



Synergistic Fe dopant and Co vacancy in CoOOH for enhanced oxygen evolution reaction
 0
0 Abstract
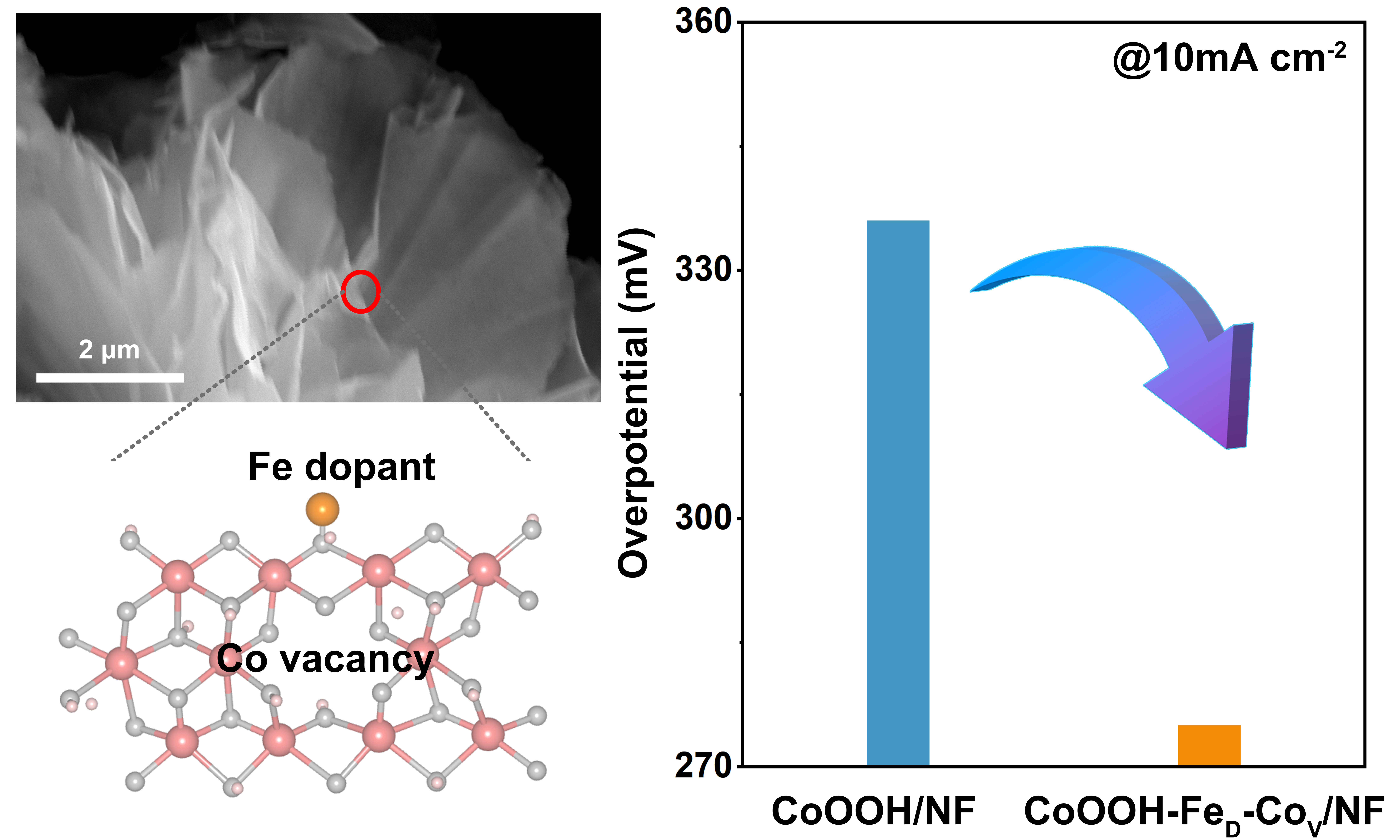
The integration of different defect engineering strategies holds significant promise for improving the performance of oxygen evolution reaction (OER). In this study, we synthesized a CoOOH-based ultrathin electrocatalyst with Fe dopants and Co vacancies. Experimental results reveal that the CoOOH-FeD-CoV demonstrates exceptional OER activity, with an overpotential of 357 mV at 100 mA cm-2. Structure characterization and theoretical calculations reveal the existence of dual defects significantly modifies the electronic configuration, optimizes the Gibbs free energy change of O* (ΔGO*), and reduces the difference between ΔGOH* and ΔGOOH*, resulting in a decreased OER overpotential. This study underscores the significance of exploring the synergistic effects of various defect structures to tailor the electronic properties for optimal intermediate binding energies, ultimately leading to enhanced OER performance.
Keywords
INTRODUCTION
In response to the global energy crisis, the emergence of a hydrogen economy marks the beginning of a new energy era. Water electrolysis is recognized as a promising method for producing high-purity hydrogen with zero carbon emissions[1]. However, the anode oxygen evolution reaction (OER) represents a significant bottleneck in enhancing the electrocatalytic water splitting efficiency, due to its slow kinetics and high reaction overpotential[2-4]. Consequently, developing high-efficiency and durable electrocatalysts for improving OER performance is crucial.
Although valuable metal catalysts, such as iridium (Ir)[5] and ruthenium (Ru)[6], demonstrate outstanding catalytic activity, their scarcity and high cost severely hinder their commercial viability. This has prompted extensive research aimed at designing cost-effective, high-performance alternatives. Transition metal-based (TM-based) materials, including cobalt (Co) and nickel (Ni), have emerged as promising candidates owing to their advantageous 3d orbital electronic structures, positioning them as optimal substitutes for Ir/Ru-based catalysts among reported non-noble metal electrocatalysts. Nevertheless, TM-based catalysts generally exhibit lower performance compared to their noble metal counterparts[7]. To enhance the effectiveness of TM-based catalysts, various strategies - including the introduction of dopants[8], the creation of vacancies[9], and interface engineering[10] - can induce charge redistribution and generate coordinatively unsaturated sites, significantly improving OER catalytic performance. Notably, integrating multiple strategies that induce different types of defects has shown promise as a synergistic approach. Although preliminary studies have reported on these synergies, further investigation at the atomic level is warranted.
In this study, we incorporated cobalt vacancies into ultrathin iron-doped CoOOH nanosheets (denoted as CoOOH-FeD) via a facile hydrothermal and wet-impregnation strategy. Benefiting from the ultrathin structure, the catalyst enhances the exposure of surface Fe dopant sites and the formation of Co vacancies, thereby facilitating a dual-defect modulation of the electronic structure. The resulting CoOOH-FeD-CoV electrocatalysts demonstrated superior electrochemical activity, achieving 100 mA cm-2 at 357 mV. Experimental and theoretical results demonstrate that Co vacancies and Fe dopants synergistically construct a dual-defect effect, which effectively regulates the local electronic structure of CoOOH, optimizes the Gibbs free energy change of ΔGO*, narrows the Gibbs free energy gap between O* and OOH*, and thus lowers the OER overpotential.
