


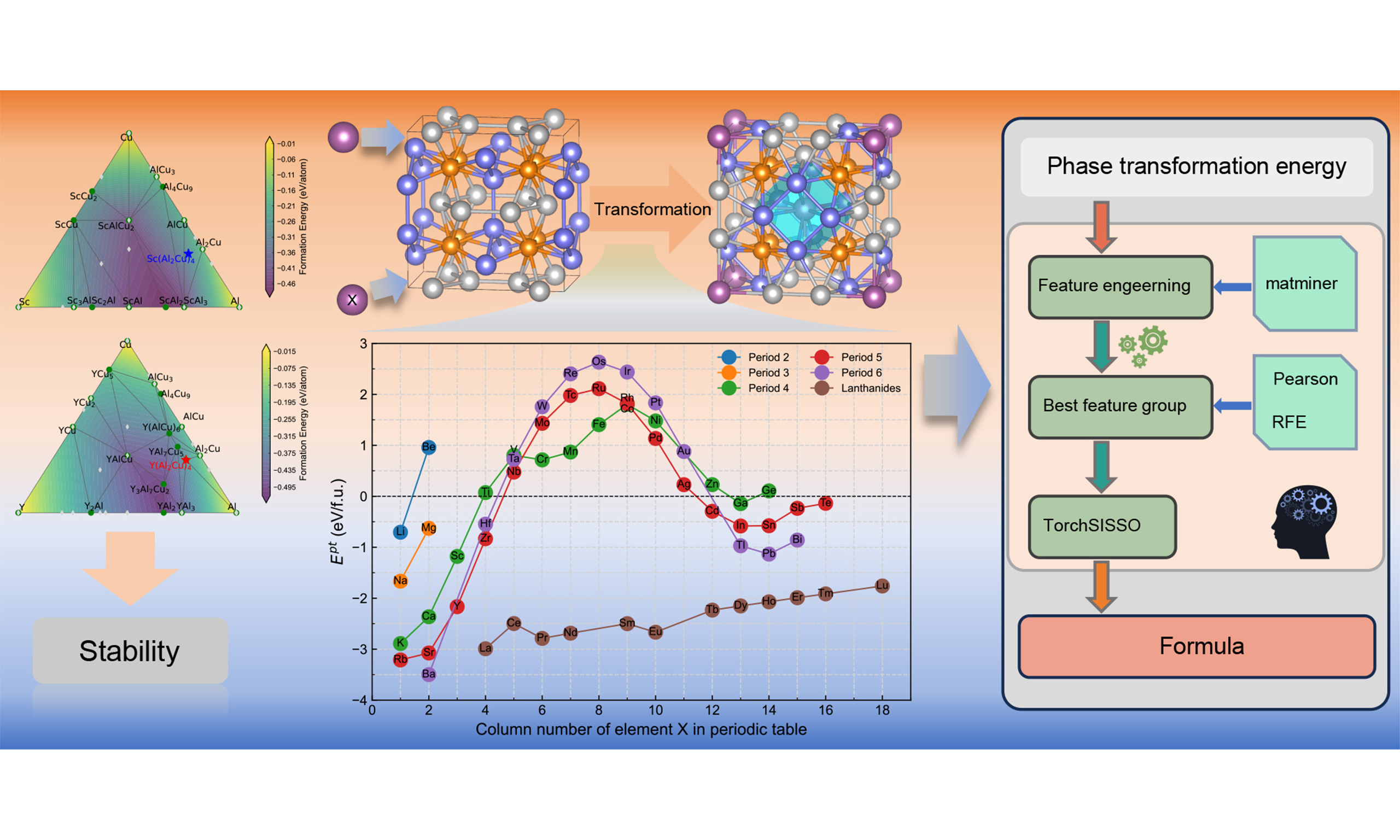
Accelerated discovery of potential heat-resistant Al8Cu4X phases via high-throughput first-principles calculations
 0
0 Abstract
The Al8Cu4X phase has emerged as a promising heat-resistant strengthening candidate for Al-Cu alloys, mitigating the instability of Ω/θ′ precipitates at elevated temperatures. In this work, we employ high-throughput first-principles calculations to systematically investigate 57 Al8Cu4X compounds, focusing on their thermodynamic stability and phase trans-formation behavior. Density functional theory calculations reveal negative formation energies for 53 compounds, and those containing rare earth elements, Ca, Sr, and Y are identified as favorable candidates for forming microscale phases at high temperatures. Phase transformation energies exhibit a pronounced periodic trend, with 33 compounds showing negative values, supporting the feasibility of forming nanoscale strengthening precipitates via high-temperature phase transformation. Symbolic regression analysis further identifies atomic volume as the primary descriptor governing the phase transformation energies, while bond order analysis demonstrates that the enhanced stability originates from a strengthened Al-Al bonding network and newly introduced Al-X bonds within the Al8Cu4X structures. Overall, this work provides a theoretical foundation for the future design and application of heat-resistant Al8Cu4X phases in aluminum alloys.
Keywords
INTRODUCTION
The growing demand for heat-resistant aluminum alloys in the aerospace and automotive industries necessitates the development of advanced alloy design strategies[1-3]. A fundamental approach to enhancing heat resistance is the incorporation of thermally stable reinforcing phases, such as ceramic phases[4-8], eutectic AlxTMy phases (where TM denotes a transition metal)[9,10], and L12-structured Al3X phases (an ordered fcc structure with the Cu3Au prototype)[11-17].
Alloying Al-Cu systems with Sc, Ca, Y, and rare earth elements has been found to promote the formation of thermally stable Al8Cu4X phases with a ThMn12-type structure (space group I4/mmm)[18-24]. At the nanoscale, as reported by Xue et al.[21], the in situ transformation of the Ω-Al2Cu phase (the characteristic age-hardening precipitate in Al-Cu-Mg-Ag alloys) into coherent Al8Cu4Sc precipitates in an Al-Cu-Mg-Ag-Sc alloy provides exceptional thermal stability up to 400 °C, dramatically elevating the alloy’s temperature limit for precipitation strengthening. Meanwhile, microscale Al8Cu4X particles, typically formed with Y and Er additions[22,23], are predominantly located along grain boundaries. These particles exert a potent grain-boundary pinning effect, thereby improving the high-temperature mechanical properties. Overall, these observations indicate that Al8Cu4X precipitates strengthen the alloy through distinct mechanisms operating at different length scales. In addition, the Al8Cu4X phase extends beyond heat-resistant Al-Cu alloys. For instance, Li et al. demonstrated that Y addition promotes fine Al8Cu4Y precipitation along grain and sub-grain boundaries in an Al-Zn-Mg-Cu alloy, effectively suppressing recrystallization[25].
Density functional theory (DFT) has been widely used to study the structural, thermodynamic, electronic, and other key properties of relevant phases in Al alloys[26,27], offering valuable insights for alloy design and microalloying strategies. However, its application to the Al8Cu4X system has been neither systematic nor comprehensive. Existing studies on Al8Cu4X (X = Y, Ce, Nd, Sm, Er and Nd)[28-30], have been confined to specific elements, primarily focusing on fundamental properties like stability, electronic structure, and lattice dynamics.
Notably, current experimental studies are predominantly focused on microscale Al8Cu4X phases, with investigations into their nanoscale counterparts being highly limited. On the computational side, efforts have primarily concentrated on elucidating the properties of known Al8Cu4X phases, lacking a systematic screening across a broader compositional space. This knowledge gap hampers the targeted design of both microscale and nanoscale Al8Cu4X precipitates.
In this study, we establish a high-throughput first-principles framework to systematically evaluate 57 Al8Cu4X compounds. Our computational workflow integrates calculations of formation energy (Ef), phase stability (via the convex hull), and phase transformation energy (Ept), thereby distinguishing elements that promote the direct formation of Al8Cu4X in the as-cast state from those that preferentially drive its precipitation from the Ω phase. Going beyond thermodynamic screening, we employ symbolic regression coupled with electronic structure analysis to identify the key electronic and structural descriptors governing structural stability and influencing Ept. This integrated approach not only provides an accelerated pathway for discovering novel heat-resistant phases but also establishes quantitative design rules for targeted microalloying in aluminum alloys.
MATERIALS AND METHODS
Structures of Al-Cu-X systems
Based on the established Al8Cu4Y lattice framework[31], we extended the X-site occupancy to cover a broader range of elements by adopting the same reported structural prototype (i.e., the same structure framework), constructing 57 initial Al8Cu4X configurations in the I4/mmm space group. The initial lattice parameters were adopted from Al8Cu4Y (a = b = 8.74 Å, c = 5.10 Å). The atomic positions within the unit cell are defined as follows: Al occupies the 8i (0.365, 0, 0) and 8j (0.275, 1/2, 0) sites, Cu occupies the 8f (1/4, 1/4, 1/4) site, and X occupies the 2a (0, 0, 0) site, resulting in a total of 26 atoms per unit cell. All structures were subjected to full structural relaxation via DFT calculations to determine their lowest-energy configurations.
For comparison, the Ω phase was modeled using the structure reported by Knowles and Stobbs[32], with space group Fmmm and lattice parameters a = 8.48 Å, b = 8.59 Å, c = 4.96 Å. The unit cell contains 24 atoms, with Al occupying 8h (0, 1/3, 0) and 8i (1/6, 0, 0), and Cu occupying 8f (1/4, 1/4, 1/4). To represent the solid-solution state of X in Al, a 3×3×3 FCC-Al supercell (108 atoms) with a single Al atom replaced by X was relaxed to obtain its equilibrium configuration.
The lattice mismatch at the interface was calculated to evaluate the coherency between the Al matrix and the precipitate phases. The interface model[21,33,34] was established with the orientation relationship of (
where U and V denote the lattice parameters along the two matching directions.
First-principles calculations
All DFT calculations were performed with the Vienna Ab initio Simulation Package (VASP)[36,37] using the Perdew-Burke-Ernzerhof (PBE) functional within the generalized gradient approximation (GGA)[38]. Brillouin zone integration employed a Monkhorst-Pack k-point mesh of 4 × 4 × 6. Electronic and ionic convergence thresholds were set to 10-5 and 10-4 eV, respectively, with full relaxation of lattice parameters and atomic positions. Bond orders (BOs) were analyzed using the DDEC6 method[39,40] implemented in the Chargemol package[41].
Energetics of phase stability and transitions
The thermodynamic stability of Al8Cu4X phases in Al-Cu alloys was evaluated using formation energies and convex hull analysis in the Al-Cu-X ternary system. The Ef per atom is defined as:
where Etot represents the total energy of the compound, ni is the number of atoms of element i and μi is the energy of element i in its stable elemental state.
