


Aging-driven organelle miscommunication in the failing heart
 0
0 
Abstract
Cardiovascular diseases are the leading cause of death in older adults worldwide, with heart failure (HF) representing one of their most serious end stages. Aging is a non-modifiable risk factor that drives a series of structural and functional changes in the heart, both at the macro and subcellular levels. This review article analyzes how intracellular organelle dysfunction and the loss of coordination between them, a process termed “interorganelle miscommunication,” contribute to the progression of HF in the context of aging. We review experimental and clinical studies on the function of mitochondria, sarcoplasmic reticulum, lysosomes, lipid droplets, and the nucleus in aging cardiomyocytes. Particular emphasis is placed on how altered interactions between these organelles affect key processes such as ATP production, calcium handling, autophagy, epigenetic regulation, and oxidative stress control. We also discuss the impact of chronic low-grade inflammation (“inflammaging”) and cellular senescence as aggravating factors in cardiac functional decline. Collectively, the evidence indicates that dysregulation of interorganelle communication not only accelerates cardiac aging but also represents a central pathogenetic mechanism in HF. In this context, the concept of a “dysfunctional interorganellar network” may serve as a new hallmark of subcellular aging and an emerging therapeutic target for preventing or delaying age-related HF.
Keywords
INTRODUCTION
Cardiovascular diseases (CVDs) are the leading cause of death worldwide, with mortality rates reaching 32%. In 2021 alone, an estimated 20 million people died from these diseases[1]. Age is a non-modifiable risk factor for CVD, and given demographic trends, an estimated 10% of the world's population is aged 60 years or older. This percentage is expected to increase to 16.6% by 2030 and to 21.4% by 2050[2]. CVDs are the most serious public health problem due to their high prevalence, which not only leads to high mortality rates but also results in considerable morbidity and disability. The American Heart Association (AHA) reports that approximately 40% of people aged 40 to 59 have CVD, a figure that rises to approximately 86% in people over 80[3].
Among the various disorders that make up this group, heart failure (HF) is characterized by persistent and severe symptoms despite maximum medical treatment, including significant limitations in physical activity and severe cardiac dysfunction[4]. The one-year mortality rate associated with this disease can reach between 50% and 75%, reflecting the severity of this condition. Patients with HF report a substantially poorer health-related quality of life (HRQoL) than the general population, largely due to physical limitations, emotional distress, and social dysfunction. Symptoms such as dyspnea, fatigue, and pain are often debilitating and directly contribute to reduced HRQoL[4,5]. The burden on health systems is also considerable, as HF often requires long-term management[6,7]. Moreover, socioeconomic disparities in healthcare access and education exacerbate the prevalence and outcomes of CVD in disadvantaged groups[6,8].
HF represents the terminal stage of multiple cardiovascular diseases and is characterized by the progressive inability of the heart to maintain adequate cardiac output. This decline begins with subtle changes in the heart and vasculature that progress to a point where cardiovascular function is insufficient to pump blood and oxygen and meet the body's needs[9,10]. Diseases that damage cardiovascular tissues accelerate this process by reducing vascular elasticity and increasing cardiac workload, typically resulting in compensatory thickening of the heart, known as cardiac hypertrophy[11]. However, because the damage is cumulative, such cardiac remodeling becomes pathological and ultimately leads to loss of function[12]. Although aging is not a disease, it accelerates the development of heart failure (HF) by contributing multiple mechanisms that exacerbate functional decline, such as arterial stiffness, endothelial dysfunction, and myocardial fibrosis[12].
This section briefly discusses the stages and factors that lead to HF. According to the American Heart Association (AHA), HF is clinically classified into two main types based on the functional status of the heart[13].
• HF with reduced ejection fraction (HFrEF): impaired heart muscle contraction (reduced ejection fraction).
• HF with preserved ejection fraction (HFpEF): impaired heart filling during diastole, but normal contraction (preserved ejection fraction).
In both cases, compensatory mechanisms initially maintain cardiac output, but ultimately cause further myocardial damage and worsening cardiac function. Pathophysiology involves a complex process of structural and functional cardiac remodeling, driven by neurohormonal mechanisms - such as activation of the renin-angiotensin-aldosterone system- and inflammatory mechanisms that exacerbate ventricular dysfunction[13].
Although there is increasing knowledge about heart failure (HF), the role of interorganelle signaling - especially communication between mitochondria and the sarcoplasmic reticulum (SR) - in HFpEF compared to HFrEF phenotypes remains poorly defined. In HFpEF, which is frequently associated with advanced age, obesity and its comorbidities, mitochondrial dysfunction, SR stress, and alterations in SR-mitochondria interactions have been described, causing an imbalance in Ca2+ homeostasis, dysfunctional lipid exchange, oxidative stress, and energy dysfunction[14,15]. On the other hand, HFrEF, typically secondary to ischemic damage and myocyte loss, is characterized by marked systolic dysfunction, eccentric ventricular remodeling, disruption of mitochondrial dynamics, and ROS production[16-18]. Given that the etiology, age, and clinical presentation differ between HFpEF and HFrEF, further investigation of interorganelle signaling differences could reveal specific pathways for differentiated therapeutic approaches.
At the macroscopic level, the most notable changes are atherosclerosis, increased arterial stiffness, ventricular hypertrophy, and myocardial fibrosis. Atherosclerosis, for example, occurs in approximately 66.9% of people over the age of 50 and contributes to pressure overload that promotes arterial stiffness, hypertrophy, and pathological fibrosis of the left ventricle, increasing the risk of myocardial infarction or stroke due to insufficient blood perfusion[19]. This process begins early in life and progresses due to several risk factors[20,21], including:
• Hyperlipidemia: elevated levels of low-density lipoproteins (LDL) increase cholesterol accumulation in the vascular system.
• High blood pressure: causes physical damage to the endothelium and promotes atheromatous plaque formation. It also increases the workload on the heart and can cause structural changes, such as left ventricular hypertrophy.
• Diabetes: high blood glucose levels damage blood vessels, leading to endothelial dysfunction, characterized by impaired vasodilation and elasticity, which increases inflammation and accelerates atherosclerosis.
• Lifestyle factors: obesity, excessive consumption of saturated fats, physical inactivity, smoking, and excessive alcohol consumption significantly increase the risk of CVD.
• Genetic factors: Genetic variants significantly influence susceptibility to CVD, especially those affecting lipid metabolism (severe dyslipidemia), blood pressure regulation, and other cardiovascular functions.
At the same time, functional and structural alterations occur at the subcellular level. Mitochondria, which are responsible for supplying 95% of the ATP needed for the heart's contractile function, show a reduction in bioenergetic efficiency, an increase in the production of reactive oxygen species (ROS), and disruption of their fusion and fission dynamics. The SR, which is key in the regulation of intracellular calcium, also suffers from dysfunction, affecting excitation-contraction coupling. Lysosomes, whose function is essential for the degradation of damaged organelles through autophagy, lose their effectiveness with age, favoring the accumulation of subcellular debris[22]. Other less studied organelles also contribute to cardiomyocyte function, such as lipid droplets (LDs) and peroxisomes, which regulate the metabolism of fatty acids, the main macronutrient for the heart under normal conditions. Both organelles are highly communicative with other organelles, forming functional contacts with mitochondria, SR, and nucleus to exchange metabolites and signals[23]. Additionally, changes in nuclear organization, loss of heterochromatin, and epigenetic alterations that compromise adaptive gene expression during cardiac aging have been documented[24].
One of the emerging mechanisms linking these subcellular dysfunctions is the alteration of communication between organelles, a phenomenon that is particularly relevant in the context of aging. The interaction between mitochondria and ER, mediated by mitochondria-associated ER membranes (MAM), is affected by age, compromising the efficient transfer of calcium and lipids, as well as the regulation of apoptosis[25]. Likewise, impaired mitonuclear communication limits the ability to respond to mitochondrial damage, affecting the biogenesis and repair of these organelles[26]. These failures are amplified by processes such as inflammaging - chronic low-grade inflammation that drives the aging process - and cellular senescence, which promote a dysfunctional tissue microenvironment, with increased fibrosis, ventricular stiffness, and loss of contractility[27].
The interconnection between these subcellular processes explains the transition from physiological aging to clinical pathology. Disruption of autophagy (due to lysosomal failure) and ROS excess (due to dysfunctional mitochondria) establish a vicious cycle of macromolecular damage, which, combined with matrix stiffness due to fibrosis, irreversibly leads to loss of contractile function[28].
The following work is structured in two main sections. Section “ORGANELLE REMODELING IN CARDIOVASCULAR DISEASES” analyzes the structural and functional changes of key organelles in CVD, while Section “ORGANELLE MISCOMMUNICATION IN THE AGING HEART” focuses on how aging compromises communication between these organelles, contributing to the progressive deterioration of the heart. Understanding these mechanisms offers new therapeutic targets for interventions that modulate organelle communication, potentially delaying the progression of HF in geriatric populations.
ORGANELLE REMODELING IN CARDIOVASCULAR DISEASES
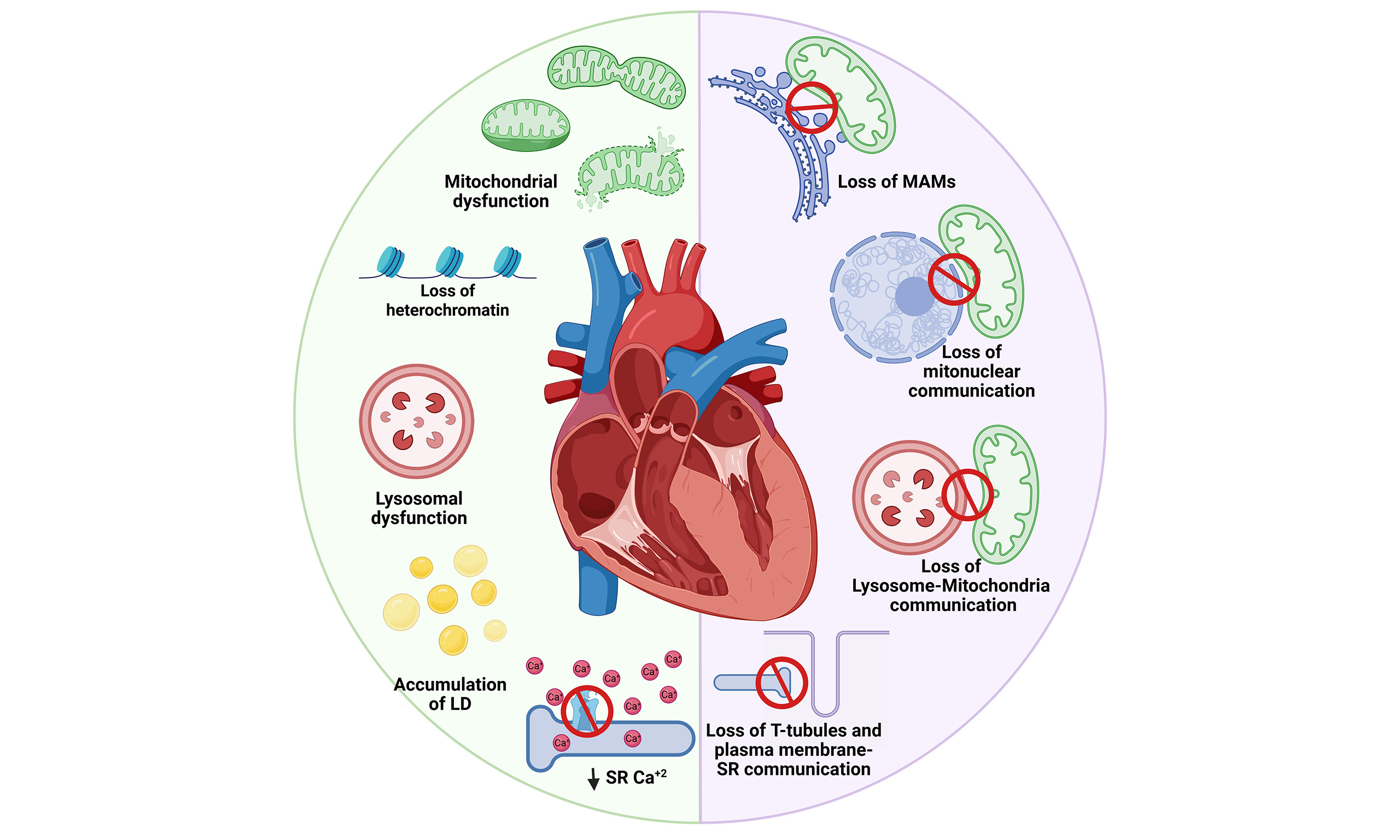
This section examines how individual organelles undergo pathological remodeling during the development of different forms of CVD, particularly HF [Figure 1]. The mechanisms underlying mitochondrial dysfunction, ER stress, chromatin remodeling damage, impaired lysosomal autophagy, and disruption of lipid droplet function were analyzed, all within the context of hypertrophy, fibrosis, ischemia, and HF.
