


EQTL Mendelian randomization to identify genetic insights and regulatory mechanisms in benign prostatic hyperplasia
 0
0 Abstract
Aim: This study aims to identify key genes and regulatory mechanisms associated with benign prostatic hyperplasia (BPH) using expression quantitative trait locus (eQTL)-based Mendelian randomization and integrative bioinformatics analyses.
Methods: Cis-expression quantitative trait locus (cis-eQTL) data were obtained from the eQTLGen Consortium. Genome-wide association study (GWAS) summary statistics for BPH were retrieved from the FinnGen biobank and the IEU OpenGWAS database. Gene expression profiles were obtained from the Gene Expression Omnibus (GEO) database. Mendelian randomization and colocalization analyses were performed to prioritize BPH-associated genes. Subsequent analyses included gene set enrichment
Results: A total of 105 and 11 BPH-associated genes were identified in the training and validation datasets, respectively. Colocalization analysis further prioritized five key genes, including C2 (Complement Component 2), GUCY1B2 (Guanylate Cyclase 1 Soluble Subunit Beta 2), OLFM4 (Olfactomedin 4), ITPR1 (Inositol 1,4,5-Trisphosphate Receptor Type 1), and KLHL36 (Kelch Like Family Member 36). Functional analyses indicated that these genes were involved in multiple BPH-related pathways, including tumor protein p53 (p53), mechanistic target of rapamycin (mTOR), transforming growth factor-beta (TGF-β), and interleukin-17 (IL-17) signaling. These genes were also associated with immune cell infiltration, immune-related factors, transcriptional regulation, miRNA interactions, metabolic pathways, and disease-related gene networks.
Conclusion: Five candidate genes associated with BPH were identified, and their potential regulatory mechanisms were characterized through integrative genetic and transcriptomic analyses. These findings provide new insights into the molecular basis of BPH and may inform future biomarker development and mechanism-driven therapeutic strategies, although further experimental validation remains necessary.
Keywords
INTRODUCTION
Benign prostatic hyperplasia (BPH) represents the most prevalent benign enlargement of the prostate gland and is a major cause of lower urinary tract symptoms (LUTS) in older men[1]. Its pathological features include histological proliferation of prostatic stromal and glandular components, progressive anatomical enlargement of the prostate, urodynamic obstruction of the bladder outlet, and clinical manifestations primarily characterized by LUTS, all of which substantially compromise patients’ quality of life[1]. The incidence of BPH rises markedly with age, typically becoming clinically evident after 40 years[1,2]. Prevalence exceeds 50% in men aged 60 years and reaches approximately 80% by 80 years of age[1,2]. Dysregulation of the balance between proliferation and apoptosis in prostatic epithelial and stromal cells is regarded as a central mechanism in BPH pathogenesis[3]. In addition, BPH development is multifactorial, involving androgens and their interaction with estrogens, stromal-epithelial crosstalk, growth factors, inflammatory cells, neurotransmitters, and genetic determinants[3]. However, the underlying molecular mechanisms remain incompletely understood. The present study identified core genes and regulatory networks associated with BPH pathogenesis through Mendelian randomization and integrative analytical approaches.
Mendelian randomization (MR) is an analytical framework that leverages genetic variants as instrumental variables (IVs) to infer potential causal relationships between exposures and outcomes[4]. Expression quantitative trait loci (eQTLs) are genetic variants associated with gene expression levels. eQTL-based MR integrates regulatory genetic variants with genome-wide association study (GWAS) data to evaluate the potential causal effects of gene expression on disease risk[5]. This approach has been widely applied to complex diseases, including cancer, endocrine disorders, metabolic diseases, and immune-related conditions[6-8]. Moreover, eQTL-based MR facilitates the prioritization of potential therapeutic targets by identifying genes whose expression levels may causally influence disease susceptibility[9]. To date, no eQTL-based MR study has systematically examined the causal relationship between gene expression and BPH. In this study, datasets from eQTLGen, FinnGen, IEU OpenGWAS, and GEO were integrated to identify BPH-associated genes through MR and colocalization analyses. Five key genes - C2 (Complement Component 2), GUCY1B2 (Guanylate Cyclase 1 Soluble Subunit Beta 2), OLFM4 (Olfactomedin 4), ITPR1 (Inositol 1,4,5-Trisphosphate Receptor Type 1), and KLHL36 (Kelch Like Family Member 36) - were further characterized, and their potential biological functions and regulatory mechanisms in BPH were explored.
MATERIALS AND METHODS
Data acquisition
Cis-expression quantitative trait locus (cis-eQTL) data were obtained from the eQTLGen Consortium (https://www.eqtlgen.org), which catalogs genetic determinants of gene expression in whole blood and their contributions to complex human traits and diseases. In its second phase, the consortium conducted large-scale genome-wide meta-analyses of cis- and trans-eQTLs using whole-blood transcriptomes derived from multiple population-based cohorts.
Outcome GWAS data were restricted to individuals of European ancestry. Summary statistics for BPH were retrieved from the FinnGen biobank (FinnGen R10, N14_PROSTHYPERPLA), where cases were defined based on corresponding International Classification of Diseases (ICD) codes extracted from registry data. This dataset comprised 32,956 cases and 130,139 controls. Replication summary statistics were obtained from the IEU OpenGWAS database (EBI GWAS Catalog; GCST90044257), in which cases were likewise identified using ICD codes, including 6,505 cases and 202,303 controls for prostate hyperplasia.
Gene expression data were retrieved from the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/info/datasets.html), a comprehensive repository of functional genomic datasets. Dataset GSE132714, generated on the GPL16791 platform, was selected for transcriptomic analysis of BPH, comprising a control group (n = 4) and a disease group (n = 18).
Mendelian randomization analysis
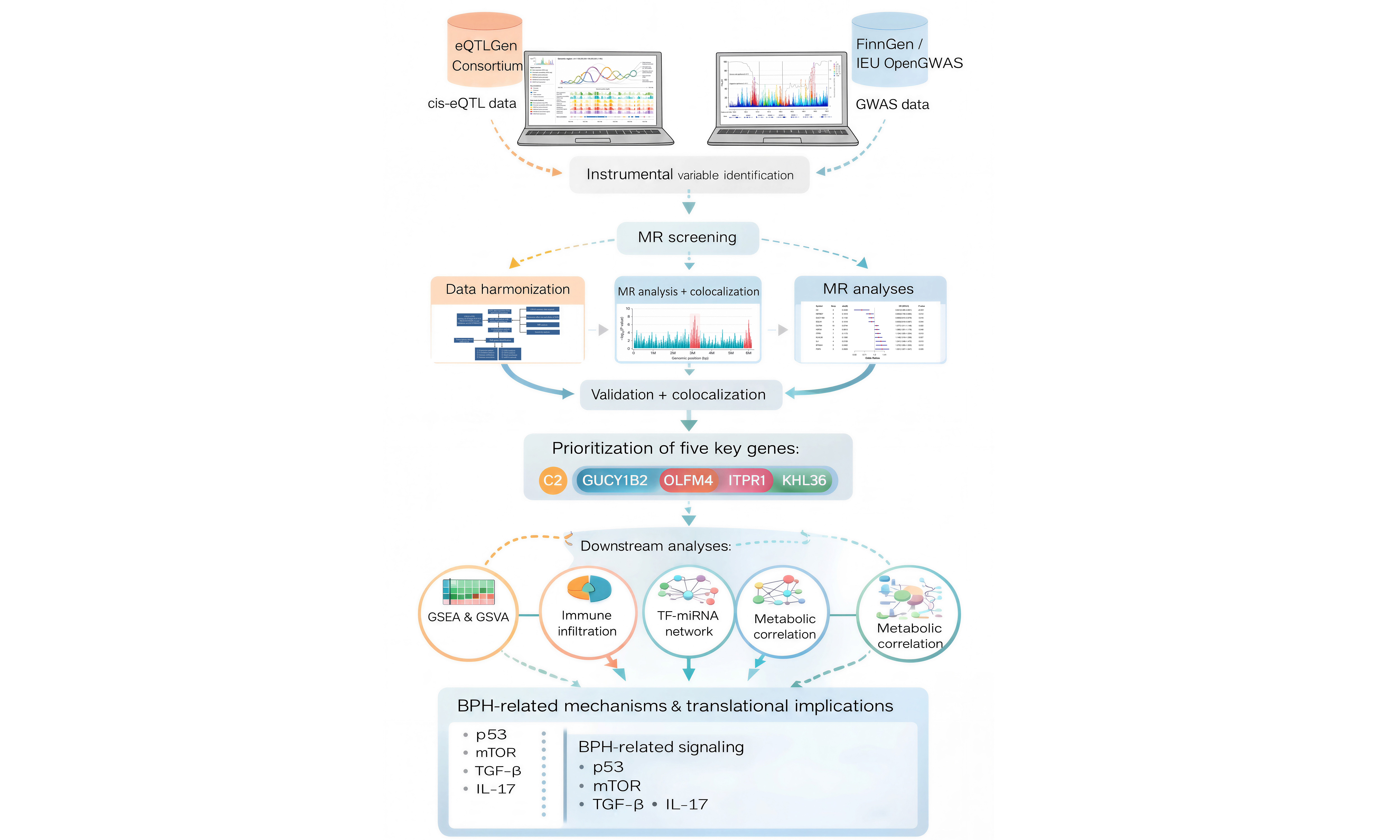
Figure 1 summarizes the analytical workflow of this MR study. Outcome identifiers were screened from the FinnGen biobank, EBI, and GWAS catalog databases. These identifiers were then used to extract relevant causal associations from eQTL GWAS summary statistics data (https://gwas.mrcieu.ac.uk/). Single nucleotide polymorphisms (SNPs) associated with each gene at a locus-wide significance threshold of P < 1 × 10-5 were selected as candidate IVs. A relaxed threshold relative to the conventional genome-wide significance level (P < 5 × 10-8) was adopted to preserve cis-eQTL instrument availability and improve coverage for gene-level MR screening. To minimize false-positive findings, linkage disequilibrium (LD) clumping, validation in an independent dataset, leave-one-out sensitivity analysis, and colocalization analysis were subsequently performed. LD clumping, validation in an independent dataset, leave-one-out sensitivity analysis, and colocalization analysis were subsequently performed. Among SNPs with R2 < 0.001 (clumping window = 10,000 kb), only variants with P < 5 × 10-5 were retained. Causal effects were estimated using four complementary MR methods, including inverse-variance weighted (IVW), MR-Egger, weighted median, and weighted mode approaches. The IVW method was used as the primary estimator due to its highest statistical efficiency under valid instrument assumptions or balanced horizontal pleiotropy, and its estimates were interpreted as the main causal effects. MR-Egger, weighted median, and weighted mode analyses were applied as sensitivity approaches to assess robustness. When only a single genetic variant was available, the Wald ratio method was used. The MR-Egger method relies on the InSIDE (Instrument Strength Independent of Direct Effect) assumption, which requires independence between instrument strength and direct pleiotropic effects. The weighted median estimator provides consistent estimates when more than 50% of instruments are valid, whereas the weighted mode method yields robust inference when the largest instrument cluster reflects the true causal effect.
