


Exploring materials data through collaboration: 2024 KRICT ChemDX Hackathon
 0
0 
Abstract
Data-driven research is in the spotlight across many science and engineering fields, including materials science, with the expectation that effective utilization of data, supported by modern artificial intelligence techniques, can lead to breakthroughs in addressing key scientific questions. Korea Research Institute of Chemical Technology (KRICT) Chemical Data Explorer platform (ChemDX), our web-based and integrated platform, including various data explorer and artificial intelligence modules, aims to enhance accessibility of chemical data for digital materials discovery. In this article, we highlight the results of the 2024 KRICT ChemDX Hackathon, an event to support data-driven research in chemistry and materials science. Hackathon participants explored ChemDX platform and developed projects ranging from machine learning models and data visualization tools to user interface improvements. These projects demonstrated the versatility and potential of data-driven research with the aid of ChemDX platform, in bridging data-driven experimental and computational research. The feedback and outcomes from this hackathon demonstrate the impressive potential of interdisciplinary data-driven research, guide further improvements to the platform, and enhance its usability and outreach.
Keywords
INTRODUCTION
Hackathons are effective tools for fostering interdisciplinary collaboration by providing an intensive environment that encourages creative problem-solving and accelerates idea development. They also support the long-term sustainability of scientific communities by attracting newcomers and strengthening collaborative networks[1,2]. Such events have been increasingly organized for chemistry and materials-related data problems. Recent examples include general materials data[3,4], electronic structure[5], Bayesian optimization[6], and large-language models (LLMs)[7,8].
The KRICT Chemical Data Explorer platform (ChemDX) is a web-based data platform developed by the Korea Research Institute of Chemical Technology (KRICT) over the past few years to facilitate data-driven research in chemistry and materials science[9]. As illustrated in Figure 1A, ChemDX comprises four specialized components: (i) Materials Data Explorer (MatDX), which enables the search and utilization of materials data; (ii) ChemAI, an AI platform specialized in materials science; (iii) Korea Chemical Bank (KCB), a chemical data platform for drug development; and (iv) Open Mixture Risk Assessment (OpenMRA) Toolkits, which predict the toxicity of chemical mixtures. The following are brief descriptions of four major components of ChemDX and their key functionalities:
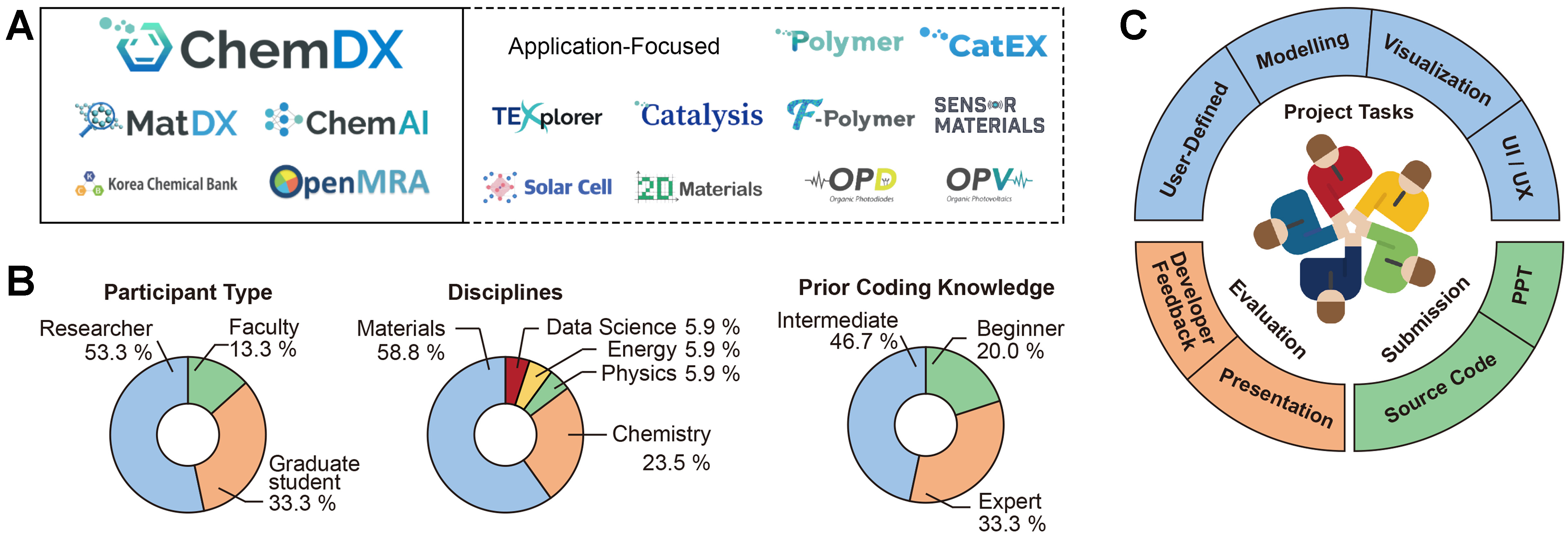
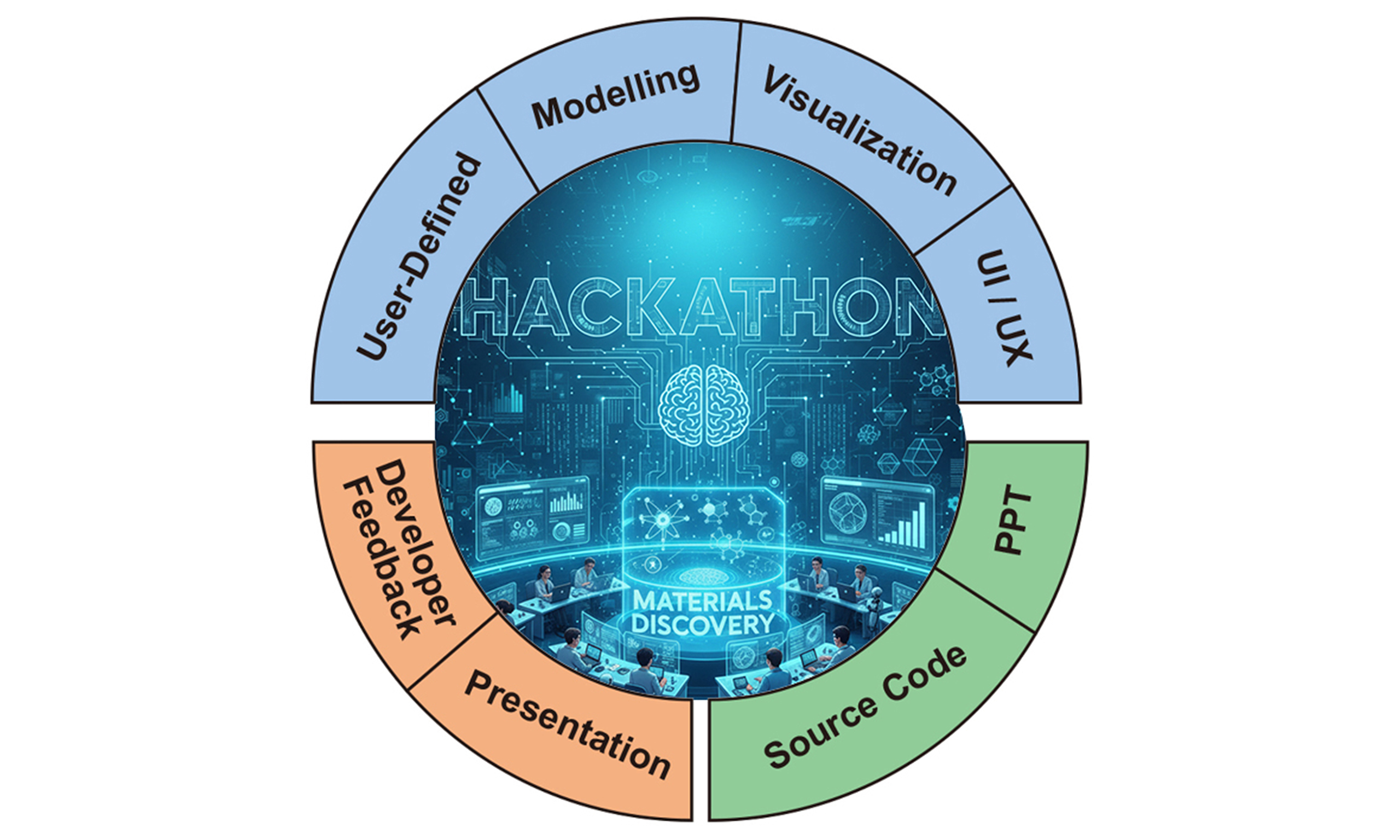
Figure 1. (A) An overview of the ChemDX platform, with four main databases shown in the solid box and application-focused modules in the dotted-lined box; (B) Participant demographics categorized by participant type, disciplines, and coding proficiency levels; (C) A workflow of the hackathon event highlighting project tasks, submission, and evaluation. ChemDX: Chemical Data Explorer platform.
