


MetALD: new insights and unraveling therapeutic potential
 0
0 Abstract
Metabolic dysfunction-associated steatotic liver disease and alcohol-associated liver disease are leading contributors to chronic liver disease globally, with incidence rates escalating in parallel with the worldwide increase in metabolic dysfunction and harmful alcohol use. A recent Delphi Consensus statement introduced the term metabolic and alcohol-associated liver disease (MetALD) to emphasize the synergistic contributions of metabolic and alcohol-related factors to liver injury. While this conceptual framework advances our understanding of heterogeneous liver disease etiologies, it also presents new diagnostic and therapeutic challenges. This review presents current evidence on the prevalence of MetALD, as well as its heterogeneous disease risk profiles, underlying pathogenesis, clinical diagnostic biomarkers, and evolving treatment strategies. Particular emphasis is placed on the necessity for robust preclinical models that accurately replicate the multifactorial pathogenesis of MetALD. Emerging therapies, including fecal microbiota transplantation and dietary supplementation, are critically evaluated, alongside perspectives on future pharmacological innovations for MetALD management.
Keywords
INTRODUCTION
Metabolic dysfunction-associated steatotic liver disease (MASLD) and alcohol-associated liver disease (ALD) are the most common causes of chronic liver disease worldwide, accounting for the majority of liver-related hospitalizations and deaths[1-3]. In recent decades, the prevalence of MASLD and ALD has been rising, owing to increasing rates of unhealthy alcohol use and obesity, as well as the growing incidence of type 2 diabetes, hypertension, and other conditions linked to the popularity of the Western diet[4].
Both ALD and MASLD share many of the pathophysiologic processes of liver impairment and exhibit comparable clinical disease progression. Traditionally, MASLD and ALD were considered to be two distinct diseases. However, recent studies have demonstrated that risk factors for both conditions frequently overlap and work in concert to promote the development of liver disease. Thus, alcohol consumption and metabolic syndrome together increase the risk of developing and worsening advanced liver disease.
For patients with nonalcoholic fatty liver disease (NAFLD), the word “fatty” may be attached to potential stigma. In addition, the term “nonalcoholic” does not accurately describe the cause of the disease. Thus, to prevent stigmatization and discrimination and to more accurately describe this condition, a Delphi Consensus Study with three sizable international liver groups recently proposed a new nomenclature for steatotic liver disease (SLD)[1]. Replacing NAFLD with the less stigmatizing term SLD can promote the assessment of alcohol’s influence, allowing for a transparent evaluation and categorization of alcohol consumption through a spectrum-based approach. Using this nomenclature, patients with SLD are divided into four groups: those with at least one cardiovascular metabolic risk factor (CMRF) and no significant alcohol consumption (less than 20 g/day for women and less than 30 g/day for men) (MASLD, alternative to NAFLD), those with at least one CMRF and increased alcohol consumption (20-50 g/day for women and 30-60 g/day for men) [metabolic and alcohol-associated liver disease (MetALD)], patients with excessive alcohol consumption (> 50/60 g/day for women/men with CMRF, or > 20/30 g/day for women/men without CMRF)[5] (ALD), and patients with steatosis but no apparent CMRFs or other distinguishable causes (cryptogenic SLD)[1,2].
MetALD, a recently developed disease entity, highlights the role of both metabolic and alcohol consumption factors in liver damage and represents a significant leap in our knowledge of liver diseases marked by both metabolic dysfunction and alcohol use[3]. At the same time, this emerging patient category presents a clinical challenge. Because its epidemiology is not yet well established, diagnosis is based on patients’ self-reported alcohol consumption, and treatment generally consists of metabolically targeted therapies[4,6], complete abstinence from alcohol, and lifestyle modifications. In this review paper, we thoroughly summarize the existing knowledge regarding the epidemiology, clinical diagnosis, and pathogenesis of MetALD to promote research and clinical interventions and offer ideas for future research directions.
EPIDEMIOLOGY AND CLINICAL OUTCOMES OF METALD
Alcohol consumption and CMRFs, the two most prominent causes of SLD, are becoming increasingly prevalent in the general population[7], thus increasing the prevalence of MetALD (ranging between 0.6% and 17%)[8-26]. Most studies on MetALD have relied on the use of extensive datasets such as the National Health and Nutrition Examination Surveys and the United Kingdom Biobank. Given the high rate of under-reporting of alcohol use in the SLD population, the actual number of MetALD cases may be higher than currently estimated. A study among teenagers aged 12 to 18 years reported the lowest prevalence at 0.6%[16]. Another study among a population with excessive drinking observed the highest prevalence of 17%[26], followed by a study in the Veterans Analysis of Liver Disease cohort, which stated a prevalence of 12.1%[10]. Additionally, several studies were conducted on a single national cohort; they included participants receiving a national health checkup in the Republic of Korea; thus, the findings were fairly comparable[8,13,18]. We believe that the variations in prevalence are strongly tied to the research population.
MetALD and intrahepatic diseases
The correlation between the newly proposed SLD and liver fibrosis is poorly understood and controversial. In a study of a Korean population, fibrosis was evaluated by magnetic resonance imaging (MRI). The mean MRI values in the MetALD group were substantially higher than those in the MASLD or ALD groups[21]. In a United States cohort, Ciardullo et al. found that patients with SLD exhibited higher liver fibrosis scores (such as the fibrosis-4 index or liver stiffness measurement) compared with non-SLD patients. However, the prevalence of liver fibrosis in patients with MASLD and MetALD was not significantly affected by changes in the new diagnostic guidelines[27]. A study conducted in a southern California cohort of obese and overweight individuals in primary care and community-based settings reached similar conclusions[28]. However, in a Korean cohort, Choe et al. found that the hazard ratios (HRs) for advanced liver fibrosis in patients with MASLD, MetALD, and ALD were 1.39 [95% confidence interval (CI): 1.25-1.55; P < 0.001], 1.75 (95%CI: 1.38-2.23; P < 0.001), and 2.00 (95%CI: 1.30-3.07; P = 0.002), respectively, compared to non-SLD patients[23]. Thus, MetALD patients have an intermediate risk of developing advanced liver fibrosis. After comparing studies such as those by Ciardullo et al.[27] and Yang et al.[28] with that of Choe et al.[23], we discuss potential reasons for these discrepancies: (1) Population differences: Variations in baseline characteristics (e.g., severity of underlying metabolic risk factors, precise levels and patterns of alcohol consumption within the MetALD definition, genetic background); (2) Methodological differences: Use of different non-invasive fibrosis scores (e.g., FIB-4, LSM) vs. MRI parameters or histology. Interestingly, only in female patients did one study find a difference between MASLD and MetALD in terms of deteriorating liver fibrosis scores[29]. This trend might result from variations in how alcohol affects liver fibrosis by gender[30]. This underscores the importance of considering demographic factors. Further research is needed to clarify the precise mechanisms involved.
Because of the widespread prevalence of obesity and alcohol use, over half of cirrhosis-related fatalities and one third of hepatocellular carcinoma (HCC)-related deaths in the United States are attributable to SLD[31]. Additionally, SLD increases the risk of HCC[8]. In a retrospective cohort study conducted at the Charité University Medical Center in Berlin between 2010 and 2020, the etiologies of 577 HCC patients were determined as MASLD (n = 158; 27.3%), MetALD (n = 51; 8.8%), and ALD (n = 68, 11.7%)[32]. Regarding the progression of HCC induced by SLD, a study from 2009 to 2010 of an SLD cohort from the Republic of Korea reported 27,118 additional HCC cases over the follow-up period (median: 13.3 years). The risk of HCC grew from MASLD (0.6%) and MetALD (0.92%) to ALD (1.26%) in a stepwise manner[8]. Similar results were reported in patients with SLD from the Korean National Health Insurance Service[11]. Remarkably, among older (aged 65-79 years) (2.6% vs. 2.7%), female (0.24% vs. 0.24%), and cirrhosis (14.5% vs. 15.1%) subgroups, MetALD and ALD patients had equivalent risks of HCC, according to a subgroup analysis of a Korean population[8].
A study by Kim et al. found that the risk of new-onset cirrhosis and decompensated cirrhosis likewise increased in the order of MASLD, MetALD, and ALD[20]. The stepwise pattern extends to cirrhosis development: the incidence of decompensated cirrhosis rises from MASLD (1.1%) to MetALD (1.3%) and ALD (2.2%); and the corresponding subdistribution HRs were 1.45 (95%CI: 1.29-1.62; P < 0.001), 1.77 (95%CI: 1.47-2.14; P < 0.001), 2.24 (95%CI: 1.74-2.89; P < 0.001)[20]. These data suggest that MetALD may be associated with an intermediate risk of liver disease severity in the SLD subtype. We acknowledge that robust head-to-head comparative data on the incidence rates of liver diseases specifically stratified by MASLD, MetALD, and ALD are still limited and represent an important area for future research.
In conclusion, current evidence suggests that among the subtypes of MASLD, MetALD, and ALD, MetALD patients typically exhibit an intermediate risk for severe liver diseases (such as advanced fibrosis, HCC, and cirrhosis) in a stepwise gradient (MASLD < MetALD < ALD)[8,20,23]. However, this risk pattern may be modulated by demographic factors and disease stage, with MetALD risk potentially equaling that of ALD in specific subgroups[8]. These findings underscore the need for future research to elucidate the precise mechanisms underlying the risk differences between subtypes, the contributing factors (including detailed patterns of alcohol consumption), and their implications for individualized risk stratification and management.