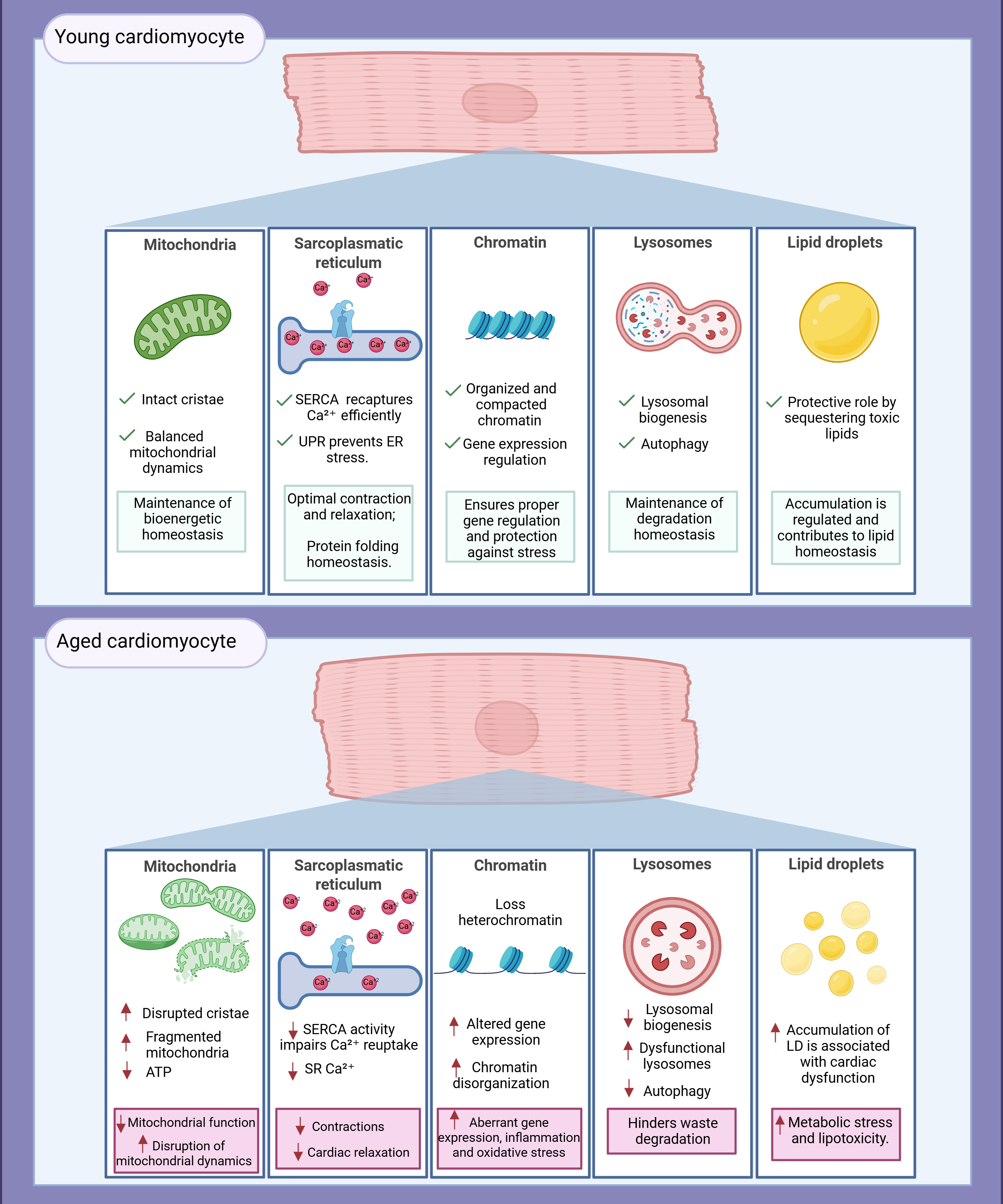
Figure 1. Organelle alterations in cardiomyocytes during aging. In young cardiomyocytes, mitochondria maintain efficient ATP production and redox homeostasis; lysosomes support effective autophagy; the SR ensures proper Ca2+ cycling; lipid droplets (LD) prevent lipotoxicity; and chromatin is preserved with regulated gene expression. In contrast, aged cardiomyocytes exhibit fragmented and dysfunctional mitochondria with increased reactive oxygen species (ROS) production and reduced bioenergetics; lysosomal activity is impaired, compromising autophagic clearance; SR Ca2+ reuptake becomes defective; LDs accumulate abnormally; and chromatin structure is altered, with loss of heterochromatin and dysregulated transcription. These alterations collectively impair cardiomyocyte function and contribute to age-related cardiac decline.
Mitochondrial dysfunction
Mitochondria are the main ATP-producing organelle in eukaryotic cells. With their specialized morphology and function, they possess an electron transport chain that synthesizes ATP necessary for cellular life and is indispensable for several biological functions[29]. Because the heart functions continuously from early development until death, it requires a constant and substantial ATP supply. Mitochondria are the main organelle responsible for the maintenance of constant energy consumption of the heart. These organelles are highly abundant in cardiomyocytes, strategically located between sarcomeres to ensure efficient ATP delivery. They occupy approximately one-third of the cell volume and produce nearly 95% of the energy the heart needs[30,31]. Mitochondrial dysfunction plays a crucial role in heart aging and failure. During aging, the heart exhibits impaired bioenergetic efficiency, increased mitochondrial DNA instability, and enhanced susceptibility to trigger apoptosis and inflammation, changes that affect cellular survival and the development of cardiac dysfunction[32,33]. Similarly, during HF, mitochondria show alterations in oxidative phosphorylation, substrate utilization, and morphology, thus contributing to chronic energy deficits and mechanical dysfunction[34]. The combination of age-related mitochondrial dysfunction and the effects of HF can disrupt cellular energy and redox balance, suggesting that the interaction between aging and HF exacerbates mitochondrial dysfunction[35]. Mitochondrial dysfunction in aged hearts involves decreased activity of respiratory complexes I, III, and IV, reduced mitochondrial content, and impaired oxidative phosphorylation[36-38]. These changes increase ROS production and ensuing oxidative damage[36,38]. Additionally, aging induces a metabolic imbalance in the myocardium, characterized by increased fatty acid uptake despite reduced lipid oxidation[29,39].
Mitochondrial morphology is regulated by dynamic fusion and fission processes - collectively termed mitochondrial dynamics - which are crucial for maintaining mitochondrial health, ultrastructure, turnover, and metabolism[29,40]. These processes affect cell viability, proliferation, and ATP production[41]. Mitochondrial fusion is primarily mediated by the proteins Mitofusin (Mfn) 1 and 2 in the outer mitochondrial membrane and Optic atrophy 1 (Opa1) in the inner mitochondrial membrane[40], whereas fission is mainly controlled by Dynamin-related protein 1 (Drp1)[42].
During aging, cardiomyocyte mitochondria undergo morphological alterations associated with functional decline, and disruption of mitochondrial dynamics contributes to the development of CVD[41]. For instance, aged C57BL/6J mice (17 months) present mitochondria with swollen and disrupted cristae, as well as broken inner membranes[43]. Similar damage has been observed when comparing young (3-month-old) to older (1- and 2-year-old) mice, with no sex dependence[44,45]. Aging hearts typically display a shift toward excessive fission, resulting in fragmented and disconnected mitochondria. Comparable alterations occur in HF models. In a rat model of pressure overload-induced HF, mitochondrial size decreased after 3 weeks[46]. Likewise, in a rat model of left anterior descending (LAD) coronary artery ligation for 9 weeks, mitochondria exhibited reduced size, abnormal distribution, and loss of cristae[47]. Genetic models further demonstrate the importance of mitochondrial dynamics. Deletion of yme1l1 (encoding OPA1) induces mitochondrial fragmentation without impairing respiration, but causes dilated cardiomyopathy with necrosis, fibrosis, and ventricular remodeling, and a metabolic switch promoting glucose use[48]. Similarly, Chen et al. showed that embryonic ablation of Mfn1/Mfn2 is lethal, while ablation in adults leads to mitochondrial fragmentation, respiratory dysfunction, and dilated cardiomyopathy, demonstrating that mitochondrial fusion is essential for maintaining cardiac mitochondrial integrity[40]. On the other hand, in a mouse model of cardiomyocyte-specific Drp1 ablation, mice died after 8 weeks of tamoxifen injection and presented elongated mitochondria in basal and postprandial states. Additionally, these animals exhibited heart hypertrophy, characterized by an increased cardiomyocyte cross-sectional area, cardiac fibrosis, lung congestion, and a decreased ejection fraction, a phenotype that aligns with HFrEF[49]. These changes were accompanied by a decrease in the activity of complexes I, II + III, and IV of the mitochondrial electron transport chain, coinciding with a decrease in ATP production[49]. Similarly, evidence suggests that age reduces the expression of proteins involved in mitochondrial dynamics, resulting in alterations in mitochondrial morphology and function. Indeed, old age mice (2 years old) show a decrease in the protein expression of Mfn2, Opa1, and Drp1 compared to young age (1 year old) animals[50]. In agreement with that, a rat model of old age (25 months) shows a decrease in Mfn1 and Mfn2 protein expression[51]. Song et al. generated a murine triple inducible knockout of Drp1/Mfn1/Mfn2 (TKO)[50]. These mice developed concentric cardiac hypertrophy and demonstrated higher survival rates compared with single-gene knockouts. In terms of mitochondrial morphology, TKO mice exhibit high mitochondrial size heterogeneity, with a predominance of smaller mitochondria, which are also accumulated in the perinuclear region, suggesting a mechanical disruption of myofilament and cell organization[50]. Moreover, the MICOS complex is essential for cristae formation and maintenance. In cardiac fibroblasts of aged mice, the mRNA levels of Opa1, mitofilin, Chchd3, and Chchd6 are decreased compared to those of young animals, suggesting a loss of the MICOS complex during the aging process[45]. The same authors found that the loss of these proteins in cardiac fibroblasts correlates with mitochondrial dysfunction, characterized by fragmented mitochondria, a reduced respiratory rate, and increased production of reactive oxygen species (ROS)[45]. These results suggest that loss of opa1 and the MICOS complex leads to oxidative stress, promoting a ROS-induced ROS mechanism associated with age-related mitochondrial functional decline[45]. Aging also impairs metabolic flexibility and mitochondrial function, inducing a decrease in cardiac function[38,52]. Aging hearts exhibit a reduction in the oxidation capacity of fatty acids and an increase in glucose metabolism[47,53]. In OPA1-knockout mice, there are decreases in Drp1 and MICOS complex proteins, as well as reduced basal, ATP-linked, and maximal respiratory rates and spare capacity, indicating that conditions of high mitochondrial bioenergetic demand may be affected by the loss of this protein[45].
Closely related to mitochondrial dynamics, mitophagy is the primary cellular process of mitochondrial quality control, which involves the autophagic removal of dysfunctional mitochondria[28]. Gao et al. showed that old mice (24-26 months) exhibit compromised myocardial function and mitochondrial morphology, characterized by decreased fractional shortening, increased apoptosis, reduced mitochondrial size, and decreased protein expression of the mitophagy machinery, namely Parkin, LC3II, phospho-p62, and TKB1[53]. All of these effects were attenuated by Parkin overexpression, which rescued cardiac aging by promoting K63-linked polyubiquitination of TBK1 to facilitate mitophagy[53]. In agreement with that,
The aging heart is characterized by an increase in the opening of the mitochondrial permeability transition pore (mPTP), which leads to the release of mitochondrial content into the cytosol. This can induce apoptosis, mitophagy, mitochondrial membrane depolarization, and mitochondrial swelling. However, the molecular mechanism that causes the mPTP opening is still unknown[54,55]. During HF, there is an increase in mPTP opening, mediated by mechanisms such as dysregulation of Ca2+ homeostasis, decrease in ATP levels, and oxidative stress[56]. A study found that, compared to young rats, the cardiac mitochondria of old rats exhibit increased opening of the mPTP, concomitant with elevated levels of superoxide and hydroxyl anion radicals, urea, and products of early lipid peroxidation, as well as iNOS activity, all of which are characteristic of oxidative and nitrosative stress[57]. Additionally, heart mitochondria from aged SIRT3-/- mice (16 months) exhibit increased mPTP opening compared to control mice, indicating that SIRT3 contributes to blocking mPTP in aging hearts[57]. Moreover, mPTP opening in the aging heart leads to the release of cytochrome c into the cytosol, which can trigger apoptosis. In rats of 16 and 24 months of age, there is an increase in cytosolic cytochrome c levels compared to young animals (6 months)[58].
Sarcoplasmic reticulum stress
The sarcoplasmic reticulum (SR) is essential for cardiac function, as it regulates Ca2+ homeostasis, lipid metabolism, and protein folding, processes that are essential for cardiac contraction and cell survival. Its dysfunction, especially in aging, triggers ER stress, inflammation, and pathological remodeling[61]. On the other hand, Ca2+-mediated electrical signaling in the heart has been reported to influence lifespan by maintaining rhythmicity and cardiac function[62].
The endoplasmic reticulum (ER) is a membranous organelle in eukaryotic cells, playing a critical role in protein synthesis, lipid metabolism, and calcium storage and signaling. In muscle cells, the ER exhibits structural and functional specialization, referred to as the SR, highlighting its unique role in regulating Ca2+ dynamics, which is essential for muscle contraction. In cardiomyocytes, the SR forms an extensive network of tubules that are closely associated with transverse tubules (T-tubules) to form dyads or triads. These structures are crucial for excitation-contraction (EC) coupling, facilitating the release and reuptake of
The SR also undergoes age-related alterations[65]. RyRs, which are responsible for Ca2+ release, exhibit reduced responsiveness and, in some cases, leak Ca2+ during resting states, resulting in the dysregulation of intracellular Ca2+ levels[66]. This negatively affects both the strength of contraction and the ability of cardiomyocytes to relax correctly between heartbeats[67]. The reduced activity of SERCA (SR Ca2+-ATPase) with aging also contributes to decreased Ca2+ reuptake into the SR, leading to Ca2+ accumulation in the cytosol and reduced heart relaxation efficiency[68]. Pathologies such as HF, dilated cardiomyopathy, and cardiac hypertrophy also exhibit low SERCA activity, further compromising Ca2+ capture[69-71].
The ER is a multifunctional organelle involved in various pathophysiological processes. One of its most important functions is the folding and translocation of transmembrane proteins[72]. When this process is disrupted, unfolded or misfolded proteins accumulate, triggering the ER unfolded protein response (UPR), which disturbs ER homeostasis and leads to what is known as ER stress[73]. In CVD, metabolic dysregulation, hypoxia, and inflammation can trigger ER stress, which induces oxidative stress and further inflammation[74,75].
As mentioned earlier, chronic inflammation has been identified as a key determinant of aging[76], a significant contributor to cardiovascular risk[77], and a marker of poor health status[78]. Inflammatory mediators can sustain or even exacerbate ER stress in tissues, and chronic ER stress, in turn, promotes chronic inflammation. Thus, a positive feedback loop may exist between ER stress and inflammation. One of the hypothesized origins of inflammaging is an imbalance in the production of damage-associated molecular patterns (DAMPs), including cellular debris, misfolded proteins, and misplaced molecules, which increase with age[78]. The accumulation of these DAMPs can be detected by the NLRP3 inflammasome, which activates its signaling pathway and subsequently leads to the production and secretion of IL-1β and IL-18. Blocking the NLRP3 inflammasome extends a healthy lifespan by attenuating multiple aging-related degenerative changes[79,80]. Several studies have identified the involvement of the Stimulator of Interferon Genes (STING) pathway in inflammation and have linked it to ER stress in models of cardiac hypertrophy, both in vivo and in vitro[80]. Activation of this signaling pathway increases type I interferon levels and NF-κB activation, and silencing this pathway attenuates the cardiotoxic effects of doxorubicin[81]. Moreover, ER stress and the UPR contribute to inflammation through multiple regulatory pathways, including NF-κB and the NLRP3 inflammasome, which impact Ca2+ homeostasis. NF-κB activation mediates myocardial and vascular inflammation in aging-related diseases[82]. Therefore, the evidence clearly shows a relationship between aging, CVD, and ER stress.
Chromatin remodeling in response to damage
Chromatin remodeling in response to cellular damage is essential for cardiac function, as it facilitates access to specific genomic regions required for activating genes involved in repair, stress response, and survival pathways. ATP-dependent chromatin remodeling complexes and epigenetic modifications such as histone methylation or acetylation are key regulators of these processes, helping to preserve cardiac homeostasis under stress conditions like ischemia or aging[83]. DNA is usually organized into a structure called chromatin, which is compacted by histones. Together, they form the nucleosome, in a structure with a high degree of condensation that regulates gene expression by restricting DNA availability to the action of transcription factors[84]. Histones can be modified through acetylation, methylation, and phosphorylation, thereby altering chromatin structure, a process termed chromatin remodeling, which is required for correct gene expression, DNA repair, and stress responses[85]. On the other hand, aging is known to reduce organ function and increase the risk of disease, which are associated with changes in chromatin structure and function, as well as shortening of telomeres[86-89]. Furthermore, it has also been linked to the progression of certain diseases such as HF[90,91].
Some DNA repair complexes are ATP-dependent. One such complex is the BRG1/BRM-associated factor (BAF) complex, which comprises the BRG1 protein, a transcription factor linked to genes involved in DNA repair. It has been associated with cardiac hypertrophy and fibrosis[90,91].
