


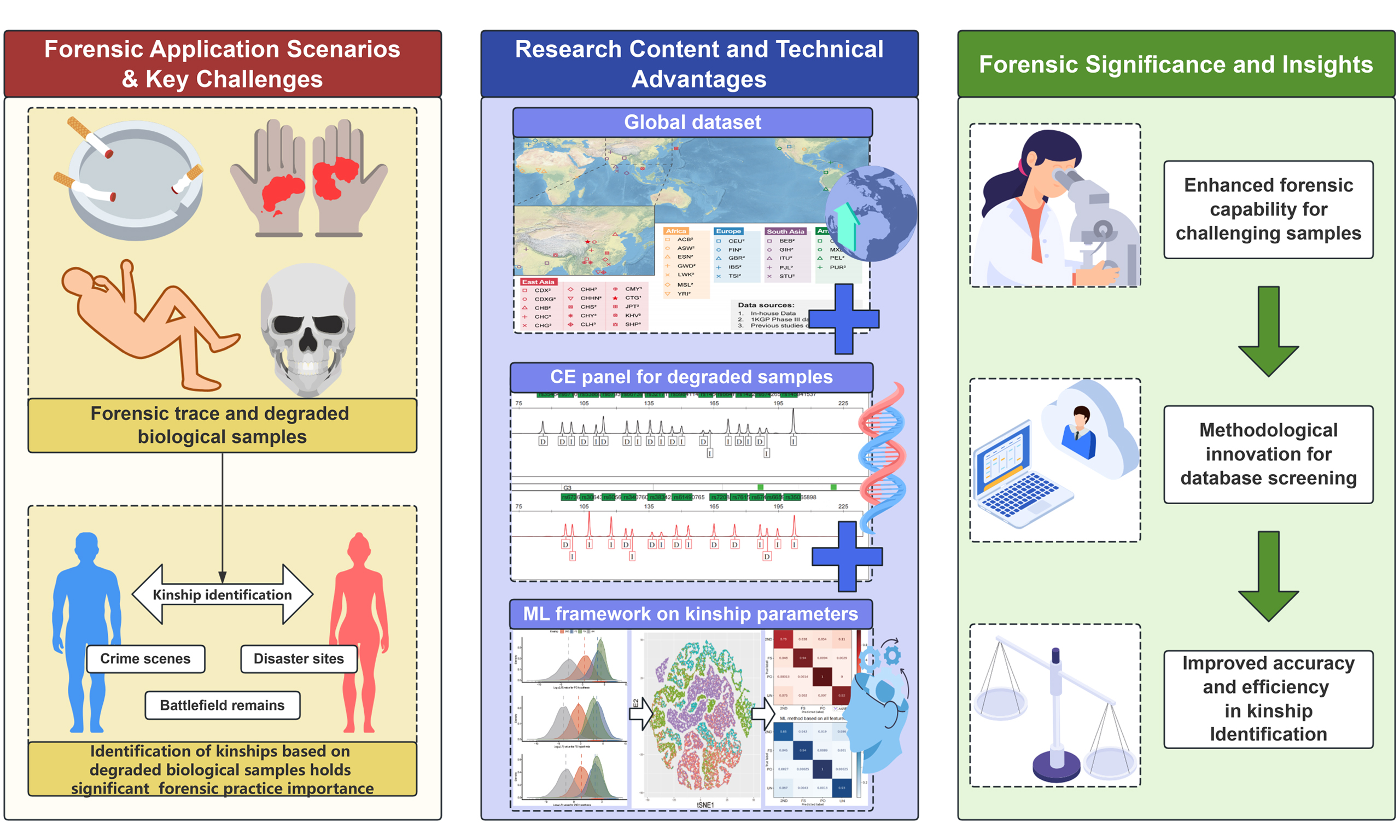
Kinship classification from a machine learning perspective: a pilot study based on genotyping data
 0
0 Abstract
Aim: Kinship analysis in trace amounts and degraded biological samples has consistently posed a challenge in forensic practice. With shorter amplicons and no stutter peak, Insertion/Deletion polymorphisms (InDels) significantly improve kinship analyses of deceased individuals and their potential living relatives. However, room for improvement remains in identifying 2nd-degree and more distant kinships. To address this issue, a kinship analysis workflow based on machine learning (ML) models was proposed.
Methods: Based on multiple kinship parameters including identity-by-state (IBS) scores, k coefficients, proportion identity-by-descent (IBD), and likelihood ratio (LR) values, this pilot study applied a recently validated InDel locus to preliminarily develop an ML workflow for forensic kinship multi-classification.
Results: In the binary classification of 2nd-degree relatives and unrelated pairs, the LR cutoff threshold workflow and the ML workflow achieved a similar accuracy of 0.9194. However, the ML method had a conclusiveness rate (CR) of 1.0, compared to 0.7066 for the LR workflow. In the multiclass task, the LR-based workflow had a macro F1 score of 0.6955/0.5212 and a CR of 0.7375/0.7046 for single and dual thresholds methods, respectively. However, the ML-based workflow showed that the optimal model - feature combination (XGBoost-IBD+LR) could classify all samples conclusively, with a macro F1 score of 0.9020.
Conclusion: In summary, the ML workflow enhanced the kinship analysis efficiency based on the InDel genotyping system by combining multiple parameters, aiming to provide a more flexible and efficient solution for large-scale database screening.
Keywords
INTRODUCTION
Forensic kinship analysis is vital and challenging for disaster victim identification and cold case investigation, particularly when dealing with highly degraded biomaterials. However, despite the strong polymorphism and widespread adoption of the short tandem repeat capillary electrophoresis as the standard approach for kinship analysis, its effectiveness is limited by longer amplicon lengths. Highly degraded biomaterials encountered at forensic scenes may result in incomplete or unreliable genotyping and artifacts. Characterized by its short amplicon, low mutation rate, and absence of stutter peaks, Insertion/Deletion polymorphism (InDel) has garnered increasing attention from forensic researchers[1-3]. Featuring smaller amplicons (< 230 bp) of the selected 57 autosomal InDel loci, the AGCU-60 InDel loci has well validated its efficacy for paternity testing in a series of studies based on numerous populations[4-9], where a complete variant call could be obtained from samples with a minimum DNA input of 125 pg. However, InDel loci, as exemplified by this panel, face challenges in kinship analysis. When analyzing highly degraded samples, prior knowledge about the genetic background of the unknown donor is often unavailable. Consequently, the only feasible approach is to conduct large-scale database matching to identify potential relatives.
In this context, kinship parameters including identity-by-state (IBS) score, method of moment (MoM), and likelihood ratio (LR) are commonly applied. The IBS score[10], assessing the similarity of DNA segments or alleles between two individuals, can be directly computed by the shared number of IBS alleles without a priori knowledge of allele frequencies and linkage disequilibrium status in the population. However, it is accompanied by drawbacks such as the need for non-generalized discriminant thresholds and a relatively low strength of evidence. MoM, grounded in the observed genotyping profile of pairwise individuals, estimates parameters such as actual kinship coefficients and k coefficients[11]. This approach simultaneously considers the count of IBS and (or) allele frequencies, yielding a higher informativeness. However, no standardized evidence interpretation system has been established on this method so far, and it is less effective at identifying 3rd-degree and more distant kinships[11,12]. The LR method is currently recommended by the International Society for Forensic Genetics[13], necessitating a priori statement of specific kinship as alternate hypotheses and the null hypothesis of unrelatedness between the two individuals. Based on the two exclusive hypotheses, LR compares the conditional probabilities of the two hypotheses. Its robustness and efficiency have been validated in numerous studies[9,14-16]. In this context, the mainstream LR cutoff threshold-based approach provides kinship identification results based on two specific hypotheses: a particular type of kinship relationship as the alternate hypothesis and unrelated individuals (UNs) as the null hypothesis. When analyzing highly degraded samples with unknown genetic backgrounds based on InDels, this requires evaluating all possible hypotheses, resulting in the accumulation of errors in the final conclusions. Considering the inherent limitation imposed by the bi-allelic nature of InDel loci, the accumulation of errors may compromise the reliability of kinship analysis.
To address these challenges, this study proposes introducing machine learning (ML) models and their evaluation systems as a novel method for interpreting kinship evidence. By eliminating the need to assume a specific kinship, this approach aims to provide a more flexible and efficient solution for large-scale database screening. In this study, a series of ML classifiers with different complexities were introduced into the InDel-based kinship classification: namely the multinomial logistic regression (MLR), multinomial Naïve Bayes (MNB), support vector machine (SVM), random forest classifier (RFC), extreme gradient boosting (XGBoost, XGB)[17], light gradient boosting machine (LightGBM, LGBM)[18] and categorical boosting (CatBoost, CATB)[11]. By comparing the common workflow, this study aims to develop a preliminary ML workflow for kinship analysis based on the AGCU-60 InDel loci and serves as a pilot exploration for integration of multiple kinship parameters in the forensic kinship classification.
METHODS
Sample collection and reference data preprocessing
A total of 175 Chinese Tibetans who claimed to be unrelated within three generations in the Tibetan Autonomous Prefecture of Gannan (CTG) were introduced into the existing population dataset of AGCU-60 InDel loci. This study was approved by the Ethics Committee of Xi’an Jiaotong University Health Science Center and Southern Medical University (NO. 2019-1039). Genotyping data of different populations were integrated. All CTG samples were collected between August 2022 and October 2023. Data sources were as follows: (1) the studied CTG group; (2) 1000 Genomes Project (1KGP) Phase III expanded[19]; (3) nine populations from the previous studies on the AGCU-60 kit[4-9,20]. When processing the 1KGP whole-genome data, we filtered samples that may have Mendelian inheritance errors (e = 0.001) using the following PLINK v2.0 (https://www.cog-genomics.org/plink/2.0) command: “--me 0.001”.
DNA extraction, polymerase chain reaction amplification, and genotyping
Genomic DNAs from the bloodstain samples were extracted based on the Chelex-100 method. Then, the Polymerase Chain Reaction (PCR) amplification conditions of the AGCU-60 kit followed the manufacturer’s protocol and previous validation study[5].
Statistical analysis of forensic parameters and genetic background
Common forensic parameters of the 57 autosomal InDel loci in the CTG group were calculated by STRAF v1.0.5[21]. Linkage disequilibrium on all pairwise loci from the 57 InDel loci was tested by Arlequin v3.5[22]. Detailed information is demonstrated in Figure 1, Supplementary Tables 1 and 2. Using PLINK v2.0 (https://www.cog-genomics.org/plink/2.0), we screened all samples unrelated within three generations with the command “--king-cutoff 0.0625”, to obtain robust allele frequencies in the population. We then merged these samples separately into a global dataset (n = 5201) and an East Asian dataset (n = 3200) according to biogeographical origins. To assess the phylogenetic relationships among the 36 global populations, the merged .vcf files were used for the Treemix analysis (https://bioconda.github.io/recipes/treemix/README.html), with the root population set as Yoruba in Ibadan, Nigeria (YRI). Finally, ADMIXTURE analyses (https://dalexander.github.io/admixture/) set at different numbers of assumed ancestry components (K = 2-10) were performed and the optimal K value was confirmed under cross-validation. The East Asian dataset was used to perform the forensic kinship analyses and the development of ML models. Genotype data of the global dataset was applied to calculate Nei’s Genetic Distance (DA) values between paired populations. Principal Component Analysis (PCA) analyses were separately performed on the allelic frequency and genotyping data of the global dataset. A locus-by-locus analysis of molecular variance was also performed to obtain the overall Fixation Index within Subpopulations (FIS), Fixation Index among Subpopulations (FST), and Total Fixation Index (FIT) values of the East Asian dataset, to reveal the latent genetic substructure within East Asian populations.