All competing phases, along with their crystal structures and formation energies, were extracted from the Materials Project database[42]. For all Al-Cu-X systems considered in this work, ternary phase diagrams were constructed using pymatgen[43,44]. A phase lying on the convex hull is thermodynamically stable, whereas a phase above the hull is unstable and may decompose into adjacent phases[45]. The energy above hull (ΔEh) quantifies the deviation from thermodynamic stability and is defined as:
where ΔEh is the formation energy of the convex hull at the same composition.
To evaluate the potential of Al8Cu4X as an aging precipitate, we calculated the phase transformation energy Ept, defined here as the reaction energy for transforming Ω into Al8Cu4X, where X is supplied from a dilute solid solution in fcc-Al. The corresponding reaction can be written as:
where the Al reservoir is bulk fcc-Al with μAl taken from pure Al. The corresponding reaction energy Ept per formula unit (f.u.) is defined as:
where
Machine learning assisted symbolic regression analysis
Machine learning assisted symbolic regression was applied to identify quantitative relationships between elemental features and Ept[46-48]. Elemental features were generated using the Magpie featurization scheme[49] via Matminer[50], covering 13 fundamental attributes (listed in Supplementary Table 1).
Before applying symbolic regression with these features, dimensionality reduction was performed to remove noise and redundant features[51,52]. Dimensionality reduction was conducted in two stages: (i) Pearson correlation coefficients filtering to remove redundant features[53], and (ii) recursive feature elimination (RFE)[54] using random forest models[55,56] with fivefold cross-validation.
The reduced feature set was used as input for symbolic regression with TorchSISSO[57]. Analytical expressions linking elemental properties to Ept were derived using a symbolic operation set {+, -, ×, ÷, exp, ln, sin, pow(2)}. This approach produced interpretable models that quantify how elemental properties govern the Ept.
RESULTS AND DISCUSSION
Lattice structure
The relaxed structure of the Ω phase is illustrated in Figure 1A. The Ω phase was initially modeled in the orthorhombic Fmmm structure. Upon structural relaxation, the lattice parameters were changed (a: 8.479 → 8.556 Å, b: 8.590 → 8.555 Å and c: 4.960 → 4.868 Å), and atomic positions underwent corresponding adjustments, leading to a transition into tetragonal I4/mcm symmetry, which coincides with the equilibrium θ phase. The crystal structure of the Al8Cu4X phase is illustrated in Figure 1B. Relative to the Ω phase, the rearrangement of Al atoms gives rise to decahedron coordinated sites, with the X atom occupying the central position.
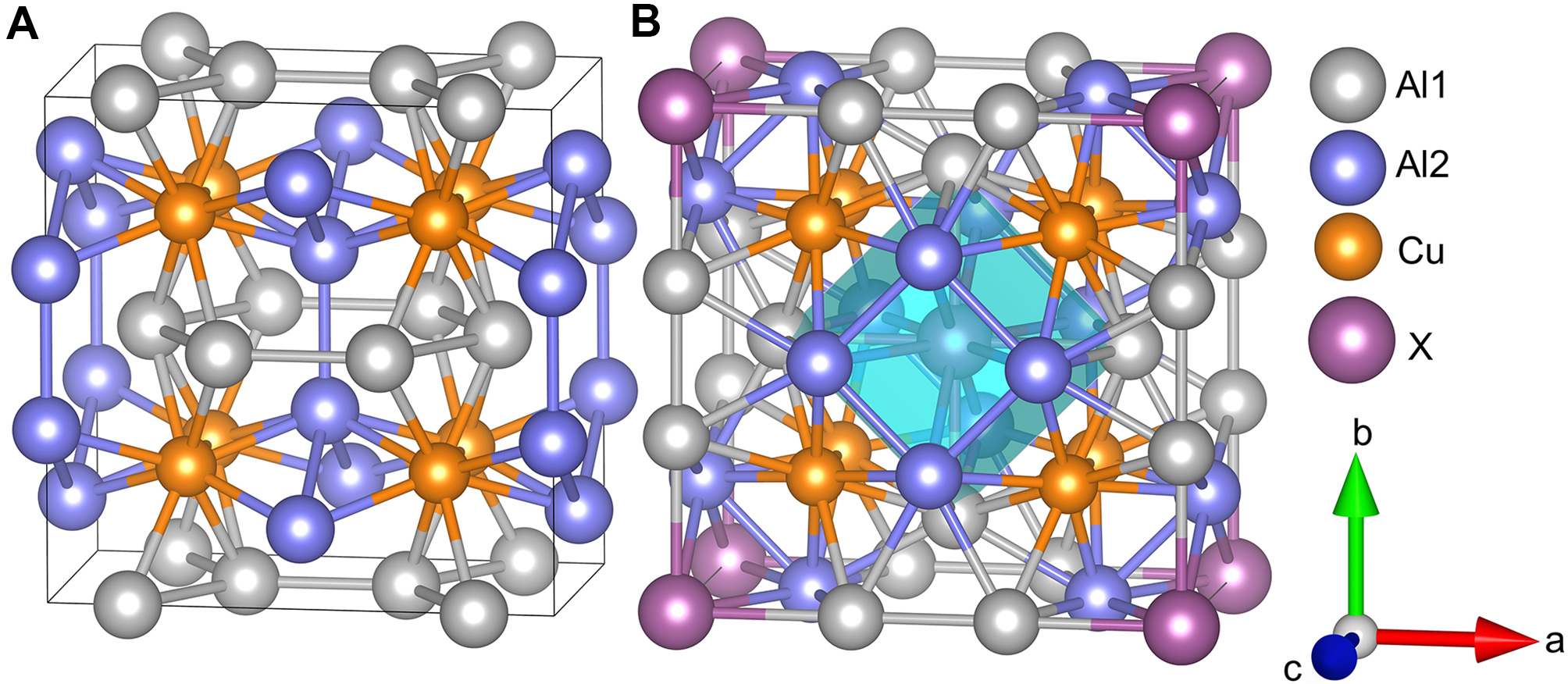
Figure 1. Crystal structures of (A) Ω and (B) Al8Cu4X produced by VESTA[58]. Insertion of element X into the Ω framework induces displacements of Al atoms, leading to the formation of decahedron coordinated sites (blue regions). VESTA: Visualization for Electronic and Structural Analysis.
The incorporation of different elements at the X site leads to variations in the lattice volume of Al8Cu4X, as shown in Figure 2. The calculated lattice volumes exhibit a periodic dependence on the atomic number of X, consistent with general periodic trends. From the fourth period onward, the lattice volume decreases and subsequently increases across each period as the atomic number of element X increases. In the second and third periods, which each contain only two Al8Cu4X data points, the element X with the higher atomic number corresponds to a smaller lattice volume. Notably, the Al8Cu4Ba phase exhibits the largest lattice volume among all studied compounds, reaching 427 Å3.
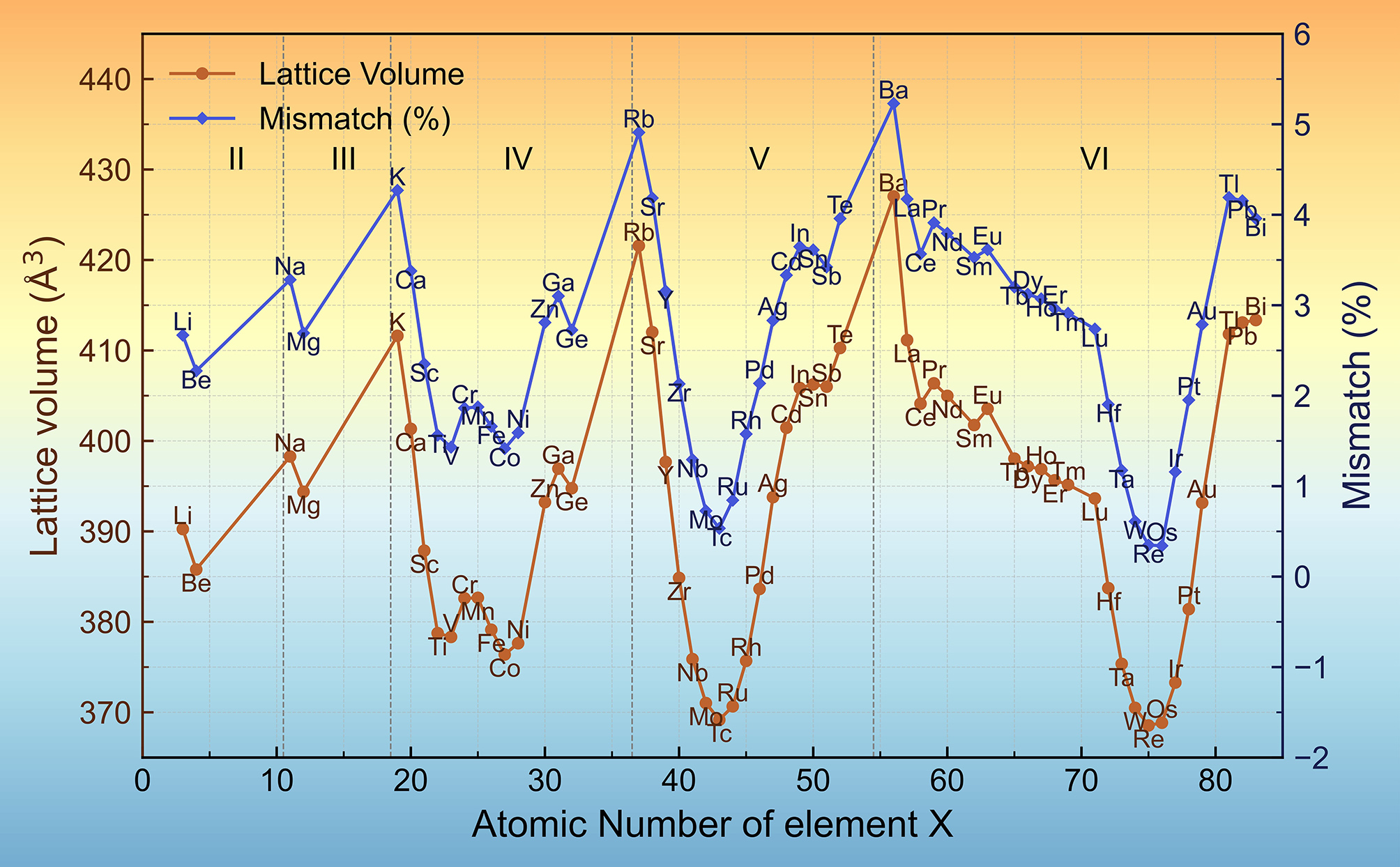
Figure 2. Lattice volume and lattice mismatch of Al8Cu4X as a function of the atomic number of element X. Vertical dashed lines separate different periods of the periodic table, labeled II-VI for the second to sixth periods.