• MatDX: MatDX is an ontology-based platform that integrates and organizes materials property data from multiple sources, including public databases (PubDX), experimental results (ExpDX) from KRICT and collaborators, computational outputs (CalcDX), and literature-based information (LitDX), which contains a total of approximately 570,000 data entries. Key features include material tagging and ontology-based classification, which enable users to efficiently search, filter, and explore data according to structure, composition, properties, applications, and methods (https://materials.chemdx.org/).
• ChemAI: ChemAI provides ready-to-use toolkits for property prediction (e.g., band gap, formation energy, thermoelectricity, absorption spectra), as well as functions for clustering, outlier detection, and model correction. Users can upload datasets in multiple formats [chemical formula, Simplified Molecular Input Line Entry System (SMILES), crystal structure, or numeric vectors], configure machine learning (ML) models with hyperparameter tuning and transfer learning, and evaluate results through error analysis and visualization. ChemAI further supports sharing of trained models and provides example datasets (https://ai.chemdx.org/).
• KCB: KCB is one of the largest national repositories for public compound collections, providing a source of chemical diversity for biology. Its mission is to systematically collect and curate compounds produced in Korea, maximize their research value through broad sharing across academia, industry, and government. Deposited compounds undergo quality verification, storage, and registration in the National Science & Technology Information Service (NTIS), ensuring accessibility to the broader research community (https://chembank.org/).
• OpenMRA: OpenMRA is a web-based platform designed to support chemical safety evaluation at the product design stage. It integrates three modules: (i) mixture toxicity prediction based on component information; (ii) combined exposure assessment drawing from domestic and international models; and (iii) mixture risk prediction that accounts for additive or synergistic effects. This open-access tool facilitates early screening, hazard reduction, and informed decision-making in chemical and consumer product development (https://www.openmra.org/).
Especially, MatDX includes four modules for exploring public datasets (PubDX), experimental data (ExpDX), computational results (CalcDX), and LitDX, which has a specialty in different branches of data-driven research in materials science. In contrast to international platforms such as the Materials Project[10] and Novel Materials Discovery Laboratory (NOMAD)[11], which provide large-scale computational data and findable, accessible, interoperable, reusable (FAIR)-compliant infrastructures, ChemDX integrates curated experimental datasets contributed by domestic consortia across diverse domains (thermoelectrics, 2D materials, solar cells, etc.). This complementary scope positions ChemDX as a bridge between experimental and computational data streams in the materials informatics community.
ChemDX also provides access to application-focused databases, such as ThermoElectric Explorer (TEXplorer) for thermoelectric (TE) materials[12,13], fluorinated polymer (F-Polymer), SolarCells for hybrid Perovskite solar cells, 2D Materials (2DMat) for managing and analyzing 2D materials data[14], SensorMaterials for experimental data of sensor materials, and so on. These datasets are curated by domestic experimentalists and theorists across diverse domains in chemistry and materials science[15-17] and applied in studies, including data-driven optimization of SnSe-based TE materials[18], catalyst design for oxidative propane dehydrogenation[19], and others[20-23]. Likewise, ChemDX bridges the gap between experimental and computational science through these efforts, enabling researchers to accelerate discoveries and optimize material properties with data-centric approaches, providing useful visualization tools such as in LitDX[24] and SolarCells[25].
Building on the trend of hackathons fostering collaboration, we organized the “2024 KRICT ChemDX Hackathon: Colaboratory” to enhance ChemDX’s usability and applicability. Especially, the hackathon was centered on the MatDX, SensorMaterials, SolarCell databases, selected from the broader ChemDX resources. The event aimed to evaluate the platform, currently in its Beta version, and identify opportunities to improve its functionality and quality. Key objectives included developing general-purpose ML models utilizing ChemDX-MatDX data to enhance its potential for predictive analytics and material property characterization. Another goal was to design creative and user-friendly data visualization features to improve user accessibility. Participants were also encouraged to propose user interface/user experience (UI/UX) improvements for seamless usability. Finally, the hackathon sought critical feedback on ChemDX datasets, including MatDX, SolarCell, and 2D Materials, to guide future development and better align the platform with user needs.
HACKATHON OVERVIEW
Event overview
The hackathon was held from November 6 to 8, 2024, at Hanbat National University and KRICT in Daejeon, South Korea. Participants were tasked with exploring the ChemDX platform, and they had the flexibility to either select their own project topics or work on suggested example datasets. The event encouraged participants to apply their expertise in ML, data visualization, or UI/UX enhancement, leveraging ChemDX datasets to develop innovative solutions. Detailed information regarding the event can be found on the official webpage (https://gitlab.chemdx.org/global-network/2024-krict-chemdx-hackathon/-/wikis/home).
In addition to free-topic exploration, participants were provided with example datasets spanning multiple domains mainly originating from MatDX and SolarCell DB: (i) MatDX for formation energy prediction [CSV (comma delimited text), 5,000 data entries]; (ii) LitDX-IPO for inorganic phosphor optical properties (Excel, 4,393 data entries); (iii) LitDX-TE for TE materials (Excel, 5,206 data entries); (iv) Sensor Materials dataset with Raman spectroscopy intensity data (Excel, 39 data entries); and (v) SolarCell DB containing photovoltaic parameters [JSON (JavaScript Object Notation), 3,572 data entries]. These resources served as common starting points for the team projects.
Participants
The event brought together 17 researchers and graduate students, ranging from beginners to experts in various disciplines including data-driven research, computational materials science, ML, and AI. As shown in Figure 1B, the participant pool comprised roughly 53% researchers, 33% graduate students, and 14% faculty members, highlighting a balance between experienced professionals and emerging talents. More than half were from materials (or related) backgrounds, followed by chemistry, physics, energy, and data science backgrounds. Additionally, coding expertise was varied, with 33% experts, 47% intermediate-level participants, and 20% beginners. This diversity of skill levels and professional backgrounds promoted an inclusive and collaborative atmosphere.
Submission and evaluation
As shown in Figure 1C, participants engaged in project tasks and were required to submit two key deliverables for evaluation. The first was a detailed oral presentation, which explained the project’s conceptual ideas, workflow, data utilization strategies, and technical implementation. The second was the source code, submitted in either Jupyter Notebook or Python file format, providing the technical foundation of the project.