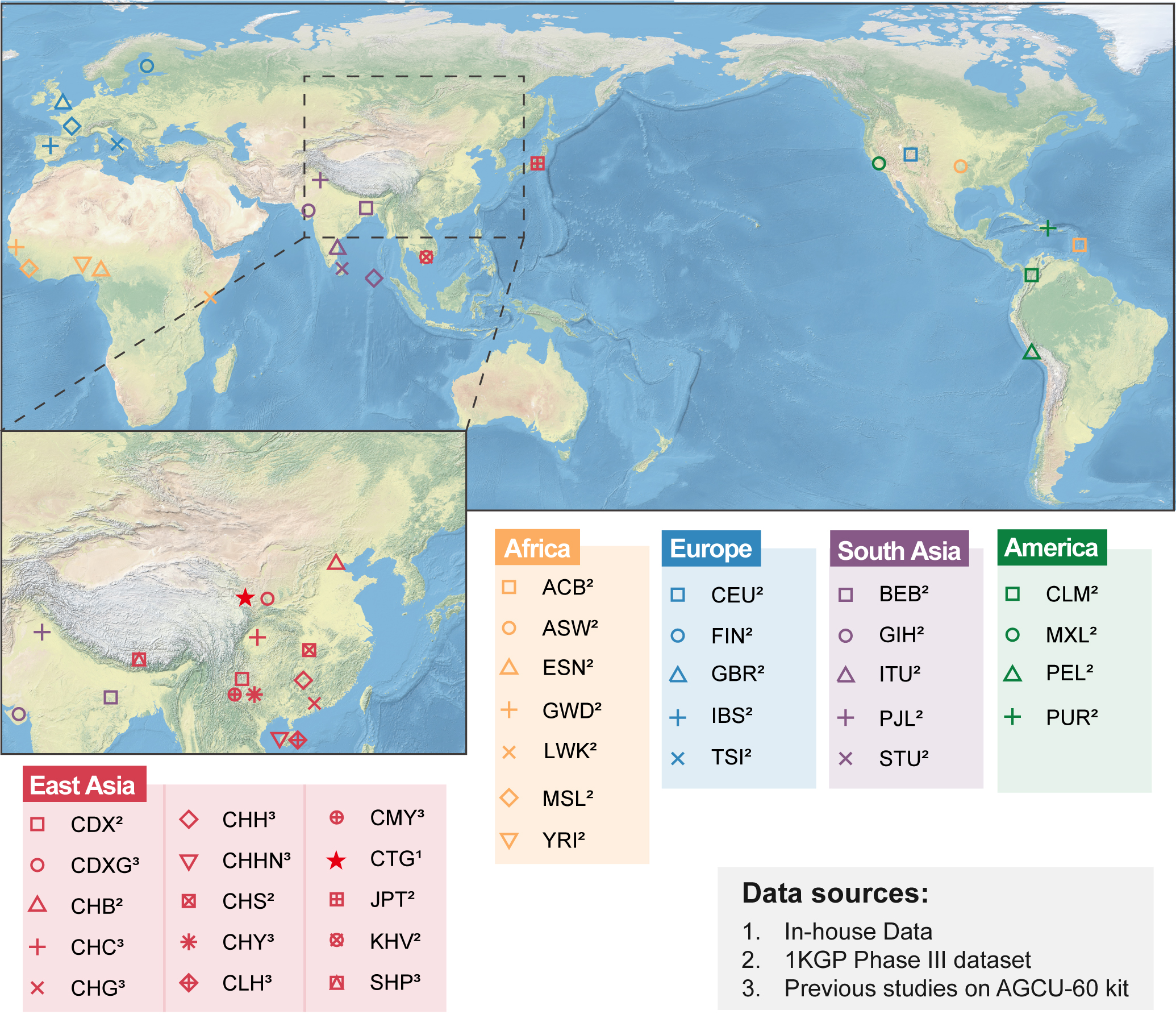
Figure 1. Geographical distributions of the 35 global populations and the studied CTG group. Populations labeled as 1, 2, and 3 are, respectively, from datasets of this study, the 1000 Genomes Project Phase III, and previous AGCU-60 InDel kit studies. The map is open access at the site of the QGIS Geographic Information System (https://www.qgis.org/). Copyright © 1989, 1991 Free Software Foundation, Inc. QGIS: Quantum Geographic Information System; ACB: African Caribbean in Barbados; ASW: African Ancestry in Southwest US; ESN: Esan in Nigeria; GWD: Gambian in Western Division, The Gambia; LWK: Luhya in Webuye, Kenya; MSL: Mende in Sierra Leone; YRI: Yoruba in Ibadan, Nigeria; CLM: Colombian in Medellin, Colombia; MXL: Mexican Ancestry in Los Angeles, California; PEL: Peruvian in Lima, Peru; PUR: Puerto Rican in Puerto Rico; CDX: Chinese Dai in Xishuangbanna, China; CDXG: Chinese Dongxiang in Gansu, China; CHB: Han Chinese in Bejing, China; CHC: Chinese Han in Chengdu, China; CHG: Chinese Han in Guangdong, China; CHH: Chinese Han in Hunan, China; CHHN: Chinese Han in Hainan, China; CHS: Southern Han Chinese, China; CHY: Chinese Hani in Yunnan, China; CLH: Chinese Li in Hainan, China; CMY: Chinese Miao in Yunnan, China; CTG: Chinese Tibetan in Tibetan Autonomous Prefecture of Gannan, China; JPT: Japanese in Tokyo, Japan; KHV: Kinh in Ho Chi Minh City, Vietnam; SHP: Dingjie Sherpa, China; CEU: Utah residents with Northern and Western European ancestry; FIN: Finnish in Finland; GBR: British in England and Scotland; IBS: Iberian populations in Spain; TSI: Toscani in Italy; BEB: Bengali in Bangladesh; GIH: Gujarati Indian in Houston, TX; ITU: Indian Telugu in the UK; PJL: Punjabi in Lahore, Pakistan; STU: Sri Lankan Tamil in the UK.
Calculation of multiple kinship parameters
Figure 2 outlines the workflow for forensic kinship classification in this study. After confirming the absence of significant genetic substructure among 15 East Asian populations, 3,201 individuals from the merged dataset were used as input for pedigree simulations in Familias[23] [Mutation options: model = equal probability (simple); rate = 0.0; range = 0.1; rate 2 = 1E-6]. Finally, 40,000 pairs of the following relatedness were simulated: (1) parent-offspring (PO); (2) full siblings (FS); (3) 2nd-degree relatives (2ND), including an equal number of half-siblings (HS), aunt-cousins (AC) and grandchild-grandparents (GP); (4) unrelated individuals (UN).
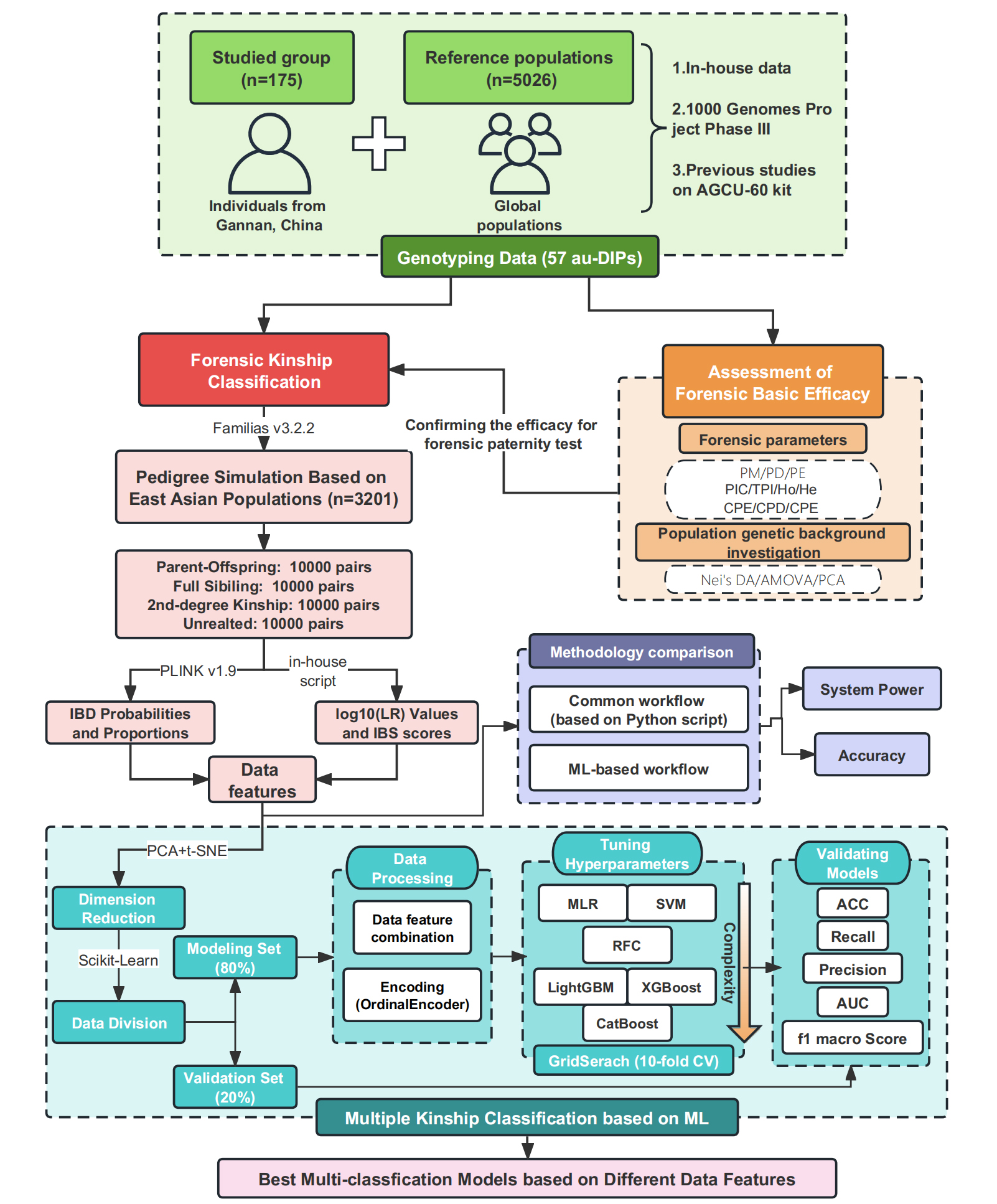
Figure 2. Workflow for the critical evaluation of forensic efficacy and kinship classification from the ML algorithm perspective. LR: Likelihood ratio; IBD: identity-by-descent; MLR: multinomial logistic regression; SVM: support vector machine; RFC: random forest classifier; ML: machine learning; LightGBM: light gradient boosting machine; PM: probability of matching; PD: power of discrimination; PE: probability of exclusion; PIC: polymorphism information content; TPI: typical paternity index; Ho: observed heterozygosity; He: expected heterozygosity; CPE: cumulative probability of exclusion; CPD: cumulative power of discrimination; Nei’s DA: Nei’s Genetic Distance; PCA: principal component analysis; t-SNE: t-distributed stochastic neighbor embedding; CV: cross-validation; ACC: accuracy; AUC: area under the curve; AMOVA: analysis of molecular variance; XGBoost: extreme gradient boosting.
