


Population genetic history of the ancient reindeer-herding Ewenki people
 0
0 Abstract
Aim: High-quality genomic resources from underrepresented populations are essential for understanding human genetic origins, population structure, and demographic history. The Ewenki, an ethnolinguistic minority mainly inhabiting the high-latitude, cold regions of Northeast China, remain insufficiently characterized at the genome-wide level. This study aimed to investigate the population structure, ancestral composition, and demographic history of the Ewenki to provide insights into human genetic evolution in Northeast Asia.
Methods: We generated genome-wide single-nucleotide polymorphism (SNP) data from 46 Ewenki individuals in Inner Mongolia and merged them with public modern and ancient genomic datasets. Population structure and demographic history were reconstructed through Principal component analysis (PCA), model-based ADMIXTURE, fineSTRUCTURE haplotype clustering, f3/f4 statistics, and qpWave/qpAdm modeling to infer ancestry composition and admixture events.
Results: The Ewenki occupy a genetically distinct position within Northeast Asia and show close genetic affinities with Chinese Mongolic, Tungusic-speaking, and ancient Northeast Asian populations. f3/f4 statistics revealed shared genetic drift and admixture signals linking the Ewenki to ancient Northeast Asian, Siberian, and Yellow River Basin-related populations. qpWave and qpAdm analyses further indicated that the Ewenki can be modeled primarily as a mixture of ancient Northeast Asian/Siberian-related ancestry and ancient Yellow River Basin farmer-related ancestry, reflecting long-term population interactions and admixture in Northeast Asia.
Conclusion: The Ewenki share significant genetic similarities with Tungusic-speaking populations, mainly resulting from admixture between ancient Northeast Asian groups and Yellow River Basin farmers.
Keywords
INTRODUCTION
The historical underrepresentation of non-European populations in human genetic studies has constrained our understanding of global population history and the genetic architecture of complex traits, particularly for ethnolinguistically diverse groups in regions of long-standing demographic complexity[1,2]. Recent expansions of non-European genomic resources have begun to address these gaps, providing new insights into the demographic history and evolutionary trajectories of underrepresented populations across Africa, the Americas, Oceania, and Asia[3-7]. Within this broader effort, Eurasia and Northeast Asia in particular represents a critical region for closing such knowledge gaps, given its central position in major prehistoric migrations, sustained interactions among Tungusic, Mongolic, Turkic, and Sinitic populations, and its long history of forest-steppe-agricultural interface dynamics. Northeast Asia is a geographically and culturally diverse region shaped over centuries by migration, conflict, and ongoing interactions[8,9]. This region generally includes northeastern China, Japan, North Korea, South Korea, Mongolia, and the Russian Far East. Key ethnolinguistic groups include Han Chinese and various regional minorities such as Manchu, Daur, Hezhen, Nanai, Oroqen, Ewenki, Mongolians, Koreans, and Japanese, among others. Genetic studies of these underrepresented populations have provided important insights into their ancestral origins, migration patterns, genetic diversity, and population-specific health issues. These groups have experienced repeated episodes of migration, admixture, and cultural exchange along steppe, forest, and maritime routes.
The Inner Mongolia Autonomous Region (IMAR) is situated in northern China and has historically been a home to the Han and various other ethnic groups. From a geographic standpoint, its boundaries span a significant area, extending from the northeast to the southwest[10-14]. This distinctive position for examining gene flow and interactions among local ethnic communities provides ample opportunities for research in population genetics. On a global scale, the Mongolian people are widely recognized for founding the Mongol Empire during the 13th century under Genghis Khan. Historical records show that the Mongolian population had a significant impact on the genetic drift of the populations they met, especially after the empire’s expansion and the conquests across a large portion of Eurasia[15]. The rise of the Mongol Empire, along with subsequent migrations and military-political integration across various periods, led to the incorporation of certain Ewenki groups into the Mongol system. The Ewenki and Mongolian peoples have historically resided in mixed settlements to the west of the Greater Khingan Mountains and within the Hulunbuir grasslands. Through their shared practices of herding, hunting, and migration, this integration enabled coexistence with Mongol tribes, thereby facilitating gene flow[16]. Today, the Ewenki people residing in Inner Mongolia retain genomic signatures of their ancient ancestry while also showing evidence of long-term genetic interaction with surrounding populations of the Mongolian Plateau. They represent a unique and invaluable genetic marker within the pluralistic and unified framework of the Chinese nation.
The Ewenki are an indigenous Tungusic-speaking people from North Asia. They mostly live in northern China (Inner Mongolia and Heilongjiang provinces), Siberia, and Mongolia. Traditionally a reindeer-herding and forest-hunting people of the densely wooded Greater Khingan Mountains, the Ewenki have long inhabited high-latitude environments characterized by extended cold winters and limited daylight-an ecological context that, combined with their distinctive subsistence strategy, makes them an informative population for investigating the demographic history of Northeast Asian forest dwellers[17]. According to the 2020 census, their population is 34,617, with most living in the Hulunbuir region of the Inner Mongolia Autonomous Region and the Greater Khingan Mountains in Heilongjiang Province[18]. The Ewenki are among the most distinctive and northernmost ethnic minorities within the Tungusic-speaking groups in China. Historically, they inhabited forests, grasslands, and wetlands, fostering long-term contact, intermarriage, and cultural exchange with Mongolians, Han Chinese, and nearby ethnic groups. Frequent contact with neighbors has led to extensive cultural and genetic mixing. As a result, their genomes reflect a history of multi-source admixture and complex population migrations. Despite the rich demographic significance of the Ewenki, previous genetic studies on this population have been largely restricted to forensic short tandem repeat/insertion-deletion (STR/InDel) marker panels or limited mitochondrial and Y-chromosomal analyses, leaving the genome-wide population structure, fine-scale ancestral composition, and admixture history of the Ewenki insufficiently characterized. To our knowledge, no prior study has systematically integrated genome-wide single-nucleotide polymorphism (SNP) data from the Ewenki of the Inner Mongolia Autonomous Region with publicly available modern and ancient genomic datasets to reconstruct their detailed demographic history within the broader Northeast Asian context. To address these gaps, this study analyzes genome-wide data from Ewenki individuals to explore their ancient origins, migration pathways, population structure, and interactions with neighboring groups[19]. The results demonstrate how both ancient lineages and more recent gene flow have shaped the current Ewenki gene pool.
MATERIALS AND METHODS
Sample collection and ethical statement
We collected saliva samples from 46 Ewenki individuals residing in the Inner Mongolia Autonomous Region. All participants met the following inclusion criteria: (i) self-reported Ewenki ancestry for at least three generations with no known intermarriage with other ethnic groups; (ii) currently residing in the Hulunbuir region of IMAR; (iii) aged 18 years or older and in general good health at sample collection; and (iv) verified as unrelated to other participants based on KING2 kinship analysis (kinship coefficients < 0.0884, corresponding to second-degree or closer relationships), resulting in 46 unrelated individuals retained from the initial 46 samples for all subsequent population genetic analyses. All subjects provided written informed consent in accordance with the Declaration of Helsinki before participating. The study protocol was approved by the Medical Ethics Committee of West China Hospital, Sichuan University (Approval No. 2024-1463).
DNA sequencing, genotyping, and quality control
DNA was extracted and purified using the PureLink Genomic DNA Mini Kit (Thermo Fisher Scientific, USA). Genome-wide SNP genotyping was performed using the Illumina Infinium Global Screening Array (GSA) v3.0 (Illumina, San Diego, CA, USA), yielding approximately 600,000 high-quality SNPs across the genomes of all 46 individuals. Genotype calling was performed against the GRCh37 (hg19) human reference genome to ensure compatibility with publicly available ancient and modern reference datasets (Human Origins, 1240K, and HGDP), all of which are coordinated on the same reference build. To ensure quality control, PLINK 1.9 was used with the following parameter settings: --mind 0.01, --geno 0.01, --hwe 0.001, and --maf 0.01. The minor allele frequency (MAF) threshold of 0.01 was chosen to retain low-frequency informative SNPs relevant for population structure analyses while excluding rare variants potentially affected by genotyping errors, following standard practice in population genomics studies of underrepresented populations[20]. Furthermore, family relationships were evaluated by calculating kinship coefficients using KING2. No pairs of individuals with kinship coefficients > 0.0884 (corresponding to second-degree or closer relationships) were identified, and one individual from each related pair was excluded from downstream analyses to ensure unrelatedness, resulting in all 46 individuals retained as unrelated samples for downstream analyses.
