


Cellular ATP levels alone do not reliably reflect overall mitochondrial bioenergetics or mitochondrial dysfunction in Barth syndrome
 0
0 Abstract
This review critically examines the question posed by Brault JF, Simon J, and Conway SJ in Journal of Translational Genetics and Genomics earlier this year: “What can ATP content tell us about Barth syndrome muscle phenotypes?”. It also offers an alternative perspective on the topic. Our answer is straightforward but warrants a detailed explanation. In the early stages of Barth syndrome, measuring adenosine triphosphate (ATP) content alone is insufficient to accurately characterize the bioenergetic profile of cells or tissues. Nevertheless, such measurements continue to attract interest - even from researchers equipped with advanced techniques - including adenosine diphosphate (ADP) and ATP assays, adenosine monophosphate (AMP) quantification, and assessments of nicotinamide adenine dinucleotide phosphate (NADPH) and nicotinamide Adenine dinucleotide (NADH) [i.e., nicotinamide adenine dinucleotide phosphate oxidized (NADP+) or nicotinamide adenine dinucleotide oxidized (NAD+)]. Assessing the rate of ATP production is a more informative and complementary approach, offering greater insight than ATP measurements alone. However, adopting a bioenergetic framework for studying Barth syndrome remains challenging. This difficulty arises largely from the profound structural and functional changes occurring within the mitochondrial compartment, which affect not only transmembrane ion transport but also the import and maturation of cytoplasmic precursors of mitochondrial proteins. Future research on Barth syndrome (BTHS) is expected to shift focus toward the central role of immature cardiolipin and monolysocardiolipin in mitochondrial membranes and their complex interactions, rather than concentrating solely on disruptions in cellular bioenergetics.
Keywords
INTRODUCTION
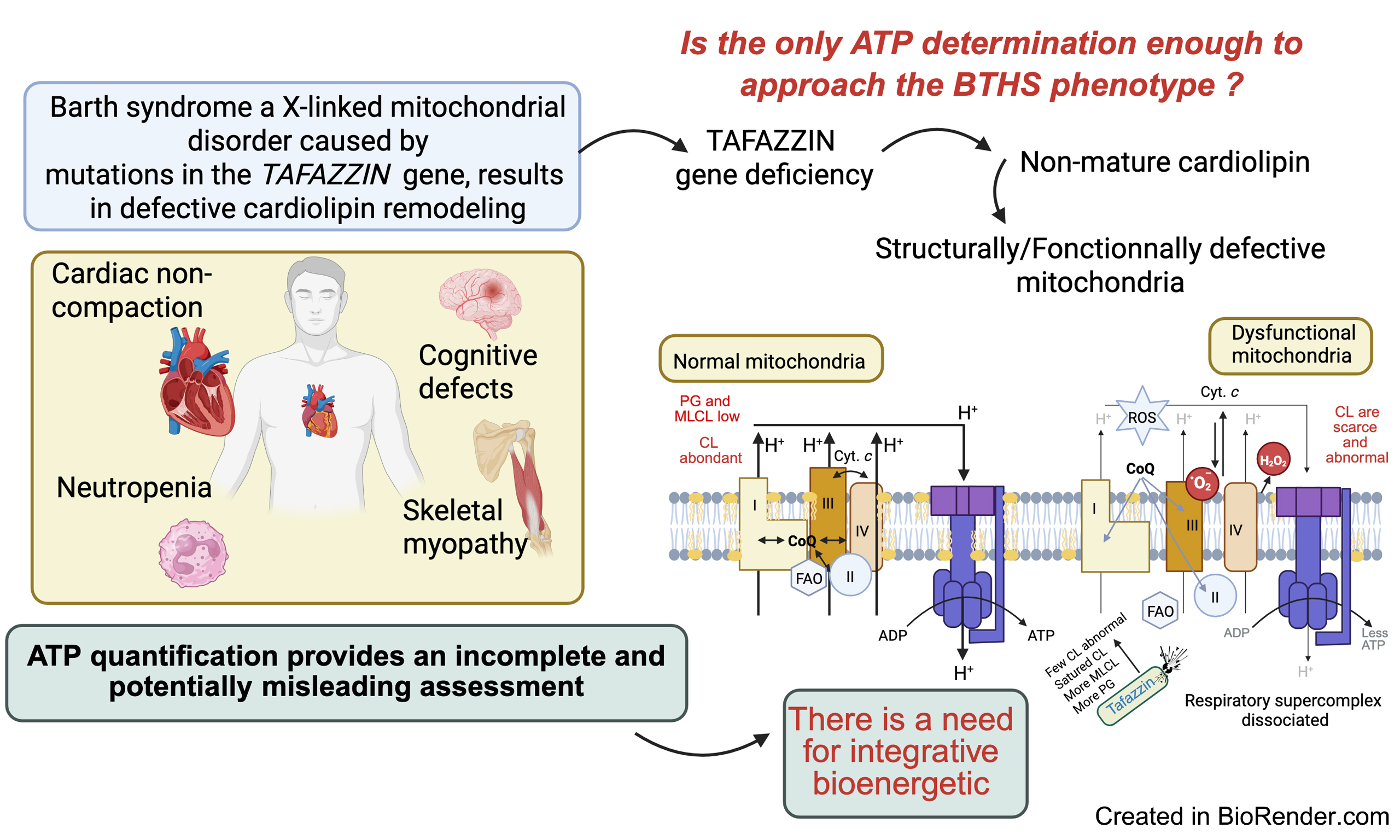
It may be surprising, but measuring cellular adenosine triphosphate (ATP) is not an effective means of assessing mitochondrial function[1], nor of identifying mitochondrial dysfunction associated with pathological conditions that disrupt bioenergetic homeostasis. A more accurate approach involves tracking metabolic intermediates of glycolysis and fatty acid oxidation, as well as markers of oxidative stress, which may be more relevant drivers of pathology than changes in ATP levels[2,3].
