


Machine learning-assisted design of carbon nanotube-based single-atom catalysts for hydrogen evolution reaction
 0
0 Abstract
Sustainable hydrogen energy offers a promising solution to the growing global energy demand associated with fossil fuel consumption. The development of efficient electrocatalysts for the hydrogen evolution reaction (HER) is important, yet the high computational cost of density functional theory (DFT) limits the rapid screening of candidate materials. In this work, a machine learning-assisted framework integrated with DFT calculations is proposed to systematically investigate the HER performance of carbon nanotube (CNT)-supported single-atom catalysts (SACs). A dataset consisting of Gibbs free energy of hydrogen adsorption (ΔGH*) was constructed from DFT calculations, including 84 M-N4-CNT(n, n) models involving 28 transition-metal centers anchored on CNTs with three different chirality indices. Based on selected intrinsic transition-metal features and the CNT chirality index, a random forest regression (RFR) model was identified as the optimal model after comparison with multiple machine learning algorithms for predicting ΔGH*. The RFR model exhibited excellent predictive accuracy, achieving a coefficient of determination (R2) of 0.98 on the test set. Notably, when applied to previously unseen M-N4-CNT(7, 7) structures, the model maintained high reliability (R2 = 0.96), demonstrating strong generalization capability. Machine learning identified Fe-N4-CNT(7, 7) as a highly promising HER electrocatalyst, with further DFT-based kinetic analysis showing that it follows a Volmer-Tafel reaction pathway. In addition, the SISSO algorithm was employed to derive an interpretable descriptor for ΔGH* based on elemental properties, achieving high fitting accuracy across different chirality indices. This descriptor provides an efficient tool for rapid catalyst screening while offering mechanistic insights into the key factors governing HER activity in M-N4-CNT systems.
Keywords
INTRODUCTION
Hydrogen has emerged as a key clean energy vector in future energy systems, owing to its zero carbon emissions and high energy density[1-3]. Electrochemical water splitting offers an ideal route for sustainable hydrogen production, in which the performance of hydrogen evolution reaction (HER) catalysts plays an important role[4]. Density functional theory (DFT), as a first-principles-based simulation approach, enables the rational design of high-performance HER catalysts at the atomic scale[5]. By calculating key parameters such as the Gibbs free energy of hydrogen adsorption (ΔGH*), DFT provides a powerful descriptor for predicting catalytic activity[6]. This has led to the successful identification of numerous highly efficient materials, including Pt-based alloys, transition metal dichalcogenides and single-atom catalysts[7-10]. Furthermore, DFT calculations reveal how defect introduction, heteroatom doping, or strain modulation affect catalytic behavior, offering insightful guidance for experimental optimization[11-13]. Nowadays, DFT calculations are increasingly focused on simulating more realistic electrochemical environments, such as interfacial electric double layers and solvation effects, to provide more practical guidance[14,15]. Among the emerging catalysts, single-atom catalysts (SACs) have attracted considerable attention due to their maximized atomic utilization, distinct electronic structures and tunable coordination environments, offering a promising platform for enhancing HER performance[16]. However, due to the diversity of environmental coordination, active site composition and reactant properties, screening single-atom HER catalysts through DFT calculations requires substantial computational resources and time.
Machine learning (ML), an efficient data-driven method, is increasingly being used to rapidly screen HER catalysts and OER/ORR with outstanding electrocatalytic performance[17-19]. Random forest regression (RFR) was used to decode key structural features from high-throughput DFT data, leading to the successful identification of biaxially strained Au-doped MoSe2 with a low overpotential of 30 mV at 10 mA cm-2[20]. Furthermore, various deep learning algorithms have been introduced to address more complex feature relationships and high-dimensional data[21]. As an example, to aid the design of complex high-entropy intermetallic compounds, Wang et al. employed a deep neural network (DNN)-based machine learning model to develop an ordered structure-activity prediction framework[22]. This framework allows for the high-throughput prediction of hydrogen adsorption energy across 20,000 microstructures for each composition. Besides regression, ML interatomic potentials (MLIPs) also enable efficient DFT-level structural relaxations, facilitating the high-throughput screening of catalysts[23,24]. For instance, Kum and Kim applied an MLIP to identify promising HER candidates, revealing key stability descriptors such as significant charge transfer and specific coordination motifs[25]. However, major issues for applying ML in catalysts design are the lack of a universal ML algorithm, a consistent database, and appropriate input features.
In this study, DFT calculations combined with machine learning techniques were employed to systematically investigate nitrogen-coordinated transition-metal single-atom catalysts supported on carbon nanotubes (CNTs). A representative dataset of M-N4-CNT(n, n) structures with different metal centers and CNT chiralities was constructed, and ΔGH* was adopted as the key descriptor for evaluating HER activity. By comparing several machine-learning algorithms, RFR was established to accurately capture the feature-activity relationships of these catalysts. The predictive capability of the model was further assessed by extrapolation to CNTs with larger diameters, enabling the identification of promising catalyst candidates. In addition, kinetic analyses under explicit solvent conditions were conducted to gain mechanistic insights into the HER process. Furthermore, the Sure Independence Screening and Sparsifying Operator (SISSO) method was applied to derive a low-dimensional descriptor from basic elemental properties, providing an interpretable framework for efficient catalyst screening and mechanistic understanding.
METHODS
DFT calculations
In this work, all DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP 6.3.2). The projector augmented wave (PAW) method was employed to describe the ion-electron interactions, with a plane-wave cutoff energy set to 500 eV[26,27]. For the exchange-correlation functional, the Perdew-Burke-Ernzerhof (PBE) formulation under the generalized gradient approximation (GGA) framework was adopted. The electronic structure was modeled with integration over the Brillouin zone sampled using a 3 × 3 × 1 Monkhorst-Pack k-point grid[28]. To ensure the accuracy of the surface model, a vacuum layer with a thickness of 18 Å was introduced along the non-periodic direction to minimize interactions between periodic images. The energy convergence criterion for geometry optimization was set to 10-5 eV, and the force convergence threshold was -0.02 eV/Å. Transition states were identified with the climbing-image nudged elastic band (CI-NEB) method[29,30], with forces on the climbing images converged to -0.03 eV/Å and an energy convergence criterion of 10-6 eV. All other parameters were the same as those used in the above calculations. The identity of each transition state was confirmed by vibrational frequency analysis, which revealed a single imaginary frequency. Ab initio molecular dynamics (AIMD) simulations were carried out using a Nosé-Hoover thermostat to maintain the temperature at 300 K with a time step of 1 fs[31,32]. All AIMD simulations were performed using Γ-point sampling of the Brillouin zone, and the POMASS value for H was set to 2.