MATERIALS AND METHODS
Material synthesis
For the preparation of atomically thin CoOOH-FeD-CoV nanomaterials, the precursor Co(OH)2-FeD-WD was fabricated first. In detail, 200 mg of the surfactant P123 was fully dissolved in a solvent mixture consisting of 3.0 g ethanol, 2.0 g DI water and 12 mL EG. Afterwards, 125 mg Co(Ac)2·6H2O, 70 mg hexamethylenetetramine (HMTA), and 6 mg WCl6 (dissolved in 2.0 g ethanol in advance) were sequentially added to the above homogeneous solution. After vigorous stirring for 2 min, the resultant mixture was transferred into a 50 mL Teflon-lined autoclave with a nickel foam (NF) substrate placed inside, followed by a hydrothermal treatment at 160 °C for 2 h to yield Co(OH)2-WD/NF. Subsequently, the Co(OH)2-WD/NF was soaked in 10 mL of 2.5 mM Fe(NO3)3·9H2O solution and subjected to constant stirring for 2 h, thus forming the intermediate Co(OH)2-FeD-WD/NF. To obtain Co(OH)2-FeD-CoV/NF, the Co(OH)2-FeD-WD/NF was dispersed in 10 mL of 1.0 M NaOH solution, with a subsequent thermal treatment at 100 °C for 1 h. To ensure the complete elimination of tungsten W atoms, the alkaline etching process was repeated twice. Thereafter, the obtained Co(OH)2-FeD-CoV/NF was subjected to 8 cycles by cyclic voltammetry (CV) test in the electrolyte, with the potential window set at 0.30-0.70 V (vs. Hg/HgO). This step was designed to drive the phase transformation of Co(OH)2 into CoOOH, thus generating CoOOH-FeD-CoV/NF. For the synthesis of control samples including CoOOH/NF, CoOOH-FeD/NF and CoOOH-CoV/NF, the same synthetic protocol was adopted, with the exclusion of WCl6 and Fe(NO3)3·9H2O for the respective samples.
Characterizations
Morphological characteristics of the synthesized catalysts were visualized using a scanning electron microscope (SEM, JSM-IT500HR, JEOL, Japan) operated at an accelerating voltage of 15 kV, as well as a transmission electron microscope (TEM, Talos F200X, Thermo Fisher Scientific, USA). A TEM-coupled energy-dispersive X-ray spectrometer (EDS) was utilized to quantify the elemental constituents and their corresponding atomic proportions. The thickness of the nanosheet architectures was determined via high-resolution scanning transmission electron microscopy (HRSTEM) and atomic force microscopy (AFM, Bruker Dimension Icon). X-ray diffraction (XRD) patterns were recorded on a Rint-Dmax2550 diffractometer with a Ni-filtered Cu Kα radiation source (λ = 0.15418 nm). Chemical valence states of the catalyst surface elements were probed by X-ray photoelectron spectroscopy (XPS) on a Physical Electronics PHI 5600 multifunctional system, where a monochromatic Al X-ray source with a power of 350 W was adopted. All acquired XPS spectra were calibrated against the C1s peak centered at 284.6 eV. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were captured using a Cs-corrected STEM instrument (Spectra 300, Thermo Fisher Scientific, USA). The existence of Co vacancies was verified by electron paramagnetic resonance (EPR) spectroscopy on a Bruker EMX PLUS spectrometer, configured with a modulation amplitude of 0.3 mT, a modulation frequency of 100 kHz and an incident power of 3.170 mW. All reported results were averaged over five independent measurements. Soft X-ray absorption spectroscopy (SXAS) measurements were conducted at the National Synchrotron Radiation Laboratory (Hefei, China) to elucidate the electronic structures of the catalysts, with particular emphasis on acquiring O K-edge spectra. The beamline applied was equipped with a bending magnet and three gratings, spanning a photon energy range of 100-1000 eV. All samples were tested in the total electron yield mode under an ultrahigh vacuum environment of 5 × 10-10 mbar. The grating was set to a resolving power of E/ΔE = 1,000, and the photon flux was maintained at 1×1010 photons per second. The raw experimental data were subsequently processed following a standardized multi-step procedure.
Electrochemical tests
Electrochemical evaluations were performed via a three-electrode configuration (CHI760E). The working electrode was the in-situ catalysts, while a Pt foil served as the counter electrode, a Hg/HgO electrode acted as the reference and the electrolyte employed for all measurements was 1.0 M NaOH solution. To complement these studies, catalysts loaded on glassy carbon electrodes (GCE) were evaluated via the RDE technique to gain additional insights into their electrochemical behavior. To prepare GCE electrode, 5 mg samples or RuO2 powder was ultrasonically dispersed in 1 mL of a homogeneous solvent blend which was composed of 500 μL DI water, 450 μL isopropy alcohol, and 50 μL of 5 wt.% Nafion solution. 5 μL ink was pipetted onto the surface of a GCE with a 2 mm radius, followed by air-drying overnight, to obtain GCE electrodes with different catalyst loadings. The pre-catalysts were initially subjected to cyclic voltammetry (CV) within a potential window of 0.20-0.65 V (0.30-0.70 V for GCE) vs. Hg/HgO at 10 mV s-1 for 8 cycles, facilitating their transition into stable catalysts. Their OER performance was examined through a series of electrochemical techniques. Linear sweep voltammetry (LSV) was subsequently conducted within the potential range of 0.30-0.80 V (vs. Hg/HgO) at 10 mV s-1. The level of current resistance compensation (iR correction) was 80% to remove the ohmic resistance. Tafel plot was created by plotting the overpotential against the logarithmic of current density. The tafel slope was calculated by the equation[9]:
CV measurements were conducted within the non-faraday region at scan rates of 10, 20, 30, 40, 50, and 60 mV s-1 to calculate Cdl, used as a proxy for the electrochemical active surface area. EIS was conducted at 0.65 V (vs. Hg/HgO) from 0.1 Hz to 100 kHz to analyze charge transfer and resistive properties.
The long-term catalytic stability was evaluated through Chronoamperometry (CA), Chronopotentiometry (CP) and amperometric i-t measurements. CA measurements were conducted at sequential applied potentials of 0.65, 0.70, 0.75, and 0.80 V versus Hg/HgO over 10 h. CP tests were conducted at various current densities of 10, 30, 50, 80, and 100 mA cm-2 over 13 h. Moreover, the stability of the catalyst was further assessed using amperometric i-t measurements conducted continuously over a duration of 120 h.
The overpotentials (η) towards OER were determined using the equation[3]:
where ηHg/HgO represents the measured potential, and the pH value of electrolyte’s was stabilized at 13.6.
Computational methods
All density functional theory (DFT) calculations were conducted using the Vienna Ab Initio Simulation Package (VASP)[11,12]. The Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation (GGA) was used, with a plane-wave basis set cutoff at 500 eV[13]. In the DFT computations, the effective Hubbard U parameter (Ueff = U-J) was set to 3.4 eV for refining the treatment of electronic correlations in the Co d-orbitals[14]. For Brillouin zone sampling during structural optimization, a 5 × 5 × 1 Monkhorst-Pack k-point grid was adopted[15]. The projector augmented wave (PAW) approach was employed to depict ion-electron interactions[16]. Convergence criteria for structural optimization were defined such that the maximum force exerted on each atom was constrained below 0.02 eV Å-1, while the energy difference was limited to less than 1 × 10-5 eV. Additionally, the semiempirical DFT-D3 correction was incorporated following Grimme’s formalism[17].