Figure 1. Flowchart of the study design. GWAS: Genome-wide association study; MR: mendelian randomization; eQTL: expression quantitative trait locus; GSEA: gene set enrichment analysis; GSVA: gene set variation analysis.
Sensitivity analysis
A leave-one-out sensitivity analysis was conducted within the MR framework to evaluate the influence of individual SNPs on causal estimates for benign prostatic enlargement. Each variant was sequentially excluded, and causal effects were re-estimated using the remaining instruments to identify SNPs exerting disproportionate influence. For each iteration, 95% confidence intervals were generated to assess the stability of single-variant contributions and the robustness of the overall estimate. All SNPs were retained in the final report, with key variant-level estimates presented. Comparison across iterations enabled identification of influential SNPs and confirmation of result robustness.
Colocalization analysis
Colocalization analysis was performed using the R package coloc, integrating whole-blood cis-eQTL summary statistics with GWAS summary data for BPH. Posterior probabilities were calculated within a 100-kb window centered on the index variant. In this framework, H3 represents the posterior probability of two traits (gene expression and prostate hyperplasia) being associated with distinct causal variants, whereas H4 represents the probability of a shared causal variant underlying both traits. A threshold of SNP.PP.H4 > 0.95 was applied to define significant colocalization.
Gene set enrichment analysis
Based on the expression levels of the five candidate genes (C2, GUCY1B2, OLFM4, ITPR1, and KLHL36), samples were stratified into high-expression and low-expression groups for each gene. Gene set enrichment analysis (GSEA) using Kyoto Encyclopedia of Genes and Genomes (KEGG) gene sets identified significantly enriched pathways (adjusted P < 0.05), highlighting biological processes associated with BPH-related expression patterns.
Gene set variation analysis
Gene set variation analysis (GSVA), an unsupervised enrichment approach, was further applied to quantify pathway activity across samples using gene sets from Molecular signatures database (MSigDB) version 7.0, enabling comparison of functional differences between samples.
Immune cell infiltration analysis
Immune cell infiltration was estimated using CIBERSORT, which deconvolutes bulk transcriptomic data to infer the relative proportions of 22 immune cell subsets, including T cells, B cells, plasma cells, and myeloid lineages, based on 547 signature genes.
Regulatory network analysis of hub genes
Transcription factor prediction was performed using the R package RcisTarget, which applies motif-based enrichment analysis. The normalized enrichment score (NES) was used to quantify motif enrichment. Motif overrepresentation within gene sets was assessed using the area under the curve (AUC) derived from gene ranking recovery, and NES values were calculated from the AUC distribution across motifs.
Construction of miRNA network
miRNA-gene regulatory relationships were constructed based on experimentally or computationally predicted interactions. Candidate miRNAs targeting key genes were retrieved from miRCode, and the resulting miRNA-gene network was visualized using Cytoscape.
Statistical analysis
MR analyses rely on three core assumptions: relevance (genetic instruments are associated with the exposure), independence (instruments are not associated with confounders), and exclusion restriction (instruments influence the outcome only through the exposure). Horizontal pleiotropy arises when instruments affect the outcome through alternative pathways. All analyses were performed in R version 4.3.0 using two-tailed tests, with P < 0.05 considered statistically significant.
RESULTS
Mendelian randomization analysis of the training set
Druggable genes were obtained from previously published literature[10]. To identify genes potentially associated with BPH within the druggable genome, GWAS summary statistics from the FinnGen biobank (finngen_R10_N14_PROSTHYPERPLA), comprising 32,956 cases and 130,139 controls, were analyzed. Using the IVW method as the primary MR estimator, 105 genes were identified as significantly associated with BPH in the training dataset (Figure 2, IVW P < 0.05). Among these, PTPN13, JAK3, KDM4C, C2, BIRC2, CD38, TGFBR1, IL1R1, RRM2B, FAM20A, XYLT1, AKR1C1, AZIN1, KBTBD7, CR1, RGS12, CD1C, IL4, ADORA3, CXCR6, LOXL3, RPS6KB2, RYR2, GNAS, CDK12, PLA2G4B, BTN3A1, MAPKAPK3, CLEC3B, CDC42, LGR6, COL6A3, VKORC1L1, AKR1C2, ULK3, CORIN, THRA, GUCY1B2, IMPA1, CA2, GPBAR1, SERPINB9, SIGLEC11, LIPN, MAPK3, APOBEC3A, IFNGR2, and SIRPB1 were associated with reduced BPH risk, whereas FPR1, FCRL5, PAM, RHD, SERPINB1, DHRS7, PRKCB, CDKL1, DHFR, SLC12A7, RIPK1, MPO, SLC22A5, OLFM4, CXCR1, SBK1, BACE1, CTSF, TMEM9B, CTSB, KLHL12, P2RY14, H3F3A, CCL3L1, CYP2U1, CD160, HLA-G, CD6, CD79B, PTK2, LAMB2, CD79A, CD68, C5, EGLN1, VEGFA, CD5, ITPR1, CRISP3, STK38, PRRT3, KLHL36, COL17A1, FCRL2, TLR6, BMP2K, CD22, MMP11, GLB1, DRD4, C5AR1, PDGFRB, AKT3, MAP3K3, FGF9, MAP4K4, and TUBB4B were associated with increased BPH risk. Leave-one-out analysis indicated that no single SNP exerted a disproportionate influence on the overall MR estimates, supporting the robustness of the training-set results.
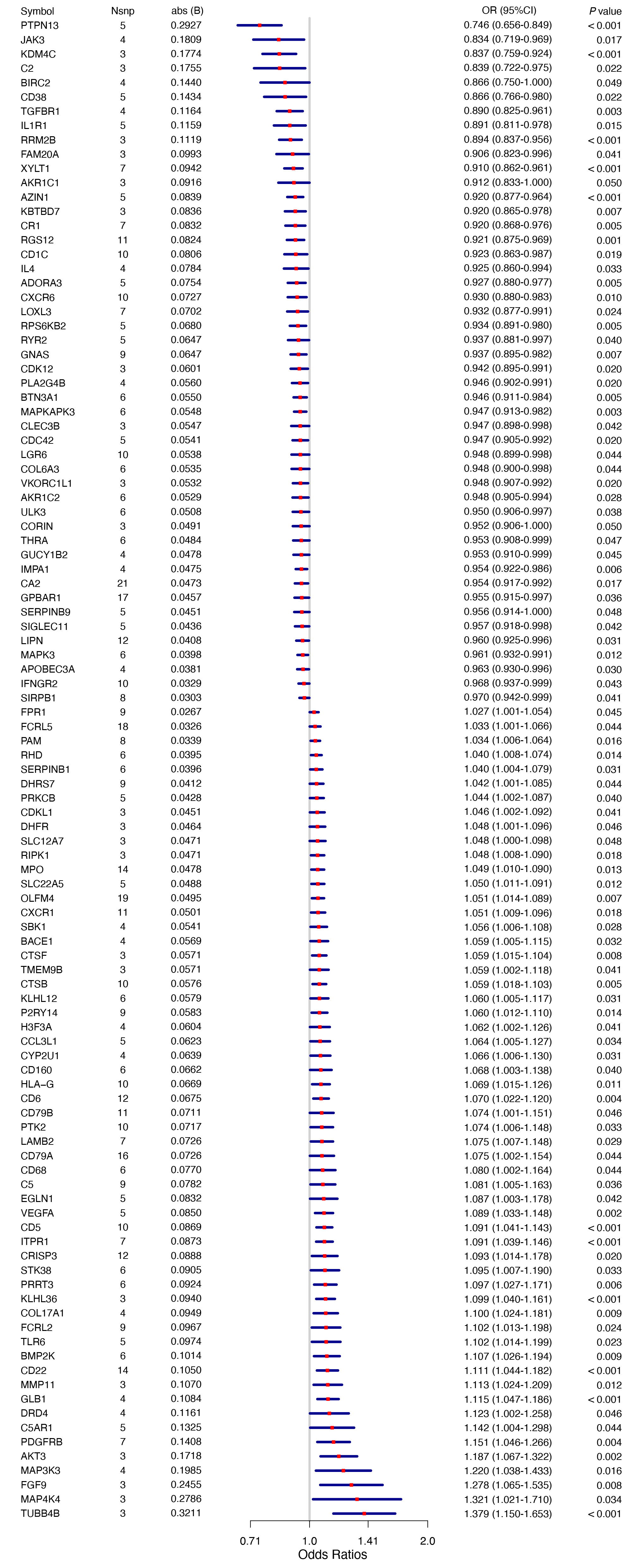
Figure 2. Mendelian randomization analysis in the training set. Mendelian randomization identified 105 causal associations between gene expression and BPH risk. BPH: Benign prostatic hyperplasia; OR: odds ratio.
Mendelian randomization analysis and colocalization analysis of the validation set
To validate these findings, an independent GWAS dataset from the IEU OpenGWAS database (GCST90044257), including 6,505 cases and 202,303 controls, was further analyzed using the same MR framework. Eleven genes were identified as significantly associated with BPH in the validation dataset (Figure 3, IVW P < 0.05). Among these, C2, KBTBD7, GUCY1B2, and EGLN1 were associated with reduced BPH risk, whereas OLFM4, H3F3A, ITPR1, KLHL36, IL4, BTN3A1, and FGF9 were associated with increased BPH risk. Leave-one-out analysis confirmed that these associations were not driven by any single instrumental SNP, supporting their stability [Figure 4]. Colocalization analysis was subsequently performed for the 11 replicated genes. Five genes - C2, GUCY1B2, OLFM4, ITPR1, and KLHL36 - showed strong evidence of colocalization (PP.H4 > 0.95) and were prioritized for downstream analyses [Figure 5]. To further evaluate their discriminatory performance, exploratory single-gene receiver operating characteristic (ROC) analyses were conducted using the GSE132714 dataset. GUCY1B2, ITPR1, and KLHL36 demonstrated moderate-to-good discrimination between BPH and control samples, whereas C2 and OLFM4 showed limited performance [Supplementary Table 1].
