


Declining nitric oxide bioavailability in cardiovascular aging: mechanistic insights and emerging interventions
 0
0 
Abstract
Nitric oxide (NO) is essential for maintaining normal cardiovascular function, and accumulating evidence suggests that its diminished bioavailability contributes to endothelial dysfunction, vascular stiffening, and impaired cardiac performance - hallmarks of cardiovascular aging. This review posits that reduced NO bioavailability with age stems from impaired endothelial and neuronal NO synthase activity, increased oxidative stress, and metabolic shifts that drive cardiovascular decline. We further discuss emerging research which highlights potential interventions, including dietary nitrate supplementation, caloric restriction, and exercise, that may restore NO signaling and counteract age-related cardiovascular dysfunction. These findings underscore the growing recognition of NO as a key regulator of cardiovascular aging and a promising therapeutic target. Addressing NO-related deficits could open new avenues for preventing and treating age-associated cardiovascular diseases, reshaping strategies for promoting healthy aging and longevity.
Keywords
INTRODUCTION
Cardiovascular aging is a leading contributor to morbidity and mortality worldwide, driven by progressive impairments in endothelial function, vascular compliance, and myocardial performance that emerge with advancing age[1,2]. Over the past few decades, significant strides have been made in understanding the cellular and molecular alterations associated with aging, such as cellular senescence, mitochondrial dysfunction, and defects in intracellular signaling. Despite these advances, age-related cardiovascular morbidities persist, underscoring the need for deeper insight into the underlying mechanisms driving cardiovascular aging.
Nitric oxide (NO) signaling plays a pivotal role in regulating cardiovascular function, and its dysregulation is central to both vascular and myocardial aging. Initially recognized for its vasodilatory effects in the endothelium, NO is essential for maintaining vascular tone, and its age-related decline contributes to vascular dysfunction, characterized by impaired vasodilation, increased oxidative stress, and endothelial cell senescence. Beyond the endothelium, NO is now also known to influence a wide array of processes in cardiomyocytes, including excitation-contraction (EC) coupling[3-5], β-adrenergic responsiveness[6], mitochondrial function[7], and extracellular matrix remodeling[8]. However, its role in age-associated myocardial dysfunction remains less well understood. Studies over the past two decades have increasingly implicated dysregulated NO signaling - resulting from alterations in nitric oxide synthase (NOS) expression, cofactor availability, and oxidative stress - as a key mechanism underlying both vascular and myocardial dysfunction. However, our mechanistic understanding of how distinct NOS isoforms contribute to age-related alterations in NO bioavailability and signaling remains incomplete. As such, defining the precise role of NO in cardiovascular aging has become an area of active investigation, with growing recognition that restoring NO signaling may hold promise for mitigating age-related cardiac decline and improving outcomes in elderly patients with cardiovascular disease.
This review highlights the critical need to understand how aging disrupts NO production and signaling in both the endothelium and myocardium, and how these disruptions converge to drive vascular and myocardial dysfunction. While much of the foundational work in the field has focused on endothelial-derived NO and its role in vascular tone and systemic hemodynamics, comparatively less attention has been given to how NO signaling is altered within the aging myocardium itself. Addressing this gap is essential, as the heart is both a target and a modulator of nitroso-redox signaling. In this review, we examine how aging disrupts NO production and signaling in both the vasculature and myocardium, emphasizing the multifaceted consequences of NO deficiency - from endothelial dysfunction and oxidative stress to impaired myocardial relaxation and cardiac reserve. We aim to move beyond traditional paradigms by integrating mechanistic and translational insights that position NO not only as a marker of cardiovascular aging but as a causal driver of cellular senescence, dysfunction, and vulnerability to disease. In doing so, we hope to reframe NO signaling as a central, modifiable axis in age-related cardiovascular decline and to highlight promising directions for future investigation and therapeutic intervention.
NO SYNTHESIS AND REGULATION
In the healthy cardiovascular system, NO is synthesized through two distinct but complementary pathways that collectively regulate vascular tone, myocardial function, and cellular homeostasis. The primary source of NO is the NOS-dependent pathway, in which NOS enzymes catalyze the oxidation of L-arginine to produce NO and L-citrulline. In parallel, a NOS-independent pathway involving the sequential reduction of dietary or endogenous nitrate to nitrite and then NO serves as an alternative source, particularly under hypoxic or acidic conditions. Together, these pathways ensure tight spatial and temporal control of NO availability and signaling under physiological conditions. This section outlines the key mechanisms governing NO production and regulation in the unaged cardiovascular system, providing a foundation for understanding how these processes become dysregulated with advancing age [Figure 1].
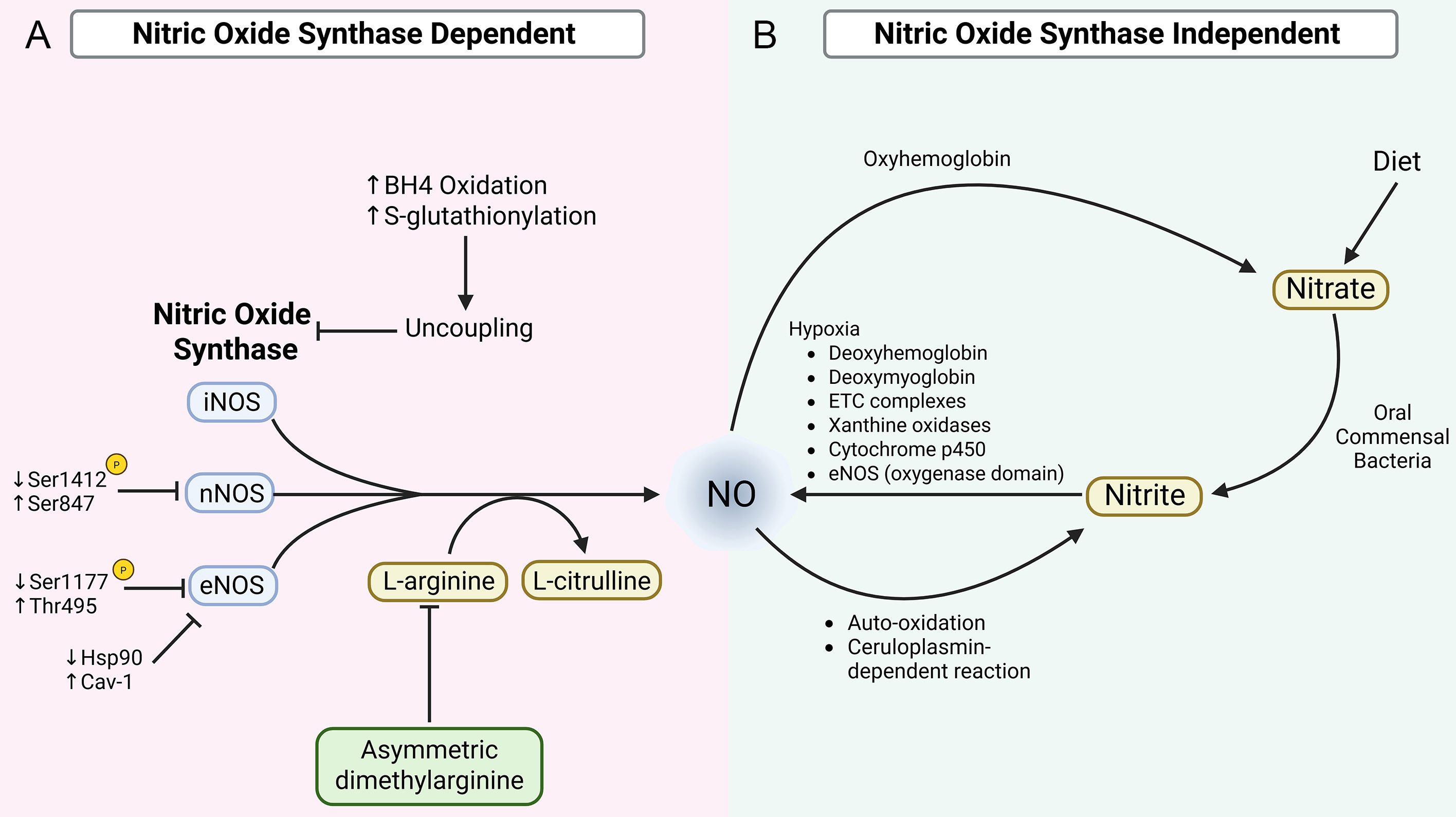
Figure 1. Synthesis and Regulation of NO. [(A): NOS-dependent NO production] NO is enzymatically synthesized through the conversion of L-arginine to L-citrulline by iNOS, nNOS, and eNOS. Each of these NOS isoforms is regulated by distinct phosphorylation sites, with phosphorylation of Ser1412 and dephosphorylation at Ser847 driving nNOS activity, and phosphorylation at Ser1177 and dephosphorylation at Thr495 driving eNOS activity. Age-related alterations of these phosphorylation states (as represented by the indicated arrows) reduce NOS activity in the cardiovascular system. Additionally, age-associated inhibitory NOS modulation by increased ADMA reduces L-arginine availability, further impairing NOS-mediated NO production. Moreover, age-associated depletion of BH4 levels and increased S-glutathionylation promote NOS uncoupling and further downregulate NO bioavailability. [(B) NOS-independent NO production] Alternatively, NO can also be produced by the reduction of nitrate and nitrite, derived from dietary sources or from the oxidation of NOS-derived NO. Circulating nitrate and nitrite act as storage forms of NO and can be reduced back to NO under hypoxic conditions. Reduction of nitrate/nitrite to NO is mediated by mechanisms involving deoxyhemoglobin, deoxymyoglobin, mitochondrial ETC complexes, xanthine oxidases, cytochrome 450, and the oxygenase domain of eNOS. Created in BioRender. Patel, P. (2025) https://BioRender.com/v5hq0f8.
NOS pathway and regulation
Three distinct isoforms of NOS exist in mammals - neuronal NOS (nNOS or NOS1), inducible NOS (iNOS or NOS2), and endothelial NOS (eNOS or NOS3) - each encoded by a separate gene. Despite their names, both nNOS and eNOS are constitutively expressed in the cardiovascular system, where they generate NO in a calcium-dependent manner[9,10]. In contrast, iNOS expression is negligible in a healthy cardiovascular system but can be transcriptionally activated in response to injury or inflammation[11-13].
All NOS isoforms function as homodimers, with each monomer consisting of an N-terminal oxygenase domain and a C-terminal reductase domain, linked together by a calmodulin (CaM)-binding sequence. These homodimers are bound and stabilized by heme, tetrahydrobiopterin (BH4), L-arginine, and zinc at the oxygenase domain[14-17]. When dimerized, electron transfer that drives NO synthesis begins at the reductase domain, where flavin cofactors (e.g., flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN)) and nicotinamide adenine dinucleotide phosphate (NADPH) initiate electron transfer to CaM[18,19]. From CaM, electrons are transferred to heme iron in the oxygenase domain, enabling the conversion of L-arginine and oxygen to L-citrulline and NO. Conversely, iNOS possesses a more tightly bound CaM, rendering it active upon expression, even at low calcium concentration[20]
In addition to dimerization, NOS activity and functionality are regulated by post-translational modifications and distinct intracellular localization. For instance, eNOS localizes to the plasma membrane caveolae and Golgi membrane upon myristoylation and palmitoylation of cysteine residues 15 and 26[21]. When eNOS is at the caveolae or the Golgi, phosphorylation of Ser1177 is required for activation and subsequent NO production[22]. Indeed, a critical mechanism underlying reduced NO bioavailability in vascular aging is the dysregulation of eNOS activity via inhibitory phosphorylation at Thr495. Phosphorylation at this residue stabilizes eNOS in an inactive conformation by sterically hindering CaM binding, a prerequisite for enzymatic activation[23]. While evidence of nNOS phosphorylation in cardiomyocytes remains limited, studies in neurons indicate that nNOS may also undergo inhibitory phosphorylation at Ser847 by calmodulin-dependent protein kinase I and II[24], whereas phosphorylation at Ser1412 by protein kinase B (also known as AKT serine/threonine kinase 1) enhances its activity[25]. Similar to eNOS and nNOS, tyrosine residues on iNOS can also be phosphorylated to increase its activity[26].
Furthermore, the allosteric binding of proteins to the NOS enzyme can also modulate functionality. At caveolae, the oxygenase domain of tyrosine-phosphorylated eNOS allosterically binds caveolin-1 (Cav-1)[27,28]. This interaction prevents the binding of CaM to eNOS, thereby inhibiting eNOS activity[29]. Both nNOS and eNOS are also inhibited by caveolin-3 (Cav-3)[30]. Conversely, heat-shock protein 90 (Hsp90), a chaperone protein that assists with protein folding and maintains protein stability, binds to eNOS and nNOS directly in a CaM-dependent manner[31,32]. This interaction releases NOS from Cav-1, thus allowing for enzyme activation. The binding of Hsp90 to both nNOS and eNOS also facilitates “re”-coupling of the enzyme and reduces superoxide (O2-) production[33].
Substrate/cofactor availability is further required for NOS activity. Among the NOS cofactors, BH4 plays an essential role in enabling electron transfer to promote L-arginine oxidation, NO formation, and release[34], in addition to its role as a homodimer stabilizer. During this reaction, electrons donated by NADPH at the carboxy-terminal reductase domain of NOS are passed to the heme catalytic center of the oxidase domain, where activation of molecular oxygen is “coupled” to NO synthesis by oxidation of L-arginine. The cofactor BH4 is required for these reactions; in its absence, electron flow to molecular oxygen becomes “uncoupled” from L-arginine oxidation, resulting in the production of O2- instead of NO[35,36]. Oxidation of BH4 to BH2 can also facilitate uncoupling, as BH2 competes with BH4 for binding to NOS[37]. Unlike BH4, BH2 lacks cofactor activity, and thus, when BH2 binds to NOS, it disrupts electron transfer, causing the enzyme to uncouple and produce O2-. The combination of increased oxidative stress and impaired NO signaling resulting from NOS uncoupling has been implicated in the pathogenesis of a wide range of disease states, including atherosclerosis[38], hypertension[39-41], diabetes[42,43] and more recently aging[44,45]. Additionally, post-translational modification of NOS by S-glutathionylation of cysteine residues in the reductase domain can also enable NOS uncoupling[46].
Nitrate-nitrite-NO
Historically, nitrate and nitrite were considered biologically inert molecules; however, extensive research over the past few decades has revealed that nitrate and nitrite serve as reserve pools of NO. Nitrate and nitrite are supplied to the body in two ways: (1) by the oxidation of endogenous NO produced by the NOS pathway; and (2) by consumption of vegetable diets rich in nitrate/nitrite. Evidence from pharmacological and genetic inhibition of eNOS demonstrating reduced nitrite level and elevated vascular resistance indicates that plasma nitrite concentration reflects NOS activity[47]. In fact, as much as 70% of nitrite is derived from NOS-dependent endogenous NO[47]. Under basal conditions, NO in the plasma reacts with oxyhemoglobin to produce methemoglobin and nitrate. NO also auto-oxidizes or undergoes ceruloplasmin-dependent reactions with copper to form nitrite[48].
Beyond endogenous production, dietary intake provides an additional source of nitrate, which undergoes sequential reduction in the body. Dietary nitrate is reduced to nitrite by commensal bacteria in the oral cavity[49]. Nitrite is then absorbed into the circulation, adding to the reserve pool in the plasma and tissues alongside NOS-dependent nitrite[48].