The oxygen evolution reaction procedure mechanisms and associated changes in Gibbs free energy can be represented as:
The calculated overpotential is determined by:
ΔGi*(i = OH, O, or, OOH) represent Gibbs free energies of OER intermediates.
RESULTS AND DISCUSSION
Synthesis and morphology characterization
As presented in Figure 1A and detailed in the Methods section, the CoOOH-FeD-CoV/NF catalyst was synthesized through a series of methodical steps. The process began with the surfactant-assisted assembly of ultrathin Co(OH)2-WD/NF, which served as the precursor catalyst. This precursor was selected for its large specific surface area and abundant surface unsaturated bonds[18], which facilitate the incorporation of defect structures. In the subsequent step, Fe3+ ions were introduced into the structure via a wet-impregnation method, resulting in the formation of Co(OH)2-FeD-WD/NF. This method ensured a uniform distribution of the Fe dopant across the matrix surface[19]. Following this, the alkaline etching method was employed to completely remove W6+ ions, which possess numerous vacant outer orbitals that interact with OH- to form soluble WO42-[20]. This step yielded the Co(OH)2-FeD-CoV/NF catalyst. Finally, Co2+ ions were oxidized through an electrochemical anodic process, leading to the production of CoOOH-FeD-CoV/NF.
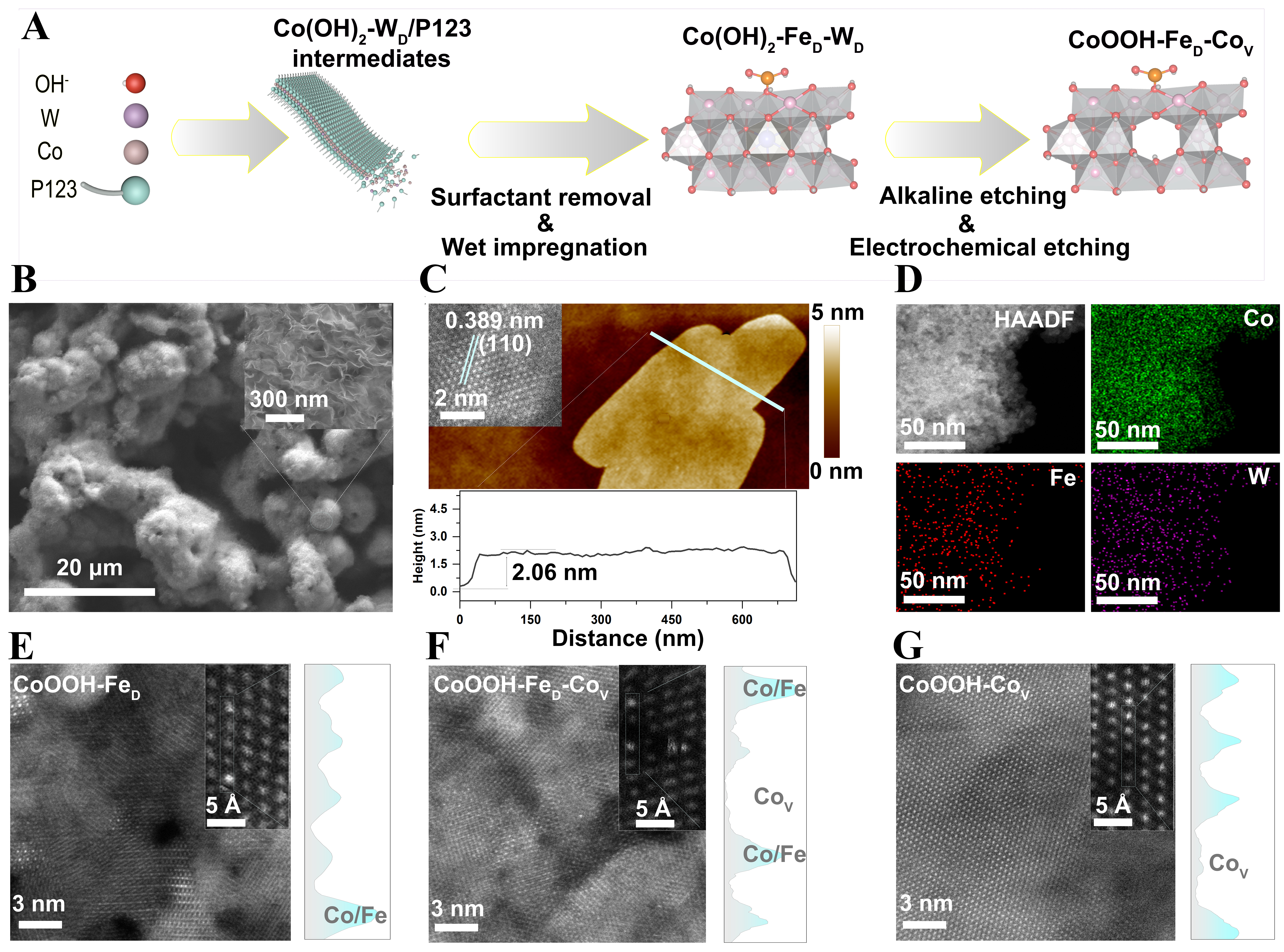
Figure 1. Synthesis and morphology characterization of materials. (A) The synthesis procedure of CoOOH-FeD-CoV; (B) SEM image of CoOOH-FeD-CoV with different magnifications; (C) AFM image of CoOOH-FeD-CoV, along the the height profile measured along the indicated straight line; (D) HAADF-STEM image of CoOOH-FeD-CoV and the corresponding mappings for different elements (Co, Fe and W); (E-G) High resolution of HAADF-STEM images and intensity profiles depicting the presence of Fe (CoOOH-FeD and CoOOH-FeD-CoV) as well as the complete removal of W dopants in CoOOH-CoV. SEM: Scanning electron microscope; AFM: atomic force microscopy; HAADF-STEM: high-angle annular dark-field scanning transmission electron microscopy.
Figure 1B demonstrates the uniform growth of CoOOH-FeD-CoV on the substrate. The morphology of the catalyst prior to phase conversion was visualized by SEM, as presented in Supplementary Figure 1. Additionally, the non-in-situ grown powder within the reaction vessel was analyzed. As shown in Supplementary Figure 2, the stretchability of these nanosheets was less favorable compared to that of the in-situ synthesized catalyst, which may negatively affect the active surface area. AFM imaging indicated that the nanosheets have an atomic thickness of approximately 2.06 nm [Figure 1C]. Furthermore, transmission electron microscopy (TEM) revealed a lattice spacing of around 3.89 Å, matching the (110) crystal plane of CoOOH (JCPDS 26-0480) as shown in the inset of Figure 1C. The X-ray diffraction (XRD) patterns displayed in Supplementary Figure 3 verify the achieved conversion of CoOOH after electrochemical activation process[21,22].
