


Podocyte lipotoxicity in diabetic kidney disease: mechanisms and targets
 0
0 Abstract
Podocyte lipotoxicity is a fundamental pathogenic mechanism in diabetic kidney disease (DKD), driven by aberrant intracellular lipid accumulation that disrupts cellular integrity and function. This review systematically examines the molecular and metabolic underpinnings of this process, focusing on dysregulated cholesterol efflux, impaired fatty acid oxidation, altered sphingolipid metabolism, and defective lipid droplet homeostasis. These disturbances converge to induce mitochondrial dysfunction, endoplasmic reticulum stress, oxidative damage, and inflammatory activation, collectively leading to podocyte apoptosis and loss. Emerging evidence further illuminates the role of epigenetic regulators, transcriptional networks, and inter-organ signaling in modulating lipotoxic responses. Mechanistic insights into podocyte lipid metabolism provide a critical foundation for developing targeted therapeutic strategies to preserve podocyte function in DKD.
Keywords
INTRODUCTION
Podocytes are highly specialized, terminally differentiated cells critical for maintaining the glomerular filtration barrier (GFB), working in concert with the glomerular endothelium and basement membrane to prevent albumin loss[1]. Podocyte injury or loss leads to reduced glomerular basement membrane coverage, initial albuminuria, and eventual glomerulosclerosis. Their reliance on anaerobic glycolysis for ~70%-80% of adenosine triphosphate (ATP) production, coupled with fewer mitochondria compared to tubular cells, renders them inherently vulnerable to metabolic stress[2-4].
Podocyte injury has been recognized as a pivotal early event in the pathogenesis of proteinuric kidney diseases, including diabetic kidney disease (DKD)[5]. In DKD, podocytes are particularly susceptible to lipotoxicity, driven by the intracellular accumulation of cholesterol esters (CE), fatty acids, triglycerides, sphingolipids, and glycerophospholipids[6]. This lipotoxicity may involve multiple mechanisms such as enhanced uptake via transporters like cluster of differentiation 36 (CD36)[7,8]; decreased cholesterol efflux[1,9-11]; increased synthesis[12]; and impaired fatty acid oxidation (FAO) in DKD[13]. Lipotoxicity may further trigger multiple pathological pathways in podocytes, including mitochondrial oxidative stress[14], endoplasmic reticulum (ER) stress[15], inflammatory responses[16,17], insulin resistance[18], actin cytoskeleton remodeling[19], and apoptosis[20], all of which contribute to the pathogenesis of DKD.
In this review, we aim to summarize current knowledge regarding podocyte lipotoxicity in DKD, tracing the pathogenic cascade from lipid accumulation and dysregulated metabolism to downstream cellular injury and death. We further discuss promising pharmacological and metabolic interventions, highlighting the challenges and future directions for translating these insights into effective reno-protective therapies.
LIPIDS AND METABOLIC PERTURBATIONS
The susceptibility of podocytes to lipid-induced injury stems from a complex interplay of metabolic disturbances. This section examines three primary axes of lipid dysregulation that converge on podocyte dysfunction in DKD: (1) altered cholesterol trafficking and membrane raft integrity; (2) disrupted fatty acid uptake and mitochondrial β-oxidation; and (3) aberrant sphingolipid signaling, with a particular focus on ceramide accumulation. Each of these pathways not only impairs cellular energetics but also serves as a critical trigger for downstream inflammatory and apoptotic cascades.
Cholesterol metabolism and lipid raft disruption in podocytes
Podocyte lipotoxicity in DKD is closely linked to dysregulated cholesterol metabolism, characterized by cholesterol accumulation, impaired cholesterol efflux, and disruption of lipid raft-associated protein dynamics. Cholesterol accumulation in podocytes can result from enhanced synthesis, reduced efflux, or altered trafficking. For instance, apolipoprotein A1 (APOL1) risk variant (RV) expression promotes cholesterol accumulation, as evidenced by significantly higher total cholesterol (TC) levels in kidney cortices of APOL1 G1 transgenic mice compared to G0 controls, with unchanged CE content[21]. In DKD, podocyte cholesterol accumulation arises from both increased uptake and decreased efflux, the latter attributed to reduced activity of the ATP-binding cassette transporter A1 (ABCA1). Podocyte-specific ABCA1 deficiency in diabetic mice correlates with increased kidney CE content and serum blood urea nitrogen (BUN), highlighting a critical role of ABCA1 in maintaining cholesterol homeostasis[22]. Mechanisms impairing ABCA1 function include coiled-coil domain-containing protein 92 (CCDC92)-mediated degradation: CCDC92, upregulated in diabetic glomeruli, interacts with proteasome activator 28 subunit α (PA28α) to enhance proteasome activity, promoting ABCA1 degradation and inhibiting cholesterol efflux, thereby increasing intracellular cholesterol[9]. Similarly, junctional adhesion molecule-like protein (JAML), elevated in DKD glomeruli, reduces ABCA1 expression, while JAML deficiency restores ABCA1 levels, ameliorating lipid accumulation[23]. NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation in diabetes also downregulates ABCA1, and inhibition with the NLRP3 inflammasome inhibitor MCC950 reverses this reduction, decreasing cholesterol accumulation in glomeruli[17]. In both Alport Syndrome (AS) and DKD, ABCA1 impairment compromises cholesterol efflux, leading to dysregulation of cholesterol and phospholipids; local tumor necrosis factor (TNF) overexpression suppresses ABCA1, further exacerbating cholesterol accumulation and mitochondrial dysfunction[14]. Additionally, sterol-O-acyltransferase-1 (SOAT1), which converts free cholesterol to CE, is upregulated in diabetic podocytes, and its inhibition increases ABCA1 expression and ABCA1/APOA1-mediated cholesterol efflux, reducing CE accumulation in DKD and AS models[11].
Lipid rafts, cholesterol-concentrated microdomains in podocyte plasma membranes, are enriched with slit diaphragm proteins such as nephrin, podocin, and CD2-associated protein, which are critical for GFB integrity[24]. Disruption of lipid rafts in DKD involves altered expression or function of lipid raft-associated proteins. Acid sphingomyelinase-like phosphodiesterase 3b (SMPDL3b), a lipid raft protein that catalyzes sphingomyelin catabolism, is upregulated in DKD, altering ceramide metabolism and promoting podocyte apoptosis; inhibiting SMPDL3b improves podocyte survival and reduces albuminuria[20]. CD36, a lipid raft-associated protein involved in fatty acid uptake, undergoes palmitoylation at cysteine 466, which is critical for its plasma membrane localization. Diabetes-induced downregulation of acyl-protein thioesterase 1 (APT1) reduces CD36 depalmitoylation, increasing membrane retention of palmitoylated CD36 and enhancing lipid uptake[7]. Ephrin-B1, localized at the slit diaphragm, interacts with nephrin to maintain podocyte structure; podocyte-specific ephrin-B1 knockout disrupts slit diaphragm molecule localization, leading to proteinuria, indicating its pivotal role in preserving lipid raft-associated signaling complexes[25]. Fat mass and obesity-associated protein (FTO), an m6A RNA demethylase upregulated in diabetic podocytes, stabilizes the mRNAs of acetyl-CoA carboxylase 1 (ACC1), increasing fatty acid synthesis and lipid droplet (LD) accumulation[26]; FTO inhibition reduces the intensity of adipophilin, a LD marker, suggesting improved lipid raft integrity[26]. Furthermore, collagen type I-induced activation of discoidin domain receptor 1 (DDR1) in AS podocytes interacts with CD36, enhancing free fatty acid (FFA) uptake and triglyceride accumulation, a process suppressed by ezetimibe, highlighting CD36’s role in lipid raft-mediated lipotoxicity[27].
While distinct from direct cholesterol synthesis, glucose-lowering agents may also intersect with lipid metabolism. Sodium-glucose cotransporter 2 (SGLT2), expressed in podocytes and upregulated in AS, contributes to LD accumulation; empagliflozin reduces LD and apoptosis in AS podocytes by shifting energy metabolism toward FAO[28]. Serine/threonine kinase 25 (STK25) overexpression in podocytes also promotes LD accumulation and oxidative stress, potentially disrupting lipid raft function[29]. Therapeutic interventions, such as Gandi capsule and its active ingredient baicalin restore podocin expression, a key slit diaphragm protein, by modulating the sirtuin 1 (SIRT1)/AMP-activated protein kinase (AMPK)/hepatocyte nuclear factor 4 alpha (HNF4A) pathway, indirectly supporting lipid raft stability[30]. Additionally, podocyte-specific disruption of protein tyrosine phosphatase 1B (PTP1B) attenuates hyperglycemia-induced renal injury by enhancing insulin signaling and autophagy, though its direct involvement in cholesterol metabolism or lipid raft disruption has not been established[31]. Collectively, these findings underscore the interplay between impaired cholesterol metabolism (via ABCA1 dysfunction) and lipid raft disruption (via altered protein dynamics) in podocyte lipotoxicity, identifying potential therapeutic targets for DKD.
FATTY ACID UPTAKE, METABOLISM, AND TOXICITY
Saturated fatty acids (SFAs), particularly palmitic acid (PA), are major contributors to podocyte lipotoxicity, with stearic acid also implicated, while monounsaturated fatty acids (MUFAs), such as oleic acid (OA), exert protective effects by mitigating SFA-induced ER stress and cell death[32]. PA induces lipotoxic damage in podocytes, characterized by insulin resistance, oxidative stress, ER stress, and apoptosis, as demonstrated in vitro through exposure of cultured podocytes to bovine serum albumin (BSA)-conjugated PA[33]. In mouse podocytes, PA stimulation increases reactive oxygen species (ROS) formation and cellular toxicity, associated with upregulation of NADPH oxidase 4 (NOX4) and pro-apoptotic protein BCL2-associated X protein (BAX)[34].
Enhanced FFA uptake by podocytes is mediated by increased expression of the scavenger receptor CD36, leading to intracellular lipid accumulation[35]. This is supported by studies in high-fat diet (HFD) mice, in which renal non-esterified fatty acid (NEFA) levels are elevated, and treatment with dehydrozingerone (DHZ) reduces lipid accumulation. Impaired fatty acid β-oxidation is a key metabolic defect in podocyte lipotoxicity. In experimental nephrosis, four enzymes of the mitochondrial β-oxidation pathway are downregulated, indicating defective FFA degradation. The rate-limiting enzyme carnitine palmitoyltransferase 1 (CPT1) is critical for β-oxidation, and its inhibition reverses the protective effects of AMPK activation against PA toxicity. Conversely, acetyl-CoA carboxylase (ACC), which generates malonyl-CoA to allosterically inhibit CPT1, contributes to impaired β-oxidation; podocyte-specific overexpression of acetyl-CoA carboxylase beta (ACCb) exacerbates podocyte injury and proteinuria in diabetic mice by reducing β-oxidation[36]. Peroxisome proliferator-activated receptor γ (PPARγ) modulates fatty acid metabolism by regulating genes such as CPT1α, with PPARγ deletion leading to dysregulated lipid oxidation, increased LD accumulation, and mitochondrial dysfunction. Sirtuin 3 (SIRT3), a mitochondrial deacetylase, also regulates β-oxidation by deacetylating and activating long-chain acyl-CoA dehydrogenase (LCAD); SIRT3 deficiency impairs β-oxidation, increasing LD accumulation in kidneys under HFD[37].