Data merging
We independently merged our newly generated Ewenki SNP data with three publicly available reference panels, resulting in three separate working datasets with distinct SNP densities and complementary analytical purposes:
(1) Human Origins (HO)_Ewenki dataset (245,657 SNPs): generated by merging with the HO panel; used for principal component analysis (PCA) and ADMIXTURE analyses, given its broad population coverage of modern Eurasian groups[21,22].
(2) 1240K_Ewenki dataset (95,353 SNPs): generated by merging with the 1240K dataset from the Allen Ancient DNA Resource (AADR v54.1.p1); used for f3, f4, qpWave, and qpAdm analyses involving ancient genomes[23,24].
(3) Human Genome Diversity Project (HGDP)_Ewenki dataset (525,456 SNPs): generated by merging with the HGDP panel; used for fineSTRUCTURE haplotype-based analyses requiring high SNP density.
All original publications contributing data to these merged panels are listed in Supplementary Table 1, in accordance with the AADR data usage guidelines.
PCA
To investigate clustering patterns within the studied and reference populations, we performed PCA using the smartPCA program in the EIGENSOFT v.6.1.4 package, based on an Illumina dataset comprising 111 modern and 77 ancient populations. Principal components were computed using the modern populations, while ancient samples were projected onto these axes by setting lsqproject: YES and shrinkmode: YES in the smartPCA parameter file. This projection-based approach mitigates potential bias from missing data and low coverage in ancient genomes, ensuring that PCA clustering accurately reflects population relationships rather than data quality differences. We applied LD pruning using the --indep-pairwise 200 25 0.4 option in PLINK 1.9. This window-based scheme (200-SNP sliding window, 25-SNP step size,
Model-based unsupervised ADMIXTURE
We combined our dataset with a wide range of modern and ancient reference populations to examine the genetic characteristics of Ewenki individuals. In this analysis, we employed ADMIXTURE in its unsupervised mode[25], utilizing the standard 10-fold cross-validation (--cv = 10) to assess genetic heritage. To handle linkage disequilibrium, we used PLINK 1.9, setting r2 > 0.4 and --indep-pairwise 200 25 0.4 to ensure strong data integrity. Subsequently, we applied the admixture models across a series of specified ancestral sources (K = 2-20), running 100 bootstrap replicates using various random seeds to evaluate consistency and reliability. We selected the optimal models based on the lowest cross-validation error estimates, ensuring the most precise depiction of ancestral genetic contributions.
fineSTRUCTURE analysis
Prior to haplotype-based analysis, genotype data were statistically phased using SHAPEIT4 v4.2.2[26] with the 1000 Genomes Project Phase 3 reference panel to ensure accurate haplotype reconstruction. The phased data were then converted to ChromoPainter format, and the co-ancestry matrix was generated using ChromoPainter v2 under the linked model with default parameters. Fine-scale population structure was inferred with fineSTRUCTURE v4 using the following core parameters: -x 100000 (burn-in), -y 100000 (sampling iterations), and -z 1000 (thinning interval), with two independent Markov chain Monte Carlo (MCMC) runs to ensure convergence. The resulting maximum a posteriori (MAP) clustering tree was visualized using the built-in fineSTRUCTURE GUI tool, with branch lengths reflecting the inferred merging patterns of population clusters.
Allele-based shared ancestry estimation
Multiple analyses were performed using ADMIXTOOLS to explore the genetic composition and admixture history of the Ewenki people. Initially, we utilized the qp3pop package with its default settings and applied a block jackknife to calculate outgroup-f3 statistics f3 (source 1, Ewenki; Mbuti) and to evaluate shared genetic drift among 27 contemporary and 46 ancient populations from East Asia. Following this, admixture-f3 statistics f3 (source 1, source 2; targeted population) were calculated to detect signals of admixture in Ewenki samples derived from various modern and ancient East Asian sources. Significant admixture was identified when negative f3 values were paired with Z scores below -3, suggesting that the targeted population was a combination of two parental groups, source 1 and source 2. Additionally, f4 statistics in the form of f4 (W, X; Y, outgroup) were used to assess potential indicators of admixture from different source populations into the targeted groups. For this purpose, we employed the qpDstat package in ADMIXTOOLS, including an extra parameter f4 Mode (f4: YES), and applied the block jackknife technique to approximate standard errors.
qpWave/qpAdm estimation
The qpWave/qpAdm software from ADMIXTOOLS was employed with default settings to determine the minimum number of ancestral populations and to compute admixture proportions. In the two-way qpAdm models, the candidate northern (Ancient Northeast Asian/Siberian) sources included AR13-10K, AR14K, Mongolia_North_N, DevilsCave_N, and Russia_Shamanka_Eneolithic, while the candidate southern (Yellow River Basin farmer-related) sources included Wadian_LN, Haojiatai_LN, Pingliangtai_LN, Lajia_LN, and Han_HGDP[27]. To improve model accuracy, we included a fixed panel of basal outgroups: Mbuti, Russia_Ust_Ishim, Russia_Kostenki14, Belgium_UP_GoyetQ116_1, Italy_North_Villabruna_HG, Israel_Natufian, Mixe, Onge, and Chokhopani. The allsnps: YES option was used to ensure comprehensive SNP inclusion.
Statistical analysis
Genome-wide SNP data were filtered using PLINK v1.9, and close genetic relatedness among individuals was evaluated using KING2. Linkage disequilibrium pruning was performed prior to PCA (smartPCA in EIGENSOFT) and unsupervised ADMIXTURE analyses, with the optimal K determined by the lowest 10-fold cross-validation error. Fine-scale population structure was further examined using ChromoCombine and fineSTRUCTURE. Population genetic statistics were computed with ADMIXTOOLS: outgroup-f3 and admixture-f3 assessed shared drift and admixture signals (Z < -3 indicating significant admixture); f4 statistics evaluated asymmetric allele sharing (|Z| > 3 considered significant); standard errors were estimated by block jackknife. qpWave assessed pairwise genetic homogeneity, and qpAdm estimated two-way ancestry proportions, with model acceptability evaluated by p-values, standard errors, and biological plausibility. All visualizations were generated using R.
RESULTS
Genetic clustering of the Ewenki
We genotyped approximately 600,000 genome-wide SNPs in 46 Ewenki individuals from IMAR and merged these data with publicly available ancient and modern genomic datasets, including the HO[28-31] resource and recently published ancient DNA data[32-35]. We grouped present-day individuals by language family: Tibeto-Burman-speaking (n = 159), Sinitic-speaking (n = 138), Tai-Kadai-speaking (n = 292), Hmong-Mien-speaking (n = 91), Austronesian-speaking (n = 120), Austroasiatic-speaking (n = 196), Mongolic-speaking (n = 144), and Tungusic-speaking (n = 64) [Supplementary Table 1]. We grouped ancient individuals by geographic origin [Figure 1A]. The map in Figure 1A was generated using ArcGIS software (Esri Inc., Redlands, CA, USA). Basemap data are derived from Esri's freely available online basemap service (© Esri and its data contributors), which permits non-commercial academic use. The Ewenki fall into a position that reflects genetic input from neighboring ethnic groups, while also preserving signals of ancient ancestry related to both Siberia and the Yellow River Basin (YRB). We performed PCA to characterize patterns of genetic structure and admixture, and to evaluate relationships between the Ewenki and other modern and ancient populations in Northeast Asia. We used 111 modern populations to compute the principal components and then projected 77 ancient populations onto these axes [Supplementary Figure 1]. PC1 separates northern groups, primarily Mongolic-speaking populations, from southern groups, including Hmong-Mien (HM)- and Tai-Kadai (TK)-speaking populations [Figure 1B]. The results reveal three major genetic clusters: one formed by Mongolic-speaking populations, one by Tungusic-speaking populations, and one by Ancient Siberian groups. These clusters are closely spaced and partially overlapping. Ancient Yellow River Basin (A-YRB) farmers cluster on the opposite side, close to the Ewenki.
