


Cancer stem cells in cholangiocarcinoma: emerging roles as therapeutic targets
 0
0 
Abstract
Cholangiocarcinoma (CCA) is a highly aggressive liver cancer associated with a poor prognosis. Cancer stem cells (CSCs) play critical roles in CCA by driving tumor initiation, metastasis, recurrence, and treatment resistance. CSCs possess unique properties, including self-renewal, quiescence, and metabolic plasticity, enabling them to evade conventional treatments such as chemotherapy and radiation. Several key CCA stem cell markers are involved in interactions with immune cells within the tumor microenvironment (TME) and in metabolic adaptations that support CSC survival and resistance through complex mechanisms. Despite advancements in chemotherapy and targeted therapies, CSCs remain a major challenge in CCA treatment. In this review, we summarize new insights, updated information, emerging therapeutic strategies, and future directions for CSC research in CCA.
Keywords
INTRODUCTION
Primary liver cancers represent a major global health problem, with nearly 900,000 new cases diagnosed annually worldwide[1]. Among these, cholangiocarcinoma (CCA) is a highly malignant cancer arising from biliary epithelial cells, known as cholangiocytes. Although CCA is relatively rare, its incidence and mortality rates have been steadily increasing globally. Over the past decade, the incidence of CCA has risen by 109%[2]. Today, CCA is the second most common type of primary liver cancer after hepatocellular carcinoma (HCC), accounting for 15% of all cases and roughly 3% of gastrointestinal cancers[3]. CCA can be classified anatomically into intrahepatic cholangiocarcinoma (iCCA) and extrahepatic cholangiocarcinoma (eCCA). eCCA is further subdivided into perihilar cholangiocarcinoma (pCCA) and distal cholangiocarcinoma (dCCA)[4]. Although eCCA is more common than iCCA, distinguishing between them can be challenging. Many cases are diagnosed at an advanced stage due to limited diagnostic tools. The high mortality rate is partly attributable to the poor performance of traditional diagnostic markers, such as CA 19-9 and carcinoembryonic antigen (CEA). Consequently, iCCA is sometimes misdiagnosed as HCC, especially in cirrhotic patients, because of similarities in treatment approaches[5]. Accurate identification of histological type and assessment of the tumor’s molecular profile are critical for selecting appropriate treatments. Furthermore, CCA is highly heterogeneous, which affects both its progression and clinical management. Several risk factors have been identified, including liver fluke infection, primary sclerosing cholangitis (PSC), choledochal cysts, Caroli disease, cirrhosis, chronic hepatitis B or C, obesity, type 2 diabetes, and chronic alcohol consumption[1]. Although CCA is rare worldwide, the highest incidence and mortality rates occur in the Greater Mekong subregion, particularly in northeastern Thailand. In this region, the primary cause is infection with the carcinogenic liver fluke Opisthorchis viverrini (OV)[6].
CCA is an aggressive, highly metastatic tumor with a poor prognosis. Surgical resection is currently the only potentially curative option for early-stage disease. Nevertheless, the 1-, 3-, and 5-year survival rates after surgery are 52.1%, 21.7%, and 11.2%, respectively, with a median survival time of 12.4 months[7,8]. For advanced or unresectable CCA, the standard first-line chemotherapy is a combination of gemcitabine and cisplatin[9]. When first-line treatment fails, there is no established standard second-line therapy. However, several clinical trials have been conducted to address this gap. Notably, as a second-line treatment following cisplatin/gemcitabine therapy, the combination of oxaliplatin and 5-fluorouracil (FOLFOX regimen)showed promising results in a phase III randomized study (UK ABC-06)[5,10]. Nevertheless, further improvements in chemotherapy are urgently needed. Recent advances in omic technologies have identified several genetic alterations in CCA, including mutations in TP53, KRAS, SMAD4, CDKN2A/B, isocitrate dehydrogenases (IDH), and members of the FGFR and EGFR families[9]. As a result, targeted therapies have been developed and evaluated in multi-phase clinical trials, including FGFR2 inhibitors, BRAF inhibitors, HER2 inhibitors, and IDH inhibitors[11]. While these precision treatments have improved survival and prognosis for some patients, only a few achieve long-term benefits. Most patients eventually develop drug resistance and experience tumor recurrence, often within 2 years of surgical resection[12].
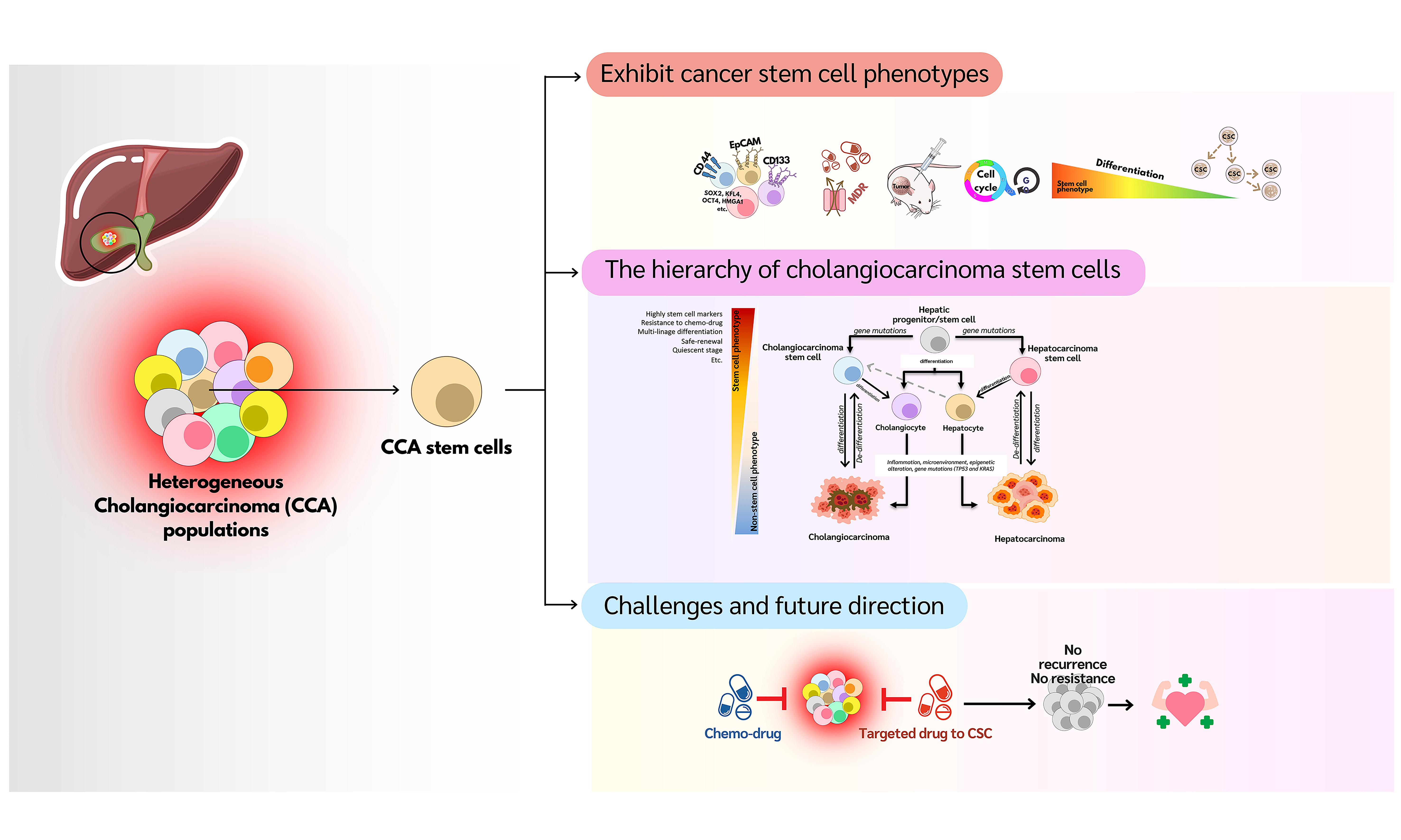
Accumulating evidence indicates that, like other malignant tumors, CCA harbors a significant population of cancer stem cells (CSCs)[13]. CSCs are a subpopulation of tumor cells with self-renew and differentiation capabilities, similar to normal stem cells[14]. In CCA, CSCs are implicated in tumor initiation, metastasis, recurrence, and chemotherapy resistance[15]. Overexpression of CSC markers in CCA tissue samples has been correlated with poor clinical outcomes and a higher likelihood of postoperative recurrence[16,17]. However, research on CSCs in CCA remains limited. This review aims to provide an updated overview of CSCs in CCA and discuss future research directions, which may contribute to the development of novel therapeutic strategies for this challenging disease.
BIOLOGICAL CHARACTERISTICS OF CSCS
The concept of CSCs was first established in human acute myeloid leukemia (AML) cells by Dick’s lab in the late 20th century. AML stem cells were identified by the presence of CD34 and absence of CD38 (CD34+CD38-). This subpopulation possesses the ability to self-renew, differentiate, and generate AML, and was thus proposed as the CSC population in human AML[18]. Over the past decades, researchers have proposed two major models for the origin of CSCs [Figure 1][19]. The first, known as the “Hierarchical Model”, suggests that CSCs originate from normal stem cells or progenitor cells that acquire genetic and epigenetic alterations over time. Accumulation of these mutations transforms them into CSCs. Through self-renewal, CSCs can asymmetrically divide to maintain their own population and produce short-lived progenitor cells with limited proliferative capacity[19]. The second, the “Stochastic Model”, proposes that any cell within a tumor has the potential to de-differentiate into a stem-like state under certain conditions. This model is based on tumor heterogeneity, emphasizing that tumor cell behavior is influenced by both intrinsic and extrinsic factors. These factors contribute to the clonal expansion of cancer cells and the relative quiescence of CSCs[20].