MetALD and extrahepatic diseases
In contrast to the risk of liver disease, cardiovascular outcomes show conflicting associations. Some studies have shown that MetALD patients have a lower risk of cardiovascular-related mortality than MASLD patients[11,33], whereas the opposite trend was observed in two other investigations[14,23]. We propose this inconsistency may reflect: (1) Population heterogeneity (e.g., higher metabolic comorbidities in U.S. cohorts[27] vs. Korean cohorts[11]); (2) Differential competing risks from liver disease. Thus, we must acknowledge that additional research is necessary to clarify the relationship between the new diagnostic criteria and the risk of cardiovascular disease.
Chronic periodontitis (CP) and MASLD are interrelated diseases[34] and are frequently linked to systemic inflammation and metabolic syndromes such as obesity and insulin resistance[35-37]. Specifically, it has been shown that CP can worsen MASLD severity by raising systemic inflammatory levels[38]. A recent study in 2024 revealed the risk of CP in the newly categorized SLD subgroups (specifically MASLD and MetALD), with higher adjusted risk ratios for MetALD compared with MASLD for both CP (1.2 vs. 1.14) and severe CP (1.28 vs. 1.22). Given that CMRFs are linked to both MASLD and MetALD, it is reasonable to hypothesize that the increased risk of periodontal disease may be the result of increased alcohol consumption.
It is becoming increasingly clear that SLD not only affects the liver but also increases the risk of non-liver diseases such as colorectal cancer (CRC) and chronic kidney disease (CKD)[39,40]. Alcohol intake and obesity are known risk factors for CRC. According to a nationwide study conducted in Japan, the 10-year cumulative incidence of CRC in the SLD subgroups was highest in ALD, followed by MetALD and MASLD (0.97%, 0.73%, and 0.48%, respectively)[39]. More research is still required to determine whether variations exist in the occurrence of other malignant tumor types under the SLD categorization.
MASLD exacerbates insulin resistance and can release several pro-inflammatory and pro-fibrotic factors, which can promote kidney injury[40]. Mori et al. clarified the relationship between CKD and each SLD type in a group of 12,138 Japanese participants who underwent annual health checkups. During the 10-year follow-up period, 1,963 of the participants developed new-onset CKD (16.2%). Multivariate Cox proportional risk model analysis showed that the HRs for developing CKD were significantly higher in MASLD [1.20 (1.08-1.33), P = 0.001] and ALD [1.41 (1.05-1.88), P = 0.022] patients than in non-SLD patients, whereas the HRs were not significantly different in MetALD subjects [1.11 (0.90-1.36), P = 0.332][19]. Further research is needed to determine whether the risk of CKD development depends on the presence of metabolic dysfunction or the amount of alcohol consumed in the new classification of SLD.
Data indicate that persons with human immunodeficiency virus (HIV) (PWH) have a high burden of SLD[41,42]; however, there have been few studies on the prevalence of SLD in PWH populations. A prospective study conducted in the United States in 2023 demonstrated that nearly half of PWH on suppressive antiretroviral medication without viral co-infection had SLD, with 90% of these cases attributable to NAFLD[42]. The following year, another study conducted by the same team revealed that 52% of PWH had SLD, 15% had clinically significant fibrosis, and 74% of SLD cases were caused by MASLD, with 19% and 6% of cases caused by MetALD and ALD, respectively[24].
Emerging evidence establishes MASLD and MetALD as significant risk factors for dementia[43], mediated through shared cardiometabolic pathologies including insulin resistance, obesity, hypertension, and dyslipidemia[44,45]. MASLD consistently increases the incidence of all-cause dementia, Alzheimer’s disease (AD), and vascular dementia (VaD)[46]. While MetALD initially demonstrates a reduced AD risk, it is associated with increased VaD risk and ultimately increases AD incidence with extended latency relative to MASLD[46]. Future research must elucidate underlying mechanisms and determine whether targeting cardiometabolic risk factors and hepatic inflammation lowers dementia risk.
PATHOGENESIS IN METALD
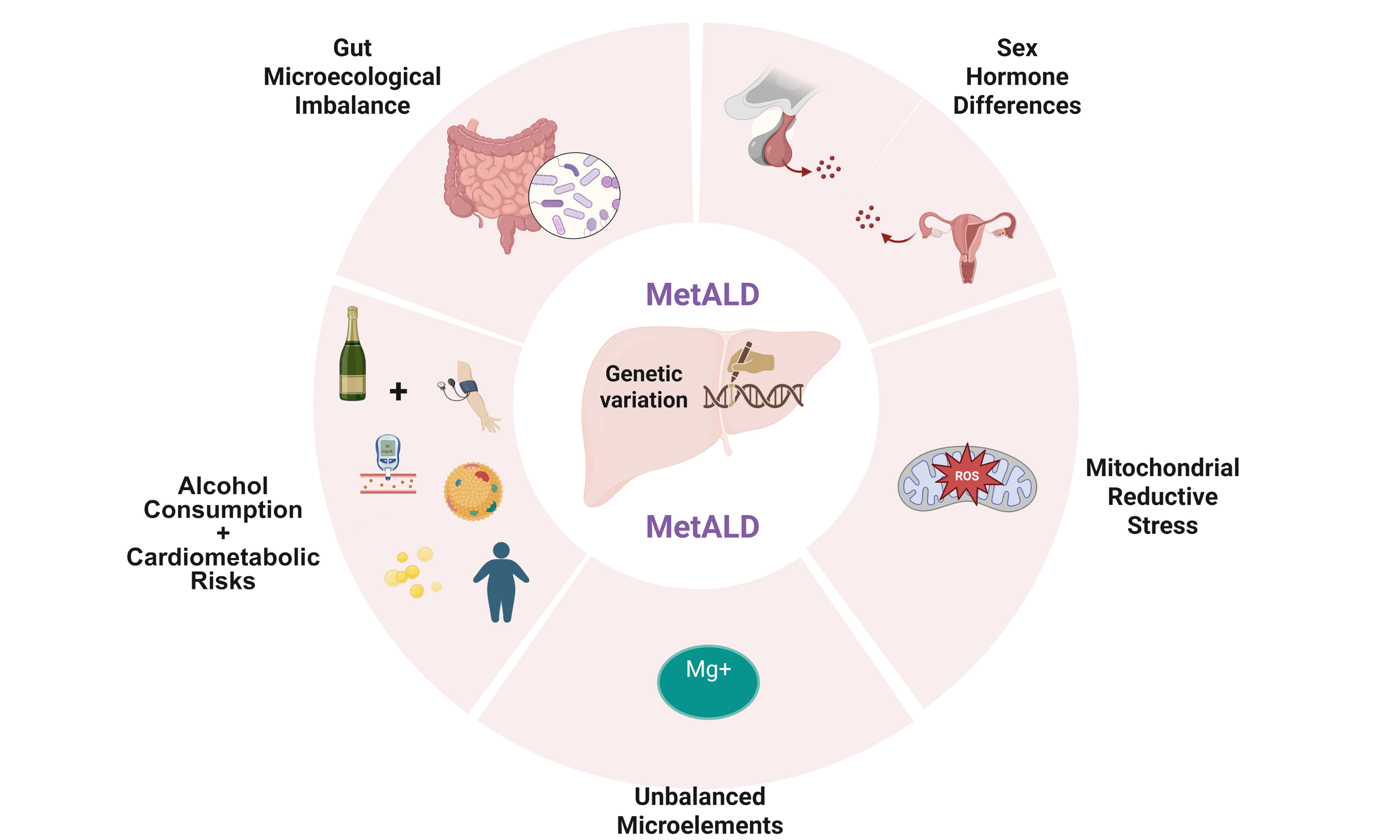
It is uncertain whether CMRFs or alcohol consumption plays the greater role in MetALD. Identifying the primary contributor is complex, and these two factors can cause liver injury independently or synergistically. Most likely, the combined effects of the two factors are the main drivers of MetALD. Although research on the mechanisms of MetALD remains limited, common mechanisms of CMRF-dominated MASLD and alcohol-consumption-dominated ALD pathogenesis may play a role in MetALD [Table 1].
The main molecular mechanisms
| Mechanisms | Consequences | Reference |
| Lipid metabolism disturbance | Adipose shrinkage and increased FFAs | [47] |
| Low serum magnesium levels | Insulin resistance and prediabetes | [48,49] |
| Mitochondrial reductive stress | High NADH/NAD+ ratio | [50] |
| Gut microecological imbalance | Increased intestinal permeability or immunity disorders | [51,52] |
| Genetic variation | Disease modifiers (higher risk of steatosis or fibrosis) | [53] |
| Sex hormone differences | Estrogen protection | [54] |
Combined effects of CMRFs and alcohol
Numerous studies have demonstrated the complex and independent relationship between alcohol use and the cardiometabolic risk that characterizes MASLD[55]. These studies emphasize that excessive alcohol consumption exacerbates hypertension, dyslipidemia, insulin resistance, and obesity, which can have a synergistic effect on the liver, accelerating liver injury and fibrosis. Alcohol consumption may lead to additional energy intake and, thus, weight gain[3,56]. However, recent research in Hefei, China, has revealed that dark tea wine has a protective effect against MASLD. In mice fed a high-fat diet, dark tea wine improved MASLD by increasing antioxidant capacity through the Nrf2/HO-1 signaling pathway, which also dramatically reduced lipid dysfunction, prevented weight gain, and improved pathological characteristics[57].