Mitochondrial dysfunction is closely linked to impaired β-oxidation and lipotoxicity. PA exposure induces mitochondrial ROS production, lipid peroxidation, and structural damage. In PPARγ-deficient podocytes, mitochondrial abnormalities are associated with increased ROS[33]. Mechanistically, accumulation of diacylglycerols (DAGs) and ceramides disrupts mitochondrial membrane permeability, leading to cytochrome c release and apoptosis[38]. Ultrastructural changes include swollen mitochondria with disarranged cristae, reduced mitochondrial mass, assessed by voltage-dependent anion channel (VDAC) expression, decreased citrate synthase activity, and downregulated ATP synthase subunit ATP5i, as observed in SIRT3-deficient kidneys. Oxidative stress is further exacerbated by impaired FAO and mitochondrial dysfunction[37]. PA also activates the DAG/protein kinase C theta (PKCθ) signaling pathway and Toll-like receptor 4 (TLR4)-mediated inflammation, contributing to mitochondrial cell death[39]. Beyond fatty acid-related mechanisms, podocyte dysfunction can involve other pathways, such as SGLT2 upregulation, which induces cytoskeletal remodeling and injury, though this is independent of direct impairment of fatty acid metabolism[40].
SPHINGOLIPIDS, CERAMIDES, AND RELATED PATHWAYS
Lipid dysregulation, characterized by defective cholesterol and FFA metabolism, is prevalent in kidney diseases and contributes to podocyte injury, with excess intracellular lipids inducing ER stress, mitochondrial dysfunction, and apoptosis[41]. Among the various lipid species involved, sphingolipids and their metabolites, particularly ceramides, are critical mediators of podocyte lipotoxicity, as their accumulation promotes mitochondrial dysfunction, oxidative stress, and apoptosis in podocytes[42]. The lipid raft enzyme SMPDL3b has been implicated in podocyte injury by altering active sphingolipid production and reducing ceramide-1-phosphate (C1P). Restoring C1P levels has been shown to normalize proteinuria in a murine model of diabetic nephropathy[43].
Ceramide accumulation significantly drives podocyte lipotoxicity by enhancing inflammation, oxidative stress, and apoptosis, which are central to the pathogenesis of diabetic nephropathy[6]. The balance between ceramides and sphingosine-1-phosphate (S1P) is crucial. In db/db mice and cultured podocytes, AdipoRon, an adiponectin receptor agonist, decreases ceramide accumulation by activating acid ceramidase. This enzyme hydrolyzes ceramide into sphingosine, which is subsequently converted to S1P, thereby normalizing the ceramide/S1P ratio and mitigating podocyte lipotoxicity[6]. Additionally, ceramide activates protein phosphatase 2A (PP2A), which inhibits AMP-activated kinase (AMPK) activity, leading to impaired lipid metabolism and increased podocyte injury; AdipoRon reverses this by downregulating PP2A activity and activating AMPK via AdipoR1[6].
Ceramide-induced oxidative stress in podocytes may be further exacerbated by PA, a saturated FFA that induces mitochondrial superoxide overproduction and hydrogen peroxide (H2O2) generation, resulting in mitochondrial damage characterized by swelling, cristolysis, and membrane disruption[44]. Moreover, ceramide-mediated podocyte inflammation can involve TLR4 signaling. Fetuin-A exacerbates PA-induced proinflammatory cytokine secretion such as monocyte chemoattractant protein-1 (MCP-1) and podocyte apoptosis through TLR4 activation, with TLR4 inhibition attenuating these inflammatory and apoptotic responses[45]. Impaired fatty acid β-oxidation, a key metabolic process disrupted in podocyte lipotoxicity, is associated with high glucose (HG)-induced reduction in the inactive phosphorylated form of acetyl-CoA carboxylase 2 (p-ACC2), and ACC2 silencing restores FAO, reduces lipid accumulation, and alleviates podocyte injury[46]. Furthermore, ceramide-induced podocyte injury can compromise slit diaphragm integrity by promoting nephrin endocytosis, a process mediated by CIN85/RukL, an adaptor protein that facilitates nephrin ubiquitination and internalization under high-glucose conditions, leading to podocyte dysfunction and proteinuria[47]. Additionally, lipid peroxidation, which impacts podocyte cytoskeletal dynamics by activating Ras homolog family member A (RhoA) through redox-sensitive cysteine residues, may represent a downstream effect of ceramide-induced oxidative stress, further contributing to podocyte motility impairment and cytoskeletal disorganization[48]. Table 1 summarizes the key molecular mediators underlying podocyte lipotoxicity in DKD.
Key molecular mediators of podocyte lipotoxicity in diabetic kidney disease
| Metabolic process | Molecules | Alteration in DKD | Pathological consequence | References |
| Cholesterol metabolism | ABCA1 | Downregulated/Decreased activity | Impaired cholesterol efflux, CE accumulation | [9-11,22,23] |
| CCDC92 | Upregulated | Promotes ABCA1 degradation, cholesterol accumulation | [9] | |
| JAML | Upregulated | Reduces ABCA1 expression, lipid accumulation | [23] | |
| SOAT1 | Upregulated | Converts free cholesterol to CE, promotes accumulation | [11] | |
| APOL1 | Risk variant | Promotes cholesterol accumulation | [21] | |
| Fatty acid uptake | CD36 | Upregulated | Enhanced FFA uptake, lipid accumulation | [7,35] |
| FAO | CPT1 | Decreased activity | Impaired β-oxidation, lipid droplet accumulation | [33,36] |
| ACC/ACCb | Upregulated | Inhibits CPT1, reduces FAO | [36] | |
| ERRα | Downregulated | Reduced ACOX1-mediated FAO, lipid accumulation | [13] | |
| ACOX1 | Downregulated | Impaired peroxisomal FAO | [13] | |
| SIRT3 | Downregulated | Impaired LCAD deacetylation, reduced FAO | [37] | |
| PPARγ | Decreased activity | Dysregulated lipid oxidation, mitochondrial dysfunction | [33] | |
| Sphingolipid metabolism | SMPDL3b | Upregulated | Alters ceramide metabolism, reduces C1P, promotes apoptosis | [43] |
| CerS6 | Upregulated | Ceramide overproduction, mitochondrial damage | [4] | |
| Lipid droplet regulation | FTO | Upregulated | Stabilizes ACC1 mRNA via m6A, promotes fatty acid synthesis | [26] |
| STK25 | Upregulated | Promotes lipid droplet accumulation, oxidative stress | [29] | |
| Lipid raft/slit diaphragm | Ephrin-B1 | Downregulated | Disrupts slit diaphragm localization, proteinuria | [25] |
| DDR1 | Increased activity | Enhances CD36-mediated FFA uptake | [27] |
PATHWAYS TRANSLATING LIPOTOXICITY INTO PODOCYTE DYSFUNCTION
The aberrant accumulation of lipids in podocytes does not act in isolation; rather, it triggers a cascade of intracellular stress responses that collectively drive cellular dysfunction and death. This section examines three interconnected pathogenic axes activated by lipotoxic insults: (1) mitochondrial dysfunction and oxidative stress; (2) ER stress and autophagic dysregulation; and (3) inflammatory signaling and immune crosstalk. These pathways are not mutually exclusive but instead amplify one another, forming a vicious cycle that culminates in podocyte apoptosis and GFB breakdown.
MITOCHONDRIAL DYSFUNCTION AND OXIDATIVE STRESS
Mitochondrial ROS generation is a central feature of podocyte lipotoxicity. PA can induce the production of mitochondrial superoxide and H2O2 in cultured podocytes[44]. Long-term PA exposure impairs antioxidant defense by significantly decreasing the protein expression of peroxidases such as peroxiredoxin 1 (Prx1), peroxiredoxin 2 (Prx2), glutathione peroxidase 1 (GPx1), and catalase, whereas OA restores these peroxidase levels, suggesting a counteractive role against PA-induced oxidative stress[16]. Moreover, ROS act as ligands for the NLRP3 inflammasome activation, linking mitochondrial oxidative stress to inflammatory signaling in podocyte lipotoxicity[49]. HG levels trigger receptor activator of nuclear factor-κB (RANK) expression, which mediates Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-p65 nuclear translocation, thereby enhancing NOX4 and P22phox expression and promoting mitochondrial oxidative stress[50]. In contrast, ginsenoside Rb1 reduces NOX4 expression and activity, decreasing ROS production and improving mitochondrial membrane potential in podocytes under high-glucose conditions[51]. Zingerone also reduces NOX4 expression, thereby reducing ROS generation in DKD[52].
Impaired mitochondrial bioenergetics is closely associated with lipotoxic podocyte injury. PA treatment decreases basal and maximal mitochondrial respiration rates and impairs ATP production in cultured podocytes, accompanied by mitochondrial structural disruption, including macropores in the mitochondrial membrane and blurry cristae, as observed in db/db and diet-induced obesity (DIO) mice[5]. Lipotoxic stress induces metabolic reprogramming in podocytes, characterized by decreased FA, increased lipid accumulation, and a shift toward glycolysis, all converging on mitochondrial impairment[4]. This is further exacerbated by a vicious cycle among mitochondrial dysfunction, inflammation, and altered lipid metabolism[12]. However, metabolic remodeling can be protective: empagliflozin induces a substrate switch from glucose to FAO in podocytes by reducing pyruvate dehydrogenase (PDH) activity and upregulating CPT1A, a rate-limiting enzyme of FAO, thereby reducing mitochondrial oxidative stress[28]. Similarly, silencing of STK25 enhances mitochondrial β-oxidation rate in kidney cells, improving mitochondrial function and reducing lipotoxic injury[29].
Mitochondrial quality control mechanisms, including autophagy/mitophagy, fusion/fission dynamics, and calcium regulation, are critical for mitigating lipotoxic stress. Lipotoxicity reduces autophagosome formation in podocytes, leading to oversaturated autophagy and the accumulation of damaged mitochondria[53]. Mitochondrial quality control proteins such as BCL2 interacting protein 3 (BNIP3) and peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1α) regulate mitophagy; knockdown of ATG7, a key autophagy protein, impairs mitophagy, increasing mitochondrial ROS and lipid peroxidation[54], suggesting a similar pathway may be operative in podocytes under lipotoxic stress. In DKD, impaired mitophagy is indicated by decreased expression of PTEN-induced kinase 1 (PINK1) and Parkin, vital for clearing damaged mitochondria, leading to mitochondrial fragmentation[8]. Mitochondrial fusion/fission is disrupted by c-Abl tyrosine kinase, which translocates to mitochondria under stress and phosphorylates mitofusin 2 (MFN2) at Y269, reducing its pro-fusion activity and causing mitochondrial fragmentation; pharmacological inhibition of c-Abl rescues mitochondrial morphology[55]. Mitochondria-associated ER membranes (MAM) integrity is also essential, as dysregulated MAM leads to mitochondrial calcium overload via the mitochondrial calcium uniporter (MCU), promoting oxidative stress and apoptosis; activation of transient receptor potential vanilloid 1 (TRPV1) channels reduces MAM-regulated mitochondrial calcium overload, ameliorating dysfunction[56]. Additionally, ceramide synthase 6 (CerS6)-derived ceramides disrupt mitochondrial function by causing mitochondrial DNA release and activating innate immune signaling, contributing to podocyte injury. Conversely, salidroside stimulates mitochondrial biogenesis by upregulating Sirt1 and PGC1α, improving mitochondrial function in diabetic nephropathy[57].
ER STRESS AND AUTOPHAGY DYSREGULATION
Lipid overload in podocytes is a key driver of ER stress activation and autophagy dysregulation, both of which contribute significantly to podocyte injury in DKD. ER stress in DKD accelerates disease progression by injuring podocytes, endothelial cells, and mesangial cells[58]. Persistent ER stress in podocytes disrupts ER function and triggers a maladaptive unfolded protein response (UPR), activating apoptotic signaling pathways and promoting podocyte apoptosis[58]. The UPR involves three major branches initiated by protein sensors: inositol-requiring enzyme 1α (IRE1α), activating transcription factor 6 (ATF6), and protein kinase RNA-like ER kinase (PERK). Under physiological conditions, these sensors are inactive due to binding to the binding immunoglobulin protein (BiP); however, accumulation of misfolded/unfolded proteins dissociates BiP, leading to their activation and podocyte damage, and even apoptosis, by disrupting slit diaphragm proteins such as podocin and nephrin[59].