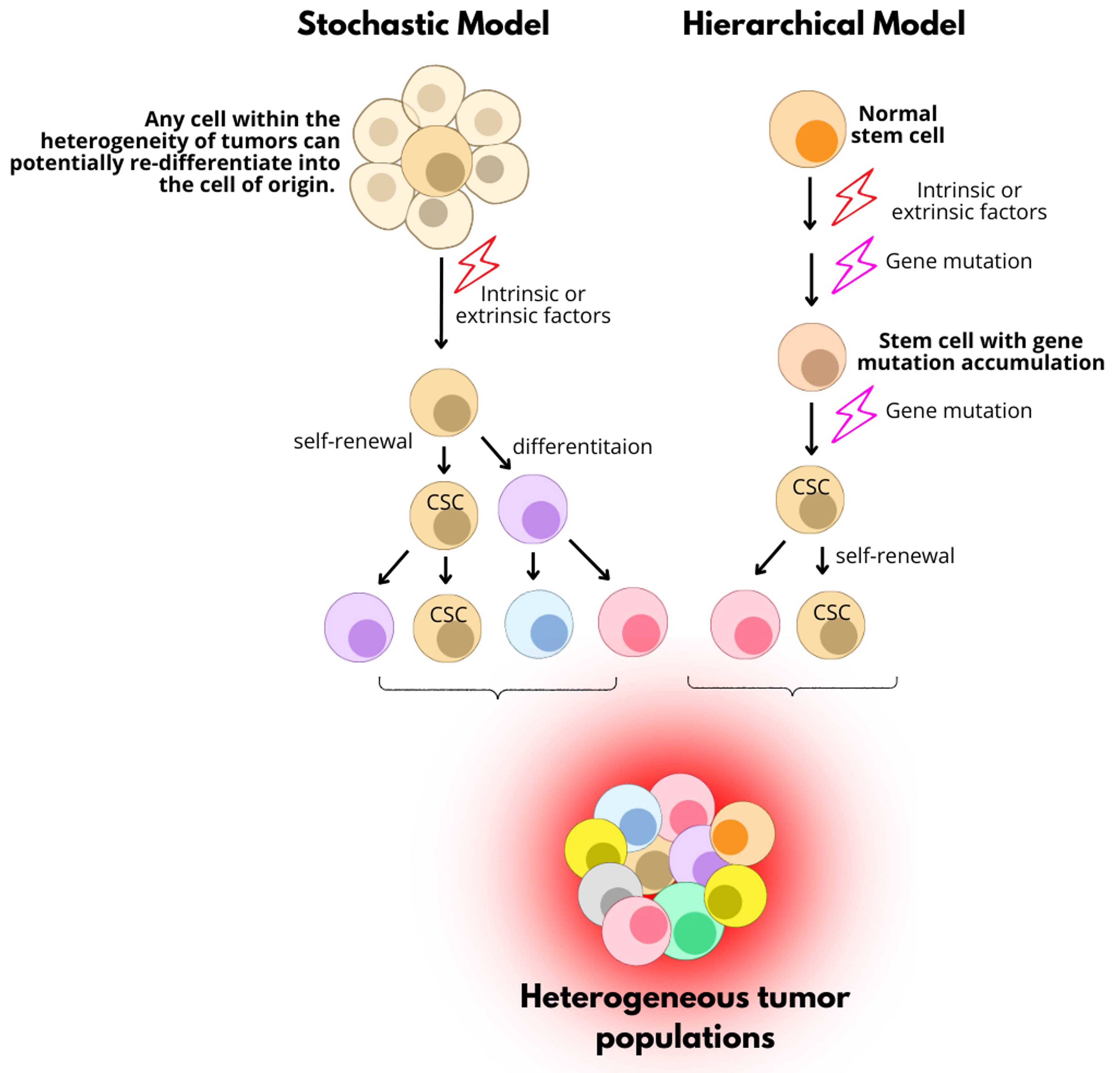
Figure 1. Two proposed models for the origin of CSCs. The “Stochastic Model” suggests that any tumor cell can become a CSC through cellular plasticity driven by intrinsic or extrinsic factors, reflecting tumor heterogeneity. In contrast, the “Hierarchical Model” posits that normal stem cells acquire genetic mutations through intrinsic or extrinsic influences, transforming them into CSCs. CSC: Cancer stem cell.
Similar to normal stem cells, CSCs share at least two defining features: self-renew and the potential to differentiate. Self-renewal allows CSCs to divide symmetrically into two identical CSCs or asymmetrically into one CSC and one more differentiated daughter cell. CSCs typically reside in a specialized microenvironment called the “niche”, which includes neighboring supportive cells, the extracellular matrix (ECM), and other factors essential for their survival. This niche helps sustain the long-term repopulation potential of CSCs[21]. Key signaling pathways are preferentially activated in CSCs, including the Wnt/β-catenin, Notch, Hedgehog (Hh), insulin-like growth factor (IGF), and nuclear factor-kappa B (NF-κB) pathways[22].
Despite advances in cancer diagnosis and treatment, recurrence and chemotherapy resistance remain major concerns. These challenges are closely related to the biological properties of CSCs. Understanding these properties is crucial for developing more effective cancer treatments. However, the unique characteristics of CSCs are not yet fully understood due to their variability across different cancer types. Nonetheless, accumulating evidence highlights several key traits of CSCs, as shown in Figure 2.
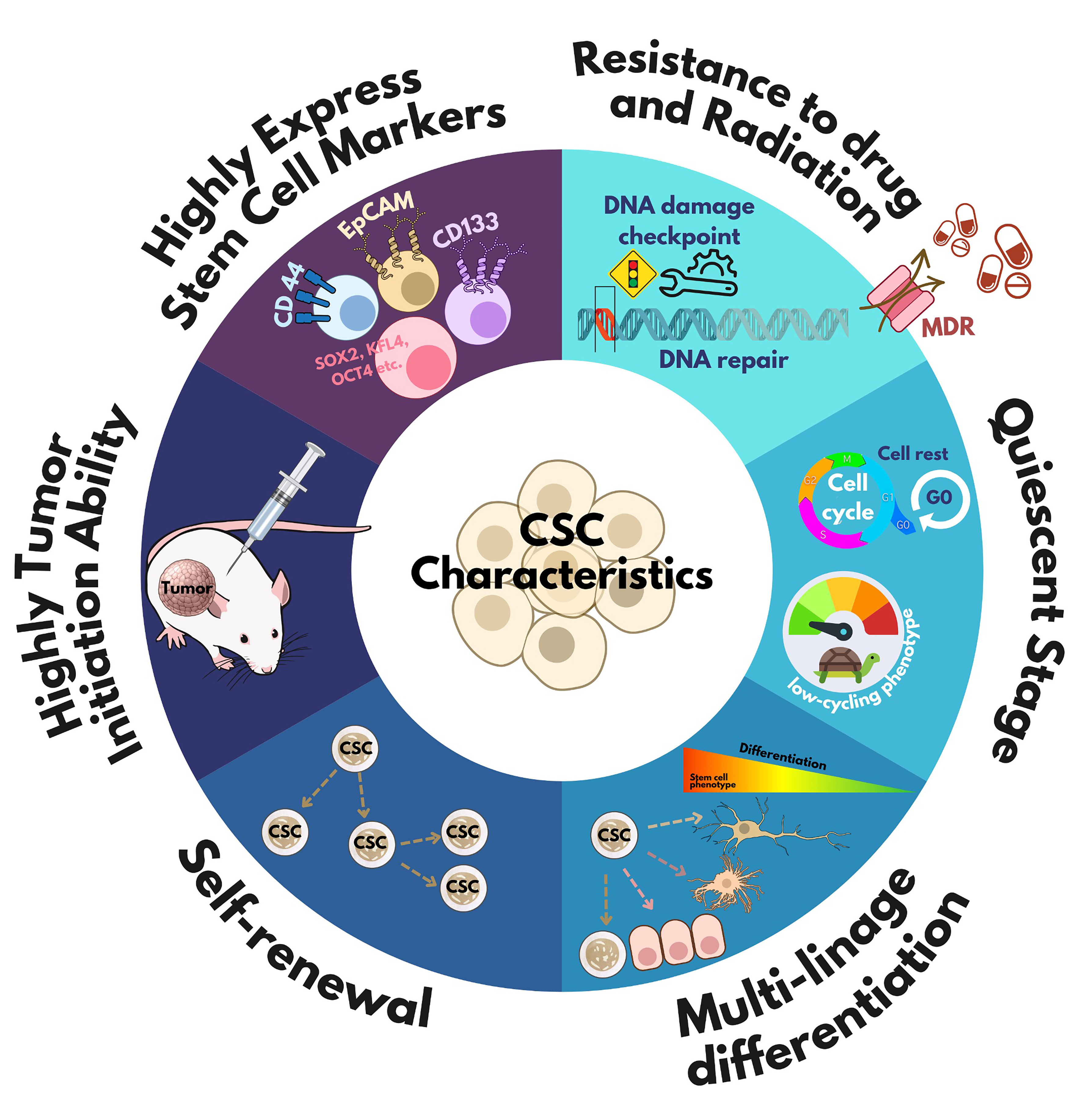
Figure 2. Cancer stem cell characteristics. The common characteristics of CSCs -such as self-renewal, expression of stem cell markers, multi-lineage differentiation, slow proliferation during quiescence, resistance to chemotherapy/radiation, and tumor-initiating ability - can be used for CSC identification. CSC: Cancer stem cell; CD: cluster of differentiation; MDR: multi-drug resistance; SOX: SRY-related HMG-box; KLF: Krüppel-like factor; oct-4: octamer-binding transcription factor 4.
CSCs highly express stem cell markers
Understanding and identifying CSC-specific markers is critical for the development of diagnostic and therapeutic strategies. Although definitive CSC markers remain elusive, similarities between CSCs and normal stem cells suggest that some stem cell markers are overexpressed in CSCs. Transcription factors such as SRY-box transcription factor 2 (SOX2), MYC, Oct4, and KLF contribute significantly to CSC characteristics by regulating self-renewal, differentiation, and tumorigenic potential[23]. Apart from transcription factors, several cell surface markers, including CD44, CD133, epithelial cell adhesion molecule (EpCAM), CD34, and CD38, can serve as diagnostic indicators of CSCs. Moreover, cytoplasmic proteins such as aldehyde dehydrogenase (ALDH) and Musashi-1/2 have also been identified as CSC markers in various cancers, where they contribute to the regulation of stem-like phenotypes[24]. Suppressing these markers can potentially impair the stemness, therapeutic resistance, and tumorigenic capacity of CSCs[15,25,26].
