

Layered double hydroxide-based single-atom catalysts: precise catalyst design on a two-dimensional ‘atomic canvas’
 0
0 Abstract
Layered double hydroxides (LDHs), with their atomically tunable layer composition, abundant surface coordination sites, and modifiable two-dimensional confined interlayer space, offer an ideal ‘atomic canvas’ for the precise design and in situ modulation of single-atom catalysts. The review elaborates on this core concept, systematically presenting synthesis strategies for the precise construction of single-atom sites on LDHs, including in situ growth, post-synthetic anchoring, and topological transformation pathways. Furthermore, the article focuses on leveraging the dynamic properties and synergistic effects of the LDH supports. Multi-dimensional strategies, such as electronic modulation, coordination engineering, interfacial synergy, and reaction pathway reconstruction, are discussed for the rational enhancement of catalytic performance and functional expansion. Despite rapid progress, challenges remain, including the stability of single atoms under high loading, elucidation of dynamic catalytic mechanisms, and scalable fabrication. Future research should integrate advanced characterization and theoretical calculations to clarify the dynamic interactions between the ‘canvas’ and single atoms, and promote the practical application of these high-performance catalysts in energy conversion and green synthesis.
Keywords
INTRODUCTION
Heterogeneous catalysis serves as a foundational technology for modern society, underpinning processes critical to efficient energy conversion, sustainable chemical synthesis and environmental remediation. The efficacy of the catalytic system is fundamentally governed by three key properties of the catalyst: the intrinsic activity, selectivity, and stability[1]. Despite their widespread use, traditional nanocatalysts are often plagued by intrinsic limitations. A substantial fraction of their expensive metal atoms is confined within the particle bulk, resulting in inherently low atom utilization efficiency. Furthermore, the non-uniform and ill-defined nature of their surface-active sites not only promotes competing side reactions but also obscures clear structure-activity relationships, hindering rational design of catalyst[2-5]. These pervasive challenges collectively constrain the development of next-generation catalytic technologies with superior efficiency and precision. Consequently, breaking through these limitations mandates a transformative leap in catalyst architectures - one that enables atomic-level precision in the design and control of active sites.
The imperative for atomic-level precision finds a compelling answer in single-atom catalysts (SACs), where isolated metal atoms are anchored onto solid supports, achieving near-theoretical 100% atomic utilization and structurally uniform active centers[6-8]. The architecture offers unparalleled opportunities to tune catalytic performance through precise coordination environment engineering, presenting a revolutionary pathway to overcome traditional bottlenecks[9-11]. However, the inherent high surface energy of isolated atoms makes them thermodynamically prone to migration and aggregation, posing significant challenges to the stability and practical loading capacities of SACs[12-14]. The selection of an optimal support is therefore critical, as it must provide robust anchoring sites forces to immobilize single atoms (SAs) while potentially synergizing in the catalytic process[15]. Current widely studied supports, such as carbon materials, metal oxides, metal-organic frameworks (MOFs) and porous organic polymers, each present a trade-off[16-24]. Carbon materials offer excellent electro-conductivity but often possess heterogeneous and complex coordination environments that are difficult to precisely tailor[25-27]. Metal oxides can provide strong well-defined coordination sites and high thermal/chemical stability, yet their functionality may be limited or lack synergistic effects[28-31]. While these conventional supports have significantly advanced SAC research, they frequently represent a compromise. It remains a formidable challenge to find a support that simultaneously fulfills three critical functions: robust stabilization of SAs, precise customization of their coordination microenvironment, and active participation in the targeted catalytic cycle. This triad of properties is essential to unlock the full, tailored potential of next-generation SACs across diverse catalytic applications.
Against this backdrop, layered double hydroxides (LDHs) have emerged as a unique class of inorganic support materials. Their distinctive structure confers a remarkable combination of properties essential for catalysis[32-34]. It features positively charged host layers built from divalent and trivalent metal cations, with charge-balancing, exchangeable anions and water molecules residing in the interlayer space. The architecture affords exceptional flexibility in tuning the layer metal composition, offers a landscape of surface hydroxyl groups, and creates a confined interlayer nano-environment[35-38]. Collectively, these characteristics render LDHs highly adaptable materials that enable the precise construction and regulation of catalytic sites at the atomic level. The exceptional tunability of LDHs arises from multi-dimensional structural control: the type, ratio, and spatial distribution of metal cations within the layers directly govern the electronic structure and available anchoring sites; the exchangeable interlayer anions allow for dynamic modulation of the local chemical environment; and the intrinsic catalytic activity of many LDHs enables them to engage in synergistic effects with anchored SAs, transcending the role of a ‘passive scaffold’.
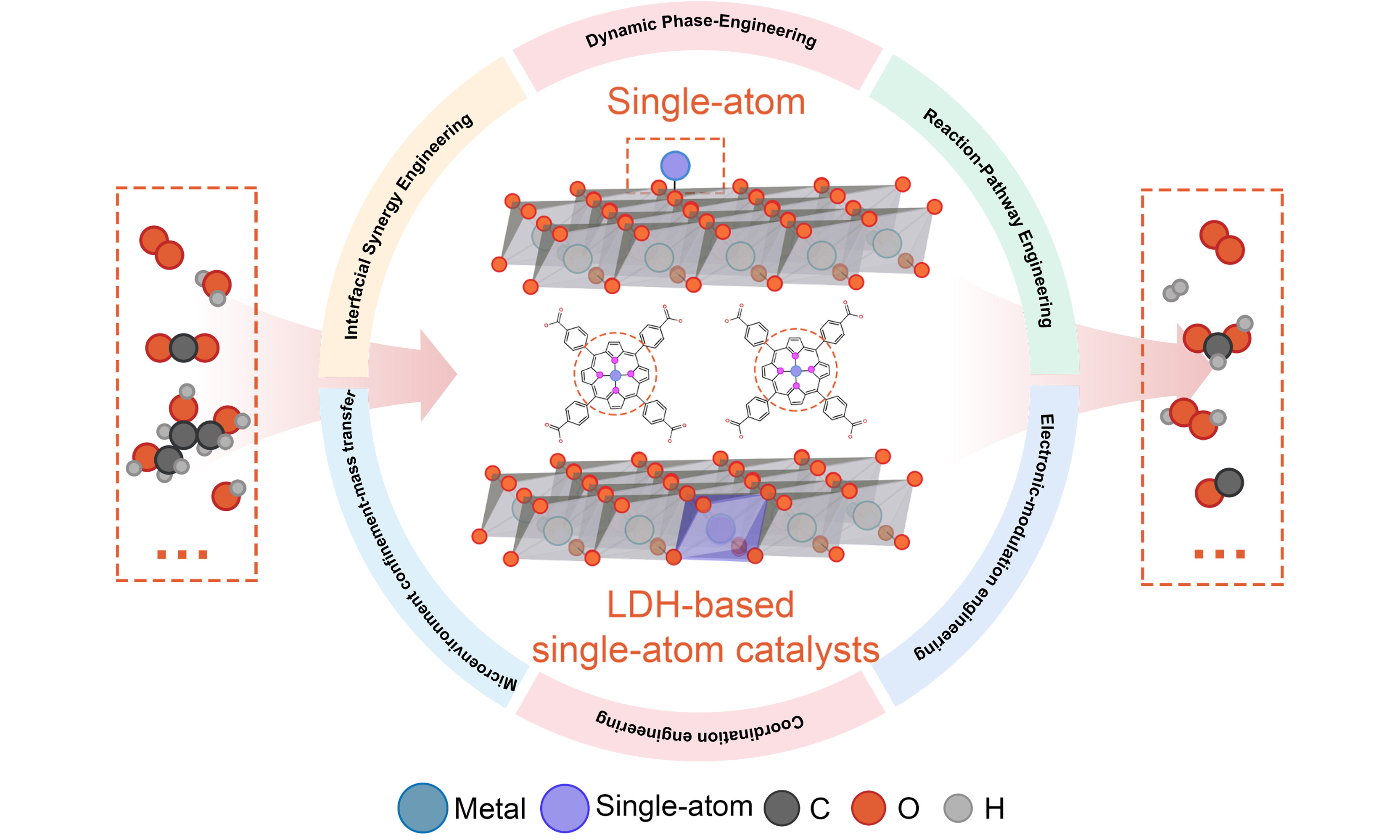
Building upon other excellent reviews that have detailed the synthesis and applications of LDH-supported SACs, the article aims to offer a focused perspective that advances the conceptual understanding of the LDH support itself. We specifically highlight that LDHs function not merely as static anchoring matrices, but as dynamic, active ‘canvases’ capable of participating in and modulating catalytic reactions [Figure 1]. The review will systematically describe how the structural and electronic properties of LDHs (e.g., their tunable composition, confined interlayer space, and surface chemistry) are exploited in synthesis to create precise single-atom sites. A central theme will be to elucidate the mechanisms through which the LDH support interacts with the anchored SACs, thereby generating synergistic effects, modulating intermediate adsorption, and in some cases, giving rise to novel reaction pathways. Finally, we will discuss the persistent challenges and future opportunities, in rationally designing and optimizing this dynamic host-guest catalytic system for enhanced performance across various energy and chemical conversion processes.
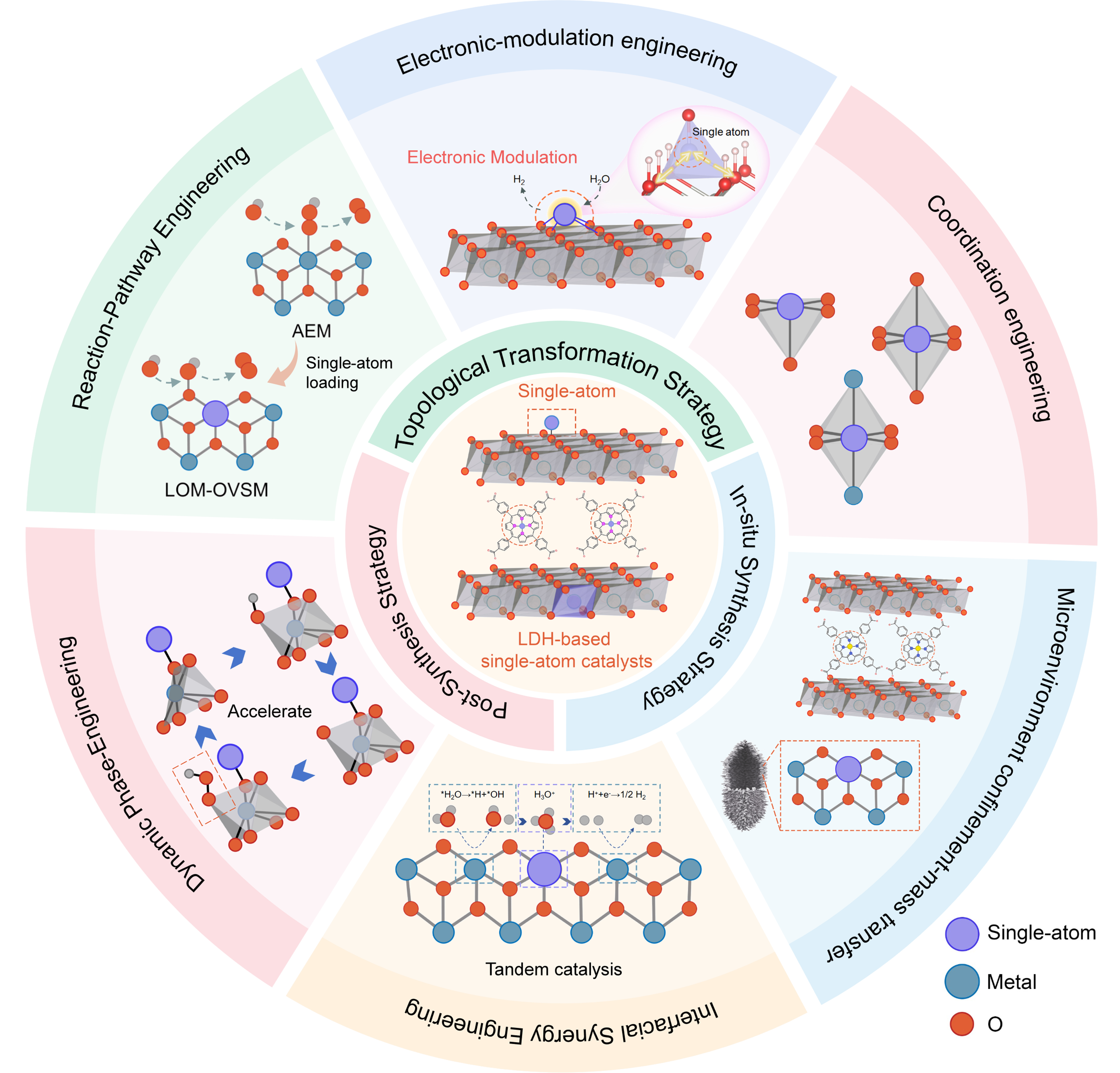
Figure 1. Schematic illustration of LDHs as a dynamic ‘atomic canvas’ for SACs. LDH: Layered double hydroxide; SAC: single-atom catalyst.
SYNTHESIS OF LDH-BASED SACS: PRECISE ‘PAINTING’ ON THE ‘ATOMIC CANVAS’
The development of effective synthesis strategies for immobilizing SACs on LDH substrates is crucial for taking full advantage of their potential in catalysis. In this section, we systematically summarize the commonly reported synthetic approaches for fabricating LDH-based SACs, which are broadly classified into two main categories: ‘in situ synthesis’ and ‘post-synthesis’ methods. Additionally, we introduce the topological transformation strategy as a distinct pathway. Each approach is described as a specialized technique for the precise immobilization of SACs onto LDHs, allowing the rational design of active sites at the atomic scale.
In situ synthesis strategy
The in situ synthesis strategy can be likened to ‘integrating pigments directly into the wet canvas during the creation’. In this approach, the single-atom metal species are incorporated concurrently with the precursor for the formation of the LDH support, enabling their direct embedding into the host lattice or anchoring onto the growing crystal surfaces.
Among various in situ routes, co-precipitation stands out for its simplicity and effectiveness. The method involves the simultaneous precipitation of metal salts, including those forming the LDH layers and the single-atom precursor, in an alkaline environment. During LDH crystallization, the target metal atoms are directly embedded within the lattice or firmly anchored on the surface. A representative example is the work by Dong et al., who incorporated single Ru atoms into a CoCr-LDH matrix via one-step co-precipitation using Na2CO3 and NaOH for pH control [Figure 2A][39]. Through comprehensive characterizations, including X-ray Diffraction (XRD), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), X-ray absorption fine structure (XAFS) and X-ray Photoelectron Spectroscopy (XPS), they confirmed the atomic dispersion and successful integration of Ru species into the LDH framework.
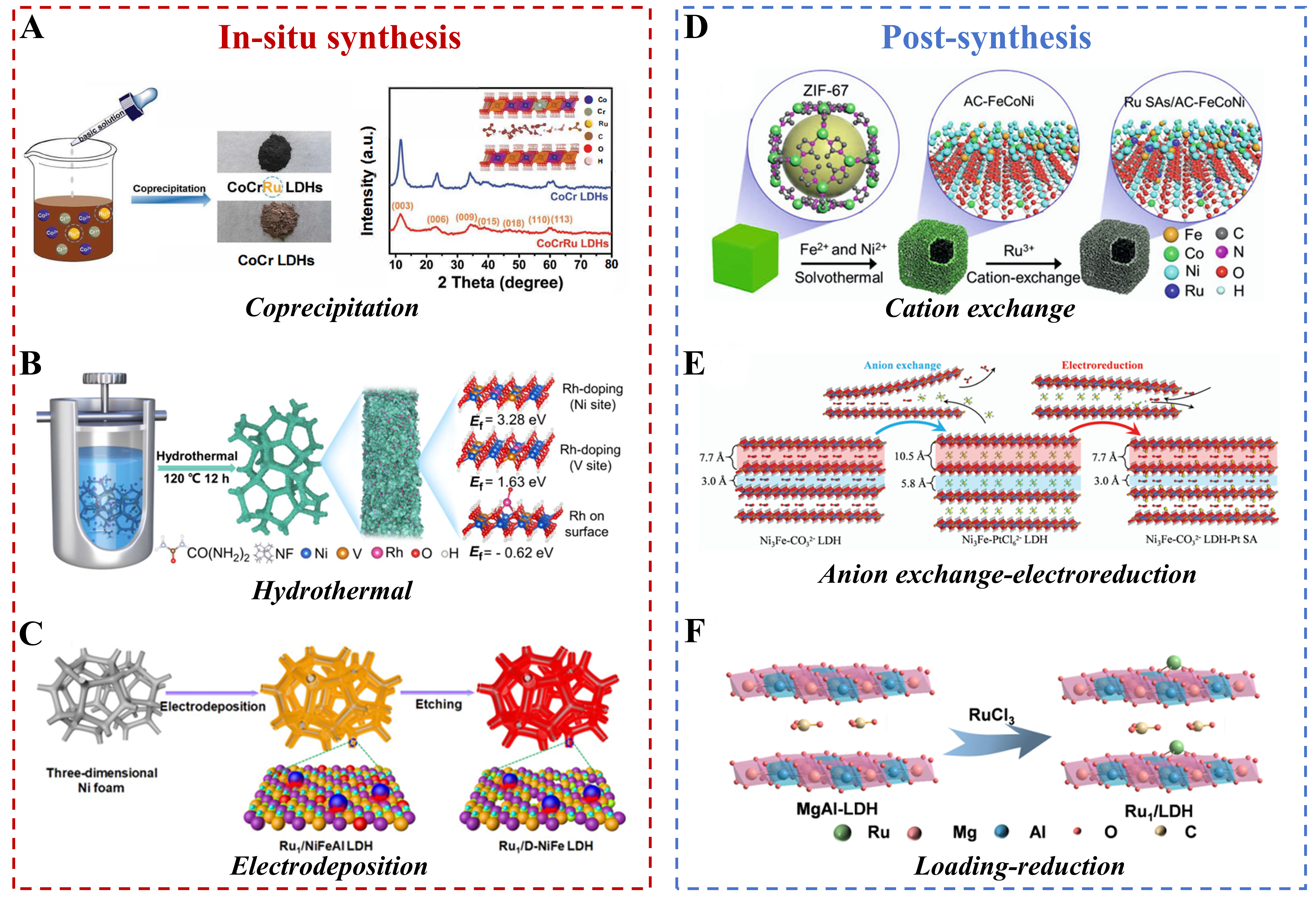
Figure 2. Techniques for ‘painting’ single atoms on the LDH canvas. The synthesis strategies illustrated are (A) coprecipitation. Reprinted with permission from Ref.[39]. Copyright 2020, Wiley-VCH; (B) hydrothermal. Reprinted with permission from Ref.[40]. Copyright 2020, Wiley-VCH; (C) electrodeposition. Reprinted with permission from Ref.[42]. Copyright 2021, Springer Nature; (D) cation exchange. Reprinted with permission from Ref.[45]. Copyright 2020, Wiley-VCH; (E) anion Exchange-electroreduction. Reprinted with permission from Ref.[48]. Copyright 2021, The Royal Society of Chemistry (F) loading-reduction. Reprinted with permission from Ref.[56]. Copyright 2023, Wiley-VCH. LDH: Layered double hydroxide.
Hydrothermal synthesis offers another effective in situ pathway, facilitating the crystallization of LDHs and the incorporation of SAs under elevated temperature and pressure in an aqueous environment. For instance, Sun et al. synthesized Rh SACs by hydrothermally treating a mixture of Rh, Ni, and V precursors on a nickel foam substrate at 120 °C [Figure 2B][40]. The obtained Rh1/NiV-LDH nanosheets were shown by XRD, Energy-Dispersive X-ray Spectroscopy (EDS), and XAFS analyses to contain uniformly dispersed Rh SAs within the LDH matrix. In a related study, Hu et al. first prepared Ru-NiFeAl-LDHs via a glycol-assisted hydrothermal process, followed by etching of Al3+ using 5 M NaOH[41]. This process generates a large number of structural defects within the NiFe-LDH host structure, these defects subsequently serve as anchoring sites for manganese atoms, thereby stabilising the isolated ruthenium atoms and ultimately leading to the preparation of the catalyst (Ru SAs/D-NiFe LDH).
Beyond solution-based methods, electrochemical deposition provides an alternative in situ route for anchoring SAs onto LDH supports. Here, metal ions from the electrolyte are electrochemically reduced and directly deposited as isolated atoms onto the LDH surface under controlled potential or current. For example, Zhai et al. employed a potentiostatic electrodeposition strategy followed by alkaline post-treatment to fabricate Ru SAs/Defect-NiFe LDH [Figure 2C][42]. XPS and XAFS analyses demonstrated strong electronic coupling between the anchored Ru SAs and the defective NiFe-LDH support, confirming the effective stabilization of Ru at the defect sites.
Post-synthesis strategy
The post-synthesis strategy can be conceptually regarded as a process of ‘secondary creation on a pre-fabricated LDH canvas’. In this approach, the LDH support is first prepared as a structural matrix, after which single-atom metal species are introduced onto or into the host material through various anchoring pathways. Compared to in situ synthesis, post-synthetic routes offer greater flexibility in selecting metal precursors and enable more precise control over single-atom loading and coordination environments. Among these, ion-exchange methods are widely employed, which typically involve replacing original metal cations or interlayer anions in pre-formed LDHs with target metal species via solution-phase or electrochemical processes[43,44].
Cation exchange is one effective pathway. For example, Hu et al. first transfomed zeolite-imidazole framework (ZIF)-67 into a hollow-structured material (AC-FeCoNi-LDH) through a simple ion-exchange process in an organic solvent[45]. Ru3+ ions were then introduced to exchange with Fe, Co, and Ni sites, leading to the anchoring of isolated Ru atoms and yielding Ru SAs/AC-FeCoNi [Figure 2D]. In another study, Israr et al. used MIL-88A as a sacrificial template to prepare NiFe-LDH hollow nanocages[46]. In a weak alkaline environment (0.02 M NaOH), Ru3+ ions coordinated with surface hydroxyl/oxygen groups and partially substituted Ni or Fe sites, forming stable Ru-O-M (M = Ni, Fe) bonds to produce Ru-SAC/NiFe-LDH. He et al. demonstrated that Rh3+, due to its higher charge density, could replace Ca2+ in CaAl-LDH via isostructural substitution, achieving a high Rh loading of 4 wt.% in the resulting Rh1CaAl-LDH catalyst[47].
Besides cation exchange, anion-exchange coupled with reduction also serves as an effective route. Chen et al. exchanged interlayer CO32- with PtCl62- to form a precursor (Ni3Fe-PtCl62- LDH), which was then electrochemically reduced to yield isolated Pt atoms confined in the interlayer regions, forming Ni3Fe-CO32- LDH-Pt SA [Figure 2E][48]. It is worth noting that the efficiency of anion-exchange is often constrained by the lateral size and layer stacking of LDH, necessitating careful control of reaction conditions.
Electrochemical deposition represents another important post-synthesis strategy that enables precise control over single-atom anchoring[49,50]. By applying constant potential, constant current, or cyclic voltammetry, metal ions in solution can be reduced and deposited as isolated atoms on LDH surfaces or within their porous structures. For instance, Wei et al.[51], Li et al.[52], and Liu et al.[53] constructed isolated Ir sites on Ni-LDH using both cathodic and anodic electrochemical deposition, denoted as Ir1/Ni-LDH-T and Ir1/Ni-LDH-V, respectively, demonstrating tunable metal-support interactions and coordination environments.
Another widely used approach is post-loading followed by reduction which typically involves three steps: preparing of the LDH support, loading of metal precursors, and reducing to achieve atomic-level dispersion[54,55]. For example, Wang et al. immersed MgAl-LDHs in a Ru3+ precursor solution for 5 h to allow adsorption via electrostatic and coordination interactions, followed by gas-phase reduction under H2/Ar at 100 °C for 2 h to form Ru1/LDH [Figure 2F][56]. Inductively Coupled Plasma Atomic Emission Spectrometry (ICP-AES) indicated a Ru loading of 1.05 wt.%, while HAADF-STEM, Fourier-transform extend X-ray absorption fine structure (FT-EXAFS), and EDS mapping confirmed the uniform dispersion of Ru SAs. Song et al. adsorbed Ir4+ on NiFe-LDHs and reduced it with NaBH4 to form a single-atom alloy (SAA) structure, yielding NiIr-SAA/NiFe-LDH[57]. In a distinct case, Shen et al. first etched NiFe-LDHs with H2O2 to generate oxygen vacancies, then introduced PtCl4 and conducted hydrothermal reduction[58]. The resulting Pt/D-NiFe-LDH exhibited Pt-M (Ni, Fe) bonds instead of typical Pt-O-M bonds, illustrating a ‘support-to-metal reverse charge transfer’ mechanism and representing the first stable negatively charged Pt species in an LDH system.
Topological transformation strategy
In addition to in situ and post-synthesis pathways, the topological transformation of LDH precursors offers a versatile and innovative route for constructing SACs[59,60]. The strategy leverages the structural memory and tunable composition of LDHs, directing their transformations via controlled heating, chemical reduction or intercalation-into derived matrices (e.g., layered double oxides, porous carbon or confined metal frameworks) while largely preserving the original morphology[61-65]. Throughout the conversion process, single metal atoms can be in situ anchored within the newly formed architecture.
A representative approach involves the transformation of MOFs into LDH-based SACs. For example, Tian et al. started with ZIF-67 (Zn/Co-MOF) as a template, introducing Ni2+ and PtCl62- in ethanol[66]. The hydroxyl groups (-OH) in ethanol partially reduced Pt4+, while the released Cl- ions triggered the framework’s disintegration and reassembly into a hollow Pt-NiCo LDH that retained the dodecahedral morphology of the parent MOFs. Subsequent calcination at 300 °C in air yielded Pt-NiCo layered double oxide (LDO) nanosheets, in which Pt was present as both SAs and sub-nanometric clusters [Figure 3A].