ATP is the main adenine nucleotide in cells, yet enhancing a cell’s bioenergetic function does not necessarily result in an increase in total ATP content. Interestingly, a rise in cellular ATP levels, which is occasionally observed, does not always reflect a corresponding expansion in the overall adenine nucleotide pool and may not accurately indicate the cell’s bioenergetic status. However, changes in the size of the adenine nucleotide pool can be indirectly linked to cellular bioenergetics through a slow cycle involving adenosine monophosphate (AMP) deaminase. This enzyme converts AMP to inosine monophosphate (IMP), thereby depleting the adenine nucleotide pool and triggering its resynthesis. When ATP/adenosine diphosphate (ADP) levels decline, adenylate kinase catalyzes the conversion of ADP to AMP. Continued energy depletion activates AMP deaminase, which degrades AMP to IMP, reducing the nucleotide pool. Conversely, when ATP/ADP levels are elevated, AMP deamination slows, allowing the pool to expand. Without further experimental investigation, it is impossible to determine whether changes in ATP levels reflect altered mitochondrial function or independent changes in adenine nucleotide metabolism. Therefore, total cellular ATP content is a poor indicator of mitochondrial dysfunction. This observation directly addresses a recent question posed by Brault JJ and Conway SJ in Journal of Translational Genetics and Genomics, “What can ATP content tell us about Barth Syndrome muscle phenotypes?”[4]. ATP quantification is subject to several limitations, most of which are technical and reduce its reliability. These include: (1) Bioluminescence assays, though highly sensitive, are vulnerable to ATP degradation if samples are not rapidly quenched[5]; (2) Colorimetric assays can be influenced by pH, salts, and other metabolites, compromising accuracy[6]; (3) Total ATP measurements cannot distinguish between mitochondrial and cytosolic pools.
It is puzzling that more accurate techniques, such as high-performance liquid chromatography (HPLC), the Seahorse XF Analyzer, or the Oroboros O2k respirometer, have not been more widely and properly applied. These methods only require careful calibration - easily achievable with modern HPLC and ultra-performance liquid chromatography (UPLC) systems - and the necessary equipment is available in most contemporary laboratories. Nonetheless, it must be acknowledged that while HPLC and mass spectrometry (MS) are highly accurate, they are labor-intensive and have low throughput[7]. Moreover, ATP quantification provides a static snapshot and lacks information on metabolic flux. It cannot distinguish between high- and low-turnover states or offer insights into ATP synthesis rates[7]. In contrast, dynamic parameters such as oxygen consumption rate (OCR), extracellular acidification rate (ECAR), and mitochondrial membrane potential (ΔΨm) provide more informative assessments of mitochondrial function[6].
We recognize that many cell and molecular biologists have distanced themselves from traditional biochemistry. However, this trend - marked by an aversion to time-consuming and technically demanding experiments - hampers the advancement of scientific knowledge.
MITOCHONDRIAL ENERGY METABOLISM: A SIMPLIFIED OVERVIEW
Mitochondrial oxidative phosphorylation - and to a lesser extent, glycolysis - supplies the large amount of ATP required to maintain proper thermogenic function. Given the constantly fluctuating energy demands of cardiomyocytes, a complex network of enzymatic and signaling pathways is required to regulate the flow of metabolic substrates into the mitochondria for oxidation and ATP production. Remarkably, the human heart synthesizes approximately 6 kg of ATP per day[8]. Energy is stored in various substrates, including fatty acids, lactate, glucose, ketone bodies, and amino acids, and is ultimately converted into mechanical work. In the heart, oxidative phosphorylation accounts for up to 95% of the total energy supply[8]. Oxidative fuel metabolism, particularly the mitochondrial Krebs cycle, generates nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide reduced form (FADH2) which feed electrons into the respiratory chain. The five complexes of the electron transport chain (ETC) transfer these electrons to molecular oxygen (O2). Complexes I, III, and IV also pump protons across the inner mitochondrial membrane (IMM), creating a proton gradient (ΔpH) and an electrochemical potential (ΔΨm). Together, these form the proton motive force (ΔμH+), which drives ATP synthesis via the F1F0-ATP synthase complex. Mitochondrial creatine kinase (MCK) then uses ATP to generate phosphocreatine (PCr), which supports myofilament contractility.
Beyond energy conversion, mitochondria are central to numerous metabolic processes, including the urea cycle, amino acid and lipid metabolism, and the biogenesis of heme and iron-sulfur clusters - all of which are energy-dependent. More recently, mitochondria have also been recognized as key players in several signaling pathways, including apoptosis, autophagy, and cellular aging.
THE LANDSCAPE OF BARTH SYNDROME
Barth et al. first described a family with an “X-linked syndrome involving the myocardium, skeletal muscle, and neutrophils”[5]. A Dutch family with cardiomyopathy and high infant mortality due to infection or heart failure has also been reported, likely exhibiting the same pathology associated with Barth syndrome (BTHS). BTHS is a rare X-linked genetic disease characterized by cardiomyopathy, skeletal myopathy, growth retardation, neutropenia, and elevated urinary excretion of 3-methylglutaconic acid[9-11]. Some authors[1,9] have proposed that the syndrome described by Neustein et al.[10] as “X-linked recessive cardiomyopathy with abnormal mitochondria” may represent the same condition. Neustein et al.[10] reported that endomyocardial biopsy from an infant with chronic congestive heart failure revealed abnormal mitochondria under electron microscopy. Similar abnormalities were found in skeletal muscle, liver, and kidney tissue at autopsy. Electron microscopy of cells and tissues from BTHS patients, or cellular models thereof, has firmly established that BTHS is a mitochondrial disease[5].
Barth syndrome is linked to structural and functional mitochondrial defects that weaken muscle tissue, resulting in skeletal myopathy and cardiomyopathy. The TAFAZZIN gene encodes a mitochondrial transacylase essential for the remodeling and maturation of cardiolipin (CL), a phospholipid enriched in the IMM and at contact sites between the IMM and outer mitochondrial membrane (OMM). CL is not only a crucial structural component of the IMM but can also redistribute to the OMM, where it interacts with numerous proteins, including respiratory chain components[1,9]. Cytoplasmic proteins that bind to the OMM also play roles in broader cellular signaling pathways[12].
Despite significant recent advances, the mechanisms that link impaired CL biogenesis to cardiomyopathy remain incompletely understood[13]. CL is critically important because of its extensive interactions with key mitochondrial and cytoplasmic proteins[14], particularly those localized at or near the OMM[12,15,16].
TAFAZZIN AND CARDIOLIPIN
The presence of immature cardiolipin species - resulting from defective transacylation - is a hallmark of cardiac dysfunction and is also implicated in ischemic heart disease and aging[17,18]. Changes in CL composition can disrupt multiple mitochondrial functions, including membrane architecture[19,20], protein import[21,22], oxidative phosphorylation[13,14,23], mitochondrial fusion and fission[24-26], iron-sulfur cluster biogenesis[27], regulation of apoptosis[15,19,28-32], autophagy[19,32], and the transport of protein precursors and metabolites across mitochondrial membranes[22,33-40].