The STEM mode of high-angle annular dark field (HAADF-STEM) images and elemental mapping results are presented in Figure 1D, illustrating a uniform distribution of Fe dopants across the nanosheets. Energy dispersive spectroscopy (EDS) results for Co(OH)2-FeD, Co(OH)2-CoV, and Co(OH)2-FeD-CoV are included in Supplementary Figure 4, showing that the concentration of W in both CoOOH-CoV and CoOOH-FeD-CoV is nearly undetectable [Supplementary Table 1]. Considering the atomic-level thickness of the nanosheets, imaging of the Fe dopants and Co vacancies was accomplished via high-resolution HAADF-STEM mode. Figure 1E-G clearly depict the presence of Fe dopants as bright spots, attributed to their adsorption on the surface and the increased atomic density within the columns, and Co vacancies as dark spots, resulting from a lower number of atoms in the column within CoOOH-FeD, CoOOH-CoV, and CoOOH-FeD-CoV. The intensity profiles obtained from specific rectangular areas further confirm the existence of the two defects in the corresponding catalysts.
Electronic structure analysis
The catalysts’ electronic structure was further investigated by X-ray photoelectron spectroscopy (XPS). As exhibited in Figure 2A, the two fitted peaks located at 780.6 and 796.1 eV can be originated from Co2+ 2p. Due to the difference in electronegativity, the introduction of Fe3+ induces electron transfer from Fe to Co, thereby increasing the electron density around Co atoms and resulting in a negative shift of the Co 2p binding energy. In addition, the electron transfer from Co atoms adjacent to cobalt vacancies to neighboring oxygen atoms also contributes to the observed negative shift in binding energy, which is shown in Supplementary Figure 5[23,24]. After activation, the peaks assigned to Co2+ 2p remain, while new peaks appear at 779.8 eV and 794.7 eV, corresponding to Co3+ 2p signals originating from CoOOH[25] [Figure 2B]. Notably, as evidenced by Supplementary Table 2, CoOOH-FeD-CoV exhibits the highest area ratio of Co3+ 2p peaks among four catalysts, indicating a greater proportion of high-valence Co species. Figure 2C presents the O 1s fine spectrum, revealing three fitted peaks corresponding to the metal-oxygen (M-O) bond, the M-OH bonds, and oxygen-containing group (H2Oad)[26]. Notably, the incorporating of Fe shifts the M-OH and M-O of CoOOH-FeD and CoOOH-FeD-CoV to higher binding energies. Generally speaking, the electronegativity of Fe is higher than that of Co, and such stronger electron-withdrawing capability reduces the electron density of adjacent O atoms, which in turn increases their binding energy. On the other hand, the CoOOH-FeD-CoV and CoOOH-CoV catalysts exhibit varying degrees of positive electric shifts, attributable to the presence of cation vacancies. Moreover, the M-O bond peak for the CoOOH-FeD-CoV is relatively more intense compared to other catalysts, indicating enhanced electronic interactions between the metal and oxygen. Figure 2D displays the Fe 2p fine spectrum for the CoOOH-FeD and CoOOH-FeD-CoV catalyst. The observed multiplet splitting is characteristic of Fe3+[27] . For CoOOH-FeD-CoV, the Fe 2p peaks show negative shifts in the range of 0.1-0.6 eV with different magnitudes, indicating a mild decrease in the valence of Fe.
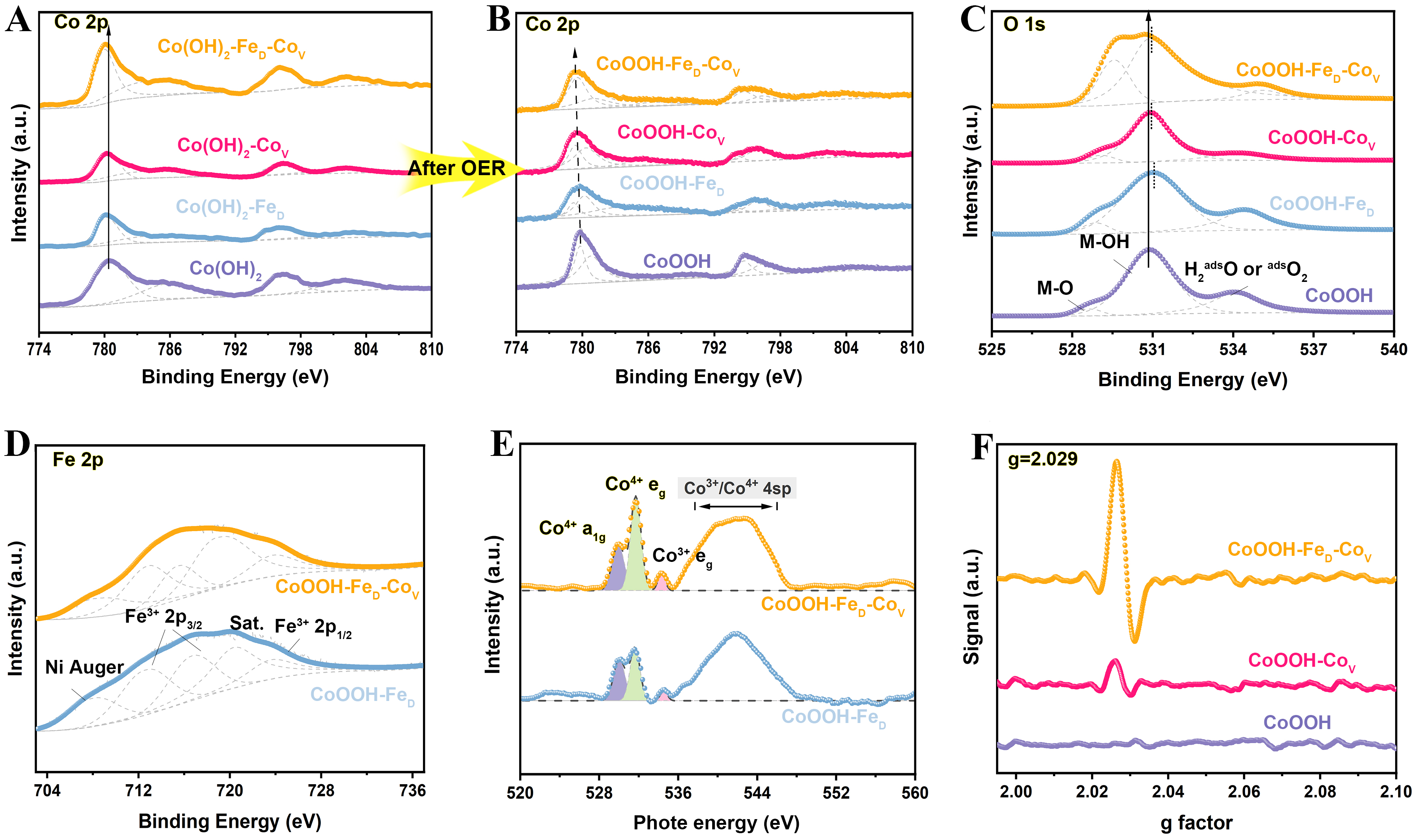
Figure 2. Structural characterization. (A and B) Co 2p XPS spectra of CoOOH-FeD-CoV and the compared cactalysts; (C) O 1s and (D) Fe 2p XPS spectra of CoOOH-FeD-CoV and the compared catalysts; (E) O K-edge NEXAFS spectra of CoOOH-FeD-CoV and CoOOH-FeD following the OER activation process; (F) EPR spectra of different catalysts after electrochemical testing, measured at 10 K. OER: Oxygen evolution reaction; XPS: X-ray photoelectron spectroscopy; NEXAFS: near-edge X-ray absorption fine structure; EPR: electron paramagnetic resonance.
