


Unveiling the new horizons of atherosclerosis: the crossroads of inflammation, pyroptosis, and immunity
 0
0 Abstract
Atherosclerosis (AS) is a chronic cardiovascular disease. Traditional lipid-lowering treatments have not fully reduced the risk of disease onset in high-risk patients. This review provides a systematic overview of immune microenvironment remodeling and the mechanisms of pyroptosis during the onset and progression of atherosclerosis. Studies have shown that AS is not only related to abnormal lipid metabolism, but also closely associated with inflammatory responses, activation of immune cells, and programmed cell death. Immune cells, including macrophages, T cells, and B cells, play key roles in plaque formation, progression, and rupture. At the same time, NLR family pyrin domain containing 3 (NLRP3) inflammasome-mediated pyroptosis exacerbates plaque instability by promoting the release of inflammatory mediators, including Interleukin-1β (IL-1β) and interleukin-18 (IL-18), We further summarize emerging therapeutic strategies targeting immune regulation and pyroptosis pathways, including NLRP3 inhibitors, cytokine-targeted agents, and nano-drug delivery systems, thereby offering new perspectives for the precise treatment of AS.
Keywords
INTRODUCTION
Atherosclerosis (AS) is a chronic cardiovascular disease characterized by unstable plaque rupture, platelet aggregation, and vessel stenosis or occlusion secondary to thrombosis. The main clinical manifestations of this condition include ischemic heart disease (IHD), peripheral arterial disease (PAD), and ischemic stroke. According to World Health Organization (WHO) estimates, cardiovascular disease is projected to cause over 23.3 million deaths annually by 2030[1]. Despite advances in lipid-lowering therapies, including statins and PCSK9 inhibitors, a significant residual cardiovascular risk remains in high-risk populations. This gap highlights the need for deeper insights beyond dyslipidemia, particularly regarding the interplay among vascular injury, immune responses, and cell death pathways in AS pathogenesis.
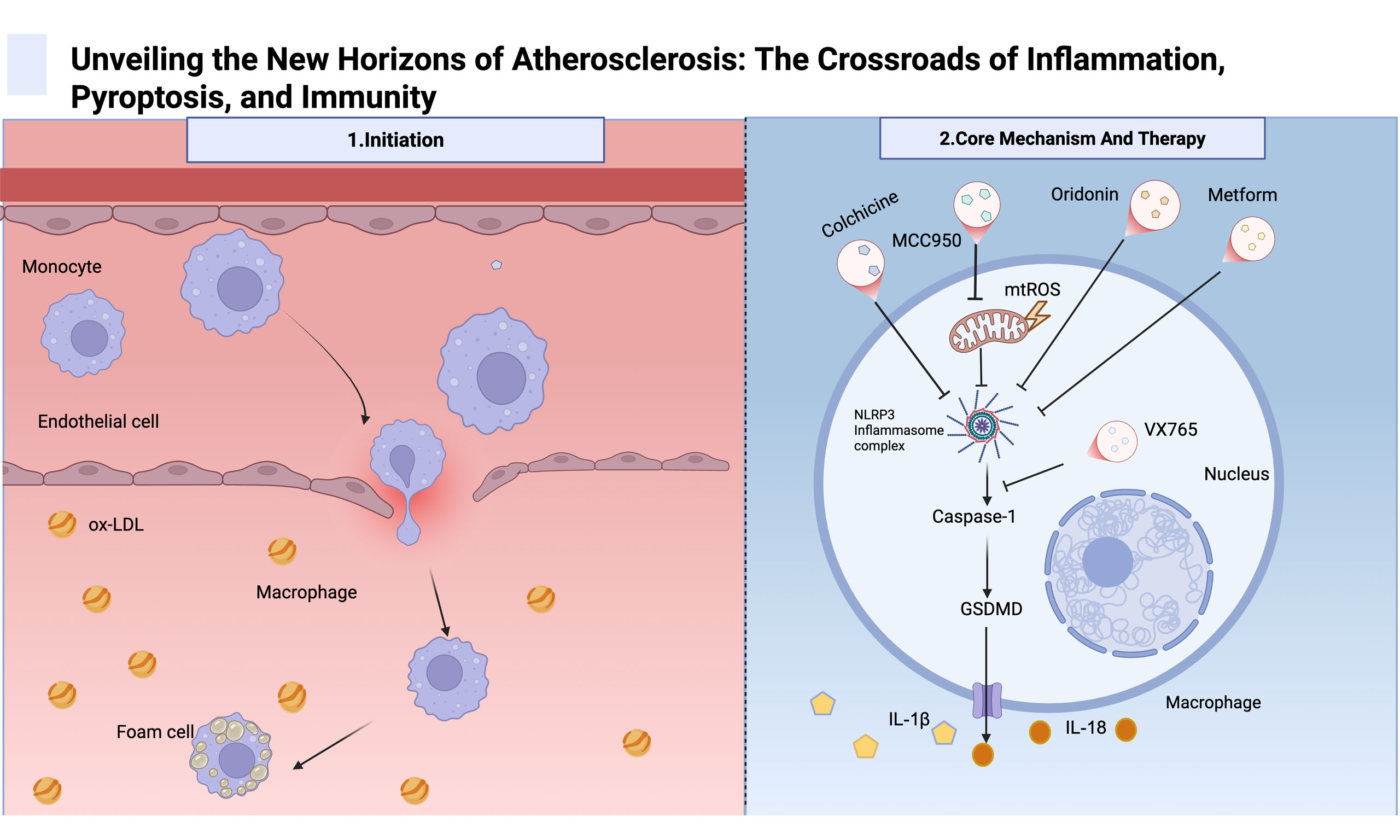
Under physiological conditions, normal endothelial cells generate nitric oxide (NO) through endothelial nitric oxide synthase (eNOS). NO can rapidly diffuse across the cell membrane and act on surrounding smooth muscle cells and platelets, maintaining vascular dilation and an anti-inflammatory state. Physiological blood flow shear stress promotes eNOS activity and NO production, which help maintain endothelial function, whereas abnormal shear stress, such as disorders or low shear stress, can lead to eNOS dysfunction, reduced NO production, and increased endothelial damage and inflammation. Under pathological conditions, eNOS uncoupling reduces NO production and increases reactive oxygen species generation, thereby exacerbating oxidative stress and vascular damage. These changes are among the key mechanisms underlying atherosclerosis[2]. These changes promote the recruitment and infiltration of monocytes, which differentiate into macrophages. The scavenger receptor CD36 on macrophages has a high affinity for oxidized low-density lipoprotein (ox-LDL), thereby promoting the formation of lipid-rich foam cells[3] [Figure 1]. Foam cells accumulate within the intima, driving chronic inflammation and plaque formation. As plaques progress, vascular smooth muscle cells (VSMCs) proliferate and produce extracellular matrix components, forming a fibrous cap over the necrotic core.
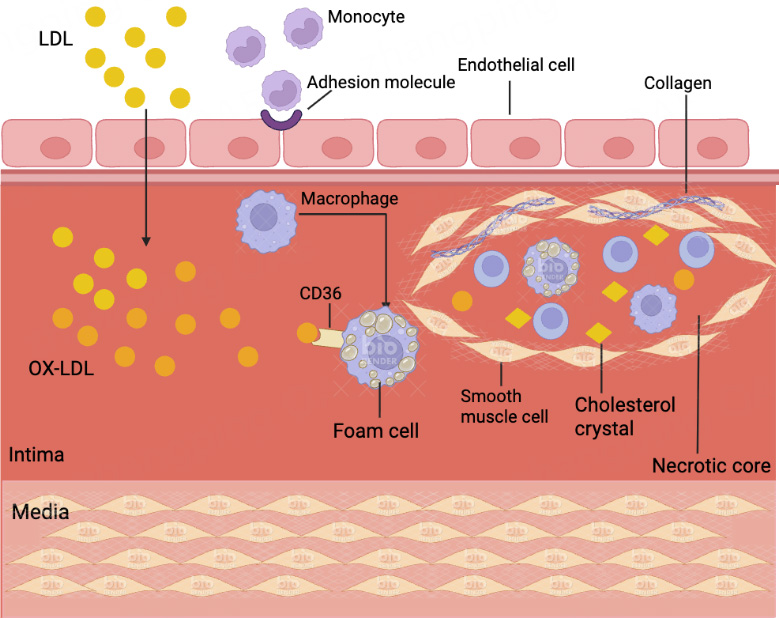
Figure 1. Schematic Diagram of the Initiation Process of Atherosclerosis. When endothelial cells are activated by cardiovascular risk factor-associated stimuli, they express leukocyte adhesion molecules and recruit monocytes. Monocytes migrate and adhere to the endothelium of blood vessels, and subsequently differentiate into macrophages. In addition, endothelial cell damage increases the permeability of arterial walls to lipoproteins and promotes the accumulation and oxidation of LDL. The scavenger receptor CD36 on inflammatory macrophages has a stronger affinity for ox-LDL, leading to the accumulation of lipids in macrophages and foam cell formation. Foam cells and immune cells accumulate within the necrotic core, and vascular smooth muscle cells form fibrous caps mainly composed of collagen. As the disease progresses, it gradually develops into a mature atherosclerotic plaque. The figure created with BioRender.com. LDL: Low-density lipoprotein; oxLDL: oxidized low-density lipoprotein.
The dynamic progression of AS ultimately leads to plaque instability, which is characterized by thinning of the fibrous cap and expansion of the necrotic core. Plaque rupture or erosion subsequently triggers acute thrombotic events, causing luminal occlusion and clinical ischemia [Figure 2]. Understanding the cellular and molecular mechanisms underlying these events is critical for developing novel therapeutic strategies.
Figure 2. Diagram illustrating the progression of atherosclerosis and plaque rupture. (A) Normal artery: The arterial wall exhibits an intact endothelium and well-defined intima, media, and adventitia layers. There is no lipid accumulation or inflammatory cell infiltration, and vascular smooth muscle cells (VSMCs) maintain a contractile phenotype ensuring vessel integrity and function; (B) Early atherosclerosis: Endothelial injury and dysfunction lead to increased intimal permeability, allowing the deposition and oxidation of low-density lipoprotein particles in the subendothelial space. This results in recruitment of monocytes and their differentiation into macrophages, which take up oxidized LDL to form foam cells. The accumulation of foam cells and inflammatory cells initiates the formation of fatty streaks and early plaques within the intimal layer, thereby causing luminal narrowing. Note that plaques are localized within the intima/subendothelial space and do not protrude freely into the vessel lumen. VSMCs proliferate and migrate from the media to the intima, promoting fibrous cap formation; (C) Advanced plaque and rupture: The plaque enlarges, characterized by a large necrotic lipid core surrounded by a fibrous cap composed mainly of collagen and VSMCs. Inflammatory macrophages secrete matrix metalloproteinases (MMPs), which degrade extracellular matrix components within the fibrous cap, causing thinning and weakening of the cap. Mechanical forces and inflammatory stimuli promote plaque rupture or erosion, exposing thrombogenic material to circulating blood. This triggers platelet aggregation and thrombus formation, which can acutely occlude the arterial lumen, leading to ischemic events. The figure illustrates the process of fibrous cap thinning and subsequent fibrous cap rupture with thrombus formation. LDL: Low-density lipoprotein.
This review highlights the remodeling of the immune microenvironment in AS and the central role of inflammatory programmed cell death, particularly pyroptosis, in the initiation and development of the disease. We systematically summarized the interactions between immune cell subsets and the pyroptosis pathway, as well as the molecular mechanisms, and reviewed emerging treatment methods targeting relevant pathways. We also revealed the current controversies and gaps in the field, aiming to provide a comprehensive perspective and theoretical basis for precision immunotherapy and clinical management of AS. Although lipid-lowering therapy remains the mainstay of clinical practice, many high-risk patients continue to face significant residual risk, suggesting the limitations of traditional approaches. Accordingly, we emphasize the crucial roles of the immune microenvironment and inflammation-driven cellular injury in AS, together with immune regulation and programmed cell death, and comprehensively review preclinical studies and the clinical application of multiple cytokines and signaling pathways, offering new insights into the identification of therapeutic targets.