With the prepared presentation, each team delivered a 10-minute presentation summarizing their project outcomes, followed by evaluations based on four key criteria:
• Data Utilization and Completeness: Assessing how effectively and creatively the ChemDX datasets were used.
• Creativity and Innovation: Evaluating the originality of the project and its potential impact or application.
• Technical Feasibility: Determining the practicality of implementing the proposed ideas.
• Teamwork and Presentation: Addressing the quality of collaboration within the team and the clarity of their presentation.
The evaluation process emphasized not only technical expertise but also creativity and effective teamwork, ensuring a well-rounded assessment of each project.
HACKATHON PROJECTS
Six interdisciplinary teams tackled distinct challenges using ChemDX data. Table 1 provides the project names, team members, and the developers’ GitHub repositories. In addition to the brief descriptions of each project in this section, each team’s repository contains additional narrative descriptions, results, and the data and code needed to reproduce their work.
Overview of hackathon projects
| Category | Details |
| Project 1 Authors Link | Generative design of inorganic thermoelectric materials A. K. Y. Low, H. Jang, S. Lee, J. Recatala-Gomez and H. Sahu https://github.com/andrelowky/KRICT-ChemDX-Hackathon |
| Project 2 Authors Link | A surrogate model for formation energy prediction using pre-trained local atomic environments H. Chun, G. Shin and C. Kim https://github.com/HojeChun/KRICT-Hackathon |
| Project 3 Authors Link | Evaluating the utility of ChemDX data for training ML models in predicting material formation enthalpies M. Minotakis and K. Kang https://github.com/Minotakm/hackathon_KRICT |
| Project 4 Authors Link | Developing visualization & rapid analysis tools for Raman spectroscopy data J. Berry and K. A. Christofidou https://github.com/bezzer365/2024-KRICT-ChemDX-Hackathon |
| Project 5 Authors Link | Data-driven approaches for accelerating inorganic crystal formation energy prediction and high-throughput Raman spectroscopy analysis K. Kim and J. F. Joung https://github.com/jfjoung/KRICT_Hackathon |
| Project 6 Authors Link | String-oriented understanding of phosphors (SOUP) J. Schrier https://github.com/jschrier/KRICT_hackathon_phosphors |
Project 1: Generative design of inorganic thermoelectric materials
Main aim: Use ML and algorithms to predict TE materials with high figure-of-merit (zT).
Description: TE materials directly convert heat into electricity and vice versa[26]. The performance of a TE material is related to a dimensionless zT, as given in zT = S2σT/κ where S is the Seebeck coefficient, σ is the electrical conductivity, κ is the thermal conductivity, and T is the temperature[26]. Participants used the LitDX dataset which comprised zT, S, σ, and κ values at different temperatures for various inorganic materials, as input to a generative design workflow [Figure 2] to perform property prediction and a physics-informed zT-T curve fitting. The structure-agnostic model CrabNet[27] algorithm was used for adjusting the property predictions based on domain expertise. Candidate materials are sampled in a combinatorial manner considering their chemical similarity based on Earth mover’s distance.
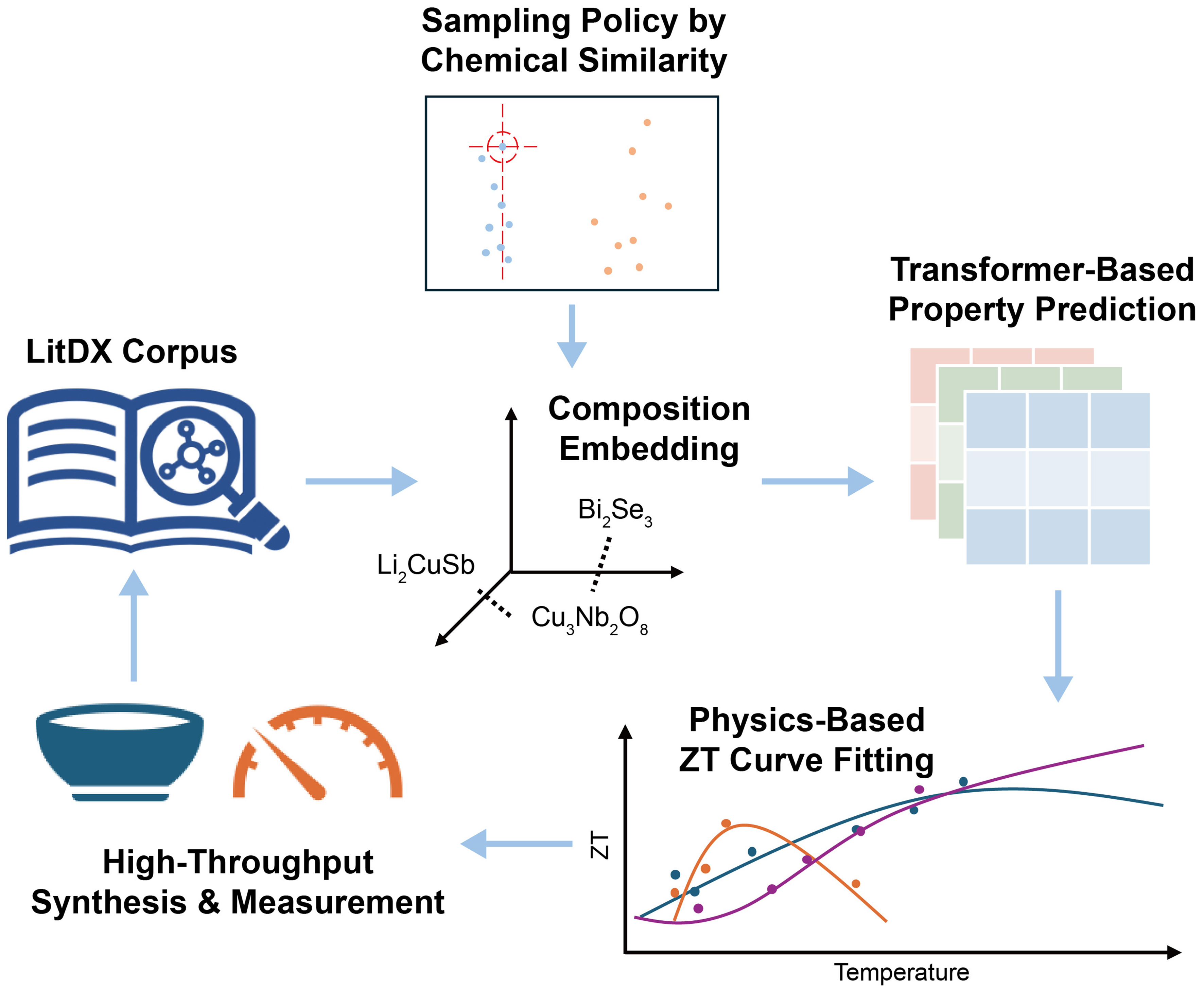
Figure 2. (Project 1) Generative design workflow to perform property prediction and a physics-informed zT-T curve fitting.
The project demonstrated reliable extrapolation of zT-T curves and provided interpretable visualizations of materials space, enabling the identification of chemically similar candidates with promising TE properties. These outcomes highlight the potential of integrating physics-informed learning with generative exploration for accelerating discovery of high-efficiency TE materials (see its GitHub page for more details in Table 1). Possible future improvements include: (i) Using the MTEncoder (multi-task pre-trained transformer encoder for materials representation learning)[28] multi-task learning approach; (ii) Developing improved optimization heuristics that balance exploration and exploitation in the search for optimal TE materials.
Project 2: A surrogate model for formation energy prediction using pre-trained local atomic environments
Main aim: Develop a surrogate model that accurately predicts formation energy by leveraging pre-trained local atomic environment representations.