For each pair in the East Asian dataset, k coefficients include three probabilities: those for the individuals having zero, one or two pairs of IBD alleles (k0, k1 and k2) coefficients were computed. Table 1 displayed the expected values of k0, k1, and k2 under different genotype combinations for the tested pair of individuals. Proportion identity-by-descent (IBD) values were derived from these k coefficients using PLINK v1.9. Mathematically, the proportion IBD value is equal to twice the kinship coefficient. Under the null and alternative hypotheses of H0 and Hi (i = 1, 2, 3), LR values for bi-allelic InDels were calculated to evaluate the underlying relationship between pairwise individuals (Eq. 1 in Supplementary Appendix A). Four different hypotheses for referring to UN, PO, FS, and 2ND relatedness are shown in Table 2. IBS scores for pairwise individuals were calculated according to Table 2. By multiplying the LR values and accumulating the IBS scores of all InDel loci in linkage equilibrium, the LR of combined LRs (CLR) and the cumulative IBS (CIBS) scores were obtained (Eq. 2 in Supplementary Appendix A).
Expected values of k coefficient and IBD proportion for different relatedness
| Type of kinship | k coefficient | IBD proportion | ||
| k 0 | k 1 | k 2 | ||
| Parent-offspring (PO) | 0 | 1 | 0 | 1/2 |
| Full siblings (FS) | 1/4 | 1/2 | 1/4 | 1/2 |
| 2ND relatives* | 1/2 | 1/2 | 0 | 1/4 |
| Unrelated (UN) | 1 | 0 | 0 | 0 |
Formulae of likelihood ratio (LR) and calculation of identity by state (IBS) score for kinship classification within 2ND-degree relatives based on different genotype pairs of biallelic InDels
| Genotype 1/Genotype 2 | Likelihood ratio | IBS score | ||
| PO/UN | FS/UN | 2ND*/UN | ||
| AA/AA | | | | 2 |
| AA/AB | | | | 1 |
| AB/AB | | | | 2 |
| AA/BB | | | | 0 |
In total, we calculated a total of 11 kinship parameters, including IBS0-2 (represent the number of genes when the shared number of IBS alleles is 0, 1, and 2, respectively), CIBS, k0-2 coefficients, proportion IBD, and LRs under three hypotheses. We then merged these parameters into 7 distinct feature combinations: (1) IBS; (2) IBD; (3) LR; (4) IBS+IBD; (5) IBS+LR; (6) IBD+LR; and (7) IBS+IBD+LR (ALL). For these feature combinations, we conducted t-distributed stochastic neighbor embedding (t-SNE) dimensionality reduction for visualization.
Common and machine learning workflows for forensic kinship analysis
The 11 kinship parameters from the 40,000 simulated pairs were split into a modeling dataset (80%) and a validation set (20%) for machine learning analysis. As the log10(LR) threshold method is widely used in kinship identification, we used this method as a reference methodology. The simplified decision-making process of the threshold-based classification is shown in Table 3. Based on the true relationship counts corresponding to different LR values described in Table 3, calculate the accuracy (ACC), conclusiveness rate (CR), and system power (SP). These are three different metrics used to evaluate the model, respectively describing the accuracy of kinship inference when the method can draw a clear conclusion, the rate at which clear conclusions can be drawn, and the overall system performance. The specific calculation methods are detailed in Eq. 3-5 in Supplementary Appendix A. A brief procedure was shown in Figure 2.
Decision-making process for traditional threshold-based kinship classification
| Classification threshold | Prediction | True relationship | ||
| Related | Unrelated | |||
| log10(LR) ≥ Ti | The two individuals are related under certain hypothesis* | K1 | U1 | |
| Ti > log10(LR) > Te | Relatedness between the two individuals are not confirmed | K2 | U2 | |
| log10(LR) ≤ Te | The two individuals are unrelated | K3 | U3 | |
| Total | M | N | ||
Developing ML models for kinship classification based on LR values and different feature combinations
We developed a series of binary models based on log10(LR) using Python v3.9.1, aiming to classify FS and 2ND relatives from UN pairs. After preprocessing the data, 7 types of ML models: MLR, MNB, SVM, RFC, XGB, LGBM, and CATB were developed. To balance bias and variance, the difference in accuracy between training and test sets was limited within 0.05[24,25]. The best model for each algorithm was evaluated using 10-fold cross-validation, comparing training time and balanced ACC. The top binary models for FS and 2ND classification were selected, and their ACC values served as benchmarks for determining baseline log10(LR) cutoff thresholds in a later section. For PO-UN classification, since the cumulative probability of exclusion (CPE) of this InDel loci had already been confirmed in the study and previous research[4-9,20], we set “0.9999” as a benchmark for the subsequent methodological comparison.
Due to the homology in the computational principles of IBD and IBS, the IBS+IBD feature combination was excluded. Multiclass models based on the remaining feature combinations were also developed using the same seven algorithms. These were converted to one-vs-rest classifiers, and their 10-fold cross-validation metrics were compared.
Methodological comparison of log10(LR) cutoff threshold-based and ML methods for kinship classification
To establish baseline LR thresholds for different kinship hypotheses, we developed a Python pipeline (Supplementary Appendix B). implementing both single- and dual-threshold methods. The single-threshold approach compares the fundamental classification performance of Bayesian and ML methods without considering confidence, excluding results between thresholds as “insufficient confidence”. The dual-threshold method incorporates confidence levels to evaluate overall methodological differences.
For the single-threshold LR method, we iterated through all possible thresholds to identify the globally optimal threshold with the highest ACC. For the LR method with dual cutoff thresholds, the ACC value corresponding to these two thresholds should be comparable to those of the best ML models. In the dual-threshold method, various thresholds are incorporated to provide a stable benchmark for comparison under different application scenarios and confidence levels. Considering that there might be multiple pairs of thresholds with the same level of ACC, baseline thresholds with the highest CR value were selected. Three sets of empirical thresholds (i.e., ±1, ±2, and ±3) were included. After determining these thresholds, a multi-classification workflow based on standard forensic practice was applied to validation set pairs with unknown relatedness. Multi-class performance was evaluated using the macro F1-score, which balances recall and precision across all classes.
RESULTS
Landscape of the basic forensic parameters for the 57 InDel loci in the CTG group
Adjusted by Bonferroni’s correction [pHWE (p values of Hardy-Weinberg equilibrium) > 8.772E-4, pLD (p values of Linkage Disequilibrium) > 3.133E-5], the 57 InDel loci in the CTG group were consistent with Hardy-Weinberg equilibrium, and had a relatively good performance in forensic individual identification [Supplementary Table 3]. No evidence of linkage disequilibrium was observed [Supplementary Table 4]. Table 4 also displays the basic forensic parameters of the 57 InDel loci. As shown in Supplementary Figure 1, most allelic frequencies in the East Asians from the 1KGP dataset fluctuated around 0.4-0.6, which was consistent with the forensic demand of performing individual identification and paternity testing in East Asian populations. Figure 3A shows the population genetic structure at different K values, with the CTG group highlighted centrally. The AGCU-60 panel identified three major ancestral components (African, East Asian, and European) across 36 global populations, clustering CTG with Han populations Chinese Han in Guangdong (CHG) and Chinese Han in Hunan (CHH). A maximum likelihood phylogenetic tree [Figure 3B] placed all 15 East Asian populations on the same major branch, with CTG and Chinese Hani in Yunnan (CHY) sharing the most recent common ancestor. Normalized fit residuals near zero for most East Asian pairs supported the tree’s reliability. However, higher residuals between Dingjie Sherpa (SHP) and CTG/Japanese in Tokyo (JPT) suggest closer genetic ties than shown, possibly due to admixture. Overall, no significant substructure was detected among the East Asian populations.