In lipotoxic environments, saturated FFA - particularly palmitate - enter podocytes predominantly via the FAT/CD36 receptor. This uptake induces substantial mitochondrial ROS production, which in turn stimulates the phospholipase C (PLC) pathway and depletes ER calcium stores. The consequent loss of calcium homeostasis promotes ER luminal dilation and upregulates stress markers, including BiP and the pro-apoptotic transcription factor C/EBP homologous protein (CHOP)[60]. A lipotoxic environment also drives chronic inflammatory responses in podocytes by activating multiple signaling pathways. TLR4 acts as a central mediator in this process. Its activation potently initiates downstream NF-κB signaling and facilitates the secretion of proinflammatory cytokines such as Tumor necrosis factor-alpha (TNF-α)[60,61]. Under hyperglycemic conditions, the TLR4/myeloid differentiation primary response 88 (MyD88)/protein kinase C delta (PKCδ)/Src homology 2 domain-containing phosphatase 1 (SHP-1) signaling cascade is further activated in podocytes, exacerbating ER stress and promoting the release of the chemokine MCP-1, thereby amplifying podocyte damage in DKD[62]. While TLR4 has also been implicated in lipotoxicity-induced β-cell dysfunction via inhibition of the transmembrane protein 24 (TMEM24)/phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathway[63], its specific role in podocyte lipotoxicity within DKD warrants further mechanistic investigation.
In addition to ER stress, lipid overload impairs autophagy, particularly lipophagy, a selective autophagic process that degrades LDs, contributing to lipotoxicity in podocytes[64]. Lipophagy deficiency is observed in renal tubular cells from DKD patients and db/db mice and is associated with ectopic lipid deposition (ELD), oxidative stress, apoptosis, and renal injury[65]. Mechanistically, lipid overload reduces expression of AdipoR1 and phosphorylated AMPK, key regulators of autophagy, thereby impairing lipophagy, leading to lipid accumulation and kidney dysfunction. Activation of the AdipoR1/AMPK pathway via AdipoRon enhances autophagy/lipophagy, reverses mTOR phosphorylation, restores Unc-51 like autophagy activating kinase 1 (ULK1)-mediated autophagosome formation, and attenuates ER stress and podocyte injury[65]. Similarly, inhibition of FTO with demethylation inhibitors such as meclofenamic acid and diacerein ameliorates lipotoxicity-induced podocyte injury by mitigating ER stress, including reduced spliced X-box binding protein 1 (XBP1s), BiP, and CHOP expression, and restoring autophagy (increased LC3I to LC3II conversion and P62 degradation). FTO mediates ACC1 mRNA stability via m6A demethylation, and its inhibition promotes YTH N6-methyladenosine RNA binding protein 2 (YTHDF2)-mediated degradation of ACC1 mRNA, thereby reducing fatty acid accumulation, ER stress, and podocyte injury[26].
Glycogen synthase kinase 3β (GSK3β) signaling is another critical link between lipotoxicity, ER stress, and autophagy dysregulation in podocytes. In DKD, lipotoxicity and hyperglycemia activate GSK3β via increased phosphorylation at the activation site (pTyr216) and decreased phosphorylation at the inhibitory site (pSer9), leading to inhibition of autophagy and podocyte injury. Treatment with saracatinib (Sar) restores podocyte autophagy by inhibiting pathological GSK3β activation, as evidenced by increased expression of autophagy-associated proteins [autophagy-related 5 (ATG5), Beclin 1 (Beclin1), and microtubule-associated protein 1 light chain 3 beta (LC3B)] and podocyte markers (podocin, nephrin, synaptopodin) in diabetic kidneys and HG-induced podocytes. This restoration of autophagy contributes to improved renal function parameters, including reduced proteinuria, BUN, creatinine, and lipids[66].
Notably, simply lowering circulating triglycerides may not be sufficient to prevent podocyte injury, as demonstrated by a study using antisense oligonucleotides (ASOs) directed against ApoC-III to reduce triglyceride levels in Black and tan brachyury (BTBR) ob/ob mice. Despite a ~70% reduction in ApoC-III and near-normal triglyceride levels, ASO treatment failed to ameliorate proteinuria, podocyte loss, or pathological manifestations of DKD, suggesting that effective treatment requires targeting multiple pathogenic pathways beyond hyperlipidemia[67]. Proteomic profiling using a fluorescence-activated cell sorting (FACS)-based tripartite isolation method (timMEP) for glomerular cells has further revealed cell-type-specific proteome changes in podocytes, including distinct phosphorylation patterns related to proteostasis at the filtration barrier and selective disruption of slit diaphragm proteins under lipotoxic stress, providing molecular insights into podocyte injury mechanisms linked to ER stress and autophagy dysregulation[68]. Additionally, asparagine endopeptidase (AEP) plays a protective role in podocytes under diabetic conditions by cleaving cofilin-1 at asparagine 138 to maintain actin cytoskeletal dynamics and exert anti-apoptotic effects, representing a unique intracellular mechanism relevant to podocyte injury, independent of direct ER stress or modulation of autophagy[69].
INFLAMMATORY SIGNALING AND IMMUNE CROSSTALK
Lipotoxicity in podocytes activates multiple inflammatory signaling pathways and immune crosstalk mechanisms that contribute to DKD progression. The NLRP3 inflammasome, a cytosolic multiprotein complex involved in innate immunity, plays a critical role, as podocytes express its components and secrete Interleukin-1 beta (IL-1β) during Diabetic nephropathy (DN) progression. HG-induced podocyte injury, including apoptosis, cytoskeletal changes, lipid accumulation, NF-κB p65 activation, and mitochondrial ROS production, is markedly inhibited by NLRP3 inflammasome inhibition (e.g., MCC950) or genetic knockout, with reduced expression of NLRP3, caspase-1 p10, and cleaved IL-1β. Additionally, IL-1β itself promotes lipid accumulation, NF-κB activation, and mitochondrial ROS generation in podocytes under both normal and HG conditions, thereby exacerbating the lipotoxic-inflammatory cycle[17].
Cytokine production in podocytes is regulated by non-coding RNAs, such as lncRNA embryonic ventral forebrain-2 (EVF-2), which is upregulated in podocytes of DN patients and correlates with inflammation[70]. EVF-2 overexpression promotes transcriptional activation of inflammatory factors such as MCP-1 and B7-1 (CD80) by interacting hnRNPU, a ribonucleoprotein that binds DNA and RNA of inflammatory cytokines such as Ccl20 to regulate their expression[70]. RNA sequencing of EVF-2-overexpressing podocytes reveals upregulated mRNAs enriched in inflammation pathways including IL-17 and TNF signaling, while podocyte-specific EVF-2 knockdown alleviates inflammation and podocyte injury[70].
NF-κB signaling is a central mediator of proinflammatory responses in podocytes under lipotoxic conditions. The stress response protein regulated in development and DNA damage responses 1 (REDD1), upregulated in diabetic podocytes, is required for NF-κB-dependent transcription of transient receptor potential canonical channel 6 (TRPC6), intracellular calcium entry, and cytoskeletal remodeling. REDD1 promotes binding of the p65 subunit to the TRPC6 promoter, sustaining proinflammatory signaling, and podocyte-specific REDD1 deletion prevents NF-κB activation, podocyte injury, and albuminuria in diabetic mice[71].
Immune crosstalk, particularly with macrophages, amplifies podocyte inflammation. In DKD, damaged renal cells recruit monocytes/macrophages, and Notch-NF-κB crosstalk in macrophages promotes M1 polarization, leading to secretion of proinflammatory cytokines (IL-1β, TNF-α) and chemokines (MCP-1) that sustain inflammation and injury. Macrophage depletion therapy reduces kidney inflammation, fibrosis, and necroptosis, improving renal function[72]. Furthermore, diabetes shifts macrophage polarization toward pro-inflammatory M1 [elevated TNF-α and nitric oxide synthase 2 (NOS2)] and reduces anti-inflammatory M2 markers (CD163, Arg-1). This shift is reversed by combined therapy with the cannabinoid receptor 1 (CB1) antagonist AM6545 and the angiotensin-converting enzyme (ACE) inhibitor perindopril. This therapy reduces macrophage infiltration (F4/80+ cells), normalizes intercellular adhesion molecule 1 (ICAM-1) expression, and increases M2 markers, with in vitro studies showing CB1 receptor activation inhibits IL-4-induced M2 polarization, an effect blocked by AM6545[73].
Protective pathways can counteract lipotoxic inflammatory signaling. AdipoRon, an adiponectin receptor agonist, interacts with AdipoR1/AdipoR2 in podocytes, activating the Ca2+/liver kinase B1 (LKB1)-AMPK/peroxisome proliferator-activated receptor alpha (PPARα) pathway[18]. This increases the expression of phosphorylated LKB1, AMPK, and PPARα, upregulating downstream effectors such as PGC-1α and phosphorylated ACC, while decreasing sterol regulatory element-binding protein 1c (SREBP-1c). The net effect includes reduced oxidative stress, apoptosis, and protective anti-inflammatory signaling, with ameliorated podocyte injury and restored podocyte number[18].
Other signaling axes have also been implicated in linking lipotoxicity to inflammation. Loss of podocyte claudin-5 (CLDN5) reduces zonula occludens-1 (ZO1) and induces nuclear translocation of ZO1-associated nucleic acid-binding protein (ZONAB), which represses WNT inhibitory factor 1 (WIF1) expression, thereby activating WNT/β-catenin signaling and promoting inflammatory and fibrotic responses[74]. Additionally, geranylgeranylation deficiency in podocytes dysregulates RhoGTPases (RhoA, Rac1) and Rap1, disrupting the actin cytoskeleton and β1 integrin clustering, which may indirectly affect inflammatory signaling by altering podocyte structure and function[75]. Collectively, these interconnected pathways - mitochondrial dysfunction, ER stress, and inflammatory signaling - converge to drive podocyte apoptosis and GFB disruption. An overview of the integrated signaling networks mediating podocyte lipotoxicity is depicted in Figure 1.