Recently, the accumulation of omics data has facilitated the identification and characterization of CSCs in different malignancies. Firdous et al. compiled CSC biomarker information into a freely accessible online resource, the Cancer Stem Cells Database (BCSCdb), which includes five categories of CSC biomarker interactions, ten clinical trial biomarkers as drug targets, and 445 CSC-related therapeutic target genes
Frequently used markers for CSC identification
| Cancer type | Markers | References |
| AML | CD34+CD38-, CD123, TIM3, CD25, CD32, CD96 | [28] |
| Bladder | Lin-CD44+CK5+CK20-, CD133, CD44, SOX2 | [29,30] |
| Breast | CD44+/CD24−/low, ALDH1+, CD24, ABCG2, CD133, CD49f, LGR5, SSEA-3, CD70, PROCR, CD44 | [31,32] |
| Glioblastoma | CD133, SSEA1, CD15, L1CAM, SOX2, Musashi-1, Nestin | [33,34] |
| Colon | CD133+, Bmi-1, CD24, CD26, CD29, CD44, CD44v6, EpCAM, LGR5 | [35] |
| Head and neck | CD44, SALL4, CD133, CD90, ALDH+, Nanog, Oct-4, SOX2 | [36] |
| Hepatocellular carcinoma | CD90+CD44+, EpCAM | [37,38] |
| Melanoma | CD133+ | [39] |
| Ovarian | CD44+CD117+, OCT4, SSEA4, ALDH1/2, LGR5, CD133, CD24 | [40,41] |
| Pancreatic | CD133, CD24, CD44, CXCR4, EpCAM, ABCG2, c-Met, ALDH-1, nestin | [42] |
| Nasopharyngeal Carcinoma | CD24 | [43] |
| Gastric carcinoma | CD44v3 | [44] |
CSCs exhibit high tumor-initiating capacity in xenotransplantation models
Xenotransplantation into immunodeficient mice has been widely used to identify CSCs based on their ability to self-renew and initiate tumors[14,45]. This approach has been applied across various cancer types, including brain, pancreatic[46], ovarian, CCA[15], and lung cancers[47]. For example, studies in human breast cancer showed that as few as 100 CD44+/CD24-/low cells - representing the stem-like population - could generate tumors in mice, whereas 10,000 cells with different phenotypes could not[48]. In colon cancer, approximately 2.5% of the CD133+ cell population possesses tumor-initiating capability. CD133+ cells generate tumors more rapidly in immunodeficient mice compared to CD133- cells, and tumor growth is further enhanced upon serial transplantation without significant phenotypic changes[49]. In CCA, CSCs demonstrated significantly faster and more robust tumor formation than their parental cancer cells when equal numbers were injected into BRJ mice[15].
CSCs can enter a quiescent state
The quiescent stage (G0 phase) is a reversible stage of cell cycle arrest in which cells exhibit low metabolic activity to maintain genomic integrity. This feature is well-documented in adult stem cells[50]. Like their normal counterparts, CSCs can transition between quiescent and proliferative states depending on tumor microenvironmental factors such as nutrient availability, intrinsic signaling, and mitochondrial membrane dynamics[50]. Quiescent CSCs are typically characterized by low RNA content, absence of proliferation markers, and minimal cell division[50]. Studies in hematopoietic stem cells (HSCs) have shown that downregulation of genes involved in DNA replication and cell cycle progression, including cyclin A2, cyclin B1, cyclin E2, and survivin, is associated with the quiescent state[51]. Importantly, quiescent CSCs can migrate to distant sites and cause tumor relapse or metastasis, even years after treatment of the primary tumor[52]. Due to their low proliferation rate, quiescence is considered a major contributor to therapeutic resistance and tumor recurrence in several cancers, such as CCA[15] and glioma[53]. Therefore, elucidating the mechanisms of CSC quiescence is a major focus in current cancer research.
CSCs resist chemotherapy and radiation
CSCs exhibit resistance to chemotherapy and radiation due to several inherent characteristics. Most current chemotherapeutic drugs are designed to target rapidly proliferating cancer cells. However, CSCs often display a slow-cycling or quiescent phenotype, allowing them to evade chemotherapy-induced cytotoxicity. The persistence of these quiescent CSCs contributes to chemotherapy resistance and tumor recurrence after standard treatments[22]. This phenomenon has been observed in various solid tumors. For example, glioma stem cells, which are relatively quiescent, can survive treatment with temozolomide (TMZ) and sustain long-term tumor growth with a high proliferative index in mouse models[54]. Similarly, in squamous cell carcinoma, slower-cycling cells exhibit increased resistance to cisplatin-based chemotherapy[55]. In addition, CSCs contribute to radioresistance. For example, CD133+ glioma cells are enriched in patients after radiotherapy. These CD133+ cells, isolated from human glioma xenografts and patient tumor tissues, showed enhanced resistance to radiation by activating DNA damage checkpoint responses and promoting more effective DNA repair compared to CD133- cells[56]. CSCs employ multiple mechanisms to resist chemo/radiotherapy. They often overexpress multidrug resistance (MDR) transporters, such as members of the ATP-binding cassette (ABC) transporter family, which enhance drug efflux and contribute to chemoresistance[57]. In CCA, CSCs exhibit high expression levels of proteins related to MDR, including ALDH3A1, ALDH1A1, and ABC transporters, thereby conferring chemoresistance[15]. High ALDH expression is also linked to increased migratory capacity and gemcitabine resistance in CCA cells[15,58]. Moreover, the CSC niche plays a critical role in mediating resistance, as CSCs rely on this specialized microenvironment for survival. Hypoxic conditions within the niche promote drug resistance by inducing the accumulation of hypoxia-inducible factors (HIF1α, HIF2α). HIF activation inhibits differentiation, regulates apoptosis, and enhances DNA repair, collectively contributing to chemotherapy resistance[57,59]. Recently, a specialized microenvironment termed the “glyco-niche”, characterized by chondroitin sulfate-modified CSPG4 (NG2) and related signaling pathways, has been identified as a key factor underlying the therapeutic resistance of glioma stem cells[60]. Targeting pathways that regulate these resistance mechanisms may enhance the sensitivity of CSCs to anticancer therapies.
CSCs possess multi-lineage differentiation ability
CSCs exhibit multipotency, enabling them to differentiate into various cell types. This unique property plays a direct role in tumorigenesis and contributes to tumor heterogeneity[14]. According to the CSC theory of cancer progression, CSCs occupy the top position in the hierarchical organization of heterogeneous tumor cell populations. From this apex, they generate the more differentiated bulk of tumor cells, which possess unlimited proliferative potential. The CSC niche mediates extracellular cues that regulate stemness and direct differentiation into diverse tumor phenotypes[14]. For example, colon CSCs have been shown to differentiate into blood cell types under chemically induced hypoxic conditions, as evidenced by high expression of erythroblast markers CD34 and TER-119. This result suggests that CSCs possess the capacity for multi-lineage differentiation into erythroblasts and erythrocytes under certain conditions.
When CCA-CSCs were cultured in adipogenic and osteogenic differentiation media, they differentiated into adipocytes and osteocytes, respectively. These findings indicate that CCA-CSC-like cells can differentiate into various cell types depending on the microenvironment and inducing factors[15]. Furthermore, in the presence of fetal bovine serum (FCS) in spheroid cell culture, CCA-CSCs became adherent cells resembling parental tumor cells. These FCS-induced differentiated cells showed reduced stem-like properties, such as decreased expression of stem cell markers and lower chemoresistance. A comprehensive mass spectrometry-based analysis revealed distinct protein expression profiles between CCA-CSCs, FCS-differentiated cells, and parental CCA cells[15]. The involvement of the integrin signaling pathway during CSC differentiation suggests that this pathway may serve as a niche for CSC differentiation[15,61,62]. This differentiation ability of CSCs provides new insights into how the tumor microenvironment (TME) influences CSC fate, contributing to tumor heterogeneity[63] and potentially attenuating aggressive behavior[64].
The differentiation ability of CSCs is often referred to as “plasticity”, which describes their capacity for phenotypic transformations, such as epithelial-mesenchymal transition (EMT), mesenchymal-epithelial transition (MET), trans-differentiation into stem/progenitor cells, or conversion between different differentiated states[13]. This plasticity plays a crucial role in cancer initiation and progression. Many intrinsic and extrinsic factors influence cellular plasticity. For example, activation of PKA can induce MET and reduce tumor-initiating ability, thereby diminishing stem cell characteristics in liver cancer[65]. Additionally, inflammatory cytokines such as TNF-α, IL-6, CCL22, and TGF-β promote the trans-differentiation of hepatocytes or non-tumor cells into stem-like phenotypes, facilitating tumor recurrence after conventional chemotherapy[13]. IL-6 can also promote the conversion of HCC cells into liver stem cells via the JAK1-STAT3-OCT4 pathway, enhancing their stemness[66]. According to the cellular plasticity model, CSCs are not necessarily the origin source of cancer. Instead, certain malignantly differentiated cells harboring oncogenic mutations can trans-differentiate into stem-like cells, thereby contributing to intratumoral heterogeneity[21]. CSCs also display highly plastic metabolic profiles, allowing them to survive under stress by dynamically switching between oxidative phosphorylation (OXPHOS) and glycolysis. The differentiation process of CSCs is highly complex due to the many factors influencing it. Therefore, understanding and controlling this process has emerged as a promising strategy to eradicate CSC populations and prevent tumor recurrence and heterogeneity.