For in-depth insight into the synergistic regulation of electronic structure by two defects, near-edge X-ray absorption fine structure (NEXAFS) spectra were collected. Figure 2E presents the O K-edge spectra of CoOOH-FeD and CoOOH-FeD-CoV. As observed, the characteristic peaks in the 529.5-535 eV range correlate with the electron transfer from O 1s to O 2p orbitals, which are hybridized with Co 3d orbitals. The two Gaussian-fitted peaks located at 529.8 and 531.7 eV are ascribed to Co4+, while the 534.2 eV peak are attributed to Co3+ eg[28]. It is further observed that the total intensity of the Co4+ peak increases progressively from CoOOH-FeD to CoOOH-FeD-CoV, suggesting that Fe doping and Co vacancies collaborate to promote the generation of Co4+ species. These high-valence Co species are well-researched as the active centers for the OER[29]. As shown in Figure 2F, EPR spectra indicate strong EPR signals for both CoOOH-CoV and CoOOH-FeD-CoV after the electrochemical tests. The g value of 2.029 signifies the presence of unpaired electrons in both catalysts. CoOOH-FeD-CoV showed a more intense signal at the g-value of 2.24 than CoOOH-CoV, implying that the Fe dopants in CoOOH-FeD-CoV results in an increased number of unpaired electrons[30].
The above analyses demonstrate that the synergistic effect between Fe doping and Co vacancies significantly modulates the electronic structure of the catalyst. Electron transfer from Fe to Co, together with the local charge redistribution induced by Co vacancies, enhances the metal-oxygen covalency and boosts the production of more high-valence Co species. These Co centers serve as the reaction sites during OER process, accelerating the generation and conversion of oxygen intermediates and thereby improving the catalyst’s charge transport and OER activity.
OER catalytic performance
The OER catalytic activity of the synthesized catalysts towards OER was assessed in 1.0 M NaOH using a three-electrode electrolyzer at room temperature. The LSV results shown in Figure 3A indicate that the CoOOH-FeD-CoV/NF catalyst exhibits an exceptionally minimal overpotential of 357 mV at 100 mA cm-2, surpassing CoOOH/NF (429 mV), CoOOH-FeD/NF (401 mV), and CoOOH-CoV/NF (435 mV). For comparative purposes, the OER performance of powder catalysts supported on glassy carbon electrodes (GCE) were also investigated under identical conditions [Supplementary Figure 6]. Upon normalizing the LSV curves [Supplementary Figure 6A] of the powder catalysts by electrochemically active surface area (ECSA) [Supplementary Figure 6B] and applying iR-correction, it was observed that although the powder catalysts displayed a similar trend [Supplementary Figure 6C], their overpotentials at the same current density are greatly higher than those of the self-supported catalysts [Supplementary Figure 6D]. This suggests that the Ni foam substrate is essential in boosting catalyst activity by promoting the growth of active materials, facilitating the charge transfer, and providing an inherent porous framework that ensures the swift release of gas bubbles.
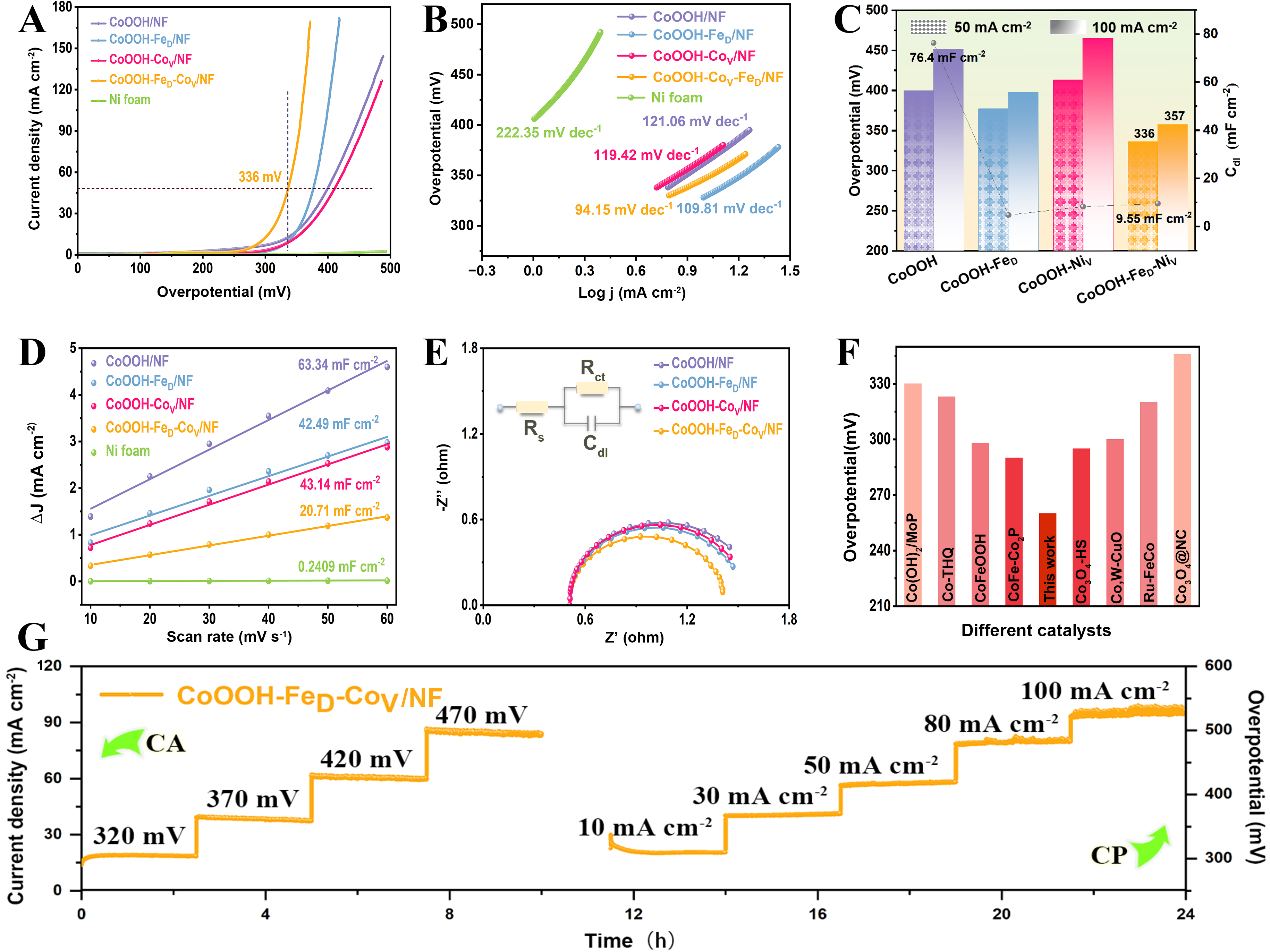
Figure 3. Evaluation of OER catalytic activity. (A) LSV curves with 80% iR correction; (B) Tafel plots generated from low-overpotential polarization data; (C) Overpotentials at j = 50 mA cm-2 and 100 mA cm-2 (left y-axis), along with the Cdl values (right y-axis); (D) Relationships between the differences of current density at 0.25 V vs. Hg/HgO and scan rates; (E) Nyquist plots of different catalysts and fitted equivalent circuit diagram; (F) Comparison of overpotentials at j = 10 mA cm-2 between CoOOH-FeD-CoV/NF and other Co-based catalysts; (G) Stability assessment through multi-potential test at overpotentials of 320, 370, 420, 470 mV for 10 h, alongside multi-current test at j = 10, 30, 50, 80, 100 mA cm-2 for 13 h. NF: Nickel foam; CA: Chronoamperometry; CP: chronopotentiometry; OER: oxygen evolution reaction; LSV: linear sweep voltammetry; Cdl: double-layer capacitance; iR: current resistance.