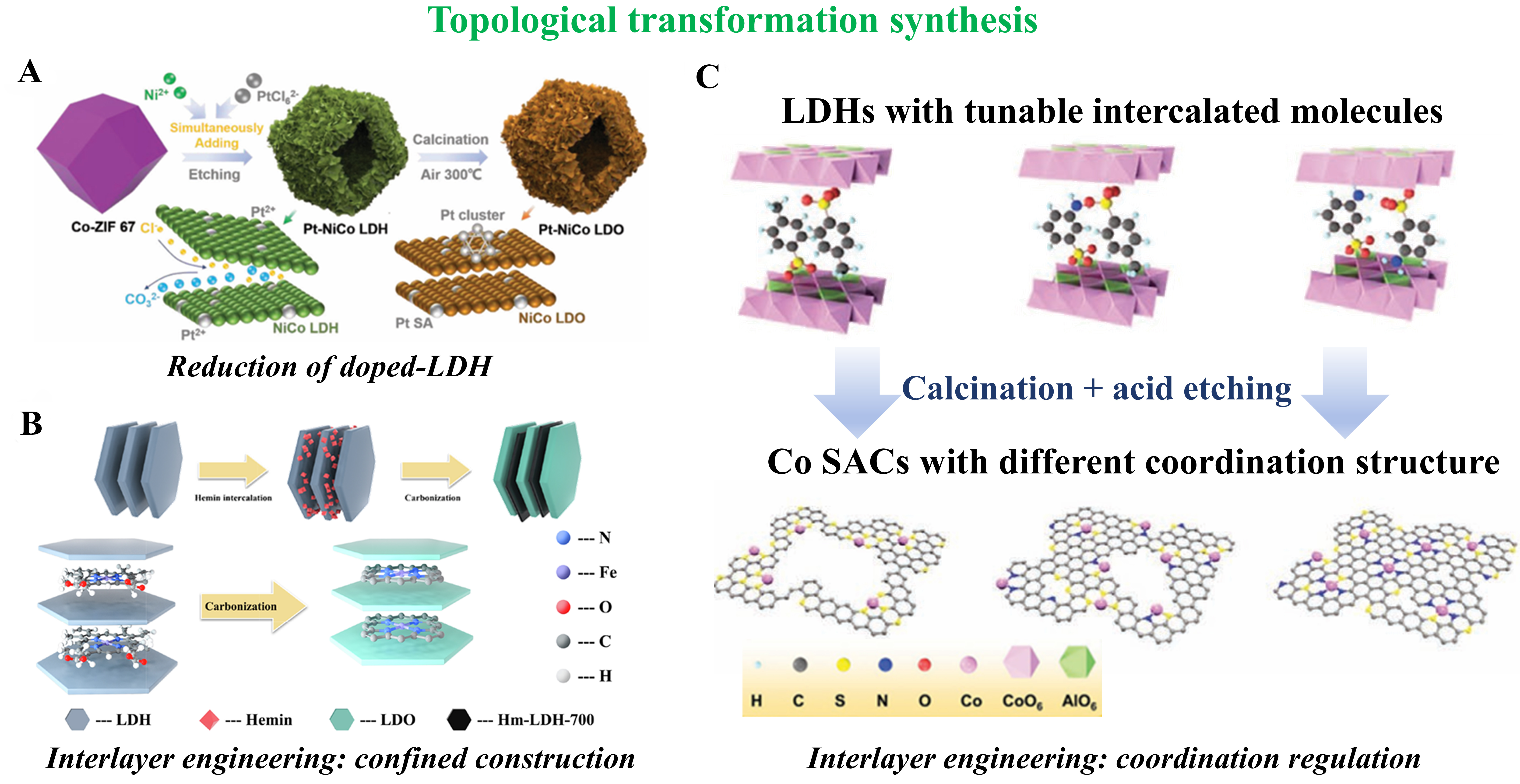
Figure 3. Schematic illustration of the topology-transformation strategy for synthesizing LDH-based SACs. The pathways include (A) reduction of doped LDH precursors. Reprinted with permission from Ref.[66]. Copyright 2024, Wiley-VCH; (B) interlayer engineering for confined construction of single-atom sites. Reprinted with permission from Ref.[67]. Copyright 2021, Elsevier; and (C) interlayer engineering for coordination regulation. Reprinted with permission from Ref.[68]. Copyright 2020, Wiley-VCH. LDH: Layered double hydroxide; SAC: single-atom catalyst.
In addition, topological transformation based on intercalation engineering offers a powerful route for the precise construction of single-atom sites with specific coordination structures within confined spaces. Yang et al. ingeniously employed this strategy by intercalating the metallomacrocycle Hemin into the interlayer gallery of LDHs[67]. Subsequent thermal treatment induced the topological conversion of the LDHs host into a LDO due to the well-known ‘memory effect’. Crucially, during the transformation process, the Hemin molecules confined between the layers were pyrolyzed, and their metal centers (Fe) coordinated in situ with nitrogen atoms originating from the Hemin ligands, leading to the formation of well-defined and confined FeN4 sites [Figure 3B]. The key to the method lies in the fact that the LDHs laminates act not merely as a convertible rigid template but, more importantly, their unique interlayer confinement effect ensures the spatial isolation and fixation of the precursors. The confinement effect effectively suppresses the migration and aggregation of metal atoms during the thermal conversion, ultimately yielding highly uniform single-atom active sites within the LDO matrix.
Furthermore, LDHs can be topologically converted into carbon-supported SACs through pyrolysis and etching. Fan et al. co-intercalated organic molecules [aniline sulfonic acid and p-toluenesulfonic acid (PA)] into CoAl-LDHs[68]. Upon heating, the LDH layers decomposed, forming Co nanoparticles and a carbon framework from the organic sources. Subsequent HCl etching removed Co nanoparticles, leaving atomically dispersed Co atoms coordinated with N or S within the carbon nanosheets, forming integrated electrode-type SACs (IE-SACs) [Figure 3C].
In summary, the topological transformation strategy enables precise reconstruction of the internal lattice, chemical composition, and coordination environment, while preserving the external geometry, layered topology, and overall morphology of LDHs. The approach not only maintains beneficial features such as ion-transport channels and gas-diffusion pathways but also offers exceptional controllability and tunability in designing SAC architecture, often leading to catalysts with enhanced activity and stability.
Scaled-up production of LDHs
While the in situ synthesis, post-synthesis, and topological transformation strategies described earlier have enabled precise immobilization of SAs on LDHs at the laboratory scale, the practical application of LDH-based SACs hinges on scalable, cost-effective, and environmentally friendly production technologies. LDHs themselves have been industrialized in fields such as flame retardants and polyvinyl chloride (PVC) heat stabilizers. This section focuses on the industrial progress, core technologies, and optimization strategies for LDH scale-up, highlighting their foundational role in mass-producing LDH-based SACs.
The separate Nucleation and Aging Steps Method (SNAS), proposed by Zhao et al. in 2002, is the only industrially validated technology for large-scale nanostructured LDHs synthesis and has been extended to pilot-scale monolayer LDH production[70]. Unlike conventional coprecipitation (which yields broad particle size distributions of 60 nm ~ 10 μm), the SNAS method decouples nucleation and crystallization: metal salt and alkali solutions are rapidly mixed in a colloid mill for instantaneous, uniform nucleation, followed by separate aging to form well-crystallized LDH nanosheets with a narrow size distribution and tunable thickness [Figure 4A][69-71]. Adjusting metal ion ratios (e.g., Mg2+/Al3+) and process parameters enables precise control over crystal phase and particle size, critical for tailoring the ‘atomic canvas’ for single-atom anchoring[72]. The SNAS method offers advantages such as simple operation (requiring only a few minutes per batch), excellent reproducibility, and compatibility with LDH compositions, enabling production at the ton-scale level Figure 4B][73]. Production lines with capacities of 10,000 tons and higher have been established in multiple regions.
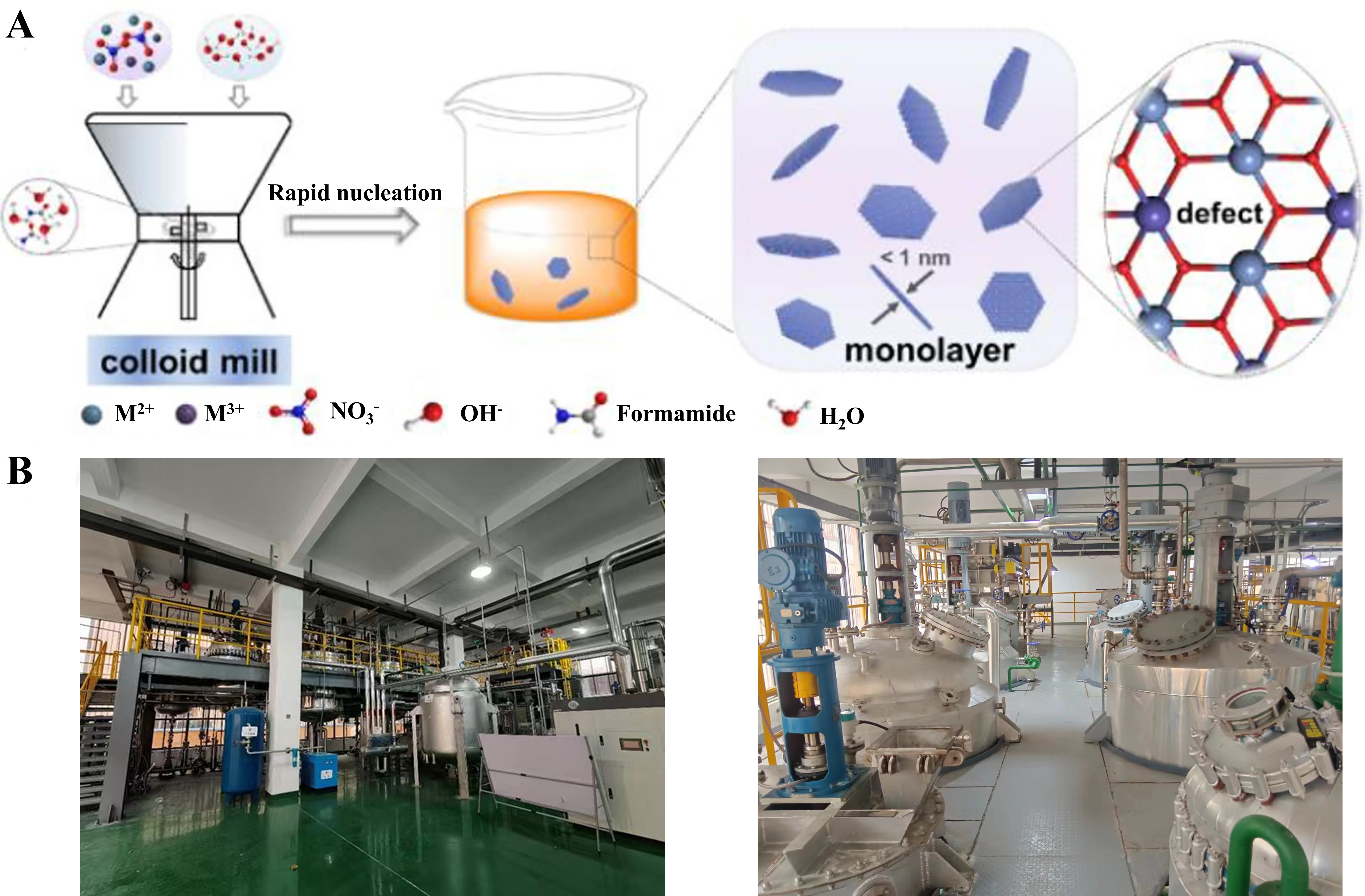
Figure 4. (A) Scale-up production of monolayer LDH nanosheets. Reprinted with permission from Ref.[69]. Copyright 2021, Elsevier. (B) The separate nucleation and aging steps method is applied in the 100-ton pilot plant and 10000-ton production line of LDHs industrial production from Quzhou Institute for Innovation in Resource Chemical Engineering. Figure 4B is an on-site photograph of the equipment used by our research group at the Quzhou Institute of Resource and Chemical Innovation to synthesize hydrotalcite on an industrial scale using the separate nucleation and aging steps method. LDH: Layered double hydroxide.
In addition, single-layer LDHs exhibit outstanding catalytic performance due to their abundant defects and large specific surface area. However, conventional ‘top-down’ and ‘bottom-up’ synthesis methods for single-layer LDHs are limited to small-scale production (g/day), severely hindering the practical large-scale application of LDH-based materials. Bai et al.[69] and Chi et al.[74] employed the SNAS method using a colloid mill reactor to develop a simplified approach for scalable synthesis (~ 50 kg/day) of monolayer LDHs [Figure 4A], yielding multiple varieties of colloidal and powdered monolayer LDHs[69,74]. The series of single-layer MgAl/CoAl/NiCo/NiFe-LDH nanosheets obtained through this method exhibit a thickness of approximately 1 nm and a high specific surface area of up to 300 m2·g-1.
In summary, the mass production of LDHs driven by SNAS technology, coupled with green optimization and exfoliation strategies, has effectively overcome bottlenecks in the transition from laboratory to industrial scale. Leveraging the established industrial foundation of LDHs, this approach enables customization of single-atom catalytic carriers, advancing practical applications in energy conversion and environmental remediation. The large-scale preparation of LDH supports the industrialization of SACs by ensuring carrier uniformity (consistent single-atom anchoring sites), structural fidelity (high specific surface area and unsaturated coordination sites), and sustainability (green synthesis meeting industrial requirements).
APPRECIATION OF THE ‘PAINTING’: HOW THE LDH ‘ATOMIC CANVAS’ RECONSTRUCTS CATALYTIC INTERFACES AND EXPLORES MULTIFUNCTIONALITY
The successful ‘painting’ of SAs onto the LDH canvas is merely the first step. The true value of this architecture is revealed during catalysis, where the dynamic interplay between the SA and the active LDH support dictates performance[75-78]. This section delves into the core mechanisms by which the LDH ‘atomic canvas’ reconstructs catalytic interfaces, transforming both the electronic landscape and the reaction pathways.
Electronic modulation engineering: precisely tuning the state of single-atom centers
The performance of SACs is largely determined by the electronic structure of the metal centers, including valence state, charge distribution, and d-band center position. Therefore, precisely tuning the electronic state of single-atom sites to optimize their adsorption strength toward reaction intermediates is a key strategy for improving catalytic activity and selectivity. Owing to their tunable layered composition, abundant surface hydroxyl groups, and unique confined environment, LDHs serve as a highly tailorable platform for stabilizing and engineering SACs. Through strong metal-support interactions (SMSI), interfacial charge transfer, defect engineering, and heteroatom doping, LDHs can effectively modulate the electron density and orbital hybridization of anchored SAs, thereby breaking the linear scaling relationships in traditional catalysis and achieving enhanced catalytic performance. This section will systematically elaborate on the electronic modulation strategies based on LDH supports and their underlying mechanisms in various catalytic reactions.
Ebitani et al. laid the cornerstone for electronic modulation engineering in LDH-based SACs through their pioneering work[79]. As early as 2005, Ebitani et al. from this team published a landmark study in which they constructed, for the first time, a structurally well-defined hetero-trimetallic single-atom catalytic system (RuMn2/HT) on a LDH (hydrotalcite, HT) surface[79]. This work pioneered the use of LDHs as a ‘macro ligand’, leveraging its surface hydroxyl sites to precisely regulate the coordination environment and electronic states of SAs through multi-metallic synergistic effects. The researchers successfully prepared RuMn2/HT by anchoring manganese Ebitani’s work cations onto the surface of a previously reported Ru/HT catalyst[79]. Using XAFS spectroscopy combined with reference spectra from RuO2 and β-MnO2 for comparative analysis [Figure 5A-D], they confirmed the high dispersion of Ru species and the formation of manganese species on the HT surface. Based on extend X-ray absorption fine structure (EXAFS) curve fitting, they proposed a refined structural model for the hetero-trimetallic species. The model features a Ru4+ center coordinated by six oxygen atoms, adjacent to Mg cations from the dolomite-like layers of HT and two Mn4+ cations. This configuration forms a unique Ru4+-Mn4+-Mn4+ structure anchored by hydroxyl groups and water molecules [Figure 5E and F].
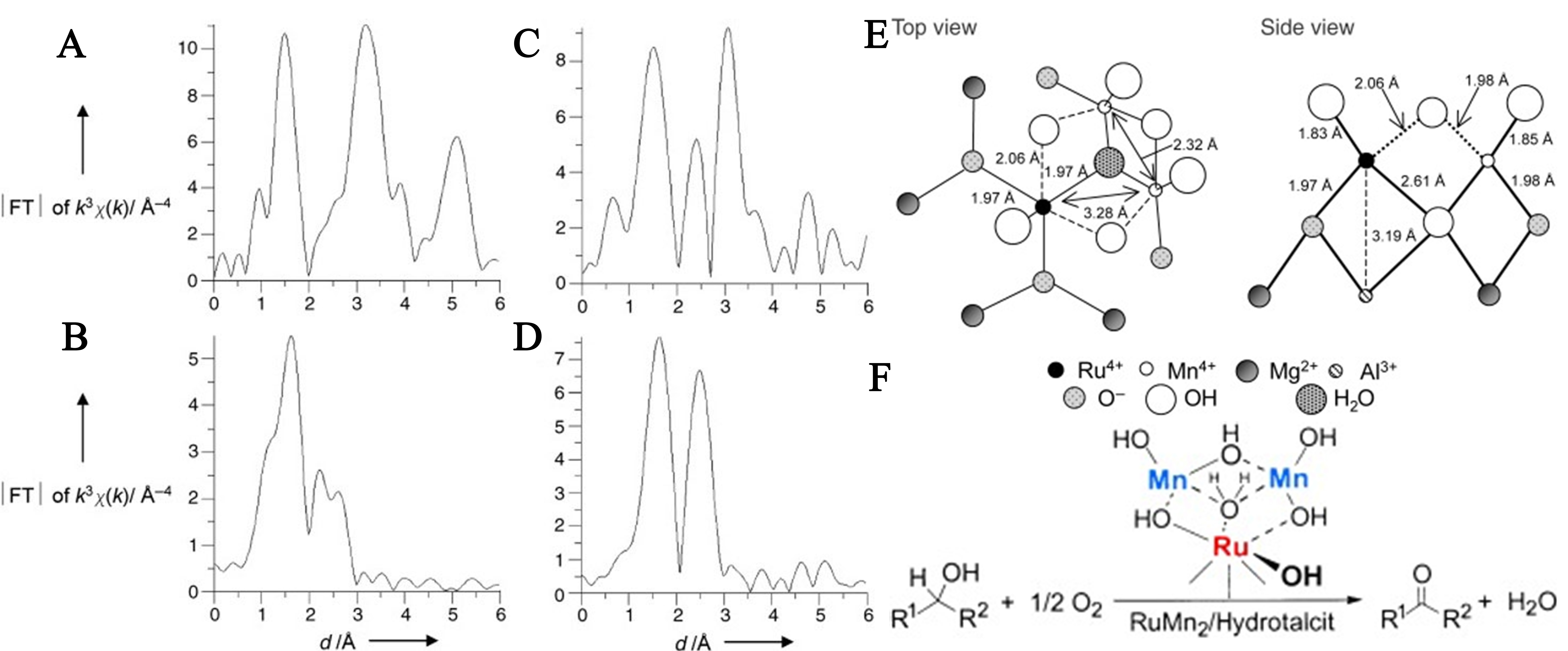
Figure 5. Fourier transformation (FT) of the k3-weighted K-edge EXAFS spectrum of (A) RuO2; (B) RuMn2/HT (Ru K-edge); (C) β-MnO2; and (D) RuMn2/HT (Mn K-edge). The phase shift was not corrected; (E) Proposed structure of the heterotrimetallic RuMnMn species on HT; (F) RuMn2/HT Catalyst for the Liquid-Phase Oxidation of Various Alcohols. (A-F) Reprinted with permission from Ref. [79]. Copyright 2005, Wiley-VCH. EXAFS: Extended X-ray absorption fine structure; HT: hydrotalcite.
This study did not exist in isolation; it was built upon the team’s prior systematic exploration. For instance, the earlier Ru/HT catalyst reported by Motokura et al. achieved the grafting of Ru4+ species onto the HT surface, validating the synergistic effect between the metal and the HT support[80]. Subsequently, the developed Rh/HT catalyst anchored Rh3+ species via Rh-O-Mg bonds, expanding the application scope of LDH-supported SACs[81]. Collectively, these studies established a research paradigm using HT as the support, employing the impregnation method, relying on XAFS as the core characterization technique, and focusing on metal-support synergy.
Therefore, Ebitani’ s work on RuMn2/HT serves as a pivotal link connecting past and future research. It not only built upon the Ru/HT framework but also innovatively introduced manganese to construct hetero-trimetallic sites, revealing for the first time at the atomic level how LDH supports can be employed to precisely regulate the electronic states of single-atom sites via multimetallic synergy. This work strongly underscores the unique merits of LDHs as a tunable platform for the rational design of precise catalysts, offering critical theoretical and experimental guidance for the development of high-performance multimetallic SACs.
Electronic modulation engineering, which precisely regulates the electronic state of active centers via strong SMSI between LDHs and SAs/composite active sites, serves as the core strategy for constructing high-efficiency water-splitting electrocatalysts[82]. As a foundational work in this domain, Sun et al. pioneered the concept of reconstruction-free dual-site catalysis by atomically dispersing Ru SAs onto a Ni3V-LDH scaffold[83]. The core mechanism, validated by in situ studies and theoretical calculations, relies on SMSI to stabilize the electronic configurations of Ru [hydrogen evolution reaction (HER), active center] and Ni [oxygen evolution reaction (OER), active center], thereby suppressing structural reconstruction during catalytic cycles. Comparative analysis of Ru oxidation state evolution during HER reveals that RuO2 undergoes a significant reduction in Ru oxidation state under cathodic potentials, while Ru/Ni3V-LDH exhibits only slight fluctuations - this direct evidence of SMSI-driven stabilization of Ru SAs lays the groundwork for understanding the catalyst’s reconstruction-free nature [Figure 6A][83]. Further investigating Ni sites, pure Ni3V-LDH shows drastic Ni oxidation state reduction during HER and structural rearrangement during OER, whereas Ru/Ni3V-LDH maintains Ni state stability with minimal bond length variation, highlighting how Ru-Ni electronic coupling enhances structural tolerance against reaction-induced distortion [Figure 6B]. Building on these observations, a schematic diagram illustrates the distinct functional roles of the dual sites: Ru sites dominate proton reduction without structural reconstruction, whereas Ni sites act as OER active centers with stabilized high oxidation states (Ni3+/Ni4+) promoted by Ru. This clearly exemplifies how LDH supports enable the targeted electronic modulation of individual active sites [Figure 6C].
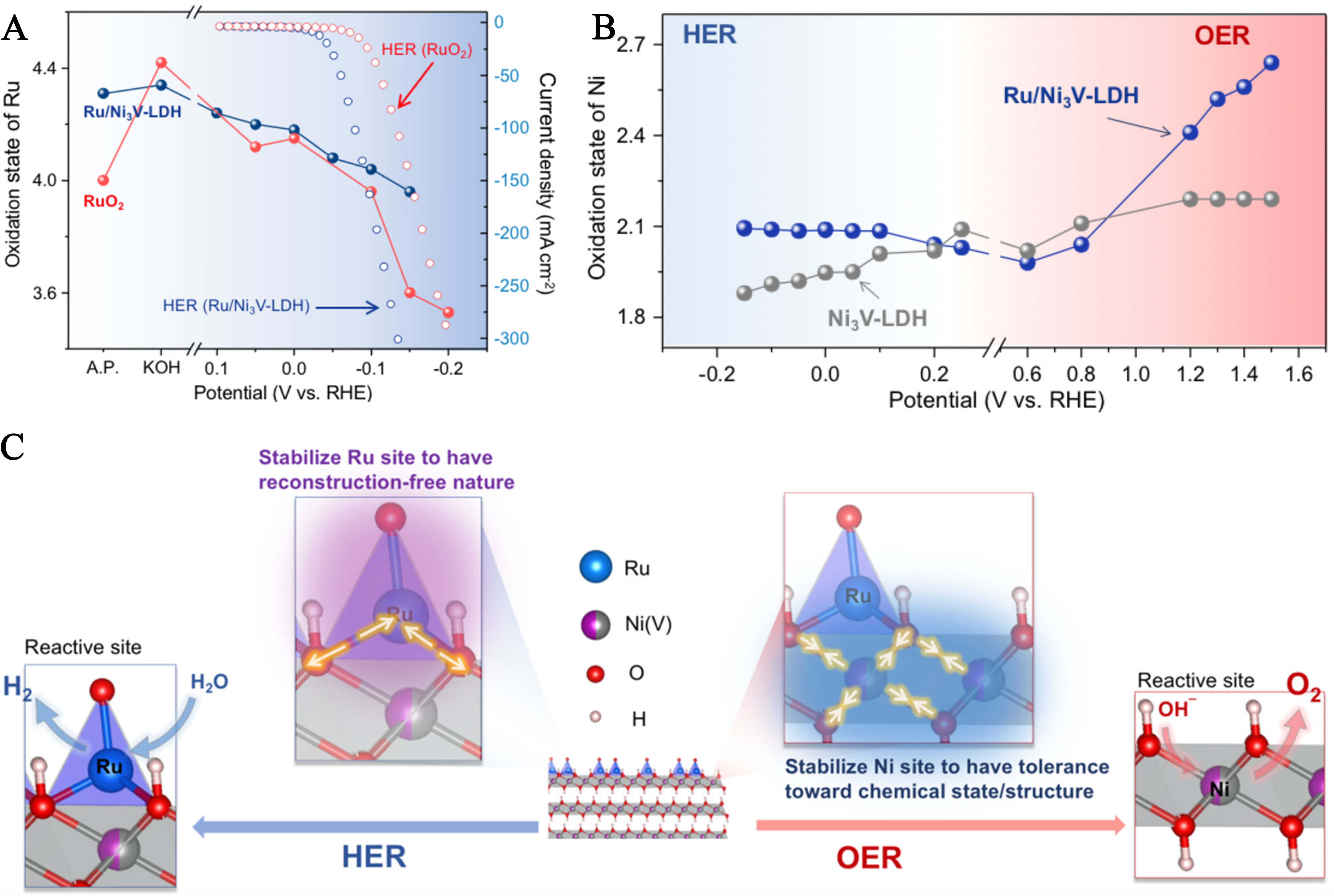
Figure 6. (A) Extracted oxidation state of the Ni site in Ni3V-LDH and Ru/Ni3V-LDH samples during HER and OER; (B) Extracted oxidation state of the Ni site in Ni3V-LDH and Ru/Ni3V-LDH; (C) Schematic models of HER and OER occurring at steady coordinated dual-active sites on the Ru/Ni3V-LDH. (A-C) Reprinted with permission from Ref.[83]. Copyright 2021, American Chemical Society. RHE: Reversible hydrogen electrode; HER: hydrogen evolution reaction; OER: oxygen evolution reaction.
Integrating organic ligand modification and metal doping into LDH-based electronic modulation, Ngo et al. developed an Ir SA anchored on phytic acid (PA)-modified Mn-doped Ni-LDH (Ir@Mn-Ni-PA) via a sequential hydrothermal and coordination assembly strategy[84]. The phosphate groups of PA form strong metal-oxygen-phosphorus coordination to stabilize Ir SAs, while Mn doping and Ir incorporation synergistically modulate the LDH matrix’s electronic structure, narrowing the bandgap and optimizing intermediate adsorption energies for both HER and OER. Density functional theory (DFT) calculations confirm that Ir SAs donate electrons to the LDH, moderating the binding strength of HER and OER intermediates to align with the Sabatier principle, realizing bidirectional electronic regulation for bifunctional water splitting.