Alcohol consumption increases the risk of hypertension[58], and reducing alcohol use can help blood pressure levels return to normal in hypertensive individuals[59]. One study has indicated that moderate drinkers are 1.45 times more likely to develop high blood pressure compared with non-drinkers, whereas heavy drinkers are 2.53 times more likely to develop high blood pressure[60]. Alcohol metabolism produces reactive oxygen species (ROS), which damage endothelial cells and raise total peripheral resistance, worsening hypertension[61]. The interaction between endothelial cell damage and total peripheral resistance may further promote the occurrence of liver injury and the development of fibrosis in MetALD.
Type 2 diabetes and alcohol use have a more complicated relationship. A recent meta-analysis revealed a J-shaped relationship between alcohol intake and type 2 diabetes in women, but no statistically significant correlation in males. Consuming 16 g/day of pure alcohol lowers the risk of type 2 diabetes in women, whereas consuming more than 49 g/day increases this risk[62]. This finding contradicts the results of earlier research. Baliunas et al. reported a U-shaped relationship between alcohol intake and diabetes and concluded that excessive drinking (> 60 g/day for males and > 50 g/day for women) is linked to an elevated risk of diabetes[63]. However, alcohol consumption may increase insulin sensitivity and aid in blood sugar regulation[64]. Yet, other research has shown that alcohol may have an impact on the survival and function of pancreatic beta cells. In vitro studies have demonstrated that alcohol induces beta-cell apoptosis and affects insulin secretion[65]. Additionally, alcohol consumption may trigger inflammation and injury to the pancreas (also called alcoholic pancreatitis), which impairs the release of digestive enzymes as well as beta cells in the pancreas, which, in turn, affects the release of insulin and the regulation of blood sugar[66]. Interestingly, alcohol use influences insulin sensitivity in individuals with various rs56221195 polymorphism genotypes[67]: alcohol intake increases hepatic insulin sensitivity in heterozygotes (TA) and pure genotypes (AA), but does not affect insulin sensitivity in individuals with wild-type genotypes (TT).
Alcohol consumption causes blood lipid levels to rise. In a meta-analysis by Brien et al., alcohol consumption was shown to considerably raise high-density lipoprotein cholesterol levels but had little or no impact on triglyceride levels[68]. In contrast, new research has demonstrated that alcohol consumption raises triglyceride levels[69].
As previously mentioned, alcohol consumption amplifies the role of metabolic risk factors for liver damage. The central aspect of liver injury caused by either metabolic risk factors or alcohol consumption is lipid metabolism disturbance, characterized by an imbalance between lipid intake, synthesis, degradation, and free fatty acid (FFA) output[70,71]. Insulin resistance and dyslipidemia raise circulating levels of FFAs. Additionally, alcohol consumption causes more adipose tissue to be lipolyzed, resulting in adipose shrinkage and increased blood levels of FFAs[47]. Through fatty acid ethyl ester (FAEE) dehydrogenases, alcohol can directly interact with FFAs to create FAEE[72], which exacerbates alcohol-induced liver injury[73]. Alcohol consumption results in hepatic steatosis by increasing the absorption of circulating fatty acids, decreasing very low-density lipoprotein secretion, and reducing mitochondrial fatty acid oxidation, which, in turn, causes steroid response-element binding protein 1-mediated lipogenesis[74].
Unbalanced microelements
A previous study found that patients with either ALD or MASLD had significantly lower mean serum magnesium levels compared to control patients without SLD[75]. Some studies have shown that low serum magnesium levels are linked to a higher risk of insulin resistance and prediabetes[48,49]. In addition, insulin resistance plays a key role in the initiation and progression of MASLD. Notably, insulin resistance and magnesium deficiency are linked in a vicious cycle. Chronic insulin resistance brought on by magnesium insufficiency worsens magnesium loss in the kidneys and impairs cellular glucose consumption, which, in turn, leads to further insulin resistance[76,77]. Although the precise mechanism is unclear, given that insulin resistance is a feature of MASLD and ALD, preliminary evidence indicates that magnesium is a contributing factor in liver disease.
Mitochondrial reductive stress
Increased hepatic mitochondrial reductive stress (i.e., high NADH/NAD+ ratio) is a common feature in the development of MASLD and ALD[50]. Over the past few decades, extensive research has focused on alcohol metabolism[78,79]. Alcohol is primarily metabolized in the liver, and the enzymes involved include aldehyde dehydrogenase (ALDH), alcohol dehydrogenase (ADH), cytochrome P450 (cytochrome P450 family 2, subfamily E, polypeptide 1, CYP2E1), and catalase. Ethanol is primarily metabolized by a variety of enzymes, such as CYP2E1 and ADH, to produce acetaldehyde. Acetaldehyde is further metabolized, primarily by mitochondrial ALDH2 and, to a considerably lesser extent, by cytosolic ALDH1, to the more innocuous acetate and acetyl coenzyme A for the metabolic process[80]. Through this mechanism, alcohol metabolism raises the NADH/NAD+ ratio, which causes reductive stress in the mitochondria. When alcohol is metabolized, CYP2E1 generates ROS, which damage the liver[81]. The main location of ROS production is complex I of the electron transport chain, which is significantly increased during mitochondrial reductive stress, thus resulting in a vicious cycle[82].
MASLD is characterized by inflammation of adipose tissue, which, in turn, leads to an increased rate of lipolysis - the release of fatty acids from adipose tissue for hepatic absorption. In the liver, fatty acids undergo a process called β-oxidation[83], which raises the NADH/NAD+ ratio in the mitochondria and leads to hepatic mitochondrial reductive stress. Mitochondrial reductive stress is associated with increased hepatic fibroproliferation via its effect on proline metabolism[84]. A recent study proved the above point, demonstrating that, during a ketogenic diet, increased hepatic fatty acid supply results in mitochondrial reductive stress[85].
Gut microecological imbalance
Gut microbiological imbalance plays a significant role in both MASLD and ALD. Alcohol consumption impairs the integrity of the intestinal barrier by disrupting tight intestinal junctions, which increases intestinal permeability, leading to an imbalance of intestinal bacteria and allowing endotoxins/lipopolysaccharides (LPS) and bacteria to enter the liver through the portal vein[51]. Thereafter, Toll-like receptor 4 (TLR4), expressed on liver parenchyma and innate immune cells, recognizes LPS. LPS/TLR4 triggers activation of nuclear factor-κB, amplifying the inflammatory response and releasing pro-inflammatory factors[52]. Recent studies have found that high alcohol-producing Klebsiella pneumoniae (HiAlc Kpn) is associated with up to 60% of patients with MASLD in China. HiAlc Kpn, a symbiotic species in the gut microbiota, can produce excess endogenous alcohol through 2,3-butanediol fermentation, initially explaining the potential role of HiAlc Kpn in the pathogenesis of MASLD[86].
Other mechanisms
Genetic variation
In addition to sharing similar clinical characteristics and natural history, MASLD and ALD are associated with the same risk factors for genetic variants, including patatin-like phospholipase domain-containing 3 (PNPLA3, rs738409), transmembrane 6 superfamily member 2 (TM6SF2, rs58542926), membrane-bound O-acyltransferase domain-containing 7 (MBOAT7, rs641738), and glucokinase regulator (GCKR, rs1260326)[53]. The overlap in genetic structure suggests that MASLD and ALD share a common pathogenesis and that a range of inherent genetic risks act as disease modifiers. It has been shown that the PNPLA3 I148M variant is associated with a higher risk of advanced fibrosis, HCC, and liver-related mortality, as well as an increase in hepatic lipid accumulation[87]. This variation, which makes up 11% of the population with cirrhosis, is thought to be the most significant genetic risk factor for MASLD[88]. Studies have also demonstrated that additional genes, including TM6SF2 and MBOAT7, play a significant role in determining the occurrence of steatosis, fibrosis, and HCC in patients with MASLD and ALD[89,90].
Sex hormone differences
As previously noted, gender plays a significant role in the pathophysiology of liver disease[30,91]. Previous studies suggest that males are more susceptible to MASLD and ALD than females[92-94]. In this context, sex hormones may play a key role. Estrogen protects against the development of MASLD by inhibiting the
CHALLENGES IN DIAGNOSING METALD
The diagnosis of MetALD relies on the existence of CMRFs and alcohol consumption. Despite being the gold standard for diagnosing MetALD, liver biopsy is invasive and limited by the histologic similarities with MASLD and ALD[96]. Thus, there remains a need for objective non-invasive markers. Furthermore, while the two-step approach (utilizing FIB-4 followed by LSM) is increasingly adopted for risk stratification in MASLD[5,97-99], its applicability and the interpretation of FIB-4 and LSM results within the MetALD population remain inadequately explored and represent a crucial evidence gap. Fewer studies have evaluated the effectiveness of the two-step approach in assessing the risk stratification of liver fibrosis in patients with suspected MetALD[100,101]. The complex interplay of metabolic dysfunction and alcohol in MetALD may alter the performance characteristics of these biomarkers compared to pure MASLD or ALD. More dedicated studies are urgently needed to validate non-invasive risk stratification strategies and establish reliable interpretation criteria for FIB-4 and LSM in patients with MetALD.
Therefore, non-invasive markers for identifying MetALD should focus on evaluating metabolic status and accurately assessing the patient’s past and current alcohol consumption. For the assessment of metabolic status, specific biomarkers can be utilized to monitor metabolism-related diseases, such as body mass index (BMI) and waist circumference for obesity; glycosylated hemoglobin, fasting and 2-h postprandial glucose, insulin, and C-peptide levels for diabetes; triglyceride and cholesterol levels for dyslipidemia; and blood pressure values for hypertension. However, due to individual recall bias, we must recognize the difficulty in accurately measuring alcohol use, as some patients may underestimate their intake. The use of other specific biomarkers, such as phosphatidyl ethanol (PEth), ethyl glucuronide (EtG), ethyl sulfate (EtS), carbohydrate-deficient transferrin (CDT), and mean corpuscular volume (MCV), to record alcohol consumption history may help with this challenge.