The Tafel slope of the CoOOH-FeD-CoV/NF catalyst was measured at 94.15 mV dec-1, lower than Ni foam (222.35 mV dec-1), CoOOH/NF (121.06 mV dec-1), CoOOH-FeD/NF (109.81 mV dec-1), and CoOOH-CoV/NF (119.42 mV dec-1) [Figure 3B]. This indicates that the CoOOH-FeD-CoV/NF catalyst displays faster OER reaction kinetics[31]. Figure 3C intuitively illustrates the double-layer capacitance (Cdl) values and overpotential of different catalysts at 50 mA cm-2 and 100 mA cm-2. The ECSA exhibits a direct proportionality to the Cdl, which was determined via CV measurements with varying scan rates, as illustrated in Figure 3D and Supplementary Figure 7. To intuitively demonstrate the intrinsic activity, we normalized the polarization curves with the double-layer capacitance [Supplementary Figure 8]. The CoOOH-FeD-CoV/NF catalyst exhibited the lowest Cdl value of 20.7 mF cm⁻2, suggesting that its high OER activity primarily originates from the inherent catalytic activity of individual active sites. To further the examination of the electrode reaction kinetics throughout the catalytic process, electrochemical impedance spectroscopy (EIS) tests were conducted in 1.0 M NaOH. The Nyquist plot shown in Figure 3E reveals that the CoOOH-FeD-CoV/NF catalyst exhibits the lowest charge transfer resistance (Rct), much lower than that of other catalysts[32]. These results indicate that the CoOOH-FeD-CoV/NF surface demonstrates enhanced charge transfer kinetics during the OER process. Finally, by comparing the performance with Co-based catalysts reported previously, it is found that our CoOOH-FeD-CoV/NF catalyst attains a well-balanced synergy between reaction kinetics and OER activity [Figure 3F][33-40].
The stability of a catalyst is a critical factor in assessing its performance in the OER. The CoOOH-FeD-CoV/NF catalyst was subjected to stability tests using various electrochemical methods. The results indicate that the catalyst maintains stability for at least 12 h during both chronoamperometry (CA) and chronopotentiometry (CP) tests [Figure 3G]. Furthermore, the current-time (i-t) curve demonstrates that the catalyst can operate stably for 150 hours at 50 mA cm-2 [Supplementary Figure 9]. The improved durability arises from the dual-defect structure created by double defects. To evaluate the catalyst’s active phase and surface morphology after catalysis, SEM and XRD analyses were conducted. The results indicate that the nanosheet morphology and phases remain unchanged [Supplementary Figures 10 and 11], suggesting high structural stability of the catalysts. Furthermore, HAADF-HRSTEM [Supplementary Figure 12] confirm the presence of Fe dopants and Co vacancies, further reinforcing the catalyst's stability during OER process. In conclusion, the CoOOH-FeD-CoV/NF catalyst exhibits superior electrocatalytic performance for OER, characterized by low overpotential, rapid reaction kinetics, high electrochemical activity, and excellent stability.
Mechanism of the synergistic effect
To further explore the synergistic effect of the duel-defect, we conducted DFT calculations to obtain the Gibbs free energies associated with the intermediates (*OH, *O, and *OOH) and d-band center of the reaction active sites. Initially, we established multiple theoretical models based on the four catalyst systems [Supplementary Figure 13] and the corresponding Gibbs free energy values for these intermediates are presented in Supplementary Table 3, which encompassed nearly all possible active sites. Figure 4A illustrates the crystal model exhibiting the most favorable OER performance, along with the OER reaction pathway that follows an adsorption-mediated evolution mechanism[41]. Taking the catalyst with the lowest overpotential as a reference, three additional catalyst models with well-defined defect types and positions were selected for further analysis. The d-band center theory elucidates that it is essential to maintain a moderate binding strength between the intermediates and the catalyst; excessively strong adsorption can cause catalyst poisoning, whereas overly weak adsorption hinders the reaction. As shown in Figure 4B, Fe doping causes a negative movement in the d-band center, weakening the ability to capture intermediates of the active sites. In contrast, introducing Co vacancies shifts the d-band center upward, resulting in overly strong adsorption of intermediates[42]. When Co vacancies are introduced into CoOOH-FeD, the dual-defect regulation leads to a moderate upward shift of the d-band center, thereby achieving an appropriate enhancement in intermediate adsorption strength.
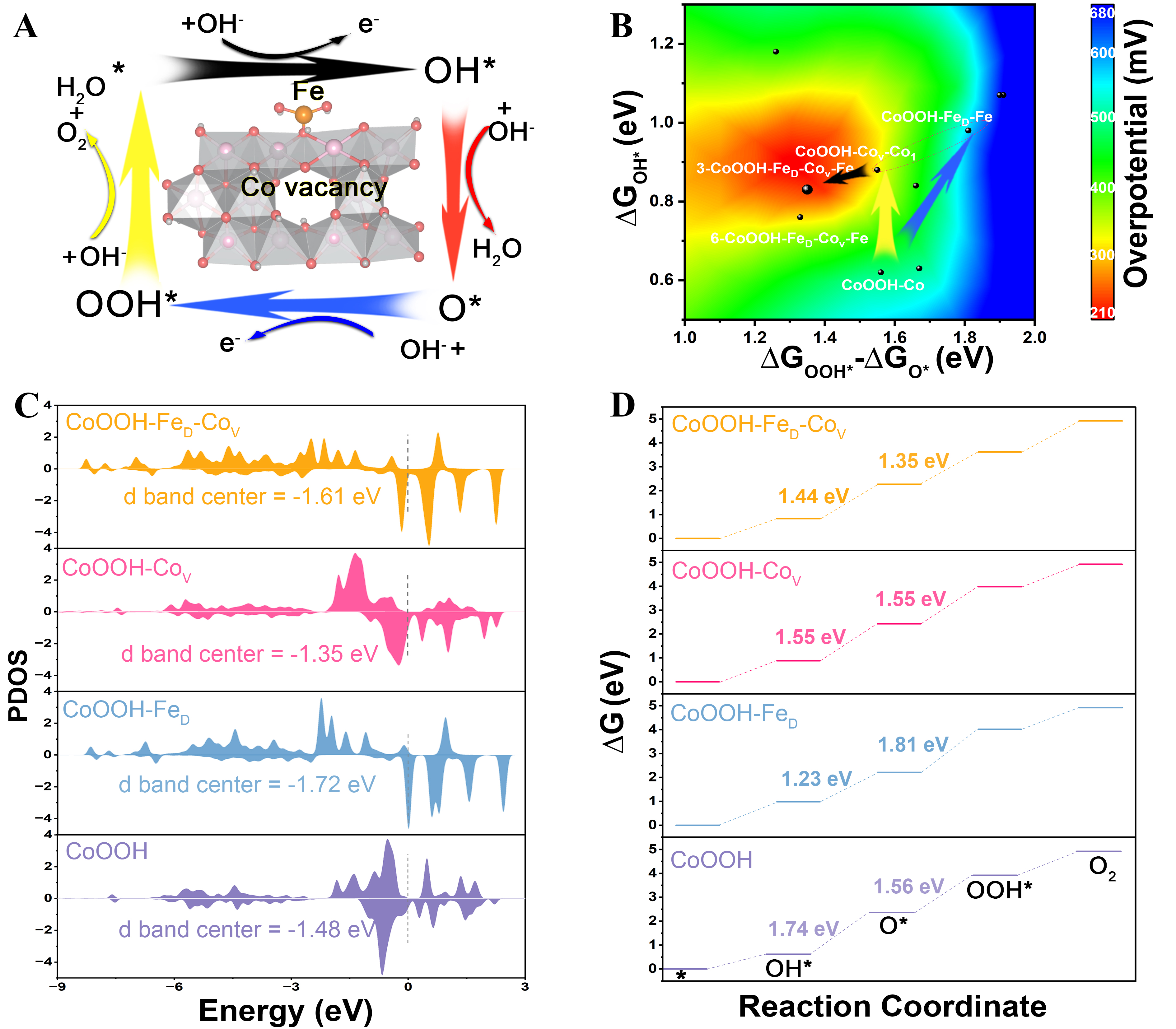
Figure 4. DFT studies of catalytic activity among different surface metal sites. (A) Atomic arrangement of 3-CoOOH-FeD-CoV-Fe and the corresponding OER reaction pathway; (B) The Gibbs free energies for OH*, O*, and OOH* at CoOOH-Co sites, CoOOH-FeD-Fe sites, 2-CoOOH-CoV-Co1 sites, 3-CoOOH-FeD-CoV-Fe sites; (C) Calculated partial density of states (PDOS) for CoOOH-Co sites, CoOOH-FeD-Fe sites, 2-CoOOH-CoV-Co1 sites, 3-CoOOH-FeD-CoV-Fe sites; (D) Contour plot of theoretical overpotential versus ΔGOOH*-ΔGO* and ΔGOH*, highlighting the near-ideal model achieved by Fe doping and Co vacancy. DFT: Density functional theory; OER: oxygen evolution reaction.