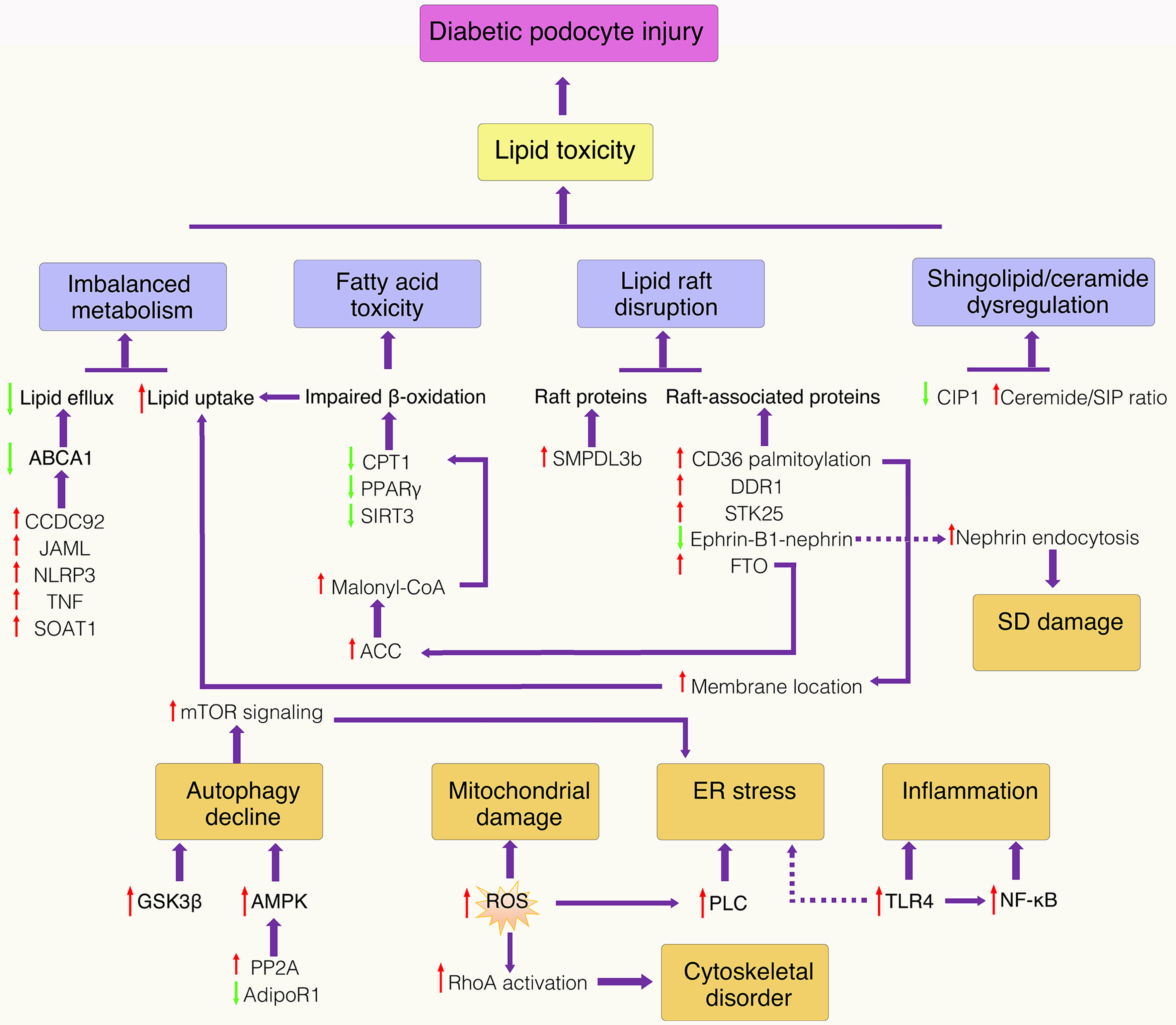
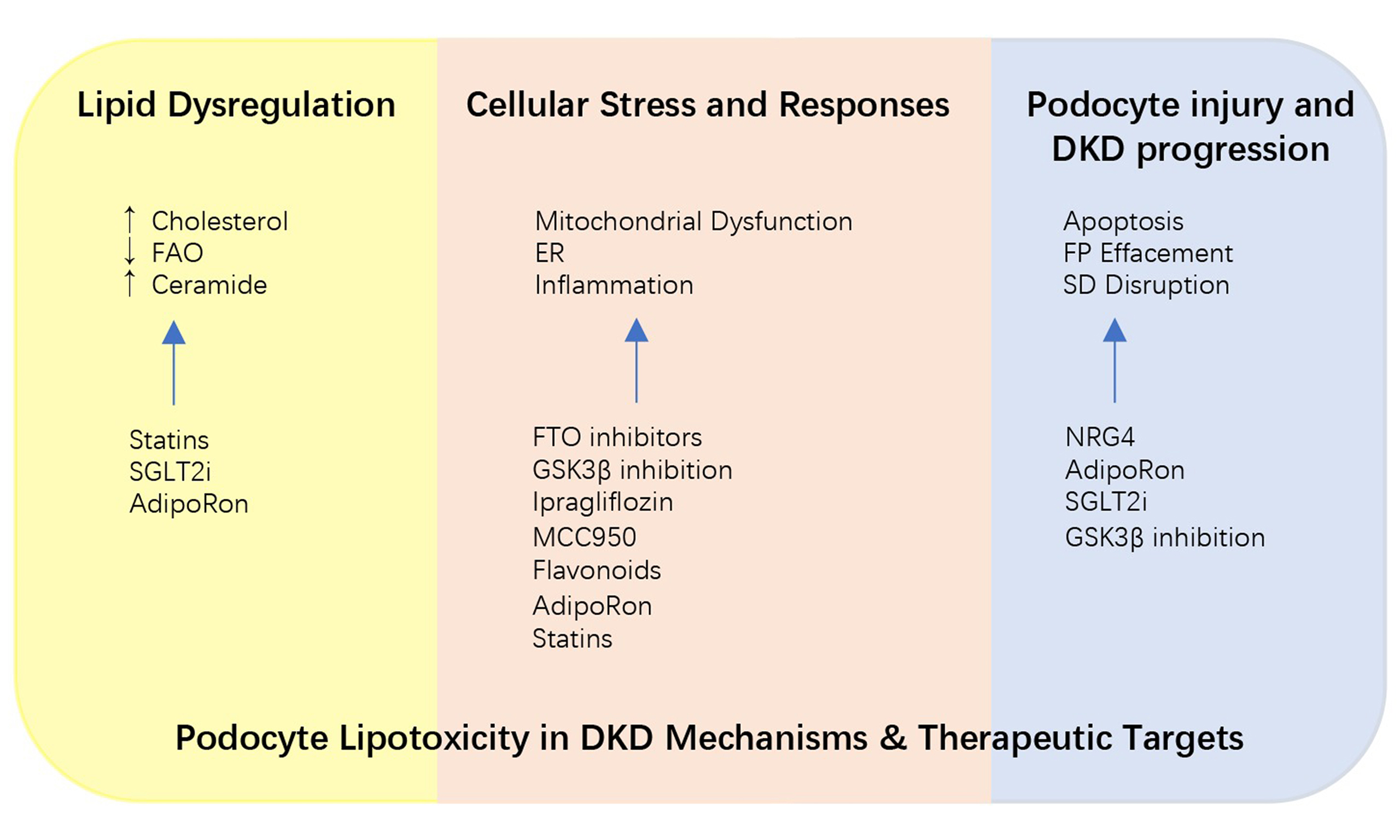
Figure 1. Schematic representation of lipotoxicity signaling pathways in podocytes in DKD. Schematic representation of integrated signaling networks mediating podocyte lipotoxicity in DKD. Dysregulated lipid metabolism (cholesterol accumulation, impaired fatty acid oxidation, ceramide overproduction) triggers mitochondrial dysfunction, ER stress, and oxidative stress, which converge to activate inflammatory pathways and apoptotic cascades, ultimately leading to podocyte loss and disruption of the glomerular filtration barrier. Red arrow: Upregulation; green arrow: downregulation. ABCA1: ATP-binding cassette transporter A1; ACC: acetyl-CoA carboxylase; JAML: junctional adhesion molecule-like protein; AMPK: AMP-activated protein kinase; CCDC92: coiled-coil domain-containing protein 92; C1P: ceramide-1-phosphate; CPT1: carnitine palmitoyltransferase 1; DDR1: discoidin domain receptor 1; ER: endoplasmic reticulum; FTO: fat mass and obesity-associated protein; GSK3β: glycogen synthase kinase 3 beta; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3: NOD-like receptor family pyrin domain containing 3; PLC: phospholipase C; PP2A: protein phosphatase 2A; PPARγ: peroxisome proliferator-activated receptor gamma; RhoA: Ras homolog family member A; ROS: reactive oxygen species; S1P: sphingosine-1-phosphate; SD: slit diaphragm; SIRT3: sirtuin 3; SMPDL3b: sphingomyelin phosphodiesterase acid-like 3b; SOAT1: sterol O-acyltransferase 1; STK25: serine/threonine kinase 25; TLR4: toll-like receptor 4; TNF: tumor necrosis factor.
INTERVENTIONS AGAINST PODOCYTE LIPOTOXICITY
Mechanistic insights have illuminated potential therapeutic targets to mitigate podocyte lipotoxicity in DKD. This section progresses from newly identified molecular regulators, to established pharmacological agents with repurposed nephroprotective effects, and finally to emerging strategies targeting epigenetic and inter-organ signaling pathways, highlighting the translational potential of targeting lipid metabolism to preserve podocyte function.
MOLECULAR REGULATORS AND THERAPEUTIC TARGETS
Studies have identified several novel molecular regulators that mediate podocyte lipotoxic injury and metabolic dysregulation in DKD. CCDC92, a novel coiled-coil domain-containing protein, is significantly upregulated in podocytes under diabetic conditions in both human DKD patients and diabetic mouse models (db/db and STZ/HFD). Podocyte-specific deletion of CCDC92 in diabetic mice ameliorates podocyte injury, reduces proteinuria, glomerular expansion, podocyte lipotoxicity, and ectopic lipid accumulation. These effects are attributed to the restoration of ABCA1 levels, a crucial mediator of lipid homeostasis, thereby reducing intracellular lipid accumulation[1].
Another key molecule is microsomal prostaglandin E synthase-2 (mPGES-2), whose expression is induced in diabetic kidneys and correlates with impaired lipolysis and renal damage, including enhanced glomerular podocyte injury and tubulointerstitial fibrosis. Genetic or pharmacological blockade of mPGES-2 in diabetic mice attenuates podocyte injury, tubulointerstitial fibrosis, lipid accumulation, and lipotoxicity, thereby improving renal function. Mechanistically, mPGES-2 competes with the nuclear receptor Rev-Erbα for heme binding, thereby limiting Rev-Erbα-mediated transcriptional repression of Fabp5, which encodes fatty acid-binding protein 5 (FABP5), a crucial mediator of fatty acid transport and renal lipid metabolism, leading to increased FABP5 expression, lipid accumulation, and renal lipotoxicity. Pharmacological inhibition of mPGES-2 with the compound SZ0232 mimics the benefits of genetic knockout, reducing proteinuria, podocyte dysfunction, tubulointerstitial fibrosis, and lipid accumulation in DKD mouse models[76].
Estrogen-related receptor alpha (ERRα) has also been identified as a critical molecular regulator of podocyte lipotoxicity in DKD by modulating FAO via transcriptional regulation of acyl-CoA oxidase 1 (ACOX1). ERRα and ACOX1 expression are significantly downregulated in podocytes under DKD conditions, leading to impaired FAO, lipid accumulation, and podocyte injury characterized by apoptosis and mitochondrial dysfunction. Podocyte-specific ERRα knock-in mice exhibit increased ACOX1 expression, enhanced fatty acid utilization, reduced lipid accumulation, improved mitochondrial function, and decreased levels of DKD injury markers, whereas targeted ACOX1 knockdown reverses these protective effects. Additionally, dapagliflozin (DAPA), a selective SGLT2 inhibitor, attenuates podocyte lipotoxicity in an ERRα-dependent manner by upregulating the ERRα-ACOX1 axis, thereby improving FAO, reducing triglyceride and FFA accumulation, and mitigating podocyte injury both in vitro and in vivo[13].
Furthermore, FTO has been shown to play a role in podocyte lipotoxic injury. FTO expression is higher in podocytes of diabetic mice and human DKD patients, correlating with lipid accumulation. Mechanistically, FTO stabilizes ACC1 mRNA through m6A modification mediated by YTH domain-containing family protein 2 (YTHDF2), leading to increased ACC1 expression and pathological fatty acid synthesis. Treatment with selective FTO demethylation inhibitors ameliorates lipotoxicity-induced podocyte injury by downregulating ACC1 levels, restoring autophagy, inhibiting apoptosis and inflammation, and mitigating ER stress and mitochondrial damage in both in vitro and in vivo DN models[26].