Description: Learning the properties of materials is not a trivial task, especially given the challenges associated with representing them. To address this, participants used representations extracted from the MACE[29] foundation model, which was pre-trained for ML interatomic potentials, as inputs to construct models for predicting the formation energy (Ef) of materials as reported in MatDX. Benchmarking was performed against a baseline model with handcrafted physical descriptors, such as density and crystal symmetry. Pre-trained representations yield a mean absolute error (MAE) of 0.11 eV/atom, outperforming the baseline model (i.e., with handcrafted features), which has an MAE of 0.18 eV/atom. The model using the pre-trained atomic features outperforms the baseline model with handcrafted physical features (e.g., density, atomic mass and crystal symmetry). This method can be transferable to other downstream tasks because it does not require additional procedures or chemical intuition to generate input features.
The project demonstrated that foundation model features can serve as strong, ready-to-use surrogates for accurate energy prediction, reducing the dependence on handcrafted descriptors. This result highlights the potential of pre-trained atomic representations as a generalizable starting point for diverse materials informatics tasks (see its GitHub page for more details in Table 1).
Project 3: Evaluating the utility of ChemDX data for training ML models in predicting material formation enthalpies
Main aim: Leverage the ChemDX material database to build a ML model to predict formation energy of materials.
Description: Akin to Project 2, Project 3 developed a ML model to predict formation energy (Ef) by leveraging data from MatDX. The project consists of three steps: (i) data extraction and cleaning; (ii) methodology experiments; and (iii) data augmentation, as depicted in Figure 3. First, the participants collected all available data from MatDX via its application programming interface (API). The dataset, comprising approximately 20,000 entries, was preprocessed by removing duplicates, entries with missing values, and significant outliers in regions of extremely high-positive and low-negative energy. The uneven distribution of material data regarding space groups and elements in the tutorial set was also characterized. Second, various regression models were evaluated, including linear regression, lasso regression, decision tree regression, gradient boosting regression, and others; random forest regression delivered the best performance. The descriptors used in this work are the most straightforward that could be made, using the available data known as Mendeleyev descriptors, enhanced by chemical information such as mean and standard deviation of electronegativity, electronegativity, and atomic number difference, an average of atomic mass, valence electrons, atomic radius, and maximum/minimum atomic radius. Initially, the trained models underperform, producing a MAE of 0.845 eV and R2 of 0.305 on the test set. This behavior can be attributed to the poor sampling of the materials space due to the limited set of available data and simplicity of the descriptors. For this reason, in the third step, the participants used pre-trained universal ML models, specifically MACE-MP0[29], SevenNet[30], and Orb v2[31], to insert information in their descriptor, effectively biasing their model into predicting the wanted property. The final ML model exhibits superior performance with an MAE of 0.549 eV and R2 of 0.910 for the test data. The result indicated the importance of the data size for ML training, acknowledging the value of establishing large databases such as ChemDX, and the continued need to improve diversity of the materials space sampling by either experiment or theory.
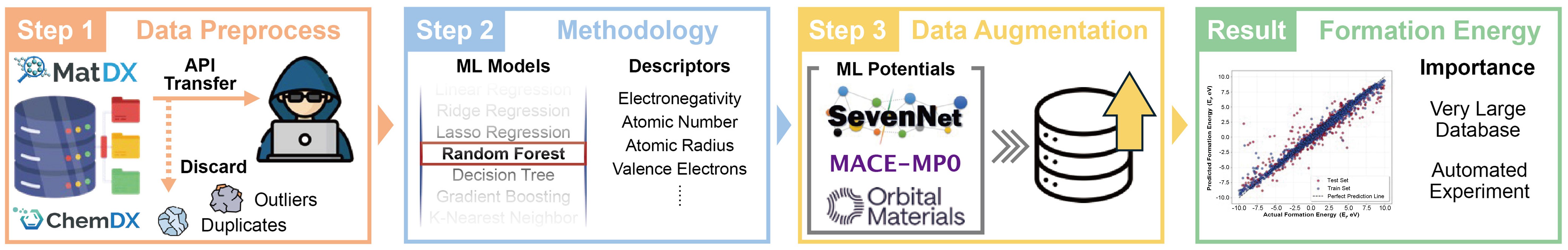
Figure 3. (Project 3) A schematic plot regarding building a ML model for formation energy predictions. ML: Machine learning.
The project not only demonstrated the utility of ChemDX as a scalable training resource but also showed how pre-trained universal models can improve the predictive accuracy of formation energy models. These results highlight the dual importance of database growth and model transferability for advancing data-driven materials discovery (see its GitHub page for more details in Table 1).
Project 4: Developing visualization & rapid analysis tools for Raman spectroscopy data
Main aim: Develop rapid visualization and analysis tools for experimental Raman spectra.
Description: This work aimed to facilitate efficient data exploration within the MatDX database by developing tools for rapid visualization and preliminary analysis of experimental data. Focusing on Raman spectroscopy, a workflow, visualized in Figure 4, that includes background subtraction, automated peak identification, and Gaussian peak fitting was implemented. Key outputs were extracted efficiently for multiple datasets, including peak positions, full width at half maximum (FWHM), peak intensities, and peak integrated intensities, and an example of a key visual output can be seen in Figure 4. While Gaussian fitting proved effective for many Raman spectra within the provided dataset, future work should investigate the implementation of more versatile peak fitting functions, such as pseudoVoigt or Lorentzian, to accommodate broader spectral features. Furthermore, the incorporation of multi-peak fitting algorithms to address overlapping peaks, which can be challenging for automated peak identification, is a key feature for building robustness into the workflow and enabling a wider range of data to be analyzed on the fly. The current workflow processes individual datasets sequentially. To enhance efficiency, the exploration of parallel processing techniques to significantly reduce analysis time for larger datasets is also a feature that could further enhance the utilization of the tools developed. This framework has the potential to be extended to other spectroscopic and diffraction techniques, such as X-ray diffraction and infrared spectroscopy, with appropriate modifications to the data processing steps. Additionally, its application could be extended to thermal analysis data by incorporating differential analysis and peak-fitting techniques. These tools can accelerate data exploration, enabling researchers to quickly identify trends, outliers, and areas of interest within the MatDX database, thus guiding further in-depth analysis and experimental design.
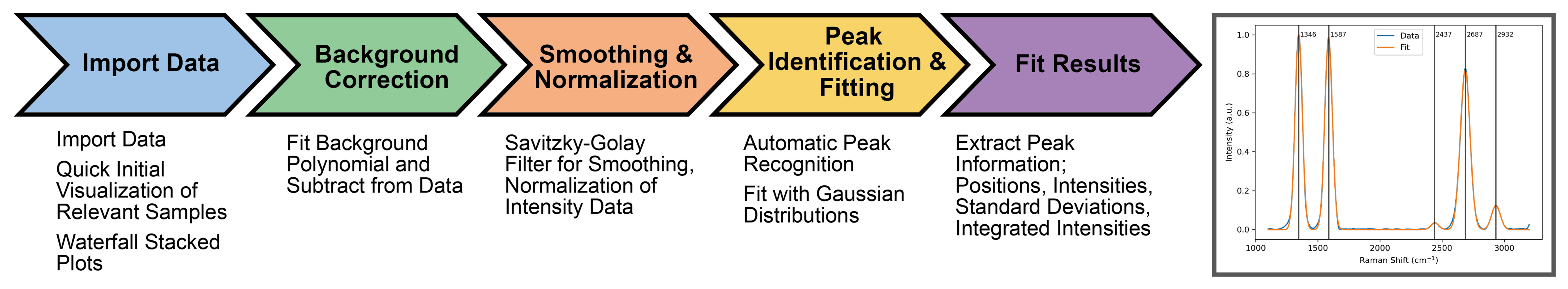
Figure 4. (Project 4) Data visualization and rapid analysis pipeline, and an example visual output of fitted data with peak identification and marked up with Raman shift positions.