Aging is accompanied by a process called loss of heterochromatin, which is associated with gene silencing. This suggests that there may be increased expression of genes that are typically repressed, contributing to cellular dysfunction. Aging also affects the structure of the nucleus, causing alterations in the organization of chromatin that impact the positioning of genes[89,92,93].
In general, specific proteins with a key role in chromatin remodeling are described, such as Sirtuin 1 (SIRT1), a member of the NAD+-dependent deacetylase sirtuin family, which deacetylates histones, thereby influencing chromatin structure and modulating the expression of transcription factors associated with genes that respond to stress, inflammation, and metabolism. The expression of SIRT1 decreases with age, which is associated with age-related CVD. Lower SIRT1 levels are linked to increased oxidative stress and inflammation, partly through its interaction with NF-κB, where SIRT1 inhibits NF-κB-mediated pro-inflammatory signaling pathway[94-97].
Lysosomes
Lysosomes play an essential role in the heart by regulating the degradation and recycling of cellular components through autophagy, a key process for maintaining homeostasis and preventing the accumulation of damaged proteins and organelles[98]. This function is especially relevant in cardiomyocytes, cells with high energy demands and low renewal capacity. Lysosomal dysfunction has been associated with cardiovascular diseases, including heart failure, due to its impact on proteome quality and mitochondrial health. In addition, the lysosome acts as a metabolic signaling center, serving as a scaffold for pathways such as mTOR, which are fundamental to cardiac function[99].
In the aging process and CVD, studies have shown decreased and abnormal autophagy. For example, decreased activity of the transcription factor EB (TFEB), a master regulator of autophagy and lysosome biogenesis, has been observed under conditions of hyperglycemia and overutilization of fatty acids, leading to glycolipotoxicity and thereby contributing to cardiotoxicity and cardiomyopathy. Conversely, overexpression of TFEB leads to atheroprotection, including reductions in plaque burden, apoptotic and necrotic areas, through decreased accumulation of ubiquitinated protein aggregates and lower IL-1β levels[100,101].
Lysosomes also regulate lipid metabolism, which is critical for maintaining healthy vascular function. Lipopolysaccharide (LPS) induces autophagy and nuclear translocation of TFEB in the heart of young but not aged mice, which may increase susceptibility to LPS-induced myocardial injury in aged mice[102].
In atherosclerotic lesions, macrophages capture oxidized LDL through the action of scavenger receptors. The proper functioning of lysosomes is crucial for the degradation of cholesterol. Dysfunctional lysosomes can lead to the accumulation of cholesterol within macrophages, contributing to the formation of foam cells and plaque instability[103].
The lysosome also regulates endothelial function and participates in transmembrane signaling of different death receptors through the formation of membrane raft redox signaling platforms, leading to endothelial dysfunction in response to various stimuli. During atherosclerosis, the lysosome-associated membrane signalosome plays a crucial role in endothelial injury, such as abnormal leukocyte adhesion, macrophage invasion or infiltration, and local oxidative stress[104].
Decreased expression of lysosome-associated membrane protein 2 (LAMP2) on the X chromosome causes Danon disease, which often leads to HF. In human cardiomyocytes, autophagosome-lysosome fusion requires the LAMP-2 B isoform; genetic correction of the LAMP-2 mutation rescues the Danon phenotype. This study provided evidence of cardiomyopathy in Danon patients and suggested that defective LAMP-2B-mediated autophagy could be a therapeutic target for treating this patient population[100,105].
Similarly, the cooperation between oxidation and chronic low-grade inflammation, which is prevalent in aging and CVD, can impact lysosomal function. This dysfunction may accumulate damaged proteins and organelles within cells, exacerbating cellular stress and promoting cardiac aging, which appears to be the key event in the development of early atherosclerotic lesions[106].
Lipid droplets
LDs are ubiquitous and dynamic cellular organelles that store lipids in cells of different organisms, such as bacteria, yeasts, and mammals[107,108]. They are delimited by a phospholipid monolayer filled with neutral lipids like triacylglycerols (TAGs) and sterols[107-109]. Mounting evidence shows that LDs can buffer toxic lipid levels, thereby mediating a cytoprotective role[109,110]. Additionally, they participate in cellular clearance of damaged and misfolded proteins[109,111], aid in cellular detoxification by sequestering xenobiotics[112], and contribute to innate immunity in infected mammalian cells through LD remodeling[113]. LD surface contains many proteins that are necessary for LD dynamics, contact with other organelles, membrane trafficking, signaling, and lipid homeostasis, such as for the synthesis of phospholipids, TAGs and their intermediates, as well as lipases and lipolytic regulators[107-109]. LDs are synthesized in the ER, maintaining an equilibrium between an embedded in the ER state and an emerged state that is regulated by the ER’s phospholipid molecular curvature, and each state is important for modulating LD functions[114].
Specifically, the heart uses a variety of carbon substrates as an energy source, with fatty acids (FAs) being the primary ones[115]. After FAs enter cardiomyocytes, they are β-oxidized by mitochondria for proper myocardial contraction[115,116]. The importance of LD in the heart is related to preventing excess ROS production from mitochondrial oxidation and free FAs-induced lipotoxicity, where excess free FAs are converted to TAG and then stored in LD[116,117].
Nevertheless, it has been established that LD function imbalance is an important player in a variety of diseases, including CVD[108]. Paradoxically, it has been reported that LD accumulation counteracts stress-associated diseases, showing a beneficial effect on aging, but, conversely, LD accumulation accentuates disease progression at advanced stages, thus promoting the aging process[109]. For example, it is well known that dietary monounsaturated fatty acids (MUFAs) extend lifespan, and their levels are positively related to lifespan[118,119]. In aging models, MUFA accumulation increases LD content in the intestine of both hermaphrodites (female-like) and male C. elegans, and cis-MUFAs, like oleic acid, enhance LDs and peroxisome formation, thereby contributing to MUFA-induced longevity in C. elegans[120].
In the cardiovascular system, LD accumulation has context-dependent effects. In some cases, it reflects lipid overload and reduced cardiac function by affecting transcriptional regulation of metabolic gene expression in failing hearts and reduced mitochondrial function. In other cases, LDs sequester toxic lipids such as cholesterol and ceramides, thereby preventing lipotoxicity[110,117]. The different metabolic states of cells could modulate the context for LD, explaining the differing effects of LDs in different cells and organs, including the heart[121].
An example of the LD’s beneficial effects in cardiovascular health is the LD-associated protein ABHD5, which acts as a protease. It has a cardioprotective effect through HDAC4 proteolysis that results in CaMKII-resistant HDAC4-NT, which in turn represses MEF2-dependent transcriptional programs that drive cardiac dysfunction through metabolic control of calcium handling[122]. Moreover, a human long non-coding RNA (lncRNA), termed Lipid-Droplet Transporter (LIPTER), acts as an RNA linker that connects LD via phosphatidic acid (PA)/phosphatidylinositol 4-phosphate (PI4P) and the cytoskeleton via binding MYH10, facilitating LD transport within human cardiomyocytes. Specifically, LIPTER deficiency impairs LD transport and metabolism as well as mitochondrial function and cardiomyocyte viability; meanwhile, its overexpression reduces cardiomyopathies and preserves cardiac function in obesity and diabetes mouse models[123]. It has also been reported that upon physical interaction with the ER membrane, LDs act as a lipid-buffer system against toxic molecules, mitigating the lipotoxicity process in foam cells of human atherosclerotic lesions[110].
On the other hand, considering the negative effect of LD on cardiovascular health, it is known that excessive cardiac LD accumulation under a hyperlipidemic state represents a negative factor for cardiac function, and evidence indicates that hyperglycemia suppresses the NKX2.5/LIPTER interaction, which induces cardiomyocyte LD deposition and cell death[121,123]. In addition, the LD-associated protein Perilipin 5 (PLIN5) is implicated in type 1 diabetes-induced cardiomyopathy, where STZ-induced type 1 diabetic PLIN5-/- mice fail to show detectable LD accumulation, but instead display decreased intramyocardial fatty acids, ceramides, sn-1,2-diacylglycerol (DAG), and lipid intermediates, and lower PKC activity, NOX subunits p47phox and p67phox levels, and ROS production in the heart, compared with WT mice[124].
ORGANELLE MISCOMMUNICATION IN THE AGING HEART
Cardiac aging entails not only structural and functional alterations at the level of individual organelles, as discussed in the previous chapter, but also a progressive loss of communication between them [Figure 2]. This disruption in subcellular coordination represents an emerging mechanism that exacerbates the functional decline of the myocardium. Unlike the changes observed exclusively in cardiovascular diseases, aging introduces a basal burden of dysfunction that predisposes the heart to fail in response to pathological stimuli. This section explores the molecular and structural pathways that underlie this miscommunication between organelles in the aging heart.
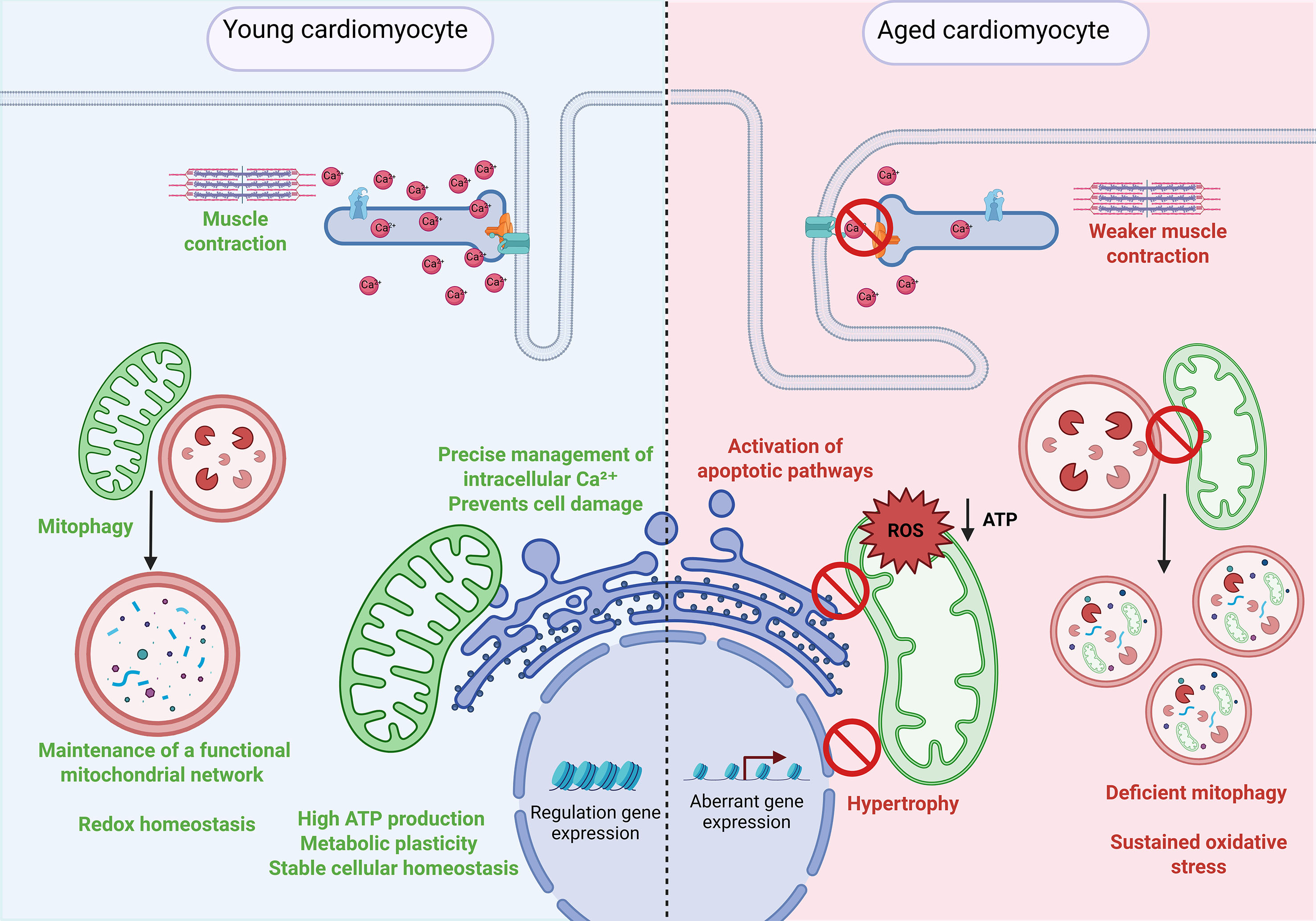
Figure 2. Cellular and subcellular alterations in aged cardiomyocytes. Comparison between young (left) and aged (right) cardiomyocytes, highlighting key age-associated changes. In young cardiomyocytes, efficient Ca2+ handling, active mitophagy, and coordinated organelle communication preserve mitochondrial function, gene expression regulation, and muscle contractility. In contrast, aged cardiomyocytes exhibit impaired Ca2+ cycling, loss of ER-mitochondria contacts, disrupted mitophagy, and mitochondrial dysfunction characterized by elevated reactive oxygen species (ROS) and reduced ATP production. These alterations lead to hypertrophy, aberrant gene expression, oxidative stress, and weaker cardiac contraction, ultimately contributing to age-related cardiac dysfunction.
Mitochondria-associated ER membranes
Mitochondria-associated ER membranes (MAMs) are specialized inter-organelle contact sites that coordinate calcium signaling, lipid transfer, and mitochondrial dynamics essential for maintaining cardiac bioenergetics. In the heart, these structures underlie excitation-contraction coupling and critically regulate apoptotic and autophagic pathways to support cardiomyocyte survival.
During aging, MAM integrity is disrupted in striated muscle, and proteomic analysis revealed age-related dysregulation of MAM-enriched proteins associated with metabolic rewiring, Ca2+ imbalance, and impaired organelle dynamics[125] [Figure 3]. The deterioration of MAM function contributes to the progression of age-related diseases, making it a potential therapeutic target[126]. Understanding MAM dysfunction in aging will provide insights into the mechanisms underlying age-associated cardiovascular and metabolic diseases, offering new avenues for developing targeted interventions.
