, driving the accumulation of pro-inflammatory B cells in the liver and exacerbating insulin resistance and fibrosis[60]. This gut-microbiota-driven B cell activation represents a distinctive feature of the MAFLD immune microenvironment.
Conversely, B cells can also exert anti-tumor effects. A distinct subset, termed tumor-associated atypical B cells (TAABs), highly expresses co-stimulatory molecules (e.g., CD80, CD86) and interacts strongly with CD8+ T cells, thereby providing immune protection in MAFLD-driven HCC[61]. The functional complexity is further highlighted by studies showing that B-cell-deficient mice exhibit only partial protection against MASH, whereas IgMi mice (which lack antibody secretion but retain B cells) show complete resistance, underscoring a crucial, antibody-independent role of B cells[55]. In summary, B cells play divergent roles in MAFLD-HCC, as they can promote disease through immunosuppressive and pro-inflammatory subsets or exert protection through specific activated subsets. Future research should further elucidate the mechanisms underlying this functional duality.
Myeloid cells
Macrophages
During MAFLD-HCC progression, bone marrow-derived macrophages, particularly MASH-associated macrophages (NAMs) with a core TREM2+APOE+GPNMB+ (TREM2: triggering receptor expressed on myeloid cells 2; APOE: apolipoprotein E; GPNMB: glycoprotein nonmetastatic melanoma protein B) phenotype, undergo dynamic reprogramming to promote disease progression and foster an immunosuppressive microenvironment[62]. Multiple factors regulate NAM dynamics and function. TGF-β signaling induces NAM formation and the secretion of pro-fibrotic factors, thereby accelerating liver fibrosis[63], whereas neuregulin 4 (NRG4) inhibits HCC development by depleting NAMs[62]. Histidine-rich glycoprotein (HRG) promotes the infiltration of NAMs, particularly TREM2+ macrophages, into both tumor and peri-tumoral tissues[64]. Hypoxia and hepatocyte-derived lipid droplets promote the polarization of macrophages towards immunosuppressive TREM2+ phenotypes[65,66]. Furthermore, deficiency of nuclear receptor coactivator 5 (Ncoa5) promotes M2-like macrophage differentiation via overexpression of platelet factor 4 (PF4)[67]. At the metabolic level, NAMs undergo reprogramming of lipid and oxidative phosphorylation, which sustains their immunosuppressive capacity and presents a potential therapeutic target[68]. Furthermore, deficiency of fibroblast growth factor 21 (FGF21) mediates the MASH-to-HCC transition by promoting the replacement of Kupffer cells with monocyte-derived macrophages via the sphingosine-1-phosphate (S1P)–Yes-associated protein (YAP) pathway[69]. Notably, although FGF21 has been shown to reverse MASH through modulation of the brain-liver axis[70], its specific role in regulating macrophages remains to be elucidated.
Macrophage function is also modulated by sex differences and the gut microbiota. In male mice, Kupffer cells drive inflammation, a process that is suppressed by estrogen in females[71,72]. Specific gut microbes, such as Akkermansia muciniphila (AKK) and Bifidobacterium bifidum, can inhibit pro-inflammatory macrophage polarization, thereby alleviating steatosis and inflammation[73,74]. Conversely, microbiota-derived glycocholic acid (GCA) activates macrophages via the Toll-like receptor 9 (TLR9)-NF-κB pathway, exacerbating liver injury through the secretion of pro-inflammatory cytokines[75]. This gut-liver axis represents a distinctive regulatory layer in MAFLD, absent in classic viral hepatitis. Therapeutic targeting of macrophage pathways shows considerable promise. Evidence from genetic models reveals key regulatory nodes: myeloid-specific deletion of activating transcription factor 3 (Atf3) exacerbates steatosis and fibrosis via the retinol-binding protein 4 (Rbp4)-stimulated by retinoic acid 6 (STRA6) axis[76], whereas deficiency of the trigger receptor 2 (Trem2) gene disrupts lipid homeostasis, establishing TREM2 as a core metabolic sensor[77]. Importantly, recent studies demonstrate that targeting TREM2 or NAMs enhances the efficacy of PD-1 checkpoint blockade, significantly improving anti-tumor immunity in preclinical models[78]. These findings, together with the recognized dual role of macrophages in both promoting disease and potentially mediating anti-tumor effects[79], underscore the importance of macrophage modulation as a compelling strategic direction for MAFLD-HCC immunotherapy.
Neutrophils
Neutrophils are the predominant myeloid cells in MASH and are significantly enriched in both tumor and adjacent tissues in MAFLD-HCC[35,80]. These cells exhibit remarkable phenotypic plasticity and functional heterogeneity, contributing to both pro-tumorigenic and anti-tumorigenic processes[81]. Unlike the acute, infection-driven infiltration seen in viral hepatitis, neutrophil accumulation in MAFLD is chronic and is primed by the metabolic milieu. A key mechanism by which neutrophils exert their pathogenic effects is through the formation of NETs. NET levels are elevated in MASH patients[82], and their formation can be driven by pathways such as the Notch signaling pathway[83]. NETs promote liver fibrosis by activating HSC and monocytes[84,85]. Additionally, neutrophil-derived proteases, such as elastase (NE) and proteinase 3 (PR3), contribute to fibrosis, a process modulated by miR-223[82].
In the immune microenvironment, NETs enhance Treg activation via metabolic reprogramming of CD4+ T cells[35], while the neutrophil chemokine C-X-C motif chemokine ligand 5 (CXCL5) can suppress CD4+ T cell recruitment while enhancing CD8+ T cell cytotoxicity[86]. Furthermore, a specific subset of tumor-associated neutrophils (TANs), termed SiglecFhi TANs, promotes tumor progression and immune escape in MAFLD-HCC by secreting TGF-β and impairing antigen presentation[87]. In summary, neutrophils in MAFLD-HCC exhibit context-dependent dual roles. NET formation represents a pivotal node for their pro-fibrotic and immunosuppressive functions, suggesting that targeting NETs could simultaneously impede fibrosis progression and enhance the efficacy of cancer immunotherapy.
DCs
In mouse models and patients with MAFLD/MASH, the number of conventional DCs (cDCs) in the liver is significantly increased[88]. This expansion may be associated with enhanced circulation of bone marrow-derived cDC precursors induced by MASH, thereby maintaining sustained antigen presentation pressure both peripherally and in the liver. cDCs not only interact directly with T cells in the liver-draining lymph nodes[88], but also migrate to other tissue-draining lymph nodes, where they interact with naïve T cells[89], thus amplifying the hepatic inflammatory response. The hepatic cDC pool is primarily composed of CD103+ type 1 conventional dendritic cells (cDC1) and CD11b+ type 2 conventional dendritic cells (cDC2) subsets[89], both of which form clusters within the MASH liver[88,90]. Different cDC subsets exhibit functional differences. X-C motif chemokine receptor 1 (XCR1)+ cDC1 cells can directly target and eliminate hepatocytes[88], highlighting their pro-inflammatory and tissue-destructive capacities in disease progression. In contrast, the role and polarization of cDCs can vary significantly by etiology; for instance, in HBV-related HCC, specific immunosuppressive DC subsets, such as CD103+PD-L1+ DCs, are enriched in peritumoral regions of relapsed patients, a phenomenon less characterized in MAFLD-HCC[91]. Meanwhile, cDC2 cells can exhibit contextual functions. While they share some effector properties, they can also alleviate inflammation by promoting Treg responses[92]. Notably, certain cDC1 populations may also possess protective functions, suppressing hepatic inflammation and maintaining immune homeostasis under specific conditions. This functional dichotomy may be closely related to the disease stage, model type, hepatic microenvironment signals, and the cellular heterogeneity of cDC populations.
Myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSCs) are pathologically activated myeloid cells with potent immunosuppressive capacity, playing crucial roles in tumor progression and immune evasion[93]. Their immunosuppressive functions span from directly inhibiting effector T and NK cells to amplifying the immunosuppressive network via the induction of Tregs and Bregs[94]. MDSCs further accelerate tumor progression by promoting angiogenesis, tissue remodeling, and metastasis[95,96]. In MAFLD-HCC, MDSCs are significantly enriched and correlate with reduced CD8+ T and NKT cell populations, suggesting their central role in impairing anti-tumor immunity[97,98], while cholesterol accumulation via squalene epoxidase (Sqle) overexpression similarly enhances MDSC-mediated immunosuppression[98].