The equilibrium volume of the Al8Cu4X may be influenced by the atomic size of element X (illustrated in Supplementary Figure 1). In the second and third periods, atomic size decreases monotonically with increasing atomic number, which is directly reflected in a reduction of the lattice volume of Al8Cu4X. From the fourth period onward, however, the trend becomes more complex. The involvement of transition elements leads to a non-monotonic change, where atomic size first decreases and then increases within the same period. Accordingly, the lattice volume of Al8Cu4X shows a similar trend of initial contraction followed by expansion.
The lattice mismatch between Al8Cu4X and the Al matrix remains very low. Except for Al8Cu4Ba, all Al8Cu4X phases exhibit a mismatch below 5% [Figure 2]. Such low misfit is typically favorable for forming coherent precipitate-matrix interfaces. Coherent nanoscale Al8Cu4X precipitates can therefore provide effective age-hardening and improve the mechanical properties of Al alloys. Moreover, coherent interfaces are often associated with relatively low interfacial energies, which is beneficial for retaining precipitate stability and resisting coarsening at elevated temperatures[59].
Thermodynamic stability
The calculated Ef of the various Al8Cu4X phases are summarized in Figure 3, revealing a strong dependence on element X. Among the 57 investigated compositions, 53 exhibit negative formation energies, indicating exothermic formation and potential stability within the Al-Cu-X system. In contrast, the phases formed by Be, W, Re, and Os display positive formation energies, suggesting endothermic and thermodynamically unstable formation. For comparison, the Ef of the Ω phase was calculated to be -0.16 eV/atom, agreeing with reported values[20,32]. It is noteworthy that the Ef of the Al8Cu4X phases formed by Li, Ca, Sc, Ti, Y, Zr, Ba, Hf, and the rare earth elements are lower than that of the Ω phase, which implies comparable or greater thermodynamic stability.
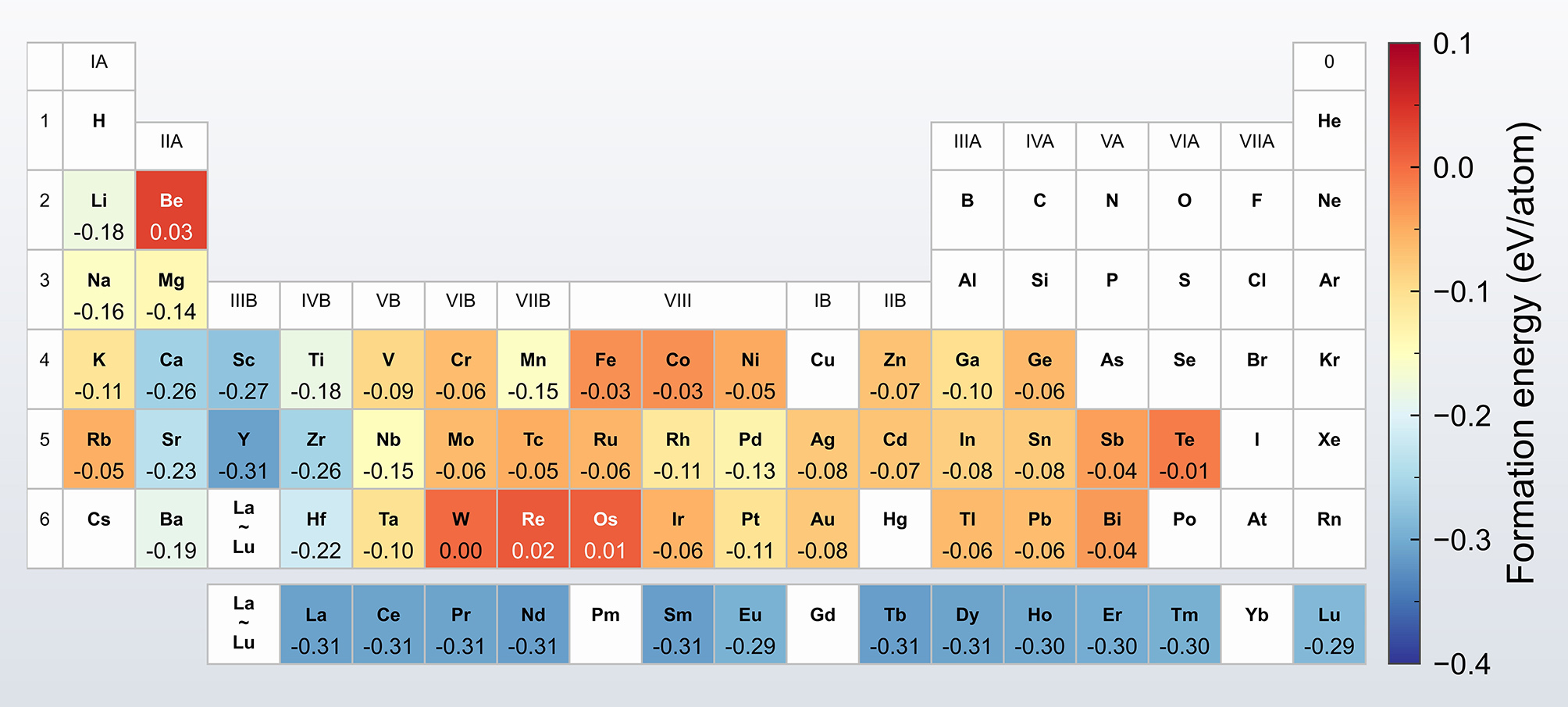
Figure 3. Ef of Al8Cu4X phases with different X elements. Ef: Formation energy.
To evaluate the relative stability of the Al8Cu4X phases, their ΔEh was calculated, as summarized in Figure 4. The results indicate that phases formed by Ca, Y, Sr and the rare earth elements are located on the convex hull (ΔEh = 0), confirming their thermodynamic stability. This computational finding is experimentally validated by the reported precipitation of Al8Cu4X phases formed by Ca, Y, and rare earth elements[18,19,22-24]. To the best of our knowledge, the thermodynamic stability of the Al8Cu4Sr phase predicted here has not been previously reported in the literature. This finding suggests that Sr could be a promising candidate for further development of heat-resistant Al-Cu alloys. Ternary phase diagrams are provided in the Supplementary Figure 2.
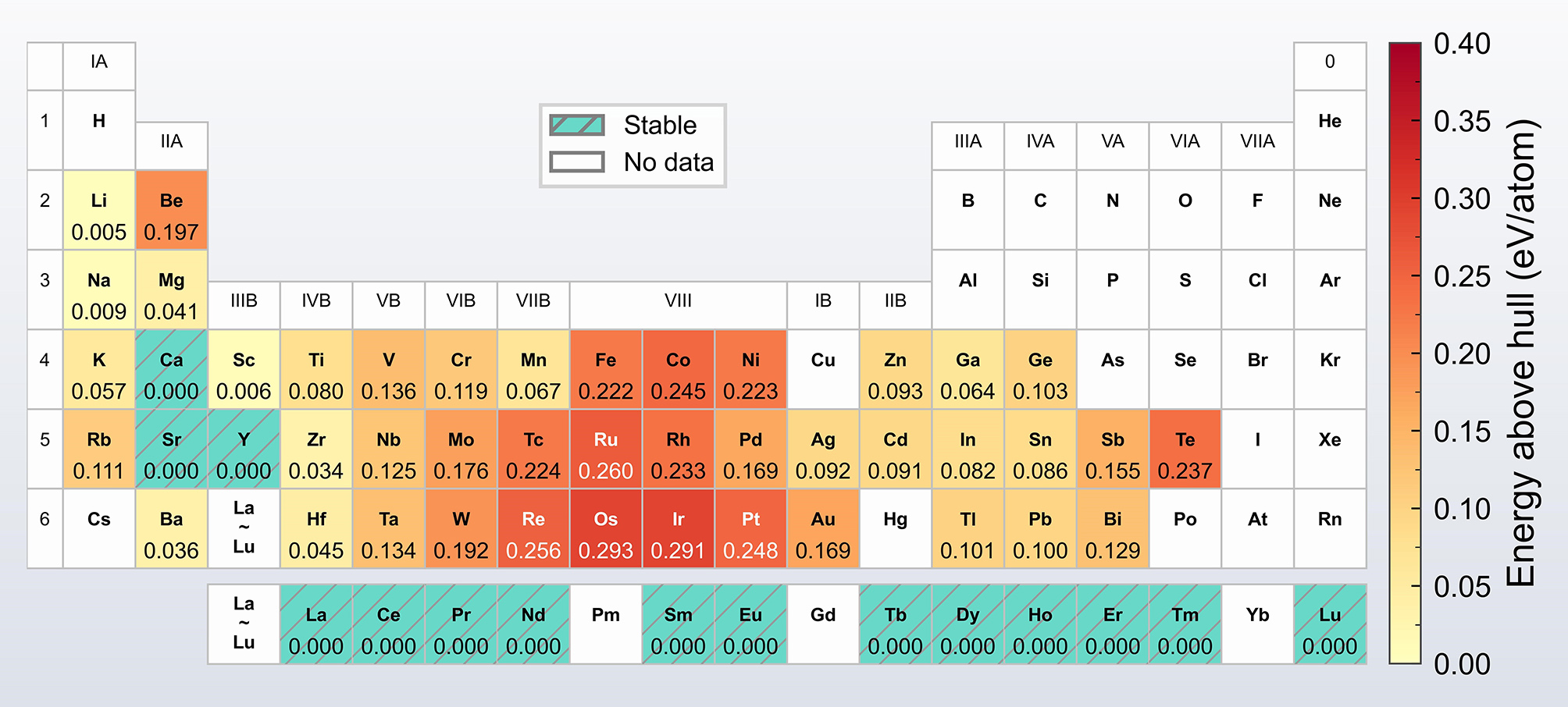
Figure 4. ΔEh of Al8Cu4X phases for different X elements. Phases highlighted with a turquoise background and diagonal hatching are thermodynamically stable. ΔEh: Energy above hull.