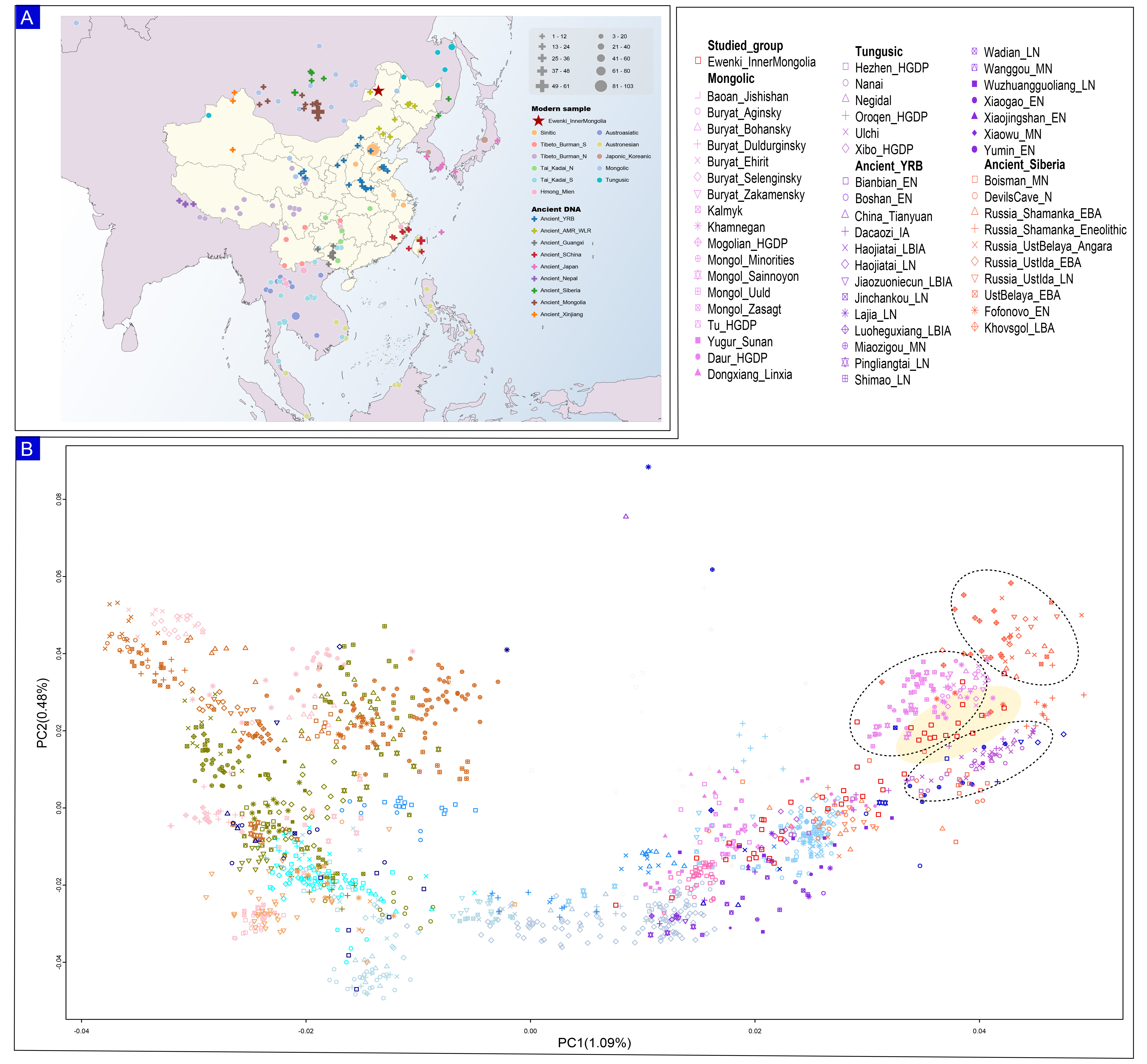
Figure 1. The geographical distribution and population patterns of the Ewenki. (A) The map displays the locations of recently gathered samples in comparison to reference populations [Supplementary Table 1]; (B) A principal component analysis (PCA) was conducted on the genetic characteristics of both contemporary and historical Northeast Asians, utilizing the combined Human Origins (HO) datasets. Distinct colors and unique shapes represent populations associated with various language families. YRB: Yellow River Basin; AMR: Amur River; WLR: West Liao River.
Population structure of the Ewenki
We investigated the ancestral composition of the Ewenki using model-based ADMIXTURE on 1,287 modern and 266 ancient individuals from Northeast Asia [Figure 2]. Cross-validation identified six ancestral sources (K = 6) as the optimal model, which showed the lowest cross-validation (CV) error [Supplementary Table 2]. The dominant ancestral components in the Ewenki were Ancient YRB (dark cyan) and Ancient Siberia (pink). These results indicate that the Ewenki carry a complex ancestry primarily derived from Ancient YRB and Ancient Siberia. In contrast, populations from Southeast Asia, southern ethnic minorities, and the Tibetan-Burmese Plateau exhibit distinct combinations of genetic components. This indicates that the genetic structure of the Ewenki primarily originates from the Siberian forest-steppe zone, with later introductions of some Central Plains agricultural lineages. This phenomenon aligns with the Ewenki's geographical distribution, their Tungusic language affiliation, and their historical nomadic-hunting pastoral lifestyle in northern regions. The target population of this study, Ewenki_InnerMongolia, is highlighted in red to distinguish it from reference populations obtained from publicly available datasets in Figure 2.
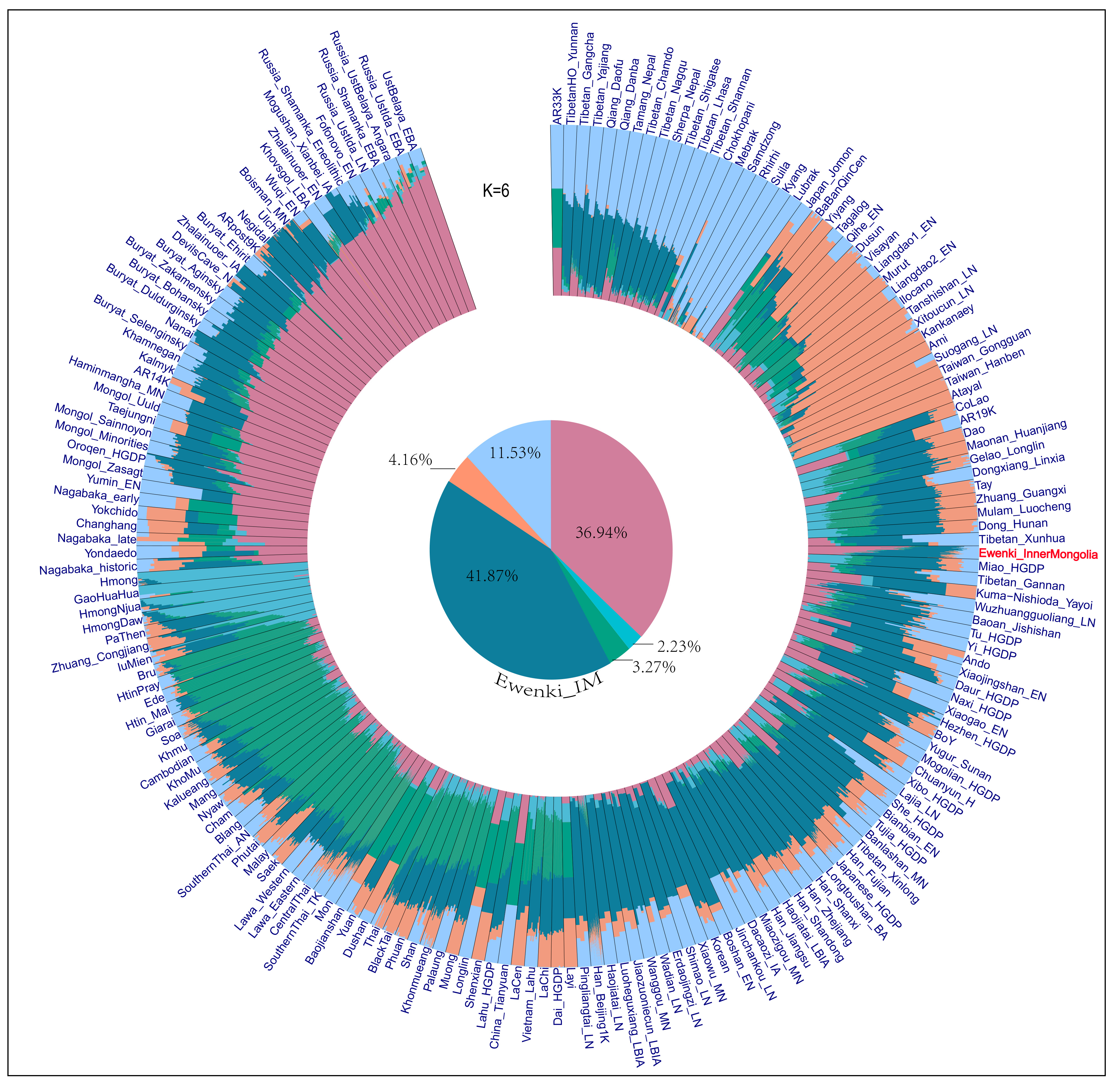
Figure 2. Model-based ADMIXTURE analysis of population structure. Ancestry proportions for K = 6 components were estimated in ancient and present-day Northeast Asian groups, with different colors indicating distinct ancestral components.