Figure 4C shows a 3D contour plot against ΔGOOH*, ΔGO*, and ΔGOH*, intuitively revealing the relationship between intermediate adsorption barrier and water oxidation activity. The overpotential gradually decreases from blue to green, yellow, and finally red. CoOOH-Co, exhibiting a relatively high overpotential of 510 mV, is located in the green area. The introduction of the duel-defects shifts the system toward the green-blue boundary and the yellow-red region, respectively. Notably, the synergistic effect between Fe doping and Co vacancies shifts the Fe sites in the 3-CoOOH-FeD-CoV-Fe catalyst to the central of the red area, achieving the minimal theoretical overpotential and optimal electrocalalytic performance. This plot provides valuable insight for designing high-performance electrocatalysts rationally by fine-tuning intermediate energetics through defect modulation. Figure 4D shows the free energy graphs of oxygen-containing intermediates. The OER kinetics are dictated by the elementary step with the highest Gibbs free energy change (ΔG1, ΔG2, ΔG3 and ΔG4), recognized as the reaction-determining step (RDS)[43]. Both Fe doping and Co vacancies can individually lower the energy barrier for the deprotonation of intermediates (OH* → O*), yet they have little effect on optimizing the formation O-O bond (O* → OOH*). Only when these two defects coexist can all reaction steps be effectively regulated simultaneously, thereby minimizing the reaction barrier of the RDS and ultimately reducing the overpotential.
CONCLUSIONS
In conclusion, the CoOOH-FeD-CoV/NF catalyst exhibits outstanding electrocatalytic performance for the OER, attributed to its dual-defect structure characterized by Fe doping and Co vacancies. The self-assembly synthesis method facilitates the uniform incorporation of Fe dopants and Co vacancies, which in turn enhances the catalyst’s activity. Experimental results reveal a low overpotential, rapid reaction kinetics, and excellent stability for the CoOOH-FeD-CoV/NF catalyst. Furthermore, DFT calculations corroborate the experimental findings, indicating that the synergistic effects of the dual defects significantly improve catalytic performance by modulating the adsorption energies of oxygen-containing intermediates. This work underscores the potential of defect engineering in the development of high-performance electrocatalysts applied in energy conversion, paving the way for further optimization and the expansion of their applications in renewable energy technologies.
DECLARATIONS
Authors’ contributions
Conceptualization, Investigation, Methodology, Data curation, Writing-Original draft: Dou X
Conceptualization, Investigation, Software: Xu, Z.; Cui, M.
Supervision, Project administration, Writing-review & editing: Yuan. D.; Dou, Y.; Dou, S.
Funding acquisition: Yuan. D.; Dou, S.
Validation, Software: Song, K.; Hu, R.; Liu, H.
Availability of data and materials
The raw data supporting the findings of this study are available within this Article and its Supplementary Materials. Further data is available from the corresponding authors upon reasonable request.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
Financial support for this work was provided by the National Oversea Postdoctoral Talent Attraction Program, the Youth Fund of the National Natural Science Foundation of China (52402288), the Young Elite Scientists Sponsorship Program of the China Association for Science and Technology (YESS20230183), the Research Fund for International Senior Scientists of the National Natural Science Foundation of China (52350710795), and the Yangfan Special Program of the Shanghai Star Project (24YF2729700).
Conflict of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Huang, J.; Clark, A. H.; Hales, N.; et al. Oxidation of interfacial cobalt controls the pH dependence of the oxygen evolution reaction. Nature. Chem. 2025, 17, 856-64.
2. He, C.; Yang, L.; Wang, J.; et al. Research progress on electronic and active site engineering of cobalt‐based electrocatalysts for oxygen evolution reaction. Carbon. Energy. 2024, 6, e573.
3. Quan, Q.; Zhang, Y.; Li, H.; et al. Atomic-scale self-rearrangement of hetero-metastable phases into high-density single-atom catalysts for the oxygen evolution reaction. Nat. Commun. 2025, 16, 2908.
4. Deng, Q.; Li, H.; Pei, K.; et al. Strategic design for high-efficiency oxygen evolution reaction (OER) catalysts by triggering lattice oxygen oxidation in cobalt spinel oxides. ACS. Nano. 2024, 18, 33718-28.
5. Lin, H. Y.; Yang, Q. Q.; Lin, M. Y.; et al. Enriched oxygen coverage localized within iR atomic grids for enhanced oxygen evolution electrocatalysis. Adv. Mater. 2024, 36, e2408045.
6. Li, J.; Zhu, Y.; Li, C.; et al. Boosting and stabilizing oxygen evolution reaction through Ru single atoms anchored amorphous NiMoOx electrocatalyst. Nat. Commun. 2025, 16, 8827.
7. Magnier, L.; Cossard, G.; Martin, V.; et al. Fe-Ni-based alloys as highly active and low-cost oxygen evolution reaction catalyst in alkaline media. Nat. Mater. 2024, 23, 252-61.