Indirect biomarkers
Alcohol affects body chemistry and various organs. Several indirect biomarkers, including MCV, alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyltranspeptidase (GGT), and CDT, can be used to assess and identify alcohol exposure. Alcohol consumption increases erythrocyte volume (i.e., MCV values), which may be related to the myelotoxic effects of alcohol and its metabolites, as well as to nutritional deficiencies associated with alcohol consumption[54]. However, factors such as age, gender, and pre-existing medical conditions must be considered[95,96]. Following alcohol consumption, AST and ALT levels rise[102], and the ALT/AST ratio can provide information to differentiate ALD from other causes of liver disease[103]. Moreover, enzymes are direct markers of liver damage and inflammation, with elevated levels indicating liver dysfunction. Increased blood levels of GGT can persist for 2-6 weeks after heavy drinking[104], but it should be noted that several medications influence GGT levels by inducing its enzymatic activity.
It has been reported that alcohol consumption inhibits glycosylation/sialylation in the Golgi apparatus of hepatocytes. This inhibition leads to reduced sialylation of transferrin and an increased proportion of sialic acid-free transferrin, also referred to as CDT, even when total transferrin levels are normal[105]. Most measurements of the ratio of CDT to total transferrin (%CDT) have been conducted in Europe[106-108], with fewer reports on the utility of %CDT measurements in Asia[109,110]. However, for an Asian cohort,
To identify new SLD subgroups, alcohol consumption must be accurately measured. For certain alcohol-consuming populations, %CDT is a valuable marker, providing more information than GGT or MCV regarding diagnostic qualities and correlation with alcohol consumption. A %CDT cutoff of 1.78% is used to identify non-drinkers or light drinkers (sensitivity: 0.72, specificity: 0.84), whereas a 2.08% cutoff is used to identify heavy drinkers (sensitivity: 0.66, specificity: 0.87)[102].
Direct biomarkers
Several biomarkers, including PEth, EtG, and EtS, measure alcohol levels or alcohol metabolites to detect alcohol usage[103], and the use of these biomarkers is increasing in clinical settings[104,105]. One study used EtG in hair to assess alcohol consumption in patients with MASLD[105]. According to this study, 10% of patients who had been previously diagnosed with MASLD had hair EtG results that suggested high-risk alcohol consumption, which met ALD criteria, and 20% had moderate alcohol consumption, which met MetALD criteria[105]. However, because of the influence of hair dyes and the requirement for an ideal quantity of hair samples, the detection of EtG in hair is not widely applicable[3].
In contrast, whole-blood PEth is a sensitive and specific biomarker that better identifies individuals with MetALD[3,103,104], but is more expensive to assess than EtG. Additionally, PEth cannot reliably identify binge alcohol consumption due to its formation mechanism requiring sustained alcohol exposure over several days[111], and PEth only reflects alcohol exposure within the past 2-4 weeks[112]. For example, a patient may experience fewer drinking days or cravings, but one binge drinking episode can result in positive PEth levels, which could affect the assessment of several dimensions of alcohol use quantification[113], so a single measurement may not fully capture longer-term drinking patterns. In both the United States and some European countries, liver transplant hospitals frequently use continuous whole-blood PEth monitoring to track alcohol use[106]. However, PEth testing faces legal barriers in several European countries where privacy laws prohibit its routine use. Regulations under the EU General Data Protection Regulation (GDPR) classify PEth as sensitive health data, restricting physician access without explicit patient consent or judicial approval {Under GDPR, health data processing requires explicit consent [Regulation (EU) 2016/679, Art 9(1)]}. Consequently, clinicians in these regions rely primarily on traditional biomarkers (e.g., liver enzymes). Interestingly, PEth is not significantly affected by gender or BMI, and a reduction of two units of alcohol (26 g of pure ethanol) per day results in a PEth concentration reduction of approximately
To date, PEth has been used in relatively few studies to measure alcohol consumption. The prevalence of MetALD was 6.4% in a prospective study in the United States that included 374 overweight or obese SLD patients with MRI-proton density fat fraction ≥ 5%. PEth demonstrated robust diagnostic accuracy in recognizing MetALD [area under the receiver operating characteristic curve (AUROC): 0.81, 95%CI: 0.73-0.89], with a Youden cutoff value of 25 ng/mL (sensitivity: 0.70, specificity: 0.88). For the diagnosis of MetALD, PEth both statistically and clinically outperformed all previously utilized indirect alcohol indicators, such as AST/ALT ratios, MCV, GGT, and the ALD/NAFLD index (ANI) (P < 0.05). Additionally, PEth performed satisfactorily in diagnosing ALD (AUROC: 0.76, 95%CI: 0.61-0.91), outperforming the AST/ALT ratio, GGT, MCV, and ANI. However, because of the small sample size of ALD patients, this difference did not achieve statistical significance[104].
POTENTIAL TREATMENTS FOR METALD
Fortunately, the new nomenclature recognizes that MetALD is a social problem and acknowledges that individuals with MetALD should be treated in the same way as persons with conditions such as obesity, diabetes, or smoking-induced chronic obstructive pulmonary disease. Currently, no medications have been developed to treat MetALD as a distinct disease. Although medications being researched for MASLD may also be helpful for MetALD, this does not mean that alcohol consumption can be condoned. Abstinence from alcohol remains the cornerstone of treatment for MetALD.
Abstinence
Approximately 54.1% of Americans use alcohol, and the prevalence of 12-month drinking, high-risk drinking, and alcohol use disorders (AUDs) in the United States increased between 2001-2002 and 2012-2013 (65.4% vs. 72.7%, 9.7% vs. 12.6%, and 8.5% vs. 12.7%, respectively)[110]. In addition, approximately 11.8% of patients with AUD develop ALD[114]. Given that alcohol consumption itself may contribute to the development of metabolic syndrome, abstinence from alcohol may, to some extent, alleviate metabolic issues and decrease medication use.
Weight management
Weight management, as a cornerstone in the management of MASLD and consequently in controlling CRMF, plays a pivotal role. A comprehensive and gradual strategy that includes exercise, bariatric surgery, dietary restrictions, and nutritional interventions can help individuals to lose weight. Hepatic steatosis is relieved in patients with MASH when they lose at least 5% of their body weight, MASH is relieved when they lose at least 7%, and the fibrosis stage is improved when they lose at least 10%[99]. Thus, effective weight reduction is a fundamental and potentially promising therapeutic option for addressing hepatic steatosis inflammation (MASH), and fibrosis in patients with MASLD, including those with MetALD.
Potential therapeutic medications
THR-β agonist
Resmetirom, an oral selective thyroid hormone receptor beta (THR-β) agonist, has demonstrated efficacy in the treatment of metabolic dysfunction-associated steatohepatitis (MASH) and is currently the only medication approved by the United States Food and Drug Administration for the treatment of non-cirrhotic MASH patients[115]. Phase-2 and phase-3 clinical trials of resmetirom demonstrated that 80 mg and 100 mg doses of resmetirom were better than placebo in improving MASH, and the incidence of serious adverse events was comparable across study groups. However, resmetirom treatment was associated with transient mild diarrhea and nausea[4,116]. A subgroup analysis of MetALD patients, screened for CDT > 2.5% and/or PEth > 20 ng/mL, revealed that resmetirom had a similar effect in alleviating hepatic fibrosis and inflammation[4].
PPAR agonist
Pemafibrate is a selective modulator of PPARα. Previous animal studies have shown that PPARα exerts anti-inflammatory activity by trans-repressing pro-inflammatory target genes[117]. Accordingly, preliminary studies have demonstrated that pemafibrate may improve liver function in patients with MASLD[118]. However, it is important to note that pemafibrate is not currently an approved therapy specifically for MASLD or MetALD, and its efficacy and safety for these indications require confirmation in large-scale, randomized controlled trials. Given that alcohol reduces the activity of the PPARα signaling pathway and decreases fatty acid β-oxidation, which promotes lipid accumulation in hepatocytes[119], and that PPARα agonists accelerate the degradation of a wide range of fatty acids, pemafibrate is a potential candidate for MetALD, pending further clinical validation[120]. Pemafibrate is metabolized in the liver and eliminated through the bile. Therefore, pemafibrate can be administered safely even in patients with poor renal function, as its blood concentration does not increase significantly in the kidneys.
The dual PPARα/PPARδ agonist GFT505 (later renamed elafibranor) has shown promising preclinical and early clinical outcomes, indicating potential for the treatment of MetALD[121,122]. In a multicenter, randomized, double-blind, placebo-controlled trial (NCT01694849), in the population that was intended to be treated, elafibranor treatment did not meet the primary endpoint of MASH remission without worsening fibrosis[123]; yet, a post hoc analysis using the revised definition revealed that the MASH pathology resolved without a worsening of stage 2 or stage 3 fibrotic status. However, elafibranor treatment caused a slight, reversible increase in serum creatinine, which prompted the other phase-2 and phase-3 studies to be terminated[123,124]. In a recent animal study by Koizumi et al., elafibranor improved intestinal barrier integrity and dramatically decreased liver fibrosis in ALD mice[122]. These preclinical findings support the potential beneficial role of elafibranor in alcohol-induced MASLD, which provides a rationale for the development of this molecular therapy in patients with MetALD[124,125].