Expanding electronic modulation from single-atom sites to composite active sites, Wu et al. constructed a NiFe LDH-based catalyst by anchoring Pt SAs in Fe vacancies and loading Pt quantum dots on the surface via a two-step strategy[85]. The dual Pt species form strong electronic interactions with the LDH matrix, and their synergy optimizes the kinetics of water dissociation and hydrogen desorption, realizing collaborative regulation of the HER process. Introducing phase engineering as a new dimension for electronic modulation, Wang et al. prepared Pt SAs supported on an amorphous/crystalline NiFe-LDH heterostructure through two-step electrodeposition[86]. Pt SAs are uniformly distributed across both phases, and their strong interaction with the heterostructure enhances water adsorption/dissociation efficiency and balances hydrogen intermediate adsorption-desorption, endowing the catalyst with adaptability to diverse electrolytes. Focusing on electronic modulation for OER optimization, Yang et al. stabilized Ru SAs on NiFe-LDH via oxygen-coordination bonds through a facile solution reduction strategy[54]. Ru SAs trigger electron rearrangement of Ni/Fe sites in the LDH matrix, optimizing the binding energy of OER intermediates and reducing the energy barrier of the rate-determining step (RDS), thereby enhancing intrinsic catalytic activity.
Focusing on site-specific regulation of SMSI without altering LDH supports, Wei et al. developed an electrochemical deposition strategy to anchor Ir SAs on Ni LDH, achieving precise control over metal-support interaction strength[51]. Cathodic deposition drives Ir atoms to three-fold face-centered cubic (fcc) hollow sites (Ir1/Ni LDH-T) with strong SMSI, while anodic deposition anchors Ir at oxygen vacancy sites (Ir1/Ni LDH-V) with weak SMSI, enabling systematic investigation of MSI-activity correlations. Structural models of the two configurations clearly show that Ir1/Ni LDH-T forms more covalent Ir-O bonds with the support compared to Ir1/Ni LDH-V. Additionally, charge density difference analysis confirms enhanced electron transfer from Ni LDH to Ir in the strong SMSI system [Figure 7A]. Projected density of states (PDOS) further reveals that strong SMSI tunes the Ir d-band center to a moderate position, balancing intermediate adsorption/desorption strength, whereas weak MSI leads to unfavorable d-band alignment [Figure 7B]. Free energy calculations for OER demonstrate that strong MSI reduces the energy barrier of the RDS (O → OOH) on Ir sites to 1.77 eV, far lower than the 2.34 eV barrier on Ir sites in the weak MSI system [Figure 7C]. Schematic OER pathways illustrate that strong MSI induces a switch of active sites from Ni to Ir [Figure 7D], while weak MSI retains Ni as the dominant active center [Figure 7E]. This site-specific MSI regulation is further corroborated by in situ Raman and isotope-labeling experiments, which confirm the absence of NiOOH redox dynamics in Ir1/Ni LDH-T during OER [Figure 7F], directly validating the active site switch. This mechanism is vividly summarized in the catalytic pathway diagram, highlighting how SMSI strength dictates active site identity and reaction kinetics.
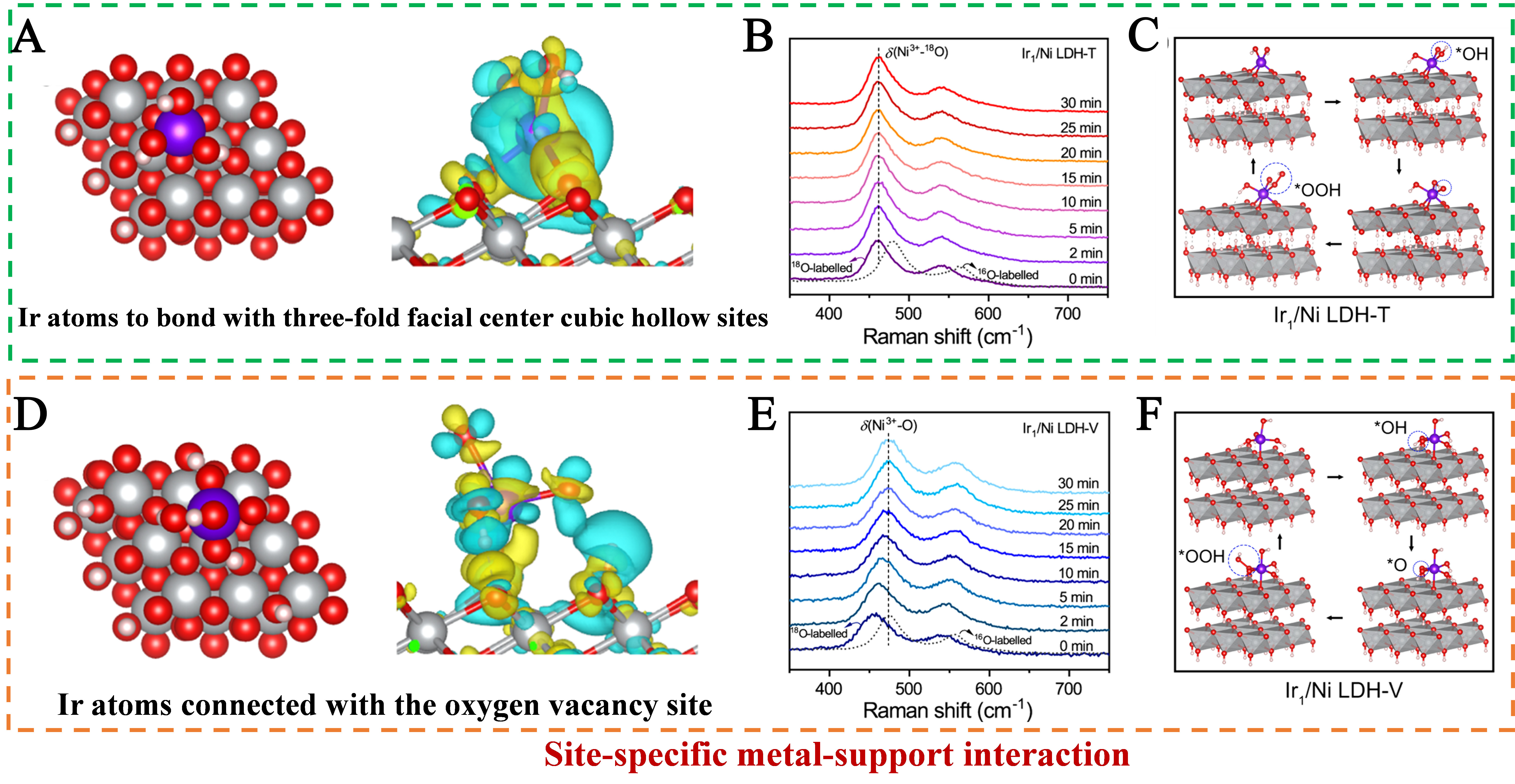
Figure 7. (A) Top-view schematic structural model of Ir1/Ni LDH-T and the charge density difference of Ir atoms on Ir1/Ni LDH-T; (B) In situ Raman spectra of 18O-labeled Ir1/Ni LDH-T; (C) Schematic diagram of the oxygen evolution reaction pathway for Ir1/Ni LDH-T; (D) The top-view schematic structural model of Ir1/Ni LDH-V and the charge density difference of Ir atoms on Ir1/Ni LDH-V; (E) In situ Raman spectra of 18O-labeled Ir1/Ni LDH-V. (F) Schematic diagram of the oxygen evolution reaction pathway for Ir1/Ni LDH-V. (A-F) Reprinted with permission from Ref.[51]. Copyright 2024, Springer Nature. LDH: Layered double hydroxide.
Extending SMSI regulation to chlorine-tolerant seawater HER: Sun et al. used a solvothermal synthesis method to anchor single platinum atoms onto NiV-LDH, where the Pt-O bond induces electronic redistribution to optimize the center of the platinum d-band, whilst vanadium doping generates oxygen vacancies, thereby synergistically regulating the adsorption of intermediates[87]. This metal-support electronic modulation reduces the energy barriers for H2O dissociation and *OH desorption, while simultaneously suppressing the adsorption of Cl- at active sites, thereby endowing the material with inherent chlorine resistance. Targeting biomass upgrading reactions, Xu et al. integrated Lewis acidic Zn clusters into 3D Ru single-atom/NiFeS-LDH, realizing synergistic electronic modulation between Ru SAs and Zn clusters[88]. Zn clusters increase Lewis acidic oxygen vacancies, regulate the Ni0/Ni2+ ratio, and stabilize high-valent Ni2+δ species, while Ru SAs enhance '5-hydroxymethylfurfural (HMF) aldehyde group adsorption - their synergy promotes proton-coupled electron transfer in 5-hydroxymethylfurfural oxidation. Zhang et al. designed a Pt/FeOOH@NiFe LDH catalyst with coexisting Pt SAs and clusters, induced by amorphous FeOOH[89]. Amorphous FeOOH facilitates H2O adsorption and activation, while Pt SAs and clusters promote H2 formation and desorption, synergistically accelerating HER kinetics via the Volmer-Heyrovsky mechanism. Hu et al. stabilized Ru SAs on defective NiFe LDH (Ru SAs/D-NiFe LDH@NF) via hydrothermal synthesis and etching[41]. Ru SAs regulate electron distribution near LDH defects, optimize Ru-NiFe interaction, and reduce the energy barrier for *OOH intermediate formation, exhibiting excellent OER performance and stability in Zn-air batteries.
Electronic modulation engineering, which regulates the electronic state of active centers via SMSI between LDHs and SACs, is pivotal for developing high-efficiency OER electrocatalysts[22,90,91]. The work by Li et al. on Ru SAs anchored on cobalt-iron LDH (Ru/CoFe-LDHs) serves as a paradigmatic study, as it systematically deciphers how LDHs modulate single-atom electronic structures through SMSI[43]. This mechanism is coherently validated by a series of correlated characterizations and theoretical calculations, forming a logical chain from structural identification to catalytic performance optimization[43]. Fourier-transformed Ru K-edge EXAFS spectra of Ru/CoFe-LDHs (alongside Ru foil, RuCl3, RuO2) reveal the absence of Ru-Ru, Ru-Cl, and Ru-O-Ru characteristic peaks, with only Ru-O bonds and weak Ru-O-M (M=Co/Fe) bonds observed. This direct evidence confirms Ru atoms are atomically dispersed on LDHs, laying the structural foundation for SMSI-induced electronic modulation [Figure 8A]. Building on this structural confirmation, model-based EXAFS fitting further quantifies the coordination environment: each Ru atom coordinates with ~ 3.9 oxygen atoms, among which ~ 2.9 Ru-O bonds are linked to Co/Fe atoms of LDHs, enabling efficient electron transfer between Ru and the support and establishing the structural basis for strong electronic coupling [Figure 8B].
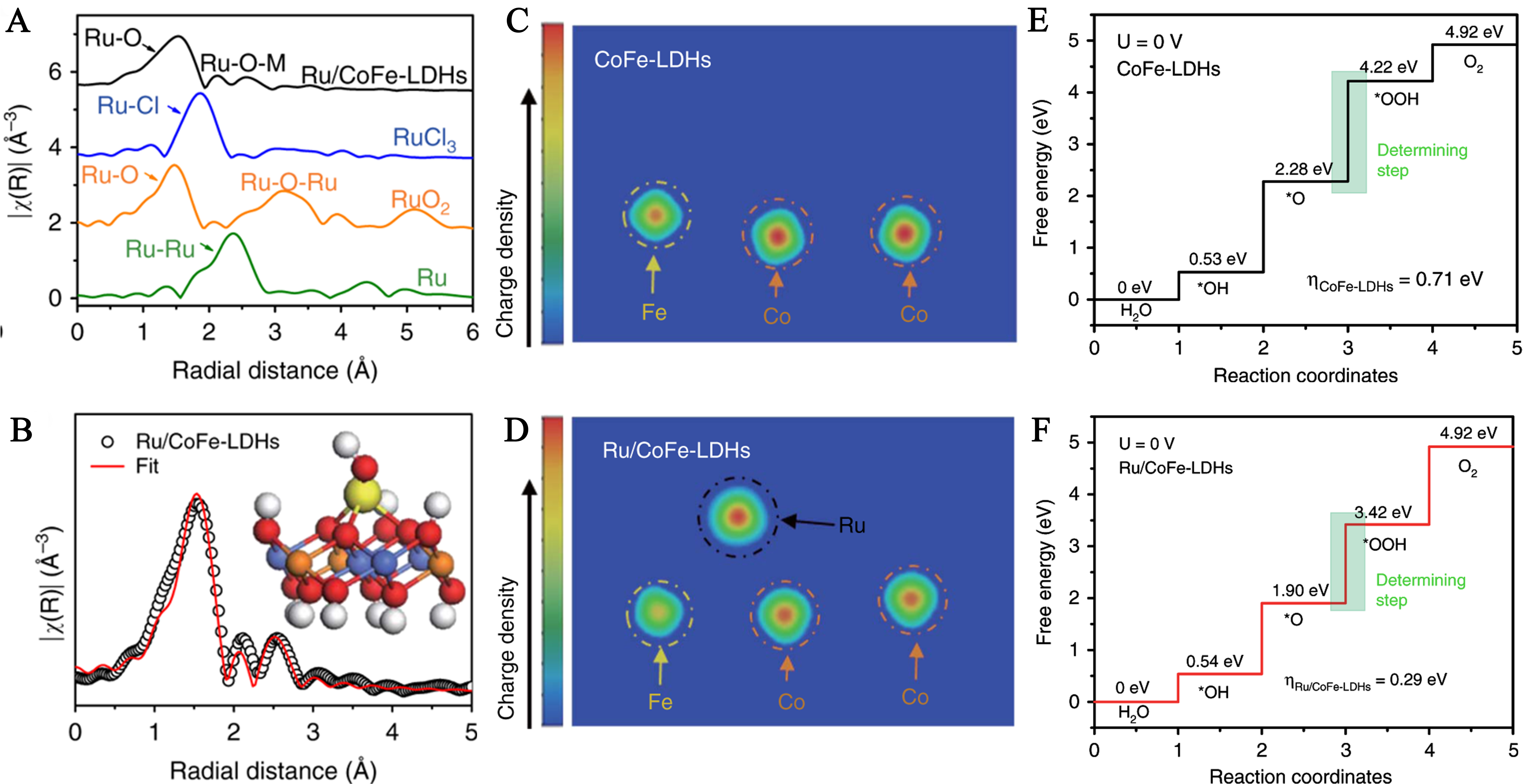
Figure 8. (A) Fourier-transformed Ru K-edge EXAFS spectra of Ru/CoFe-LDHs, RuCl3, RuO2, and Ru metal; (B) Corresponding model-based fittings of Ru EXAFS for Ru/CoFe-LDHs and simulated EXAFS spectra from Ru-O and Ru-O-M (M = Co or Fe) bonds (the inset is the magnifying local structure of Ru/CoFe-LDHs), showing the exclusive existence of Ru-O-M bonds in Ru/CoFe-LDHs sample. The differential charge density of elements in CoFe-LDHs (C) and Ru/CoFe-LDHs (D) from computational simulation, revealing electron donation from LDHs to Ru. Gibbs free-energy diagram for the four steps of OER on CoFe-LDHs (E) and Ru/CoFe-LDHs (F). (A-F) Reprinted with permission from Ref.[43]. Copyright 2019, Springer Nature. EXAFS: Extend X-ray absorption fine structure; LDH: layered double hydroxide.
To unravel the essence of electronic modulation, differential charge density comparisons of CoFe-LDHs and Ru/CoFe-LDHs provide critical insights: pristine CoFe-LDHs exhibit uniform electron distribution across the matrix [Figure 8C], while Ru anchoring induces significant electron density reduction at Co/Fe sites and accumulation at Ru sites. This observation verifies electron transfer from LDHs to Ru via Ru-O-M bridging bonds, a core driver for optimizing catalytic activity [Figure 8D]. The impact of electronic modulation on OER kinetics is ultimately clarified by Gibbs free energy diagrams: for pristine CoFe-LDHs, the RDS of (*O → *OOH) exhibits a high energy barrier of 1.94 eV and a theoretical overpotential of 0.71 eV [Figure 8E]. In contrast, Ru/CoFe-LDH retains the same RDS but reduces the barrier to 1.52 eV and overpotential to 0.29 eV [Figure 8F]. This improvement originates from SMSI-tuned d-band centers of Ru, which balance the adsorption/desorption of OER intermediates (*OH, *O, *OOH) and break the scaling relation limitation.
Integrating heterostructure coupling and interface engineering, dual strategies have been developed to enhance electronic modulation for water splitting. Chen et al. constructed a Ru single-atom/NiFe-LDH/MoNiS core-shell heterostructure (RuSA/NiFe-LDH/MoNiS) via sequential hydrothermal and immersion methods, where Ru atoms form Ru-O-Ni motifs on NiFe-LDH[92]. SMSI-induced electron redistribution between Ru, NiFe-LDH, and MoNiS optimizes the d-band center of Ru, accelerating water dissociation and balancing *H/*OH adsorption-desorption for efficient alkaline HER. Deng et al. anchored single Ru atoms on CoFe-LDH (Ru0.51-CoFe-LDH) and integrated it with BiVO4 to form an n-n heterojunction photoanode[93]. Ru SAs trigger electron rearrangement in CoFe-LDH, inducing a negative shift of its band edge to enhance interfacial band bending, which facilitates photogenerated charge separation and transfer for photoelectrochemical water splitting.
Extending electronic modulation to light-responsive systems, light-induced dynamic regulation of single-atom states has emerged as a novel strategy for photocatalytic CO2 reduction. Fan et al. anchored Pt SAs on ZnNiTi-LDHs with metal ion vacancies (Pt1/ZnNiTi-LDHs-E) via defect immobilization after selective Zn2+ etching[94]. Under different light intensities, Pt SAs capture photoelectrons to form Pt(IV), electron-rich Pt(II), and near-neutral Ptδ+ species. They drive CO generation, C2H6 formation via C-C coupling (with Ni2+/Ti4+ sites), and CH4 production, respectively, via direct hydrogenation, realizing structure-dependent reaction pathway control[94].
Extending this framework to lattice doping strategies, Dong et al. fabricated Ru lattice-doped CoCr LDHs (CoCrRu LDHs), where single Ru atoms downshift the d states of adjacent Co atoms and enhance electron donation from Cr to oxygenates[39]. This synergistic charge transfer weakens Co’s adsorption of *O/*OH and strengthens its binding to *OOH, breaking the scaling relation and boosting OER activity. Focusing on iridium single-atom systems, Biswal et al. reported Ir1/NiCr LDH by anchoring Ir atoms on NiCr LDH via surface hydroxyl groups[95]. SMSI-induced electron redistribution between Ir and Ni/Cr optimizes Ir’s coordination environment (tetracoordinate Ir-O bonds), reduces the energy barrier of the OER RDS, and endows the catalyst with excellent activity and stability in both alkaline and seawater electrolytes.
Electronic modulation engineering, which regulates the electronic state of active centers via SMSI between LDHs and SACs/dual-atomic catalysts (DACs), remains a core strategy for developing high-efficiency bifunctional oxygen electrocatalysts [oxygen reduction reaction (ORR)/OER] for zinc-air batteries (ZABs)[96]. Liu et al. constructed a NiFe-LDH/ Fe1-N-C heterostructure, revealing how LDH nanodots modulate the electronic structure of single-atom Fe1-N-C through interfacial coupling[96]. Optimized atomic configurations of ORR elemental steps on NiFe-LDH/Fe1-N-C clearly show the spatial arrangement of NiFe-LDH nanodots and Fe1-N-C matrix, with Fe-N4 moieties identified as ORR active sites and NiFe-LDH as OER active centers. This structural design ensures intimate interfacial contact, laying the foundation for electron transfer between the two components [Figure 9A][96]. Building on this structural basis, calculated energy barriers of ORR reveal that the RDS for Fe1-N-C (hydrogenation of *O2 to *OOH) has a high energy barrier of 2.59 eV, while NiFe-LDH modification reduces this barrier to 1.96 eV - this reduction originates from the enhanced adsorption and activation of *O2 by interfacial electronic coupling [Figure 9B]. To decipher the essence of this modulation, differential charge density analysis shows significant electron redistribution: NiFe-LDH donates 1.25 e- to Fe1-N-C, with electron accumulation around Fe-N4 sites and depletion in NiFe-LDH. This charge transfer directly optimizes the binding strength between Fe active sites and ORR intermediates [Figure 9C]. This electronic modulation is further quantified by the d-band center of Fe 3d orbitals: Fe1-N-C alone has a d-band center of -2.13 eV, while NiFe-LDH coupling upshifts it to -1.45 eV. This upshift increases the number of unoccupied orbitals in Fe 3d, strengthening the interaction with *O2 and facilitating its activation [Figure 9D].
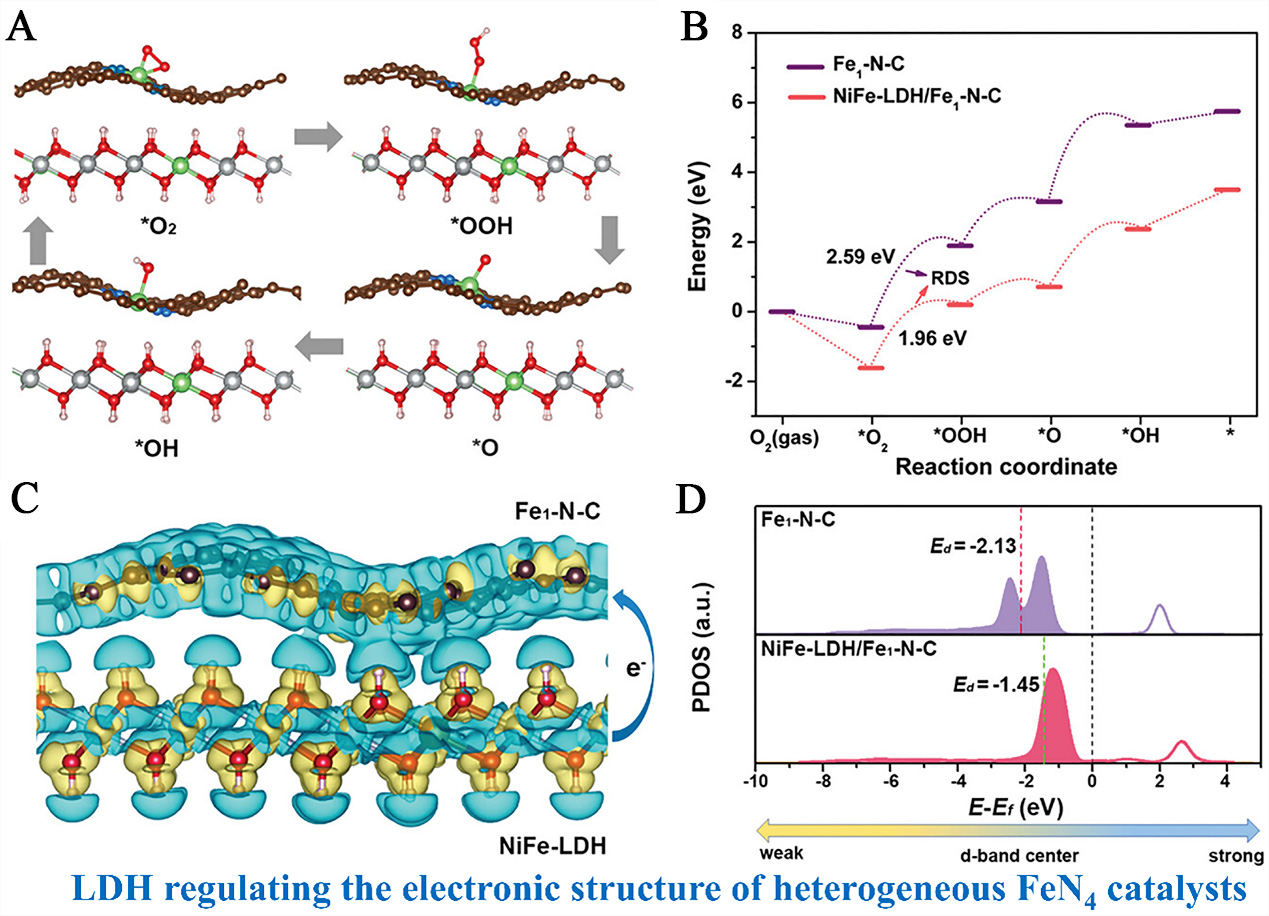
Figure 9. (A) The optimized atomic configurations of ORR elemental steps on the NiFe-LDH/Fe1-N-C; (B) Calculated energy barrier of ORR on NiFe-LDH/Fe1-N-C and Fe1-N-C; (C) Side view of differential charge density by Löwdin charge analysis of NiFe-LDH/Fe1-N-C heterostructures with an isosurface value of 0.005 e Å-3. (D) d-Band centers for Fe 3d of NiFe-LDH/Fe1-N-C and Fe1-N-C. (A-D) Reprinted with permission from Ref.[96]. Copyright 2023, Wiley-VCH. LDH: Layered double hydroxide.
Extending this heterostructure coupling strategy to DACs, Qin et al. constructed FeNi-LDH@DACs with a ‘sesame-ball-like’ architecture by anchoring FeNi-LDH nanodots on FeNi DACs[62]. Electron transfer from LDH to DACs modulates the d-band center of FeNi atomic sites, optimizing oxygen intermediate adsorption energies, and the porous carbon framework of DACs enhances conductivity - this synergy endows the catalyst with a peak power density of 211.6 mW cm-2 and 500 h cycling stability in ZABs. Focusing on size-confined coupling, Wang et al. fabricated NiFe-ND/FeCo-NC by depositing ultrasmall NiFe-LDH nanodots on 3D flower-like FeCo-NC[97]. The 3D mesoporous structure of FeCo-NC confines NiFe-LDH growth to ~ 4 nm, and strong interfacial coupling induces electron depletion in Ni sites - this enhances the formation of Ni (Fe)OOH active phases during OER, delivering a small discharge-charge voltage gap of 0.87 V in ZABs.
Coordination engineering
The catalytic performance of SACs is intrinsically linked to the local coordination environment of the active metal centers. Coordination engineering therefore represents a powerful strategy for optimizing SACs by precisely tailoring this microenvironment, including the type and number of coordinating atoms, bond lengths/angles, and the presence of vacancies. For LDH-based SACs, this confers an exceptional capability to precisely regulate the interaction between the SACs and the LDH support. The layered structure of LDHs, with its abundant hydroxyl groups and tunable cation vacancies, provides a unique platform for such engineering. By manipulating the type and distribution of these vacancies (e.g., M2+ vs. M3+ sites), one can directionally design both the primary coordination shell (directly bonded atoms) and the secondary coordination sphere of the anchored SA[55]. This precise control over the coordination geometry directly modulates the electronic structure (e.g., oxidation state, d-band center) and, consequently, the adsorption/desorption behavior of key reaction intermediates, enabling rational optimization of catalytic activity and selectivity[98-101]. Among various approaches, the regulation of cation vacancies to construct well-defined anchoring sites stands out as a pivotal method for stabilizing SAs while fine-tuning their catalytic microenvironments.
