


TGF-β stimulation of EMT programs elicits non-genomic ER-α activity and anti-estrogen resistance in breast cancer cells


Abstract
Aim: Estrogen receptor-α (ER-α) activation drives the progression of luminal breast cancers. Signaling by transforming growth factor-β (TGF-β) typically opposes the actions of ER-α; it also induces epithelial-mesenchymal transition (EMT) programs that promote breast cancer dissemination, stemness and chemoresistance. The impact of EMT programs on nongenomic ER-α signaling remains unknown and was studied herein.
Methods: MCF-7 and BT474 cells were stimulated with TGF-β to induce EMT programs, at which point ER-α expression, localization, and nongenomic interactions with receptor tyrosine kinases and MAP kinases (MAPKs) were determined. Cell sensitivity to anti-estrogens both before and after traversing the EMT program was also investigated.
Results: TGF-β-stimulated MCF-7 and BT474 cells to acquire EMT phenotypes, which enhanced cytoplasmic accumulation of ER-α without altering its expression. Post-EMT cells exhibited: (1) elevated expression of EGFR and IGF1R, which together with Src formed cytoplasmic complexes with ER-α; (2) enhanced coupling of EGF, IGF-1 and estrogen to the activation of MAPKs; and (3) reduced sensitivity to tamoxifen, an event reversed by administration of small molecule inhibitors against the receptors for TGF-β, EGF, and IGF-1, as well as those against MAPKs.
Conclusion: EMT stimulated by TGF-β promotes anti-estrogen resistance by activating EGFR-, IGF1R-, and MAPK-dependent nongenomic ER-α signaling.
Keywords
Introduction
Transforming growth factor-β (TGF-β) normally acts as a suppressor of mammary tumorigenesis by inducing cellular arrest, apoptosis, or differentiation, and by creating a cell microenvironment that inhibits cell motility, invasion and metastasis. However, during breast cancer development, the tumor suppressing functions of TGF-β is frequently subverted, thus converting TGF-β from a tumor suppressor to a promoter of breast cancer growth and metastasis.[1-3] Although the molecular mechanisms responsible for eliciting the dichotomous activities of TGF-β remain to be fully elucidated, accumulating evidence implicates canonical Smad2/3-dependent signaling in mediating tumor suppression by TGF-β and noncanonical Smad2/3-independent signaling in mediating its tumor promoting activities.[1-3] Amongst the best characterized noncanonical signaling pathways operant in coupling TGF-β to tumorigenesis are: (1) integrins and components of focal adhesion complexes; (2) MAP kinase and small GTP-binding protein family members; and (3) PI3K/AKT and NF-κB pathways;[4] they also function to drive epithelial-mesenchymal transitions (EMT) stimulated by TGF-β, thereby promoting breast cancer dissemination, stemness, and chemoresistance.[5]
Within the mammary gland, signaling by estrogen receptor (ER-α) plays an essential role not only during glandular development and differentiation, but also during the initiation and progression of luminal breast cancers.[6-8] Indeed, the oncogenic activities of dysregulated ER-α signaling underlie the clinical success of anti-estrogen drugs (e.g. tamoxifen) as first-line therapies to treat ER-positive breast cancers. However, despite their initial efficacy, anti-estrogen drugs often become ineffective as patient tumors develop resistance and undergo disease recurrence.[9,10] At present, the mechanisms resulting in acquired anti-estrogen resistance are not fully understood. However, compelling evidence implicates nongenomic ER-α signaling as a major culprit of resistance to anti-estrogen-based therapies.[9,11-13] Likewise, aberrant expression of a truncated metastasis tumor antigen 1 (MTA1) mutant was found to bind and sequester ER-α in the cytoplasm, thus enhancing the nongenomic actions of ER-α and disease progression in breast cancers.[14]
Given the pathophysiologic parallels that exist between nongenomic ER-α and noncanonical TGF-β signaling in driving breast cancer progression, we speculated that EMT programs induced by TGF-β may elicit nongenomic ER-α signaling and endocrine resistance in luminal breast cancers. The aim of this study was to test this hypothesis and further our understanding of how EMT programs drive disease progression and acquired resistance to anti-estrogen-based therapies in human breast cancers.
Methods
Cell lines and chemical inhibitors
Human luminal A MCF-7 cells were obtained from ATCC (USA) and cultured as previously described,[15] while human luminal B BT474 cells were kindly provided by Dr. Mark W. Jackson (Case Western Reserve University, USA) and propagated in DMEM supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, USA) and 1% Pen/Strep (Invitrogen, USA). Pharmacological agonists and inhibitors used herein are described in the Supplementary Table 1.
Pharmacological agonists and inhibitors
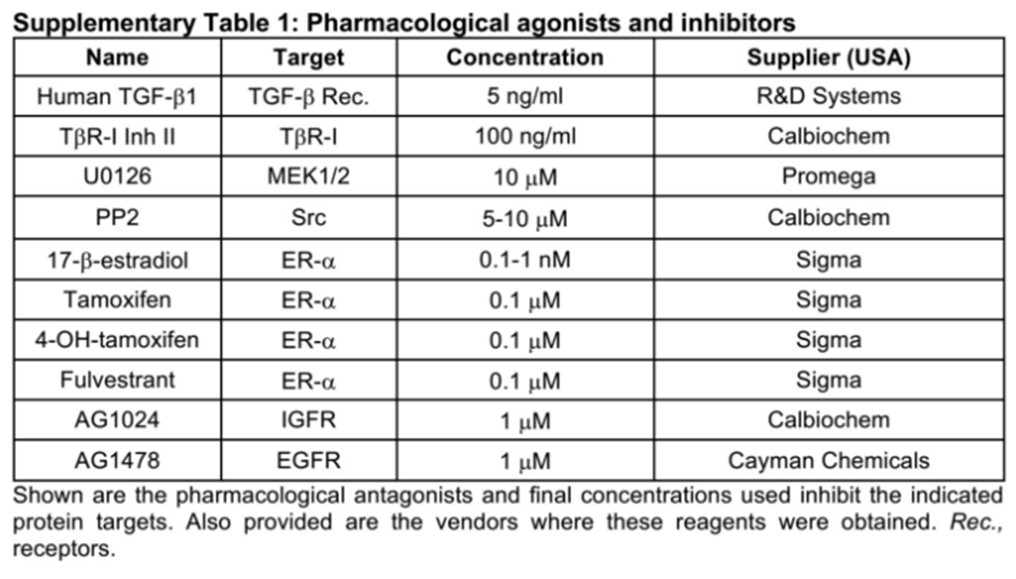 |
Cell biological assays