GLP-1 receptor agonist
Liraglutide is a glucagon-like peptide 1 (GLP-1) receptor agonist. A 12-week clinical trial (NCT01237119) demonstrated that liraglutide reduced BMI, high- and low-density lipoprotein cholesterol levels, and ALT levels[126]. Another GLP-1 receptor agonist, semaglutide, has been proven in two recent clinical trials to have weight-loss effects[127,128]. In addition, several studies have shown that GLP-1 receptor agonists can reduce alcohol intake, indirectly improve inflammation and hepatic steatosis, and eventually reverse fibrosis[129-132]. This combination of weight loss, inflammation reduction, and alcohol intake reduction can have a significant impact on MetALD. Notably, a 72-week phase 2 trial revealed that daily subcutaneous semaglutide (0.1-0.4 mg) significantly increased NASH resolution rates versus placebo (up to 59%), though without significant fibrosis improvement[129]. In contrast, the phase 3 ESSENCE trial demonstrated that weekly 2.4 mg semaglutide improved liver histology in MASH patients with moderate or advanced fibrosis[133]. Thus, GLP-1 receptor agonists are anticipated as a targeted medication for preventing major components of disease progression.
Farnesoid X receptor agonist
Obeticholic acid, an endogenous agonist of the Farnesoid X receptor (FXR), improves insulin sensitivity and reduces the expression of inflammation and liver fibrosis markers in individuals with MASLD and type 2 diabetes[134]. Cilofexor, a synthetic FXR agonist, has been shown to significantly reduce hepatic steatosis in MASH patients[135]. Recently, two clinical trials (NCT03987074, NCT03449446) investigated the use of FXR agonists in combination with other drugs for the treatment of MASH[136-138]. However, further research is required to determine their safety and effectiveness.
These potential treatments investigate the targeted pharmacotherapies for MetALD in non-transplant clinical contexts. Generalization to transplant recipients or marginal donor livers should be cautious. Yakubu et al. demonstrated that GLP-1 receptor agonist, in addition to glycemic control, reduced body weight and resulted in a lower prevalence of hepatic steatosis in liver transplant recipients[139]. FXR agonists have been shown in animal studies to reduce hepatic ischemia-reperfusion injury[140], indicating that some medications may still have protective potential for the pathologic liver, even though cold ischemic injury to the donor liver during transplantation may superimpose metabolic abnormalities. Therefore, more studies should explore their efficacy in transplant settings in the future, especially for grafts with pre-existing metabolic diseases.
Other possible therapies
Fecal microbiota transplantation
Alcohol consumption has a profound effect on the diversity of gut microbes, and fecal microbiota transplantation (FMT) is an emerging treatment for related diseases. Although FMT has not been studied in MetALD, some of the strongest evidence for altered microbiomes comes from studies in severe alcoholic hepatitis[141,142]. FMT is safe and is associated with reduced short-term alcohol cravings and consumption as well as favorable microbiome changes compared with placebo treatment[141]. FMT is a crucial area of therapeutic research in MetALD, owing to its effects on alcohol consumption, capacity to enhance intestinal barrier function, and potential to improve metabolic patterns.
Dietary supplementation
Glucosamine, a natural amino monosaccharide, is frequently used as a dietary supplement to support healthy joints[143]. New research suggests that glucosamine possesses advantageous anti-inflammatory and antioxidant properties, which may benefit liver disease. One study showed that glucosamine enhanced intestinal barrier function, reduced LPS transfer, and lessened hepatic inflammation in a mouse model of hepatic steatosis[144]. Notably, in a Chinese cohort, Lai et al. found that glucosamine reduced oxidative stress in the liver and could attenuate acute liver damage caused by alcohol by inhibiting inflammation and oxidative stress[145]. A large cohort study indicated that glucosamine may lower the risk of cardiovascular disease in MetALD[146]. Therefore, glucosamine supplementation may be a beneficial therapeutic option for patients with MetALD. Preclinical studies on other dietary supplement formulations are still in progress[147].
Estrogen supplementation
Sex hormone therapy for MetALD has attracted considerable interest. Given its protective effects[54,95], estrogen supplementation has become the primary strategy for hormone therapy. However, when using oral contraceptives or hormone replacement therapy, excessive alcohol consumption should be avoided, as alcohol interferes with the liver’s ability to metabolize hormones. Excessive alcohol usage can lead to a dramatic increase in circulating hormone levels, which raises a woman’s risk of developing hormone-sensitive breast and reproductive tract malignancies.
METALD-RELATED MODELS
MetALD is a complex condition involving an intricate interplay of multiple factors, including obesity, insulin resistance, dyslipidemia, and chronic alcohol consumption, and replicating this complexity in an animal model is a great challenge. Thus, researchers are still in the early stages of creating mouse models for MetALD. In the DUAL (ALD plus MAFLD) model developed by Benedé-Ubieto’s research team[148], the experimental group of mice received a 23-week treatment with the DUAL diet, corresponding to a Western-style diet consisting of 40% fat, 22% fructose, and 2% cholesterol, along with 10% v/v ethanol added to 6.75% D-glucose-sweetened drinking water. This diet is straightforward and manageable, and it is linked to minimal mortality even after prolonged feeding, making it a fairly safe feeding method. However, because of their quick metabolism, estimating daily alcohol intake from the amount of water consumed by the mice overestimates their blood alcohol levels.
Babuta et al. created a preclinical model to investigate MetALD[149]; this model replicates the main characteristics of alcoholic hepatitis in humans, including severe liver inflammation, increased immune cell infiltration, impaired hepatic synthesis function, raised serum bilirubin, and enhanced liver damage. The experimental group of mice received a high-fat, high-cholesterol, high-sucrose diet (MASH diet) and
While current models provide valuable insights into the mechanisms underlying MetALD, their translation to human trials faces substantial barriers. Designing and conducting clinical studies for MetALD involves unique complexities: (1) Quantifying alcohol consumption reliably is challenging due to dependence on self-reporting and societal stigma leading to underreporting[1]; (2) Regulatory restrictions vary globally regarding interventions in populations with active alcohol consumption; (3) Ethical considerations arise when enrolling metabolically compromised individuals with concurrent alcohol exposure. These hurdles necessitate innovative trial designs and multidisciplinary collaboration. Cross-laboratory research is required to develop and test complex models that can simulate the chronic part of this disease.
CONCLUSIONS
MetALD is a unique disease entity within the SLD spectrum that has had a significant impact since its introduction, garnering the attention of hepatologists and other scholars. Numerous studies based on large databases have explored the prevalence of MetALD and found that it is associated with a risk of different liver diseases (e.g., cirrhosis, HCC) as well as other diseases. The prevalence of MetALD may be underestimated due to individual recall bias and underreporting of alcohol use. Thus, there is an urgent need to identify a reliable, easy-to-test biomarker to recognize different levels and patterns of alcohol use. A threshold for alcohol consumption to diagnose MetALD must be validated in a prospective study cohort.
Undoubtedly, an appropriate animal model that accurately replicates the many forms of liver damage caused by MetALD would enhance our understanding of its pathophysiologic mechanisms. Future studies should give special attention to the pathophysiology of liver damage shared by CMRFs and alcohol consumption. Because of the lack of animal models that accurately replicate the pathogenesis of MetALD, treatment options are relatively limited. Elucidating the mechanisms driving MetALD will help researchers better understand how this condition occurs and will facilitate the development of novel medications to treat MetALD. However, potential therapeutic medications will require further validation in specific human clinical trials.
DECLARATIONS
Authors’ contributions
Contributed to the study’s conception and design: Feng Y, Han P, Liu T, Gao Y
Designed the review outline: Gao Y
Drafted the manuscript: Gao Y, Feng Y
Revised and edited the paper: Gao Y, Feng Y, Han P, Liu T
Read and approved the final manuscript: Feng Y, Han P, Liu T, Gao Y
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was sponsored by the National Natural Science Foundation of China (grants No. U24A20654; 82170602), the Natural Science Foundation for self-exploration research of Jilin Province (YDZJ202401427ZYTS), Jilin Provincial Key Laboratory of Metabolic Liver Diseases (YDZJ202502CXJD002), and the National Key Research and Development Program of China (No. 2024YFE0213800).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Rinella ME, Lazarus JV, Ratziu V, et al; NAFLD Nomenclature consensus group. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol. 2023;79:1542-56.
3. Arab JP, Díaz LA, Rehm J, et al. Metabolic dysfunction and alcohol-related liver disease (MetALD): position statement by an expert panel on alcohol-related liver disease. J Hepatol. 2025;82:744-56.
4. Harrison SA, Bedossa P, Guy CD, et al; MAESTRO-NASH Investigators. A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. N Engl J Med. 2024;390:497-509.
5. Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO clinical practice guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J Hepatol. 2024;81:492-542.
6. Leggio L, Hendershot CS, Farokhnia M, et al. GLP-1 receptor agonists are promising but unproven treatments for alcohol and substance use disorders. Nat Med. 2023;29:2993-5.
7. Manthey J, Shield KD, Rylett M, Hasan OSM, Probst C, Rehm J. Global alcohol exposure between 1990 and 2017 and forecasts until 2030: a modelling study. Lancet. 2019;393:2493-502.
8. Yun B, Park H, Ahn SH, Oh J, Kim BK, Yoon JH. Liver cancer risk across metabolic dysfunction-associated steatotic liver disease and/or alcohol: a nationwide study. Am J Gastroenterol. 2025;120:410-9.
