




Stem-cell-derived extracellular vesicles in neurodegeneration and neuroaging: therapeutic potential and challenges
 0
0 Abstract
Neuroaging is a complex biological process in which the brain undergoes progressive functional decline marked by synaptic loss, neuroinflammation, and cognitive decline. At the molecular and cellular level, aging is driven by multiple interconnected hallmarks, including genomic instability, telomere attrition, epigenetic alterations, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication. Among these, cellular senescence, a state of irreversible cell cycle arrest, has emerged as a critical contributor to brain aging. Senescent cells accumulate with age, driven by the p53-p21 and p16-pRb pathways, and secrete pro-inflammatory factors via senescence-associated secretory phenotype (SASP), thereby exacerbating neurodegeneration, vascular dysfunction, and cognitive decline. Extracellular vesicles (EVs) are natural nanocarriers of proteins, lipids, and nucleic acids, and have emerged as key mediators of intercellular communication and therapeutics for aging and age-related conditions. EVs derived from various cell types, such as mesenchymal stem cells (MSCs), neural stem cells (NSCs), and induced pluripotent stem cells (iPSCs), can modulate senescence-related pathways, reduce inflammation, and promote tissue repair. Preclinical studies demonstrate that stem-cell-derived EVs can improve cognitive performance, enhance neurogenesis, reduce senescence phenotype, improve neuronal survival through neuroprotective miRNAs (miR-181a-2-3p), suppress neuroinflammation via inhibition of NLRP3 inflammasome, and support synaptic plasticity. Stem cell EVs possess natural biocompatibility, the ability to cross the blood-brain barrier (BBB), and targeted delivery mechanisms, making them promising candidates for anti-aging interventions. This review elaborates on the multifaceted role of stem cell EVs in mitigating brain aging, senescence, and age-associated chronic disease phenotype.
Keywords
INTRODUCTION
Aging, broadly defined as organismal/chronological aging, is a complex, time-dependent biological process[1] characterized by gradual loss of tissue homeostasis, metabolic dysfunction, extracellular matrix alterations, chronic inflammation, stem cell depletion, and cellular senescence[2]. At the cellular level, aging reflects underlying telomere attrition, mitochondrial dysfunction, oxidative stress, and epigenetic alterations[3]. For instance, telomeres, the protective caps on chromosomes, progressively shorten with each cell division due to the "end-replication problem" and insufficient telomerase activity. This shortening triggers replicative arrest (a key mechanism of cellular senescence, even in young organisms under stress) and compromises genomic integrity[4,5]. Simultaneously, mitochondrial efficiency declines with age, reducing ATP production and increasing the generation of reactive oxygen species (ROS). This elevated ROS creates oxidative stress, which further damages mitochondria, establishing a self-amplifying vicious cycle of mitochondrial dysfunction and oxidative stress[6]. Furthermore, aging involves epigenetic dysregulation such as alterations in chromatin remodeling, histone modifications, DNA methylation, and noncoding RNA expression, which disrupts the precise control of gene expression essential for cell function and overall cellular health[7]. Collectively, the cumulative damage from telomere shortening, mitochondrial/oxidative stress, and epigenetic drift drives cellular senescence, a state of irreversible cell cycle arrest[4,8-11], characterized by multiple hallmarks such as increased expression of p16, p21, lysosomal mass, multinucleation, senescence-associated heterochromatic foci (SAHFs), persistent DNA damage foci (e.g., γH2AX), lamin B1 loss, lipofuscin accumulation, and a pathogenic senescence-associated secretory phenotype (SASP)[12]. Although cellular senescence contributes to organismal/chronological aging (natural, time-dependent functional decline observed in older individuals)[13,14], it can also be induced independently of chronological age in different contexts, such as stress-induced premature senescence (triggered by factors such as oxidative stress, DNA damage, or metabolic dysfunction)[15], oncogene-induced senescence (aberrant oncogene activation that serves as a tumor-suppressive mechanism)[16], and therapy- or injury-induced senescence (develops in response to radiation, chemotherapeutics, or tissue damage)[17,18]. Over time, senescent cell accumulation across organs such as the brain, heart, and kidneys promotes the onset and progression of age-related chronic diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD)[19-22]. Therefore, it is essential to distinguish between organismal aging, cellular senescence, and age-associated diseases (which are more prevalent in older individuals but are not exclusive to aging itself)[23,24].
In the brain, aging manifests as neuroaging, a gradual structural and functional decline marked by synaptic loss, reduced neurogenesis, and impaired homeostasis, leading to mild cognitive impairment (MCI), a preclinical stage of cognitive decay[25-27]. Within the brain, a complex network of glial cells and non-neuronal cells exhibits a classical senescence phenotype, contributing to neurodegeneration[19,28-31]. Emerging data demonstrate that the accumulation of senescent cells in the central nervous system (CNS) directly contributes to neurodegenerative diseases, and vice versa, by promoting inflammation, disrupting neurovascular units, and impairing synaptic communication[32-36]. Critically, post-mitotic neurons, once considered senescence-resistant, have been shown to demonstrate a distinct senescence phenotype in response to aging or injury[37,38]. A recent study reported that impaired autophagy in microglia promotes cellular senescence, leading to the emergence of SASP and exacerbation of AD neuropathology in a mouse model[39]. Furthermore, in the PD mouse model, nuclear inclusions containing α-synuclein (α-syn) species have been shown to induce transcriptional alterations involving an increase in p21 expression and additional genes related to SASP. These transcriptional changes are linked to DNA damage, lysosomal dysfunction, oxidative stress, chronic inflammation, and gliosis, ultimately accelerating neuronal loss and neurodegeneration[40]. Thus, cellular senescence represents not just a biomarker but a mechanistic driver of brain aging and neurodegeneration.
Extracellular vesicles (EVs) are heterogeneous lipid-bilayer particles released by all cells and serve as critical mediators of intercellular communication by transferring proteins, nucleic acids, and lipids between cells[41]. EVs are classified into three primary subtypes based on biogenesis pathway and diameter: (1) Exosomes (30-150 nm), formed through the endosomal pathway via inward budding of multivesicular bodies (MVBs) and released upon MVB-plasma membrane fusion, enriched in tetraspanins (CD63, CD81) and endosomal sorting complex proteins (ALIX, TSG101); (2) Microvesicles (100-500 nm), generated by direct outward budding and fission of the plasma membrane, carrying surface phosphatidylserine and selectins; and (3) Apoptotic bodies (500-5,000 nm), produced during programmed cell death through plasma membrane blebbing, containing nuclear fragments and histones[42-44]. In the context of senescence and neurodegeneration, EVs play a dual role involving propagating pathology and offering therapeutic promise[45]. During aging or pathological insult, neurons and glial cells release EVs enriched with SASP[30,46-48], neurotoxic protein aggregates (such as prion and mutant huntingtin protein)[49,50], and regulatory microRNAs (e.g., G3BP1)[51], ultimately accelerating neurodegenerative processes. Conversely, EVs have emerged as promising diagnostic and therapeutic tools[52]. Plasma and brain-derived EVs circulating in biofluids contain disease-specific biomarkers, such as Aβ, p-Tau217, and p-Tau181 for AD[53-55], oligomeric α-syn for PD[56], and TDP-43 in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)[57], enabling non-invasive disease monitoring. Moreover, engineered EVs can deliver therapeutic cargoes, including siRNAs targeting mutant mRNA[58], miRNA targeting senescent cells[59,60], neurotrophic factors[61], or even functional mitochondria[62], thereby rejuvenating aged or damaged neural cells[63,64]. Recent evidence supports the promising role of stem cell-derived EVs in counteracting senescence and age-related phenotypes involving multiple brain-related diseases and disorders[65-67]. Therefore, this review emphasizes the stem cell-derived EVs’ influence on brain aging, senescence, and age-associated brain diseases, and how they can be leveraged to restore brain homeostasis and cognitive function.