ATHEROSCLEROSIS IS AN INFLAMMATORY DISEASE
In 1858, Rudolf Virchow, a founder of cellular pathology, first recognized that atherosclerosis is an inflammatory disease of the arterial wall and embraced the concept that it is a chronic inflammatory disease of the arterial intima. Emerging evidence underscores the pivotal involvement of the immune system in both the initiation and progression of atherosclerosis. Moreover, the development of AS is initially triggered by excessive ox-LDL and damaged endothelial cells (ECs)[4]. Dynamic shifts within the lesional microenvironment coincide with the recruitment and infiltration of diverse immune populations. Cellular components of both innate and adaptive immunity, including monocytes, macrophages, T lymphocytes, and B cells, orchestrate the balance that influences plaque progression, maturation, and susceptibility to rupture. Advances in high-resolution analytical techniques, particularly single-cell transcriptomics and proteomic profiling, have recently delineated a strong correlation between the phenotypic and functional states of plaque-resident immune cells and indices of plaque instability. Notably, various stimuli driving AS inflammation and pyroptosis, such as ox-LDL, cholesterol crystals, and hemodynamic stress, are essentially cellular stressors. They can establish a permissive molecular milieu conducive to NLR family pyrin domain containing 3 (NLRP3) inflammasome formation, the subsequent synthesis of proinflammatory cytokines and inflammatory cell death (pyroptosis) by activating conserved pathways such as the integrated stress response, thereby remodeling cellular metabolism and gene expression programs.
Genetic differences among individuals in stress-sensing and response genes, such as general control nonderepressible 2 (GCN2) and C/EBP homologous protein (CHOP), may be important intrinsic factors contributing to the heterogeneity of AS susceptibility and pathological manifestations[5]. Variants in these genes can influence the cellular stress response, leading to variations in inflammation, apoptosis, and cellular survival during atherosclerotic plaque development. For instance, GCN2 is known to play a critical role in detecting amino acid deprivation, activating pathways that regulate inflammation and cell survival[6,7]. In AS, CHOP is closely related to Interleukin-1 alpha (IL-1α) and the inflammatory response of macrophages. IL-1α can enhance CHOP expression, and increased expression of CHOP promotes the proliferation and migration of VSMCs, thereby accelerating AS progression[8,9].
Monocytes and macrophages represent the predominant inflammatory cell populations within atherosclerotic plaques, where they localize centrally and play a critical role in lesion pathology[10]. Therefore, elucidating the cellular composition and spatial organization of the systemic immune microenvironment in affected individuals is of paramount importance. In this section, we focus on the changes in these cells during the disease, particularly how these cells contribute to atherosclerosis through inflammation-driven processes. In the following, we focus on immune cells to introduce the role of immunity in atherosclerosis.
Monocyte and macrophage: the center of AS
Ross Gerrity confirmed the role of monocytes in atherosclerotic lesions and provided relevant electron microscopic evidence in 1981. Based on this theory, scientists have found that monocytes, driven by other differentiation factors, may differentiate into macrophages in the subendothelial space. Several studies have shown that foam cells are hallmarks of AS. Thus, the scientists speculate that removing excess cholesterol from macrophage-derived foam cells could be one strategy for treating AS in the future. Elucidating the multifaceted functions of monocytes holds promise for uncovering novel therapeutic strategies directed against atheromatous plaques. Investigations into the specific contributions of monocytes and macrophages during atherosclerotic pathogenesis, exemplified by the work of Victoria Stoneman and colleagues, have established the indispensable role these immune cells play in disease development. The following is a detailed description of the exact influence of macrophages on atherosclerotic lesions.
Macrophages contribute to plaque necrosis
The causes of atherosclerotic plaque rupture remain a subject of debate. However, most scientists hold the idea that the high-risk plaques are characterized by two factors: a thinned fibrous cap and a necrotic core, and macrophages serve as critical mediators during this pathological period. Macrophages can transform into subtypes, such as M1 macrophages, M2 macrophages, M (Hb), Mox, and M4. M1 macrophages, induced by lipopolysaccharide (LPS) and interferon-gamma (IFN-γ), secrete pro-inflammatory cytokines including IL-1, IL-6, and tumor necrosis factor (TNF). In contrast, M2 macrophages, which are activated by IL-4, can play an important role in counteracting the inflammatory environment by releasing anti-inflammatory cytokines, such as IL-10 and transforming growth factor β (TGF-β) [Figure 3]. To be specific, M1 macrophages are primarily found in the rupture core of vulnerable atherosclerotic plaques, and are markedly relevant to plaque necrosis. Moreover, IFN-γ, released by Th1 cells, has been found to activate macrophages to polarize toward the M1 phenotype. Studies have further shown that macrophages are enabled to cause thinning of the fibrous cap, especially through M1 macrophage-derived matrix metalloproteinases (MMPs). MMPs possess the enzymatic capacity to degrade constituents of the extracellular matrix (ECM), with specific implications for the integrity of the plaque’s fibrous cap. This proteolytic activity is a principal contributor to cap thinning and eventual plaque rupture[11]. Thus, in the clinical setting, paternal pleiotropic acidic phosphatase α (PPAPα) agonists can be used to inhibit AS by reducing the production of MMP subtypes[12].
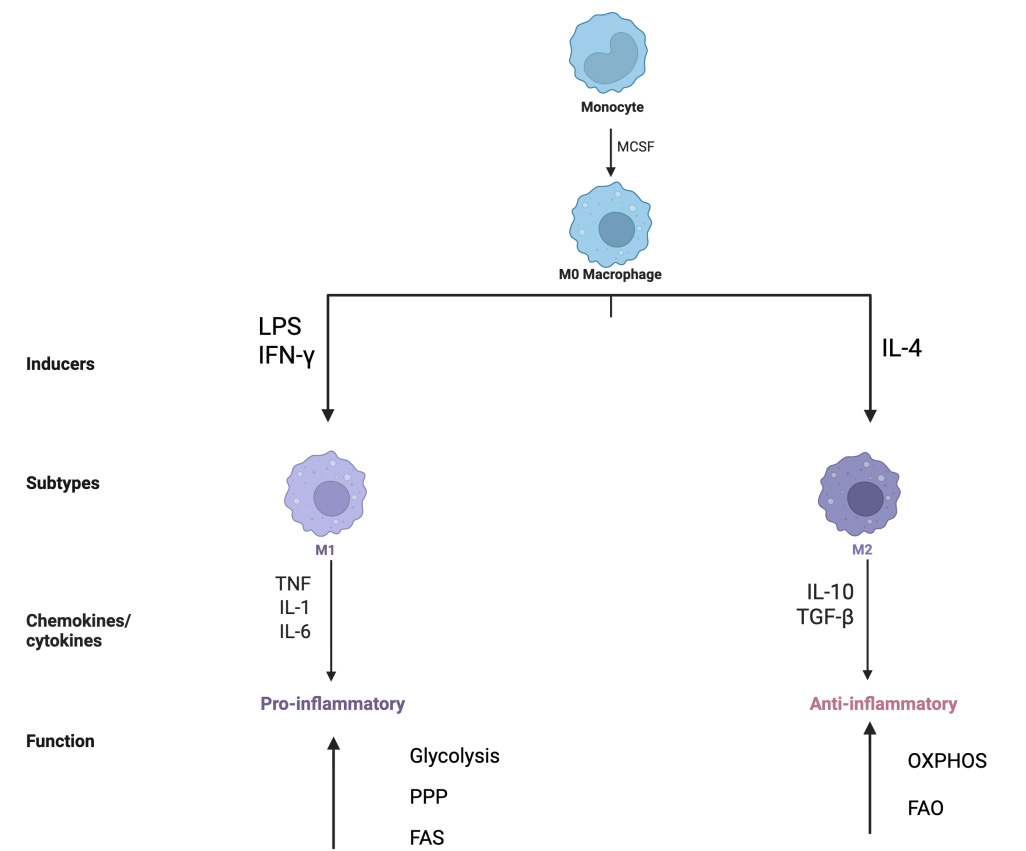
Figure 3. Diagram of Macrophage Polarization and Metabolic Reprogramming Macrophages have the potential to differentiate into different subtypes, and metabolic reprogramming occurs. Classical activation of macrophages, driven by stimuli such as LPS and IFN-γ, promotes a pro-inflammatory phenotype associated with the production of cytokines like IL-1, IL-6, and tumor necrosis factor (TNF). Alternatively, activation via IL-4 induces an anti-inflammatory state characterized by the secretion of mediators including IL-10 and transforming growth factor-beta (TGF-β). Beyond the primary, macrophages undergo metabolic reprogramming in the AS lesion environment, including glycolysis, oxidative phosphorylation (OXPHOS), pentose phosphate pathway (PPP), fatty acid oxidation (FAO), and fatty acid synthesis (FAS). The M1 subtype exhibited enhanced glycolysis, increased PPP, and enhanced FAS. The M2 subtype showed increased OXPHOS and FAO. The figure created with BioRender.com. LPS: Lipopolysaccharide; IFN-γ: interferon-gamma; IL-1: interleukin-1; IL-6: interleukin-6; TNF: tumor necrosis factor; IL-4: interleukin-4; IL-10: interleukin-10; MCSF: macrophage colony-stimulating factor.
Additionally, apoptotic foam cells derived from macrophages that fail to undergo timely efferocytosis may progress to secondary necrosis, contributing to the expansion of the necrotic core within the plaque. This process is another factor leading to the formation of vulnerable plaques. Even living macrophages can contribute to plaque rupture by secreting cytokines, proteases, and procoagulant or thrombotic factors[13]. In recent years, numerous studies have shown that regulating M1/M2 macrophage polarization can inhibit AS. A study utilized an anti-human CD147 antibody to inhibit the polarization of M1 macrophages and promote M2 macrophage polarization, thereby suppressing inflammation and enhancing cell proliferation. Prior to this, CD147 derived from the bone marrow system was believed to exacerbate AS by enhancing macrophage infiltration and polarization, leading to increased cell apoptosis and impaired function of the lipid-rich cells within the plaque, and further increasing plaque instability[14]. Macrophage activation is accompanied by metabolic changes[15]. M2 macrophages mainly rely on oxidative phosphorylation (OXPHOS) to produce Adenosine Triphosphate (ATP), while M1 macrophages induce pro-inflammatory metabolic changes through aerobic glycolysis. Monocarboxylate transporters (MCTs) mediate intercellular lactate shuttling[16]. Among them, MCT4 is mainly expressed in cells with high glycolysis and participates in the efflux of lactate, thereby limiting lactate accumulation within cells[17]. The latest research has found that macrophage MCT4 deficiency induces local activation of M2 genes through histone lactylation, promoting the transition from a damaged to reparative phenotype and enhancing self-renewal and homeostasis[18]. This highlights the crucial role of macrophages in atherosclerotic disease. By regulating the balance between M1 and M2 macrophages, the progression of AS can be effectively inhibited.
Macrophages undergo metabolic reprogramming in AS
Macrophages are among the innate immune cells characterized by the ability to generate immune memory and to acquire heightened reactivity through metabolic reprogramming upon re-exposure to the same stimulus. This process, called trained immunity, can also be found in other cells associated with AS. For example, ox-LDL-trained macrophages may take up more ox-LDL and have a greater potential to become foam cells. Metabolic reprogramming refers to changes in the requirements of energy in response to different environments, which are achieved by altering metabolic mechanisms to increase or attenuate synthetic reactions. The Warburg effect is one of the most widely studied forms of metabolic reprogramming during tumorigenesis. In the AS lesion environment, macrophages undergo significant metabolic reprogramming[19,20]. Specifically, pathways such as glycolysis and the pentose phosphate pathway (PPP) are upregulated, facilitating rapid ATP production and providing essential precursors for lipid biosynthesis and reactive oxygen species (ROS) generation[21,22]. In contrast, the tricarboxylic acid (TCA) cycle and OXPHOS are often downregulated, reflecting a metabolic shift that promotes a pro-inflammatory state[23,24]. Fatty acid oxidation (FAO) may also be decreased, leading to an accumulation of lipids and foam cell formation[23,25,26]. Such metabolic alterations are crucial in driving the inflammatory processes associated with plaque instability and progression.