The hydrogen evolution reaction (HER) proceeds as follows[33]:
Under standard conditions, the chemical potential of H+/e- equals half the chemical potential of H2. The Gibbs free energy of atomic hydrogen adsorption (ΔGH*)[33] can be calculated by:
where ΔEH* is the hydrogen adsorption energy, and ΔEZPE and ΔSH* represent the differences in zero-point energy and entropy, respectively, between adsorbed hydrogen and gaseous hydrogen under standard conditions. Solvent effects on the surface and adsorbates are described using explicit solvation models. Here, explicit water layers are modelled by 20 H2O molecules surrounding the adsorbate and cation.
The theoretical exchange current density at pH = 0 was calculated in accordance with Norskov’s assumption[34], using the following equation:
Here, k0, kb and T represent the proton transfer rate constant, Boltzmann constant and environment temperature, respectively. And, k0 and T were set to 200 s-1·site-1 and 298.15 K, respectively.
Machine learning
All programming tasks were carried out in a Python 3.8 environment using Jupyter Notebook[35]. The Pearson correlation coefficient (r)[36] was employed to quantify the linear relationships between the features, as calculated in Equation 4:
where xi and yi are the compared features,
Gradient Boosting Regression (GBR) and Linear Regression (LR) were implemented using the scikit-learn library[37]. Convolutional Neural Networks (CNN) and Multilayer Perceptrons (MLP) models were developed using the PyTorch deep learning framework[38]. The dataset was divided into a training set and a test set, with 80% of the data used for training and the remaining 20% reserved for testing. The performance of the machine learning models was evaluated using the coefficient of determination (R2)[39] and the root mean square error (RMSE)[40], as calculated in Equations 5 and 6, respectively:
where
RESULTS AND DISCUSSION
To accelerate the discovery of HER electrocatalysts, a machine learning-assisted workflow was employed to screen potential M-N4-CNT(n, n)-based materials. As illustrated in Figure 1, this workflow integrates DFT calculations, data collection, feature engineering, machine learning model construction and application, and descriptor extraction.
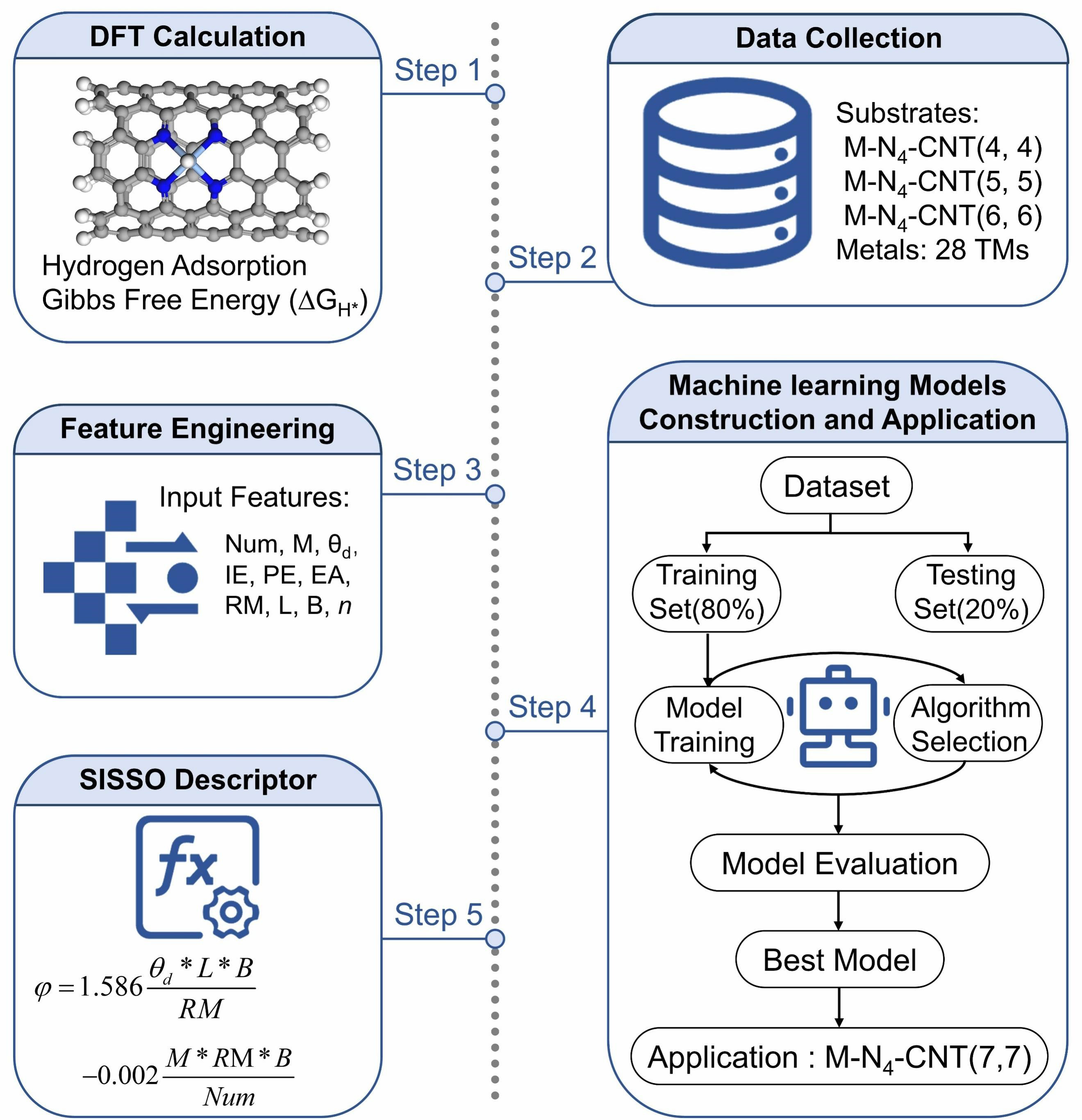
Figure 1. Flowchart of machine learning framework for HER studies on M-N4-CNT(n, n) catalysts. Here, Num, M, θd, PE, IE, EA, RM, L, and B denote the atomic number, atomic mass, d-electron count, Pauling electronegativity, first ionization energy, electron affinity, covalent radius, period, and group number of the transition metal, respectively, while n represents the carbon nanotube chirality index. DFT: Density functional theory; Num: atomic number; M: atomic mass; θd: d-electron count; PE: Pauling electronegativity; IE: first ionization energy; EA: electron affinity; RM: covalent radius; L: period; B: group number; SISSO: Sure Independence Screening and Sparsifying Operator; HER: hydrogen evolution reaction; CNT: carbon nanotube.