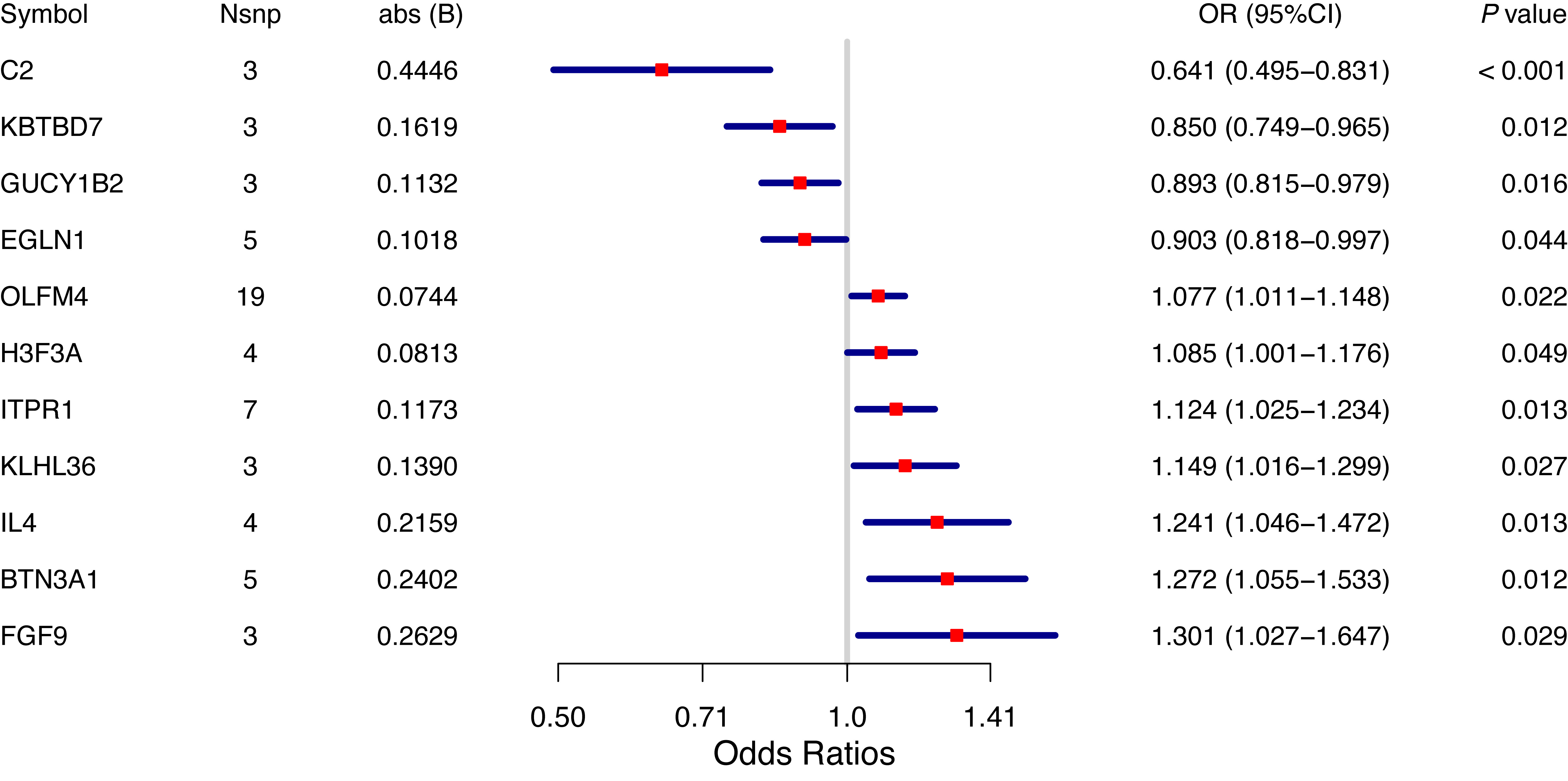
Figure 3. Mendelian randomization analysis in the validation set. The IVW method was used as the primary estimator to identify genes significantly associated with BPH in the validation dataset (IVW P < 0.05). MR-Egger, weighted median, and weighted mode methods were applied as complementary sensitivity analyses. BPH: Benign prostatic hyperplasia; IVW: Inverse-variance weighted; MR: mendelian randomization; OR: odds ratio.
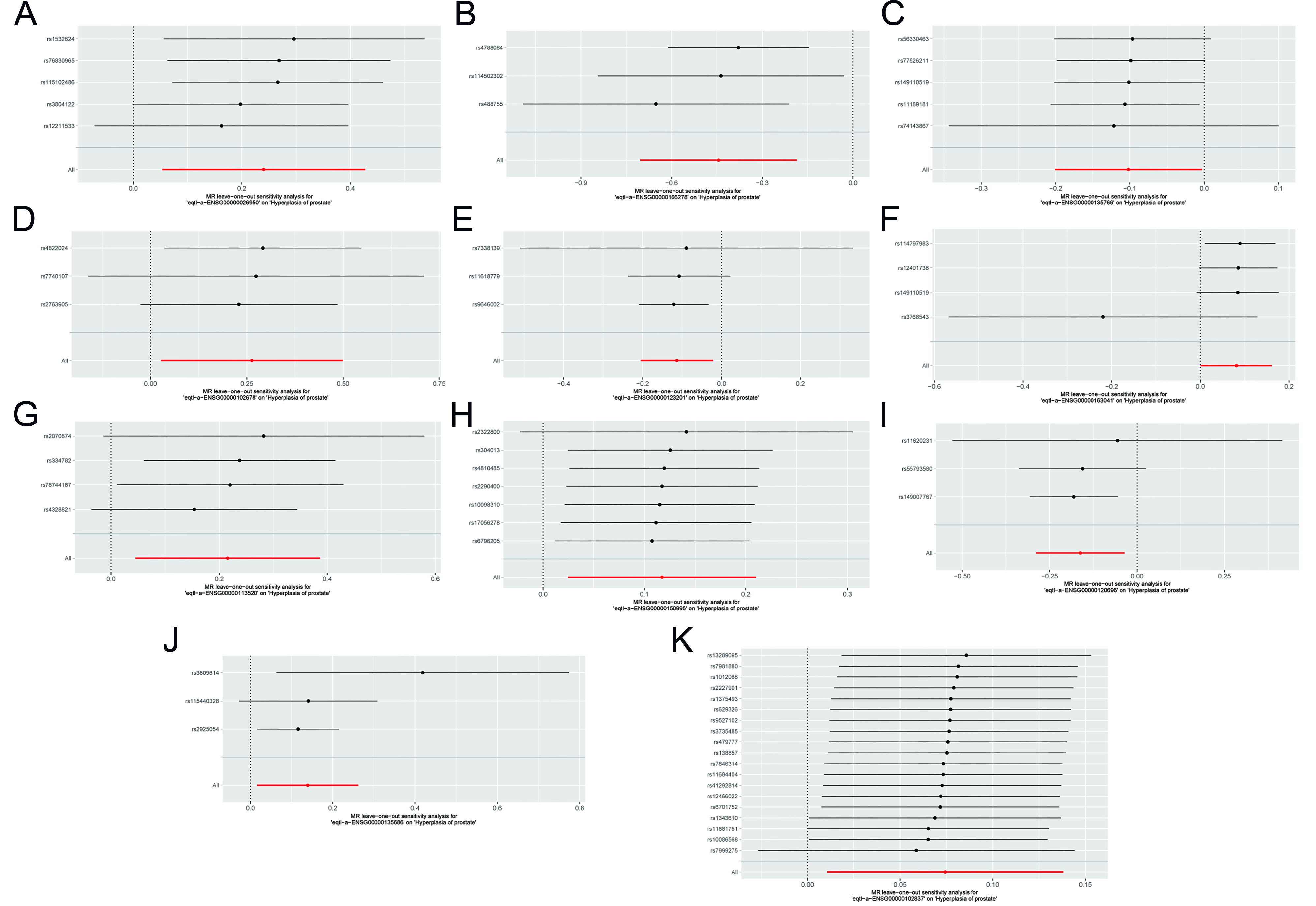
Figure 4. Leave-one-out sensitivity analysis. (A-K) Forest plots of leave-one-out analyses for SNPs corresponding to key genes. MR: Mendelian randomization; SNPs: single nucleotide polymorphisms.