Notably, the gut microbiota modulates the dynamics of MDSC. AKK reduces monocytic MDSCs and M2 macrophages by inhibiting cholesterol and bile acid metabolism, thereby partially reversing immunosuppression[73]. Therapeutically, targeting YTH N6-methyladenosine RNA binding protein F1 (Ythdf1) or Sqle via lipid nanoparticles, or AKK supplementation, significantly enhances the efficacy of PD-1 blockade in MAFLD-HCC models[73,97,98]. These findings establish MDSCs as metabolically regulated, microbiota-sensitive cellular hubs in MAFLD-HCC, offering promising targets for combination immunotherapy.
Mregs
A recent study has identified a unique myeloid cell population in the HCC immune microenvironment that is closely associated with fatty liver-related HCC: thrombospondin-1 (THBS1)+ regulatory myeloid cells (THBS1+ Mregs). The study demonstrated that the abundance of THBS1+ myeloid cells positively correlates with poor patient prognosis and that THBS1+ Mregs are significantly enriched in fatty liver-related HCC[99]. Spatial transcriptomics analysis further revealed that these THBS1+ Mregs are primarily located in the tumor fibrotic areas and spatially co-localize and interact with cancer-associated fibroblasts (CAFs). In animal models, inhibition of triggering receptor expressed on myeloid cells 1 (TREM1) increased the CD8+/forkhead box P3 (Foxp3)+ T cell ratio and enhanced the cytotoxic activity of CD8+ T cells in fatty liver-related HCC tumors[99]. In conclusion, THBS1+ Mregs, a recently defined myeloid subset in the MAFLD-HCC immune microenvironment, are characterized by dual immunosuppressive and stromal regulatory functions. Their TREM1-mediated signaling pathway may serve as a potential target to improve immune therapy responsiveness in fatty liver-related HCC.
Collectively, myeloid cells are not passive bystanders but active architects of the immunosuppressive niche in MAFLD-HCC. TREM2+ macrophages, C-X-C motif chemokine receptor 2 (CXCR2)+ neutrophils/NETs, THBS1+ Mregs, and MDSCs do not operate in isolation; they form a synergistic network that restrains both lymphoid anti-tumor activity (e.g., exhausting CD8+ T cells) and amplifies immunosuppressive circuits (e.g., expanding Tregs). Critically, this network is intricately regulated by the metabolic milieu (e.g., cholesterol, lipids) and gut-liver axis signals, making it a defining and therapeutically targetable feature that distinguishes MAFLD-HCC from other etiologies. This unique immunopathology constitutes a defining and therapeutically targetable feature of MAFLD-HCC. The subsequent sections will explore the therapeutic strategies emerging from this understanding, including cell therapy, metabolic interventions, and small-molecule inhibitors [Figure 2].
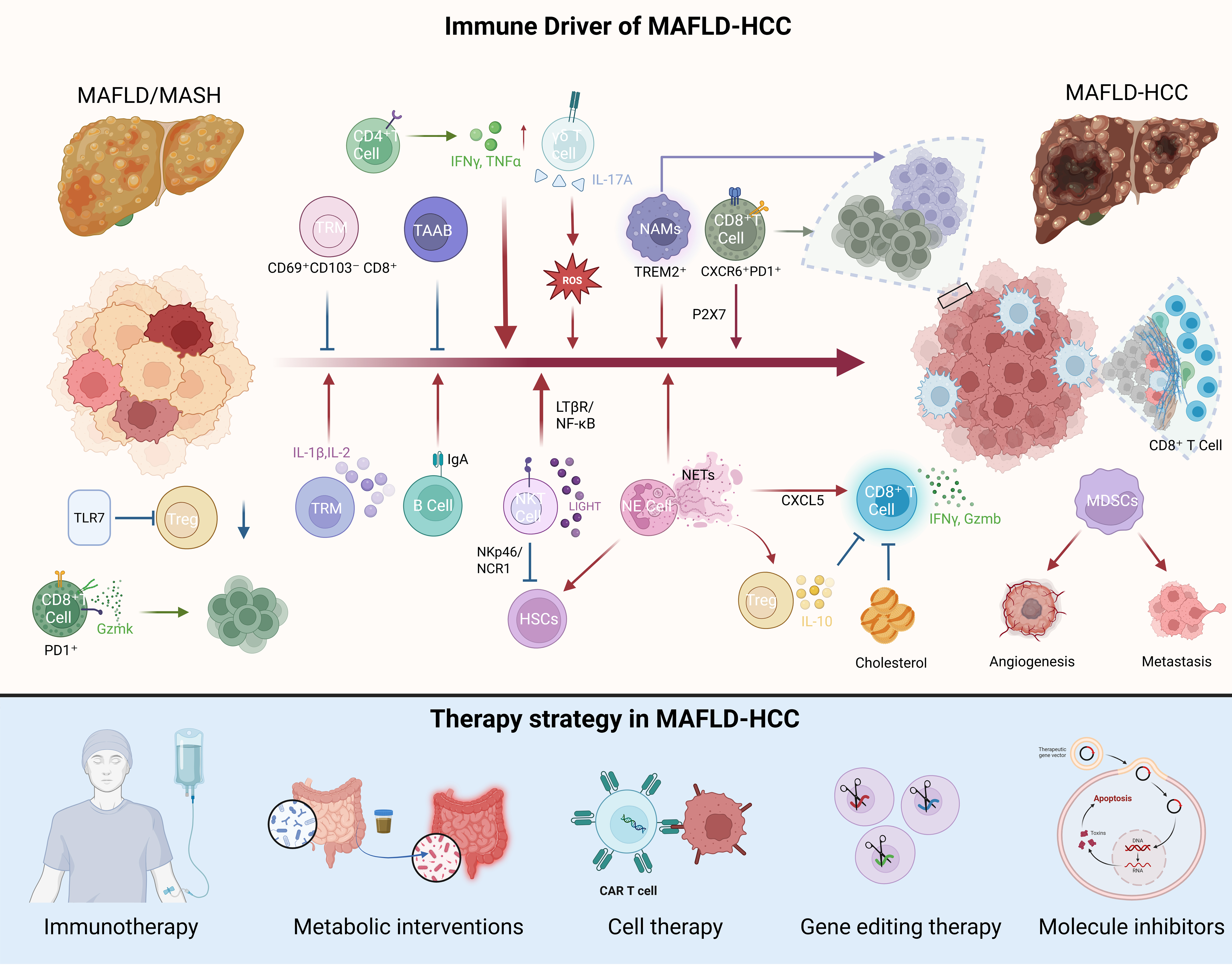
Figure 2. Schematic summary of the immunopathogenesis and therapeutic landscape in MAFLD-HCC. Created in BioRender. Han, H. (2025) https://BioRender.com/0xpwfeh. MAFLD-HCC: Metabolic-associated fatty liver disease-related hepatocellular carcinoma; MASH: metabolic-associated steatohepatitis; CD4+: cluster of differentiation 4 positive; IFNγ: interferon gamma; TNFα: tumor necrosis factor alpha; γδ: gamma delta T cells; IL-17A: interleukin-17A; TRM: Tissue-resident memory T; TAAB: tumor-associated atypical B cell; ROS: reactive oxygen species; NAMs: MASH-associated macrophage; TREM2+: triggering receptor expressed on myeloid cells 2; CD8+: cluster of differentiation 8 positive; CXCR6+: C-X-C motif chemokine receptor 6; PD1+: programmed cell death protein 1; P2X7: P2X purinoceptor 7; LTβR: lymphotoxin-beta receptor; NF-κB: nuclear factor-kappa B; TLR7: Toll-like receptor 7; Treg: regulatory T cell; IL-1β: interleukin-1 beta; IL-2: interleukin-2; IgA: immunoglobulin A; NKT: natural killer T; LIGHT: tumor necrosis factor superfamily member 14; NKp46: natural killer cell p46; NCR1: natural cytotoxicity triggering receptor 1; HSCs: hepatic stellate cells; NE: elastase; NETs: neutrophil extracellular traps; CXCL5: C-X-C motif chemokine ligand 5; IL-10: interleukin-10; MDSCs: myeloid-derived suppressor cells; CAR: chimeric antigen receptor.
IMMUNOTHERAPY IN MAFLD-RELATED HCC
In recent years, the clinical application of ICIs has achieved significant success in the treatment of various malignancies, particularly in the field of advanced HCC. The use of ICIs in clinical practice has fundamentally altered the treatment landscape of advanced HCC[100]. Inhibiting immune checkpoints such as PD-1, PD-L1, and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) can relieve tumor-mediated immunosuppression, thereby restoring anti-tumor immune responses. In unresectable or recurrent metastatic HCC, several studies have confirmed that ICIs provide more durable survival benefits compared to traditional tyrosine kinase inhibitors (TKIs), shifting the focus of systemic treatment from TKIs to immune or immunotherapy combination strategies based on ICIs[101-103]. As a result, immunotherapy has gradually become one of the foundational treatment options for HCC, with several drugs already approved as first- or second-line therapies for the disease [Table 1]. However, despite the success of immunotherapy in HCC, its effectiveness in treating MAFLD-related HCC still faces challenges. This review focuses on the efficacy of ICIs in non-viral HCC patients.