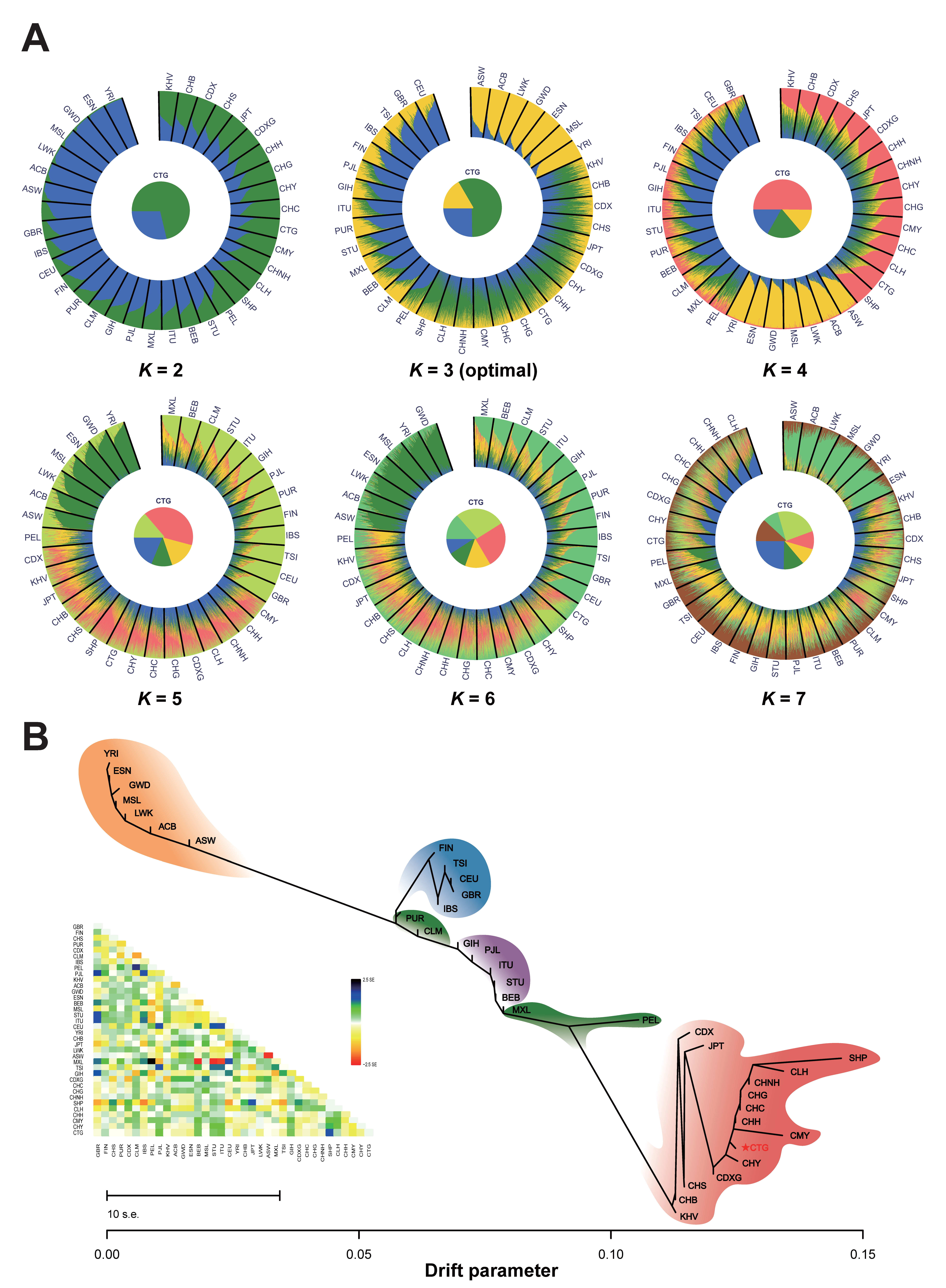
Figure 3. ADMIXTURE and Treemix analyses based on the 57 InDel loci genotyping data of individuals from different continental regions; (A) shows the ADMIXTURE analysis results when the K value is 2 to 7, and the CTG group is consistently displayed in the center; (B) is the maximum likelihood tree constructed by Treemix software and populations from the same intercontinental region were labeled by a specific color. A residual heatmap is also shown in the bottom left corner of the plot, and the color of each cell, as per the color scale legend, represents the normalized fit residual value between the pairwise populations. CTG: Tibetan Autonomous Prefecture of Gannan; ACB: African Caribbean in Barbados; ASW: African Ancestry in Southwest US; ESN: Esan in Nigeria; GWD: Gambian in Western Division, The Gambia; LWK: Luhya in Webuye, Kenya; MSL: Mende in Sierra Leone; YRI: Yoruba in Ibadan, Nigeria; CLM: Colombian in Medellin, Colombia; MXL: Mexican Ancestry in Los Angeles, California; PEL: Peruvian in Lima, Peru; PUR: Puerto Rican in Puerto Rico; CDX: Chinese Dai in Xishuangbanna, China; CDXG: Chinese Dongxiang in Gansu, China; CHB: Han Chinese in Bejing, China; CHC: Chinese Han in Chengdu, China; CHG: Chinese Han in Guangdong, China; CHH: Chinese Han in Hunan, China; CHHN: Chinese Han in Hainan, China; CHS: Southern Han Chinese, China; CHY: Chinese Hani in Yunnan, China; CLH: Chinese Li in Hainan, China; CMY: Chinese Miao in Yunnan, China; CTG: Chinese Tibetan in Tibetan Autonomous Prefecture of Gannan, China; JPT: Japanese in Tokyo, Japan; KHV: Kinh in Ho Chi Minh City, Vietnam; SHP: Dingjie Sherpa, China; CEU: Utah residents with Northern and Western European ancestry; FIN: Finnish in Finland; GBR: British in England and Scotland; IBS: Iberian populations in Spain; TSI: Toscani in Italy; BEB: Bengali in Bangladesh; GIH: Gujarati Indian in Houston, TX; ITU: Indian Telugu in the UK; PJL: Punjabi in Lahore, Pakistan; STU: Sri Lankan Tamil in the UK.
Frequency data of 57 InDels in unrelated East Asian individuals
| Rs ID | Deletion | Insertion | Insertion frequency |
| rs3067397 | G | GTATCT | 0.4191 |
| rs10607699 | C | CCCT | 0.6870 |
| rs71852971 | T | TACTC | 0.4008 |
| rs139764906 | C | CCTAA | 0.4985 |
| rs67487831 | T | TTCAA | 0.6137 |
| rs11277697 | T | TTTAGG | 0.4061 |
| rs113011930 | C | CTTCT | 0.3886 |
| rs144941014 | T | TGAA | 0.6198 |
| rs146875868 | C | CTCTT | 0.4534 |
| rs145191158 | T | TTTTG | 0.5473 |
| rs66477007 | T | TAAGA | 0.5481 |
| rs10590825 | C | CCCT | 0.5092 |
| rs3834231 | C | CTAGG | 0.5229 |
| rs145577149 | C | CAAAT | 0.3626 |
| rs35309403 | T | TACTG | 0.6336 |
| rs140820428 | G | GAGA | 0.3748 |
| rs66595817 | T | TCTTTC | 0.5198 |
| rs66879403 | T | TTGA | 0.4542 |
| rs60867863 | G | GATTA | 0.4298 |
| rs567292477 | T | TTATAAC | 0.4710 |
| rs79225518 | C | CAAG | 0.4802 |
| rs3217112 | C | CTAATA | 0.5511 |
| rs67426579 | A | AGTG | 0.5069 |
| rs151335218 | C | CAAGT | 0.3916 |
| rs142221201 | C | CAAAG | 0.4198 |
| rs66649248 | G | GATC | 0.6557 |
| rs57981446 | T | TAGGAG | 0.5015 |
| rs67365630 | C | CACT | 0.5565 |
| rs35464887 | C | CTTTA | 0.5473 |
| rs76158822 | T | TTTAAG | 0.4878 |
| rs5897566 | T | TTAAC | 0.6427 |
| rs67405073 | C | CTGA | 0.3870 |
| rs140683187 | G | GAAC | 0.4206 |
| rs5787309 | A | ATTATT | 0.4763 |
| rs34287950 | G | GTTT | 0.3710 |
| rs67100350 | G | GTAGT | 0.6137 |
| rs769299 | G | GTATC | 0.6053 |
| rs3076465 | C | CTTAT | 0.3328 |
| rs67939200 | C | CTCA | 0.4076 |
| rs145941537 | C | CAATT | 0.6382 |
| rs67264216 | A | ATGTCG | 0.3595 |
| rs35453727 | G | GAGA | 0.5405 |
| rs35065898 | A | AACTT | 0.5832 |
| rs561160795 | C | CTGG | 0.3779 |
| rs61490765 | C | CTTAAT | 0.5580 |
| rs34419736 | T | TAAG | 0.4817 |
| rs77635204 | G | GAGAA | 0.4603 |
| rs34421865 | A | ACTCT | 0.4198 |
| rs66739142 | C | CTCTTT | 0.6588 |
| rs557813049 | G | GTGTGC | 0.6389 |
| rs145010051 | T | TGGA | 0.4824 |
| rs72031009 | C | CTAGAG | 0.4664 |
| rs77206391 | C | CACAA | 0.4626 |
| rs33971783 | C | CTGTT | 0.5725 |
| rs72085595 | T | TTGTC | 0.2519 |
| rs34529638 | A | ACCT | 0.4298 |
| rs538690481 | G | GTCTGAA | 0.4649 |
Genetic background exploration of the East Asian populations
Based on the subpopulation frequency data, population-level PCA analyses [Supplementary Figure 2A] showed that 36 populations from the global dataset were roughly divided into three hierarchical clusters in the dimensions of the first two principal components (PCs), where no significant genetic substructure was found among the East Asians. It was worth noting that, although the SHP population was also distributed within the East Asian cluster, its average levels of PC2 and PC3 were higher compared to other East Asian populations [Supplementary Figure 2B]. As shown in Supplementary Table 5, no significant genetic differentiation was observed among 29 East Asian populations, and the FIS value showed a low level of inbreeding among individuals within these populations. Supplementary Table 6 shows the pairwise Nei’s DA values among the CTG group and 35 reference populations based on 57 InDel loci in the AGCU-60 panels. The above results all indicate that the East Asian dataset could serve as an input dataset for the subsequent kinship classifications.
Comprehensive assessment of multiple forensic kinship parameters
We filtered 655 individuals with no kinship within five degrees of kinship from the East Asian dataset of 3,200 individuals. Based on these individuals, the allele frequencies of 57 InDels were calculated [Table 4]. Supplementary Table 7 shows the pairwise kinship coefficient calculation results based on these 655 individuals. Using these data, we performed pedigree simulations with Familias. Based on the 40,000 simulated pairs, comprising different kinship parameters were calculated for the individual pairs, including IBS (IBS0-2 and CIBS); IBD (k0 coefficients and proportion IBD), and LR values under hypotheses of PO-UN, FS-UN and 2ND-UN. For k coefficients, proportion IBD, and IBS0 with relatively discrete distributions, we applied data binning to transform them into histograms for a better visualization [Figure 4A and B]. As shown in Figure 4A, most kinship parameters exhibited significant skewness in their distributions. Among the IBD-related parameters, FS pairs showed distributions that were closest to the theoretical values presented in Table 1, while the observed values for the UN pairs differed the most. Although k0 could distinguish between four types of relationship pairs [Supplementary Figure 3A]; k1 primarily distinguished between PO and UN pairs; k2 was mainly used to identify FS pairs; and IBD proportions for 2ND and UN pairs exhibited unique distributions, respectively. Most of the IBS-related parameters followed a normal distribution (except for the IBS0 value). As shown in Figure 4B and Supplementary Figure 3B, IBS0 could distinguish between PO and UN pairs; IBS1 showed a unique distribution for FS pairs; IBS2 and CIBS exhibited distribution differences among individual pairs with four types of relatedness. The LR-related parameters showed a normal distribution and appeared as three clusters: (1) UN pairs, (2) 2ND pairs, and (3) PO and FS pairs [Figure 4C]. Overall, LR values for specific kinship were generally highest under their corresponding hypotheses.