Different macrophage subtypes use sugars in different ways. The metabolic handling of glucose differs markedly between polarized macrophage subsets: classically activated (M1) macrophages predominantly rely on glycolytic pathways for energy, whereas alternatively activated (M2) macrophages preferentially utilize oxidative phosphorylation for glucose metabolism. Increased glycolysis in macrophages drives their transformation to a pro-inflammatory subtype. It was found that under the condition of AS, most macrophages transformed to the M1 subtype and produced their required energy through glycolysis, which was consistent with the results of previous studies. However, glycolysis produces ATP at a lower but faster rate than OXPHOS[27]. Additionally, it is reported that glycolysis-related genes are highly active in AS patients[28]. As research has deepened, more explanations have been proposed for the mechanism underlying increased glycolysis. Evidence has revealed that the expression of hypoxia-inducible factor-1α (HIF-1α) and Glucose Transporter Type 1 (GLUT-1) glucose transporter is increased at atherosclerotic plaque sites, which are often hypoxic or anaerobic. In addition to hypoxia, ox-LDL-induced macrophages also activate HIF-1α, leading to increased glycolytic activity[29]. Wang et al. proved that the 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) inhibitor PFK158 can reduce the activity of glycolysis, suggesting that PFKFB3 is one of the key factors in macrophage inflammatory activation in AS[30]. Recent studies have identified PFKFB3-driven glycolysis as an upstream regulator of NLRP3 inflammasome activation in hypoxic macrophages[30,31]. Pharmacological inhibition of PFKFB3 with PFK158 suppresses the HIF-1α/PFKFB3/NLRP3 axis, reducing Interleukin-1β (IL-1β) secretion and atherosclerotic plaque burden in mouse models. Notably, PFK158 has completed phase I clinical trials for cancer (NCT02044861) with acceptable safety profiles, supporting its potential repurposing for atherosclerosis[32]. However, caution is warranted as complete myeloid PFKFB3 deletion impairs macrophage efferocytosis and exacerbates necrotic core formation, highlighting the need for dose optimization in future clinical applications. Moreover, iron loading enhances glycolysis in macrophages and exacerbates AS. Additionally, iron overload promotes ROS production, which plays a pro-inflammatory role through oxidative stress.
Increased PPP in M1 macrophages can promote the production of Nicotinamide Adenine Dinucleotide Phosphate Hydrogen (NADPH), which can further lead to the release of inflammatory factors. Moreover, increased PPP and glycolysis promote the production of NO and ROS. Moreover, several studies have found that TCA cycle flux is disrupted in M1 macrophages, leading to the accumulation of cycle intermediates that play a role in macrophage activation and exhibit pro-inflammatory properties[33]. In addition, the accumulated products of the blocked TCA cycle, namely the substrate during fatty acid synthesis (FAS), lead to FAS enhancement. This bridges the gap between lipid metabolism and glucose metabolism, which are linked to each other. The activity of the cholesterol transporters ABCA1 and ABCG1 in M1 macrophages is reduced, leading to excessive cholesterol accumulation in these cells, which activates the immune response[34,35]. IL-10 released by M2 macrophages can contribute to upregulating the transporters ABCA1, thereby promoting the efflux of cholesterol. This is one of the anti-inflammatory mechanisms in which M2 macrophages play a role in the progression of atherosclerosis. In M2 macrophages, the increase in OXPHOS promotes ATP production, which helps provide energy for the anti-inflammatory effects of M2 subtypes and is an important part of tissue repair. In addition, the increase in FAO can also promote its anti-inflammatory effect, which may be leveraged to inhibit AS. FAO is another important source of energy for IL-4/IL-13 activated anti-inflammatory macrophages. This is also one of the crucial mechanisms that lead to their different roles.
T Cells in atherogenesis
Within atherosclerotic plaques, T lymphocytes, specifically the CD4+ and CD8+ subsets, are recognized as vital components of adaptive immunity. Following antigen recognition, antigen-presenting cells stimulate CD4+ T lymphocytes, triggering their differentiation into T helper and regulatory T cell populations.
Th1 cells can secrete IFN-γ and tumor necrosis factor α (TNF-α), which can be identified by the defining transcription factor T-bet, and further contribute to plaque instability and the inflammatory environment. Moreover, the proinflammatory macrophage subtype observed within plaques is hypothesized to release cytokines that can recruit T cells and promote their inflammatory transformation, which is then amplified by IFN-γ secreted by pro-inflammatory cells. IFN-γ signaling is largely mediated by Janus kinase (JAK) and signal transducers and activator of transcription 1 (STAT1) cytosolic factor pathway[36]. The latest research has found that m6A modification plays a crucial role in IFN-γ-induced upregulation and is subsequently essential for the development of atherosclerosis through macrophage activation mediated by downstream STAT1[37]. In another experiment, AAV-6 highly expressing mice were subjected to carotid artery constriction and ligation. The researchers then obtained mice with unstable carotid artery plaques for subsequent experiments. By reducing the IFN-γ levels in T cells, the growth of plaques can be inhibited, and stability can be achieved[38]. TNF-α is an inflammatory cytokine that participates in the pathogenesis of atherosclerosis and is known as an endothelial cell activator. TNF-α can induce the expression of intracellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) in human umbilical vein endothelial cells (HUVECs). A previous study has shown that inhibiting TNF-α can reduce atherosclerosis in ApoE-/- mice[39].
On the contrary, Tregs are thought to possess an anti-atherosclerotic effect, because they can release anti-inflammatory cytokines like TGF-β and IL-10. Endothelial-to-mesenchymal transition (EndMT) has been identified as a critical driver of vascular inflammation and atherosclerosis, and TGF-β is a key mediator of EndMT[40]. In endothelial cells, activation of TGF-β signaling is a key driver of atherosclerosis progression. It induces endothelial-to-mesenchymal transition, converting endothelial cells into fibroblast-like cells, while upregulating adhesion molecules such as VCAM-1, ICAM-1, and pro-inflammatory chemokines, thereby exacerbating vascular inflammation and plaque instability[41,42]. Studies have confirmed that TGF-β2 is the most effective subtype in inducing EndMT, and oxidative stress and hypoxia can synergistically enhance this process[43]. At the same time, an imbalance between collagen and matrix metalloproteinases results in a thinner fibrous cap and an easier rupture of the plaque, and the quantity is positively correlated with human unstable plaques[44,45]. Therefore, regulatory T cells can inhibit effector T cell activity, whereas enhanced effector T cell activity promotes the pathological process of atherosclerosis[46]. In addition, single-cell sequencing techniques have shown that the number of Tregs in enlarged atherosclerotic plaques is greatly reduced. This, in turn, could help explain the protective role of Tregs in atherosclerotic disease. Th17 (T helper type 17) cells are characterized by their ability to secrete the inflammatory mediator IL-17 through several pathways to promote AS, in contrast to the protective effects of Tregs. High expression of IL-17 is associated with increased inflammation within the plaque, reduced smooth muscle cell content, and thinning of the fibrous cap, leading to an increased risk of plaque rupture. However, some studies have also found that IL-17 can stimulate smooth muscle cells to produce type I collagen, promoting thickening of the fibrous cap and thereby stabilizing the plaque [Figure 4]. The impact of IL-17 on plaque stability may be regulated by the local immune environment, such as the levels of IFN-γ and Treg cells[47]. Specifically, in an inflammatory environment characterized by high IFN-γ, high IL-23, and low IL-10, IL-17 mainly exerts a pathogenic role: it induces neutrophil and monocyte recruitment into the plaque by inducing chemokines[48]; it synergistically enhances the production of pro-inflammatory factors such as IL-6 and Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) in combination with TNF-α and IFN-γ; it upregulates matrix metalloproteinases to degrade collagen, weaken the fibrous cap, and increase the risk of plaque rupture[49]; and it collaboratively promotes neovascularization and hemorrhage within the plaque with TNF-α[50]. IL-17/IFN-γ double-positive T cells are present in human coronary artery plaques and synergistically enhance the inflammatory response; in ApoE-/- mice, IL-17 deficiency can reduce vascular inflammation and lesions[51]. Conversely, in a regulatory microenvironment characterized by high TGF-β, high IL-10, and low IFN-γ, IL-17 exerts a protective role: it stimulates smooth muscle cells to synthesize type I collagen, increases the thickness of the fibrous cap, and enhances plaque stability[52]; it inhibits the Th1 response through negative regulation with IFN-γ, indirectly reducing inflammation; it inhibits the expression of VCAM-1 in endothelial cells and reduces the recruitment of T cells and monocytes to the plaque; and, under the combined action of TGF-β and IL-6, certain Th17 cell subsets can simultaneously produce IL-10 and exert anti-inflammatory effects[53,54]. Key evidence includes: IL-17 expression in human carotid artery plaques is positively correlated with smooth muscle cell and collagen content[55]; in Low-density lipoprotein receptor knockout (LDLR-/-) mice, supplementation with recombinant IL-17 can reduce plaque area, accompanied by a decrease in IFN-γ and an increase in IL-10; and in patients with acute myocardial infarction, the combination of low IL-17 and high VCAM-1 is associated with a higher 2-year risk of major cardiovascular events, suggesting that IL-17 may exert a protective effect by inhibiting VCAM-1[56] [Table 1]. The CC chemokine ligand 18 (CCL18) is considered to be a T-cell chemokine, which is produced by monocytes and CD4+T cells. Its receptor, CCR6, is expressed in T-cell subtypes such as Treg and Th17 cells[57]. Moreover, CCL18 promotes IL-6 secretion, which in turn drives Tregs to differentiate into pathogenic Th17 cells. Therefore, maintaining the balance of Th17/Treg provides insight into the treatment of AS. Moreover, this is the first time that the CCL18/CCR6 chemokine pathway has been considered as a target for atherosclerosis intervention[57].
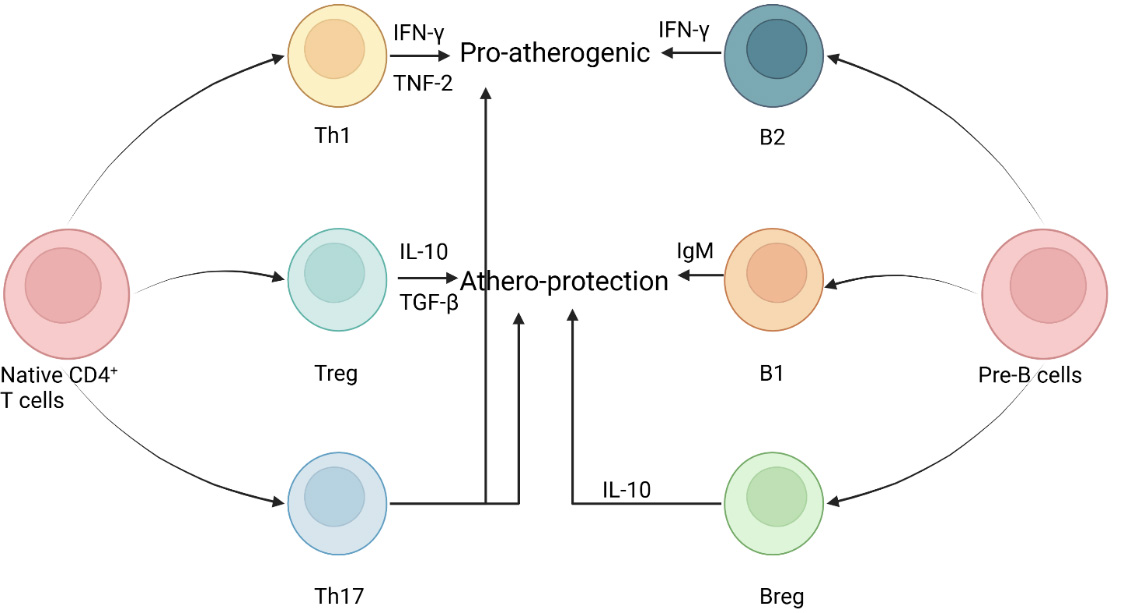
Figure 4. Schematic diagram illustrating the roles of T cells and B cells in atherosclerosis. CD4+ T lymphocytes give rise to T helper cells and regulatory T cells. Th1 cells can secrete IFN-γ and TNF-α, further leading to plaque instability and the body’s inflammatory environment. Tregs are considered to have anti-atherosclerotic effects because they can release anti-inflammatory cytokines, including TGF-β and IL-10. Th17 cells are characterized by their ability to secrete the inflammatory mediator IL-17 through multiple pathways to promote AS, which is the opposite of the protective effect of Tregs. In addition, Th17 cells also have pathogenic effects that contribute to the deterioration of AS. Like T cells, B cells express different functions through various subtypes, such as B1, B2, and Breg. B1 cells are characterized by secreting IgM antibodies, which can act on ox-LDL and protect against atherosclerosis. Breg cells can help stabilize plaques and inhibit inflammatory processes by releasing the anti-inflammatory cytokine IL-10. On the contrary, B2 cells can produce pro-inflammatory cytokines, such as IFN-γ and pathogenic IgG, which can intensify atherosclerosis activity. The figure created with BioRender.com. IFN-γ: Interferon-gamma; TNF-α: tumor necrosis factor-alpha; Th1: T helper type 1 cell; B2: conventional B cell; IL-10: interleukin-10; IgM: immunoglobulin M; TGF-β: transforming growth factor-beta; Treg: regulatory T cell; B1: innate-like B cell; Th17: T helper type 17 cell; Breg: regulatory B cell; OxLDL: oxidized low-density lipoprotein; IgG: immunoglobulin G; ox-LDL: oxidized low-density lipoprotein.