We further performed a fineSTRUCTURE analysis among Tungusic speakers and their neighbors, comprising 110 individuals from different Tungusic-speaking groups, to explore the fine-scale genetic structure of the Ewenki [Figure 3]. The results revealed two primary genetic clusters: one encompassing Ewenki-related ethnic minority groups residing in Northeast China, and the other consisting of Ulchi, Negidal, and Nanai groups dwelling in the Amur River Basin. Notably, the Ewenki, Oroqen, and Hezhen formed a tightly clustered subgroup at a shallow branching level, indicating a high degree of genetic similarity among these populations. In contrast, the Amur River Basin groups (Ulchi, Negidal, Nanai) joined this cluster only at a deeper, more basal branching level, reflecting greater genetic differentiation between the two subgroups within the broader Tungusic-speaking complex.
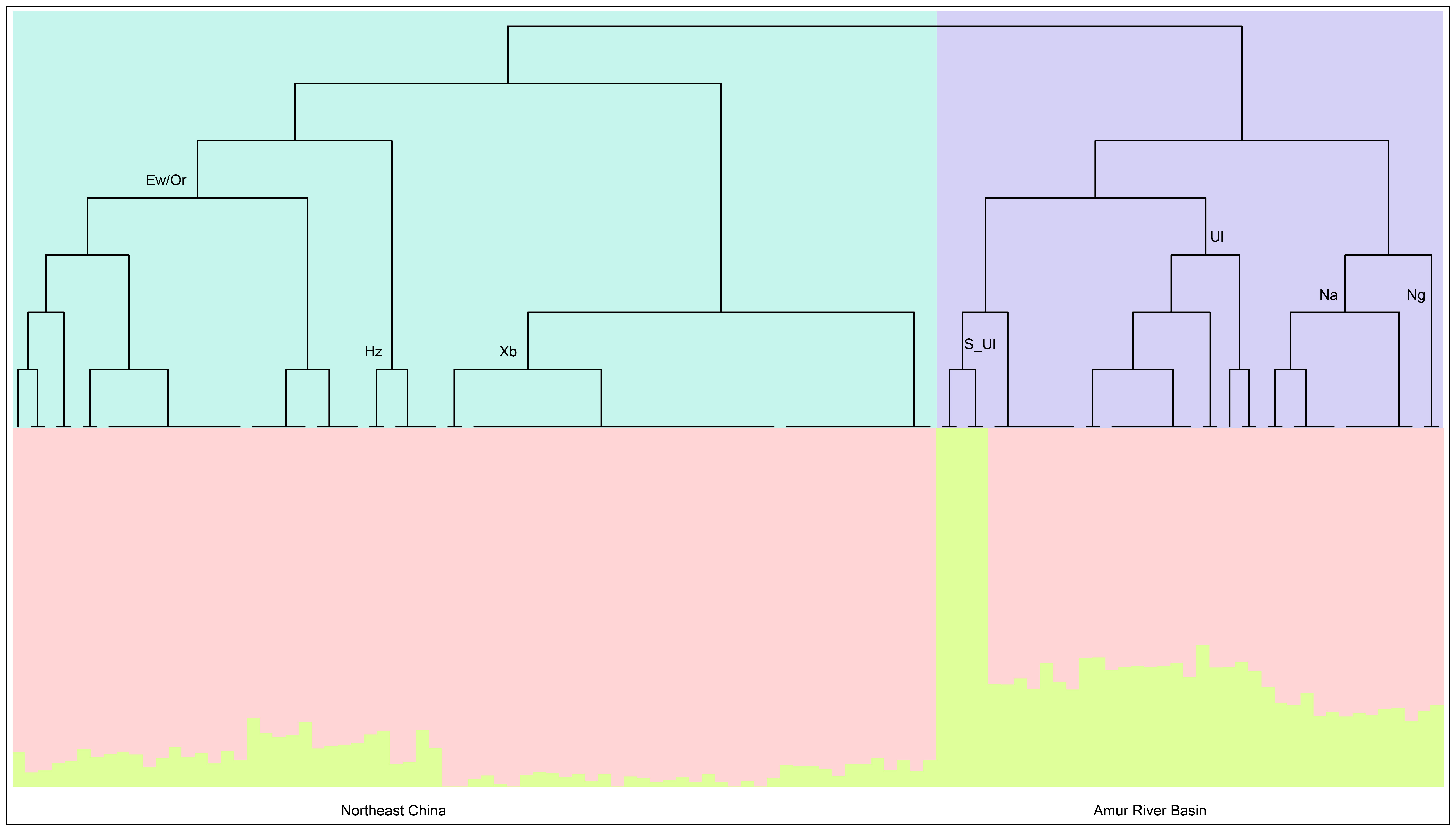
Figure 3. Population structure of the model-based fineSTRUCTURE. The fineSTRUCTURE-generated maximum a posteriori (MAP) tree illustrates clustering trends among 110 individuals from seven distinct groups in Northeast Asia. Adjacent to the tree, the ADMIXTURE plot for the same datasets represents two predefined ancestral sources. Abbreviations are as follows: Ew: Ewenki; Or: Oroqen; Hz: Hezhen; Xi: Xibo; Ul: Ulchi; Na: Nanai; Ng: Negidal.
Shared genetic drift and genetic composition of the Ewenki
We characterized the genetic relationships and ancestry composition of the Ewenki using outgroup f3 statistics of the form f3(source 1, Ewenki; Mbuti), which measured drift and thus allele sharing with a wide panel of modern and ancient reference populations. The Ewenki show elevated affinity with Tungusic and Mongolian populations [Figure 4A]. We then evaluated potential ancestral sources of the Ewenki using admixture-f3 statistics of the form f3 (source 1, source 2; Ewenki), testing different modern and ancient Northeast Asian populations as sources. We detect significant admixture signals (Z < -3) for pairs involving Sino-Tibetan and Altaic-speaking groups [Figure 4B]. Among ancient references, they share more alleles with the Ancient Siberian and Ancient Yellow River Basin Farmer groups [Figure 4C]. Using ancient populations, we also observe substantial signals (Z < -3) for admixture between Ancient Siberian herders and Ancient Yellow River Basin Farmers [Figure 4D]. Together, the admixture-f3 results indicate that the Ewenki derived from multiple ancestral sources and experienced complex historical interactions with neighboring groups. The detailed f3-statistics, including f3 values, standard errors, Z-scores, and P-values, are provided [Supplementary Tables 3 and 4].