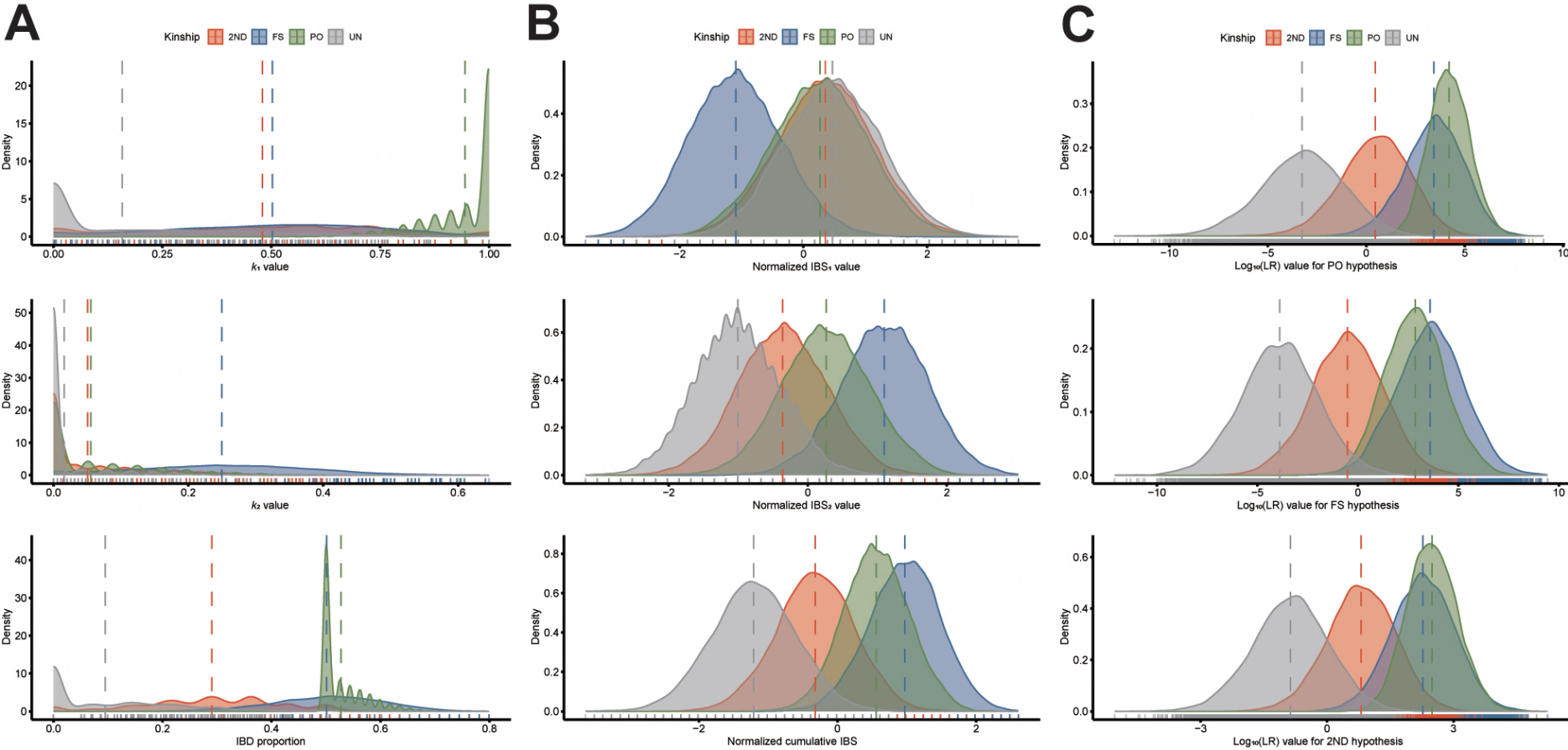
Figure 4. Density distribution maps of different kinship parameters. (A) presents the density distributions of IBD parameters (with bin width of 0.1), including k0, k1, k2, and proportion IBD; (B) demonstrates the density distributions of IBS parameters, including IBS0 (with bin width of 0.1), IBS1, IBS2, and normalized cumulative IBS; (C) displays the distributions of log10(LR) values under different hypotheses. The vertical dashed lines represent the median value of the correspondence relationships. IBD: Identity-by-descent; IBS: identity-by-state; FS: full siblings; PO: parent-offspring; 2ND: 2nd-degree relatives; UN: unrelated individuals.
Figure 5A-C presents the t-SNE results based on data feature combinations for a single category of kinship parameters. Among the features of IBD, IBS and IBS+IBD [Figure 5A, B and D], despite evident non-linear boundaries between different kinship pairs, there were no discernible structural patterns. In the dimensionality reduction based on LR values [Figure 5C], a majority of PO and FS pairs clustered distinct from UN, but part of the 2ND pairs was still mixed with UN, FS, and PO pairs. When involving LR values in the feature combinations [Figure 5E-G]. In IBS+LR [Figure 5F], LR values and IBS values jointly influenced the data structure, reducing the mixing of UN with other kinship pairs to some extent. In the ALL combination [Figure 5G], the data structure was still dominated by LR parameters, and the mixing of UN and 2ND kinship pairs with other kinship pairs was further diminished. Even though the above visualizations provided insights into the distribution structure of the data, kinship classification efficiencies of these feature combinations still required the development of fine-tuned models for validation.
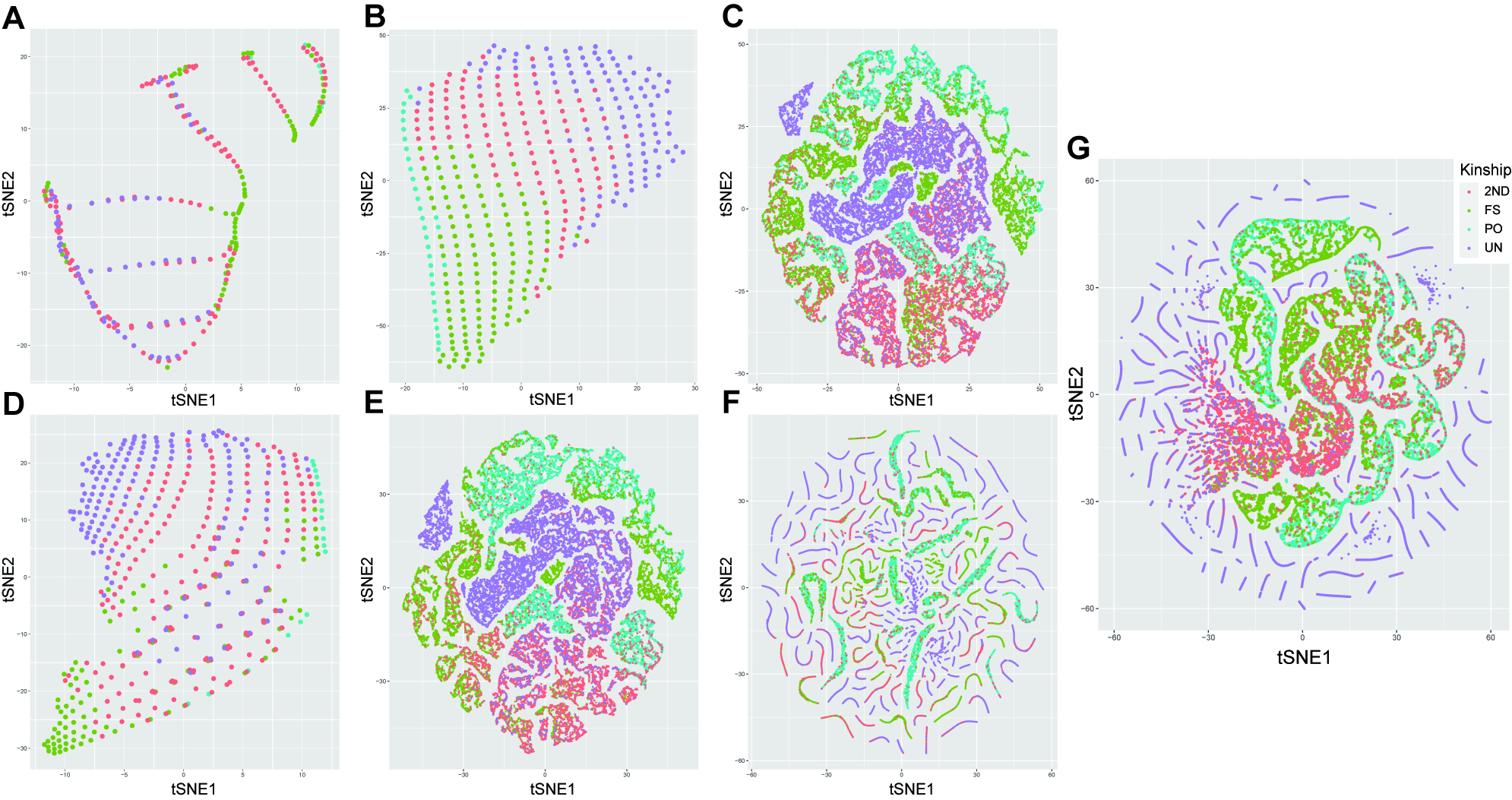
Figure 5. Visualizations of dimension reduction for different feature combinations based on the t-SNE algorithm. (A) and (B) are the t-SNE dimension reduction results for IBD parameters (k0, k1, k2, and IBD proportions) and IBS parameters (IBS0, IBS1, IBS2, and normalized cumulative IBS), respectively; (C) displays the results for log10(LR) values under different hypotheses; (D-G) show the results for feature combinations of IBD+IBS, IBD+log10(LR), IBS+log10(LR) and all mentioned features, respectively. Different colored dots represent different types of kinship pairs. IBD: Identity-by-descent; IBS: identity-by-state; FS: full siblings; PO: parent-offspring; 2ND: 2nd-degree relatives; UN: unrelated individuals; t-SNE: t-distributed stochastic neighbor embedding.
Efficacy evaluations of the binary and multi-classification ML models for kinship classifications
Hyperparameter configurations and corresponding model performance metrics can be found in Supplementary Table 8. As for the FS-UN classification, the LGBM model demonstrated the best performance (ACC = 0.9902). For the 2ND-UN task, the XGB model exhibited the best performance (ACC = 0.9194). Overall, despite its good interpretability in forensic practice, MNB had the lowest performance in both FS-UN and 2ND-UN binary classifications, with ACC values of 0.9474 and 0.7960, respectively. Although in the FS-UN classification, there was little difference in performance among models with different complexities; in the more challenging 2ND-UN classification, the ACC of three ensemble learning models (XGB, LGBM, and CATB) were all above 0.91, significantly higher than the average ACC of 0.84 for MLR and SVM models.
Performance metrics and hyperparameter configurations for fine-tuned models under different feature combinations were provided in Supplementary Table 9. Overall, the IBD, IBD+LR, and ALL feature combinations exhibited higher performance, with ACC values all exceeding 0.92, while optimal models under other combinations displayed ACCs below 0.90. We selected the IBD combination, which had the best performance among feature combinations based on a single kind of kinship parameter, and feature combinations with multiple kinship parameters for further evaluation. Figure 6A-D shows the Receiver Operating Characteristic (ROC) curves and learning curves for the optimal models under the IBD, IBS+LR, IBD+LR, and ALL feature combinations. It could be observed that the ALL combination had the best performance, followed by IBD+LR, IBD, and IBS+LR. Learning curves shown in Figure 6E-H indicated that IBD demonstrated the best fitting, followed by IBS+LR and ALL. For the IBD+LR combination, a difference of approximately 0.05 between the training set and test set in 10-fold cross-validation suggested that further increasing model complexity may lead to overfitting.