CCA STEM CELLS
Cellular origin of CCA
In 1989, Sell et al. proposed, using a rat model of chemical hepatocarcinogenesis, that HCC and CCA originate from pluripotent liver stem cells capable of developing into CCA[67]. Since then, multiple studies have demonstrated that HCC and CCA may arise from common bipotent hepatic progenitor cells. These progenitor cells, which highly express specific stem cell markers, can differentiate into either hepatocytes or cholangiocytes[68]. Genetic mutations in TP53 and KRAS - commonly found in both HCC and CCA - can drive these progenitor cells to undergo malignant transformation, leading to HCC and CCA, respectively
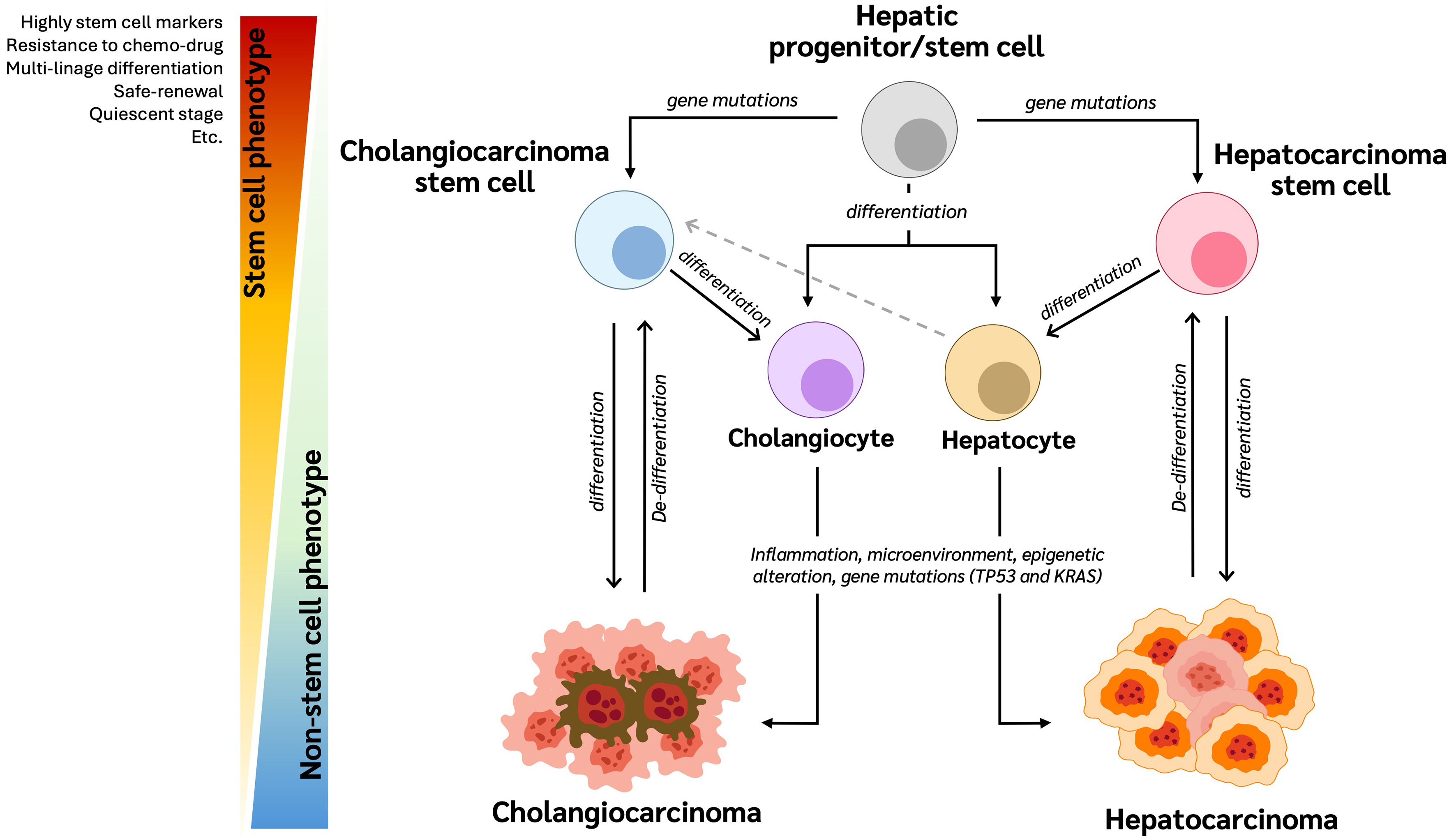
Figure 3. The hierarchy of cholangiocarcinoma stem cells. Hepatic progenitor/stem cells differentiate into hepatocytes and cholangiocytes. Through carcinogenesis involving various factors, these cells can develop into HCC and CCA, respectively. Moreover, hepatic progenitor/stem cells with genetic abnormalities can reprogram into CCA-CSCs or HCC cells. These abnormal cells then proliferate and differentiate to form CCA and HCC cells. Additionally, hepatocytes can act as stem-like cells in the liver and possess the ability to reprogram into CCA-CSCs, suggesting that mature hepatocytes may serve as an origin of CCA through genetic and epigenetic alterations, as well as influences from the microenvironment. CSC: Cancer stem cell; CCA: cholangiocarcinoma; HCC: hepatocellular carcinoma.
Building on this, genomic alterations, including gene mutations and chromosomal aberrations such as gains, losses, or rearrangements, are recognized as key drivers of cellular transformation and play a central role in cancer initiation and progression[71]. The differential identification of genetic alterations between iCCA and eCCA holds important implications for classification, diagnosis, prognosis, and treatment[72]. Recent integrative molecular profiling studies have identified several genetic alterations: mutations in FGFR1-3, IDH1, IDH2, BAP1, RNF43, and EPHA2 occur more frequently in iCCA, whereas mutations in ARID1B and ELF3, and fusions involving PRKACA and PRKACB are more common in eCCA[73]. Moreover, mutations in TP53, KRAS, ARID1A, PIK3CA, and loss of CDKN2A/B are common to both iCCA and eCCA[74]. These molecular distinctions are crucial for developing targeted therapies. Apart from these mutations, genetic heterogeneity across ethnic populations has also been highlighted. Comparative analyses between Asian and Western cohorts suggest that variations in genetic profiles may reflect differences in underlying etiological risk factors. Notably, Asian iCCA patients - more often exposed to liver fluke infections and hepatitis B virus (HBV) - tend to have a higher frequency of DNA repair gene mutations than Western patients, whose risk factors include PSC, metabolic syndrome, and inflammatory bowel disease[75]. These population-specific genetic patterns underscore the importance of considering regional etiological factors. PSC is a major risk factor for both iCCA and eCCA, although iCCA commonly develops in the context of PSC[76]. However, the molecular mechanisms underlying PSC-associated carcinogenesis remain incompletely understood. PSC-related iCCA shares several clinical and pathological features with eCCA, particularly with the perihilar subtype (pCCA), and differs from cirrhosis-associated iCCA[77]. In a study of 10 CCA patients with PSC involving the perihilar region, gallbladder, and intrahepatic areas, genetic analysis revealed frequent loss of chromosome 9p21 and inactivation of the p16 tumor suppressor gene. This highlights the importance of distinguishing PSC-associated CCA from CCA of other etiologies, particularly when considering therapeutic interventions and targeted therapies[77].
Chromosomal abnormalities are closely linked to disease etiology and have significant implications for targeted therapy, potentially serving as predictive markers in treatment planning. For instance, in a cohort of 194 CCA patients, chromosomal aberrations such as gains of chromosomes 3 and 7, polysomy of chromosomes 3, 7, and 17, and heterozygous loss of 9p21 were identified. Patients with polysomy of chromosomes 3 + 7 exhibited significantly shorter overall survival, and those with polysomy of chromosomes 3, 7, and 17, along with heterozygous 9p21 loss, had even poorer survival outcomes. Notably, these alterations were not associated with the anatomical subtypes of CCA (iCCA vs. eCCA)[78]. These findings suggest that genetic alterations may serve as prognostic markers for poor outcomes in CCA patients. Moreover, distinct chromosomal patterns have been observed between gemcitabine-responsive and non-responsive CCA patients, indicating that chromosomal signatures may predict chemotherapy responses. This knowledge could enhance personalized treatment strategies and guide chemotherapy selection[79]. The association between chemotherapy resistance and CSC characteristics suggests that chromosomal abnormalities may contribute to therapeutic resistance by promoting stemness-related phenotypes. Although the roles of chromosomal aberrations in CCA are well recognized, the precise origins of CCA and CCA-CSCs remain unclear. Several hypotheses propose that increased chromosomal instability, arising from cell transformation, cell-cell fusion, and horizontal gene transfer in normal stem cells, progenitor cells, or even differentiated cells, may represent important sources of CSCs and key drivers of cancer progression[71,80]. Supporting this, previous studies have demonstrated that circulating HSCs can fuse with resident cells in various tissues, potentially contributing to the generation of CSCs and tumorigenesis in organs such as the intestine[81].
Collectively, genomic alterations at both the gene and chromosomal levels play crucial roles in tumor initiation and progression, as well as in the generation of CSCs. Therefore, a comprehensive understanding of both chromosomal and genetic aberrations, along with their resulting protein products, is essential for developing more effective targeted therapies and improving clinical outcomes for CCA patients.
CCA stem cell markers
CSC populations have been identified in CCA tissues for over a decade. Numerous CSC markers have been characterized in CCA-CSCs and these stem cell markers play vital roles in CCA progression. High expression levels of CSC markers such as CD44, CD44v6, CD44v8-10, and EpCAM in CCA tissues are associated with poor clinical outcomes and a higher likelihood of postoperative recurrence[16]. Moreover, CD44v9, when highly expressed, significantly contributes to stem-like phenotypes and promotes CCA progression through modulation of the Wnt-β-catenin signaling pathway[25]. In addition, ALDH-high CCA cells exhibit significantly enhanced migratory capacity and resistance to gemcitabine[58]. The ALDH family comprises intracellular enzymes involved in detoxification, differentiation, and resistance to chemotherapeutic drugs, and ALDH enzymes are commonly used as stem cell markers in various cancers[24]. However, the specific CCA subtypes related to these markers remain under investigation.
Several challenges complicate the identification of CCA-CSCs. For instance, iCCA and HCC share certain features. Some iCCA populations are enriched with liver-specific stem cells and molecular signatures linked to poor prognosis in HCC, suggesting a possible shared origin and similar expression of stem cell markers such as EpCAM, SALL4, CK19, and CD44[13]. While HCC shows greater heterogeneity with a smaller CSC fraction, CCA is highly invasive and tends to have a larger CSC proportion[13]. To date, the differences in CSC biology between HCC and CCA have not been fully clarified, and precise stem cell markers for each CCA subtype remain unidentified. Oishi et al. performed transcriptomic profiling on 23 iCCA and cHCC-CCA tumor samples and found that several hepatic stem/progenitor cell-specific genes, including POU5F1 (Oct4), NANOG, MYC, TGFB1, NCAM1, and PROM1, were more abundantly expressed in stem-like iCCA cells than in mature hepatocytes[82]. Their results also showed that miR-200c contributes to the EMT pathway and maintains stem-like features in iCCA; its expression is associated with poor prognosis[82].
Separately, Lin et al. used small RNA sequencing to identify microRNA (miRNA) expression patterns in CCA. The let-7c/miR-99a/miR-125b cluster plays an important role in regulating stem-like properties by downregulating CD133 and CD44. This evidence highlights the potential of miRNA as therapeutic targets and supports the development of miRNA-based therapies for CCA[83].
Accumulating evidence suggests that CCA is a stem cell-driven disease. Therefore, further investigation is necessary to elucidate the role of CSCs in CCA development and progression. In 2023, Panawan et al. established and characterized CCA-CSC clones as long-term, sphere-forming stem cells that may serve as representative CSC models for further in vitro and in vivo studies[15]. These CCA-CSCs exhibited stem cell phenotypes, including high expression of stem cell markers, multi-lineage differentiation potential, and strong tumorigenic capacity in mouse xenograft models. To explore the molecular mechanisms regulating CSCs, proteomic analyses were performed by comparing CCA-CSCs with their differentiated progenitor cells and parental CCA cells. Approximately 6,000 proteins were quantitatively identified, with 1,123 proteins significantly upregulated in CCA-CSCs. These included stem cell markers such as CD44, CD38, EpCAM, Klf4, ALDH3A1, ALDH1A1, and ABCB1, as well as chemoresistance-related proteins including ALDH3A1, ALDH1A1, and ATP-binding cassette (ABC) transporters. Further clustering and network analyses revealed that HMGA1 is selectively highly expressed and plays an important role in CCA-CSC biology, representing a novel stem cell marker in CCA[15]. HMGA proteins (HMGA1 and HMGA2) interact with the transcriptional machinery to regulate gene expression, playing key roles in stem cell self-renewal and differentiation[84,85]. Suppression of HMGA1 induced differentiation of CCA-CSCs, increasing their sensitivity to chemotherapeutic agents. This effect was particularly pronounced when HMGA1 suppression was coupled with chemotherapy[15]. Additionally, aurora kinase A (Aurora-A) was enriched in the CCA stem cell model and is also considered a novel CSC marker. Aurora-A, a serine/threonine kinase, plays a crucial role during meiosis[86]. In glioma stem cells, Aurora-A supports self-renewal and tumorigenicity by interacting with AXIN and inhibiting the AXIN/GSK3β/β-catenin complex, leading to Wnt pathway activation[87]. Aurora-A can also translocate to the nucleus to activate the MYC promoter, enhancing CSC traits in breast CSCs[88]. Inhibition of Aurora-A reduces the viability of both CCA stem cells and parental CCA cells. v, simultaneous suppression of HMGA1 and Aurora-A has a synergistic effect on specifically targeting CCA-CSCs, emphasizing their roles in CCA-CSC biology[15]. Although the exact mechanism by which HMGA1 influences AURKA transcription remains unclear, strong correlations in mRNA levels of HMGA1 and AURKA have been observed in CCA patient tissues. Based on current evidence, HMGA1 and Aurora-A are proposed as potential therapeutic targets in CCA, with a focus on eradicating CSC populations. Recent studies have also indicated that the upregulation of STAT3 modulates HMGA1 and AURKA expression, which may control the stem-like cell phenotypes of CCA [Figure 4][15]. Further research is needed to understand the mechanisms regulating CCA-CSCs. Several important CSC markers in CCA are summarized in Table 2.