ESTABLISHED AND REPURPOSED PHARMACOLOGICAL AGENTS
Statins function by competitively inhibiting HMG-CoA reductase (HMGCR), suppressing cholesterol synthesis and directly protecting renal function. For example, simvastatin induces heme oxygenase-1 (HO-1) via the extracellular signal-regulated kinase (ERK)/protein kinase A (PKA)-cAMP response element-binding protein (CREB) axis, scavenging ROS and mitigating oxidative damage, while atorvastatin inhibits NLRP3 inflammasome activation, promotes autophagic degradation of apoptosis-associated speck-like protein containing a CARD (ASC) specks, reduces cholesterol-induced inflammation, restores aquaporin 2 (AQP2) expression, and improves renal water and sodium reabsorption function[77]. Clinical observations suggest statins reduce cardiovascular risk and systemic inflammation with modest renal benefits, though their effects in advanced kidney disease are limited and controversial in DKD[78].
SGLT2 inhibitors (SGLT2i) have demonstrated significant nephroprotective effects. Empagliflozin reduces proximal tubular FFA uptake by downregulating CD36 via the PPARγ pathway[79], shifts podocyte energy metabolism from glucose to fatty acids[28], activates the HO-1-adiponectin axis, and suppresses NLRP3 inflammasome activity, thereby alleviating lipid accumulation, inflammation, fibrosis, and apoptosis[80]. Ipragliflozin reduces ELD and ER stress[81], while dapagliflozin promotes cholesterol efflux via Krüppel-like factor 5 (KLF5) -ABCA1 and enhances FAO via ERRα-ACOX1 signaling, thereby reducing lipid accumulation[82]. Large clinical trials validate the nephroprotective effects of SGLT2i, slowing CKD progression partially by reducing FFA levels[83].
Flavonoids, a class of natural compounds, exert therapeutic effects in DKD mainly by regulating oxidative stress and inflammation, involving pathways such as nuclear factor erythroid 2-related factor 2 (Nrf-2)/glutathione (GSH), ROS production, HO-1, transforming growth factor beta 1 (TGF-β1), advanced glycation end products (AGEs)/receptor for advanced glycation end products (RAGE), and pro-inflammatory cytokines (IL-1, IL-6, TNF-α)[84]. Specific flavonoids including quercetin, apigenin, baicalin, luteolin, hesperidin, genistein, and kaempferol have been demonstrated to reduce oxidative stress and inflammation, promote podocyte autophagy, and inhibit renin-angiotensin-aldosterone system (RAAS) overactivity[85]. For instance, quercetin reduces oxidative stress markers, inhibits the NLRP3 inflammasome, and downregulates lipid metabolism-related pathways [SREBP cleavage-activating protein (SCAP), sterol regulatory element-binding protein 2 (SREBP2), and low-density lipoprotein receptor (LDLr)] in animal models[86]. Baicalin increases SIRT1 expression and inhibits the NF-κB and TGF-β/SMAD family member 3 (Smad3) pathways to prevent renal fibrosis[87]; and kaempferol exerts antioxidant and anti-inflammatory effects linked to TNF receptor-associated factor 6 (TRAF6) downregulation and increased glucagon-like peptide-1 (GLP-1) release[88]. Despite promising preclinical data, clinical studies on flavonoids in DKD are limited, and poor bioavailability restricts their clinical application, suggesting a need for improved formulations and structural modifications[84].
EMERGING THERAPEUTIC STRATEGIES AND NOVEL PATHWAYS
Epigenetic regulation and transcriptional reprogramming have also emerged as key mechanisms in podocyte lipotoxicity, with histone modifiers and transcription factors serving as potential therapeutic targets. Lysine-specific demethylase 6A (KDM6A), a histone demethylase, is upregulated in diabetic podocytes, and its inhibition restores podocyte markers and H3K27me3 levels[89].
KDM6A and its downstream effector, Krüppel-like factor 10 (KLF10), form a pathogenic positive feedback loop: KLF10 represses nephrin expression, and the two molecules reciprocally enhance each other’s activity. Genetic ablation of either KDM6A or KLF10 protects against podocyte injury and proteinuria in diabetic models[89]. Clinically, elevated levels of both proteins/mRNAs are found in human DKD tissues and urinary exosomes[90], underscoring their translational relevance.
Recent evidence has also implicated dedicator of cytokinesis 5 (DOCK5) in podocyte lipotoxicity[91]. DOCK5, a guanine nucleotide exchange factor predominantly expressed in podocytes, was identified through transcriptomic screening as critically related to podocyte lipid metabolism[91]. Its expression is significantly reduced in both patients with proteinuric kidney disease and mouse models. Podocyte-specific DOCK5 deficiency exacerbates podocyte injury and glomerular pathology in proteinuric kidney disease, primarily through enhanced fatty acid uptake via the liver X receptor α (LXRα)/CD36 signaling pathway. Mechanistically, DOCK5 deficiency upregulates LXRα in an m6A-dependent manner, thereby enhancing CD36-mediated fatty acid uptake and promoting lipid accumulation in podocytes. Conversely, restoration of Dock5 expression ameliorates podocyte injury and attenuates proteinuric kidney disease progression, suggesting that Dock5 may serve as a promising therapeutic target for mitigating podocyte lipotoxicity in DKD[91].
In the context of lipid metabolism modulation and inter-organ crosstalk, brown adipose tissue (BAT)-derived neuregulin 4 (NRG4) has emerged as a molecular regulator linking lipid metabolism modulation (through BAT function) and metabolic reprogramming of podocytes[92]. BAT-specific NRG4 deficiency exacerbates podocyte apoptosis, albuminuria, and glomerular damage in mouse models of diabetic nephropathy, while NRG4 supplementation via adeno-associated virus (AAV) or BAT transplantation reverses these effects. Recombinant NRG4 (rNRG4) treatment reduces high-glucose-induced podocyte apoptosis in vitro by increasing anti-apoptotic Bcl-2 and decreasing pro-apoptotic Bax and cleaved caspase-3 levels, while also enhancing podocyte viability and preserving nephrin and podocin expression[92]. The Akt-GSK-3β signaling pathway is essential for the protective effect of NRG4 on podocytes; NRG4 restores phosphorylation of Akt and GSK-3β impaired by diabetic conditions, and inhibition of this pathway diminishes NRG4’s anti-apoptotic effect[93]. These findings suggest NRG4 and its downstream Akt-GSK-3β pathway as promising therapeutic targets for preventing or reversing lipotoxicity-induced apoptosis in DKD.
Maintaining podocyte LD homeostasis and structural integrity through protein stabilization mechanisms represents another critical area of research. CH-ILK-binding protein (CHILKBP), also known as α-parvin or actopaxin, a focal adhesion protein highly expressed in podocytes, is essential for maintaining podocyte structure and function[94]. Podocyte-specific CHILKBP conditional knockout (cKO) mice develop massive proteinuria, podocyte injury including foot process effacement, podocyte loss, glomerulosclerosis, and kidney failure. Mechanistically, CHILKBP directly associates with the tight junction protein ZO-1, stabilizing ZO-1 by preventing its lysosome-dependent degradation in podocytes. Loss of CHILKBP decreases ZO-1 protein levels and mobility at cell junctions, leading to disrupted F-actin cytoskeleton organization and decreased expression and localization of slit diaphragm protein Nephrin, thereby compromising podocyte filtration barrier function[94]. Importantly, restoring CHILKBP or ZO-1 expression in CHILKBP-deficient podocytes in vitro and in vivo reverses podocyte dysfunction, improves cytoskeletal organization, rescues slit diaphragm integrity, reduces albuminuria, and attenuates podocyte foot process effacement and glomerulosclerosis[95]. This emphasizes the critical role of the CHILKBP-ZO-1 interaction and suggests that targeting the CHILKBP-ZO-1 pathway could be a therapeutic strategy to prevent or reverse podocyte lipotoxicity and injury in DKD.
Beyond these classes, other pharmacological strategies have been explored. Redox modulators, including natural compounds such as astragaloside IV (AS-IV), an active component of Astragalus membranaceus, suppress podocyte apoptosis in diabetic nephropathy via the miR-378/TNF receptor-associated factor 5 (TRAF5) signaling pathway. AS-IV treatment decreases albuminuria, serum creatinine, blood urea nitrogen, blood glucose, and blood pressure, and increases nephrin protein levels in diabetic rats, indicating protection against diabetic nephropathy progression[96]. Additionally, targeting novel molecular pathways, such as Metadherin (MTDH)-mediated signaling, represents a potential approach. MTDH activates β-catenin signaling via a Wnt ligand-independent mechanism involving PKA-mediated phosphorylation of β-catenin at Ser675, and interacts with pyruvate kinase M2 (PKM2) to promote β-catenin-dependent gene transcription leading to podocyte injury. Pharmacological inhibition of PKA (with H-89), blockade of β-catenin transcriptional activity (by ICG-001), or prevention of PKM2 deaggregation (by TEPP-46) disrupts this cascade, attenuating podocyte injury and proteinuria[97].
Direct targeting of LD accumulation represents an emerging therapeutic strategy for podocyte lipotoxicity. LD accumulation exacerbates podocyte injury and albuminuria in DKD[91,98]. A recent phenotypic screen identified a novel small molecule (compound 2726.007) that activates lipophagy, selective autophagic degradation of LDs, thereby reducing LD accumulation and ameliorating renal injury in db/db mice[98,99]. Additionally, molecules such as FTO and STK25 modulate LD dynamics by regulating fatty acid synthesis and mitochondrial β-oxidation, respectively[26,29]. These findings highlight the therapeutic potential of directly targeting LD accumulation in DKD. Figure 2 provides a schematic overview of the key pathogenic processes and corresponding therapeutic interventions in podocyte lipotoxicity, organized from lipid metabolic perturbations through cellular stress responses to podocyte damage and DKD progression. Table 2 provides a comprehensive overview of pharmacological and molecular interventions targeting podocyte lipotoxicity in DKD.