Phase transformation behavior
The Ept quantifies the tendency for the Ω phase to transform into the Al8Cu4X phase. As shown in Figure 5, 33 Al8Cu4X phases exhibit negative Ept values. Al8Cu4Sc exhibits a negative Ep (-1.18 eV/f.u.), indicating that in Sc-containing alloys, the Ω phase has a tendency to transform into Al8Cu4Sc, which is consistent with experimental observations of the formation of Al8Cu4Sc from the Ω phase under thermal exposure[21]. Additionally, the Ept of Al8Cu4X phases formed by 33 elements, including Li, Na, K, Rb, Mg, Ca ,Sr, Ba, Sc, Y, Zr, Hf, Cd, Ga, In, Tl, Sn ,Pb, Sb, Bi, Te and the rare earth elements are all negative, suggesting that these phases may also arise from the Ω phase.
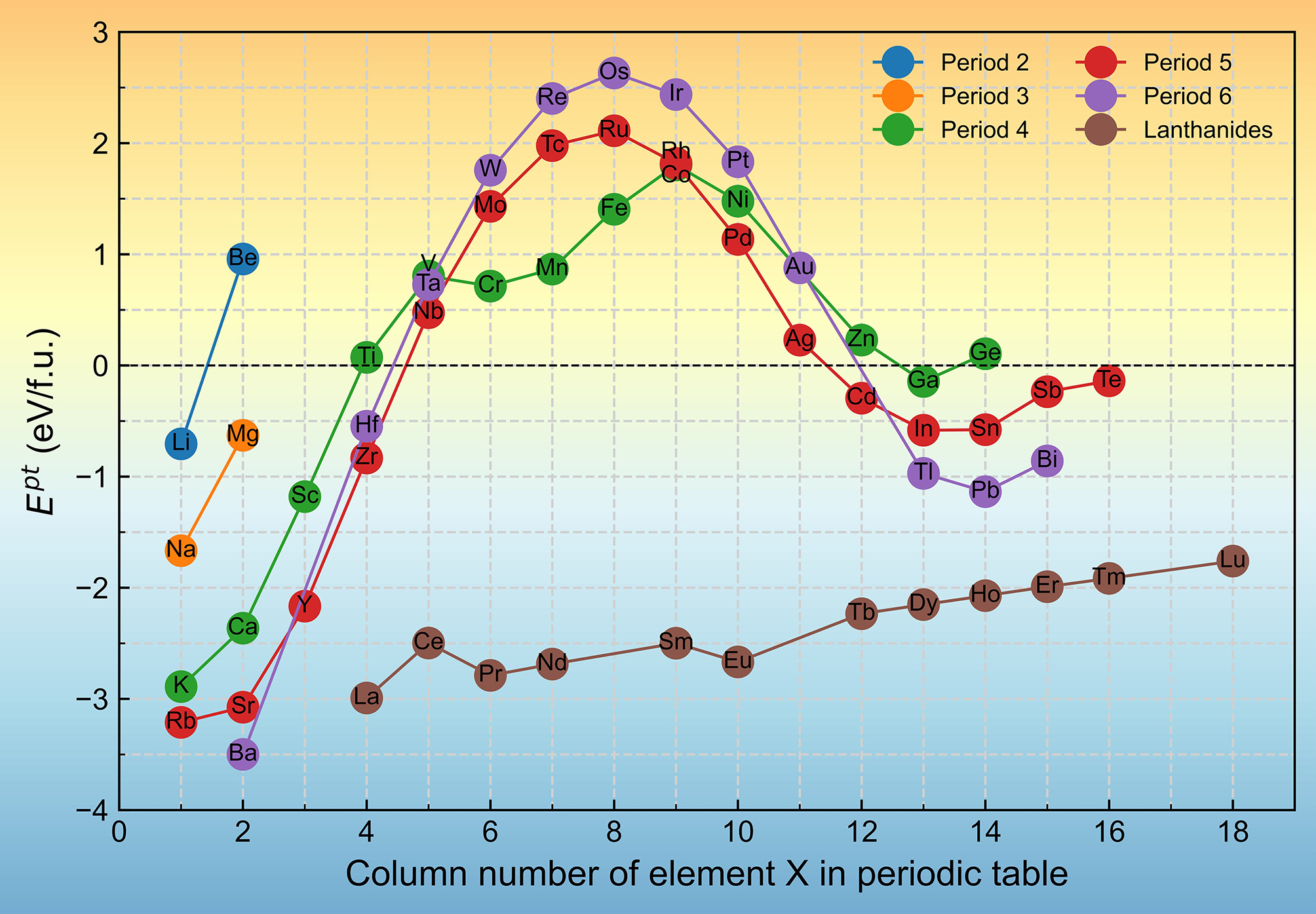
Figure 5. Ept of Al8Cu4X as a function of the column number of element X in the periodic table. Ept: Phase transformation energy.
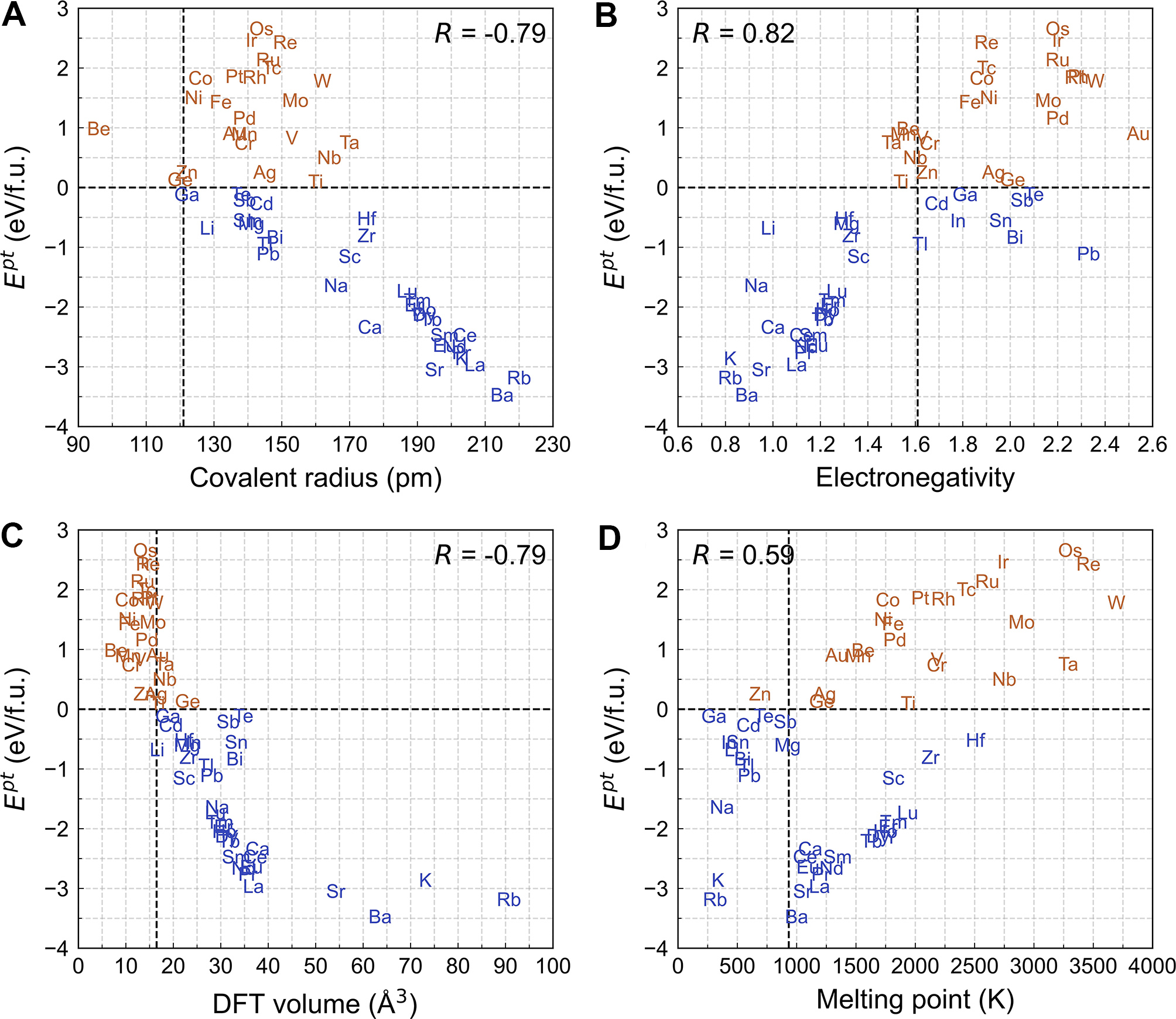
Figure 6. Relationship between element features and Ept: (A) covalent radius, (B) electronegativity, (C) DFT volume and (D) melting point. Blue elements correspond to negative Ept values, whereas brown elements correspond to positive Ept. The vertical dashed line in each plot represents the corresponding value for Al. Ept: Phase transformation energy; DFT: density functional theory.
