



fig1
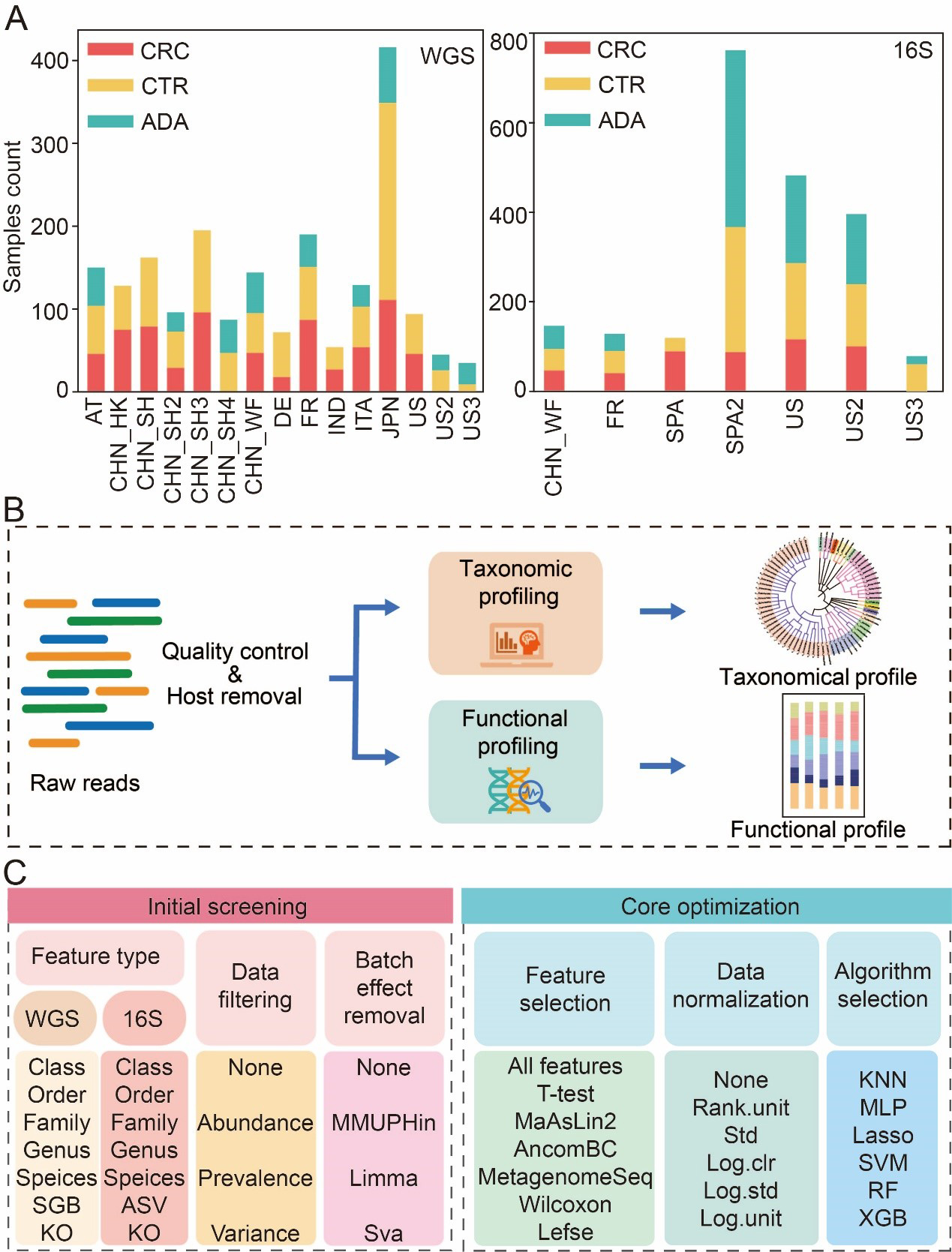
Figure 1. Data collection statistics and benchmark process. (A) Summary of sample aggregation and distribution; (B) A standardized bioinformatics pipeline for processing raw metagenomic data to achieve cross-cohort data harmonization; (C) Two-stage benchmarking workflow. The initial screening phase (left) evaluates various feature types, filtering methods, and batch correction strategies to identify optimal feature types and promising preprocessing strategies. Building on these findings, the core optimization phase (right) systematically evaluates combinatorial pipelines of core modules, including feature selection, data normalization, and classification algorithms. CRC: Colorectal cancer; CTR: control; ADA: adenoma; WGS: whole genome sequencing; 16S: 16S rRNA gene sequencing; AT: Austria; CHN: China; DE: Germany; ITA: Italy; JPN: Japan; IND: India; FR: France; SPA: Spain; US: United States; ASV: amplicon sequence variant; SGB: species-level genome bin; KO: Kyoto Encyclopedia of Genes and Genomes Orthology; MMUPHin: meta-analysis methods with uniform pipeline for heterogeneity integration; Limma: linear models for microarray data; SVA: surrogate variable analysis; MaAsLin2: multivariable association with linear models 2; AncomBC: analysis of composition of microbiomes with bias correction; MetagenomeSeq: MetagenomeSeq (Zero-inflated Gaussian model toolkit); Lefse: linear discriminant analysis effect size; Std: standardization; Log.clr: log centered log-ratio transformation; Log.std: log transformation with standardization; Log.unit: log transformation with unit scaling; Rank.unit: rank normalization with unit scaling; KNN: k-nearest neighbors; MLP: multilayer perceptron; Lasso: least absolute shrinkage and selection operator; SVM: support vector machine; RF: random forest; XGB: eXtreme Gradient Boosting.