9. Younossi ZM, de Avila L, Racila A, et al. Prevalence and predictors of cirrhosis and portal hypertension in the United States. Hepatology. 2025;Epub ahead of print.
10. John BV, Bastaich D, Mezzacappa C, et al; Veterans Analysis of Liver Disease (VALID) group of investigators. Identifying metabolic dysfunction-associated steatotic liver disease using natural language processing in a US national cohort. Am J Gastroenterol. 2025; Epub ahead of print.
11. Oh R, Kim S, Cho SH, et al. Metabolic dysfunction-associated steatotic liver disease and all-cause and cause-specific mortality. Diabetes Metab J. 2025;49:80-91.
12. Shine BK, Son M, Moon SY, Han SH. Metabolic dysfunction-associated steatotic liver disease and the risk of chronic periodontitis: a nationwide cohort study. Nutrients. 2024;17:125.
13. Baek SU, Yoon JH. High-sensitivity C-reactive protein levels in metabolic dysfunction-associated steatotic liver disease (MASLD), metabolic alcohol-associated liver disease (MetALD), and alcoholic liver disease (ALD) with metabolic dysfunction. Biomolecules. 2024;14:1468.
14. Oh R, Kim G, Lee KN, et al. Metabolic dysfunction-associated steatotic liver disease in patients with type 2 diabetes: risk of heart failure. Cardiovasc Diabetol. 2024;23:391.
15. Díaz LA, Lazarus JV, Fuentes-López E, et al. Disparities in steatosis prevalence in the United States by race or ethnicity according to the 2023 criteria. Commun Med. 2024;4:219.
16. Shi GX, Qian YS, Jiang CM, et al. Prevalence of steatotic liver disease (MASLD, MetALD, ALD) and clinically significant fibrosis in US adolescents: Authors’ name. Sci Rep. 2024;14:25724.
17. Ogawa Y, Tomeno W, Imamura Y, et al. Distribution of Fibrosis-4 index and vibration-controlled transient elastography-derived liver stiffness measurement for patients with metabolic dysfunction-associated steatotic liver disease in health check-up. Hepatol Res. 2024;Epub ahead of print.
18. Park HJ, Lee S, Lee JS. Differences in the prevalence of NAFLD, MAFLD, and MASLD according to changes in the nomenclature in a health check-up using MRI-derived proton density fat fraction. Abdom Radiol. 2024;49:3036-44.
19. Mori K, Tanaka M, Sato T, et al. Metabolic dysfunction-associated steatotic liver disease (SLD) and alcohol-associated liver disease, but not SLD without metabolic dysfunction, are independently associated with new onset of chronic kidney disease during a 10-year follow-up period. Hepatol Res. 2024;Epub ahead of print.
20. Kim GA, Jeong S, Jang H, Lee DH, Joo SK, Kim W. Metabolic dysfunction-associated steatotic liver disease and metabolic dysfunction-associated steatotic liver disease with increased alcohol intake increase the risk of developing hepatocellular carcinoma and incident or decompensated cirrhosis: a Korean nationwide study. Liver Cancer. 2024;13:426-37.
21. Lee CM, Yoon EL, Kim M, et al. Prevalence, distribution, and hepatic fibrosis burden of the different subtypes of steatotic liver disease in primary care settings. Hepatology. 2024;79:1393-400.
22. Schneider CV, Schneider KM, Raptis A, Huang H, Trautwein C, Loomba R. Prevalence of at-risk MASH, MetALD and alcohol-associated steatotic liver disease in the general population. Aliment Pharmacol Ther. 2024;59:1271-81.
23. Choe HJ, Moon JH, Kim W, Koo BK, Cho NH. Steatotic liver disease predicts cardiovascular disease and advanced liver fibrosis: a community-dwelling cohort study with 20-year follow-up. Metabolism. 2024;153:155800.
24. Gawrieh S, Vilar-Gomez E, Woreta TA, et al. Prevalence of steatotic liver disease, MASLD, MetALD and significant fibrosis in people with HIV in the United States. Aliment Pharmacol Ther. 2024;59:666-79.
25. Miwa T, Tajirika S, Imamura N, et al. Prevalence of steatotic liver disease based on a new nomenclature in the japanese population: a health checkup-based cross-sectional study. J Clin Med. 2024;13:1158.
26. Israelsen M, Torp N, Johansen S, et al; GALAXY consortium. Validation of the new nomenclature of steatotic liver disease in patients with a history of excessive alcohol intake: an analysis of data from a prospective cohort study. Lancet Gastroenterol Hepatol. 2024;9:218-28.
27. Ciardullo S, Carbone M, Invernizzi P, Perseghin G. Exploring the landscape of steatotic liver disease in the general US population. Liver Int. 2023;43:2425-33.
28. Yang AH, Tincopa MA, Tavaglione F, et al. Prevalence of steatotic liver disease, advanced fibrosis and cirrhosis among community-dwelling overweight and obese individuals in the USA. Gut. 2024;73:2045-53.
29. Sogabe M, Okahisa T, Kagawa M, et al. The association between alcohol consumption and cardiometabolic factors and liver fibrosis in metabolic dysfunction-associated steatotic liver disease and metabolic dysfunction and alcohol-associated liver disease. Aliment Pharmacol Ther. 2024;60:1587-98.
30. Wegermann K, Garrett ME, Zheng J, et al. Sex and menopause modify the effect of single nucleotide polymorphism genotypes on fibrosis in NAFLD. Hepatol Commun. 2021;5:598-607.
31. Paik JM, Golabi P, Biswas R, Alqahtani S, Venkatesan C, Younossi ZM. Nonalcoholic fatty liver disease and alcoholic liver disease are major drivers of liver mortality in the United States. Hepatol Commun. 2020;4:890-903.
32. Uluk D, Pein J, Herda S, et al. Metabolic dysfunction-associated steatotic liver disease (MASLD) impacts long-term outcomes after curative-intent surgery for hepatocellular carcinoma. Aliment Pharmacol Ther. 2025;61:1318-32.
33. Tamaki N, Kimura T, Wakabayashi SI, et al. Long-term clinical outcomes in steatotic liver disease and incidence of liver-related events, cardiovascular events and all-cause mortality. Aliment Pharmacol Ther. 2024;60:61-9.
34. Joo K, Kang YW, Moon SY, Baek YH, Son M. Association between nonalcoholic fatty liver disease scores and chronic periodontitis: a retrospective cohort study. J Periodontol. 2025;96:490-8.
35. Blasco-Baque V, Garidou L, Pomié C, et al. Periodontitis induced by porphyromonas gingivalis drives periodontal microbiota dysbiosis and insulin resistance via an impaired adaptive immune response. Gut. 2017;66:872-85.
36. Khan S, Barrington G, Bettiol S, Barnett T, Crocombe L. Is overweight/obesity a risk factor for periodontitis in young adults and adolescents? Obes Rev. 2018;19:852-83.
37. Ahmad A, Furuta M, Shinagawa T, et al. Association of periodontal status with liver abnormalities and metabolic syndrome. J Oral Sci. 2015;57:335-43.
38. Akinkugbe AA, Avery CL, Barritt AS, et al. Do genetic markers of inflammation modify the relationship between periodontitis and nonalcoholic fatty liver disease? J Dent Res. 2017;96:1392-9.
39. Kimura T, Tamaki N, Wakabayashi SI, et al. Colorectal cancer incidence in steatotic liver disease (MASLD, MetALD, and ALD). Clin Gastroenterol Hepatol. 2025;Epub ahead of print.
40. Targher G, Byrne CD. Non-alcoholic fatty liver disease: an emerging driving force in chronic kidney disease. Nat Rev Nephrol. 2017;13:297-310.
41. Lake JE, Overton T, Naggie S, et al. Expert panel review on nonalcoholic fatty liver disease in persons with human immunodeficiency virus. Clin Gastroenterol Hepatol. 2022;20:256-68.
42. Gawrieh S, Lake JE, Debroy P, et al. Burden of fatty liver and hepatic fibrosis in persons with HIV: a diverse cross-sectional US multicenter study. Hepatology. 2023;78:578-91.
43. Bao X, Kang L, Yin S, et al. Association of MAFLD and MASLD with all-cause and cause-specific dementia: a prospective cohort study. Alzheimers Res Ther. 2024;16:136.
44. Cheon SY, Song J. Novel insights into non-alcoholic fatty liver disease and dementia: insulin resistance, hyperammonemia, gut dysbiosis, vascular impairment, and inflammation. Cell Biosci. 2022;12:99.
45. de la Monte SM. Insulin resistance and neurodegeneration: progress towards the development of new therapeutics for Alzheimer’s disease. Drugs. 2017;77:47-65.
46. Shin WY, Kang ES, Oh YH, et al. Metabolic dysfunction-associated steatotic liver disease, metabolic alcohol-related liver disease, and incident dementia: a nationwide cohort study: MASLD, MetALD, and dementia risk. BMC Gastroenterol. 2025;25:308.
47. Li Y, Chao X, Wang S, Williams JA, Ni HM, Ding WX. Role of mechanistic target of rapamycin and autophagy in alcohol-induced adipose atrophy and liver injury. Am J Pathol. 2020;190:158-75.
48. Li W, Zhu X, Song Y, et al. Intakes of magnesium, calcium and risk of fatty liver disease and prediabetes. Public Health Nutr. 2018;21:2088-95.
49. Barbagallo M, Veronese N, Dominguez LJ. Magnesium in type 2 diabetes mellitus, obesity, and metabolic syndrome. Nutrients. 2022;14:714.