Analyzing the effects of TGF-β and EMT programs on ER-α signaling in MCF-7 and BT474 cells was determined as follows: (1) cell proliferation assays: cells were treated in the absence or presence of TGF-β1 (5 ng/mL; R&D Systems, USA) for 72 h to induce EMT, at which point they were subcultured in 96-well plates (10,000 cells/well) for 5 days with either diluent or inhibitors to the TGF-β type I receptor (TβR-I; 100 ng/mL), the epidermal growth factor receptor (EGFR; 1 mol/L), the insulin-like growth factor 1 receptor (IGF1R; 1 mol/L), mitogen-activated protein kinase kinase (MEK; 10 mol/L), or ER-α (0.1 mol/L; Supplementary Table 1). Differences in cell growth and survival were determined by incubating the cells with MTS Plus reagent (20 µL; Promega, USA) for 1 h at 37 ˚C, followed by measuring absorbance at 490 nm on a Promega Modulus II Microplate Multimode instrument (Promega, USA); (2) 3-dimensional (3D) growth assays: 3D-cultures were prepared by diluting pre- or post-EMT MCF-7 and BT474 cells in complete media supplemented with 5% Cultrex (Trevigen, Gaithersburg, USA), which subsequently were seeded onto solidified Cultrex cushions (500 µL/well) contained in 6-well plates (150,000 cells/well). Afterward, the cells were cultured in the absence or presence of TGF-β1 (5 ng/mL), estradiol (1 nmol/L), tamoxifen (0.1 nmol/L), or fulvestrant (0.1 mol/L; Supplementary Table 1) for 8 days, during which time they were fed every 3 days with full growth media supplemented with 5% Cultrex and pharmacological agents. Differences in organoid growth were calculated using NIH Image J; (3) luciferase reporter gene assays: pre- and post-EMT MCF-7 and BT474 cells were allowed to adhere overnight to 24-well plates (40,000 cells/well). The cells were transiently transfected as described previously[16,17] with the following reporter plasmids: (a) pSBE-luciferase, which contains 4 copies of the Smad3/4-binding element (4X-CAGA) and serves as a direct measure of canonical TGF-β signaling; (b) p3TP-lux,[18] which contains 3 copies of TPA-responsive elements and 96 bp of the PAI-1 promoter and responds to both canonical (i.e. Smad3/4) and noncanonical (i.e. AP-1) TGF-β signaling; (c) pERE-TATA-luciferase,[19] which contains 3 copies of the estrogen response element (3X-GGTCACAGTGACC) and responds to estrogen; (d) pTopFlash-luciferase,[20] which contains 7 copies of the TCF/LEF-binding sites (7X-AGATCAAAGGgggta) and responds to β-catenin activation; and (e) pCMV-β-gal, which was used to control for differences in transfection efficiency. Afterward, the cells were incubated in the absence or presence of TGF-β1 (5 ng/mL), estradiol (0.1 nmol/L), 4-OHT (0.1 mol/L), or TβRI inhibitor II (100 ng/mL; Supplementary Table 1) as described previously;[16,17] (4) direct and indirect immunofluorescence: pre- and post-EMT MCF-7 and BT474 cells were cultured overnight in 8-well chamber slides (80,000 cells/well) prior to being treated for 96 h in serum-free media supplemented with TGF-β1 (5 ng/mL), estradiol (0.1 nmol/L), 4-OHT (0.1 µmol/L), or fulvestrant (0.1 µmol/L; Supplementary Table 1). Afterwards, the cells were fixed in 4% paraformaldehyde and processed for direct immunofluorescence with phalloidin (0.25 mol/L; Molecular Probes, USA), or for indirect immunofluorescence with either anti-E-cadherin (E-cad) or anti-β-catetnin antibodies (1:250 dilution; BD Biosciences, San Jose, USA), followed by sequential incubation with biotinylated secondary antibodies (5 g/mL; Jackson ImmunoResearch, USA) and Alexa-streptavidin (1.2 g/mL; Molecular Probes, USA). Following extensive washing with PBS, the slides were mounted on glass slides with Prolong mounting medium (Molecular Probes) as described previously.[16,17]
Semi-quantitative real-time PCR analysis
Total RNA from MCF-7 or BT474 cells were purified using the RNeasy Plus Mini kit (Qiagen, USA) according to the manufacturer’s instruction. Afterwards, cDNAs were synthesized by iScript reverse transcription kit (Bio-Rad, USA), diluted 5-fold in H2O, and employed in semi-quantitative real-time PCR reactions (20 µL) using SYBR Green system (Bio-Rad, USA) that contained 5 µL of diluted cDNA and 0.1 µmol/L of oligonucleotide pairs listed in Supplementary Table 2. Differences in RNA concentration were controlled by normalizing individual gene signals to their corresponding β-actin or GAPDH as indicated.
Real-time PCR primer pairs
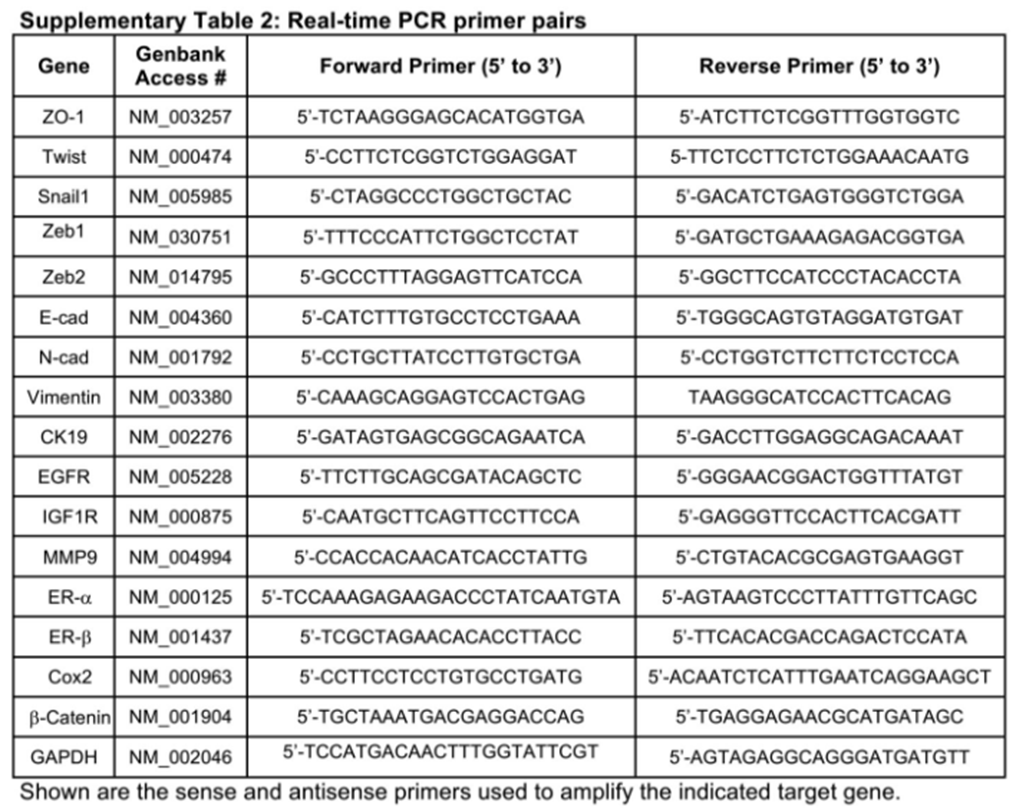 |
Western blotting analyses
Pre- and post-EMT MCF-7 and BT474 cells were lysed in Buffer H/1% Triton X-100[21] and solubilized for 60 min on ice. After microcentrifugation, the clarified cell extracts were resolved through 10% SDS-PAGE electrophoresis gels, transferred electrophoretically to nitrocellulose membranes, and blocked in 5% milk prior to incubation with primary antibodies listed in Supplementary Table 3. The resulting immunocomplexes were visualized by enhanced chemiluminescence reactions, and differences in protein loading were monitored by reprobing stripped membranes with antibodies against either β-actin, ERK1/2, or p38 MAPK as indicated.