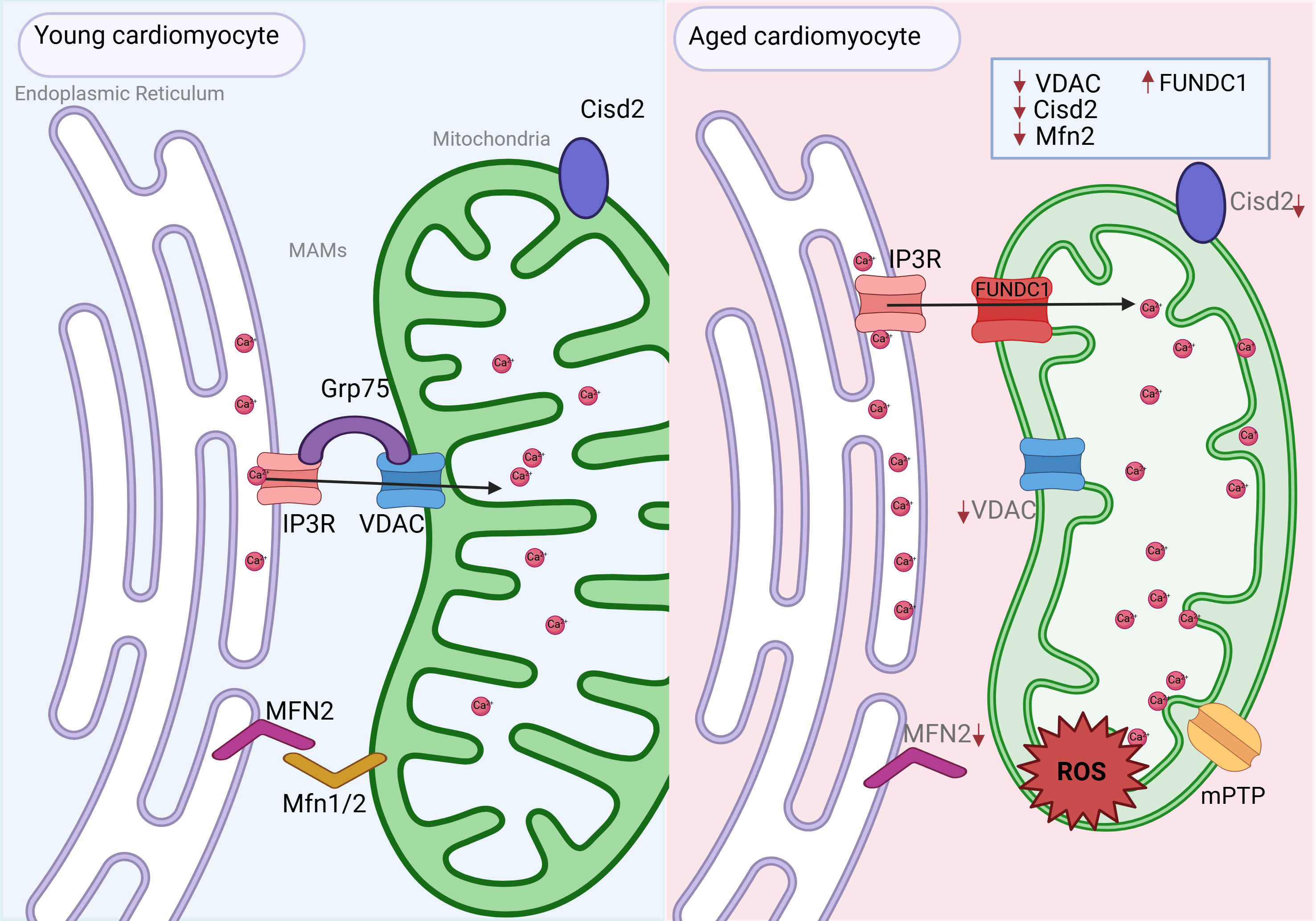
Figure 3. Age-associated alterations in mitochondria-associated ER membranes (MAMs) in cardiomyocytes. Under physiological conditions (young heart), MAMs facilitate proper communication between the sarcoplasmic reticulum (SR) and mitochondria, enabling efficient Ca2+ transfer, lipid exchange, and mitochondrial dynamics. This supports optimal ATP production, controlled autophagy, and cardiomyocyte survival. Key structural and regulatory proteins such as MFN2, VDAC1, and CISD2 maintain MAM integrity and mitochondrial function. In contrast, aged cardiomyocytes exhibit disrupted MAM architecture, characterized by reduced MFN2 and CISD2 expression and increased levels of FUNDC1. These changes lead to impaired SR-mitochondria communication, excessive mitochondrial Ca2+ uptake, oxidative stress, mitochondrial fragmentation, and bioenergetic failure. The resulting mitochondrial dysfunction and accumulation of damaged organelles contribute to cellular senescence and increase the heart’s susceptibility to age-related diseases such as heart failure and diabetic cardiomyopathy.
Under normal physiological conditions, MAMs are characterized by dynamic interactions that maintain Ca2+ homeostasis and support mitochondrial bioenergetics[127]. These interactions enable efficient ATP production and regulate apoptosis and autophagy, which are crucial for cardiomyocyte survival and function. The close apposition of ER and mitochondrial membranes enables efficient Ca2+ transfer, which is vital for excitation-contraction coupling in heart muscle cells. Under normal physiological conditions, this Ca2+ transfer supports mitochondrial ATP production and regulates apoptotic pathways, ensuring cellular homeostasis and survival[128,129].
MAMs also play a significant role in lipid metabolism by mediating the exchange of lipids between the ER and mitochondria. This interaction is crucial for fatty acid oxidation, providing the cardiac function with energy. Proteins such as MFN2 and Voltage-dependent anion channel 1 (VDAC1) are essential to maintain MAM structure and function, enabling optimal metabolic coupling between these organelles[130,131].
As the heart ages, significant structural and functional changes occur at MAMs, disrupting their regular communication. Research indicates that aging is associated with reduced abundance of key proteins involved in MAM formation, leading to impaired Ca2+ signaling and altered lipid metabolism. For instance, the CDGSH iron-sulfur domain 2 (CISD2) gene, which promotes longevity and is abundant within MAM, has been shown to preserve energy metabolism and reduce oxidative stress in aged hearts. Elevated levels of CISD2 can reverse age-related cardiac dysfunction; conversely, its knockout leads to mitochondrial dysfunction and increased ROS production[132]. Notably, CISD2 has been linked to reduced lipofuscin accumulation in aging hearts, and its downregulation has been associated with premature aging phenotypes[133-135].
Additionally, aging cardiomyocytes exhibit a decline in mitochondrial dynamics characterized by increased fission and decreased fusion events. This imbalance contributes to mitochondrial fragmentation and dysfunction, further impairing MAM integrity. The accumulation of damaged mitochondria exacerbates oxidative stress and promotes cellular senescence - key features of aging that significantly impact cardiac function[136].
The miscommunication at MAM becomes more pronounced in various cardiac diseases associated with aging, such as HF and diabetic cardiomyopathy. Structural changes to MAM are linked to mitochondrial dysfunction and impaired cellular signaling pathways in these conditions. For example, in diabetic cardiomyopathy, elevated levels of FUN14 domain-containing 1 (FUNDC1) have been observed in cardiac tissues, which affects MAM structure and function by altering Ca2+ handling and energy metabolism[137,138]. There is also evidence of altered mitochondrial dynamics in aged hearts characterized by increased fission and reduced fusion processes. This imbalance can lead to dysfunctional mitochondria accumulating within cardiomyocytes, further impairing energy production and increasing susceptibility to ischemic injury. Moreover, the loss of effective communication at MAM during disease states results in diminished ATP production and compromised cellular survival mechanisms. This miscommunication not only accelerates cardiac aging but also heightens the risk of developing HF as individuals age.
Plasma membrane in cardiac aging
The plasma membrane of cardiomyocytes, particularly their T-tubule system, is essential for the heart, as it ensures the propagation of action potentials and the coordinated release of Ca2+, which are fundamental for contraction. With aging, its disorganization compromises signaling between calcium channels and the SR, contributing to cardiac dysfunction[139] [Figure 4].
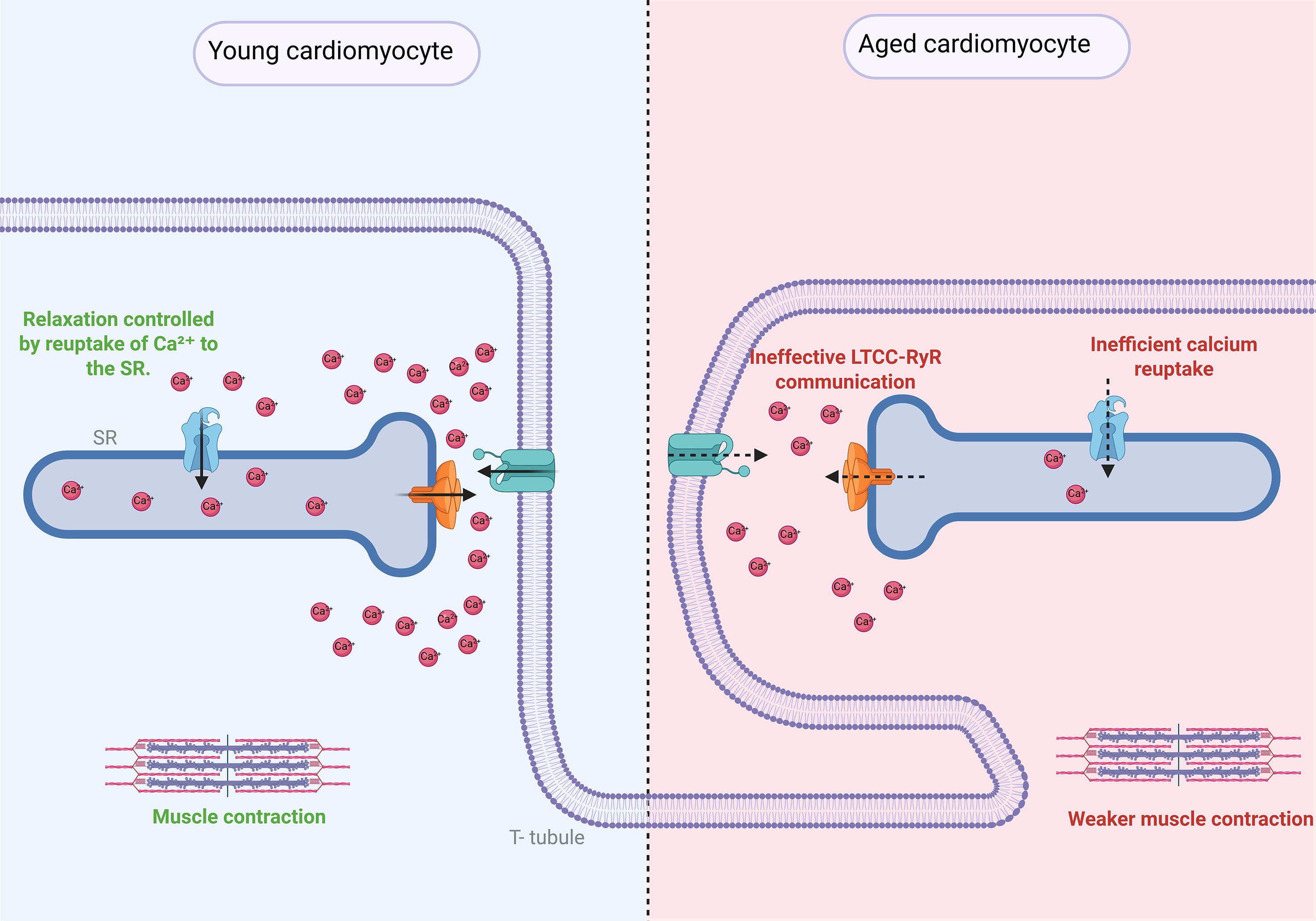
Figure 4. Structural and functional coupling between T-tubules and the sarcoplasmic reticulum (SR) in cardiomyocytes and its disruption during aging. In young cardiomyocytes, T-tubules invaginate from the plasma membrane and align precisely with the sarcoplasmic reticulum (SR), allowing for a rapid propagation of action potentials and synchronized Ca2+ release for excitation-contraction (E-C) coupling. L-type Ca2+ channels (LTCCs) located in T-tubules interact closely with ryanodine receptors (RyR) on the SR to trigger Ca2+-induced Ca2+ release. During aging, T-tubule architecture becomes disorganized and less dense, leading to misalignment with the SR, reduced LTCC-RyR coupling efficiency, delayed Ca2+ transients, and impaired contractile function. These structural and functional alterations contribute to the development of cardiac dysfunction, including heart failure with preserved ejection fraction (HFpEF), arrhythmias, and impaired relaxation.
The plasma membrane of cardiomyocytes not only delimits the cell, but is also a key component in the regulation of the flow of ions such as sodium (Na+), potassium (K+), and Ca2+, which are essential for the mechanical function of the heart muscle[140,141]. Additionally, cardiomyocyte membrane receptors respond to extracellular cues, such as hormones and neurotransmitters, activating signaling cascades that regulate cellular function[63]. Cardiomyocytes are further specialized by plasma membrane invaginations known as T-tubules, which interact with the ER, termed the SR in muscle cells. This structural arrangement enables rapid propagation of action potentials throughout the cardiomyocyte[142].
The proximity between T-tubules and the SR facilitates Ca2+ release from the SR in response to depolarization, a crucial process for cardiac muscle contraction[143]. T-tubules facilitate the propagation of electrical signals into the cell, allowing the action potential to reach the SR quickly[144]. The SR stores and releases Ca2+ in a controlled manner, an essential step in muscle contraction[145]. The process of excitation-contraction coupling depends on the precise interaction between L-type Ca2+ channels (LTCCs) in the T-tubules and RyRs[144].
LTCCs mediate the entry of extracellular Ca2+, which triggers Ca2+ release from the SR[67]. T-tubules were first described in 1881 by Retzius, who suggested the existence of electrical depolarization in muscle cells[146]. One of the most significant changes with aging is the disorganization and loss of T-tubules in cardiomyocytes[68]. This disorganization disrupts the signaling synchronization between the plasma membrane and the SR, affecting the coordinated Ca2+ release[68]. The reduced density and irregular distribution of T-tubules in the aged heart slow the propagation of the action potential, resulting in less efficient Ca2+ release and, consequently, weaker muscle contraction[147]. The disruption in communication between T-tubules and the SR is also associated with increased muscle relaxation time, contributing to heart failure commonly seen in cardiac aging[148]. Some associated pathologies include HFpEF, cardiac arrhythmias, dilated cardiomyopathy, and systolic HF, where T-tubule disorganization contributes to diastolic dysfunction, leading to significant consequences for cardiovascular health[149-151].
In the aged heart, a decrease in the functionality of LTCC has been described, leading to reduced Ca2+ entry during the depolarization phase, which affects the contractile strength of cardiac muscle[152]. Additionally, alterations in the communication between LTCC and RyR reduce the efficacy of excitation-contraction coupling, contributing to a reduced capacity of the heart to generate effective contractions[152].
The disruption in communication between the plasma membrane, T-tubules, and SR has profound implications for cardiac function[153]. The disorganization of T-tubules and dysfunction of the SR and LTCC result in inefficient Ca2+ release and a decrease in the heart's ability to generate strong, coordinated contractions[144]. These changes contribute to the reduced capacity of the aging heart to maintain efficient pumping, which can lead to the development of HF and arrhythmias[154].
Mitonuclear communication in cardiac aging
Mitonuclear communication is essential for maintaining cellular homeostasis, particularly in tissues with high energy demands such as the myocardium. Under physiological conditions, this crosstalk regulates key processes including mitochondrial biogenesis, cellular differentiation, and metabolic function. However, during aging, this interaction becomes dysregulated, leading to impaired mitochondrial biogenesis and the activation of pathways associated with cellular senescence[155] [Figure 5]. These alterations promote cardiomyocyte hypertrophy, contributing to pathological cardiac remodeling and the progression toward heart failure.