EVs-BASED THERAPEUTICS FOR BRAIN AGING
Neuroaging is mediated by interconnected biological phenomena, including three main pathophysiological mechanisms: chronic neuroinflammation, synaptic dysfunction, and impaired neurogenesis[25]. These mechanisms collectively undermine cognitive resilience and increase susceptibility to neurodegeneration, synergistically driving brain aging[25,68]. Previous studies have demonstrated that aging is accompanied by brain atrophy, synaptic loss, degeneration of white matter and myelin, hippocampal shrinkage, and progressive cognitive deficits[69-74]. Notably, a recent study demonstrated that EVs derived from antler blastema progenitor cells [EVABPC: skeletal mesenchymal stem cells (MSCs) found in regenerating deer antlers] improved brain function in aged models. Specially, administration of these EVsABPC in aged C57BL/6J mice (18 months old) via tail vein injection (40 µg, 3 times weekly for 4 weeks) and to aged rhesus macaques (16-18 years old) via intravenous injection (15 mg, every two weeks for 20 weeks) significantly enhanced physical and cognitive performance, reduced microlgial activation, and reversed epigenetic age by over 3 months in aged mice and more than 2 years[75]. Likewise, in aged male Wistar rats (12-13 months old), a single stereotactic injection of adipose tissue stem cell-derived extracellular vesicles (ADSC-EVs; 40 or 100 μg per rat for either 3 or 7 days) conjugated with Exo-pep-11, into the lateral ventricle significantly enhanced neural stem cell (NSC) (cells capable of differentiation into neurons, astrocytes, and oligodendrocytes) self-renewal. Mechanistically, Exo-pep-11 targets EphA4, a receptor tyrosine kinase that normally upregulates phosphatase and tension homolog (PTEN) to suppress protein kinase B (PKB/AKT) phosphorylation, thereby limiting NSC proliferation[76]. Blocking EphA4 signaling reduced PTEN expression and activated AKT signaling, which promoted NSC proliferation, survival, and self-renewal[77]. These effects are marked by an increase in NSC proliferation and elevated expression of Nestin (an intermediate filament protein essential for NSC cytoskeletal integrity and self-renewal) and DNA-binding protein ID1 (a transcriptional regulator that maintains NSC self-renewal by inhibiting differentiation). Furthermore, these EVs also promotes neurogenesis (the generation of new neurons from NSCs) in the subventricular zone-olfactory bulb axis in these rats, as evidenced by increases in tyrosine hydroxylase (TH: a key enzyme in dopamine synthesis and a marker of dopaminergic neuron differentiation) and Tuj1, a neuron-specific class III
EV-BASED THERAPEUTICS FOR CELLULAR SENESCENCE-ASSOCIATED NEURODEGENERATION
While neurodegeneration and cognitive decline are hallmarks of organismal aging, they are also mirrored by cellular senescence, a phenomenon that can occur independently of age in response to stress, injury, or genetic predisposition[92,93]. For example, neurological insults such as stroke (significant interruption in cerebral blood flow that leads to severe focal brain injuries), chronic hypoxia/ischemia (a sustained reduction in oxygen and nutrient supply that disrupts neuronal metabolism and accelerates neurodegeneration), metabolic stress (e.g., insulin resistance), and neurodegenerative diseases accelerate senescence across multiple neural populations, including NSCs, neurons, and microglia[94-97]. Senescent neural cells not only lose their functional potential but also promote neurodegeneration through the SASP[98]. Recent evidence highlights the therapeutic potential of EVs derived from various stem cell sources in mitigating neural senescence and restoring normal functioning. For instance, in an induced neural stem cell (iNSC) model of ischemic stroke, EVs from hypoxic-preconditioned human embryonic stem cells (hESCs-HypoxEVs, 40 ug/mL for 24 h), enriched with glutathione (GSH) redox system, effectively attenuated cellular senescence by significantly reducing expression of the senescence markers p16 and SA-β-Gal, while also suppressing key SASP factors including IL-6, CXCL8, and MMP9[95]. Complementing these findings, a study using primary NSCs from neonatal C57BL/6J mice demonstrated that NSCs subjected to oxygen-glucose deprivation (OGD) undergo senescence, leading to a decline in both NSC differentiation and neurogenesis[63]. Likewise, in an in vitro insulin-resistant neural stem and progenitor cell (NSPC) model, insulin and palmitic acid (IPA) induced senescence characterized by upregulation of p16INK4a and p21, and impairment of IRS-1/Forkhead box O (FoxO) signaling, which reduced FoxO1/3a recruitment to proliferation-related genes such as Cyclin D and SRY-box transcription factor 2 (SOX2). In contrast, treatment of cultured NSPCs with NSPC-derived exosomes (2.5 μg of vesicles per 1 × 106 cells) for 3 days significantly mitigated IPA-induced alterations and reversed proliferation deficits, thereby counteracting the senescent phenotype[96]. Furthermore, in a D-galactose-induced BV2 microglial senescence model, induced pluripotent stem cell (iPSC)-derived EVs (iPSC-EVs, 1 × 109 particles/mL) significantly reduced senescence markers (p16, p21, p53, γ-H2AX, and SA-β-Gal) and attenuated classically activated M1 microglia pro-inflammatory polarization, while promoting an anti-inflammatory phenotype. Mechanistically, these EVs deliver TGF-β1, which upregulates Rictor and activates AKT phosphorylation, thereby suppressing both inflammation and senescence[93]. Moreover, in vivo studies in senescence-accelerated mouse prone 8 (SAMP8) and the C57BL/6 mouse models of varying ages identified age-related senescence in hippocampal NSCs (H-NSCs), linked to reduced hippocampal neurogenesis, a critical process for maintaining and restoring hippocampus-dependent cognitive functions. This reduction contributes to cognitive deficits often observed in aging and neurodegenerative disorders. In contrast, intravenous administration of ESCs-EVs counteracts H-NSCs senescence in both SAMP8 (6-month treatment) and C57BL/6 (8-month treatment) mice. This effect is mediated through the upregulation of myelin transcription factor 1 (MYT1), which activates downstream signaling pathways involving SIRT1, nicotinamide phosphoribosyl transferase (NAMPT), and hypoxia-inducible factor 2 subunit α (HIF-2α)[99-101]. Additionally, MSC-derived exosomes (MSCs-Exos) have been shown to reduce oxidative stress, exert anti-apoptotic effects, and increase SIRT1 levels, thereby preventing brain aging in SAMP8 mice[66]. Mechanistically, SIRT1, NAMPT, and HIF-2α form a coordinated axis that regulates neurogenesis by linking metabolic and hypoxic signaling. NAMPT maintains NAD+ levels, enabling SIRT1 activation, which deacetylates HIF-2α to enhance its stability and transcriptional activity, thereby promoting NSC survival, differentiation, and vascular support[102-105].
EV-BASED THERAPEUTICS FOR AGE-RELATED CHRONIC DISEASES
Age-related chronic diseases, including AD and PD, result from progressive cellular and molecular impairments[106,107]. These impairments overlap with those of brain aging, thereby accelerating cognitive decline and neurodegeneration[108-111]. Emerging evidence highlights stem cell-derived EVs as a promising therapeutic agent, capable of delivering bioactive molecules that restore neural homeostasis and attenuate disease progression. Understanding these mechanisms is essential for developing targeted strategies to preserve brain health during aging.
EV-BASED THERAPEUTICS FOR AD
AD represents a prime example of accelerated neuroaging, pathologically characterized by the accumulation of Aβ plaques and neurofibrillary tangles composed of hyperphosphorylated tau protein[41,112]. Epidemiological studies demonstrate that the risk of developing AD doubles every decade after age 65[113]. Mounting evidence indicates senescent cell accumulation in the brain of AD patients[114-116], suggesting their potential role in disease progression. Supporting this, pharmacological clearance of senescent cells in AD mouse models has been shown to reduce Aβ deposition, tau pathology, and improve cognitive function[117-120]. In parallel, recent studies demonstrated that exposing human iPSC-derived neurons to human iPSC-NSC-EVs (6 × 109 EVs) effectively counteracts Aβ42-induced neurotoxicity through multifaceted mechanisms, including antioxidant, anti-apoptotic, pro-autophagic, and anti-tauopathy effects, positioning them as a promising cell-free therapeutic approach for AD[97]. Likewise, mesenchymal stem cell-derived EVs enriched with tyrosine phosphatase-2 (SHP2) (MSCs-EVs-SHP2) have been shown to induce mitophagy in SH-SY5Y neuronal cells in vitro and an experimental model of AD [intracerebroventricular injection of Aβ1-42 (5 mg/mL)] with a treatment regimen of 100 µg of EVs per mouse every two days, for 2 weeks. These EVs enhanced mitochondrial function, reduced NLRP3 inflammasome activation, and decreased neuronal apoptosis in the cerebral cortex, ultimately rescuing synaptic loss and improving cognitive function[121]. Additionally, both MSCs-EVs and iNSCs-EVs exhibit anti-aging effects by mitigating AD-like phenotypes. In four-month-old 5XFAD mice, intravenous administration of MSCs-EVs and iNSCs-EVs (0.5 μg/μL, every three days for one month) has been shown to reduce amyloid plaques, inflammatory responses, and improve dendritic spine density and arborization in the prefrontal cortex and hippocampus, thereby enhancing learning and memory functions[122]. In another study using two- or six-month-old 5XFAD mice, intravenous (retro-orbital vein) administration of hNSCs-EVs (once or twice over 4-6 weeks) has been shown to reduce microglial activation, lower pro-inflammatory cytokine levels, decrease amyloid-beta plaque accumulation, mitigate synaptic loss, and improve cognitive function[123]. Similarly, in 3-month-old 5xFAD mice, intranasally delivered hiPSCs-NSCs-EVs (30 × 109 EVs, once weekly for 2 weeks) were internalized by astrocytes and microglia, surrounding the plaques. Moreover, these EVs diminish microglia activation by downregulating genes associated with NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome components and IFN-1 signaling pathways. Concurrently, these EVs inhibit Interleukin 6 (IL-6) and IFN-1 signaling in astrocytes. Overall, EV therapy reduced hippocampal amyloid-beta plaques, p-tau accumulation, astrocyte hypertrophy, and microglial clustering, hence enhancing cognitive functions[124].