The determinants of the direction of IL-17 action
| Pro-atherogenic (pathogenic) | Anti-atherogenic (protective) | |
| IFN-γ | High | Low |
| IL-10 | Low | High |
| Treg | Low | High |
| Microenvironment | High IL-23, high IL-1β, high TNF-α | High TGF-β, high IL-6 |
| Main effect | Inflammatory cell recruitment, MMP ↑, collagen ↓ | Collagen ↑, VCAM-1 ↓, IFN-γ ↓ |
Cytotoxic CD8+ T cells exert a dual effect on AS depending on disease stage: in early atherosclerosis, they may promote plaque vulnerability by reducing monocyte recruitment; in advanced disease, they can reduce macrophage and Th1 cell populations and exert protective effects. Thus, it is crucial to elucidate the dominant mechanisms by which CD8+ T cells influence atherosclerosis. Wang et al.[58] carried out a series of controlled experiments and suggested that, compared with stable plaques, the amount of CD8+ T cells was significantly lower in unstable plaques, which is instructive to support van Duijn’s conclusion[59] mentioned above. Vos et al. have found that Casitas B-lymphoma proto-oncogene-B (CBL-B), which exists in macrophages and T cells, can specifically increase the activation of pro-inflammatory CD8+ T cells; however, excessive activation leads to T cell depletion. Thus, CD8+ T cells may lose their pro-atherogenic role, which is the reason why plaque was reduced in CBL-B deficiency mice[60]. This makes CBL-B a potential therapeutic target.
This evidence shows that AS is a complex immune disease, and the complex interactions among various immune cells lead to the persistent balance of protective and pro-atherogenic effects during the atherogenic process. Based on the theories mentioned above, T cell regulation is another major therapeutic strategy for AS. A better understanding of the exact function of T cells in atherosclerotic lesions and their mechanisms of action will provide a new basis for the future development of therapies that specifically target T cell subsets to combat atherosclerosis.
B cells in atherogenesis
Like T cells, B cells express diverse roles through various subtypes such as B1, B2, and regulatory B cells (Breg). However, B cells are less abundant than T cells in atherosclerotic lesions. B1 cells are characterized by the secretion of IgM antibodies in a T-cell-independent manner. Subsequently, studies have found that antibodies against ox-LDL are produced by B1 cell-derived plasma cells. This finding provides another link between B cells involved in humoral immunity and atherosclerotic disease. These antibodies specifically bind to oxidized LDL, resulting in decreased macrophage uptake, diminished foam cell generation, and suppression of inflammatory pathways, ultimately exerting a protective effect in atherosclerosis. Similarly, Bregs can contribute to plaque stabilization and inhibit inflammatory processes by releasing the anti-inflammatory cytokine IL-10. This enables them to regulate the inflammatory process, thereby inhibiting the formation of atherosclerotic plaques. In contrast, B2 cells secrete pro-inflammatory cytokine interferon-γ and pathogenic IgG antibodies, which may exacerbate atherosclerotic mechanisms at the lesion site. In summary, the role of B cells is subset-specific and can be divided into subsets that promote atherosclerosis and subsets that inhibit AS.
The role of natural killer cells in atherosclerosis
Natural Killer (NK) cells, traditionally known for their role in antiviral and antitumor immunity, have been increasingly implicated in the pathogenesis of atherosclerosis. NK cells have been detected within human and murine atherosclerotic plaques, predominantly in advanced lesions, necrotic cores, and plaque shoulder regions[61]. Moreover, elevated circulating NK cell counts have been reported in patients with advanced atherosclerosis, suggesting their active involvement in disease progression.
NK cell recruitment is mediated by chemokines and cytokines and involves interactions with macrophages and dendritic cells. Activating receptor ligands expressed by macrophages and endothelial cells enhance NK cytotoxicity. Ox-LDL and cholesterol crystals also influence NK cell function[62].
However, different experimental models have yielded conflicting results regarding the exact role of NK cells. Some studies report that NK cell deficiency or depletion in mice reduces plaque size and inflammation, supporting a pro-atherogenic role. For example, NK depletion in ApoE-/- mice attenuated atherosclerosis. Conversely, other models, such as mice lacking NK function, showed no significant difference or even increased lesion development. This discrepancy may arise from differences in mouse strains, methods of NK cell depletion, or compensatory immune mechanisms[63,64].
In conclusion, while strong evidence supports a pro-inflammatory and plaque-destabilizing role for NK cells in atherosclerosis, their full contribution remains incompletely defined due to contradictory experimental findings. Clarifying these divergent results is important for developing NK cell-targeted therapies.
Other immune cells
Beyond monocytes, macrophages, T/B lymphocytes, and NK cells, several innate immune cell types, including neutrophils, mast cells, and dendritic cells (DCs), play crucial roles in the pathogenesis of atherosclerosis. DCs serve as pivotal bridging cells between innate and adaptive immunity by internalizing ox-LDL, presenting antigens to T cells, and secreting pro-inflammatory mediators that promote lesion progression[65,66].
Neutrophils infiltrate plaques predominantly during acute phases, releasing ROS, proteases, and neutrophil extracellular traps (NETs)[67,68]. While these processes aid in the clearance of necrotic debris, they also degrade elastic fibers within the fibrous cap through elastase activity, leading to cap thinning and increased plaque vulnerability. Pharmacological inhibitors of NET formation, such as cilastatin sodium (SVT), have thus been proposed as novel strategies for plaque stabilization[69].
Mast cells accumulate progressively within plaques and degranulate to release a variety of cytokines, directly exacerbating local inflammation and promoting thrombotic events. Clinical correlations indicate that heightened mast cell activation is associated with parameters of plaque vulnerability, suggesting that mast cell stabilizers hold therapeutic promise[70].
The role of extracellular vesicles and miRNA in atherosclerosis
Emerging evidence positions extracellular vesicles (EVs) and their non-coding RNA (ncRNA) cargo as novel mediators in the pathogenesis of atherosclerosis, bridging the gut-heart axis and liver-vascular crosstalk. Bacterial EVs (BEVs) from pathogens such as Helicobacter pylori and Porphyromonas gingivalis deliver lipopolysaccharide and virulence factors directly into arterial cells, triggering endothelial dysfunction, NLRP3 inflammasome activation, foam cell formation, and vascular calcification[71]. Conversely, BEVs from commensal Akkermansia muciniphila and probiotics reinforce intestinal barrier integrity and promote anti-inflammatory M2 macrophage polarization, thereby limiting systemic inflammation and atherogenesis. Beyond the gut, hepatocyte-derived EVs enriched with miR-1 under non-alcoholic fatty liver disease conditions transfer this miRNA to endothelial cells, where it suppresses KLF4 and activates nuclear factor kappa B (NF-κB), driving vascular inflammation and plaque progression[72]; inhibition of miR-1 markedly reduces atherosclerosis in mice. Collectively, these findings establish EV-ncRNA axes as critical regulators of AS, offering novel diagnostic biomarkers and therapeutic targets independent of traditional lipid pathways[73,74].
PYROPTOSIS
Pyroptosis is a novel form of programmed cell death that promotes the release of inflammatory cytokines by increasing cell membrane permeability. In other words, pyroptosis is an inflammatory form of cell death and has been proven to be related to atherosclerosis.
Although pyroptosis and apoptosis are both specific forms of cell death, there are differences between them. In contrast to apoptosis, inflammasome formation can be observed during pyroptosis. Second, the caspases required for activation are also different. For example, pyroptosis can be divided into canonical and noncanonical pathways according to the dependent caspase involved. In addition, pyroptosis is characterized by the rupture of the cell membrane and the outflow of its contents, while the morphological characteristics of apoptosis mainly show that apoptotic bodies are present on the surface of the cell membrane but the cell membrane is intact[75,76] [Table 2]. Recently, scientists have found that this mechanism is likely closely related to atherosclerotic lesions. Several studies have given evidence to prove that pyroptosis is involved in the inflammatory process of atherosclerosis. Scientists have found that pyroptosis occurs in effector cells associated with atherosclerosis, such as endothelial cells, macrophages, and smooth muscle cells. Inhibiting pyroptosis in these cells may reduce the inflammatory response of the arterial wall. NLRP3, GSDMD, and caspase-1 are associated with the development and accumulation of atherosclerotic plaques. In addition, high-risk factors of atherosclerosis, including ox-LDL and cholesterol crystals, have also been shown to promote pyroptosis through multiple mechanisms. In the following, the exact mechanisms by which pyroptosis promotes atherosclerosis will be described in detail.
Differentiation of pyroptosis and apoptosis
| Inflammasome | Caspase | Ruptured cell membrane | Inflammatory response | |
| Pyroptosis | Present | 1/4/5/11 | Present | Present |
| Apoptosis | Absent | 3/7/9 | Absent | Absent |
This section of the table explains pyroptosis and apoptosis, both of which are specific forms of cell death, but there are various differences between them. The formation of inflammasomes can be observed during pyroptotic development. In contrast, there is no involvement of inflammasomes in apoptosis. In addition, the caspases required to activate pyroptosis and apoptosis are also different. Moreover, pyroptosis is characterized by cell membrane rupture and release of cellular contents, whereas apoptosis is characterized by the presence of apoptotic bodies on the cell surface while the cell membrane remains intact.
Caspase and GSDMD in pyroptosis
The inflammatory caspase subfamily includes caspases1, 4 and 5 in human and caspases11 in rodents[77,78]. According to the dependent caspase, pyroptosis-related signaling pathways can be divided into classical pathways mediated by caspase-1 and non-classical pathways mediated by caspase-4/5/11.
Gasdermin D (GSDMD) is a member of the gasdermin protein family that forms membrane pores. GSDMD is the common substrate of caspases in both of the above pathways. Thus, it is considered to be a key effector of pyroptosis, inducing cell rupture, the release of large amounts of Interleukin-18 (IL-18) and IL-1β, and inflammation.
In the classical pathway, activated caspase-1 can cleave GSDMD to generate GSDMD-NT, which inserts into cell membranes to form pores, increases membrane permeability, induces cell swelling and rupture, and promotes the release of cellular contents [Figure 5]. Recent studies have shown that GSDMD-NT palmitoylation can control a key regulatory mechanism for the properties of GSDMD, namely membrane localization and activation. This provides a possible target for the treatment of inflammatory diseases. On the other hand, activated caspase-1 can cleave pro-IL-1β and pro-IL-18, activating them to form mature IL-1β and IL-18, which are released into the extracellular space through membrane pores to amplify the inflammatory response and eventually lead to pyroptosis. IL-1β may have both beneficial and adverse effects on the development of atherosclerotic plaques. However, it is generally accepted that its adverse effects predominate, which warrants further study. Compared with IL-1β, although fewer studies have examined IL-18, it also has pro-atherogenic effects in mice and humans. In contrast, Shao Feng’s lab conducted a series of experiments and found that human caspase-4/5 and mouse caspase-11 bind bacterial LPS, which is responsible for further activation of the pyroptosis pathway [Figure 5]. This pathway is not associated with the cleavage of inflammatory cytokines pro-IL-1β and pro-IL-18.