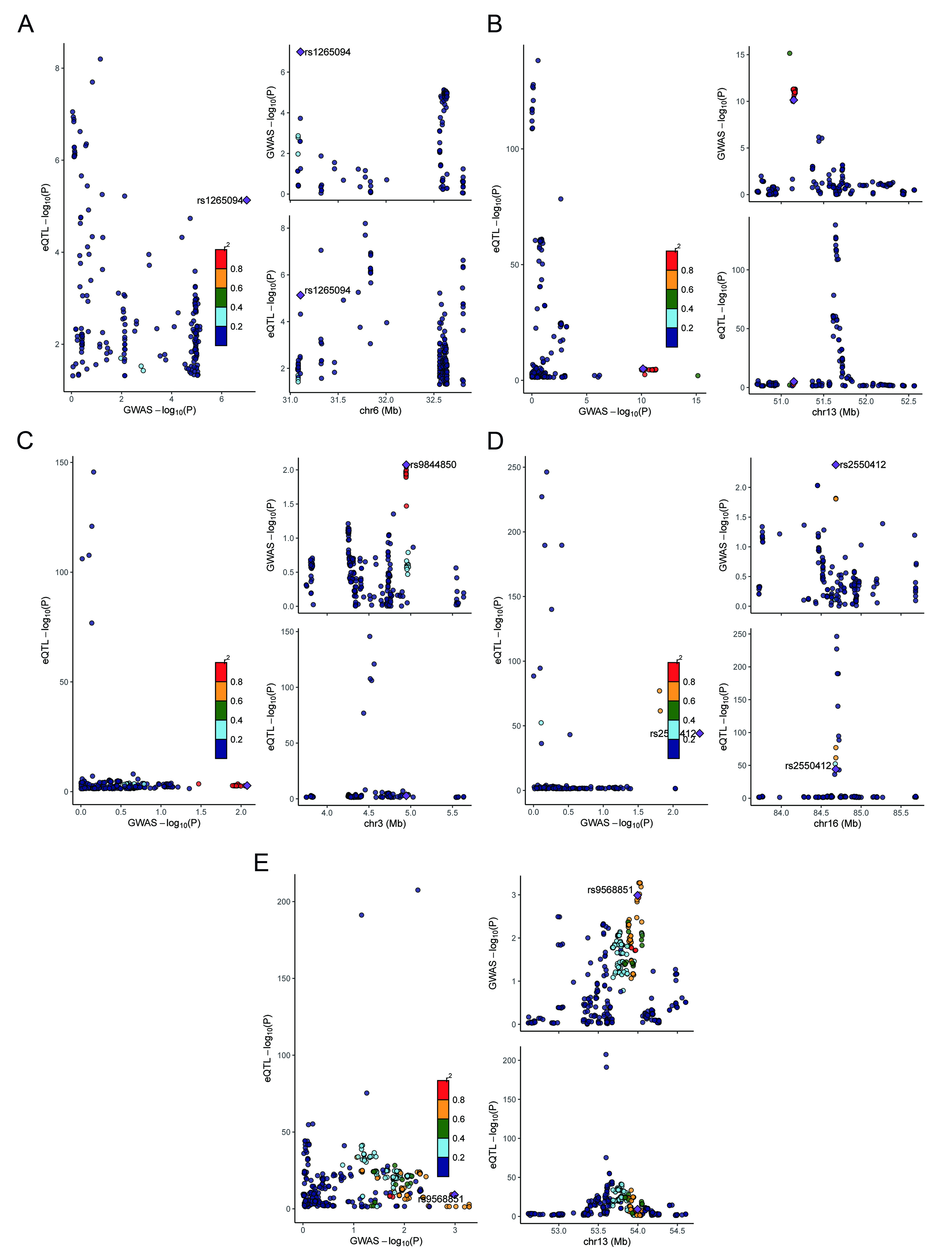
Figure 5. Colocalization analysis. (A-E) Regional association plots comparing GWAS and eQTL signals for C2, GUCY1B2, OLFM4, ITPR1, and KLHL36. Left panels display the relationship between GWAS and eQTL association signals, whereas right panels show regional association patterns across genomic coordinates. eQTL: Expression quantitative trait locus; GWAS: Genome-wide association study; C2: complement component 2; GUCY1B2: guanylate cyclase 1 soluble subunit beta 2; OLFM4: olfactomedin 4; ITPR1: inositol 1,4,5-trisphosphate receptor type 1; KLHL36: kelch like family member 36.
GSEA
To investigate the biological functions of the five prioritized genes, GSEA was performed based on their expression profiles. C2 was mainly enriched in ribosome, pentose phosphate pathway (PPP), and fatty acid metabolism [Figure 6A]. GUCY1B2 was enriched in mechanistic target of rapamycin (mTOR), mitogen-activated protein kinase (MAPK), and Wnt signaling pathways [Figure 6B]. IITPR1 was enriched in tumor protein p53 (p53), transforming growth factor-beta (TGF-β), and tumor necrosis factor (TNF) signaling pathways
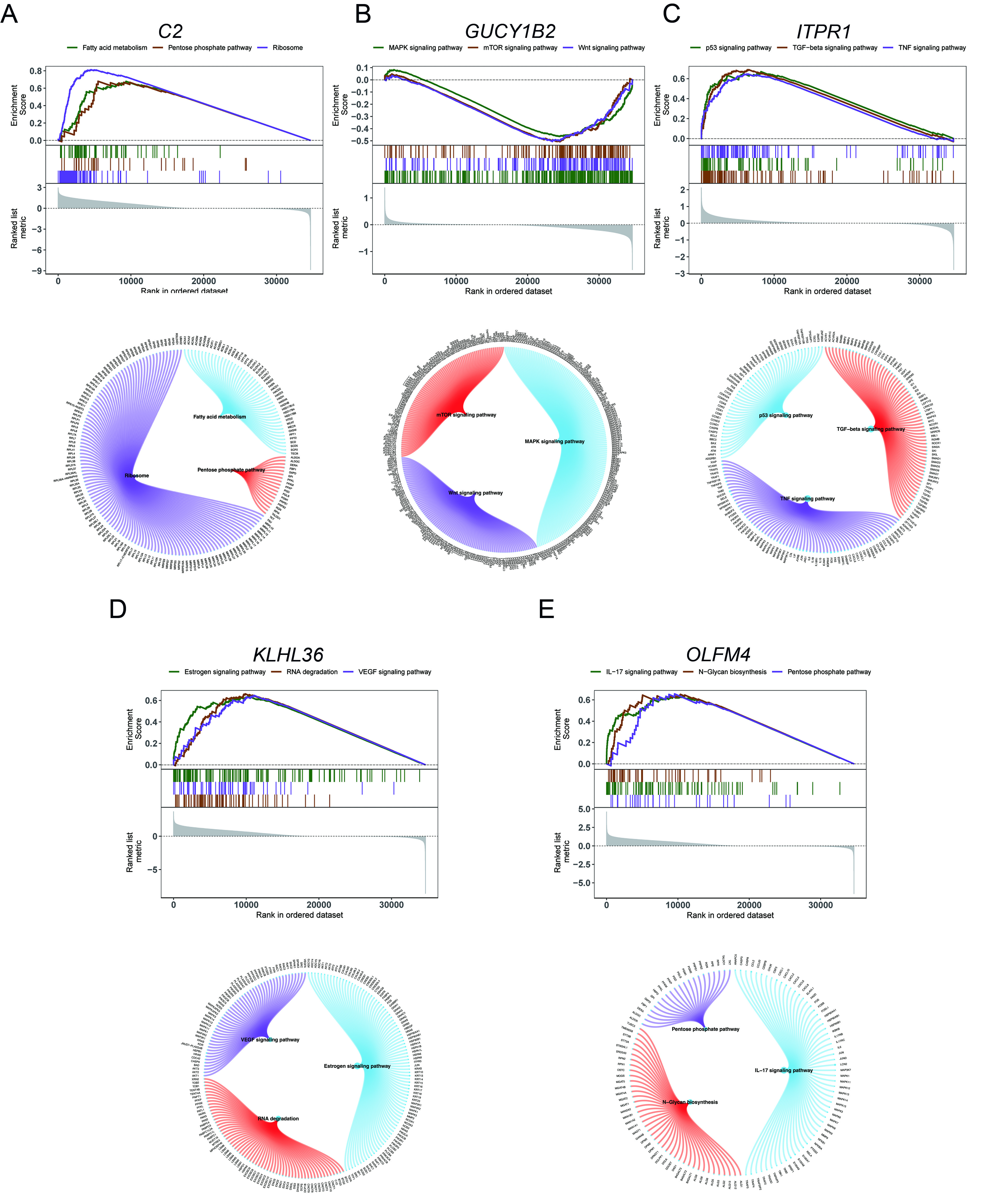
Figure 6. GSEA of key genes. (A-E) KEGG pathway enrichment analysis of key genes, including associated signaling pathways and contributing genes. GSEA: Gene set enrichment analysis; C2: complement component 2; GUCY1B2: guanylate cyclase 1 soluble subunit beta 2; OLFM4: olfactomedin 4; ITPR1: inositol 1,4,5-trisphosphate receptor type 1; KLHL36: kelch like family member 36; KEGG: kyoto encyclopedia of genes and genomes; mTOR: mechanistic target of rapamycin; MAPK: mitogen-activated protein kinase; TNF: tumor necrosis factor; TGF: transforming growth factor; IL-17: interleukin-17.
GSVA
GSVA was performed to compare pathway activity between the high- and low-expression groups for each prioritized gene. C2 was associated with p53 signaling and related pathways [Figure 7A]. GUCY1B2 showed enrichment in reactive oxygen species signaling and interleukin-6 (IL-6)/Janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) signaling [Figure 7B]. ITPR1 was associated with IL-2/STAT5 signaling, Hedgehog signaling, and related pathways [Figure 7C]. KLHL36 was enriched in mTORC1 signaling, Notch signaling, and additional cancer-related pathways [Figure 7D]. OLFM4 was associated with phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mTOR signaling, Wnt/β-catenin signaling, and other proliferation-related pathways [Figure 7E]. Overall, GSVA results further supported the involvement of these genes in key biological processes relevant to BPH.
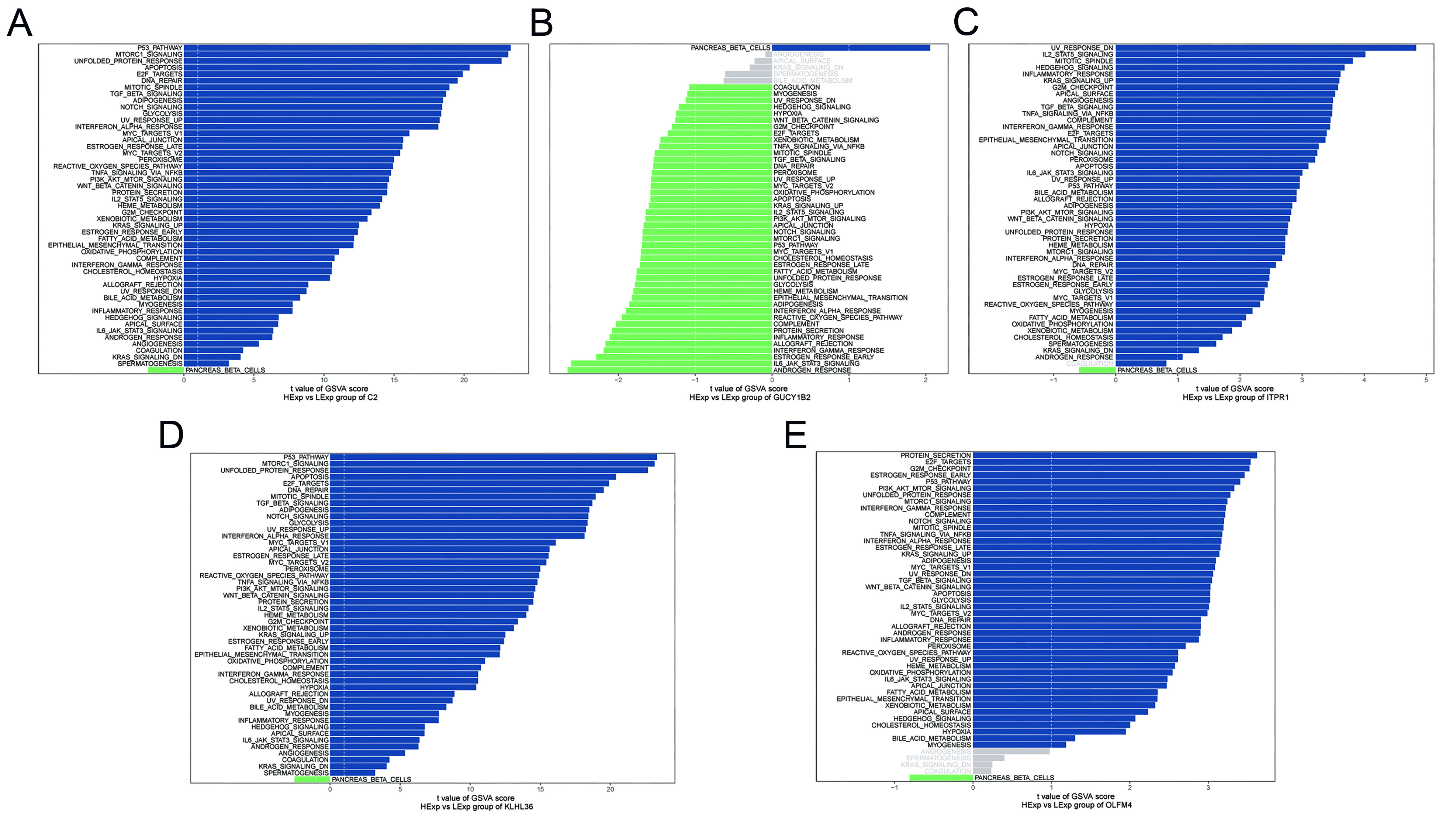
Figure 7. GSVA of key genes. (A-E) GSVA results for key genes. Blue indicates pathways enriched in the high-expression group, whereas green indicates pathways enriched in the low-expression group. Hallmark gene sets were used as the reference. C2: complement component 2; GUCY1B2: guanylate cyclase 1 soluble subunit beta 2; OLFM4: olfactomedin 4; ITPR1: inositol 1,4,5-trisphosphate receptor type 1; KLHL36: kelch like family member 36; GSVA: gene set variation analysis.