Jin et al. utilized NiFe-LDH as a support to construct a series of Ru SACs by regulating M3+/M2+ cation vacancies[55]. Employing alkali etching-induced vacancy formation as the core coordination control strategy, NiFeAl-LDH and NiZnFe-LDH were treated with NaOH to selectively remove Al3+ and Zn2+, respectively, generating M3+ vacancies (LDH-VIII) and M2+ vacancies (LDH-VII). Subsequent RuCl3 impregnation and H2/Ar reduction anchored Ru SAs at vacancy sites or surface hydroxyl sites of pristine NiFe-LDH, successfully yielding three Ru SACs (Ru1/LDH-VIII, Ru1/LDH-VII, Ru1/LDH) with distinctly different coordination environments, establishing a foundation for precise coordination structure control [Figure 10A].
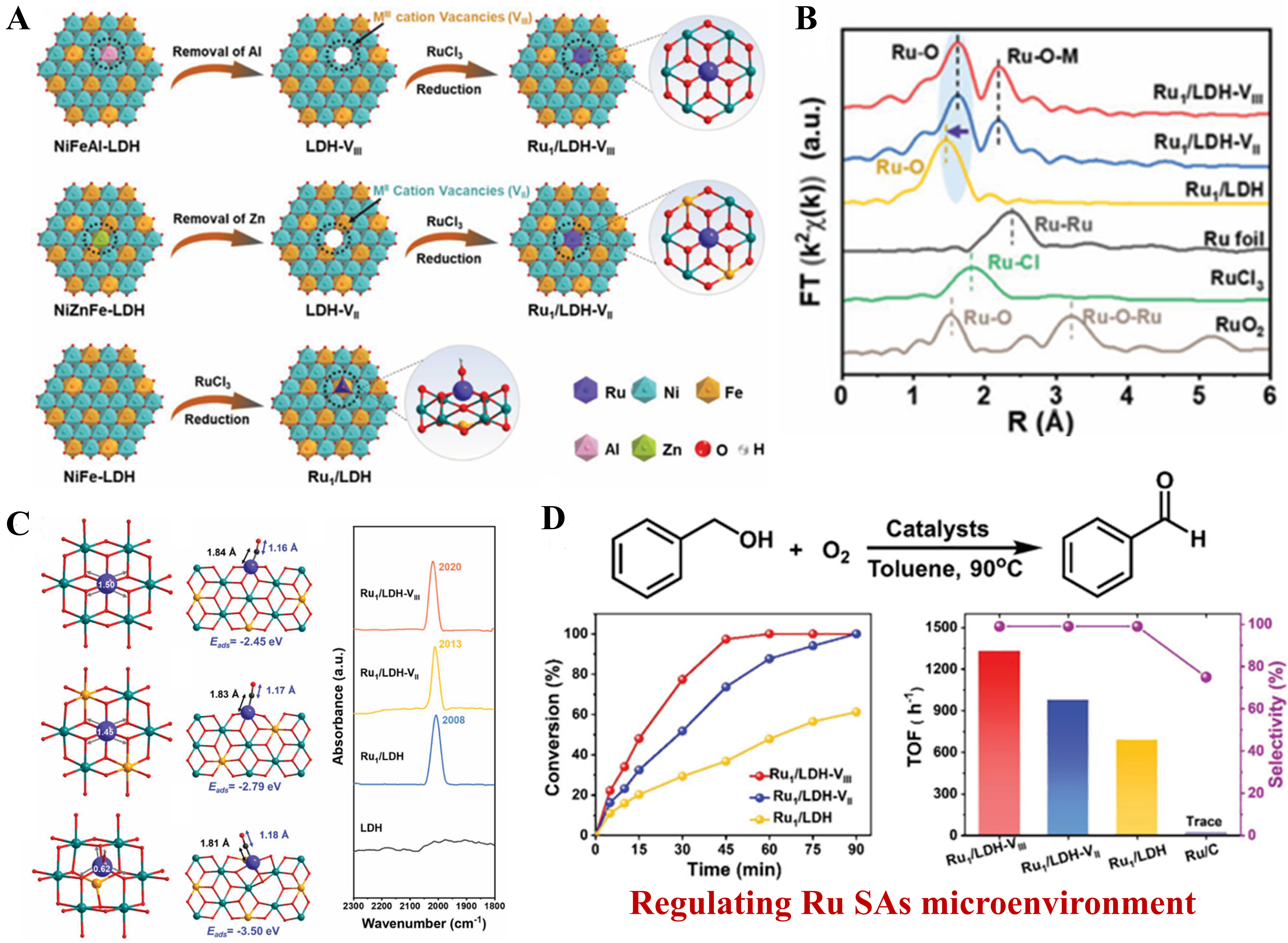
Figure 10. (A) Schematic of Ru1/LDH-VIII, Ru1/LDH-VII, Ru1/LDH synthesis via defect engineering; (B) FT-EXAFS spectra of Ru1/LDH-VIII/VII, and refs (Ru foil, RuCl3, RuO2); (C) Electron transfer between Ru and NiFe-LDH (MIII/MII vacancies or none); CO adsorption on (110) surfaces of Ru1/LDH-VIII/VII; in situ FT-IR of saturated CO chemisorption; (D) Aerobic oxidation of benzyl alcohol (chemical equation) and plots of conversion-time, TOF, and selectivity. (A-D) Reprinted with permission from Ref. [55]. Copyright 2021, Wiley-VCH. LDH: Layered double hydroxide; FT-EXAFS: Fourier-transform extend X-ray absorption fine structure; FT-IR: Fourier transform infrared spectroscopy; TOF: turnover frequency.
Differences in coordination structures are central to regulating catalytic performance. Both Ru1/LDH-VIII and Ru1/LDH-VII exhibit strong Ru-O coordination peaks at 1.62 Å, with Ru-O-M (M = Ni/Fe) secondary coordination peaks detected at 2.2 Å[55]. This indicates Ru SAs are embedded within LDH layers to form a hexacoordinated configuration (Ru-O6), where Ru1/LDH-VIII exhibits a single Ru-O-Ni secondary coordination, while Ru1/LDH-VII shows coexisting Ru-O-Ni/Fe coordination; in contrast, the Ru-O peak of Ru1/LDH appears at 1.44 Å without a secondary coordination peak, confirming its surface tetracoordinate adsorption configuration (Ru-O4). This difference in coordination number and coordinating atom directly modulates the electronic state of the SA [Figure 10B]. The electronic structure changes further validate the coordination engineering effect. XPS and DFT calculations reveal that in the hexacoordinated Ru1/LDH-VIII, the Ru 3p3/2 binding energy reaches 463.6 eV in the Ru1/LDH-VIII system, exhibiting a +4 oxidation state and transferring 1.50 e- to the carrier[55]. This is significantly higher than in the tetracoordinate Ru1/LDH system (463.1 eV, 0 ~ +3 oxidation state, transferring 0.62 e-). In the CO chemisorption Fourier transform infrared spectroscopy (FT-IR) spectrum, the characteristic peak of Ru1/LDH-VIII appears at 2,020 cm-1, indicating the lowest d orbital electron occupancy. This shift originates from the precise modulation of the Ru atom’s electron density by the hexacoordinated environment [Figure 10C]. Catalytic performance testing directly validated the coordination engineering. With the selective aerobic oxidation of benzyl alcohol employed as the probe reaction, the hexacoordinated Ru1/LDH-VIII catalyst delivered a benzyl alcohol conversion of 99.0% and a turnover frequency (TOF) of 1,331 h-1 under the reaction conditions of toluene as solvent (3 mL), a Ru-to-benzyl alcohol molar ratio of 1:500, 1 bar O2 pressure and 90 °C reaction temperature, which was significantly superior to the performance of Ru1/LDH-VII (87.7% conversion, 976 h-1 TOF) and Ru1/LDH (47.9% conversion, 659 h-1 TOF)[55]. Notably, all three Ru SAC systems achieved nearly 100% selectivity for the target product benzaldehyde in this reaction. DFT calculations confirmed that the hexacoordinated Ru-O-Ni environment reduced the benzaldehyde desorption energy barrier to 1.20 eV, whereas the tetracoordinated environment exhibited a barrier as high as 1.91 eV, highlighting the decisive role of coordination geometry in catalytic kinetics [Figure 10D].
Vacancy engineering can be synergistically combined with LDH size control to tailor coordination configurations suitable for photocatalytic applications. Yu et al. extended vacancy-mediated coordination to ultrathin LDHs, anchoring Ru at Al vacancies in ultrathin CoAl-LDH (~ 1.0 wt.% Ru) via formamide-assisted hydrothermal synthesis and wet impregnation, thereby preparing a Ru SAC[102]. The exfoliation process of ultrathin LDHs preferentially generates Al vacancies (whose formation energy is lower than that of Co vacancies); the Ru SA forms an unsaturated Ru-O5 coordination with neighboring oxygen atoms, thereby avoiding the aggregation phenomenon observed in bulk LDH-supported Ru catalysts. This coordination environment enhances the adsorption and activation of CO2 and stabilizes the COOH* intermediate, thereby lowering the protonation energy barrier for photocatalytic CO2 reduction.
Doping-induced symmetry breaking has emerged as an innovative coordination engineering strategy for creating unsaturated active sites for electrocatalysis. Mu et al. employed symmetry-breaking doping as a coordination control strategy, anchoring Ru SAs onto FeCo-LDH via a mixed-solvent method (RuxSACs@FeCo-LDH), thereby breaking the symmetry inherent in the ordered atomic arrangement of FeCo-LDH[103]. Ru doping at the symmetry-broken interface induced the in situ formation of Ru-O-TM (Fe/Co/Ni) nanocomposites, in which Ru SAs in an unsaturated coordination environment facilitated O-O coupling in the OER reaction. X-ray absorption near edge structure (XANES) and EXAFS analyses confirmed the positive oxidation state of Ru (0 < δ < 4) and the absence of Ru-Ru bonds, whilst DFT calculations indicated that this symmetry-broken structure optimizes the adsorption of intermediates by modulating the d-band centers of the active sites.
Axial ligand modification provides a method for precisely regulating the microenvironment of single-atom coordination without altering the main coordination shell. By engineering the axial ligands to finely tune the coordination microenvironment, Zhang et al. utilized electrodeposition and irradiation-assisted ligand exchange to prepare Pt SACs on NiFe-LDH (X-Pt/LDH, where X = -F, -Cl, -Br, -I, -OH)[104]. The Pt SA forms a tetrahedral coordination with three surface oxygen atoms of the LDH and one axial ligand; the electronegativity of the ligand modulates the oxidation state of Pt (2.08 for HO-Pt/LDH and 2.78 for Cl-Pt/LDH) and the d-band center. DFT calculations indicate that Cl- is the optimal axial ligand, as it optimizes the adsorption energies of *H and *OH, thereby accelerating hydrolysis in the alkaline HER while maintaining the dispersion of the Pt atoms.
Extending coordination engineering to defective LDH supports, Zhai et al. stabilized Ru SAs on defective NiFe LDH (Ru1/D-NiFe LDH) via electrodeposition and etching, where defects provide unsaturated coordination sites to strengthen Ru-O bonding. This coordination environment optimizes H adsorption energies for HER and promotes O-O coupling for OER, enabling an ultralow HER overpotential of 18 mV at 10 mA cm-2[42]. Focusing on heterointerface coordination, Dao et al. fabricated defect-rich Ru-doped NiFe LDH on NiCo2O4 nanowires (def-Ru-NiFe LDH/NCO), where Ru SAs coordinate with oxygen atoms at the heterointerface[105]. This unique coordination structure induces electronic redistribution, reduces charge transfer resistance, and lowers the OER overpotential to 225 mV at 10 mA cm-2, with excellent stability over 60 h of operation. Targeting Zn-air battery applications, Hu et al. anchored Ru SAs on defective NiFe LDH (Ru SAs/D-NiFe LDH@NF) via hydrothermal synthesis and etching[41]. The Ru-O coordination at defect sites regulates electron distribution near vacancies, optimizing the formation of OER intermediates and endowing the catalyst with a maximum power density of 170 mW cm-2 and 350 h cycling stability in Zn-air batteries.
Using LDH as a catalyst support, precise modulation of electronic structures is achieved by controlling the coordination environment of single-atom and dual-atom catalysts, thereby optimizing the adsorption - desorption behavior of electrocatalytic reaction intermediates. By leveraging the phase structure of LDHs to modulate the coordination environment, Hu et al. developed a Ru SAC supported on a hybrid amorphous/crystalline FeCoNi LDH[45]. The amorphous outer layer of LDH provides abundant defects and unsaturated coordination sites to stabilize Ru SAs, while the crystalline inner layer ensures carrier rigidity. Electron rearrangement occurs between Ru and Fe/Co/Ni (Ru attracts electrons from Co/Ni and transfers electrons to Fe), lowering the energy barrier for the RDS of OER (Ru-O* formation).
Targeting the coordination advantages of heterointerfaces, Wu et al. anchored Ir single atoms at the NiFe LDH/NiMo heterointerface[106]. Ir is stabilized through dual Ir-O and Ir-Ni coordination, with its electronic modulation optimizing adsorption strength for key HER/OER intermediates. This catalyst exhibits structural stability in both alkaline water and seawater, as the heterointerface coordination environment effectively suppresses Ir aggregation and dissolution. Employing an innovative dual-single-atom coordination strategy, Liu et al. constructed a CoFe-LDH-based catalyst by anchoring Ru/Ir dual single atoms via oxygen vacancies[107]. Oxygen vacancies provide stable coordination sites for the dual single atoms. Synergistic electronic regulation between Ru and Ir brings their d-band centers closer to the Fermi level, balancing the adsorption-desorption kinetics of OER intermediates. Electrons transfer from CoFe-LDH to Ru/Ir, forming stable metal-oxygen coordination structures.
By employing LDH as a catalyst support, the coordination environment between single atoms and LDH can be precisely regulated via the construction of specific coordination bonds[51,53]. This enables precise modulation of the electronic structure at active sites, thereby optimizing the adsorption behavior of electrocatalytic reaction intermediates. The core mechanism relies on the atomic-level control capabilities of coordination engineering.
Focusing on coordination bond regulation and electronic interactions between single atoms and LDH, Hu et al. synthesized Ir single-atom/NiFe LDH systems (IrSAC-NiFe-LDH and NiIrSAA-NiFe-LDH)[57]. Analysis of charge density differences clearly reveals electron transfer patterns. In NiIrSAA-NiFe-LDH, Ir atoms exhibit electron enrichment (-0.15 e-), while Ni atoms show electron deficiency (0.15 e-), confirming directed electron transfer from Ni to Ir [Figure 11A]. Conversely, in IrSAC-NiFe-LDH, both Ir and its neighboring Ni atoms lose electrons, which accumulate toward oxygen species, forming a unique electron redistribution pattern [Figure 11B]. Based on electronic structure characteristics, adsorption site preferences were further clarified: Ni sites in NiIrSAA-NiFe-LDH are the preferred adsorption sites for HER. In IrSAC-NiFe-LDH, Ni sites adjacent to Ir are the optimal adsorption sites for OER, with the coordination environment determining the functional division of active centers [Figure 11C]. Free energy calculations quantified this coordination-driven effect: NiIrSAA-NiFe-LDH exhibits hydrogen adsorption free energy (-0.17 eV) closer to the ideal value, while Ir SAC-NiFe-LDH reduces the energy barrier at the RDS for OER. Both improvements stem from coordination-induced optimization of electronic states [Figure 11D].
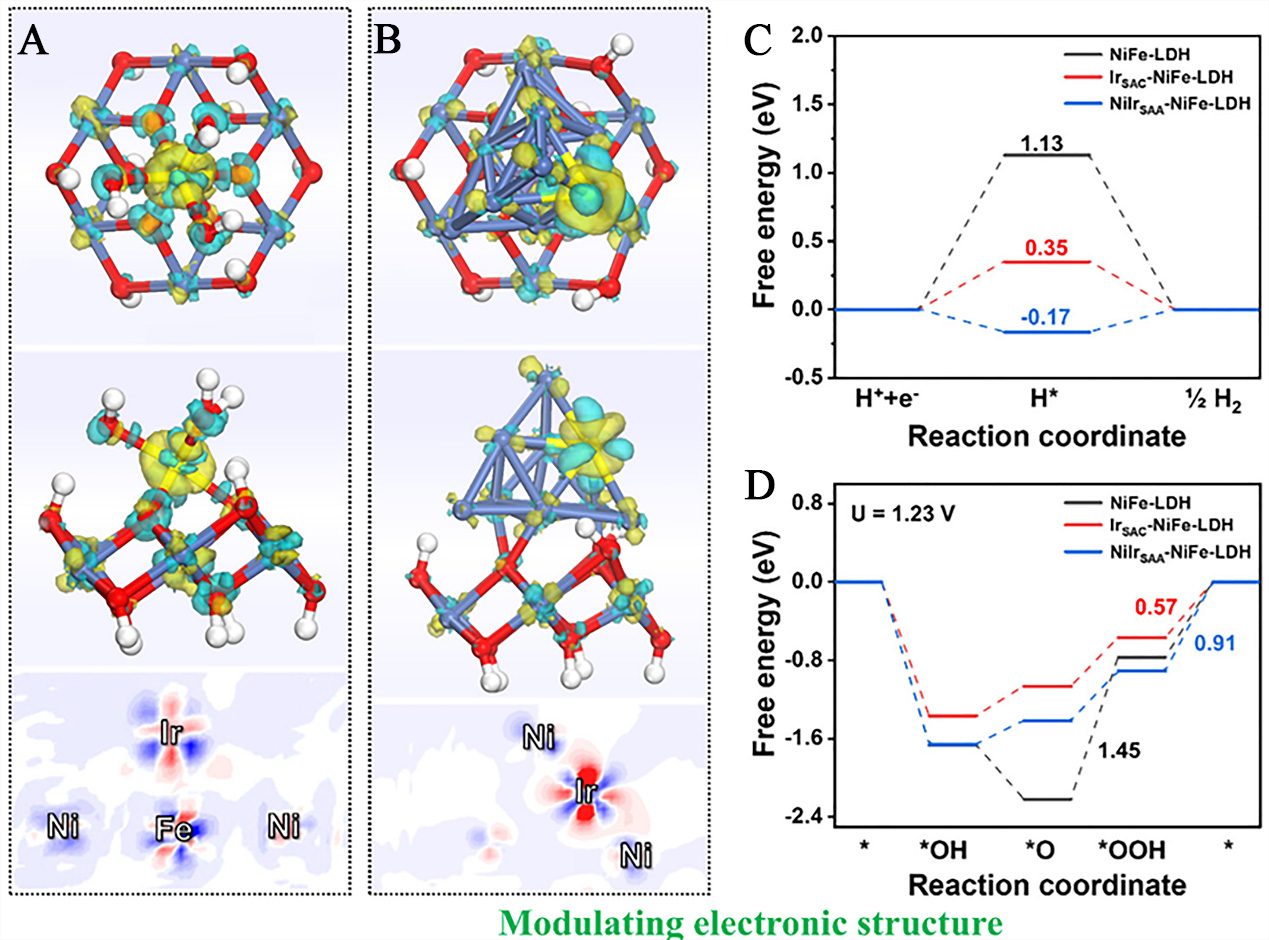
Figure 11. Top and side views of differential charge densities of (A) NilrSAA-NiFe-LDH and (B) lrSAC-NiFe-LDH. Gibbs free energy dagrams for (c) the HER at U = 0 and (D) OER at U =1.23 V on NilrSAA-NiFe-LDH, lrSAC-NiFe-LDH, and NiFe-LDH. (A-D) Reprinted with permission from Ref. [57]. Copyright 2023, American Chemical Society. LDH: Layered double hydroxide; SAA: single-atom alloy; SAC: single-atom catalyst; HER: hydrogen evolution reaction; OER: oxygen evolution reaction.
Extending the coordination engineering strategy of Pt and LDH, Hu et al. constructed the ePt/NiFe LDH@e-nf catalyst via electrodeposition[53]. This unique Pt-Ni/Fe coordination bond replaces the conventional Pt-O-M bond, enabling direct electronic regulation of the metal centers. This coordination pattern elevates the valence state of Ni/Fe while optimizing the electron density at Pt sites. Electron transfer from Ni/Fe to Pt enhances interactions between intermediates and active sites, aligning closely with the core logic of Pt-Ni coordination-driven electronic structure regulation.
Building upon defect-assisted coordination innovation, Shen et al. designed a Pt SAC supported on oxygen-defect-doped NiFe LDH (D-NiFe-LDH)[58]. By substituting conventional Pt-O bonds with Pt-Ni electronic bridges, oxygen defects further promote electron transfer from Ni to Pt, inducing Pt to adopt a negative valence state. This coordination environment regulation not only optimizes electron distribution but also shifts the d-band center position of Pt, weakening hydrogen adsorption strength. Its mechanism aligns with the Pt-Ni/Fe coordination electron regulation logic, representing an extension of Pt-LDH coordination engineering exploration.
Focusing on multi-coordination regulation of Ir with LDH, Duan et al. proposed a dynamic Cl- adsorption-assisted coordination strategy, forming unique Ir-OH/Cl mixed coordination in
Focusing on single-atom-cluster synergy and intra-layer coordination strategies, Tian et al. precisely introduced Pt SAs and Pt clusters into NiCo LDO layers via a dual-ion etching-phase transition method[66]. Pt SAs insert into LDO layers, occupying partial Ni sites to form Pt-Co bonds. This synergizes with surface Pt clusters, enhancing Pt-NiCo LDO electronic interactions. It promotes preferential adsorption of H* species onto O sites above Pt SAs, accelerating water-splitting kinetics in the Volmer step. The core mechanism lies in intra-layer coordination and synergistic electronic regulation between single atoms and clusters. Leveraging the unique advantages of interfacial coordination engineering, Wu et al. anchored Ir single atoms at the NiFe LDH/NiMo interface to construct a bifunctional catalyst[106]. The Ir single atom is stabilized by dual Ir-O and Ir-Ni coordination, inducing electron transfer from NiMo to NiFe LDH while establishing directed electron redistribution among Ir, Ni, and Fe sites. This precisely optimizes the adsorption strength of key HER/OER intermediates, with the core mechanism originating from heterointerfacial coordination-induced multi-component electronic synergy modulation.
Microenvironment confinement-mass transfer
Beyond simply anchoring active sites, the true power of LDHs as an ‘atomic canvas’ lies in their ability to orchestrate both the local chemical environment of single atoms and the macroscopic transport of reactants and products. Microenvironment confinement-mass transfer synergy has thus emerged as a key strategy for reconstructing catalytic interfaces and unlocking multifunctional performance in LDH-based catalysts[109,110]. This dual-control paradigm leverages the structural versatility of LDHs. On one hand, their designable architectures (e.g., monolayer nanosheets, porous frameworks, and heterojunction couplings) can be engineered to optimize mass transport pathways, reduce diffusion resistance, and expose abundant accessible surfaces. On the other hand, their unique layered and confined spaces (e.g., interlayer galleries, defect sites, and interfacial domains) enable precise tailoring of the local microenvironment surrounding the active sites, including coordination geometry, electronic density, and intermediate stabilization[111,112]. By synergistically integrating confinement effects (which stabilize single atoms and modulate reaction pathways) with optimized mass transfer (which ensures efficient supply of reactants and removal of products), this approach significantly enhances catalytic kinetics, selectivity, and long-term stability, as evidenced by a growing body of representative studies.
Wang et al. loaded single-atom Ru (Ru1/mono-NiFe) onto monolayer NiFe LDH[113]. Transmission electron microscopy (TEM) images reveal that both monolayer NiFe and Ru1/mono-NiFe exhibit uniform monolayer nanosheet morphology with lateral dimensions of approximately 30 nm and thickness of about 1 nm, retaining the layered structure of LDH after ruthenium anchoring [Figure 12A and B][113]. High-resolution transmission electron microscopy (HRTEM) confirmed a lattice fringe spacing of 0.15 nm, corresponding to the (110) plane of LDH, verifying the structural integrity of the support [Figure 12C]. HAADF-STEM revealed uniformly distributed isolated bright spots on the monolayer LDH surface, confirming atomically dispersed Ru without aggregation [Figure 12D].
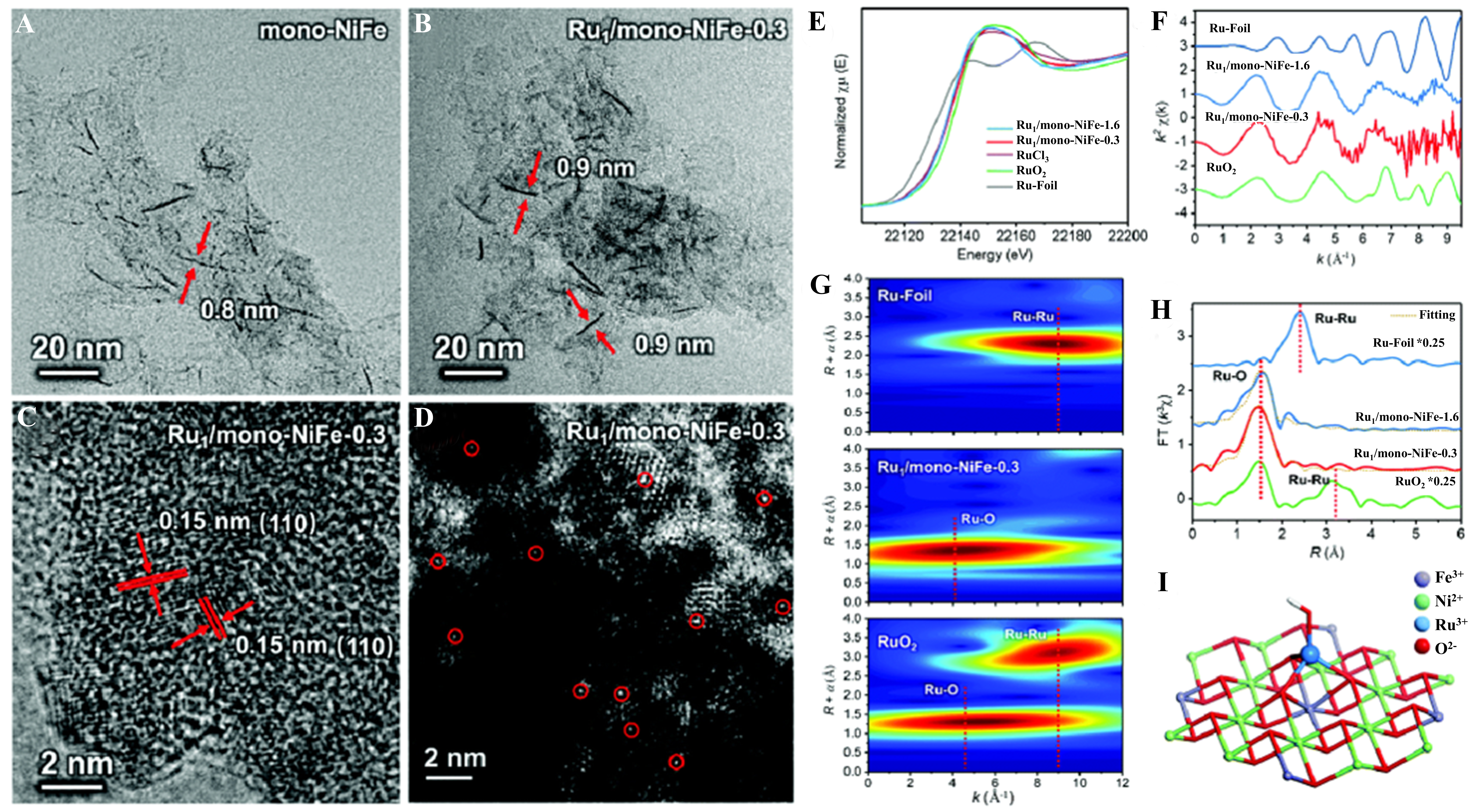
Figure 12. TEM images of (A) mono-NiFe and (B) Ru1/mono-NiFe-0.3; (C) HRTEM image of Ru1/mono-NiFe-0.3; (D) HAADF-STEM image of Ru1/mono-NiFe-0.3; (E) The Ru K-edge XANES spectra; (F) the Ru K-edge EXAFS k2χ functions for Ru1/mono-NiFe-1.6, Ru1/mono-NiFe-0.3, RuCl3, RuO2 and Ru Foil; (G) the wavelet transforms for the k2-weighted EXAFS signals of Ru1/mono-NiFe-0.3, RuO2 and Ru Foil; (H) the magnitude of k2-weighted Fourier transforms of the Ru K-edge EXAFS spectra for Ru1/mono-NiFe-0.3, Ru1/mono-NiFe-1.6, RuO2 and Ru Foil; (I) schematic illustration of Ru1/mono-NiFe-x. (A-I) Reprinted with permission from Ref.[113] Copyright 2019, The Royal Society of Chemistry. TEM: Transmission electron microscopy; HRTEM: high-resolution transmission electron microscopy; HAADF-STEM: high-angle annular dark-field scanning transmission electron microscopy; XANES: X-ray absorption near edge structure; EXAFS: extend X-ray absorption fine structure.