M-N4-CNT(n, n) (n = 4, 5, 6) catalyst structures were constructed by embedding transition metal atoms into hydrogen-terminated armchair carbon nanotubes, following the method of Qin et al. [Supplementary Figure 1 and Figure 2A][41]. A database of 84 catalysts was systematically constructed by combining 28 transition metals with three different nanotube substrates [Figure 2B]. The ∆GH* is widely recognized as a suitable descriptor for evaluating HER activity, as it reflects the binding strength of hydrogen atoms on the catalyst surface[42]. According to the Sabatier principle, an excessively strong ∆GH* impedes hydrogen desorption, while an overly weak value indicates difficulty in the initial hydrogen adsorption step[43]. Optimal HER performance is achieved when ∆GH* is close to 0 eV, which ensures an efficient balance between the proton/electron transfer steps and the subsequent hydrogen desorption process[44,45].
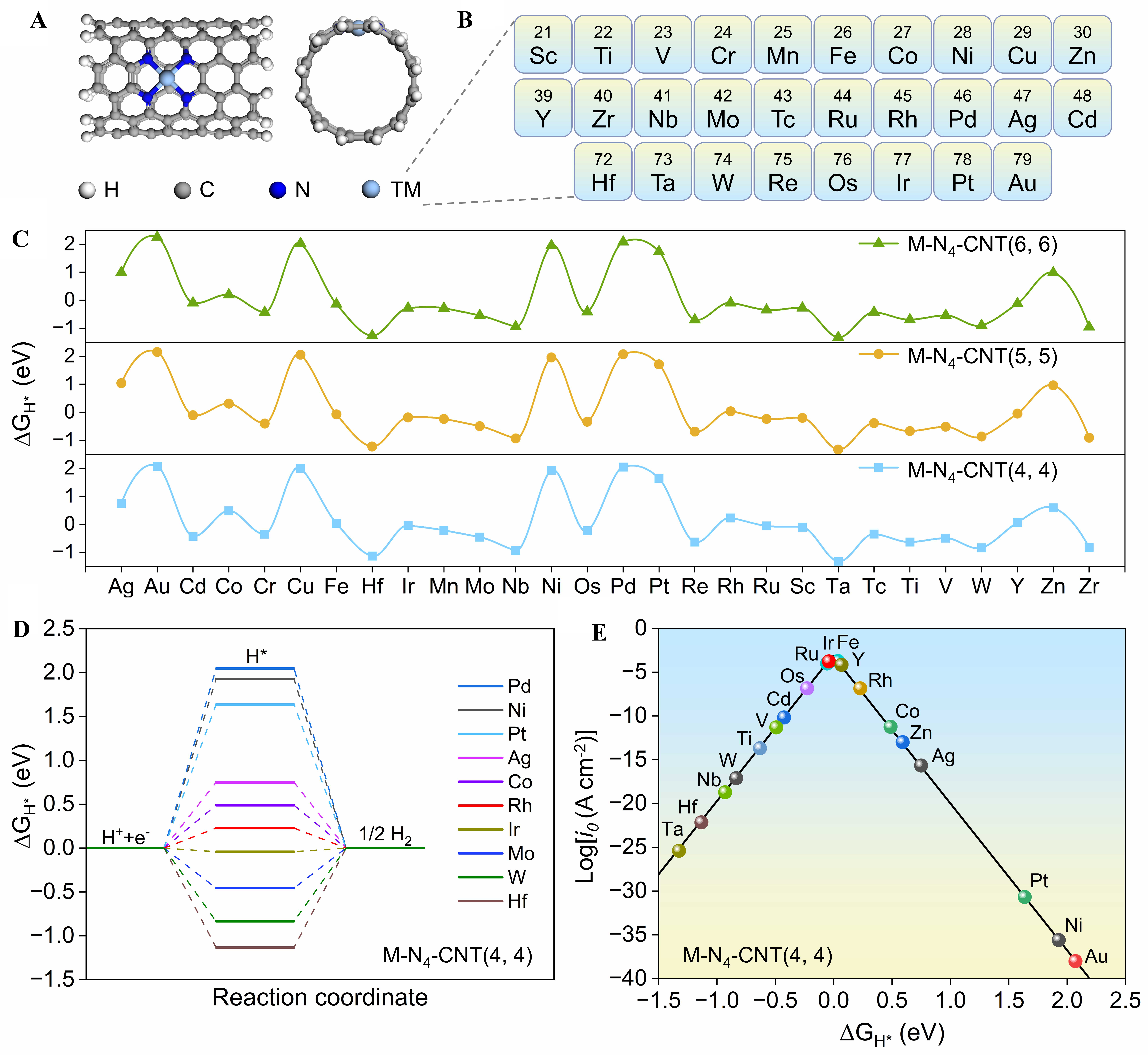
Figure 2. (A) Top and side views of M-N4-CNT(6, 6); (B) The 28 transition metal atoms considered in this study; (C) Comparison of the ∆GH* values for M-N4-CNT(4, 4), M-N4-CNT(5, 5) and M-N4-CNT(6, 6); (D) Gibbs free energy diagram of the HER on M-N4-CNT(4, 4); (E) Volcano plot of the exchange current density as a function of the ∆GH* on M-N4-CNT(4, 4). ∆GH*: Gibbs free energy of hydrogen adsorption; CNT: carbon nanotube; HER: hydrogen evolution reaction.
As illustrated in Figure 2C, the ΔGH* values were systematically evaluated for M-N4-CNT(n, n) (n = 4, 5, 6). The results reveal a consistent trend of ΔGH* across the metal series, with values ranging from -1.4 to 2.3 eV. In most M-N4-CNT(n, n) (M = Co, Mn, Mo, etc.) systems, the ΔGH* decreases as the radius of curvature increases. The Gibbs free energy diagrams for the HER on M-N4-CNT(4, 4), M-N4-CNT(5, 5), and M-N4-CNT(6, 6) are shown in Figure 2D and Supplementary Figure 2. Iridium exhibits highly desirable ΔGH* values across all three carbon nanotube substrates. In particular, Ir-N4-CNT(4, 4) shows a ΔGH* value of only -0.04 eV. To quantitatively describe the catalytic activity, the relationship between ΔGH* and the exchange current (i0) was investigated. For M-N4-CNT(4, 4) (M = Ir, Ru, Fe, Y, Rh), the points corresponding to these metals are located near the peak of the volcano plot [Figure 2E], indicating relatively high exchange current densities and thus superior HER performance. A similar trend of elements is observed in the volcano plots of exchange current density for hydrogen adsorption on M-N4-CNT(5, 5) [Supplementary Figure 3A] and M-N4-CNT(6, 6) [Supplementary Figure 3B].