Nitrite is also readily reduced to NO under acidic and hypoxic conditions[50]. Rat hearts subjected to
Several other mammalian enzymes involved in nitrite reduction have been identified. Among these are xanthine oxidoreductases[55,56], cytochrome p450 enzymes[53], and eNOS. Most notably, under hypoxic conditions, eNOS does not convert L-arginine to NO[57], as there is little oxygen available. Rather, the oxygenase domain of eNOS serves as a location for nitrite reduction[57,58] and maintains NO levels to sustain vasodilation.
NO AND VASCULAR AGING
In the late 1970s and 1980s, a series of groundbreaking studies identified that NO stimulated soluble guanylyl cyclase (sGC) to produce cyclic guanosine 3′,5′-monophosphate (cGMP), subsequently activating protein kinase G (PKG) to induce smooth muscle relaxation and vasodilation[59,60]. Following this discovery, NO quickly became recognized as a critical signaling molecule in the vasculature, playing a key role in maintaining endothelial function, vascular tone, and overall cardiovascular health. The age-related decline of vascular health, coined "vascular aging", is characterized by increases in arterial stiffness, endothelial dysfunction, intima thickening, and chronic inflammation, all contributing to the pathogenesis of cardiovascular disease[61]. Given NO’s central role in vascular homeostasis, researchers began investigating whether age-related vascular dysfunction might be driven, at least in part, by alterations in NO signaling.
Changes in NO bioavailability in vascular aging
Many studies have directly measured NO bioavailability in aged models and have obtained mixed results, with some studies reporting elevated circulating NO levels[62-64], while others report reductions[65-67]. However, this can be explained given the dynamic and multifaceted roles of NO, as evaluating circulating NO levels may not be sufficient to fully understand the role of NO in aging: NO can either be released as a signaling agent through its interaction with thiol groups or be stored in tissues as nitrate/nitrite[68,69]. Despite evidence that nitrate/nitrite metabolism is maintained with age in rats, a recent study in young and old rats found that plasma and tissue nitrate levels increased with age, whereas nitrite levels decreased[70]. These findings suggest that nitrate and nitrite levels may be regulated independently, potentially via distinct pathways, and can vary with age and across different tissues.
Metabolites
Metabolites that can augment NO production, such as L-arginine, or inhibit NOS (e.g., asymmetric dimethylarginine (ADMA)) are reported to change with age. Serum collected from aging rats had a 70% decrease in L-arginine[71] and a ~47% increase in ADMA[72]. Likewise, healthy men, but not women, showed an increase in ADMA with age, which was negatively correlated with flow-mediated dilation in the popliteal artery[73]. L-arginine levels in women were shown to change throughout menopause, with a statistically nonsignificant decrease during pre-menopause and an increase post-menopause[74]. More studies that examine NO reservoirs and associated metabolites may lend more insight into NO bioavailability changes that may occur during aging and explain the differences observed in levels of NO bioavailability.
NOS expression and activity
Variations in NOS expression have also been observed with aging and may contribute to the differences in NO bioavailability reported. As with circulating NO, studies on age-related changes in NOS expression and activity have yielded inconsistent results. In the aortas of aged rats, iNOS expression and activity rise, whilst eNOS expression is elevated, yet activity is decreased[75,76]. Another study found reduced Ser1177 phosphorylation and elevated phospho-Thr495 levels, supporting the finding of decreased eNOS activity observed in aortic endothelium from aged rats[77]. Contrary to these studies, van der Loo et al. (2000) found increased eNOS expression and NOS activity in aged rat aortas[67], although the levels of BH4 and its oxidation status were not measured. In humans, aging has been associated with increased eNOS Ser1177 phosphorylation, indicating increased eNOS activity[78], although the balance between activating (Ser1177) and inhibitory (Thr495) phosphorylation sites was not determined. Aged human umbilical vein endothelial cells (HUVECs), however, do exhibit a 6-fold increase in Thr495 phosphorylation alongside reduced Ser1177 phosphorylation, directly impairing NO synthesis[79,80]. This imbalance arises from age-related upregulation of Rho-kinase (ROCK), which directly phosphorylates Thr495, and from diminished protein kinase B (Akt/PKB) signaling responsible for Ser1177 activation[80]. Co-morbidities, such as hypertension, may further influence age-associated changes in NOS isoform expression and activity. Hypertension in aging rats shifts eNOS and iNOS expression by decreasing eNOS levels and raising iNOS[81]. Furthermore, Sirtuin 1 (SIRT1), a NAD+-dependent deacetylase, enhances eNOS activity by deacetylating and upregulating its expression. As NAD+ levels decline with age, SIRT1 expression and activity decrease, resulting in a decrease in eNOS activity[82-85]. Studies have demonstrated that activation of SIRT1 reduces vascular aging in aged mice[86-88]. Thus, despite extensive study, the full complexity of age-related NOS regulation remains unresolved, and investigations into more nuanced, context-specific changes may offer deeper insight. Moreover, observed differences in NOS expression may reflect species-specific variability or compensatory responses aimed at preserving NO bioavailability - hypotheses that warrant further investigation.
NOS uncoupling
In addition to changes in the expression of NOS, NO bioavailability becomes attenuated by the uncoupling of electron flow from arginine oxidation in eNOS. This mechanism results in a decreased production of NO and instead generates O2-[89]. Studies have examined age-induced eNOS uncoupling and have reported elevations in BH2 and diminished BH4 levels and significant O2- production[90-92]. Similarly, in the mesenteric arteries of aged mice, the eNOS monomer/dimer ratio was elevated in old mice, and N(G)-nitro-L-arginine methyl ester (L-NAME) inhibitable O2- production was elevated[91], findings that were replicated in the aortas of aged rats[93,94]. Importantly, in all these studies of eNOS uncoupling in aged animal models, O2- levels and vascular function were restored following supplementation with either BH4 or its precursor, sepiapterin, demonstrating the importance of BH4 and maintenance of eNOS coupling in the vasculature. Moreover, a reduction in the binding of eNOS to Hsp90, which facilitates the coupling of eNOS, has been observed in aged piglets[95]. Additionally, Cav-1 expression is increased in aged endothelial cells, resulting in the sequestration of eNOS within the caveolae[32]. Together, the reduction in Hsp90 and the increase in Cav-1 further exacerbate the uncoupling and decrease the activity of eNOS. The age-related uncoupling of eNOS can lead to an increase in O2-, which can react with NO to form peroxynitrite[96]. This new free radical can then react with tyrosine residues in proteins to produce 3-nitrotyrosine, a marker of nitrosative stress[96]. Consistent with this idea, an increase in total protein nitrotyrosine is observed in the vasculature of aged mice[91]. Specifically, an increase in nitrotyrosine and eNOS colocalization in aged mesenteric arteries was observed. This post-translational modification of eNOS by nitrotyrosine not only impairs its enzymatic activity but also perpetuates a deleterious cycle of reduced NO production and increased oxidative stress in the aging vasculature.
NOS cofactor availability
Changes in NOS cofactor availability with age may also indirectly affect NO bioavailability. Arginase isoforms (Arg-I & II) convert L-arginine into L-ornithine as part of the urea cycle to dispose of ammonia[97]. This enzyme is an important regulator of NO production as it competes with NOS for L-arginine. As Arginase expression or activity increases, endothelial dysfunction also rises, and NO bioavailability decreases[98]. This relationship between arginase activity, endothelial dysfunction, and NO bioavailability mirrors the patterns observed in vascular aging[99]. Despite these parallels, investigations into arginase expression and activity in aged tissues have yielded conflicting findings, where some studies show that Arginase activity and expression rise in aged animals[100-102], while others observe no difference[103,104] or have reported a decrease[105,106]. Together, these findings underscore the multifactorial regulation of NO signaling in the aging vasculature, involving alterations in substrate/cofactor availability, enzyme expression and activity, and redox balance.
Sex-specific differences in NO signaling and vascular aging
Sex-specific differences in NO signaling emerge as a critical determinant of vascular aging patterns between men and women, with estrogen and testosterone exerting distinct modulatory effects on NO bioavailability and cardiovascular health. Premenopausal women demonstrate superior vascular protection through estrogen-mediated enhancement of eNOS expression and activity, resulting in greater NO synthesis, improved endothelium-dependent vasodilation, and lower arterial blood pressure compared to age-matched men[107-109]. This protective effect involves multiple mechanisms including estrogen receptor-α activation that upregulates eNOS gene expression, enhanced phosphorylation at activating sites (Ser1177), and preservation of BH4 cofactor availability, collectively maintaining eNOS coupling and preventing O2- production[109,110]. The menopause transition marks a critical inflection point where women experience an accelerated decline in endothelial function that surpasses the gradual deterioration observed in men, coinciding with estrogen deficiency-induced reductions in NO bioavailability, increased oxidative stress, and loss of anti-inflammatory protection[109,111,112]. Conversely, testosterone’s role in male vascular aging presents a more complex picture, with physiological concentrations promoting NO synthesis through androgen receptor-mediated activation of extracellular signal-regulated protein kinase (ERK)1/2 and phosphatidylinositol 3 kinase (PI3K)/Akt pathways[113], yet age-related testosterone decline potentially contributes to endothelial dysfunction through increased endothelin-1 signaling and reduced antioxidant capacity[111,113,114]. The differential reliance on basal NO bioavailability also exhibits sex-specific patterns, with females demonstrating greater dependence on endogenous NO production while males show enhanced responsiveness to exogenous NO supplementation, suggesting fundamental differences in vascular NO regulation that may inform targeted therapeutic strategies for age-related cardiovascular dysfunction[115]. These sex-specific differences in NO signaling underscore the importance of personalized approaches to cardiovascular aging interventions, recognizing that optimal strategies for preserving vascular health may vary significantly between men and women across different life stages.
Impact of changes in NO bioavailability on vascular aging
Vasodilation
Although reported changes in NO bioavailability in aged animals have been inconsistent, there is a clearer consensus regarding the functional consequences of NO signaling on vascular function with age. In fact, a decrease in NO-dependent endothelial vasodilation is now considered a hallmark characteristic of aging vasculature. Studies using vascular rings (aortic or mesenteric) from older animals consistently demonstrate reduced relaxation responses to NO donors compared to those from young animals[116,117]. Furthermore, acetylcholine-induced vasodilation, which is inhibited by NOS blockers in young animals and humans, becomes NOS-independent or is abolished altogether in older counterparts[117,118]. Supporting this, BH4 infusion has been shown to restore vasodilation in healthy older men by enhancing NO production[119]. Interestingly, vascular function appears better preserved in aging females[120,121]. Overall, these studies have largely shown that aging is associated with a decline in NO-mediated vascular responsiveness.
Oxidative stress
Another NO-driven hallmark of aging is the increase in oxidative and nitrosative stress[122,123]. With age, NO is scavenged by reactive oxygen species (ROS), specifically O2-. NO reacts rapidly with O2- to form peroxynitrite[96]. This new free radical can then react with tyrosine residues in proteins to produce 3-nitrotyrosine, a marker of nitrosative stress. For example, the peroxynitrite nitration of manganese O2- dismutase (MnSOD) inhibits its function as a ROS scavenger and further exacerbates oxidative stress[67]. Changes in NO bioavailability in turn may also be influenced by the rise in ROS. ROS promotes eNOS uncoupling, further exacerbating the reduced bioavailability of NO[124]. A study found that aortic tissue isolated from aged rats had elevated O2-production and 3-nitrotyrosine modifications compared to the young rats[67]. This indicates that an overall increase in oxidative stress may influence the loss of NO bioavailability with aging. Increased oxidative stress accelerates vascular aging by inducing molecular and pathological changes. One significant consequence is the deterioration of arterial structural integrity with aging[125]. Conversely, anti-oxidative treatments have been shown to mitigate these effects, helping to prevent large artery stiffening and preserve vascular function[126].
Metabolism
The functional integrity of the heart and vasculature is dependent on normal cellular metabolism. A growing body of research highlights the impact of NO on energy metabolism in vascular aging through mitochondria. Mitochondria are critical organelles involved in energy metabolism, and their decline in function has been associated with aging. NO can impair mitochondrial function both directly, through interactions with the energy transport chain, and indirectly, through increased nitrosative and oxidative stress. NO can directly impact the ETC by either binding, nitration, or s-nitrosylation of various complexes, impairing their function and inhibiting mitochondrial respiration[127]. As the efficacy of the ETC decreases, electron leakage increases, resulting in elevated ROS. The proximity of ROS to mitochondrial DNA (mtDNA) increases mtDNA mutations, which further accelerates mitochondrial decline. Mitochondrial oxidative stress is a key characteristic of aging that promotes vascular dysfunction[128-131]. Studies have shown that treatment with mitochondrial-targeted antioxidants, such as mitoquinol mesylate (MitoQ), resveratrol, and SS-31 (d-Arg-2′,6′-dimethyltyrosine-Lys-Phe-NH2), has improved endothelial function in aged arteries of rodents[87,130,132]. Furthermore, increased peroxynitrite availability promotes post-translational modifications, such as tyrosine nitration, altering the structure and function of critical mitochondrial proteins[127,133,134]. NO promotes mitochondrial fission by nitrosating Parkin, which upregulates dynamin-related protein 1 (Drp1)[135], the primary driver of mitochondrial fission. NO also increases Drp1 phosphorylation at Ser616, further activating mitochondrial fission[135]. Increased S-nitrosated Drp1 is often observed in aged cells and is associated with impaired energy metabolism[136]. Overall, impaired NO signaling in aging disrupts mitochondrial bioenergetics and function, further exacerbating vascular dysfunction.
Cellular senescence
Cellular senescence, the cessation of cell proliferation after a cell has reached its maximum division potential, is a natural component of aging. Studies accelerating senescence in aging models have shown a progression of heart failure and endothelial dysfunction[137-139]. Moreover, chronic senolytic treatment improved endothelial dysfunction in aged mice[140]. NO has been shown to regulate cellular senescence by enhancing telomerase activity, which is essential for maintaining chromosomal stability and prolonging cell lifespan[141]. In HUVECs, treatment with an NO donor decreased the proportion of senescent cells, whereas inhibition with L-NAME increased the proportion of senescent cells[141]. This suggests that NO plays a protective role against endothelial senescence and vascular aging. Furthermore, the expression of SIRT1 in senescent cells is also reduced, which subsequently leads to decreased activity of eNOS and further diminishes the protective effects of NO[83].
Inflammation
Aging is associated with chronic low-grade inflammation, often referred to as “inflammaging”[142]. Studies have shown that aging is correlated with a shift in gene expression favoring pro-inflammatory cytokines[131,143,144]. The consequences of chronic inflammation on vascular dysfunction and formation of atherosclerotic plaques are well-studied. NO can exert anti-inflammatory effects in the vasculature by inhibiting leukocyte adhesion and vascular smooth muscle cell (VSMC) proliferation[145,146]. However, with aging, reduced NO bioavailability diminishes these protective effects and exacerbates vascular inflammation. Moreover, NO-dependent elevated ROS levels activate nuclear factor kappa B (NF-κB), which promotes the expression of pro-inflammatory mediators. Both endothelial cells and VSMCs exhibit increased NF-κB activity with aging[86,147]. Additionally, in aged rat arterioles, an increase in expression of iNOS and formation of peroxynitrite is observed[148]. The increase in peroxynitrite promotes vascular dysfunction in addition to iNOS-induced regulation of inflammatory cytokines, such as tumor necrosis factor-alpha (TNFα). Inhibition of NO-induced apoptosis in coronary arteries by activating TNFα; however, treatment of aged arteries with NO donor or TNFα inhibitor decreased this apoptosis[149].