EV-BASED THERAPEUTICS FOR PD
PD is another age-related neurodegenerative disorder characterized by the progressive loss of dopaminergic neurons in the substantia nigra (SN) and accumulation of α-syn protein aggregates, driven by interconnected cellular pathologies including mitochondrial dysfunction, impaired protein clearance, chronic neuroinflammation, and selective neuronal vulnerability to oxidative stress and calcium dysregulation[125,126]. This degenerative cascade is initiated by the downregulation of key regulatory molecules such as the neuroprotective proteins (Parkin), the synaptic regulator [DJ-1, also known as Parkinson’s disease protein 7 (PARK7)], and dysregulated microRNAs (including miR-181a-2-3p)[127-129]. miR-181a-2-3p, which plays a key role in regulating autophagy and apoptosis, has been consistently found to be downregulated in PD, both in patient tissues and experimental animal models[129,130]. This downregulation has been further confirmed in SH-SY5Y cells and PD mouse models following exposure to 6-hydroxydopamine (6-OHDA), a neurotoxin commonly used to stimulate PD pathology[131]. However, treatment with MSC-EVs (200 µg EV-protein, every 3 days for 8 weeks) effectively restored miR-181a-2-3p expression in both SH-SY5Y cells and mouse brain tissue (administered via tail vein). Restoration of this microRNA led to enhanced neuronal viability and antioxidant defenses, marked by increased superoxide dismutase (SOD) levels, and concurrently reduced apoptosis and oxidative stress indicators such as ROS and malondialdehyde (MDA). Mechanistically, miR-181a-2-3p directly targets and suppresses early growth response 1 (EGR1), a transcription factor that upregulates NOX4, a major driver of oxidative stress through activation of the p38 MAPK pathway, thus alleviating oxidative damage and supporting neuronal cell survival[131]. Furthermore, in the same mouse model, administration of human umbilical cord MSC-derived exosomes (hucMSCs-Exos; 200 µg EV protein, every 3 days for 8 weeks, tail vein injection) has been demonstrated to reach the SN, significantly reduce dopaminergic neuronal loss, and upregulate dopamine levels and its metabolites[132]. Likewise, in the rotenone-induced rat model of PD, repeated tail vein administration of neural-induced human adipose tissue-derived stem cell exosomes (NI-hADSC-Exos,
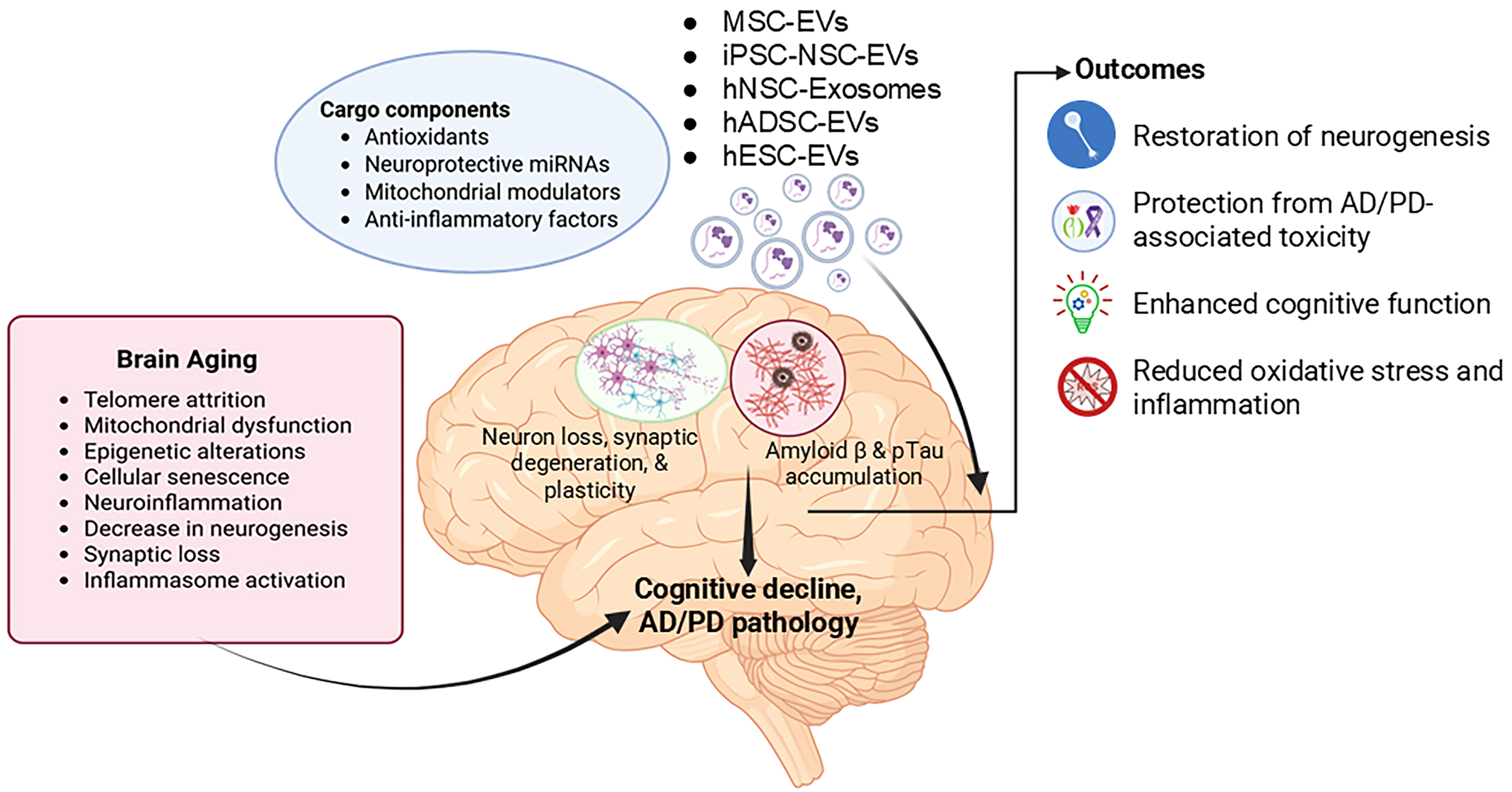
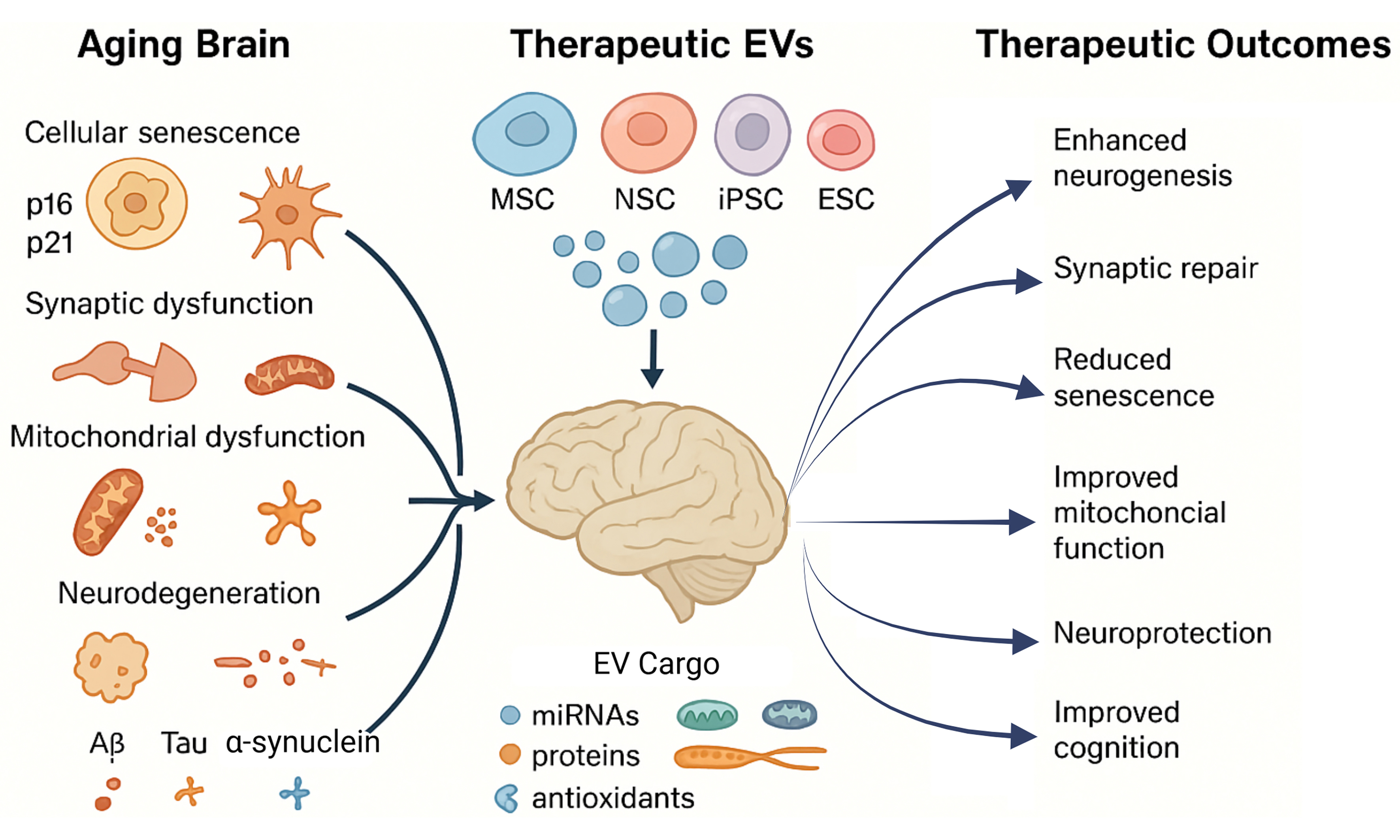
Figure 1. Schematic representation of therapeutic intervention by stem cell-derived EVs in Aging. Aging triggers cellular senescence, neuroinflammation, and synaptic dysfunction, driving cognitive decline and contributing to Alzheimer’s and Parkinson’s disease pathology. Stem cell-derived extracellular vesicles carry antioxidants, neuroprotective microRNAs, mitochondrial regulators, and anti-inflammatory molecules. These vesicles enhance neurogenesis, protect against neurotoxicity, alleviate oxidative stress and inflammation, and improve cognitive function. (Created in BioRender. https://BioRender.com/u7czgbc). EVs: Extracellular vesicles; MSC: mesenchymal stem cell; iPSC-NSCs: induced pluripotent stem cell-derived neural stem cells; hNSCs: human neural stem cells; hADSCs: human adipose-derived stem cells; AD: Alzheimer’s disease; PD: Parkinson’s disease.
Therapeutic effects of stem cell-derived extracellular vesicles on aging and neurodegenerative disorders
| Stem cell type | EV source | Administration | Mechanism/Target | Disease model | Reference |
| Antler blastema stem cells | Antler pedicle periosteum and regenerating antler tissue of sika deer | Tail vein injection | Intravenous EVs improved physical performance, enhanced cognitive function, and reduced systemic inflammation in aged mice, while reversing epigenetic age by over 3 months. In macaques, EV treatment was also neuroprotective, reduced inflammation, improved locomotor function, and reduced epigenetic age by over 2 years | Aged macaques and mice | Hao et al. (2025)[75] |
| Mesenchymal stem cells | Bone marrow-derived MSCs of a single young monkey | Intravenous | EV treatment promoted debris clearing, anti-inflammatory C1q+ microglia in the perilesional motor cortex, aiding recovery. In premotor regions, EVs reduced C1q+ synaptic tagging and microglia-spine contacts, suggesting improved synaptic plasticity through local damage clearance and prevention of chronic inflammation | Aged rhesus macaques with cortical injury | Zhou et al. (2023)[87] |
| Mesenchymal stem cells | Bone marrow-derived MSCs of a single young monkey | Intravenous | EV treatment reduced oligodendrocyte damage and enhanced myelin maintenance in sublesional white matter. These effects correlated with motor recovery, suggesting EVs promote repair and functional improvement in the aged brain | Aged rhesus macaques | Go et al. (2021)[89] |
| hESCs | Hypoxia-conditioned hESC line BG01 (WiCell Research Institute) | Intravenous injection | hESC-EVs reversed neural stem cell senescence after ischemic injury by suppressing SASP factors and restoring neuroprotection, with hypoxia-preconditioned EVs showing enhanced GSH-dependent anti-senescence activity. MFGE-8-coated hESC-HypoxEVs further promoted neurogenesis and improved sensorimotor recovery | MCAO model from eight-week-old male SD rats | Lee et al. (2025)[95] |
| Postnatal hippocampal NSPC | Newborn (0-1 days) C57bl/6 mice | Intranasal administration of exosomes from NSPC | NSPC-derived EVs reverse insulin resistance-induced senescence by restoring IRS-1/FoxO signaling, reducing p21, and preserving hippocampal neurogenesis | Thirty- to 35-day old C57BL/6 mice | Natale et al. (2022)[96] |