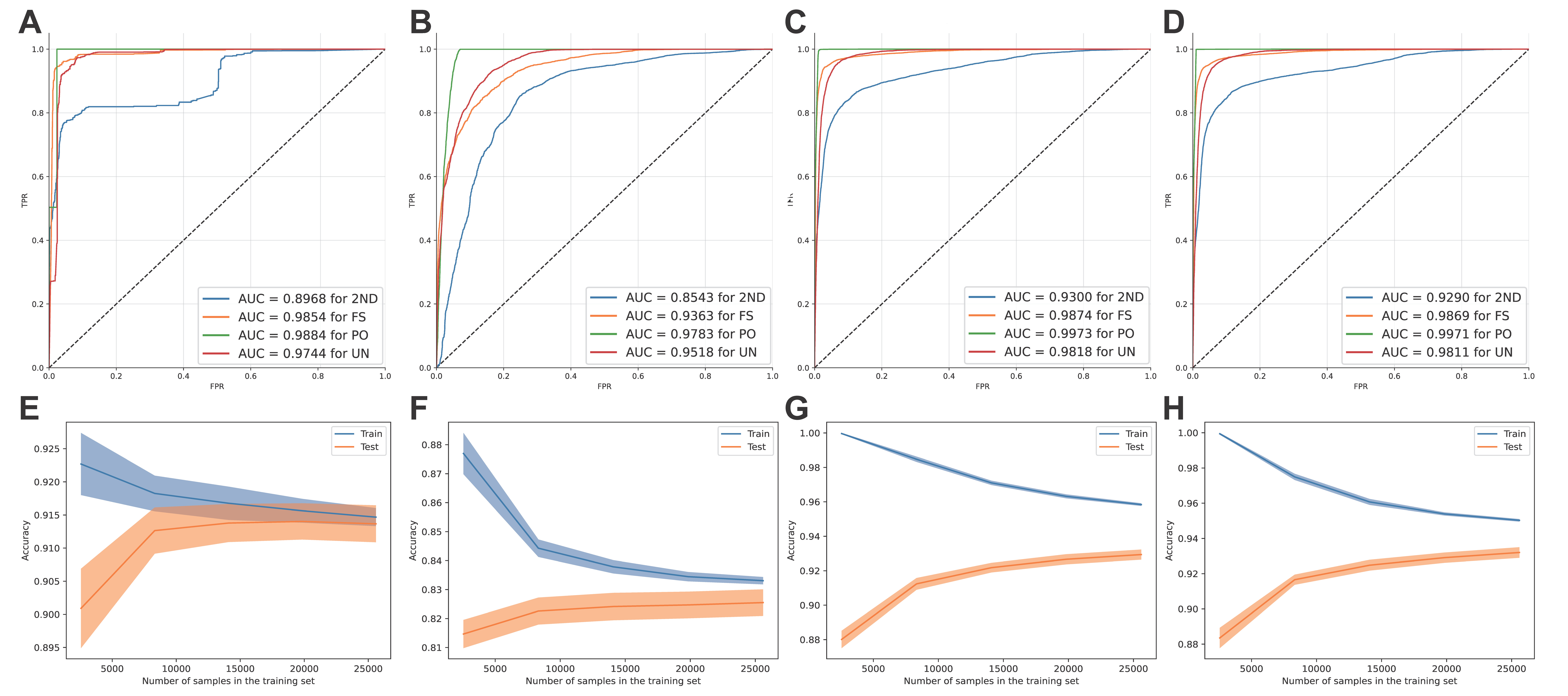
Figure 6. ROC curves and learning curves of the top four optimal models based on IBD, IBS+LR, IBD+LR, and all features. (A-D) show the ROC curves of the top four optimal models based on feature combinations of IBD, IBS+LR values, IBD+LR values, and all features, and AUC values of the one vs. rest classifiers for each class are listed in the bottom right corner; (E-H) display the learning curves of the optimal models based on feature combinations of IBD, IBS+LR values, IBD+LR values, and all features. IBD: Identity-by-descent; IBS: identity-by-state; LR: likelihood ratio; 2ND: 2nd-degree relatives; FS: full siblings; PO: parent-offspring; ROC: receiver operating characteristic; AUC: area under the curve; UN: unrelated individuals.
To enhance the interpretability of kinship classification models, we selected the model of ALL-LGBM for decision process visualization and feature importance analysis [Figure 7]. Figure 7A displays one of the decision trees of the model, where samples were progressively classified in multiple nodes based on different thresholds of kinship parameters. Figure 7B illustrates the distribution of feature importance in the optimal model of ALL-LGBM. Results indicated that the LR parameters played a dominant role in kinship multi-classifications, with LRPO-UN being the most important one. Following that, the k1 of IBD was the most crucial kinship parameter.
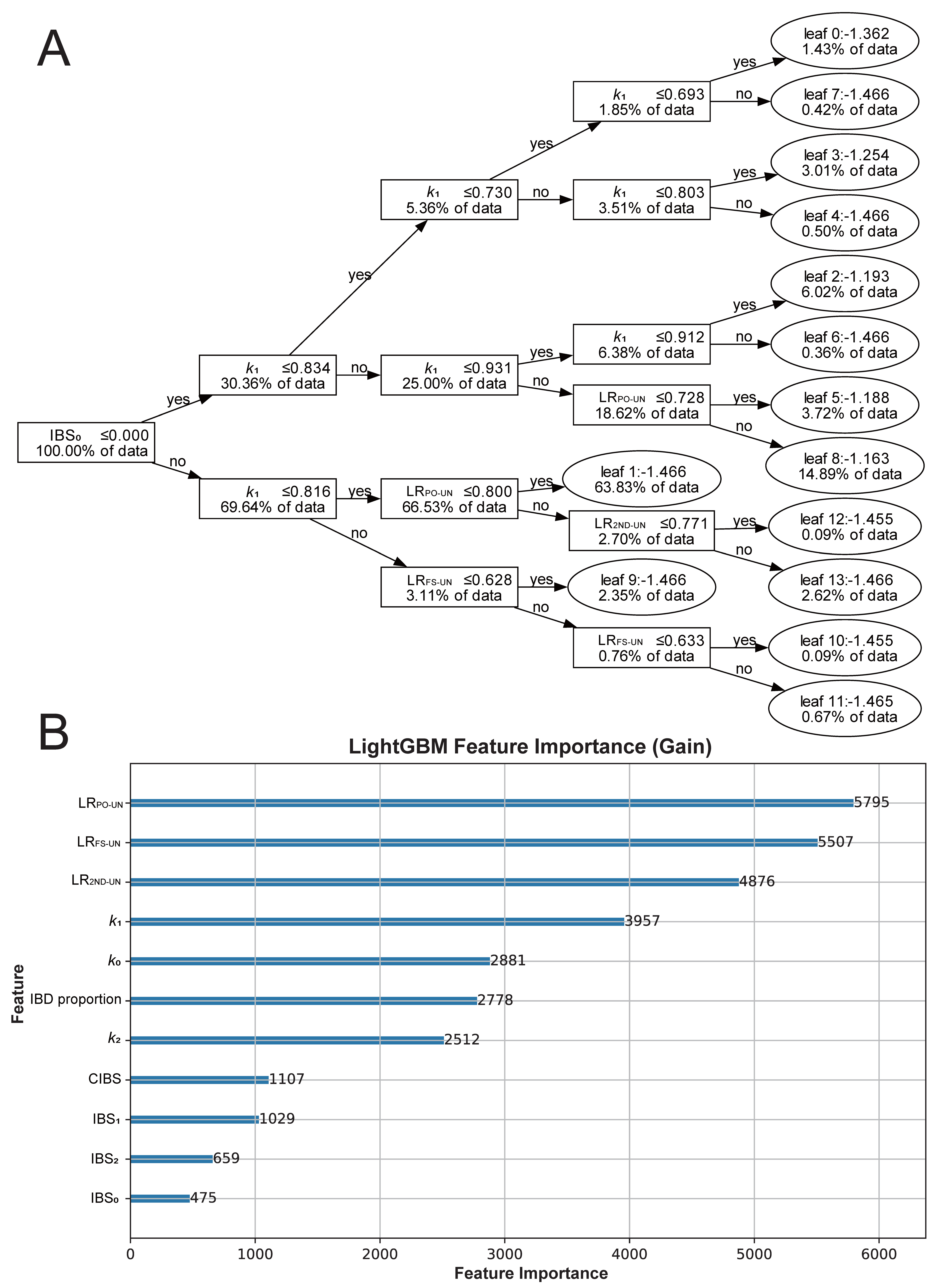
Figure 7. Decision tree visualization and feature importance of the best model (LGBM) based on all features in the modeling set. (A) shows a visualization for one of the 620 decision tree estimators in the developed LGBM model based on all features in the modeling set. In addition to showcasing the specific process of classification, the distribution ratio of samples is also displayed in each node and leaf, along with the data features and their corresponding thresholds upon which the classification is made; (B) displays the corresponding importance rank of each data feature based on the total gain of this feature's splits. LRPO-UN, LRFS-UN, and LR2ND-UN are referred to as the LR value under hypotheses of PO-UN, FS-UN, and 2ND-UN. LR: Likelihood ratio; PO: parent-offspring; FS: full siblings; 2ND: 2nd-degree relatives; UN: unrelated individuals.
Comparing the methodologies of cutoff threshold-based and ML classification
We used an exhaustive search with a step size of 0.01 to find the single cutoff thresholds with the highest ACCs: PO-UN (1.54), FS-UN (-0.03), and 2ND-UN (-0.01). Considering the calculated CPE values and the ACCs of optimal models developed for binary classification tasks of FS-UN and 2ND-UN, log10(LR) dual-thresholds were obtained: PO-UN (0.58/3.11), FS-UN (-1.30/0.22), and 2ND-UN (-0.49/0.53). Common empirical thresholds, such as ±1, ±2, ±3, etc., were also applied for methodological comparison. Performance metrics corresponding to these thresholds in the modeling set are presented in Table 5. When the ML workflow based on LGBM achieved the highest accuracy (0.9902), in the traditional workflow at the same accuracy level, dual thresholds were selected as -1.30 and 0.22, respectively. At this point, the threshold-based method had a CR of 0.9553 and an SP of 0.9459, both lower than the ML method’s values of 1.0 and 0.9902, respectively. Compared to the single-threshold method, binary kinship classifications based on dual thresholds gained a significant increase in ACC values, but at the cost of sacrificing CR values. This trade-off was particularly evident in the empirical threshold-based 2ND-UN classification. While the ML workflow based on XGB achieved the highest accuracy (0.9194), in the common workflow at the same accuracy level, dual thresholds were selected as -0.49 and 0.53, respectively. Herein, the common and ML workflows presented CRs of 0.7066 and 1.0, as well as SPs of 0.6528 and 0.9194, respectively. Despite having clear interpretability and numerous practical applications, this methodology led to a substantial decrease in CR and SP values.
Comparison of kinship binary classification methodologies based on log10(LR) values in the modeling set
| Kinship pair | Methodology | Threshold value | ACC | CR | SP |
| PO/UN | Single threshold1 | 1.54 | 0.9952 | 1 | 0.9952 |
| Dual threshold | 0.58/3.11 | 0.9999 | 0.9120 | 0.9119 | |
| FS/UN | Single threshold1 | -0.03 | 0.9811 | 1 | 0.9811 |
| Dual thresholds | -1.30/0.22 | 0.99022 | 0.9553 | 0.9459 | |
| ±1 | 0.9950 | 0.9386 | 0.9339 | ||
| ±2 | 0.9993 | 0.8324 | 0.8318 | ||
| ±3 | 0.9999 | 0.6618 | 0.6617 | ||
| ML model (LGBM) | / | 0.9902 | 1 | 0.9902 | |
| 2ND/UN | Single threshold1 | -0.01 | 0.8386 | 1 | 0.8386 |
| Dual thresholds | -0.49/0.53 | 0.91943 | 0.7066 | 0.6528 | |
| ±1 | 0.9593 | 0.4406 | 0.4227 | ||
| ±2 | 0.9950 | 0.0865 | 0.0861 | ||
| ±3 | 0.9999 | 0.0070 | 0.0070 | ||
| ML model (XGB) | / | 0.9194 | 1 | 0.9194 |
Subsequently, we conducted a kinship multiclassification in the validation set based on the baseline dual-thresholds [Table 6]. Detailed metrics for the remaining ML models can be found in Supplementary Table 10. Performance metrics for the threshold-based methods were significantly lower than those for ML methods. In the multiclassification without assumption on relatedness, the methods of single-threshold and dual thresholds displayed macro F1 values of 0.6955 and 0.5212, as well as CRs of 1.0 and 0.70464. Although the dual thresholds method exhibited higher precision (0.8271) compared to the single-threshold method, the overall recall and macro F1 value of the dual-threshold method was significantly lower (0.5740) due to the existence of the “gray zone”. Among the ML models, the IBD+LR-XGB model exhibits the optimal classification performance (F1 = 0.9020), while the ALL-XGB model ranks second (F1 = 0.9008). Additionally, these two models not only demonstrated higher performances but also had shorter fitting times (0.4615 s and 0.6484 s).