50. Jokinen MJ, Luukkonen PK. Hepatic mitochondrial reductive stress in the pathogenesis and treatment of steatotic liver disease. Trends Pharmacol Sci. 2024;45:319-34.
51. Jew MH, Hsu CL. Alcohol, the gut microbiome, and liver disease. J Gastroenterol Hepatol. 2023;38:1205-10.
52. Raya Tonetti F, Eguileor A, Mrdjen M, et al. Gut-liver axis: recent concepts in pathophysiology in alcohol-associated liver disease. Hepatology. 2024;80:1342-71.
53. Romeo S, Sanyal A, Valenti L. Leveraging human genetics to identify potential new treatments for fatty liver disease. Cell Metab. 2020;31:35-45.
54. Duan H, Gong M, Yuan G, Wang Z. Sex hormone: a potential target at treating female metabolic dysfunction-associated steatotic liver disease? J Clin Exp Hepatol. 2025;15:102459.
55. Åberg F, Byrne CD, Pirola CJ, Männistö V, Sookoian S. Alcohol consumption and metabolic syndrome: clinical and epidemiological impact on liver disease. J Hepatol. 2023;78:191-206.
56. Traversy G, Chaput JP. Alcohol consumption and obesity: an update. Curr Obes Rep. 2015;4:122-30.
57. Zhai X, Li S, Wang T, Bai J, Xu F, Zhou W. Dark tea wine protects against metabolic dysfunction-associated steatotic liver disease in vivo through activating the Nrf2/HO-1 antioxidant signaling pathway. J Med Food. 2024;27:912-21.
58. Cecchini M, Filippini T, Whelton PK, et al. Alcohol intake and risk of hypertension: a systematic review and dose-response meta-analysis of nonexperimental cohort studies. Hypertension. 2024;81:1701-15.
59. Puddey IB, Beilin LJ, Vandongen R. Regular alcohol use raises blood pressure in treated hypertensive subjects. A randomised controlled trial. Lancet. 1987;1:647-51.
60. Masengere P, Halbesma N, Ndejjo R, et al. Additive interaction of conjoint tobacco smoking and heavy drinking on hypertension prevalence in rural Uganda: a community-based cross-sectional study. BMC Public Health. 2025;25:201.
61. Mukhopadhyay P, Yokus B, Paes-Leme B, et al. Chronic alcohol consumption accelerates cardiovascular aging and decreases cardiovascular reserve capacity. Geroscience. 2025;Epub ahead of print.
62. Llamosas-Falcón L, Rehm J, Bright S, et al. The relationship between alcohol consumption, BMI, and type 2 diabetes: a systematic review and dose-response meta-analysis. Diabetes Care. 2023;46:2076-83.
63. Baliunas DO, Taylor BJ, Irving H, et al. Alcohol as a risk factor for type 2 diabetes: a systematic review and meta-analysis. Diabetes Care. 2009;32:2123-32.
64. Schrieks IC, Heil AL, Hendriks HF, Mukamal KJ, Beulens JW. The effect of alcohol consumption on insulin sensitivity and glycemic status: a systematic review and meta-analysis of intervention studies. Diabetes Care. 2015;38:723-32.
65. Kim JY, Song EH, Lee HJ, et al. Chronic ethanol consumption-induced pancreatic β-cell dysfunction and apoptosis through glucokinase nitration and its down-regulation. J Biol Chem. 2010;285:37251-62.
66. Rasineni K, Srinivasan MP, Balamurugan AN, et al. Recent advances in understanding the complexity of alcohol-induced pancreatic dysfunction and pancreatitis development. Biomolecules. 2020;10:669.
67. Fu Q, Dai H, Shen S, et al. Interactions of genes with alcohol consumption affect insulin sensitivity and beta cell function. Diabetologia. 2025;68:116-27.
68. Brien SE, Ronksley PE, Turner BJ, Mukamal KJ, Ghali WA. Effect of alcohol consumption on biological markers associated with risk of coronary heart disease: systematic review and meta-analysis of interventional studies. BMJ. 2011;342:d636.
69. Thiele M, Suvitaival T, Trošt K, et al; MicrobLiver Consortium, GALAXY Consortium. Sphingolipids are depleted in alcohol-related liver fibrosis. Gastroenterology. 2023;164:1248-60.
71. Gluchowski NL, Becuwe M, Walther TC, Farese RV Jr. Lipid droplets and liver disease: from basic biology to clinical implications. Nat Rev Gastroenterol Hepatol. 2017;14:343-55.
72. Williams JA, Ding WX. A mechanistic review of mitophagy and its role in protection against alcoholic liver disease. Biomolecules. 2015;5:2619-42.
73. Wu H, Cai P, Clemens DL, Jerrells TR, Ansari GA, Kaphalia BS. Metabolic basis of ethanol-induced cytotoxicity in recombinant HepG2 cells: role of nonoxidative metabolism. Toxicol Appl Pharmacol. 2006;216:238-47.
75. Turecky L, Kupcova V, Szantova M, Uhlikova E, Viktorinova A, Czirfusz A. Serum magnesium levels in patients with alcoholic and non-alcoholic fatty liver. Bratisl Lek Listy. 2006;107:58-61.
76. Gommers LM, Hoenderop JG, Bindels RJ, de Baaij JH. Hypomagnesemia in type 2 diabetes: a vicious circle? Diabetes. 2016;65:3-13.
77. Fan L, Zhu X, Zhang X, et al. Magnesium depletion score and mortality in individuals with metabolic dysfunction associated steatotic liver disease over a median follow-up of 26 years. Nutrients. 2025;17:244.
78. Zakhari S. Overview: how is alcohol metabolized by the body? Alcohol Res Health. 2006;29:245-54.
80. Crabb DW, Matsumoto M, Chang D, You M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc Nutr Soc. 2004;63:49-63.
81. Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol. 2009;83:519-48.
83. McGarry JD, Foster DW. Regulation of hepatic fatty acid oxidation and ketone body production. Annu Rev Biochem. 1980;49:395-420.
84. Ding Z, Ericksen RE, Escande-Beillard N, et al. Metabolic pathway analyses identify proline biosynthesis pathway as a promoter of liver tumorigenesis. J Hepatol. 2020;72:725-35.
85. Luukkonen PK, Dufour S, Lyu K, et al. Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A. 2020;117:7347-54.
86. Yuan J, Chen C, Cui J, et al. Fatty liver disease caused by high-alcohol-producing klebsiella pneumoniae. Cell Metab. 2019;30:675-88.e7.
87. Luukkonen PK, Porthan K, Ahlholm N, et al. The PNPLA3 I148M variant increases ketogenesis and decreases hepatic de novo lipogenesis and mitochondrial function in humans. Cell Metab. 2023;35:1887-96.e5.
88. Jamialahmadi O, Mancina RM, Ciociola E, et al. Exome-wide association study on alanine aminotransferase identifies sequence variants in the GPAM and APOE associated with fatty liver disease. Gastroenterology. 2021;160:1634-46.e7.
89. Liu YL, Reeves HL, Burt AD, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. 2014;5:4309.
90. Buch S, Stickel F, Trépo E, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet. 2015;47:1443-8.
91. Nataraj K, Schonfeld M, Rodriguez A, Sharma M, Weinman S, Tikhanovich I. Androgen effects on alcohol-induced liver fibrosis are controlled by a notch-dependent epigenetic switch. Cell Mol Gastroenterol Hepatol. 2025;19:101414.
92. Teng ML, Ng CH, Huang DQ, et al. Global incidence and prevalence of nonalcoholic fatty liver disease. Clin Mol Hepatol. 2023;29:S32-42.
93. Danpanichkul P, Díaz LA, Suparan K, et al. Global epidemiology of alcohol-related liver disease, liver cancer, and alcohol use disorder, 2000-2021. Clin Mol Hepatol. 2025;31:525-47.
94. Danpanichkul P, Pang Y, Mahendru T, et al. Sex disparities in alcohol-associated liver disease and subtype differences in alcohol-attributable cancers in the United States. Clin Mol Hepatol. 2025;31:1058-70.
95. Nataraj K, Schonfeld M, Rodriguez A, Tikhanovich I. Protective role of 17β-estradiol in alcohol-associated liver fibrosis is mediated by suppression of integrin signaling. Hepatol Commun. 2024;8:e0428.
96. Malnick SDH, Alin P, Somin M, Neuman MG. Fatty liver disease-alcoholic and non-alcoholic: similar but different. Int J Mol Sci. 2022;23:16226.
97. Yip TC, Lee HW, Lin H, et al. Prognostic performance of the two-step clinical care pathway in metabolic dysfunction-associated steatotic liver disease. J Hepatol. 2025;83:304-14.
98. Wattacheril JJ, Abdelmalek MF, Lim JK, Sanyal AJ. AGA clinical practice update on the role of noninvasive biomarkers in the evaluation and management of nonalcoholic fatty liver disease: expert review. Gastroenterology. 2023;165:1080-8.
99. Rinella ME, Neuschwander-Tetri BA, Siddiqui MS, et al. AASLD practice guidance on the clinical assessment and management of nonalcoholic fatty liver disease. Hepatology. 2023;77:1797-835.
100. Díaz LA, Tavaglione F, Mittal N, et al. Noninvasive pathway for stratifying fibrosis in suspected metabolic dysfunction and alcohol-associated liver disease (MetALD). Hepatol Commun. 2025;9:e0718.
101. Oh JH, Ahn SB, Cho S, Nah EH, Yoon EL, Jun DW. Diagnostic performance of non-invasive tests in patients with MetALD in a health check-up cohort. J Hepatol. 2024;81:772-80.