| Human ESC-sEVs | Human ESC (H9) provided by the Institute of Biochemistry and Cell Biology of the Chinese Academy of Sciences (Shanghai, China) | Intravenous injection | ESC-sEVs rejuvenate aging hippocampal NSCs by transferring SMAD4/5 to activate the MYT1-Egln3-SIRT1 axis, restoring neurogenesis and cognition | SAMP8 & C57BL/6 mice | Hu et al. (2021)[99] |
| MSC-Exos | Mouse BM-MSCs | Tail vein injection | MSC-derived exosomes delay brain aging by activating SIRT1, reducing oxidative stress and apoptosis, and improving cognition | SAMP8 senescent mice | Zhang et al. (2023)[66] |
| hiPSC-NSC-EVs | hiPSC-NSC | In vitro | hiPSC-NSC-EVs protect neurons from Aβ-42-induced senescence-like changes by reducing oxidative stress, restoring mitochondrial function, and suppressing tau pathology | Mature human neurons differentiated from human NSCs | Rao et al. (2025)[97] |
| Mesenchymal stem cells | Engineered mesenchymal stem cell-derived extracellular vesicles with tyrosine phosphatase-2 (SHP2) high expression (MSC-EVs-SHP2) | Intravenous injection | SHP2-enriched MSC-EVs induce mitophagy, reducing mitochondrial damage, neuroinflammation, and cognitive decline in the AD mice model | AD was established in C57BL/6 mice by injecting 5 mg/mL-1 β-amyloid (1-42) into the lateral ventricle of the cerebrum | Xu et al. (2022)[121] |
| Mouse NSCs and iNSCs | Conditioned medium of cultured mouse NSCs and iNSCs | intravenous injection | NSC-EVs mitigate AD-related aging phenotypes by reducing amyloid and inflammation, restoring synapses, and improving cognition | 5XFAD mice | Gao et al. (2023)[122] |
| hNSC | EVs from conditioned medium of cultured hNSCs | RO sinus injection | hNSC-EVs mitigate Alzheimer’s hallmarks by reducing amyloid and tau pathology, restoring synapses, and improving cognition | 5XFAD mice | Apodaca et al. (2021)[123] |
| Mesenchymal stem cells | EVs from hucMSCs | Tail vein injection | MSC-EVs deliver miR-181a-2-3p to inhibit the EGR1/NOX4 pathway, reducing oxidative stress and protecting dopaminergic neurons in PD | 6-OHDA-induced PD model (SH-SY5Y cells and C57BL/6 mice) | Ma et al. (2022)[131] |
| Mesenchymal stem cells | EVs from hucMSCs | Intravenous injection | hucMSC-Exos induce autophagy, protect dopaminergic neurons, restore dopamine, and improve motor function in PD models. | 6-OHDA-induced PD model (SH-SY5Y cells and adult male SD rats) | Chen et al. (2020)[132] |
| NI-hADSC | NI-hADSC-Exo from adipose tissues of human donors | Intravenous injection | NI-hADSC-Exos clear α-syn aggregates, restore autophagy, reduce inflammation, and protect dopaminergic neurons in PD rats | Rotenone (subcutaneous)-induced Parkinson’s disease rat model | Ramalingam et al. (2025)[133] |
CONCLUSION AND FUTURE PERSPECTIVES
Stem cell-derived EVs have gained increasing interest as a potential non-cellular therapeutic platform to counteract brain aging, cellular senescence, and age-related brain disorders, including AD and PD. These nanosized lipid bilayer vesicles are endogenous carriers of proteins, nucleic acids, lipids, and metabolites, which parallels the molecular signature of their parent cells. In contrast to stem cell transplantations, EV-based therapy avoids risks such as tumorigenicity, immune rejection, and ethical concerns, while retaining the same important properties in tissue repair and immune modulation. As discussed in this article, EVs from MSCs, NSCs, hESCs, and iPSCs have demonstrated significant potential in rejuvenating senescent neural cell populations, promoting neurogenesis, restoring synaptic plasticity, and ameliorating behavioral deficits in diverse models of aging and neurodegeneration[59,64,75,90]. For instance, in aged mice and rhesus macaques, EVs isolated from antler blastema progenitor cells significantly reversed epigenetic age, improved cognitive function, and reduced systemic inflammation[75]. Additionally, EVs from adipose-derived stem cells conjugated with Exo-pep-11 enhanced neural stem cell proliferation by targeting the EphA4/PTEN/AKT axis, thereby improving self-renewal and neurogenesis[78]. Hypoxia-preconditioned hESC-EVs enriched with GSH suppressed senescence markers and SASP in ischemia-induced NSCs[95], while iPSC-derived EVs significantly lowered markers of cellular aging and shifted microglia from a pro-inflammatory (M1) state toward a more anti-inflammatory profile[93]. Similar regenerative effects have been observed across PD models, where stem-cell EVs restored dopaminergic neuronal function, reduced α-syn aggregates, and modulated neuroinflammation via microRNA- and mitophagy-based mechanisms[131-133].