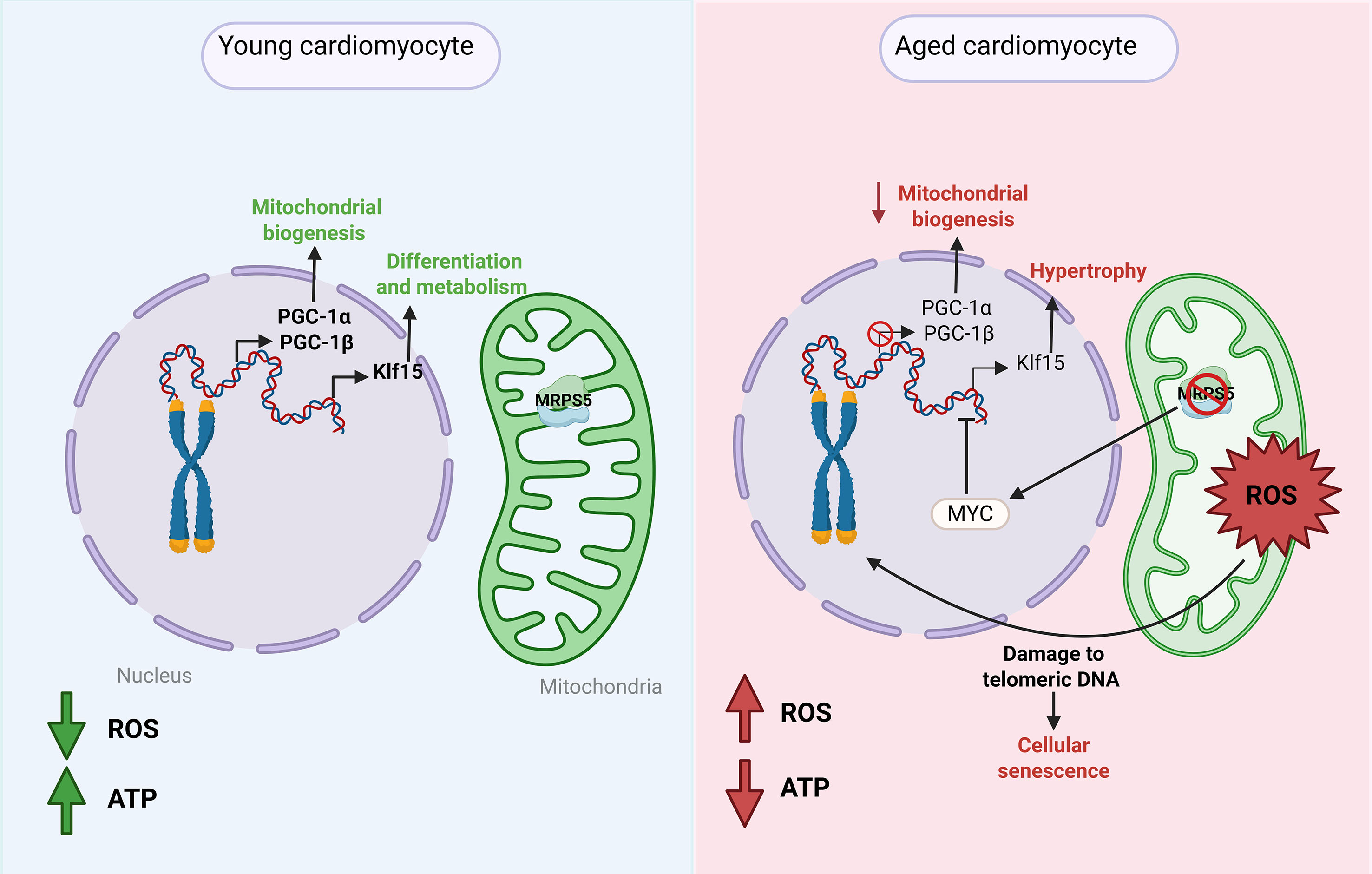
Figure 5. Alterations in mitonuclear communication during cardiac aging. In young cardiomyocytes, mitonuclear signaling - mediated by the coactivators PGC-1α and PGC-1β and the transcription factor KLF15 - supports mitochondrial biogenesis, oxidative metabolism, and cellular homeostasis. In contrast, aged cardiomyocytes exhibit disrupted mitonuclear communication, characterized by downregulation of PGC-1α/β and a shift toward Klf5 expression. This imbalance contributes to mitochondrial dysfunction, increased production of reactive oxygen species (ROS), cellular senescence, and pathological hypertrophy, ultimately promoting adverse cardiac remodeling and age-related functional decline.
The relationship between the nucleus and mitochondria has recently been characterized in the context of cardiac aging. The purpose of mitonuclear communication is to maintain cellular homeostasis under both basal conditions and in response to various stresses[156]. Mutations in genes involved in mitonuclear communication and imbalanced signaling resulting from altered mitonuclear responses are often associated with developmental defects and human diseases[156]. Anderson et al. described how mitochondria-derived ROS trigger telomeric DNA damage, leading to senescence in aged cardiomyocytes, suggesting new avenues for improved cardiac regeneration therapies[157]. Along the same lines, research has established that telomere dysfunction compromises cardiomyocyte function by repressing PGC-1α and PGC-1β, and this simultaneous repression is associated with impaired mitochondrial biogenesis, thereby hindering cardiac ATP production[158,159].
Moreover, Gao et al. provide evidence that the transcription factor Kruppel 15 (KLF15) can transmit signals via L-phenylalanine/c-myc and AMP/p-CREB from mitochondria to nuclei to activate gene expression that can modulate cardiac metabolism and hypertrophy, potentially leading to age-related CVD[156].
It has also been reported that under conditions of mitochondrial stress, such as myocardial infarction, mitonuclear signaling plays a critical role in cardiac regeneration. Gao et al. demonstrated that in a heterozygous Mitochondrial ribosomal protein S5 (MRPS5) knockout model (Mrps5+/-), cardiomyocyte proliferation is enhanced. However, this effect is attenuated when one allele of the transcription factor ATF4 is also genetically deleted (Mrps5+/-/Atf4+/-), leading to a loss of the heart’s regenerative capacity. These findings highlight the essential role of MRPS5/ATF4 mitonuclear communication in regulating cardiomyocyte proliferation and cardiac regeneration[155].
Mitonuclear communication plays a crucial role in cardiac aging and overall longevity. Mitochondrial dysfunction, a hallmark of aging, involves oxidative stress, damage, and impaired biogenesis[160]. Age-related decline in NAD+ levels disrupts nucleus-mitochondria communication, leading to a pseudohypoxic state and reduced mitochondrial function, which can be reversed by raising NAD+ levels[161]. Mitochondrial metabolites and stress signals modulate epigenetic changes, affecting aging processes and homeostasis[162]. Key regulators of mitochondrial function and aging include PGC-1α, SIRT1, AMPK, and mTOR[155]. Potential therapeutic strategies to improve mitochondrial function in aging and CVD include mitochondrial-targeted antioxidants, calorie restriction, calorie restriction mimetics, and exercise training[160].
Lysosome-mitochondria communication
Communication between lysosomes and mitochondria is essential for regulating cellular stress, particularly through autophagy mechanisms, a process that is indispensable for preserving cardiac function and activating cardioprotective responses. It has been shown that the loss of functional contacts between these two organelles contributes to mitochondrial dysfunction and an alteration in lysosomal dynamics, which is associated with the development of various CVDs, such as acute myocardial infarction (MI)[163] and ischemia/reperfusion (I/R) injury[164] [Figure 6]. In addition, disorders such as mucopolysaccharidosis type II (Hunter syndrome) also present alterations in these contacts, which affect cellular bioenergetics and contribute to their pathophysiology.
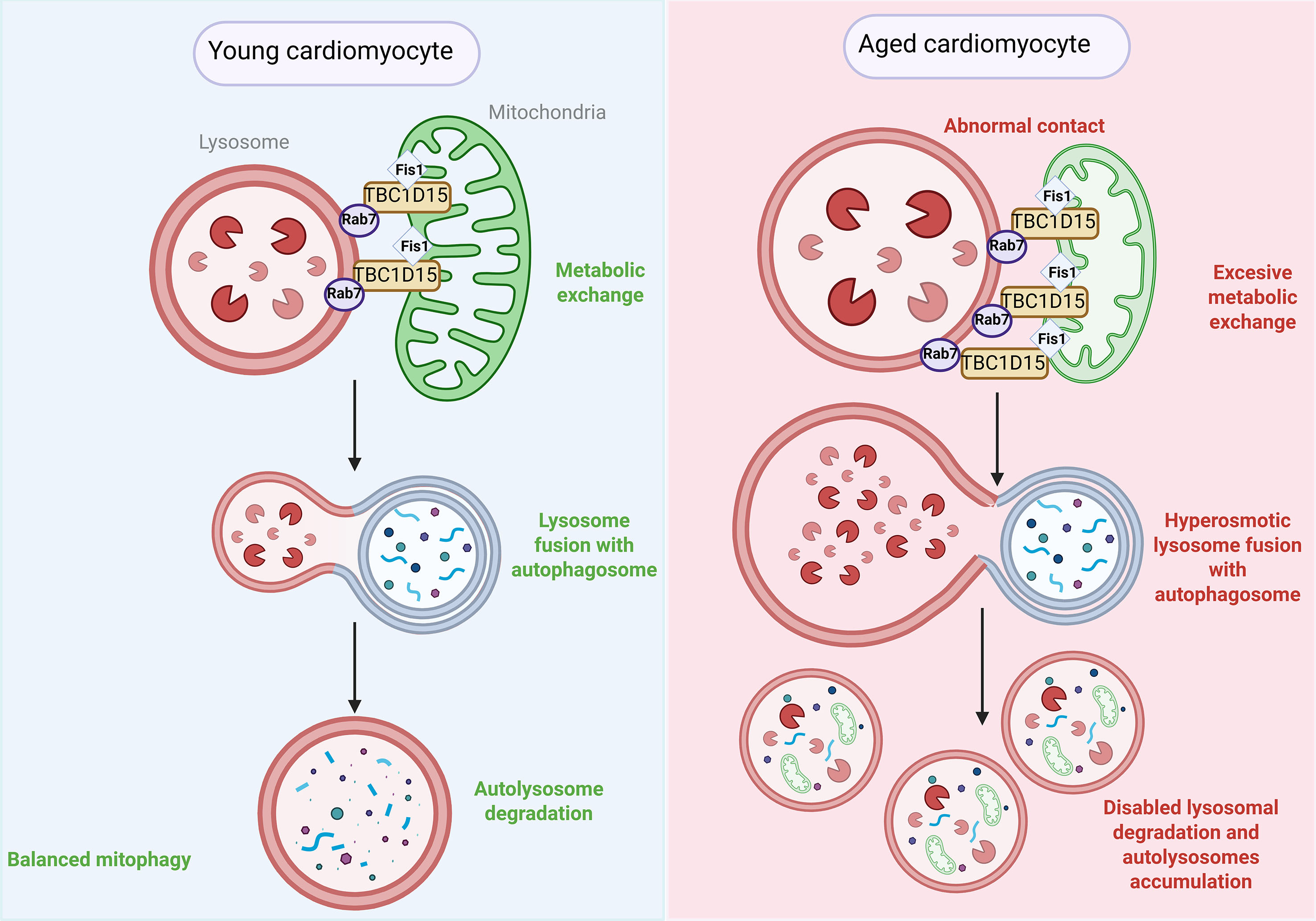
Figure 6. Lysosome-mitochondria communication in cardiomyocytes under physiological and aging conditions. In healthy cardiomyocytes, transient contact sites between mitochondria and lysosomes are regulated by Rab7 in its GTP-bound active state and its effector TBC1D15, which is recruited to the mitochondrial surface via the fission protein Fis1. These contacts contribute to mitochondrial dynamics, mitophagy, and cellular homeostasis. Conversely, in aged or stressed cardiomyocytes, impaired recruitment of TBC1D15 or dysregulation of Rab7 activity leads to prolonged or unstable contacts, defective mitophagy, and altered mitochondrial fission, resulting in the accumulation of dysfunctional mitochondria and elevated oxidative stress. These alterations contribute to mitochondrial dysfunction, impaired cardiomyocyte contractility, and the progression of age-related cardiovascular diseases.
An additional process that significantly affects mitochondria-lysosome communication is aging, a physiological condition characterized by progressive deterioration of cellular homeostasis mechanisms, including the autophagic-lysosomal system and mitochondrial function. During aging, there is a reduction in autophagy efficiency, an accumulation of dysfunctional mitochondria, and a decreased ability to adapt to cellular stress, processes that have been associated with a loss of functional contacts between mitochondria and lysosomes[165-167]. These changes directly impact highly energetic tissues such as the heart, contributing to the progression of CVD and heart failure with preserved ejection fraction (HFpEF)[168]. Age-related mitochondrial dysfunction is also linked to an increase in reactive oxygen species (ROS), deterioration in mitochondrial biogenesis, and alterations in organelle dynamics, which can further compromise the integrity of these interorganelle contacts[167-169].
Both mitochondria and lysosomes play central roles in cellular homeostasis and are involved in the storage of key metabolites such as calcium, iron, and lipids, as well as in the regulation of cell death pathways[170-175]. Recent studies have shown that there is a direct structural and functional interaction between these two organelles, independent of classical degradation processes such as mitophagy or the elimination of mitochondria-derived vesicles (MDVs)[171].
This mitochondria-lysosome communication occurs predominantly under conditions of cellular stress[176,177] and can occur through degradative processes - such as PINK1/Parkin-dependent mitophagy via receptors such as optineurin or NDP52[178,179] - or through the formation of MDVs (~100 nm) that transport specific subsets of mitochondrial proteins to lysosomes for degradation[176,180]. However, there are also non-degradative contacts, with an approximate distance of ~10 nm between the membranes, in which there is no exchange of content between the two compartments[181]. In these cases, it has been observed that such contacts do not promote either autophagosome biogenesis or mitophagy[172].
The regulation of these contacts is mediated by the lysosomal GTPase Rab7, which controls lysosomal dynamics. The effector protein TBC1D15, a member of the TBC domain protein family, is recruited to the outer mitochondrial membrane via the Fis1 protein and binds to Rab7 in its active form (GTP) to facilitate its hydrolysis to the inactive form (GDP)[181-184]. This conversion induces the dissociation of contacts between mitochondria and lysosomes. Mutations in TBC1D15 (such as D397A or R400K), which inhibit its GAP activity, prevent this dissociation, prolonging contacts without affecting their formation[184], suggesting the existence of alternative coupling mechanisms between the two organelles[172,184]. Similarly, the Fis1 mutation that prevents the recruitment of TBC1D15 also causes a prolongation of the interaction time between mitochondria and lysosomes[184].
These contacts not only enable physical interaction between the two organelles, but also act as signaling platforms to modulate lysosomal dynamics through Rab7-GTP activity[172]. TBC1D15-mediated Rab7-GTP hydrolysis enables the dissociation of contacts and the release of Rab7 effectors from the lysosomal membrane[185]. It has been observed that the loss of TBC1D15 GAP activity causes lysosomal elongation[181], while mitochondrial fission is regulated by vesicles positive for LAMP1, a lysosome marker, in the absence of endosomes or peroxisomes[181]. Disruption of these contacts affects the architecture of the mitochondrial network, reducing the rate of mitochondrial fission[181]. In addition, a brain-specific isoform of the Drp1 protein (Drp1 ABCD) has been described, which participates in mitochondrial fission and is associated with LAMP1-positive vesicles at the mitochondria-lysosome contacts[186].