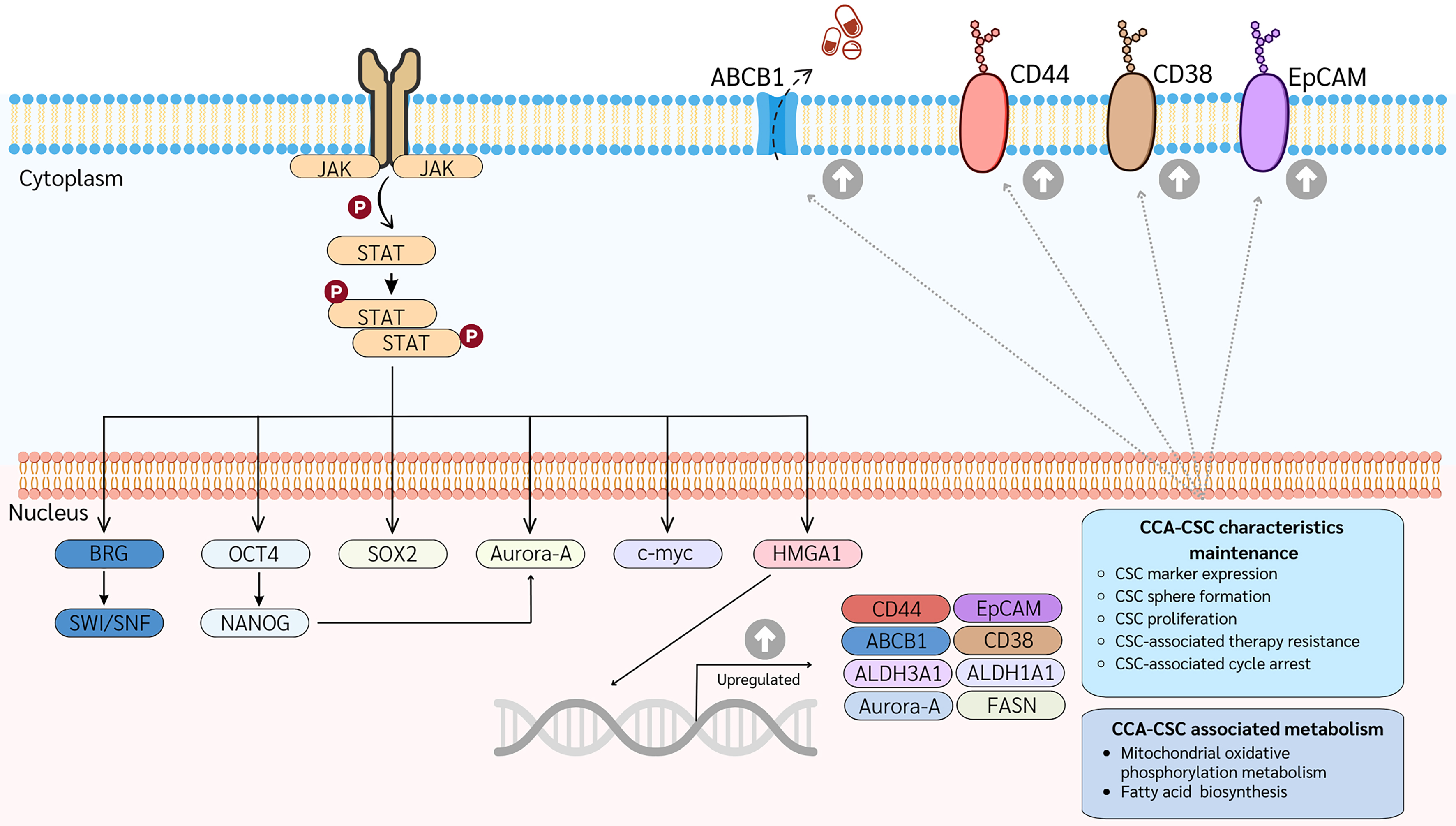
Figure 4. Schematic signaling pathways controlling CCA-CSCs. The STAT3-HMGA1-Aurora A network is highly enriched in CCA-CSCs, regulating their stem-like characteristics and metabolism. All proteins depicted are upregulated in CCA-CSCs. CSC: Cancer stem cell; CCA: cholangiocarcinoma; JAK: janus Kinase; STAT: signal transducer and activator of transcription; ABC: ATP-binding cassette transporter; CD: cluster of differentiation; EpCAM: epithelial cell adhesion molecule; BRG: brahma-related gene; SWI/SNF: SWItch/sucrose non-fermentable; OCT4: octamer-binding transcription factor 4; NANOG: homeobox protein NANOG; FASN: fatty acid synthase.
Cancer stem cell markers in cholangiocarcinoma
| Markers | Functional roles in CCA |
| CD44/CD44v9 | Play important roles in cell migration, differentiation, and survival. Suppression of CD44v9 induces apoptosis and cell cycle arrest, and inhibits migration/invasion and tumor growth in mouse xenografts[25] |
| CD38 | Highly expressed in CCA stem cell models compared to parental CCA cells[15] |
| CD147 | Promotes metastasis in CCA by modulating EMT[89]. Highly expressed in CCA stem cell models compared to parental cells[15] |
| CD133 | High expression correlates with increased metastasis and invasiveness[90] and with shorter overall and disease-free survival compared to CD133- patients[91,92] |
| CD24 | Overexpression predicts poor survival and is associated with resistance to chemotherapy and radiotherapy[93] |
| EpCAM | Highly expressed in CCA, associated with poor prognosis and high recurrence rates[16,70,92] |
| ALDH | Responsible for aldehyde detoxification[58]. ALDH1A1 and ALDH3A1 are highly expressed in CCA stem cell models[15]. ALDHhigh CCA cells show enhanced migration and gemcitabine resistance. High ALDH1A3 expression is linked to poor prognosis[58] |
| ABC transporter | Play roles in drug efflux and resistance[94]. ABC transporters (ABCB1, ABCG2, ABCA3) are upregulated in 5-FU-resistant cells at the mRNA level[94], and ABCB1 is upregulated at the protein level in CCA stem cell models[15] |
| SOX2 | Important for pluripotency and self-renewal[95]. Overexpression is associated with aggressive behavior, poor survival, and recurrence[16,96]. Suppression reduces aggressiveness[97]. Upregulated at both mRNA and protein levels in CCA-CSCs[15] |
| c-myc | Regulates growth and proliferation. Inhibition induces G0/G1 cell cycle arrest[98]. Upregulated at the mRNA level in CCA-CSCs[15] |
| Oct3/4 (POU5F1) | Key for pluripotency and reprogramming[99]. Upregulation is associated with gemcitabine resistance[100]. Upregulated at the mRNA level in CCA-CSCs[15] |
| HMGA1 | Highly expressed in CCA, linked to poor prognosis. Promotes radioresistance via DNA repair gene activation. Suppression reduces viability, induces differentiation, and sensitizes cells to chemotherapy [15,101] |
| AURKA | Controls mitosis and centrosome function[88]. Suppression reduces the viability of both CSCs and parental cells, with synergistic effects when combined with HMGA1 inhibition[15] |
Microenvironment for CCA-CSC
The TME refers to the ecosystem surrounding a tumor. This ecosystem comprises various cell types, including immune cells, fibroblasts, blood vessels (particularly vascular endothelial cells), as well as the ECM and soluble factors. The TME plays a crucial role in cancer progression and has been proposed as a potential target for cancer treatment[102]. Similar to its role in other cancers, the TME also critically modulates CSC behavior, forming what is referred to as the ‘‘CSC niche’’. The CSC niche represents the specific microenvironment surrounding CSCs, which includes tumor-associated macrophages (TAMs), cancer-associated fibroblasts (CAFs), and various small molecules such as soluble factors, cytokines, and growth factors. This specialized microenvironment controls CSC self-renewal, regulates stemness and differentiation, and enhances drug resistance[103]. Consequently, eliminating CSCs by targeting a single molecular marker or signaling pathway remains challenging due to the complex crosstalk between CSCs and different cells within the TME. When CCA-CSCs were cocultured with macrophages, both M1 (CD80 and HLA-DR) and M2 (CD163) markers were induced, indicating polarization toward a tumor-associated macrophage phenotype. This polarization promotes the self-renewal ability of CCA-CSCs[104]. CSCs require a specific microenvironment to maintain their stemness properties. This was self-renewal ability in studies using 3D culture conditions to generate spheres mimicking the CCA-CSC environment, where secreted molecules such as IL13, osteoactivin (OA), and IL34 were shown to recruit infiltrating monocytes and prime macrophage activation[95]. These findings suggest that CCA-CSCs are closely associated with macrophages, providing a rationale for developing combination therapies targeting both CCAs and their niche in CCA. Moreover, the CSC niche not only modulates CSC behavior but also plays an important role in regulating other surrounding cell populations within the tumor.