To construct a machine learning model for predicting the HER performance of M-N4-CNT(n, n) catalysts, ΔGH* was selected as the target output, as it serves as a key descriptor of hydrogen adsorption behavior. A total of ten input features were initially considered, including nine intrinsic properties of the transition metal - atomic number (Num), atomic mass (M), d-electron count (θd), Pauling electronegativity (PE), first ionization energy (IE), electron affinity (EA), covalent radius (RM), period (L), and group number (B). In addition, the carbon nanotube chirality index (n) was included as a structural descriptor. The Pearson correlation coefficient was employed to quantify the strength and direction of linear relationships between the input features and the target variable, as well as the intercorrelations among the features. This coefficient ranges from -1 to 1, where -1 indicates a perfect negative linear correlation, 0 represents no linear correlation, and 1 corresponds to a perfect positive linear correlation[46,47]. As shown in Supplementary Figure 4, the Pearson correlation matrix for all input features and ΔGH* is presented. In the matrix, color intensity reflects the magnitude of the correlation, with yellow and blue denoting positive and negative correlations, respectively, and darker shades of either color indicating stronger correlations. With respect to correlations with the target output ΔGH*, θd exhibits the highest positive correlation coefficient (r = 0.76), indicating that the electronic configuration of the transition metal plays a dominant role in determining hydrogen adsorption behavior. B also shows a strong positive correlation with ΔGH* (r = 0.72), while RM displays a pronounced negative correlation (r = -0.62). Analysis of inter-feature correlations reveals several cases of strong collinearity. Notably, Num and L are highly correlated (r = 0.98), reflecting the intrinsic periodic trends of the periodic table. Similarly, θd and B show an extremely strong correlation (r = 0.98), consistent with the systematic variation of d-electron filling across transition-metal groups. To reduce feature redundancy and improve model robustness, Num and B were removed from the input feature set. L was retained despite its high correlation with M because it provides distinct physical information related to the period and slightly improves model performance. After feature selection, the refined input features consist of M, θd, PE, IE, EA, RM, L, and n [Figure 3A].
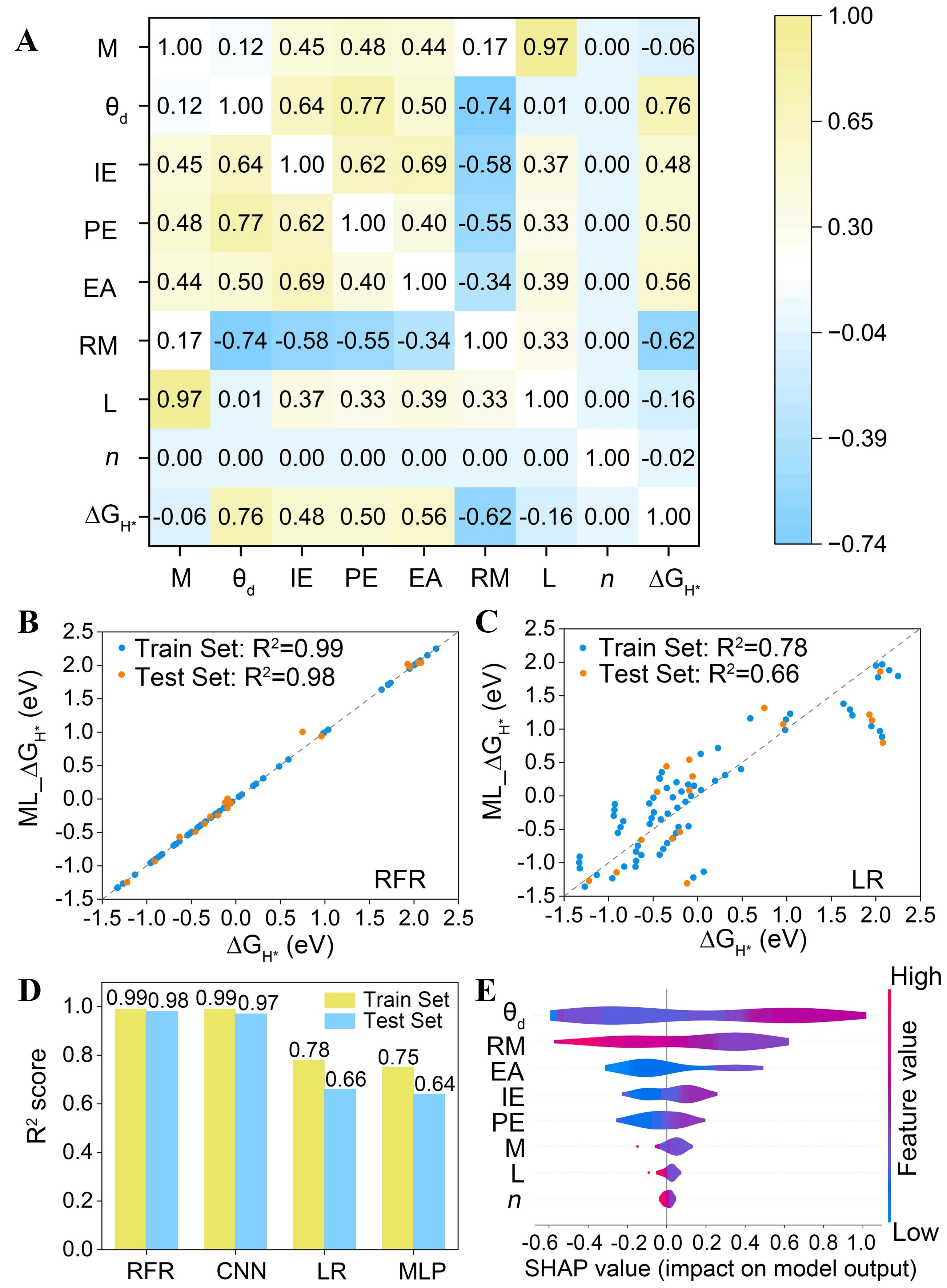
Figure 3. (A) Heatmap of Pearson correlation coefficients between the selected features and ∆GH*; (B) Comparison of the ∆GH* values predicted by the RFR model versus the DFT calculated values; (C) Comparison of the ∆GH* values predicted by the LR model versus the DFT calculated values; (D) R2 for different models; (E) Density map of SHAP values for all features. M: Atomic mass; θd: d-electron count; PE: Pauling electronegativity; IE: first ionization energy; EA: electron affinity; RM: covalent radius; L: period; n: carbon nanotube chirality index; ∆GH*: Gibbs free energy of hydrogen adsorption; R2: coefficient of determination; ML: machine learning; RFR: Random Forest Regression; LR: Linear Regression; SHAP: SHapley Additive exPlanations; CNN: Convolutional Neural Networks; MLP: Multilayer Perceptrons; DFT: density functional theory.