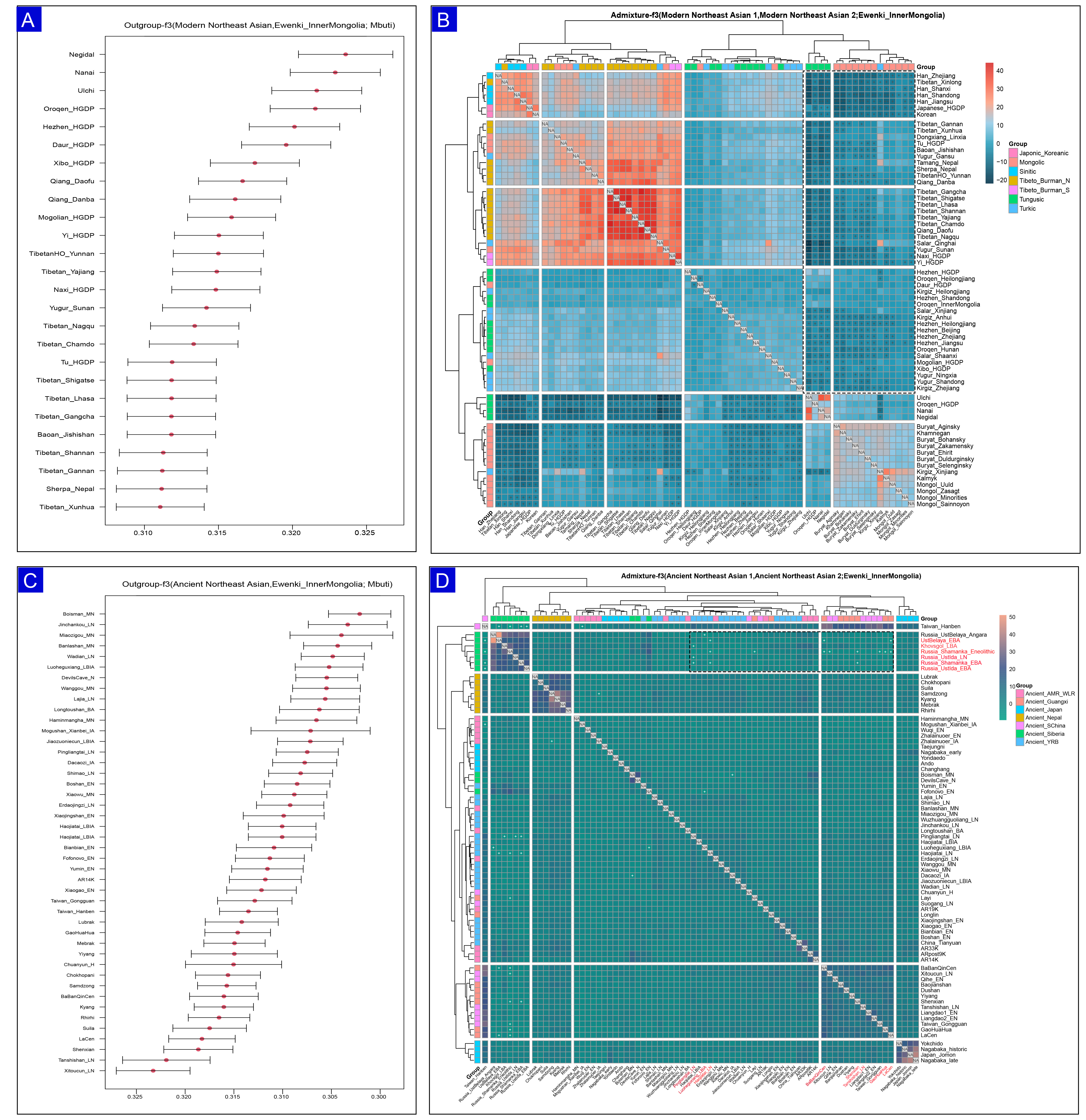
Figure 4. Outgroup and admixture-f3 statistics for the Ewenki. (A) Outgroup-f3 values of the form f3 (Modern Northeast Asian, Ewenki; Mbuti) were calculated using the HO_Illumina datasets to quantify the shared genetic drift with contemporary Northeast Asians; (B) Admixture-f3 tests of the form f3 (Modern Northeast Asian 1, Modern Northeast Asian 2; Ewenki) were employed to detect signals of Ewenki ancestry derived from pairs of modern Northeast Asian populations; (C) Outgroup-f3 statistics of the form f3 (Ancient Northeast Asian, Ewenki; Mbuti) were computed to evaluate the genetic affinities between the Ewenki and ancient Northeast Asian groups; (D) Admixture-f3 analyses of the form f3 (Ancient Northeast Asian 1, Ancient Northeast Asian 2; Ewenki) identify significant mixture events; notably negative values (Z < -3) suggest that the Ewenki can be modeled as a blend of two ancestral sources (source 1 and source 2), which are highlighted with an asterisk (*) to denote statistically significant admixture.
To detect the difference in gene flow between Ewenki and northern Han Chinese/Mongolia, we calculated f4 (Han_North/Mongolia, Ewenki_InnerMongolia; Tungusic, Mbuti). The results indicated that the Ewenki exhibit a stronger affinity to Tungusic populations, such as the Ulchi, Negidal, and Nanai, compared to either Han North or Mongolian groups [Supplementary Tables 5-7]. Overall, the data support a genetic profile for the Ewenki that is firmly rooted in Tungusic ancestry, with an additional affinity for Sino-Tibetan groups when contrasted with Mongolians. To detect the difference in gene flow between Ewenki and ancient_YRB/ancient_Siberia/ancient_Northeast Asia (NEA), we calculated f4 (ancient_YRB/ancient_Siberia, Ewenki_InnerMongolia; ancient_NEA, Mbuti). The results indicated that the Ewenki exhibit significantly greater genetic drift with Ancient Northeast Asian (ANA) and ancient Siberian populations than with ancient YRB farmers [Supplementary Tables 8-10]. We performed f4 statistics in the form of f4 (ancient_YRB, Ewenki_InnerMongolia; ancient_Siberia, Mbuti) to formally explore possible differentiated gene flow between Ewenki and ancient Siberia/ancient_YRB. Numerous tests showed strongly negative Z-scores, indicating a pronounced affinity of the Ewenki towards ancient Siberia relative to Neolithic Age farmers from the YRB. Conversely, when Y was set to ANA, comparisons with ancient Siberian groups often showed significantly negative values. This indicates that the Ewenki are genetically closer to ANA sources than to ancient Siberian populations. Collectively, these findings support substantial ANA ancestry in the Ewenki, stronger than that observed in ancient YRB farmers and ancient Siberia.
Admixture scenarios and gene flow in the Ewenki
Based on the evidence of gene flow within Ewenki populations, we initially applied qpWave on paired groups to formally test whether each pair of populations could be modeled as descending from the same number of ancestral source populations relative to a fixed set of outgroups, thereby assessing pairwise genetic homogeneity [Figure 5A]. The pairwise qpWave analysis was then expanded to include 72 populations to investigate the demographic relationships between the Ewenki and other ancient and modern groups. The results showed a strong genetic affinity between the Ewenki and ANA populations. Additionally, populations labeled as A-Siberia groups appeared genetically uniform, supporting either a recent common origin or significant admixture among ANA groups. Next, we applied qpAdm to estimate admixture proportions using predefined northern (Ancient Northeast Asian/Siberian) and southern (Yellow River Basin farmer) ancestral sources [Figure 5B and Supplementary Table 11]. In the model pairing Mongolia_North_N (northern source) with Han_HGDP (southern source), the Ewenki derived 79.8% of their ancestry from the northern source and 20.2% from the southern source. Similarly, in the model pairing AR13-10K (northern source) with Han_HGDP (southern source), the northern contribution reached 74.7%, with the remaining 25.3% from the southern source. When the later Neolithic Wadian_LN was used as the southern source (paired with Russia_UstIda_EBA as the northern source), the southern contribution ranged from 67.3% to 71.6%, with the remainder attributable to the northern source. Together, these results suggest that the Ewenki primarily originated from interactions between Ancient Northeast Asian/Siberian populations and Yellow River Basin farmer-related ancestries. The detailed qpWave results, including p-values and ranks, are provided [Supplementary Table 12].