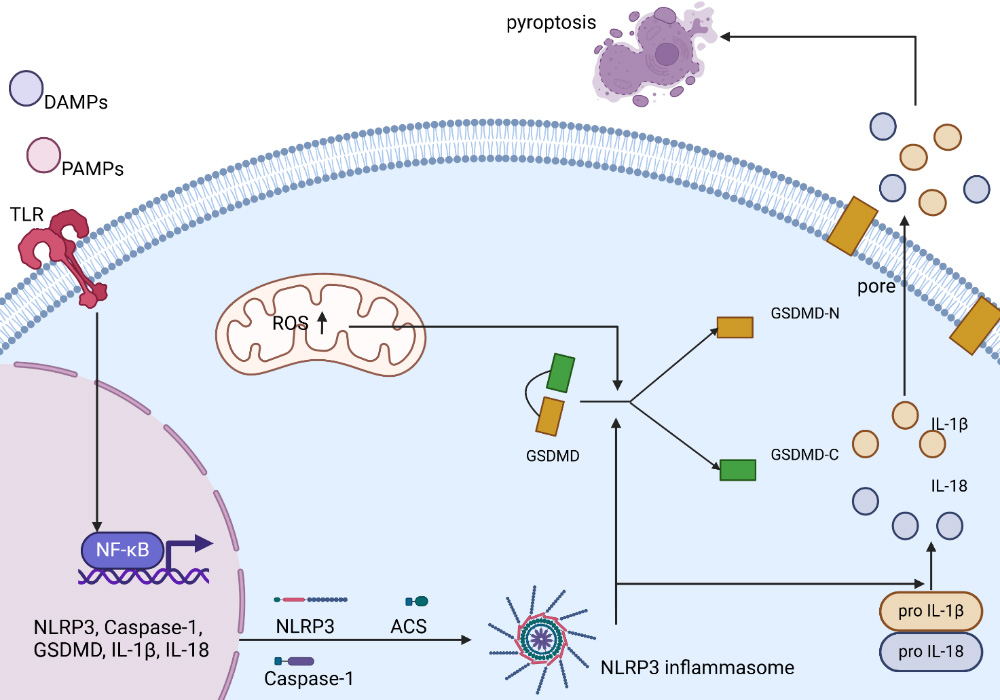
Figure 5. Schematic diagram of NLRP3 inflammasome activation and the cell pyroptosis pathway. The mechanism of activation of the classic NLRP3 inflammasome pathway is shown in the figure. The activation of the NLRP3 inflammasome first depends on TLRs recognizing PAMPs or DAMPs and inducing NF-κB activation. Subsequently, NF-κB mediates the transcription of NLRP3, ASC, pro-Caspase-1, pro-IL-1β, and pro-IL-18. Other signals that trigger NLRP3 inflammasome assembly include increased ROS generation in mitochondria. The activation of inflammasomes promotes the cleavage of caspase-1, thereby promoting the cleavage of GSDMD. The N-terminus of GSDMD forms pores in the cell membrane, promoting the release of mature IL-1β and IL-18. The release of IL-1β and IL-18 can induce various forms of inflammatory responses, which are related to the occurrence and progression of cell apoptosis. The figure created with BioRender.com. DAMP: Damage-associated molecular pattern; PAMP: pathogen-associated molecular pattern; TLR: toll-like receptor; ROS: reactive oxygen species; GSDMD: gasdermin D; GSDMD-N: gasdermin D N-terminal domain; GSDMD-C: gasdermin D C-terminal domain; IL-1β: interleukin-1 beta; IL-18: interleukin-18; NF-κB: nuclear factor kappa-B; pro IL-1β: pro-interleukin-1 beta; pro IL-18: pro-interleukin-18; NLRP3: NOD-like receptor family pyrin domain-containing protein 3; ASC: apoptosis-associated speck-like protein containing a CARD; Caspase-1: cysteine-aspartic acid protease 1.
Previous studies have confirmed that caspase-1-induced increase in IL-1β and IL-18 can promote atherosclerosis and increase plaque instability. The important role of caspase in pyroptosis has attracted considerable attention and has become a new therapeutic target for many human diseases[79,80]. In murine models of atherosclerosis (e.g., ApoE-/- or Ldlr-/- mice), genetic deletion of GSDMD[81] or pharmacological inhibition of NLRP3[82-84] significantly reduced plaque burden and inflammation.
In vitro studies have demonstrated that caspase-1-mediated cleavage of GSDMD and pro-IL-1β/pro-IL-18 is sufficient to induce pyroptosis in human macrophages, endothelial cells, and VSMCs upon stimulation with ox-LDL or cholesterol crystals. Although GSDMD-mediated pyroptosis is closely related to atherosclerosis, current studies remain limited in their exploration of its cellular pathways and molecular mechanisms. To further investigate the localization of GSDMD in various atherosclerotic cell components, Fan et al. conducted relevant research. In 2024, they provided a novel therapeutic target mechanism for GSDMD-associated atherosclerosis[81]. They established two groups of mice, including ApoE-/- mice and GSDMD-/- ApoE-/- mice, which demonstrated that GSDMD exists in macrophages by using single-cell RNA sequencing[85] and found that GSDMD inhibitor Y1 can also be used to prohibit the promotion of atherosclerosis. However, whether GSDMD affects pyroptosis in other cell types, such as ECs and VSMCs, remains to be investigated.
NLRP3 inflammasome
The nucleotide-binding oligomerization domain-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome is widely expressed in human immune cells, such as monocytes, macrophages, T cells, and B cells. To demonstrate the expression of pyroptosis-related proteins in human coronary arteries and their relationship with AS, Zhou et al. performed immunohistochemical analysis of 40 human coronary artery specimens and showed that AS severity was positively correlated with caspase-1 and NLRP3 expression[86]. NLRP3 is involved in inflammation that leads to atherosclerosis. Moreover, the main components of the NLRP3 inflammasome are cytosolic pattern recognition receptors (PRRs), the apoptosis-associated spec-like protein containing a CARD (adaptor ASC), and caspase-1[87]. In response to various endogenous or exogenous stimuli, the NLRP3 inflammasome in different tissues and organs recognizes pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), including ox-LDL and cholesterol crystals [Figure 5]. When the NF-κB transcription factor is activated, NLRP3 expression is upregulated. This inflammasome activates downstream inflammation-related signaling pathways to promote the production of pro-inflammatory cytokines, which can further increase the risk of plaque rupture. The FAO pathway reduces NLRP3 activation, thereby inhibiting macrophage production of IL-1β, a cytokine that promotes the inflammatory response. Recently, it has been found that the NLRP3 inflammasome pathway is dependent on the activation signal IκB kinase β (IKKβ) and can be independent of NIMA-related kinase 7 (NEK7)[88]. With further study, researchers have demonstrated that under low-shear stress (LSS), the phosphorylation level of Inhibitor of Nuclear Factor Kappa-B Kinase Subunit Epsilon (IKKε) in endothelial cells increases, thereby promoting activation of signal transducer and activator of STAT1[89]. Activated STAT1 binds to the NLRP3 promoter region, resulting in cellular pyroptosis. In addition, STAT1 activation promotes the secretion of pro-inflammatory factors by driving macrophage polarization toward the M1 subtype, leading to unstable plaque formation and aggravation of atherosclerotic lesions.
In addition to promoting AS, NLRP3 may also contribute to neurological disorders such as Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis. Therefore, MCC950 may offer therapeutic potential for these diseases and broaden its clinical application. NLRP3 inhibitors are entering clinical trials, but their best indications are still being explored. NLRP3 inhibitors may serve as anti-inflammatory drugs for inflammatory diseases that currently lack effective treatment[90].
ROS
Energy metabolism occurs primarily in mitochondria, which are involved in the production of ROS. However, excessive accumulation of ROS can induce mitochondrial dysfunction, which plays an important role in the pathogenesis of many diseases. Reactive oxygen species are further involved in pyroptosis and foam cell formation in atherosclerosis. Oxidative stress is also considered to be one of the pathogenic mechanisms of AS. Increased ROS promotes the expression of inflammatory and adhesion molecules and promotes ox-LDL formation. Recently, more and more studies have proved that excessive ROS can contribute to EC pyroptosis by upregulating the expression of NLRP3 [Figure 5]. With further research, it has been found that pathological stimuli, such as ox-LDL, hyperglycemia, nicotine, and Ang II, can increase ROS in endothelial cells, thereby activating the NLRP3 inflammasome. In addition, M1 macrophages, which have pro-inflammatory effects, can also produce ROS and promote the development of an inflammatory response, then lead to damaged ECs[85].
The mechanism by which ox-LDL and cholesterol crystals contribute to pyroptosis
One of the key mechanisms by which cholesterol crystals induce pyroptosis is that lysosomes rupture, releasing their contents into the cytoplasm, thereby activating the NLRP3 inflammasome. However, recent studies have found that Endoplasmic Reticulum (ER) cholesterol accumulation promotes inflammasome activation, underscoring the importance of elevated ER cholesterol in activating the NLRP3 inflammasome. In addition, cholesterol crystals can stimulate excessive ROS production, thereby inducing the activation of the NLRP3 inflammasome and caspase-1.
Early results showed that cholesterol crystals accumulate in ABCA1/ABCG1-deficient macrophages, where the expression of NLRP3, caspase-1, and IL-1β is increased, and inflammasome activation is initiated. In addition, the lack of ABCA1/ABCG1 in macrophages also leads to excessive accumulation of cholesterol[91]. Based on this mechanism, Hou et al. found that Xinmaikang (XMK) can down-regulate the expression of NLRP3, IL-1β and Caspase-1, and can alleviate AS in ApoE-/- mice[92]. The liver X receptor (LXR) is the most common nuclear receptor that can regulate cholesterol. It is characterized by not only regulating cholesterol metabolism but also exerting anti-inflammatory effects and reducing intracellular cholesterol accumulation. LXRs can effectively prevent AS by inhibiting the NLRP3 inflammasome and NF-κB. In addition, LXR agonists have been shown to increase the expression of ABCA1 to reduce pyroptosis, thereby reducing atherosclerosis in ApoE-/- mice[93]. 7-Ketocholesterol (7KC) is the oxidized product of cholesterol oxidation in low-density lipoprotein. 7KC has been shown to induce macrophage reprogramming and foam cell formation; therefore, it can be developed as a potential therapeutic target for AS.
Ox-LDL can directly or indirectly trigger pyroptosis. On the one hand, ox-LDL can be directly recognized and activate downstream signaling pathways that promote the transcription of pro-IL-1β and pro-caspase-1. Secondly, ox-LDL can induce oxidative stress and mitochondrial dysfunction through multiple mechanisms, resulting in increased ROS production; elevated ROS further exacerbates mitochondrial dysfunction.
NF-κB
Activated NF-κB has been shown to promote pyroptosis through a variety of mechanisms, such as ROS production, ER stress, and mitochondrial dysfunction. NF-κB signaling is thought to be a key factor in atherosclerotic complications and has a connection with CVD. At the cellular level, NF-κB is involved in monocyte differentiation, transformation of macrophage subtypes, and transformation into foam cells. In response to pro-atherosclerotic factors such as ox-LDL, it promotes NF-κB activation, induces endothelial cell dysfunction and inflammation, and promotes monocyte recruitment to the subendothelial intima. Moreover, this EC dysfunction in turn leads to the activation of NF-κB signaling. NF-κB can be detected to increase in VSMCs in atherosclerotic lesions, thereby mediating the decrease of VSMC contractility[94,95]. Therefore, targeting NF-κB has gradually become a major focus of research worldwide.
Artemisinin has been found to be widely used in the treatment of malaria, but it has also recently been found to be useful in inflammatory diseases by inhibiting NF-κB either through direct binding or by suppressing upstream signaling pathways. Doublecortin-like kinase 1 (DCLK1) has been shown to promote activation of the NF-κB pathway and subsequently the secretion of inflammatory mediators. DCLK1 deficiency or pharmacological inhibition can block the progression of AS. Scutellarin reduces NLRP3 inflammasome activation via the NF-κB/NLRP3 signaling pathway and has a protective effect on the heart through its anti-inflammatory properties. Rnd3 has a protective effect against endothelial cell pyroptosis by inhibiting tumor necrosis factor receptor-associated factor 6 (TRAF6) ubiquitination, thereby suppressing NF-κB activation and promoting TRAF6 degradation. In addition, TRAF6-specific deficiency counteracts Rnd3-induced exacerbation of endothelial cell pyroptosis both in vivo and in vitro. These findings establish a strong link between Rnd3 and the TRAF6/NF-κB/NLRP3 signaling pathway in endothelial cells, suggesting that Rnd3 plays an important role in preventing pyroptosis. He et al.[96], have demonstrated that MgCl2 can inhibit the expression of pyroptosis-related molecules and the secretion of IL-1β and IL-18 by downregulating the NF-κB signaling pathway, which can further reduce ox-LDL-induced pyroptosis of vascular smooth muscle cells. These findings provide a promising basis for targeting NF-κB in AS.
THERAPY
Traditional treatment method: lipid-lowering
As scientists continue to study AS, they are gradually realizing that the immune system is closely linked to atherosclerosis. As such, pharmaceutical companies can use these theories as targets to develop new drugs for patients with atherosclerosis. The role of immunomodulatory strategies in the treatment of atherosclerosis has attracted great attention, offering novel therapeutic approaches that are quite different from traditional lipid-lowering therapies. In traditional treatment of AS, clinicians focus on lowering LDL, with typical medications such as statins[97] and PCSK9 inhibitors[98].