Angiogenesis
Angiogenesis is an essential physiological process for tissue repair and homeostasis. However, aberrations in angiogenesis contribute to a variety of age-associated pathologies, including stroke, myocardial infarction, and cancer. Aging is associated with a decline in angiogenic capacity, which has been linked to reduced vascular NO bioavailability[150]. Multiple studies have demonstrated that various NO delivery methods enhance angiogenesis in animal models[151-154]. NO influences angiogenesis through several mechanisms, including endothelial cell proliferation, tip cell formation, and migration. One study found that NO enhances endothelial cell proliferation by modulating DNA synthesis. This effect was abolished following pretreatment with the NO inhibitor, L-NAME[155]. NO modulates the formation of tip cells via the regulation of the sGC pathway. In human endothelial cells, L-NAME decreases tip cell formation, whereas the presence of an NO donor increases tip cell formation[156]. Furthermore, NO has been shown to enhance endothelial cell migration, another essential step in angiogenesis. Studies using HUVECs demonstrated increased cell migration upon treatment with NO donors. Similarly, in ex vivo models, NO donors promoted angiogenesis in rat aortic rings, further emphasizing the pro-angiogenic properties of NO[157]. Moreover, NO can control angiogenesis through the vascular endothelial growth factor (VEGF) signaling pathway. NO activates hypoxia-inducible factor 1-alpha (HIF-1α), which upregulates VEGF expression[158]. The addition of NO donors or cofactors increases VEGF synthesis, while inhibition of NO production via L-NAME decreases VEGF synthesis in VSMCs[159]. Moreover, VEGF-induced endothelial cell proliferation is attenuated by L-NAME[160]. NO is a critical regulator of angiogenesis, and thus, the limited bioavailability with age greatly impacts the vascular angiogenic capacity.
Changes in NO bioavailability are a critical mechanism of vascular aging, contributing to endothelial dysfunction, inflammation, arterial stiffening, and culminating in the progression of atherosclerosis. Over time, atherosclerosis and endothelial dysfunction will increase pulse pressure and cardiac afterload, compounding the risk of cardiovascular diseases. Collectively, NO-related changes drive an atherosclerotic phenotype in aging, resulting in an increased risk for cardiovascular disease.
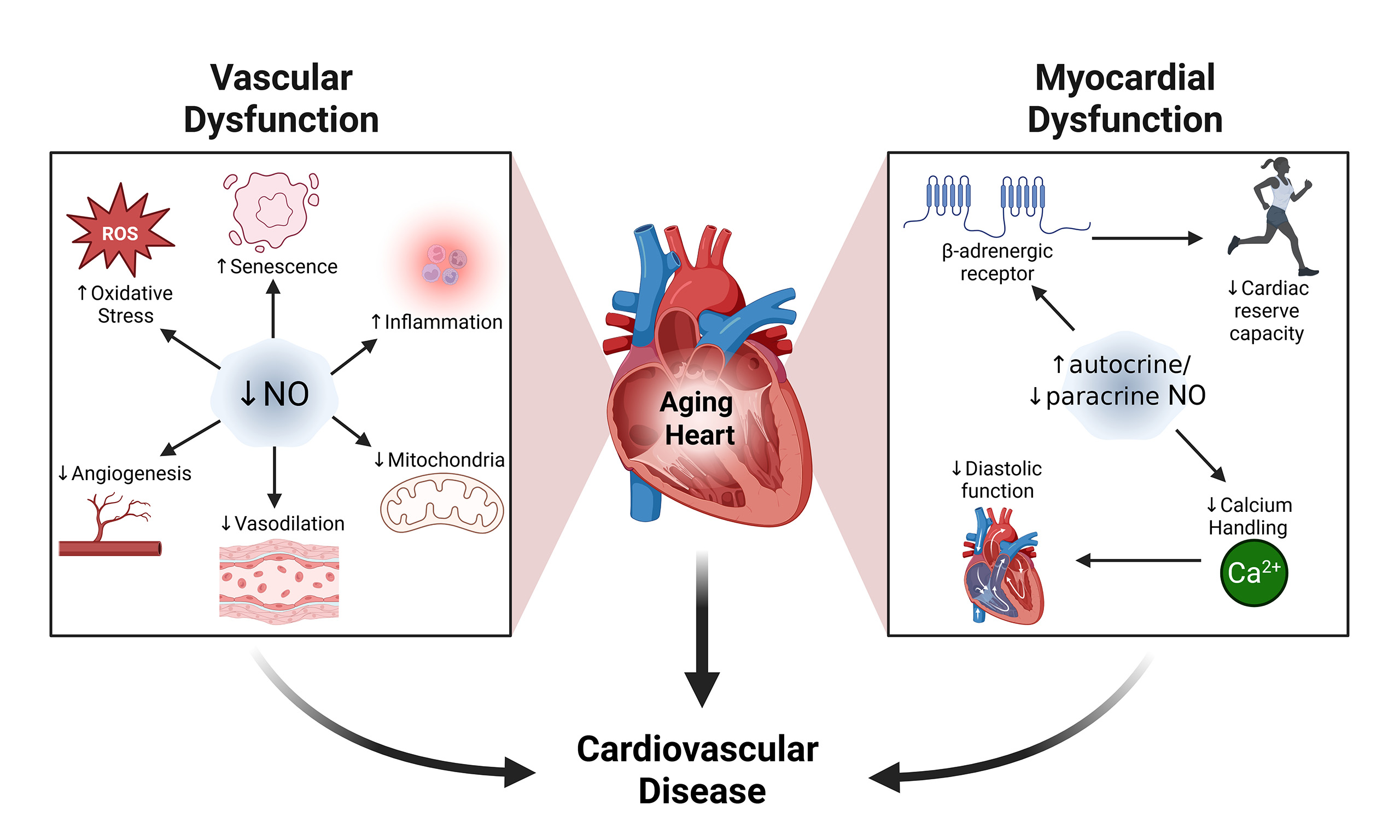
In summary, age-related changes in NO signaling are multifactorial and affect vascular health through complex and interconnected mechanisms involving altered NOS expression and activity, cofactor availability, increased oxidative stress, and disrupted mitochondrial metabolism. These disruptions contribute to hallmark features of vascular aging, including endothelial dysfunction, arterial stiffness, and impaired metabolic resilience [Figure 2]. Critically, the processes of vasodilation, oxidative stress, metabolism, cellular senescence, inflammation and angiogenesis are not distinct processes but rather are interconnected dynamically, with reduced NO availability acting as a central link. Reduced NO availability limits vasodilatory capacity and promotes oxidative stress, which in turn amplifies inflammatory signaling and accelerates endothelial dysfunction. These changes further reduce NO availability which stimulates cellular senescence, further impairing NO signaling and metabolic flexibility within the vascular wall. Viewing these mechanisms as interconnected nodes of a dynamic system highlights the crucial role of NO in cardiovascular aging and its potential as a target to decelerate cardiovascular aging. Given the central role of NO in cardiovascular physiology, many of these mechanisms may also extend beyond the vasculature to influence myocardial aging - a topic explored in the next section.
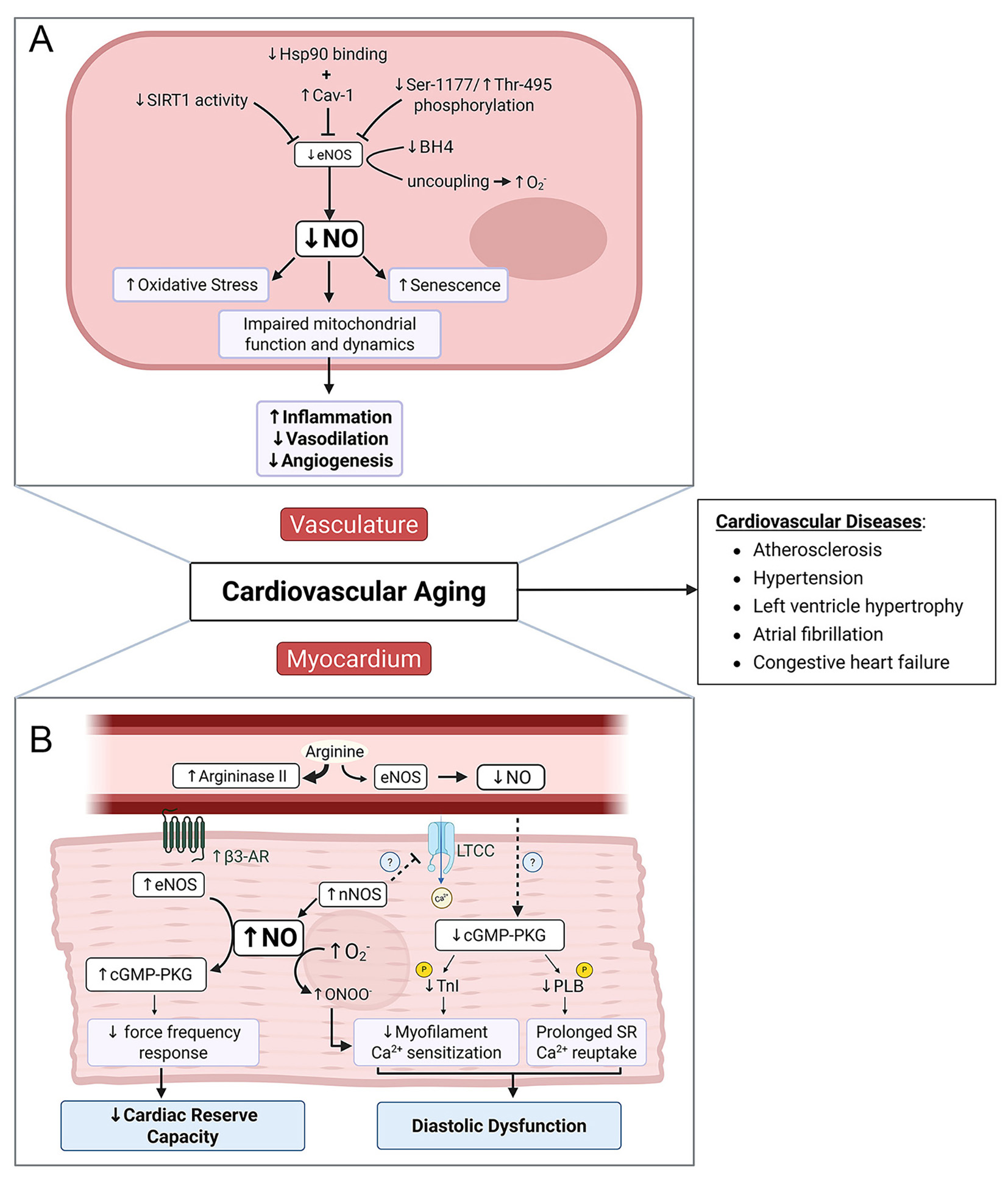
Figure 2. Mechanisms of Age-Associated Decline in NO Signaling and Its Contribution to Cardiovascular Dysfunction. [(A): Vasculature] In the vascular endothelium, aging-associated declines in eNOS activity and NO bioavailability are driven by reduced SIRT1 activity, elevated Cav-1 expression, and diminished Hsp90-eNOS interaction. Post-translational modifications - specifically, reduced phosphorylation at Ser1177 and increased phosphorylation at Thr495 - further suppress eNOS activity. Concurrently, age-related depletion of BH4 levels promotes eNOS uncoupling, shifting its activity toward O2- production. The resulting reduction in NO bioavailability contributes to increased oxidative stress, mitochondrial dysfunction, and endothelial cell senescence. Collectively, these changes impair vascular function by promoting inflammation, reducing vasodilation, and attenuating angiogenesis. [(B) Myocardium] With advancing age, diminished NO bioavailability in the endothelium reduces paracrine signaling to cardiomyocytes, likely contributing to the downregulation of the cGMP-PKG pathway and, ultimately, diastolic dysfunction. This decline is further aggravated by the formation of peroxynitrite, generated from the interaction between nNOS-derived NO and O2-. In contrast, heightened β3-adrenergic receptor activity stimulates cardiomyocyte-associated eNOS-cGMP-PKG signaling, which leads to reduced inotropy, particularly at elevated heart rates, thus lowering cardiac reserve capacity. Plasmalemmal translocation of nNOS may further act to inhibit L-type calcium channels and negatively impact cardiac EC coupling, although this remains to be fully established. Together, age-associated declines in NO bioavailability in both the vascular endothelium and myocardium contribute to cardiovascular dysfunction and elevate the risk of life-threatening cardiovascular diseases. Created in BioRender. Patel, P. (2025) https://BioRender.com/v5hq0f8.
NO AND MYOCARDIAL AGING
In conjunction with vascular impairment, normal cardiovascular aging is associated with negative alterations in myocardial structure and function, including impaired diastolic filling, reduced cardiac reserve capacity, left ventricular (LV) hypertrophy, atrial enlargement, and changes in cardiac rhythm or rate[161]. While these changes do not constitute overt cardiovascular disease, they significantly reduce exercise tolerance, increase frailty, and lower the threshold for clinical symptoms, ultimately diminishing quality of life. Moreover, age-related cardiac remodeling alters the myocardial substrate in ways that amplify the severity of heart disease and contribute to poorer outcomes in older adults. Indeed, among all co-morbidities, aging-related cardiovascular dysfunction is a key contributor to the disproportionately higher incidence of LV hypertrophy, atrial fibrillation, and congestive heart failure observed in older adults[162-164], underscoring the need to identify underlying molecular mechanisms and potential therapeutic targets.
Changes in NO bioavailability in myocardial aging
Although numerous studies have examined age-related changes in NOS expression and activity in the aging myocardium, conclusions regarding NO bioavailability and the specific origins of NOS-associated effects - whether from the coronary endothelium or cardiomyocytes - remain discrepant and incomplete. In general, nNOS expression within the heart has been shown to increase with age[165-167], whereas eNOS has been reported to be unaltered[100,167], elevated[168], or reduced[166,169]. For those few studies that have, in turn, evaluated calcium-dependent NOS activity in the aged left ventricle, most suggest an overall increase[166,168]. However, increased NOS activity does not necessarily equate to greater NO production or signaling. Indeed, several studies report an age-associated upregulation of Arg-II[100,170], which competes with NOS for the shared substrate L-arginine, potentially limiting NO synthesis and reducing its bioavailability.
Supporting this mechanism, pharmacological inhibition of Arg-II has been shown to enhance myocardial NO bioavailability in aged rat hearts and in isolated cardiomyocytes from senescent hearts Arg-II inhibition restored the force-frequency relationship[100] - a biophysical property known to be regulated by NO[171,172]. Similarly, L-arginine supplementation improves diastolic function in aged rat hearts[168], although its direct effects on myocardial NO levels were not assessed. Beyond limitations in NO synthesis, aging is also associated with increased oxidative stress, which further depletes NO. As earlier noted, elevated O2- levels - driven by upregulated iNOS[100] and enhanced NADPH oxidase activity[167] - can react with NO to form the more reactive and cytotoxic free radical, peroxynitrite[167,173], leading to reduced NO signaling. Thus, while the precise effects of aging on myocardial nitroso-redox signaling remain unresolved, mounting evidence suggests that altered NOS activity, substrate competition, and oxidative stress collectively reduce NO bioavailability in the aging heart.
Functional consequences of reduced NO in the aging myocardium
Calcium handling and diastolic dysfunction
Age-associated diastolic dysfunction and diminished contractile reserve are hallmark features of myocardial aging that limit cardiac performance, particularly during exertion. These functional impairments are driven by delayed calcium transient decay rates, reduced contraction amplitude, and prolonged time to peak contraction[174-176]. Mechanistically, these effects stem from reduced peak L-type calcium currents, sarcoplasmic reticulum (SR) calcium uptake rates, and slower cross-bridge cycling, which occur independently of broad changes in the expression of EC coupling proteins, with the exception of consistently reported reductions in sarco (endo) plasmic reticulum calcium ATPase (SERCA) expression and activity[174,177,178]. Rather, emerging evidence suggests that these functional deficits occur secondary to age-associated alterations in cellular signaling and impaired post-translational regulation of core EC coupling proteins.