To characterize the microenvironment and electronic structure of Ru SAs, the XANES and extended EXAFS analyses were performed. The Ru K-edge XANES spectrum of the single-atom NiFe system exhibits an absorption position consistent with RuCl3, indicating Ru exists in the +3 oxidation state [Figure 12E]. The k²-weighted EXAFS oscillation spectrum of the single-atom NiFe system shows significant differences compared to Ru foil and RuO2, reflecting its unique coordination environment [Figure 12F]. Wavelet transform (WT) analysis of the EXAFS signal reveals only characteristic peaks, indicating the Ru atom possesses a distinctive coordination environment. WT analysis reveals that the EXAFS signal exhibits only a characteristic Ru-O shell peak at ~ 1.5 Å, with no Ru-Ru bond signal detected, further confirming the single-atom dispersion [Figure 12G]. The FT-EXAFS spectrum exhibits only a Ru-O peak at ~ 1.5 Å without a metallic Ru-Ru peak, validating the Ru-O coordination configuration [Figure 12H]. A schematic summarizes the precise positioning of the Ru SA: anchored atop the Fe atom by three oxygen atoms, forming a well-defined coordination microenvironment [Figure 12I]. This microenvironmental constraint (precise Ru-O-Fe coordination) stabilizes the hydrazine electrooxidation intermediates (*N2H3, *N2H) while reducing the bandgap energy. Concurrently, the monolayer LDH structure and high specific surface area (~ 358 m2 g-1) optimize mass transfer between reactants and products. The synergistic effect of microenvironment confinement and enhanced mass transfer endows the Ru1/mono NiFe catalyst with exceptional activity and stability.
Monolayer LDHs offer unique advantages for microenvironment regulation and mass transfer optimization, with heteroatom doping or single-atom anchoring further enhancing catalytic microenvironment modulation. Zeng et al. anchored Ce single atoms on monolayer NiV LDH (Ce SAs/m-NiV LDH) via vanadium vacancy trapping during hydrothermal synthesis, forming stable Ce-O-Ni coordination configurations[114]. The monolayer structure reduces mass transfer resistance and exposes abundant active sites, while Ce single atoms narrow the LDH bandgap and strengthen density of states near the Fermi level, synergistically optimizing charge transfer and OH- adsorption for OER. Wang et al. decorated non-precious Na single atoms on monolayer NiFe-LDH (Na/NiFe-LDH) via electrochemical insertion, constructing unique Na-O3 coordination motifs on the LDH surface[115]. The monolayer microenvironment facilitates rapid electrolyte diffusion, while Na single atoms induce spin-state inversion of Fe 3d orbitals by modulating Fe-O bond strength, optimizing OER intermediate adsorption kinetics without altering intrinsic Fe active sites.
Sun et al. constructed Ni SACs using organically intercalated LDH as a precursor[63]. The intercalated microenvironment induces uniform dispersion of Ni active sites, while the hierarchical porous structure of carbon nanosheets enhances mass transfer, significantly improving 2e- ORR performance and boosting a substantial increase in H2O2 yield. Liu et al. fabricated NiFe-LDH/Fe1-N-C heterostructured hollow nanorods, where the hollow microenvironment reduced mass transfer resistance, while interfacial microenvironment regulation (electron transfer from LDH to Fe1-N-C) optimized ORR/OER intermediate adsorption, achieving dual-function catalytic performance for ZABs[96]. Focusing on ‘sesame ball’-like heterostructure design, Lu et al. anchored ultrasmall FeNi-LDH nanoparticles onto ZIF-8-derived FeNi-DACs[62]. The porous carbon framework of DACs not only provides spatial confinement for LDH growth (inhibiting agglomeration and exposing more active sites) but also constructs a unique microenvironment through interfacial electronic coupling - transferring approximately 0.61 e- from LDH to DACs modulates the d-band center of FeNi atom pairs, optimizing adsorption energy for ORR/OER intermediates. This architecture simultaneously addresses the insufficient conductivity of LDH and the weak OER activity of DACs. The porous structure shortens mass transport pathways and enhances charge transfer efficiency, enabling the catalyst to exhibit a peak power density of 211.6 mW cm-2 and long-term cycling stability of 500 h in ZABs, significantly outperforming precious metal composite catalysts.
LDHs enable precise control over the microenvironment of active sites (e.g., coordination structure, defect distribution) via two-dimensional interlayer confinement, three-dimensional hollow confinement, or synergistic confinement - mass transfer strategies. Simultaneously, optimizing reactant/product diffusion efficiency enables synergistic enhancement of catalytic performance.
Two-dimensional interlayer confinement: Atomic-scale spatial constraints and microenvironment customization. Focusing on the two-dimensional confinement effect within LDH layers, atomic-scale constrained spaces are constructed via ion exchange or intercalation strategies to stabilize active sites and regulate reaction microenvironments. Chen et al. developed 5, 10, 15, 20-tetrakis(4-carboxyphenyl) porphyrin Co(II) (TCPP-Co) intercalated MgAl-LDH-supported Co SACs [Figure 13A][116]. The positively charged LDH layers stabilize negatively charged peroxymonosulfate (PMS) ions. The interlayer confinement space induces selective generation of surface-bound ·OH and SO4·- radicals, suppressing radical self-quenching and PMS decomposition while shortening the diffusion distance between pollutants and radicals. Co atom is anchored via Co-N4 coordination. The two-dimensional confinement of LDH both fixes the active site and optimizes mass transfer efficiency, enabling efficient degradation of organic pollutants[116]. Extending the concept of precise regulation through two-dimensional intercalation confinement, Fan et al. proposed a confinement synthesis strategy for LDH co-intercalated with organic molecules[68]. Using CoAl-LDH as a precursor, they co-intercalated m-aminobenzoic acid (MA) and PA. MA provided N species to form Co-N active sites, while PA regulated pore structure to enhance active site exposure, while the two-dimensional interlayer confinement suppresses Co atom agglomeration, achieving uniform single-atom dispersion. Leveraging the advantages of flexible interlayer confinement in LDH, Yang et al. intercalated heme (Hm) between MgAl-LDH layers[67]. High-temperature carbonization yielded the Fe-N-C catalyst (Hm-LDH-700). The two-dimensional interlayer space of LDH restricts heme aggregation. After carbonization, a graphene-like two-dimensional porous structure with a specific surface area of 1,065.79 m2 g-1 is formed. Hierarchical pore channels accelerate O2 transport, uniformly dispersing Fe-N4 active sites and significantly enhancing ORR catalytic performance. This represents an extended application of the two-dimensional confinement strategy. Interlayer confinement can be synergistically combined with LDH’s ‘memory effect’ to construct catalytic membranes, realizing convection-enhanced mass transfer for pollutant purification. Wang et al. embedded Fe single atoms into MgAl-LDH interlayers via the ‘memory effect’ (calcination-reconstruction) and fabricated a catalytic membrane (FeSA-in-LDH/M), with Fe single atoms stabilized as Fe-N4 coordination configurations[117]. The two-dimensional interlayer confinement enriches phenoxyl radicals and PMS intermediates, while the membrane’s flow-through mode compresses the diffusion boundary layer, achieving convection-enhanced mass transfer that promotes pollutant polymerization and separation.
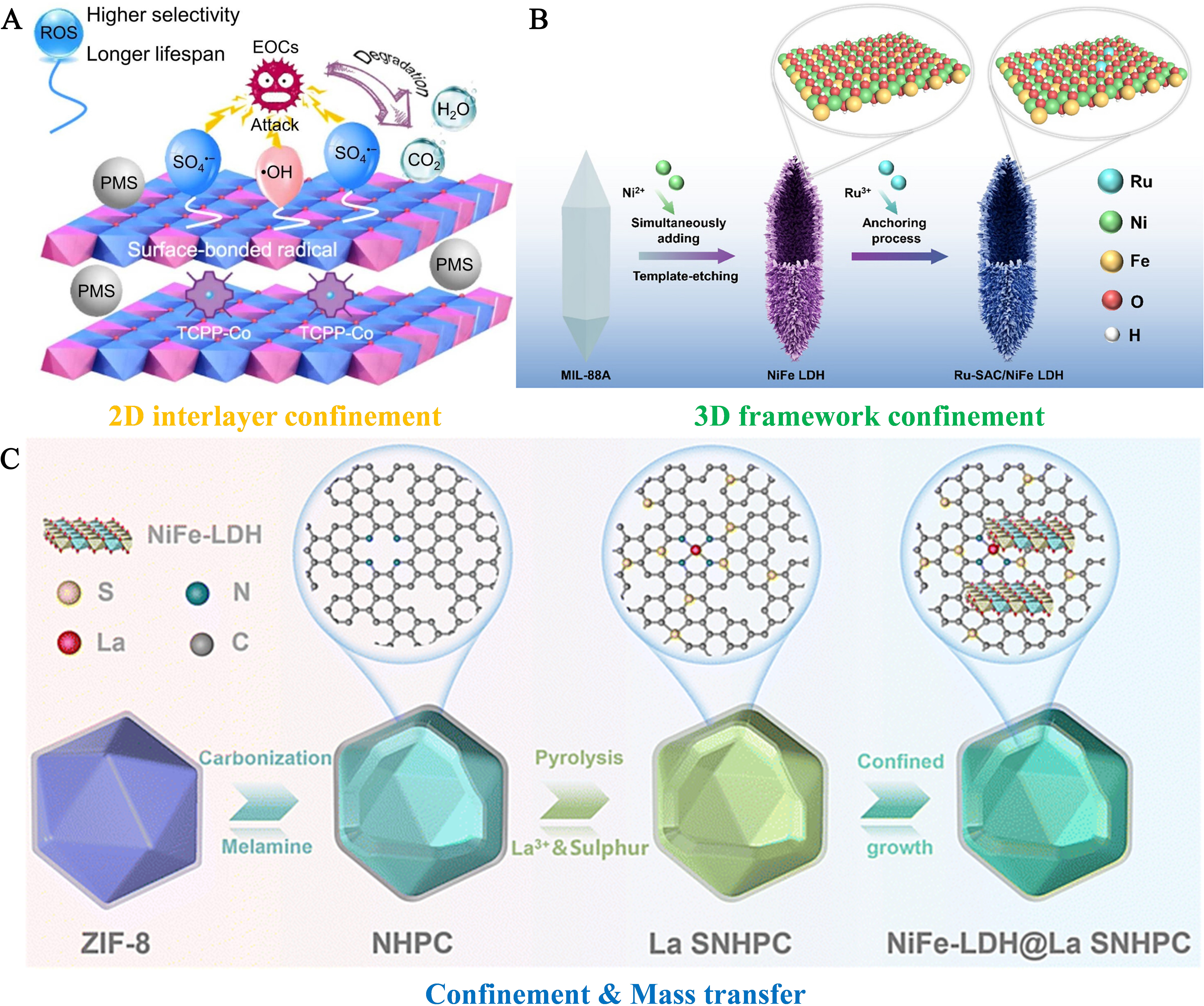
Figure 13. (A) In layered double hydroxides, single-atom covalent sites can be activated by persulfate to selectively generate surface-bound free radicals that degrade EOCs. Reprinted with permission from Ref.[116]. Copyright 2024, Elsevier; (B) Schematic illustration for the fabrication of Ru-SAC/NiFe LDH Reprinted with permission from Ref.[46]. Copyright 2025, Wiley-VCH; (C) Schematic illustration of the synthesis route to NiFe-LDH@La@SNHPC. Reprinted with permission from Ref.[61]. Copyright 2025, The Royal Society of Chemistry. 2D: Two-dimensional; 3D: three-dimensional; ROS: reactive oxygen species; PMS: peroxymonosulfate; LDH: layered double hydroxide; ZIF: zeolitic imidazolate framework; NHPC: N-doped hollow porous carbon; SNHPC: S/N co-doped hollow hierarchical porous carbon; EOC: emerging organic contaminant.
Three-Dimensional Hollow Confinement, Macro-Structural Design and Mass Transfer Optimization. Leveraging LDH’s topological transformation properties, a three-dimensional hollow structure was engineered. Spatial confinement stabilizes active sites while shortening mass transfer pathways and reducing diffusion resistance. Israr et al. synthesized Ru SACs supported on NiFe LDH hollow nanocages (Ru-SAC/NiFe LDH) via MIL-88A template etching-coprecipitation [Figure 13B][46]. The three-dimensional hollow nanocage provides abundant mesopores (~ 2.19 nm) and a large specific surface area (~ 157.5 m2 g-1). Ru atoms are anchored via Ru-O-M (M = Ni/Fe) bonds on cage surfaces. The hollow structure reduces reactant diffusion resistance, while electron transfer from Ni/Fe stabilizes Ru’s +3.7 oxidation state, synergistically enhancing OER kinetics.
Synergy of confinement and mass transfer, multidimensional regulation for synergistic enhancement. Integrating confinement regulation with mass transfer optimization through multi-level structural design achieves dual enhancement of microenvironment control and mass transfer efficiency. Yin et al. designed a nanoreactor [NiFe-LDH@La S/N co-doped hollow hierarchical porous carbon (SNHPC)] featuring hollow porous carbon loaded with La single atoms and NiFe-LDH nanoplates, corresponding to the structural concept in Figure 13C[61]. The hollow cavities and multi-level channels of the carbon carrier enrich local OH- concentration and enhance electrostatic field strength. La atoms are coordinatively anchored via La-N4 bonds, forming a localized coexistence structure with NiFe-LDH nanoparticles. The confinement effect modulates electronic structure and lowers the OER rate-limiting step energy barrier, while the porous structure accelerates mass transfer at the gas-liquid-solid triple phase boundary. This catalyst achieves 350 h long-term cycling stability in ZABs. Focusing on a systematic summary of confinement mechanisms, Fan et al. provide a review that systematically outlines synthesis strategies for LDH-based confined SACs (spatial, coordination, and ionic confinement)[110]. They clarify that confinement methods such as two-dimensional interlayer and three-dimensional hollow structures stabilize single atoms through size effects and electronic effects while optimizing mass transfer, providing theoretical support for the application of various confinement strategies.
Interfacial synergy engineering: enabling functional division of labor and relay catalysis
Interface design serves as a core strategy by which LDH supports enable the rational reconstruction of catalytic systems and unlock multifunctional catalytic performance. Single active sites are often inadequate to address the multi-step demands of complex reactions[118-121]. By strategically constructing interfaces between LDHs and various active components (single atoms, clusters, etc.), interfacial synergy engineering creates systems where multiple sites cooperate through functional division of labor and relay catalysis. The approach modulates electronic interactions and local microenvironments at the interfaces, allowing distinct sites to specialize in specific reaction steps while working in concert to efficiently drive the overall process[121,122]. This section elaborates on the design principles, mechanistic insights, and catalytic applications of such synergistic interfaces, revealing how this strategy expands the functional potential of LDH-based catalysts.
Using CoV-LDH as the catalyst support, a synergistic three-active-site system was constructed via Pt single-atom anchoring. Leveraging interfacial synergy engineering, functional division and relay catalysis were achieved in alkaline HER: water adsorption and decomposition → H3O+ migration → H2 generation. Li et al. designed Pt single-atom-anchored CoV-LDHs catalysts (Pt/CoV-LDHs)[52]. They synthesized needle-like CoV-LDHs via hydrothermal synthesis, followed by electrochemical deposition to anchor Pt SAs on its surface. This formed triple active sites (V, Co, Pt) enabling a three-step relay reaction for alkaline HER.
The relay catalysis schematic clearly illustrates the functional division among the three active sites: the V site adsorbs water, the Co site synergistically decomposes water and migrates H, while the Pt site generates H2 [Figure 14A]. Computational modeling of Pt/CoV-LDHs confirmed Pt SAs are anchored to the CoV-LDH surface via dioxygen coordination [Figure 14B]. Free energy calculations indicate that the V site (adjacent to an oxygen vacancy) exhibits the lowest water adsorption energy (-0.025 eV), making it the optimal water adsorption site [Figure 14C]. The water splitting energy barrier diagram reveals that the synergistic action of Co and V sites reduces the water splitting energy barrier to only 0.043 eV, making the reaction spontaneous [Figure 14D]. The hydrogen atom migration pathway from the Co site to the Pt site is also depicted [Figure 14E]. The energy diagram confirms the thermodynamic spontaneity of this migration process [Figure 14F]. Hydrogen adsorption energy calculations indicate that the Pt site exhibits an adsorption energy of -0.111 eV, close to the ideal value, making it the primary active site for H2 formation, while the Co site primarily facilitates hydrogen migration [Figure 14G].
Figure 14. (A) Schematic design diagram for the tandem catalysis. (B) Computational model of designed Pt/CoV-LDHs; (C) The H2O adsorption free energy at Pt site and V site on Pt/CoV-LDHs; (D) Water dissociation barrier on catalyst; (E) The migration path of hydrogen, and (F) corresponding energies diagram; (G) Calculated adsorption energies of H on Co1, Co2, and Pt sites. (A-G) Reprinted with permission from Ref.[52]. Copyright 2024, Elsevier. LDH: Layered double hydroxide.
Experimental and theoretical validation demonstrates that the V site adsorbs and activates water via oxygen vacancies, while Co and V synergistically decompose water to form H3O+. The Co site transfers H to the Pt site, where H2 is rapidly generated via the Volmer-Tafel mechanism. In situ Raman spectroscopy confirms the presence of the H3O+ intermediate, establishing a localized acid-like microenvironment that significantly enhances alkaline HER activity.
Using LDHs as catalyst supports, multi-active-site systems can be constructed via interfacial coordination engineering to achieve synergistic effects characterized by functional division and relay catalysis. These include synergy between metal single atoms and acidic sites, alternating synergy between single atoms and LDH supports, and bidirectionally stable synergy between single atoms and LDH-based active sites.
Focusing on the electronic regulation at the interface between LDH-derived carriers and single atoms, a metal-acid dual active site was constructed to achieve efficient relay of multi-step hydrogenation-deoxygenation (HDO) reactions through functional division of labor. Wei et al. used CoAl-LDH as a precursor to prepare a Co2AlO4-supported Pt SAC (0.4%Pt/Co2AlO4) via a topological transformation[123]. They achieved synergistic interaction between Pt SAs and Brønsted acid sites on the carrier surface through interfacial engineering: Pt SAs activate adsorbed 5-hydroxymethylbenzene, while Brønsted acid sites facilitate its oxidation. Through interfacial engineering, Wei et al.[123] achieved synergistic interaction between Pt SAs and Brønsted acid sites on the support surface; Pt atoms activate the C=O bond of adsorbed 5-hydroxymethylfurfural (5-HMF), lowering the energy barrier for the RDS of C-OH bond cleavage, while Brønsted acid sites accelerate the second-step C-OH bond dissociation. This relay mechanism completes the HDO reaction, enabling efficient synthesis of 2,5-dimethylfuran (DMF). This functional division between metal and acid sites exemplifies the application of interfacial synergistic engineering in multi-step organic transformations.
By leveraging the interlayer confinement effect of LDH to anchor single atoms, an alternating synergistic system of single atoms and LDH carriers was constructed. Functional division between HER and OER enhanced overall water splitting performance. Chen et al. employed an anion exchange-spatial confinement strategy to embed Pt SAs into the interlayer of Ni3Fe-LDH (Ni3Fe-CO32--LDH-Pt SA), revealing an alternating synergistic mechanism of ‘single-atom-empowered carrier + carrier-assisted single atom’[48]. HER polarization curves demonstrate that this catalyst achieves 10 mA cm-2 at just 45 mV overpotential, outperforming commercial 20 wt.% Pt/C, clearly illustrating the synergistic enhancement effect between Pt SACs and LDH [Figure 15A]. This enhancement stems from dual effects: Pt SACs significantly boost LDH’s electron transport capacity, while Ni3Fe-LDH accelerates water dissociation, enabling HER to follow a hybrid Heyrovsky-Volmer/Tafel-Volmer mechanism [Figure 15B]. Schematic diagrams clearly illustrate the functional division between single atoms as active sites and LDH as a synergistic carrier [Figure 15C]. For the OER, Pt SACs not only promote the transformation of Ni3Fe-LDH into the active phase Ni2+δFe3+ζOxHy, but also optimize its intrinsic activity. The OER polarization curve reveals an overpotential of only 218 mV at 10 mA cm-2 [Figure 15D]. The mechanism of promoting phase transformation is clarified by schematic diagrams, Pt SACs reduce the phase transition energy barrier by modulating the Ni electron density [Figure 15E]. Differential charge density maps further validate the electronic interaction between Pt and Ni/Fe-O bonds [Figure 15F]. Bidirectionally stable synergy between SACs and LDHs enables active site protection and enhances OER performance.
Figure 15. (A) HER polarization curves in 1 M KOH; (B) Schematic representations of HER mechanism for Pt/C in 1 M KOH and (C) Ni3Fe - CO32- LDH-Pt SA in 1 M KOH; (D) OER polarization curves for Ni3Fe - CO32- LDH and Ni3Fe - CO32- LDH-Pt SA. (E) Schematic of promotion effect on active phase transition for Pt single - atom in Ni3Fe - CO32- LDH; (F) Differential charge densities of Ni3Fe - CO32- LDH-Pt SA. (A-F) Reprinted with permission from Ref.[48]. Copyright 2021, The Royal Society of Chemistry. LDH: Layered double hydroxide; RDS: rate-determining step; HER: hydrogen evolution reaction; OER: oxygen evolution reaction; SA: single atom.
The bidirectional synergistic logic of interface coordination enables mutual stabilization between the single atom and LDH active sites, achieving simultaneous enhancement of catalytic activity and stability. Cao et al. prepared an Ir single-atom-doped CoFe-LDH/rGO catalyst (Ir/CoFe-LDH/rGO) via a hydrothermal-impregnation method[44]. They discovered a bidirectional synergistic effect between Ir SACs and CoFe-LDH, LDH modulates Ir’s electronic state via Ir-O-M (M = Co/Fe) coordination bonds, enhancing its OER activity. Conversely, Ir single atoms stabilize Fe active sites on LDH through two mechanisms: Fe sites adjacent to Ir exhibit reduced activity and enhanced stability due to electronic coupling with Ir, while distant Fe sites experience decreased local OH- concentration caused by Ir’s rapid consumption of OH-, thereby minimizing Fe dissolution and phase transitions. This achieves bidirectional stabilizing synergy between ‘active sites and carrier’, refining the bidirectional enabling mechanism of interfacial synergy.
Dynamic phase engineering: directing in situ reconstruction toward active phases
Dynamic phase engineering represents an advanced paradigm for catalyst design that goes beyond static active-site optimization. This strategy specifically employs LDH supports to direct and regulate the in situ reconstruction of the matrix under catalytic operating conditions. Through targeted doping with single atoms or heteroatoms, the inherent phase transformation of LDHs into more active (oxy)hydroxide phases (e.g., NiFeOOH, FeNiOOH) is not merely allowed but precisely steered[124,125]. The core mechanism hinges on the dopants modulating the host’s electronic structure and lowering the energy barrier for phase transition. This deliberate intervention optimizes the adsorption strength of reaction intermediates on the newly formed active phase, thereby synergistically enhancing both catalytic activity and operational stability.
Focusing on single atoms as ‘phase-transition catalysts’ that guide the in situ reconstruction of LDH into the active NiFeOOH phase, Zhang et al. synthesized single-atom Au-doped NiFe LDH (sAu/NiFe LDH) via a simple strategy, revealing the core mechanism of single-atom-induced dynamic LDH restructuring[126]. They discovered that LDH does not retain its original structure during the OER process. Instead, it undergoes irreversible in situ restructuring induced by single-atom Au, forming a stable NiFeOOH active phase [Figure 16A]. During this reconstruction, the Au atom bonds to LDH via an Au-O bond, transferring 0.32 e- to the Fe active site and surrounding atoms. This regulates charge distribution and optimizes the adsorption energies of OER intermediates (OH*, O*, OOH*) [Figure 16B]. Free energy calculations indicate that the overpotential for the RDS (O* → OOH*) on the reconstructed sAu/NiFeOOH-NiFe LDH is only 0.18 V, significantly lower than that of the unreconstructed NiFe LDH (0.26 V). Experimentally, this catalyst exhibits an overpotential of 237 mV at 10 mA cm-2, demonstrating a sixfold increase in activity with no significant decay after 2,000 cycles [Figure 16C].