Several machine learning regression models, including Random Forest Regression (RFR), Linear Regression (LR), Convolutional Neural Networks (CNN) and Multilayer Perceptrons (MLP), were employed to establish feature-target relationships and predict ∆GH*. Detailed hyperparameters of the four models are provided in Supplementary Table 1. The dataset was randomly divided into a training set (80%) and a testing set (20%) to ensure reliable model training and an unbiased performance evaluation. A comparison between the ground truth ΔGH* values and the predictions obtained from the RFR, CNN, LR, and MLP models is presented in Figure 3B and C and Supplementary Figure 5, respectively. The predictive performance of each model was evaluated using the R2 and the RMSE. The R2 value reflects the goodness of fit, with values closer to unity indicating better agreement between predicted and actual values. The RMSE measures the average prediction error, with lower values corresponding to higher prediction accuracy. The R2 and RMSE values for all models are summarized in Figure 3D and Supplementary Figure 6. Among the four models, RFR exhibited the best predictive performance, achieving an R2 of 0.99 on the training set and 0.98 on the testing set, along with low RMSE values of 0.04 eV and 0.10 eV, respectively. The CNN model also demonstrated strong predictive capability, yielding an R2 of 0.99 for training and 0.97 for testing, which is comparable to that of RFR. In contrast, both LR and MLP showed significantly inferior performance. Specifically, LR achieved an R2 of 0.78 with an RMSE of 0.46 eV in the training set and an R2 of 0.66 with an RMSE of 0.61 eV in the testing set. Similarly, MLP yielded an R2 of 0.75 (RMSE = 0.49 eV) for training and an R2 of 0.64 (RMSE = 0.64 eV) for testing, making it the least accurate model among the four candidates. Overall, these results demonstrate that the RFR model is capable of accurately predicting ∆GH* for M-N4-CNT(n, n) catalysts and outperforms the other machine learning algorithms considered in this study. SHapley Additive exPlanations (SHAP) analysis was conducted to further elucidate the predictive mechanism of the RFR model and identify the key descriptors governing ∆GH*, as shown in Figure 3E. The SHAP analysis indicates that the θd is the most influential descriptor, followed by RM, highlighting the significant roles of transition-metal electronic structure and atomic size in governing hydrogen adsorption.
The trained RFR model was subsequently applied to predict ΔGH* values for M-N4-CNT(7, 7) structures, with the corresponding top and side views of the computational model [Supplementary Figure 7]. Figure 4A displays a diagonal scatter plot comparing the RFR-model-predicted values with those from direct DFT calculations. The majority of data points lie closely along the diagonal, indicating a strong linear correlation between the predicted and DFT-calculated results. The RFR model accurately replicates the DFT calculations, as evidenced by a high R2 of 0.96. Notably, the machine learning model completes ΔGH* predictions for all M-N4-CNT(7, 7) systems within one minute, drastically reducing computational cost and time compared to conventional DFT approaches. Supplementary Figures 8 and 9 present a direct comparison of the HER Gibbs free energy diagrams from DFT and machine-learning predictions for different metal centers (e.g., Fe, Ir, Co) in M-N4-CNT(7, 7), demonstrating the high accuracy of the RFR model. This is further supported by the volcano plot in Figure 4B, which shows the exchange current density as a function of ΔGH* derived from both DFT and RFR predictions. Structures such as Fe-N4-CNT(7, 7), Ir-N4-CNT(7, 7) and Co-N4-CNT(7, 7) are located near the volcano apex, indicating hydrogen adsorption free energies close to the optimal value. Notably, the value of log(i0) based on ML_ΔGH* in the Fe-N4-CNT(7, 7) system is -6.19 A·cm-2, outperforming -6.99 A·cm-2 for Ir-N4-CNT(7, 7). Meanwhile, Supplementary Figure 10 shows that during the molecular dynamics simulations performed at 300 K for 5 ps, the total energy fluctuates around the equilibrium value, confirming the stability of the Fe-N4-CNT(7, 7) catalyst. To elucidate the dominant reaction pathway for the HER on Fe-N4-CNT(7, 7) in an explicit aqueous environment, the energy barriers for the Tafel and Heyrovsky steps were computed using the CI-NEB method. The Tafel step, which involves the recombination of two adsorbed H atoms, exhibits a barrier of 0.39 eV [Figure 4C]. In contrast, the Heyrovsky step, corresponding to the reaction between an adsorbed H atom and a solvated proton, presents a significantly higher barrier of 0.80 eV [Figure 4D]. Therefore, the HER on Fe-N4-CNT(7, 7) preferentially follows the Volmer-Tafel mechanism, as the lower kinetic barrier of the Tafel step governs the overall reaction pathway.