Clinical studies have shown that statins are HMG-CoA reductase inhibitors and play a key role in preventing atherosclerosis by improving endothelial function and stabilizing plaques. Statins were originally marketed as powerful lipid-lowering drugs, but recent studies have shown that they can also reduce pyroptosis. In addition, clinical practice has shown that the risk of AS does not always decrease after statin use, prompting further investigation. In 2015, Altaf demonstrated that a high dose of rosuvastatin can suppress the inflammatory development of atherosclerosis by downregulating NLRP3 expression, which helped improve our understanding of the pathogenesis and management of atherosclerosis. Recent studies have shown that atorvastatin inhibits upstream components of the NLRP3 activation pathway, reducing the secretion of IL-1β, IL-18, and GSDMD. Additionally, it can reduce the immune response and the accumulation of lipid in macrophage[99]. Notably, simvastatin and mevastatin can also inhibit NLRP3 in ECs, which is activated by ox-LDL. These findings have indicated that statins are gradually gaining the attention of researchers. While statins are widely believed to have cholesterol-lowering effects, they have also been linked to an increased likelihood of developing diabetes and myopathy[100].
Some studies have shown that adding a Proprotein convertase subtilisin/Kexin 9 (PCSK9) inhibitor to existing statin treatment can not only improve lipid levels but also accelerate the regression of coronary atherosclerosis[101]. PCSK9 can bind to the LDL receptor (LDLR) and induce its degradation, thereby increasing circulating LDL levels. Therefore, PCSK9 inhibitors have become an important pillar in the treatment of cardiovascular-related diseases due to their ability to reduce LDL. However, further research has shown that PCSK9 is also closely related to immunity and oxidative stress.
Treatment for NLRP3
Metformin can improve macrophage dysfunction, inhibit foam cell formation, and even suppress NLRP3 inflammasome formation[102]. Therefore, the clinical application of metformin has expanded from a first-line hypoglycemic drug for diabetes to a potential agent for delaying AS. Previous studies have shown that activation of mTORC1 (mTOR complex 1) can promote atherosclerosis. However, Studies have shown that the mTORC2 pathway inhibits AS, suggesting that treatment aimed at regulating mTOR needs to pay attention to this pair of balanced but opposite signaling pathways. In addition, the nuclear factor of activated T-cells (NFAT) plays an important role in T cells and has increasingly been implicated in cardiovascular diseases. Liu et al. showed that t foam cell formation increased in NFATC3-deficient mice, and this increase was negatively correlated with plaque instability[103]. C-type natriuretic peptide (CNP) has been shown to degrade HIF-1α and enhance plaque stability, suggesting a novel protective effect against atherosclerosis[104]. Additionally, Li et al. had shown that Immune Responsive Gene 1 (IRG1), which is mainly expressed in macrophages and neutrophils, is upregulated in atherosclerotic plaques. 4-octyl itaconate (4-OI), a metabolite produced by IRG1, can be considered a promising treatment for atherosclerosis by establishing beneficial plaques[105].
Sulfonylureas, such as glyburide, which are used to treat type 2 diabetes mellitus (T2DM), have been shown to inhibit NLRP3 signaling. Oridonin, as a common Chinese medicine, has been demonstrated to inhibit the assembly and activation of the NLRP3 inflammasome[90]. In addition to MCC950, various NLRP3 inhibitors (such as oridonin and VX765) and HINT2 overexpression can inhibit pyroptosis. However, clinical application still requires toxicity assessment.
The pro-inflammatory M1 subtype plays a key role in AS progression; thus, inhibiting macrophage polarization toward anti-inflammatory subtypes may provide a new therapeutic strategy. In fact, SNX10 (sorting nexin10) accumulates in arterial plaques. You et al.[106] had demonstrated that SNX10 deficiency can reduce foam cell formation, promote macrophage polarization toward an anti-inflammatory phenotype through the Lck/Yes-related novel protein tyrosine kinase (Lyn)-protein kinase B (Akt)-transcription factor EB (TFEB) signaling pathway, and disrupt the balance between glycolysis and mitochondrial oxidative phosphorylation.
Reduce inflammatory response in the progression of cardiovascular disease
Canakinumab
The Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS) was the first to demonstrate that canakinumab can block the action of IL-1β, highlighting its role in inflammation regulation rather than just lipid reduction. In addition, it was also the first trial of anti-inflammatory therapy for AS. However, in this trial, cholesterol levels were not reduced, and mortality was not changed, likely due to the high incidence of infections. Therefore, the idea of inhibiting the production of ROS and the activation of the NLRP3 inflammasome inspired researchers to slow the progression of AS by inhibiting pyroptosis. For example, anakinra competitively inhibits IL-1 and is known as a recombinant IL-1 receptor (IL-1R) antagonist. Ku et al. found a 30% reduction in atherosclerotic plaque area in ApoE-/- mice that were treated with Anakinra, compared to controls, confirming its anti-inflammatory effect in the treatment of AS[107].
Colchicine
Colchicine has been approved by the US Food and Drug Administration (FDA) for widespread use in autoimmune and inflammatory diseases for the first time. In recent years, accumulating evidence has shown that low-dose colchicine improves cardiovascular outcomes. In 2023, the FDA approved 0.5mg colchicine for reducing CVD events in atherosclerotic cardiovascular disease (ASCVD) patients. Its specific mechanism of action is as follows. Firstly, colchicine can alter the composition of inflammatory cells in plaques and reduce plaque size by preventing inflammatory monocytes and neutrophils from entering the plaques from the bloodstream. Second, colchicine has the potential to inhibit NLRP3 inflammasome activation and suppress the production of IL-1β and IL-18. Furthermore, colchicine can inhibit the uptake of cholesterol crystals by endothelial cells, thereby avoiding pyroptotic cell death induced by cholesterol crystals[108].
Melatonin
Melatonin (MLT) is a neuroendocrine hormone primarily secreted by the pineal gland and various organs. The role of MLT in inhibiting the progression of AS has been explored through several possible pathways and experimentally validated[109].
In animal experiments, mice treated with MLT showed a significant reduction in aortic arch plaque compared with the control group. At the same time, a new mechanism was identified, providing experimental evidence for the clinical application of MLT in the treatment of AS. MLT inhibits AS by reducing oxidative stress and suppressing macrophage pyroptosis, which suggests the therapeutic potential of the SIRT3/FOXO3α/ROS axis in AS[110]. In addition, MLT regulates macrophage polarization and oxidative stress homeostasis by inhibiting the activation of downstream inflammatory pathways mediated by NADPH oxidase 2 (NOX2) in macrophages.
Galectin-3 (Gal-3), a potential biomarker of cardiovascular inflammation, is associated with poor prognosis and promotes inflammation, thereby exacerbating endothelial damage. Recent studies indicate that MLT inhibits Gal-3 by suppressing the NF-κB signaling pathway and subsequently downregulating Gal-3 expression.
The change in medication administration is also another major advancement in the treatment of cardiovascular diseases
In addition to the drugs mentioned above, a new treatment method, vaccines, has also attracted the attention of researchers. Rheumatoid arthritis and multiple sclerosis are inflammatory diseases with complex autoimmune processes that can be targeted by vaccination strategies. Atherosclerosis has also been shown in recent studies to be an autoimmune disease, similar to the two diseases mentioned above. Therefore, vaccination is considered to prevent atherosclerosis[111].
As research continues to advance, scientists are focusing not only on drug innovation but also on more effective drug delivery methods. Current routes of administration can cause drug side effects in tissues outside the lesion site. To enable more precise diagnosis and drug delivery, nanoparticles have attracted widespread attention. Compared with traditional drug delivery means, this novel technology can reduce adverse effects related to medicine. It can also maximize drug efficacy with fewer doses by targeting the drug specifically to sites of inflammation. In particular, studies have shown that nanoparticles can specifically target sites of macrophage aggregation, accurately locate lesions, and release nanoparticle-loaded drugs precisely where they are needed. However, this has only been demonstrated in animal models, and its effect on human clinical treatment remains unknown. Thus, translating these findings from theory to practice remains challenging. Beyond vaccines and nanoparticles, bispecific T-cell engager (BiTE) technology has emerged as a novel immunotherapeutic strategy for atherosclerosis. Amrute et al.[112] identified fibroblast activation protein (FAP) as a specific marker of modulated smooth muscle cells (SMCs) in human coronary plaques using single-cell multiomics and spatial transcriptomics. They developed a half-life-extended anti-FAP BiTE that redirects T-cell cytotoxicity to eliminate FAP+ modulated SMCs. In ApoE-/- mouse models, anti-FAP BiTE treatment significantly reduced plaque burden, increased fibrous cap thickness and collagen content, and remodeled the plaque cellular landscape. This proof-of-concept study demonstrates that immunotherapeutic depletion of pathogenic stromal cells, independent of lipid lowering, represents a promising precision medicine approach for coronary artery disease.
CONCLUSION AND DISCUSSION
Atherosclerosis-related cardiovascular disease remains the leading cause of death worldwide, and research on the mechanism of AS is still an evolving field. There are thousands of papers in the literature on the pathogenesis of atherosclerosis, which is associated with a high mortality rate from cardiovascular disease caused by atherosclerosis. This review emphasizes pyroptosis and changes in the immune microenvironment during atherosclerosis. As expected, these findings correlate with macrophages, T cells, B cells, and other immune cells, supporting the view that atherosclerosis is an inflammatory disease. This review systematically elucidated the distribution of a variety of immune cells in atherosclerosis and laid a foundation for further understanding the immune mechanisms underlying the occurrence and development of AS. Moreover, it is of great significance to comprehensively summarize the formation and mechanisms of the atherosclerotic inflammatory microenvironment. Furthermore, significant progress has been made in treating AS by specifically inhibiting the NLRP3 inflammasome, and many novel therapeutic approaches can inhibit NLRP3 activation without affecting other inflammasomes. In addition, other pyroptosis-related molecules have attracted attention, and blocking any molecule in the pyroptosis activation pathway can reduce the release of inflammatory factors, thereby delaying the progression and deterioration of AS [Figure 6].
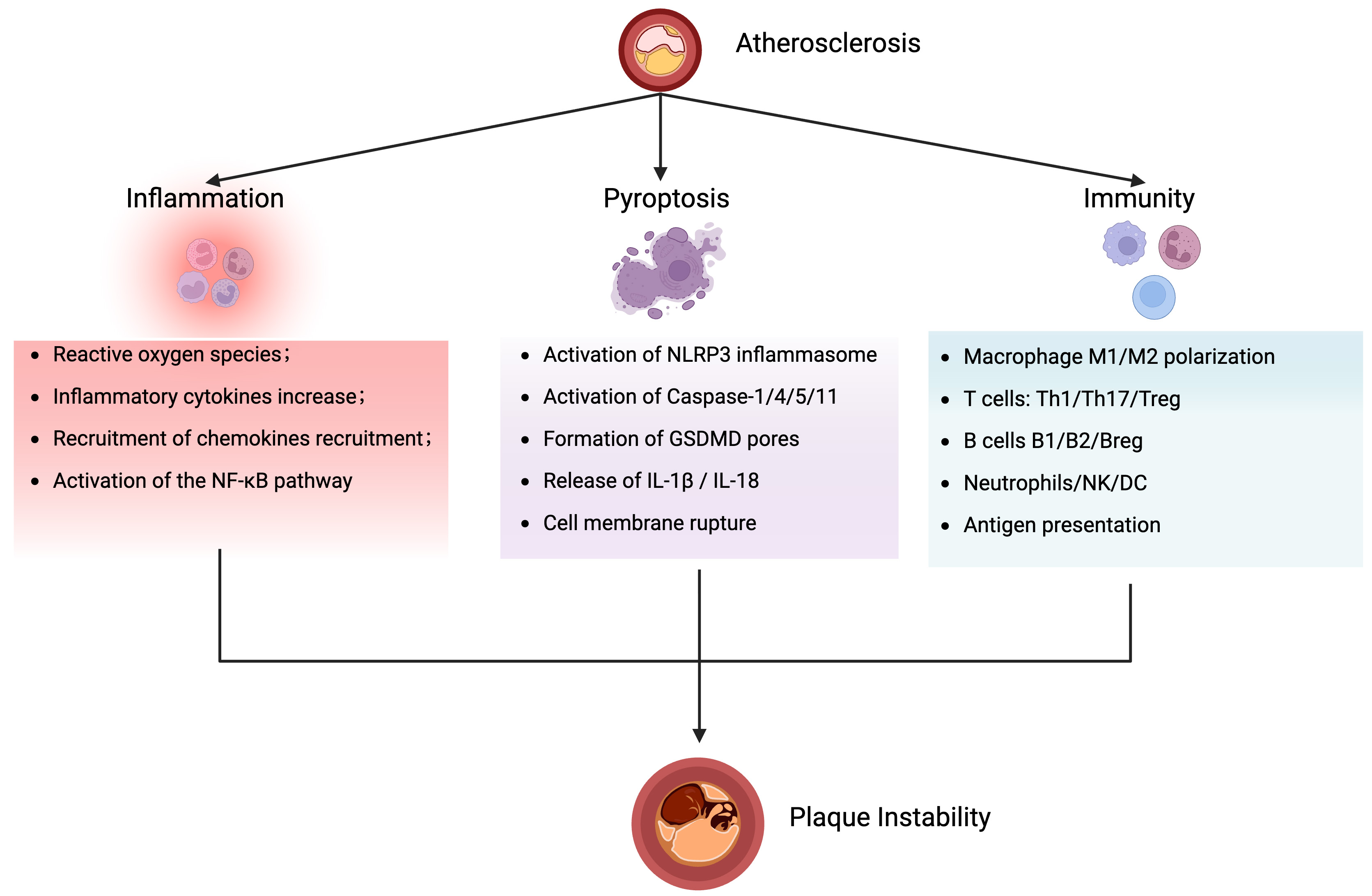
Figure 6. Schematic overview of the crossroads of atherosclerosis, inflammation, pyroptosis, and immunity. Atherosclerosis is the central pathology, where inflammation, pyroptosis, and immunity form a mutually reinforcing network that drives plaque instability and cardiovascular events. The figure created with BioRender.com. NF-κB: Nuclear factor kappa-B; IL-1β: interleukin-1 beta; IL-18: interleukin-18; Th17: T helper type 17 cell; Breg: regulatory B cell; B1: innate-like B cell; B2: conventional B cell; GSDMD: gasdermin D; NLRP3: NOD-like receptor family pyrin domain-containing protein 3; NK: natural killer; DC: dendritic cells.