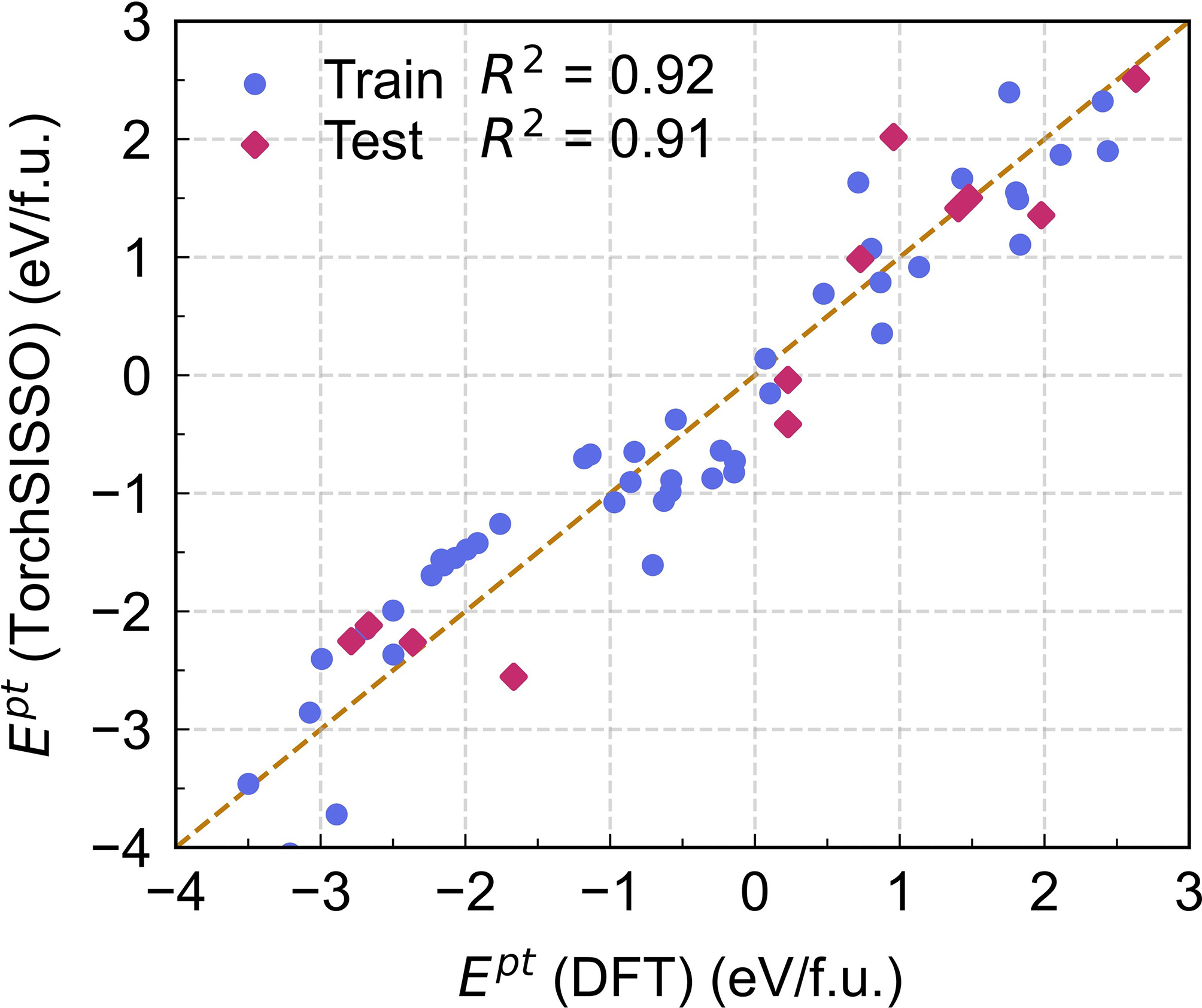
Figure 7. Comparison between Ept predicted by TorchSISSO and those obtained from DFT calculations for both training and testing sets. Ept: Phase transformation energy; SISSO: Sure Independence Screening and Sparsifying Operator; R2: coefficient of determination; DFT: density functional theory.
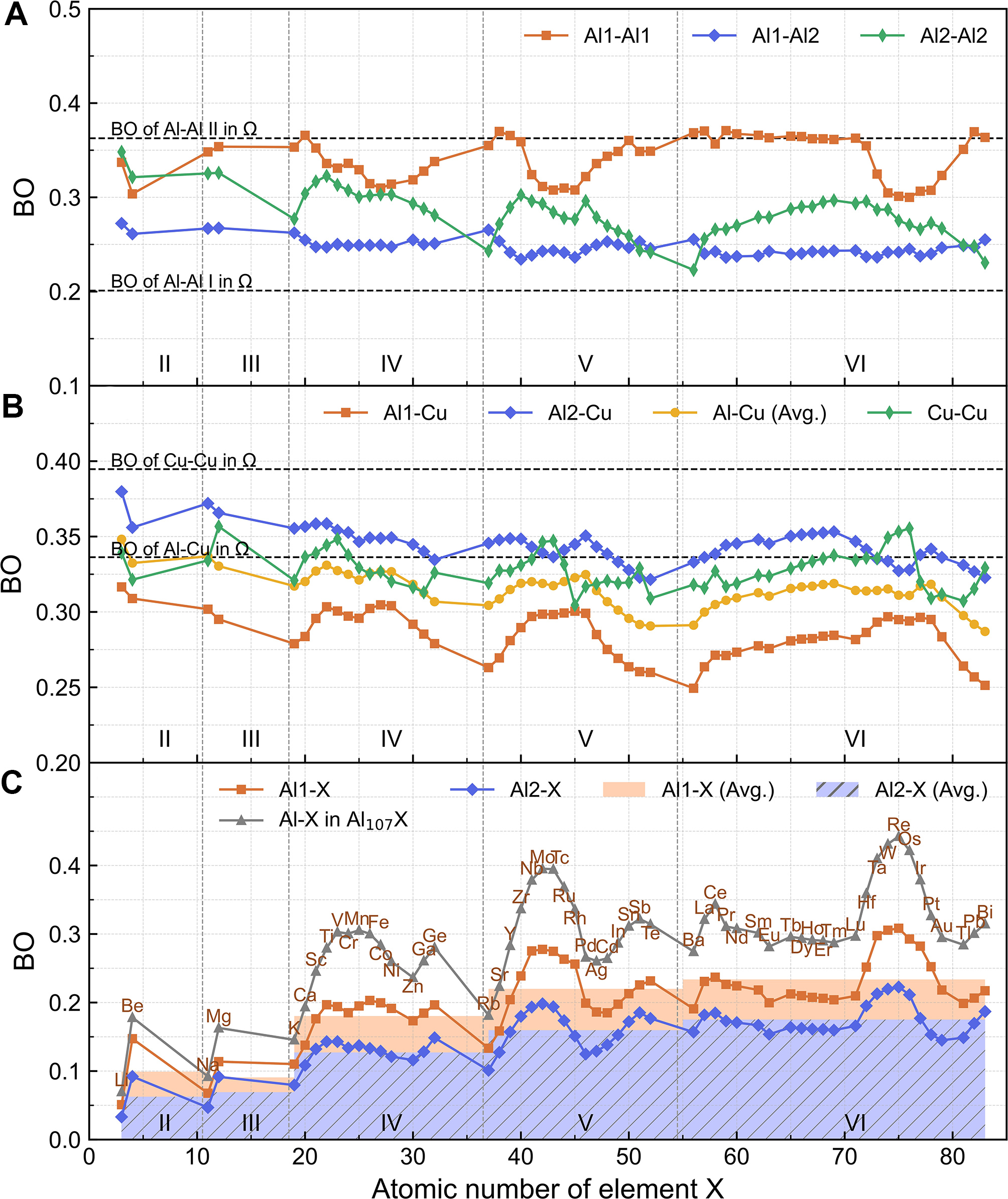
Figure 8. (A) BOs of Al1-Al1, Al1-Al2 and Al2-Al2 in Al8Cu4X vs. atomic number of X; (B) BOs of Al1-Cu, Al2-Cu and Cu-Cu in Al8Cu4X vs. atomic number of X; (C)Al1-X, Al2-X in Al8Cu4X and Al-X in solid solution (Al107X) vs. atomic number of X. The vertical dashed line indicates the separation between periods in the periodic table, with II to VI representing the first through sixth periods. BOs: Bond orders; BO: bond orders.
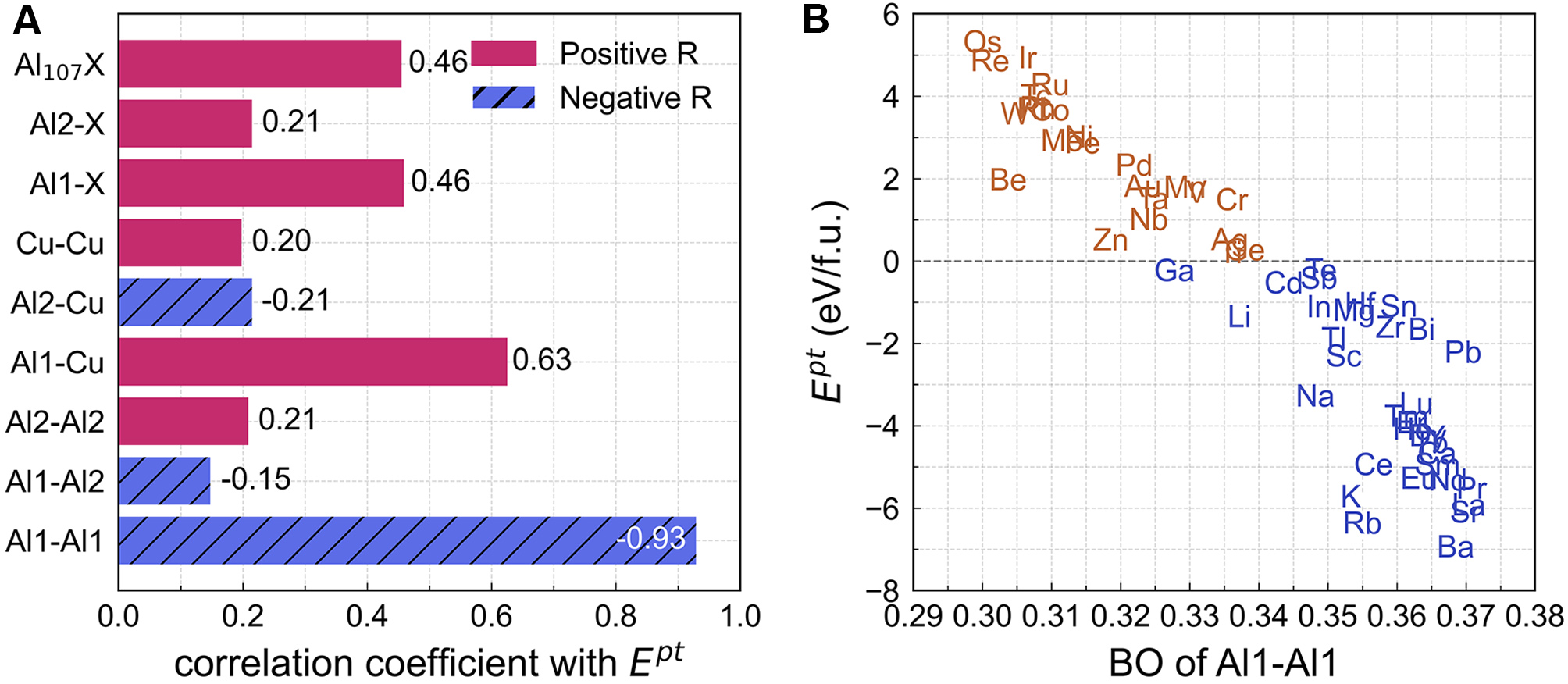
Figure 9. (A) Pearson correlation coefficients (R) between BO and Ept; (B) Al1-Al1 BOs vs. Ept for Al8Cu4X phases, where each symbol denotes the X element; blue indicates Ept < 0 and brown indicates Ept > 0. BO: Bond order; BOs: bond orders; Ept: phase transformation energy.