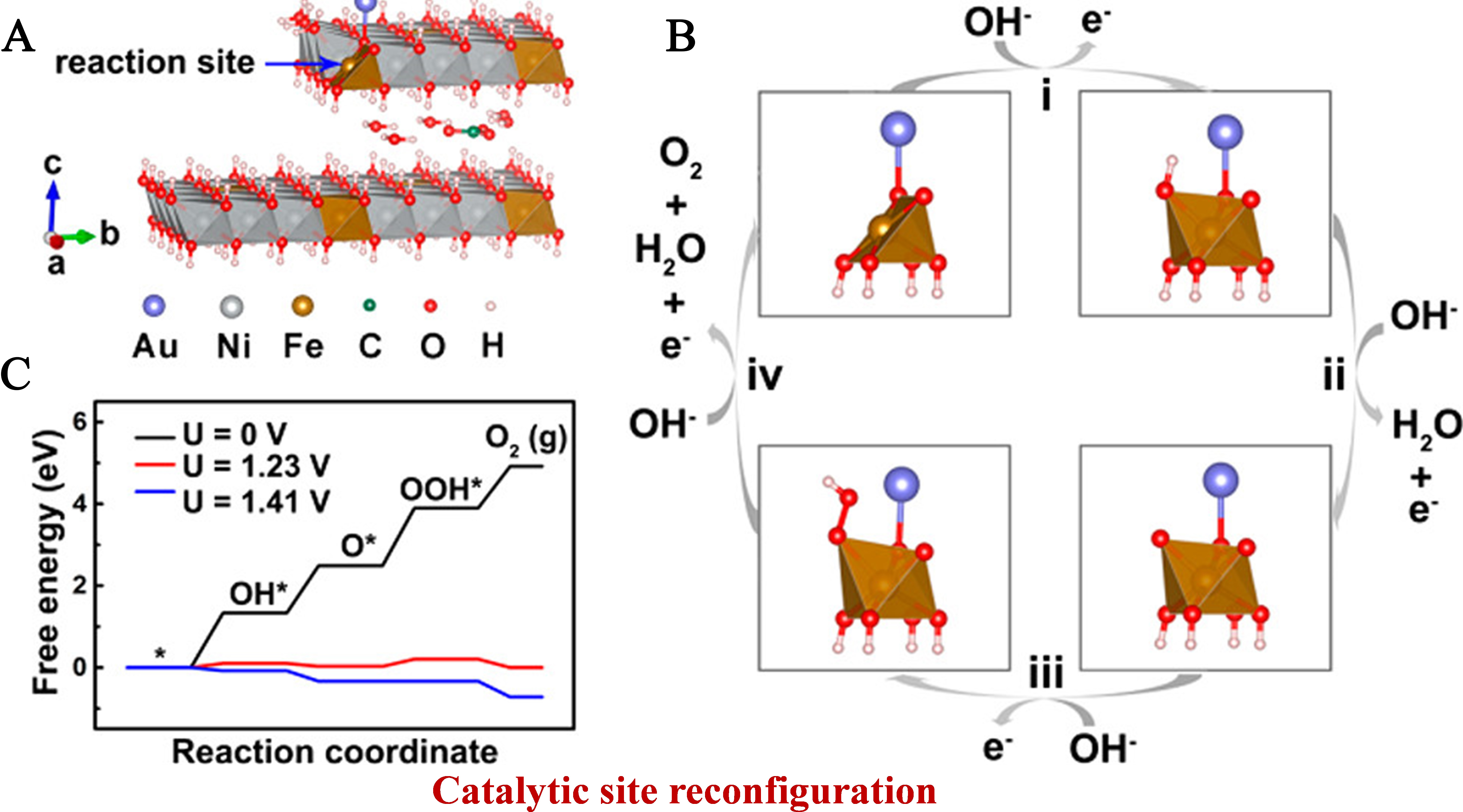
Figure 16. (A) Two-layer slab model for sAu/NiFe LDH with interlayer CO32- anions and water molecules; (B) Proposed OER pathways with OH*, O* and OOH* intermediates for sAu/NiFe LDH; (C) Free energy diagram for the OER at different potentials on the surface of sAu/NiFe LDH model. (A-C) Reprinted with permission from Ref.[126]. Copyright 2018, American Chemical Society. LDH: Layered double hydroxide; OER: oxygen evolution reaction; sAu/NiFe LDH: single-atom Au-doped NiFe LDH.
Single-Atom Ir-Induced LDH Reconfiguration, Directional Formation of Active Sites and Carrier Activation. Building upon the logic of single-atom-induced reconfiguration, Li et al. employed an in situ electrostatic adsorption anchoring strategy to load Ir single atoms onto NiFe LDH (Ir/NiFe LDH)[127]. They discovered that the Ir SACs (with a +5.5 oxidation state) bind to LDH via Ir-O bonds, not only activating the inert LDH carrier but also guiding LDH to undergo in situ reconstruction into the Ni2+δFe3+ζOxHy active phase during the OER process. During this reconstruction, bidirectional electron transfer occurs between Ir and Ni/Fe, shifting Ni’s binding energy positively and Fe’s negatively. This optimizes the d-band center and lowers the energy barrier of the RDS in OER. In situ Raman spectroscopy confirms the formation of the active phase. X-ray absorption spectroscopy (XAS) and TEM characterization reveal uniform dispersion of Ir single atoms without agglomeration or detachment post-reaction, with strong metal-carrier interactions ensuring the stability of the reconstruction process.
Ru-Doped-Induced LDH Reconfiguration, Mechanism regulation of SACs vs. Double-Doped Reconfiguration. To optimize reconfiguration for multi-reaction scenarios, Xu et al. prepared single-atom Ru-doped NiFe LDH (Ru0.3/NiFe) via two-step electrodeposition[49]. They found that introducing Ru SAs guided LDH reconfiguration into the active FeNiOOH phase. Ru atoms bind to LDH via Ru-O-M (M = Ni/Fe) bonds, facilitating electron transfer from Ni to Ru and electron accumulation from O to Ru, thereby optimizing the adsorption energy of HMF oxidation intermediates. XAS characterization confirms Ru exists in the +2.7 oxidation state, atomically dispersed in a RuO4 structure, while Raman spectroscopy verifies the transformation of LDH into the active NiOOH phase during reaction. DFT calculations indicate that Ru introduction reduces Ni dehydrogenation free energy, promoting Ni(OH)2 conversion to NiOOH while enhancing HMF-catalyst surface interactions to accelerate reaction pathways. Extending the strategy of dual-doping-induced reconstruction, Zhu et al. designed Ru single-atom and S anion dual-doped NiFe LDH (Ru-S-NiFe LDH)[128]. They discovered that the S anion accelerates the reconstruction of LDH into the active NiFeOOH phase by modulating lattice parameters and electron distribution, thereby lowering the reconstruction energy barrier. The Ru SA, dispersed in Ru-O/S coordination forms, optimizes the electronic structure of the active phase through electron transfer while mitigating Ru’s excessive oxidation. Operando XAS characterization revealed that the valence states of Ru and Ni increased with rising reaction potential, while Fe showed no significant change, confirming Ru and Ni as primary active sites with Fe primarily contributing structural stability. DFT calculations validated that dual doping synergistically reduces the adsorption energy difference of OER intermediates, optimizing reaction kinetics.
Reaction-pathway engineering: switching mechanisms for performance breakthrough
Reaction-pathway engineering represents a transformative strategy that extends beyond tuning individual active sites to actively reprogram the reaction network itself. By utilizing LDHs as a dynamic catalytic platform, this approach takes advantage of their structural and electronic versatility to direct and switch catalytic mechanisms. By strategic modifications such as single-atom doping, defect creation, or topological transformation, the intrinsic scaling relationships and thermodynamic limitations of conventional pathways can be circumvented[129,130]. The key lies in the ability of LDHs - through their tunable composition, confinement effects, and strong interfacial interactions - to precisely modulate intermediate adsorption energies, alter charge transfer routes, and even activate lattice oxygen species. This capacity enables a deliberate shift from one dominant mechanism to another (e.g., from adsorbate evolution to lattice oxygen oxidation in OER, or from partial to deep hydrogenation), providing a fundamental driving force for breakthroughs in catalytic selectivity and efficiency.
OER remains the kinetic bottleneck of water splitting, with traditional mechanisms facing inherent trade-offs: the adsorbate evolution mechanism (AEM) is constrained by the linear scaling relationship of intermediate adsorption energies, while the lattice oxygen mechanism (LOM) - though capable of overcoming this scaling limitation - often suffers from structural instability. LDHs paired with single-atom engineering address this dual challenge, leveraging LDH’s structural flexibility and electronic tunability to enable OER pathway switching (AEM-to-LOM) or dual-pathway synergy, while inducing directional active site remodeling that balances catalytic kinetics and structural robustness. Wang et al. synthesized high-entropy MnFeCoNiCu LDH modified with Au SAs and oxygen vacancies, where the synergistic effect of Au SAs and vacancies weakens Ni-O bonds to direct OER pathway conversion from AEM to LOM[131]. This modulation reconstructs Ni sites into the core active centers of LOM, circumventing AEM’s intrinsic scaling limitation. Figure 17A presents the pH dependence of OER activity [ρRHE (the proton reaction orders on reversible hydrogen electrode scale) = 0.89] derived from linear sweep voltammetry (LSV) tests, which is significantly higher than that of pure high-entropy LDH (0.36), providing direct evidence for non-proton-coupled electron transfer-dominated LOM characteristics. Figure 17B and C demonstrate a notable overpotential at a current density of 100 mA cm-2 (Δη100 = 0.102 V) in tetramethylammonium hydroxide (TMAOH) electrolyte, as TMA+ strongly binds to O22- intermediates generated via LOM, hindering the reaction process and further validating LOM dominance. In situ Raman spectroscopy detects the O22- stretching vibration peak at 1,089 cm-1, and 18O isotope labeling experiments verify extensive lattice oxygen participation in O2 generation, solidifying the AEM-to-LOM pathway shift [Figure 17D-F].
Figure 17. (A) LSV curves in KOH and log j vs. pH plot at 1.45 V vs. RHE for proton reaction order; (B) Polarization curves of AuSA-MnFeCoNiCu LDH and MnFeCoNiCu LDH in 1.0 M KOH/TMAOH, with corresponding Δη100 and Tafel slope variations; (C) Raman spectra of AuSA-MnFeCoNiCu LDH and MnFeCoNiCu LDH after 30 min electrolysis at 1.45 V vs. RHE in 1.0 M KOH/TMAOH; (D) In situ Raman spectra of MnFeCoNiCu LDH; (E) In situ Raman spectra of AuSA-MnFeCoNiCu LDH; (F) 18O isotope-labeling mass spectra; (G) Adsorbate evolution mechanism and lattice oxygen oxidation mechanism on MnFeCoNiCuOOH. (A-G) Reprinted with permission from Ref.[131]. Copyright 2023, Springer Nature. AEM: Adsorbate evolution mechanism; LOM: lattice oxygen mechanism; LSV: linear sweep voltammetry; SA: single atom; RHE: reversible hydrogen electrode; Δη100: refers to the overpotential at a current density of 100 mA cm-2.
Wang et al. synthesized cerium-substituted NiFe-LDH (NiFe-Ce LDH)[132]. Ce single-atom doping modulates NiFe LDH via Ce 4f-O 2p electronic interaction, which shifts the O 2p band center upward to controllably activate local LOM while retaining AEM, forming a synergistic dual reaction pathway. LDH’s interlayer structure stabilizes the in situ reconstructed NiOOH active layer, and in situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) and Raman spectroscopy separately detect *OOH (AEM-specific) and OO* (LOM-specific) intermediates, with Fe sites stabilized by Ce-induced electronic regulation. This dual-pathway design integrates the kinetic advantage of LOM and the stability of AEM, realizing balanced catalytic performance. Duan et al. precisely anchored Ir single atoms to the Fe sites of NiFe LDH, forming a distinct Ir-O-Fe coordination configuration[133]. This configuration enhances metal-oxygen covalent interaction and shifts the O 2p band center upward, enabling complete AEM-to-LOM conversion that avoids the high-energy *OOH intermediate. LDH serves as a stable anchoring platform for Ir single atoms and induces remodeling of active sites into Ir-O-Fe/γ-NiOOH composite centers, where the synergistic interaction between Ir single atoms and LDH-derived γ-NiOOH further optimizes catalytic dynamics.
Single-atom charge density modulation: Pathway-selective switching in HDO reactions. Dai et al. successfully synthesized two Pt SACs - Pt1/NiAl-LDH and Pt1/NiAl-IMC - using NiAl-LDH as a precursor via an anion intercalation-two-dimensional confinement thermal reduction strategy[60]. PtCl62- intercalated between LDH layers is confined by electrostatic interactions. After low-temperature reduction at 200 °C, the LDH support structure is retained, with Pt SAs existing in a Pt-Ox coordination state and an electron-deficient state. High-temperature reduction at 400 °C induces a topological transformation of LDH into NiAl intermetallic compounds (IMC), where Pt atoms form Pt-Nix coordination and exhibit an electron-rich state [Figure 18A]. This charge density difference directly switches the HDO pathway of 4-propylguaiacol, electron-deficient Pt sites enhance C=O bond adsorption while inhibiting C-OH bond cleavage, favoring selective HDO to yield 4-propylguaiacol Propylcyclohexanol. Electron-rich Pt sites inject electrons into the C-O bond to weaken its bond energy, shifting the pathway toward deep deoxygenation to form propylcyclohexane. This achieves precise control over product selectivity. By modifying the support structure to finely regulate the local charge density of platinum SACs, the product selectivity of the lignin hydrodeoxygenation reaction can be tuned[60].
Figure 18. (A) Schematic illustration of the confinement strategy to synthesize Pt1/NiAl-LDH and Pt1/NiAl-IMC catalysts. Reprinted with permission from Ref.[60]. Copyright 2025, Wiley-VCH; (B) Schematic illustration of the fabrication of the catalysts and the main products of CO photo-hydrogenation. Reprinted with permission from Ref.[65]. Copyright 2023, Springer Nature. LDH: Layered double hydroxide; IMC: intermetallic compound; NP: nanoparticle; NA: nanoalloy; SAA: single-atom alloy.
By leveraging the topological transformation properties of LDH to construct SAA systems, the competition between C-C coupling and over-hydrogenation in CO hydrogenation is regulated, enabling the switching of reaction pathways to produce either short-chain hydrocarbons or long-chain liquid fuels. Zhao et al. synthesized the Ru1CoAl-LDH precursor via a one-step hydrothermal method[65]. Hydrogen reduction at 650°C induced the LDH topological transition, yielding an aluminum oxide-supported ruthenium-cobalt SAA catalyst (Ru1Co-SAA) [Figure 18B]. The layered structure of LDH provides confined space for atomically dispersed Ru and Co. After reduction, Ru atoms replace Co lattice sites to form a Ru-Co coordinated SAA structure without Ru-Ru agglomeration. This unique structure alters the reaction pathway for CO photohydrogenation. The single-atom Ru sites enhance CO dissociative adsorption and lower the C-C coupling energy barrier, while the Co sites suppress excessive hydrogenation of CHx intermediates to methane, thereby synergistically promoting the formation of highly unsaturated C2 intermediates. This significantly increases the probability of carbon chain growth, enabling the catalyst to efficiently produce C5+ liquid fuels near atmospheric pressure (0.1 ~ 0.5 MPa). This breakthrough overcomes the limitation of traditional Fischer-Tropsch synthesis (FTS) requiring high pressure to promote long-chain formation.
Versatile functional expansion: beyond catalysis via the LDH ‘atomic canvas’
The ‘atomic canvas’ essence of LDHs transcends mere catalytic interface reconstruction, leveraging their inherently tunable layered architecture, flexible topological transformation capabilities, and controllable interfacial interactions to drive functional expansion into a diverse array of fields. Beyond traditional electrocatalysis, this platform has penetrated nanozymes, organic synthesis, electrochemical conversion, environmental remediation, plastic waste upcycling, and biomedical applications[134-142]. Through atomically precise engineering - including single-atom/nanoparticle synergistic anchoring, interlayer molecule intercalation, defect modulation, and topological derivation - LDHs act as versatile carriers, structural precursors, or active components. They enable multifunctional capabilities ranging from radical scavenging and biomass valorization to selective chemical bond formation, pollutant abatement, cyclic alcohol/ketone synthesis, and antitumor/antibacterial therapies. This broad applicability stems from LDHs’ core advantage of ‘structurally designable-functionally customizable’, allowing targeted regulation of active-site structure and electronic environment to address field-specific challenges via tailored structural engineering.
LDHs serve as a versatile platform for multi-scenario functional extension, enabling precise regulation of active-site structure and electronic environment via component doping, topological transformation, synergistic loading of single atoms or nanoparticles, and organic molecule intercalation. This platform extends to diverse fields including CO2 electroreduction, biocatalysis, energy storage, and multi-pathway electrocatalysis, addressing field-specific challenges via targeted structural engineering and electronic regulation.
For CO2 electroreduction, Zhang et al. developed a novel, straightforward method for preparing SACs for Electrochemical carbon dioxide reduction reaction (CO2RR) by utilizing NiZn-LDHs as the metal source[143]. NiZn-LDH serves as a Ni source and structural template, polyhydroxy compound-induced monolayer exfoliation mitigates Ni atom aggregation during calcination, while Zn volatilization constructs mesoporous architectures to ensure uniform active site dispersion and facilitated mass transport. Topological transformation of LDH enables synergistic loading of Ni single atoms and nanoparticles. Ni nanoparticles promote H2O dissociation to generate H* and reduce the energy barrier for *COOH formation, while Ni SAC accelerates CO desorption, balancing proton supply and hydrogenation kinetics. Self-supporting nanoarrays derived from LDH topological transformation anchor atomically dispersed active sites uniformly, enhancing electron conduction efficiency and shortening CO2 diffusion pathways to adapt to high-current-density reaction requirements. Integrating CO2 capture with in situ conversion expands the application of the LDH ‘canvas’ in resource recycling. Gao et al. fabricated thickness-tunable Pt/MgFe-LDHs by capturing atmospheric CO2 as interlayer CO32-, constructing Ptδ+-Vo-Fe2+ interfacial sites via Pt single-atom anchoring on oxygen vacancies[144]. This design realizes photocatalytic coupling of CO32- reduction (to CO/CH4) and glycerol oxidation (to dihydroxyacetone/lactic acid), achieving simultaneous utilization of low-valued CO2 and biomass derivatives.
In the realm of biocatalysis (nanozymes), LDHs capitalize on biocompatibility and single-atom anchoring capability to construct nanozyme systems. Liu et al. synthesized a ruthenium single-atom nanozyme (SAE) characterized by atomically dispersed ruthenium atoms on a biocompatible magnesium-aluminum LDH (Ru1/LDH)[56]. The prepared Ru1/LDH SAE exhibits high intrinsic peroxidase (POD)-like catalytic activity, with performance 20 times that of Ru nanocluster (NC) nanoenzymes. This nanozyme, possessing both POD and superoxide dismutase (SOD) activities, mediates the heterolytic cleavage of hydrogen peroxide (H2O2) (free energy: 0.43 eV), thereby efficiently scavenging superoxide anion (O2-·) and hydroxyl radical (·OH). Optimization of the single-atom coordination environment (e.g., Ru-O3 configuration on MgAl-LDH) enhances nanozyme catalytic efficiency and cycling stability, enabling continuous free radical scavenging over multiple cycles while sustaining long-term antioxidant effects in cellular environments [Figure 19A]. To investigate the intracellular antioxidant activity of Ru1/LDH, 2’,7’-dichlorofluorescein (DCFH-DA) was employed as a fluorescent probe to detect reactive oxygen species levels. RAW264.7 cells (mouse monocyte-macrophage leukaemia cells) exhibited elevated fluorescence signals after 12 h of Rosup stimulation. Following the addition of LDH or Ru1/LDH, fluorescence intensity decreased. Notably, the fluorescence signal exhibited a marked reduction for Ru1/LDH. Flow cytometric quantification revealed that nearly 83% of reactive oxygen species (ROS) could be effectively eliminated. This demonstrates that Ru1/LDH efficiently scavenges intracellular ROS, thereby protecting cells from oxidative stress [Figure 19B].
Figure 19. (A) Biocompatible MgAl-LDH-supported Ru single-atom nanozyme with multi-enzyme mimetic activity and high intrinsic peroxidase-like activity; (B) ROS-scavenging in RAW 264.7 cells by Ru1/LDH and LDH. (A and B) Reprinted with permission from Ref.[56]. Copyright 2023, Wiley-VCH; (C) Optimized atomic configurations of ORR/OER elementary steps on FeNi-LDH@DACs; (D) Top/side views of FeNi-LDH@DACs charge density difference; (E) Electron density map of FeNi-LDH@DACs; (F) Structures and charge transfer directions of FeNi-LDH@DACs and FeNi-DACs; (G) Schematic of liquid Zn-air batteries; (H) Discharge-charge cycling of FeNi-LDH@DACs and 20% Pt/C + RuO2 in liquid ZABs. (C-H) Reprinted with permission from Ref.[62]. Copyright 2025, American Chemical Society. SOD: Superoxide dismutase; CAT: catalase; DPPH: 1,1-diphenyl-2-picrylhydrazyl radical; LDH: layered double hydroxide; DAC: dual-atomic catalyst; PCD: peroxidase; ROS: reactive oxygen species; ORR: oxygen reduction reaction; OER: oxygen evolution reaction; ZAB: zinc-air battery.
Expanding the biomedical potential of LDH-based single-atom systems, multi-functional nanozymes and sonosensitizers are innovatively developed. Wang et al. constructed DT-ZnFe-LDH@Cu by anchoring Cu single-atom nanozymes on ZnFe-LDH and modifying with aminated dextran, endowing it with POD, oxidase, and catalase-like activities for bacterial keratitis treatment[145]. The Cu-N coordination sites generate reactive oxygen species to eradicate biofilm-embedded bacteria, while surface modification facilitates biofilm penetration and macrophage polarization from M1 to M2 to alleviate inflammation. Wang et al. coupled Pt SAs with amorphous CoMgMo-LDH to fabricate a sonosensitizer (Pt/a-LDH), leveraging defect-single atom synergy to narrow the bandgap and suppress electron-hole recombination[146]. The Pt-O coordination sites enhance oxygen adsorption/activation, promoting singlet oxygen generation for sonodynamic therapy and inducing immunogenic cell death to trigger anti-tumor immune responses.
Extending LDH-based single-atom systems to environmental remediation, nonradical catalytic pathways are innovatively developed. Wang et al. constructed a Fe single-atom modified LDH membrane (Fe-LDH) that activates PMS via a surface-bound PMS intermediate-driven electron transfer pathway[147]. The Fe-N4 coordination sites on LDH induce phenolic pollutant polymerization, leveraging nanochannel confinement to enhance reactant proximity and phenoxyl radical enrichment for efficient water purification. Ma et al. derived a Pt single-atom/Cu NC synergistic catalyst from NiFeCu-LDH precursors, where Pt SAs (Pt-N coordination) enhance methyl mercaptan (CH3SH) adsorption and Cu NCs activate O2[148]. This Langmuir-Hinshelwood mechanism-driven system achieves efficient CH3SH oxidation, expanding LDH’s application in volatile sulfur compound abatement.
Advancing plastic waste upcycling and aromatic hydrogenation, LDH-based single/dual-atom catalysts enable selective conversion to high-value cyclic alcohols/ketones. Liu et al. anchored Pd single atoms on calcined MgAl-LDH (Pd/CHT-800) to form Pd-O-MgAl active sites, realizing selective hydrogenation of phenols to cyclohexanones in aqueous media[149]. The electron-deficient Pd single atoms activate benzene rings while the LDH’s surface basicity inhibits carbonyl group adsorption, avoiding over-hydrogenation to cyclohexanols. Wang et al. developed a RuLa dual-atom catalyst from La-doped CoAl-LDH precursors via anion intercalation and thermal reduction, constructing isolated Ru-La atomic pairs on CoAl oxide[150]. This catalyst mediates tandem hydropyrolysis-hydrogenation of polycarbonate waste, preserving intrinsic oxygen functionalities to produce cyclohexanol via non-oxidative pathways, with Ru activating H2 and La facilitating hydrogen spillover.
Using LDHs as electrode substrates, the OER/ORR bifunctional catalytic performance can be optimized via single-atom loading, defect engineering, or heterogeneous coupling, meeting the requirements of energy storage devices such as ZABs. By employing defective LDHs as an electronic modulation platform, single atoms can be anchored to tailor the electron distribution and boost reaction activity. For example, Hu et al. developed a defect-type nickel-iron LDH catalyst anchored by Ru SAs (Ru SAs/D-NiFe LDH@NF)[41]. This material was synthesized via a hydrothermal reaction combined with an alkaline etching strategy. Cation vacancies generated by the alkaline etching anchored atomically dispersed Ru SAs, forming a stable and highly active catalytic interface. The Ru SAs transfer electrons to the LDH, optimizing the formation of an energy barrier (1.60 eV) at *OOH. By coupling LDH with SAC, synergistic active sites can be constructed on a ‘composite canvas’, improving the bifunctional catalytic performance of ZABs. Wang et al. synthesized a bifunctional catalyst by using atomically dispersed FeCo-NC materials with a three-dimensional floral morphology as a unique substrate for the controlled deposition of ultrafine NiFe LDH nanodots (NiFe-NDs)[97]. The LDH nanoparticles provide highly active OER sites, while the iron-cobalt-nitrogen-carbon (FeCo-NC) nanoflowers optimize ORR kinetics. The interfacial coupling between the two accelerates charge transfer, achieving a balance between activity and stability.
Using LDH as a structural template, hierarchical porous bifunctional electrodes can be constructed to balance the catalytic activity and mass transfer efficiency in ZABs. Qin et al. proposed a novel interface-coupled ‘sesame ball’ catalyst, featuring FeNi-LDH nanoparticles densely grown on porous, conductive FeNi-DAC[62]. This structure leverages potent synergistic effects to optimize intermediate adsorption, reduce activation energy, and enhance electron transfer, thereby achieving stable bifunctional ORR/OER performance. Within the FeNi-LDH@DACs composite structure, the tight interfacial coupling between LDH nanodots and the FeNi diatomic catalyst enables significant charge redistribution. This interfacial charge transfer effectively modulates the d-band centers of Fe and Ni active atoms, bringing their adsorption strengths for oxygen intermediates (*OOH, *OH) closer to the optimal range defined by the Sabatier principle. Consequently, ORR kinetics are enhanced by lowering the energy barrier for *OOH formation and accelerating electron transfer. In this system, spontaneous interfacial electron redistribution substantially boosts charge transfer and intrinsic ORR activity. Overall, strong electronic coupling at the LDH-DAC interface coordinates the favorable electronic structures of active FeNi sites, thereby improving ORR catalytic performance [Figure 19C-F]. The battery achieves a discharge voltage of 1.35 V and a charging voltage of 1.58 V at 10 mA cm-2, with an energy efficiency of 85%, and exhibits stable operation for 500 h without significant performance degradation [Figure 19G].
As a versatile platform for the molecular-level control of multi-pathway ORR catalysis, Sun et al. synthesized a hierarchical carbon nanosheet array electrode with single-atom nickel sites (Ni-SAC) via calcination and etching of Mn-doped NiAl-LDH intercalated with m-diaminobenzenesulfonic acid (MA). MA tailors the microenvironment to promote the selective adsorption of O2 and H* generation by Ni single atoms, enabling 2e- ORR for H2O2 production[63].