Immunoblotting antibodies
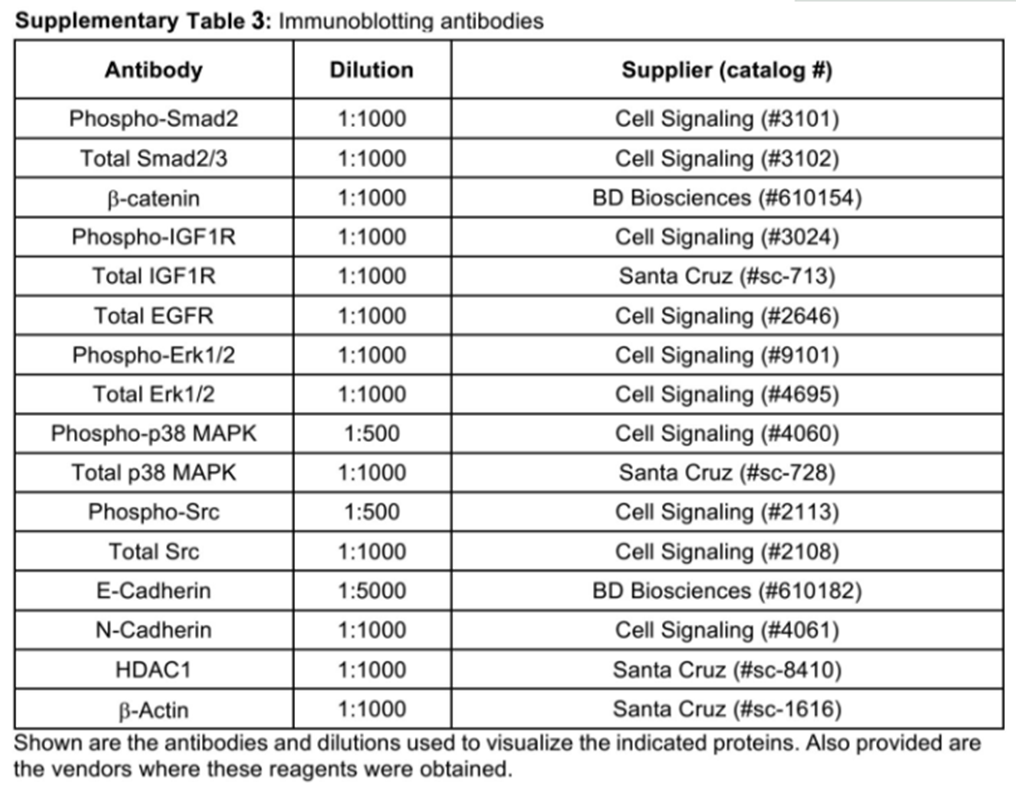 |
Co-immunoprecipitation assays
Clarified cell extracts (1 mg/tube; see above) prepared from pre- and post-EMT MCF-7 cells were incubated under continuous rotation with 1 µg of antibodies against either β-catenin, E-cad, ER-α, or Src overnight at 4 ˚C. The resulting immunocomplexes were collected by microcentrifugation, washed, and fractionated through 10% SDS-PAGE gels prior to their immobilization to nitrocellulose membranes, which subsequently were probed with antibodies against either phospho-tyrosine, β-catenin, EGFR, IGF1R, or ER-α [Supplementary Table 3]. Differences in protein loading and/or input were monitored by immunoblotting with corresponding antibodies as indicated.
Statistical analysis
Statistical values were defined using an unpaired Student’s T-test, where a P value < 0.05 was considered significant. P values for all experiments analyzed are indicated.
Results
TGF-β induces EMT in MCF-7 cells and potentiates noncanonical TGF-β signaling
EMT programs stimulated by TGF-β have been associated with the acquisition of motile and metastatic phenotypes, and with the generation of cancer stem cells and chemoresistance.[4,22,23] With respect to cancers of the breast, these features of EMT are more commonly associated with basal-like/TNBC subtypes, with little evidence related to how TGF-β and its stimulation of EMT programs impact ER-α signaling in luminal breast cancers. To address this question, we administered TGF-β1 to human MCF-7 cells (i.e. luminal A subtype) to induce an EMT program under 2D- and 3D-culture conditions. Figure 1A shows that MCF-7 cells readily transitioned from epithelial to mesenchymal morphologies when stimulated with TGF-β1. Moreover, these morphological alterations exhibited by MCF-7 cells coincided with their (1) downregulated expression of the epithelial marker E-cadherin in 3D-cultures [Supplementary Figure 1A], and (2) upregulated expression of mesenchymal markers vimentin, N-cadherin, Cox-2, and MMP-9 [Supplementary Figure 1B-D], as well as that of the master EMT transcription factors, Snail, Twist, Zeb1, and Zeb2 [Figure 1B and C]. Likewise, post-EMT MCF-7 cells showed a striking loss of β-catenin from the plasma membrane [Supplementary Figure 1E] that reflected its enhanced phosphorylation on tyrosine residue(s) and reduced capacity to bind E-cadherin [Figure 1D].