Despite these promising results, clinical translation of stem cell-derived EV therapies is limited by a number of unresolved biological and technical barriers. One of the biggest concerns is that of immunocompatibility. While EVs are commonly described as immune-evasive due to the lack of MHC class II molecules, emerging evidence suggests that allogeneic EVs could still carry immune-surface proteins or membrane-bound antigens that could lead to an immune response, especially upon repeated systemic exposure[134,135]. Another obstacle is the heterogeneity of EV populations. Even within EVs secreted by a specific cell type, significant heterogeneity can be observed with respect to size, content, and surface markers, as determined by culture conditions, passage number, and EV biogenesis pathway[136,137]. This heterogeneity presents challenges to the reproducibility of therapeutic effects, as well as the standardization of manufacturing processes for clinical use. Moreover, the pharmacokinetics of EVs are suboptimal. Stem cell-derived EVs exhibit rapid clearance and poor brain penetration. For example, radiolabeling studies of human umbilical cord MSC-EVs with Zirconium-89 revealed a biphasic circulation pattern with a distribution phase half-life of ~28 min and an elimination phase half-life of ~12 h, with predominant accumulation in the liver and spleen, and < 0.05% of the injected dose reaching the brain[138]. Similarly, technetium-99m-labeled hUC-MSC-EVs showed a distribution phase half-life of ~1.13 min and an elimination phase half-life of ~45.98 min, with most vesicles sequestered in the liver and spleen, and only trace amounts (< 0.1%) detected in the brain[139]. These findings significantly limit their therapeutic efficacy in neurodegenerative settings.
The targeting of cells and functional delivery also further restricts the scope of therapeutic application of these EVs. The majority of EVs are taken up into cells by non-specific endocytosis, subsequently undergoing lysosomal degradation. Endosomal escape is a significant hurdle due to inefficient cytoplasmic delivery of the therapeutic RNA and protein cargo of the EVs[140]. Although still under preclinical investigation, some engineering strategies, such as the use of fusogenic peptides and pH-responsive coatings, are being examined[141]. Off-target effects remain another major concern, especially as EVs have been shown to accumulate in peripheral organs. For immunotherapy, tumor-promoting miRNAs or growth factors could, in principle, be passed through pluripotent stem cell-derived EVs[142]. These risks emphasize the importance of rigorous preclinical safety assessments and long-term toxicity studies. Additionally, the successful translation of EVs to clinical use will require the scalable and reproducible manufacturing of GMP-grade EVs through well-benchmarked protocols for isolating, characterizing, and testing for potency of EVs[143]. Storage conditions, such as cryopreservation and lyophilization, need to be optimized to preserve their bioactivity[144].
In summary, EVs from stem cells represent an exciting new therapeutic platform that has the potential to rejuvenate neural systems, intervene in senescence, and fight neurodegeneration. However, their efficient transfer from laboratory to clinic needs to overcome several barriers, including immunogenicity, heterogeneity, pharmacokinetics, biodistribution, targeting, endosomal escape, and large-scale production. Such challenges will require continued interdisciplinary collaboration across neuroscience, bioengineering, immunology, and regulatory science. With continuous research to overcome these obstacles, EVs derived from stem cells have the potential to emerge as a breakthrough treatment for neurodegenerative disorders and in the promotion of healthy brain aging.
DECLARATIONS
Acknowledgment
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The graphic abstract is created in Biorender (https://BioRender.com/lhz6dk0).
Authors’ contributions
Writing, reviewing, and editing: Kumar M, Ray S, Sil S
All authors have read and agreed to the published version of the manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
The authors are grateful to NIH for funding support (R21AG069541, RO1DA060755, RO1DA060753).
Conflicts of interest
Sil S is a Junior Editorial Board member of the journal Extracellular Vesicles and Circulating Nucleic Acids. Sil S was not involved in any steps of editorial processing, notably including reviewer selection, manuscript handling, or decision making. The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Tenchov R, Sasso JM, Wang X, Zhou QA. Aging hallmarks and progression and age-related diseases: a landscape view of research advancement. ACS Chem Neurosci. 2024;15:1-30.
2. Li Y, Tian X, Luo J, Bao T, Wang S, Wu X. Molecular mechanisms of aging and anti-aging strategies. Cell Commun Signal. 2024;22:285.
3. Chakravarti D, LaBella KA, DePinho RA. Telomeres: history, health, and hallmarks of aging. Cell. 2021;184:306-22.
4. Rossiello F, Jurk D, Passos JF, d’Adda di Fagagna F. Telomere dysfunction in ageing and age-related diseases. Nat Cell Biol. 2022;24:135-47.
5. Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22:75-95.
6. Lima T, Li TY, Mottis A, Auwerx J. Pleiotropic effects of mitochondria in aging. Nat Aging. 2022;2:199-213.
7. Wang K, Liu H, Hu Q, et al. Epigenetic regulation of aging: implications for interventions of aging and diseases. Signal Transduct Target Ther. 2022;7:374.
8. Li Y, Lu J, Cao X, et al. A newly synthesized rhamnoside derivative alleviates alzheimer’s amyloid-β-induced oxidative stress, mitochondrial dysfunction, and cell senescence through upregulating SIRT3. Oxid Med Cell Longev. 2020;2020:7698560.
9. Choo KB, Tai L, Hymavathee KS, et al. Oxidative stress-induced premature senescence in Wharton’s jelly-derived mesenchymal stem cells. Int J Med Sci. 2014;11:1201-7.
10. Dasgupta N, Arnold R, Equey A, Gandhi A, Adams PD. The role of the dynamic epigenetic landscape in senescence: orchestrating SASP expression. NPJ Aging. 2024;10:48.
11. Martínez-Zamudio RI, Stefa A, Nabuco Leva Ferreira Freitas JA, et al. Escape from oncogene-induced senescence is controlled by POU2F2 and memorized by chromatin scars. Cell Genom. 2023;3:100293.