Both autocrine and paracrine NO signaling are essential for tuning EC coupling at baseline and in response to stress[12]. Yet, while few studies have explored whether changes in NO bioavailability or the broader nitroso-redox balance feed into age-related cardiac dysfunction, there is reason to believe this may play a prominent role. Indeed, NO is known to modulate SR calcium uptake and, thus, cardiac relaxation via both paracrine signaling from endothelial-derived eNOS[179-181] and autocrine effects of nNOS-derived NO within cardiomyocytes[182-184]. As mentioned above, studies using L-arginine supplementation or Arg-II inhibition have shown improvements in diastolic dysfunction in aged rat hearts owing to restored NOS-derived NO production[100,168]. Although speculative, cursory evidence suggests this reduction in NO bioavailability is driven by a reduction in eNOS-derived NO from the microcirculatory endothelium within the heart. This reduction appears to be linked to Arg-II upregulation[100,170], which competes with NOS for the shared substrate L-arginine[185], thus limiting NO synthesis. As this Arg-II upregulation is present in non-myocytic cells within the heart[170], it suggests the loss of NO occurs outside of the cardiomyocyte and, thus, contributes to cardiac dysfunction via diminished paracrine NO signaling. Such paracrine NO-dependent regulation of relaxation is reported to occur through eNOS-cGMP-PKG signaling, which increases phospholamban (PLB) phosphorylation at Ser17, leading to enhanced SERCA activity and increased SR calcium uptake[40,186]. Studies have also shown that eNOS-cGMP-PKG signaling can directly alter troponin I phosphorylation and cross-bridge cycling kinetics, thus speeding contraction and relaxation, especially at higher heart rates[40,180,181]. Age-associated loss of such eNOS-cGMP-PKG signaling would, therefore, be expected to not only contribute to impaired relaxation but also limit the cardiac reserve capacity by diminishing the force-frequency relationship. Consistent with this, Arg-II inhibition not only improves NO bioavailability but also restores the force-frequency relationship in aged myocardium[100].
While reductions in eNOS-derived NO may contribute to impaired vascular-cardiac signaling and myocardial relaxation with age, increasing evidence suggests that alterations in cardiomyocyte-intrinsic nNOS activity could also play a critical yet complex role in diastolic dysfunction. In young hearts, nNOS-derived NO supports efficient calcium handling and relaxation by promoting PLB phosphorylation and enhancing SR calcium reuptake via cAMP-dependent protein kinase A (PKA)[182,183,187]. In contrast, aging is associated with heightened nNOS activity in the context of increased oxidative stress, leading to elevated peroxynitrite formation and protein nitration. These redox-driven changes impair SR calcium cycling and increase myofilament stiffness, thereby exacerbating diastolic dysfunction[167,173]. Notably, genetic ablation of nNOS in aged mice reverses calcium transient prolongation and improves myocardial relaxation - an effect attributed to reduced O2- and peroxynitrite levels - highlighting a detrimental shift in nNOS signaling with age[167]. These contrasting findings underscore the need for greater spatial and temporal resolution in assessing NOS isoform activity and redox dynamics in the aging heart. Additionally, age-related disruptions in NO-mediated regulation of nearby oxidases (e.g., Xanthine Oxidoreductase) and their downstream targets[3,12] remain poorly defined. Thus, although altered NO signaling is a central contributor to diastolic dysfunction in the aged heart, the molecular mechanisms remain incompletely understood and may very well be context dependent.
Beta-adrenergic stimulation and cardiac reserve capacity
While persistent changes in EC coupling machinery are likely to diminish the inotropic capacity of the heart over time, much of the reduced cardiac reserve capacity noted in aging humans has been ascribed to attenuated beta-adrenergic responsiveness[188-190], despite elevated levels of circulating catecholamines[191]. In senescent hearts from rats and mice, beta1-adrenergic receptor (β1-AR) expression has consistently been shown to be reduced, leading to a decrease in cAMP/PKA-dependent signaling and a diminished inotropic response[165,188-190]. However, in parallel, beta3-adrenergic receptor (β3-AR) expression is reportedly upregulated in the aging heart, where it contributes significantly to the attenuated inotropic response to β-adrenergic stimulation observed in both isolated muscle preparations and intact aging animals[165]. Both L-NAME and N5-(1-Imino-3-butenyl)-l-ornithine (L-VNIO), two pan-NOS inhibitors, partially restored the positive inotropic effect of β-adrenergic stimulation in isolated LV papillary muscles from aged hearts, indicating the involvement of NOS-derived NO. Nevertheless, it remains unclear whether the functional effects are primarily driven by eNOS-dependent coupling with β3-AR, or by nNOS relocalization to the plasmalemmal membrane, as seen in senescent post-myocardial infarction hearts[169]. Support for the former comes from studies in healthy, young hearts, where eNOS is known to mediate β-adrenergic signaling through both autocrine and paracrine effects of NO[171,192,193]. In these models, NO from eNOS has been shown to attenuate the inotropic response to adrenergic stimulation, largely through the β3-AR-NOS signaling axis[194-196]. This pathway involves NO’s inhibition of the L-type Ca2+ current and phosphorylation of cardiac troponin I, which suppresses myocardial contraction[193,197]. Conversely, findings in isolated ventricular cardiomyocytes suggest that nNOS-derived NO may also contribute to blunted β-adrenergic responsiveness[187,198,199], specifically through its association with sarcoplasmic Cav-3[169,198]. Notably, sarcolemmal translocation of nNOS would not only provide a mechanistic basis for the age-related attenuation of β-adrenergic responsiveness but could also contribute to reduced myofilament calcium sensitivity and impaired force-frequency relationships, potentially explaining the decline in early diastolic filling observed in aging hearts[3,12,200]. While further research is needed to delineate the dominant mechanism, the interplay between NOS isoforms and β-adrenergic signaling appears likely to contribute to impaired contractile function and reduced cardiac reserve with age.
While beyond the scope of this review, it is worth noting that the development and severity of several age-associated cardiovascular diseases, including atrial fibrillation and heart failure, are strongly correlated with the extent of myocardial NOS-derived NO production (see[12,201,202] for further details). Moreover, studies using nNOS knockout mice have demonstrated that loss of nNOS-derived NO alone is sufficient to increase AF susceptibility[203] and promote age-dependent maladaptive LV hypertrophy, even in the absence of arterial hypertension[171,204,205]. Additionally, dysregulated NO signaling has been implicated in impaired lusitropy (as detailed above), mitochondrial dysfunction, and adverse extracellular matrix remodeling[206,207] - key factors that exacerbate myocardial stiffness and diastolic dysfunction in aging hearts. Thus, altered NO bioavailability may not merely contribute to age-associated cardiac dysfunction but also fundamentally reshape the myocardial substrate on which heart disease is superimposed, likely worsening pathology and clinical outcomes for older patients.
ENHANCEMENT OF NO TO PROMOTE HEALTHY CARDIOVASCULAR AGING
Across a range of model organisms, studies aimed at uncovering the molecular basis of healthy aging and extended lifespan have repeatedly converged on a common mechanism: enhanced NO signaling. From yeast to nematodes to mammals, boosting NO levels - whether through increased enzymatic production or exogenous sources such as commensal bacteria - has been linked to improved mitochondrial function, stress resistance, and longevity[208-210]. These findings, though diverse in model and mechanism, underscore the conserved and fundamental role of NO in promoting organismal health across the lifespan. Building on this foundation, the following section explores strategies to enhance NO bioavailability and promote cardiovascular health in humans. These include both endogenous and exogenous approaches, such as dietary nitrate supplementation, stimulation of NOS activity, and lifestyle interventions such as caloric restriction (CR) and exercise. By examining these interventions, we highlight promising approaches to counteract cardiovascular aging through targeted enhancement of NO signaling, with the hope of encouraging further testing of similar therapeutic strategies to improve longevity and quality of life in the future.
Dietary supplementation to enhance nitrate-nitrite
As previously noted, an alternate method for nitrate and nitrite to enter circulation is through the consumption of nitrate-rich foods that are reduced by commensal oral bacteria[48,49]. This results in increased nitrite concentration in the saliva, some of which is then absorbed into circulation[211]. Following this accumulation, nitrite is physiologically reduced into NO by heme-containing proteins, including hemoglobin[68]. Elevating circulating nitrite levels has, in turn, been shown to improve endothelial function and reduce oxidative stress markers in healthy, older patients and aged animals[65,212-219]. These studies indicate that supplementation of exogenous NO sources might counteract the decreasing NO bioavailability with age. Given these results, providing nitrate/nitrite-based therapies to increase NO bioavailability for aging populations has been appealing, especially for those at risk of disease.
Beetroot juice is rich in nitrate and has been a promising, low-cost intervention strategy to reduce hypertension and subsequent cardiovascular disease risk[220-222]. In a recent randomized, controlled trial, the effects of beetroot juice on oxidative stress and inflammation were assessed in patients between 56-71 years old with diagnosed hypertension. Four weeks of beetroot juice mildly improved antioxidant levels and reduced some inflammatory markers[223]; however, consumption of beetroot juice for four weeks did not significantly alter vascular function or blood pressure in older hypertensive patients[224]. These results have been observed by other groups as well, although some studies do report changes in blood pressure following nitrate consumption[225-228]. Co-morbidities may be a key factor to consider with nitrate/nitrite therapeutics. Age and sex discrepancies, for example, may be important attributes for the effect of these therapeutics[121,229]. Nevertheless, nitrate/nitrite therapeutics are likely to improve age-related vascular function in healthy populations, but co-morbidities may mitigate these effects, perhaps by potential differences in NO production mechanisms[229].
Dietary supplementation to boost NOS-derived NO
Apart from dietary supplementation, interventions have also focused on increasing NO bioavailability through the stimulation of NOS production or reduction of NO scavenging by ROS. Increasing BH4 availability has been shown by multiple clinical studies to improve endothelial function, especially endothelium-dependent vasodilation, via increased NOS coupling and NO production[119,230-233]. Interestingly, however, acute BH4 supplementation was found to have no effect on improving vascular dysfunction in subjects older than 65[234].
With age, L-arginine levels are reportedly reduced, so ingestion of L-citrulline, a precursor of L-arginine in the de novo arginine synthesis pathway, may stimulate endogenous de novo L-arginine synthesis. A study found that in the fasted state, patients > 60 years old with heart failure had significantly lower de novo arginine synthesis and NO synthesis rates compared to adults between 21-40, and citrulline ingestion resulted in an increase in NO synthesis in both groups[235]. While some studies have shown that L-arginine supplementation can be beneficial due to the high activity levels of arginase in the small intestine, nitrate/nitrite is considered a more effective method of increasing NO bioavailability[218]. However, many studies examining the effects of supplements on enhancing NOS-mediated NO bioavailability have been conducted on healthy populations, and further research is needed in populations with co-morbidities.
Caloric restriction
CR, defined as reduced caloric intake without malnutrition or starvation, has recently garnered recognition as a promising non-pharmacological intervention to mitigate age-associated diseases and promote longevity[236-238]. Evidence from mice, rats, and humans subjected to varying degrees of CR has collectively demonstrated that CR alleviates age-induced increases in blood pressure[239,240], lowers pulse wave velocity[239], improves arterial dilation[241], and reduces metabolic syndrome score[240] (an indicator of cardiometabolic risk). These effects are mediated, in part, through the impact of CR on NO-dependent pathways. Indeed, CR has been shown to upregulate eNOS expression, which then promotes mitochondrial biogenesis[242] and improves endothelial function in the aging vascular systems[242,243]. CR also supports LV function recovery in aged hearts following ischemia/reperfusion[244]. These beneficial effects of CR are significantly attenuated by genetic ablation or pharmacological inhibition of NOS expression[242,243,245,246], thus highlighting the necessity of NOS enzymes in CR-mediated cardioprotection.
The timing of CR initiation is also a critical determinant of its efficacy, as late-onset short-term CR in older mice shows no influence on eNOS expression[76,247]. Interestingly, however, late-onset CR decreases iNOS expression, subsequently reducing NO levels[76]. While reduced iNOS expression indicates a less inflammatory state, a low NO level implies that NO replenishment might be limited with late-onset CR. Conversely, Rippe et al. (2010) demonstrated that short-term CR in older mice led to a slight increase in eNOS expression and a significant increase in NO bioavailability. Interestingly, this increase in NO bioavailability was primarily attributed to the CR-dependent decrease in O2- bioactivity[241]. Although consensus exists regarding the involvement of NOS enzymes in CR-mediated cardioprotection, the precise duration of CR and the downstream molecular mechanisms affecting NOS activity remain to be fully elucidated.
Much of the research on CR-induced changes in NO highlights the role of SIRT1 as a key regulatory factor. Increased expression and nuclear localization of SIRT1 have been identified as pivotal upstream mechanisms driving CR-dependent increase in NOS expression and activity[243,246]. Deacetylation of eNOS by SIRT1 increases eNOS activity and thus, NO bioavailability[85]. Interestingly, elevated eNOS expression can also facilitate nuclear localization of SIRT1[246], suggesting that eNOS and SIRT1 engage in a positive feedback loop to mutually regulate each other's activity. Further investigation into the SIRT1-NOS interaction under CR is warranted to unravel the molecular basis of CR-induced vascular and metabolic improvements. Such insights could also reveal novel therapeutic targets in the CR-SIRT1-NOS axis to combat age-associated diseases and promote healthy aging.
In addition to SIRT1, several other nutrient-signaling pathways have also been implicated in modulating NOS activity under CR. For example, activation of insulin signaling, marked by activating phosphorylation of Akt, has been associated with increased adiponectin levels in serum from CR-subjected rats. This signaling cascade promotes eNOS phosphorylation, thereby enhancing eNOS activity and elevating NO production[248]. Moreover, CR reduces mTOR phosphorylation, rendering it inactive[239], while simultaneously increasing adenosine monophosphate-activated protein kinase (AMPK) phosphorylation[244]. This dual modulation of the two nutrient sensing pathways may help improve cardiovascular function and prevent age-associated decline, potentially through an interaction between these nutrient sensors and NO production pathways discussed above.
While significant progress has been made in understanding the role of NO-dependent pathways in CR-mediated benefits, many critical questions remain unanswered. For instance, although CR-mediated changes in NO levels in aging vascular systems are extensively studied, the precise molecular mechanisms underlying the interplay between NOS enzymes and nutrient-sensing pathways in response to CR require further exploration. Furthermore, while the role of NO in vascular health is well-established, its role in the myocardium is less clear. Whether CR-driven improvements in myocardial function following age-related cardiac disease, such as ischemia/reperfusion[244], are mediated by alterations in the NO signaling pathway remains largely unexplored. Addressing these gaps in knowledge is essential for advancing our understanding of how CR-driven modulation of NO signaling pathways contributes to protecting the aging vascular system.
Exercise
Along with diet, physical activity is a key modifiable factor influencing cardiovascular disease risk and may exert protective effects in part by improving endothelial function and vascular health with aging[249,250]. Several studies have shown that sedentary individuals are more likely to develop cardiovascular diseases, whereas increased physical activity significantly reduces this risk[251]. Notably, NO bioavailability is lower in sedentary adults as compared to their physically active counterparts[226]. Recent evidence further demonstrates that older individuals who consistently engaged in more than five hours of high-intensity exercise per week over three decades had higher circulating NO levels than those who performed less than two hours of moderate-intensity exercise per week over the same period[252]. In contrast to the long-term effects of regular exercise, acute bouts of exercise have been shown to transiently increase NO production and reduce resting blood pressure in elderly women, while also improving vascular NO responsiveness[253-256].