Figure 1. TGF-β induces EMT in MCF-7 cells and potentiates noncanonical TGF-β signaling. (A) MCF-7 cells were treated with TGF-β1 (5 ng/mL) for 0-72 h to induce an EMT program. Photomicrographs depict accompanying alterations in cell morphology (×400); (B and C) MCF-7 cells were stimulated with TGF-β1 (5 ng/mL) for 0-96 h, at which point total RNA was harvested and subjected to real-time PCR to monitor differences in the expression of Snail and Twist (B), and of Zeb1 and Zeb2 (C). Data are the mean fold-changes (± SE; n = 3; *P < 0.05; Student's t-test); (D) MCF-7 cells were treated with TGF-β1 (5 ng/mL) for 0-96 h, at which point detergent-solubilized extracts were immunoblotted for phospho-Tyr (pY), β-catenin (β-cat), or β-actin as indicated (top). Additionally, E-cad immunocomplexes were captured and immunoblotted for β-catenin (β-cat) and E-cad as indicated (bottom). Images are representative of 3-independent experiments. (E-G) Pre- and post-EMT MCF-7 cells were transiently transfected overnight with the pSBE-(E), TopFlash-(F), or p3TP-(G) luciferase reporter genes, as well as the pCMV-β-galactosidase reporter gene to control for differences in transfection efficiency. Afterward, the transfectants were stimulated overnight with TGF-β1 (5 ng/mL) prior to measuring luciferase and β-gal activities. Data are the mean fold-changes (± SE; n = 3;*P < 0.05; Student's t-test). TGF: transforming growth factor; EMT: epithelial-mesenchymal transition; SE: standard error

Supplementary Figure 1. TGF-β induces EMT phenotypes in MCF-7 cells in both 2D- and 3D cultures. (A-C) MCF-7 cells were propagated in either 2D- or 3D-culture systems and treated with TGF-b1 (5 ng/mL) for either 72 h (A and B) or 96 h (C) in 2D-cultures, or for either 8 days (A and B) or 96 h (C) as indicated. Afterward, total RNA has harvested and subjected to real-time PCR analyses to monitor the expression of the epithelial markers E-cad and CK19 (A), of the mesenchymal markers vimentin and N-cad (B), or of Cox-2 (C). (D) MCF-7 cells were stimulated with TGF-b1 for 0-96 h as indicated, at which point the levels of MMP-9 were monitored by realtime PCR. Data are the mean fold-changes (± SE; n = 3; *P < 0.05; Student's t-test). (E) MCF-7 cells were stimulated with TGF-b1 (5 ng/mL) for 96 h, and subsequently were fixed in paraformaldehyde and processed for indirect immunofluorescence of b-catenin, as well as for direct immunofluorescence of actin. Images are representative of 2-independent experiments (×400). TGF: transforming growth factor; EMT: epithelial-mesenchymal transition; SE: standard error
We also investigated the impact of EMT programs on MCF-7 cell behavior and intracellular signaling. To do so, we incubated MCF-7 cells in the absence (i.e. pre-EMT) or presence (i.e. post-EMT) of TGF-β1 for 72-96 h to induce an EMT program, at which point pre- and post-EMT cells were subcultured and transiently transfected with the following reporter genes: (1) pSBE-luciferase, which monitors canonical Smad3/4 signaling stimulated by TGF-β; (2) pTopFlash-luciferase, which monitors noncanonical β-catenin signaling stimulated by TGF-β; and (3) p3TP-luciferase, which monitors canonical Smad2/3/4 and noncanonical AP-1 signaling stimulated by TGF-β. Figure 1E shows that luciferase expression driven by the synthetic SBE promoter was significantly inhibited in post-EMT cells, suggesting that EMT programs suppress canonical signaling Smad-based signaling in response to TGF-β. Accordingly, the coupling of TGF-β to noncanonical β-catenin [Figure 1F] and AP-1 (Figure 1G; p3TP) activation was dramatically augmented in a manner reminiscent of EMT-induced signaling alterations observed previously in basal-like/TNBCs.[4,24] Interestingly, EMT-associated events transpired in both culture systems (i.e. 2D- and 3D-cultures), although the magnitude of EMT response was typically greater in 2D-cultures, suggesting that rigid microenvironments potentiate EMT programs stimulated by TGF-β.[25-27]
Similar induction of EMT programs was observed in human BT474 cells (i.e. luminal B subtype) stimulated with TGF-β. For instance, BT474 cells displayed a more mesenchymal morphology in response to TGF-β [Supplementary Figure 2A]; they also exhibited robust cadherin switching as the cells transitioned from pre- (e.g. E-cadherin dominant) to post-EMT (e.g. N-cadherin dominant) states [Supplementary Figure 2B]. Likewise, BT474 cells undergoing EMT programs exhibited a time-dependent loss of epithelial markers (e.g. β-catenin, CK19, and ZO-1; Supplementary Figure 2C) and gain of mesenchymal markers (e.g. MMP9, Twist, and vimentin; Supplementary Figure 2D). Collectively, these events culminated in the redistribution of E-cadherin [Supplementary Figure 2E] and β-catenin [Supplementary Figure 2F] from the plasma membrane to the cytoplasm, consistent with the ability of TGF-β to induce EMT programs in luminal breast cancers.

Supplementary Figure 2. TGF-β induces EMT phenotypes in BT474 cells. (A) BT474 cells were treated with TGF-β1 (5 ng/mL) for 0-72 h to induce an EMT program. Photomicrographs depict accompanying alterations in cell morphology (× 400). (B) BT474 cells were stimulated with TGF-β1 (5 ng/mL) for 0-96 h as indicated, at which point differences in the expression of E-cad and N-cad mRNA (top) and protein (bottom) were determined. mRNA data are the mean foldchanges (± SE; n = 3; *P < 0.05; Student's t-test), while protein data are representative images from 3-independent experiments. (C and D) BT474 cells were treated with TGF-β1 (5 ng/mL) for 0-96 h, at which point the expression of b-catenin, CK19, and ZO-1 (C) or of MMP-9, Twist-1, and vimentin (D) were monitored by real-time PCR. Data are the mean fold-changes (± SE; n = 3;*P < 0.05; Student's t-test). (E and F) BT474 cells were stimulated with TGF-β1 (5 ng/mL) for 72 h, and subsequently were fixed in paraformaldehyde and processed for indirect immunofluorescence of either E-cad (E) or b-catenin (F), as well as for direct immunofluorescence of actin (E and F). Nuclei were visualized by DAPI staining. Images are representative from 3-independent experiments (× 400). TGF: transforming growth factor; EMT: epithelial-mesenchymal transition; SE: standard error
Induction of EMT programs by TGF-β promotes ER-α accumulation in the cytoplasm of MCF-7 cells
We next investigated the interplay between TGF-β and ER-α in regulating EMT programs in MCF-7 cells, especially in light of the ability of TGF-β to inhibit ER-α signaling and vice versa.[28] In doing so, we first determined whether ER-α signaling impacted the coupling of TGF-β to EMT programs in MCF-7 cells. Although estradiol treatment had no obvious effect on E-cad localization in MCF-7 cells, hormone administration did suppress the ability of TGF-β to induce the redistribution of E-cadherin from the plasma membrane [Figure 2A]. ER-α was the predominant estrogen receptor expressed in MCF-7 [Figure 2B] and BT474 [Supplementary Figure 3A], indicating that the actions of estrogen are mediated entirely by ER-α in these breast cancer cells. As such, we also monitored the impact of TGF-β on ER-α expression in MCF-7 and BT474 cells. Figure 2C shows that TGF-β transiently increased the synthesis of ER-α transcripts when MCF-7 cells were propagated in 2D-cultures, an event that also trended to occur in BT474 cells [Supplementary Figure 3B]. Conversely, ER-α transcript levels were dramatically repressed when MCF-7 organoids were stimulated with TGF-β in 3D-cultures [Figure 2D]. Although TGF-β clearly regulated ER-α transcription, activation of the TGF-β pathway had little-to-no effect on ER-α protein expression in both MCF-7 and BT474 cells [Figure 2E] despite remaining competent to inhibit luciferase expression driven by an estrogen-response element (ERE; Figure 2F and Supplementary Figure 3C). The antagonist activities of TGF-β on ER-α signaling are magnified in post-EMT MCF-7 cells as compared to their pre-EMT counterparts [Figure 2G]. Mechanistically, we observed TGF-β stimulation of EMT programs to rapidly promote ER-α translocation from the nucleus to the cytoplasm, resulting in ER-α being entirely excluded from the nucleus by 72 h [Figure 2H]. Collectively, these findings suggest that TGF-β and its stimulation of EMT programs inhibit the genomic functions of ER-α by inducing its cytoplasmic sequestration.