Pyroptosis is a form of programmed cell death with pro-inflammatory properties, and its association with AS has been the focus of research in recent years. Pyroptosis is closely related to the occurrence and development of AS, and related inhibitors provide a new way to treat AS by inhibiting pyroptosis.
However, this review has several limitations. The limitations of this review are as follows: first, most studies remain limited to correlating pyroptosis-related molecules, such as NLRP3 and GSDMD, with plaque instability, without a clear causal chain proving that pyroptosis directly drives the progression of AS. Second, the summarized conclusions are mainly based on animal models and in vitro experiments, with insufficient clinical evidence. There is a lack of data from human cohorts or intervention trials, which limits the reliability of clinical translation. Third, the research focuses on a single cell type and a single pathway, lacking systematic analysis of multicellular interactions and of overall signaling network regulation. Fourth, there is a lack of stratified studies based on population heterogeneity, making it difficult to identify differences in pyroptosis pathway activity across genetic backgrounds and disease stages, and limiting consideration of individual variation.
Although great efforts have been made to determine the exact mechanism of atherosclerosis, many fundamental issues remain unresolved. To address these issues, investigators need to develop more effective methods.
DECLARATIONS
Acknowledgments
The Graphical Abstract and figures were created with BioRender.com.
Authors’ contributions
Collected relevant papers and drafted the manuscript: Wang Y
Participated in revising the review: Dong G
Involved in the initiation and revision of this review: Wang Y, Dong G, Wang Y, Zhang L, Wang M
Finalized the manuscript: Zhang L, Wang M
Provided financial support for this review: Wang M
All authors read and approved the final manuscript.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool DeepSeek (version DeepSeek-R1, released 2025-1-20) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by the Natural Science Foundation of Shandong Province (Major Basic Research) (ZR2024ZD18); Supported by the National Administration of Traditional Chinese Medicine (GZY-KJS-SD-2023-068) and the Natural Science Foundation of Shandong Province (ZR2021MC003)
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095-128.
2. Nguyen TD, Rahman NT, Sessa WC, Lee MY. Endothelial nitric oxide synthase (eNOS) S1176 phosphorylation status governs atherosclerotic lesion formation. Front Cardiovasc Med. 2023;10:1279868.
3. Wang K, Sun C, Zhuang H, Jiang XC, Chen Y. AFM reveals differential effects of acidification on LDL- and oxidized LDL-receptor interactions: biomechanical implications in atherogenesis. Cell Mol Biol Lett. 2025;30:32.
4. Wu J, Liu T, Xie W, Zhuo Y, Feng Y. Ox-LDL promotes M1-like polarization of macrophages through the miR-21-5p/SKP2/EP300 pathway. J Biochem Mol Toxicol. 2024;38:e23516.
5. Yashin AI, Wu D, Arbeev K, et al. Roles of interacting stress related genes in lifespan regulation: insights for translating experimental findings to humans. J Transl Genet Genom. 2021;5:357-79.
6. Cheong JE, Sun L. Targeting the IDO1/TDO2-KYN-AhR pathway for cancer immunotherapy - challenges and opportunities. Trends Pharmacol Sci. 2018;39:307-25.
7. St Paul M, Saibil SD, Kates M, et al. Ex vivo activation of the GCN2 pathway metabolically reprograms T cells, leading to enhanced adoptive cell therapy. Cell Rep Med. 2024;5:101465.
8. Almog T, Keshet R, Kandel-Kfir M, et al. Gene deletion of Interleukin-1α reduces ER stress-induced CHOP expression in macrophages and attenuates the progression of atherosclerosis in apoE-deficient mice. Cytokine. 2023;167:156212.
9. Chen R, Zhang Y, Zhao C. CHOP increases TRIB3-dependent miR-208 expression to potentiate vascular smooth muscle cell proliferation and migration by downregulating TIMP3 in atherosclerosis. Cardiovasc Drugs Ther. 2022;36:575-88.
10. Hong JG, Zheng HL, Wang P, Huang P, Gong DP, Zeng ZY. Hsa_ circ_0006867 regulates ox-LDL-induced endothelial injury via the miR-499a-3p/ADAM10 axis. Clin Hemorheol Microcirc. 2024;88:115-27.
11. Badimon L, Vilahur G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J Intern Med. 2014;276:618-32.
12. Liu SL, Bajpai A, Hawthorne EA, et al. Cardiovascular protection in females linked to estrogen-dependent inhibition of arterial stiffening and macrophage MMP12. JCI Insight. 2019:4.
14. Lv JJ, Wang H, Zhang C, et al. CD147 sparks atherosclerosis by driving M1 phenotype and impairing efferocytosis. Circ Res. 2024;134:165-85.
15. Yuan Y, Fan G, Liu Y, et al. The transcription factor KLF14 regulates macrophage glycolysis and immune function by inhibiting HK2 in sepsis. Cell Mol Immunol. 2022;19:504-15.
16. Felmlee MA, Jones RS, Rodriguez-Cruz V, Follman KE, Morris ME. Monocarboxylate transporters (SLC16): function, regulation, and role in health and disease. Pharmacol Rev. 2020;72:466-85.
17. Tassinari M, Tanzi G, Maggiore F, et al. Molecular mechanism of thyroxine transport by monocarboxylate transporters. Nat Commun. 2025;16:4493.
18. Zhang Y, Jiang H, Dong M, et al. Macrophage MCT4 inhibition activates reparative genes and protects from atherosclerosis by histone H3 lysine 18 lactylation. Cell Rep. 2024;43:114180.
19. Vellasamy DM, Lee SJ, Goh KW, et al. Targeting immune senescence in atherosclerosis. Int J Mol Sci. 2022;23:13059.
20. Zhao L, Lv X, Chen W, et al. Athero-oncology perspective: identifying hub genes for atherosclerosis diagnosis using machine learning. Front Immunol. 2025;16:1616096.
21. Zhan J, Chen Y, Liu Y, et al. IDO1-mediated AhR activation up-regulates pentose phosphate pathway via NRF2 to inhibit ferroptosis in lung cancer. Biochem Pharmacol. 2025;236:116913.
22. Zhen X, Zhang M, Hao S, Sun J. Glucose-6-phosphate dehydrogenase and transketolase: key factors in breast cancer progression and therapy. Biomed Pharmacother. 2024;176:116935.
23. Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771-84.
24. Soto-Heredero G, Gómez de Las Heras MM, Gabandé-Rodríguez E, Oller J, Mittelbrunn M. Glycolysis-a key player in the inflammatory response. FEBS J. 2020;287:3350-69.
25. Huang SC, Everts B, Ivanova Y, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15:846-55.
26. Ma C, Hua Y, Yang S, et al. Wogonin attenuates atherosclerosis via KLF11-mediated suppression of PPARα-YAP1-driven glycolysis and enhancement of ABCA1/G1-mediated cholesterol efflux. Adv Sci. 2025;12:e2500610.
27. Petit PX. Cellular ATP levels alone do not reliably reflect overall mitochondrial bioenergetics or mitochondrial dysfunction in Barth syndrome. J Transl Genet Genom. 2025;9:194-206.
28. Bekkering S, van den Munckhof I, Nielen T, et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis. 2016;254:228-36.
29. Li M, Liu X, Yu X, Yin A, Fu X, Guan X. FBP1/HIF-1α Axis mediates macrophage metabolic reprogramming and serves as diagnostic biomarkers in atherosclerosis. Int Immunopharmacol. 2026;169:116012.
30. Wang X, Liu X, Wu W, et al. Hypoxia activates macrophage-NLRP3 inflammasome promoting atherosclerosis via PFKFB3-driven glycolysis. FASEB J. 2024;38:e23854.
31. Guo S, Li A, Fu X, et al. Gene-dosage effect of Pfkfb3 on monocyte/macrophage biology in atherosclerosis. Br J Pharmacol. 2022;179:4974-91.
32. Poels K, Schnitzler JG, Waissi F, et al. Inhibition of PFKFB3 hampers the progression of atherosclerosis and promotes plaque stability. Front Cell Dev Biol. 2020;8:581641.
33. Groh L, Keating ST, Joosten LAB, Netea MG, Riksen NP. Monocyte and macrophage immunometabolism in atherosclerosis. Semin Immunopathol. 2018;40:203-14.
34. Yuan P, Hu X, Zhou Q. The nanomaterial-induced bystander effects reprogrammed macrophage immune function and metabolic profile. Nanotoxicology. 2020;14:1137-55.
35. Kieler M, Hofmann M, Schabbauer G. More than just protein building blocks: how amino acids and related metabolic pathways fuel macrophage polarization. FEBS J. 2021;288:3694-714.
36. Galán M, Fernández-Méndez L, Núñez V, et al. cDC1s promote atherosclerosis via local immunity and are targetable for therapy. Circ Res. 2025;137:400-16.
37. Huangfu N, Li F, Wang C, et al. METTL3/RBM15 augments the stability of Kdm6b mRNA and promotes STAT1-mediated macrophage activation and atherosclerosis. Exp Mol Med. 2025;57:2916-29.
38. Chen B, Zhu L, Lin X, et al. SLC4A10 impedes atherosclerosis by diminishing IFN-γ/GZMB levels of CD8+ T cells via the MAPK pathway. Front Immunol. 2025;16:1568999.
39. Wang Y, Huang H, Liu Z, et al. Talin1 modulates the Piezo1-YAP axis to regulate endothelial cell inflammation and atherosclerosis. Cell Mol Life Sci. 2025;83:40.
40. Liang G, Wang S, Shao J, et al. Tenascin-X mediates flow-induced suppression of EndMT and atherosclerosis. Circ Res. 2022;130:1647-59.
41. Zhu X, Wang Y, Soaita I, et al. Acetate controls endothelial-to-mesenchymal transition. Cell Metab. 2023;35:1163-1178.e10.
42. Wang X, Abraham S, McKenzie JAG, et al. LRG1 promotes angiogenesis by modulating endothelial TGF-β signalling. Nature. 2013;499:306-11.
43. Liuizė A, Mongirdienė A. TGF-β isoforms and GDF-15 in the development and progression of atherosclerosis. Int J Mol Sci. 2024;25:2104.
44. Guo S, Zhou Y, Xie X. Resveratrol inhibiting TGF/ERK signaling pathway can improve atherosclerosis: backgrounds, mechanisms and effects. Biomed Pharmacother. 2022;155:113775.
45. Chen PY, Qin L, Li G, et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat Metab. 2019;1:912-26.
47. Wang Y, Zang J, Liu C, Yan Z, Shi D. Interleukin-17 links inflammatory cross-talks between comorbid psoriasis and atherosclerosis. Front Immunol. 2022;13:835671.
48. Dubash S, Bridgewood C, McGonagle D, Marzo-Ortega H. The advent of IL-17A blockade in ankylosing spondylitis: secukinumab, ixekizumab and beyond. Expert Rev Clin Immunol. 2019;15:123-34.