In aging animal models, eNOS protein expression increases despite unchanged eNOS messenger RNA (mRNA) levels; however, exercise elevates both mRNA and protein expression, leading to improved NO-mediated vasodilation and reduced arterial stiffness[257,258]. Enhancing NO bioavailability with age is thought not only to support vascular function but also to enhance exercise tolerance and performance in older adults[259]. A systematic review evaluated the effects of nitrite supplementation on exercise performance in both young and older individuals. A meta-analysis of over 100 studies and reviews revealed that while outcomes varied based on biological factors, the most substantial benefits were observed in young, healthy, and physically active men[260]. Thus, while the cardiovascular benefits of exercise are well established, emerging evidence suggests that increased NO bioavailability may be a key mediator of these effects[261].
Current therapeutics and future strategies
Apart from lifestyle interventions, many advances have been made towards enhancing NO bioavailability in the aging cardiovascular system. Pharmacological agents, such as sodium nitroprusside, act as direct NO donors and have historically been used for the acute treatment of hypertensive emergencies[262]. More recently, their safety and efficacy have also been demonstrated in clinical trials involving patients with acute heart failure[263]. On the other hand, nitrate esters such as nitroglycerin, isosorbide dinitrate, and isosorbide mononitrate, which are metabolized to liberate NO, are widely used for treating angina[264], heart failure[265], and other acute coronary syndromes[266]. Interestingly, however, the use of nitroglycerin in conjunction with adjunctive therapies, including antithrombotic or lipid-lowering agents, has been associated with an increased incidence of major adverse cardiovascular events in patients over the age of 75, while no measurable benefit or harm was observed in younger patients[267]. Similarly, a recent preclinical study in rat models found no cardioprotective benefit of nitroglycerin administration in limiting infarct size, either at baseline or in the context of metabolic syndrome[268]. In contrast, another study demonstrated that nitroglycerin, but not isosorbide dinitrate, improved post-ischemia/reperfusion outcomes[269]. These variable responses to traditional direct NO donors may stem from common co-morbidities in older patients, the lack of tissue- or pathway-specific targeting, and the heightened oxidative stress in the aging cardiovascular system, where NO rapidly reacts with O2- to form peroxynitrite. Collectively, these limitations underscore the imminent need to better understand how NO-mediated pathways are regulated in different biological and clinical settings, and to develop more targeted, personalized therapeutic strategies that safely and efficiently elicit NO-dependent benefits in age-associated cardiovascular diseases.
Recent insights into the SIRT1-NOS-NO pathway have created such opportunities for targeted therapeutics. SIRT1-activating compounds such as resveratrol[270] have garnered extensive interest as potential cardioprotective agents[271,272], given that aging is associated with reduced SIRT1 activity, lower NO levels, and increased NOS uncoupling. Notably, preclinical studies have demonstrated resveratrol’s ability to reduce NOS uncoupling, enhance NO production, and limit O2- accumulation through upregulation of guanosine triphosphate (GTP) cyclohydrolase, the rate-limiting enzyme in the BH4 synthesis pathway[273]. Clinical trials have further shown promising outcomes of resveratrol in diabetes[274], ischemia/reperfusion[275], hypertension[276,277], and related cardiovascular diseases[278]. More recently, novel SIRT1 activators, such as SRT2104, have entered clinical trials with growing evidence showing their efficacy in treating cardiovascular diseases[279].
Nicotinamide adenine dinucleotide (NAD+) serves as a key co-substrate for SIRT1. Thus, studies have also focused on NAD+-boosting approaches to increase SIRT1 activity. NAD+ precursors, such as niacin and nicotinamide riboside, have been explored for their potential to enhance SIRT1 activity and support NO signaling[88,280]. Preclinical studies have demonstrated their cardioprotective effects in the context of aortic aneurysms, ischemia/reperfusion, and other aging-associated cardiovascular complications[281,282].
Altogether, these advances highlight the therapeutic potential of selectively enhancing SIRT1 signaling to preserve NO bioavailability in the aging heart. More broadly, these advances emphasize the promise of precision therapeutics that modulate NO signaling through pathway-specific mechanisms.
Beyond SIRT1 agonists, other classes of drugs such as peroxisome proliferator-activated receptor-alpha (PPAR) agonists[283,284] and β-adrenergic receptor blockers[285], are currently in clinical use and have been reported to enhance NOS activity; however, the specific pathway underlying this increase and the extent to which they contribute to NO-dependent benefits in patients with age-associated cardiovascular diseases remain unclear. A deeper focus on the upstream substrates, enzymes, and cofactors that govern NO bioavailability will be essential for ensuring consistent, clinically meaningful NO-dependent benefits in aging cardiovascular diseases.
CONCLUSION
NO is a critical regulator of cardiovascular homeostasis, and its dysregulation represents a hallmark of vascular and myocardial aging. This review has outlined how both NOS-dependent and nitrate-nitrite pathways of NO synthesis are altered with age, contributing to a spectrum of deleterious effects - from impaired vasodilation and increased oxidative stress to reduced myocardial performance and diminished cardiac reserve. These age-associated changes in NO signaling are mechanistically linked to endothelial dysfunction, cellular senescence, inflammation, and metabolic decline, core features of cardiovascular aging. Moreover, compelling evidence across model organisms and human studies suggests that restoring NO bioavailability can counteract these effects and promote healthier aging. Interventions such as dietary nitrate supplementation, NOS-boosting nutrients, CR, and regular exercise offer promising strategies for reactivating endogenous NO pathways.
Emerging evidence highlights several promising targets for preserving NO bioavailability and mitigating cardiovascular aging. These include enzymes regulating substrate and cofactor availability (e.g., arginase II, NAD+ metabolism), signaling pathways such as SIRT1 that maintain eNOS activity, and mitochondrial sources of oxidative stress that contribute to NO depletion. Strategies to modulate these targets, in parallel with lifestyle interventions, represent important avenues for future translational research. Further mechanistic studies are needed to determine how these pathways interact and whether their combined modulation can enhance resilience of the aging cardiovascular system. Ultimately, targeting NO pathways represents a promising and increasingly actionable approach to extending cardiovascular health span to reduce the burden of age-related cardiovascular disease.
DECLARATIONS
Acknowledgment
The graphical abstract was created with BioRender.com (Created in BioRender. Patel, P. (2025) https://BioRender.com/v5hq0f8).
Authors’ contributions
Wrote the first draft of the manuscript: Adhikari H, Patel P, Javvaji N
Provided critical revisions to the manuscript: Crabtree MJ, Simon JN
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the W. W. Smith Charitable Trust Fund grant H1206 (to Simon JN), a National Institutes of Health T32 grant 5T32HL091804-15 (to Adhikari H), and a Surrey University PhD studentship and faculty research support (to Crabtree MJ).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a "set up" for vascular disease. Circulation. 2003;107:139-46.
2. Vakka A, Warren JS, Drosatos K. Cardiovascular aging: from cellular and molecular changes to therapeutic interventions. J Cardiovasc Aging. 2023;3:23.
3. Khan SA, Lee K, Minhas KM, et al. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci USA. 2004;101:15944-8.
4. Sears CE, Bryant SM, Ashley EA, et al. Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ Res. 2003;92:e52-9.
5. Hegyi B, Shimkunas R, Jian Z, Izu LT, Bers DM, Chen-Izu Y. Mechanoelectric coupling and arrhythmogenesis in cardiomyocytes contracting under mechanical afterload in a 3D viscoelastic hydrogel. Proc Natl Acad Sci USA. 2021;118:e2108484118.
6. Irie T, Sips PY, Kai S, et al. S-Nitrosylation of calcium-handling proteins in cardiac adrenergic signaling and hypertrophy. Circ Res. 2015;117:793-803.
7. Loke KE, Laycock SK, Mital S, et al. Nitric oxide modulates mitochondrial respiration in failing human heart. Circulation. 1999;100:1291-7.
8. Cuadrado I, Castejon B, Martin AM, et al. Nitric oxide induces cardiac protection by preventing extracellular matrix degradation through the complex caveolin-3/EMMPRIN in cardiac myocytes. PLoS One. 2016;11:e0162912.
9. Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc Natl Acad Sci USA. 1999;96:657-62.
10. Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J Biol Chem. 1996;271:22810-4.
11. Kinugawa K, Shimizu T, Yao A, Kohmoto O, Serizawa T, Takahashi T. Transcriptional regulation of inducible nitric oxide synthase in cultured neonatal rat cardiac myocytes. Circ Res. 1997;81:911-21.
12. Simon JN, Duglan D, Casadei B, Carnicer R. Nitric oxide synthase regulation of cardiac excitation-contraction coupling in health and disease. J Mol Cell Cardiol. 2014;73:80-91.
13. Roy R, Wilcox J, Webb AJ, O'Gallagher K. Dysfunctional and dysregulated nitric oxide synthases in cardiovascular disease: mechanisms and therapeutic potential. Int J Mol Sci. 2023;24:15200.
14. Raman CS, Li H, Martásek P, Král V, Masters BS, Poulos TL. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell. 1998;95:939-50.
15. Hemmens B, Goessler W, Schmidt K, Mayer B. Role of bound zinc in dimer stabilization but not enzyme activity of neuronal nitric-oxide synthase. J Biol Chem. 2000;275:35786-91.
16. Zhao Y, Vanhoutte PM, Leung SW. Vascular nitric oxide: beyond eNOS. J Pharmacol Sci. 2015;129:83-94.
17. Crane BR, Rosenfeld RJ, Arvai AS, et al. N-terminal domain swapping and metal ion binding in nitric oxide synthase dimerization. EMBO J. 1999;18:6271-81.
18. Fleming I. Molecular mechanisms underlying the activation of eNOS. Pflugers Arch. 2010;459:793-806.
19. Carlström M, Weitzberg E, Lundberg JO. Nitric oxide signaling and regulation in the cardiovascular system: recent advances. Pharmacol Rev. 2024;76:1038-62.
20. Piazza M, Guillemette JG, Dieckmann T. Dynamics of nitric oxide synthase-calmodulin interactions at physiological calcium concentrations. Biochemistry. 2015;54:1989-2000.
21. García-Cardeña G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci USA. 1996;93:6448-53.
22. Fulton D, Fontana J, Sowa G, et al. Localization of endothelial nitric-oxide synthase phosphorylated on serine 1179 and nitric oxide in Golgi and plasma membrane defines the existence of two pools of active enzyme. J Biol Chem. 2002;277:4277-84.
23. Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr495 regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:E68-75.
24. Komeima K, Hayashi Y, Naito Y, Watanabe Y. Inhibition of neuronal nitric-oxide synthase by calcium/calmodulin-dependent protein kinase IIalpha through Ser847 phosphorylation in NG108-15 neuronal cells. J Biol Chem. 2000;275:28139-43.
25. Sharma NM, Patel KP. Post-translational regulation of neuronal nitric oxide synthase: implications for sympathoexcitatory states. Expert Opin Ther Targets. 2017;21:11-22.
26. Tyryshkin A, Gorgun FM, Abdel Fattah E, et al. Src kinase-mediated phosphorylation stabilizes inducible nitric-oxide synthase in normal cells and cancer cells. J Biol Chem. 2010;285:784-92.
27. García-Cardeña G, Fan R, Stern DF, Liu J, Sessa WC. Endothelial nitric oxide synthase is regulated by tyrosine phosphorylation and interacts with caveolin-1. J Biol Chem. 1996;271:27237-40.
28. Ju H, Zou R, Venema VJ, Venema RC. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J Biol Chem. 1997;272:18522-5.
29. Chen Z, D S Oliveira S, Zimnicka AM, et al. Reciprocal regulation of eNOS and caveolin-1 functions in endothelial cells. Mol Biol Cell. 2018;29:1190-202.
30. García-Cardeña G, Martasek P, Masters BS, et al. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem. 1997;272:25437-40.
31. García-Cardeña G, Fan R, Shah V, et al. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821-4.
32. Gratton JP, Fontana J, O'Connor DS, Garcia-Cardena G, McCabe TJ, Sessa WC. Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro. Evidence that hsp90 facilitates calmodulin stimulated displacement of eNOS from caveolin-1. J Biol Chem. 2000;275:22268-72.
33. Zheng H, Li J, Feng C. Heat shock protein 90 enhances the electron transfer between the FMN and heme cofactors in neuronal nitric oxide synthase. FEBS Lett. 2020;594:2904-13.
34. Alkaitis MS, Crabtree MJ. Recoupling the cardiac nitric oxide synthases: tetrahydrobiopterin synthesis and recycling. Curr Heart Fail Rep. 2012;9:200-10.
35. Crabtree MJ, Hale AB, Channon KM. Dihydrofolate reductase protects endothelial nitric oxide synthase from uncoupling in tetrahydrobiopterin deficiency. Free Radic Biol Med. 2011;50:1639-46.
36. McNeill E, Channon KM. The role of tetrahydrobiopterin in inflammation and cardiovascular disease. Thromb Haemost. 2012;108:832-9.
37. Crabtree MJ, Smith CL, Lam G, Goligorsky MS, Gross SS. Ratio of 5,6,7,8-tetrahydrobiopterin to 7,8-dihydrobiopterin in endothelial cells determines glucose-elicited changes in NO vs. superoxide production by eNOS. Am J Physiol Heart Circ Physiol. 2008;294:H1530-40.
38. Li L, Chen W, Rezvan A, Jo H, Harrison DG. Tetrahydrobiopterin deficiency and nitric oxide synthase uncoupling contribute to atherosclerosis induced by disturbed flow. Arterioscler Thromb Vasc Biol. 2011;31:1547-54.
39. Landmesser U, Dikalov S, Price SR, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201-9.
40. Silberman GA, Fan TH, Liu H, et al. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation. 2010;121:519-28.
41. Elbatreek MH, Sadegh S, Anastasi E, et al. NOX5-induced uncoupling of endothelial NO synthase is a causal mechanism and theragnostic target of an age-related hypertension endotype. PLoS Biol. 2020;18:e3000885.
42. Roe ND, Thomas DP, Ren J. Inhibition of NADPH oxidase alleviates experimental diabetes-induced myocardial contractile dysfunction. Diabetes Obes Metab. 2011;13:465-73.
43. Wu HE, Baumgardt SL, Fang J, et al. Cardiomyocyte GTP cyclohydrolase 1 protects the heart against diabetic cardiomyopathy. Sci Rep. 2016;6:27925.
44. Nakhaee S, Azadi R, Salehinia H, et al. The role of nitric oxide, insulin resistance, and vitamin D in cognitive function of older adults. Sci Rep. 2024;14:30020.
45. Phua TJ. Hallmarks of aging: middle-aging hypovascularity, tissue perfusion and nitric oxide perspective on healthspan. Front Aging. 2024;5:1526230.
46. Chen CA, Wang TY, Varadharaj S, et al. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468:1115-8.
47. Kleinbongard P, Dejam A, Lauer T, et al. Plasma nitrite reflects constitutive nitric oxide synthase activity in mammals. Free Radic Biol Med. 2003;35:790-6.
48. Kapil V, Khambata RS, Jones DA, et al. The noncanonical pathway for in vivo nitric oxide generation: the nitrate-nitrite-nitric oxide pathway. Pharmacol Rev. 2020;72:692-766.
49. Duncan C, Dougall H, Johnston P, et al. Chemical generation of nitric oxide in the mouth from the enterosalivary circulation of dietary nitrate. Nat Med. 1995;1:546-51.
50. Zweier JL, Wang P, Samouilov A, Kuppusamy P. Enzyme-independent formation of nitric oxide in biological tissues. Nat Med. 1995;1:804-9.