Clinical trials of target inhibitors or immune checkpoints in the tumor microenvironment
| Clinical trial | Treatment | Phase | Targets | Efficacy | |
| First-line | IMBrave150[104,105] | Atezolizumab: 1,200 mg IV, every 3 weeks; bevacizumab: 15 mg/kg IV, every 3 weeks | III | PD-L1; VEGF | Median OS: 17.0 m vs. 18.1 m Median PFS: 7.1 m vs. 5.6 m |
| HIMALAYA[106,107] | Tremelimumab: 300 mg IV plus durvalumab: 1,500 mg, IV once, followed by durvalumab: 1,500 mg, IV; every 4 weeks | III | CTLA-4; PD-L1 | Not yet posted | |
| ORIENT-32[108] | Sintilimab: 200 mg IV, every 3 weeks; IBI305: 15 mg/kg IV, every 3 weeks | II/III | PD-1; VEGF | Not yet posted | |
| CheckMate459[109] | Nivolumab: 240 mg IV, every 2 weeks; sorafenib: 400 mg PO BID | III | PD-1 | ORR: 12% (7-18) Median OS: 16.4 m vs. 14.7 m | |
| Second-line | KEYNOTE-224[110] | Pembrolizumab: 200 mg IV, every 3 weeks plus best supportive care | II | PD-1 | ORR: 20% |
| KEYNOTE-240[111] | Pembrolizumab: 200 mg IV every 3 weeks | III | PD-1 | PFS (HR): 0.75 (0.56-1.01) | |
| KEYNOTE-394[112] | Pembrolizumab: 200 mg IV every 3 weeks | III | PD-1 | ORR: 15.6% PFS (HR): 0.72% (0.55-0.93) | |
| CheckMate 040[113,114] | Nivolumab: 1 mg/kg IV plus ipilimumab: 3 mg/kg IV every 3 weeks for 4 cycles, followed by nivolumab: 240 mg IV every 2 weeks | I/II | PD-1 | ITT: ORR: 22% Asian: ORR: 14% | |
| CheckMate9DW[115] | Nivolumab: 1 mg/kg IV plus ipilimumab: 3 mg/kg IV every 3 weeks for 4 cycles, followed by nivolumab: 240 mg IV every 2 weeks | III | PD-1 | Median OS: 19.3 m vs. 18.4 m Unstratified HR: 0.84 (0.63-1.14) |
Effectiveness of immunotherapy in MAFLD-related HCC
Although ICIs have demonstrated notable efficacy in many solid tumors and are established as first-line treatment for HCC, their application in MAFLD-related HCC remains nascent. The efficacy of ICIs in MAFLD-HCC is significantly influenced by the etiological heterogeneity of the disease. Previous clinical trials often conducted subgroup analyses comparing “viral (HBV/HCV) HCC vs. non-viral HCC” without independently analyzing patients with MAFLD-HCC. This broad non-viral category includes various etiologies such as MASH, ALD, autoimmune hepatitis, primary biliary cholangitis (PBC), and primary sclerosing cholangitis (PSC), making it difficult to separate the impact of each underlying cause on the treatment response. Therefore, there is still a lack of independent, prospective validation specifically for the MAFLD-HCC patient population.
Existing clinical evidence shows that compared to viral HCC, the immune benefits in the non-viral HCC subgroup are less stable, with differences observed between various treatment regimens. The CheckMate-459 trial (NCT02576509) performed a subgroup analysis to explore the impact of HCC etiology on the efficacy of ICIs. The analysis showed that in the non-viral subgroup, the OS with nivolumab was similar to that with sorafenib [16.0 vs. 17.4 months; hazard ratio (HR) = 0.91, 95% confidence interval (CI): 0.72-1.16][116]. In contrast, compared to non-viral HCC patients, those with HBV-related (HR = 0.77; 95%CI: 0.56-1.05) and HCV-related (HR = 0.71; 95%CI: 0.49-1.01) HCC showed a greater trend toward a survival benefit from immunotherapy[110]. In the IMbrave 150 phase III clinical trial (NCT03434379), atezolizumab combined with bevacizumab (Atez/Bev) demonstrated superior results compared to sorafenib (OS HR = 0.58, 95%CI: 0.42-0.79). Subgroup analysis showed that this advantage was mainly observed in HBV-related patients, with an OS HR of 0.58 (95%CI: 0.40-0.83), and HCV-related patients, with an OS HR of 0.43 (95%CI: 0.25-0.73), whereas non-viral etiology patients showed no survival benefit with an OS HR of 1.05 (95%CI: 0.68-1.63)[104]. In the KEYNOTE-240 trial (NCT02702401), non-viral HCC patients had limited survival benefit from pembrolizumab treatment. Although the risk ratio showed a tendency for benefit (HR = 0.88), the 95%CI (0.64-1.20) included 1.0, indicating that the efficacy did not reach statistical significance. This contrasts with the clear clinical benefit in HBV-HCC patients (HR = 0.57, 95%CI: 0.35-0.94)[104].
Although clinical trial data on immunotherapy for MAFLD-HCC are limited, some studies suggest that under specific immunotherapy regimens, non-viral populations may still experience significant survival benefits [Table 2]. A breakthrough finding from the HIMALAYA study (NCT03298451) confirmed that non-viral HCC patients could derive clear survival benefits from the STRIDE (NCT03298451) regimen (durvalumab single high dose combined with tremelimumab + durvalumab maintenance). In the sorafenib-controlled trial (NCT01908426), this subgroup showed a significant 26% reduction in the risk of death (HR = 0.74, 95%CI: 0.57-0.95), with efficacy intensity only second to HBV-related HCC patients (HR = 0.64). This result contrasts sharply with HCV-related HCC patients (HR = 1.06, 95%CI: 0.76-1.49)[106,120]. The significant benefit (P < 0.05) in the non-viral subgroup provides an important first-line immunotherapy option for late-stage HCC patients without a viral etiology.
Clinical trials of immunotherapy for non-viral HCC
| Trial/study | HCC etiology | OS [HR (95%CI)] | Survival benefit |
| Checkmate 459[109] | Non-viral HCC | 0.95 (0.74-1.22) | No |
| IMbrave150[104] | Non-viral HCC | 1.05 (0.68-1.63) | No |
| KEYNOTE-240[111] | Non-viral HCC | 0.88 (0.64-1.20) | No |
| COSMIC-312[117] | Non-viral HCC | 1.18 (0.78-1.79) | No |
| HIMALAYA[106] | Non-viral HCC | 0.74 (0.57-0.95) | Yes |
| REACH-2[118] | Non-viral HCC | 0.6 (0.40-0.79) | Yes |
| CELESTIAL[119] | Non-viral HCC | 0.72 (0.54-0.96) | Yes |
Additionally, a retrospective study has shown that for patients with advanced HCC related to MAFLD/MASH, single-agent lenvatinib demonstrated significant survival benefits compared to the atezolizumab plus bevacizumab (Atez/Bev) regimen. Specifically, in multivariate analysis, patients in the MAFLD/MASH subgroup treated with lenvatinib had significantly better OS and progression-free survival (PFS) than those treated with Atez/Bev (OS HR = 0.46, P = 0.011; PFS HR = 0.55, P = 0.031). This advantage was not observed in non-MAFLD/MASH patients. This study is the first to suggest that lenvatinib may be a more effective treatment option than Atezolizumab/bevacizumab, especially in patients with MAFLD-HCC within the non-viral HCC population[121]. Larger-scale evidence comes from a meta-analysis integrating several key Phase III studies [including cabozantinib combined with nivolumab, COSMIC-312 (NCT03755791), HIMALAYA (NCT03298451), LEAP-002 (NCT03713593), RATIONALE-301 (NCT03412773), ORIENT-32 (NCT03794440), and others], which shows that ICIs provide OS improvements in various etiology subgroups, but the degree of improvement varies[122]. These results collectively indicate that, although MAFLD-HCC has not achieved survival improvements comparable to those of viral HCC in the current immunotherapy framework, some regimens may have advantages in specific subgroups. There is an urgent need for prospective clinical studies with sufficient etiological stratification to determine the optimal treatment strategy and the patient populations who would benefit the most.