Ultrastructural abnormalities of mitochondria were noted in early reports of BTHS[5] [Figure 1] and have been linked to defective mitochondrial complex III function[11], although without alteration in complex III binding[15]. In yeast, early studies suggested defects in energy coupling and membrane stability[41], while research using patient-derived lymphoblasts revealed abnormal proliferation, altered membrane potential, and normal ATP production, indicating partial uncoupling and compensatory expansion of the mitochondrial compartment[33]. The extent of ultrastructural changes varies depending on the model system and organ examined and may be more pronounced in differentiated tissues than in embryonic ones. It has been hypothesized that this effect is more significant in mitochondria with a higher density of stacked cristae[42]. Notably, cardiac mitochondria have twice the diameter and a higher proportion of lamellar cristae compared to those in other organs. Cristae increase the inner membrane surface area, facilitating cardiolipin assembly, which may explain the predominance of cardiovascular defects in BTHS patients[42].
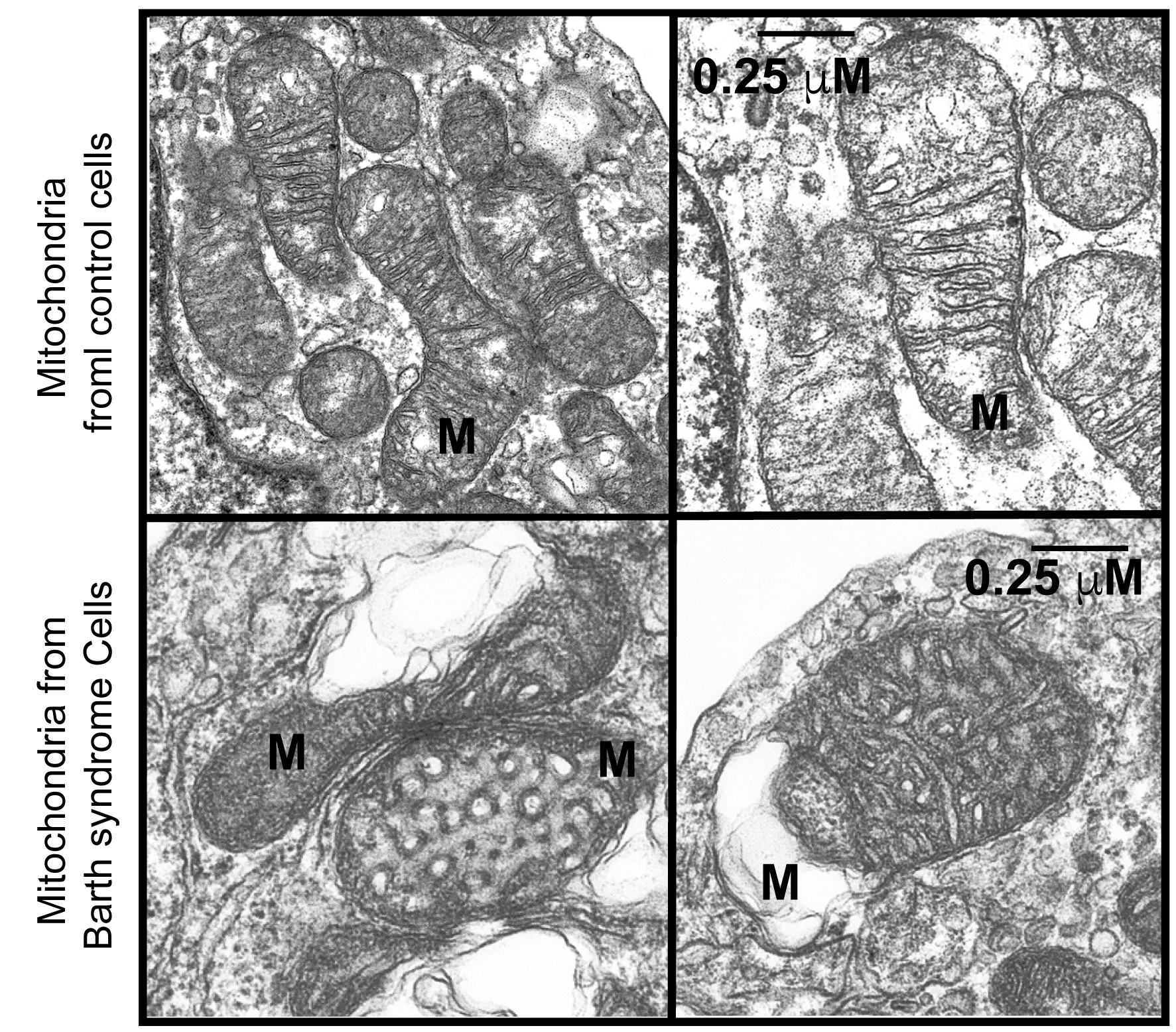
Figure 1. Electron microscopy of BTHS lymphoblastic mitochondria. M: mitochondria; scale bars for all four panels represent 0.25 μm. Reproduced from Ref.15[15] with permission from Biochimica et Biophysica Acta. BTHS: Barth syndrome.
The discovery of TAFAZZIN followed the identification of genetic mutations in patients with BTHS[43]. Bioinformatic analyses identified TAFAZZIN as encoding an acyltransferase, and Xu et al. demonstrated that Tafazzin specifically affects mitochondrial lipids[41]. This discovery confirmed the central role of mitochondria in Barth syndrome, a conclusion that had already been suggested through human biopsy analysis[44], and highlighted the critical importance of lipid metabolism in the disease. CL is the most severely affected lipid in Barth syndrome. Not only is the total amount of CL reduced[5,11,41,42,44], but its molecular species composition is also profoundly altered[44]. Furthermore, a CL derivative, monolysocardiolipin (MLCL), accumulates, although it is typically undetectable in healthy tissues. An abnormal CL profile, characterized by increased MLCL and phosphatidylglycerol (PG) content, can impair the activity of several mitochondrial proteins, including components of the ETC and oxidative phosphorylation (OXPHOS) complexes, thereby disrupting ATP production. The MLCL/cytochrome c complex has been shown to exhibit peroxidase activity, which damages polyunsaturated fatty acids in phospholipids and ATP-generating enzymes.
TAFAZZIN mutations result in abnormal CL remodeling, which destabilizes mitochondrial inner membrane complexes in yeast[34], disrupts respiratory supercomplex formation in patient-derived lymphoblasts[43], and interferes with supercomplex formation in other models[45]. Despite these cardiolipin-related changes and respiratory supercomplex destabilization, metabolic flux through the tricarboxylic acid (TCA) cycle appears largely unaffected in patient-derived skin fibroblasts[44]. In a Drosophila flight muscle model of BTHS, a reduction in F1F0 ATP synthase dimer density within the IMM was observed. This was due to a decrease in high-curvature regions of the membrane, resulting in less elongated and more scattered dimer rows[46]. In TAFAZZIN knockdown mice, a broad spectrum of mitochondrial abnormalities was observed[47,48].