Figure 2. Induction of EMT programs by TGF-β promotes ER-α accumulation in the cytoplasm of MCF-7 cells. (A) MCF-7 cells were treated with either TGF-β1 (5 ng/mL), estradiol (1 nmol/L), or both agonists for 96 h as indicated. Afterward, the cells were fixed in paraformaldehyde and processed for indirect E-cad immunofluorescence. Images are representative of 3-independent experiments (×400). (B and C) MCF-7 cells were treated with TGF-β1 (5 ng/mL) in either 2D- (B) or 3D- (C) cultures for 0-96 h, at which point differences in ER-α expression were measured by real-time PCR. Data are the mean fold-changes (± SE; n = 3; *P < 0.05; Student's t-test). (D) Differential mRNA expression of ER-α and ER-β in quiescent MCF-7 cells. Data are the mean (± SE; n = 3; *P < 0.05; Student's t-test). (E) MCF-7 (top) and BT474 (bottom) cells were stimulated with TGF-β1 (5 ng/mL) for 0-96 h. Afterward, ER-α expression levels were monitored by immunoblotting. Images are representative of 4-independent experiments. (F) MCF-7 cells were transiently transfected overnight with either the pSBE- or pERE-luciferase reporter genes, as well as with the pCMV-β-galactosidase reporter gene to control for differences in transfection efficiency. Afterward, the transfectants were stimulated overnight either singly or in combination with TGF-β1 (T; 5 ng/mL) or estradiol (E; 0.1 nmol/L) prior to measuring luciferase and β-gal activities. Data are the mean fold-changes (± SE; n = 3; *P < 0.05; Student's t-test). (G) Pre- and post-EMT MCF-7 cells were transiently transfected overnight with pERE-luciferase and pCMV-β-gal-luciferase cDNAs, followed by 24 h treatment with either TGF-β1 (5 ng/mL) or estradiol (0.1 nmol/L). Data are the mean fold-changes (± SE; n = 3; *P < 0.05; Student's t-test). (H) MCF-7 cells were treated with TGF-β1 (5 ng/mL) for 0-96 h, at which point the expression of ER-α in cytoplasmic and nuclear cell fractions was determined by immunoblotting. Stripped blots were reprobed with antibodies to either HDAC1 or β-actin to monitor integrity of nuclear and cytoplasmic fractions, respectively. Images are representative of at least 3-independent experiments. TGF: transforming growth factor; EMT: epithelial-mesenchymal transition; ER: estrogen receptor; SE: standard error

Supplementary Figure 3. TGF-β regulates estrogen signaling and EGFR expression in BT474 cells. (A) Differential mRNA expression of ER-a and ER-b in quiescent BT474 cells. (B) BT474 cells were stimulated with TGF-β1 (5 ng/mL) for 0-96 h as indicated. Afterward, total RNA was harvested and subjected to real-time PCR to monitor differences in ER-a expression. (C) BT474 cells were transiently transfected overnight with p3TP-, pSBE-, or pERE-luciferase and pCMV-bgal cDNAs prior to their stimulation for 24 h with TGF-β1 (5 ng/mL). Afterward, luciferase and bgal activities contained in detergent-solubilized cell extracts were measured. Data are the mean fold-changes (± SE; n = 3;*P < 0.05; Student's t-test). TGF: transforming growth factor; EGFR: epidermal growth factor receptor; SE: standard error
TGF-β stimulation of EMT promotes the interaction of ER-α with EGFR, IGF1R and Src in MCF-7 and BT474 cells
Previous studies have shown the ability of cytoplasmic ER-α to interact physically with EGFR and IGF1R, leading to the activation of MAP kinases and resistance to tamoxifen.[9,11-13] Because EMT programs stimulated by TGF-β resulted in ER-α exclusion from the nucleus, we speculated that EMT programs may underlie the associations of ER-α with receptor tyrosine kinases (RTKs). To test this hypothesis, we determined the impact of TGF-β on the expression of EGFR and IGF1R in luminal breast cancer cells. As shown in Figure 3A-C, TGF-β significantly stimulated the synthesis of EGFR and IGF1R mRNA in MCF-7 cells propagated in either 2D- or 3D-culture. Similar increases in EGFR transcript levels were also observed in BT474 cells stimulated with TGF-β [Supplementary Figure 4A], while the abnormally high levels of IGF1R mRNA in BT474 cells [Supplementary Figure 4B] masked any apparent effects of TGF-β on IGF1R transcript levels. We also observed TGF-β stimulation of MCF-7 and BT474 cells to induce the production of EGFR proteins, which were readily captured in ER-α immunocomplexes [Figure 3D-F]. Likewise, IGF1R was also readily captured in ER-α immunocomplexes isolated from MCF-7 and BT474 cells stimulated with TGF-β [Figure 3F and G]. Finally, Src has been reported to facilitate the extranuclear activities and localization of ER-α in breast cancers.[29] Accordingly, we also found Src to associate with ER-α in a TGF-β-dependent manner in MCF-7 cells [Figure 3H], suggesting a potential role for Src in facilitating the extranuclear localization and function of ER-α. Collectively, these findings suggest that TGF-β and EMT programs result in the upregulation of EGFR and IGF1R, which form cytoplasmic complexes with ER-α and Src.