51. Cosby K, Partovi KS, Crawford JH, et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498-505.
52. Shiva S, Huang Z, Grubina R, et al. Deoxymyoglobin is a nitrite reductase that generates nitric oxide and regulates mitochondrial respiration. Circ Res. 2007;100:654-61.
53. Feelisch M, Fernandez BO, Bryan NS, et al. Tissue processing of nitrite in hypoxia: an intricate interplay of nitric oxide-generating and -scavenging systems. J Biol Chem. 2008;283:33927-34.
54. Castello PR, David PS, McClure T, Crook Z, Poyton RO. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab. 2006;3:277-87.
55. Millar TM, Stevens CR, Benjamin N, Eisenthal R, Harrison R, Blake DR. Xanthine oxidoreductase catalyses the reduction of nitrates and nitrite to nitric oxide under hypoxic conditions. FEBS Lett. 1998;427:225-8.
56. Li H, Samouilov A, Liu X, Zweier JL. Characterization of the effects of oxygen on xanthine oxidase-mediated nitric oxide formation. J Biol Chem. 2004;279:16939-46.
57. Gautier C, van Faassen E, Mikula I, Martasek P, Slama-Schwok A. Endothelial nitric oxide synthase reduces nitrite anions to NO under anoxia. Biochem Biophys Res Commun. 2006;341:816-21.
58. Mikula I, Durocher S, Martasek P, Mutus B, Slama-Schwok A. Isoform-specific differences in the nitrite reductase activity of nitric oxide synthases under hypoxia. Biochem J. 2009;418:673-82.
59. Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3':5'-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci USA. 1977;74:3203-7.
60. Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524-6.
61. Cheng DCY, Climie RE, Shu M, Grieve SM, Kozor R, Figtree GA. Vascular aging and cardiovascular disease: pathophysiology and measurement in the coronary arteries. Front Cardiovasc Med. 2023;10:1206156.
62. Maurya PK, Rizvi SI. Alterations in plasma nitric oxide during aging in humans. Ind J Biochem Biophys. 2009;46:130-2.
63. Ozdemir S, Yargiçoğlu P, Ağar A, Gümüşlü S, Bîlmen S, Hacioğlu G. Role of nitric oxide on age-dependent alterations: investigation of electrophysiologic and biochemical parameters. Int J Neurosci. 2002;112:263-76.
64. Toprakçi M, Ozmen D, Mutaf I, et al. Age-associated changes in nitric oxide metabolites nitrite and nitrate. Int J Clin Lab Res. 2000;30:83-5.
65. Rossman MJ, Gioscia-Ryan RA, Santos-Parker JR, et al. Inorganic nitrite supplementation improves endothelial function with aging: translational evidence for suppression of mitochondria-derived oxidative stress. Hypertension. 2021;77:1212-22.
66. Siervo M, Hussin AM, Calella P, et al. Associations between aging and vitamin D status with whole-body nitric oxide production and markers of endothelial function. J Nutr. 2024;154:469-78.
67. van der Loo B, Labugger R, Skepper JN, et al. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731-44.
68. DeMartino AW, Kim-Shapiro DB, Patel RP, Gladwin MT. Nitrite and nitrate chemical biology and signalling. Br J Pharmacol. 2019;176:228-45.
69. Hess DT, Stamler JS. Regulation by S-nitrosylation of protein post-translational modification. J Biol Chem. 2012;287:4411-8.
70. Piknova B, Park JW, Thomas SM, Tunau-Spencer KJ, Schechter AN. Nitrate and nitrite metabolism in aging rats: a comparative study. Nutrients. 2023;15:2490.
71. Moretto J, Guglielmetti AS, Tournier-Nappey M, et al. Effects of a chronic l-arginine supplementation on the arginase pathway in aged rats. Exp Gerontol. 2017;90:52-60.
72. Xiong Y, Yuan LW, Deng HW, Li YJ, Chen BM. Elevated serum endogenous inhibitor of nitric oxide synthase and endothelial dysfunction in aged rats. Clin Exp Pharmacol Physiol. 2001;28:842-7.
73. Nakamura A, Kajitani S, Sato K, et al. Decline of popliteal artery flow-mediated dilation with aging and possible involvement of asymmetric dimethylarginine in healthy men. J Med Ultrason. 2019;46:503-11.
74. Klawitter J, Hildreth KL, Christians U, Kohrt WM, Moreau KL. A relative L-arginine deficiency contributes to endothelial dysfunction across the stages of the menopausal transition. Physiol Rep. 2017;5:e13409.
75. Karaś A, Bar A, Pandian K, et al. Functional deterioration of vascular mitochondrial and glycolytic capacity in the aortic rings of aged mice. Geroscience. 2024;46:3831-44.
76. Zanetti M, Gortan Cappellari G, Burekovic I, Barazzoni R, Stebel M, Guarnieri G. Caloric restriction improves endothelial dysfunction during vascular aging: effects on nitric oxide synthase isoforms and oxidative stress in rat aorta. Exp Gerontol. 2010;45:848-55.
77. Smith AR, Visioli F, Frei B, Hagen TM. Age-related changes in endothelial nitric oxide synthase phosphorylation and nitric oxide dependent vasodilation: evidence for a novel mechanism involving sphingomyelinase and ceramide-activated phosphatase 2A. Aging Cell. 2006;5:391-400.
78. Donato AJ, Gano LB, Eskurza I, et al. Vascular endothelial dysfunction with aging: endothelin-1 and endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2009;297:H425-32.
79. Lee HY, Zeeshan HMA, Kim HR, Chae HJ. Nox4 regulates the eNOS uncoupling process in aging endothelial cells. Free Radic Biol Med. 2017;113:26-35.
80. Sugimoto M, Nakayama M, Goto TM, Amano M, Komori K, Kaibuchi K. Rho-kinase phosphorylates eNOS at threonine 495 in endothelial cells. Biochem Biophys Res Commun. 2007;361:462-7.
81. Chou TC, Yen MH, Li CY, Ding YA. Alterations of nitric oxide synthase expression with aging and hypertension in rats. Hypertension. 1998;31:643-8.
82. Gomes AP, Price NL, Ling AJ, et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624-38.
83. Gong H, Pang J, Han Y, et al. Age-dependent tissue expression patterns of Sirt1 in senescence-accelerated mice. Mol Med Rep. 2014;10:3296-302.
84. Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One. 2012;7:e42357.
85. Mattagajasingh I, Kim CS, Naqvi A, et al. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2007;104:14855-60.
86. Gano LB, Donato AJ, Pasha HM, Hearon CM Jr, Sindler AL, Seals DR. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am J Physiol Heart Circ Physiol. 2014;307:H1754-63.
87. Pearson KJ, Baur JA, Lewis KN, et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157-68.
88. Ganji S, Kamanna S, Kamanna VS, Kashyap ML. Niacin increases human aortic endothelial Sirt1 activity and nitric oxide: effect on endothelial function and vascular aging. Am J Transl Res. 2023;15:6771-8.
89. Bendall JK, Douglas G, McNeill E, Channon KM, Crabtree MJ. Tetrahydrobiopterin in cardiovascular health and disease. Antioxid Redox Signal. 2014;20:3040-77.
90. Perrier E, Fournet-Bourguignon MP, Royere E, et al. Effect of uncoupling endothelial nitric oxide synthase on calcium homeostasis in aged porcine endothelial cells. Cardiovasc Res. 2009;82:133-42.
91. Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. 2009;297:H1829-36.
92. Bouly M, Bourguignon MP, Roesch S, et al. Aging increases circulating BH2 without modifying BH4 levels and impairs peripheral vascular function in healthy adults. Transl Res. 2021;238:36-48.
93. Kim JH, Bugaj LJ, Oh YJ, et al. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol. 2009;107:1249-57.
94. Delp MD, Behnke BJ, Spier SA, Wu G, Muller-Delp JM. Ageing diminishes endothelium-dependent vasodilatation and tetrahydrobiopterin content in rat skeletal muscle arterioles. J Physiol. 2008;586:1161-8.
95. Aschner JL, Zeng H, Kaplowitz MR, Zhang Y, Slaughter JC, Fike CD. Heat shock protein 90-eNOS interactions mature with postnatal age in the pulmonary circulation of the piglet. Am J Physiol Lung Cell Mol Physiol. 2009;296:L555-64.
96. Piacenza L, Zeida A, Trujillo M, Radi R. The superoxide radical switch in the biology of nitric oxide and peroxynitrite. Physiol Rev. 2022;102:1881-906.
97. Li Z, Wang L, Ren Y, et al. Arginase: shedding light on the mechanisms and opportunities in cardiovascular diseases. Cell Death Discov. 2022;8:413.
98. Wernly B, Pernow J, Kelm M, Jung C. The role of arginase in the microcirculation in cardiovascular disease. Clin Hemorheol Microcirc. 2020;74:79-92.
99. Santhanam L, Christianson DW, Nyhan D, Berkowitz DE. Arginase and vascular aging. J Appl Physiol. 2008;105:1632-42.
100. Khan M, Steppan J, Schuleri KH, et al. Upregulation of arginase-II contributes to decreased age-related myocardial contractile reserve. Eur J Appl Physiol. 2012;112:2933-41.
101. Masi S, Colucci R, Duranti E, et al. Aging modulates the influence of arginase on endothelial dysfunction in obesity. Arterioscler Thromb Vasc Biol. 2018;38:2474-83.
102. Pandya CD, Lee B, Toque HA, et al. Age-dependent oxidative stress elevates arginase 1 and uncoupled nitric oxide synthesis in skeletal muscle of aged mice. Oxid Med Cell Longev. 2019;2019:1704650.
103. Liu P, Smith PF, Appleton I, Darlington CL, Bilkey DK. Age-related changes in nitric oxide synthase and arginase in the rat prefrontal cortex. Neurobiol Aging. 2004;25:547-52.
104. Liu P, Smith PF, Appleton I, Darlington CL, Bilkey DK. Hippocampal nitric oxide synthase and arginase and age-associated behavioral deficits. Hippocampus. 2005;15:642-55.
105. Liu P, Smith PF, Appleton I, Darlington CL, Bilkey DK. Regional variations and age-related changes in nitric oxide synthase and arginase in the sub-regions of the hippocampus. Neuroscience. 2003;119:679-87.
106. Liu P, Smith PF, Appleton I, Darlington CL, Bilkey DK. Nitric oxide synthase and arginase in the rat hippocampus and the entorhinal, perirhinal, postrhinal, and temporal cortices: regional variations and age-related changes. Hippocampus. 2003;13:859-67.
107. Kittnar O. Selected sex related differences in pathophysiology of cardiovascular system. Physiol Res. 2020;69:21-31.
108. Majmudar NG, Robson SC, Ford GA. Effects of the menopause, gender, and estrogen replacement therapy on vascular nitric oxide activity. J Clin Endocrinol Metab. 2000;85:1577-83.
109. Somani YB, Pawelczyk JA, De Souza MJ, Kris-Etherton PM, Proctor DN. Aging women and their endothelium: probing the relative role of estrogen on vasodilator function. Am J Physiol Heart Circ Physiol. 2019;317:H395-404.
110. Novella S, Dantas AP, Segarra G, Medina P, Hermenegildo C. Vascular aging in women: is estrogen the fountain of youth? Front Physiol. 2012;3:165.
111. Moreau KL. Modulatory influence of sex hormones on vascular aging. Am J Physiol Heart Circ Physiol. 2019;316:H522-6.
112. Moreau KL, Gavin KM, Plum AE, Seals DR. Ascorbic acid selectively improves large elastic artery compliance in postmenopausal women. Hypertension. 2005;45:1107-12.
113. Lopes RA, Neves KB, Carneiro FS, Tostes RC. Testosterone and vascular function in aging. Front Physiol. 2012;3:89.
114. Babcock MC, DuBose LE, Witten TL, et al. Oxidative stress and inflammation are associated with age-related endothelial dysfunction in men with low testosterone. J Clin Endocrinol Metab. 2022;107:e500-14.
115. Craig JC, Colburn TD, Hirai DM, Schettler MJ, Musch TI, Poole DC. Sex and nitric oxide bioavailability interact to modulate interstitial Po2 in healthy rat skeletal muscle. J Appl Physiol. 2018;124:1558-66.
116. Lee TJ, Shirasaki Y, Nickols GA. Altered endothelial modulation of vascular tone in aging and hypertension. Blood Vessels. 1987;24:132-6.
117. Perrin-Sarrado C, Dahboul F, Leroy P, Lartaud I. Aging and hypertension decrease endothelial NO-related dilating function and gamma-glutamyl transferase activity but not S-nitrosoglutathione-induced aortic vasodilation. Fundam Clin Pharmacol. 2018;32:134-40.
118. Barton M, Cosentino F, Brandes RP, Moreau P, Shaw S, Lüscher TF. Anatomic heterogeneity of vascular aging: role of nitric oxide and endothelin. Hypertension. 1997;30:817-24.
119. Higashi Y, Sasaki S, Nakagawa K, et al. Tetrahydrobiopterin improves aging-related impairment of endothelium-dependent vasodilation through increase in nitric oxide production. Atherosclerosis. 2006;186:390-5.
120. Alvares TS, Maturana FM, Soares RN. Sex differences in the predictors of skeletal muscle microvascular reactivity in older individuals. Maturitas. 2024;189:108115.
121. Ruediger SL, Pizzey FK, Koep JL, Coombes JS, Askew CD, Bailey TG. Comparison of peripheral and cerebral vascular function between premenopausal, early and late postmenopausal females. Exp Physiol. 2023;108:518-30.
122. Guzik TJ, Touyz RM. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension. 2017;70:660-7.
123. Pagan LU, Gomes MJ, Gatto M, Mota GAF, Okoshi K, Okoshi MP. The role of oxidative stress in the aging heart. Antioxidants. 2022;11:336.
124. Wang C, Nie X, Zhang Y, et al. Reactive oxygen species mediate nitric oxide production through ERK/JNK MAPK signaling in HAPI microglia after PFOS exposure. Toxicol Appl Pharmacol. 2015;288:143-51.
125. Mahoney SA, Bloom SI, Seals DR, Donato AJ, Rossman MJ, Clayton ZS. Mechanisms of cellular senescence-induced vascular aging: evidence of senotherapeutic strategies. J Cardiovasc Aging. 2025;5:6.
126. Fleenor BS, Eng JS, Sindler AL, Pham BT, Kloor JD, Seals DR. Superoxide signaling in perivascular adipose tissue promotes age-related artery stiffness. Aging Cell. 2014;13:576-8.
127. Giacomo G, Rizza S, Montagna C, Filomeni G. Established principles and emerging concepts on the interplay between mitochondrial physiology and S-(De)nitrosylation: implications in cancer and neurodegeneration. Int J Cell Biol. 2012;2012:361872.
128. Csiszar A, Sosnowska D, Wang M, Lakatta EG, Sonntag WE, Ungvari Z. Age-associated proinflammatory secretory phenotype in vascular smooth muscle cells from the non-human primate Macaca mulatta: reversal by resveratrol treatment. J Gerontol A Biol Sci Med Sci. 2012;67:811-20.
129. Springo Z, Tarantini S, Toth P, et al. Aging exacerbates pressure-induced mitochondrial oxidative stress in mouse cerebral arteries. J Gerontol A Biol Sci Med Sci. 2015;70:1355-9.
130. Tarantini S, Valcarcel-Ares NM, Yabluchanskiy A, et al. Treatment with the mitochondrial-targeted antioxidant peptide SS-31 rescues neurovascular coupling responses and cerebrovascular endothelial function and improves cognition in aged mice. Aging Cell. 2018;17:e12731.
131. Ungvari Z, Orosz Z, Labinskyy N, et al. Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am J Physiol Heart Circ Physiol. 2007;293:H37-47.