These morphological defects include a mixture of swollen, honeycomb-like, and expanded, collapsed, or absent cristae [Figure 1], with variability in mitochondrial network size. A comprehensive bioenergetic and lipidomic analysis of these mice revealed altered substrate utilization and reduced activities of complexes III and V[49]. Independent evaluations of mitochondrial function in the same mouse model[48] and in induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) from BTHS patients identified tissue-specific reductions in complex II (succinate dehydrogenase) activity[50]. Another study using BTHS iPSC-CMs demonstrated reduced F1F0 ATP synthase activity and overall ATP levels when cells were cultured in galactose, which inhibits glycolytic ATP generation[50]. Interestingly, basal oxygen consumption is elevated in BTHS iPSC-CMs, likely due to compensatory mechanisms, although overall respiratory capacity is reduced[50]. TAFAZZIN-deficient mitochondria also exhibit increased reactive oxygen species (ROS) production in yeast[33], TAFAZZIN knockdown mice[31], and human iPSC-CMs[50]. Collectively, these findings form a detailed picture of the complex bioenergetic landscape of Barth syndrome, highlighting the departure from normal mitochondrial homeostasis [Figure 2][51].
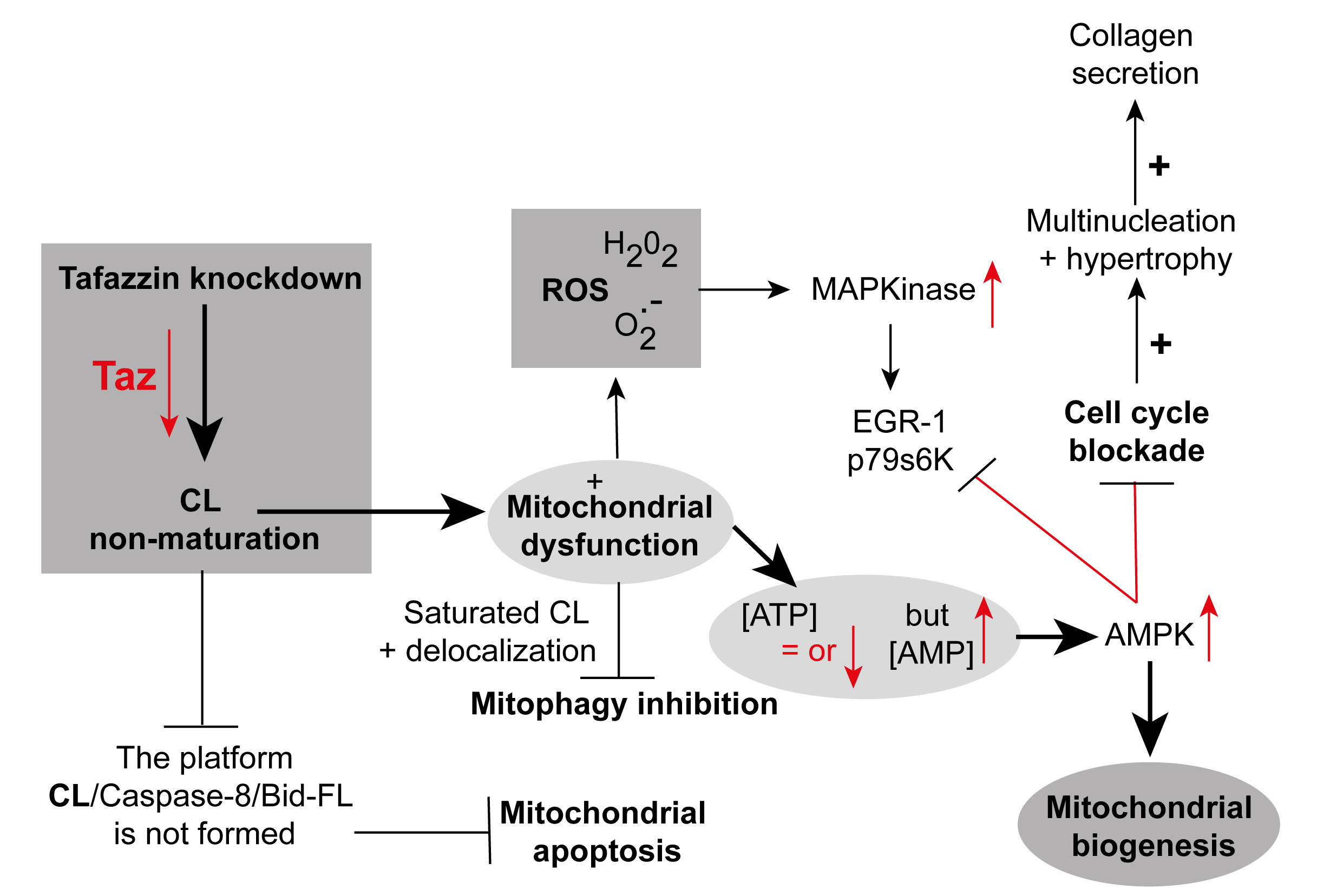
Figure 2. TAFAZZIN knockdown in neonatal cardiac fibroblasts. Knockdown of the TAFAZZIN gene disrupts mitochondrial homeostasis and alters mitochondrial membrane structure, leading to slightly reduced ATP levels and increased AMP concentrations. This schematic is adapted primarily from the work of He et al.[51] and reproduced with permission from American Journal of Physiology, Heart and Circulatory Physiology, courtesy of the American Physiological Society, USA. Elements have also been adapted from Ref.9[9], with permission from Frontiers in Genetics. ATP: adenosine triphosphate; AMP: adenosine monophosphate.
WHAT ABOUT THE PHOSPHORYLATED ADENOSINE NUCLEOTIDES?
The levels of ATP, ADP, and AMP reflect the energy status of living cells and are closely tied to mitochondrial function[52,53], with some ATP also produced via glycolysis. These phosphorylated adenosine nucleotides are essential molecules that provide immediate chemical energy, support signal transduction, and serve as metabolic precursors[53]. However, measuring ATP alone is insufficient to fully assess the energy status of cells and tissues in Barth syndrome.
A more accurate approach is to calculate the total cellular ATP production rate, rather than relying solely on ATP concentration.
Newly developed systems that simultaneously measure oxygen consumption (i.e., changes in O2 over time in absolute terms) and extracellular acidification (i.e., changes in pH over time) enable accurate calculation of ATP production rates. For example, the Agilent Seahorse XF Analyzer[54] simultaneously quantifies extracellular acidification and oxygen consumption, enabling estimation of total ATP production[55-57]. The Oroboros instrument can also be adapted for such analyses[53] and may include pH measurements. To further improve accuracy, the Real-Time ATP Rate Assay Report Generator software enables offline analysis by automatically converting experimental OCR and ECAR into total ATP production rates. It can also calculate pathway-specific ATP production - ATP derived from oxidative phosphorylation (OxPhos ATP) and ATP derived from glycolysis (Glyco ATP). This system provides a clear and efficient means of visualizing ATP production dynamics[52,55].