Single atoms synergize with Lewis acid sites to achieve highly selective lignin depolymerization. Meng et al. prepared the Mo1Al/MgO catalyst by interlayer confinement anchoring MoO42- using MgAl-LDH as a precursor, followed by thermal reduction[151]. This catalyst features coexisting Mo1-O5 single atoms and Al Lewis acid sites. The two-dimensional confinement effect of LDH suppresses Mo species agglomeration. The Al Lewis acid sites formed after calcination synergize with Mo single atoms to selectively cleave the β-aromatic ether bond in lignin under an inert atmosphere. Methanol solvent participates in the reaction process: Mo1-O5 single atoms activate ether bonds, while Al Lewis acid sites stabilize reaction intermediates. This ultimately yields monophenolic products containing Cα = Cβ double bonds with high selectivity, achieving yields close to theoretical values.
Topological transformation constructs SAAs, enabling highly selective anti-Marsch hydrogenation. Ma et al. used ZnAl-LDH as a precursor, undergoing thermal reduction-induced topological transformation to obtain Pt SACs supported on Zn(Al)O[152]. The topological transformation of LDH provides stable anchoring sites for Pt atoms, enabling coexistence of Pt10 and Pt1δ+ species by regulating Pt loading. These two Pt species form a synergistic catalytic system: Pt10 activates the N-H bond of amines to render it electrophilic, while Pt1δ+ activates the C=C bond of alkenes via π-bonding to make the β-C nucleophilic. The nucleophilic β-C attacks the electrophilic amine, achieving anti-Marcy addition highly selectively (up to 92%) forming the C-N bond.
Lattice confinement engineering in LDHs enables precise modulation of single-atom dispersion and electronic state for thermal catalysis. Ma et al. developed pseudo-single-atom Pt catalysts by confining Sn/Zr promoters in LDH brucite-like lattices, where lattice confinement induces electron-rich (Sn) or electron-defected (Zr) sites to guide selective Pt dispersion[153]. This strategy modulates Pt’s electronic properties for n-hexane reforming, realizing controlled formation of single atoms and sub-nanometer clusters without aggregation.
In summary, LDH’s ‘atomic canvas’ nature enables versatile functional expansion across electrocatalysis, biocatalysis, environmental remediation, plastic upcycling, biomedicine, and organic synthesis. Via atomically precise engineering (single-atom loading, intercalation, defect modulation, topological transformation), LDHs tailor active-site structure and electronic state to address field-specific demands. Their ‘structurally designable-functionally customizable’ advantage underpins synergistic enhancements between the canvas and anchored species. Table 1 summarizes recent advances in LDH-based SACs, including synthesis methods, single-atom loadings, bonding modes, and application fields. These advancements establish LDHs as a cross-disciplinary platform for high-performance multifunctional materials.
Recent advances in the synthesis and applications of LDH-based single-atom catalysts
| Strategy | Catalysts | Methods | Metal (Loading) | Bonding mode | Application | Ref. |
| In situ synthesis | CoCrRu LDHs | Co-precipitation | Ru(4.4 wt.%) | Ru-O-Co | OER | [39] |
| Rh1/NiV-LDH | Hydrothermal | Rh(6.59 wt.%) | Rh-O | UOR | [40] | |
| Ru SAs/D-NiFe LDH@NF | Hydrothermal | Ru(2.403 wt.%) | Ru-O | OER | [41] | |
| NiFe-LDH@La SNHPC | Hydrothermal | La(0.5 wt.%) | La-N | OER | [61] | |
| Ru/Ni3V-LDH | Hydrothermal | Ru(9.45 wt.%) | Ru-O-Ni | OER HER | [83] | |
| Ru-S-NiFe LDH | Hydrothermal | Ru(1.9 wt.%) | Ru-O Ru-S | OER | [128] | |
| Ru1/D-NiFe LDH | Electrochemical deposition | Ru(1.2 wt.%) | Ru-O | HER | [42] | |
| Ir/NiFe LDH | Electroreduction | Ir(0.56 wt.%) | Ir-O | OER | [127] | |
| Post-synthesis | Ru/CoFe-LDHs | Cation-exchange | Ru(0.45 wt.%) | Ru-O | OER | [45] |
| Ru-SAC/NiFe-LDH | Cation-exchange | Ru(2.34 wt.%) | Ru-O-Ni Ru-O-Fe | OER | [46] | |
| Rh1CaAl-LDH | Cation-exchange | Rh(4 wt.%) | Rh-O | QHY | [47] | |
| Ni3Fe-CO32- LDH-Pt SA | Anion-exchange | Pt(not given) | Pt-O | HER OER | [48] | |
| SA-Co-LDH | Anion-exchange | Co(0.5 wt.%) | Co-N | PMS | [116] | |
| Ru0.3/NiFe | Electrochemical deposition | Ru(0.3 wt.%) | Ru-O-NiRu-O-Fe | HMFOR,OER | [49] | |
| Ir1/Ni-LDH-T Ir1/Ni-LDH-V | Electrochemical deposition | Ir(2.18 wt.%) Ir(2.54 wt.%) | Ir-O | OER | [51] | |
| ePt/NiFe LDH@e-nf | Electrochemical deposition | Pt(0.39 wt.%) | Pt-Ni Pt-Fe | HER | [53] | |
| PtSA/QD-NiFeV9 LDH | Electrochemical deposition | Pt(1.3 wt.%) | Pt-O | HER | [85] | |
| IrSA-NiFe LDH/NiMo | Electrochemical deposition | Ir(3.8 wt.%) | Ir-O Ir-Ni | HER OER | [106] | |
| Pt/CoV-LDHs | Electrochemical deposition | Pt(0.71 wt.%) | Pt-O | HER | [52] | |
| sAu/NiFe LDH | Electrochemical deposition | Au(0.4 wt.%) | Au-O | OER | [126] | |
| Ru1/LDH | Post-loading followed by reduction | Ru(1.05 wt.%) | Ru-O | RONS | [56] | |
| Ru1/LDH-VIII | Post-loading followed by reduction | Ru(1.1 wt.%) | Ru-O Ru-O-Ni | Benzyl alcohol oxidation | [55] | |
| NiIrSAA/NiFe-LDH | Post-loading followed by reduction | Ir(8.31 wt.%) | Ir-O Ir-Ni | HER OER | [57] | |
| Pt/D-NiFe-LDH | Post-loading followed by reduction | Pt(1.27 wt.%) | Pt-Ni, Pt-Fe | HER | [58] | |
| Pt/FeOOH@NiFe LDH | Post-loading followed by reduction | Pt(2 wt.%) | Pt-O | HER | [89] | |
| Ir1/NiCr LDH | impregnation | Ir(1.59 wt.%) | Ir-O | OER | [95] | |
| Ru, Ir@CoFe-LDH/IF | Impregnation | Ru(0.065 wt.%) Ir(1.405 wt.%) | Ru-O Ir-O | OER | [107] | |
| Ir/CoFe-LDH | Impregnation | Ir(0.5 wt.%) | Ir-OH Ir-Cl | OER | [108] | |
| def-Ru-NiFe LDH/NCO | Hydrothermal | Ru(0.3 wt.%) | Ru-O | OER HER | [105] | |
| topological transformation | Pt-NiCo LDO | The transformation of MOFs into LDH-based SACs | Pt(5.06 wt.%) | Pt-Co | HER | [66] |
| Hm-LDH-700 | The topological conversion of the LDHs host into a LDO | Fe(N.A) | Fe-N | ORR | [67] | |
| IE-SACs | LDHs can be topologically converted into carbon-supported SACs through pyrolysis and etching | Co(3.22 wt.%) | Co-N Co-S | ORR OER | [68] |
CONCLUSION AND OUTLOOK
This review has established LDHs as a uniquely versatile two-dimensional ‘atomic canvas’ for the precision design of SACs. Their intrinsic advantages, including a tunable layered structure, abundant and well-defined coordination sites, and a remarkable capacity for topological transformation, provide an unparalleled platform for the atomically precise construction of catalytic centers. We have systematically summarized the synthetic strategies that enable this precision, encompassing in situ synthesis, post-synthesis modification, topological transformation, and scalable preparation, which collectively form the foundation for the controllable construction of single-atom active sites. Moving beyond synthesis, we have elaborated on how active LDH supports dynamically reconstruct catalytic interfaces and enable multifunctional performance via seven core engineering strategies. Electronic modulation tailors the electronic state of single atoms, while coordination engineering optimizes the local atomic environment. Microenvironment confinement-mass transfer synergy enhances both site activity and reactant flux, and interfacial synergy engineering enables functional division and relay catalysis. Dynamic phase engineering guides in situ reconstruction into highly active states, reaction-pathway engineering switches fundamental mechanisms for performance breakthroughs, and versatile functional expansion extends applications from traditional catalysis to biomedicine and environmental technology.
Despite significant progress, key challenges persist. Achieving both high single-atom loading and uniform dispersion remains difficult, as excessive loading often leads to aggregation. The dynamic evolution of single-atom sites under working conditions is not fully understood, limited by the integration of advanced operando characterization tools. Scaling up the synthesis of LDH-based SACs without compromising structural integrity and catalytic activity is a major hurdle for industrial adoption. Furthermore, the multifunctional integration of these catalysts for complex, cross-disciplinary applications is still in its nascent stages.
Looking beyond single-atom sites, the future development of LDH-based catalysts may evolve towards more sophisticated multi-atom cooperative systems, such as precisely constructed diatomic or triatomic catalysts. As evidenced in other 2D materials such as VS2 and TiTe2 [Figure 20][154], leveraging atomic orbital coupling and electronic synergy between neighboring metal sites is a promising strategy to further regulate activity and stability. Future research on LDHs should thus focus on the precise design of such multi-atom centers and the elucidation of their structure-activity relationships, pushing the frontier towards higher-order active-site engineering.
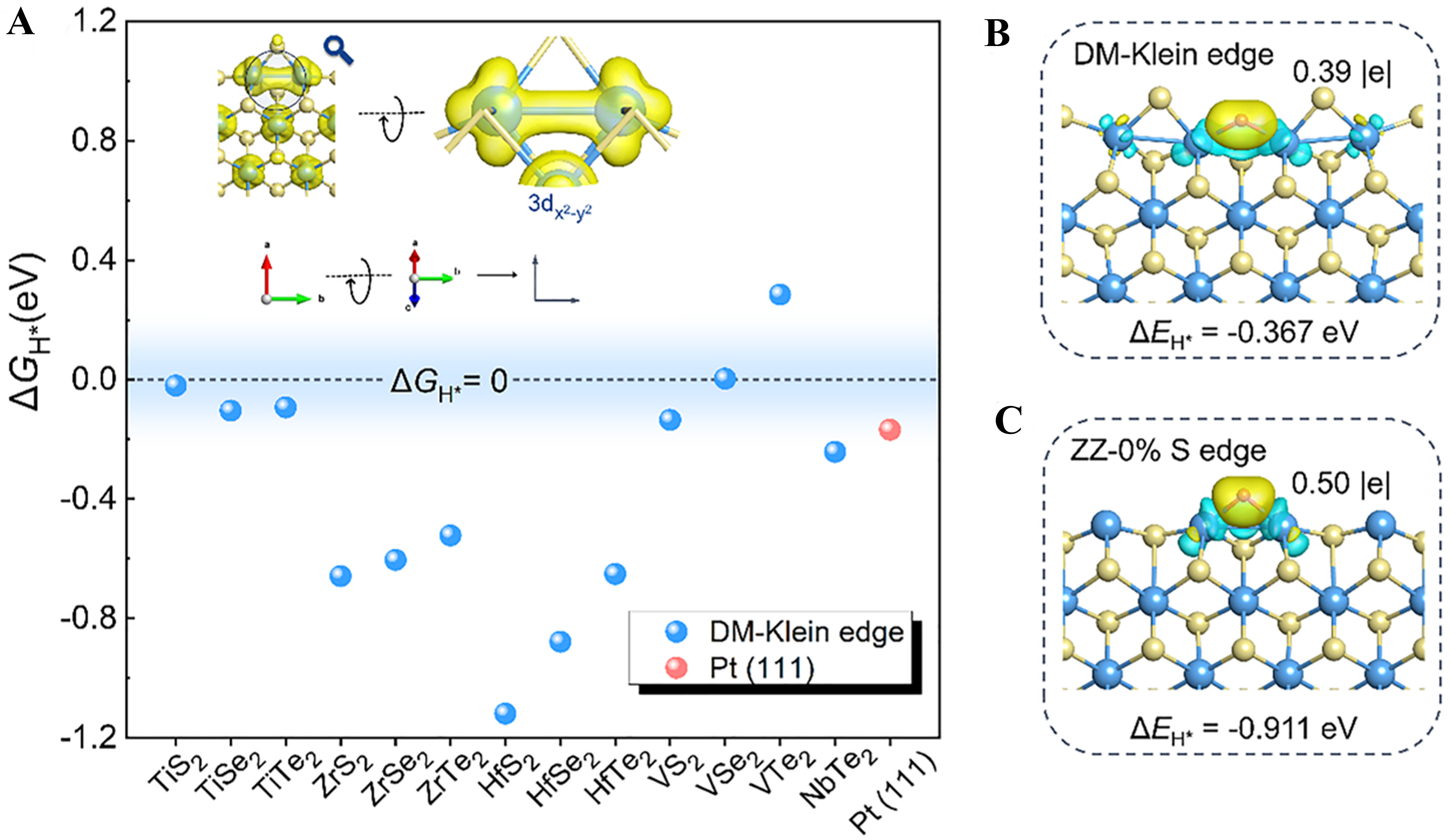
Figure 20. (A) Calculated adsorption free energy of hydrogen ΔGH* values of thirteen DM-Klein edge and Pt (111) surface. The blue shaded region indicates that |ΔGH*| < 0.15 eV. The band decomposed charge density distributions of the 1T-VS2 nanoribbon with DM-Klein edges in the energy interval between EF-1.0 eV to EF are shown in the inset. Adsorption energy (ΔEH*), differential charge density, and Bader charge analysis of (B) DM-Klein edge and (C) ZZ-0% S edge of 1T-VS2. (A-C) Reprinted with permission from Ref.[154]. Copyright 2022, Wiley-VCH. ΔGH*: The adsorption free energy of hydrogen; DM: dimer-like metal; EF: the edge formation energy; ZZ: the zigzag edge.
Looking ahead, the advancement of LDH-based SACs will focus on overcoming existing limitations while pursuing greater precision, deeper mechanistic understanding, and enhanced practicality. Firstly, regarding synthesis, novel strategies, such as combining atomic layer deposition (ALD) with LDH interlayer confinement or utilizing template-guided topological transformation, should be developed to achieve atomic-level precision in single-atom positioning, thereby overcoming the conventional trade-off between loading and dispersion. We note that while ALD enables precise single-atom placement, its high cost and low throughput limit scalability. In contrast, the hypothesis that LDH interlayer confinement can act as a ‘molecular template’ to restrict nucleation to specific lattice sites is presented as an attractive but unvalidated concept requiring in-situ characterization. Secondly, integrating advanced in situ characterization techniques (e.g., in situ XAFS, in situ TEM, operando Raman) with multi-scale theoretical calculations [e.g., Density Functional Theory + Hubbard U (DFT+U), machine learning] will provide deeper insights into dynamic catalytic mechanisms, particularly elucidating the nature of synergistic interactions between single atoms and LDH supports during reactions. We critically appraise that current studies rely heavily on ex-situ characterization, which fails to capture dynamic evolution under reaction conditions; thus, integrating operando techniques is presented as a validated necessity rather than merely an optional enhancement. Thirdly, multifunctional integration represents a key future direction. By tailoring LDH composition, interlayer species, and interface structures, these catalysts can be endowed with dual or multi-catalytic functionalities or extended into emerging fields such as biomedicine (e.g., reactive oxygen species scavenging) and environmental remediation (e.g., pollutant degradation). The hypothesis that heteroatom doping can introduce synergistic multi-functional catalysis is clearly stated as unvalidated, since existing work focuses on single-function performance without systematic multi-task evaluation. Fourthly, efforts must be dedicated to optimizing scaled-up preparation processes (e.g., continuous co-precipitation, solvent-free synthesis) and enhancing the structural stability of catalysts under harsh reaction conditions to facilitate the transition from laboratory research to industrial application. We critically appraise that most LDHbased SACs show poor stability under harsh conditions due to singleatom leaching or structural collapse, which is a major barrier that must be prioritized, as laboratory tests often do not replicate industrial conditions. Finally, exploring LDH-based SACs incorporating unconventional single-atom species (e.g., rare-earth metals, non-metals) and novel coordination configurations (e.g., heteroatomic coordination, defect-induced coordination) will open new avenues for designing high-performance catalysts, further harnessing the unique advantages of LDHs as a versatile structural platform in advanced materials. design. We critically appraise that rareearth single atom, despite unique electronic properties, are limited by cost and abundance; we then propose the plausible hypothesis that nonmetal single atoms (e.g., P, S) could serve as lowcost alternatives, while acknowledging that their activity and stability in LDH systems remain largely unstudied.
DECLARATIONS
Authors’ contributions
Writing original draft: Song, X.
Methodology: Song, X.
Formal analysis: Song, X.
Writing Section SYNTHESIS OF LDH-BASED SACS: PRECISE ‘PAINTING’ ON THE ‘ATOMIC CANVAS’: Zhang, Y.
Drawing the Figure: Sun, Q.
Writing Outlook: Liu, Y.
Writing and Conceptualization: Bai, X. J.
Review & editing: Zhao, Y.
Project administration: Zhao, Y.
Funding acquisition: Zhao, Y.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This research was supported by the National Natural Science Foundation of China (Grant Nos. 22288102, 22438007, 12504002), the Scientific and Technological Development Project of Jilin Province (Grant No. YDZJ202601ZYTS011), and the CAS Youth Interdisciplinary Team.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Liu, S.; Meyer, Q.; Xu, D.; et al. Breaking the activity and stability trade-off of platinum-free catalysts for the oxygen reduction reaction in hydrogen fuel cells. ACS. Nano. 2025, 19, 19524-51.
2. Zhuang, L.; Gao, Q.; Chen, L.; et al. Catalytic immunotherapy via ultrasmall Ir supported metal clusters on defect-engineered nanoplatforms for tumor metabolic disruption and enhanced immunogenic cell death. Nano. Today. 2025, 65, 102878.
3. Ma, Y.; Liu, S.; Mao, J.; et al. Ligand-restricted synthesis of highly paired dual-atom catalysts. Nat. Mater. 2026, 25, 80-90.
4. Liu, Y.; Niu, R.; Wang, Y.; Zhang, H.; Zhao, Y. Preparation and biomedical applications of single-metal atom catalysts. Nat. Protoc. 2026, 21, 775-807.
5. Gu, C. H.; Du, M.; Han, R. Y.; Zhang, A. Y.; Yu, H. Q.; Xing, M. Ultrafast water purification by template-free nanoconfined catalysts derived from municipal sludge. Angew. Chem. Int. Ed. Engl. 2025, 64, e202423629.
6. Zhu, C.; Fu, S.; Shi, Q.; Du, D.; Lin, Y. Single-atom electrocatalysts. Angew. Chem. Int. Ed. Engl. 2017, 56, 13944-60.
7. Liang, X.; Yao, S.; Li, Z.; Li, Y. Challenge and chance of single atom catalysis: the development and application of the single atom site catalysts toolbox. Acc. Chem. Res. 2025, 58, 1607-19.
8. Liang, X.; Fu, N.; Yao, S.; Li, Z.; Li, Y. The progress and outlook of metal single-atom-site catalysis. J. Am. Chem. Soc. 2022, 144, 18155-74.
9. Lin, X.; Zhen, S.; Wang, X.; et al. Data-driven design of single-atom electrocatalysts with intrinsic descriptors for carbon dioxide reduction reaction. Trans. Tianjin. Univ. 2024, 30, 459-69.
10. Shangguan, Y.; Wang, R.; Zhou, Y.; Feng, X.; Chen, H. Design and engineering of single-atom catalysts for environmental remediation: principles and structural-property relationship. Nano. Res. 2025.
11. Xu, C.; Zhang, Y. P.; Zheng, T. L.; et al. Contracted Fe-N5-C11 sites in single-atom catalysts boosting catalytic performance for oxygen reduction reaction. ACS. Appl. Mater. Interfaces. 2023, 15, 32341-51.
12. Qiao, Y.; Zhan, S.; He, Q.; et al. Dynamic geminal-atom coordination for highly efficient photo-fenton catalysis. Adv. Mater. 2026, 38, e18940.
13. Liu, H.; Liu, C.; Zong, X.; Wang, Y.; Hu, Z.; Zhang, Z. Role of the support effects in single-atom catalysts. Chem. Asian. J. 2023, 18, e202201161.
14. Lang, Z.; Wang, X.; Jabeen, S.; et al. Destabilization of single-atom catalysts: characterization, mechanisms, and regeneration strategies. Adv. Mater. 2025, 37, e2418942.
15. Yu, Y.; Zhu, Z.; Huang, H. Surface engineered single-atom systems for energy conversion. Adv. Mater. 2024, 36, e2311148.
16. Bai, X.; Zhai, X.; Zhang, L.; Fu, Y.; Qi, W. Site-directed reduction engineering within bimetal-organic frameworks for efficient size-selective catalysis. Matter 2021, 4, 2919-35.
17. Bai, X. J.; Yang, C.; Tang, Z. Enabling long-distance hydrogen spillover in nonreducible metal-organic frameworks for catalytic reaction. Nat. Commun. 2024, 15, 6263.
18. Song, X.; Zheng, H.; Zhang, J.; et al. Lanthanide-O-Ru on RuO2 mediates the binding behavior of OH, enabling efficient and stable water oxidation in acidic solution. ACS. Catal. 2025, 15, 11366-76.
19. Tang, Y.; Wang, H.; Guo, C.; et al. Synergies between atomically dispersed Ru single atoms and nanoparticles on CeAlOx for enhanced photo-thermal catalytic CO2 hydrogenation. Adv. Mater. 2026, 38, e12793.
20. Zhou, Y.; Mao, Y.; Ye, C.; et al. Ru single atoms anchored on Co3O4 nanorods for efficient overall water splitting under pH‐universal conditions. Adv. Energy. Mater. 2025, 15, 2500700.
21. Liu, W.; Liu, Q.; Wan, X.; Shui, J. Boosting the oxygen reduction performance of Fe-N-C catalyst using zeolite as an oxygen reservoir. Trans. Tianjin. Univ. 2024, 30, 428-35.
22. Kaiser, S. K.; Chen, Z.; Faust Akl, D.; Mitchell, S.; Pérez-Ramírez, J. Single-atom catalysts across the periodic table. Chem. Rev. 2020, 120, 11703-809.
23. Gusmão, R.; Veselý, M.; Sofer, Z. Recent developments on the single atom supported at 2D materials beyond graphene as catalysts. ACS. Catal. 2020, 10, 9634-48.
24. Lang, R.; Du, X.; Huang, Y.; et al. Single-atom catalysts based on the metal-oxide interaction. Chem. Rev. 2020, 120, 11986-2043.
25. Wang, Y.; Ma, X. Ayeza; et al. A review of carbon-supported single-atom catalysts for electrochemical reactions. Carbon 2024, 228, 119343.
26. Han, G.; Zhang, W.; Li, L.; et al. Progress in the high-temperature synthesis of atomically dispersed metal on carbon and understanding of their formation mechanism. Energy. Mater. 2023.
27. Liu, K.; Wang, L.; Chen, W.; et al. The roadmap of carbon‐based single‐atom catalysts: rational design and electrochemical applications. Rare. Metals. 2025, 44, 7987-8132.
28. Ling, Y.; Ge, H.; Chen, J.; et al. General strategy toward hydrophilic single atom catalysts for efficient selective hydrogenation. Adv. Sci. (Weinh). 2022, 9, e2202144.
29. Wang, C.; Han, Y.; Tian, M.; et al. Main-group catalysts with atomically dispersed in sites for highly efficient oxidative dehydrogenation. J. Am. Chem. Soc. 2022, 144, 16855-65.
30. Xu, L.; Lu, L.; Xu, N.; et al. Progressive learning-guided discovery of single-atom metal oxide catalysts for acidic oxygen evolution reaction. Angew. Chem. Int. Ed. Engl. 2025, 64, e202510965.
31. Tukur, P.; Tukur, F.; Mo, Y.; Yan, Q.; Dun, C.; Wei, J. High entropy oxides: new superior supports for single atom catalysts. Next. Materials. 2024, 3, 100192.
32. Ren, J.; Li, Z.; Wang, R.; et al. Lattice distortion promotes dehydrogenation of layered double hydroxide for enhanced electrocatalytic oxidation activity. AIChE. J. 2025, 72, e70104.
33. Ren, J.; Wang, J.; Li, Z.; et al. Electrocatalytic hydrogenation coupling oxidation using water in a highly efficient paired cell enabled by an oxygen defect-rich layered double hydroxide. J. Mater. Chem. A. 2024, 12, 4333-42.
34. Wang, Y.; Zhang, M.; Liu, Y.; et al. Recent advances on transition-metal-based layered double hydroxides nanosheets for electrocatalytic energy conversion. Adv. Sci. (Weinh). 2023, 10, e2207519.
35. Shuai, S.; Peng, F.; Zhao, Y.; Kong, X.; Shi, W.; Shao, M. Confinement-deconfinement engineering induces ultrastrong dipole moments in metastable assemblies: driving leap in photocatalytic hydrogen evolution efficiency. Adv. Funct. Mater. 2025, 36, e16910.
36. Li, S.; Li, Z.; Yue, J.; et al. Photocatalytic CO2 reduction by near-infrared-light (1200 nm) irradiation and a ruthenium-intercalated NiAl-layered double hydroxide. Angew. Chem. Int. Ed. Engl. 2024, 63, e202407638.
37. Wang, R.; Ma, D.; Kong, X.; et al. Metastable supramolecular assembly of simple monomers enabled by confinement: towards aqueous phase room temperature phosphorescence. Angew. Chem. Int. Ed. Engl. 2024, 63, e202409162.
38. Gao, Z.; Ma, H.; Wang, Q.; Li, D.; Feng, J.; Duan, X. Structure engineering of layered double hydroxides (LDHs) for heterogeneous catalysis. Chem. Res. Chin. Univ. 2024, 40, 590-610.
39. Dong, C.; Zhang, X.; Xu, J.; et al. Ruthenium-doped cobalt-chromium layered double hydroxides for enhancing oxygen evolution through regulating charge transfer. Small 2020, 16, e1905328.
40. Sun, H.; Li, L.; Chen, H. C.; et al. Highly efficient overall urea electrolysis via single-atomically active centers on layered double hydroxide. Sci. Bull. (Beijing). 2022, 67, 1763-75.
41. Hu, X.; Cai, C.; Wang, Y.; et al. Ru single atoms regulate electron distribution in defective NiFe LDH for enhanced oxygen evolution reaction. Nano. Res. 2025, 18, 94908120.
42. Zhai, P.; Xia, M.; Wu, Y.; et al. Engineering single-atomic ruthenium catalytic sites on defective nickel-iron layered double hydroxide for overall water splitting. Nat. Commun. 2021, 12, 4587.
43. Li, P.; Wang, M.; Duan, X.; et al. Boosting oxygen evolution of single-atomic ruthenium through electronic coupling with cobalt-iron layered double hydroxides. Nat. Commun. 2019, 10, 1711.