The alteration of this interorganelle communication has been implicated in various neurodegenerative and metabolic pathologies. For example, in Charcot-Marie-Tooth disease type 2 (CMT2), Parkinson's disease (PD), and several lysosomal storage disorders (LSDs), dysregulation of mitochondria-lysosome contacts has been reported[187-190]. In CMT2B, mutations in Rab7 increase its active form (GTP), which may promote the overformation of pathological contacts[185,191,192]. In PD, both mitochondrial and lysosomal dysfunction have been observed in human dopaminergic neurons[188,189], while in lysosomal diseases such as Gaucher, Niemann-Pick disease type C, and neuronal ceroid lipofuscinosis, reduced mitochondrial membrane potential and elevated ROS levels have been documented[171,192-194]. This evidence positions mitochondrial dysfunction as a common feature of LSDs, possibly mediated by alterations in mitochondria-lysosome contacts[181]. In the context of aging, these same mechanisms are progressively exacerbated and could amplify cellular vulnerability to various degenerative pathologies[167,168]. In the cardiac context, mitochondrial dysfunction directly contributes to the development of multiple CVDs[195]. Recent studies have highlighted the cardioprotective role of mitochondria-lysosome interactions in models of acute MI and I/R[163,164]. The decrease in TBC1D15 in these models is associated with an increase in infarct area, greater fibrosis, cardiomyocyte apoptosis, and mitochondrial damage[163]. Conversely, its overexpression improves these parameters by promoting its interaction with Fis1 and Rab7-GTP and by recruiting Drp1 to the mitochondria-lysosome contacts, facilitating asymmetric mitochondrial fission[164].
Together, these observations reinforce the idea that mitochondria-lysosome contacts are not only essential for mitochondrial quality control, but also represent a key regulatory platform for cellular homeostasis and cardiac tissue protection. Emerging evidence also highlights that their dysfunction is a common mechanism underlying both physiological aging and multiple chronic diseases. Despite advances, the precise role of these contacts in the pathophysiology of MI, heart failure (HF), and aging requires further experimental exploration.
CONCLUSION AND FUTURE DIRECTIONS
Regarding architecture and function, the heart is a remarkable biological machine that begins beating on days 21-23 post-fertilization and continues until death. Accordingly, cardiomyocyte organelles display an extraordinary level of specialization. The SR provides mechanical support and regulates the Ca2+ signaling that drives contraction, working in conjunction with the plasma membrane. Mitochondria, on the other hand, serve as efficient ATP sources to sustain a heart rate of 60-100 beats per minute in an adult. Thus, mitochondrial bioenergetics must attune to the rhythmic contractile activity. Meanwhile, the nucleus and lysosomes regulate mitochondrial turnover by driving mitogenesis and mitophagy, respectively. During aging, not only does organelle function gradually decline, but also the coordination of their activities, thereby rendering organelle communication a potential therapeutic target for heart diseases [Figure 2]. Other lesser-studied organelles also contribute to cardiomyocyte function, such as LDs and peroxisomes, which regulate fatty acid metabolism, the primary macronutrient for the heart under normal conditions. Both organelles are rather “talkative” with other organelles[165,166], and thus, their communication within cardiomyocytes requires further research. In the case of the heart, rather than focusing on the communication between pairs of organelles, future evidence will enable us to define a communication network of organelles that harmonizes fat metabolism with cardiomyocyte energy demands. Interestingly, organelles themselves are networks, either continuous like the SR or discontinuous like mitochondria. Therefore, the concept of an “organelle internetwork” is more descriptive.
A key approach in geroscience is the “hallmarks of aging”, which are conserved mechanisms that appear with age, and their modulation can slow down or accelerate the aging process[167]. Mitochondrial dysfunction is one of these hallmarks, while ER stress and lysosomal alterations correspond to “loss of proteostasis” and “disabled macroautophagy”, respectively[168]. Indeed, this triumvirate of organelles is essential for defining cell function and dysfunction. However, disabled macroautophagy not only affects lysosomal function, but is a general quality control mechanism for all organelles. Additionally, these hallmarks are highly intertwined, since decreased organelle quality control also promotes mitochondrial dysfunction and ER stress[168]. Thus, further insights into the function of the lesser-studied organelles might result in the definition of “disrupted organelle internetworking” as an overarching subcellular hallmark of aging.
As discussed, ER-mitochondria communication during cardiomyocyte aging decreases, and the same happens in other quiescent cell types such as neurons and skeletal myocytes, which contributes to neurodegeneration and insulin resistance, respectively[140]. By contrast, in hepatocytes, which can proliferate[169], obesity is associated with chronic enrichment of ER-mitochondria contact sites, leading to mitochondrial Ca2+ overload and impaired oxidative capacity[196]. Similarly, in endothelial cells, which can also proliferate, aging correlates with increased ER-mitochondria Ca2+ transfer that renders cells more susceptible to death by Ca2+ overload[197]. Thus, the modulation of MAM during aging and pathogenesis depends on cell type, and evidences suggest that proliferative cells are more prone to excess ER-mitochondria Ca2+ transfer compared to the post-mitotic. This difference might stem from the cell fates that depend on Ca2+ signaling. The aforementioned quiescent cells are more complex in terms of architecture and their regeneration is limited. Concomitantly, they downregulate ER-mitochondria communication during aging and pathogenesis, which might be a strategy to prevent excess mitochondria-derived oxidative stress and cell death. After all, both neurons and cardiomyocytes are nearly irreplaceable in the adult. In the case of hepatocytes and endothelial cells, in turn, ER-mitochondria communication increases, which might be a compensatory mechanism to cope with stress. In these cells, the ensuing cell death is not as problematic as in quiescent cells. Furthermore, in proliferative cells, mitochondrial Ca2+ overload might also lead to cell senescence[198,199], which arrests the cell cycle and recruits immune cells through inflammatory factors to promote the removal and replacement of the damaged cells[200]. In contrast, quiescent cells by definition cannot undergo cell senescence, but attain a “senescence-like” phenotype characterized by structural damage and a pro-inflammatory secretome that contributes to neurodegeneration[201] or muscle aging[202]. Since organelles play a key role in determining cell fates (especially the ER and mitochondria), it is expected that they behave distinctly in cell types with such diverging trajectories in terms of proliferation, dysfunction, death, and survival.
Among organelles, mitochondria are unique in that they have their own DNA (mitochondrial DNA, mtDNA), and thus have partial autonomy in propagating inside the cell and defining their own components, namely proteins and RNAs. Accordingly, mitochondrial transplantation has lately emerged as a therapeutic approach for aged or diseased organs, including the heart[203]. Administering healthy mitochondria to damaged cells has been reported to improve bioenergetics performance[203]. Thus, mitochondria are promising candidates as biological therapeutic agents against mitochondrial dysfunction[204]. However, because mitochondria interact extensively with other organelles, this raises the question: if mitochondrial transplantation alleviates mitochondrial dysfunction, can this strategy be used to “treat” the dysfunction of other organelles? Certainly, transplanted mitochondria not only propagate within the recipient cell but also have to interact with the existing organelles. This opens the possibility to “engineer” therapeutic mitochondria with a desirable interaction with other organelles. However, the mtDNA only codes a handful of proteins of the electron transport chain, which are not directly involved in organelle communication. Therefore, the use of mitochondria as vectors is complex and still has substantial progress to make[205].
Regarding the translation of basic research on organelle “internetworking” into clinical applications, this field still requires further development. Chemical modulators of ER-mitochondria communication have been reviewed elsewhere, proposing three classes according to their mechanism[206,207]. Class I corresponds to direct modulators of tethering proteins, such as LDC3/Dynarrestin, which increases PTPIP51-VAPB interaction, or TAT-MP1Gly, which targets Mfn2-, or pridopidine, which binds to Sigma-1 receptor - all of them potentially modulating organelle communication[208-210]. Class II drugs regulate the expression of the tethering proteins at the transcriptional level, such as metformin and breviscapine, which have protective actions against cardiac remodeling and I/R injury, respectively[211,212]. Class III comprises regulators of upstream signaling, like quercetin, which modulates organelle contacts through AMPK[213]. In terms of plasma membrane-SR communication, this signaling axis is targeted by common drugs, such as dihydropyridines and dantrolene, which alleviate hypertension through LTCC inhibition or malignant hyperthermia via RyR inhibition, respectively[214,215]. Both can be considered modulators of the plasma membrane-SR junction at the functional rather than physical level, because they depress Ca2+-mediated communication between both compartments; however, to our knowledge, neither of them has been reported to prevent sarcolemmal or SR pathological remodeling. Regarding mitochondria-nucleus communication, available strategies are largely limited to “mitotoxics”, such as doxycycline, which promotes pathological mitonuclear signaling through mitochondrial stress[216]. Doxycycline is an antibiotic that targets bacterial translation; however, because of the similarity of mitochondrial and bacterial translation systems, it also alters OXPHOS stoichiometry[217]. Mitochondria-lysosome interplay corresponds mainly to mitophagy, an area of active drug development. Among the drugs targeting this axis, rapamycin has been reported to increase mitophagy through enhancement of general autophagy[218]. Interestingly, rapamycin and its derivatives (“rapalogs”) are known anti-aging drugs[219]. Similarly, urolithins, which are metabolites of the ellagitannins abundant in the pomegranate, are promising mitophagy inducers[220]. Of note, they display cardioprotective capacity[221]. Finally, in the area of mitochondria-LD interaction, statins, which are largely known as cardiovascular protectors, reduce the expression of PLIN5 in the liver[222].
In summary, organelle communication and internetworking represent a promising field for further investigation, which will enable us to tackle age-related CVD and extend the human health span, thereby improving our quality of life.
DECLARATIONS
Acknowledgment
All figures are original and were created in BioRender. Maracaja-Coutinho, V. (2025) https://BioRender.com/vcui0ou. The authors thank Dr. Vinicius Maracaja-Coutinho for generously providing access to the license.
Authors’ contributions
Conception, writing, revision, and final approval: Ortega-Muñoz A, Bascuñan-Ortiz E, Troncoso MF,
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by Universidad de Chile Enlace ENL09/24 (to Bravo-Sagua R) and Puente 2024P1DID (to Bravo-Sagua R), FONDECYT grants 1240443 (to Lavandero S), 1220392 (Chiong M) and 3240492 (to Troncoso MF), and FONDAP Apoyo 1523A0008 (to Lavandero S, Chiong M and Bravo-Sagua R).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
2. Gómez LA. [Cardiovascular diseases: a public health problem and a global challenge]. Biomedica. 2011;31:469-73.
3. Yazdanyar A, Newman AB. The burden of cardiovascular disease in the elderly: morbidity, mortality, and costs. Clin Geriatr Med. 2009;25:563-77, vii.
4. Niklasson A, Maher J, Patil R, et al. Living with heart failure: patient experiences and implications for physical activity and daily living. ESC Heart Fail. 2022;9:1206-15.
5. Mulugeta H, Sinclair PM, Wilson A. The experience of people living with heart failure in Ethiopia: a qualitative descriptive study. PLoS One. 2024;19:e0310600.
6. Kreatsoulas C, Anand SS. The impact of social determinants on cardiovascular disease. Can J Cardiol. 2010;26 Suppl C:8C-13C.
7. Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. 2006;367:1747-57.
8. Alizadeh G, Gholipour K, Azami-Aghdash S, et al. Social, economic, technological, and environmental factors affecting cardiovascular diseases: a systematic review and thematic analysis. Int J Prev Med. 2022;13:78.
9. Ziaeian B, Fonarow GC. The prevention of hospital readmissions in heart failure. Prog Cardiovasc Dis. 2016;58:379-85.
10. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling-concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35:569-82.
12. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a "set up" for vascular disease. Circulation. 2003;107:139-46.
13. McDonagh TA, Metra M, Adamo M, et al. 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2021;42:3599-726.
14. Del Campo A, Perez G, Castro PF, Parra V, Verdejo HE. Mitochondrial function, dynamics and quality control in the pathophysiology of HFpEF. Biochim Biophys Acta Mol Basis Dis. 2021;1867:166208.
15. Paraskevaidis I, Kourek C, Farmakis D, Tsougos E. Mitochondrial dysfunction in cardiac disease: the fort fell. Biomolecules. 2024;14:1534.
16. Schwartz B, Gjini P, Gopal DM, Fetterman JL. Inefficient batteries in heart failure: metabolic bottlenecks disrupting the mitochondrial ecosystem. JACC Basic Transl Sci. 2022;7:1161-79.
17. Calle X, Garrido-Moreno V, Lopez-Gallardo E, et al. Mitochondrial E3 ubiquitin ligase 1 (MUL1) as a novel therapeutic target for diseases associated with mitochondrial dysfunction. IUBMB Life. 2022;74:850-65.
18. Liu H, Mao H, Ouyang X, Lu R, Li L. Intercellular mitochondrial transfer: the novel therapeutic mechanism for diseases. Traffic. 2024;25:e12951.
19. Ávila PM, Morales HF, Quirós-Meza G, Salazar-Nassar J, Castillo-Rivas J, Castillo-Rivas J. Prevalencia y factores de riesgo de enfermedad ateroesclerótica sistémica. Acta Méd Costarricense. 2014;56:6-11.
20. Frąk W, Wojtasińska A, Lisińska W, Młynarska E, Franczyk B, Rysz J. Pathophysiology of cardiovascular diseases: new insights into molecular mechanisms of atherosclerosis, arterial hypertension, and coronary artery disease. Biomedicines. 2022;10:1938.
21. Papazoglou AS, Kyriakoulis KG, Barmpagiannos K, et al. Atherosclerotic risk factor prevalence in adults with congenital heart disease: a meta-analysis. JACC Adv. 2024;3:101359.
22. Kim S, Khoriaty R, Li L, et al. ER-to-golgi transport and SEC23-dependent COPII vesicles regulate T cell alloimmunity. J Clin Invest. 2021;131:136574.
23. Lodhi IJ, Semenkovich CF. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab. 2014;19:380-92.
24. Nikolich-Žugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. 2018;19:10-9.
25. Paillusson S, Stoica R, Gomez-Suaga P, et al. There's something wrong with my MAM; the ER-mitochondria axis and neurodegenerative diseases. Trends Neurosci. 2016;39:146-57.
26. Quirós PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol. 2016;17:213-26.
27. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14:576-90.
28. Chung JY, Ain QU, Song Y, Yong SB, Kim YH. Targeted delivery of CRISPR interference system against Fabp4 to white adipocytes ameliorates obesity, inflammation, hepatic steatosis, and insulin resistance. Genome Res. 2019;29:1442-52.
29. Mone P, Agyapong ED, Morciano G, et al. Dysfunctional mitochondria elicit bioenergetic decline in the aged heart. J Cardiovasc Aging. 2024;4:13.
30. Murphy E, Ardehali H, Balaban RS, et al. Mitochondrial function, biology, and role in disease: a scientific statement from the american heart association. Circ Res. 2016;118:1960-91.
31. Nguyen BY, Ruiz-Velasco A, Bui T, Collins L, Wang X, Liu W. Mitochondrial function in the heart: the insight into mechanisms and therapeutic potentials. Br J Pharmacol. 2019;176:4302-18.