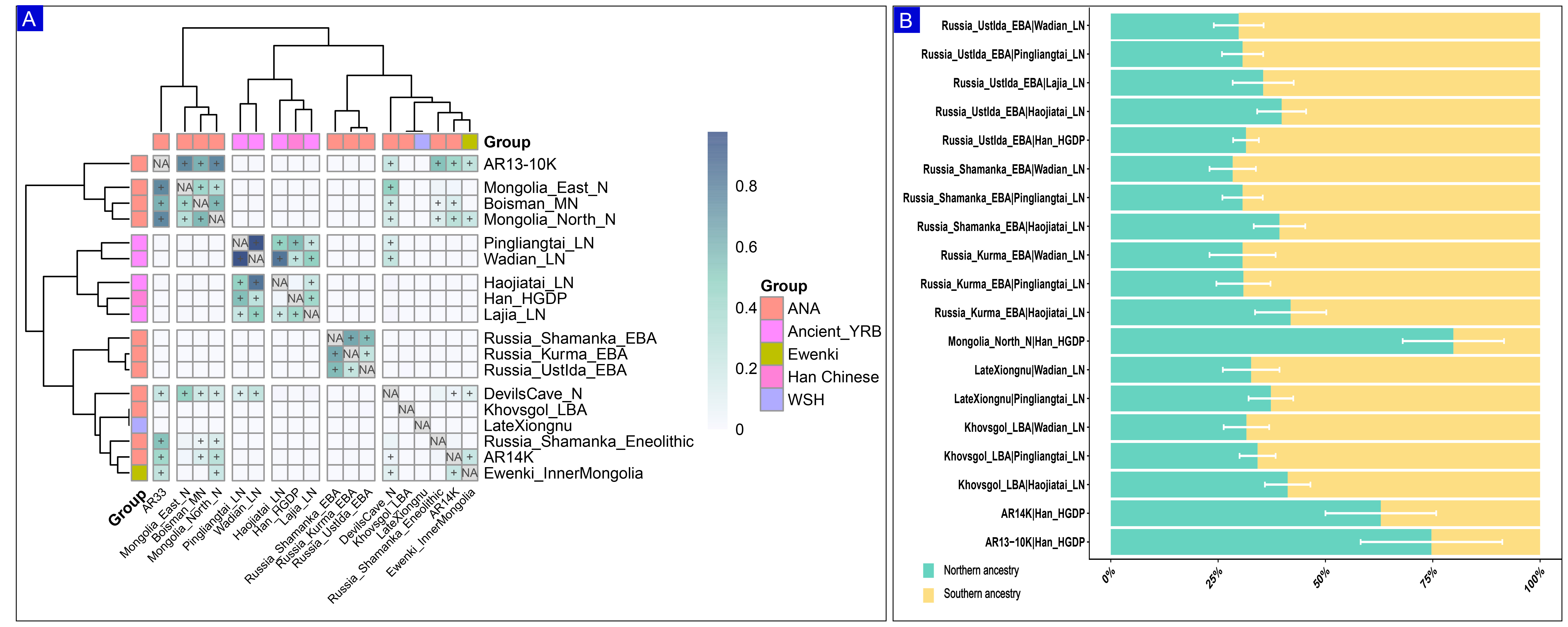
Figure 5. Population relationships and genetic affinity. (A) Pairwise qpWave tests highlight both genetic differentiation and shared ancestry among ancient populations. Rank-0 P-values greater than 0.05 indicate that two reference groups are genetically homogeneous (“++”), while p-values exceeding 0.01 are marked with “+”; (B) The qpAdm-inferred ancestry proportions for the Ewenki are presented. Two-way models support contributions from both northern and southern ancestry, with white error bars representing the standard error of each estimate. YRB: Yellow River Basin; ANA: Ancient Northeast Asian; WSH: Western Steppe Herders.
DISCUSSION
Genome-wide SNP genotyping and whole-genome sequencing have emerged as powerful complementary approaches for reconstructing population history and genetic structure
To investigate the population structure and admixture history of the Ewenki ethnic group, we conducted PCA and Admixture analyses
To further validate the genetic relationships between the Ewenki and other ethnic groups, we conducted f3-statistics [Figure 4]. In the outgroup-f3 analysis, compared with other modern populations, the Ewenki showed higher f3 values with Negidal, Nanai, Ulchi, Oroqen, and Daur, indicating closer genetic relationships with these groups. This shows that the Ewenki belong to the Tungusic-speaking populations, which share common ancestral origins. Additionally, the higher f3 value with Daur suggests a certain degree of genetic affinity between the Ewenki and the Mongolic-speaking populations. Compared with other ancient populations, the Ewenki showed higher f3 values with Boisman_MN, Jinchankou_LN, and Miaozigou_MN, indicating closer genetic affinities with these groups
The genetic admixture patterns inferred in this study align well with multiple lines of cross-disciplinary evidence concerning Northeast Asian population dynamics[76]. The deep ancestral component shared between the Ewenki and ancient Siberian/Northeast Asian populations is consistent with archaeological evidence of continuous boreal forest hunter-gatherer occupation since the late Pleistocene, exemplified by sites such as Devil’s Cave and the Boisman culture, paralleling the traditional Ewenki subsistence economy preserved into recent centuries[77]. The Ancient Northeast Asian-Yellow River Basin farmer admixture detected in our qpAdm models corresponds to the Neolithic-to-Bronze Age expansion (~6,000-4,000 BP) of millet agriculture from the Central Plains into the West Liao River and Mongolian Plateau regions-a process also reflected in the linguistic record, as the Tungusic homeland has been traced to the West Liao River basin. Subsequent historical episodes including the Xiongnu, Mongol, and Manchu confederations facilitated population movement and intermarriage between forest-dwelling Tungusic groups and steppe pastoralists, accounting for the genetic affinity observed between the Ewenki and Mongolic-speaking populations on the Hulunbuir grasslands[78].
This study enhances our understanding of human population history in Northeast Asia and highlights the Ewenki’s unique genetic profile. Their distinctive combination of ANA and Ancient Yellow River Basin Farmer ancestry demonstrates how human diversity remains in the genome and can be uncovered through modern sequencing technologies. mtDNA data indicate that the Ewenki share haplogroups with northern Chinese populations, a pattern resembling that seen in Mongolian groups and pointing to close genetic ties among them
Limitations
This study has several limitations that must be acknowledged. First, the sample size of the target population is relatively small, which limits the statistical power and presents challenges in comprehensively addressing the genetic diversity within this ethnic minority. the geographical scope of the sampling within the Ewenki population is limited, as it includes only individuals from the Inner Mongolia Autonomous Region. While these data offer valuable insights into the genetic structure of this underrepresented group, they may not comprehensively capture the genetic diversity present among all Ewenki subgroups, particularly those residing in Heilongjiang and Siberia
CONCLUSIONS
The Ewenki are a genetically unique Tungusic-speaking group in Northeast Asia whose current ancestry reflects both deep regional roots and repeated contact episodes. By combining genome-wide SNP data from Ewenki individuals with modern and ancient Eurasian references, we demonstrate that the Ewenki primarily derive from Ancient Northeast Asian lineages, with additional ancestry related to early Yellow River Basin farmers. Formal tests (f3, f4), qpWave, and qpAdm analyses support a model where the Ewenki resulted from admixture between Ancient Siberian/Ancient Northeast Asian sources and agricultural populations from the Yellow River Basin, followed by long-term interactions with neighboring Tungusic and Mongolic groups. These findings refine the demographic history of the Ewenki, emphasize their strong genetic ties to other Tungusic-speaking populations, and show that small and understudied ethnolinguistic minorities hold crucial insights into population structure, migration, and adaptation in Northeast Asia.
DECLARATIONS
Acknowledgments
We thank all the volunteers who generously participated in this study.