49. Tsiogka A, Gregoriou S, Stratigos A, et al. The impact of treatment with IL-17/IL-23 inhibitors on subclinical atherosclerosis in patients with plaque psoriasis and/or psoriatic arthritis: a systematic review. Biomedicines. 2023;11:318.
50. Chiricozzi A, Guttman-Yassky E, Suárez-Fariñas M, et al. Integrative responses to IL-17 and TNF-α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol. 2011;131:677-87.
51. Ryu H, Chung Y. Regulation of IL-17 in atherosclerosis and related autoimmunity. Cytokine. 2015;74:219-27.
52. Hawkes JE, Yan BY, Chan TC, Krueger JG. Discovery of the IL-23/IL-17 signaling pathway and the treatment of psoriasis. J Immunol. 2018;201:1605-13.
53. Wu TJ, Chang SL, Lin CY, et al. IL-17 Facilitates VCAM-1 production and monocyte adhesion in osteoarthritis synovial fibroblasts by suppressing miR-5701 synthesis. Int J Mol Sci. 2022;23:6804.
54. Chen J, Dai AX, Tang HL, et al. Increase of ALCAM and VCAM-1 in the plasma predicts the Alzheimer’s disease. Front Immunol. 2022;13:1097409.
55. Shi TY, Wen XH, Meng J, Lu YW. Effect of IL-17 on pulmonary artery smooth muscle cells and connective tissue disease-associated pulmonary arterial hypertension. Immun Inflamm Dis. 2024;12:e1243.
56. Chen C, Zhang Q, Liu S, et al. IL-17 and insulin/IGF1 enhance adhesion of prostate cancer cells to vascular endothelial cells through CD44-VCAM-1 interaction. Prostate. 2015;75:883-95.
57. Singh A, Kraaijeveld AO, Curaj A, et al. CCL18 aggravates atherosclerosis by inducing CCR6-dependent T-cell influx and polarization. Front Immunol. 2024;15:1327051.
58. Wang J, Kang Z, Liu Y, Li Z, Liu Y, Liu J. Identification of immune cell infiltration and diagnostic biomarkers in unstable atherosclerotic plaques by integrated bioinformatics analysis and machine learning. Front Immunol. 2022;13:956078.
59. van Duijn J, Kritikou E, Benne N, et al. CD8+ T-cells contribute to lesion stabilization in advanced atherosclerosis by limiting macrophage content and CD4+ T-cell responses. Cardiovasc Res. 2019;115:729-38.
60. Vos WG, van Os BW, den Toom M, et al. T cell specific deletion of Casitas B lineage lymphoma-b reduces atherosclerosis, but increases plaque T cell infiltration and systemic T cell activation. Front Immunol. 2024;15:1297893.
61. Liu X, Li L, Yin Y, Zhang L, Wang W. Single-cell transcriptomic, transcriptomic, and metabolomic characterization of human atherosclerosis. Ann Transl Med. 2022;10:1215.
62. Wang Y, Zou Y, Jiang Q, et al. Ox-LDL-induced CD80+ macrophages expand pro-atherosclerotic NKT cells via CD1d in atherosclerotic mice and hyperlipidemic patients. Am J Physiol Cell Physiol. 2024;326:C1563-72.
63. Whitman SC, Rateri DL, Szilvassy SJ, Yokoyama W, Daugherty A. Depletion of natural killer cell function decreases atherosclerosis in low-density lipoprotein receptor null mice. Arterioscler Thromb Vasc Biol. 2004;24:1049-54.
64. Nour-Eldine W, Joffre J, Zibara K, et al. Genetic depletion or hyperresponsiveness of natural killer cells do not affect atherosclerosis development. Circ Res. 2018;122:47-57.
65. Zernecke A. Dendritic cells in atherosclerosis: evidence in mice and humans. Arterioscler Thromb Vasc Biol. 2015;35:763-70.
66. Liu D, Zhao J, Li L, et al. CD73: agent development potential and its application in diabetes and atherosclerosis. Front Immunol. 2024;15:1515875.
67. McAlpine CS, Kiss MG, Rattik S, et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature. 2019;566:383-7.
68. Keeter WC, Moriarty AK, Galkina EV. Role of neutrophils in type 2 diabetes and associated atherosclerosis. Int J Biochem Cell Biol. 2021;141:106098.
69. Zhang M, Montroy J, Sharma R, et al. The effects of biological sex on sepsis treatments in animal models: a systematic review and a narrative elaboration on sex- and gender-dependent differences in sepsis. Crit Care Explor. 2021;3:e0433.
70. Dileepan KN, Raveendran VV, Sharma R, et al. Mast cell-mediated immune regulation in health and disease. Front Med. 2023;10:1213320.
71. Lin Y, Wang J, Bu F, et al. Bacterial extracellular vesicles in the initiation, progression and treatment of atherosclerosis. Gut Microbes. 2025;17:2452229.
72. Jiang F, Chen Q, Wang W, Ling Y, Yan Y, Xia P. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J Hepatol. 2020;72:156-66.
73. Páramo JA, Cenarro A, Civeira F, Roncal C. Extracellular vesicles in atherosclerosis: current and forthcoming impact. Clin Investig Arterioscler. 2025;37:100718.
74. Jiapaer Z, Li C, Yang X, et al. Extracellular non-coding RNAs in cardiovascular diseases. Pharmaceutics. 2023;15:155.
75. Ketelut-Carneiro N, Fitzgerald KA. Apoptosis, pyroptosis, and necroptosis-Oh My! J Mol Biol. 2022;434:167378.
76. Fan TJ, Han LH, Cong RS, Liang J. Caspase family proteases and apoptosis. Acta Biochim Biophys Sin. 2005;37:719-27.
77. Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity. 2019;50:1352-64.
78. Wei X, Xie F, Zhou X, et al. Role of pyroptosis in inflammation and cancer. Cell Mol Immunol. 2022;19:971-92.
79. Sharma BR, Kanneganti TD. NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol. 2021;22:550-9.
80. Yin Z, Zhang J, Shen Z, Qin JJ, Wan J, Wang M. Regulated vascular smooth muscle cell death in vascular diseases. Cell Prolif. 2024;57:e13688.
81. Fan X, Han J, Zhong L, et al. Macrophage-derived GSDMD plays an essential role in atherosclerosis and cross talk between macrophages via the mitochondria-STING-IRF3/NF-κB axis. Arterioscler Thromb Vasc Biol. 2024;44:1365-78.
82. Jiang X, Ma C, Gao Y, et al. Tongxinluo attenuates atherosclerosis by inhibiting ROS/NLRP3/caspase-1-mediated endothelial cell pyroptosis. J Ethnopharmacol. 2023;304:116011.
83. Pan X, Xu H, Ding Z, et al. Guizhitongluo tablet inhibits atherosclerosis and foam cell formation through regulating Piezo1/NLRP3 mediated macrophage pyroptosis. Phytomedicine. 2024;132:155827.
84. Zeng W, Wu D, Sun Y, et al. The selective NLRP3 inhibitor MCC950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci Rep. 2021;11:19305.
85. Chen R, Zhang H, Tang B, et al. Macrophages in cardiovascular diseases: molecular mechanisms and therapeutic targets. Signal Transduct Target Ther. 2024;9:130.
86. Zhou Y, Zhou H, Hua L, et al. Verification of ferroptosis and pyroptosis and identification of PTGS2 as the hub gene in human coronary artery atherosclerosis. Free Radic Biol Med. 2021;171:55-68.
87. Qiu Y, Shi YN, Zhu N, et al. A lipid perspective on regulated pyroptosis. Int J Biol Sci. 2023;19:2333-48.
88. Schmacke NA, O’Duill F, Gaidt MM, et al. IKKβ primes inflammasome formation by recruiting NLRP3 to the trans-Golgi network. Immunity. 2022;55:2271-2284.e7.
89. Lv Y, Jiang Z, Zhou W, et al. Low-shear stress promotes atherosclerosis via inducing endothelial cell pyroptosis mediated by IKKε/STAT1/NLRP3 pathway. Inflammation. 2024;47:1053-66.
90. Coll RC, Schroder K, Pelegrín P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci. 2022;43:653-68.
91. Tall AR, Bornfeldt KE. Inflammasomes and atherosclerosis: a mixed picture. Circ Res. 2023;132:1505-20.
92. Hou C, Jiang X, Sheng W, et al. Xinmaikang (XMK) tablets alleviate atherosclerosis by regulating the SREBP2-mediated NLRP3/ASC/Caspase-1 signaling pathway. J Ethnopharmacol. 2024;319:117240.
93. Vieira CP, Fortmann SD, Hossain M, et al. Selective LXR agonist DMHCA corrects retinal and bone marrow dysfunction in type 2 diabetes. JCI Insight. 2020;5:137230.
94. Lu QB, Wan MY, Wang PY, et al. Chicoric acid prevents PDGF-BB-induced VSMC dedifferentiation, proliferation and migration by suppressing ROS/NFκB/mTOR/P70S6K signaling cascade. Redox Biol. 2018;14:656-68.
95. Francis GA. The greatly under-represented role of smooth muscle cells in atherosclerosis. Curr Atheroscler Rep. 2023;25:741-9.
96. He X, Bai Q, Zhang X, Zhang L. MgCl2 attenuates ox-LDL-induced vascular smooth muscle-derived foam cells pyroptosis by downregulating the TLR4/NF-κB Signaling pathway. Biol Trace Elem Res. 2023;201:5242-56.
97. Okuyama H, Langsjoen PH, Hamazaki T, et al. Statins stimulate atherosclerosis and heart failure: pharmacological mechanisms. Expert Rev Clin Pharmacol. 2015;8:189-99.
98. Ragusa R, Basta G, Neglia D, De Caterina R, Del Turco S, Caselli C. PCSK9 and atherosclerosis: looking beyond LDL regulation. Eur J Clin Invest. 2021;51:e13459.
99. Peng S, Xu LW, Che XY, et al. Atorvastatin inhibits inflammatory response, attenuates lipid deposition, and improves the stability of vulnerable atherosclerotic plaques by modulating autophagy. Front Pharmacol. 2018;9:438.
100. Guo C, Chi Z, Jiang D, et al. Cholesterol homeostatic regulator SCAP-SREBP2 integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity. 2018;49:842-856.e7.
101. Bodapati AP, Hanif A, Okafor DK, et al. PCSK-9 inhibitors and cardiovascular outcomes: a systematic review with meta-analysis. Cureus. 2023;15:e46605.
102. Feng X, Chen W, Ni X, et al. Metformin, macrophage dysfunction and atherosclerosis. Front Immunol. 2021;12:682853.
103. Liu X, Guo JW, Lin XC, et al. Macrophage NFATc3 prevents foam cell formation and atherosclerosis: evidence and mechanisms. Eur Heart J. 2021;42:4847-61.
104. Bao Q, Zhang B, Zhou L, et al. CNP ameliorates macrophage inflammatory response and atherosclerosis. Circ Res. 2024;134:e72-91.
105. Li W, Li Y, Kang J, et al. 4-octyl itaconate as a metabolite derivative inhibits inflammation via alkylation of STING. Cell Rep. 2023;42:112145.
106. You Y, Bao WL, Zhang SL, et al. Sorting nexin 10 mediates metabolic reprogramming of macrophages in atherosclerosis through the lyn-dependent TFEB signaling pathway. Circ Res. 2020;127:534-49.
107. Ku EJ, Kim BR, Lee JI, et al. The anti-atherosclerosis effect of anakinra, a recombinant human interleukin-1 receptor antagonist, in apolipoprotein E knockout mice. Int J Mol Sci. 2022;23:4906.
108. Yang M, Lv H, Liu Q, et al. Colchicine alleviates cholesterol crystal-induced endothelial cell pyroptosis through activating AMPK/SIRT1 pathway. Oxid Med Cell Longev. 2020;2020:9173530.
109. Rahmani S, Roohbakhsh A, Pourbarkhordar V, Hayes AW, Karimi G. Melatonin regulates mitochondrial dynamics and mitophagy: cardiovascular protection. J Cell Mol Med. 2024;28:e70074.
110. Cong L, Liu X, Bai Y, et al. Melatonin alleviates pyroptosis by regulating the SIRT3/FOXO3α/ROS axis and interacting with apoptosis in atherosclerosis progression. Biol Res. 2023;56:62.
111. Wu R, Sun F, Zhang W, Ren J, Liu GH. Targeting aging and age-related diseases with vaccines. Nat Aging. 2024;4:464-82.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].