The project demonstrated the ability to rapidly process and extract peak parameters from Raman spectra, improving accessibility of raw experimental data. These outputs illustrate the potential of lightweight, reproducible pipelines to accelerate characterization across broader spectroscopic and diffraction datasets (see its GitHub page for more details in Table 1).
Project 5: Data-driven approaches for accelerating inorganic crystal formation energy prediction and high-throughput Raman spectroscopy analysis
Main aim: Develop efficient data-driven methods for predicting inorganic crystal formation energy and automating high-throughput Raman spectroscopy analysis [Figure 5].
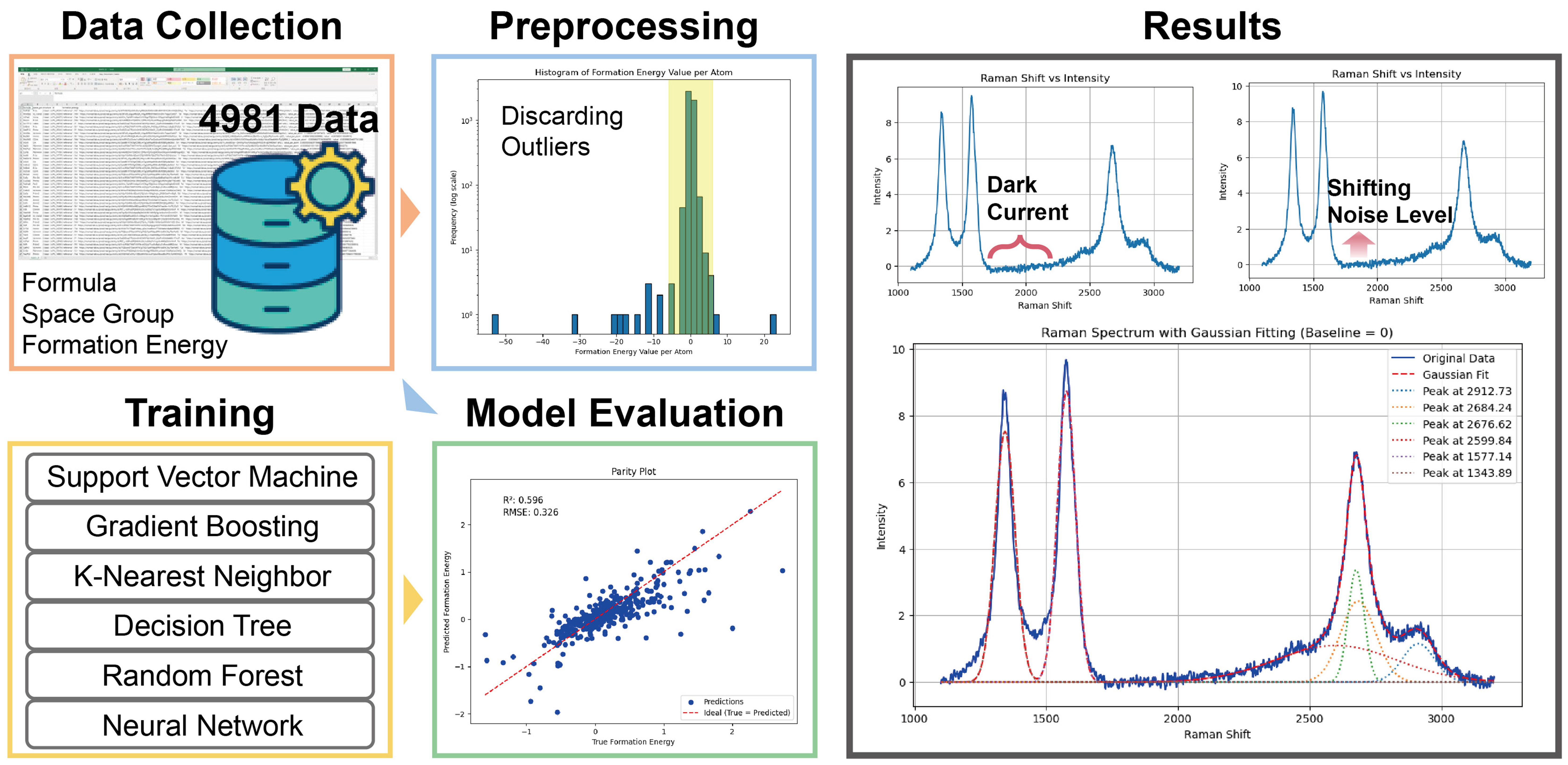
Figure 5. (Project 5) Workflow to perform formation energy prediction and Raman spectroscopy analysis.
Description - Formation Energy Prediction of Inorganic Crystals: Similar to Projects 2 and 3, Project 5 used ML to predict formation energy (Ef) using simplified input representations. Participants employed atom-wise stoichiometric vectorization and one-hot encoding for space groups to encode inorganic crystals, avoiding the costly process of obtaining precise 3D structural data. Participants evaluated various ML models, including gradient boosting, support vector machines, k-nearest neighbors, decision trees, random forests, and a simple multi-layer perceptron neural network. A 9-fold cross-validation strategy was applied to each method; the final prediction performance was calculated as the average of these models’ outputs. Among the approaches, the neural network demonstrated the best performance, achieving root mean square error (RMSE) of 0.326 eV and R2 = 0.596. These results show that one-hot encoding combined with a simple neural network can deliver competitive performance, offering a practical alternative to traditional computational methods for materials discovery.
Description - High-Throughput Raman Spectroscopy Analysis: Similar to Project 4, participants developed a program to automate Raman spectrum analysis. First, the dark current was removed by subtracting the intensity values from the noise regions. A peak detection function was then used to identify peaks with signal-to-noise ratios greater than 3, where the threshold for peaks is defined as three times the standard deviation of the dark current. This ensures that only analytically meaningful peaks are detected. This automated program accelerates the analysis of Raman spectra obtained from high-throughput experiments, allowing researchers to efficiently identify spectra containing peaks of interest and, consequently, the relevant samples.
The aforementioned two projects demonstrated that simple representations coupled with neural networks can achieve reliable formation energy predictions, while automated Raman analysis enables rapid screening of large spectral datasets. These tools provide a scalable framework for integrating computational prediction with experimental high-throughput characterization (see its GitHub page for more details in Table 1).
Project 6: String-oriented understanding of phosphors (SOUP)
Main aim: Predict the emission color of a phosphor given only its chemical formula provided as a string [Figure 6].