8. Jia, Z.; Yuan, Y.; Zhang, Y.; et al. Optimizing 3d spin polarization of CoOOH by in situ Mo doping for efficient oxygen evolution reaction. Carbon. Energy. 2023, 6, e418.
9. Yan, L.; Dong, G.; Huang, X.; Zhang, Y.; Bi, Y. Unraveling oxygen vacancy changes of WO3 photoanodes for promoting oxygen evolution reaction. Appl. Catal. B. Environ. 2024, 345, 123682.
10. Xiao, Y.; Fu, J.; Pihosh, Y.; et al. Interface engineering for photoelectrochemical oxygen evolution reaction. Chem. Soc. Rev. 2025, 54, 1268-317.
11. Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15-50.
12. Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. Condens. Matter. 1996, 54, 11169-86.
13. Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865-8.
14. Wan, X.; Liu, X.; Li, Y.; et al. Fe-N-C electrocatalyst with dense active sites and efficient mass transport for high-performance proton exchange membrane fuel cells. Nat. Catal. 2019, 2, 259-68.
15. Monkhorst, H. J.; Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B. 1976, 13, 5188-92.
16. Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B. Condens. Matter. 1994, 50, 17953-79.
17. Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104.
18. Liu, J.; Ji, Y.; Nai, J.; et al. Ultrathin amorphous cobalt-vanadium hydr(oxy)oxide catalysts for the oxygen evolution reaction. Energy. Environ. Sci. 2018, 11, 1736-41.
19. Uchida, H.; Suzuki, H.; Watanabe, M. High-performance electrode for medium-temperature solid oxide fuel cells: effects of composition and microstructures on performance of ceria-based anodes. J. Electrochem. Soc. 2019, 145, 615-20.
20. Armstrong, R.; Edmondson, K.; Firman, R. The anodic dissolution of tungsten in alkaline solution. J. Electroanal. Chem. Interfacial. Electrochem. 1972, 40, 19-28.
21. Jung, H.; Ma, A.; Abbas, S. A.; et al. A new synthetic approach to cobalt oxides: Designed phase transformation for electrochemical water splitting. Chem. Eng. J. 2021, 415, 127958.
22. Guo, Q.; Li, Y.; Xu, Z.; Liu, R. CeO2‐Accelerated Surface Reconstruction of CoSe2 Nanoneedle Forms Active CeO2@CoOOH Interface to Boost Oxygen Evolution Reaction for Water Splitting (Adv. Energy Mater. 4/2025). Adv. Energy. Mater. 2025, 15, 2570021.
23. Dou, Y.; Yuan, D.; Yu, L.; et al. Interpolation between W dopant and Co vacancy in CoOOH for enhanced oxygen evolution catalysis. Adv. Mater. 2022, 34, e2104667.
24. Zhang, J.; Yang, D.; Yang, Z.; Wang, L. Utilizing cationic vacancy defects to switch oxygen evolution mechanisms on atomically dispersed Ru for enhanced acidic catalytic performance. Appl. Catal. B. Environ. 2025, 364, 124845.
25. Wang, B.; Chen, X.; He, Y.; et al. Fe2O3/P-doped CoMoO4 electrocatalyst delivers efficient overall water splitting in alkaline media. Appl. Catal. B. Environ. 2024, 346, 123741.
26. Fan, J.; Xia, J.; Wang, H.; et al. Unveiling the role of single atomic ruthenium decorated cactus‐like bimetallic phosphides for alkaline water electrolysis. Adv. Energy. Mater. 2025, 15, 2501995.
27. Zhao, P.; Fu, S.; Luo, Y.; Peng, C.; Cheng, L.; Jiao, Z. Deciphering the space charge effect of the CoNiLDH/FeOOH n-n heterojunction for efficient electrocatalytic oxygen evolution. Small 2023, 19, e2305241.
28. Mizokawa, T.; Wakisaka, Y.; Sudayama, T.; et al. Role of oxygen holes in LixCoO2 revealed by soft X-ray spectroscopy. Phys. Rev. Lett. 2013, 111, 056404.
29. Kang, W.; Wei, R.; Yin, H.; et al. Unraveling sequential oxidation kinetics and determining roles of multi-cobalt active sites on Co3O4 catalyst for water oxidation. J. Am. Chem. Soc. 2023, 145, 3470-7.
30. Golden, E. M.; Giles, N. C.; Maniego, E.; et al. Identification of native defects (vacancies and antisites) in CdSiP2 crystals. J. Appl. Phys. 2015, 118, 185702.
31. Xia, Y.; Chi, J.; Tang, J.; et al. Research progress of anionic vacancies in electrocatalysts for oxygen evolution reaction. Chin. J. Catal. 2024, 66, 110-38.
32. Ahmed, M. G.; Tay, Y. F.; Chi, X.; et al. Cation migration-induced lattice oxygen oxidation in spinel oxide for superior oxygen evolution reaction. Angew. Chem. Int. Ed. Engl. 2025, 64, e202416757.
33. Shen, Y.; Zhu, Y.; Wang, X.; et al. Activating lattice oxygen redox of Co3O4 through rare-earth-excited electron self-donation for improving electrocatalytic oxygen evolution. Adv. Funct. Mater. 2025, 36, e25303.
34. Kim, N. I.; Kim, Y.; Lee, J.; et al. Architecting layered CoFeOOH for the oxygen evolution reaction: engineering structure for an anion exchange membrane water electrolyzer. ACS. Nano. 2025, 19, 41704-17.
35. Chen, Y.; Mao, J.; Zhou, H.; et al. Coordination shell dependent activity of CuCo diatomic catalysts for oxygen reduction, oxygen evolution, and hydrogen evolution reaction. Adv. Funct. Mater. 2023, 34, 2311664.
36. Wang, Y.; Bai, X.; Huang, J.; et al. Metal-oxygen bonding-induced structural transition regulation in Co-THQ for high-performance OER. ACS. Catal. 2025, 15, 17040-53.
37. Kim, S.; Lee, J.; Kim, Y. B.; et al. Enhanced alkaline water electrolysis by the rational decoration of RuOx with the in situ-grown CoFe nanolayer. ACS. Nano. 2025, 19, 10026-37.
38. Yu, X.; Wang, X.; He, P.; et al. CoFe alloy realizing enhanced Fe-bridged electron superhighways in Mott-Schottky heterojunctions for efficient water and urea electrolysis. J. Mater. Sci. Technol. 2026, 260, 298-308.
39. Nickel, C.; Troglauer, D. L.; Dallos, Z.; et al. Self-optimizing cobalt tungsten oxide electrocatalysts toward enhanced oxygen evolution in alkaline media. Angew. Chem. Int. Ed. Engl. 2025, 64, e202424074.
40. Zhang, K.; Li, N.; Weng, Y.; et al. Navigating covalency of cobalt oxides for enhanced oxygen evolution. Adv. Funct. Mater. 2025, 35, 2507212.
41. Gao, T.; Jiao, D.; Wang, L.; et al. Switchable acidic oxygen evolution mechanisms on atomic skin of ruthenium metallene oxides. J. Am. Chem. Soc. 2025, 147, 4159-66.
42. Shi, B. C.; Jin, M.; Zou, Y.; et al. Cathodic electrodeposition activation of NiFe‐based metal-organic frameworks for enhanced oxygen evolution reaction. Rare. Metals. 2025, 44, 10144-54.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].