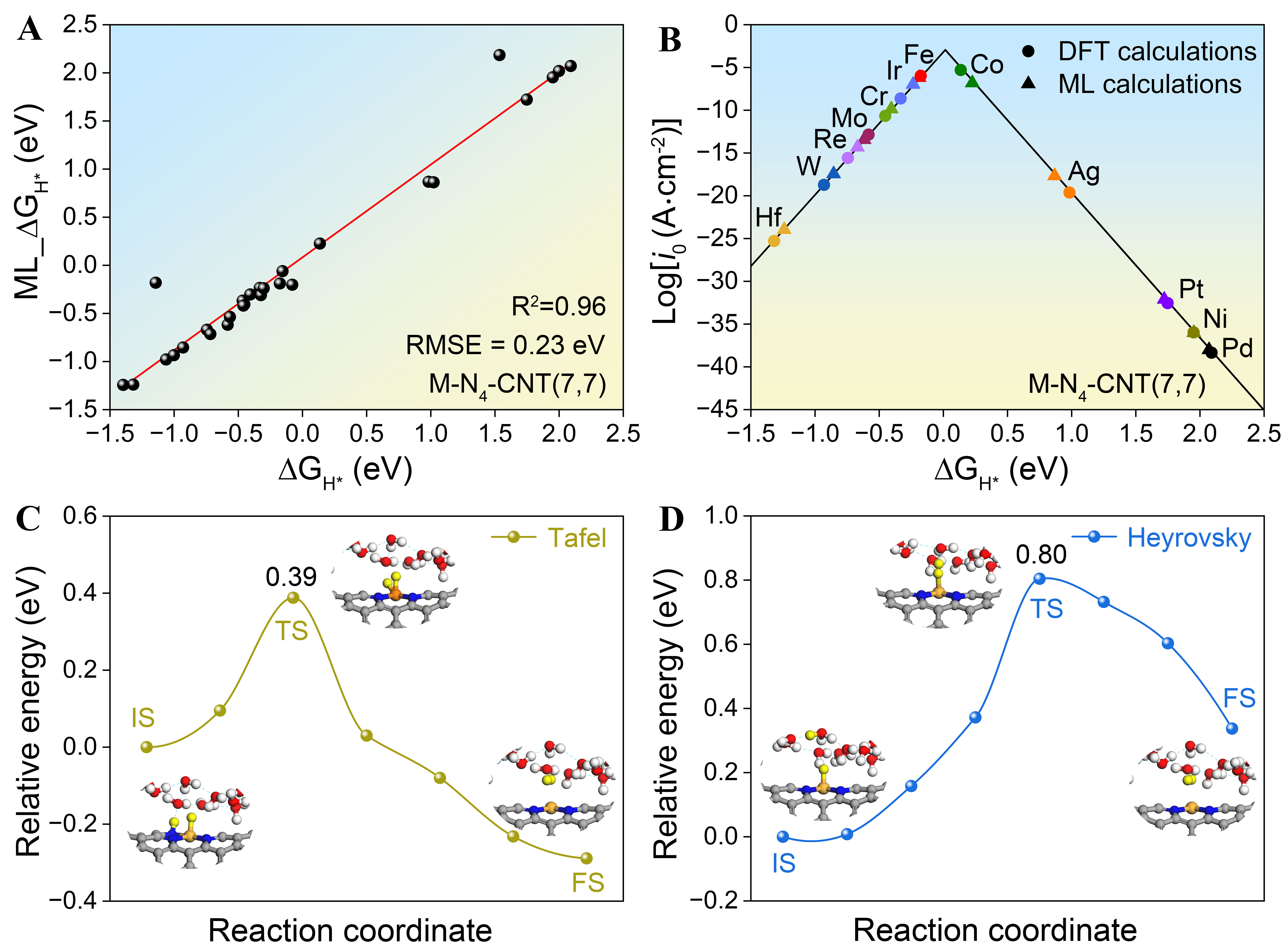
Figure 4. (A) Diagonal scatter plot of DFT-calculated versus machine learning predicted ∆GH* on M-N4-CNT(7, 7); (B) Exchange current density for DFT-calculated and machine learning predicted ∆GH* on M-N4-CNT(7, 7); (C) CI-NEB path and corresponding configurations for the Tafel mechanism on Fe-N4-CNT(7, 7) with explicit water molecules; (D) CI-NEB path and corresponding configurations for the Heyrovsky mechanism on Fe-N4-CNT(7, 7) with explicit water molecules. ML: Machine learning; ∆GH*: Gibbs free energy of hydrogen adsorption; R2: coefficient of determination; RMSE: root mean square error; CNT: carbon nanotube; DFT: density functional theory; IS: Initial State; TS: Transition State; FS: Final State; CI-NEB: climbing-image nudged elastic band.
To enhance model interpretability and reveal the underlying physical mechanisms, the SISSO algorithm was employed to construct compact and interpretable descriptors from fundamental features via iterative screening and sparse regression[48,49]. It should be noted that, in contrast to the Pearson correlation analysis, SISSO does not impose constraints on the linear independence of the input variables. Instead, SISSO aims to identify low-dimensional and physically interpretable descriptors by combining features through nonlinear combinations. Moreover, the descriptor is not necessarily unique, and alternative expressions with similar predictive performance may exist. Based on the ten previously selected input features, a descriptor φ describing ΔGH* was derived using the SISSO algorithm, and its explicit mathematical form is given in Equation 7:
Here, θd, L, B, RM, M, and Num denote the d-electron count, period number, group number, covalent radius, atomic mass, and atomic number of the transition metals, respectively. The derived descriptor φ exhibits a strong linear correlation with ΔGH* across various nanotube structures, with R2 values reaching up to 0.92 for M-N4-CNT(4, 4), (5, 5), and (6, 6) [Figure 5A-C]. Remarkably, when applied to M-N4-CNT(7,7), the descriptor still exhibits a strong correlation with ΔGH* (R2 = 0.89, Figure 5D), highlighting its generalizability across nanotube structures of different curvatures. Despite the inherent difficulty in interpreting many machine learning models, the SISSO-derived φ serves as a highly effective and practical tool for the rapid screening of ΔGH*.
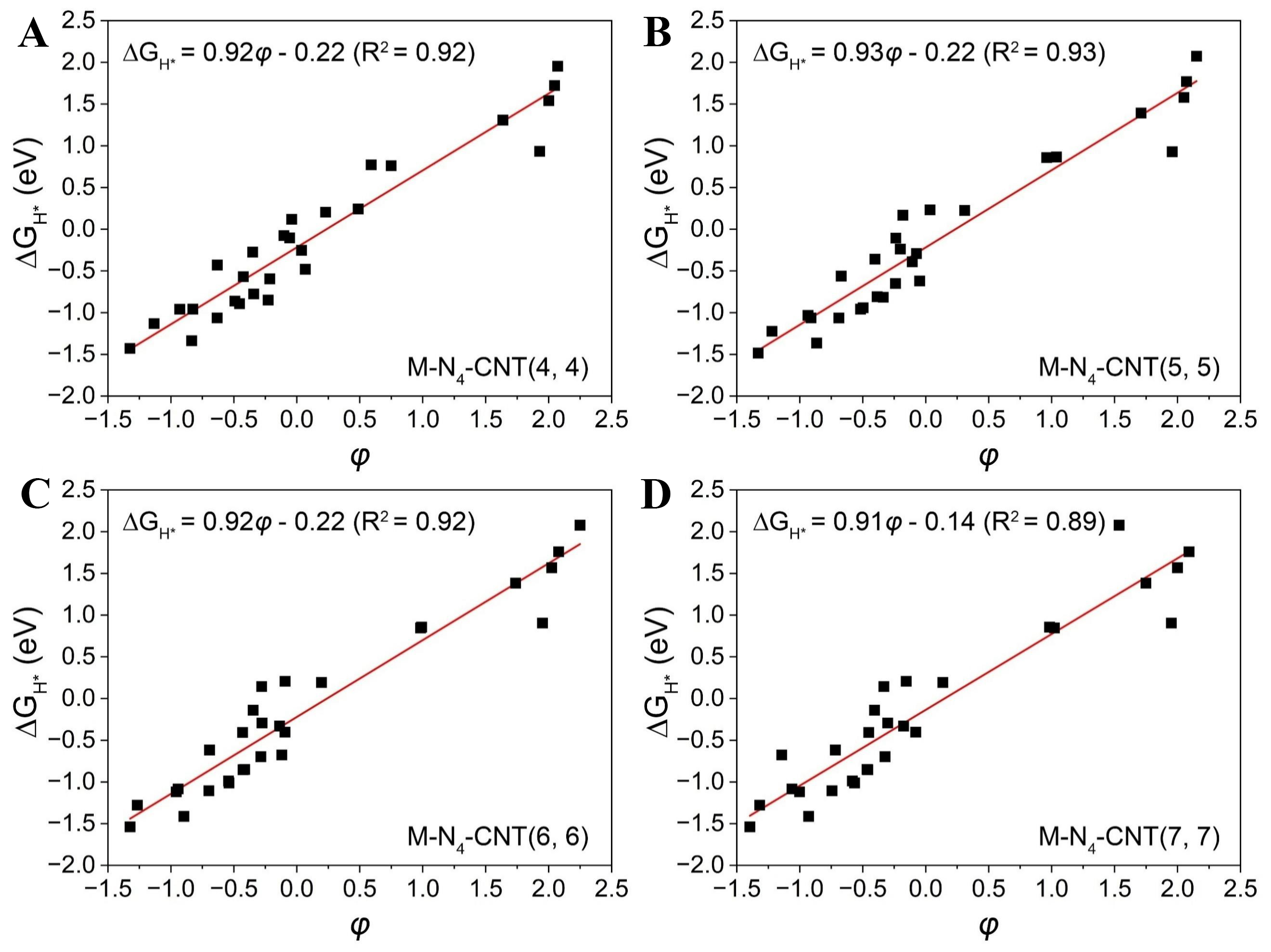
Figure 5. Relationship between ∆GH* and φ on (A) M-N4-CNT(4, 4); (B) M-N4-CNT(5, 5); (C) M-N4-CNT(6, 6) and (D) M-N4-CNT(7, 7). ∆GH*: Gibbs free energy of hydrogen adsorption; R2: coefficient of determination; CNT: carbon nanotube.
CONCLUSIONS
In summary, this work establishes a systematic computational framework that integrates DFT calculations with machine learning for the efficient screening and rational design of M-N4-CNT catalysts for the HER. A highly accurate RFR model was developed to predict ΔGH* using intrinsic properties of the transition-metal center, significantly reducing the computational cost of conventional DFT-based screening, while achieving an R2 of 0.98 on the test set and strong transferability to unseen M-N4-CNT(7,7) systems (R2 = 0.96). Based on the machine learning predictions, Fe-N4-CNT(7, 7) was identified as a highly promising HER electrocatalyst. In addition, a compact and physically interpretable SISSO-derived descriptor for ΔGH* was obtained, providing mechanistic insights into the key factors governing catalytic activity. Overall, this DFT-machine learning-descriptor framework offers a general strategy for accelerating the discovery and understanding of efficient HER electrocatalysts.