44. Cao, J.; Mou, T.; Mei, B.; et al. Improved electrocatalytic activity and stability by single iridium atoms on iron-based layered double hydroxides for oxygen evolution. Angew. Chem. Int. Ed. Engl. 2023, 62, e202310973.
45. Hu, Y.; Luo, G.; Wang, L.; et al. Single Ru atoms stabilized by hybrid amorphous/crystalline FeCoNi layered double hydroxide for ultraefficient oxygen evolution. Adv. Energy. Mater. 2020, 11, 2002816.
46. Israr, M.; Ali, S.; Zhang, J.; et al. NiFe LDH hollow nanocages confined Ru single atoms for remarkable oxygen evolution reaction. Small 2025, 21, e2500828.
47. He, J.; Liang, Y.; Zhao, B.; et al. Isomorphous substitution in CaAl-hydrotalcite to construct high density single-atom catalysts for selective N-Heteroarene hydrogenation. Chin. J. Catal. 2025, 70, 353-62.
48. Chen, W.; Wu, B.; Wang, Y.; et al. Deciphering the alternating synergy between interlayer Pt single-atom and NiFe layered double hydroxide for overall water splitting. Energy. Environ. Sci. 2021, 14, 6428-40.
49. Xu, H.; Xin, G.; Hu, W.; et al. Single-atoms Ru/NiFe layered double hydroxide electrocatalyst: Efficient for oxidation of selective oxidation of 5-hydroxymethylfurfural and oxygen evolution reaction. App. Catal. B:. Environ. 2023, 339, 123157.
50. Fan, B.; Wang, H.; Han, X.; Deng, Y.; Hu, W. Single atoms (Pt, Ir and Rh) anchored on activated NiCo LDH for alkaline hydrogen evolution reaction. Chem. Commun. (Camb). 2022, 58, 8254-7.
51. Wei, J.; Tang, H.; Sheng, L.; et al. Site-specific metal-support interaction to switch the activity of Ir single atoms for oxygen evolution reaction. Nat. Commun. 2024, 15, 559.
52. Li, X.; Yan, Y.; Yao, Y.; Liu, Y. Three-in-one tandem catalysis for alkaline hydrogen evolution reaction on Pt/CoV-LDHs. Chem. Eng. J. 2024, 489, 151237.
53. Liu, Z. Y.; Ge, J. J.; Yu, L.; Ong, S. H.; Zhang, L. J.; Hu, J. M. Tailoring Pt-Ni/Fe coordination in single-atom Pt/NiFe LDH with facile synthesis for efficient and long-term alkaline water electrolysis. Small 2025, 21, e2504076.
54. Yang, Y.; Yang, Q.; Yang, Y.; et al. Enhancing water oxidation of Ru single atoms via oxygen-coordination bonding with NiFe layered double hydroxide. ACS. Catal. 2023, 13, 2771-9.
55. Jin, J.; Han, X.; Fang, Y.; et al. Microenvironment engineering of Ru single‐atom catalysts by regulating the cation vacancies in NiFe‐layered double hydroxides. Adv. Funct. Mater. 2021, 32, 2109218.
56. Wang, B.; Fang, Y.; Han, X.; et al. Atomization-induced high intrinsic activity of a biocompatible MgAl‐LDH supported Ru single-atom nanozyme for efficient radicals scavenging. Angew. Chem. Int. Ed. Engl. 2023, 62, e202307133.
57. Hu, Y.; Shen, T.; Song, Z.; et al. Atomic modulation of single dispersed Ir species on self-supported NiFe layered double hydroxides for efficient electrocatalytic overall water splitting. ACS. Catal. 2023, 13, 11195-203.
58. Shen, S.; Li, Q.; Zhang, H.; et al. Negative-valent platinum stabilized by Pt-Ni electron bridges on oxygen-deficient NiFe-LDH for enhanced electrocatalytic hydrogen evolution. Adv. Mater. 2025, 37, e2500595.
59. Zhang, T.; Han, X.; Yang, H.; et al. Atomically dispersed nickel(I) on an alloy-encapsulated nitrogen-doped carbon nanotube array for high-performance electrochemical CO2 reduction reaction. Angew. Chem. Int. Ed. Engl. 2020, 59, 12055-61.
60. Dai, Q.; Xu, Z.; Wang, S.; et al. Fine-tuned charge density of pt single-atom sites for controllable hydrodeoxygenation of lignin. Angew. Chem. Int. Ed. Engl. 2025, 64, e202504347.
61. Yin, L.; Liu, Y.; Zhang, S.; et al. Hollow carbon nanoreactors integrating NiFe-LDH nanodots with adjacent La single atoms for efficient oxygen electrocatalytic reactions. Mater. Horiz. 2025, 12, 5400-9.
62. Qin, T.; Zheng, L.; Pei, Z.; et al. Electron redistribution via LDH-to-DACs coupling enhances d-band regulation and stability for zinc-air battery electrocatalysis. ACS. Nano. 2025, 19, 32231-45.
63. Sun, Y.; Fan, K.; Li, J.; et al. Boosting electrochemical oxygen reduction to hydrogen peroxide coupled with organic oxidation. Nat. Commun. 2024, 15, 6098.
64. Li, P.; Xie, Q.; Zheng, L.; et al. Topotactic reduction of layered double hydroxides for atomically thick two-dimensional non-noble-metal alloy. Nano. Res. 2017, 10, 2988-97.
65. Zhao, J.; Liu, J.; Li, Z.; et al. Ruthenium-cobalt single atom alloy for CO photo-hydrogenation to liquid fuels at ambient pressures. Nat. Commun. 2023, 14, 1909.
66. Tian, Y.; Luo, Y.; Wu, T.; et al. Coupling interaction between precisely located Pt single-atoms/clusters and NiCo-layered double oxide to boost hydrogen evolution reaction. Adv. Funct. Mater. 2024, 34, 2405919.
67. Yang, Z.; Yan, X.; Tang, Z.; et al. Facile synthesis of hemin-based Fe-N-C catalyst by MgAl-LDH confinement effect for oxygen reduction reaction. Appl. Surf. Sci. 2022, 573, 151505.
68. Fan, K.; Li, Z.; Song, Y.; Xie, W.; Shao, M.; Wei, M. Confinement synthesis based on layered double hydroxides: a new strategy to construct single-atom-containing integrated electrodes. Adv. Funct. Mater. 2020, 31, 2008064.
69. Bai, S.; Li, T.; Wang, H.; Tan, L.; Zhao, Y.; Song, Y. Scale-up synthesis of monolayer layered double hydroxide nanosheets via separate nucleation and aging steps method for efficient CO2 photoreduction. Chem. Eng. J. 2021, 419, 129390.
70. Zhao, Y.; Li, F.; Zhang, R.; Evans, D. G.; Duan, X. Preparation of layered double-hydroxide nanomaterials with a uniform crystallite size using a new method involving separate nucleation and aging steps. Chem. Mater. 2002, 14, 4286-91.
71. Li, T.; Hao, X.; Bai, S.; Zhao, Y.; Song, Y. Controllable synthesis and scale-up production prospect of monolayer layered double hydroxide nanosheets. Acta. Phys. -Chim. Sin. 2020, 36, 1912005.
72. Zhao, Y.; Zhao, Y.; Waterhouse, G. I. N.; et al. Layered-double-hydroxide nanosheets as efficient visible-light-driven photocatalysts for dinitrogen fixation. Adv. Mater. 2017, 29, 1703828.
73. Evans, D. G.; Duan, X. Preparation of layered double hydroxides and their applications as additives in polymers, as precursors to magnetic materials and in biology and medicine. Chem. Commun. (Camb). 2006, 485-96.
74. Chi, H.; Dong, J.; Li, T.; et al. Scaled-up synthesis of defect-rich layered double hydroxide monolayers without organic species for efficient oxygen evolution reaction. Green. Energy. Environ. 2022, 7, 975-82.
75. Shi, X.; Wen, Z.; Gu, Q.; et al. Metal-support frontier orbital interactions in single-atom catalysis. Nature 2025, 640, 668-75.
76. Qi, K.; Chhowalla, M.; Voiry, D. Single atom is not alone: metal-support interactions in single-atom catalysis. Mater. Today. 2020, 40, 173-92.
77. Yang, J.; Li, W.; Wang, D.; Li, Y. Electronic metal-support interaction of single-atom catalysts and applications in electrocatalysis. Adv. Mater. 2020, 32, e2003300.
78. Lu, S.; Li, X.; Zhang, G.; Wang, S. Unlocking single-atom induced electronic metal-support interactions in electrocatalytic one-electron water oxidation for wastewater purification. Nat. Commun. 2025, 16, 4346.
79. Ebitani, K.; Motokura, K.; Mizugaki, T.; Kaneda, K. Heterotrimetallic RuMnMn species on a hydrotalcite surface as highly efficient heterogeneous catalysts for liquid-phase oxidation of alcohols with molecular oxygen. Angew. Chem. Int. Ed. Engl. 2005, 44, 3423-6.
80. Motokura, K.; Nishimura, D.; Mori, K.; Mizugaki, T.; Ebitani, K.; Kaneda, K. A ruthenium-grafted hydrotalcite as a multifunctional catalyst for direct alpha-alkylation of nitriles with primary alcohols. J. Am. Chem. Soc. 2004, 126, 5662-3.
81. Motokura, K.; Hashimoto, N.; Hara, T.; et al. Rhodium-grafted hydrotalcite catalyst for heterogeneous 1,4-addition reaction of organoboron reagents to electron deficient olefins. Green. Chem. 2011, 13, 2416.
82. Zhou, H.; Ban, J.; Shen, Y.; et al. Strategies to maximize the oxygen evolution reaction in layered double hydroxides by electronic defect engineering. eScience 2025, 5, 100380.
83. Sun, H.; Tung, C. W.; Qiu, Y.; et al. Atomic metal-support interaction enables reconstruction-free dual-site electrocatalyst. J. Am. Chem. Soc. 2022, 144, 1174-86.
84. Ngo, Q. P.; Paek, S. Y.; Lee, J. Y.; et al. Tailored design of iridium single atoms on Mn-Ni‐phytate with robust bifunctionality for enhanced anion exchange membrane water electrolysis. Adv. Energy. Mater. 2026, 16, e06645.
85. Wu, Y.; Chen, M.; Liu, D.; et al. Modulating the synergy of Pt single atoms and quantum dots on NiFe LDH for efficient and robust hydrogen evolution. J. Mater. Sci. Technol. 2025, 215, 111-20.
86. Wang, B.; Luo, Z.; Sun, H.; et al. Electronic modulation of amorphous/crystalline NiFe LDH by atomic Pt loading enabling industrial hydrogen production in alkaline water and seawater. J.Energy. Chem. 2025, 107, 427-39.
87. Sun, H.; Chen, H. C.; Humayun, M.; et al. Unlocking the catalytic potential of platinum single atoms for industry-level current density chlorine tolerance hydrogen generation. Adv. Funct. Mater. 2024, 34, 2408872.
88. Xu, H.; Lu, L.; Yu, Z.; et al. Synergistic interaction between Zn clusters and single-atom Ru in 3D NiFeS-LDH enhance 5-hydroxymethylfurfural electrocatalytic oxidation. Appl. Catal. B. Environ. 2025, 375, 125439.
89. Zhang, Y.; Zhao, M.; Wu, J.; et al. Construction of Pt single-atom and cluster/FeOOH synergistic active sites for efficient electrocatalytic hydrogen evolution reaction. ACS. Catal. 2024, 14, 7867-76.
90. Zhou, J.; Jiang, J.; Li, Y.; et al. Unveiling long-range wavelike alternately competitive and cooperative bond coupling modes in single-atom catalysis. J. Am. Chem. Soc. 2025, 147, 30239-47.
91. Shan, J.; Ye, C.; Jiang, Y.; Jaroniec, M.; Zheng, Y.; Qiao, S. Z. Metal-metal interactions in correlated single-atom catalysts. Sci. Adv. 2022, 8, eabo0762.
92. Chen, Y.; Chen, X.; Chen, M.; et al. Cross-scale manipulation of Ru single-atom catalyst for enhanced hydrogen evolution and overall water splitting at industrial current density. Chin. Chem. Lett. 2025, 111813.
93. Deng, W.; He, G.; Zhou, H.; et al. Single-atom Ru in CoFe-LDH drives efficient charge separation on BiVO4 for solar water splitting. Nanomicro. Lett. 2026, 18, 212.
94. Fan, J.; Zhao, Y.; Du, H.; et al. Light-induced structural dynamic evolution of Pt Single atoms for highly efficient photocatalytic CO2 reduction. ACS. Appl. Mater. Interfaces. 2022, 14, 26752-65.
95. Biswal, S. Divya, .; Mishra, B.; et al. Electronic modulation of iridium single atomic sites on NiCr layered double hydroxide for an improved electrocatalytic oxygen evolution reaction. J. Mater. Chem. A. 2024, 12, 2491-500.
96. Liu, Z. Q.; Liang, X.; Ma, F. X.; et al. Decoration of NiFe‐LDH nanodots endows lower Fe‐d band center of Fe1‐N‐C hollow nanorods as bifunctional oxygen electrocatalysts with small overpotential gap. Adv. Energy. Mater. 2023, 13, 2203609.
97. Wang, Y.; Zhang, G.; Ma, M.; et al. Ultrasmall NiFe layered double hydroxide strongly coupled on atomically dispersed FeCo-NC nanoflowers as efficient bifunctional catalyst for rechargeable Zn-air battery. Sci. China. Mater. 2020, 63, 1182-95.
98. Kumar, A.; Gil-Sepulcre, M.; Fandré, J. P.; et al. Regulating local coordination sphere of Ir single atoms at the atomic interface for efficient oxygen evolution reaction. J. Am. Chem. Soc. 2024, 146, 32953-64.
99. Qian, S.; Jiang, T.; Wang, J.; et al. Surface nanosteps modulate the local environment of Co single atoms to boost the electrocatalytic hydrogen evolution reaction. ACS. Catal. 2024, 14, 18690-700.
100. Guan, X.; Han, R.; Asakura, H.; et al. Subsurface single-atom catalyst enabled by mechanochemical synthesis for oxidation chemistry. Angew. Chem. Int. Ed. Engl. 2024, 63, e202410457.
101. Tang, C.; Jin, R.; Xia, Y.; et al. Asymmetric low-coordination tailoring of single-atom cobalt catalysts enabling efficient oxygen reduction reaction. Nano. Energy. 2025, 137, 110776.
102. Yu, T.; Xu, L.; Guo, P.; Xu, J.; Shen, B. Confinement of atomically dispersed Ru sites via metal defects over ultrathin layered double hydroxide for efficient photocatalytic CO2 reduction. Small 2026, 22, e11495.
103. Mu, X.; Gu, X.; Dai, S.; et al. Breaking the symmetry of single-atom catalysts enables an extremely low energy barrier and high stability for large-current-density water splitting. Energy. Environ. Sci. 2022, 15, 4048-57.
104. Zhang, T.; Jin, J.; Chen, J.; et al. Pinpointing the axial ligand effect on platinum single-atom-catalyst towards efficient alkaline hydrogen evolution reaction. Nat. Commun. 2022, 13, 6875.
105. Dao, H. T.; Hoa, V. H.; Sidra, S.; Mai, M.; Zharnikov, M.; Kim, D. H. Dual efficiency enhancement in overall water splitting with defect-rich and Ru atom-doped NiFe LDH nanosheets on NiCo2O4 nanowires. Chem. Eng. J. 2024, 485, 150054.
106. Wu, Y.; Chen, M.; Sun, H.; et al. Coupling Ir single atom with NiFe LDH/NiMo heterointerface toward efficient and durable water splitting at large current density. Appl. Catal. B:. Environ. 2025, 360, 124548.
107. Liu, Z.; Dai, Y.; Li, K.; et al. Dual single-atom anchored on oxygen vacancy of CoFe-LDH for highly stable electrochemical water oxidation. Small 2026, 22, e12240.
108. Duan, X.; Sha, Q.; Li, P.; et al. Dynamic chloride ion adsorption on single iridium atom boosts seawater oxidation catalysis. Nat. Commun. 2024, 15, 1973.
109. Dai, J.; Zhang, H. Recent Advances in catalytic confinement effect within micro/meso-porous crystalline materials. Small 2021, 17, e2005334.
110. Fan, X.; Li, D.; Shu, Y.; Feng, Y.; Li, F. Confinement effect and application in catalytic oxidation-reduction reaction of confined single-atom catalysts. ACS. Catal. 2024, 14, 12991-3014.
111. Li, X.; Pereira-Hernández, X. I.; Chen, Y.; et al. Functional CeOx nanoglues for robust atomically dispersed catalysts. Nature 2022, 611, 284-8.
112. Zhang, S.; Hedtke, T.; Zhou, X.; Elimelech, M.; Kim, J. environmental applications of engineered materials with nanoconfinement. ACS. EST. Eng. 2021, 1, 706-24.
113. Wang, Z.; Xu, S. M.; Xu, Y.; et al. Single Ru atoms with precise coordination on a monolayer layered double hydroxide for efficient electrooxidation catalysis. Chem. Sci. 2019, 10, 378-84.
114. Zeng, K.; Chao, M.; Tian, M.; et al. Atomically dispersed cerium sites immobilized on vanadium vacancies of monolayer nickel-vanadium layered double hydroxide: accelerating water splitting kinetics. Adv. Funct. Mater. 2023, 34, 2308533.
115. Wang, J.; Wen, N.; Wang, Y.; Jiao, X.; Chen, D.; Xia, Y. Spin-state inversion in non-precious Na single atoms decorated NiFe-LDH monolayer for efficient electrocatalytic water oxidation. Appl. Surf. Sci. 2023, 611, 155751.
116. Chen, C.; Yan, M.; Li, Y.; et al. Single-atom co sites confined in layered double hydroxide for selective generation of surface-bound radicals via peroxymonosulfate activation. Appl. Catal. B:. Environ. 2024, 340, 123218.
117. Wang, Y.; Yu, X.; Wu, W.; Sun, Z.; Wang, H.; Guo, J. Constructing the purification-permeability balance via polymerization route: a novel confined layered double hydroxide catalytic membrane incorporating single-atom iron. Water. Res. 2026, 292, 125319.
118. Li, X.; Xie, Z.; Roy, S.; et al. Amorphous high-entropy phosphide nanosheets with multi-atom catalytic sites for efficient oxygen evolution. Adv. Mater. 2025, 37, e2410295.
119. Lu, J.; Shen, C.; Tian, Y.; et al. An atomically precise Ru1Au6(TBBT)6(PPh3)6 cluster catalyst for ammonia production. Angew. Chem. Int. Ed. Engl. 2025, 64, e202516398.
120. Wu, Y.; Tang, X.; Yuan, K.; Chen, Y. Single-atom sites combined with metal nano-aggregates for efficient electrocatalysis. Energy. Environ. Sci. 2023, 16, 5663-87.
121. Zhang, J.; Gu, Y.; Lu, Y.; et al. Each performs its own functions: Nickel oxide supported ruthenium single-atoms and nanoclusters relay catalysis with multi-active sites for efficient alkaline hydrogen evolution reaction. Appl. Catal. B:. Environ. 2023, 325, 122316.
122. Liu, C.; Qiao, B.; Zhang, T. Integration of single atoms for tandem catalysis. JACS. Au. 2024, 4, 4129-40.
123. Wang, L.; Yang, Y.; Shi, Y.; et al. Single-atom catalysts with metal-acid synergistic effect toward hydrodeoxygenation tandem reactions. Chem. Catalysis. 2023, 3, 100483.
124. Won, Y. S.; Kirubasankar, B.; Kim, H. J.; et al. Phase transformation of needle-like Fe-Co0.85Se to hexagonal Fe-Co3O4 for enhanced high-current-density oxygen evolution via lattice oxygen redox. Small 2025, 21, e2505220.
125. Manivelan, N.; Piao, J.; Kim, J.; et al. Unveiling the aluminum doping effects of in-situ transmogrified dual-LDH heterostructure and its fermi-level alignment to water splitting potentials. Adv. Energy. Mater. 2024, 15, 2403889.
126. Zhang, J.; Liu, J.; Xi, L.; et al. Single-atom Au/NiFe layered double hydroxide electrocatalyst: probing the origin of activity for oxygen evolution reaction. J. Am. Chem. Soc. 2018, 140, 3876-9.
127. Li, D.; Ding, L.; Zhang, S.; et al. In-situ electrostatic anchoring of highly oxidized Ir single atoms on NiFe LDH for efficient water oxidation. Chem. Eng. J. 2025, 519, 165662.
128. Zhu, Y.; Wang, J.; Weiser, G.; et al. Ru single atoms and sulfur anions dual-doped NiFe layered double hydroxides for high-current-density alkaline oxygen evolution reaction. Adv. Energy. Mater. 2025, 15, 2500554.
129. Lv, S.; Zhou, Y.; Yang, W.; et al. The defects of single atom Co1‐N‐C regulate the H‐binding energy to achieve divergent hydrogen evolution or transfer hydrogenation with HCOOH. Adv. Funct. Mater. 2025, 35, 2423864.
130. Yan, Q.; Chen, Y.; Tang, B.; et al. Precise engineering of asymmetric tri-active sites by symbiotic strategy for photocontrolled directional reforming of biomass. Angew. Chem. Int. Ed. Engl. 2025, 64, e202505718.
131. Wang, F.; Zou, P.; Zhang, Y.; et al. Activating lattice oxygen in high-entropy LDH for robust and durable water oxidation. Nat. Commun. 2023, 14, 6019.
132. Wang, S.; Lin, B.; Qi, M.; et al. Cerium-doped NiFe hydroxides enabling hybrid pathways for durable alkaline water oxidation under fluctuating power. ACS. Nano. 2025, 19, 31915-28.
133. Duan, Z.; Cui, Z.; Gao, Z.; et al. Single-atom iridium orchestrates a reaction pathway shift to activate lattice oxygen for efficient oxygen evolution. ACS. Catal. 2025, 15, 16882-92.
134. Bai, Y.; Fang, Z.; Jia, K.; et al. Cascade design and facile fabrication of Cu/Cu2O/CuAl-layered double hydroxides as efficient nitrate reduction electrocatalysts. Small 2025, 21, e2408546.
135. Mavvaji, M.; Senan, A. M.; Akkoc, S. Magnetic layered double hydroxide nanocomposites: synthesis, catalytic performances, environmental and biological applications. Energy. Environ. Mater. 2025, 8, e70015.
136. Wang, C.; Yuan, F.; Yan, Z.; et al. High entropy 2D layered double hydroxide nanosheet toward cascaded nanozyme-initiated chemodynamic and immune synergistic therapy. J. Am. Chem. Soc. 2025, 147, 136-48.
137. Hou, H.; Lei, G.; Huo, H.; et al. CuCo-layered double hydroxide nanosheets grown on hierarchical carbonized wood as bifunctional electrode for supercapacitor and hydrogen evolution reaction. Adv. Sci. (Weinh). 2025, 12, e08630.
138. Kumar, J.; Solangi, N. H.; Neiber, R. R.; et al. Chemical perspectives on synthesis, functionalization, artificial intelligence, and energy storage applications of layered double hydroxides-based nanomaterials: a comprehensive review. Coord. Chem. Rev. 2025, 537, 216705.
139. Huang, Y.; Xie, R.; Li, K.; Tian, R.; Lin, Y.; Lu, C. Addressing the origin of single-atom-activated supports monitored by electrochemiluminescence. ACS. Appl. Mater. Interfaces. 2023, 15, 1610-8.
140. Ding, G.; Li, C.; Ni, Y.; Chen, L.; Shuai, L.; Liao, G. Layered double hydroxides and their composites as high-performance photocatalysts for CO2 reduction. EES. Catal. 2023, 1, 369-91.
141. Wang, K.; Wang, T.; Islam, Q. A.; Wu, Y. Layered double hydroxide photocatalysts for solar fuel production. Chin. J. Catal. 2021, 42, 1944-75.
142. Li, P.; Lyu, J.; Zhao, Y.; et al. Towards highly efficient selective hydrogenation: the role of single-atom catalysts. Chin. J. Catal. 2026, 81, 69-96.
143. Zhang, P.; Chen, H.; Chen, L.; et al. Atomically dispersed Ni-N-C catalyst derived from NiZn layered double hydroxides for efficient electrochemical CO2 reduction. Chin. J. Catal. 2023, 45, 152-61.
144. Gao, M.; Fan, J.; Fan, J.; Wang, Q.; Li, D.; Feng, J. Process coupling of CO2 and glycerol comprehensive utilization based on anionic 2D nanomaterial. AIChE. J. 2022, 69, e17976.
145. Wang, K.; Yuan, M. S.; Dai, P.; et al. ZnFe layered double hydroxide nanosheets loaded with Cu single-atom nanozymes with multi-enzyme-like catalytic activities as an effective treatment for bacterial keratitis. Adv. Sci. (Weinh). 2025, 12, e2411999.
146. Wang, T.; Hu, T.; Wang, S.; et al. Coupling Pt single-atoms with amorphous LDH nanosheets as a high-efficiency sonosensitizer for sonodynamic immunotherapy. Adv. Mater. 2026, 38, e72457.
147. Wang, Y.; Wu, W.; Yu, X.; Sun, Z.; Wang, H.; Guo, J. Ultraefficient water purification with low peroxymonosulfate input by a polymerization-driven single-atom iron dispersed layered double hydroxide membrane. J. Membr. Sci. 2025, 732, 124250.
148. Ma, Y.; Kang, D.; Wen, Y.; et al. Synergistic effects of Pt single atomic site and Cu nanoclusters for catalytic oxidation of methyl mercaptan. Appl. Surf. Sci. 2024, 657, 159718.
149. Liu, Y.; Meng, Q.; Zhang, K.; et al. Selective hydrogenation of phenols to cyclohexanones over hydrotalcite-supported Pd single-atom catalyst. Angew. Chem. Int. Ed. Engl. 2025, 64, e202504260.
150. Wang, J.; Li, D.; Zhang, Z.; et al. Selective upcycling of polycarbonate waste to cyclohexanol via RuLa dual-atom catalysis. Angew. Chem. Int. Ed. Engl. 2026, 65, e24681.
151. Meng, G.; Lan, W.; Zhang, L.; et al. Synergy of single atoms and Lewis acid sites for efficient and selective lignin disassembly into monolignol derivatives. J. Am. Chem. Soc. 2023, 145, 12884-93.
152. Ma, X.; An, Z.; Song, H.; Shu, X.; Xiang, X.; He, J. Atomic Pt-catalyzed heterogeneous anti-Markovnikov C-N formation: Pt10 Activating N-H for Pt1δ+-activated C=C attack. J. Am. Chem. Soc. 2020, 142, 9017-27.
153. Ma, X.; An, Z.; Zhu, Y.; Wang, W.; He, J. Pseudo-single-atom platinum induced by the promoter confined in brucite-like lattice for catalytic reforming. ChemCatChem 2016, 8, 1773-7.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0









Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].