Figure 3. TGF-β stimulation of EMT promotes the interaction of ER-α with EGFR, IGF1R, and Src in MCF-7 and BT474 cells. (A and B) MCF-7 cells were treated with TGF-β1 (5 ng/mL) for 0-96 h, at which point EGFR (A) and IGF1R (B) transcript levels were measured by real-time PCR. Data are the mean fold-changes (± SE; n = 3; *P < 0.05; Student's t-test). (C) MCF-7 cells were propagated in 2D and 3D cultures. Afterward, total RNA was isolated and subjected to real-time PCR to monitor the expression of EGFR and IGF1R. Data are the mean fold-changes (± SE; n = 3; *P < 0.05; Student's t-test); (D) MCF-7 cells were treated with TGF-β1 (5 ng/mL) for 0-96 h, at which point detergent-solubilized extracts were immunoblotted with antibodies against either EGFR or β-actin as indicated (top). Additionally, ER-α immunocomplexes were captured and immunoblotted for EGFR and ER-α as indicated (bottom). Images are representative of 4-independent experiments; (E) BT474 cells were stimulated with TGF-β1 (5 ng/mL) as indicated, and subsequently were subjected to immunoblot analyses to monitor the expression of EGFR, IGF1R, and β-actin. Images are representative of 3-individual experiments; (F and G) BT474 cells were stimulated with TGF-β1 (5 ng/mL) for 0-72 h, while MCF-7 cells were stimulated with TGF-β1 for 0-96 h. Afterward, detergent-solubilized extracts were immunoprecipitated with antibodies against ER-α and subsequently were immunoblotted with antibodies against IGF1R and ER-α. Images are representative of at least 3-individual experiments; (H) MCF-7 cells were treated with TGF-β1 (5 ng/mL) for 0-96 h, at which point detergent-solubilized extracts were immunoblotted for Src or β-actin as indicated (top). Additionally, Src immunocomplexes were captured and immunoblotted for ER-α and Src as indicated (bottom). Images are representative of 4-independent experiments. TGF: transforming growth factor; EMT: epithelial-mesenchymal transition; IGF: insulin-like growth factor; ER: estrogen receptor; EGFR: epidermal growth factor receptor; SE: standard error

Supplementary Figure 4. TGF-β regulates EGFR and IGF-1R expression in BT474 cells. (A) BT474 cells were stimulated with TGF-β1 (5 ng/mL) for 0-96 h as indicated, at which point differences in EGFR expression were monitored by real-time PCR. (B) Differential mRNA expression of EGFR and IGF-1R in quiescent BT474 cells. Data are the mean fold-changes (± SE; n = 3; *P < 0.05; Student's t-test). TGF: transforming growth factor; IGF: insulin-like growth factor; EGFR: epidermal growth factor receptor; SE: standard error
TGF-β stimulation of EMT programs enhances EGFR, IGF1R, and MAP kinase signaling in MCF-7 and BT474 cells
The activation of MAP kinases by TGF-β is essential for its stimulation of EMT programs.[4,30] Interestingly, nongenomic ER-α signaling is mediated in part through its ability to activate MAP kinases, thereby contributing to the acquisition of tamoxifen resistance in ER-positive breast cancer cells.[31] Given these parallels and reliance upon MAP kinases, we speculated that TGF-β and EMT programs would elicit the hyperactivation of MAP kinases downstream of upregulated expression of EGFR and IGF1R in post-EMT cells, leading to the initiation of nongenomic ER-α signaling. In testing this hypothesis, we first monitored the activation status of MAP kinases in MCF-7 and BT474 cells when stimulated by TGF-β. Although TGF-β did indeed elicit a modest and transient activation of ERK1/2 in MCF-7 cells [Figure 4A], its ability to stimulate both ERK1/2 and p38 MAPK was greatly potentiated as MCF-7 and BT474 cells transitioned through the EMT program [Figure 4B and C]. These events were specific for MAP kinases as no alterations in AKT phosphorylation were detected under both transient and prolonged TGF-β stimulations (data not shown). Interestingly, Figure 4D shows that administration of the TβR-I inhibitor II to inactivate TβR-I prevented both the upregulated expression of EGFR and the activation of MAP kinases (i.e. ERK1/2 and p38 MAPK) in MCF-7 cells stimulated with TGF-β. Moreover, administration of AG1478 to inactivate EGFR abrogated ERK1/2 activity in post-EMT MCF-7 cells [Figure 4E]. Similar potentiation of ERK1/2 activity was also observed in post-EMT MCF-7 cells stimulated with either IGF-1, estradiol, or EGF [Figure 4F], a reaction partially dependent upon the greatly magnified activation of IGF1R in these post-EMT cells [Figure 4G]. Collectively, these findings demonstrate that EMT programs induced by TGF-β not only result in the robust stimulation of MAP kinases, but also elicit increased sensitivity and activation of post-EMT breast cancer cells to IGF1, estrogen, and EGF.

Figure 4. TGF-β stimulation of EMT programs enhances EGFR, IGF1R, and MAP kinase signaling in MCF-7 and BT474 cells. (A and B) MCF-7 cells were stimulated with TGF-β1 (5 ng/mL) as indicated. Afterward, the activation status of ERK1/2 and p38 MAPK was determined by immunoblotting; (C) BT474 cells were treated with TGF-β1 (5 ng/mL) for 0-96 h prior to monitoring the activation status ERK1/2 and p38 MAPK by immunoblotting. (D) MCF-7 cells were stimulated with TGF-β1 (5 ng/mL) in the absence or presence of the TβR-I inhibitor II (100 ng/mL) for 72 h. Afterward, the expression levels of EGFR and activation status of p38 MAPK and ERK1/2 were determined by immunoblotting; (E-G) pre- and post-EMT MCF-7 cells were treated with AG1478 (1 µmol/L; E), with IGF-1 (100 ng/mL; top), estradiol (0.1 nmol/L; middle), and EGF (100 ng/mL; bottom; F), or with IGF-1 (100 ng/mL) in the absence or presence of AG1024 (1 µmol/L; G) as indicated. Afterward, the expression levels of EGFR and activation status of ERK1/2 and IGF1R were determined by immunoblotting as indicated. Data are representative images from at least 3-independent experiments. TGF: transforming growth factor; EMT: epithelial-mesenchymal transition; IGF: insulin-like growth factor; EGFR: epidermal growth factor receptor
TGF-β stimulation of EMT programs promotes tamoxifen resistance in MCF-7 cells
The aforementioned findings showed that TGF-β and its stimulation of EMT programs engendered the nuclear exclusion of ER-α, leading to its (1) physical interaction with EGFR, IGF1R, and Src, and (2) enhanced activation of MAP kinases [Figures 2-4]. We next examined the functional consequences of these events on luminal breast cancer growth and their sensitivity to tamoxifen. In doing so, we first propagated pre- and post-EMT MCF-7 organoids in the absence of presence of ER-α modulators. Figure 5A shows that post-EMT MCF-7 organoids grew more robustly as compared to their pre-EMT counterparts; they were also significantly more sensitive to growth inhibition by administration of small molecule antagonists to either IGF1R (i.e. AG1024; Supplementary Figure 5A) or EGFR (i.e. AG1478; Supplementary Figure 5B), findings consistent with the ability of post-EMT cells to upregulate their expression of IGF1R and EGFR and activation of ERK1/2 [Figures 3 and 4]. Additionally, post-EMT MCF-7 cells also exhibited significantly increased cell growth and decreased sensitivity to tamoxifen-induced cell death [Figure 5B]. Importantly, co-administration of tamoxifen with small molecule inhibitors against either TβR-I (i.e. TβR-I inhibitor II), IGF1R (i.e. AG1024), EGFR (i.e. AG1478), or MEK1/2 (i.e. U0126) restored MCF-7 cell sensitivity to tamoxifen as determined by MTS [Figure 5B] or clonogenic [Figure 5C and D] assays. Taken together, these results demonstrated that post-EMT cells acquire resistance to tamoxifen by upregulating EGFR and IGF1R expression and MAP kinase activation, culminating in extranuclear localization and nongenomic signaling of ER-α in MCF-7 cells.