DECLARATIONS
Authors’ contributions
Conception, design, and manuscript drafting: Xue, M.
Data analysis, visualization and manuscript review & editing: Mei, Z.
Investigation, formal analysis, and supervision: Hu, C.; Tian, Z.
Revision, funding acquisition, and supervision: Ren, Y.; Liu, C.
Availability of data and materials
The data supporting the findings can be obtained from the corresponding author upon reasonable request.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
Liu, C. greatly appreciates financial provided by the National Natural Science Foundation of China (Grant no. 22473108), the foundation of China’s National Key R&D Programme (Grant no. 2023YFB3810601), and the Hundred Talents Program of CAS.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Qian, S.; Jiang, T.; Wang, J.; et al. Surface nanosteps modulate the local environment of Co single atoms to boost the electrocatalytic hydrogen evolution reaction. ACS. Catal. 2024, 14, 18690-700.
2. Yan, Y.; Yu, R.; Liu, M.; et al. General synthesis of neighboring dual-atomic sites with a specific pre-designed distance via an interfacial-fixing strategy. Nat. Commun. 2025, 16, 334.
3. Liu, C.; Li, Q.; Wu, C.; et al. Single-boron catalysts for nitrogen reduction reaction. J. Am. Chem. Soc. 2019, 141, 2884-8.
4. Li, B.; Nie, K.; Wang, K.; et al. Phase-dependent reverse electronic metal-support interaction to boost alkaline hydrogen evolution. Adv. Mater. 2026, 38, e18017.
5. Zhang, T.; Yu, Y. H.; Liu, C. W.; Qin, G. W.; Li, S. Constructing Ni4W/WO3/NF with strongly coupled interface for hydrogen evolution in alkaline media. Rare. Metals. 2023, 42, 3945-51.
6. Zhou, X.; Tamtaji, M.; Zhou, W.; Goddard Iii, W. A.; Chen, G. DFT screening of dual-atom catalysts on carbon nanotubes for enhanced oxygen reduction reaction and oxygen evolution reaction: comparing dissociative and associative mechanisms. J. Mater. Chem. A. 2024, 12, 28381-9.
7. Liu, C.; Dai, Z.; Zhang, J.; Jin, Y.; Li, D.; Sun, C. Two-dimensional boron sheets as metal-free catalysts for hydrogen evolution reaction. J. Phys. Chem. C. 2018, 122, 19051-5.
8. Chen, Z. W.; Li, J.; Ou, P.; et al. Unusual Sabatier principle on high entropy alloy catalysts for hydrogen evolution reactions. Nat. Commun. 2024, 15, 359.
9. Gong, F.; Liu, Y.; Zhao, Y.; et al. Universal sub-nanoreactor strategy for synthesis of yolk-shell MoS2 supported single atom electrocatalysts toward robust hydrogen evolution reaction. Angew. Chem. Int. Ed. Engl. 2023, 62, e202308091.
10. Jiang, B.; Zhu, J.; Xia, Z.; et al. Correlating single-atomic ruthenium interdistance with long-range interaction boosts hydrogen evolution reaction kinetics. Adv. Mater. 2024, 36, e2310699.
11. Hua, Z.; Wang, J.; Wu, X.; et al. Enhancing hydrogen evolution reaction via heterophase boundaries and stacking faults in molten salt electrodeposited Mg‐Ni alloys. Adv. Energy. Mater. 2025, 15, e03249.
12. Xiong, Y.; Li, H.; Liu, C.; et al. Single-atom Fe catalysts for Fenton-like reactions: roles of different N species. Adv. Mater. 2022, 34, e2110653.