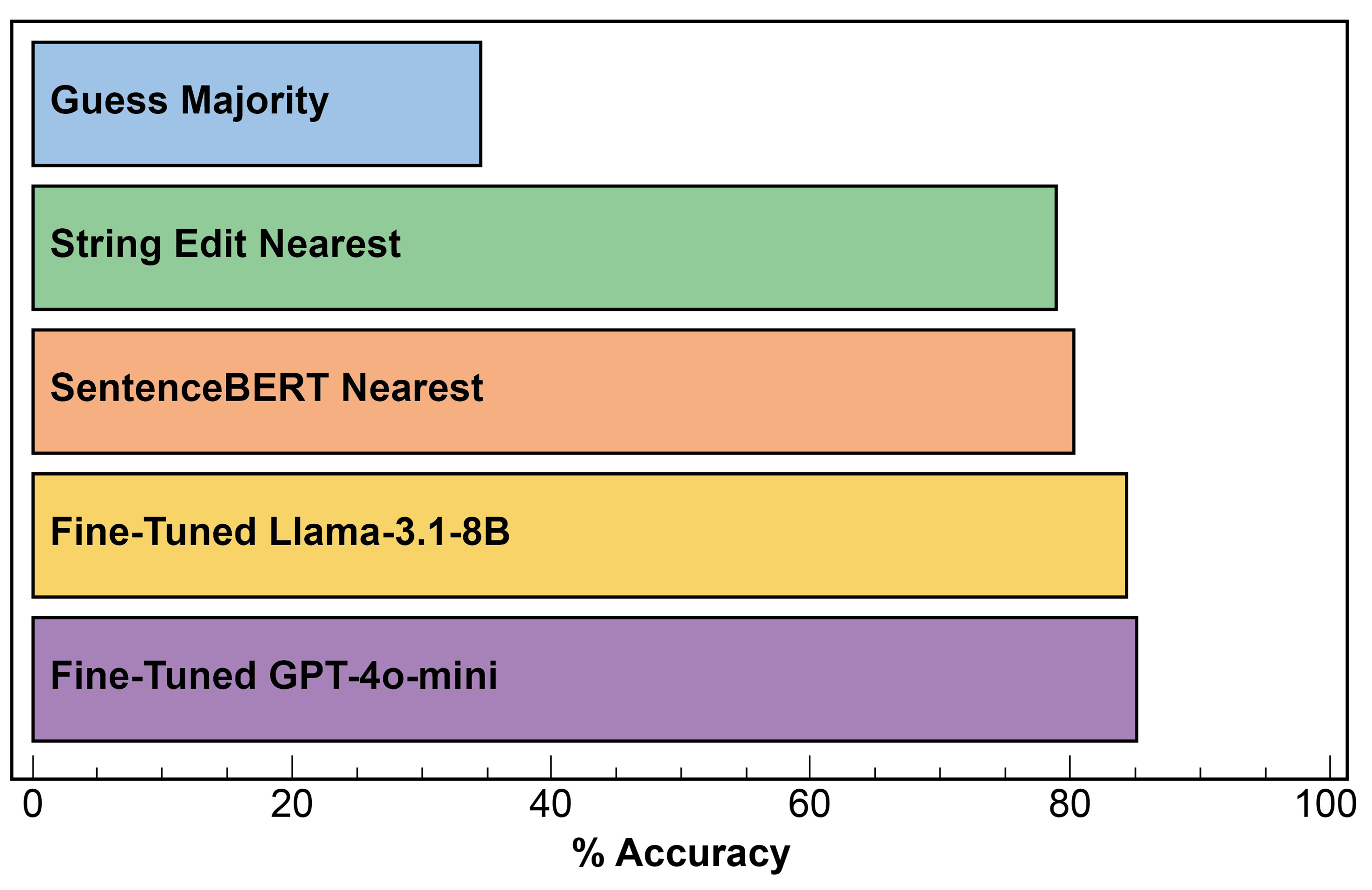
Figure 6. (Project 6) String oriented understanding of phosphors (SOUP): accuracy of predicting the color (8 classes) given a chemical formula for an inorganic phosphor, as evaluated on 752 test compositions.
Description: Project 6 used the KRICT LitDX Inorganic Phosphors Optical Property dataset[32]. Motivated by recent work on fine-tuned LLMs[33-40] and text embedding vectors[41,42] for chemistry and materials prediction tasks, the goal was to demonstrate and compare different methods for predicting the emission color (e.g., “Orange”) given only the chemical formula string as an input (e.g., “Ba2La6.4Si6O26Eu1.6”). Four different methods were compared: (i) nearest-neighbor classification using the Levenshtein string-edit distance; (ii) nearest-neighbor classification using the cosine distance of the SentenceBERT embeddings[43]; (iii) a fine-tuned Llama-3.1.8B LLM[44]; and (iv) a fine-tuned gpt-4o-mini LLM[45]. All approaches are substantially better than guessing the majority class, and the fine-tuned LLMs achieve accuracies as high as 85% on predicting the emission color.
The project demonstrated that purely string-based approaches can capture chemically meaningful patterns for optical property prediction, with fine-tuned LLMs substantially outperforming string- or embedding-based baselines. These results highlight the promise of LLM-guided text modalities as lightweight, transferable surrogates for rapid screening in optical materials discovery (see its GitHub page for more details in Table 1).
OBSERVATIONS AND DISCUSSIONS
The hackathon had four notional goals: (i) Fostering education and collaboration; (ii) Collecting feedback and potential improvements to the ChemDX platform; (iii) Observing current trends and opportunities in materials ML research; (iv) Addressing broader challenges in materials discovery. Using a combination of participant exit survey responses and observations/discussions of the organizers, we reflect on these goals.
Hackathon Organization: Participants valued the hackathon for networking, hands-on learning, and professional development. They came with a wide range of programming and data science skill levels, from beginners to experts. Mixing experts and beginners within each team proved effective in fostering participants’ learning and networking. This aligns with recommendations from the literature, which emphasize the importance of involving experienced professionals as mentors and judges to ensure the success of hackathons[1,2]. One recommendation is to create asynchronous preparatory materials on basic Python skills, ML workflows, and database navigation to provide necessary background knowledge. Students suggested incentives such as certificates, for greater participation. In addition, they proposed extending the event to 3-4 days and increasing team sizes to 3-4 members with a balanced mix of students and senior researchers to improve collaboration (We allowed teams to form spontaneously after initial introductions and descriptions of the datasets).
Scaling to a global audience remains a consideration. While this hackathon emphasized gathering participants together in space and time, other models, such as “online” or “hybrid” hackathons that combine online presentations and local “hubs” (with the option to collaborate locally or online) have been conducted for the Chemistry and Materials community[7,8,46]. While both online and hybrid events have the advantage of enabling global participation, they also reduce the opportunity for informal networking and information exchanges during meals and breaks, which is especially valuable for both postdoctoral and graduate student participants.
Insights from the ChemDX hackathon: While the scale of participation was modest and primarily centered on ChemDX datasets, the outcomes revealed broadly relevant insights: (i) the utility of foundation models for property prediction, (ii) persistent challenges in data quality such as sparsity and outliers, and (iii) opportunities for automated analysis workflows. These observations are consistent with recent reviews on materials databases[47] which emphasize the importance of robust data aggregation and reproducibility in accelerating materials discovery. These insights, while derived from a relatively small event, are transferable to broader domains, highlighting the potential of hackathon models for addressing data challenges across disciplines.
Feedback on ChemDX: We received valuable feedback from participants, which we believe will contribute to the advancement of materials informatics. Key suggestions included enriched metadata and search functionalities, expanded API support for interoperability, and bulk download functionality. Participants also emphasized the importance of data standardization, providing detailed explanations with references to assist non-domain experts. While these aspects represent important limitations and directions for future development of ChemDX or other data platforms, it should be noted that enabling bulk downloads may pose psychological burdens for experimental data providers, making their implementation a practical challenge.
Trends in Materials ML: Hackathons provide an opportunity to observe new ideas and trends. We highlight a few general emerging themes here: (i) The combination of open-source ML ecosystem (e.g., scikit-learn[48]) and materials-specific foundation models (e.g., MACE[29], SevenNet[30], and OrbNet[31]) made it easy for teams to perform modeling tasks in a compressed time period. The former allows for rapid model training and testing; the latter provides useful representations that generally outperform naïve physicochemical representations; (ii) LLMs lower the barrier for programming tasks. The value of LLMs for materials and chemistry programming has been previously discussed[49-51]. All participants were given access to ChatGPT Plus, allowing them to make gpt-4o-mini and gpt-4o queries related to their programming tasks. LLM-assistance facilitated coding and debugging by novice programmers, and even expert programmers expressed how it helped them work with unfamiliar software libraries and packages. Towards that end, more deliberate training in good practices for using LLMs in programming would be valuable[52]; (iii) Domain expertise remains important. Even in highly curated datasets such as MatDX, there are still duplicates, outliers, and uneven distributions of data that limit ML modeling unless properly accounted for by the user (This was particularly explored by Project 3). Hackathons that bring together teams with diverse expertise can be one way to identify and document these issues.