Figure 5. TGF-β stimulation of EMT programs promotes tamoxifen resistance in MCF-7 cells. (A) Pre- and post-EMT MCF-7 organoids were treated with estradiol (0.1 nmol/L), tamoxifen (0.1 µmol/L), or fulvestrant (0.1 µmol/L) for 8 days, at which point photomicrographs were captured and analyzed on Image J to assess differences in organoid growth. Images are representative of 3-independent experiments, while data are the mean fold-changes (± SE; n = 3; *P < 0.05; Student's t-test; ×400); (B) pre- and post-EMT MCF-7 cells were treated with tamoxifen (0.1 µmol/L) in the absence or presence of TβR-I Inhibitor II (100 ng/mL; left), of AG1024 (1 µmol/L; middle), or of AG1478 (1 µmol/L; right) as indicated. Afterward, differences in cell growth and survival were analyzed by MTS assays. Data are the mean (± SE; n = 3; *P < 0.05; Student's t-test) growth relative to untreated pre-EMT cells (*P < 0.05; Student's t-test); (C-E) pre- and post-EMT MCF-7 cells were treated with tamoxifen (0.1 µmol/L) in the absence or presence of U0126 (10 µmol/L; left), of TβR-I Inhibitor II (100 ng/mL; middle), or AG1024 (1 µmol/L; right) for 10 days, at which point the number of surviving colonies in 11 random fields/plate was enumerated. Data are the mean (± SE; n = 3; *P < 0.05; Student's t-test). TGF: transforming growth factor; EMT: epithelial-mesenchymal transition; SE: standard error

Supplementary Figure 5. Post-EMT cells are sensitized to inhibitors against IGF-1R and EGFR. Pre- and post-EMT MCF-7 cells were cultured in the absence (diluent) or presence of AG1024 (10 µmol/L; A) or AG1478 (10 µmol/L; B) for 5 days prior to analyzing differences in cell growth and survival by MTS assays. Data are the mean growth (± SE; n = 3; *P < 0.05; Student's t-test). EMT: epithelial-mesenchymal transition; IGF: insulin-like growth factor; EGFR: epidermal growth factor receptor; SE: standard error
Discussion
The induction of EMT programs by TGF-β plays important roles in driving the progression, dissemination, and recurrence of human breast cancers; these events also underlie the development, expansion, and self-renewal of cancer stem cells, as well as the acquisition of chemoresistant phenotypes.[4,22,23] Although EMT programs are more commonly associated with basal-like/TNBCs, we show herein that luminal A (e.g. MCF-7) and luminal B (e.g. BT474) breast cancer cells not only undergo EMT in response to TGF-β, but that they also exhibit diminished sensitivity to tamoxifen that results from the: (1) upregulated expression of EGFR and IGF1R, which interact physically with ER-α; (2) hyperactivation states of MAP kinases (e.g. ERK1/2 and p38 MAPK); and (3) extranuclear exclusion and nongenomic functions of ER-α. Importantly, the enhanced tumorigenic activities displayed by post-EMT cells were readily reversed by co-administration of tamoxifen with small molecule inhibitors against either TβR-I (i.e. TβR-I inhibitor II), IGF1R (i.e. AG1024), EGFR (i.e. AG1478), or MEK1/2 (i.e. U0126), thereby laying the foundation for future investigations related to how inactivating EMT programs can be harnessed to improve the clinical outcomes of breast cancer patients.
Several studies previously implicated the nongenomic actions of ER-α in promoting tamoxifen resistance and disease progression.[9,11-13,32] For instance, the aberrant expression of truncated MTA1 mutants that remain competent to bind ER-α and sequester it in the cytoplasm clearly contribute to the generation of nongenomic ER-α activity.[14] Indeed, MTA1 expression inactivates hormone-induced nuclear translocation of ER-α, an event that enhances tumor progression and correlates with a loss of ER-α in the nucleus.[14] Along these lines, ER-α expression and activity also require interactions with growth factor signaling systems to enable luminal breast cancer cells to become insensitive to ER-α modulating agents. For instance, tamoxifen-resistant MCF-7 cells house cytoplasmic complexes comprised of ER-α, EGFR, and Src that elicit hyperactivation of MAP kinases by either EGF or estradiol. Consequently, targeted inactivation of either Src, EGFR, or MAPK not only restores ER-α to the nucleus, but also reestablishes the antitumor activities of tamoxifen in ER-positive tumors.[12,32] Besides its ability to bind EGFR, ER-α also complexes with the IGF1R at the cell membrane, an interaction involving the adaptor protein Shc.[31,32] Moreover, tamoxifen-resistant breast cancer cells can also undergo RTK switching as a means to evade targeted inactivation of either EGFR or IGF1R, thereby preserving nongenomic ER-α signaling.[12] It should be noted that RTK switching is a mechanism associated TGF-β and its stimulation of EMT programs.[33] Given the parallels between the aforementioned findings and those presented herein, it is tempting to speculate that EMT programs function as essential drivers of nongenomic ER-α signaling and disease progression in luminal breast cancer cells. Future studies need to determine the exact molecular mechanisms whereby TGF-β and EMT programs elicit extranuclear exclusion of ER-α; they also need to explore the linkages between EMT programs and ER-α mutations in regulating metastasis and disease recurrence.
As mentioned previously, EMT programs are closely associated with the acquisition of drug-resistant phenotypes.[4,22,23] Indeed, tamoxifen-resistant MCF-7 cells exhibit post-EMT morphologies (i.e. mesenchymal-/fibroblastoid-like) that reflect a loss of E-cadherin expression and a gain of β-catenin signaling, events that were readily reversed following administration of EGFR inhibitors.[34] A similar dependence upon EMT programs was observed in pancreatic cancers as they acquired resistance to gemcitabine, 5-fluorouracil, and cisplatin, a reaction driven by the upregulated expression of Zeb1. Indeed, genetic inactivation of Zeb1 in post-EMT, chemoresistant pancreatic cancer cells was sufficient to induce a mesenchymal-to-epithelial transition (MET) that reinstated cellular sensitivity to conventional chemotherapeutic agents.[35] Likewise, resistance to EGFR and FGFR3 inhibitors in cancers of the bladder coincides with their completion of EMT programs engendered by the loss of miR-200 family member expression. Importantly, restoring miR-200 expression in post-EMT bladder cancer cells not only induced their undertaking of MET programs, but also reactivated their sensitivity to EGFR and FGFR3 inhibitors.[36] Our studies herein reinforce the central importance of EMT programs to elicit chemoresistance in developing and recurring cancers, particularly those arising in the breast. Moreover, we established TGF-β and EMT programs as drivers of tamoxifen resistance and nongenomic ER-α signaling in luminal breast cancers; we also provide a new and potentially impactful approach to eliminate tamoxifen-resistant, post-EMT breast cancer cells through the combined administration of tamoxifen with inhibitors against either TβR-I, EGFR, IGF1R, or MEK1/2, thereby restoring the effectiveness of anti-hormone therapies in ER-positive tumors.