Adenine nucleotides (ATP, ADP, and AMP) are key organic molecules that supply immediate energy, mediate signaling, and serve as metabolic intermediates[58]. In the context of Barth syndrome, TAFAZZIN gene mutations result in defective CL maturation and, notably, a partial reduction in total CL levels. CL deficiency affects multiple aspects of cellular signal transduction, particularly those occurring at the mitochondrial membrane, where CL plays a crucial role.
For example, a cell that primarily generates ATP through OxPhos may be described as having a “preference” for OxPhos-derived ATP over glycolysis-derived ATP. In cardiac tissue, this ratio may be as high as 95:5, suggesting a metabolic hierarchy. However, this simplification may overlook the complexity of metabolic network homeostasis. If an OxPhos-dominant cell shifts toward glycolysis, can it still be said to “prefer” OxPhos ATP? This line of reasoning becomes problematic when such “preferences” are interpreted as drivers of biological processes. These conceptual issues can be resolved by reframing ATP production observations in terms of network homeostasis. In systems where glycolysis and OxPhos operate simultaneously, quantifying ATP output from each pathway is essential for understanding overall metabolic behavior [Figure 3].
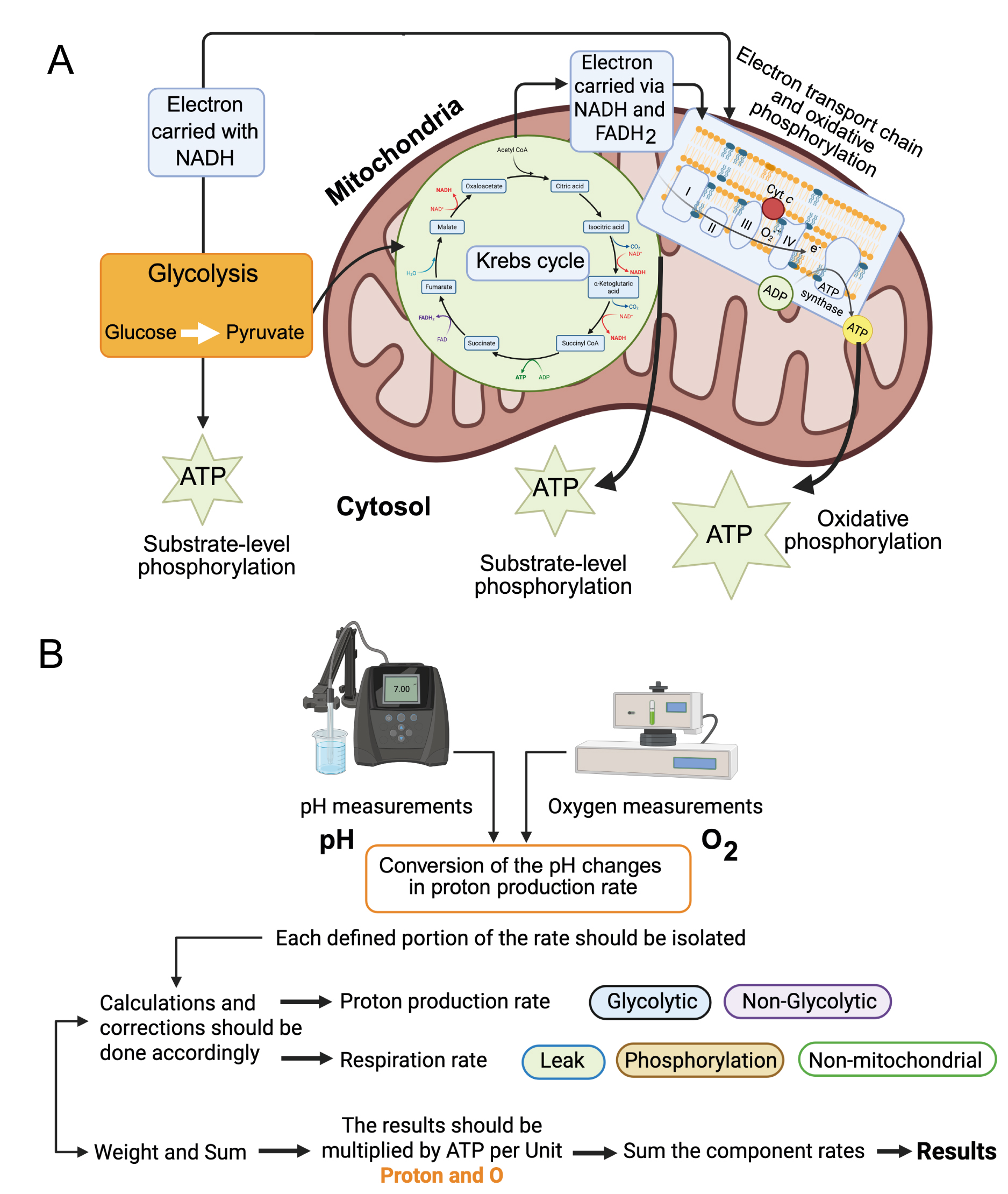
Figure 3. Extracellular Flux Measurements and ATP production. (A) Schematic representation of the pathways leading to ATP production. Created in BioRender.com; (B) Glucose is metabolized to lactic acid, CO2 and H2O. Glycolysis produces electrons (e-), glycolytic ATP (ATPglyc), and pyruvate. Pyruvate can be reduced to lactate + H+. The TCA cycle generates electrons, substrate-level oxidative ATP (part of ATPoxid.), and CO2. CO2 hydration produces HCO3- and H+. Electron transport enables oxidative phosphorylation, generating the remaining ATPoxid. and consuming O2 to produce H2O. Glyc: Glycolytic; Phosphor: phosphorylating; Mito: mitochondrial; Oxid: oxidized. Created in BioRender.com. ATP: adenosine triphosphate;
At this point, it is important to consider Crabtree’s hypothesis[59,60], which posits that the introduction of glycolytic ATP into a system that normally relies on OxPhos triggers a homeostatic response. The well-documented Crabtree effect describes how the addition of sugar to glycolysis-competent cells increases glycolytic flux while reducing respiration. The glycolytically produced ATP supplements the total ATP supply, reducing ADP levels and increasing the ATP/ADP ratio. This, in turn, decreases the cellular demand for ATP from respiration, resulting in a slowdown of mitochondrial respiration-a direct kinetic response to elevated ATP/ADP ratios[60]. Metabolic reprogramming and mitochondrial bioenergetic defects play critical roles in various physiological and pathological processes[60], including Barth syndrome. While cell culture is a widely used method for investigating cellular metabolism and disease mechanisms, standard culture protocols often interfere with the Crabtree effect. Therefore, this phenomenon cannot be overlooked when studying cellular energetics in Barth syndrome.