Cell therapy for MAFLD-related HCC
Immunocellular therapy is a treatment method in which immune cells are extracted from the patient’s body, cultured, expanded, and genetically modified in vitro to enhance their anti-cancer capabilities. These modified cells are then reinfused into the body to precisely recognize and attack cancer cells. This therapy offers high efficiency, targeting, and specificity, effectively reducing damage to normal tissues while improving treatment outcomes[123]. Research in this field has expanded to encompass various cancer types, leveraging immune effectors such as chimeric antigen receptor T (CAR-T) cells, other engineered T cells, tumor-infiltrating lymphocytes (TILs), NK cells, cytokine-induced killer (CIK) cells, and B cells. In recent years, specific T-cell immunotherapies have gained increasing attention in the treatment of HCC, particularly CAR-T-cell therapy and T cell receptor-engineered T-cell (TCR-T) therapy. Both technologies have demonstrated promising clinical potential in cancer immunotherapy[124]. Although CAR-T cell therapy has shown good results in hematological malignancies, its efficacy in solid tumors is relatively poor, particularly in HCC. This is partly due to the immunosuppressive microenvironment and tumor heterogeneity characteristic of solid tumors[125]. Current CAR-T targets under investigation for HCC include Glypican-3 (GPC3), alpha-fetoprotein (AFP), cellular mesenchymal-epithelial transition factor (c-Met), Mucin 1 (MUC1), epithelial cell adhesion molecule (EpCAM), CD133, CD147, NKG2D, and carcinoembryonic antigen (CEA). Among these, GPC3 is one of the most commonly used targets, with many clinical trials currently underway. In a study combining local treatment with chimeric antigen receptor (CAR)-GPC3 T-cell therapy in advanced HCC patients, two GPC3-positive patients achieved more than five years of disease-free survival (DFS) and over eight years of OS after treatment, demonstrating the potential benefits of CAR-GPC3-T-cell therapy for HCC patients[126]. Additionally, GPC3-CAR-T cells co-expressed the cytokine IL-15 and were equipped with a safety switch, effectively reducing cytokine storms during treatment. A Phase I trial (NCT05103631 and NCT04377932) of these GPC3-CAR-T cells in pediatric patients with refractory HCC demonstrated effective control of CRS and yielded a 66% disease control rate (DCR) with a 33% ORR[127]. These data suggest that although CAR-T therapy in HCC is still in preclinical and early clinical stages, its potential should not be underestimated, especially when combined with immune microenvironment modulation. CAR-T therapy remains predominantly in preclinical and early clinical research for HCC. Studies have shown that modulating NK cell function in MASH liver can inhibit the progression of MASH, suggesting that using NKT cells in MAFLD-HCC immunocellular therapy may have significant therapeutic potential[42]. For MAFLD-HCC patients, NKT cell therapy holds promise in regulating the immune microenvironment, restoring the body’s anti-tumor immune response, and providing a more precise treatment approach for HCC immunotherapy. As our understanding of the mechanisms of immunocellular therapy in MAFLD-HCC advances, future developments may lead to more precise, multi-targeted immunocellular therapy regimens.
THERAPEUTIC STRATEGIES FOR IMMUNOTHERAPY SENSITIZATION IN MAFLD-RELATED HCC
Metabolic interventions in MAFLD-related HCC
MAFLD is a liver disease driven by metabolic dysfunction, highlighting the key role of metabolic abnormalities in disease onset and progression. As a result, most current therapeutic strategies primarily target metabolic processes, aiming to alleviate liver cell stress and damage caused by metabolic disorders, as well as to address progressive changes such as liver fibrosis. In recent years, the role of the gut microbiome in host health and disease has gained significant attention. Studies have shown that the composition, metabolites, and toxins released by the gut microbiome can influence immune cell function through the bloodstream and the gut-liver axis, thereby driving oxidative stress, inflammation, and tumorigenesis[128]. Notably, the gut-liver axis plays a critical role in the development of MAFLD. Research has shown that before liver steatosis occurs, the intestinal barrier function is compromised, and pro-inflammatory factors and intestinal metabolites can enter the liver via the portal vein, triggering inflammation and promoting progression to HCC[129]. In response to this phenomenon, gut microbiome interventions, including antibiotics, probiotics, prebiotics, and fecal microbiota transplantation (FMT), have been proposed as new strategies to improve the efficacy of immunotherapy in HCC. Additionally, increasing evidence suggests that the liver receives microbial metabolites from the gut via the bloodstream and that alterations in the gut microbiome can influence the infiltration and function of immune cells in the tumor microenvironment, potentially impacting the effectiveness of immunotherapy. For example, an ongoing clinical trial (NCT04130763) is investigating the use of FMT capsules in combination with ICIs to enhance the efficacy of anti-PD-1 therapy. This study is still in its early stages of development. Furthermore, multiple clinical trials targeting the gut microbiome are underway to evaluate the efficacy of probiotics, prebiotics, and FMT in MAFLD/HCC[130]. For instance, in a study of 21 patients with MAFLD, 3 grams of fecal material were transplanted into the duodenum via endoscopy and compared with a control group (probiotic group). The results indicated that FMT reduced small bowel permeability in MAFLD patients, potentially improving the condition[131]. Additionally, in another study, 75 patients with MAFLD received FMT through colonoscopy, with the FMT group showing reduced hepatic fat accumulation by improving gut microbiome dysbiosis, thus alleviating fatty liver disease[132]. In addition to FMT, studies on probiotics for MAFLD are increasing. Several clinical trials have investigated the effects of probiotics and their combinations on liver health[130,133,134]. For example, a study on 22 patients with MAFLD using a 4-month probiotic cocktail treatment showed some efficacy[135]. Other microbial interventions, such as prebiotics, synbiotics, and postbiotics, have also been applied in multiple studies, particularly in the context of antibiotic and microbial-derived metabolic product interventions, which are expanding[130]. These studies, although demonstrating the potential of gut microbiome interventions in MAFLD-HCC treatment, are still limited by small sample sizes, a lack of long-term follow-up, and a lack of histological validation. Future large-scale, long-term clinical trials are needed to further confirm their effectiveness.
Furthermore, recent studies have identified specific microbial metabolites in the gut microbiome that are closely linked to immune responses, offering new therapeutic avenues targeting these metabolites. For instance, IL-33 released by intestinal epithelial cells (IECs) has been shown to induce gut microbiome dysbiosis and the synthesis of trimethylamine-N-oxide (TMAO), which in turn causes liver damage through oxidative stress mechanisms[136]. A key microbial metabolite, phenylacetylglutamine (PAGln), has also been identified as a factor that reduces the efficacy of anti-PD-1 therapy, suggesting that targeting PAGln and its associated metabolic pathways may become a potential strategy for combination therapy in future immunotherapy[137]. In terms of metabolic pathways, fatty acid metabolism has been identified as a potential target for drug intervention[138]. Studies have shown that acetyl-coenzyme A (CoA) carboxylase (ACC) and its downstream pathways for fatty acid synthesis play a crucial role in the development of MAFLD/HCC[139]. Additionally, grapefruit-derived cationic lipid-modified nanoparticles [grapefruit-derived cationic lipid-modified naringenin-loaded nanoparticles (NP-NAR)] have demonstrated effects in animal models, alleviating MAFLD progression by promoting fatty acid oxidation and regulating the gut microbiome[140]. Further exploration of this mechanism may provide new clinical strategies for treating MAFLD-HCC through fatty acid metabolic reprogramming. Moreover, the gut microbiome also protects against MAFLD through the production of 2-hydroxy-4-methylpentanoic acid (HMP)[141]. These studies collectively suggest that targeting the gut-liver axis to treat MAFLD-related liver inflammation holds great promise. Repurposing existing metabolic drugs offers another strategic avenue. Metformin, a first-line therapy for type 2 diabetes, activates adenosine monophosphate-activated protein kinase (AMPK). This pleiotropic action reduces lipogenesis, inhibits tumor proliferation and metastasis, remodels the tumor immune microenvironment, and synergizes with anti-PD-1 therapy[142,143]. Additionally, amino acid metabolism has been found to be closely linked to immune responses. For example, targeting the cancer-specific arginine-binding factor RNA-binding motif protein 39 (RBM39) can rejuvenate T-cell proliferation and function, thereby enhancing immunotherapy[144]. Furthermore, overexpression of the lysine transporter solute carrier family 3 member 2 (SLC3A2) in HCC cells creates a lysine-depleted microenvironment, which compromises T cell fitness and blunts immunotherapy response. Therefore, lysine supplementation or targeting SLC3A2 may enhance the sensitivity of HCC patients to lenvatinib and immune checkpoint blockade therapy[145]. Overall, the role of the gut-liver axis in MAFLD-HCC and its impact on immunotherapy is becoming a research hotspot.