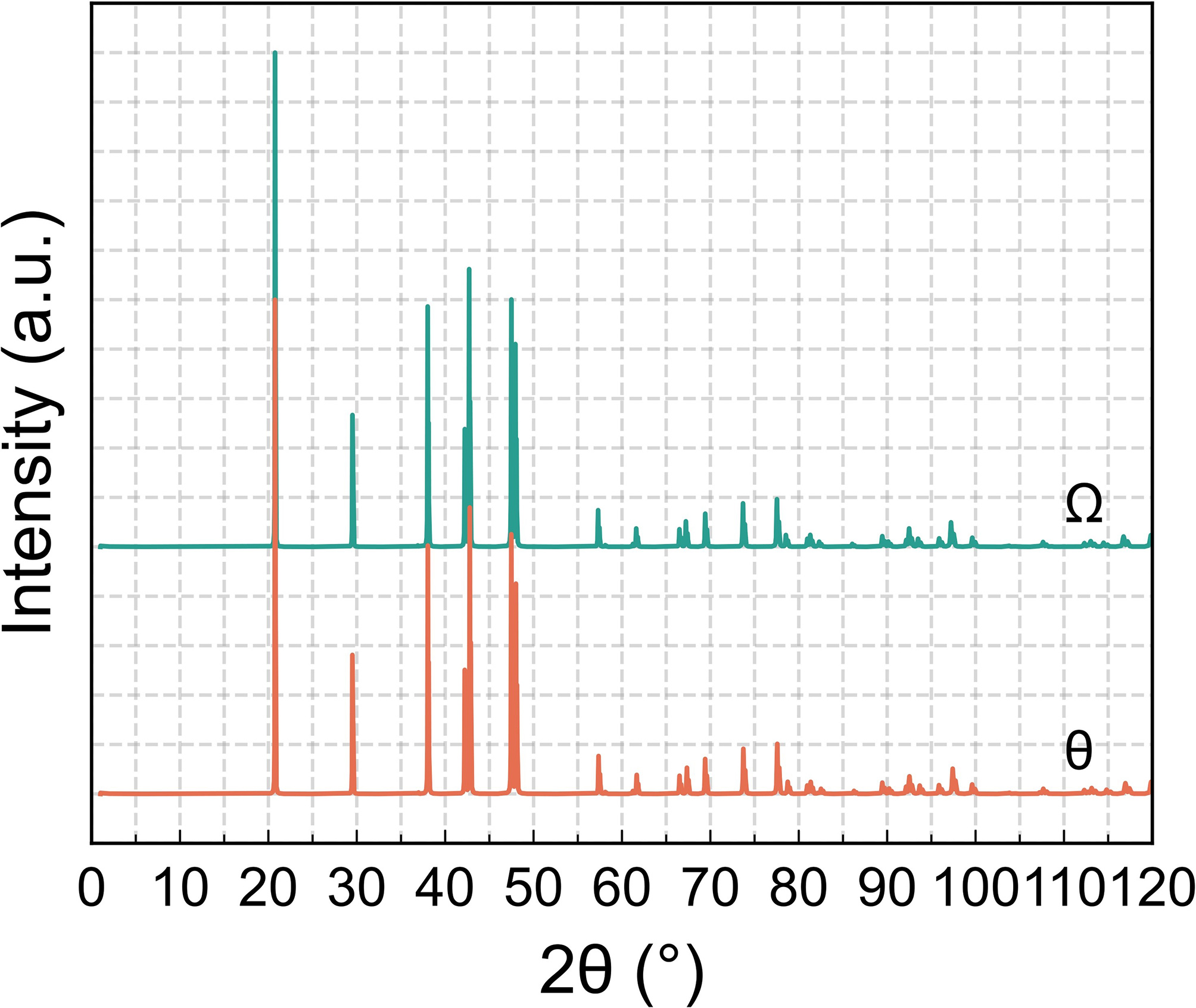
Figure 10. Simulated powder XRD patterns demonstrating the high structural similarity between the relaxed Ω and θ phases, as evidenced by their identical peak positions and shapes. XRD: X-ray diffraction.
The trend of Ept shows a well-defined periodic variation. Specifically, In the second and third periods, where only two data points are available, Ept decreases monotonically with increasing atomic number. From the fourth period onward, it generally increases and then decreases across each period. It varies inversely with the lattice volume of Al8Cu4X. This suggests that the atomic size of X, a primary determinant of the lattice volume, also significantly influences Ept.
Correlation between phase transformation behavior and elemental features
The variation of Ept with atomic number shown in Figure 5 indicates that intrinsic elemental properties strongly influence the Ω-to-Al8Cu4X transformation. To elucidate this relationship, machine learning assisted symbolic regression was employed.
Initially, Pearson correlation filtering was applied to remove features with correlation coefficients exceeding 0.8, yielding 8 candidate descriptors. RFE further identified an optimal subset of 5 features. Detailed information regarding the Pearson correlation filtering and RFE procedures is provided in Supplementary Figures 3 and 4.
The five features retained after RFE were used as input for symbolic regression via TorchSISSO, which ultimately selected four features for predicting Ept: covalent radius (X1), electronegativity (X2), DFT-derived atomic volume (X3), and melting point (X4). This selection suggests that these descriptors capture the dominant factors governing Ept. Covalent radius represents half the interatomic distance in a covalent bond, electronegativity is defined according to the Pauling scale, and DFT volume corresponds to the average volume occupied per atom in a fully relaxed crystal. As shown in Figure 6, Pearson correlation coefficients between these features and Ept are -0.79, 0.82, -0.79, and 0.59, respectively.
The equation derived from TorchSISSO fitting is as follows:
The TorchSISSO-derived equation indicates that Ept correlates positively with X2 and X4, and negatively with X1 and X3, consistent with the Pearson analysis. Comparison with DFT-calculated formation energies [Figure 7] demonstrates excellent agreement on both training and test sets (R2 = 0.92 and 0.91, respectively), confirming that the model accurately captures the dominant elemental properties controlling Ept. Moreover, the sparse and explicit closed-form expression highlights nonlinear relationships between descriptors and Ept and reveals coupled descriptor effects through ratio-type terms (e.g., X4/X3 and X1/X2).
Among these features, the DFT-derived atomic volume demonstrates the strongest correlation with Ept, as illustrated in Figure 6C. Al8Cu4X phases exhibiting negative Ept generally incorporate X atoms with larger atomic volumes than aluminum, suggesting that larger atomic sizes facilitate the stabilization of Al8Cu4X structures. In contrast, most elements associated with positive Ept values possess smaller atomic volumes than Al, with the exceptions of Ge, Ti, Nb, Ta, and Au. This finding corresponds to the previous explanation for the periodic variation of Al8Cu4X with atomic number, namely, that the atomic size of X significantly influences Ept.
Therefore, it can be concluded that the Ept for the Ω-to-Al8Cu4X transformation is governed by a synergistic interplay of elemental descriptors identified through symbolic regression. Atomic volume shows the strongest influence on phase stability, with larger X-atom volumes generally favoring Al8Cu4X formation. The derived model [Equation 5] effectively integrates these features, achieving high predictive accuracy (R2 > 0.91) and demonstrating that the transformation behavior is controlled by a multi-descriptor mechanism rooted in atomic size and electronic structure.
BO analysis of bonding characteristics and their relation to Ept
Furthermore, BO analysis was employed to assess the structural stability of the Al8Cu4X phases, and its correlations with Ept are also discussed. Generally, the higher the bond order, the stronger the bond, and stronger bonding is qualitatively associated with higher structural stability. After structural relaxation, the Ω phase transforms into a structure identical to the θ phase, which makes the Al1 and Al2 atomic sites indistinguishable, as shown in Figure 1A. In this configuration, each Al atom forms three Al-Al bonds with BO values of 0.20 (connected to the same Cu atom at a bond length of 2.9 Å) and 0.36 (connected to a neighboring Cu atom at 2.7 Å). The Al-Cu and Cu-Cu bonds exhibit BO values of 0.34 and 0.40, respectively.
In Al8Cu4X, as shown in Figure 8A the BOs of Al1-Al2 and Al2-Al2 are intermediate between those of the two distinct Al-Al bonds in the Ω phase, with certain Al1-Al1 bonds being slightly strengthened. As illustrated in Figure 1B, each Al2 atom resides at the top or bottom face of an octahedron and is bonded to four Al1 and two Al2 atoms. In contrast, each Al1 atom is coordinated with four Al2 and one Al1 atom. This configuration results in a higher number of strong Al-Al bonds, forming a more robust Al-Al network than that in the Ω phase. This enhanced connectivity improves structural stability and supports the role of Al8Cu4X as a high temperature strengthening phase.
In addition, as shown in Figure 8B, BO analysis further shows that Al1-Cu and Cu-Cu bonds in Al8Cu4X are weaker than those in the Ω phase. Although a few Al2-Cu bonds exhibit higher BOs, the average BOs of all Al-Cu bonds (considering both Al1-Cu and Al2-Cu) is lower in all Al8Cu4X phases except for Al8Cu4Li and Al8Cu4Na when compared to the Ω phase. This net weakening of the Al-Cu and Cu-Cu bonding partially counteracts the stabilizing effect of the enhanced Al-Al network, thereby influencing the overall structural stability of the Al8Cu4X phase.
Regarding Al-X bonding, the average BOs of Al1-X and Al2-X generally increase from the third period onward, as shown in Figure 8C. This trend can be attributed to the increased contribution of d and f orbitals from higher-period X elements, which enhances covalent interaction with Al according to atomic orbital bonding theory[60]. It is noteworthy that although the variation trend of Al-X bonds within Al8Cu4X follows a similar trend to that in the corresponding solid solution, the bonds in the Al8Cu4X are consistently weaker relative to the solid-solution state. Nevertheless, the introduction of Al-X bonds results in a more complex bonding network in Al8Cu4X compared to the Ω phase, thereby enhancing its structural stability.
The correlations between BO metrics and the Ept were systematically examined. As illustrated in Figure 9A, among all BO quantities considered, the strongest correlation with Ept is exhibited by the Al1-Al1 BO, with a Pearson correlation coefficient of R = -0.93. In contrast, considerably weaker correlations are observed for other BO metrics, with absolute values of the correlation coefficient not exceeding 0.63. The relationship between Al1-Al1 BO and Ept is further depicted in Figure 9B. A threshold behavior is observed: Al8Cu4X phases characterized by Al1-Al1 BO values exceeding 0.34 are generally associated with negative Ept values. Conversely, for phases with Al1-Al1 BO below this threshold, predominantly positive Ept values are obtained, with Al8Cu4Li and Al8Cu4Ga identified as exceptions.