Authors’ contributions
Contributed equally to this work: Wei D, Zhang L
Conceived and designed the study: Liu C, Wang M, Hai X, He G
Developed the methodology: Mengge Wang, Guanglin He
Conducted analysis manuscript: Duan S, Feng Y, Chen J, Sun Q, Luo L
Availability of data and materials
The supplementary materials include all the information. The genome-wide SNP genotype data generated in this study, together with the corresponding processed VCF and PLINK-format files, have been deposited in the Genome Sequence Archive (GSA) for Human at the National Genomics Data Center, China National Center for Bioinformation. For further information and requests for resources, please contact the corresponding authors.
AI and AI-assisted tools statement
Not applicable.
Conflicts of interest
Wang M and He G are members of the Junior Editorial Board of the Journal of Translational Genetics and Genomics. They were not involved in any aspect of the editorial process for this manuscript, including reviewer selection, manuscript handling, and decision-making. The other authors declare that they have no conflicts of interest.
Financial support and sponsorship
This study was supported by the Sichuan Science and Technology Program (2024NSFSC1518).
Ethical approval and consent to participate
The present study strictly adhered to the ethical guidelines of the Declaration of Helsinki and was approved by the Medical Ethics Committees of West China Hospital, Sichuan University (2024-1463). All subjects provided informed consent before participating.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Taliun D, Harris DN, Kessler MD, et al. ; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature. 2021;590:290-9.
2. All of Us Research Program Genomics Investigators. Genomic data in the all of us research program. Nature. 2024;627:340-6.
3. He G, Yao H, Duan S, et al. ; 10K_CPGDP Consortium. Pilot work of the 10K Chinese people genomic diversity project along the silk road suggests a complex east-west admixture landscape and biological adaptations. Sci China Life Sci. 2025;68:914-33.
4. He G, Duan S, Chen G, et al. Human population genetic history and evolutionary dynamics on the Eastern Tibetan plateau. Mol Biol Evol. 2025;42:msaf258.
5. He G, Chen J, Duan S, et al. ; 10K_CPGDP Consortium. Largest-scale genomic resource reconstructing the genetic origin, population structure, and biological adaptations of the hui people. Mol Biol Evol. 2025;42:msaf225.
6. Lea AJ, Caldas IV, Garske KM, et al. Adaptations to water stress and pastoralism in the Turkana of northwest Kenya. Science. 2025;389:1246-51.
7. He Y, Zhang X, Peng MS, et al. ; Consortium of Anthropological Research in Southeast Asia and Southwest China (CASEAC). Genome diversity and signatures of natural selection in mainland Southeast Asia. Nature. 2025;643:417-26.
8. Silcocks M, Farlow A, Hermes A, et al. ; National Centre for Indigenous Genomics. Indigenous Australian genomes show deep structure and rich novel variation. Nature. 2023;624:593-601.
9. Reis ALM, Rapadas M, Hammond JM, et al. ; National Centre for Indigenous Genomics. The landscape of genomic structural variation in Indigenous Australians. Nature. 2023;624:602-10.
10. Jeong C, Wang K, Wilkin S, et al. A dynamic 6,000-year genetic history of Eurasia’s Eastern steppe. Cell. 2020;183:890-904.e29.
11. Gnecchi-Ruscone GA, Szécsényi-Nagy A, Koncz I, et al. Ancient genomes reveal origin and rapid trans-Eurasian migration of 7th century Avar elites. Cell. 2022;185:1402-1413.e21.
12. Lee J, Miller BK, Bayarsaikhan J, et al. Genetic population structure of the Xiongnu Empire at imperial and local scales. Sci Adv. 2023;9:eadf3904.
13. Gnecchi-Ruscone GA, Rácz Z, Samu L, et al. Network of large pedigrees reveals social practices of Avar communities. Nature. 2024;629:376-83.
14. Wang K, Tobias B, Pany-Kucera D, et al. Ancient DNA reveals reproductive barrier despite shared Avar-period culture. Nature. 2025;638:1007-14.
15. Bai H, Guo X, Zhang D, et al. The genome of a Mongolian individual reveals the genetic imprints of Mongolians on modern human populations. Genome Biol Evol. 2014;6:3122-36.
16. Zerjal T, Xue Y, Bertorelle G, et al. The genetic legacy of the Mongols. Am J Hum Genet. 2003;72:717-21.
17. Li Q, Dong K, Xu L, et al. The distribution of three candidate cold-resistant SNPs in six minorities in North China. BMC Genomics. 2018;19:134.
18. Lan Q, Zhao C, Wei C, Xu H, Shen C, Zhu B. Genetic insights and evaluation of forensic features in Mongolian and Ewenki groups using the InDel variations. Front Biosci. 2022;27:67.
19. Wang M, Huang Y, Liu K, et al. ; 10K_CPGDP Consortium. Multiple human population movements and cultural dispersal events shaped the landscape of Chinese paternal heritage. Mol Biol Evol. 2024;41.
20. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
21. Wang CC, Yeh HY, Popov AN, et al. Genomic insights into the formation of human populations in East Asia. Nature. 2021;591:413-9.
22. Ning C, Li T, Wang K, et al. Ancient genomes from northern China suggest links between subsistence changes and human migration. Nat Commun. 2020;11:2700.
23. Bergström A, McCarthy SA, Hui R, et al. Insights into human genetic variation and population history from 929 diverse genomes. Science. 2020;367:eaay5012.
24. Choin J, Mendoza-Revilla J, Arauna LR, et al. Genomic insights into population history and biological adaptation in Oceania. Nature. 2021;592:583-9.
25. Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655-64.
26. Lawson DJ, Hellenthal G, Myers S, Falush D. Inference of population structure using dense haplotype data. PLoS Genet. 2012;8:e1002453.
27. Patterson N, Moorjani P, Luo Y, et al. Ancient admixture in human history. Genetics. 2012;192:1065-93.
28. Mallick S, Micco A, Mah M, et al. The Allen Ancient DNA Resource (AADR) a curated compendium of ancient human genomes. Sci Data. 2024;11:182.
29. Tao L, Yuan H, Zhu K, et al. Ancient genomes reveal millet farming-related demic diffusion from the Yellow River into southwest China. Curr Biol. 2023;33:4995-5002.e7.
30. Du P, Zhu K, Wang M, et al. Genomic dynamics of the Lower Yellow River Valley since the Early Neolithic. Curr Biol. 2024;34:3996-4006.e11.
31. Li S, Wang R, Ma H, et al. Ancient genomic time transect unravels the population dynamics of Neolithic middle Yellow River farmers. Sci Bull. 2024;69:3365-70.
32. Wang F, Wang R, Ma H, et al. Neolithization of Dawenkou culture in the lower Yellow River involved the demic diffusion from the Central Plain. Sci Bull. 2024;69:3677-81.
33. Xiong J, Wang R, Chen G, et al. Inferring the demographic history of Hexi Corridor over the past two millennia from ancient genomes. Sci Bull. 2024;69:606-11.
34. Zhu K, Hu C, Yang M, et al. The demic diffusion of Han culture into the Yunnan-Guizhou plateau inferred from ancient genomes. Natl Sci Rev. 2024;11:nwae387.
35. Fang H, Liang F, Ma H, et al. Dynamic history of the Central Plain and Haidai region inferred from Late Neolithic to Iron Age ancient human genomes. Cell Rep. 2025;44:115262.
36. UK Biobank Whole-Genome Sequencing Consortium. Whole-genome sequencing of 490,640 UK Biobank participants. Nature. 2025;645:692-701.
37. Verma A, Huffman JE, Rodriguez A, et al. Diversity and scale: genetic architecture of 2068 traits in the VA million veteran program. Science. 2024;385:eadj1182.
38. Yang Q, Sun Y, Duan S, et al. High-quality Population-specific Haplotype-resolved Reference Panel in the Genomic and Pangenomic Eras. Genom Proteom Bioinform. 2025;23:qzaf022.
39. Wang Z, Liu K, Yuan H, et al. YanHuang paternal genomic resource suggested a weakly-differentiated multi-source admixture model for the formation of Han’s founding ancestral lineages. Genom Proteom Bioinform. 2025;23:qzaf049.
40. Wang M, Duan S, Sun Q, et al. Genomic insights into population structure, adaptation, and archaic introgression at the Himalayan-East Asian crossroads. J Genet Genomics. 2026;53:414-32.