Advances in small-molecule inhibitors targeting MAFLD-related HCC
In addition to immunotherapy, recent years have seen a growing focus on screening and developing small-molecule inhibitors targeting MAFLD-HCC to enhance the efficacy of immunotherapy and improve patient prognosis. Significant progress has been made, with several targets identified that intervene in the onset and progression of MAFLD-HCC. Targeting TREM2+ macrophages has emerged as a promising strategy. These cells, whose abundance correlates with TGF-β1 levels, promote glycolysis and pyruvate kinase M2 (PKM2) expression through IL-1β secretion, thereby suppressing CD8+ T cell infiltration and fostering a pro-tumorigenic environment[146]. The TREM2 antagonist PY314 has demonstrated favorable tolerability and minimal side effects in an early-phase clinical trial (NCT04691375), underscoring its therapeutic potential[147]. These findings suggest that targeting the TREM2 pathway in immune cells could provide new treatment strategies for MAFLD-HCC. Activation of the adenosine A1 receptor (A1R) exerts protective effects in MAFLD/MASH. A1R agonists, including the natural compound timosaponin A3 (TA3), identified via molecular docking, inhibit sterol regulatory element-binding protein (SREBP) maturation and translocation by promoting SCAP-sequestosome-1 (SQSTM1) interaction. This mechanism reduces de novo lipogenesis (DNL) and inflammation, demonstrating significant efficacy in preclinical models and providing a rationale for its application in MAFLD-HCC[148]. Other key targets include signal transducer and activator of transcription 3 (STAT3) and EF-hand domain family member D2 (EFHD2). The STAT3 inhibitor HJC0152 reduces HCC incidence in MAFLD mouse models[149], while EFHD2 has been identified as a critical regulator of immune and inflammatory responses in MASH, marking it as a promising therapeutic target[150]. Targeting EFHD2 reduces chronic liver inflammation, thereby delaying the progression from MASH to HCC. Drug repurposing offers a complementary approach. The angiotensin II receptor inhibitor losartan reduces hepatic and peri-tumoral fibrosis, thereby enhancing CD8+ T cell infiltration and synergizing with anti-PD-1 therapy by modulating the TGF-β axis and suppressing immunosuppressive fibroblasts[151]. Aspirin has also demonstrated utility in preventing or treating HCC in high-risk populations[152]. Bioinformatic analyses have further illuminated the landscape, nominating core genes such as TREM2, growth differentiation factor 15 (GDF15), tricarboxylic acid cycle 39A (TTC39A), and annexin A2 (ANXA2) as key drivers of MASH-to-HCC progression and potential diagnostic or therapeutic targets[153]. In conclusion, small-molecule inhibitors targeting specific drivers of MAFLD-HCC hold immense potential as adjuvants to immunotherapy. By precisely intervening in metabolic pathways, immune cell functions, and the tumor microenvironment, these agents are poised to overcome resistance mechanisms and significantly enhance therapeutic efficacy. The continued development and clinical validation of these targeted drugs are expected to make them a cornerstone of combination therapy for patients with MAFLD-HCC.
DISCUSSION
The future of treating MAFLD-HCC lies in the development of more personalized and precision medicine approaches. With the application of multi-omics technologies, we can now gain a deeper understanding of the immune microenvironment and identify specific immune cell subsets and metabolic features that drive MAFLD-HCC. Building on this, emerging strategies targeting the metabolic-immune axis show great promise. For instance, pioneering nanoplatforms that co-deliver a cisplatin prodrug and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) plasmids targeting PKM2 can simultaneously induce DNA damage and suppress the Warburg effect, thereby reversing lactate-mediated immunosuppression and activating T-cell immunity[154]. This exemplifies a powerful chemo-immunometabolic combination strategy. Furthermore, the integration of epigenetic modulation is another key frontier. The use of ROS-responsive poly(β-amino ester) vectors to co-deliver a CRISPR interference (CRISPRi) system for PD-L1 knockdown and the epigenetic drug azacytidine has demonstrated enhanced immunotherapy by improving antigen presentation[155].
In the future, combining these studies with artificial intelligence (AI) can help design more personalized treatment strategies, integrating ICIs, metabolic interventions, cell therapies, epigenetic modulators, and small-molecule inhibitors in a highly customized manner. The exploration of antibody-drug conjugates (ADCs) and their combination with the aforementioned modalities should also be prioritized. Translating scientific discoveries into clinical practice is crucial, and a key challenge is to convert the targets and mechanisms into effective clinical treatment plans. We need to develop reliable predictive biomarkers and optimize preclinical models, including those utilizing advanced polymeric gene vectors[156], to more accurately assess the efficacy of new therapies.
Although significant progress has been made, optimizing patient prognosis remains a major challenge. Overcoming this bottleneck depends on deeply analyzing disease heterogeneity, promoting innovative combination therapies, and accelerating the translation of cutting-edge modalities. By integrating multi-dimensional strategies such as immunotherapy, metabolic intervention, epigenetic regulation, and gene therapy, and leveraging the power of multi-omics and AI, we hope to achieve more effective and durable control of MAFLD-HCC, ultimately improving patient survival.
DECLARATIONS
Acknowledgments
The graphical abstract was created with BioRender.com [Created in BioRender. Han, H. (2025) https://BioRender.com/9vkibbd].
Authors’ contributions
Manuscript conception and design: Liu Y, Wang JB
Manuscript draft and revision: Han HN, Ma JH
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the Noncommunicable Chronic Diseases–National Science and Technology Major Project (Grant No. 2023ZD0507500), the National Natural Science Foundation of China (Grant Nos. 82272787 and 82473189), and the Clinical and Translational Research Project of Anhui Province (Grant No. 202204295107020022).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copy right
© The Author(s) 2025.
REFERENCES
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229-63.
2. Rumgay H, Ferlay J, de Martel C, et al. Global, regional and national burden of primary liver cancer by subtype. Eur J Cancer. 2022;161:108-18.
3. Allemani C, Matsuda T, Di Carlo V, et al.; CONCORD Working Group. Global surveillance of trends in cancer survival 2000-14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet. 2018;391:1023-75.
4. Wong GL, Wong VW. Risk prediction of hepatitis B virus-related hepatocellular carcinoma in the era of antiviral therapy. World J Gastroenterol. 2013;19:6515-22.
5. Liu Y, Liu L. Changes in the epidemiology of hepatocellular carcinoma in Asia. Cancers. 2022;14:4473.
6. Eslam M, Newsome PN, Sarin SK, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73:202-9.
7. Rinella ME, Lazarus JV, Ratziu V, et al.; NAFLD Nomenclature consensus group. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology. 2023;78:1966-86.
8. Habibullah M, Jemmieh K, Ouda A, Haider MZ, Malki MI, Elzouki AN. Metabolic-associated fatty liver disease: a selective review of pathogenesis, diagnostic approaches, and therapeutic strategies. Front Med. 2024;11:1291501.
9. Hong S, Sun L, Hao Y, et al. From NAFLD to MASLD: when metabolic comorbidity matters. Ann Hepatol. 2024;29:101281.
10. Hagström H, Vessby J, Ekstedt M, Shang Y. 99% of patients with NAFLD meet MASLD criteria and natural history is therefore identical. J Hepatol. 2024;80:e76-7.
11. Crane H, Gofton C, Sharma A, George J. MAFLD: an optimal framework for understanding liver cancer phenotypes. J Gastroenterol. 2023;58:947-64.
12. Costante F, Airola C, Santopaolo F, Gasbarrini A, Pompili M, Ponziani FR. Immunotherapy for nonalcoholic fatty liver disease-related hepatocellular carcinoma: lights and shadows. World J Gastrointest Oncol. 2022;14:1622-36.
13. Chan SL, Sun HC, Xu Y, et al. The Lancet Commission on addressing the global hepatocellular carcinoma burden: comprehensive strategies from prevention to treatment. Lancet. 2025;406:731-78.
14. Pinter M, Pinato DJ, Ramadori P, Heikenwalder M. NASH and hepatocellular carcinoma: immunology and immunotherapy. Clin Cancer Res. 2023;29:513-20.
15. Yahoo N, Dudek M, Knolle P, Heikenwälder M. Role of immune responses in the development of NAFLD-associated liver cancer and prospects for therapeutic modulation. J Hepatol. 2023;79:538-51.
16. Llovet JM, Pinyol R, Yarchoan M, et al. Adjuvant and neoadjuvant immunotherapies in hepatocellular carcinoma. Nat Rev Clin Oncol. 2024;21:294-311.