12. González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021;288:56-80.
13. Tuttle CSL, Waaijer MEC, Slee-Valentijn MS, Stijnen T, Westendorp R, Maier AB. Cellular senescence and chronological age in various human tissues: a systematic review and meta-analysis. Aging Cell. 2020;19:e13083.
14. Ajoolabady A, Pratico D, Bahijri S, et al. Hallmarks and mechanisms of cellular senescence in aging and disease. Cell Death Discov. 2025;11:364.
15. Keshavjee B, Lambelet V, Coppola H, et al. Stress-induced premature senescence related to oxidative stress in the developmental programming of nonalcoholic fatty liver disease in a rat model of intrauterine growth restriction. Antioxidants. 2022;11:1695.
16. Larsson LG. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin Cancer Biol. 2011;21:367-76.
17. Luo J, Sun T, Liu Z, et al. Persistent accumulation of therapy-induced senescent cells: an obstacle to long-term cancer treatment efficacy. Int J Oral Sci. 2025;17:59.
18. Saleh T, Bloukh S, Carpenter VJ, et al. Therapy-induced senescence: an “old” friend becomes the enemy. Cancers. 2020;12:822.
19. Dehkordi SK, Walker J, Sah E, et al. Profiling senescent cells in human brains reveals neurons with CDKN2D/p19 and tau neuropathology. Nat Aging. 2021;1:1107-16.
20. Herdy JR, Mertens J, Gage FH. Neuronal senescence may drive brain aging. Science. 2024;384:1404-6.
21. Valentijn FA, Falke LL, Nguyen TQ, Goldschmeding R. Cellular senescence in the aging and diseased kidney. J Cell Commun Signal. 2018;12:69-82.
22. Salerno N, Marino F, Scalise M, et al. Pharmacological clearance of senescent cells improves cardiac remodeling and function after myocardial infarction in female aged mice. Mech Ageing Dev. 2022;208:111740.
23. Li Z, Zhang Z, Ren Y, et al. Aging and age-related diseases: from mechanisms to therapeutic strategies. Biogerontology. 2021;22:165-87.
25. Yang Z, Wen J, Erus G, et al. Brain aging patterns in a large and diverse cohort of 49,482 individuals. Nat Med. 2024;30:3015-26.
26. Ho NCW, Bethlehem RAI, Seidlitz J, et al; Lifespan Brain Chart Consortium. Atypical brain aging and its association with working memory performance in major depressive disorder. Biol Psychiatry Cogn Neurosci Neuroimaging. 2024;9:786-99.
27. Antoniades M, Srinivasan D, Wen J, et al. Relationship between MRI brain-age heterogeneity, cognition, genetics and Alzheimer’s disease neuropathology. EBioMedicine. 2024;109:105399.
28. Ya J, Bayraktutan U. Senolytics and senomorphics targeting p38MAPK/NF-κB pathway protect endothelial cells from oxidative stress-mediated premature senescence. Cells. 2024;13:1292.
29. Rezagholizadeh N, Datta G, Hasler WA, et al. SLC38A9 is directly involved in Tat-induced endolysosome dysfunction and senescence in astrocytes. Life Sci Alliance. 2025;8:e202503231.
30. Gross PS, Durán-Laforet V, Ho LT, et al. Senescent-like microglia limit remyelination through the senescence associated secretory phenotype. Nat Commun. 2025;16:2283.
31. Luo N, Zhu W, Li X, et al. Defective autophagy of pericytes enhances radiation-induced senescence promoting radiation brain injury. Neuro Oncol. 2024;26:2288-304.
32. Budamagunta V, Kumar A, Rani A, et al. Effect of peripheral cellular senescence on brain aging and cognitive decline. Aging Cell. 2023;22:e13817.
33. Zhang W, Sun HS, Wang X, Dumont AS, Liu Q. Cellular senescence, DNA damage, and neuroinflammation in the aging brain. Trends Neurosci. 2024;47:461-74.
34. Cuanalo-Contreras K, Schulz J, Mukherjee A, Park KW, Armijo E, Soto C. Extensive accumulation of misfolded protein aggregates during natural aging and senescence. Front Aging Neurosci. 2022;14:1090109.
35. Herdy JR, Traxler L, Agarwal RK, et al. Increased post-mitotic senescence in aged human neurons is a pathological feature of Alzheimer’s disease. Cell Stem Cell. 2022;29:1637-52.e6.
36. Jiang SY, Tian T, Yao H, et al. The cGAS-STING-YY1 axis accelerates progression of neurodegeneration in a mouse model of Parkinson’s disease via LCN2-dependent astrocyte senescence. Cell Death Differ. 2023;30:2280-92.
37. Jurk D, Wang C, Miwa S, et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell. 2012;11:996-1004.
38. :2024.01.20.576299. Donovan LJ, Brewer CL, Bond SF, et al. Aging and injury drive neuronal senescence in the dorsal root ganglia. bioRxiv 2024; bioRxiv:2024.01.20.576299.
39. Choi I, Wang M, Yoo S, et al. Autophagy enables microglia to engage amyloid plaques and prevents microglial senescence. Nat Cell Biol. 2023;25:963-74.
40. Du T, Li G, Zong Q, Luo H, Pan Y, Ma K. Nuclear alpha-synuclein accelerates cell senescence and neurodegeneration. Immun Ageing. 2024;21:47.
41. Chemparathy DT, Ray S, Ochs C, et al. Neuropathogenic role of astrocyte-derived extracellular vesicles in HIV-associated neurocognitive disorders. J Extracell Vesicles. 2024;13:e12439.
42. van der Pol E, Böing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. 2012;64:676-705.
43. Doyle LM, Wang MZ. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells. 2019;8:727.
44. Odegaard KE, Chand S, Wheeler S, et al. Role of extracellular vesicles in substance abuse and HIV-related neurological pathologies. Int J Mol Sci. 2020;21:6765.
45. Scuteri A, Donzelli E. Dual role of extracellular vesicles in neurodegenerative diseases. World J Stem Cells. 2024;16:1002-11.
46. Schnatz A, Müller C, Brahmer A, Krämer-Albers EM. Extracellular vesicles in neural cell interaction and CNS homeostasis. FASEB Bioadv. 2021;3:577-92.
47. Wallis R, Mizen H, Bishop CL. The bright and dark side of extracellular vesicles in the senescence-associated secretory phenotype. Mech Ageing Dev. 2020;189:111263.
48. Guix FX, Capitán AM, Casadomé-Perales Á, et al. Increased exosome secretion in neurons aging in vitro by NPC1-mediated endosomal cholesterol buildup. Life Sci Alliance. 2021;4:e202101055.
49. Soukup J, Moško T, Kereïche S, Holada K. Large extracellular vesicles transfer more prions and infect cell culture better than small extracellular vesicles. Biochem Biophys Res Commun. 2023;687:149208.
50. Jeon I, Cicchetti F, Cisbani G, et al. Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol. 2016;132:577-92.
51. Neueder A, Nitzschner P, Wagner R, et al. Huntington disease alters the actionable information in plasma extracellular vesicles. Clin Transl Med. 2024;14:e1525.
52. You B, Yang Y, Zhou Z, et al. Extracellular vesicles: a new frontier for cardiac repair. Pharmaceutics. 2022;14:1848.
53. Boyer E, Deltenre L, Dourte M, et al. Comparison of plasma soluble and extracellular vesicles-associated biomarkers in Alzheimer’s disease patients and cognitively normal individuals. Alzheimers Res Ther. 2024;16:141.
54. Fowler SL, Behr TS, Turkes E, et al. Tau filaments are tethered within brain extracellular vesicles in Alzheimer’s disease. Nat Neurosci. 2025;28:40-8.
55. Sattarov R, Havers M, Orbjörn C, et al. Phosphorylated tau in cerebrospinal fluid-derived extracellular vesicles in Alzheimer’s disease: a pilot study. Sci Rep. 2024;14:25419.
56. Gilboa T, Ter-Ovanesyan D, Wang SC, et al. Measurement of α-synuclein as protein cargo in plasma extracellular vesicles. Proc Natl Acad Sci U S A. 2024;121:e2408949121.
57. Chatterjee M, Özdemir S, Fritz C, et al. Plasma extracellular vesicle tau and TDP-43 as diagnostic biomarkers in FTD and ALS. Nat Med. 2024;30:1771-83.
58. Rufino-Ramos D, Albuquerque PR, Leandro K, et al. Extracellular vesicle-based delivery of silencing sequences for the treatment of Machado-Joseph disease/spinocerebellar ataxia type 3. Mol Ther. 2023;31:1275-92.