Metabolism of CCA-CSCs
The metabolic phenotype of CSCs has been extensively studied for decades and appears to vary depending on tumor type. For instance, stem-like cells from breast cancer, nasopharyngeal carcinoma, and HCC preferentially rely on glycolysis for their energy metabolism[105]. Ragg et al. showed that mitochondrial oxidative metabolism contributes to the CSC phenotype in CCA, as evidenced by high mitochondrial membrane potential, increased mitochondrial mass, and overexpression of the master regulator of mitochondrial biogenesis (PGC-1α). Conversely, glucose uptake and lactate production - both metabolic hallmarks of glycolysis - were reduced in cells exhibiting a stem-like phenotype, suggesting that CCA stem cells primarily depend on mitochondrial oxidative phosphorylation[17]. Another study revealed that iCCA stem cells produce high levels of free fatty acids (FAs) and unsaturated triglycerides (TAGs), supported by the upregulation of fatty acid synthase (FASN), a key enzyme in de novo fatty acid biosynthesis. Elevated FASN expression correlated with poor prognosis in iCCA patients. Inhibition of FASN reduced stem-like properties and suppressed tumor growth in both in vitro and in vivo models. These results suggest that alterations in fatty acid metabolism are crucial for maintaining the stem-like characteristics of iCCA and that targeting FASN could represent a potential therapeutic approach for CCA[106]. Moreover, many CSCs exhibit enhanced lipid metabolism, which supports their energy needs and provides essential components for cell membranes and signaling molecules[107]. Therefore, targeting lipid metabolism may offer an effective therapeutic approach. Overall, metabolism plays a crucial role in regulating both cancer cells and CSCs. CSCs exhibit high metabolic plasticity, enabling them to adapt to diverse microenvironments by switching between glycolysis and OXPHOS to generate ATP. A deeper understanding of CSC metabolism could pave the way for more effective strategies to target these cells and improve cancer treatment outcomes.
THERAPEUTIC IMPLICATIONS OF CSCs
The growing recognition of CSCs as a crucial component in carcinogenesis has led to increasing efforts to eliminate these cells to prevent tumor recurrence and metastasis, as well as to overcome chemo- and radioresistance[14]. Several strategies have been proposed to specifically target and eliminate CSCs, thereby enhancing the effectiveness of cancer treatments and reducing the risk of relapse. These strategies include inhibiting key CSC signaling pathways, disrupting pathways that regulate CSC maintenance and differentiation, altering the pro-tumorigenic microenvironment, and targeting CSCs through specific markers. Currently, many CSC-targeted therapeutic strategies are under evaluation in clinical trials for various tumor types, including oral squamous cell carcinoma, breast cancer, lung cancer, pancreatic cancer, head and neck cancer, ovarian cancer, esophageal squamous cell carcinoma, and glioblastoma[22]. However, information on stem-like cells in CCA remains limited. Since several studies have reported that CSCs contribute to cancer progression, metastasis, and chemoresistance, there are ongoing clinical trials exploring potential CSC-targeted therapies. Targeting CSC differentiation pathways is considered a promising approach. This strategy aims to either induce CSC quiescence or promote their differentiation into more mature, less tumorigenic cell types. Suppression of HMGA1, for example, can induce differentiation in CCA-CSCs, as demonstrated by increased cell proliferation and reduced expression of CSC markers. Differentiated CCA-CSC show decreased tumor-initiating potential, reduced drug resistance (including to 5-FU, cisplatin, and gemcitabine), and a diminished ability to cause recurrence[15]. Several targeted therapeutic approaches are shown in Table 3. However, to date, clinical trials specifically targeting CCA-CSCs have not yet been conducted.
Therapeutic strategies targeting CCA-CSCs
| Therapeutic strategies | Target | Mechanism |
| Targeting unique stem cell markers | CD44v9 | Inhibition of CD44v9 induces apoptosis and cell cycle arrest in CCA, and also reduces migration, invasion, and tumor growth in mouse xenograft models[25] |
| CD147 | CD147 enhances metastatic potential in CCA by regulating the EMT pathway[89] | |
| CD133 | Elevated CD133 expression in CCA tissues is associated with highly metastatic and invasive forms of the disease[90]. In another study, engineered anti-CD133-CAR4 T cells demonstrated strong effectiveness against CD133-positive CCA cells, showing potential for future in vivo research and clinical applications[108] | |
| ALDH1A3 | ALDHhigh CCA cells display enhanced migratory ability and increased resistance to gemcitabine. Elevated ALDH1A3 expression in patient tissues is associated with poor prognosis[58] | |
| HMGA1 | HMGA1 knockdown promotes stem cell differentiation and enhances sensitivity to chemotherapeutic drugs[15] | |
| Targeting metabolic reprogramming | Lipid metabolism | Inhibition of FASN reduces stem-like characteristics and suppresses tumor growth[106] |
| Oxidative phosphorylation metabolism | CCA stem cells rely on mitochondrial oxidative phosphorylation. Inhibition of PGC-1α and related OXPHOS genes impairs tumor-initiating capacity, decreases stem cell marker expression, and induces EMT[17] | |
| Inhibition of key CSC signaling pathways | IGF-1R/PI3K/AKT axis | The IGF-1R/PI3K/AKT pathway supports the survival of iCCA stem-like cells; IGF-1R inhibition disrupts tumorsphere formation[109] |
| Combined therapies | HMGA1 | HMGA1 knockdown combined with Cisplatin, Gemcitabine, or 5-FU enhances the sensitivity of CCA cells to these chemotherapies[15] |
| Targeting of quiescent CSCs | c-myc | c-myc regulates cell growth and proliferation; inhibition induces cell cycle arrest at the G0/G1 phase in CCA cell lines[98] |
| AURKA | AURKA suppression reduces the viability of both CCA stem-like and parental cells and enhances the synergistic effect of HMGA1 knockdown[15] | |
| CDK4/6 | Inhibition of CDK4/6 activity by palbociclib or abemaciclib reduces the CCA-CSC population and slows disease progression[110] |
Despite the promise of these strategies, the mechanisms regulating CSC maintenance and differentiation remain poorly understood. Ongoing research is essential to clarify these pathways and refine therapeutic approaches. Zhang et al. analyzed RNA-seq data from three public CCA cohorts and proposed treatment guidelines for iCCA patients based on eight CSC-associated genes. Their findings suggest that patients with low-risk scores may benefit more from immunotherapy, while those with high-risk scores are more suitable candidates for anti-tumor drug therapies. These strategies highlight the significant potential of CSCs in personalized risk assessment, chemotherapy, and immunotherapy for individuals with iCCA[111]. Overall, the data demonstrate that CSCs influence CCA progression and prognosis through complex mechanisms and may serve as an important risk factor for evaluating the clinical prognosis of iCCA patients.
CHALLENGES AND FUTURE DIRECTIONS
CCA is a highly heterogeneous tumor composed of diverse cell populations. This heterogeneity likely arises from the varied origins of CCA cells, including hepatic stem or progenitor cells, hepatocytes, cholangiocytes, and peribiliary gland stem cells. Advanced sequencing techniques, particularly single-cell sequencing, have been used to identify distinct cellular subgroups within CCA[112]. Bian et al. analyzed scRNA-seq data from public databases of 24 CCA patients, identifying 11,993 cells and revealing substantial differences in stemness and differentiation among malignant cells. Notably, high-stemness malignant CCA cells expressed cytokines such as CCL2, CCL20, CXCL1, CXCL2, CXCL6, CXCL8, TNFRSF12A, and IL6ST, indicating interactions with surrounding immune cells. These interactions may facilitate the escape of CSCs from immune surveillance[113]. Additionally, Song et al. performed scRNA-seq on 144,878 cells from 14 paired iCCA tumors and non-tumor liver tissues, identifying 13 clusters with distinct marker expression profiles. These included stem cell markers such as EpCAM and various CD family markers, indicating the presence of CSCs within CCA heterogeneity[114]. Targeting specific subpopulations within tumors and accurately identifying CCA subtypes remain major challenges. In recent years, CSCs have been shown to influence CCA progression and prognosis significantly. However, the biology of CCA-CSCs remains poorly understood. One major obstacle is the lack of established CSC models for CCA, compounded by the heterogeneity among CSC populations, with different clones exhibiting varying degrees of stemness, phenotypes, and aggressiveness. The CSC population typically constitutes only 0.01%-2% of the total tumor mass, making their isolation and identification in heterogeneous tumor tissues difficult. Nevertheless, CSCs can be isolated from many solid tumors and their derived cell lines. Isolating CSCs directly from solid tumors might uncover novel subtypes not present in established cell lines. However, there are disadvantages, such as the need for careful and rapid processing of fresh biopsy specimens to maintain cell viability and integrity[115], as well as the necessity of efficient separation techniques to prevent contamination with non-cancerous cells[115]. Due to their low abundance, isolating and enriching CSC populations remains challenging[116]. CSCs are often cultured as free-floating spheres in serum-free conditions supplemented with specific growth factors that mimic the stem cell niche[117]. This is the most widely used method for isolating and enriching CSCs from mixed populations and for assessing their stemness potential[118]. More recently, this method has been adapted to enrich CSCs from primary cell lines. These established protocols are generally easier to implement in vitro, allowing expansion and manipulation in various cancer cell lines. Cell lines provide a relatively homogeneous cancer cell population, which can simplify CSC studies within a given cancer type. In comparison, the parental adherent monolayer cells can be used as a conventional cancer cell model alongside CSC models[15,119]. However, the process of establishing cell lines may select certain subpopulations that do not fully represent the original tumor heterogeneity or the CSC population[15]. Both primary tumor tissue and cell lines serve as valuable sources for isolating and studying CSCs, each with unique advantages and limitations. Authentication of CSC models requires confirmation of the CSC phenotype using multiple features, including sphere formation assays, expression of specific cell surface markers, tumor initiation and metastatic potential, differentiation ability, and drug resistance. To deepen our understanding of CSCs and develop effective therapeutic strategies for CCA, it is essential to isolate and study more CSC-like clones with diverse phenotypes and levels of stemness. The lack of accurate models contributes to the gap between promising laboratory results and clinical success. Future research should focus on establishing precise CSC models for better identification and characterization, offering insights into tumor heterogeneity and the TME, and facilitating the development of new approaches to target CSCs beyond traditional chemotherapy. Furthermore, the development of precise biomarkers could help predict patient responses to CSC-targeted therapies. Ultimately, personalized drug screens designed to identify the most effective treatments against both cancer cells and CSCs hold promise for improving patient outcomes and advancing therapeutic innovations in CCA.
CONCLUSION
CCA is a complex and aggressive malignancy characterized by significant heterogeneity. Advancements in single-cell sequencing have enhanced our understanding of its cellular diversity, revealing distinct subpopulations with varying degrees of stemness and differentiation. The identification of CSCs within CCA has been linked to tumor progression, metastasis, recurrence, and resistance to conventional therapies. However, isolating and characterizing CSCs remains challenging due to their low abundance and the lack of reliable markers. Future research on CCA-CSCs should focus on developing robust CSC models to clarify their roles in CCA progression and to identify novel therapeutic targets directed at both CSCs and bulk tumor cells. Additionally, establishing reliable biomarkers for CSCs could facilitate the prediction of patient responses, leading to more personalized and effective treatment strategies. Ultimately, a deeper understanding of CSC biology holds the potential to improve patient outcomes and advance the development of innovative therapeutic approaches for CCA.