Performance metrics of optimal classifiers based on different feature combinations for kinship multiple classification tasks in the validation set
| Feature combination | Optimal classifier | Macro F1 score | Precision | Recall | CR | Fit time(s) |
| log10(LR) | Single threshold | 0.6955 | 0.7375 | 0.7069 | 1 | - |
| Dual thresholds | 0.5212 | 0.8271 | 0.5740 | 0.7046 | ||
| XGB | 0.7998 | 0.8005 | 0.8009 | 1 | 0.7981 | |
| IBS | LGBM | 0.8003 | 0.8024 | 0.8029 | 1 | 0.0529 |
| IBD | RFC | 0.8982 | 0.8989 | 0.8991 | 3.5254 | |
| IBS+ log10(LR) | CATB | 0.8204 | 0.8217 | 0.8223 | 3.4871 | |
| IBD+ log10(LR) | XGB | 0.9020 | 0.9023 | 0.9023 | 0.4615 | |
| ALL | XGB | 0.9008 | 0.9009 | 0.9011 | 0.6484 |
Figure 8 presents the normalized confusion matrices for the cutoff threshold methods and ML models. It could be concluded that the single-threshold method was suitable for the PO-UN classification task but struggled to differentiate between FS and 2ND pairs. Despite moderate improvement observed in the dual cutoff thresholds, FS pairs still tended to be misclassified as PO. Faced with the challenging task of 2ND pairs classification with the InDel capillary electrophoresis panel, IBS+LR-CATB, and IBD-RFC models exhibited lower accuracies. In contrast, models that incorporated both IBD and LR kinship parameters, such as IBD+LR-XGB and ALL-XGB, showed significant improvements. Overall, the incorporation of various kinship parameters and ML models was considered beneficial for forensic kinship analysis based on the InDel capillary electrophoresis system, especially evident in terms of multiclassification.
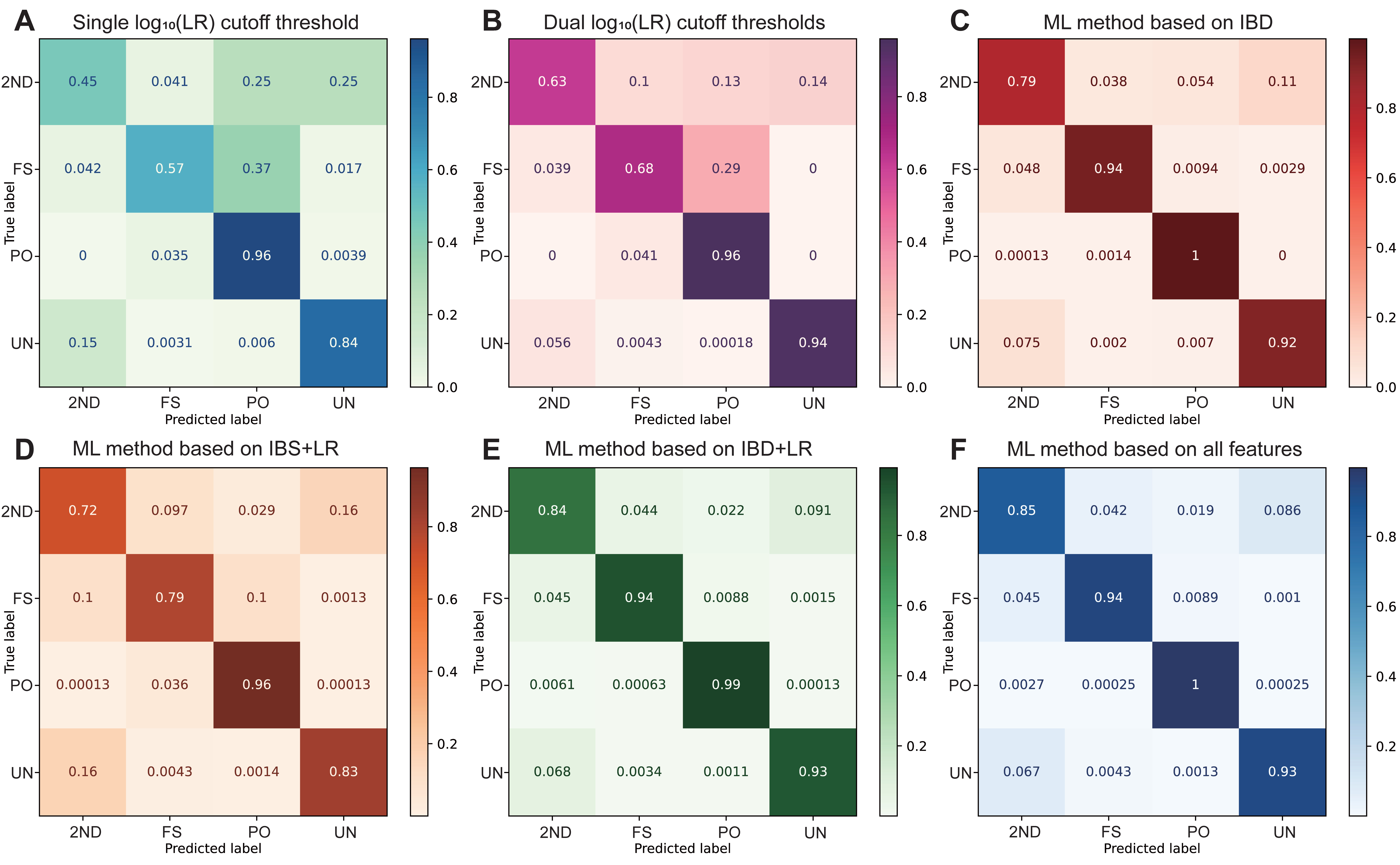
Figure 8. Normalized confusion matrices based on the thresholding method and four machine learning models with better performance under kinship multi-classification tasks in the modeling set. (A and B) are confusion matrices of single-threshold and dual-threshold methods; (C-F) show the confusion matrices of the top four optimal models based on IBD, IBS+LR, IBD+LR, and all features, respectively. IBD: Identity-by-descent; IBS: identity-by-state; LR: likelihood ratio; ML: machine learning; FS: full siblings; PO: parent-offspring; 2ND: 2nd-degree relatives; UN: unrelated individuals.
DISCUSSION
Overview of the forensic basic parameters and genetic background
In the CTG group, the 57 InDel loci panel was capable of forensic individual identification and paternity testing in East Asian populations more than other existing commercial systems[26,27]. Population genetic investigations indicate that the performance of individual identification and paternity testing is relatively robust in the studied East Asian populations. As mentioned in Section 3.2, we observed a latent population substructure among the SHP group and other East Asian populations in PCA, forming two clusters in the PC1-PC3 and PC2-PC3 dimensions. However, the analysis of molecular variance did not reveal statistically significant inter-population genetic differences within these East Asian populations. Furthermore, the latent population substructure was also excluded by the results of Nei’s DA distances among the studied East Asian populations. These results also suggested that the potential nonsignificant population substructure may not be attributed to genetic differences. Possible explanations included potential batch effects arising from the 1KGP data derived from whole-genome sequencing, differing from the CE-platform-based 57 InDel loci data. Regarding the SHP population, the observed results in PCA might be influenced by the larger sample size (n = 628). In summary, the merged data of 15 populations in the East Asian dataset represent a consistent genetic distribution and it is acceptable to conduct kinship simulations based on this merged dataset. In the results section, we observed that for IBD-related parameters, the observed values for UN pairs showed the greatest variation. The cause of this issue may be one of the limitations of this study: the relatively small number of genetic markers led to an increased false positive rate in MoM algorithm which is typically calculated based on high-density markers. Additionally, among the 655 individuals used to simulate UN pairs in this study, approximately 0.82% (1761) of the resulting 214,185 kinship coefficient values fell within the range of 0-0.0625. This may have caused the simulation of UN pairs to deviate from real-world scenarios.
Forensic kinship classifications from the machine learning perspective
Even though many forensic genomics studies combining ML algorithms were intended to directly input raw genotyping data[28-30], models developed after data preprocessing have been proven more cost-effective and accurate. Compared to the existing dimensionality reduction methods, we believe that kinship parameters of IBD, IBS, and LR are more advisable to use, since these parameters are designed according to Mendel’s Law. ML model developed based on this can be more interpretable and able to be compared directly with the LR cutoff threshold-based method. Therefore, based on the merged East Asian dataset, we constructed a series of binary kinship classification models under LR values and then trained the all-in-one multi-classification models by using 6 different feature combinations. As discussed in Section 3.3, the distribution characteristics of various types of kinship parameters might impact forensic kinship analysis and ML model construction. For example, assuming no Mendelian errors, parameters such as IBS0 and k0 could differentiate between PO and other relatedness pairs. k2 displayed a significant impact on classifying FS pairs, which LR values cannot solely achieve under certain hypotheses. Visualization results of multiple kinship parameters showed that, with the introduction of different types of kinship parameters, the mixing between different relatedness pairs gradually decreased. Based on these results, we believe that combining multiple parameters can increase the information about the relationship between two individuals, which provides a theoretical foundation for subsequent ML model construction based on various feature combinations.
Methodology comparison in the kinship binary classification of FS-UN from the modeling set showed that the ML method did not demonstrate an evident advantage when compared to the single-threshold method, with differences between SP values less than 0.01. For example, there were minor performance differences between the mathematical model of MLR and the optimal model of LGBM. This can be ascribed to the fact that the scale and dimensions of the input dataset are rather small (< 50 k objects). Even though this task can be properly tackled by mathematical models such as MLR and SVM, according to Occam’s Razor Principle, the simpler method of cutoff threshold should be finally applied in such cases. However, due to the bi-allelic nature of the studied InDel loci, the classification task for the 2ND kinship is more difficult. In such a scenario, the application of ML algorithms, especially the ensemble algorithms, was observed to effectively improve the SP values in the classification of 2ND pairs including HS, AC, and GP relatedness. By comparing the performances of the cutoff threshold-based method and ML models based on log10(LR2ND-UN) values in the 2ND kinship binary classification, we found that the ML models achieved equivalent ACCs without sacrificing CR values, and the XGB model was generally considered the best-performing. However, due to the No Free Lunch Theorem[31], there is no such perfect model that could universally fit all kinds of data and prediction tasks and only the optimal fitting model for the particular dataset. In the following multi-kinship analysis based on different data feature combinations, the most suitable models were respectively LR-XGB, IBS-XGB, IBD-RFC, IBS+LR-XGB, IBD+LR-LGBM, and ALL-LGBM. At the same time, increasing the data feature dimensions could improve the efficiency of ML models in distinguishing individual pairs with different kinship, especially for 2ND and UN pairs. However, this improvement was achieved at the cost of a higher model variance close to 0.05, which might be caused by the increased model complexity. Moreover, with different feature combinations used in the dataset, the fitting speed of SVM and RFC decreased with the increase of dataset features. As for the CATB model, it was the slowest-fitting model among all ensemble models, with its speed largely dependent on the structure of the dataset, making its training time significantly longer in the small dataset[32]. In general, under the 6 different data feature combinations, ensemble learning algorithms of RFC, XGB, and LGBM constantly showed promising classifying abilities and fast fitting speeds. These ensemble learning algorithms, especially boosting algorithms, had greater application potential in the scenario of forensic kinship multi-classification.