ATP MEASUREMENTS: NECESSARY BUT NOT SUFFICIENT
ATP levels are most commonly measured using assay kits based on bioluminescence or fluorescence detection. However, kits for measuring ADP and AMP are less widely available and can become prohibitively expensive for routine use. As noted in Section 5, Seahorse analyzers are a relatively new and increasingly popular technology[52]. They enable real-time measurement of ATP flux in living cells, offering valuable insights into cellular bioenergetics, including mitochondrial respiration and glycolysis. Nonetheless, Seahorse analyzers are costly and cannot quantify ADP and AMP.
Several analytical techniques can be used to quantify ADP and AMP, including chromatographic methods such as ion exchange chromatography, thin-layer chromatography, and HPLC, all of which offer high sensitivity and efficiency[61,62]. Moving forward, efforts must be directed toward developing more advanced and sensitive methodologies capable of simultaneously measuring ATP, ADP, and AMP, as well as redox-related metabolites like NADPH, NADH, NADP+, and NAD+[63]. We concur with our colleagues that quantifying the energy status of cells or tissues is challenging. However, this challenge does not stem from a lack of available techniques. Rather, the difficulty lies in ensuring scientific and technical rigor throughout the experimental process. ATP is a labile molecule that degrades rapidly during sample collection and preparation[61].
Although difficult to assess, it is likely that many existing ATP measurements are unreliable because they lack quantitative accuracy. Various methods have been used to measure ATP in muscle tissue, including magnetic resonance spectroscopy (MRS)[62], fluorescent reporters[63], luciferase-based assays[64,65], and HPLC or UPLC on tissue samples[58,59]. However, only HPLC, when used with a solid calibration system, enables the accurate quantification of ATP in cells or tissues[66]. Importantly, measuring ATP alone is insufficient. A meaningful assessment of a cell or tissue’s energy status requires quantification of both ATP and ADP to calculate the ADP/ATP ratio. The focus should be on the absolute quantification of all three adenine nucleotides-ATP, ADP, and AMP-expressed in absolute units (e.g., nmol/mg of protein).
WHAT ELSE COULD IT BE IF ATP PRODUCTION IS NOT THE MAIN PROBLEM?
We have discussed the cellular energetic disturbances resulting from defective cardiolipin maturation due to impaired transacylase activity. However, could the primary issue in Barth syndrome be the broader consequences of TAFAZZIN mutations and altered cardiolipin levels, rather than impaired ATP production per se? Under normal conditions, cardiolipin interacts with various mitochondrial proteins to optimize numerous mitochondrial processes (see above). Surprisingly, in a recent publication[67], we demonstrated that modulated inhibition of cytosolic protein synthesis - either by cycloheximide at picomolar concentrations or by specific gene mutations - can restore functional oxidative phosphorylation in TAFAZZIN1Δ yeast and TAFAZZIN-deficient human cells. This downregulation of cytosolic protein translation appears to alleviate the cytoplasmic accumulation of misfolded or excess mitochondrial precursor proteins, which may otherwise be toxic to the cell. Such treatments, which reduce mitochondrial protein precursor overload, are beneficial for cells affected by BTHS. This effect was previously described in a yeast model of autosomal dominant progressive external ophthalmoplegia (adPEO), linked to mutations in the adenine nucleotide translocator (ANT)[68]. That model revealed a condition termed “mitochondrial precursor over-accumulation stress (mPOS)”, a form of protein stress. mPOS activates the unfolded protein response triggered by mitochondrial precursor misfolding (UPRam), leading to reduced cytosolic protein synthesis and enhanced proteasome activity, thereby promoting increased clearance of proteins by the proteasome[67].
Extending from these observations, it is plausible that immature cardiolipin and the accumulation of MLCL alter mitochondrial membrane properties[69]. Membrane proteins bind less efficiently to MLCL and immature CLs, which negatively impacts their function[70]. Conversely, disruptions in the respiratory chain can enhance MLCL accumulation[71], creating a feedback loop in which a high MLCL/CL ratio destabilizes mitochondrial membranes - particularly by impairing the tight packing of respiratory supercomplexes. Moreover, MLCL accumulation has been shown to inhibit translation of mitochondrial protein precursors via effects on the mitochondrial membranes[67]. Interestingly, the downstream consequences of this inhibition parallel the effects of reduced cytosolic protein synthesis: both lead to mitochondrial benefits that extend beyond considerations of ATP levels.
Another important aspect is the increased ROS production associated with mitochondrial dysfunction in Barth syndrome. Beyond bioenergetics, cardiolipin deficiency also impacts mitochondrial ROS generation, calcium handling, and sensitivity to apoptosis[5]. These dysfunctions have profound physiological consequences that occur independently of measured ATP levels.
CONCLUSION
Barth syndrome is a severe disorder caused by mutations in the TAFAZZIN gene, leading to defects in mitochondrial structure and function. Alterations in the biosynthesis and remodeling of CL particularly affect tissues with high energy demands, such as the heart and skeletal muscle, though other tissues can also be involved. In this context, a clearer understanding of mitochondrial bioenergetics is highly desirable. However, as clearly demonstrated, measuring cellular ATP levels alone does not reliably reflect mitochondrial function or dysfunction[1]. At this stage, it is important to consider the likelihood that compensatory mechanisms are at play[7,13].
This hypothesis presents one of the key challenges for researchers, especially given that such mechanisms are not permanent. Another major issue requiring detailed investigation is the complex impact of CL non-maturation and MLCL accumulation. It is worth noting that the addition of a small amount of cycloheximide, in the context of non-mature CL and MLCL accumulation, alters the translation of mitochondria-targeted pre-proteins and extends cell survival[68]. This raises the possibility that reduced protein synthesis may, in fact, lower overall energy requirements. A more comprehensive understanding necessitates precise quantification of all adenine nucleotides (ATP, ADP, and AMP), as well as redox cofactors such as NADPH/NADP+ and NADH/NAD+. These measurements are essential for constructing a detailed bioenergetic profile that can inform how signal transduction networks are modulated. Furthermore, the ability to assess ATP production rates using systems that simultaneously measure absolute values of oxygen consumption and acidification should become standard practice.