17. Kudo M, Finn RS, Galle PR, et al. IMbrave150: efficacy and safety of atezolizumab plus bevacizumab versus sorafenib in patients with barcelona clinic liver cancer stage b unresectable hepatocellular carcinoma: an exploratory analysis of the phase III study. Liver Cancer. 2023;12:238-50.
18. Peng Y, Wong CC, Yu J. The paradox of immunotherapy in NASH-HCC. Signal Transduct Target Ther. 2021;6:228.
19. Pfister D, Núñez NG, Pinyol R, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592:450-6.
20. Cheu JWS, Wong CCL. The immune microenvironment of steatotic hepatocellular carcinoma: current findings and future prospects. Hepatol Commun. 2024;8:e0516.
21. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. 2019;37:457-95.
22. Dudek M, Pfister D, Donakonda S, et al. Auto-aggressive CXCR6+ CD8 T cells cause liver immune pathology in NASH. Nature. 2021;592:444-9.
23. Li M, Wang L, Cong L, et al. Spatial proteomics of immune microenvironment in nonalcoholic steatohepatitis-associated hepatocellular carcinoma. Hepatology. 2024;79:560-74.
24. Pan Y, Chen H, Zhang X, et al. METTL3 drives NAFLD-related hepatocellular carcinoma and is a therapeutic target for boosting immunotherapy. Cell Rep Med. 2023;4:101144.
25. Valenzuela-Pérez L, Kim Lee HS, Bayer RL, et al. CD4+ T cells promote fibrosis during metabolic dysfunction-associated steatohepatitis. bioRxiv. 2025.
26. Brown ZJ, Fu Q, Ma C, et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4+ T cell apoptosis promoting HCC development. Cell Death Dis. 2018;9:620.
27. Schneider C, Teufel A, Yevsa T, et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut. 2012;61:1733-43.
28. Miao Y, Li Z, Feng J, et al. The role of CD4+T cells in nonalcoholic steatohepatitis and hepatocellular carcinoma. Int J Mol Sci. 2024;25:6895.
29. Ohkura N, Sakaguchi S. Transcriptional and epigenetic basis of Treg cell development and function: its genetic anomalies or variations in autoimmune diseases. Cell Res. 2020;30:465-74.
30. Ma X, Hua J, Mohamood AR, Hamad AR, Ravi R, Li Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology. 2007;46:1519-29.
31. Roh YS, Kim JW, Park S, et al. Toll-like receptor-7 signaling promotes nonalcoholic steatohepatitis by inhibiting regulatory T cells in mice. Am J Pathol. 2018;188:2574-88.
32. Zhang C, Li L, Feng K, Fan D, Xue W, Lu J. ‘Repair’ Treg cells in tissue injury. Cell Physiol Biochem. 2017;43:2155-69.
33. Katz SC, Ryan K, Ahmed N, et al. Obstructive jaundice expands intrahepatic regulatory T cells, which impair liver T lymphocyte function but modulate liver cholestasis and fibrosis. J Immunol. 2011;187:1150-6.
34. Fabregat I, Moreno-Càceres J, Sánchez A, et al.; IT-LIVER Consortium. TGF-β signalling and liver disease. FEBS J. 2016;283:2219-32.
35. Wang H, Zhang H, Wang Y, et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol. 2021;75:1271-83.
36. Behary J, Amorim N, Jiang XT, et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat Commun. 2021;12:187.
37. Wang H, Tsung A, Mishra L, Huang H. Regulatory T cell: a double-edged sword from metabolic-dysfunction-associated steatohepatitis to hepatocellular carcinoma. EBioMedicine. 2024;101:105031.
38. Hindson J. T cells in NASH and liver cancer: pathology and immunotherapy. Nat Rev Gastroenterol Hepatol. 2021;18:367.
39. Wolf MJ, Adili A, Piotrowitz K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26:549-64.
40. Clària J. Natural killer cell recognition and killing of activated hepatic stellate cells. Gut. 2012;61:792-3.
41. Tang W, Zhou J, Yang W, et al. Aberrant cholesterol metabolic signaling impairs antitumor immunosurveillance through natural killer T cell dysfunction in obese liver. Cell Mol Immunol. 2022;19:834-47.
42. Xu J, Xia Q, Wu T, et al. Prophylactic treatment with Bacteroides uniformis and Bifidobacterium bifidum counteracts hepatic NK cell immune tolerance in nonalcoholic steatohepatitis induced by high fat diet. Gut Microbes. 2024;16:2302065.
43. Martínez-Chantar ML, Delgado TC, Beraza N. Revisiting the role of natural killer cells in non-alcoholic fatty liver disease. Front Immunol. 2021;12:640869.
44. Hunter S, Willcox CR, Davey MS, et al. Human liver infiltrating γδ T cells are composed of clonally expanded circulating and tissue-resident populations. J Hepatol. 2018;69:654-65.
45. Harley IT, Stankiewicz TE, Giles DA, et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology. 2014;59:1830-9.
46. Zhan C, Peng C, Wei H, Wei K, Ou Y, Zhang Z. Diverse subsets of γδT cells and their specific functions across liver diseases. Int J Mol Sci. 2025;26:2778.
47. Li F, Hao X, Chen Y, et al. The microbiota maintain homeostasis of liver-resident γδT-17 cells in a lipid antigen/CD1d-dependent manner. Nat Commun. 2017;7:13839.
48. Marinović S, Lenartić M, Mladenić K, et al. NKG2D-mediated detection of metabolically stressed hepatocytes by innate-like T cells is essential for initiation of NASH and fibrosis. Sci Immunol. 2023;8:eadd1599.
49. Li Y, You Z, Tang R, Ma X. Tissue-resident memory T cells in chronic liver diseases: phenotype, development and function. Front Immunol. 2022;13:967055.
50. Koda Y, Teratani T, Chu PS, et al. CD8+ tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat Commun. 2021;12:4474.
51. Gupta PK, Godec J, Wolski D, et al. CD39 expression identifies terminally exhausted CD8+ T cells. PLoS Pathog. 2015;11:e1005177.
52. Li Y, Huang B, Jiang X, et al. Mucosal-associated invariant T cells improve nonalcoholic fatty liver disease through regulating macrophage polarization. Front Immunol. 2018;9:1994.
53. Toubal A, Kiaf B, Beaudoin L, et al. Mucosal-associated invariant T cells promote inflammation and intestinal dysbiosis leading to metabolic dysfunction during obesity. Nat Commun. 2020;11:3755.
54. Deschler S, Pohl-Topcu J, Ramsauer L, et al. Polyunsaturated fatty acid-induced metabolic exhaustion and ferroptosis impair the anti-tumour function of MAIT cells in MASLD. J Hepatol. 2025;83:1364-78.
55. Karl M, Hasselwander S, Zhou Y, et al. Dual roles of B lymphocytes in mouse models of diet-induced nonalcoholic fatty liver disease. Hepatology. 2022;76:1135-49.
56. Xie Y, Huang Y, Li ZY, et al. Interleukin-21 receptor signaling promotes metabolic dysfunction-associated steatohepatitis-driven hepatocellular carcinoma by inducing immunosuppressive IgA+ B cells. Mol Cancer. 2024;23:95.
57. Huang Y, Xie Y, Zhang Y, et al. Single-cell transcriptome reveals the reprogramming of immune microenvironment during the transition from MASH to HCC. Mol Cancer. 2025;24:177.
58. Petriv N, Suo H, Hochnadel I, et al. Essential roles of B cell subsets in the progression of MASLD and HCC. JHEP Rep. 2024;6:101189.
59. Kotsiliti E, Leone V, Schuehle S, et al. Intestinal B cells license metabolic T-cell activation in NASH microbiota/antigen-independently and contribute to fibrosis by IgA-FcR signalling. J Hepatol. 2023;79:296-313.
60. Barrow F, Khan S, Fredrickson G, et al. Microbiota-driven activation of intrahepatic B cells aggravates NASH through innate and adaptive signaling. Hepatology. 2021;74:704-22.
61. Wang H, Herman A, Barrow F, et al. Single-cell RNA sequencing reveals a reprogramming of hepatic immune cells and a protective role for B cells in MASH-driven HCC. Hepatol Commun. 2025;9:e0668.
62. Zhang P, Chen Z, Kuang H, et al. Neuregulin 4 suppresses NASH-HCC development by restraining tumor-prone liver microenvironment. Cell Metab. 2022;34:1359-76.e7.
63. Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity. 2019;50:924-40.
64. Foglia B, Sutti S, Cannito S, et al. Histidine-rich glycoprotein in metabolic dysfunction-associated steatohepatitis-related disease progression and liver carcinogenesis. Front Immunol. 2024;15:1342404.