Immune infiltration analysis
To characterize the immune microenvironment associated with BPH, CIBERSORT was applied to estimate the relative proportions of 22 immune cell types. The overall immune landscape is presented in Figure 8A, and correlations among immune cell subsets are shown in Figure 8B. Comparative analysis between control and BPH groups revealed significant differences in specific immune cell populations, particularly natural killer (NK) cells and plasma cells [Figure 8C], suggesting that altered immune infiltration may contribute to BPH pathophysiology.
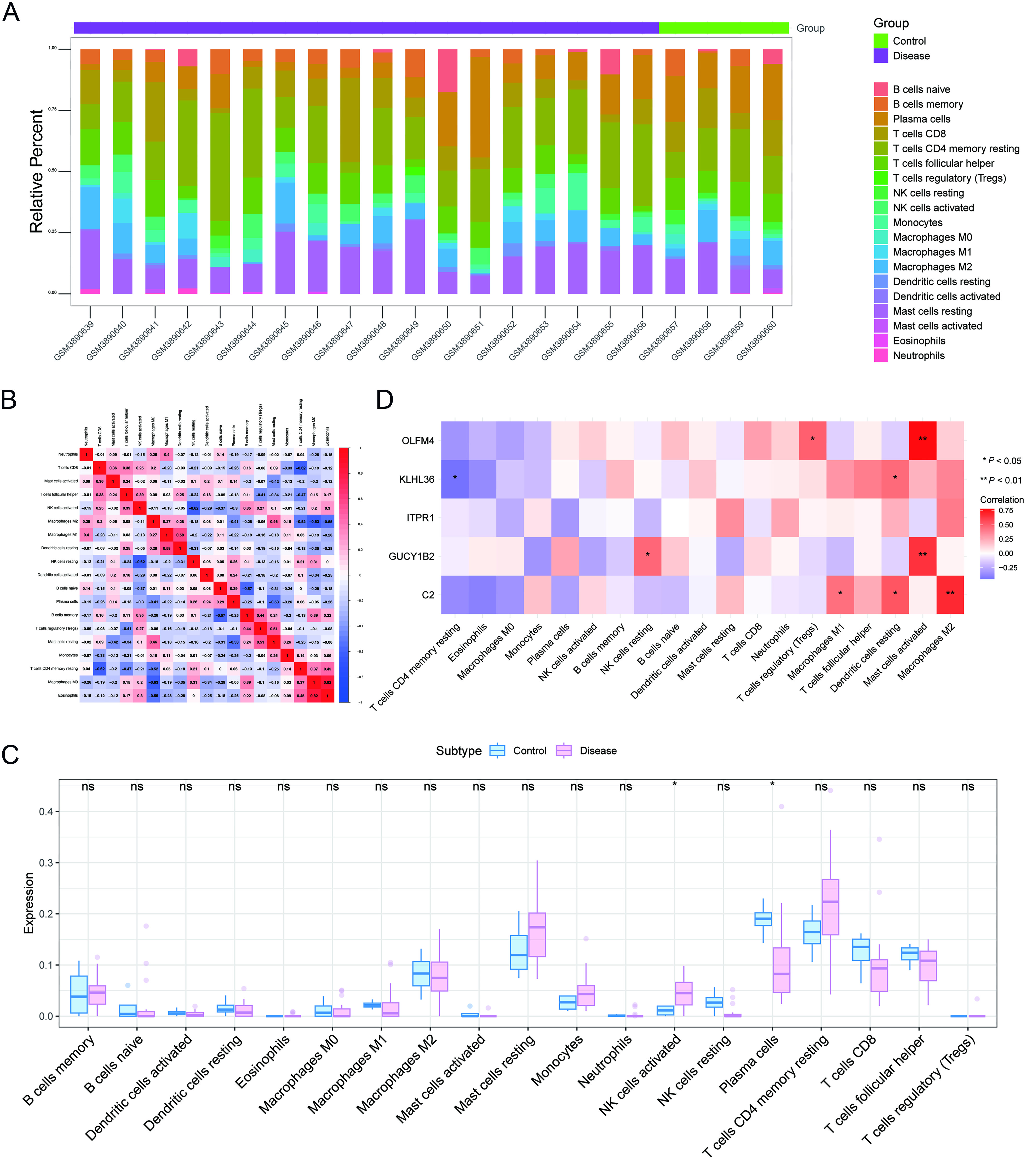
Figure 8. Immune infiltration analysis. (A) Relative proportions of immune cell subpopulations; (B) Pearson correlations among immune cell types (blue: negative correlation; red: positive correlation); (C) Differences in immune cell composition between control and BPH groups, assessed using the Wilcoxon rank-sum test; (D) Correlations between prioritized genes and immune cell infiltration, evaluated using Pearson correlation analysis. *P < 0.05; **P < 0.01; ***P < 0.001. BPH: Benign prostatic hyperplasia.
The relationship between key genes, immune cells, and immune factors
Associations between the five prioritized genes and immune cell infiltration were further evaluated. Significant correlations were observed across multiple immune cell subsets [Figure 8D]. Notably, C2 showed a strong association with M1 macrophages, whereas GUCY1B2, KLHL36, and OLFM4 were also linked to several immune cell populations, indicating potential interactions between these genes and the immune microenvironment in BPH. In addition, the Tumor and Immune System Interaction Database (TISIDB) was used to assess the relationships between the prioritized genes and immune-related factors, including chemokines, immune inhibitors, immune stimulators, major histocompatibility complex (MHC) molecules, and receptors [Figure 9]. Collectively, these results suggest that the prioritized genes may contribute to BPH progression through immune-related regulatory mechanisms.

Figure 9. Associations between key genes and immune-related factors. (A-E) Correlations between key genes and chemokines, immune inhibitors, immune stimulators, MHC molecules, and receptors. MHC: Major histocompatibility complex; C2: complement component 2; GUCY1B2: guanylate cyclase 1 soluble subunit beta 2; OLFM4: olfactomedin 4; ITPR1: inositol 1,4,5-trisphosphate receptor type 1; KLHL36: kelch like family member 36.
Transcriptional regulation and miRNA network related to key genes
To further explore upstream regulatory mechanisms, transcription factor enrichment analysis was performed using RcisTarget. Coordinated regulation of the five genes by multiple transcription factors was observed, and significantly enriched motifs and corresponding transcription factors are presented in Figure 10A and B. Among them, nuclear factor kappa B subunit 1 (NFKB1) exhibited the highest normalized enrichment score (NES) (6.3), followed by RELA proto-oncogene, NF-kappa B subunit (RELA) (NES = 5.67), suggesting that inflammatory transcriptional programs may be involved in regulating these genes. A miRNA-mRNA regulatory network was also constructed using the miRCode database (https://mirdb.org/). A total of 81 candidate miRNAs targeting the five prioritized genes were identified, generating 183 miRNA-mRNA interaction pairs [Figure 10C]. These results indicate that the prioritized genes may be regulated through both transcription factor- and miRNA-mediated mechanisms.