59. Xiao X, Xu M, Yu H, et al. Mesenchymal stem cell-derived small extracellular vesicles mitigate oxidative stress-induced senescence in endothelial cells via regulation of miR-146a/Src. Signal Transduct Target Ther. 2021;6:354.
60. Mas-Bargues C, Sanz-Ros J, Román-Domínguez A, et al. Extracellular vesicles from healthy cells improves cell function and stemness in premature senescent stem cells by miR-302b and HIF-1α activation. Biomolecules. 2020;10:957.
61. Larrow DR, Cohen MS. Endoscopic approach for tympanostomy tube insertion in patients with trisomy 21. Otol Neurotol. 2025;46:e206.
62. Dave KM, Venna VR, Rao KS, et al. Mitochondria-containing extracellular vesicles from mouse vs. human brain endothelial cells for ischemic stroke therapy. J Control Release. 2024;373:803-22.
63. Liu J, Peng L, He L, et al. Induced mesenchymal stem cells-small extracellular vesicles alleviate post-stroke cognitive impairment by rejuvenating senescence of neural stem cells. J Mol Neurosci. 2024;74:29.
64. Yu L, Wen H, Liu C, et al. Embryonic stem cell-derived extracellular vesicles rejuvenate senescent cells and antagonize aging in mice. Bioact Mater. 2023;29:85-97.
65. Chun C, Smith AST, Kim H, et al. Astrocyte-derived extracellular vesicles enhance the survival and electrophysiological function of human cortical neurons in vitro. Biomaterials. 2021;271:120700.
66. Zhang X, Liu T, Hou X, et al. Exosomes secreted by mesenchymal stem cells delay brain aging by upregulating SIRT1 expression. Sci Rep. 2023;13:13213.
67. Li Q, Niu X, Yi Y, et al. Inducible pluripotent stem cell-derived small extracellular vesicles rejuvenate senescent blood-brain barrier to protect against ischemic stroke in aged mice. ACS Nano. 2023;17:775-89.
69. Head D, Rodrigue KM, Kennedy KM, Raz N. Neuroanatomical and cognitive mediators of age-related differences in episodic memory. Neuropsychology. 2008;22:491-507.
70. Mungas D, Harvey D, Reed BR, et al. Longitudinal volumetric MRI change and rate of cognitive decline. Neurology. 2005;65:565-71.
71. Fjell AM, Walhovd KB. Structural brain changes in aging: courses, causes and cognitive consequences. Rev Neurosci. 2010;21:187-221.
72. Rajah MN, Kromas M, Han JE, Pruessner JC. Group differences in anterior hippocampal volume and in the retrieval of spatial and temporal context memory in healthy young versus older adults. Neuropsychologia. 2010;48:4020-30.
73. Bethlehem RAI, Seidlitz J, White SR, et al; 3R-BRAIN, AIBL, Alzheimer’s Disease Neuroimaging Initiative, Alzheimer’s Disease Repository Without Borders Investigators, CALM Team, Cam-CAN, CCNP, COBRE, cVEDA, ENIGMA Developmental Brain Age Working Group, Developing Human Connectome Project, FinnBrain, Harvard Aging Brain Study, IMAGEN, KNE96, Mayo Clinic Study of Aging, NSPN, POND, PREVENT-AD Research Group, VETSA. Brain charts for the human lifespan. Nature. 2022;604:525-33.
74. Bennett IJ, Madden DJ. Disconnected aging: cerebral white matter integrity and age-related differences in cognition. Neuroscience. 2014;276:187-205.
75. Hao Y, Yu B, Qin M, et al. Extracellular vesicles from antler blastema progenitor cells reverse bone loss and mitigate aging-related phenotypes in mice and macaques. Nat Aging. 2025;5:1790-809.
76. Chen Q, Liu J, Sawada T, Wei C, Wu S, Han F. Possible role of EphA4 and VEGFR2 interactions in neural stem and progenitor cell differentiation. Exp Ther Med. 2020;19:1789-96.
77. Yokote H, Fujita K, Jing X, et al. Trans-activation of EphA4 and FGF receptors mediated by direct interactions between their cytoplasmic domains. Proc Natl Acad Sci U S A. 2005;102:18866-71.
78. Ghosh S, Roy R, Mukherjee N, et al. EphA4 targeting peptide-conjugated extracellular vesicles rejuvenates adult neural stem cells and exerts therapeutic benefits in aging rats. ACS Chem Neurosci. 2024;15:3482-95.
79. Jankovic V, Ciarrocchi A, Boccuni P, DeBlasio T, Benezra R, Nimer SD. Id1 restrains myeloid commitment, maintaining the self-renewal capacity of hematopoietic stem cells. Proc Natl Acad Sci U S A. 2007;104:1260-5.
80. Harischandra DS, Rokad D, Ghaisas S, et al. Enhanced differentiation of human dopaminergic neuronal cell model for preclinical translational research in Parkinson’s disease. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165533.
81. Lee S, Choi K, Ahn H, Song K, Choe J, Lee I. TuJ1 (class III beta-tubulin) expression suggests dynamic redistribution of follicular dendritic cells in lymphoid tissue. Eur J Cell Biol. 2005;84:453-9.
82. Park D, Xiang AP, Mao FF, et al. Nestin is required for the proper self-renewal of neural stem cells. Stem Cells. 2010;28:2162-71.
83. Winship IR, Murphy TH. Remapping the somatosensory cortex after stroke: insight from imaging the synapse to network. Neuroscientist. 2009;15:507-24.
84. Singh AD, Kumar M, Swathi BH, Bhargavi P, Godbole A, Khushu S. Age-related cortical changes and cognitive performance in healthy adults. Brain Cogn. 2025;187:106306.
85. Floel A, Cohen LG. Recovery of function in humans: cortical stimulation and pharmacological treatments after stroke. Neurobiol Dis. 2010;37:243-51.
86. Festa LK, Grinspan JB, Jordan-Sciutto KL. White matter injury across neurodegenerative disease. Trends Neurosci. 2024;47:47-57.
87. Zhou Y, Bhatt H, Mojica CA, et al. Mesenchymal-derived extracellular vesicles enhance microglia-mediated synapse remodeling after cortical injury in aging Rhesus monkeys. J Neuroinflammation. 2023;20:201.
88. Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. 2010;112:733-43.
89. Go V, Sarikaya D, Zhou Y, et al. Extracellular vesicles derived from bone marrow mesenchymal stem cells enhance myelin maintenance after cortical injury in aged rhesus monkeys. Exp Neurol. 2021;337:113540.
90. Medalla M, Chang W, Calderazzo SM, et al. Treatment with mesenchymal-derived extracellular vesicles reduces injury-related pathology in pyramidal neurons of monkey perilesional ventral premotor cortex. J Neurosci. 2020;40:3385-407.
91. Go V, Bowley BGE, Pessina MA, et al. Extracellular vesicles from mesenchymal stem cells reduce microglial-mediated neuroinflammation after cortical injury in aged Rhesus monkeys. Geroscience. 2020;42:1-17.
92. Chang J, Li Y, Shan X, et al. Neural stem cells promote neuroplasticity: a promising therapeutic strategy for the treatment of Alzheimer’s disease. Neural Regen Res. 2024;19:619-28.
93. Niu X, Xia Y, Luo L, et al. iPSC-sEVs alleviate microglia senescence to protect against ischemic stroke in aged mice. Mater Today Bio. 2023;19:100600.
94. Kodali M, Madhu LN, Reger RL, et al. A single intranasal dose of human mesenchymal stem cell-derived extracellular vesicles after traumatic brain injury eases neurogenesis decline, synapse loss, and BDNF-ERK-CREB signaling. Front Mol Neurosci. 2023;16:1185883.
95. Lee Y, Lee J, Kim J, et al. hESC-derived extracellular vesicles enriched with MFGE-8 and the GSH redox system act as senotherapeutics for neural stem cells in ischemic stroke. Free Radic Biol Med. 2025;229:333-49.
96. Natale F, Leone L, Rinaudo M, et al. Neural stem cell-derived extracellular vesicles counteract insulin resistance-induced senescence of neurogenic niche. Stem Cells. 2022;40:318-31.
97. Rao S, Madhu LN, Babu RS, et al. Extracellular vesicles from hiPSC-derived NSCs protect human neurons against Aβ-42 oligomers induced neurodegeneration, mitochondrial dysfunction and tau phosphorylation. Stem Cell Res Ther. 2025;16:191.