DECLARATIONS
Acknowledgments
The authors would like to thank the support from the Fundamental Fund of Khon Kaen University to Silsirivanit A and the National Research Council of Thailand to Panawan O and Silsirivanit A. Thanks also goes to Prof. Yukifumi Nawa for English Editing via the KKU publication clinic.
Authors’ contributions
Conceptualized and designed the review, performed the literature search, wrote the original draft, handled revisions, and coordinated the approval of the final manuscript: Panawan O
Provided supervision, contributed to the synthesis and analysis of the literature, and revised the manuscript for intellectual content: Silsirivanit A
Provided supervision, secured funding for the review, and helped with project administration: Araki N
Availability of data and materials
Not applicable.
Financial support and sponsorship
This study was supported by grants from the National Research Council of Thailand (N42A680057 to Panawan O and N42A650238 to Silsirivanit A).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Guest RV, Goeppert B, Nault JC, Sia D. Morphomolecular pathology and genomic insights into the cells of origin of cholangiocarcinoma and combined hepatocellular-cholangiocarcinoma. Am J Pathol. 2025;195:345-61.
2. Elvevi A, Laffusa A, Scaravaglio M, et al. Clinical treatment of cholangiocarcinoma: an updated comprehensive review. Ann Hepatol. 2022;27:100737.
3. Vij M, Puri Y, Rammohan A, et al. Pathological, molecular, and clinical characteristics of cholangiocarcinoma: a comprehensive review. World J Gastrointest Oncol. 2022;14:607-27.
4. Banales JM, Marin JJG, Lamarca A, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17:557-88.
5. Colangelo M, Di Martino M, Polidoro MA, et al. Management of intrahepatic cholangiocarcinoma: a review for clinicians. Gastroenterol Rep. 2025;13:goaf005.
6. Sripa B, Pairojkul C. Cholangiocarcinoma: lessons from Thailand. Curr Opin Gastroenterol. 2008;24:349-56.
7. Sriputtha S, Khuntikeo N, Promthet S, Kamsa-Ard S. Survival rate of intrahepatic cholangiocarcinoma patients after surgical treatment in Thailand. Asian Pac J Cancer Prev. 2013;14:1107-10.
8. Tawarungruang C, Khuntikeo N, Chamadol N, et al. Survival after surgery among patients with cholangiocarcinoma in Northeast Thailand according to anatomical and morphological classification. BMC Cancer. 2021;21:497.
9. Valle JW, Lamarca A, Goyal L, Barriuso J, Zhu AX. New horizons for precision medicine in biliary tract cancers. Cancer Discov. 2017;7:943-62.
10. Lamarca A, Palmer DH, Wasan HS, et al; Advanced Biliary Cancer Working Group. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): a phase 3, open-label, randomised, controlled trial. Lancet Oncol. 2021;22:690-701.
11. Li Y, Yu J, Zhang Y, Peng C, Song Y, Liu S. Advances in targeted therapy of cholangiocarcinoma. Ann Med. 2024;56:2310196.
12. Luvira V, Eurboonyanun Ch, Bhudhisawasdi V, et al. Patterns of recurrence after resection of mass-forming type intrahepatic cholangiocarcinomas. Asian Pac J Cancer Prev. 2016;17:4735-9.
13. Liang N, Yang T, Huang Q, et al. Mechanism of cancer stemness maintenance in human liver cancer. Cell Death Dis. 2022;13:394.
15. Panawan O, Silsirivanit A, Chang CH, et al. Establishment and characterization of a novel cancer stem-like cell of cholangiocarcinoma. Cancer Sci. 2023;114:3230-46.
16. Padthaisong S, Thanee M, Namwat N, et al. Overexpression of a panel of cancer stem cell markers enhances the predictive capability of the progression and recurrence in the early stage cholangiocarcinoma. J Transl Med. 2020;18:64.
17. Raggi C, Taddei ML, Sacco E, et al. Mitochondrial oxidative metabolism contributes to a cancer stem cell phenotype in cholangiocarcinoma. J Hepatol. 2021;74:1373-85.
18. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730-7.
20. Daniela FQ, Meghan JT, Lynne-Marie P. Microenvironmental regulation of cancer stem cell phenotypes. Current Stem Cell Research & Therapy. 2012;7:197-216.
21. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16:225-38.
22. Chu X, Tian W, Ning J, et al. Cancer stem cells: advances in knowledge and implications for cancer therapy. Signal Transduct Target Ther. 2024;9:170.
23. Yang L, Shi P, Zhao G, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5:8.
24. Mcgrath NA, Fu J, Gu SZ, Xie C. Targeting cancer stem cells in cholangiocarcinoma (Review). Int J Oncol. 2020;57:397-408.
25. Suwannakul N, Ma N, Midorikawa K, et al. CD44v9 induces stem cell-like phenotypes in human cholangiocarcinoma. Front Cell Dev Biol. 2020;8:417.
26. Liu CL, Chen YJ, Fan MH, Liao YJ, Mao TL. Characteristics of CD133-sustained chemoresistant cancer stem-like cells in human ovarian carcinoma. Int J Mol Sci. 2020;21:6467.
27. Firdous S, Ghosh A, Saha S. BCSCdb: a database of biomarkers of cancer stem cells. Database. 2022;2022:baac082.
28. Ding Y, Gao H, Zhang Q. The biomarkers of leukemia stem cells in acute myeloid leukemia. Stem Cell Investig. 2017;4:19.
29. Chan KS, Espinosa I, Chao M, et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc Natl Acad Sci U S A. 2009;106:14016-21.
30. Li Y, Lin K, Yang Z, et al. Bladder cancer stem cells: clonal origin and therapeutic perspectives. Oncotarget. 2017;8:66668-79.
31. Tomar R, Rakheja G, Verma N, Thakur S, Khurana N, Ghuliani D. Role of CD44 as cancer stem cell marker in triple-negative breast cancer and its association with histological grade and angiogenesis. Indian J Pathol Microbiol. 2023;66:258-63.
32. Zhang X, Powell K, Li L. Breast cancer stem cells: biomarkers, identification and isolation methods, regulating mechanisms, cellular origin, and beyond. Cancers. 2020;12:3765.
33. Rodriguez SMB, Staicu GA, Sevastre AS, et al. Glioblastoma stem cells-useful tools in the battle against cancer. Int J Mol Sci. 2022;23:4602.
34. Prager BC, Bhargava S, Mahadev V, Hubert CG, Rich JN. Glioblastoma stem cells: driving resilience through chaos. Trends Cancer. 2020;6:223-35.
35. Hervieu C, Christou N, Battu S, Mathonnet M. The role of cancer stem cells in colorectal cancer: from the basics to novel clinical trials. Cancers. 2021;13:1092.
36. Neal ME, Brenner JC, Prince MEP, Chinn SB. Advancement in cancer stem cell biology and precision medicine-review article head and neck cancer stem cell plasticity and the tumor microenvironment. Front Cell Dev Biol. 2021;9:660210.
37. Yang ZF, Ho DW, Ng MN, et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153-66.
38. Terris B, Cavard C, Perret C. EpCAM, a new marker for cancer stem cells in hepatocellular carcinoma. J Hepatol. 2010;52:280-1.
39. Jamal SME, Alamodi A, Wahl RU, et al. Melanoma stem cell maintenance and chemo-resistance are mediated by CD133 signal to PI3K-dependent pathways. Oncogene. 2020;39:5468-78.
40. Parte SC, Batra SK, Kakar SS. Characterization of stem cell and cancer stem cell populations in ovary and ovarian tumors. J Ovarian Res. 2018;11:69.
41. Klemba A, Purzycka-Olewiecka JK, Wcisło G, et al. Surface markers of cancer stem-like cells of ovarian cancer and their clinical relevance. Contemp Oncol. 2018;22:48-55.
42. Ishiwata T, Matsuda Y, Yoshimura H, et al. Pancreatic cancer stem cells: features and detection methods. Pathol Oncol Res. 2018;24:797-805.
43. Yang CH, Wang HL, Lin YS, et al. Identification of CD24 as a cancer stem cell marker in human nasopharyngeal carcinoma. PLoS One. 2014;9:e99412.
44. Giraud J, Seeneevassen L, Rousseau B, et al. CD44v3 is a marker of invasive cancer stem cells driving metastasis in gastric carcinoma. Gastric Cancer. 2023;26:234-49.
45. Schatton T, Frank MH. The in vitro spheroid melanoma cell culture assay: cues on tumor initiation? J Invest Dermatol. 2010;130:1769-71.
46. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-8.
47. Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111-5.
48. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946-51.
49. Eramo A, Lotti F, Sette G, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504-14.
50. Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. 2013;14:329-40.
51. Forsberg EC, Passegué E, Prohaska SS, et al. Molecular signatures of quiescent, mobilized and leukemia-initiating hematopoietic stem cells. PLoS One. 2010;5:e8785.
53. Putthisen S, Silsirivanit A, Panawan O, et al. Targeting alpha2,3-sialylated glycan in glioma stem-like cells by Maackia amurensis lectin-II: a promising strategy for glioma treatment. Exp Cell Res. 2022;410:112949.
54. Chen J, Li Y, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522-6.
55. Oshimori N, Oristian D, Fuchs E. TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell. 2015;160:963-76.
56. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756-60.
57. Zhou HM, Zhang JG, Zhang X, Li Q. Targeting cancer stem cells for reversing therapy resistance: mechanism, signaling, and prospective agents. Signal Transduct Target Ther. 2021;6:62.
58. Chen MH, Weng JJ, Cheng CT, et al. ALDH1A3, the major aldehyde dehydrogenase isoform in human cholangiocarcinoma cells, affects prognosis and gemcitabine resistance in cholangiocarcinoma patients. Clin Cancer Res. 2016;22:4225-35.
59. O’Reilly D, Johnson P, Buchanan PJ. Hypoxia induced cancer stem cell enrichment promotes resistance to androgen deprivation therapy in prostate cancer. Steroids. 2019;152:108497.
60. Niibori-Nambu A, Yamasaki Y, Kobayashi D, et al. Chondroitin sulfate modification of CSPG4 regulates the maintenance and differentiation of glioma-initiating cells via integrin-associated signaling. J Biol Chem. 2024;300:105706.
61. Narushima Y, Kozuka-Hata H, Koyama-Nasu R, et al. Integrative network analysis combined with quantitative phosphoproteomics reveals transforming growth factor-beta receptor type-2 (TGFBR2) as a novel regulator of glioblastoma stem cell properties. Mol Cell Proteomics. 2016;15:1017-31.
62. Niibori-Nambu A, Midorikawa U, Mizuguchi S, et al. Glioma initiating cells form a differentiation niche via the induction of extracellular matrices and integrin αV. PLoS One. 2013;8:e59558.