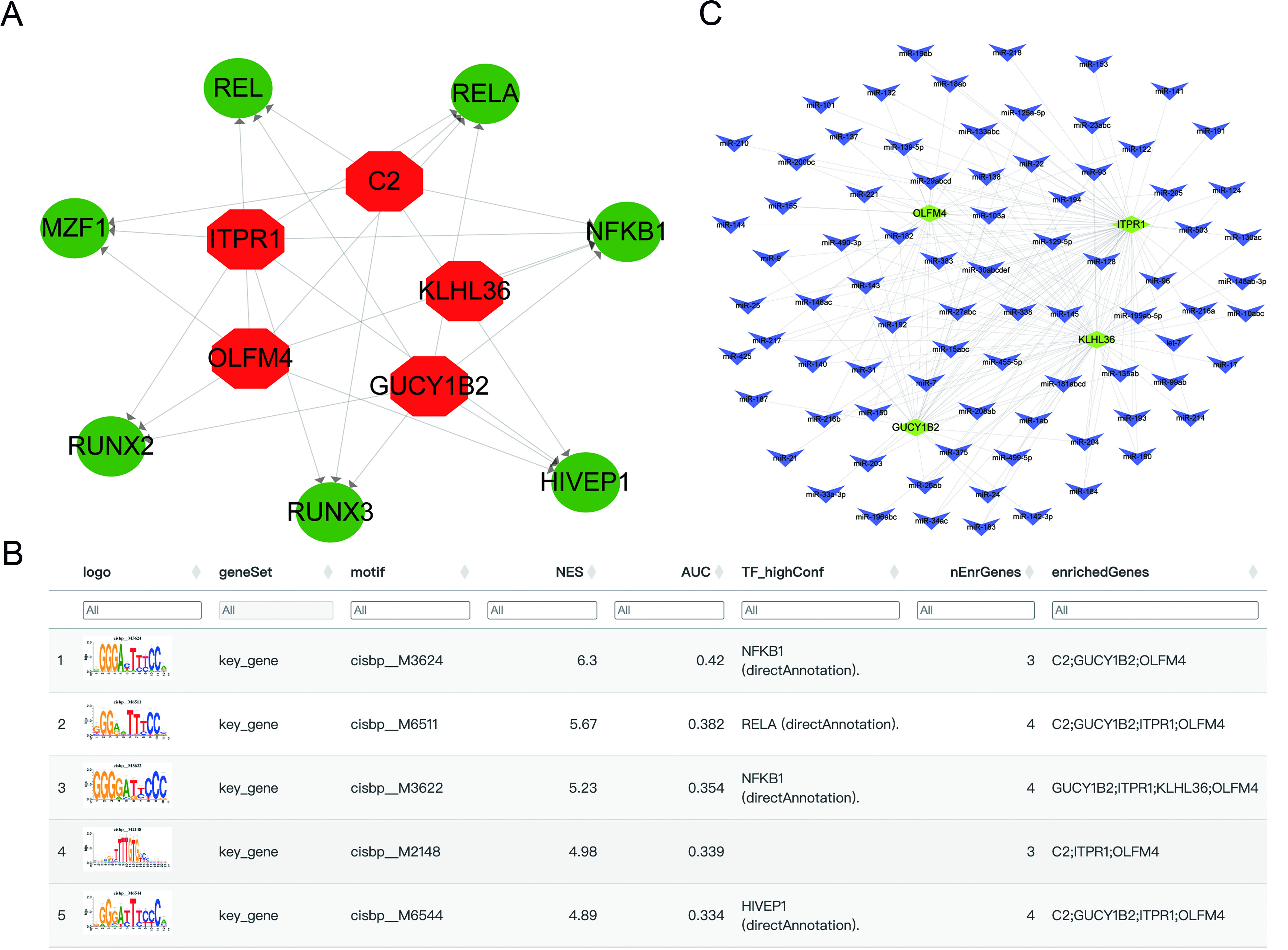
Figure 10. Transcriptional and miRNA regulatory networks associated with key genes. (A) Transcriptional regulatory network (red: key genes; green: transcription factors); (B) Enriched motifs and corresponding transcription factors; (C) miRNA-gene interaction network (yellow: mRNA; blue: miRNA). C2: Complement component 2; GUCY1B2: guanylate cyclase 1 soluble subunit beta 2; OLFM4: olfactomedin 4; ITPR1: inositol 1,4,5-trisphosphate receptor type 1; KLHL36: kelch like family member 36; REL: REL proto-oncogene, NF-kB subunit; MZF1: Myeloid zinc finger 1; RUNX2: RUNX family transcription factor 2; RUNX3: RUNX family transcription factor 3; HIVEP1: HIVEP zinc finger 1; NFKB1: nuclear factor kappa B subunit 1; RELA: RELA proto-oncogene, NF-kB subunit.
Correlation between key genes, metabolic pathways, and disease progression genes
Associations between the prioritized genes and metabolic pathways were further investigated. Heatmap analysis indicated that these genes were linked to multiple metabolic processes, including amino acid metabolism, lipid metabolism, drug metabolism, and other progression-related pathways [Figure 11A]. BPH-related genes were subsequently retrieved from the GeneCards database, and the top 20 candidates were selected for comparison of expression levels between control and BPH groups. Among these, AKT1, BRCA2, CDH1, GNAS, KLK3, POR, and STAR exhibited significant differential expression [Figure 11B]. Correlation analysis further revealed close associations between the prioritized genes and established disease-related genes [Figure 11C]. Notably, C2 showed a significant negative correlation with CYP11B1 (Pearson’s r = -0.539), whereas KLHL36 displayed a strong positive correlation with GNAS (Pearson’s r = 0.874). These results further support the involvement of the prioritized genes in metabolic alterations and disease progression in BPH.
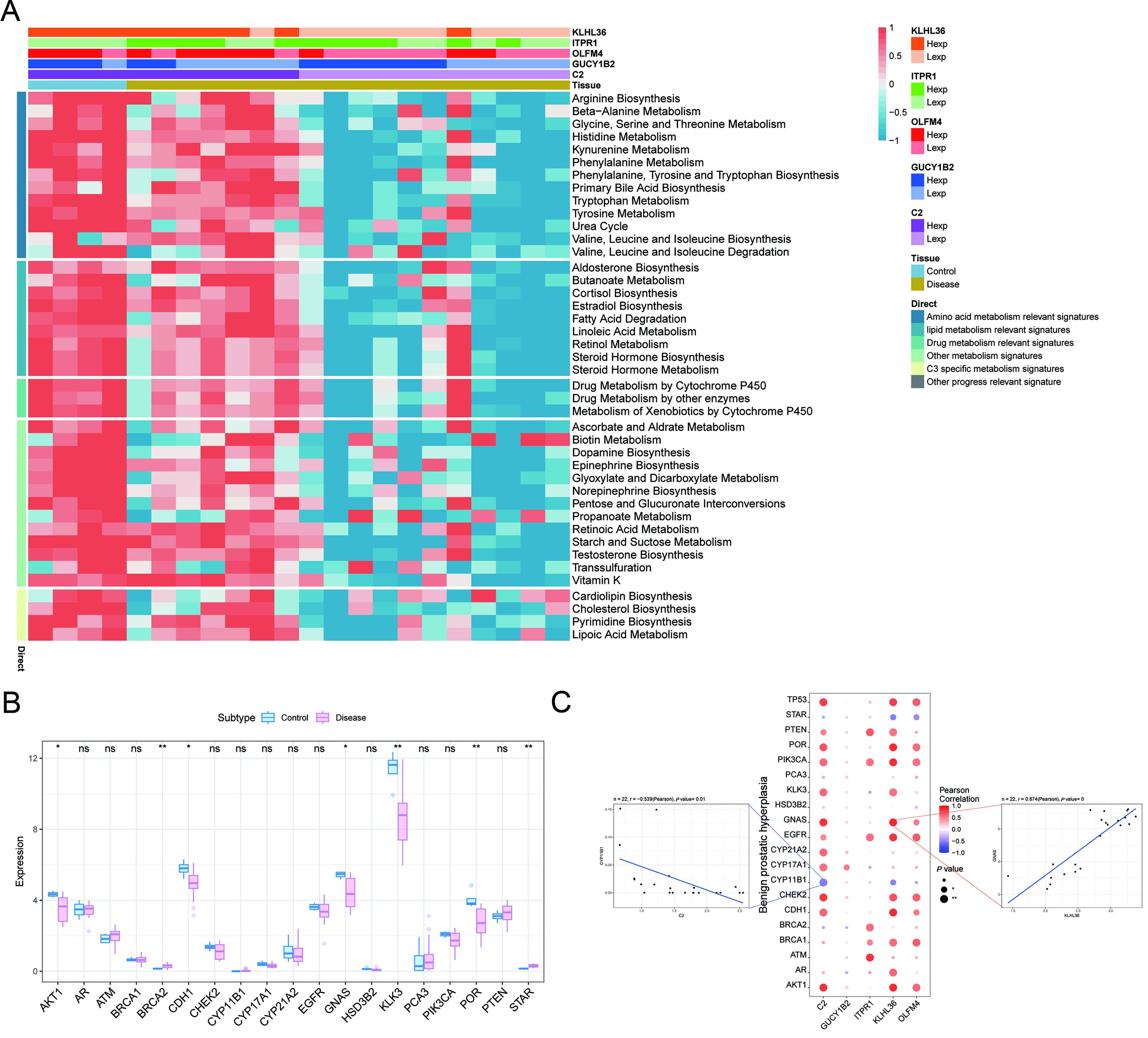
Figure 11. Correlations between key genes, representative pathways, and disease-related genes. (A) Heatmap of correlations between prioritized genes and representative metabolic pathways; blue indicates lower values and red indicates higher values; (B) Differential expression of representative BPH-related genes between control and BPH samples; blue represents controls and pink represents patients; (C) Correlation analysis between prioritized genes and representative BPH-related genes. The central bubble plot shows Pearson correlation coefficients, with color indicating direction and magnitude and dot size reflecting statistical significance. The left and right scatter plots illustrate representative correlations for C2 versus CYP11B1 and KLHL36 versus GNAS, respectively. BPH: Benign prostatic hyperplasia; C2: Complement component 2; GUCY1B2: guanylate cyclase 1 soluble subunit beta 2; OLFM4: olfactomedin 4; ITPR1: inositol 1,4,5-trisphosphate receptor type 1; KLHL36: kelch like family member 36; GNAS: GNAS complex locus.
DISCUSSION
BPH is a common age-related disorder that primarily affects older men[11]. Its pathogenesis is multifactorial and is influenced by age-associated metabolic dysregulation, hormonal imbalance, and chronic inflammation. BPH can be chronic and debilitating when inadequately managed, substantially impairing quality of life in affected men worldwide[12]. Long-term prognosis largely depends on treatment response and timely management of complications. Pharmacological therapy, including α-blockers and 5α-reductase inhibitors, is generally effective in relieving symptoms and maintaining long-term disease control[13]. 5α-reductase inhibitors reduce prostate volume by suppressing dihydrotestosterone (DHT) synthesis, thereby lowering the risk of urinary retention[14,15]. However, prolonged or lifelong administration is often required, and adverse effects such as dizziness, orthostatic hypotension, and sexual dysfunction may occur, thereby reducing quality of life and treatment adherence. In some cases, surgical intervention, including transurethral resection of the prostate (TURP) or laser-based procedures, is required depending on disease severity. Although these procedures provide substantial symptom relief, they are also associated with complications such as bleeding, urinary incontinence, retrograde ejaculation, and prolonged recovery[16-18].