65. Liang Y, Zhang R, Biswas S, et al. Integrated single-cell transcriptomics reveals the hypoxia-induced inflammation-cancer transformation in NASH-derived hepatocellular carcinoma. Cell Prolif. 2024;57:e13576.
66. Zhou L, Qiu X, Meng Z, et al. Hepatic danger signaling triggers TREM2+ macrophage induction and drives steatohepatitis via MS4A7-dependent inflammasome activation. Sci Transl Med. 2024;16:eadk1866.
67. Zhang Y, Luo Y, Liu X, et al. NCOA5 haploinsufficiency in myeloid-lineage cells sufficiently causes nonalcoholic steatohepatitis and hepatocellular carcinoma. Cell Mol Gastroenterol Hepatol. 2024;17:1-27.
68. Clement CC, Nanaware PP, Yamazaki T, et al. Pleiotropic consequences of metabolic stress for the major histocompatibility complex class II molecule antigen processing and presentation machinery. Immunity. 2021;54:721-36.e10.
69. Shi X, Zheng Q, Wang X, et al. Compromised macrophages contribute to progression of MASH to hepatocellular carcinoma in FGF21KO mice. Sci Adv. 2024;10:eado9311.
70. Rose JP, Morgan DA, Sullivan AI, et al. FGF21 reverses MASH through coordinated actions on the CNS and liver. Cell Metab. 2025;37:1515-29.e6.
71. Mohammed S, Thadathil N, Ohene-Marfo P, et al. Absence of either Ripk3 or Mlkl reduces incidence of hepatocellular carcinoma independent of liver fibrosis. Mol Cancer Res. 2023;21:933-46.
72. Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121-4.
73. Wu XQ, Ying F, Chung KPS, et al. Intestinal Akkermansia muciniphila complements the efficacy of PD1 therapy in MAFLD-related hepatocellular carcinoma. Cell Rep Med. 2025;6:101900.
74. Min BH, Devi S, Kwon GH, et al. Gut microbiota-derived indole compounds attenuate metabolic dysfunction-associated steatotic liver disease by improving fat metabolism and inflammation. Gut Microbes. 2024;16:2307568.
75. Su T, He Y, Wang M, et al. Macrophage-hepatocyte circuits mediated by grancalcin aggravate the progression of metabolic dysfunction associated steatohepatitis. Adv Sci. 2024;11:e2406500.
76. Hu S, Li R, Gong D, et al. Atf3-mediated metabolic reprogramming in hepatic macrophage orchestrates metabolic dysfunction-associated steatohepatitis. Sci Adv. 2024;10:eado3141.
77. Jaitin DA, Adlung L, Thaiss CA, et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell. 2019;178:686-98.e14.
78. Vennin C, Cattaneo CM, Bosch L, et al. Taxanes trigger cancer cell killing in vivo by inducing non-canonical T cell cytotoxicity. Cancer Cell. 2023;41:1170-85.e12.
79. Kohlhepp MS, Liu H, Tacke F, Guillot A. The contradictory roles of macrophages in non-alcoholic fatty liver disease and primary liver cancer - challenges and opportunities. Front Mol Biosci. 2023;10:1129831.
80. Leslie J, Mackey JBG, Jamieson T, et al. CXCR2 inhibition enables NASH-HCC immunotherapy. Gut. 2022;71:2093-106.
81. Kwak JW, Houghton AM. Targeting neutrophils for cancer therapy. Nat Rev Drug Discov. 2025;24:666-84.
82. Zhang P, Liu D, Wu L, et al. Neutrophil serine proteases NE and PR3 controlled by the miR-223/STAT3 axis potentiate MASH and liver fibrosis. Hepatology. 2025.
83. Xu M, Xu H, Ling YW, et al. Neutrophil extracellular traps-triggered hepatocellular senescence exacerbates lipotoxicity in non-alcoholic steatohepatitis. J Adv Res. 2025:S2090-1232(25)00175-4.
84. Xia Y, Wang Y, Xiong Q, et al. Neutrophil extracellular traps promote MASH fibrosis by metabolic reprogramming of HSC. Hepatology. 2025;81:947-61.
85. Babuta M, Morel C, de Carvalho Ribeiro M, et al. Neutrophil extracellular traps activate hepatic stellate cells and monocytes via NLRP3 sensing in alcohol-induced acceleration of MASH fibrosis. Gut. 2024;73:1854-69.
86. Tu T, Hong H, Alhousari D, et al. Proinflammatory macrophages release CXCL5 to regulate T cell function and limit effects of αPD-1 in steatosis-driven liver cancer. JHEP Rep. 2025;7:101385.
87. Teo JMN, Chen Z, Chen W, et al. Tumor-associated neutrophils attenuate the immunosensitivity of hepatocellular carcinoma. J Exp Med. 2025:222.
88. Deczkowska A, David E, Ramadori P, et al. XCR1+ type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat Med. 2021;27:1043-54.
89. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563-604.
90. Haas JT, Vonghia L, Mogilenko DA, et al. Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat Metab. 2019;1:604-14.
91. Seyhan D, Allaire M, Fu Y, et al. Immune microenvironment in hepatocellular carcinoma: from pathogenesis to immunotherapy. Cell Mol Immunol. 2025;22:1132-58.
92. Heier EC, Meier A, Julich-Haertel H, et al. Murine CD103+ dendritic cells protect against steatosis progression towards steatohepatitis. J Hepatol. 2017;66:1241-50.
93. Gabrilovich DI, Bronte V, Chen SH, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425; author reply 426.
95. Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med. 2015;66:97-110.
96. Safarzadeh E, Orangi M, Mohammadi H, Babaie F, Baradaran B. Myeloid-derived suppressor cells: important contributors to tumor progression and metastasis. J Cell Physiol. 2018;233:3024-36.
97. Wang L, Zhu L, Liang C, et al. Targeting N6-methyladenosine reader YTHDF1 with siRNA boosts antitumor immunity in NASH-HCC by inhibiting EZH2-IL-6 axis. J Hepatol. 2023;79:1185-200.
98. Wen J, Zhang X, Wong CC, et al. Targeting squalene epoxidase restores anti-PD-1 efficacy in metabolic dysfunction-associated steatohepatitis-induced hepatocellular carcinoma. Gut. 2024;73:2023-36.
99. Giraud J, Chalopin D, Ramel E, et al. THBS1+ myeloid cells expand in SLD hepatocellular carcinoma and contribute to immunosuppression and unfavorable prognosis through TREM1. Cell Rep. 2024;43:113773.
100. Zheng C, Zheng L, Yoo JK, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169:1342-56.e16.
101. Finn RS, Qin S, Ikeda M, et al.; IMbrave150 Investigators. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894-905.
102. Zayac A, Almhanna K. Hepatobiliary cancers and immunotherapy: where are we now and where are we heading? Transl Gastroenterol Hepatol. 2020;5:8.
103. Bruix J, Chan SL, Galle PR, Rimassa L, Sangro B. Systemic treatment of hepatocellular carcinoma: an EASL position paper. J Hepatol. 2021;75:960-74.
104. Cheng AL, Qin S, Ikeda M, et al. Updated efficacy and safety data from IMbrave150: atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol. 2022;76:862-73.
105. Galle PR, Finn RS, Qin S, et al. Patient-reported outcomes with atezolizumab plus bevacizumab versus sorafenib in patients with unresectable hepatocellular carcinoma (IMbrave150): an open-label, randomised, phase 3 trial. Lancet Oncol. 2021;22:991-1001.
106. Abou-Alfa GK, Lau G, Kudo M, et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evid. 2022;1:EVIDoa2100070.
107. Rimassa L, Chan SL, Sangro B, et al. Five-year overall survival update from the HIMALAYA study of tremelimumab plus durvalumab in unresectable HCC. J Hepatol. 2025;83:899-908.
108. Ren Z, Xu J, Bai Y, et al.; ORIENT-32 study group. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): a randomised, open-label, phase 2-3 study. Lancet Oncol. 2021;22:977-90.
109. Yau T, Park JW, Finn RS, et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022;23:77-90.
110. Zhu AX, Finn RS, Edeline J, et al.; KEYNOTE-224 investigators. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 2018;19:940-52.
111. Finn RS, Ryoo BY, Merle P, et al.; KEYNOTE-240 investigators. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase III trial. J Clin Oncol. 2020;38:193-202.
112. Finn RS, Gu K, Chen X, et al. Second-line pembrolizumab for advanced HCC: meta-analysis of the phase III KEYNOTE-240 and KEYNOTE-394 studies. JHEP Rep. 2025;7:101350.