Addressing Challenges in Materials Discovery: Recent reviews and perspectives have highlighted challenges related to the lack of interoperability in materials datasets[47], computational reproducibility[53-55], and more generally, the need for training materials scientists in relevant computational/informatics skills[56,57]. In parallel, emerging platforms such as AlphaMat[58] and MatGPT[59] illustrate the development of integrated materials informatics toolkits. These challenges are equally applicable to all areas of science, technology, engineering, and mathematics (STEM), not just materials science. Short-duration hackathons are an informal complement to other training efforts by providing early graduate students with practical, first-hand experience of the promises and challenges in working with real experimental data. Student participants reported that the hackathon experience motivated them to pursue formal coursework and other training opportunities in data science. For many students, this was their first use of GitHub version management, and the requirement to publish their data and code with the guidance of more experienced postdoc mentors. This is a step towards encouraging students to adopt better computational reproducibility practices in their subsequent research.
CONCLUSION
The 2024 KRICT ChemDX Hackathon provided a platform to test datasets for anomalies and develop general ML models using ChemDX datasets. Participants proposed creative data visualization features, UI/UX improvements, and provided practical feedback on the ChemDX DB platform. The event encouraged collaboration among participants and laid the groundwork for an annual hackathon. Although the participant pool was relatively small and the scope of the projects focused on ChemDX datasets, the lessons are transferable to other domains. Also, while the team projects remain prototypes to require systematic benchmarking in follow-up studies, they illustrate diverse methodological possibilities.
DECLARATIONS
Acknowledgments
The authors gratefully acknowledge the support from National Research Foundation of Korea (NRF) and U.S. National Science Foundation (NSF).
Authors’ contributions
Hackathon participation, project development, data analysis:
Project 1: Low, A. K. Y.; Jang, H.; Lee, S.; Recatala-Gomez, J.; Sahu, H.
Project 2: Chun, H.; Shin, G.; Kim, C.
Project 3: Minotakis, M.; Kang, K.
Project 4: Berry, J.; Christofidou, K. A.
Project 5: Kim, K.; Joung, J. F.
Project 6: Schrier, J.
Conceptualization, organization of hackathon, supervision, critical revision of the manuscript, manuscript drafting: Yoo, S. H.; Schrier, J.; Jang, W.
Conceptualization, organization of hackathon: Soon, A.; Park, M.; Kim, B. H.; Shin, K.; Shin, J.
Availability of data and materials
All source codes and project repositories are publicly available at the KRICT ChemDX Hackathon GitHub repository (https://github.com/KRICT-DATA/2024-KRICT-ChemDX-Hackathon).
Financial support and sponsorship
This research was supported by a major research project from the Korea Research Institute of Chemical Technology (KK2451-10) and the Nano & Material Technology Development Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (RS-2024-00446683). Jang, W. acknowledges support from the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2022R1C1C200856713). Schrier, J. acknowledges support from the U.S. National Science Foundation (PHY-2226511, CHE-2320718).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Nolte, A.; Hayden, L. B.; Herbsleb, J. D. How to support newcomers in scientific hackathons - an action research study on expert mentoring. Proc. ACM. Hum. Comput. Interact. 2020, 4, 1-23.
2. Heller, B.; Amir, A.; Waxman, R.; Maaravi, Y. Hack your organizational innovation: literature review and integrative model for running hackathons. J. Innov. Entrep. 2023, 12, 6.
3. NC State. MATDAT18: materials and data science hackathon. https://matdat18.wordpress.ncsu.edu/. (accessed 2025-11-07).
4. Sparks, T. D.; Curtis, F. E.; Fredrickson, D. C.; Benedek, N. A. Insights and innovations from the SSMCDAT 2023: bridging solid-state materials chemistry and data science. Chem. Mater. 2024, 36, 5293-6.
5. University of Latvia. Hackathon 2022. https://www.quantumtheory.lu.lv/events/hackathon-2022/. (accessed 2025-11-07).
6. BO Hackathon with Acceleration Consortium. Hackathon agenda. https://ac-bo-hackathon.github.io/agenda/. (accessed 2025-11-07).
7. Jablonka, K. M.; Ai, Q.; Al-Feghali, A.; et al. 14 examples of how LLMs can transform materials science and chemistry: a reflection on a large language model hackathon. Digit. Discov. 2023, 2, 1233-50.
8. Zimmermann, Y.; Bazgir, A.; Afzal, Z.; et al. Reflections from the 2024 large language model (LLM) hackathon for applications in materials science and chemistry. arXiv 2024, arXiv:2411.15221. Available online: https://doi.org/10.48550/arXiv.2411.15221. (accessed 7 Nov 2025).
9. KRICT Chemical Data Explorer (ChemDX). https://www.chemdx.org/. (accessed 2025-11-07).
10. Horton, M. K.; Huck, P.; Yang, R. X.; et al. Accelerated data-driven materials science with the Materials Project. Nat. Mater. 2025, 24, 1522-32.
11. Scheidgen, M.; Himanen, L.; Ladines, A. N.; et al. NOMAD: a distributed web-based platform for managing materials science research data. J. Open. Source. Softw. 2023, 8, 5388.
12. ThermoElectric Materials Explorer (TEXplorer). https://texplorer.org/about. (accessed 2025-11-07).
13. Lee, Y. L.; Lee, H.; Jang, S.; et al. TEXplorer.org: thermoelectric material properties data platform for experimental and first-principles calculation results. APL. Mater. 2023, 11, 041111.
14. Yang, J. H.; Kang, H.; Kim, H. J.; et al. https://2DMat.ChemDX.org: experimental data platform for 2D materials from synthesis to physical properties. Digit. Discov. 2024, 3, 573-85.
15. Mok, D. H.; Back, S. Atomic structure-free representation of active motifs for expedited catalyst discovery. J. Chem. Inf. Model. 2021, 61, 4514-20.
16. Na, G. S.; Chang, H. A public database of thermoelectric materials and system-identified material representation for data-driven discovery. npj. Comput. Mater. 2022, 8, 897.
17. Jang, S.; Na, G. S.; Choi, Y.; Chang, H. Optical property dataset of inorganic phosphor. Sci. Rep. 2024, 14, 7639.
18. Lee, Y. L.; Lee, H.; Kim, T.; et al. Data-driven enhancement of ZT in SnSe-based thermoelectric systems. J. Am. Chem. Soc. 2022, 144, 13748-63.
19. Kim, J. S.; Chung, I.; Oh, J.; et al. Closed-loop optimization of catalysts for oxidative propane dehydrogenation with CO2 using artificial intelligence. J. CO2. Util. 2023, 78, 102620.
20. Kim, H. W.; Lee, S. W.; Na, G. S.; et al. Reaction condition optimization for non-oxidative conversion of methane using artificial intelligence. React. Chem. Eng. 2021, 6, 235-43.
21. Park, J.; Oh, J.; Kim, J.; et al. Catalyst discovery for propane dehydrogenation through interpretable machine learning: leveraging laboratory-scale database and atomic properties. ACS. Sustainable. Chem. Eng. 2024, 12, 10376-86.