102. Morinaga M, Kon K, Uchiyama A, et al. Carbohydrate-deficient transferrin is a sensitive marker of alcohol consumption in fatty liver disease. Hepatol Int. 2022;16:348-58.
103. Nguyen VL, Paull P, Haber PS, Chitty K, Seth D. Evaluation of a novel method for the analysis of alcohol biomarkers: ethyl glucuronide, ethyl sulfate and phosphatidylethanol. Alcohol. 2018;67:7-13.
104. Tavaglione F, Amangurbanova M, Yang AH, et al. Head-to-head comparison between phosphatidylethanol versus indirect alcohol biomarkers for diagnosis of MetALD versus MASLD: a prospective study. Aliment Pharmacol Ther. 2025;61:1043-54.
105. Staufer K, Huber-Schönauer U, Strebinger G, et al. Ethyl glucuronide in hair detects a high rate of harmful alcohol consumption in presumed non-alcoholic fatty liver disease. J Hepatol. 2022;77:918-30.
106. Arab JP, Izzy M, Leggio L, Bataller R, Shah VH. Management of alcohol use disorder in patients with cirrhosis in the setting of liver transplantation. Nat Rev Gastroenterol Hepatol. 2022;19:45-59.
107. Finanger T, Melby K, Spigset O, Andreassen TN, Lydersen S, Skråstad RB. Relationship between alcohol intake based on daily smartphone-reported consumption and PEth concentrations in healthy volunteers. Alcohol Alcohol. 2024;59:agae040.
108. Aradottir S, Asanovska G, Gjerss S, Hansson P, Alling C. PHosphatidylethanol (PEth) concentrations in blood are correlated to reported alcohol intake in alcohol-dependent patients. Alcohol Alcohol. 2006;41:431-7.
109. Viel G, Boscolo-Berto R, Cecchetto G, Fais P, Nalesso A, Ferrara SD. Phosphatidylethanol in blood as a marker of chronic alcohol use: a systematic review and meta-analysis. Int J Mol Sci. 2012;13:14788-812.
110. Grant BF, Chou SP, Saha TD, et al. Prevalence of 12-month alcohol use, high-risk drinking, and DSM-IV alcohol use disorder in the United States, 2001-2002 to 2012-2013: results from the national epidemiologic survey on alcohol and related conditions. JAMA Psychiatry. 2017;74:911-23.
111. Piano MR, Tiwari S, Nevoral L, Phillips SA. Phosphatidylethanol levels are elevated and correlate strongly with AUDIT scores in young adult binge drinkers. Alcohol Alcohol. 2015;50:519-25.
112. Tavaglione F, Díaz LA, Ajmera V, et al. Clinical utility of phosphatidylethanol to detect underreported alcohol use and enhance steatotic liver disease subclassification. J Hepatol. 2025;Epub ahead of print.
113. Díaz LA, Collier S, Yin J, Loomba R. Safety and tolerability of injectable extended-release naltrexone for the management of alcohol use disorder in advanced alcohol-associated liver disease. Aliment Pharmacol Ther. 2025:Epub ahead of print.
114. Vannier AGL, Shay JES, Fomin V, et al. Incidence and progression of alcohol-associated liver disease after medical therapy for alcohol use disorder. JAMA Netw Open. 2022;5:e2213014.
116. Harrison SA, Bashir MR, Guy CD, et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2019;394:2012-24.
117. Delerive P, De Bosscher K, Besnard S, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem. 1999;274:32048-54.
118. Sugimoto R, Iwasa M, Eguchi A, et al. Effect of pemafibrate on liver enzymes and shear wave velocity in non-alcoholic fatty liver disease patients. Front Med. 2023;10:1073025.
119. Yu S, Rao S, Reddy JK. Peroxisome proliferator-activated receptors, fatty acid oxidation, steatohepatitis and hepatocarcinogenesis. Curr Mol Med. 2003;3:561-72.
120. Iwasa M, Sugimoto R, Eguchi A, et al. Effectiveness of 1-year pemafibrate treatment on steatotic liver disease: the influence of alcohol consumption. Eur J Gastroenterol Hepatol. 2024;36:793-801.
121. Cariou B, Zaïr Y, Staels B, Bruckert E. Effects of the new dual PPAR α/δ agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care. 2011;34:2008-14.
122. Koizumi A, Kaji K, Nishimura N, et al. Effects of elafibranor on liver fibrosis and gut barrier function in a mouse model of alcohol-associated liver disease. World J Gastroenterol. 2024;30:3428-46.
123. Ratziu V, Harrison SA, Francque S, et al; GOLDEN-505 Investigator Study Group. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150:1147-59.e5.
124. Pirola L. Elafibranor, a dual PPARα and PPARδ agonist, reduces alcohol-associated liver disease: lessons from a mouse model. World J Gastroenterol. 2025;31:99312.
125. Cheng CH, Hao WR, Cheng TH. Elafibranor: a promising therapeutic approach for liver fibrosis and gut barrier dysfunction in alcohol-associated liver disease. World J Gastroenterol. 2025;31:98783.
126. Armstrong MJ, Hull D, Guo K, et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J Hepatol. 2016;64:399-408.
127. Knop FK, Aroda VR, do Vale RD, et al; OASIS 1 Investigators. Oral semaglutide 50 mg taken once per day in adults with overweight or obesity (OASIS 1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2023;402:705-19.
128. Rubino D, Abrahamsson N, Davies M, et al; STEP 4 Investigators. Effect of continued weekly subcutaneous semaglutide vs placebo on weight loss maintenance in adults with overweight or obesity: the STEP 4 randomized clinical trial. JAMA. 2021;325:1414-25.
129. Newsome PN, Buchholtz K, Cusi K, et al; NN9931-4296 Investigators. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384:1113-24.
130. Armstrong MJ, Gaunt P, Aithal GP, et al; LEAN trial team. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387:679-90.
131. Loomba R, Abdelmalek MF, Armstrong MJ, et al; NN9931-4492 investigators. Semaglutide 2·4 mg once weekly in patients with non-alcoholic steatohepatitis-related cirrhosis: a randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol Hepatol. 2023;8:511-22.
132. Newsome PN, Sanyal AJ, Engebretsen KA, et al. Semaglutide 2.4 mg in participants with metabolic dysfunction-associated steatohepatitis: baseline characteristics and design of the phase 3 ESSENCE trial. Aliment Pharmacol Ther. 2024;60:1525-33.
133. Sanyal AJ, Newsome PN, Kliers I, et al; ESSENCE Study Group. Phase 3 trial of semaglutide in metabolic dysfunction-associated steatohepatitis. N Engl J Med. 2025;392:2089-99.
134. Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145:574-82.e1.
135. Patel K, Harrison SA, Elkhashab M, et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology. 2020;72:58-71.
136. Fiorucci S, Biagioli M, Baldoni M, et al. The identification of farnesoid X receptor modulators as treatment options for nonalcoholic fatty liver disease. Expert Opin Drug Discov. 2021;16:1193-208.
137. Alkhouri N, Herring R, Kabler H, et al. Safety and efficacy of combination therapy with semaglutide, cilofexor and firsocostat in patients with non-alcoholic steatohepatitis: a randomised, open-label phase II trial. J Hepatol. 2022;77:607-18.
138. Loomba R, Noureddin M, Kowdley KV, et al; for the ATLAS Investigators. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology. 2021;73:625-43.
139. Yakubu I, Spengler J, Taylor P, et al. Impact of glucagon-like peptide-1 receptor agonists on metabolic health in liver transplant recipients. Transplantation. 2025;Epub ahead of print.
140. Zhao Q, Wang X, Liu K, et al. Activation of farnesoid X receptor enhances the efficacy of normothermic machine perfusion in ameliorating liver ischemia-reperfusion injury. Am J Transplant. 2024;24:1610-22.
141. Bajaj JS, Gavis EA, Fagan A, et al. A randomized clinical trial of fecal microbiota transplant for alcohol use disorder. Hepatology. 2021;73:1688-700.
142. Pande A, Sharma S, Khillan V, et al. Fecal microbiota transplantation compared with prednisolone in severe alcoholic hepatitis patients: a randomized trial. Hepatol Int. 2023;17:249-61.
143. Derfoul A, Miyoshi AD, Freeman DE, Tuan RS. Glucosamine promotes chondrogenic phenotype in both chondrocytes and mesenchymal stem cells and inhibits MMP-13 expression and matrix degradation. Osteoarthritis Cartilage. 2007;15:646-55.
144. Li F, Zhang Z, Bai Y, et al. Glucosamine improves non-alcoholic fatty liver disease induced by high-fat and high-sugar diet through regulating intestinal barrier function, liver inflammation, and lipid metabolism. Molecules. 2023;28:6918.
145. Lai W, Zhou S, Bai Y, et al. Glucosamine attenuates alcohol-induced acute liver injury via inhibiting oxidative stress and inflammation. Curr Res Food Sci. 2024;8:100699.
146. Ryu T, Chang Y, Yoo JJ, et al. Glucosamine supplementation attenuates progression of metabolic dysfunction-associated steatotic liver disease and related comorbidities. Clin Nutr. 2025;47:119-28.
147. Dou JY, Liu SH, Guo J, et al. Dietary supplementation of pterostilbene, a major component in small berries, prevents alcohol-induced liver injury associated with lipid accumulation and inflammation. Food Funct. 2024;15:11206-19.
148. Benedé-Ubieto R, Estévez-Vázquez O, Guo F, et al. An experimental DUAL model of advanced liver damage. Hepatol Commun. 2021;5:1051-68.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].