Given these limitations, identification of genes involved in the initiation and progression of prostate enlargement is of considerable importance. Such genes may serve as actionable targets for more precise therapeutic strategies. They provide insights into the molecular pathogenesis of BPH, improve understanding of disease progression, and support the development of novel interventions. Targeting key genes may reduce reliance on conventional pharmacotherapy, minimize long-term adverse effects, and support more individualized treatment approaches.
eQTL data were obtained from the eQTLGen Consortium, GWAS data were retrieved from the FinnGen biobank and EBI databases, and BPH-related gene expression profiles were downloaded from the NCBI GEO database. These datasets were integrated to construct training and validation cohorts for gene prioritization using MR and colocalization analyses. In the training cohort, 163,095 samples were analyzed, leading to the identification of 105 BPH-associated genes. In the validation cohort, 208,808 samples were analyzed, confirming 11 BPH-associated genes. Colocalization analysis further prioritized five candidate genes: C2, GUCY1B2, ITPR1, KLHL36, and OLFM4.
GSEA indicated that C2 was primarily enriched in ribosomal pathways, suggesting a potential role in protein synthesis within prostatic cells. GSVA further suggested involvement of C2 in activation of the p53 signaling pathway. Accumulating evidence indicates that upregulation of functional p53 and activation of the p53 pathway suppress proliferation and promote apoptosis in BPH tissues and cells. Pharmacological agents such as captopril and cetrorelix have been shown to upregulate p53 expression in experimental models, thereby attenuating androgen receptor-induced prostatic hyperplasia. Accordingly, C2 may contribute to delayed BPH progression through modulation of p53 pathway activity.
GUCY1B2 is regarded as a frameshift-mutated pseudogene that does not encode a functional protein[19]. Nevertheless, it may still have regulatory relevance in certain human biological processes. Functional enrichment analysis indicated that GUCY1B2 is associated with mTOR, MAPK, and Wnt signaling pathways. Accumulating evidence suggests that AKT/mTOR signaling promotes stromal cell proliferation in BPH[20,21]. Activation of the AKT pathway disrupts the Bax/Bcl-2 balance in prostatic cell lines, thereby suppressing apoptosis in prostate cells[22]. Conversely, inhibition of the AKT/mTOR pathway attenuates cell proliferation and inflammatory responses[23,24]. In addition, the p38/MAPK pathway regulates FOXO3a activity, thereby inhibiting apoptosis and reactive oxygen species accumulation in BPH-1 epithelial cells[25]. Increased expression of Wnt/β-catenin and cyclin D1 has been observed in BPH specimens, indicating that activation of Wnt/β-catenin signaling is associated with disease progression. Inhibition of this pathway has been shown to alleviate BPH progression in vivo and in vitro by inducing apoptosis, causing G0/G1 cell cycle arrest, reducing tissue fibrosis, and suppressing epithelial-mesenchymal transition (EMT)[26]. These findings suggest that GUCY1B2 may attenuate BPH progression through modulation of the mTOR, MAPK, and Wnt signaling pathways.
ITPR1-related expression patterns were enriched in the p53, TGF-β, and TNF signaling pathways. In addition, inflammatory cell infiltration within the BPH microenvironment is associated with elevated levels of growth factors and proinflammatory cytokines[27]. Notably, TGF-β1 is predominantly secreted by Leydig cells into the prostatic stromal microenvironment, promoting both epithelial and stromal cell differentiation[28]. Inhibition of TGF-β1/Smad signaling has been shown to suppress BPH progression and EMT in BPH-1 cells[29]. EMT contributes to excessive accumulation of myofibroblasts and extracellular matrix deposition in the prostate, resulting in increased tissue stiffness, reduced urethral compliance, and exacerbation of LUTS. Vickman et al. demonstrated that TNF-α antagonism alters BPH pathogenesis and significantly reduces disease incidence[30]. ITPR1 may regulate intracellular Ca2+ signaling and thereby influence multiple pathways, including mTOR, MAPK, and Wnt signaling, as well as TNF-α- and TGF-β1-mediated inflammatory responses. Accordingly, ITPR1 may play a pivotal role in BPH development, and its targeting, along with associated pathways, may represent a potential therapeutic strategy.
KLHL36 is enriched in VEGF, estrogen response, and RNA degradation pathways. VEGF is markedly overexpressed in BPH tissues and plays a central role in disease initiation and progression[31-33]. Inhibition of vascular endothelial growth factor receptor 2 (VEGFR-2) signaling and endothelial tube formation has been shown to effectively suppress prostate tissue growth and neovascularization[34]. Abdel-Aziz et al. demonstrated that febuxostat attenuates testosterone-induced BPH in rats through suppression of inducible nitric oxide synthase (iNOS)/cyclooxygenase-2 (COX-2) and VEGF/TGF-β signaling pathways[35]. Trujillo-Rojas et al. further suggested that BPH development may be associated with altered expression profiles of prostate chemokines, particularly VEGF[36]. Although the prostate is classically regarded as an androgen-dependent organ, androgen signaling alone is insufficient to fully account for disease progression. Estrogen-related pathways also contribute to BPH pathogenesis. Recent evidence indicates that G protein-coupled estrogen receptor (GPER)-mediated signaling regulates epithelial proliferation and survival in BPH[37], while estradiol promotes EMT in benign prostatic epithelial cells[38]. More broadly, estrogen signaling and the androgen-to-estrogen balance shift have been implicated in tissue remodeling, LUTS development, and BPH progression[39-41]. Collectively, KLHL36 may contribute to BPH pathogenesis through regulation of angiogenesis, epithelial proliferation, and extracellular matrix remodeling.
OLFM4 is enriched in the PPP, IL-17 signaling, and N-glycan biosynthesis pathways. OLFM4 is known to regulate cell adhesion and migration through interactions with adhesion molecules, cytoskeletal components, and the extracellular matrix[42]. By modulating the PPP, OLFM4 may support proliferative demands and enhance antioxidant capacity, thereby facilitating BPH progression. Emerging evidence suggests that stromal proliferation in BPH can occur in an androgen-independent manner and is closely linked to microenvironmental remodeling[43]. In addition, IL-17 levels are elevated in patients with BPH and correlate with prostate volume[44,45]. IL-17 signaling can also activate the TGF-β1/ERK pathway. OLFM4 may amplify these effects by modulating extracellular matrix dynamics and cell adhesion, thereby promoting fibrosis and glandular hyperplasia and contributing to BPH progression.
Inflammation plays a central role in the initiation and progression of BPH by driving persistent tissue injury, dysregulated repair, and chronic immune activation. This process involves repeated tissue damage, aberrant wound healing, immune dysregulation, and interaction with androgen signaling. Immune infiltration was evaluated using the CIBERSORT algorithm, which deconvolutes bulk transcriptomic data to estimate the relative proportions of 22 immune cell subsets. Correlation analyses were then performed to assess associations between gene expression and inferred immune cell fractions. Significant differences were observed in NK cells and plasma cells between patient groups [Figure 8C]. NK cells are critical mediators of immune surveillance and cytotoxic responses, contributing to the elimination of abnormal cells; however, excessive activation may exacerbate local inflammation and promote prostatic hyperplasia and tissue remodeling. Plasma cells may contribute to chronic inflammatory maintenance and tissue repair through antibody production and immunomodulatory functions. Further investigation is required to clarify the immune regulatory mechanisms underlying BPH progression.
Finally, associations between the prioritized genes and metabolic pathways were examined [Figure 11A]. Distinct genes showed differential involvement in amino acid metabolism and lipid metabolism pathways, with additional associations to disease status through other metabolic and signaling routes. In addition, expression analyses of BPH-related genes revealed consistent correlations between the prioritized genes and established disease-associated genes, further supporting their potential involvement in the pathological mechanisms of BPH.
Although integrative analyses consistently prioritized C2, GUCY1B2, OLFM4, ITPR1, and KLHL36 as candidate BPH-associated genes, the present study is primarily computational and hypothesis-generating. Accordingly, interpretation of their biological roles should remain cautious pending experimental validation. Future studies are warranted to characterize their expression patterns and functional roles in BPH tissues, cellular systems, and animal models. In particular, quantitative polymerase chain reaction (PCR), immunohistochemistry, loss- and gain-of-function assays, and pathway perturbation experiments may clarify whether these genes directly regulate epithelial proliferation, stromal remodeling, inflammation, or other pathogenic processes in BPH.
Motif-based transcription factor enrichment analysis suggested that NFKB1 and RELA may serve as key upstream regulators of the identified genes, indicating that inflammatory transcriptional programs may contribute to BPH pathogenesis. In addition, 81 miRNAs targeting the five key genes were identified, forming 183 miRNA-mRNA interaction pairs. These results support a complex multilayer regulatory network underlying BPH progression and provide candidate regulatory molecules for future experimental validation. From a translational perspective, the prioritized genes may have potential value in BPH stratification and target discovery. In particular, C2 and OLFM4 may represent promising biomarker or intervention candidates due to their consistent prioritization across genetic analyses and their involvement in inflammation-, metabolism-, and microenvironment-related pathways. Although further validation is required, these findings provide a basis for translating genetic prioritization into clinically relevant biomarker panels and mechanism-informed therapeutic strategies for BPH.
Limitations
This study has several limitations. First, eQTL data were derived from whole-blood samples rather than prostate tissue, which may not fully capture regulatory mechanisms in prostatic epithelial or stromal compartments. Second, both discovery and validation GWAS datasets were restricted to individuals of European ancestry, limiting generalizability to other populations. Third, despite supportive colocalization results for the five key genes, residual bias from horizontal pleiotropy or linkage disequilibrium cannot be fully excluded. Fourth, functional interpretations of C2, GUCY1B2, OLFM4, ITPR1, and KLHL36 were based primarily on bioinformatic inference and require experimental validation in relevant BPH models. Finally, the relatively small sample size of the GEO dataset may limit statistical power in exploratory transcriptomic analyses.
Conclusion
In conclusion, five candidate genes associated with BPH - C2, GUCY1B2, OLFM4, ITPR1, and KLHL36 - were identified through eQTL-based MR and colocalization analyses. Integrated downstream analyses suggested that these genes may contribute to BPH pathogenesis through coordinated regulation of signaling pathways, immune microenvironment interactions, transcriptional networks, miRNA-mediated regulation, and metabolic reprogramming. Among them, C2 and OLFM4 may represent promising candidates for future biomarker development and mechanism-based therapeutic strategies, although further experimental and clinical validation remains essential. These findings may also support future efforts toward genetically informed stratification and precision medicine approaches in BPH.
DECLARATIONS
Authors’ contributions
Conceived the study: Yao X, Guo Y, Zhang Y, Song W
Drafted the manuscript: Gu Z
Prepared the figures: Pan X, Huang D
Interpreted the data: Ni P
Polished the article: Zou L
Collected the references: Zhang Y
Participated in the discussion and gave constructive comments: Chen Y
All authors have read and agreed to the published version of the manuscript.