Design principles for heat-resistant Al-Cu alloys strengthened by Al8Cu4X
Symmetry analysis further confirms that the relaxed Ω phase and the equilibrium θ phase are crystallographically equivalent. This structural equivalence is further corroborated by their nearly identical simulated X-ray diffraction (XRD) peak positions and line profiles, as shown in Figure 10. Moreover, their formation energies are nearly indistinguishable (approximately -0.16 eV/atom), indicating that the relaxed Ω phase essentially converges to the equilibrium θ phase. This observation is consistent with the earlier findings of Yang[61]. Previous studies have also suggested that, in Al-Cu-Mg-Ag alloys, Mg-Ag layers introduce lattice distortions into the precipitated Al-Cu phase, thereby promoting the formation and stabilization of the Ω phase[62]. Given the close crystallographic similarity between θ and Ω, the Ω phase is generally regarded as a θ-derived structure whose stability relies on the presence of Mg-Ag layers. Once these layers are disrupted, the Ω phase tends to revert to the θ structure, thereby diminishing its strengthening contribution, as also noted by Lu et al.[63].
The enhanced heat resistance of Al8Cu4X phases is attributed to two principal factors: intrinsic thermodynamic stability and retarded coarsening kinetics. First, Al8Cu4X phases possess a more complex bonding network compared to the Ω phase and, unlike the Ω phase which is a lattice-distorted equilibrium phase, they exhibit a higher energy barrier for phase transformation. Furthermore, the Al8Cu4X formed by Ca, Y, Sr, and rare earth elements are themselves thermodynamically stable, as they reside directly on the convex hull (ΔEh = 0). Second, and critically, when X is a slow-diffusing element such as Er, Sc, or Zr (with reported diffusion coefficients of 9.7 × 10-20, 4 × 10-19, and 1.2 × 10-20 m2/s at 300 °C, respectively[64-66]), the Ostwald ripening process of the precipitates becomes governed by the sluggish diffusion of X. Given that the diffusion coefficients of these elements are orders of magnitude lower than those of Al and Cu (7.1 × 10-17 and 5.2 × 10-17 m2/s at 300 °C, respectively[67,68]), the resulting Al8Cu4X phases possess exceptionally low coarsening rates. This intrinsic resistance to coarsening, which does not rely on Mg-Ag stabilization as in the case of the Ω phase, ensures excellent microstructural stability at elevated temperatures.
For the design of microscale phases, priority should be given to elements that form Al8Cu4X phases with zero energy above the convex hull (ΔEh = 0), such as Ca, Sr, Y, and the rare earth elements, to ensure thermodynamic stability. Beyond strictly stable phases, those with ΔEh below 0.01 eV/atom, such as Al8Cu4Li, Al8Cu4Na, and Al8Cu4Sc can be considered effectively stable according to established criteria[69,70]. Nevertheless, the practical formation and stability of such metastable phases still require experimental validation. This is exemplified by the Al-Cu-Sc system, where Sc tends to form the W-phase (Al5-8Cu7-4Sc), which shares a similar crystal structure with Al8Cu4X but has a different elemental stoichiometry.
To achieve nanoscale precipitation control in Al-Cu-X systems, a hierarchical screening strategy for alloying element selection is proposed. The primary criterion requires a negative Ept, a condition satisfied by main-group metals (except Be and Ge), along with Zr, Hf, Cd, Sc, Y, and the rare earth elements. To further enhance stability, elements that form Al8Cu4X phases with zero or near-zero ΔEh values, such as Li, Na, Sc, Ca, Sr, Y, and the rare earth elements, are prioritized due to their superior thermodynamic stability.
Limitations of the current work
The conclusions of this work are primarily based on 0 K DFT calculations within a fixed Al8Cu4X structural prototype (derived from the Al8Cu4Y framework). As such, they are subject to uncertainties associated with the chosen computational approximations, the use of static (0 K) energies, and idealized structural models. Therefore, the predicted stability trends and candidate elements require targeted experimental validation under realistic processing and service conditions.
CONCLUSIONS
By integrating high-throughput first-principles calculations with machine learning, we develop a design strategy for heat-resistant Al alloys centered on 57 Al8Cu4X phases to identify stable compounds capable of forming either micro-scale or nanoscale strengthening precipitates within the matrix. This work provides theoretical support for the design of Al8Cu4X strengthened heat-resistant alloys and broadens the potential application of rare-earth elements in Al-Cu alloys. The key findings are summarized as follows:
Screening based on Ef identifies 53 Al8Cu4X compounds with negative values, signifying their thermodynamic favorability for formation. Convex hull analysis further reveals that Al8Cu4X phases containing rare earth elements, as well as Ca, Sr, and Y, are thermodynamically stable and are suitable for precipitation as microscale phases, thus contributing to high-temperature strengthening.
Screening based on Ept identifies 33 Al8Cu4X compounds with negative values, indicating a thermodynamic driving force for transformation from the Ω phase. Additionally, Al8Cu4X phases formed by Li, Na, Sc, Ca, Sr, Y, and rare earth elements exhibit zero or near-zero transformation energies, highlighting their significant potential for forming nanoscale strengthening precipitates through high-temperature phase transformation.
Machine learning assisted symbolic regression analysis was employed to investigate the relationship between elemental features and Ept. The analysis reveals that the atomic volume of X is the primary factor influencing transformation energies. Additionally, BO analysis provides further insights into the stability mechanisms of Al8Cu4X phases, demonstrating that the enhanced structural stability of these phases results from a more complex Al-Al bonding network compared to the Ω phase and newly introduced Al-X bonds.
DECLARATIONS
Authors’ contributions
Made substantial contributions to conception and design of the study and performed data analysis and interpretation: Zhou, B.; Li, X.; Xiao, W.
Performed computational modeling and data acquisition: Zhou, B.; Liu, Q.; Zhu, K.
Supervision and funding acquisition: Li, X.; Xiao, W.; Li, Z.; Wen, K.; Yan, L.; Yan, H.; Zhang, Y.; Xiong, B.
Wrote the manuscript: Zhou, B.; Xiao, W.
Review and editing: Zhou, B.; Li, X.; Xiao, W.
Availability of data and materials
The data and code that support the findings of this study are publicly available on GitHub: https://github.com/RuneoFox/Al8Cu4XFilter.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (version GPT-5.2, released 2025-12-11) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was financially supported by the Advanced Materials-National Science and Technology Major Project (No. 2025ZD0619700) and additional related initiatives.
Conflicts of interest
All authors are affiliated with China GRINM Group Co., Ltd. Zhou, B.; Li, X.; Xiao, W.; Liu, Q.; Zhu, K.; Wen, K.; Yan, L.; Yan, H.; and Zhang, Y. are affiliated with GRIMAT Engineering Institute Co., Ltd., a subsidiary of China GRINM Group Co., Ltd. The authors declare no competing interests.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Wadsworth, J.; Nieh, T. G.; Stephens, J. J. Recent advances in aerospace refractory metal alloys. Int Mater Rev 1988, 33, 131-50.
2. Gayle, F. W.; Goodway, M. Precipitation hardening in the first aerospace aluminum alloy: the wright flyer crankcase. Science 1994, 266, 1015-7.
3. Zhu, L.; Li, N.; Childs, P. Light-weighting in aerospace component and system design. Propuls Power Res 2018, 7, 103-19.
4. Msebawi, M. S.; Leman, Z.; Shamsudin, S.; et al. The effects of CuO and SiO2 on aluminum AA6061 hybrid nanocomposite as reinforcements: a concise review. Coatings 2021, 11, 972.
5. Li, J.; Zhao, K.; Ren, L.; et al. Microstructure evolution, mechanical properties, and strengthening mechanisms of heat-resistant Al-based composite reinforced by a combination of AlN and TiN particles. J Mater Res Technol 2023, 24, 5628-41.
6. Shorowordi, K.; Laoui, T.; Haseeb, A.; Celis, J.; Froyen, L. Microstructure and interface characteristics of B4C, SiC and Al2O3 reinforced Al matrix composites: a comparative study. J Mater Process Technol 2003, 142, 738-43.
7. Xiu, Z.; Yang, W.; Chen, G.; Jiang, L.; Ma, K.; Wu, G. Microstructure and tensile properties of Si3N4p/2024Al composite fabricated by pressure infiltration method. Mater Des 2012, 33, 350-5.
8. Bian, Y.; Gao, T.; Liu, L.; Liu, G.; Liu, X. Liquid-solid reaction mechanism in Al-ZrO2(-B2O3) system and the preparation of (α-Al2O3+ZrB2/ZrAl3)/Al composites. J Alloys Compd 2020, 842, 155926.
9. Yi, M.; Zhang, P.; Yang, C.; et al. Improving creep resistance of Al-12 wt.% Ce alloy by microalloying with Sc. Scr Mater 2021, 198, 113838.
10. Zhang, M.; Lewis, R.; Gibeling, J. Mechanisms of creep deformation in a rapidly solidified Al-Fe-V-Si alloy. Mater Sci Eng A 2021, 805, 140796.
11. Knipling, K. E. Core/triple shell precipitates in Al-Er-Sc-Zr-(V,Nb,Ta) alloys. Microsc Microanal 2018, 24, 2204-5.
12. Nasim, W.; Yazdi, S.; Santamarta, R.; et al. Structure and growth of core-shell nanoprecipitates in Al-Er-Sc-Zr-V-Si high-temperature alloys. J Mater Sci 2019, 54, 1857-71.
13. Yang, C.; Cao, L.; Gao, Y.; et al. Nanostructural Sc-based hierarchy to improve the creep resistance of Al-Cu alloys. Mater Des 2020, 186, 108309.
14. Poplawsky, J. D.; Michi, R. A.; Allard, L. F.; Bahl, S.; Plotkowski, A. J.; Shyam, A. Using θ′ interfaces as templates for planar L12 precipitation in AlCuMnZr alloys. Addit Manuf Lett 2022, 3, 100086.
15. Wang, W.; Pan, Q.; Lin, G.; et al. Internal friction and heat resistance of Al, Al-Ce, Al-Ce-Zr and Al-Ce-(Sc)-(Y) aluminum alloys with high strength and high electrical conductivity. J Mater Res Technol 2021, 14, 1255-74.
16. Lu, Q.; Wang, J.; Li, H.; et al. Synergy of multiple precipitate/matrix interface structures for a heat resistant high-strength Al alloy. Nat Commun 2023, 14, 2959.
17. Poplawsky, J. D.; Milligan, B. K.; Allard, L. F.; et al. The synergistic role of Mn and Zr/Ti in producing θ′/L12 co-precipitates in Al-Cu alloys. Acta Mater 2020, 194, 577-86.
18. Akopyan, T.; Belov, N.; Letyagin, N.; Sviridova, T.; Cherkasov, S. New quaternary eutectic Al-Cu-Ca-Si system for designing precipitation hardening alloys. J Alloys Compd 2024, 993, 174695.