DECLARATIONS
Acknowledgments
The author gratefully acknowledges the following collaborators who have contributed to this work on Barth syndrome over the years: Anne-Sophie Armand (INEM, France), Laeticia Arnauné-Pelloquin (University of Toulouse, Toulouse, France), Pascale Bellenguer (University of Toulouse, Toulouse, France), Marie Boutant (Institut Cochin, Paris, France), Eyal Gottlieb (Beatson Institute, Glasgow, Scotland), Riekelt H. Houtkooper (AMC, Amsterdam, The Nederlands), Olivier Jalmar (Institut Cochin, Paris, France), François Gonzalvez (Institut Cochin, Paris, France), Marilena D’Aurelio (Weill Medical College of Cornell University, New York, USA), Thomas Landes (University of Grenoble, France), Giovanni Manfredi (Weill Medical College of Cornell University, New York, USA), Ian Max Møller (Department of Molecular Biology and Genetics, Aarhus, Demark), Aoula Moustapha (University of Pharmacy, Latakia, Syria) , Jean-Philippe Puech (Institut Cochin, Paris, France), Christian Slomianny (INSERM, Lille, France), Nellie Taleux (University of Toulouse, Toulouse, France), Guillaume Vial (University of Grenoble, Grenoble, France), Frederic M. Vaz (Amsterdam Medical Center, Amsterdam, The Nederlands), and Ronald J. Wanders (Amsterdam Medical Center, Amsterdam, The Netherlands). The author also acknowledges the stimulating discussions with Prof. Peter G. Barth (Amsterdam Medical Center, Amsterdam, The Netherlands). Special thanks to Dr. Anne-Sophie Armand for creating the illustration used in Figure 3 and for obtaining permission to publish it under a BioRender license attributed to INEM. The author also thanks her for producing the graphical abstract, also created using a BioRender license granted to Institut Necker Enfants Malades (INSERM U1151, Université Paris Cité, 160 rue de Vaugirard, 75015 Paris, France).
Authors’ contributions
The author contributed solely to the article.
Availability of data and materials
The data supporting the conclusions of this article are available from the author upon reasonable request.
Financial support and sponsorship
PETIT XP is employed by the CNRS. This work was partially supported by CNRS institutional funding. Additional funding was provided by AFM grants (Association Française contre les Myopathies), grant numbers AFM 15137 and AFM 15661, awarded to PETIT XP. The funders had no role in study design, data collection, or data analysis.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
2. Chatham JC, Young ME. Metabolic remodeling in the hypertrophic heart: fuel for thought. Circ Res. 2012;111:666-8.
3. Nickel A, Löffler J, Maack C. Myocardial energetics in heart failure. Basic Res Cardiol. 2013;108:358.
4. Brault JJ, Conway SJ. What can ATP content tell us about Barth syndrome muscle phenotypes? J Transl Genet Genom. 2025;9:1-10.
5. Barth PG, Scholte HR, Berden JA, et al. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci. 1983;62:327-55.
6. Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297-312.
7. Wu M, Neilson A, Swift AL, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125-36.
8. Yurista SR, Nguyen CT, Rosenzweig A, de Boer RA, Westenbrink BD. Ketone bodies for the failing heart: fuels that can fix the engine? Trends Endocrinol Metab. 2021;32:814-26.
9. Saric A, Andreau K, Armand AS, Møller IM, Petit PX. Barth syndrome: from mitochondrial dysfunctions associated with aberrant production of reactive oxygen species to pluripotent stem cell studies. Front Genet. 2015;6:359.
10. Neustein HB, Lurie PR, Dahms B, et al. An X-linked recessive cardiomyopathy with abnormal mitochondria. Pediatrics. 1979;64:24-9.
11. Barth PG, Valianpour F, Bowen VM, et al. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am J Med Genet A. 2004;126A:349-54.
12. Gonzalvez F, Pariselli F, Dupaigne P, et al. tBid interaction with cardiolipin primarily orchestrates mitochondrial dysfunctions and subsequently activates Bax and Bak. Cell Death Differ. 2005;12:614-26.
13. Bazán S, Mileykovskaya E, Mallampalli VK, Heacock P, Sparagna GC, Dowhan W. Cardiolipin-dependent reconstitution of respiratory supercomplexes from purified Saccharomyces cerevisiae complexes III and IV. J Biol Chem. 2013;288:401-11.
14. Pfeiffer K, Gohil V, Stuart RA, et al. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003;278:52873-80.
15. Gonzalvez F, D’Aurelio M, Boutant M, et al. Barth syndrome: cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim Biophys Acta. 2013;1832:1194-206.
16. Gonzalvez F, Schug ZT, Houtkooper RH, et al. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol. 2008;183:681-96.
17. Saini-Chohan HK, Holmes MG, Chicco AJ, et al. Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J Lipid Res. 2009;50:1600-8.
18. Khuchua Z, Yue Z, Batts L, Strauss AW. A zebrafish model of human Barth syndrome reveals the essential role of tafazzin in cardiac development and function. Circ Res. 2006;99:201-8.
19. Li XX, Tsoi B, Li YF, Kurihara H, He RR. Cardiolipin and its different properties in mitophagy and apoptosis. J Histochem Cytochem. 2015;63:301-11.
21. Gebert N, Joshi AS, Kutik S, et al. Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr Biol. 2009;19:2133-9.
22. Jiang F, Ryan MT, Schlame M, et al. Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J Biol Chem. 2000;275:22387-94.
23. Zhang M, Mileykovskaya E, Dowhan W. Gluing the respiratory chain together: Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem. 2002;277:43553-6.
24. Ban T, Heymann JA, Song Z, Hinshaw JE, Chan DC. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum Mol Genet. 2010;19:2113-22.
25. DeVay RM, Dominguez-Ramirez L, Lackner LL, Hoppins S, Stahlberg H, Nunnari J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J Cell Biol. 2009;186:793-803.
26. Joshi AS, Thompson MN, Fei N, Hüttemann M, Greenberg ML. Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J Biol Chem. 2012;287:17589-97.
27. Patil VA, Greenberg ML. Cardiolipin-mediated cellular signaling. In: Capelluto DG, editor. Lipid-mediated protein signaling. Dordrecht: Springer Netherlands; 2013. pp. 195-213.
28. Ikon N, Su B, Hsu FF, Forte TM, Ryan RO. Exogenous cardiolipin localizes to mitochondria and prevents TAZ knockdown-induced apoptosis in myeloid progenitor cells. Biochem Biophys Res Commun. 2015;464:580-5.
29. Heit B, Yeung T, Grinstein S. Changes in mitochondrial surface charge mediate recruitment of signaling molecules during apoptosis. Am J Physiol Cell Physiol. 2011;300:C33-41.
30. Kim TH, Zhao Y, Ding WX, et al. Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome C release. Mol Biol Cell. 2004;15:3061-72.
31. Manganelli V, Capozzi A, Recalchi S, et al. Altered Traffic of cardiolipin during apoptosis: exposure on the cell surface as a trigger for “antiphospholipid antibodies”. J Immunol Res. 2015;2015:847985.