Availability of data and materials
The datasets analyzed in this study are publicly available from the eQTLGen Consortium, FinnGen, the IEU OpenGWAS database, and the Gene Expression Omnibus (GEO) under accession number GSE132714.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (version GPT-5.4, released 2025-03-05) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship:
This work was supported in part by the National Natural Science Foundation of China, Youth Project (No. 82203150).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
This study was based exclusively on publicly available summary-level datasets from eQTLGen, FinnGen, IEU OpenGWAS, and GEO (GSE132714). No new human participants, human samples, or animal experiments were involved; therefore, additional ethical approval and informed consent were not required.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Chughtai B, Forde JC, Thomas DD, et al. Benign prostatic hyperplasia. Nat Rev Dis Primers. 2016;2:16031.
2. Chen X, Yang S, He Z, et al. Comprehensive analysis of the global, regional, and national burden of benign prostatic hyperplasia from 1990 to 2021. Sci Rep. 2025;15:5644.
3. Bushman W. Etiology, epidemiology, and natural history of benign prostatic hyperplasia. Urol Clin North Am. 2009;36. 403-15, v.
4. Zou J, Talluri R, Shete S. Approaches to estimate bidirectional causal effects using Mendelian randomization with application to body mass index and fasting glucose. PLoS One. 2024;19:e0293510.
5. Varathan P, Gorijala P, Jacobson T, et al. Integrative analysis of eQTL and GWAS summary statistics reveals transcriptomic alteration in Alzheimer brains. BMC Med Genomics. 2022;15:93.
6. Porcu E, Rüeger S, Lepik K, Santoni FA, Reymond A, Kutalik Z; eQTLGen Consortium, BIOS Consortium. Mendelian randomization integrating GWAS and eQTL data reveals genetic determinants of complex and clinical traits. Nat Commun. 2019;10:3300.
7. Sun Y, Liu Y, Dian Y, Zeng F, Deng G, Lei S. Association of glucagon-like peptide-1 receptor agonists with risk of cancers-evidence from a drug target Mendelian randomization and clinical trials. Int J Surg. 2024;110:4688-94.
8. Mullin BH, Tickner J, Zhu K, et al. Characterisation of genetic regulatory effects for osteoporosis risk variants in human osteoclasts. Genome Biol. 2020;21:80.
9. Chauquet S, Zhu Z, O'Donovan MC, Walters JTR, Wray NR, Shah S. Association of antihypertensive drug target genes with psychiatric disorders: a Mendelian randomization study. JAMA Psychiatry. 2021;78:623-31.
10. Finan C, Gaulton A, Kruger FA, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med. 2017;9:eaag1166.
11. Xu G, Dai G, Huang Z, Guan Q, Du C, Xu X. The etiology and pathogenesis of benign prostatic hyperplasia: the roles of sex hormones and anatomy. Res Rep Urol. 2024;16:205-14.
12. Wang SY, Cai Y, Hu X, et al. P. gingivalis in oral-prostate axis exacerbates benign prostatic hyperplasia via IL-6/IL-6R pathway. Mil Med Res. 2024;11:30.
13. Lerner LB, McVary KT, Barry MJ, et al. Management of lower urinary tract symptoms attributed to benign prostatic hyperplasia: AUA GUIDELINE PART I-initial work-up and medical management. J Urol. 2021;206:806-17.
14. McConnell JD, Bruskewitz R, Walsh P, et al. The effect of finasteride on the risk of acute urinary retention and the need for surgical treatment among men with benign prostatic hyperplasia. Finasteride Long-Term Efficacy and Safety Study Group. N Engl J Med. 1998;338:557-63.
15. Roehrborn CG, Bruskewitz R, Nickel JC, et al.; Proscar Long-Term Efficacy and Safety Study Group. Sustained decrease in incidence of acute urinary retention and surgery with finasteride for 6 years in men with benign prostatic hyperplasia. J Urol. 2004;171:1194-8.
18. Yang Q, Peters T, Donovan J, Wilt T, Abrams P. Transurethral incision compared with transurethral resection of the prostate for bladder outlet obstruction: a systematic review and meta-analysis of randomized controlled trials. J Urol 2001;165:1526-32.
19. Wu X, Lv D, Lei M, et al. A 10-gene signature as a predictor of biochemical recurrence after radical prostatectomy in patients with prostate cancer and a Gleason score ≥7. Oncol Lett. 2020;20:2906-18.
20. Shi YF, Yu DJ, Jiang CY, et al. TRAF6 regulates proliferation of stromal cells in the transition and peripheral zones of benign prostatic hyperplasia via Akt/mTOR signaling. Prostate. 2018;78:193-201.
21. Liu J, Zhou W, Yang L, et al. STEAP4 modulates cell proliferation and oxidative stress in benign prostatic hyperplasia. Cell Signal. 2024;113:110933.
22. Binmahfouz LS, Almukadi H, Alamoudi AJ, et al. 6-paradol alleviates testosterone-induced benign prostatic hyperplasia in rats by inhibiting AKT/mTOR axis. Plants. 2022;11:2602.
23. Jiang X, Wang J, Chen P, et al. [6]-Paradol suppresses proliferation and metastases of pancreatic cancer by decreasing EGFR and inactivating PI3K/AKT signaling. Cancer Cell Int. 2021;21:420.
24. El-Maadawy WH, Hassan M, Abdou RM, et al. 6-paradol alleviates diclofenac-induced acute kidney injury via autophagy enhancement-mediated by AMPK/AKT/mTOR and NLRP3 inflammasome pathways. Environ Toxicol Pharmacol. 2022;91:103817.
25. Li C, Hu WL, Lu MX, Xiao GF. Resveratrol induces apoptosis of benign prostatic hyperplasia epithelial cell line (BPH-1) through p38 MAPK-FOXO3a pathway. BMC Complement Altern Med. 2019;19:233.
26. Wang Z, Yang S, Li Y, et al. Simvastatin improves benign prostatic hyperplasia: role of peroxisome-proliferator-activated receptor-γ and classic WNT/β-catenin pathway. Int J Mol Sci. 2023;24:4911.
27. Funahashi Y, Wang Z, O’Malley KJ, et al. Influence of E. coli-induced prostatic inflammation on expression of androgen-responsive genes and transforming growth factor beta 1 cascade genes in rats. Prostate. 2015;75:381-9.
28. Chen Y, Xu H, Liu C, et al. LncRNA DIO3OS regulated by TGF-β1 and resveratrol enhances epithelial mesenchymal transition of benign prostatic hyperplasia epithelial cells and proliferation of prostate stromal cells. Transl Androl Urol. 2021;10:643-53.
29. Kim HJ, Jin BR, An HJ. Hesperidin ameliorates benign prostatic hyperplasia by attenuating cell proliferation, inflammatory response, and epithelial-mesenchymal transition via the TGF-β1/Smad signaling pathway. Biomed Pharmacother. 2023;160:114389.
30. Vickman RE, Aaron-Brooks L, Zhang R, et al. TNF is a potential therapeutic target to suppress prostatic inflammation and hyperplasia in autoimmune disease. Nat Commun. 2022;13:2133.
31. Zhang T, Mao C, Chang Y, Lyu J, Zhao D, Ding S. Hypoxia activates the hypoxia-inducible factor-1α/vascular endothelial growth factor pathway in a prostatic stromal cell line: a mechanism for the pathogenesis of benign prostatic hyperplasia. Curr Urol. 2024;18:185-93.
32. Consoli V, Burò I, Gulisano M, et al. Evaluation of the antioxidant and antiangiogenic activity of a pomegranate extract in BPH-1 prostate epithelial cells. Int J Mol Sci. 2023;24:10719.
33. Lekas AG, Lazaris AC, Chrisofos M, et al. Finasteride effects on hypoxia and angiogenetic markers in benign prostatic hyperplasia. Urology. 2006;68:436-41.
34. Kim EY, Jin BR, Chung TW, et al. 6-sialyllactose ameliorates dihydrotestosterone-induced benign prostatic hyperplasia through suppressing VEGF-mediated angiogenesis. BMB Rep. 2019;52:560-5.
35. Abdel-Aziz AM, Gamal El-Tahawy NF, Salah Abdel Haleem MA, Mohammed MM, Ali AI, Ibrahim YF. Amelioration of testosterone-induced benign prostatic hyperplasia using febuxostat in rats: The role of VEGF/TGFβ and iNOS/COX-2. Eur J Pharmacol. 2020;889:173631.
36. Trujillo-Rojas L, Fernández-Novell JM, Blanco-Prieto O, Rigau T, Rivera Del Álamo MM, Rodríguez-Gil JE. The onset of age-related benign prostatic hyperplasia is concomitant with increased serum and prostatic expression of VEGF in rats: potential role of VEGF as a marker for early prostatic alterations. Theriogenology. 2022;183:69-78.
37. Yang Y, Sheng J, Hu S, et al. Estrogen and G protein-coupled estrogen receptor accelerate the progression of benign prostatic hyperplasia by inducing prostatic fibrosis. Cell Death Dis. 2022;13:533.
38. Liu Z, Li S, Chen S, et al. YAP-mediated GPER signaling impedes proliferation and survival of prostate epithelium in benign prostatic hyperplasia. iScience. 2024;27:109125.
39. Shi X, Peng Y, Du X, et al. Estradiol promotes epithelial-to-mesenchymal transition in human benign prostatic epithelial cells. Prostate. 2017;77:1424-37.
40. Wynder JL, Nicholson TM, DeFranco DB, Ricke WA. Estrogens and male lower urinary tract dysfunction. Curr Urol Rep. 2015;16:61.
41. Wang Z, Hu L, Salari K, et al. Androgenic to oestrogenic switch in the human adult prostate gland is regulated by epigenetic silencing of steroid 5α-reductase 2. J Pathol. 2017;243:457-67.
42. Nicholson TM, Ricke WA. Androgens and estrogens in benign prostatic hyperplasia: past, present and future. Differentiation. 2011;82:184-99.
43. Liu W, Rodgers GP. Olfactomedin 4 in cancer development and progression. Biochim Biophys Acta Rev Cancer. 2025;1880:189423.
44. Hata J, Harigane Y, Matsuoka K, et al. Mechanism of androgen-independent stromal proliferation in benign prostatic hyperplasia. Int J Mol Sci. 2023;24:11634.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].