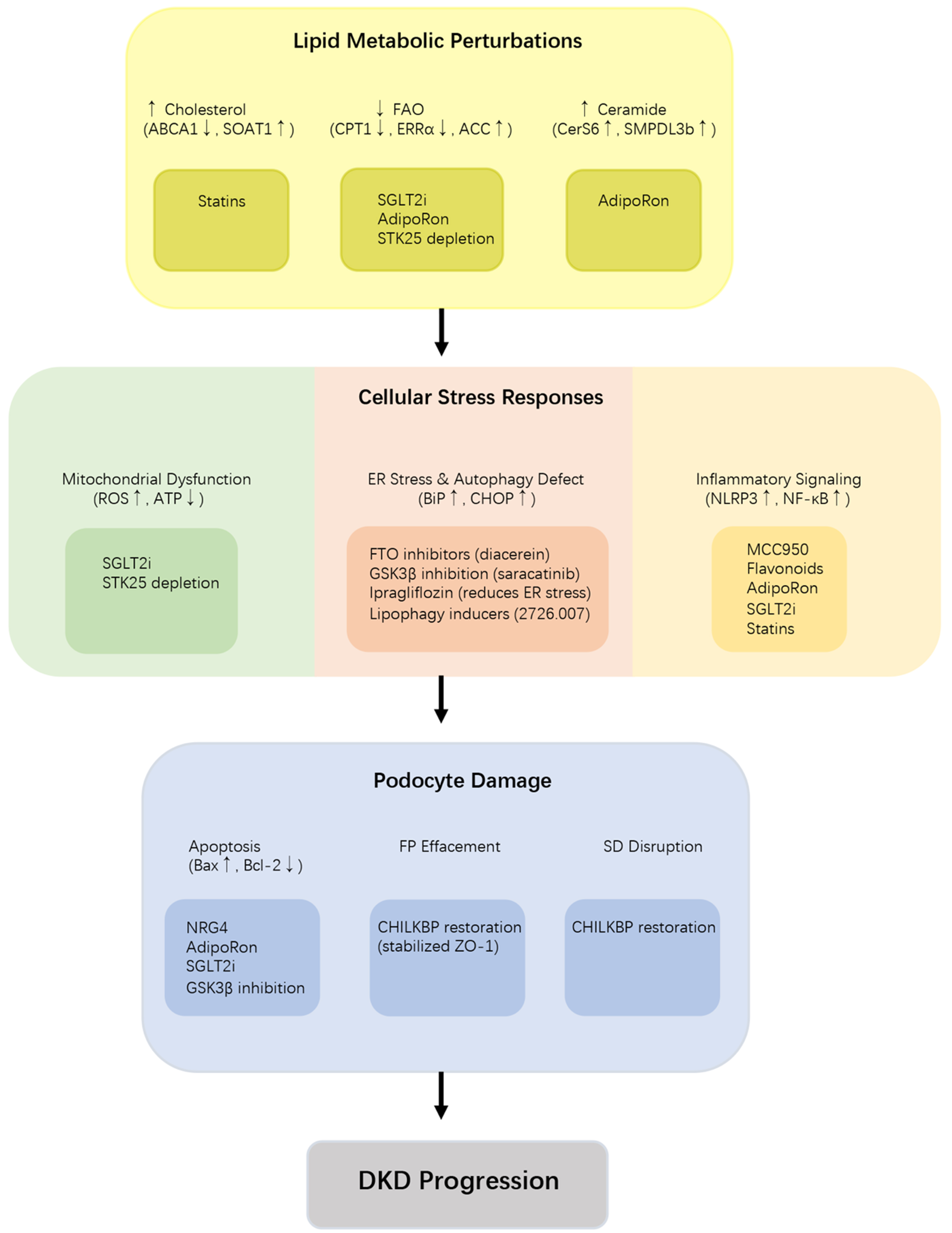
Figure 2. Key pathogenic processes and therapeutic interventions in podocyte lipotoxicity in DKD. Schematic overview of the hierarchical pathogenic cascade from lipid dysregulation to podocyte injury in DKD. Lipid dysregulation (cholesterol accumulation, impaired FAO, ceramide overproduction triggers cellular stress responses (mitochondrial dysfunction, ER stress/autophagy defect, inflammation), leading to podocyte damage (apoptosis, FP effacement, SD disruption). Corresponding therapeutic interventions are annotated at each stage, including statins, SGLT2i, AdipoRon, STK25 depletion, FTO inhibitors, GSK3β inhibition, ipragliflozin, MCC950, flavonoids, NRG4, and CHILKBP restoration. Bold arrows: Activation or promotion; upward small arrows: upregulation; downward small arrows: downregulation. CHILKBP: CH-ILK-binding protein; DKD: diabetic kidney disease; ER: endoplasmic reticulum; FAO: fatty acid oxidation; FP: foot process; FTO: fat mass and obesity-associated protein; GSK3β: glycogen synthase kinase 3 beta; MCC950: NLRP3 inflammasome inhibitor; NRG4: neuregulin 4; SD: slit diaphragm; SGLT2i: sodium-glucose cotransporter 2 inhibitors; STK25: serine/threonine kinase 25; ZO-1: zonula occludens-1.
Therapeutic interventions targeting podocyte lipotoxicity in DKD
| Therapeutic strategies | Potential drugs | Mechanisms | Effect on lipids | Stage of evidence | References |
| SGLT2 inhibitors | Empagliflozin | Shifts metabolism from glucose to FAO, reduces CD36-mediated FFA uptake | Reduces LD, improves mitochondrial function | Preclinical (AS/DKD models) + Clinical trials | [28,81] |
| Dapagliflozin | Activates ERRα-ACOX1 axis, promotes KLF5-ABCA1 cholesterol efflux | Enhances FAO, reduces TG and FFA, reduces lipid accumulation | Preclinical + Clinical trials | [13,83] | |
| Ipragliflozin | Reduces ectopic lipid deposition and ER stress | Reduces lipid accumulation | Preclinical | [82] | |
| Statins | Simvastatin | Inhibits HMGCR, induces HO-1 via ERK/PKA-CREB axis | Scavenges ROS, reduces oxidative damage | Preclinical + Clinical observations | [78] |
| Atorvastatin | Inhibits NLRP3 inflammasome, promotes ASC degradation | Reduces cholesterol-induced inflammation | Preclinical | [78] | |
| PPAR agonists | AdipoRon | Activates AdipoR1/R2, activates AMPK/PPARα pathway | Reduces ceramide, normalizes ceramide/S1P ratio | Preclinical (db/db mice, cultured podocytes) | [6,18] |
| NLRP3 inhibitors | MCC950 | Pharmacological inhibition of NLRP3 inflammasome | Reduces lipid accumulation, reduces IL-1β, restores ABCA1 | Preclinical | [17] |
| mPGES-2 inhibitors | SZ0232 | Blocks mPGES-2, restores Rev-Erbα-mediated repression of Fabp5 | Reduces FABP5, reduces lipid accumulation | Preclinical (DKD mouse models) | [77] |
| FTO inhibitors | Meclofenamic acid, Diacerein | Inhibits FTO demethylation activity | Reduces ACC1, reduces fatty acid synthesis, restores autophagy | Preclinical (in vitro and in vivo DN models) | [26] |
| Flavonoids | Baicalin | Activates SIRT1/AMPK/HNF4A pathway | Improves podocyte lipid metabolism | Preclinical | [30] |
| Quercetin | Inhibits NLRP3, downregulates SCAP-SREBP2-LDLr pathway | Reduces oxidative stress, reduces lipid pathway | Preclinical (animal models) | [87] | |
| Kaempferol | Downregulates TRAF6, increases GLP-1 | Antioxidant, anti-inflammatory | Preclinical | [89] | |
| Epigenetic modulators | KDM6A inhibition | Restores H3K27me3 levels, disrupts KDM6A-KLF10 feedback loop | Protects against podocyte injury, reduces proteinuria | Preclinical (diabetic models) | [90] |
| Inter-organ signals | NRG4 | BAT-derived, activates Akt-GSK-3β pathway | Reduces apoptosis, increases Bcl-2, preserves nephrin/podocin | Preclinical (DKD mouse models, in vitro) | [92,93] |
| Cytoskeletal stabilizers | CHILKBP restoration | Stabilizes ZO-1, prevents lysosomal degradation | Restores slit diaphragm integrity, reduces albuminuria | Preclinical (podocyte-specific cKO mice) | [94,95] |
| Kinase inhibitors | STK25 depletion | Enhances mitochondrial β-oxidation | Reduces LD, reduces oxidative stress | Preclinical | [29] |
| GSK3β inhibition | Restores autophagy (increases ATG5, Beclin1, LC3B) | Reduces podocyte injury, improves renal function | Preclinical | [67] | |
| Lipophagy inducers | Compound 2726.007 | Activates lipophagy, reduces LD accumulation | Reduces LD, reduces apoptosis, reduces albuminuria | Preclinical (db/db mice, in vitro) | [99,100] |
BIOMARKERS AND FUTURE PERSPECTIVES
Biomarkers for podocyte lipotoxicity
Recent research has identified several novel biomarkers for podocyte lipotoxicity in DKD. One recently defined molecule is ABCA1[20], the expression of which is negatively correlated with podocyte lipid accumulation and injury, implicating it as a key biomarker of podocyte lipotoxicity. Modulation by therapeutic agents (e.g., A30) reduces albuminuria and improves glomerular histopathology, further supporting its biomarker potential.
CCDC92 is specifically expressed in podocytes and upregulated under diabetic conditions, with increased expression correlating with DKD severity, podocyte lipid accumulation, and proteinuria[1]. CCDC92 promotes ABCA1 degradation through ubiquitin-proteasomal pathways, linking its expression to impaired cholesterol efflux and lipotoxicity, thus emerging as a potential biomarker[9].
Ceramide metabolism is also implicated, as podocyte CerS6 expression is elevated in DKD and focal segmental glomerulosclerosis (FSGS) biopsies, and correlates with reduced kidney function. Its overexpression induces proteinuria, directly linking ceramide synthesis to podocyte lipotoxicity[4]. FTO, a regulator of N6-methyladenosine RNA modification, is upregulated in podocytes of DN models and patients, correlating with lipid accumulation and podocyte loss, making FTO a candidate biomarker[26].
Fatty acid-handling proteins represent another class of biomarkers. Soluble CD36, a fatty acid transporter, has been proposed as a prognostic indicator for DKD, while fatty acid-binding proteins (FABPs) show altered expression: FABP1 is downregulated and may serve as an early urinary biomarker, whereas FABP4 is upregulated in podocytes[8]. Additionally, serine/threonine protein kinase 25 (STK25), a regulator of ectopic lipid storage, correlates with renal lipid accumulation and DKD severity, with its inhibition ameliorating lipotoxic injury[29]. Furthermore, decreased expression of adiponectin receptors AdipoR1 and AdipoR2 has been identified in diabetic kidneys, along with their downstream signaling components including calcium/calmodulin-dependent protein kinase beta (CaMKKb), LKB1 and AMPK, suggesting them as biomarkers of podocyte lipotoxicity in early DKD[18].
Translational gap and future perspectives
Despite the identification of promising biomarkers and mechanistic targets, a persistent gap remains between preclinical discoveries and clinical application. Translating the wealth of preclinical findings on podocyte lipotoxicity into effective human therapies for DKD presents significant challenges. A similar gap persists between the promising results observed in animal models and the outcomes of clinical trials. For example, while PPARα agonists (fibrates) demonstrated clear renoprotective effects in diabetic rodent models, their clinical translation has been inconsistent, sometimes associated with initial rises in serum creatinine[100]. This discrepancy underscores fundamental differences in species-specific lipid metabolism, disease progression timelines, and the complexity of human comorbidities.
Several promising targets identified in mechanistic studies await robust clinical validation. The protective role of recombinant klotho protein (rKL) against palmitate-induced podocyte injury and its efficacy in diabetic mice highlight its potential, yet human trials are necessary to confirm its therapeutic relevance[101]. Similarly, interventions targeting novel pathways, such as stimulator of interferon genes (STING) inhibition[5], the DOT1-like histone lysine methyltransferase (DOT1L)-phospholipase C-like 1 (PLCL1) epigenetic axis[15], or specific regulators like STK25[29] have shown efficacy in preclinical models but require the development of specific pharmacological agents and proof-of-concept studies in human cohorts.
Future clinical trial design must evolve to address these translational hurdles. Given the cell-type specificity of lipotoxic mechanisms, which differ significantly between podocytes and proximal tubules, therapies may need to be combined or tailored to target distinct nephron segments[20]. Trial endpoints should extend beyond the traditional marker of albuminuria to include more direct measures of podocyte health, preservation of glomerular filtration rate (GFR), and fibrosis regression. Incorporating dynamic molecular biomarkers, such as those derived from quantitative proteomics and degradomics, could provide a more sensitive and mechanistic assessment of drug efficacy at the cellular level[102].