32. Chu CT, Bayır H, Kagan VE. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: implications for Parkinson disease. Autophagy. 2014;10:376-8.
33. Gu Z, Valianpour F, Chen S, et al. Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol Microbiol. 2004;51:149-58.
34. Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. 2000;39:257-88.
35. Koshkin V, Greenberg ML. Oxidative phosphorylation in cardiolipin-lacking yeast mitochondria. Biochem J. 2000;347:687-91.
36. Kadenbach B, Mende P, Kolbe HV, Stipani I, Palmieri F. The mitochondrial phosphate carrier has an essential requirement for cardiolipin. FEBS Lett. 1982;139:109-12.
37. Robinson NC. Functional binding of cardiolipin to cytochrome c oxidase. J Bioenerg Biomembr. 1993;25:153-63.
38. Noël H, Pande SV. An essential requirement of cardiolipin for mitochondrial carnitine acylcarnitine translocase activity. Lipid requirement of carnitine acylcarnitine translocase. Eur J Biochem. 1986;155:99-102.
39. Vaz FM, Houtkooper RH, Valianpour F, Barth PG, Wanders RJ. Only one splice variant of the human TAZ gene encodes a functional protein with a role in cardiolipin metabolism. J Biol Chem. 2003;278:43089-94.
40. Brandner K, Mick DU, Frazier AE, Taylor RD, Meisinger C, Rehling P. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth Syndrome. Mol Biol Cell. 2005;16:5202-14.
41. Xu Y, Kelley RI, Blanck TJ, Schlame M. Remodeling of cardiolipin by phospholipid transacylation. J Biol Chem. 2003;278:51380-5.
42. Acehan D, Xu Y, Stokes DL, Schlame M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab Invest. 2007;87:40-8.
43. Xu Y, Malhotra A, Ren M, Schlame M. The enzymatic function of tafazzin. J Biol Chem. 2006;281:39217-24.
44. McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J Mol Biol. 2006;361:462-9.
45. Xu Y, Sutachan JJ, Plesken H, Kelley RI, Schlame M. Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab Invest. 2005;85:823-30.
46. Xu Y, Condell M, Plesken H, et al. A Drosophila model of Barth syndrome. Proc Natl Acad Sci U S A. 2006;103:11584-8.
47. Phoon CK, Acehan D, Schlame M, et al. Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J Am Heart Assoc. 2012:1.
48. Acehan D, Vaz F, Houtkooper RH, et al. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J Biol Chem. 2011;286:899-908.
49. Zhu S, Chen Z, Zhu M, et al. Cardiolipin remodeling defects impair mitochondrial architecture and function in a murine model of Barth syndrome cardiomyopathy. Circ Heart Fail. 2021;14:e008289.
50. Wang G, McCain ML, Yang L, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20:616-23.
51. He Q. Tafazzin knockdown causes hypertrophy of neonatal ventricular myocytes. Am J Physiol Heart Circ Physiol. 2010;299:H210-6.
52. Desousa BR, Kim KK, Jones AE, et al. Calculation of ATP production rates using the seahorse XF analyzer. EMBO Rep. 2023;24:e56380.
53. Doerrier C, Garcia-souza LF, Krumschnabel G, Wohlfarter Y, Mészáros AT, Gnaiger E. High-resolution FluoRespirometry and OXPHOS protocols for human cells, permeabilized fibers from small biopsies of muscle, and isolated mitochondria. In: Palmeira CM, Moreno AJ, editors. Mitochondrial bioenergetics. New York: Springer; 2018. pp. 31-70.
54. Gu X, Ma Y, Liu Y, Wan Q. Measurement of mitochondrial respiration in adherent cells by seahorse XF96 cell mito stress test. STAR Protoc. 2021;2:100245.
55. Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem. 2017;292:7189-207.
56. Pelletier M, Billingham LK, Ramaswamy M, Siegel RM. Extracellular flux analysis to monitor glycolytic rates and mitochondrial oxygen consumption. Methods Enzymol. 2014;542:125-49.
57. Divakaruni AS, Jastroch M. A practical guide for the analysis, standardization and interpretation of oxygen consumption measurements. Nat Metab. 2022;4:978-94.
58. Miller SG, Hafen PS, Brault JJ. Increased adenine nucleotide degradation in skeletal muscle atrophy. Int J Mol Sci. 2019;21:88.
59. de Kok MJC, Schaapherder AF, Wüst RCI, et al. Circumventing the crabtree effect in cell culture: a systematic review. Mitochondrion. 2021;59:83-95.
60. Handel ME, Brand MD, Mookerjee SA. The whys and hows of calculating total cellular ATP production rate. Trends Endocrinol Metab. 2019;30:412-6.
61. Tullson PC, Whitlock DM, Terjung RL. Adenine nucleotide degradation in slow-twitch red muscle. Am J Physiol. 1990;258:C258-65.
62. Law AS, Hafen PS, Brault JJ. Liquid chromatography method for simultaneous quantification of ATP and its degradation products compatible with both UV-Vis and mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2022;1206:123351.
63. Petit PX, Gendron M, Schrantz N, et al. Oxidation of pyridine nucleotides during Fas- and ceramide-induced apoptosis in Jurkat cells: correlation with changes in mitochondria, glutathione depletion, intracellular acidification and caspase 3 activation. Biochem J. 2001;353:357-67.
64. Hancock CR, Brault JJ, Wiseman RW, Terjung RL, Meyer RA. 31P-NMR observation of free ADP during fatiguing, repetitive contractions of murine skeletal muscle lacking AK1. Am J Physiol Cell Physiol. 2005;288:C1298-304.
65. Marvin JS, Kokotos AC, Kumar M, et al. iATPSnFR2: A high-dynamic-range fluorescent sensor for monitoring intracellular ATP. Proc Natl Acad Sci U S A. 2024;121:e2314604121.
66. Stanley PE, Williams SG. Use of the liquid scintillation spectrometer for determining adenosine triphosphate by the luciferase enzyme. Anal Biochem. 1969;29:381-92.
67. Yang NC, Ho WM, Chen YH, Hu ML. A convenient one-step extraction of cellular ATP using boiling water for the luciferin-luciferase assay of ATP. Anal Biochem. 2002;306:323-7.
68. Tullson PC, Terjung RL. Adenine nucleotide degradation in striated muscle. Int J Sports Med. 1990;11 Suppl 2:S47-55.
69. de Taffin de Tilques M, Lasserre JP, Godard F, et al. Decreasing cytosolic translation is beneficial to yeast and human Tafazzin-deficient cells. Microb Cell. 2018;5:220-32.
70. Wang X, Chen XJ. A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature. 2015;524:481-4.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].