41. Ioannidis AG, Blanco-Portillo J, Sandoval K, et al. Paths and timings of the peopling of Polynesia inferred from genomic networks. Nature. 2021;597:522-6.
42. Ioannidis AG, Blanco-Portillo J, Sandoval K, et al. Native American gene flow into Polynesia predating Easter Island settlement. Nature. 2020;583:572-7.
43. Malaspinas AS, Westaway MC, Muller C, et al. A genomic history of Aboriginal Australia. Nature. 2016;538:207-14.
44. Fan S, Spence JP, Feng Y, et al. Whole-genome sequencing reveals a complex African population demographic history and signatures of local adaptation. Cell. 2023;186:923-939.e14.
45. Gurdasani D, Carstensen T, Fatumo S, et al. Uganda genome resource enables insights into population history and genomic discovery in Africa. Cell. 2019;179:984-1002.e36.
46. Lachance J, Vernot B, Elbers CC, et al. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell. 2012;150:457-69.
47. Skoglund P, Thompson JC, Prendergast ME, et al. Reconstructing prehistoric African population structure. Cell. 2017;171:59-71.e21.
48. Joshi E, Biddanda A, Popoola J, et al. ; 54gene Team, NCD-GHS Consortium. Whole-genome sequencing across 449 samples spanning 47 ethnolinguistic groups provides insights into genetic diversity in Nigeria. Cell Genom. 2023;3:100378.
49. Choudhury A, Aron S, Botigué LR, et al. ; TrypanoGEN Research Group, H3Africa Consortium. High-depth African genomes inform human migration and health. Nature. 2020;586:741-8.
50. Fortes-Lima CA, Burgarella C, Hammarén R, et al. The genetic legacy of the expansion of Bantu-speaking peoples in Africa. Nature. 2024;625:540-7.
51. Gurdasani D, Carstensen T, Tekola-Ayele F, et al. The African Genome Variation Project shapes medical genetics in Africa. Nature. 2015;517:327-32.
52. Lipson M, Ribot I, Mallick S, et al. Ancient West African foragers in the context of African population history. Nature. 2020;577:665-70.
53. Lipson M, Sawchuk EA, Thompson JC, et al. Ancient DNA and deep population structure in sub-Saharan African foragers. Nature. 2022;603:290-6.
54. Hanotte O, Bradley DG, Ochieng JW, Verjee Y, Hill EW, Rege JE. African pastoralism: genetic imprints of origins and migrations. Science. 2002;296:336-9.
55. Patin E, Lopez M, Grollemund R, et al. Dispersals and genetic adaptation of Bantu-speaking populations in Africa and North America. Science. 2017;356:543-6.
56. Prendergast ME, Lipson M, Sawchuk EA, et al. Ancient DNA reveals a multistep spread of the first herders into sub-Saharan Africa. Science. 2019;365:eaaw6275.
57. Zhang P, Luo H, Li Y, et al. ; Han100K Initiative. NyuWa Genome resource: a deep whole-genome sequencing-based variation profile and reference panel for the Chinese population. Cell Rep. 2021;37:110017.
58. Cao Y, Li L, Xu M, et al. ; ChinaMAP Consortium. The ChinaMAP analytics of deep whole genome sequences in 10,588 individuals. Cell Res. 2020;30:717-31.
59. Yang MY, Zhong JD, Li X, et al. SEAD reference panel with 22,134 haplotypes boosts rare variant imputation and genome-wide association analysis in Asian populations. Nat Commun. 2024;15:10839.
60. Zhou D, Wu M, Tan Q, et al. Non-coding genetic elements of lung cancer identified using whole genome sequencing in 13,722 Chinese. Nat Commun. 2025;16:7365.
61. Jiang T, Guo H, Liu Y, et al. A comprehensive genetic variant reference for the Chinese population. Sci Bull. 2024;69:3820-5.
62. Lipson M, Cheronet O, Mallick S, et al. Ancient genomes document multiple waves of migration in Southeast Asian prehistory. Science. 2018;361:92-5.
63. McColl H, Racimo F, Vinner L, et al. The prehistoric peopling of Southeast Asia. Science. 2018;361:88-92.
64. Sun Y, Wang M, Sun Q, et al. Distinguished biological adaptation architecture aggravated population differentiation of Tibeto-Burman-speaking people. J Genet Genomics. 2024;51:517-30.
65. Huang Y, Wang M, Wang Z, et al. Short tandem repeats in populations of the Qinghai-Tibet Plateau and adjacent regions provide insights into high-altitude adaptation. Sci Adv. 2025;11:eadx1590.
66. Lin X, Liu Y, Zhang J, et al. Genomic resources from the Yunnan-Guizhou Plateau reveal the linguistic‐linked demographic history and insights into human adaptation and disease of multi‐ancestry populations. J Syst Evol. 2025;63:1370-89.
67. He G, Sun Y, Duan S, et al. Ancient genomes give insight into 160,000 years of East Asian population dynamics and biological adaptation. Genome Biol. 2025;26:420.
68. Robbeets M, Bouckaert R, Conte M, et al. Triangulation supports agricultural spread of the Transeurasian languages. Nature. 2021;599:616-21.
69. Ma H, Zhou Y, Wang R, et al. Ancient genomes shed light on the long-term genetic stability in the Central Plain of China. Sci Bull. 2025;70:333-7.
70. Yang T, He J, Li C, et al. Ancient DNA reveals the population interactions and a Neolithic patrilineal community in Northern Yangtze Region. Nat Commun. 2025;16:8728.
71. Yang MA, Fan X, Sun B, et al. Ancient DNA indicates human population shifts and admixture in northern and southern China. Science. 2020;369:282-8.
72. Zhang D, Sun B, Li F, et al. Neolithic genomes reveal long distance interactions in agropastoral border zone of Yan Mountain Region. Sci Bull 2026;71:148-58.
73. Xiong J, Xu Y, Chen G, et al. The genomic history of East Asian Middle Neolithic millet- and rice-agricultural populations. Cell Genom. 2025;5:100976.
74. Zou Y, Tan J, Zhou J, et al. Ancient genomes from the Yellow River Bend reveal long-distance population interactions between the Central Plains, Steppe, and southern China. Cell Rep. 2025;44:116034.
75. Liu J, Liu Y, Zhao Y, et al. East Asian Gene flow bridged by northern coastal populations over past 6000 years. Nat Commun. 2025;16:1322.
76. Damgaard PB, Marchi N, Rasmussen S, et al. 137 ancient human genomes from across the Eurasian steppes. Nature. 2018;557:369-74.
77. Flegontov P, Altınışık NE, Changmai P, et al. Palaeo-Eskimo genetic ancestry and the peopling of Chukotka and North America. Nature. 2019;570:236-40.
78. Sikora M, Pitulko VV, Sousa VC, et al. The population history of northeastern Siberia since the Pleistocene. Nature. 2019;570:182-8.
79. Luo L, Wang M, Liu Y, et al. Sequencing and characterizing human mitochondrial genomes in the biobank-based genomic research paradigm. Sci China Life Sci. 2025;68:1610-25.
80. Moltke I, Grarup N, Jørgensen ME, et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature. 2014;512:190-3.
81. Skoglund P, Mallick S, Bortolini MC, et al. Genetic evidence for two founding populations of the Americas. Nature. 2015;525:104-8.
82. Sohail M, Palma-Martínez MJ, Chong AY, et al. Mexican Biobank advances population and medical genomics of diverse ancestries. Nature. 2023;622:775-83.
83. Stæger FF, Andersen MK, Li Z, et al. Genetic architecture in Greenland is shaped by demography, structure and selection. Nature. 2025;639:404-10.
84. Ziyatdinov A, Torres J, Alegre-Díaz J, et al. ; Regeneron Genetics Center, Mexico City Prospective Study. Genotyping, sequencing and analysis of 140,000 adults from Mexico City. Nature. 2023;622:784-93.
85. Gusareva ES, Ghosh AG, Kharkov VN, et al. From North Asia to South America: Tracing the longest human migration through genomic sequencing. Science. 2025;388:eadk5081.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].