132. Gioscia-Ryan RA, LaRocca TJ, Sindler AL, Zigler MC, Murphy MP, Seals DR. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J Physiol. 2014;592:2549-61.
133. Li J, Billiar TR, Talanian RV, Kim YM. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem Biophys Res Commun. 1997;240:419-24.
134. Pourbagher-Shahri AM, Farkhondeh T, Talebi M, Kopustinskiene DM, Samarghandian S, Bernatoniene J. An overview of NO signaling pathways in aging. Molecules. 2021;26:4533.
135. Zhang Z, Liu L, Jiang X, Zhai S, Xing D. The essential role of Drp1 and its regulation by S-nitrosylation of parkin in dopaminergic neurodegeneration: implications for parkinson's disease. Antioxid Redox Signal. 2016;25:609-22.
136. Wang L, Yang Z, He X, et al. Mitochondrial protein dysfunction in pathogenesis of neurological diseases. Front Mol Neurosci. 2022;15:974480.
137. Bhayadia R, Schmidt BM, Melk A, Hömme M. Senescence-induced oxidative stress causes endothelial dysfunction. J Gerontol A Biol Sci Med Sci. 2016;71:161-9.
138. Gevaert AB, Shakeri H, Leloup AJ, et al. Endothelial senescence contributes to heart failure with preserved ejection fraction in an aging mouse model. Circ Heart Fail. 2017;10:e003806.
139. Rossman MJ, Kaplon RE, Hill SD, et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol. 2017;313:H890-5.
140. Roos CM, Zhang B, Palmer AK, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15:973-7.
141. Hayashi T, Matsui-Hirai H, Miyazaki-Akita A, et al. Endothelial cellular senescence is inhibited by nitric oxide: implications in atherosclerosis associated with menopause and diabetes. Proc Natl Acad Sci USA. 2006;103:17018-23.
142. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14:576-90.
143. Song Y, Shen H, Schenten D, Shan P, Lee PJ, Goldstein DR. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2012;32:103-9.
144. Tavenier J, Rasmussen LJH, Houlind MB, et al. Alterations of monocyte NF-κB p65/RelA signaling in a cohort of older medical patients, age-matched controls, and healthy young adults. Immun Ageing. 2020;17:25.
145. Gao F, Lucke-Wold BP, Li X, et al. Reduction of endothelial nitric oxide increases the adhesiveness of constitutive endothelial membrane ICAM-1 through Src-mediated phosphorylation. Front Physiol. 2017;8:1124.
146. Kim SA, Sung JY, Woo CH, Choi HC. Laminar shear stress suppresses vascular smooth muscle cell proliferation through nitric oxide-AMPK pathway. Biochem Biophys Res Commun. 2017;490:1369-74.
147. Ungvari Z, Bailey-Downs L, Gautam T, et al. Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-{kappa}B activation in the nonhuman primate Macaca mulatta. J Gerontol A Biol Sci Med Sci. 2011;66:866-75.
148. Csiszar A, Ungvari Z, Edwards JG, et al. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90:1159-66.
149. Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol Genomics. 2004;17:21-30.
150. Bach MH, Sadoun E, Reed MJ. Defects in activation of nitric oxide synthases occur during delayed angiogenesis in aging. Mech Ageing Dev. 2005;126:467-73.
151. Khan M, Dhammu TS, Matsuda F, et al. Promoting endothelial function by S-nitrosoglutathione through the HIF-1α/VEGF pathway stimulates neurorepair and functional recovery following experimental stroke in rats. Drug Des Devel Ther. 2015;9:2233-47.
152. Yang C, Hwang HH, Jeong S, et al. Inducing angiogenesis with the controlled release of nitric oxide from biodegradable and biocompatible copolymeric nanoparticles. Int J Nanomedicine. 2018;13:6517-30.
153. Zacharek A, Chen J, Zhang C, et al. Nitric oxide regulates Angiopoietin1/Tie2 expression after stroke. Neurosci Lett. 2006;404:28-32.
154. Ziche M, Morbidelli L, Masini E, et al. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest. 1994;94:2036-44.
155. Jeong H, Choi D, Oh Y, Heo J, Hong J. A nanocoating co-localizing nitric oxide and growth factor onto individual endothelial cells reveals synergistic effects on angiogenesis. Adv Healthc Mater. 2022;11:e2102095.
156. Priya MK, Sahu G, Soto-Pantoja DR, et al. Tipping off endothelial tubes: nitric oxide drives tip cells. Angiogenesis. 2015;18:175-89.
157. Yang Z, Yang Y, Xiong K, et al. Nitric oxide producing coating mimicking endothelium function for multifunctional vascular stents. Biomaterials. 2015;63:80-92.
158. Yamamoto N, Oyaizu T, Enomoto M, et al. VEGF and bFGF induction by nitric oxide is associated with hyperbaric oxygen-induced angiogenesis and muscle regeneration. Sci Rep. 2020;10:2744.
159. Dulak J, Józkowicz A, Dembinska-Kiec A, et al. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20:659-66.
160. Morbidelli L, Chang CH, Douglas JG, Granger HJ, Ledda F, Ziche M. Nitric oxide mediates mitogenic effect of VEGF on coronary venular endothelium. Am J Physiol. 1996;270:H411-5.
161. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation. 2003;107:346-54.
162. Cheng S, Xanthakis V, Sullivan LM, et al. Correlates of echocardiographic indices of cardiac remodeling over the adult life course: longitudinal observations from the Framingham Heart Study. Circulation. 2010;122:570-8.
163. Ho KK, Pinsky JL, Kannel WB, Levy D. The epidemiology of heart failure: the Framingham study. J Am Coll Cardiol. 1993;22:6A-13A.
164. Magnani JW, Wang N, Benjamin EJ, et al. Atrial fibrillation and declining physical performance in older adults: the health, aging, and body composition study. Circ Arrhythm Electrophysiol. 2016;9:e003525.
165. Birenbaum A, Tesse A, Loyer X, et al. Involvement of beta 3-adrenoceptor in altered beta-adrenergic response in senescent heart: role of nitric oxide synthase 1-derived nitric oxide. Anesthesiology. 2008;109:1045-53.
166. Hayashi H, Hess DT, Zhang R, et al. S-nitrosylation of β-arrestins biases receptor signaling and confers ligand independence. Mol Cell. 2018;70:473-87.e6.
167. Villmow M, Klöckner U, Heymes C, Gekle M, Rueckschloss U. NOS1 induces NADPH oxidases and impairs contraction kinetics in aged murine ventricular myocytes. Basic Res Cardiol. 2015;110:506.
168. Zieman SJ, Gerstenblith G, Lakatta EG, et al. Upregulation of the nitric oxide-cGMP pathway in aged myocardium: physiological response to l-arginine. Circ Res. 2001;88:97-102.
169. Damy T, Ratajczak P, Robidel E, et al. Up-regulation of cardiac nitric oxide synthase 1-derived nitric oxide after myocardial infarction in senescent rats. FASEB J. 2003;17:1934-6.
170. Potenza DM, Cheng X, Ajalbert G, et al. Cell-autonomous and non-cell-autonomous effects of arginase-II on cardiac aging. bioRxiv. 2024.
171. Barouch LA, Harrison RW, Skaf MW, et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337-9.
172. Khan SA, Skaf MW, Harrison RW, et al. Nitric oxide regulation of myocardial contractility and calcium cycling: independent impact of neuronal and endothelial nitric oxide synthases. Circ Res. 2003;92:1322-9.
173. Thomas MM, Vigna C, Betik AC, Tupling AR, Hepple RT. Cardiac calcium pump inactivation and nitrosylation in senescent rat myocardium are not attenuated by long-term treadmill training. Exp Gerontol. 2011;46:803-10.
174. Feridooni HA, Dibb KM, Howlett SE. How cardiomyocyte excitation, calcium release and contraction become altered with age. J Mol Cell Cardiol. 2015;83:62-72.
175. Howlett SE, Grandy SA, Ferrier GR. Calcium spark properties in ventricular myocytes are altered in aged mice. Am J Physiol Heart Circ Physiol. 2006;290:H1566-74.
176. Kane AE, Bisset ES, Keller KM, Ghimire A, Pyle WG, Howlett SE. Age, sex and overall health, measured as frailty, modify myofilament proteins in hearts from naturally aging mice. Sci Rep. 2020;10:10052.
177. Lompré AM, Lambert F, Lakatta EG, Schwartz K. Expression of sarcoplasmic reticulum Ca2+-ATPase and calsequestrin genes in rat heart during ontogenic development and aging. Circ Res. 1991;69:1380-8.
178. Schmidt U, del Monte F, Miyamoto MI, et al. Restoration of diastolic function in senescent rat hearts through adenoviral gene transfer of sarcoplasmic reticulum Ca2+-ATPase. Circulation. 2000;101:790-6.
179. Grocott-Mason R, Fort S, Lewis MJ, Shah AM. Myocardial relaxant effect of exogenous nitric oxide in isolated ejecting hearts. Am J Physiol. 1994;266:H1699-705.
180. Layland J, Li JM, Shah AM. Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol. 2002;540:457-67.
181. Shah AM, Spurgeon HA, Sollott SJ, Talo A, Lakatta EG. 8-bromo-cGMP reduces the myofilament response to Ca2+ in intact cardiac myocytes. Circ Res. 1994;74:970-8.
182. Carnicer R, Hale AB, Suffredini S, et al. Cardiomyocyte GTP cyclohydrolase 1 and tetrahydrobiopterin increase NOS1 activity and accelerate myocardial relaxation. Circ Res. 2012;111:718-27.
183. Zhang YH, Zhang MH, Sears CE, et al. Reduced phospholamban phosphorylation is associated with impaired relaxation in left ventricular myocytes from neuronal NO synthase-deficient mice. Circ Res. 2008;102:242-9.
184. Carnicer R, Duglan D, Ziberna K, et al. BH4 increases nNOS activity and preserves left ventricular function in diabetes. Circ Res. 2021;128:585-601.
185. Steppan J, Ryoo S, Schuleri KH, et al. Arginase modulates myocardial contractility by a nitric oxide synthase 1-dependent mechanism. Proc Natl Acad Sci USA. 2006;103:4759-64.
186. Chuaiphichai S, Chu SM, Carnicer R, et al. Endothelial cell-specific roles for tetrahydrobiopterin in myocardial function, cardiac hypertrophy, and response to myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2023;324:H430-42.
187. Ashley EA, Sears CE, Bryant SM, Watkins HC, Casadei B. Cardiac nitric oxide synthase 1 regulates basal and beta-adrenergic contractility in murine ventricular myocytes. Circulation. 2002;105:3011-6.
188. Jiang MT, Moffat MP, Narayanan N. Age-related alterations in the phosphorylation of sarcoplasmic reticulum and myofibrillar proteins and diminished contractile response to isoproterenol in intact rat ventricle. Circ Res. 1993;72:102-11.
189. Xiao RP, Spurgeon HA, O'Connor F, Lakatta EG. Age-associated changes in beta-adrenergic modulation on rat cardiac excitation-contraction coupling. J Clin Invest. 1994;94:2051-9.
190. Xiao RP, Tomhave ED, Wang DJ, et al. Age-associated reductions in cardiac beta1- and beta2-adrenergic responses without changes in inhibitory G proteins or receptor kinases. J Clin Invest. 1998;101:1273-82.
191. Kaye D, Esler M. Sympathetic neuronal regulation of the heart in aging and heart failure. Cardiovasc Res. 2005;66:256-64.
192. Takimoto E, Champion HC, Belardi D, et al. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res. 2005;96:100-9.
193. Wang H, Kohr MJ, Traynham CJ, Ziolo MT. Phosphodiesterase 5 restricts NOS3/soluble guanylate cyclase signaling to L-type Ca2+ current in cardiac myocytes. J Mol Cell Cardiol. 2009;47:304-14.
194. Belge C, Hammond J, Dubois-Deruy E, et al. Enhanced expression of β3-adrenoceptors in cardiac myocytes attenuates neurohormone-induced hypertrophic remodeling through nitric oxide synthase. Circulation. 2014;129:451-62.
195. Gauthier C, Leblais V, Kobzik L, et al. The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J Clin Invest. 1998;102:1377-84.
196. Moniotte S, Kobzik L, Feron O, Trochu JN, Gauthier C, Balligand JL. Upregulation of β3-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation. 2001;103:1649-55.
197. Lee DI, Vahebi S, Tocchetti CG, et al. PDE5A suppression of acute beta-adrenergic activation requires modulation of myocyte beta-3 signaling coupled to PKG-mediated troponin I phosphorylation. Basic Res Cardiol. 2010;105:337-47.
198. Carnicer R, Suffredini S, Liu X, et al. The subcellular localisation of neuronal nitric oxide synthase determines the downstream effects of NO on myocardial function. Cardiovasc Res. 2017;113:321-31.
199. Martin SR, Emanuel K, Sears CE, Zhang YH, Casadei B. Are myocardial eNOS and nNOS involved in the beta-adrenergic and muscarinic regulation of inotropy? A systematic investigation. Cardiovasc Res. 2006;70:97-106.
200. Jeong EM, Monasky MM, Gu L, et al. Tetrahydrobiopterin improves diastolic dysfunction by reversing changes in myofilament properties. J Mol Cell Cardiol. 2013;56:44-54.
201. Simon JN, Ziberna K, Casadei B. Compromised redox homeostasis, altered nitroso-redox balance, and therapeutic possibilities in atrial fibrillation. Cardiovasc Res. 2016;109:510-8.
202. Zhao S, Johnston AM, Yiu CHK, Moreira LM, Reilly S, Wehrens XHT. Aging-associated mechanisms of atrial fibrillation progression and their therapeutic potential. J Cardiovasc Aging. 2024;4:22.
203. Reilly SN, Liu X, Carnicer R, et al. Up-regulation of miR-31 in human atrial fibrillation begets the arrhythmia by depleting dystrophin and neuronal nitric oxide synthase. Sci Transl Med. 2016;8:340ra74.
204. Dawson D, Lygate CA, Zhang MH, Hulbert K, Neubauer S, Casadei B. nNOS gene deletion exacerbates pathological left ventricular remodeling and functional deterioration after myocardial infarction. Circulation. 2005;112:3729-37.
205. Jumrussirikul P, Dinerman J, Dawson TM, et al. Interaction between neuronal nitric oxide synthase and inhibitory G protein activity in heart rate regulation in conscious mice. J Clin Invest. 1998;102:1279-85.
206. Brown GC, Borutaite V. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc Res. 2007;75:283-90.
207. Tyagi SC, Hayden MR. Role of nitric oxide in matrix remodeling in diabetes and heart failure. Heart Fail Rev. 2003;8:23-8.
208. García-Prieto CF, Gil-Ortega M, Plaza A, et al. Caloric restriction induces H2O2 formation as a trigger of AMPK-eNOS-NO pathway in obese rats: Role for CAMKII. Free Radic Biol Med. 2019;139:35-45.
209. Hariharan A, Jing Y, Collie ND, Zhang H, Liu P. Altered neurovascular coupling and brain arginine metabolism in endothelial nitric oxide synthase deficient mice. Nitric Oxide. 2019;87:60-72.
210. Rudgalvyte M, Atzei P, de Brito Francisco R, Naef R, Glauser DA. Dual-acting nitric oxide donor and phosphodiesterase inhibitor TOP-N53 increases lifespan and health span of Caenorhabditis elegans. MicroPubl Biol. 2024:10.17912.
211. Miller GD, Collins S, Ives J, et al. Efficacy and variability in plasma nitrite levels during long-term supplementation with nitrate containing beetroot juice. J Diet Suppl. 2023;20:885-910.