Declarations
Authors’ contributionsConceived and designed the study: M. Tian, W.P. Schiemann
Performed molecular and cellular analyses: M. Tian
Performed literature search and prepared the manuscript: M. Tian
Revised the manuscript: W.P. Schiemann
Financial support and sponsorshipResearch support was provided in part by the National Institutes of Health to W.P.S. (CA129359, CA177069, and CA194518).
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalThe study was approved by Institutional Scientific and Ethical Committees.
REFERENCES
2. Tian M, Neil JR, Schiemann WP. Transforming growth factor-b and the hallmarks of cancer. Cell Signal 2011;23:951-62.
4. Parvani JG, Taylor MA, Schiemann WP. Noncanonical TGF-b signaling during mammary tumorigenesis. J Mammary Gland Biol Neoplasia 2011;16:127-46.
5. Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-b in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia 2010;15:169-90.
6. Arendt LM, Kuperwasser C. Form and function: how estrogen and progesterone regulate the mammary epithelial hierarchy. J Mammary Gland Biol Neoplasia 2015;20:9-25.
7. Finlay-Schultz J, Sartorius CA. Steroid hormones, steroid receptors, and breast cancer stem cells. J Mammary Gland Biol Neoplasia 2015;20:39-50.
8. Ali S, Coombes RC. Estrogen receptor a in human breast cancer: occurrence and significance. J Mammary Gland Biol Neoplasia 2000;5:271-81.
9. Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer 2002;2:101-12.
10. Osborne CK, Fuqua SA. Mechanisms of tamoxifen resistance. Breast Cancer Res Treat 1994;32:49-55.
11. Song RX, Barnes CJ, Zhang Z, Bao Y, Kumar R, Santen RJ. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor a to the plasma membrane. Proc Natl Acad Sci U S A 2004;101:2076-81.
12. Song RX, Chen Y, Zhang Z, Bao Y, Yue W, Wang JP, Fan P, Santen RJ. Estrogen utilization of IGF-1-R and EGF-R to signal in breast cancer cells. J Steroid Biochem Mol Biol 2010;118:219-30.
13. Song RX, Zhang Z, Chen Y, Bao Y, Santen RJ. Estrogen signaling via a linear pathway involving insulin-like growth factor I receptor, matrix metalloproteinases, and epidermal growth factor receptor to activate mitogen-activated protein kinase in MCF-7 breast cancer cells. Endocrinology 2007;148:4091-101.
14. Kumar R, Wang RA, Mazumdar A, Talukder AH, Mandal M, Yang Z, Bagheri-Yarmand R, Sahin A, Hortobagyi G, Adam L, Barnes CJ, Vadlamudi RK. A naturally occurring MTA1 variant sequesters oestrogen receptor-a in the cytoplasm. Nature 2002;418:654-7.
15. Parvani JG, Davuluri G, Wendt MK, Espinosa C, Tian M, Danielpour D, Sossey-Alaoui K, Schiemann WP. Deptor enhances triple-negative breast cancer metastasis and chemoresistance through coupling to survivin expression. Neoplasia 2015;17:317-28.
16. Tian M, Schiemann WP. Preclinical efficacy of cystatin C to target the oncogenic activity of transforming growth factor b in breast cancer. Transl Oncol 2009;2:174-83.
17. Tian M, Schiemann WP. PGE2 receptor EP2 mediates the antagonistic effect of COX-2 on TGF-b signaling during mammary tumorigenesis. FASEB J 2010;24:1105-16.
18. Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, Wang XF, Massague J. TGFb signals through a heteromeric protein kinase receptor complex. Cell 1992;71:1003-14.
19. Hall JM, McDonnell DP. The estrogen receptor b-isoform (ERb) of the human estrogen receptor modulates ERa transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 1999;140:5566-78.
20. Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol 2003;13:680-5.
21. Schiemann WP, Pfeifer WM, Levi E, Kadin ME, Lodish HF. A deletion in the gene for transforming growth factor b type I receptor abolishes growth regulation by transforming growth factor b in a cutaneous T-cell lymphoma. Blood 1999;94:2854-61.
22. Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev 2016;35:645-54.
23. Wendt MK, Tian M, Schiemann WP. Deconstructing the mechanisms and consequences of TGF-b-induced EMT during cancer progression. Cell Tissue Res 2012;347:85-101.
24. Morrison CD, Parvani JG, Schiemann WP. The relevance of the TGF-b paradox to EMT-MET Programs. Cancer Lett 2013;341:30-40.
25. Taylor MA, Amin J, Kirschmann DA, Schiemann WP. Lysyl oxidase contributes to mechanotransduction-mediated regulation of transforming growth factor-b signaling in breast cancer cells. Neoplasia 2011;13:406-18.
26. Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, Chen AC, Sah RL, Taylor SS, Engler AJ, Yang J. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol 2015;17:678-88.
27. Gilbert PM, Weaver VM. Cellular adaptation to biomechanical stress across length scales in tissue homeostasis and disease. Semin Cell Dev Biol 2017;67:141-152.
28. Band AM, Laiho M. Crosstalk of TGF-b and estrogen receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia 2011;16:109-15.
29. Song RX, Zhang Z, Santen RJ. Estrogen rapid action via protein complex formation involving ERa and Src. Trends Endocrinol Metab 2005;16:347-53.
30. Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF-b1-induced EMT in vitro. Neoplasia 2004;6:603-10.
31. Santen RJ, Fan P, Zhang Z, Bao Y, Song RX, Yue W. Estrogen signals via an extra-nuclear pathway involving IGF-1R and EGFR in tamoxifen-sensitive and -resistant breast cancer cells. Steroids 2009;74:586-94.
32. Fan P, Wang J, Santen RJ, Yue W. Long-term treatment with tamoxifen facilitates translocation of estrogen receptor a out of the nucleus and enhances its interaction with EGFR in MCF-7 breast cancer cells. Cancer Res 2007;67:1352-60.
33. Thomson S, Petti F, Sujka-Kwok I, Mercado P, Bean J, Monaghan M, Seymour SL, Argast GM, Epstein DM, Haley JD. A systems view of epithelial-mesenchymal transition signaling states. Clin Exp Metastasis 2011;28:137-55.
34. Hiscox S, Jiang WG, Obermeier K, Taylor K, Morgan L, Burmi R, Barrow D, Nicholson RI. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of b-catenin phosphorylation. Int J Cancer 2006;118:290-301.
35. Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey DJ, Choi W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res 2009;69:5820-8.
Cite This Article

How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright














Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].