98. Deng S, Xie H, Xie B. Cell-based regenerative and rejuvenation strategies for treating neurodegenerative diseases. Stem Cell Res Ther. 2025;16:167.
99. Hu G, Xia Y, Chen B, et al. ESC-sEVs rejuvenate aging hippocampal NSCs by transferring SMADs to regulate the MYT1-Egln3-Sirt1 axis. Mol Ther. 2021;29:103-20.
100. Sahay A, Scobie KN, Hill AS, et al. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature. 2011;472:466-70.
101. Babcock KR, Page JS, Fallon JR, Webb AE. Adult hippocampal neurogenesis in aging and Alzheimer’s disease. Stem Cell Reports. 2021;16:681-93.
102. Wang SN, Xu TY, Li WL, Miao CY. Targeting nicotinamide phosphoribosyltransferase as a potential therapeutic strategy to restore adult neurogenesis. CNS Neurosci Ther. 2016;22:431-9.
103. Leu T, Fandrey J, Schreiber T. (H)IF applicable: promotion of neurogenesis by induced HIF-2 signalling after ischaemia. Pflugers Arch. 2021;473:1287-99.
104. Mormone E, Iorio EL, Abate L, Rodolfo C. Sirtuins and redox signaling interplay in neurogenesis, neurodegenerative diseases, and neural cell reprogramming. Front Neurosci. 2023;17:1073689.
105. Oh H, Kwak JS, Yang S, et al. Reciprocal regulation by hypoxia-inducible factor-2α and the NAMPT-NAD+-SIRT axis in articular chondrocytes is involved in osteoarthritis. Osteoarthritis Cartilage. 2015;23:2288-96.
106. Guo J, Huang X, Dou L, et al. Aging and aging-related diseases: from molecular mechanisms to interventions and treatments. Signal Transduct Target Ther. 2022;7:391.
107. Liu Y, Tan Y, Zhang Z, Yi M, Zhu L, Peng W. The interaction between ageing and Alzheimer’s disease: insights from the hallmarks of ageing. Transl Neurodegener. 2024;13:7.
108. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018;4:575-90.
109. Pajares M, I Rojo A, Manda G, Boscá L, Cuadrado A. Inflammation in Parkinson’s disease: mechanisms and therapeutic implications. Cells. 2020;9:1687.
110. Aarsland D, Batzu L, Halliday GM, et al. Parkinson disease-associated cognitive impairment. Nat Rev Dis Primers. 2021;7:47.
111. Testo AA, Roundy G, Dumas JA. Cognitive decline in Alzheimer’s disease. Curr Top Behav Neurosci. 2025;69:181-95.
112. Yeapuri P, Machhi J, Foster EG, et al. Amyloid precursor protein and presenilin-1 knock-in immunodeficient mice exhibit intraneuronal Aβ pathology, microgliosis, and extensive neuronal loss. Alzheimers Dement. 2025;21:e70084.
113. Safiri S, Ghaffari Jolfayi A, Fazlollahi A, et al. Alzheimer’s disease: a comprehensive review of epidemiology, risk factors, symptoms diagnosis, management, caregiving, advanced treatments and associated challenges. Front Med. 2024;11:1474043.
114. Musi N, Valentine JM, Sickora KR, et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 2018;17:e12840.
116. Koh SH, Choi SH, Jeong JH, et al. Telomere shortening reflecting physical aging is associated with cognitive decline and dementia conversion in mild cognitive impairment due to Alzheimer’s disease. Aging. 2020;12:4407-23.
117. Zhang P, Kishimoto Y, Grammatikakis I, et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci. 2019;22:719-28.
118. Gonzales MM, Garbarino VR, Marques Zilli E, et al. Senolytic therapy to modulate the progression of Alzheimer’s disease (SToMP-AD): a pilot clinical trial. J Prev Alzheimers Dis. 2022;9:22-9.
119. Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018;562:578-82.
120. Ogrodnik M, Evans SA, Fielder E, et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell. 2021;20:e13296.
121. Xu F, Wu Y, Yang Q, et al. Engineered extracellular vesicles with SHP2 high expression promote mitophagy for Alzheimer’s disease treatment. Adv Mater. 2022;34:e2207107.
122. Gao G, Li C, Ma Y, et al. Neural stem cell-derived extracellular vesicles mitigate Alzheimer’s disease-like phenotypes in a preclinical mouse model. Signal Transduct Target Ther. 2023;8:228.
123. Apodaca LA, Baddour AAD, Garcia C Jr, et al. Human neural stem cell-derived extracellular vesicles mitigate hallmarks of Alzheimer’s disease. Alzheimers Res Ther. 2021;13:57.
124. Madhu LN, Kodali M, Upadhya R, et al. Extracellular vesicles from human-induced pluripotent stem cell-derived neural stem cells alleviate proinflammatory cascades within disease-associated microglia in Alzheimer’s disease. J Extracell Vesicles. 2024;13:e12519.
125. Morris HR, Spillantini MG, Sue CM, Williams-Gray CH. The pathogenesis of Parkinson’s disease. Lancet. 2024;403:293-304.
126. Krawczuk D, Groblewska M, Mroczko J, Winkel I, Mroczko B. The role of α-synuclein in etiology of neurodegenerative diseases. Int J Mol Sci. 2024;25:9197.
127. Pereira SL, Grossmann D, Delcambre S, Hermann A, Grünewald A. Novel insights into Parkin-mediated mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Curr Opin Neurobiol. 2023;80:102720.
128. Björkblom B, Adilbayeva A, Maple-Grødem J, et al. Parkinson disease protein DJ-1 binds metals and protects against metal-induced cytotoxicity. J Biol Chem. 2013;288:22809-20.
129. Liu Y, Song Y, Zhu X. MicroRNA-181a regulates apoptosis and autophagy process in Parkinson’s disease by inhibiting p38 Mitogen-Activated Protein Kinase (MAPK)/c-Jun N-Terminal Kinases (JNK) Signaling Pathways. Med Sci Monit. 2017;23:1597-606.
130. Ding H, Huang Z, Chen M, et al. Identification of a panel of five serum miRNAs as a biomarker for Parkinson’s disease. Parkinsonism Relat Disord. 2016;22:68-73.
131. Ma J, Shi X, Li M, et al. MicroRNA-181a-2-3p shuttled by mesenchymal stem cell-secreted extracellular vesicles inhibits oxidative stress in Parkinson’s disease by inhibiting EGR1 and NOX4. Cell Death Discov. 2022;8:33.
132. Chen HX, Liang FC, Gu P, et al. Exosomes derived from mesenchymal stem cells repair a Parkinson’s disease model by inducing autophagy. Cell Death Dis. 2020;11:288.
133. Ramalingam M, Jang S, Hwang J, Cho HH, Kim BC, Jeong HS. Neural-induced human adipose tissue-derived stem cell secretome exerts neuroprotection against rotenone-induced Parkinson’s disease in rats. Stem Cell Res Ther. 2025;16:193.
134. Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213-28.
135. Chen R, Powell JS, Shufesky WJ, et al. Transplants foster B cell alloimmunity by relaying extracellular vesicles to follicular dendritic cells. Cell Rep. 2025;44:115832.
136. Yáñez-Mó M, Siljander PR, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066.
137. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367:eaau6977.
138. Ding L, Guo Q, Ren Y, et al. Construction of [89Zr]Zr-labeled human umbilical cord mesenchymal stem cell-derived extracellular vesicles for noninvasive detection of tumors. ACS Appl Mater Interfaces. 2025;17:30589-99.
139. Chung YH, Ho YP, Farn SS, et al. In vivo SPECT imaging of Tc-99m radiolabeled exosomes from human umbilical-cord derived mesenchymal stem cells in small animals. Biomed J. 2024;47:100721.
140. Elsharkasy OM, Nordin JZ, Hagey DW, et al. Extracellular vesicles as drug delivery systems: why and how? Adv Drug Deliv Rev. 2020;159:332-43.
141. Walker S, Busatto S, Pham A, et al. Extracellular vesicle-based drug delivery systems for cancer treatment. Theranostics. 2019;9:8001-17.
142. Barilani M, Peli V, Manzini P, et al. Extracellular vesicles from human induced pluripotent stem cells exhibit a unique microRNA and circRNA signature. Int J Biol Sci. 2024;20:6255-78.
143. Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].