Ultimately, the path forward lies in embracing a precision medicine approach. This involves leveraging multi-omics technologies to define patient-specific "lipotoxic signatures" that can guide the selection of targeted therapies. Furthermore, developing innovative drug delivery systems that specifically target podocytes will be crucial for enhancing efficacy and minimizing systemic side effects. By integrating deep mechanistic understanding with advanced clinical trial methodologies, the field can bridge the translational gap and transform DKD management from passive observation to active, mechanism-driven intervention.
Conclusion
Podocyte lipotoxicity is a critical and modifiable driver of DKD progression, arising from a multifaceted breakdown in lipid homeostasis, characterized by increased lipid uptake, impaired lipid efflux, and defective lipid oxidation. These disturbances trigger interconnected cascades of mitochondrial dysfunction, ER stress, impaired autophagy, and sustained pro-inflammatory signaling, culminating in podocyte apoptosis and disruption of the GFB. Preclinical studies have delineated key molecular regulators, including CD36, ABCA1, the ERRα-ACOX1 axis, and the NLRP3 inflammasome, and identified promising interventions such as SGLT2i, statins, flavonoids, and emerging epigenetic modulators. However, a significant translational gap persists. Bridging this gap will require integrative multi-omics approaches to define patient-specific lipotoxic signatures, the development of podocyte-selective drug delivery systems, and the design of clinical trials that evaluate hard renal outcomes beyond albuminuria. Moving forward, collaborative efforts integrating mechanistic insights with innovative trial design are essential to transform these findings into effective, targeted therapies that preserve podocyte function and halt DKD progression.
DECLARATIONS
Authors’ contributions
Conceptualization: Hu M
Writing-original draft: Hu M, Tian F, Yan C
Writing-review and editing: Hu M, Tian F
Visualization: Tian F
Resources: Yan C
Funding acquisition: Hu M
All authors have read and agreed to the published version of the manuscript.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This study was funded by National Natural Science Foundation of China (Grant No. 82100768), Natural Science Foundation of Shandong Province (Grant Nos. ZR2025MS1368 and ZR2020QH062).
Conflicts of interest
Hu M is a Junior Editorial Board member of the journal Metabolism and Target Organ Damage. Hu M was not involved in any steps of the editorial process, including reviewer selection, manuscript handling, and decision-making. The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Zuo F, Wang Y, Xu X, et al. CCDC92 deficiency ameliorates podocyte lipotoxicity in diabetic kidney disease. Metabolism. 2024;150:155724.
2. Brinkkoetter PT, Bork T, Salou S, et al. Anaerobic glycolysis maintains the glomerular filtration barrier independent of mitochondrial metabolism and dynamics. Cell Rep. 2019;27:1551-66.e5.
3. Nakamichi R, Hayashi K, Itoh H. Effects of high glucose and lipotoxicity on diabetic podocytes. Nutrients. 2021;13:241.
4. Hu H, Liang W, Ding G. Podocyte metabolic reprogramming and targeted therapy. J Am Soc Nephrol. 2026;37:619-33.
5. Zang N, Cui C, Guo X, et al. cGAS-STING activation contributes to podocyte injury in diabetic kidney disease. iScience. 2022;25:105145.
6. Choi SR, Lim JH, Kim MY, et al. Adiponectin receptor agonist AdipoRon decreased ceramide, and lipotoxicity, and ameliorated diabetic nephropathy. Metabolism. 2018;85:348-60.
7. Wang J, Hu J, Hu H, et al. APT1-derived depalmitoylation of CD36 alleviates diabetes-induced lipotoxicity in podocytes. Int J Biol Sci. 2025;21:3852-66.
8. Chae SY, Kim Y, Park CW. Oxidative stress induced by lipotoxicity and renal hypoxia in diabetic kidney disease and possible therapeutic interventions: targeting the lipid metabolism and hypoxia. Antioxidants. 2023;12:2083.
9. Zuo FW, Liu ZY, Wang MW, et al. CCDC92 promotes podocyte injury by regulating PA28α/ABCA1/cholesterol efflux axis in type 2 diabetic mice. Acta Pharmacol Sin. 2024;45:1019-31.
10. Nishi H, Nangaku M. Podocyte lipotoxicity in diabetic kidney disease. Kidney Int. 2019;96:809-12.
11. Liu X, Ducasa GM, Mallela SK, et al. Sterol-O-acyltransferase-1 has a role in kidney disease associated with diabetes and Alport syndrome. Kidney Int. 2020;98:1275-85.
12. Fornoni A, Merscher S. Lipid metabolism gets in a JAML during kidney disease. Cell Metab. 2020;32:903-5.
13. Hu H, Wang J, Peng Z, Fan Y, Yang Q, Hu J. Dapagliflozin attenuates diabetes-induced podocyte lipotoxicity via ERRα-Mediated lipid metabolism. Free Radic Biol Med. 2025;234:178-91.
14. Requena B, Shabaka A, Lanzon B, et al. Lipidomics unveils critical lipid pathway shifts in Alport syndrome. Kidney Int Rep. 2025;10:2805-20.
15. Hu Y, Ye S, Kong J, et al. DOT1L protects against podocyte injury in diabetic kidney disease through phospholipase C-like 1. Cell Commun Signal. 2024;22:519.
16. Lee E, Lee HS. Peroxidase expression is decreased by palmitate in cultured podocytes but increased in podocytes of advanced diabetic nephropathy. J Cell Physiol. 2018;233:9060-9.
17. Wu M, Yang Z, Zhang C, et al. Inhibition of NLRP3 inflammasome ameliorates podocyte damage by suppressing lipid accumulation in diabetic nephropathy. Metabolism. 2021;118:154748.
18. Kim Y, Lim JH, Kim MY, et al. The adiponectin receptor agonist AdipoRon ameliorates diabetic nephropathy in a model of type 2 diabetes. J Am Soc Nephrol. 2018;29:1108-27.
19. Luo Z, Chen Z, Hu J, Ding G. Interplay of lipid metabolism and inflammation in podocyte injury. Metabolism. 2024;150:155718.
21. Ryu JH, Ge M, Merscher S, et al. APOL1 renal risk variants promote cholesterol accumulation in tissues and cultured macrophages from APOL1 transgenic mice. PLoS One. 2019;14:e0211559.
22. Ge M, Molina J, Ducasa GM, et al. APOL1 risk variants affect podocyte lipid homeostasis and energy production in focal segmental glomerulosclerosis. Hum Mol Genet. 2021;30:182-97.
23. Fu Y, Sun Y, Wang M, et al. Elevation of JAML promotes diabetic kidney disease by modulating podocyte lipid metabolism. Cell Metab. 2020;32:1052-62.e8.
24. Loreth D, Sachs W, Meyer-Schwesinger C. The life of a kidney podocyte. Acta Physiol. 2025;241:e70081.
25. Fukusumi Y, Zhang Y, Yamagishi R, et al. Nephrin-binding Ephrin-B1 at the slit diaphragm controls podocyte function through the JNK pathway. J Am Soc Nephrol. 2018;29:1462-74.
26. Chang K, Hong F, Liu H, et al. FTO aggravates podocyte injury and diabetic nephropathy progression via m6A-dependent stabilization of ACC1 mRNA and promoting fatty acid metabolism. Biochem Pharmacol. 2025;235:116819.
27. Kim JJ, David JM, Wilbon SS, et al. Discoidin domain receptor 1 activation links extracellular matrix to podocyte lipotoxicity in Alport syndrome. EBioMedicine. 2021;63:103162.
28. Ge M, Molina J, Kim JJ, et al. Empagliflozin reduces podocyte lipotoxicity in experimental Alport syndrome. Elife. 2023;12:e83353.
29. Cansby E, Caputo M, Gao L, et al. Depletion of protein kinase STK25 ameliorates renal lipotoxicity and protects against diabetic kidney disease. JCI Insight. 2020;5:140483.
30. Zhang Y, Yao H, Li C, et al. Gandi capsule improved podocyte lipid metabolism of diabetic nephropathy mice through SIRT1/AMPK/HNF4A pathway. Oxid Med Cell Longev. 2022;2022:6275505.
31. Ito Y, Hsu MF, Bettaieb A, et al. Protein tyrosine phosphatase 1B deficiency in podocytes mitigates hyperglycemia-induced renal injury. Metabolism. 2017;76:56-69.
32. Sieber J, Jehle AW. Free fatty acids and their metabolism affect function and survival of podocytes. Front Endocrinol. 2014;5:186.
33. Carrasco AG, Izquierdo-Lahuerta A, Valverde ÁM, et al. The protective role of peroxisome proliferator-activated receptor gamma in lipotoxic podocytes. Biochim Biophys Acta Mol Cell Biol Lipids. 2023;1868:159329.
34. Lee ES, Kang JS, Kim HM, et al. Dehydrozingerone inhibits renal lipotoxicity in high-fat diet-induced obese mice. J Cell Mol Med. 2021;25:8725-33.
35. Nosadini R, Tonolo G. Role of oxidized low density lipoproteins and free fatty acids in the pathogenesis of glomerulopathy and tubulointerstitial lesions in type 2 diabetes. Nutr Metab Cardiovasc Dis. 2011;21:79-85.
36. Tanaka Y, Kume S, Maeda S, et al. Overexpression of acetyl CoA carboxylase β exacerbates podocyte injury in the kidney of streptozotocin-induced diabetic mice. Biochem Biophys Res Commun. 2018;495:1115-21.
37. Locatelli M, Macconi D, Corna D, et al. Sirtuin 3 deficiency aggravates kidney disease in response to high-fat diet through lipotoxicity-induced mitochondrial damage. Int J Mol Sci. 2022;23:8345.
38. Nicholson RJ, Pezzolesi MG, Summers SA. Rotten to the cortex: ceramide-mediated lipotoxicity in diabetic kidney disease. Front Endocrinol. 2020;11:622692.
39. Kume S, Maegawa H. Lipotoxicity, nutrient-sensing signals, and autophagy in diabetic nephropathy. JMA J. 2020;3:87-94.
40. Cassis P, Locatelli M, Cerullo D, et al. SGLT2 inhibitor dapagliflozin limits podocyte damage in proteinuric nondiabetic nephropathy. JCI Insight. 2018;3:98720.
41. Park HS, Lim JH, Kim MY, et al. Resveratrol increases AdipoR1 and AdipoR2 expression in type 2 diabetic nephropathy. J Transl Med. 2016;14:176.
42. Opazo-Ríos L, Mas S, Marín-Royo G, et al. Lipotoxicity and diabetic nephropathy: novel mechanistic insights and therapeutic opportunities. Int J Mol Sci. 2020;21:2632.
43. Mitrofanova A, Mallela SK, Ducasa GM, et al. SMPDL3b modulates insulin receptor signaling in diabetic kidney disease. Nat Commun. 2019;10:2692.
44. Lee E, Choi J, Lee HS. Palmitate induces mitochondrial superoxide generation and activates AMPK in podocytes. J Cell Physiol. 2017;232:3209-17.
45. Orellana JM, Kampe K, Schulze F, Sieber J, Jehle AW. Fetuin-A aggravates lipotoxicity in podocytes via interleukin-1 signaling. Physiol Rep. 2017;5:e13287.
46. Wang Q, Zhao B, Zhang J, et al. Faster lipid β-oxidation rate by acetyl-CoA carboxylase 2 inhibition alleviates high-glucose-induced insulin resistance via SIRT1/PGC-1α in human podocytes. J Biochem Mol Toxicol. 2021;35:e22797.
47. Teng B, Schroder P, Müller-Deile J, et al. CIN85 deficiency prevents nephrin endocytosis and proteinuria in diabetes. Diabetes. 2016;65:3667-79.
48. Kruger C, Burke SJ, Collier JJ, Nguyen TT, Salbaum JM, Stadler K. Lipid peroxidation regulates podocyte migration and cytoskeletal structure through redox sensitive RhoA signaling. Redox Biol. 2018;16:248-54.