113. Kudo M, Matilla A, Santoro A, et al. CheckMate 040 cohort 5: a phase I/II study of nivolumab in patients with advanced hepatocellular carcinoma and Child-Pugh B cirrhosis. J Hepatol. 2021;75:600-9.
114. Yau T, Kang YK, Kim TY, et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the CheckMate 040 randomized clinical trial. JAMA Oncol. 2020;6:e204564.
115. Yau T, Galle PR, Decaens T, et al.; CheckMate 9DW investigators. Nivolumab plus ipilimumab versus lenvatinib or sorafenib as first-line treatment for unresectable hepatocellular carcinoma (CheckMate 9DW): an open-label, randomised, phase 3 trial. Lancet. 2025;405:1851-64.
116. Yau T, Park J, Finn R, et al. CheckMate 459: a randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann Oncol. 2019;30:v874-5.
117. Kelley R, Yau T, Cheng A, et al. VP10-2021: Cabozantinib (C) plus atezolizumab (A) versus sorafenib (S) as first-line systemic treatment for advanced hepatocellular carcinoma (aHCC): results from the randomized phase III COSMIC-312 trial. Ann Oncol. 2022;33:114-6.
118. Zhu AX, Kang YK, Yen CJ, et al.; REACH-2 study investigators. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20:282-96.
119. Abou-Alfa GK, Meyer T, Cheng AL, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med. 2018;379:54-63.
120. Abou-Alfa GK, Chan SL, Kudo M, et al. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. J Clin Oncol. 2022;40:379.
121. Rimini M, Rimassa L, Ueshima K, et al. Atezolizumab plus bevacizumab versus lenvatinib or sorafenib in non-viral unresectable hepatocellular carcinoma: an international propensity score matching analysis. ESMO Open. 2022;7:100591.
122. Meyer T, Galani S, Lopes A, Vogel A. Aetiology of liver disease and response to immune checkpoint inhibitors: an updated meta-analysis confirms benefit in those with non-viral liver disease. J Hepatol. 2023;79:e73-6.
123. Akce M, Zaidi MY, Waller EK, El-Rayes BF, Lesinski GB. The potential of CAR T cell therapy in pancreatic cancer. Front Immunol. 2018;9:2166.
124. Wu X, Jin B, Liu X, Mao Y, Wan X, Du S. Research trends of cellular immunotherapy for primary liver cancer: a bibliometric analysis. Hum Vaccin Immunother. 2024;20:2426869.
125. Tian Y, Li Y, Shao Y, Zhang Y. Gene modification strategies for next-generation CAR T cells against solid cancers. J Hematol Oncol. 2020;13:54.
126. Shi Y, Shi D, Chi J, et al. Combined local therapy and CAR-GPC3 T-cell therapy in advanced hepatocellular carcinoma: a proof-of-concept treatment strategy. Cancer Commun. 2023;43:1064-8.
127. Steffin D, Ghatwai N, Montalbano A, et al. Interleukin-15-armoured GPC3 CAR T cells for patients with solid cancers. Nature. 2025;637:940-6.
128. Liu L, Shah K. The potential of the gut microbiome to reshape the cancer therapy paradigm: a review. JAMA Oncol. 2022;8:1059-67.
129. Guo Z, Yao Z, Huang B, et al. MAFLD-related hepatocellular carcinoma: exploring the potent combination of immunotherapy and molecular targeted therapy. Int Immunopharmacol. 2024;140:112821.
130. Lau HCH, Zhang X, Yu J. Gut microbiome in metabolic dysfunction-associated steatotic liver disease and associated hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2025;22:619-38.
131. Craven L, Rahman A, Nair Parvathy S, et al. Allogenic fecal microbiota transplantation in patients with nonalcoholic fatty liver disease improves abnormal small intestinal permeability: a randomized control trial. Am J Gastroenterol. 2020;115:1055-65.
132. Xue L, Deng Z, Luo W, He X, Chen Y. Effect of fecal microbiota transplantation on non-alcoholic fatty liver disease: a randomized clinical trial. Front Cell Infect Microbiol. 2022;12:759306.
133. Aller R, De Luis DA, Izaola O et al. Effect of a probiotic on liver aminotransferases in nonalcoholic fatty liver disease patients: a double blind randomized clinical trial. Eur Rev Med Pharmacol Sci. 2011;15:1090-5.
134. Zhu W, Yan M, Cao H, Zhou J, Xu Z. Effects of Clostridium butyricum capsules combined with rosuvastatin on intestinal flora, lipid metabolism, liver function and inflammation in NAFLD patients. Cell Mol Biol. 2022;68:64-9.
135. Loguercio C, Federico A, Tuccillo C, et al. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J Clin Gastroenterol. 2005;39:540-3.
136. Hai S, Li X, Xie E, et al. Intestinal IL-33 promotes microbiota-derived trimethylamine N-oxide synthesis and drives metabolic dysfunction-associated steatotic liver disease progression by exerting dual regulation on HIF-1α. Hepatology. 2025;82:184-98.
137. Zhu X, Hu M, Huang X, et al. Interplay between gut microbial communities and metabolites modulates pan-cancer immunotherapy responses. Cell Metab. 2025;37:806-23.e6.
138. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908-22.
139. Bian H, Liu YM, Chen ZN. New avenues for NASH therapy by targeting ACC. Cell Metab. 2022;34:191-3.
140. Dong L, Lou W, Xu C, Wang J. Naringenin cationic lipid-modified nanoparticles mitigate MASLD progression by modulating lipid homeostasis and gut microbiota. J Nanobiotechnology. 2025;23:168.
141. Zhang Y, Wang X, Lin J, et al. A microbial metabolite inhibits the HIF-2α-ceramide pathway to mediate the beneficial effects of time-restricted feeding on MASH. Cell Metab. 2024;36:1823-38.e6.
142. Bao J, Zhao Y, Xu X, Ling S. Advances in the use of metformin for liver disease. Curr Med Chem. 2025;32:3591-605.
143. Wabitsch S, McCallen JD, Kamenyeva O, et al. Metformin treatment rescues CD8+ T-cell response to immune checkpoint inhibitor therapy in mice with NAFLD. J Hepatol. 2022;77:748-60.
144. Mossmann D, Müller C, Park S, et al. Arginine reprograms metabolism in liver cancer via RBM39. Cell. 2023;186:5068-83.e23.
145. Chang Y, Wang N, Li S, et al. SLC3A2-mediated lysine uptake by cancer cells restricts T-cell activity in hepatocellular carcinoma. Cancer Res. 2025;85:2250-67.
146. Guo H, Wang M, Ni C, et al. TREM2 promotes the formation of a tumor-supportive microenvironment in hepatocellular carcinoma. J Exp Clin Cancer Res. 2025;44:20.
147. Qiu H, Shao Z, Wen X, et al. TREM2: keeping pace with immune checkpoint inhibitors in cancer immunotherapy. Front Immunol. 2021;12:716710.
148. Allard B, Jacoberger-Foissac C, Cousineau I, et al. Adenosine A2A receptor is a tumor suppressor of NASH-associated hepatocellular carcinoma. Cell Rep Med. 2023;4:101188.
149. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798-809.
150. Fu JT, Liu J, Wu WB, et al. Targeting EFHD2 inhibits interferon-γ signaling and ameliorates non-alcoholic steatohepatitis. J Hepatol. 2024;81:389-403.
151. Gu L, Zhu Y, Lee M, et al. Angiotensin II receptor inhibition ameliorates liver fibrosis and enhances hepatocellular carcinoma infiltration by effector T cells. Proc Natl Acad Sci U S A. 2023;120:e2300706120.
152. Qiu X, Gao F, Wang K, Zhang Z, Shao C, Xu X. Aspirin in hepatocellular carcinoma: is it an out-of-date or promising treatment? ILIVER. 2022;1:55-64.
153. Fan H, Wang R, Wen B, Xiong J. Biomarkers and potential therapeutic targets driving progression of non-alcoholic steatohepatitis to hepatocellular carcinoma predicted through transcriptomic analysis. Front Immunol. 2024;15:1502263.
154. Zou W, Huo B, Tu Y, et al. Metabolic reprogramming by chemo-gene co-delivery nanoparticles for chemo-immunotherapy in head and neck squamous cell carcinoma. Acta Biomater. 2025;199:361-73.
155. Deng H, Li Q, Wang B, et al. Reshaping tumor immune microenvironment through ROS-responsive prodrug polyplexes via synergistic effect of CRISPRi system and epigenetic inhibitor for breast cancer therapy. Mater Today Bio. 2025;35:102285.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].