212. DeVan AE, Johnson LC, Brooks FA, et al. Effects of sodium nitrite supplementation on vascular function and related small metabolite signatures in middle-aged and older adults. J Appl Physiol. 2016;120:416-25.
213. Sindler AL, Fleenor BS, Calvert JW, et al. Nitrite supplementation reverses vascular endothelial dysfunction and large elastic artery stiffness with aging. Aging Cell. 2011;10:429-37.
214. Sindler AL, Devan AE, Fleenor BS, Seals DR. Inorganic nitrite supplementation for healthy arterial aging. J Appl Physiol. 2014;116:463-77.
215. Hayes E, Dent E, Shannon OM, et al. Higher plant-derived nitrate intake is associated with lower odds of frailty in a cross-sectional study of community-dwelling older women. Eur J Nutr. 2024;63:2281-90.
216. Justice JN, Gioscia-Ryan RA, Johnson LC, et al. Sodium nitrite supplementation improves motor function and skeletal muscle inflammatory profile in old male mice. J Appl Physiol. 2015;118:163-9.
217. Benjamim CJR, Lopes da Silva LS, da Silva Gonçalves L, et al. The effects of dietary nitrate ingestion on physical performance tests in 50-65 years old postmenopausal women: a pilot randomized, double-blind, placebo-controlled, and crossover study. Clin Nutr. 2024;43:1642-6.
218. Kurhaluk N. Supplementation with l-arginine and nitrates vs age and individual physiological reactivity. Nutr Rev. 2024;82:1239-59.
219. Carvalho LRRA, Guimarães DD, Flôr AFL, et al. Effects of chronic dietary nitrate supplementation on longevity, vascular function and cancer incidence in rats. Redox Biol. 2021;48:102209.
220. Ocampo DA, Paipilla AF, Marín E, Vargas-Molina S, Petro JL, Pérez-Idárraga A. Dietary nitrate from beetroot juice for hypertension: a systematic review. Biomolecules. 2018;8:134.
221. Siervo M, Lara J, Ogbonmwan I, Mathers JC. Inorganic nitrate and beetroot juice supplementation reduces blood pressure in adults: a systematic review and meta-analysis. J Nutr. 2013;143:818-26.
222. Zamani H, de Joode MEJR, Hossein IJ, et al. The benefits and risks of beetroot juice consumption: a systematic review. Crit Rev Food Sci Nutr. 2021;61:788-804.
223. Fejes R, Pilat N, Lutnik M, et al. Effects of increased nitrate intake from beetroot juice on blood markers of oxidative stress and inflammation in older adults with hypertension. Free Radic Biol Med. 2024;222:519-30.
224. Fejes R, Lutnik M, Weisshaar S, et al. Increased nitrate intake from beetroot juice over 4 weeks affects nitrate metabolism, but not vascular function or blood pressure in older adults with hypertension. Food Funct. 2024;15:4065-78.
225. Baranauskas MN, Blechschmid TH, Long EB, Coggan AR, Carter SJ. Dietary NO3- does not enhance endothelial dependent cutaneous vascular conductance in older women. Microvasc Res. 2024;155:104706.
226. Siervo M, Scialò F, Shannon OM, Stephan BCM, Ashor AW. Does dietary nitrate say NO to cardiovascular ageing? Current evidence and implications for research. Proc Nutr Soc. 2018;77:112-23.
227. Zoughaib WS, Hoffman RL, Yates BA, Moorthi RN, Lim K, Coggan AR. Short-term beetroot juice supplementation improves muscle speed and power but does not reduce blood pressure or oxidative stress in 65-79 y old men and women. Nitric Oxide. 2023;138-9:34-41.
228. Stanaway L, Rutherfurd-Markwick K, Page R, et al. Acute supplementation with nitrate-rich beetroot juice causes a greater increase in plasma nitrite and reduction in blood pressure of older compared to younger adults. Nutrients. 2019;11:1683.
229. Capper T, Clifford T, Taylor G, et al. Ageing modifies acute resting blood pressure responses to incremental consumption of dietary nitrate: a randomised, cross-over clinical trial. Br J Nutr. 2023;129:442-53.
230. Pierce GL, Jablonski KL, Walker AE, et al. Tetrahydrobiopterin supplementation enhances carotid artery compliance in healthy older men: a pilot study. Am J Hypertens. 2012;25:1050-4.
231. DuBose LE, Ozemek C, Wick T, Richardson V, Hildreth KL, Moreau KL. Role of BH4 deficiency as a mediator of oxidative stress-related endothelial dysfunction in menopausal women. Am J Physiol Heart Circ Physiol. 2022;323:H975-82.
232. Moreau KL, Meditz A, Deane KD, Kohrt WM. Tetrahydrobiopterin improves endothelial function and decreases arterial stiffness in estrogen-deficient postmenopausal women. Am J Physiol Heart Circ Physiol. 2012;302:H1211-8.
233. Eskurza I, Myerburgh LA, Kahn ZD, Seals DR. Tetrahydrobiopterin augments endothelium-dependent dilatation in sedentary but not in habitually exercising older adults. J Physiol. 2005;568:1057-65.
234. Bisconti AV, Garten RS, Broxterman RM, et al. No effect of acute tetrahydrobiopterin (BH4) supplementation on vascular dysfunction in the old. J Appl Physiol. 2022;132:773-84.
235. Kim IY, Schutzler SE, Schrader A, et al. Acute ingestion of citrulline stimulates nitric oxide synthesis but does not increase blood flow in healthy young and older adults with heart failure. Am J Physiol Endocrinol Metab. 2015;309:E915-24.
236. Mattison JA, Colman RJ, Beasley TM, et al. Caloric restriction improves health and survival of rhesus monkeys. Nat Commun. 2017;8:14063.
237. Guo Z, Mitchell-Raymundo F, Yang H, et al. Dietary restriction reduces atherosclerosis and oxidative stress in the aorta of apolipoprotein E-deficient mice. Mech Ageing Dev. 2002;123:1121-31.
238. Colman RJ, Anderson RM, Johnson SC, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201-4.
239. Donato AJ, Walker AE, Magerko KA, et al. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell. 2013;12:772-83.
240. Kraus WE, Bhapkar M, Huffman KM, et al. 2 years of calorie restriction and cardiometabolic risk (CALERIE): exploratory outcomes of a multicentre, phase 2, randomised controlled trial. Lancet Diabetes Endocrinol. 2019;7:673-83.
241. Rippe C, Lesniewski L, Connell M, LaRocca T, Donato A, Seals D. Short-term calorie restriction reverses vascular endothelial dysfunction in old mice by increasing nitric oxide and reducing oxidative stress. Aging Cell. 2010;9:304-12.
242. Nisoli E, Tonello C, Cardile A, et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314-7.
243. Ungvari Z, Parrado-Fernandez C, Csiszar A, de Cabo R. Mechanisms underlying caloric restriction and lifespan regulation: implications for vascular aging. Circ Res. 2008;102:519-28.
244. Shinmura K, Tamaki K, Bolli R. Short-term caloric restriction improves ischemic tolerance independent of opening of ATP-sensitive K+ channels in both young and aged hearts. J Mol Cell Cardiol. 2005;39:285-96.
245. Shinmura K, Tamaki K, Ito K, et al. Indispensable role of endothelial nitric oxide synthase in caloric restriction-induced cardioprotection against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2015;308:H894-903.
246. Shinmura K, Tamaki K, Bolli R. Impact of 6-mo caloric restriction on myocardial ischemic tolerance: possible involvement of nitric oxide-dependent increase in nuclear Sirt1. Am J Physiol Heart Circ Physiol. 2008;295:H2348-55.
247. Yang H, Shi M, Story J, Richardson A, Guo Z. Food restriction attenuates age-related increase in the sensitivity of endothelial cells to oxidized lipids. J Gerontol A Biol Sci Med Sci. 2004;59:316-23.
248. Cerqueira FM, Brandizzi LI, Cunha FM, Laurindo FR, Kowaltowski AJ. Serum from calorie-restricted rats activates vascular cell eNOS through enhanced insulin signaling mediated by adiponectin. PLoS One. 2012;7:e31155.
249. Gioscia-Ryan RA, Clayton ZS, Zigler MC, et al. Lifelong voluntary aerobic exercise prevents age- and Western diet- induced vascular dysfunction, mitochondrial oxidative stress and inflammation in mice. J Physiol. 2021;599:911-25.
250. Chen Q, Gao X, Wang C, Zhang P. Influence of different exercise types on vascular endothelial function in middle-aged and older adults - A systematic review and network meta-analysis. Arch Gerontol Geriatr. 2025;128:105624.
251. Liang ZD, Zhang M, Wang CZ, Yuan Y, Liang JH. Association between sedentary behavior, physical activity, and cardiovascular disease-related outcomes in adults-A meta-analysis and systematic review. Front Public Health. 2022;10:1018460.
252. Nyberg M, Blackwell JR, Damsgaard R, Jones AM, Hellsten Y, Mortensen SP. Lifelong physical activity prevents an age-related reduction in arterial and skeletal muscle nitric oxide bioavailability in humans. J Physiol. 2012;590:5361-70.
253. Reia TA, da Silva RF, Jacomini AM, et al. Acute exercise, plasma nitric oxide, and blood pressure in older adults with different levels of training status: the influence of polymorphisms of endothelial nitric oxide synthase. J Phys Act Health. 2021;18:516-23.
254. Lauer T, Heiss C, Balzer J, et al. Age-dependent endothelial dysfunction is associated with failure to increase plasma nitrite in response to exercise. Basic Res Cardiol. 2008;103:291-7.
255. Maeda S, Tanabe T, Otsuki T, et al. Moderate regular exercise increases basal production of nitric oxide in elderly women. Hypertens Res. 2004;27:947-53.
256. Freeberg KA, Craighead DH, Heinbockel TC, et al. Time-efficient, high-resistance inspiratory muscle strength training increases cerebrovascular reactivity in midlife and older adults. Am J Physiol Heart Circ Physiol. 2023;325:H1059-68.
257. Spier SA, Delp MD, Meininger CJ, Donato AJ, Ramsey MW, Muller-Delp JM. Effects of ageing and exercise training on endothelium-dependent vasodilatation and structure of rat skeletal muscle arterioles. J Physiol. 2004;556:947-58.
258. Lesniewski LA, Zigler ML, Durrant JR, et al. Aging compounds western diet-associated large artery endothelial dysfunction in mice: prevention by voluntary aerobic exercise. Exp Gerontol. 2013;48:1218-25.
259. Shannon OM, Clifford T, Seals DR, Craighead DH, Rossman MJ. Nitric oxide, aging and aerobic exercise: sedentary individuals to Master's athletes. Nitric Oxide. 2022;125-6:31-9.
260. Senefeld JW, Wiggins CC, Regimbal RJ, Dominelli PB, Baker SE, Joyner MJ. Ergogenic effect of nitrate supplementation: a systematic review and meta-analysis. Med Sci Sports Exerc. 2020;52:2250-61.
261. Nystoriak MA, Bhatnagar A. Cardiovascular effects and benefits of exercise. Front Cardiovasc Med. 2018;5:135.
263. Garatti L, Frea S, Bocchino PP, et al. Sodium nitroprusside in acute heart failure: a multicenter historic cohort study. Int J Cardiol. 2022;369:37-44.
264. Kim KH, Adnan G, Schaller DJ. Nitroglycerin. Treasure Island (FL): StatPearls Publishing; 2025.
265. Taylor AL, Sabolinski ML, Tam SW, et al. Effect of fixed-dose combined isosorbide dinitrate/hydralazine in elderly patients in the African-American heart failure trial. J Card Fail. 2012;18:600-6.
266. Rao SV, O'Donoghue ML, Ruel M, et al. 2025 ACC/AHA/ACEP/NAEMSP/SCAI guideline for the management of patients with acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Joint Committee on clinical practice guidelines. Circulation. 2025;151:e771-862.
267. Komaki S, Matsuura Y, Tanaka H, et al. Nitroglycerin use and adverse clinical outcomes in elderly patients with acute coronary syndrome. Open Heart. 2024;11:e002494.
268. Naryzhnaya NV, Mukhomedzyanov AV, Kilin M, Voronkov NS, Kurbatov BK, Maslov LN. Infarct-limiting efficacy of KATP channel activators and nitric oxide donors under conditions of their chronic administration and diet-induced metabolic syndrome conditions in rats. Bull Exp Biol Med. 2025;179:1-5.
269. Gatzke N, Hillmeister P, Dülsner A, et al. Nitroglycerin application and coronary arteriogenesis. PLoS One. 2018;13:e0201597.
270. Salehi B, Mishra AP, Nigam M, et al. Resveratrol: a double-edged sword in health benefits. Biomedicines. 2018;6:91.
271. Brown K, Theofanous D, Britton RG, et al. Resveratrol for the management of human health: how far have we come? A systematic review of resveratrol clinical trials to highlight gaps and opportunities. Int J Mol Sci. 2024;25:747.
272. Gordish KL, Beierwaltes WH. Resveratrol induces acute endothelium-dependent renal vasodilation mediated through nitric oxide and reactive oxygen species scavenging. Am J Physiol Renal Physiol. 2014;306:F542-50.
273. Xia N, Daiber A, Habermeier A, et al. Resveratrol reverses endothelial nitric-oxide synthase uncoupling in apolipoprotein E knockout mice. J Pharmacol Exp Ther. 2010;335:149-54.
274. Movahed A, Raj P, Nabipour I, et al. Efficacy and safety of resveratrol in type 1 diabetes patients: a two-month preliminary exploratory trial. Nutrients. 2020;12:161.
275. Fourny N, Lan C, Sérée E, Bernard M, Desrois M. Protective effect of resveratrol against ischemia-reperfusion injury via enhanced high energy compounds and eNOS-SIRT1 expression in type 2 diabetic female rat heart. Nutrients. 2019;11:105.
276. DiNatale JC, Crowe-White KM. Effects of resveratrol supplementation on nitric oxide-mediated vascular outcomes in hypertension: a systematic review. Nitric Oxide. 2022;129:74-81.
277. Wong RH, Howe PR, Buckley JD, Coates AM, Kunz I, Berry NM. Acute resveratrol supplementation improves flow-mediated dilatation in overweight/obese individuals with mildly elevated blood pressure. Nutr Metab Cardiovasc Dis. 2011;21:851-6.
278. Abolfazli S, Karav S, Johnston TP, Sahebkar A. Regulatory effects of resveratrol on nitric oxide signaling in cardiovascular diseases. Pharmacol Rep. 2025;77:355-74.
279. Chang N, Li J, Lin S, et al. Emerging roles of SIRT1 activator, SRT2104, in disease treatment. Sci Rep. 2024;14:5521.
280. Cantó C, Houtkooper RH, Pirinen E, et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838-47.
281. Horimatsu T, Blomkalns AL, Ogbi M, et al. Niacin protects against abdominal aortic aneurysm formation via GPR109A independent mechanisms: role of NAD+/nicotinamide. Cardiovasc Res. 2020;116:2226-38.
282. Wu NC, Wang JJ. Niacin pretreatment attenuates lung ischemia and reperfusion-induced pulmonary barrier function impairment by reducing oxidative stress and activating SIRT1 in an isolated-perfused rat lung model. Transplant Proc. 2018;50:2834-8.
283. Goya K, Sumitani S, Xu X, et al. Peroxisome proliferator-activated receptor alpha agonists increase nitric oxide synthase expression in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:658-63.
284. Linz W, Wohlfart P, Baader M, et al. The peroxisome proliferator-activated receptor-alpha (PPAR-alpha) agonist, AVE8134, attenuates the progression of heart failure and increases survival in rats. Acta Pharmacol Sin. 2009;30:935-46.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].