49. Xin R, Sun X, Wang Z, et al. Apocynin inhibited NLRP3/XIAP signalling to alleviate renal fibrotic injury in rat diabetic nephropathy. Biomed Pharmacother. 2018;106:1325-31.
50. Ke G, Chen X, Liao R, et al. Receptor activator of NF-κB mediates podocyte injury in diabetic nephropathy. Kidney Int. 2021;100:377-90.
51. He JY, Hong Q, Chen BX, et al. Ginsenoside Rb1 alleviates diabetic kidney podocyte injury by inhibiting aldose reductase activity. Acta Pharmacol Sin. 2022;43:342-53.
52. Cui Y, Shi Y, Bao Y, Wang S, Hua Q, Liu Y. Zingerone attenuates diabetic nephropathy through inhibition of nicotinamide adenine dinucleotide phosphate oxidase 4. Biomed Pharmacother. 2018;99:422-30.
53. Stanigut AM, Tuta L, Pana C, et al. Autophagy and mitophagy in diabetic kidney disease-a literature review. Int J Mol Sci. 2025;26:806.
54. Baechler BL, Bloemberg D, Quadrilatero J. Mitophagy regulates mitochondrial network signaling, oxidative stress, and apoptosis during myoblast differentiation. Autophagy. 2019;15:1606-19.
55. Martinez A, Lamaizon CM, Valls C, et al. c-Abl phosphorylates MFN2 to regulate mitochondrial morphology in cells under endoplasmic reticulum and oxidative stress, impacting cell survival and neurodegeneration. Antioxidants. 2023;12:2007.
56. Ni L, Yuan C. The mitochondrial-associated endoplasmic reticulum membrane and its role in diabetic nephropathy. Oxid Med Cell Longev. 2021;2021:8054817.
57. Xue H, Li P, Luo Y, et al. Salidroside stimulates the Sirt1/PGC-1α axis and ameliorates diabetic nephropathy in mice. Phytomedicine. 2019;54:240-7.
58. Wang X, Zhao J, Li Y, Rao J, Xu G. Epigenetics and endoplasmic reticulum in podocytopathy during diabetic nephropathy progression. Front Immunol. 2022;13:1090989.
59. Wang M, Chen Z, Tang Z, Tang S. Natural products derived from traditional Chinese medicines targeting ER stress for the treatment of kidney diseases. Ren Fail. 2024;46:2396446.
60. Liu Q, Guo S, Tong Y, et al. Renal lipotoxicity in diabetic and obesity-associated kidney diseases: Molecular mechanisms and therapeutic targeting. Diabetes Obes Metab. 2026;28:60-82.
61. Cha JJ, Hyun YY, Lee MH, et al. Renal protective effects of toll-like receptor 4 signaling blockade in type 2 diabetic mice. Endocrinology. 2013;154:2144-55.
62. Li Z, Tan D, Lin J, et al. Podocyte TLR4 deletion alleviates diabetic kidney disease through prohibiting PKCδ/SHP-1-dependent ER stress and relieving podocyte damage and inflammation. J Adv Res. 2026;82:845-62.
63. Lan C, Li Y, Weng Z, et al. TLR4 mediates lipotoxic β-cell dysfunction by inhibiting the TMEM24/PI3K/AKT pathway. Acta Biochim Biophys Sin. 2025;57:1684-95.
64. Gao J, Wu Y, Cai X, Zhao H, Xing J. Therapeutic potential of natural medicines in diabetic kidney disease: restoring lipid homeostasis via lipophagy modulation. Front Pharmacol. 2025;16:1665339.
65. Han Y, Xiong S, Zhao H, et al. Lipophagy deficiency exacerbates ectopic lipid accumulation and tubular cells injury in diabetic nephropathy. Cell Death Dis. 2021;12:1031.
66. Li XZ, Jiang H, Xu L, et al. Sarsasapogenin restores podocyte autophagy in diabetic nephropathy by targeting GSK3β signaling pathway. Biochem Pharmacol. 2021;192:114675.
67. Attie AD, Schueler KM, Keller MP, et al. Reversal of hypertriglyceridemia in diabetic BTBR ob/ob mice does not prevent nephropathy. Lab Invest. 2021;101:935-41.
68. Hatje FA, Wedekind U, Sachs W, et al. Tripartite separation of glomerular cell types and proteomes from reporter-free mice. J Am Soc Nephrol. 2021;32:2175-93.
69. Lei C, Li M, Qiu Y, et al. Asparaginyl endopeptidase protects against podocyte injury in diabetic nephropathy through cleaving cofilin-1. Cell Death Dis. 2022;13:184.
70. Zhang C, Zhao H, Yan Y, et al. LncRNA evf-2 exacerbates podocyte injury in diabetic nephropathy by inducing cell cycle re-entry and inflammation through distinct mechanisms triggered by hnRNPU. Adv Sci. 2024;11:e2406532.
71. Sunilkumar S, Yerlikaya EI, Toro AL, et al. Podocyte-specific expression of the stress response protein REDD1 is necessary for diabetes-induced podocytopenia. Diabetes. 2025;74:398-408.
72. Alonazi AS, Aloraini RM, Albulayhi LM, et al. Macrophage depletion alleviates immunosenescence in diabetic kidney by modulating GDF-15 and Klotho. Int J Mol Sci. 2025;26:3990.
73. Barutta F, Bellini S, Mastrocola R, et al. Reversal of albuminuria by combined AM6545 and perindopril therapy in experimental diabetic nephropathy. Br J Pharmacol. 2018;175:4371-85.
74. Sun H, Li H, Yan J, et al. Loss of CLDN5 in podocytes deregulates WIF1 to activate WNT signaling and contributes to kidney disease. Nat Commun. 2022;13:1600.
75. Boi R, Bergwall L, Ebefors K, Bergö MO, Nyström J, Buvall L. Podocyte geranylgeranyl transferase type-I is essential for maintenance of the glomerular filtration barrier. J Am Soc Nephrol. 2023;34:641-55.
76. Zhong D, Chen J, Qiao R, et al. Genetic or pharmacologic blockade of mPGES-2 attenuates renal lipotoxicity and diabetic kidney disease by targeting Rev-Erbα/FABP5 signaling. Cell Rep. 2024;43:114075.
77. Kong Y, Feng W, Zhao X, et al. Statins ameliorate cholesterol-induced inflammation and improve AQP2 expression by inhibiting NLRP3 activation in the kidney. Theranostics. 2020;10:10415-33.
78. Chikatimalla R, Trivedi YV, Ruhela N, et al. Statins and kidney health: exploring cardiovascular benefits, renal protection, and risks in chronic kidney disease. Postgrad Med. 2025;137:588-600.
79. Huang CC, Chou CA, Chen WY, et al. Empagliflozin ameliorates free fatty acid induced-lipotoxicity in renal proximal tubular cells via the PPARγ/CD36 pathway in obese mice. Int J Mol Sci. 2021;22:12408.
80. Ye T, Zhang J, Wu D, et al. Empagliflozin attenuates obesity-related kidney dysfunction and NLRP3 inflammasome activity through the HO-1-adiponectin axis. Front Endocrinol. 2022;13:907984.
81. Hosokawa K, Takata T, Sugihara T, et al. Ipragliflozin ameliorates endoplasmic reticulum stress and apoptosis through preventing ectopic lipid deposition in renal tubules. Int J Mol Sci. 2019;21:190.
82. Sun J, Zhang X, Wang S, et al. Dapagliflozin improves podocytes injury in diabetic nephropathy via regulating cholesterol balance through KLF5 targeting the ABCA1 signalling pathway. Diabetol Metab Syndr. 2024;16:38.
83. Di Costanzo A, Esposito G, Indolfi C, Spaccarotella CAM. SGLT2 inhibitors: a new therapeutical strategy to improve clinical outcomes in patients with chronic kidney diseases. Int J Mol Sci. 2023;24:8732.
84. Hu Q, Qu C, Xiao X, et al. Flavonoids on diabetic nephropathy: advances and therapeutic opportunities. Chin Med. 2021;16:74.
85. Sulaiman MK. Molecular mechanisms and therapeutic potential of natural flavonoids in diabetic nephropathy: Modulation of intracellular developmental signaling pathways. Curr Res Pharmacol Drug Discov. 2024;7:100194.
86. Zheng Y, Ye X, Han X, Geng W, Zhao L, Meng D. Immunomodulatory roles of quercetin in diabetic nephropathy: targeting inflammation, oxidative stress, and ferroptosis. Front Pharmacol. 2025;16:1687677.
87. Ren G, Jiao P, Yan Y, Ma X, Qin G. Baicalin exerts a protective effect in diabetic nephropathy by repressing inflammation and oxidative stress through the SphK1/S1P/NF-κB signaling pathway. Diabetes Metab Syndr Obes. 2023;16:1193-205.
88. Cao Y-L, Lin J-H, Hammes H-P, et al. Flavonoids in treatment of chronic kidney disease. Molecules. 2022;27:2365.
89. Lin CL, Hsu YC, Huang YT, et al. A KDM6A-KLF10 reinforcing feedback mechanism aggravates diabetic podocyte dysfunction. EMBO Mol Med. 2019;11:e9828.
90. Liebisch M, Wolf G. Role of epigenetic changes in the pathophysiology of diabetic kidney disease. Glomerular Dis. 2024;4:211-26.
91. Qu H, Liu X, Zhu J, et al. Dock5 deficiency promotes proteinuric kidney diseases via modulating podocyte lipid metabolism. Adv Sci. 2024;11:e2306365.
92. Ding S, Xu JL, Tong JY, et al. Brown adipose tissue alleviates podocyte apoptosis through NRG4 in a male mouse model of diabetic kidney disease. Diabetologia. 2025;68:1057-75.
93. Chen M, Fang Y, Ge Y, Qiu S, Dworkin L, Gong R. The redox-sensitive GSK3β is a key regulator of glomerular podocyte injury in type 2 diabetic kidney disease. Redox Biol. 2024;72:103127.
94. Guo C, Ding Y, Yang A, et al. CHILKBP protects against podocyte injury by preserving ZO-1 expression. Cell Mol Life Sci. 2022;80:18.
95. Rogg M, Maier JI, Van Wymersch C, et al. α-Parvin defines a specific integrin adhesome to maintain the glomerular filtration barrier. J Am Soc Nephrol. 2022;33:786-808.
96. Lei X, Zhang BD, Ren JG, Luo FL. Astragaloside suppresses apoptosis of the podocytes in rats with diabetic nephropathy via miR-378/TRAF5 signaling pathway. Life Sci. 2018;206:77-83.
97. Chen X, Xiao J, Tao D, et al. Metadherin orchestrates PKA and PKM2 to activate β-catenin signaling in podocytes during proteinuric chronic kidney disease. Transl Res. 2024;266:68-83.
98. Njeim R, Gye H, Kelly CB, et al. A therapeutic lead compound for diabetic kidney disease with a unique lipophagy activation mechanism: SA-OR29. J Am Soc Nephrol. 2024;35:10.1681/asn.20248p613cev.
99. Njeim R, Awada B, Donow H, et al. Novel lipophagy inducers as potential therapeutics for lipid metabolism disorders. ACS Chem Biol. 2025;20:1406-16.
100. Li Q, Shi Z, Shi X, et al. Peroxisome proliferator-activated receptors in kidney diseases: a promising therapeutic target. Biomed Pharmacother. 2026;195:118983.
101. Suk Kang J, Son SS, Lee JH, et al. Protective effects of klotho on palmitate-induced podocyte injury in diabetic nephropathy. PLoS One. 2021;16:e0250666.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].