32. Martín-Fernández B, Gredilla R. Mitochondria and oxidative stress in heart aging. Age. 2016;38:225-38.
33. Muthu S, Tran Z, Thilagavathi J, et al. Aging triggers mitochondrial, endoplasmic reticulum, and metabolic stress responses in the heart. J Cardiovasc Aging. 2025;5:4.
35. Liu SZ, Marcinek DJ. Skeletal muscle bioenergetics in aging and heart failure. Heart Fail Rev. 2017;22:167-78.
36. Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial metabolism in aging heart. Circ Res. 2016;118:1593-611.
37. Tatarková Z, Kuka S, Račay P, et al. Effects of aging on activities of mitochondrial electron transport chain complexes and oxidative damage in rat heart. Physiol Res. 2011;60:281-9.
38. Tocchi A, Quarles EK, Basisty N, Gitari L, Rabinovitch PS. Mitochondrial dysfunction in cardiac aging. Biochim Biophys Acta. 2015;1847:1424-33.
40. Chen Y, Liu Y, Dorn GW 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011;109:1327-31.
41. Scheffer DDL, Garcia AA, Lee L, Mochly-Rosen D, Ferreira JCB. Mitochondrial fusion, fission, and mitophagy in cardiac diseases: challenges and therapeutic opportunities. Antioxid Redox Signal. 2022;36:844-63.
42. Zerihun M, Sukumaran S, Qvit N. The Drp1-mediated mitochondrial fission protein interactome as an emerging core player in mitochondrial dynamics and cardiovascular disease therapy. Int J Mol Sci. 2023;24:5785.
43. Łysek-Gładysińska M, Wieczorek A, Jóźwik A, et al. Aging-related changes in the ultrastructure of hepatocytes and cardiomyocytes of elderly mice are enhanced in ApoE-deficient animals. Cells. 2021;10:502.
44. Fernández-Ortiz M, Sayed RKA, Fernández-Martínez J, et al. Melatonin/Nrf2/NLRP3 connection in mouse heart mitochondria during aging. Antioxidants. 2020;9:1187.
45. Vue Z, Neikirk K, Vang L, et al. Three-dimensional mitochondria reconstructions of murine cardiac muscle changes in size across aging. Am J Physiol Heart Circ Physiol. 2023;325:H965-82.
46. Chaanine AH, Jeong D, Liang L, et al. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012;3:265.
47. Liu T, Chen L, Kim E, Tran D, Phinney BS, Knowlton AA. Mitochondrial proteome remodeling in ischemic heart failure. Life Sci. 2014;101:27-36.
48. Wai T, García-Prieto J, Baker MJ, et al. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science. 2015;350:aad0116.
49. Ikeda Y, Shirakabe A, Maejima Y, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264-78.
50. Song M, Franco A, Fleischer JA, Zhang L, Dorn GW 2nd. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab. 2017;26:872-83.e5.
51. Zhao L, Zou X, Feng Z, et al. Evidence for association of mitochondrial metabolism alteration with lipid accumulation in aging rats. Exp Gerontol. 2014;56:3-12.
52. Wang Y, Li Y, He C, Gou B, Song M. Mitochondrial regulation of cardiac aging. Biochim Biophys Acta Mol Basis Dis. 2019;1865:1853-64.
53. Gao B, Yu W, Lv P, Liang X, Sun S, Zhang Y. Parkin overexpression alleviates cardiac aging through facilitating K63-polyubiquitination of TBK1 to facilitate mitophagy. Biochim Biophys Acta Mol Basis Dis. 2021;1867:165997.
54. Hafner AV, Dai J, Gomes AP, et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging. 2010;2:914-23.
55. Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Changes in the mitochondrial permeability transition pore in aging and age-associated diseases. Mech Ageing Dev. 2013;134:1-9.
56. Kwong JQ, Molkentin JD. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015;21:206-14.
57. Strutynska NA, Kotsiuruba AV, Budko AY, Mys LA, Sagach VF. Mitochondrial dysfunction in the aging heart is accompanied by constitutive no-synthases uncoupling on the background of oxidative and nitrosative stress. Fiziol Zh (1994). 2016;62:3-11.
58. Phaneuf S, Leeuwenburgh C. Cytochrome c release from mitochondria in the aging heart: a possible mechanism for apoptosis with age. Am J Physiol Regul Integr Comp Physiol. 2002;282:R423-30.
59. Hofer T, Servais S, Seo AY, et al. Bioenergetics and permeability transition pore opening in heart subsarcolemmal and interfibrillar mitochondria: effects of aging and lifelong calorie restriction. Mech Ageing Dev. 2009;130:297-307.
60. Chen Q, Thompson J, Hu Y, et al. High-dose metformin treatment to inhibit complex I during early reperfusion protects the aged mouse heart via decreased mitochondrial permeability transition pore opening. J Pharmacol Exp Ther. 2024;392:100529.
61. Zhang J, Zhao Y, Gong N. Endoplasmic reticulum stress signaling modulates ischemia/reperfusion injury in the aged heart by regulating mitochondrial maintenance. Mol Med. 2024;30:107.
62. Lam A, Karekar P, Shah K, et al. Drosophila voltage-gated calcium channel α1-subunits regulate cardiac function in the aging heart. Sci Rep. 2018;8:6910.
64. Eisner DA, Caldwell JL, Kistamás K, Trafford AW. Calcium and excitation-contraction coupling in the heart. Circ Res. 2017;121:181-95.
65. Mehdizadeh M, Aguilar M, Thorin E, Ferbeyre G, Nattel S. The role of cellular senescence in cardiac disease: basic biology and clinical relevance. Nat Rev Cardiol. 2022;19:250-64.
66. Györke S, Carnes C. Dysregulated sarcoplasmic reticulum calcium release: potential pharmacological target in cardiac disease. Pharmacol Ther. 2008;119:340-54.
67. Brette F, Sallé L, Orchard CH. Differential modulation of L-type Ca2+ current by SR Ca2+ release at the T-tubules and surface membrane of rat ventricular myocytes. Circ Res. 2004;95:e1-7.
68. Guo A, Zhang C, Wei S, Chen B, Song LS. Emerging mechanisms of T-tubule remodelling in heart failure. Cardiovasc Res. 2013;98:204-15.
69. Gil-Hernández A, Silva-Palacios A. Relevance of endoplasmic reticulum and mitochondria interactions in age-associated diseases. Ageing Res Rev. 2020;64:101193.
70. Wisneski JA, Stanley WC, Neese RA, Gertz EW. Effects of acute hyperglycemia on myocardial glycolytic activity in humans. J Clin Invest. 1990;85:1648-56.
71. Koss KL, Kranias EG. Phospholamban: a prominent regulator of myocardial contractility. Circ Res. 1996;79:1059-63.
72. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21:421-38.
73. Bravo R, Parra V, Gatica D, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol. 2013;301:215-90.
74. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900-17.
75. Wang X, Xu L, Gillette TG, Jiang X, Wang ZV. The unfolded protein response in ischemic heart disease. J Mol Cell Cardiol. 2018;117:19-25.
76. Liberale L, Badimon L, Montecucco F, Lüscher TF, Libby P, Camici GG. Inflammation, aging, and cardiovascular disease: JACC review topic of the week. J Am Coll Cardiol. 2022;79:837-47.
77. Weber C, Habenicht AJR, von Hundelshausen P. Novel mechanisms and therapeutic targets in atherosclerosis: inflammation and beyond. Eur Heart J. 2023;44:2672-81.
78. Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and 'garb-aging'. Trends Endocrinol Metab. 2017;28:199-212.
79. Ferrucci L, Corsi A, Lauretani F, et al. The origins of age-related proinflammatory state. Blood. 2005;105:2294-9.
80. Youm YH, Grant RW, McCabe LR, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18:519-32.
81. Luo W, Zou X, Wang Y, et al. Critical role of the cGAS-STING pathway in doxorubicin-induced cardiotoxicity. Circ Res. 2023;132:e223-42.
82. Seals DR, Jablonski KL, Donato AJ. Aging and vascular endothelial function in humans. Clin Sci. 2011;120:357-75.
83. Gilsbach R, Schwaderer M, Preissl S, et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat Commun. 2018;9:391.
84. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381-95.
85. Lans H, Marteijn JA, Vermeulen W. ATP-dependent chromatin remodeling in the DNA-damage response. Epigenetics Chromatin. 2012;5:4.
87. Liu B, Yip RKh, Zhou Z. Chromatin remodeling, DNA damage repair and aging. Curr Genomics. 2012;13:533-47.
88. Muñoz-Lorente MA, Cano-Martin AC, Blasco MA. Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat Commun. 2019;10:4723.
89. Paneni F, Diaz Cañestro C, Libby P, Lüscher TF, Camici GG. The aging cardiovascular system: understanding it at the cellular and clinical levels. J Am Coll Cardiol. 2017;69:1952-67.
90. McKinsey TA, Vondriska TM, Wang Y. Epigenomic regulation of heart failure: integrating histone marks, long noncoding RNAs, and chromatin architecture. F1000Res. 2018;7:1713.
91. Han P, Hang CT, Yang J, Chang CP. Chromatin remodeling in cardiovascular development and physiology. Circ Res. 2011;108:378-96.
92. Mahmoud SA, Poizat C. Epigenetics and chromatin remodeling in adult cardiomyopathy. J Pathol. 2013;231:147-57.
93. Mathiyalagan P, Keating ST, Du XJ, El-Osta A. Chromatin modifications remodel cardiac gene expression. Cardiovasc Res. 2014;103:7-16.
94. Alcendor RR, Gao S, Zhai P, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512-21.
95. Matsushima S, Sadoshima J. The role of sirtuins in cardiac disease. Am J Physiol Heart Circ Physiol. 2015;309:H1375-89.
96. Ministrini S, Puspitasari YM, Beer G, Liberale L, Montecucco F, Camici GG. Sirtuin 1 in endothelial dysfunction and cardiovascular aging. Front Physiol. 2021;12:733696.
97. Rodriguez F, Seta F. Editorial: the role of sirtuin-1 in cardiovascular and renal pathophysiology. Front Physiol. 2021;12:770386.
98. Sciarretta S, Maejima Y, Zablocki D, Sadoshima J. The role of autophagy in the heart. Annu Rev Physiol. 2018;80:1-26.
99. Liu Y, Shoji-Kawata S, Sumpter RM Jr, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci USA. 2013;110:20364-71.
100. Li F, Lang F, Zhang H, Xu L, Wang Y, Hao E. Role of TFEB mediated autophagy, oxidative stress, inflammation, and cell death in endotoxin induced myocardial toxicity of young and aged mice. Oxid Med Cell Longev. 2016;2016:5380319.
101. Okura Y, Brink M, Itabe H, Scheidegger KJ, Kalangos A, Delafontaine P. Oxidized low-density lipoprotein is associated with apoptosis of vascular smooth muscle cells in human atherosclerotic plaques. Circulation. 2000;102:2680-6.
102. Zhang AY, Yi F, Zhang G, Gulbins E, Li PL. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension. 2006;47:74-80.
103. Tanaka Y, Guhde G, Suter A, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406:902-6.
104. Wang J, Cheng X, Xiang MX, et al. IgE stimulates human and mouse arterial cell apoptosis and cytokine expression and promotes atherogenesis in Apoe-/- mice. J Clin Invest. 2011;121:3564-77.
105. Terman A, Kurz T, Gustafsson B, Brunk UT. The involvement of lysosomes in myocardial aging and disease. Curr Cardiol Rev. 2008;4:107-15.
106. Bhat OM, Li PL. Lysosome function in cardiovascular diseases. Cell Physiol Biochem. 2021;55:277-300.
107. Wilfling F, Haas JT, Walther TC, Farese RV Jr. Lipid droplet biogenesis. Curr Opin Cell Biol. 2014;29:39-45.
109. Bresgen N, Kovacs M, Lahnsteiner A, Felder TK, Rinnerthaler M. The Janus-faced role of lipid droplets in aging: insights from the cellular perspective. Biomolecules. 2023;13:912.
110. Perrotta I. Interaction between lipid droplets and endoplasmic reticulum in human atherosclerotic plaques. Ultrastruct Pathol. 2017;41:1-9.
111. Moldavski O, Amen T, Levin-Zaidman S, et al. Lipid droplets are essential for efficient clearance of cytosolic inclusion bodies. Dev Cell. 2015;33:603-10.
112. Hammoudeh N, Soukkarieh C, Murphy DJ, Hanano A. Involvement of hepatic lipid droplets and their associated proteins in the detoxification of aflatoxin B1 in aflatoxin-resistance BALB/C mouse. Toxicol Rep. 2020;7:795-804.
113. Bosch M, Sánchez-Álvarez M, Fajardo A, et al. Mammalian lipid droplets are innate immune hubs integrating cell metabolism and host defense. Science. 2020;370:eaay8085.
114. Choudhary V, Golani G, Joshi AS, et al. Architecture of lipid droplets in endoplasmic reticulum is determined by phospholipid intrinsic curvature. Curr Biol. 2018;28:915-26.e9.
115. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207-58.
116. Osumi T, Kuramoto K. Heart lipid droplets and lipid droplet-binding proteins: biochemistry, physiology, and pathology. Exp Cell Res. 2016;340:198-204.
117. Goldberg IJ, Reue K, Abumrad NA, et al. Deciphering the role of lipid droplets in cardiovascular disease: a report from the 2017 national heart, lung, and blood institute workshop. Circulation. 2018;138:305-15.
118. Chen Y, Yang L, Wang K, et al. Relationship between fatty acid intake and aging: a Mendelian randomization study. Aging. 2024;16:5711-39.
119. Valencak TG, Ruf T. Feeding into old age: long-term effects of dietary fatty acid supplementation on tissue composition and life span in mice. J Comp Physiol B. 2011;181:289-98.
120. Papsdorf K, Miklas JW, Hosseini A, et al. Lipid droplets and peroxisomes are co-regulated to drive lifespan extension in response to mono-unsaturated fatty acids. Nat Cell Biol. 2023;25:672-84.
121. Guo D, Zhang M, Qi B, et al. Lipid overload-induced RTN3 activation leads to cardiac dysfunction by promoting lipid droplet biogenesis. Cell Death Differ. 2024;31:292-308.
122. Jebessa ZH, Shanmukha KD, Dewenter M, et al. The lipid droplet-associated protein ABHD5 protects the heart through proteolysis of HDAC4. Nat Metab. 2019;1:1157-67.
123. Han L, Huang D, Wu S, et al. Lipid droplet-associated lncRNA LIPTER preserves cardiac lipid metabolism. Nat Cell Biol. 2023;25:1033-46.
124. Kuramoto K, Sakai F, Yoshinori N, et al. Deficiency of a lipid droplet protein, perilipin 5, suppresses myocardial lipid accumulation, thereby preventing type 1 diabetes-induced heart malfunction. Mol Cell Biol. 2014;34:2721-31.
125. Poston CN, Krishnan SC, Bazemore-Walker CR. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J Proteomics. 2013;79:219-30.
126. Li J, Zhang D, Brundel BJJM, Wiersma M. Imbalance of ER and mitochondria interactions: prelude to cardiac ageing and disease? Cells. 2019;8:1617.