19. Tian, W.; Hu, M.; Chen, X.; et al. Effect of Ce addition on microstructure, mechanical properties and corrosion behavior of Al-Cu-Mn-Mg-Fe alloy. Mater Res Express 2020, 7, 036532.
20. Barkov, M. V.; Mamzurina, O. I.; Glavatskikh, M. V.; Barkov, R. Y.; Pozdniakov, A. V. Structure and properties of Al-Cu-Yb Alloy with iron and silicon impurities. Russ J Non-ferrous Met 2022, 63, 434-40.
21. Xue, H.; Yang, C.; De, Geuser. F.; et al. Highly stable coherent nanoprecipitates via diffusion-dominated solute uptake and interstitial ordering. Nat Mater 2023, 22, 434-41.
22. Mei, Z.; Liu, Z.; Bai, S.; Wang, J.; Cao, J. Effects of yttrium additions on microstructures and mechanical properties of cast Al-Cu-Mg-Ag alloys. J Alloys Compd 2021, 870, 159435.
23. Xie, H.; Zhao, J.; Cao, J.; et al. Effect of minor Er additions on the microstructures and mechanical properties of cast Al-Cu-Mg-Ag alloys. Materials 2021, 14, 4212.
24. Felner, I.; Nowik, I. Magnetism and hyperfine interactions of 57Fe, 151Eu, 155Gd, 161Dy, 166Er and 170Yb in RM4Al8 compounds (R = rare earth or Y, M = Cr, Mn, Fe, Cu). J Phys Chem Solids 1979, 40, 1035-44.
25. Li, J.; Zhang, Y.; Cao, X.; et al. Accelerated discovery of high-strength aluminum alloys by machine learning. Commun Mater 2020, 1, 74.
26. Wang, S.; Yang, X.; Wang, J.; Zhang, C.; Xue, C. Identifying the crystal structure of T1 precipitates in Al-Li-Cu alloys by ab initio calculations and HAADF-STEM imaging. J Mater Sci Technol 2023, 133, 41-57.
27. Xue, B.; Xiao, W.; Li, X.; et al. Comprehensive investigation on the structural, electronic and mechanical properties of T-Mg32(Al, Zn)49 phases in Al-Mg-Zn alloys. J Mater Sci Technol 2024, 173, 237-46.
28. Kang, Y.; Chen, N.; Shen, J. Atomistic simulation of the lattice constants and lattice vibrations in RT4Al8 (R = Nd, Sm; T = Cr, Mn, Cu, Fe). J Alloys Compd 2003, 352, 26-33.
29. Yang, W.; Pang, M.; Tan, Y.; Zhan, Y. A comparative first-principles study on electronic structures and mechanical properties of ternary intermetallic compounds Al8Cr4Y and Al8Cu4Y: pressure and tension effects. J Phys Chem Solids 2016, 98, 298-308.
30. Guo, Y.; Wang, Y.; Chen, H.; Xu, H.; Hu, M.; Ji, Z. First-principles study on stability, electronic, mechanical and thermodynamic properties of Al-Cu-RE ternary compounds. Solid State Commun 2019, 287, 63-7.
31. Zarechnyuk, O. S. Ternary compounds with a thmn12 superstructure in the systems yttrium-transition metal-aluminium. Dopovidi Akad. Nauk Ukr. RSR 1966, 6, 767. (in Russian) https://www.osti.gov/biblio/4517568 (accessed 2026-04-13).
32. Knowles, K. M.; Stobbs, W. M. The structure of {111} age-hardening precipitates in Al-Cu-Mg-Ag alloys. Acta Crystallogr B Struct Sci 1988, 44, 207-27.
33. Sun, L.; Irving, D. L.; Zikry, M. A.; Brenner, D. First-principles investigation of the structure and synergistic chemical bonding of Ag and Mg at the Al|Ω interface in a Al-Cu-Mg-Ag alloy. Acta Mater 2009, 57, 3522-8.
34. Kang, S. J.; Kim, Y.; Kim, M.; Zuo, J. Determination of interfacial atomic structure, misfits and energetics of Ω phase in Al-Cu-Mg-Ag alloy. Acta Mater 2014, 81, 501-11.
35. Hirel, P. Atomsk: a tool for manipulating and converting atomic data files. Comput Phys Commun 2015, 197, 212-9.
36. Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci 1996, 6, 15-50.
37. Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys Rev B Condens Matter 1994, 49, 14251-69.
38. Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys Rev Lett 1996, 77, 3865-8.
39. Manz, T. A.; Limas, N. G. Introducing DDEC6 atomic population analysis: part 1. Charge partitioning theory and methodology. RSC Adv 2016, 6, 47771-801.
40. Limas, N. G.; Manz, T. A. Introducing DDEC6 atomic population analysis: part 2. Computed results for a wide range of periodic and nonperiodic materials. RSC Adv 2016, 6, 45727-47.
41. Manz, T. A. Introducing DDEC6 atomic population analysis: part 3. Comprehensive method to compute bond orders. RSC Adv 2017, 7, 45552-81.
42. Ong, S. P.; Cholia, S.; Jain, A.; et al. The materials Application Programming Interface (API): a simple, flexible and efficient API for materials data based on REpresentational State Transfer (REST) principles. Comput Mater Sci 2015, 97, 209-15.
43. Ong, S. P.; Wang, L.; Kang, B.; Ceder, G. Li-Fe-P-O2 phase diagram from first principles calculations. Chem Mater 2008, 20, 1798-807.
44. Ong, S. P.; Jain, A.; Hautier, G.; Kang, B.; Ceder, G. Thermal stabilities of delithiated olivine MPO4 (M = Fe, Mn) cathodes investigated using first principles calculations. Electrochem Commun 2010, 12, 427-30.
45. Beck, H. P.; Zhou, M.; Hasanovic, P.; Gießelmann, E.; Springborg, M. Course on the use of DFT calculations to improve understanding of phase diagrams in solid-state chemistry. J Chem Educ 2021, 98, 3207-17.
46. Koza, J. Genetic programming as a means for programming computers by natural selection. Stat Comput 1994, 4, BF00175355.
47. Wang, Y.; Wagner, N.; Rondinelli, J. M. Symbolic regression in materials science. MRS Communications 2019, 9, 793-805.
48. La, Cava. W.; Burlacu, B.; Virgolin, M.; et al. Contemporary symbolic regression methods and their relative performance. Adv Neural Inf Process Syst 2021, 2021, 1-16.
49. Ward, L.; Agrawal, A.; Choudhary, A.; Wolverton, C. A general-purpose machine learning framework for predicting properties of inorganic materials. npj Comput Mater 2016, 2, 16028.
50. Ward, L.; Dunn, A.; Faghaninia, A.; et al. Matminer: an open source toolkit for materials data mining. Comput Mater Sci 2018, 152, 60-9.
51. Li, J.; Cheng, K.; Wang, S.; et al. Feature selection: a data perspective. ACM Comput Surv 2017, 50, 1-45.
52. Cai, J.; Luo, J.; Wang, S.; Yang, S. Feature selection in machine learning: a new perspective. Neurocomputing 2018, 300, 70-9.
53. Pearson, K. Note on regression and inheritance in the case of two parents. Proc R Soc London 1895, 58, 240-2.
54. Furlanello, C.; Serafini, M.; Merler, S.; Jurman, G. An accelerated procedure for recursive feature ranking on microarray data. Neural Netw 2003, 16, 641-8.
55. Pedregosa, F.; Varoquaux, G.; Gramfort, A.; et al. Scikit-learn: machine learning in python. J. Mach. Learn. Res. 2011, 12, 2825-30. https://www.jmlr.org/papers/volume12/pedregosa11a/pedregosa11a.pdf?source=post_page (accessed 2026-04-13).
57. Muthyala, M.; Sorourifar, F.; Paulson, J. A. TorchSISSO: a PyTorch-based implementation of the sure independence screening and sparsifying operator for efficient and interpretable model discovery. Digit Chem Eng 2024, 13, 100198.
58. Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J Appl Crystallogr 2011, 44, 1272-6.
59. Xue, H.; Yang, C.; Zhang, P.; Wu, S. H.; Liu, G.; Sun, J. Heat-resistant Al alloys: microstructural design and microalloying effect. J Mater Sci 2024, 59, 9749-67.
60. Zhang, D.; Liu, Y. Concise derivation of formulas for calculating the bonding ability of hybrid orbitals and the angle between hybrid orbitals. Daxue Huaxue 2022, 0, 202208064-0.
61. Yang, S.; Wilson, N.; Nie, J. Revisit of the structure of Ω precipitate in Al-Cu-Mg-Ag alloys. Scripta Materialia 2021, 205, 114204.
62. Reich, L.; Murayama, M.; Hono, K. Evolution of Ω phase in an Al-Cu-Mg-Ag alloy - a three-dimensional atom probe study. Acta Mater 1998, 46, 6053-62.
63. Lu, Q.; Hu, J.; Yang, T.; et al. Revisiting the effect of Ag additions on Ω precipitation and heat resistance of Al-Cu-Mg-Si-Ag alloys. Mater Sci Eng A 2023, 885, 145539.
64. Kharakterova, M. L. Phase composition of aluminum-copper-scandium alloys at 450 and 500 °C. Metally 1991, 4, 191-4. https://inis.iaea.org/records/zt3px-z3342 (accessed 2026-04-13).
65. Norman, A. F.; Prangnell, P. B.; Mcewen, R. S. The solidification behaviour of dilute aluminium-scandium alloys. Acta Mater 1998, 46, 5715-32.
66. Kairy, S.; Rouxel, B.; Dumbre, J.; et al. Simultaneous improvement in corrosion resistance and hardness of a model 2xxx series Al-Cu alloy with the microstructural variation caused by Sc and Zr additions. Corros Sci 2019, 158, 108095.
67. Tang, Y.; Xiao, D.; Huang, L.; et al. Effect of minor Sc addition on the microstructure evolution of Al-Cu-Li-Mg alloy during homogenization with different cooling modes. Met Mater Int 2022, 28, 2422-33.
68. Qin, J.; Dai, W.; Ren, X.; Liu, Z.; Wang, B. Investigation of W phase crystal structure and evolution mechanism based on Al-Cu-Sc alloy during homogenization. Mater Charact 2024, 207, 113536.
69. Emery, A. A.; Wolverton, C. High-throughput DFT calculations of formation energy, stability and oxygen vacancy formation energy of ABO3 perovskites. Sci Data 2017, 4, 170153.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].