22. Yang, J. H.; Lee, J.; Kwon, H.; Sohn, E. H.; Chang, H.; Jang, S. High glass transition temperature fluorinated polymers based on transfer learning with small experimental data. Macromol. Rapid. Commun. 2024, 45, e2400161.
23. Kim, J.; Noh, J.; Im, J. Machine learning-enabled chemical space exploration of all-inorganic perovskites for photovoltaics. npj. Comput. Mater. 2024, 10, 1270.
24. LitDX. DB’s visualization function. https://litdx.materials.chemdx.org/. (accessed 2025-11-07).
25. Solar Cell. DB’s visualization function. https://solar.chemdx.org/statistics. (accessed 2025-11-07).
27. Wang, A. Y.; Kauwe, S. K.; Murdock, R. J.; Sparks, T. D. Compositionally restricted attention-based network for materials property predictions. npj. Comput. Mater. 2021, 7, 545.
28. Prein, T.; Pan, E.; Dörr, T.; Olivetti, E.; Rupp, J. L. M. MTENCODER: a multi-task pretrained transformer encoder for materials representation learning. 2023. https://rgdoi.net/10.13140/RG.2.2.20897.79202. (accessed 7 Nov 2025).
29. Batatia, I.; Benner, P.; Chiang, Y.; et al. A foundation model for atomistic materials chemistry. arXiv 2024, arXiv:2401.00096. Available online: https://doi.org/10.48550/arXiv.2401.00096. (accessed 7 Nov 2025).
30. Park, Y.; Kim, J.; Hwang, S.; Han, S. Scalable parallel algorithm for graph neural network interatomic potentials in molecular dynamics simulations. J. Chem. Theory. Comput. 2024, 20, 4857-68.
31. ORB forcefield models from Orbital Materials. https://github.com/orbital-materials/orb-models. (accessed 2025-11-07).
32. ChemDX - LitDX. https://litdx.materials.chemdx.org. (accessed 2025-11-07).
33. Jablonka, K. M.; Schwaller, P.; Ortega-Guerrero, A.; Smit, B. Leveraging large language models for predictive chemistry. Nat. Mach. Intell. 2024, 6, 161-9.
34. Xie, Z.; Evangelopoulos, X.; Omar, ÖH.; Troisi, A.; Cooper, A. I.; Chen, L. Fine-tuning GPT-3 for machine learning electronic and functional properties of organic molecules. Chem. Sci. 2024, 15, 500-10.
35. Zhong, S.; Guan, X. Developing quantitative structure–activity relationship (QSAR) models for water contaminants’ activities/properties by fine-tuning GPT-3 models. Environ. Sci. Technol. Lett. 2023, 10, 872-7.
36. Kim, S.; Jung, Y.; Schrier, J. Large language models for inorganic synthesis predictions. J. Am. Chem. Soc. 2024, 146, 19654-9.
37. Song, Z.; Lu, S.; Ju, M.; Zhou, Q.; Wang, J. Is large language model all you need to predict the synthesizability and precursors of crystal structures? arXiv 2024, arXiv:2407.07016. Available online: https://doi.org/10.48550/arXiv.2407.07016. (accessed 7 Nov 2025).
38. Jacobs, R.; Polak, M. P.; Schultz, L. E.; Mahdavi, H.; Honavar, V.; Morgan, D. Regression with large language models for materials and molecular property prediction. arXiv 2024, arXiv:2409.06080. Available online: https://doi.org/10.48550/arXiv.2409.06080. (accessed 7 Nov 2025).
39. Rubungo, A. N.; Li, K.; Hattrick-Simpers, J.; Dieng, A. B. LLM4Mat-bench: benchmarking large language models for materials property prediction. Mach. Learn. Sci. Technol. 2025, 6, 020501.
40. Van Herck, J.; Gil, M. V.; Jablonka, K. M.; et al. Assessment of fine-tuned large language models for real-world chemistry and material science applications. Chem. Sci. 2025, 16, 670-84.
41. Sayeed, H. M.; Baird, S. G.; Sparks, T. D. Structure feature vectors derived from Robocrystallographer text descriptions of crystal structures using word embeddings. ChemRxiv 2023. Available online: http://dx.doi.org/10.26434/chemrxiv-2023-3q8wj. (accessed 7 Nov 2025).
42. Kim, S.; Schrier, J.; Jung, Y. Explainable synthesizability prediction of inorganic crystal polymorphs using large language models. Angew. Chem. Int. Ed. 2025, 64, e202423950.
43. Reimers, N.; Gurevych, I. Sentence-BERT: sentence embeddings using siamese BERT-networks. arXiv 2019, arXiv:1908.10084. Available online: https://doi.org/10.48550/arXiv.1908.10084. (accessed 7 Nov 2025).
44. Grattafiori, A.; Dubey, A.; Jauhri, A.; et al. The Llama 3 herd of models. arXiv 2024, arXiv:2407.21783. Available online: https://doi.org/10.48550/arXiv.2407.21783. (accessed 7 Nov 2025).
45. OpenAI Platform. GPT-4o mini. https://platform.openai.com/docs/models#gpt-4o-mini. (accessed 2025-11-07).
46. Baird, S.; Ansari, M.; Afzal, Z.; et al. Bayesian optimization hackathon for chemistry and materials. ChemRxiv 2025. Available online: https://doi.org/10.26434/chemrxiv-2025-dzh5z. (accessed 7 Nov 2025).
47. Ottomano, F.; De Felice, G.; Gusev, V. V.; Sparks, T. D. Not as simple as we thought: a rigorous examination of data aggregation in materials informatics. Digit. Discov. 2024, 3, 337-46.
48. Pedregosa, F.; Varoquaux, G.; Gramfort, A.; et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825-30. http://jmlr.org/papers/v12/pedregosa11a.html. (accessed 7 Nov 2025).
49. Hocky, G. M.; White, A. D. Natural language processing models that automate programming will transform chemistry research and teaching. Digit. Discov. 2022, 1, 79-83.
50. White, A. D.; Hocky, G. M.; Gandhi, H. A.; et al. Assessment of chemistry knowledge in large language models that generate code. Digit. Discov. 2023, 2, 368-76.
51. Microsoft Research AI4Science, Microsoft Azure Quantum. The impact of large language models on scientific discovery: a preliminary study using GPT-4. arXiv 2023, arXiv:2311.07361. Available online: https://doi.org/10.48550/arXiv.2311.07361. (accessed 7 Nov 2025).
52. Hare, P. M. Coding with AI in the Physical Chemistry Laboratory. J. Chem. Educ. 2024, 101, 3869-74.
53. Coudert, F. Reproducible research in computational chemistry of materials. Chem. Mater. 2017, 29, 2615-7.
54. Persaud, D.; Ward, L.; Hattrick-Simpers, J. Reproducibility in materials informatics: lessons from ‘A general-purpose machine learning framework for predicting properties of inorganic materials’. Digit. Discov. 2024, 3, 281-6.
55. Butler, K. T.; Choudhary, K.; Csanyi, G.; Ganose, A. M.; Kalinin, S. V.; Morgan, D. Setting standards for data driven materials science. npj. Comput. Mater. 2024, 10, 1411.
56. McDowell, D. L. Gaps and barriers to successful integration and adoption of practical materials informatics tools and workflows. JOM 2021, 73, 138-48.
57. The Minerals, Metals & Materials Society. MGI workforce. https://www.tms.org/MGIworkforce. (accessed 2025-11-07).
58. Wang, Z.; Chen, A.; Tao, K.; et al. AlphaMat: a material informatics hub connecting data, features, models and applications. npj. Comput. Mater. 2023, 9, 1086.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].