13. Yan, Y.; Wen, B.; Liu, M.; et al. Orienting electron fillings in d orbitals of cobalt single atoms for effective zinc-air battery at a subzero temperature. Adv. Funct. Mater. 2024, 34, 2316100.
14. Tian, Y.; Huang, B.; Song, Y.; et al. Effect of ion-specific water structures at metal surfaces on hydrogen production. Nat. Commun. 2024, 15, 7834.
15. Wu, J.; Wang, X.; Zheng, W.; et al. Manipulating interfacial charge distribution for water reduction. J. Am. Chem. Soc. 2025, 147, 39181-91.
16. Jin, H.; Chen, X.; Da, Y.; et al. Identifying the bifunctional mechanism in alkaline water electrolysis by Lewis pairs at the single-atom scale. J. Am. Chem. Soc. 2025, 147, 3874-84.
17. Liu, M.; Fu, Q.; Zhong, W.; Peera, S. G.; Liu, C. Machine learning high-throughput screening of rare earth SACs with different coordination environments for the HER. Chem. Commun. (Camb). 2026, 62, 506-9.
18. Fu, Q.; Xu, T.; He, C.; Wang, D.; Liu, M.; Liu, C. Machine learning-assisted study of RENxC6-x-doped graphene as potential electrocatalysts for oxygen electrode reactions. Langmuir 2024, 40, 10726-36.
19. Fu, Q.; Xu, T.; Wang, D.; Liu, C. Rare earth modified carbon-based catalysts for oxygen electrode reactions: a machine learning assisted density functional theory investigation. Carbon 2024, 223, 119045.
20. Zhang, T.; Ye, Q.; Liu, Y.; et al. Data-driven discovery of biaxially strained single atoms array for hydrogen production. Nat. Commun. 2025, 16, 3644.
21. Yu, Q.; Ma, N.; Leung, C.; Liu, H.; Ren, Y.; Wei, Z. AI in single-atom catalysts: a review of design and applications. J. Mater. Inf. 2025, 5, 9.
22. Wang, Z.; Chen, X.; Lin, T.; et al. Machine learning-guided design of L12-type Pt-based high-entropy intermetallic compound for electrocatalytic hydrogen evolution. Adv. Mater. 2026, 38, e10424.
23. Li, W.; Chen, D.; Lou, Z.; et al. Inhibiting overoxidation of dynamically evolved RuO2 to achieve a win-win in activity-stability for acidic water electrolysis. J. Am. Chem. Soc. 2025, 147, 10446-58.
24. Yang, C.; Wu, C.; Xie, W.; Xie, D.; Hu, P. General reactive element-based machine learning potentials for heterogeneous catalysis. Nat. Catal. 2025, 8, 891-904.
25. Kum, H.; Kim, J. High-throughput screening of Ru-based MOF-supported single-atom catalysts for hydrogen evolution reaction via machine learning interatomic potential. ACS. Catal. 2025, 15, 19756-67.
26. Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B. Condens. Matter. 1993, 47, 558-61.
27. Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 1999, 59, 1758-75.
28. Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865-8.
29. Henkelman, G.; Uberuaga, B. P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901-4.
30. Sheppard, D.; Xiao, P.; Chemelewski, W.; Johnson, D. D.; Henkelman, G. A generalized solid-state nudged elastic band method. J. Chem. Phys. 2012, 136, 074103.
31. Wang, T.; Wu, Q.; Han, Y.; Guo, Z.; Chen, J.; Liu, C. Advanced theoretical modeling methodologies for electrocatalyst design in sustainable energy conversion. Appl. Phys. Rev. 2025, 12, 011316.
32. Hoover, W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. Gen. Phys. 1985, 31, 1695-7.
33. Nørskov, J. K.; Rossmeisl, J.; Logadottir, A.; et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B. 2004, 108, 17886-92.
34. Nørskov, J. K.; Bligaard, T.; Logadottir, A.; et al. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, J23.
36. Pearson, K. VII. Mathematical contributions to the theory of evolution. - III. Regression, heredity, and panmixia. Philos. Trans. A. Math. Phys. Eng. Sci. 1896, 253-318.
37. Kramer, O. Scikit-Learn. In Machine Learning for Evolution Strategies; Studies in Big Data, Vol. 20; Springer International Publishing, 2016; pp 45-53. DOI: 10.1007/978-3-319-33383-0_5.
38. Paszke, A.; Gross, S.; Massa, F.; et al. PyTorch: an imperative style, high‑performance deep learning library. arXiv 2019;arXiv:1912.01703. Available online: https://doi.org/10.48550/arXiv.1912.01703. [accessed 9 Apr 2026].
40. Hyndman, R. J.; Koehler, A. B. Another look at measures of forecast accuracy. Int. J. Forecast. 2006, 22, 679-88.
41. Qin, Y.; Li, Y.; Zhao, W.; Chen, S.; Wu, T.; Su, Y. Computational study of transition metal single-atom catalysts supported on nitrogenated carbon nanotubes for electrocatalytic nitrogen reduction. Nano. Res. 2022, 16, 325-33.
42. Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; et al. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt interfaces. Science 2011, 334, 1256-60.
43. Nørskov, J.; Bligaard, T.; Logadottir, A.; et al. Universality in heterogeneous catalysis. J. Catal. 2002, 209, 275-8.
44. Fan, J.; Wang, H.; Liao, G.; Song, C.; Zou, J. Potassium-doped g-C3N4/nitrogen-doped g-C3N4 step-scheme homojunction for enhanced H2 evolution photocatalysis. Sci. China. Technol. Sci. 2025, 68, 1620206.
45. Feng, Y.; Xie, Y.; Yu, Y.; et al. Electronic metal-support interaction induces hydrogen spillover and platinum utilization in hydrogen evolution reaction. Angew. Chem. Int. Ed. Engl. 2025, 64, e202413417.
46. Saxena, S.; Khan, T. S.; Jalid, F.; Ramteke, M.; Haider, M. A. In silico high throughput screening of bimetallic and single atom alloys using machine learning and ab initio microkinetic modelling. J. Mater. Chem. A. 2020, 8, 107-23.
47. Toyao, T.; Suzuki, K.; Kikuchi, S.; Takakusagi, S.; Shimizu, K.; Takigawa, I. Toward effective utilization of methane: machine learning prediction of adsorption energies on metal alloys. J. Phys. Chem. C. 2018, 122, 8315-26.
48. Ouyang, R.; Curtarolo, S.; Ahmetcik, E.; Scheffler, M.; Ghiringhelli, L. M. SISSO: a compressed-sensing method for identifying the best low-dimensional descriptor in an immensity of offered candidates. Phys. Rev. Mater. 2018, 2, 083802.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].