63. Kumon K, Afify SM, Hassan G, et al. Differentiation of cancer stem cells into erythroblasts in the presence of CoCl2. Sci Rep. 2021;11:23977.
64. Arima Y, Nobusue H, Saya H. Targeting of cancer stem cells by differentiation therapy. Cancer Sci. 2020;111:2689-95.
65. Pattabiraman DR, Bierie B, Kober KI, et al. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science. 2016;351:aad3680.
66. Dubois-Pot-Schneider H, Fekir K, Coulouarn C, et al. Inflammatory cytokines promote the retrodifferentiation of tumor-derived hepatocyte-like cells to progenitor cells. Hepatology. 2014;60:2077-90.
67. Sell S, Dunsford HA. Evidence for the stem cell origin of hepatocellular carcinoma and cholangiocarcinoma. Am J Pathol. 1989;134:1347-63.
68. Moeini A, Haber PK, Sia D. Cell of origin in biliary tract cancers and clinical implications. JHEP Rep. 2021;3:100226.
69. Alison MR, Vig P, Russo F, et al. Hepatic stem cells: from inside and outside the liver? Cell Prolif. 2004;37:1-21.
70. Kokuryo T, Yokoyama Y, Nagino M. Recent advances in cancer stem cell research for cholangiocarcinoma. J Hepatobiliary Pancreat Sci. 2012;19:606-13.
72. Testa U, Pelosi E, Castelli G. Cholangiocarcinoma: molecular abnormalities and cells of origin. Technol Cancer Res Treat. 2023;22:15330338221128689.
73. Clements O, Eliahoo J, Kim JU, Taylor-Robinson SD, Khan SA. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: a systematic review and meta-analysis. J Hepatol. 2020;72:95-103.
74. Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet. 2015;47:1003-10.
75. Cao J, Hu J, Liu S, et al. Intrahepatic cholangiocarcinoma: genomic heterogeneity between eastern and western patients. JCO Precis Oncol. 2020;4:PO.18.00414.
76. Miyazu T, Ishida N, Asai Y, et al. Intrahepatic cholangiocarcinoma in patients with primary sclerosing cholangitis and ulcerative colitis: two case reports. World J Gastrointest Surg. 2023;15:1224-31.
77. Ahrendt SA, Eisenberger CF, Yip L, et al. Chromosome 9p21 loss and p16 inactivation in primary sclerosing cholangitis-associated cholangiocarcinoma. J Surg Res. 1999;84:88-93.
78. Ainthachot S, Sa-Ngiamwibool P, Thanee M, et al. Chromosomal aberrations, visualized using UroVysion® fluorescence in-situ hybridization assay, can predict poor prognosis in formalin-fixed paraffin-embedded tissues of cholangiocarcinoma patients. Hum Pathol. 2022;126:31-44.
79. Techa-Ay S, Watcharadetwittaya S, Deenonpoe R, et al. Identifying a unique chromosomal pattern to predict the gemcitabine response in patients with cholangiocarcinoma. Sci Rep. 2025;15:11984.
80. Damelin M, Sun YE, Sodja VB, Bestor TH. Decatenation checkpoint deficiency in stem and progenitor cells. Cancer Cell. 2005;8:479-84.
81. Rizvi AZ, Swain JR, Davies PS, et al. Bone marrow-derived cells fuse with normal and transformed intestinal stem cells. Proc Natl Acad Sci U S A. 2006;103:6321-5.
82. Oishi N, Kumar MR, Roessler S, et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology. 2012;56:1792-803.
83. Lin KY, Ye H, Han BW, et al. Genome-wide screen identified let-7c/miR-99a/miR-125b regulating tumor progression and stem-like properties in cholangiocarcinoma. Oncogene. 2016;35:3376-86.
84. Parisi S, Piscitelli S, Passaro F, Russo T. HMGA proteins in stemness and differentiation of embryonic and adult stem cells. Int J Mol Sci. 2020;21:362.
85. Kim DK, Seo EJ, Choi EJ, et al. Crucial role of HMGA1 in the self-renewal and drug resistance of ovarian cancer stem cells. Exp Mol Med. 2016;48:e255.
86. Li M, Gao K, Chu L, Zheng J, Yang J. The role of Aurora-A in cancer stem cells. Int J Biochem Cell Biol. 2018;98:89-92.
87. Xia Z, Wei P, Zhang H, et al. AURKA governs self-renewal capacity in glioma-initiating cells via stabilization/activation of β-catenin/Wnt signaling. Mol Cancer Res. 2013;11:1101-11.
88. Zheng F, Yue C, Li G, et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat Commun. 2016;7:10180.
89. Dana P, Kariya R, Vaeteewoottacharn K, et al. Upregulation of CD147 promotes metastasis of cholangiocarcinoma by modulating the epithelial-to-mesenchymal transitional process. Oncol Res. 2017;25:1047-59.
90. Leelawat K, Thongtawee T, Narong S, Subwongcharoen S, Treepongkaruna SA. Strong expression of CD133 is associated with increased cholangiocarcinoma progression. World J Gastroenterol. 2011;17:1192-8.
91. Cai X, Li J, Yuan X, et al. CD133 expression in cancer cells predicts poor prognosis of non-mucin producing intrahepatic cholangiocarcinoma. J Transl Med. 2018;16:50.
92. Wakizaka K, Yokoo H, Kamiyama T, et al. CD133 and epithelial cell adhesion molecule expressions in the cholangiocarcinoma component are prognostic factors for combined hepatocellular cholangiocarcinoma. Hepatol Res. 2020;50:258-67.
93. Agrawal S, Kuvshinoff BW, Khoury T, et al. CD24 expression is an independent prognostic marker in cholangiocarcinoma. J Gastrointest Surg. 2007;11:445-51.
94. Pote MS, Gacche RN. ATP-binding cassette efflux transporters and MDR in cancer. Drug Discov Today. 2023;28:103537.
95. Raggi C, Correnti M, Sica A, et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J Hepatol. 2017;66:102-15.
96. Sun Q, Li J, Wang G, Xie Y. Role of the embryonic protein SOX2 in cholangiocarcinoma. Cell Biochem Biophys. 2014;70:1311-6.
97. Yu A, Zhao L, Kang Q, Li J, Chen K, Fu H. SOX2 knockdown slows cholangiocarcinoma progression through inhibition of transcriptional activation of lncRNA PVT1. Biochem J. 2020;477:3527-40.
98. Luo G, Li B, Duan C, et al. c-Myc promotes cholangiocarcinoma cells to overcome contact inhibition via the mTOR pathway. Oncol Rep. 2017;38:2498-506.
99. Pei F, Tao Z, Lu Q, Fang T, Peng S. Octamer-binding transcription factor 4-positive circulating tumor cell predicts worse treatment response and survival in advanced cholangiocarcinoma patients who receive immune checkpoint inhibitors treatment. World J Surg Oncol. 2024;22:110.
100. Choodetwattana P, Proungvitaya S, Jearanaikoon P, Limpaiboon T. The upregulation of OCT4 in acidic extracellular pH is associated with gemcitabine resistance in cholangiocarcinoma cell lines. Asian Pac J Cancer Prev. 2019;20:2745-8.
101. Song J, Cui D, Wang J, et al. Overexpression of HMGA1 confers radioresistance by transactivating RAD51 in cholangiocarcinoma. Cell Death Discov. 2021;7:322.
102. Carloni R, Rizzo A, Ricci AD, et al. Targeting tumor microenvironment for cholangiocarcinoma: opportunities for precision medicine. Transl Oncol. 2022;25:101514.
103. Guo Q, Zhou Y, Xie T, et al. Tumor microenvironment of cancer stem cells: perspectives on cancer stem cell targeting. Genes Dis. 2024;11:101043.
104. Wang X, Golino JL, Hawk NV, Xie C. Reciprocal interaction of cancer stem cells of cholangiocarcinoma with macrophage. Stem Cell Rev Rep. 2023;19:2013-23.
106. Lori G, Pastore M, Navari N, et al. Altered fatty acid metabolism rewires cholangiocarcinoma stemness features. JHEP Rep. 2024;6:101182.
107. Liu H, Zhang Z, Song L, Gao J, Liu Y. Lipid metabolism of cancer stem cells. Oncol Lett. 2022;23:119.
108. Sangsuwannukul T, Supimon K, Sujjitjoon J, et al. Anti-tumour effect of the fourth-generation chimeric antigen receptor T cells targeting CD133 against cholangiocarcinoma cells. Int Immunopharmacol. 2020;89:107069.
109. Rattanasinchai C, Navasumrit P, Chornkrathok C, Ruchirawat M. Kinase library screening identifies IGF-1R as an oncogenic vulnerability in intrahepatic cholangiocarcinoma stem-like cells. Biochim Biophys Acta Mol Basis Dis. 2025;1871:167521.
110. Kongtanawanich K, Prasopporn S, Jamnongsong S, et al. A live single-cell reporter system reveals drug-induced plasticity of a cancer stem cell-like population in cholangiocarcinoma. Sci Rep. 2024;14:22619.
111. Zhang J, Cui T, Xu J, Wang P, Lv C, Pan G. The potential of cancer stem cells for personalized risk assessment and therapeutic intervention in individuals with intrahepatic cholangiocarcinoma. Discov Oncol. 2024;15:306.
112. Chen W, Xu D, Liu Q, Wu Y, Wang Y, Yang J. Unraveling the heterogeneity of cholangiocarcinoma and identifying biomarkers and therapeutic strategies with single-cell sequencing technology. Biomed Pharmacother. 2023;162:114697.
113. Bian J, Fu J, Wang X, et al. Characterization of immunogenicity of malignant cells with stemness in intrahepatic cholangiocarcinoma by single-cell RNA sequencing. Stem Cells Int. 2022;2022:3558200.
114. Song G, Shi Y, Meng L, et al. Single-cell transcriptomic analysis suggests two molecularly subtypes of intrahepatic cholangiocarcinoma. Nat Commun. 2022;13:1642.
115. Dobbin ZC, Landen CN. Isolation and characterization of potential cancer stem cells from solid human tumors--potential applications. Curr Protoc Pharmacol. 2013;63:14.28.1-9.
116. Correia C, Weiskittel TM, Ung CY, et al. Uncovering pharmacological opportunities for cancer stem cells-a systems biology view. Front Cell Dev Biol. 2022;10:752326.
117. Ishii H, Mimura Y, Zahra MH et al. Isolation and characterization of cancer stem cells derived from human glioblastoma. Am J Cancer Res. 2021;11:441-57.
118. Gao W, Wu D, Wang Y, et al. Development of a novel and economical agar-based non-adherent three-dimensional culture method for enrichment of cancer stem-like cells. Stem Cell Res Ther. 2018;9:243.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0










Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].