In this study, the common kinship analysis workflow refers to calculating LR values for all kinship hypotheses based on two individuals with unknown relatedness. Then, the assumption of kinship should be confirmed based on the maximum LR value, and the predefined thresholds are used to validate the final conclusion. However, this method has the following limitations: (1) One of the most essential parts of the common workflow is to estimate the most possible relationship based on the maximum LR value. Although research based on sequencing data suggested that the LR values for specific kinship pairs were generally the highest under their respective hypotheses[15], our study based on 57 InDel loci did not consistently support this, introducing a risk of misjudgment for kinship identification in InDel-CE panels with lower polymorphism; (2) Individuals between the dual thresholds are considered “undetermined”. Their potential relatedness information is typically ignored in routine practice, leaving a risk of false negative results; (3) Without a priori range of the relatedness between individual pairs, kinship analysis requires calculating LR values under every possible kinship hypothesis. Although this method is acceptable for case analyses, its computation load and time complexity may be significantly increased in the real-time parallel comparisons within large forensic genotyping databases. Fortunately, by introducing multiple feature combinations and ML algorithms, the above limitations can be preliminarily tackled. As shown in Section 3.3.1, the mutual confirmation of various kinship parameters enabled the ML classifiers to learn patterns between different kinship pairs from a more comprehensive perspective, resulting in higher values of ACC. The advantage of ML classifiers is that the kinship multi-classification could be performed without suffering from inconclusiveness, leading to a higher CR value. Moreover, the developed ML method does not require an exhaustive search for all potential kinships, allowing for a fast preliminary family or pedigree screening in the existing database.
With relatively short amplification fragments, InDel-CE panels were suitable for the detection of degraded biomaterials, and it was easier to popularize in front-line forensic practice. In the future, supplementary ML models developed on InDel-CE panels can effectively improve the ACC and SP of the identification of the 2ND kinship. However, it is essential to emphasize that in this study, the main intention of ML models is to improve CR values, facilitating the deployment of models in the large-scale preliminary screening of relatedness within forensic genotyping databases. However, in the context of forensic practice, there is still a need to develop a more comprehensive system for evidence interpretation. Compared with the common workflow, the ML classifiers used in this study did not allow for improving ACC values by sacrificing CR values, resulting in a classification accuracy that was difficult to meet the forensic practice requirement of 0.9999 and a weaker strength of evidence for forensic practice. We believe that incorporating classification probabilities might be a direction worth exploring. By setting a decision threshold based on the classification probability for each type of relatedness, the accuracy might be increased accordingly. However, this classification probability is determined by the input dataset and will not faithfully reflect the real-world situation. Large-scale validation and more real-world pedigree samples are still necessary to establish relatively reliable prior probabilities and further validate the ML methodology.
DECLARATIONS
Acknowledgments
Great contributions of forensic researchers for expanding the AGCU-60 population database are much appreciated, and we also gratefully thank the participants in this study.
Authors’ contributions
Conceptualization: Lei F, Wu X, Zhu B
Data curation, formal analysis, project administration, validation: Lei F, Liu Q
Funding acquisition, supervision: Xie T, Zhu B
Investigation: Liu Q, Wu X
Methodology: Lei F, Liu Q, Xie T
Resources: Zhu B
Software: Lei F, Wu X
Visualization, writing - original draft: Lei F
Writing - review & editing: Wu X, Xie T, Zhu B
Availability of data and materials
The raw data for this article are available upon reasonable request to the corresponding authors with the permission of the authorities.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This study was funded by the National Natural Science Foundation of China (Nos. 82293650, 82293652, and 82572152) and the National Key R&D Program of China (2022YFC3302004-1).
Conflicts of interest
Zhu B is a Section Editor of Journal of Translational Genetics and Genomics. Zhu B is also the Guest Editor of the Special Issue entitled “Topic: Molecular Innovation in Forensic Genetics” in the Journal of Translational Genetics and Genomics. Zhu B was not involved in any steps of the editorial process, notably including reviewers’ selection, manuscript handling, or decision-making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
The present study strictly adhered to the ethical guidelines of the Helsinki Declaration and was approved by the Ethics Committee of Xi’an Jiaotong University Health Science Center and Southern Medical University (NO. 2019-1039). All subjects voluntarily signed the informed consent form.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Pereira R, Phillips C, Alves C, Amorim A, Carracedo A, Gusmão L. A new multiplex for human identification using insertion/deletion polymorphisms. Electrophoresis. 2009;30:3682-90.
2. Manta F, Caiafa A, Pereira R, et al. Indel markers: genetic diversity of 38 polymorphisms in Brazilian populations and application in a paternity investigation with post mortem material. Forensic Sci Int Genet. 2012;6:658-61.
3. Zhang YD, Shen CM, Jin R, et al. Forensic evaluation and population genetic study of 30 insertion/deletion polymorphisms in a Chinese Yi group. Electrophoresis. 2015;36:1196-201.
4. Fan H, He Y, Li S, et al. Systematic evaluation of a novel 6-dye direct and multiplex PCR-CE-based InDel typing system for forensic purposes. Front Genet. 2021;12:744645.
5. Liu J, Du W, Jiang L, et al. Development and validation of a forensic multiplex InDel assay: the AGCU InDel 60 kit. Electrophoresis. 2022;43:1871-81.
6. Chen X, Nie S, Hu L, et al. Forensic efficacy evaluation and genetic structure exploration of the Yunnan Miao group by a multiplex InDel panel. Electrophoresis. 2022;43:1765-73.
7. Fang Y, Zhao C, Jin X, et al. Genetic characterization evaluation of a novel multiple system containing 57 deletion/insertion polymorphic loci with short amplicons in Hunan Han population and its intercontinental populations analyses. Gene. 2022;809:146006.
8. Chen M, Cui W, Bai X, et al. Comprehensive evaluations of individual discrimination, kinship analysis, genetic relationship exploration and biogeographic origin prediction in Chinese Dongxiang group by a 60-plex DIP panel. Hereditas. 2023;160:14.
9. Xu H, Nie S, Hu L, et al. Comprehensive understanding the forensic systematic effectiveness in Chinese Yunnan Hani group and intercontinental population Architecture differentiation analyses via a novel set of autosomal InDel markers. Front Biosci. 2023;28:5.
10. Chakraborty R, Jin L. Determination of relatedness between individuals using DNA fingerprinting. Hum Biol. 1993;65:875-95.
11. Weir BS, Anderson AD, Hepler AB. Genetic relatedness analysis: modern data and new challenges. Nat Rev Genet. 2006;7:771-80.
12. Kling D, Tillmar A. Forensic genealogy-A comparison of methods to infer distant relationships based on dense SNP data. Forensic Sci Int Genet. 2019;42:113-24.
13. Coble MD, Buckleton J, Butler JM, et al. DNA Commission of the International Society for Forensic Genetics: recommendations on the validation of software programs performing biostatistical calculations for forensic genetics applications. Forensic Sci Int Genet. 2016;25:191-7.
14. Heinrich V, Kamphans T, Mundlos S, Robinson PN, Krawitz PM. A likelihood ratio-based method to predict exact pedigrees for complex families from next-generation sequencing data. Bioinformatics. 2017;33:72-8.
15. Galván-Femenía I, Barceló-Vidal C, Sumoy L, Moreno V, de Cid R, Graffelman J. A likelihood ratio approach for identifying three-quarter siblings in genetic databases. Heredity. 2021;126:537-47.
16. Xu Q, Wang Z, Kong Q, et al. Improving the system power of complex kinship analysis by combining multiple systems. Forensic Sci Int Genet. 2022;60:102741.
17. Chen T, Guestrin C. XGBoost: A scalable tree boosting system. In: KDD '16: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; Association for Computing Machinery, New York, NY, USA, 2016: pp. 785-94.
18. Ke G, Meng Q, Finley T, et al. LightGBM: a highly efficient gradient boosting decision tree. In: Proceedings of the 31st International Conference on Neural Information Processing Systems. Red Hook, NY, USA: Curran Associates Inc.; 2017. p. 3149-57. Available from https://hal.science/hal-03953007/ [accessed 30 March 2026].
19. Byrska-Bishop M, Evani US, Zhao X, et al. ; Human Genome Structural Variation Consortium. High-coverage whole-genome sequencing of the expanded 1000 genomes project cohort including 602 trios. Cell. 2022;185:3426-3440.e19.
20. Wang M, Du W, Tang R, et al. Genomic history and forensic characteristics of Sherpa highlanders on the Tibetan Plateau inferred from high-resolution InDel panel and genome-wide SNPs. Forensic Sci Int Genet. 2022;56:102633.
21. Gouy A, Zieger M. STRAF-A convenient online tool for STR data evaluation in forensic genetics. Forensic Sci Int Genet. 2017;30:148-51.
22. Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564-7.
23. Kling D, Tillmar AO, Egeland T. Familias 3 - extensions and new functionality. Forensic Sci Int Genet. 2014;13:121-7.
24. Geman S, Bienenstock E, Doursat R. Neural networks and the bias/variance dilemma. Neural Comput. 1992;4:1-58.
26. LaRue BL, Ge J, King JL, Budowle B. A validation study of the Qiagen Investigator DIPplex® kit; an INDEL-based assay for human identification. Int J Legal Med. 2012;126:533-40.
27. Pereira R, Gusmão L. Capillary electrophoresis of 38 noncoding biallelic mini-Indels for degraded samples and as complementary tool in paternity testing. Methods Mol Biol. 2012;830:141-57.
28. Alladio E, Poggiali B, Cosenza G, Pilli E. Multivariate statistical approach and machine learning for the evaluation of biogeographical ancestry inference in the forensic field. Sci Rep. 2022;12:8974.
29. Sun K, Yao Y, Yun L, et al. Application of machine learning for ancestry inference using multi-InDel markers. Forensic Sci Int Genet. 2022;59:102702.
30. Pilli E, Morelli S, Poggiali B, Alladio E. Biogeographical ancestry, variable selection, and PLS-DA method: a new panel to assess ancestry in forensic samples via MPS technology. Forensic Sci Int Genet. 2023;62:102806.
31. Wolpert D, Macready W. No free lunch theorems for optimization. IEEE Trans Evol Comput. 1997;1:67-82.
32. Prokhorenkova L, Gusev G, Vorobev A, Dorogush AV, Gulin A. CatBoost: unbiased boosting with categorical features. In: Proceedings of the 32nd International Conference on Neural Information Processing Systems; Red Hook, NY, USA: Curran Associates Inc.; 2018. p. 6639-49. Available from https://proceedings.neurips.cc/paper_files/paper/2018/file/14491b756b3a51daac41c24863285549-Paper.pdf. [accessed 30 March 2026].
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].