127. Lopez-Crisosto C, Pennanen C, Vasquez-Trincado C, et al. Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat Rev Cardiol. 2017;14:342-60.
128. Carpio MA, Means RE, Brill AL, Sainz A, Ehrlich BE, Katz SG. BOK controls apoptosis by Ca2+ transfer through ER-mitochondrial contact sites. Cell Rep. 2021;34:108827.
129. Rasola A, Bernardi P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium. 2011;50:222-33.
130. Hu Y, Chen H, Zhang L, et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy. 2021;17:1142-56.
131. Shoshan-Barmatz V, De S, Meir A. The mitochondrial voltage-dependent anion channel 1, Ca2+ transport, apoptosis, and their regulation. Front Oncol. 2017;7:60.
132. Loncke J, de Ridder I, Kale J, et al. CISD2 counteracts the inhibition of ER-mitochondrial calcium transfer by anti-apoptotic BCL-2. Biochim Biophys Acta Mol Cell Res. 2025;1872:119857.
133. Wang CH, Chen YF, Wu CY, et al. Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2+ homeostasis. Hum Mol Genet. 2014;23:4770-85.
134. Yeh CH, Chou YJ, Kao CH, Tsai TF. Mitochondria and calcium homeostasis: Cisd2 as a big player in cardiac ageing. Int J Mol Sci. 2020;21:9238.
135. Chen YF, Kao CH, Chen YT, et al. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 2009;23:1183-94.
136. Janikiewicz J, Szymański J, Malinska D, et al. Mitochondria-associated membranes in aging and senescence: structure, function, and dynamics. Cell Death Dis. 2018;9:332.
137. Wang C, Dai X, Wu S, et al. FUNDC1-dependent mitochondria-associated endoplasmic reticulum membranes are involved in angiogenesis and neoangiogenesis. Nat Commun. 2024;15:4572.
138. Wu S, Lu Q, Wang Q, et al. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation. 2017;136:2248-66.
139. Lagendijk AK, Szabó A, Merks RM, Bakkers J. Hyaluronan: a critical regulator of endothelial-to-mesenchymal transition during cardiac valve formation. Trends Cardiovasc Med. 2013;23:135-42.
140. Morgado-Cáceres P, Liabeuf G, Calle X, et al. The aging of ER-mitochondria communication: a journey from undifferentiated to aged cells. Front Cell Dev Biol. 2022;10:946678.
141. Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82:503-68.
142. Li D, Li Y, Ding H, Wang Y, Xie Y, Zhang X. Cellular senescence in cardiovascular diseases: from pathogenesis to therapeutic challenges. J Cardiovasc Dev Dis. 2023;10:439.
143. Setterberg IE, Le C, Frisk M, Perdreau-Dahl H, Li J, Louch WE. The physiology and pathophysiology of T-tubules in the heart. Front Physiol. 2021;12:790227.
144. Kostin S, Scholz D, Shimada T, et al. The internal and external protein scaffold of the T-tubular system in cardiomyocytes. Cell Tissue Res. 1998;294:449-60.
145. Louch WE, Mørk HK, Sexton J, et al. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol. 2006;574:519-33.
146. Retzius G. Zur Kenntnis der quergestreiften Muskelfaser. Biol Untersuch. 1881:1-11. Available from: https://www.zobodat.at/pdf/Biolog-Untersuchungen_1_0001-0026.pdf [Last accessed on 10 Oct 2025].
147. Heinzel FR, Bito V, Biesmans L, et al. Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res. 2008;102:338-46.
148. Louch WE, Bito V, Heinzel FR, et al. Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res. 2004;62:63-73.
149. Zhang HB, Li RC, Xu M, et al. Ultrastructural uncoupling between T-tubules and sarcoplasmic reticulum in human heart failure. Cardiovasc Res. 2013;98:269-76.
150. Zorn-Pauly K, Schaffer P, Pelzmann B, et al. Oxidized LDL induces ventricular myocyte damage and abnormal electrical activity-role of lipid hydroperoxides. Cardiovasc Res. 2005;66:74-83.
151. Bertic M, Worme M, Foroutan F, et al. Predictors of survival and favorable neurologic outcome in patients treated with eCPR: a systematic review and meta-analysis. J Cardiovasc Transl Res. 2022;15:279-90.
152. Kho C, Lee A, Hajjar RJ. Altered sarcoplasmic reticulum calcium cycling-targets for heart failure therapy. Nat Rev Cardiol. 2012;9:717-33.
153. Rog-Zielinska EA, Scardigli M, Peyronnet R, et al. Beat-by-beat cardiomyocyte T-tubule deformation drives tubular content exchange. Circ Res. 2021;128:203-15.
154. Anderson R, Prolla T. PGC-1alpha in aging and anti-aging interventions. Biochim Biophys Acta. 2009;1790:1059-66.
155. Gao F, Liang T, Lu YW, et al. Reduced mitochondrial protein translation promotes cardiomyocyte proliferation and heart regeneration. Circulation. 2023;148:1887-906.
156. Gao F, Liang T, Lu YW, et al. A defect in mitochondrial protein translation influences mitonuclear communication in the heart. Nat Commun. 2023;14:1595.
157. Anderson R, Lagnado A, Maggiorani D, et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019;38:e100492.
158. Lai L, Leone TC, Zechner C, et al. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22:1948-61.
159. Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359-65.
160. Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110:1109-24.
161. Gomes AP, Price NL, Ling AJ, et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624-38.
162. Zhu D, Li X, Tian Y. Mitochondrial-to-nuclear communication in aging: an epigenetic perspective. Trends Biochem Sci. 2022;47:645-59.
163. Yu W, Sun S, Xu H, Li C, Ren J, Zhang Y. TBC1D15/RAB7-regulated mitochondria-lysosome interaction confers cardioprotection against acute myocardial infarction-induced cardiac injury. Theranostics. 2020;10:11244-63.
164. Sun S, Yu W, Xu H, et al. TBC1D15-Drp1 interaction-mediated mitochondrial homeostasis confers cardioprotection against myocardial ischemia/reperfusion injury. Metabolism. 2022;134:155239.
165. Liao PC, Yang EJ, Borgman T, et al. Touch and Go: membrane contact sites between lipid droplets and other organelles. Front Cell Dev Biol. 2022;10:852021.
166. Kim J, Bai H. Peroxisomal stress response and inter-organelle communication in cellular homeostasis and aging. Antioxidants. 2022;11:192.
167. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186:243-78.
168. Abdellatif M, Rainer PP, Sedej S, Kroemer G. Hallmarks of cardiovascular ageing. Nat Rev Cardiol. 2023;20:754-77.
169. Chen F, Jimenez RJ, Sharma K, et al. Broad distribution of hepatocyte proliferation in liver homeostasis and regeneration. Cell Stem Cell. 2020;26:27-33.e4.
170. Burté F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. 2015;11:11-24.
171. Plotegher N, Duchen MR. Mitochondrial dysfunction and neurodegeneration in lysosomal storage disorders. Trends Mol Med. 2017;23:116-34.
172. Wong YC, Kim S, Peng W, Krainc D. Regulation and function of mitochondria-lysosome membrane contact sites in cellular homeostasis. Trends Cell Biol. 2019;29:500-13.
173. Deus CM, Yambire KF, Oliveira PJ, Raimundo N. Mitochondria-lysosome crosstalk: from physiology to neurodegeneration. Trends Mol Med. 2020;26:71-88.
174. Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. 2020;21:101-18.
175. Cartes-Saavedra B, Ghosh A, Hajnóczky G. The roles of mitochondria in global and local intracellular calcium signalling. Nat Rev Mol Cell Biol. 2025;26:456-75.
176. Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014;33:2142-56.
177. Hamacher-Brady A, Choe SC, Krijnse-Locker J, Brady NR. Intramitochondrial recruitment of endolysosomes mediates Smac degradation and constitutes a novel intrinsic apoptosis antagonizing function of XIAP E3 ligase. Cell Death Differ. 2014;21:1862-76.
178. Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci USA. 2014;111:E4439-48.
179. Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309-14.
180. McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014;33:282-95.
181. Wong YC, Ysselstein D, Krainc D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature. 2018;554:382-6.
182. Zhang XM, Walsh B, Mitchell CA, Rowe T. TBC domain family, member 15 is a novel mammalian Rab GTPase-activating protein with substrate preference for Rab7. Biochem Biophys Res Commun. 2005;335:154-61.
183. Peralta ER, Martin BC, Edinger AL. Differential effects of TBC1D15 and mammalian Vps39 on Rab7 activation state, lysosomal morphology, and growth factor dependence. J Biol Chem. 2010;285:16814-21.
184. Onoue K, Jofuku A, Ban-Ishihara R, et al. Fis1 acts as a mitochondrial recruitment factor for TBC1D15 that is involved in regulation of mitochondrial morphology. J Cell Sci. 2013;126:176-85.
185. Meggouh F, Bienfait HM, Weterman MA, de Visser M, Baas F. Charcot-marie-tooth disease due to a de novo mutation of the RAB7 gene. Neurology. 2006;67:1476-8.
186. Itoh K, Adachi Y, Yamada T, et al. A brain-enriched Drp1 isoform associates with lysosomes, late endosomes, and the plasma membrane. J Biol Chem. 2018;293:11809-22.
187. Rossor AM, Tomaselli PJ, Reilly MM. Recent advances in the genetic neuropathies. Curr Opin Neurol. 2016;29:537-48.
188. Burbulla LF, Song P, Mazzulli JR, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science. 2017;357:1255-61.
189. Plotegher N, Duchen MR. Crosstalk between lysosomes and mitochondria in parkinson's disease. Front Cell Dev Biol. 2017;5:110.
190. Cisneros J, Belton TB, Shum GC, Molakal CG, Wong YC. Mitochondria-lysosome contact site dynamics and misregulation in neurodegenerative diseases. Trends Neurosci. 2022;45:312-22.
191. Verhoeven K, De Jonghe P, Coen K, et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet. 2003;72:722-7.
192. Cleeter MW, Chau KY, Gluck C, et al. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem Int. 2013;62:1-7.
193. Osellame LD, Rahim AA, Hargreaves IP, et al. Mitochondria and quality control defects in a mouse model of Gaucher disease-links to Parkinson's disease. Cell Metab. 2013;17:941-53.
194. Li H, Ham A, Ma TC, et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy. 2019;15:113-30.
195. Yang J, Guo Q, Feng X, Liu Y, Zhou Y. Mitochondrial dysfunction in cardiovascular diseases: potential targets for treatment. Front Cell Dev Biol. 2022;10:841523.
196. Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. 2014;20:1427-35.
197. Madreiter-Sokolowski CT, Waldeck-Weiermair M, Bourguignon MP, et al. Enhanced inter-compartmental Ca2+ flux modulates mitochondrial metabolism and apoptotic threshold during aging. Redox Biol. 2019;20:458-66.
198. Ziegler DV, Martin N, Bernard D. Cellular senescence links mitochondria-ER contacts and aging. Commun Biol. 2021;4:1323.
199. Ziegler DV, Vindrieux D, Goehrig D, et al. Calcium channel ITPR2 and mitochondria-ER contacts promote cellular senescence and aging. Nat Commun. 2021;12:720.
200. Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22:75-95.
201. Chou SM, Yen YH, Yuan F, Zhang SC, Chong CM. Neuronal senescence in the aged brain. Aging Dis. 2023;14:1618-32.
202. Englund DA, Zhang X, Aversa Z, LeBrasseur NK. Skeletal muscle aging, cellular senescence, and senotherapeutics: current knowledge and future directions. Mech Ageing Dev. 2021;200:111595.
203. Sun M, Jiang W, Mu N, Zhang Z, Yu L, Ma H. Mitochondrial transplantation as a novel therapeutic strategy for cardiovascular diseases. J Transl Med. 2023;21:347.
204. Yamada Y, Maruyama M, Kita T, Usami SI, Kitajiri SI, Harashima H. The use of a MITO-Porter to deliver exogenous therapeutic RNA to a mitochondrial disease's cell with a A1555G mutation in the mitochondrial 12S rRNA gene results in an increase in mitochondrial respiratory activity. Mitochondrion. 2020;55:134-44.
205. Renteln M. A synthetic mitochondrial-based vector for therapeutic purposes. Med Hypotheses. 2018;117:28-30.
206. Magalhães Rebelo AP, Dal Bello F, Knedlik T, et al. Chemical modulation of mitochondria-endoplasmic reticulum contact sites. Cells. 2020;9:1637.
207. Dentoni G, Castro-Aldrete L, Naia L, Ankarcrona M. The potential of small molecules to modulate the mitochondria-endoplasmic reticulum interplay in Alzheimer's disease. Front Cell Dev Biol. 2022;10:920228.
208. Dietel E, Brobeil A, Delventhal L, Tag C, Gattenlöhner S, Wimmer M. Crosstalks of the PTPIP51 interactome revealed in Her2 amplified breast cancer cells by the novel small molecule LDC3/Dynarrestin. PLoS One. 2019;14:e0216642.
209. Franco A, Kitsis RN, Fleischer JA, et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature. 2016;540:74-9.
210. Naia L, Ly P, Mota SI, et al. The Sigma-1 receptor mediates pridopidine rescue of mitochondrial function in huntington disease models. Neurotherapeutics. 2021;18:1017-38.
211. Salvatore T, Galiero R, Caturano A, et al. Effects of metformin in heart failure: from pathophysiological rationale to clinical evidence. Biomolecules. 2021;11:1834.
212. Gao J, Chen G, He H, et al. Therapeutic effects of breviscapine in cardiovascular diseases: a review. Front Pharmacol. 2017;8:289.
213. Zhang QY, Pan Y, Wang R, et al. Quercetin inhibits AMPK/TXNIP activation and reduces inflammatory lesions to improve insulin signaling defect in the hypothalamus of high fructose-fed rats. J Nutr Biochem. 2014;25:420-8.
214. Wang AL, Iadecola C, Wang G. New generations of dihydropyridines for treatment of hypertension. J Geriatr Cardiol. 2017;14:67-72.
215. Kobayashi S, Yano M, Suetomi T, et al. Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing interdomain interactions within the ryanodine receptor. J Am Coll Cardiol. 2009;53:1993-2005.
216. Wüst RCI, Coolen BF, Held NM, et al. The antibiotic doxycycline impairs cardiac mitochondrial and contractile function. Int J Mol Sci. 2021;22:4100.
217. Houtkooper RH, Mouchiroud L, Ryu D, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451-7.
218. Chiao YA, Kolwicz SC, Basisty N, et al. Rapamycin transiently induces mitochondrial remodeling to reprogram energy metabolism in old hearts. Aging. 2016;8:314-27.
219. Lee DJW, Hodzic Kuerec A, Maier AB. Targeting ageing with rapamycin and its derivatives in humans: a systematic review. Lancet Healthy Longev. 2024;5:e152-62.
220. Andreux PA, Blanco-Bose W, Ryu D, et al. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab. 2019;1:595-603.
221. Liu S, Faitg J, Tissot C, et al. Urolithin A provides cardioprotection and mitochondrial quality enhancement preclinically and improves human cardiovascular health biomarkers. iScience. 2025;28:111814.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].